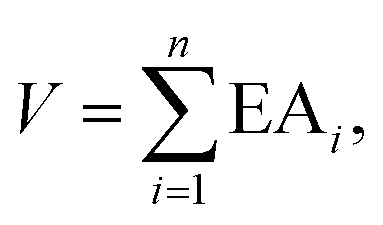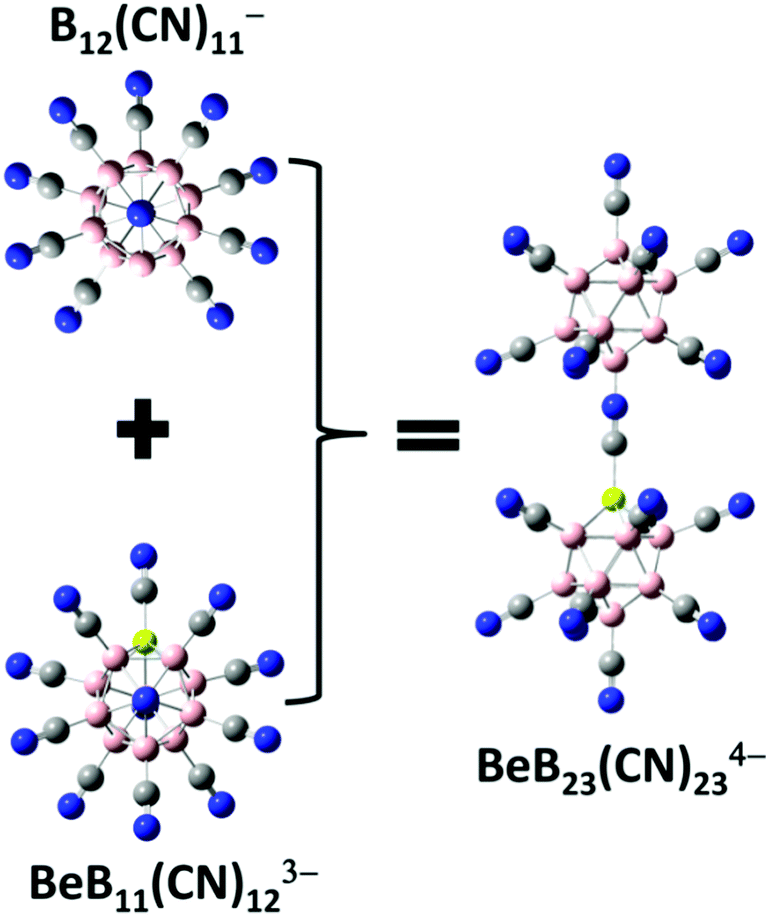Super-electrophiles of tri- and tetra-anions stabilized by selected terminal groups and their role in binding noble gas atoms†
Received
3rd May 2021
, Accepted 16th July 2021
First published on 19th July 2021
Abstract
Stabilization of multiply-charged atomic clusters in the gas phase has been a topic of great interest not only because of their potential applications as weakly-coordinating anions, but also for their ability to promote unusual reactions and serve as building blocks of materials. Recent experiments have shown that, after removing one terminal ligand from the closo-dodecacyano-borate, B12(CN)122−, the cluster can strongly bind an argon atom at room temperature. Bearing this in mind, here, we have developed more than a dozen highly stable tri- and tetra-anions using density functional theory (DFT) calculations with hybrid functional (B3LYP) and semi-empirical dispersion corrections. The interactions between the clusters and noble gas atoms, including Ne, Ar and Kr, are studied. The resulting super-electrophilic sites embedded in these charged clusters can bind noble gas atoms with binding energies up to 0.7 eV. This study enriches the database of highly-charged clusters and provides a viable design rule for super-electrophiles that can strongly bind noble gas atoms.
Stability of multiply-charged negative ions has been a topic of great interest for a long time.1–5 While these highly-negative anions can be stabilized in solutions or in matrices protected by solvation shells or counter ions, respectively, they are seldom stable in the gas phase, particularly if their sizes are small. One of the well-known stable gas-phase dianions is closo-dodecaborate, B12H122−, whose second electron is bound by 0.9 eV. Using density functional theory Zhao et al.6 recently showed that the stability of B12H122− can be greatly enhanced by using CN instead of H as terminal groups, with the resulting B12(CN)122− dianion exhibiting a second electron affinity of 5.3 eV. Warneke et al.7 had subsequently shown, using both theory and experiment, that the stability of B12H122− can also be enhanced by replacing H by halogens. Later, Moon et al.8 predicted that the second electron affinity of B12(CN)122− can be further enhanced by replacing CN with BO moieties. The extraordinary stability of B12(CN)122− has been recently experimentally confirmed by Mayer et al.9 and the measured second electron affinity of 5.5 eV is in good agreement with the calculated value. Zhao et al.10 then showed that stable gas-phase tri-anions can be formed by tailoring the composition of the boron core of the dianion. They predicted a stable gas-phase tri-anion, BeB11(CN)123−, whose third electron is bound by an extraordinarily large value of 2.65 eV. Fang and Jena have11 prescribed a systematic rule by which small clusters can carry even more extra electrons, forming stable gas-phase tetra- and penta-anions with −4 and −5 charge states, respectively. The above studies clearly established the important role of terminal groups in the stability of highly-charged anions.
