
Ultrafast photochemistry of a molybdenum carbonyl–nitrosyl complex with a triazacyclononane coligand†
Niklas
Gessner
a,
Anna K.
Bäck
c,
Johannes
Knorr
 b,
Christoph
Nagel
d,
Philipp
Marquetand
c,
Ulrich
Schatzschneider
d,
Leticia
González
c and
Patrick
Nuernberger
*a
b,
Christoph
Nagel
d,
Philipp
Marquetand
c,
Ulrich
Schatzschneider
d,
Leticia
González
c and
Patrick
Nuernberger
*a
aa Physikalische Chemie II, Ruhr-Universität Bochum, Universitätsstraße 150, D-44801 Bochum, Germany; Institut für Physikalische und Theoretische Chemie, Universität Regensburg, Universitätsstraße 31, D-93053 Regensburg, Germany. E-mail: patrick.nuernberger@ur.de; Tel: +49-941-943-4487
bPhysikalische Chemie II, Ruhr-Universität Bochum, Universitätsstraße 150, D-44801 Bochum, Germany; Erlangen Graduate School in Advanced Optical Technologies (SAOT), Friedrich-Alexander University Erlangen-Nürnberg (FAU), Paul-Gordan-Straße 6, 91052 Erlangen, Germany
cInstitute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 17, A-1090 Wien, Austria
dInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, D-97074 Würzburg, Germany
First published on 16th September 2021
Abstract
Transition metal complexes capable of releasing small molecules such as carbon monoxide and nitric oxide upon photoactivation are versatile tools in various fields of chemistry and biology. In this work, we report on the ultrafast photochemistry of [Mo(CO)2(NO)(iPr3tacn)]PF6 (iPr3tacn = 1,4,7-triisopropyl-1,4,7-triazacyclononane), which was characterized under continuous illumination and with femtosecond UV-pump/UV-probe and UV-pump/MIR-probe spectroscopy, as well as with stationary calculations. The experimental and theoretical results demonstrate that while the photodissociation of one of the two CO ligands upon UV excitation can be inferred both on an ultrafast timescale as well as under exposure times of several minutes, no evidence of NO release is observed under the same conditions. The binding mode of the diatomic ligands is impacted by the electronic excitation, and transient intermediates are observed on a timescale of tens of picoseconds before CO is released from the coordination sphere. Furthermore, based on calculated potential energy scans, we suggest that photolysis of NO could be possible after a subsequent excitation of an electronically excited state with a second laser pulse, or by accessing low-lying excited states that otherwise cannot be directly excited by light.
Introduction
As diatomic molecules, carbon monoxide and nitric oxide are probably the smallest natural products and signaling factors in biological systems.1,2 Both are generated endogenously by enzymatic processes,3 with CO produced by heme oxygenase (HO) activity on heme4,5 while NO results from nitric oxide synthase (NOS) conversion of L-arginine.6 The two molecules have an important physiological function, in particular in response to oxidative stress, and their signaling pathways are heavily intertwined.7,8 While synthetic nitric oxide donors have been explored for their therapeutic potential long before the endogenous production of NO was even known – dating back about 150 years to the discovery of the beneficial activity of organic nitrates and nitrite esters in the treatment of angina pectoris –9,10 it has been only recently that CO-releasing molecules (CORMs) have been developed as prodrugs for carbon monoxide delivery for therapeutic applications in human medicine.11,12 Various mechanisms have been explored for spatial and temporal control of carbon monoxide release from carrier systems,13,14 mostly based on ligand exchange-mediated and photochemical processes.15–18 Recently, also an innovative class of enzyme-triggered CO-releasing molecules has been introduced.19,20 A light-controlled delivery of either CO or NO from the same molecule would be also beneficial, yet the design and mechanistic comprehension of a corresponding system is still a desideratum. This has motivated us to pursue a research program aimed at identifying mixed-ligand carbonyl/nitrosyl complexes from which either the CO or NO ligand or both can be released in a carefully controlled manner. Such an endeavor would return novel tools for inorganic chemical biology and augment the study of the biological activity of these two important signaling mediators. Based on our long-standing interest in transition metal-based photoactivated CO-releasing molecules (PhotoCORMs),21 a light-triggered liberation of CO and/or NO from the coordination sphere of a metal-coligand fragment was selected as the primary development target.With regard to the primary dynamics of photoinduced ligand release from metal complexes, the vast majority of femtosecond mid-infrared (MIR) studies has investigated metal-carbonyl compounds, with pioneering work focused at the ultrafast light-induced cleavage of metal–metal or metal–CO bonds and subsequent cooling and recombination dynamics.22–25 Further studies have also addressed possible chelation26 as well as catalytic activity following CO photorelease27 and have applied advanced time-resolved methods such as two-dimensional,27 pump–repump–probe,28 or X-ray femtosecond laser spectroscopy.29 In contrast, the photodynamics of metal-nitrosyl compounds in solution have only more recently moved into the focus of femtosecond studies, and so far are mostly directed at NO linkage isomerism.30–33 For example, sodium nitroprusside (Na2[Fe(CN)5(NO)]) showed nitrosyl linkage isomerization induced by ultraviolet laser pulses which occurs on a sub-picosecond timescale; also ultrafast NO release induced by these pulses was shown by Khalil et al.,31 whereas Orr-Ewing and colleagues demonstrated that at room temperature, cobalt-nitrosyl complexes of the general formula [CoX2(NO)(PCH3Ph2)2] with X = Cl, Br feature a dynamic interconversion between linear and bent NO.32 In the last few years, also the photodynamics of ruthenium-nitrosyl compounds were explored theoretically,34–39 elucidating the competition between photoinduced linkage isomerization and ligand release.
Time-resolved studies on mixed carbonyl–nitrosyl complexes have mostly been focused on [Co(CO)3(NO)]. Wang and coworkers studied the photochemistry of this compound in the gas phase using time-resolved IR spectroscopy.40 Their analysis of the branching ratio for NO vs. CO loss revealed that 355 nm excitation predominantly leads to the coordinatively unsaturated species [Co(CO)3] and [Co(CO)2(NO)] via loss of one nitrosyl or carbonyl ligand whereas 266 nm light eventually leads to the formation of [Co(CO)2] as well as NO-retaining products. Nakata et al. later substantiated the generation of [Co(CO)3] to be most prominent upon resonance-enhanced multiphoton ionization at around 230 nm.41 Harris and coworkers excited the same compound in n-hexane with 400 nm pulses and reported excited-state species with bent NO, both with and without additional CO release, while a photoproduct with only carbonyl ligands remaining was also identified.30 Mann and colleagues unveiled a strong influence of the environment on the photoproduct distribution by studying the thermal and photolytic liquid-phase reactions of mixed-ligand nitrosyl-carbonyl complexes of rhodium and iridium with triphenylphosphine.42
In a previous study of the photochemistry of the water-soluble manganese tricarbonyl complex [Mn(CO)3(tpm)]Cl with tpm = tris(pyrazolyl)methane using femtosecond spectroscopy combined with density functional theory (DFT) calculations, we found that several electronic transitions in the UV may lead to CO loss, but always just one CO ligand is released on an ultrafast timescale.28 In the present study, this approach was extended to the mixed CO/NO ligand complex [Mo(CO)2(NO)(iPr3tacn)]PF6 (1) with iPr3tacn = 1,4,7-triisopropyl-1,4,7-triazacyclononane, as depicted in Scheme 1. Its photolytic dynamics were studied in acetonitrile with UV-pump/UV-probe and UV-pump/IR-probe ultrafast spectroscopy. In particular, we were interested in how the photodynamics of this mixed carbonyl–nitrosyl compound differ from systems with only carbonyl ligands. McPhail et al. studied the reaction of [Mo(Cp)(CO)2(NO)] with triphenylphosphine in benzene and found that the simple substitution product [Mo(Cp)(CO)(NO)(PPh3)] as well as the isocyanato complex [Mo(Cp)(CO)(NCO)(PPh3)2] form in equal amounts.43 In the present joint experimental and theoretical study, we explore which of the small ligands in compound 1 is released, if any, and discuss the role of NO isomerism in this process.