Applications of the mono- and di-anions in the synthesis of halogen-free electrolytes for Li, Na, and Mg batteries have since been discussed, both theoretically and experimentally.12–14 Mayer et al.9 have demonstrated yet another important functionality of the di-anions, such as B12(CN)122−. Upon removal of one CN moiety from the cluster, they found that the exposed B atom in the B12 cage becomes electron deficient and positively charged. The exposed B can then interact with Ar atom through both charge transfer and covalent bonding with a binding energy of 0.61 eV. Recently, Meenakshi and Ghanty15,16 showed that the use of electronegative ligands as well as replacement of one B atom by Be in the cluster can further enhance the binding energy of noble gas atoms. Experiment has been carried out on binding Ne atoms using the same procedure.17 Here, by density functional calculations with hybrid functional (B3LYP) and semi-empirical dispersion corrections, we have developed a number of stable tri-anions using a set of linearly configured ligand clusters and found that the species can interact with noble gas atoms (Ng) strongly.
Design of stable tri- and tetra-anions
Inspired by the above works and realizing the importance of enlarging the pool of multiply-charged anions, we followed a rational design approach proposed earlier by Fang and Jena11 and later by Zhong et al.18 and examined their potential to bind noble gas atoms (e.g., Ne, Ar and Kr). It was demonstrated11 that the stability of a charged cluster can be measured by the sum (V) of the adiabatic electron affinities (EAi) of a cluster with n extra electrons, namely,| | 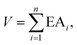 | (1) |
wherewith Ei being the energy of the cluster in the ith-charge state. Note that only a very limited number of clusters are known to be stable as tri-anions so far, including BeB11(CN)123−, BeB11(SCN)123−, BeB11(BO)123−, B12(CN)10(C2)23− and B12(CN)12(OBeS)23−.10,11,18 Among these, BeB11(CN)123− has the highest third electron affinity, EA3 = 2.65 eV8 and B12(CN)10(C2)23− is the smallest stable tri-anion with a radius of 4.85 Å.18 It is found that the stability of these charged clusters is determined by their closeness to the closed-shell model (all-paired electrons), the electron affinity of the terminal groups in the structures, and their sizes.11 The general prescription for designing highly charged clusters is to assemble them from known stable clusters with lesser charges. We chose mono-anions having large electron affinities as the terminal groups to the boron-cage skeleton whose stability is governed by the Wade–Mingos rule.19 To avoid steric hindrance between the neighboring groups,20 these terminal groups are all in linear configurations as shown in Fig. S1 and Table S1 of the ESI.† Each of these ligands has a magnetic moment of 1 μB in the ground state, since their closed shell corresponds to a ‘−2’ charge state. Among them, C2−, SBN−, OBeO−, OMgO−, NB2N−, O3Be2− and O3Mg2− all have larger EA1 than that of CN− (4.10 eV). Previously, CN− as the ligand has led to many di-, tri-, tetra- and even penta-anions11 with great stability (e.g., B12(CN)122−). Thus, it is expected that these mono-anions can also form stable gas-phase tri-anions. Super-electrophilic sites that strongly bind noble gas (Ng) atoms can then be created based on the newly developed tri- and tetra-anions.