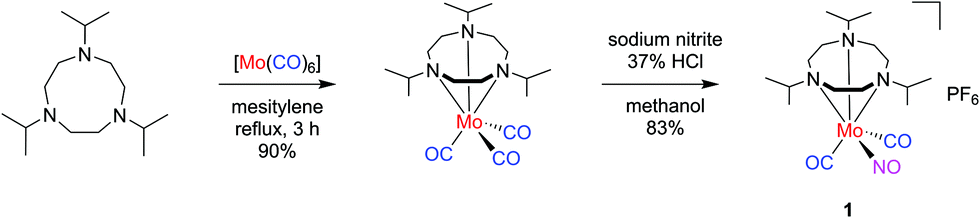 | ||
| Scheme 1 Two-step synthesis of mixed-ligand carbonyl/nitrosyl complex 1. | ||
Results and discussion
Synthesis and initial characterization
The mixed-ligand carbonyl/nitrosyl complex 1 was prepared in a two-step procedure from 1,4,7-triisopropyl-1,4,7-triazacyclononane and molybdenum hexacarbonyl, as described by Wieghardt et al.44 Along these lines, the Mo tricarbonyl complex [Mo(CO)3(iPr3tacn)] resulting from the first step45 was treated with sodium nitrite in methanol. Addition of hydrochloric acid followed by precipitation with potassium hexafluorophosphate led to isolation of 1 in good yield (Scheme 1). The analytical data is fully in line with the published values44 and in particular shows the characteristic antisymmetrical and symmetrical carbonyl C–O stretching vibrations of the cis-Mo(CO)2 moiety together with the N–O stretch (vide infra).Single crystals of the compound were grown by slow diffusion of n-hexane into a dichloromethane solution of the compound. The resulting molecular structure is shown in Fig. 1. The molybdenum center is in a pseudooctahedral coordination environment with a fac-Mo(CO)2(NO) moiety on one and the tacn ligand occupying the opposite triangular face. The Mo1–N1–O1 angle of 175.27(16)° is in line with a {Mo–NO}6 formulation and an electronic configuration of (e1)4(b2)2(e2)0, which translates to a low-spin Mo(0) 4d6 center coordinated to a NO+ ligand.46 In line with the considerations of Bau, Liu, and Shiu, an unequivocal assignment of the NO ligand position can be assumed.47–49 Such absence of CO/NO positional disorder was also observed in [Mo(tpz)(CO)2(NO)] with tpz = tris(pyrazolyl)borate50 while other Mo(CO)2(NO) complexes did not give X-ray crystal structures which allowed a clear assignment of the three diatomic ligands.51–56
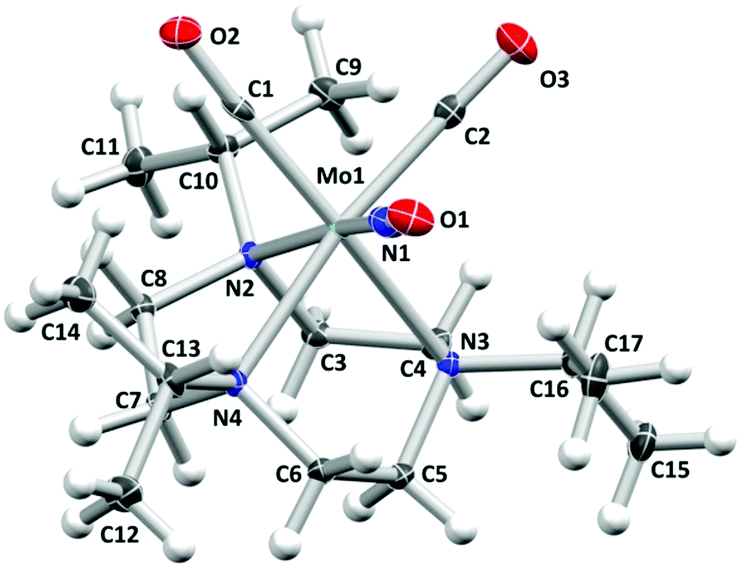 | ||
| Fig. 1 Molecular structure of the cation of 1. Atomic displacement ellipsoids are shown at the 50% probability level. The hexafluorophosphate counterion is omitted for clarity. Selected bond lengths (Å) and angles (°): Mo1–C1 1.954(2), Mo1–C2 1.963(2), Mo1–N1 1.8808(19), Mo1–N2 2.3023(15), Mo1–N3 2.3102(16), Mo1–N4 2.3010(16), C1–O2 1.160(2), C2-O3 1.161(2), N1–O1 1.188(2), Mo1–C1–O2 177.27(17), Mo1–C2–O3 174.88(17), Mo1–N1–O1 175.27(16), C1–Mo1–N3 169.33(7), C2–Mo1–N4 170.86(7), N1–Mo1–N2 168.32(7). | ||
Continuous illumination of 1 at 285 nm under the conditions of the myoglobin assay revealed spectral changes that are indicative of the liberation of CO, while non-illuminated samples did not show any spectral changes, thus substantiating photoinduced CO release.57 However, the number of CO molecules released per complex unit could not be determined reliably due to the instability of myoglobin under UV illumination. In the following, the prospects of ligand release are thus addressed both by theoretical and experimental spectroscopy approaches.