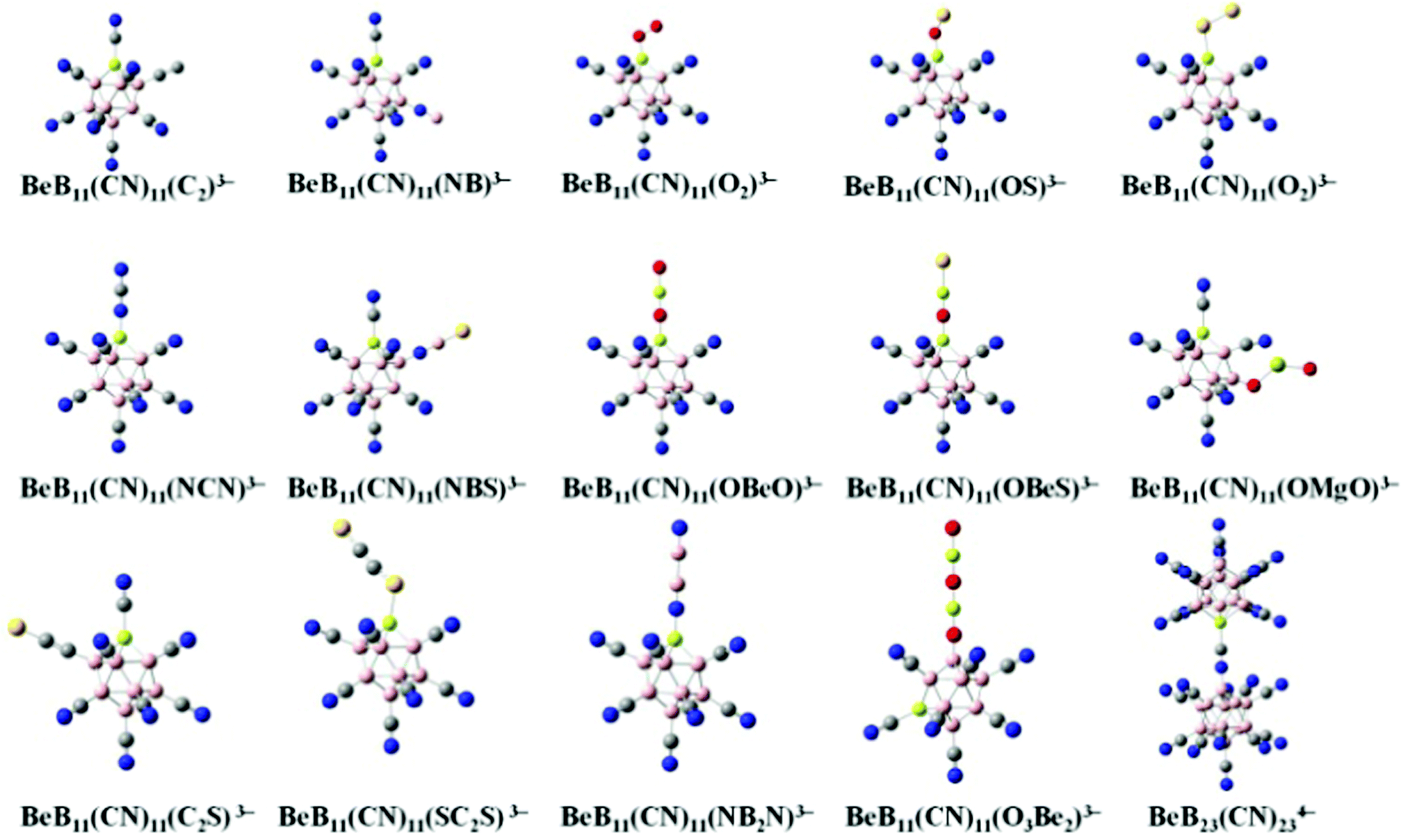
We start with BeB11(CN)123− and replace one of the CN moieties with one of the ligands, including C2, NB, O2, OS, S2, NCN, NBS, OBeO, OBeS, OMgO, C2S, SC2S, NB2N and O3B2, to form new stable tri-anions. The optimized geometries of these tri-anions are given in Fig. 1. Their basic properties such as the molar volume, ionic radius, multiplicity (M), binding energies of successive electrons, EAi, and the total energy gain by adding all the extra electrons, V (eV) are given in Table 1. For each of them, the ground state configuration is identified by comparing the energies of all possible structural isomers with non-equivalent CN ligand relative to the Be site substituted by the linear mono-anion, as shown in Table S2 of ESI.† No general rule is found to identify which site is preferred for the ground state. Previously, there were only 5 stable gas-phase tri-anions that have been reported. These include BeB11(CN)123−, BeB11(BO)123− and BeB11(SCN)123− whose EA3 are 2.65, 1.30 and 0.59 eV, respectively.10 In addition, two stable tri-anions without beryllium (Be) insertion into the boron cage were also found in the previous study, including B12(CN)10(C2)23− and B12(CN)10(OBeS)23− with EA3 of 0.92 and 1.43 eV, respectively.18 Compared to these, the new stable tri-anions in the current study all show 1 μB magnetic moment in their ground states and have similar sizes as well as similar values of EA3. Among them, BeB11(CN)11(OMgO)3− and BeB11(CN)11(O3Be2)3− exhibit the largest EA3 values of 2.40 and 2.16 eV, respectively, which are comparable to that of BeB11(CN)123−. It is noted that, whenever p-electrons are involved in the bonding, the terminal groups become tilted when attached to the boron site.12
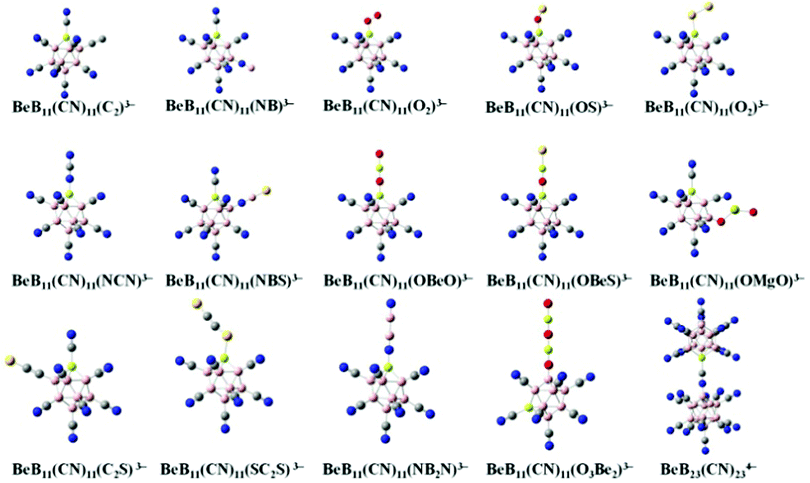 |
| | Fig. 1 Optimized structures of the new gas-phase tri-anions that are stabilized by replacing one of the CN ligands in BeB11(CN)123− with selected mono-anions shown in Fig. S1 and Table S1 of ESI.† The basic properties of these tri-anions are given in Table 1. Boron atoms are in pink, carbon in grey, nitrogen in blue, beryllium in light yellow, oxygen in red, sulfur in yellow and magnesium in yellow green. | |
Table 1 Calculated basic properties, including the molar volume (MV in cm3 mol−1), ionic radius (R in Å), multiplicity (M), EAi (eV) and V (eV) value, of the developed stable tri- and tetra-anions in their ground states. Their corresponding optimized structures are shown in Fig. 1. In each case, the ionic radius is estimated as half of the longest dimension of the tri-anion structure. The parameters of the previously known stable tri-anion BeB11(CN)123−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 are given for comparison. BeB11(CN)11(OBeS)− is found to have a distorted optimized structure compared to its “−3” charge state, resulting in an abnormally small EA2 value
10 are given for comparison. BeB11(CN)11(OBeS)− is found to have a distorted optimized structure compared to its “−3” charge state, resulting in an abnormally small EA2 value
| Tri-anion |
MV |
R
|
M
|
EA1 |
EA2 |
EA3 |
V
|
| BeB11(CN)123− |
317.125 |
6.11 |
1 |
8.44 |
4.76 |
2.65 |
15.85 |
| BeB11(CN)11(C2)3− |
313.16 |
6.11 |
2 |
4.75 |
4.36 |
0.55 |
9.66 |
| BeB11(CN)11(NB)3− |
364.04 |
6.11 |
2 |
8.67 |
3.28 |
1.86 |
13.17 |
| BeB11(CN)11(O2)3− |
251.17 |
5.89 |
2 |
7.78 |
4.55 |
1.03 |
13.36 |
| BeB11(CN)11(OS)3− |
307.94 |
6.24 |
2 |
6.98 |
3.95 |
0.13 |
11.06 |
| BeB11(CN)11(S2)3− |
398.62 |
6.90 |
2 |
7.70 |
3.99 |
0.50 |
12.19 |
| BeB11(CN)11(NCN)3− |
324.15 |
6.69 |
2 |
6.03 |
3.85 |
1.31 |
11.19 |
| BeB11(CN)11(NBS)3− |
369.33 |
6.94 |
2 |
5.61 |
3.98 |
0.58 |
10.17 |
| BeB11(CN)11(OBeO)3− |
331.34 |
6.83 |
2 |
9.12 |
3.23 |
1.87 |
14.22 |
| BeB11(CN)11(OBeS)3− |
382.18 |
7.29 |
2 |
11.01 |
0.84 |
1.62 |
13.47 |
| BeB11(CN)11(OMgO)3− |
325.84 |
6.71 |
2 |
9.09 |
2.83 |
2.40 |
14.32 |
| BeB11(CN)11(C2S)3− |
250.01 |
6.92 |
2 |
7.64 |
3.77 |
0.60 |
12.01 |
| BeB11(CN)11(SC2S)3− |
366.61 |
7.54 |
2 |
7.42 |
3.93 |
0.61 |
11.96 |
| BeB11(CN)11(NB2N)3− |
410.55 |
7.55 |
2 |
8.61 |
3.80 |
1.76 |
14.17 |
| BeB11(CN)11(O3Be2)3− |
322.39 |
8.04 |
2 |
9.30 |
3.17 |
2.16 |
14.63 |
| Tetra-anion |
MV |
R
|
M
|
EA1 |
EA2 |
EA3 |
EA4 |
V
|
| BeB23(CN)234− |
586.46 |
9.92 |
2 |
9.10 |
6.32 |
4.08 |
0.94 |
20.44 |
Besides the stable tri-anions, a stable gas-phase tetra-anion BeB23(CN)234− is also developed. As demonstrated by Fang and Jena, it is possible to stabilize a tetra-anion by assembling a stable mono-anion and a stable tri-anion.11 First, removal of one CN ligand from the stable di-anion B12(CN)122− results in a stable mono-anion, B12(CN)11−, with an EA1 value of 8.49 eV. Combining this to the known tri-anion BeB11(CN)123−, the tetra-anion BeB23(CN)234− is formed as shown in Fig. 2. The basic properties of this tetra-anion are given in Table 1. It has a large EA4 value of 0.94 eV, only second to the tetra-anion Be2B22(CN)234− which has the highest EA4 value of 1.48 eV11 known so far.