Characterization by experimental optical spectroscopy
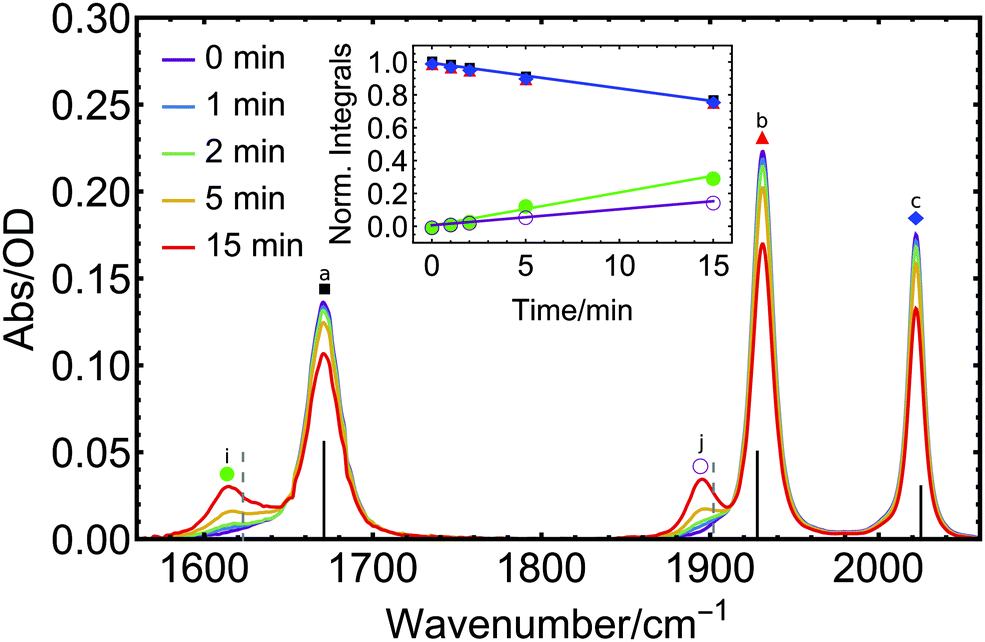 | ||
| Fig. 2 The FTIR spectra of 1 in acetonitrile upon illumination with a 285 nm diode for different exposure times (0–15 min) showing vibrational signals of electronic ground-state species. Vertical lines indicate calculated IR spectra of intact complex 1 (black) and the CO-loss product [Mo(CO)(NO)(CH3CN)(iPr3tacn)]+ (gray) (DFT B3LYP-D3(BJ)/def2-SVP; implicit solvent acetonitrile, redshifted by 101 cm−1 for ≤1905 cm−1 and by 70 cm−1 for >1905 cm−1). The inset shows the linearly modelled kinetics of all five main signals over the course of the experiment (note that the blue, black, and red traces are more or less fully overlapping). | ||
This decrease is accompanied by the rise of two new distinct signals at 1617 and 1895 cm−1 (Fig. 2i and j), one red-shifted with respect to the NO peak and the other one red-shifted relative to the CO vibration of the parent molecule, with clear isosbestic points observed at 1653 and 1913 cm−1. This indicates the formation of a single product in the primary photoprocess. The new species formed exhibits two bands, one in the CO and one in the NO spectral region, respectively. Multiple potential photoproducts were explored by DFT calculations to provide an explanation for both rising signals by considering three scenarios: (i) CO, NO and CO + NO dissociation with subsequent introduction of an acetonitrile solvent molecule to the metal coordination sphere in each case, (ii) NO linkage isomerization with and without dissociation, as well as (iii) species with coordinated water molecules which might originate from water traces (see Fig. S1–S4 and Table S1, ESI† for structures, spectra and raw peak data of all considered potential photoproducts). When comparing the spectral position of the initial photoproduct with our DFT calculations, excellent accordance is only found for the species where one CO ligand has dissociated and is subsequently replaced by a solvent molecule (Fig. 2 and Fig. S1–S4, ESI† for other potential photoproducts). From this analysis, we can clearly identify [Mo(CO)(NO)(CH3CN)(iPr3tacn)]+ as the most likely photoproduct among all considered species.
For a quantitative description, a sum of four Voigt profiles (the NO absorption has an asymmetric shape which requires inclusion of an additional Voigt profile at the lower-energy side) was fitted to the initial FTIR spectrum of 1 (see Fig. S5 and S6, ESI†). Subsequent spectra obtained after illumination are well reproduced from this initial spectrum modified by a joint scaling factor – to model the decrease in concentration of the intact complex – and two further Voigt profiles to model the product formation. The photoinduced changes in the peak heights can be expressed through a rate constant k. To determine k, integrals of the individual absorption bands of the starting material, as obtained from the Voigt fits, were normalized to their value at time t = 0 min when illumination was started (red, blue, and black curves in the inset of Fig. 2), to give k = −0.016 min−1 for this particular illumination geometry.
The use of an identical k value for the formation of the product signals is justified from the clear conversion of starting material to product, as indicated by the presence of isosbestic points. The integrals of the NO (green) and CO (purple) product peaks were divided by the integrals of NO and CO peaks of 1 at t = 0, yielding the rising curves in the inset of Fig. 2. The corresponding slopes are the product of k and the relative ratio of the absorption strengths
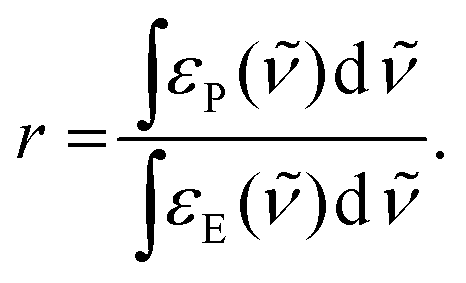 | (1) |
| Complex 1 | Product | Ratio | |||||
|---|---|---|---|---|---|---|---|
| NO | COanti | COsym | NO | CO | r NO | r CO | |
| a Absorption band integrals in OD cm−1. b Ratio of the CO signal of the product and the COanti signal of the educt. c Calculated intensity on DFT B3LYP-D3(BJ)/def2-SVP; implicit solvent acetonitrile. d For product [Mo(CO)(NO)(CH3CN)(iPr3tacn)]+. e For product [Mo(CO)(NO)(H2O)(iPr3tacn)]+. | |||||||
| Experiment | 3.84a | 4.36a | 2.60a | 4.95a | 2.75a | 1.29 | 0.63 |
| 1671 | 1931 | 2022 | 1617 | 1895 | |||
| Theory | 2181c | 1966c | 1192c | 1939cd | 1704cd | 0.89 d | 0.87d |
| 1671 | 1928 | 2025 | 1623 | 1902 | |||
| 553ce | 1884ce | 0.25e | 0.97e | ||||
| 1377 | 1699 | ||||||
The steady-state UV-absorption spectrum of 1 in CH3CN (Fig. 3, purple trace) exhibits a strong absorption band at 208 nm accompanied by a weak shoulder at 235 nm and another distinct band at 265 nm. The same 285 nm diode used in the IR experiments described above was also employed to study changes in the steady-state UV absorption spectrum of 1 upon long-term UV exposure. With increasing illumination time, the two initial absorption bands of 1 decrease in intensity while a new weak band grows in at 330 nm (Fig. 3). Such a red-shift of the UV absorption upon CO loss is known from other metal carbonyl complexes.58 Isosbestic points are discernible, e.g. at approximately 308 nm, indicating that only two species that vary in concentration contribute to the absorption spectrum. The absorption band that grows results from a stable product, as no relaxation back to the initial spectrum is found when repeating the measurement after several hours without further sample treatment.