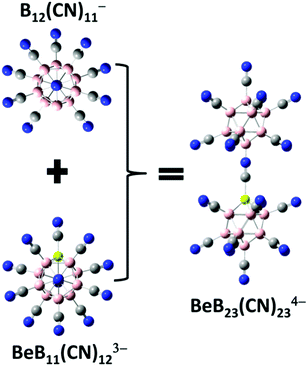 |
| | Fig. 2 Optimized structure of the new stable gas-phase tetra-anion BeB23(CN)234− that is developed by assembling a known mono-anion B12(CN)11− and a tri-anion BeB11(CN)123−. Boron atoms are in pink, beryllium in light yellow, carbon in grey, and nitrogen in blue. | |
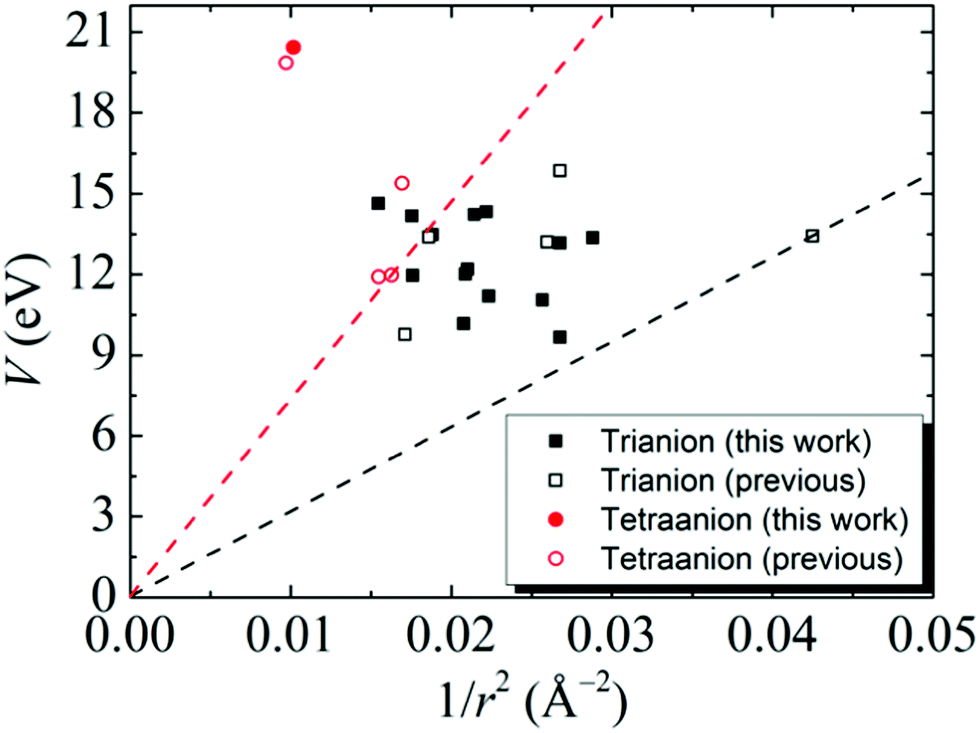
The results of tri- and tetra-anions in Table 1 can be well described by a phenomenological model in which each additional electron is in a bound state of a general 3D potential well.11 According to this model, the key quantities to stabilize a multiply-charged cluster are the V value and the cluster radius (r). A plot of V vs. 1/r2 for the studied clusters is given in Fig. 3, showing that the all the data points associated with the tri- and tetra-anions lie above a threshold line
where
n = 3 and 4 for tri- and tetra-anions, respectively. The threshold line (black dashed line in
Fig. 3) of the tri-anions is determined by B
12H
10(C
2)
23− which holds the record as the smallest stable tri-anion known so far.
18 The threshold line (red dashed line in
Fig. 3) of tetra-anions is determined by BeB
11(CN)
11Sr(CN)
44−.