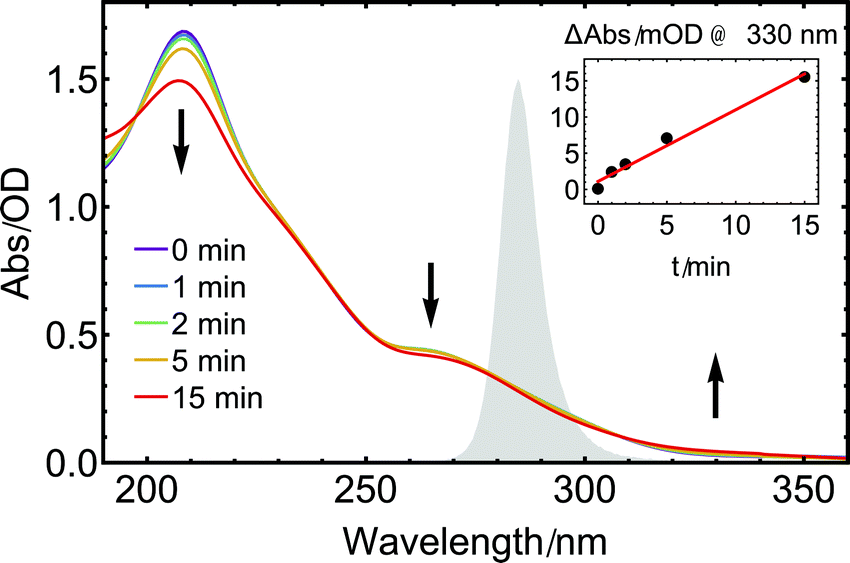 | ||
| Fig. 3 UV absorption spectra of 1 in acetonitrile under illumination with a 285 nm diode for different exposure times (0–15 min). The spectrum of the UV diode is shown in the background (grey shaded area). Arrows indicate rising and decaying absorption bands. In the inset, the change in intensity at 330 nm is plotted versus the illumination time. | ||
As illustrated by the inset of Fig. 3, the initial absorption band at longer wavelengths increases linearly with illumination time, in accordance with the measurements performed in the MIR range. For illumination times beyond 15 min, the initial clean interconversion is lost, indicating that additional species are formed by further reactions of the monocarbonyl species, presumably involving a further photoexcitation step or follow-up dark reactions.59,60
 | ||
| Fig. 4 Ultrafast dynamics of 1 in acetonitrile under 285 nm excitation probed in the MIR: (a) early dynamics covering the CO signal region. The decaying positive signals can be assigned to transient intermediates, while the constant positive signal below 1915 cm−1 is caused by a product of the photoreaction. (b) Decay Associated Difference Spectra (DADS) resulting from global target analysis comprising a parallel model of two exponential functions (blue and red) and a constant offset (black). | ||
In this DADS corresponding to 17 ps (red curve), two negative signals at the position of the GSB indicate a pronounced recovery of the electronic ground state of 1. Together with a distinct product absorption at 1995 cm−1, these findings may indicate the existence of the intermediate 5-coordinated species [Mo(CO)(NO)(iPr3tacn)]+ that either reverts back to the electronic ground state of intact complex 1 by geminate recombination or forms [Mo(CO)(NO)(CH3CN)(iPr3tacn)]+ by coordination of a solvent molecule. Furthermore, the distinctive peak at 1995 cm−1 rises with a lifetime of 1.9 ps as the respective DADS (blue line) indicates a lack of positive contribution in this specific region. The broad positive signal of this DADS might originate from relaxation of vibrationally hot molecules and possibly relaxation to the lowest-lying excited state (vide infra).
A different explanation for the 17 ps dynamics is conceivable, in which the signal at 1995 cm−1 is connected to the antisymmetric CO stretch of an intact yet electronically excited molecule 1. The observation of a further peak around 1885 cm−1, which then would correspond to the symmetric CO stretch of excited 1, would support this assignment. Furthermore, these two peaks decay on a 17 ps timescale (we however note that some signal remains in the black DADS at the low wavenumber edge of the measurement of Fig. 4), with a substantial fraction of the GSB of 1 recovering simultaneously. Since the product peak at 1895 cm−1 (black DADS) rises with the same 17 ps dynamics, one may infer that electronically excited 1 cools on a 1.9 ps timescale and then the excited state is depopulated via two competing reaction channels. These are relaxation back to the electronic ground state (reflected in the GSB recovery) and the loss of a CO ligand (seen in the rise of the product signal), yielding an excited-state lifetime of 17 ps. In the excited state, the two CO ligands exhibit a red-shifted vibration and thus a smaller force constant, which in turn points towards a stronger bond to the metal center. This stronger bonding of the CO ligands might be a consequence of weaker bonding of the nitrosyl ligand, for example due to linkage isomerization41 as already reported for [Co(CO)3(NO)].30 Hence, our data suggest that for the goal of NO release by repumping with a further laser pulse, the re-excitation would have to occur within less than 17 ps.
Transient excited-state signals and GSB recovery were also monitored by probing electronic transitions. Particularly, in UV pump/UV probe studies (Fig. 5a), a broad positive absorption signal spanning from 300 to 380 nm was found after photoexcitation and is assigned to excited-state absorption (ESA). It decays within the first 20 ps while a small positive contribution around 325 nm remains for at least 4 ns constituting the longest pump–probe delay accessible with our transient absorption spectrometer. The spectral properties of the remaining species are in line with the results shown in Fig. 3 and further substantiate the finding of a stable photoproduct formed after photolysis of one CO ligand from the metal center. The observation that only minor changes in the absorption spectrum arise upon UV illumination (Fig. 3) indicate that only a small percentage of excited molecules of 1 releases CO. This interpretation is supported by the GSB recovery displayed in Fig. 4 and is complemented by the transient UV data (Fig. 5) which features a strong decay of the ESA within the first few picoseconds but only a rather small remaining product absorption. Direct observation of GSB recovery in the UV (for expected spectral range, compare Fig. 3) is obscured by the strong ESA signal and the detection range of our transient absorption spectrometer towards shorter wavelengths.
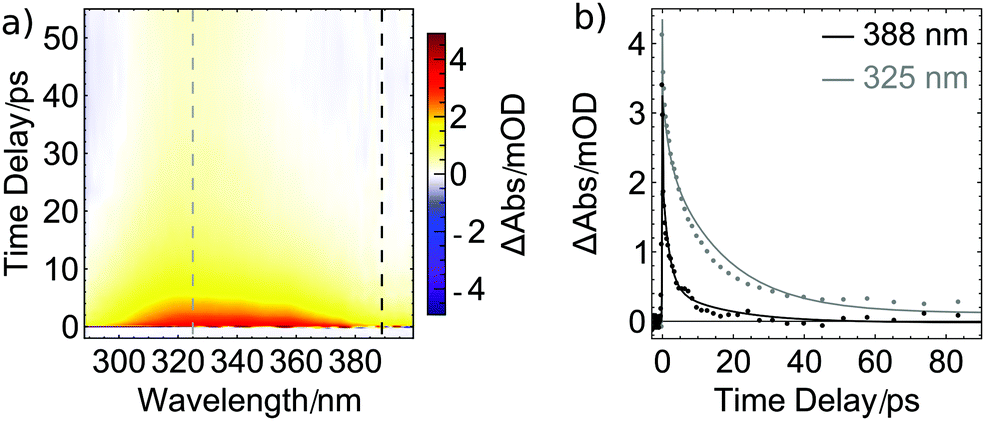 | ||
| Fig. 5 Ultrafast dynamics of 1 in acetonitrile under 285 nm excitation: (a) broadband transient absorption in the ultraviolet regime showing early dynamics. A broad excited-state absorption (ESA, red) is followed by a more narrow product absorption (PA, yellow) centered at 325 nm. (b) Transients at 325 nm (superimposed ESA and PA dynamics) and 388 nm (ESA dynamics) for time delays up to 90 ps (dashed lines in (a)). Solid lines represent the fitted model also used for the MIR results with fixed lifetimes. | ||
In Fig. 5b, the decay of the ESA signal is plotted for two probe wavelengths. The solid lines are fits with the time constants obtained from the ultrafast MIR data shown in Fig. 4b, supporting the assumption that the same dynamics are monitored. Consequently, the ESA dynamics observed in the UV can also be approximated by the two lifetimes of 1.9 and 17 ps. For the assignment of the former, vibrational cooling or transitions to other excited states might be inferred from the MIR data. Since also in the UV the signal is very pronounced, we tentatively assign it to a transition from the initially excited state to a lower-lying excited state. For [Co(CO)3(NO)], it was found that a triplet state with a bent NO ligand is reached within less than one pico-second.30 In the present compound, a corresponding state might be accessible which is stable for 17 ps before it further relaxes or eventually fully releases a CO ligand, causing the constant signal at 325 nm.
Computational results
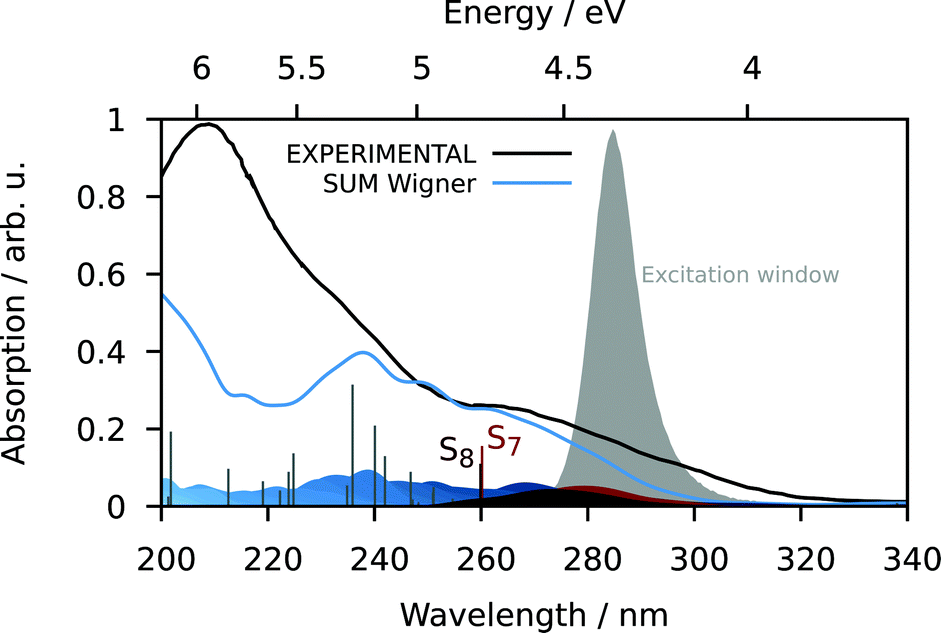 | ||
| Fig. 6 UV-vis absorption spectrum with the experimental spectrum in continuous black, the simulated spectrum from the excitation of the ground state geometry as stick diagram and from the excitation of the 100 Wigner-sampled geometries in blue color (convolution with FWHM = 0.15 eV; contributions of the different states indicated as blue-colored Gaussians at the bottom). The bands for states S7 and S8 stretch until about 300 nm and thus, these states absorb predominantly in the experimental excitation window. | ||
S7 and S8 exhibit an oscillator strength at the ground-state equilibrium geometry of 0.014 and 0.016 a.u., respectively. Both are primarily characterized by excitations from a π(MoNO) towards a π*(MoCO) orbital with additional contributions from a π(MoNO) towards a π*(MoNOCO) for S7 and a π*(MoCONO) for S8 (Fig. 7). A more detailed analysis of the electronically excited states is given in Fig. 8 in terms of charge transfer numbers.64 In this fragment-based charge transfer analysis, correlated electron–hole pairs of the charge transfer are localized on predefined fragments. For this purpose, 1 was dissected into four fragments, the molybdenum metal center (M) and three ligands (LCO, LNO, and Ltacn). Charge transfer interactions between these fragments were identified and give rise to 16 distinct excited states classes such as metal-to-ligand charge transfer states (MLCT), metal-centered (MC) or ligand-centered (LC) states (Fig. 8).
 | ||
| Fig. 7 Natural transition orbitals (NTOs) of the two dominant singlet states S7 at 4.76 eV (260.5 nm) and S8 at 4.77 eV (259.9 nm) with the corresponding weights of the NTO pair given inside the arrow (TD-DFT B3LYP-D3(BJ)/def2-SVP; implicit acetonitrile); labelling includes state, excitation energy and PRNTO in parenthesis (the participation ratio indicates how many NTO pairs are representative for a state). | ||
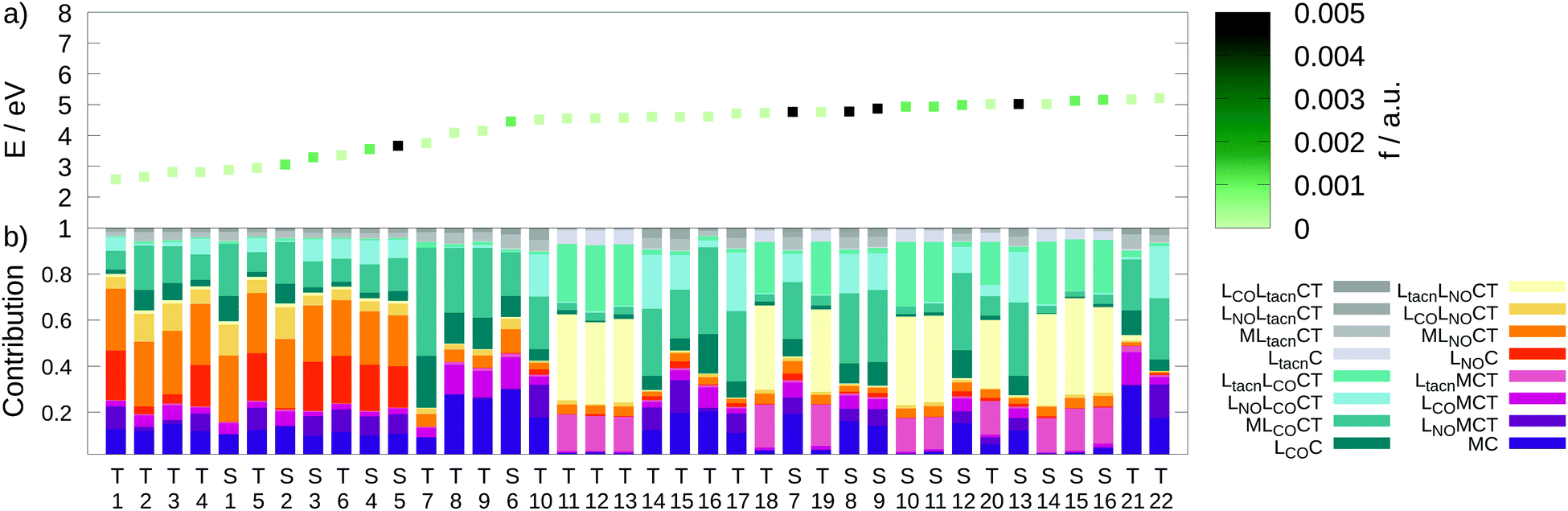 | ||
| Fig. 8 (a) Excitation energies E (eV) of the lowest 16 singlet and 22 triplet states colored by the oscillator strengths f (a.u.). (b) Charge transfer analysis with color-coded decomposition (TD-DFT B3LYP-D3(BJ)/def2-SVP; implicit acetonitrile). | ||
Excited states of transition metal complexes are usually characterized by different contributions of different character, as can be clearly seen in Fig. 8. Even though there seems to be a large variety of states contributing to the absorption spectrum, several groups of states can be identified. The low-energy region (excitation energy <4.0 eV; wavelength >310 nm) is dominated by CT excitations towards the NO-ligand, originating either from the metal (MLNOCT), or the CO (LCOLNOCT), or ligand-centered on the NO itself (LNOC). This result is of particular significance because a directed CT at lower excitation energies indicates a possibility for NO dissociation if suitable antibonding orbitals along the Mo–N-bond become occupied so that it is weakened. At higher excitation energies, different states with increased CT towards the CO-ligand (MLCOCT) and metal-centered excitations (MC) as well as a progressively higher involvement of the tacn ligand in LtacnLNOCT, LtacnLCOCT and LtacnMCT at energetically higher states are found.