11 This suggests that all the newly-developed multiply-charged clusters adhere to the general potential model.
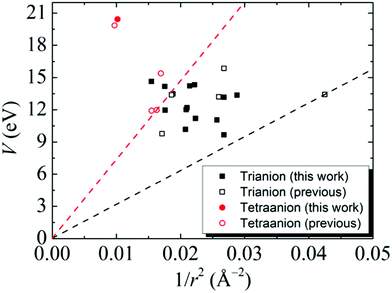 |
| | Fig. 3 Calculated V vs. 1/r2 data (eqn (1)) for the stable gas-phase tri- (black) and tetra- (red) anions developed in this study (solid square) and from previous studies in ref. 11 (open square). The threshold lines (eqn (3)) for the tri- (black dashed line) and tetra- (red dashed line) anions are given. | |
Binding of noble gas atoms to electrophilic sites in multiply charged anions
Earlier studies have shown that the mono-anion B12(CN)11− is a super-electrophilic cluster. Upon removal of one CN ligand from B12(CN)122−, the exposed B atom becomes electron deficient and binds strongly to Ar.9 Further research has shown that more super-electrophilic clusters can be developed by replacing one of the B atoms in the core by a Be atom as well as using other multiply charged clusters.15–17 These highly charged clusters are often formed by terminal ligands with large EA values. When a ligand is removed, the exposed boron or beryllium site in the cluster becomes highly electron deficient and can, therefore, bind Ng atoms strongly via chemical bonding.
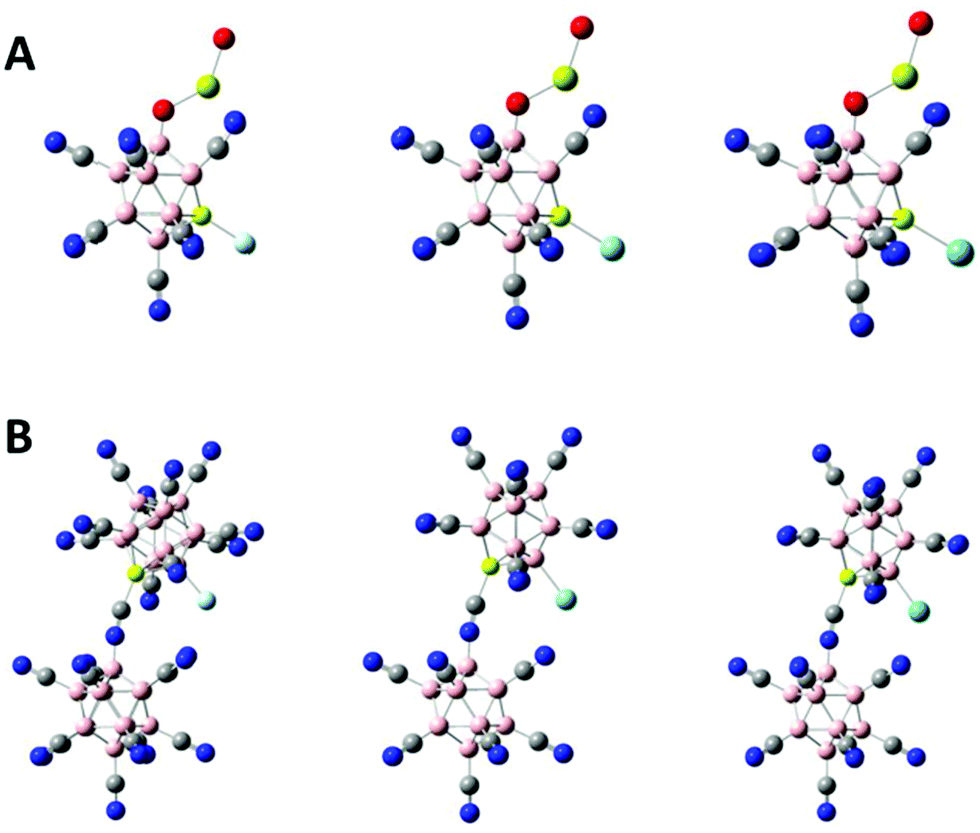
We have studied the potential of the newly developed tri- and tetra-anions to bind Ng atoms. For demonstration, we chose the tetra-anion BeB23(CN)234− and the tri-anion BeB11(CN)11(OMgO)3− which has the largest EA3 value of all the tri-anions in this study. After removing one CN ligand from BeB11(CN)11(OMgO)3−, the ground-state of BeB11(CN)10(OMgO)2− is the one in which the Be site is exposed (see Fig. S2A of ESI†). Fig. 4A shows the optimized structures of the ground states [BeB11(CN)10(OMgO)–Ng]2− (Ng = Ne, Ar, Kr). As given in Table 2, the exposed Be site is highly electron-deficient. There is significant charge transfer from the Ng atoms to the cluster when the two binds. The ground-state configuration of BeB23(CN)223− corresponds to the one in which the B site adjacent to Be is exposed (see Fig. S2B of ESI†). The optimized structures of [BeB23(CN)22–Ng]3− (Ng = Ne, Ar, Kr) are shown in Fig. 4B. As listed in Table 2, there is also significant charge transfer from the Ng atoms to the cluster when the two binds. The charge transfer of 0.34e out of Ar is comparable to that (0.41e) in the case of [B12(CN)11–Ar]−.9 The binding energies between the Be site and the Ng atoms are considered quite large, especially for the case of [BeB23(CN)22–Ng]3−, where the binding energies are higher than those in the case of [BeB11(CN)11–Ng]2−,17 as shown in Table 2.
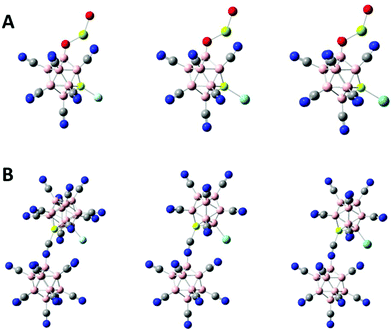 |
| | Fig. 4 Optimized ground-state structures of (A) [BeB11(CN)10(OMgO)–Ng]2− and (B) [BeB23(CN)22–Ng]3− with Ng = Ne, Ar and Kr from left to right. Boron atom are in pink, carbon in grey, nitrogen in blue, beryllium in light yellow, oxygen in red, magnesium in green yellow and Ng atoms in light blue. | |
Table 2 Calculated binding parameters between the di-anion BeB11(CN)10(OMgO)2− and the noble gas (Ng) atoms, including the bond length (BL) between Ng and the exposed Be, the charge state of Be, the charge transfer (e) out of the Ng atom and the binding energy (BE) between the Be site and the Ng atom. For comparison, in each case of [BeB23(CN)22–Ng]3− (Ng = Ne, Ar, Kr), the binding energy in the parenthesis corresponds to the case of [BeB11(CN)11–Ng]2−15
|
|
BL (Å) |
q
Be
|
q
Ng
|
BE (eV) |
| [BeB11(CN)10(OMgO)–Ng]2− |
| Ne |
1.942 |
1.051 |
0.106 |
0.14 |
| Ar |
2.177 |
0.853 |
0.264 |
0.22 |
| Kr |
2.258 |
0.755 |
0.346 |
0.27 |
|
|
| [BeB23(CN)22–Ng]3− |
| Ne |
2.003 |
0.433 |
0.114 |
0.40 (0.14) |
| Ar |
2.166 |
0.185 |
0.343 |
0.59 (0.47) |
| Kr |
2.193 |
0.024 |
0.489 |
0.71 (0.57) |
To understand the nature of the chemical bonding between Ng and the cluster, we conducted energy decomposition analysis on Ar–Be in [BeB11(CN)10(OMgO)–Ar]2−. It is found that, out of the total interaction energy of ΔE (−3.42 kcal mol−1), the repulsion (ΔErep = 29.52 kcal mol−1) and polarization (ΔEpol = −13.03 kcal mol−1) terms are dominant, indicating increasing orbital overlap as well as significant changes in the orbital shape when Ar interacts with Be in the cluster. The dispersion energy (ΔEdisp = −5.17 kcal mol−1) also contributes significantly to the total interaction energy. Thus, the Ar–Be interaction can be characterized as a mixture of ionic and covalent bonding as well as the interaction between polarized electron clouds, which is similar to the cases of [BeB11(CN)11Ng]2− dianions.15 Although the energy decomposition analysis of Ar–B in [BeB23(CN)22–Ar]3− is computationally prohibitive due to its large size, it is expected that chemical bonding will have a similar nature as found in the above cases.
In conclusion, using a set of stable mono-anions with linear configuration as the terminal groups in boron-based polyhedral clusters, we have developed more than a dozen of tri-anions (in the “−3” charge state) with compact sizes that are stable in the gas phase. In addition, a stable gas-phase tetra-anion (in the “−4” charge state) is also found by assembling one stable mono-anion together with another stable tri-anion. It is found that the relationship between the electron-holding ability of these tri- and tetra-anions (quantified by V in eqn (1)) and their sizes (quantified by their radii) are consistent with the phenomenological model of electronic bound states in a general 3D potential well.11 Further studies show that these stable clusters can serve as super-electrophiles, strongly interacting with noble gas atoms via chemical bonding.