In order to obtain further insight into possible NO and CO dissociation mechanisms, scans of the potential energies along a Mo–C and Mo–N bond elongation were performed using the multiconfigurational method SA(8)-CASSCF(8,10) (state-averaged over 8 singlet states complete active space self-consistent field with 8 electrons in 10 orbitals) in gas phase. The orbitals comprising the active space are shown in Fig. 9 and are primarily localized on Mo, NO and CO. The orbital choice was guided by the requirement to describe both dissociation reactions on the same footing. The selected active space fulfils this criterion as can be seen by the smooth potentials shown in Fig. 10. Among the states computed with CASSCF, the singlet states S7 and S6 exhibit the highest oscillator strengths for the vertical excitation at the ground state minimum geometry (Table 2). Based on the obtained potential energy curves (Fig. 10), an excitation into either of these two bright states is expected to lead to CO dissociation, in agreement with the experimental observations. A possible pathway is indicated by violet arrows in Fig. 10. Note that excitation energies are overestimated in CASSCF and the excitation arrow has been accordingly adjusted. Interestingly, an excitation at lower energies, to the S4 or S5 states might result in NO cleavage. As can be seen from Table 2, both states are of predominant ππ* character with the π* orbital located along the Mo–NO bond, which corroborates the conclusions from the CT analysis (see above). In this respect, S4 seems most favourable due to its character with partial charge transfer from CO to NO (Table 2). However, a direct excitation to this and other low-lying states is impeded by their small oscillator strengths.
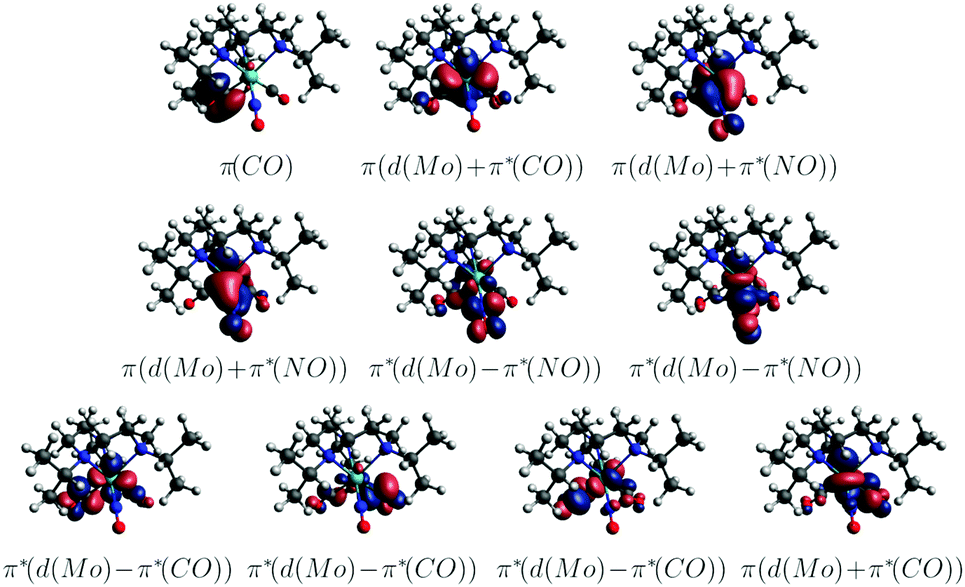 | ||
| Fig. 9 Active space of the CASSCF(8,10) computations (CASSCF(8,10)/ANO-RCC-MB; gas phase; natural orbitals). | ||
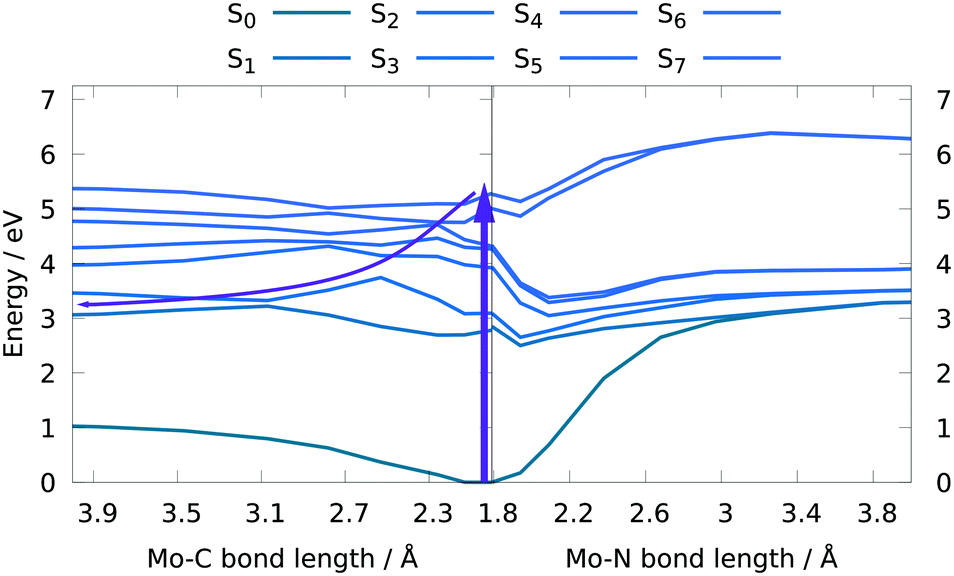 | ||
| Fig. 10 Potential energy curves along a Mo–CO (left) and a Mo-NO (right) bond elongation yielding a bond dissociation energy of 1.12 eV for CO and 3.38 eV for NO dissociation (CASSCF(8,10)/ANO-RCC-MB; gas phase). Arrows indicate a possible photo-induced dissociation pathway. | ||
| State | ΔE (eV) | f (a.u.) | Character | Weight (%) |
|---|---|---|---|---|
| S1 | 2.78 | <0.001 | πMoCOπ*MoNO | 61 |
| πMoCOπ*MoNO | 15 | |||
| S2 | 3.09 | <0.001 | πMoCOπ*MoNO | 62 |
| πMoCOπ*MoNO | 14 | |||
| S3 | 3.92 | <0.001 | πMoNOπ*MoNO | 32 |
| πMoCONOπ*MoNO | 26 | |||
| πMoCONOπ*MoNO | 25 | |||
| S4 | 4.26 | 0.002 | πMoCONOπ*MoNO | 50 |
| πMoNOπ*MoNO | 13 | |||
| πMoNOπ*MoNO | 10 | |||
| πMoCOπ*MoCO | 7 | |||
| S5 | 4.31 | 0.002 | πMoNOπ*MoNO | 34 |
| πMoNOπ*MoNO | 31 | |||
| πMoCONOπ*MoNO | 16 | |||
| S6 | 5.03 | 0.005 | πMoCOπMoCO | 66 |
| S7 | 5.28 | 0.149 | πMoCOπ*MoCO | 37 |
| πMoCONOπMoCO | 17 | |||
| πMoNOπ*MoNO | 7 |
Conclusion
Employing a combination of theory, stationary, and femtosecond time-resolved spectroscopy, the primary photodynamics of the mixed CO/NO complex [Mo(CO)2(NO)(iPr3tacn)]PF6 with iPr3tacn = 1,4,7-triisopropyl-1,4,7-triazacyclononane was explored. Findings deduced from all spectroscopic approaches employed in the present study support an exclusive ultrafast CO release from 1 upon UV excitation. The transient intermediate survives for 17 ps (see orange box in Fig. 11 for putative species), but the loss of CO is rather inefficient. Thus, although the complex carries both carbonyl and nitrosyl ligands in the coordination sphere, it solely acts as a photoactivatable CO-releasing molecule. The calculations confirm this picture but also unveil that an excitation to low-lying excited states might enable NO release. However, a direct excitation to these states is difficult due to their small oscillator strengths. Still, we conjecture that cleavage of the metal–NO bond might be achievable by an additional excitation within the first picoseconds. This route will be pursued in further experiments towards dual-functional CO/NO photoreleasing molecules.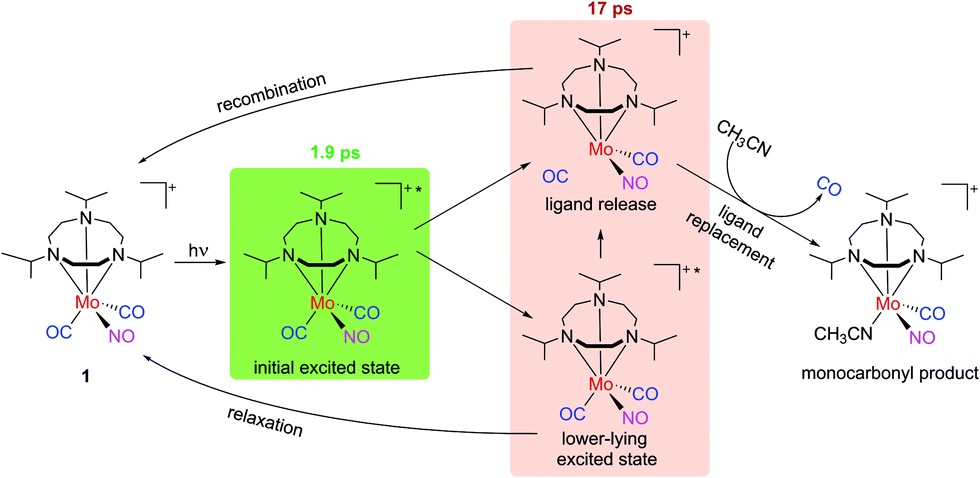 | ||
| Fig. 11 Sketch of the photodynamics: molecule 1 (PF6− not shown for simplicity) is excited and then, with a 1.9 ps decay, turns into a transient intermediate. As described in the text, this might be due to a species in a lower-lying excited state or with a photolyzed CO but a free coordination site. With a time constant of 17 ps, the transient intermediate signal decays, both leading back to 1 and to a monocarbonyl product with a solvent molecule coordinated to it. | ||
Experimental section
General synthetic procedures
All reactions were carried out in Schlenk flasks under an atmosphere of dry dinitrogen using anhydrous solvents purified according to standard procedures. The apparatus was wrapped in aluminium foil to prevent exposure to light and samples were kept in the dark whenever possible. All chemicals were purchased from commercial sources and used as received. The iPr3tacn ligand was prepared by reaction of 1,4,7-triazacyclonane with isopropyl bromide in toluene following a published procedure.45 NMR spectra were recorded on Bruker Avance 200 and Avance 500 spectrometers (1H at 199.93 and 500.13 MHz; 13C at 50.27 and 125.77 MHz). The 1H and 13C spectra were calibrated against the residual signal of the solvent serving as an internal reference.65 Infrared spectra were recorded on pure solid samples using a Nicolet 380 FT-IR spectrometer equipped with a SMART iTR ATR unit. Intensities of the stretching vibrations are marked as strong (s), medium (m), or weak (w). The elemental composition of the compounds was determined with a Vario MICRO cube analyzer.![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2966 (w), 1888 (vs), 1727 (vs), 1443 (w), 1388 (w), 1123 (w), 1065 (w) cm−1; 1H NMR (199.93 MHz, DMSO-d6): δ = 3.28 (m, 3H, CH, partially obscured by solvent water signal), 2.70 (s, 12H, CH2), 1.29 (d, 18H, 3J = 6.4 Hz, CH3) ppm; 13C NMR (50.27 MHz, DMSO-d6): δ = 229.0 (CO), 61.7 (CH), 54.4 (CH2), 19.2 (CH3) ppm. Due to the presence of some impurities, no correct CHN analysis could be obtained. However, since these did not interfere with the next synthetic step, no attempt was made for further purification.
= 2978 (w), 2018 (w), 1939 (vs), 1912 (vs), 1672 (m), 1447 (w), 1116 (w), 1065 (w), 829 (vs) cm−1; 1H NMR (500.13 MHz, CD2Cl2): δ = 3.53 (septet, 2H, 3J = 6.5 Hz, CH), 3.21 (septet, 1H, 3J = 6.5 Hz, CH), 3.07–3.13 (m, 6H, CH2), 3.00–3.06 (m, 4H, CH2), 2.88–2.94 (m, 2H, CH2), 1.42 (d, 6H, 3J = 6.5 Hz, CH3), 1.40 (d, 6H, 3J = 6.5 Hz, CH3), 1.30 (d, 6H, 3J = 6.5 Hz, CH3) ppm; 13C NMR (125.27 MHz, CD2Cl2): δ = 223.3 (CO), 62.93 (CH and CH2), 19.72 (CH3), 19.63 (CH3) ppm; anal. calcd for C17H33F6MoN4O3P·H2O (%): C 34.01, H 5.88, N 9.33. Found (%): C 34.34, H 5.63, N 9.26.