Methods
The calculations are carried out using density functional theory (DFT) with the B3LYP hybrid exchange–correlation functional.21,22 We used the Gaussian 03 with 6-31+G(d,p) basis sets for all atoms.23 Although DFT with standard basis sets has been commonly used to calculate electron affinities for anions, it can suffer from the self-interaction error due to the use of approximate exchange functionals.24,25 Being a hybrid functional with empirical parameters, the B3LYP functional used here can cancel some self-interaction error and has shown relatively small mean absolute errors (<0.1 eV) in the calculated adiabatic electron affinities of anions.25 The exchange–correlation energy functional and basis sets used in this paper have been proven to be capable of providing reliable results in previous works.6,10,11 Moreover, some theoretical results have been validated by experimental study.9,13,14 Dispersive interactions are considered in all calculations using the D3 version of Grimme's dispersion with the original D3 damping function.26 In all cases, structures are fully optimized without any symmetry constraints. All considered structures have real vibrational frequencies and, therefore, correspond to the minima in the potential energy surface. The charge analysis is carried out using the natural bond orbital (NBO) method in each case. The energy decomposition analysis based on localized molecular orbitals is carried out using the method in ref. 27 with DFT and the same basis sets. The optimized geometries of the two clusters that can bind strongly with Ng are given in Tables S3 and S4 in the ESI.† Other data are available upon request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 11504301), Fundamental Research Funds for the Central Universities (XDJK2019 C108). P. J. acknowledges support of the U.S. DOE, Office of Basic Energy Sciences, Division of Material Sciences and Engineering under Award No. DE-FG02-96ER45579.
References
- A. Dreuw and L. S. Cederbaum, Chem. Rev., 2002, 102, 181–200 CrossRef CAS PubMed.
- A. A. Tuinman and R. N. Compton, J. Phys. Chem. A, 1998, 102, 9791–9796 CrossRef CAS.
- N. Zint, A. Dreuw and L. S. Cederbaum, J. Am. Chem. Soc., 2002, 124, 4910–4917 CrossRef CAS PubMed.
- W. B. Wang, A. P. Sergeeva, X. P. Xing, M. Massaouti, T. Karpuschkin, O. Hampe, A. I. Boldyrev, M. M. Kappes and L. S. Wang, J. Am. Chem. Soc., 2009, 131, 9836–9842 CrossRef PubMed.
- L. S. Wang and X. B. Wang, J. Phys. Chem. A, 2000, 104, 1978–1990 CrossRef CAS.
- H. Zhao, J. Zhou and P. Jena, Angew. Chem., 2016, 128, 3768–3772 CrossRef.
- J. Warneke, G. L. Hou, E. Apra, C. Jenne, Z. Yang, Z. Qin, K. Kowalski, X. B. Wang and S. S. Xantheas, J. Am. Chem. Soc., 2017, 139, 14749–14756 CrossRef CAS PubMed.
- J. Moon, H. Baek and J. Kim, Chem. Phys. Lett., 2018, 698, 72–76 CrossRef CAS.
- M. Mayera, V. Lessen, M. Rohdenburg, G. L. Hou, Z. Yang, R. M. Exner, E. Apràe, V. A. Azov, S. Grabowsky, S. S. Xantheas, K. R. Asmisa, X. B. Wang, C. Jenne and J. Warneke, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8167–8172 CrossRef PubMed.
- T. Zhao, J. Zhou, Q. Wang and P. Jena, Angew. Chem., 2017, 129, 13606–13610 CrossRef.
- H. Fang and P. Jena, Angew. Chem., Int. Ed., 2019, 58, 11248–11252 CrossRef CAS PubMed.
- H. Fang and P. Jena, J. Phys. Chem. C, 2017, 121, 7697–7702 CrossRef.
- T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., 2014, 126, 3237–3241 CrossRef.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900–7904 CrossRef CAS PubMed.
- K. Meenakshi and T. K. Ghanty, Chem. Commun., 2019, 55, 14379–14382 RSC.
- K. Meenakshi and T. K. Ghanty, Phys. Chem. Chem. Phys., 2020, 22, 13368–13372 RSC.
- M. Martin, R. Markus, L. Valentin van, C. N. Marc, G. Simon, R. A. Kunt, J. Carsten and W. Jonas, Chem. Commun., 2020, 56, 4591–4594 RSC.
- M. Zhong, H. Fang and P. Jena, Phys. Chem. Chem. Phys., 2020, 22, 4880–4883 RSC.
- D. M. P. A. Mingos, Nat. Phys. Sci., 1972, 236, 99–102 CrossRef CAS.
- A. I. Boldyrev and J. Simons, J. Chem. Phys., 1993, 98, 4745–4752 CrossRef CAS.
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785–788 CrossRef PubMed.
-
M. J. Frisch, et al., Gaussian 03, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- M.-C. Kim, E. Sim and K. Burke, Communication: Avoiding unbound anions in density functional calculations, J. Chem. Phys., 2011, 134, 171103 CrossRef PubMed.
- O. A. Vydrov and G. E. Scuseria, Ionization potentials and electron affinities in the Perdew–Zunger self-interaction corrected density-functional theory, J. Chem. Phys., 2005, 122, 184107 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01969d |
| ‡ Co-first authors. |
|
| This journal is © the Owner Societies 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Deepika
*b,
Deepika