= 2966 (w), 1888 (vs), 1727 (vs), 1443 (w), 1388 (w), 1123 (w), 1065 (w) cm−1; 1H NMR (199.93 MHz, DMSO-d6): δ = 3.28 (m, 3H, CH, partially obscured by solvent water signal), 2.70 (s, 12H, CH2), 1.29 (d, 18H, 3J = 6.4 Hz, CH3) ppm; 13C NMR (50.27 MHz, DMSO-d6): δ = 229.0 (CO), 61.7 (CH), 54.4 (CH2), 19.2 (CH3) ppm. Due to the presence of some impurities, no correct CHN analysis could be obtained. However, since these did not interfere with the next synthetic step, no attempt was made for further purification.
= 2978 (w), 2018 (w), 1939 (vs), 1912 (vs), 1672 (m), 1447 (w), 1116 (w), 1065 (w), 829 (vs) cm−1; 1H NMR (500.13 MHz, CD2Cl2): δ = 3.53 (septet, 2H, 3J = 6.5 Hz, CH), 3.21 (septet, 1H, 3J = 6.5 Hz, CH), 3.07–3.13 (m, 6H, CH2), 3.00–3.06 (m, 4H, CH2), 2.88–2.94 (m, 2H, CH2), 1.42 (d, 6H, 3J = 6.5 Hz, CH3), 1.40 (d, 6H, 3J = 6.5 Hz, CH3), 1.30 (d, 6H, 3J = 6.5 Hz, CH3) ppm; 13C NMR (125.27 MHz, CD2Cl2): δ = 223.3 (CO), 62.93 (CH and CH2), 19.72 (CH3), 19.63 (CH3) ppm; anal. calcd for C17H33F6MoN4O3P·H2O (%): C 34.01, H 5.88, N 9.33. Found (%): C 34.34, H 5.63, N 9.26.
Ultrafast time-resolved pump–probe spectroscopy
With a Ti:Sa amplifier system (Solstice; Spectra Physics: 1 kHz, 800 nm, 100 fs), a non-collinear optical parametric amplifier (TOPAS-White; Light Conversion) is pumped, and 285 nm UV pump pulses (mechanically chopped at 500 Hz) are spatially and temporally overlapped with probe pulses in a flow cell providing a liquid film of 200 μm thickness between Suprasil windows. Sample flow rates were kept sufficiently high to ensure a complete sample exchange between subsequent pump–probe pairs. Supercontinuum probe pulses were generated in a continuously moving CaF2 plate.66 A motorized stage enables scanning a pump–probe delay range of 4 ns. At each time delay, the average of 1000 consecutive transient absorption spectra was recorded by a CCD detector (Pixis 2k; Princeton Instruments) after passing a polychromator (Acton SP2500i; Princeton Instruments), whereas long-term fluctuations were minimized by averaging over several data sets. The UV pump pulses were compressed to 40 fs at the sample position (determined by a X-FROG measurement) using an acousto-optic programmable dispersive filter (Dazzler; Fastlite). The polarizations of both beams were held at the magic angle configuration.66,67The UV pump/MIR probe experiments were performed at a different setup. MIR probe pulses stem from a collinear optical parametric amplifier (TOPAS Prime; Light Conversion) pumped by another amplifier system (Spitfire Ace; Spectra Physics: 1 kHz, 800 nm, 120 fs) and are detected with an identical CCD camera after upconversion in a MgO(5%):LiNbO3 crystal with a strongly chirped 800 nm beam.68,69 Details on the design and the specifications of this chirped-pulse upconversion setup are described elsewhere.70 The employed cuvette comprised two CaF2 windows and a sample thickness of 200 μm.
UV/vis-absorption spectra were recorded with a Jasco V-670 spectrophotometer, whereas a Jasco FT/IR-4100 spectrometer was used to measure linear absorption spectra in the MIR. We equipped both spectrometers with the same 285 nm UV diode to detect changes in the respective absorption signatures upon continuous UV-exposure. Compound 1 was dissolved in acetonitrile (spectroscopic grade) at concentrations of approximately 5 mM both for the static UV- and FTIR-absorption measurements as well as for transient absorption experiments.
X-ray crystallography
A suitable crystal of [Mo(CO)2(NO)(iPr3tacn)]PF6 was selected and soaked in perfluoro polyether oil, mounted on a MiTeGen sample holder and transferred to a stream of cold nitrogen on the diffractometer. Diffraction data were collected on a Bruker FR591 Apex II with a rotating anode using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The crystal was kept at 100 K during data collection. Using Olex2,71 the structure was solved with the olex2.solve structure solution program,72 using the Charge Flipping solution method. The model was refined with the olex2.refine refinement package using Gauss–Newton minimisation.73 For the structure, all non-hydrogen atoms were refined anisotropically, all hydrogen atoms assigned to idealised geometric positions, and the latter also included in the structure factors calculation. Crystallographic data (excluding structure factors) has been deposited with the CCDC 1414615.†Computational details
The geometry of 1 was optimized using the hybrid functional B3LYP,74,75 with a def2-SVP76,77 basis set together with the Grimme78 D3 dispersion correction and Becke–Johnson damping scheme (BJ) to account for long-range interactions, as implemented in the Gaussian09 suite of programs.79 We chose B3LYP because it exhibits relatively low mean absolute errors especially in the subsequently performed TDDFT calculations and performs well for similar transition metal systems.80–82 An effective core potential for the Mo central atom was employed assuming 28 electrons frozen and 14 valence electrons.78 Furthermore, second-order Douglas–Kroll–Hess integrals were used to include scalar relativistic effects.83 Solvent effects were implicitly accounted for by the use of the polarized continuum model (acetonitrile ε = 35.6).84–86The excited-state calculations were performed with TD-DFT level and the same level of theory including the Tamm–Dancoff approximation (TDA),87 as implemented in ORCA.88,89 The RIJCOSX (resolution of identity for Coulomb and chain of spheres for exchange terms) approximation was employed to accelerate the calculations.90 For implicit solvent effects,91 the implemented SMD (solvation model based on density) was utilized. The Wigner ensemble92 included a set of 100 geometries generated from a harmonic frequency calculation of the ground state equilibrium structure, using the program package SHARC2.1.93–95 The TD-DFT vertical excitations were carried out for a total of 60 singlet and 60 triplet states in each geometry. The CT analysis was performed using the program toolbox TheoDORE.96–98
For the investigation of the dissociation behavior, [Mo(CO)2(NO)(iPr3tacn)]+ was re-optimized in gas phase using the def2-TZVPP basis set. CASSCF(8,10) calculations averaged over 7 states with equal weights were carried out using the MOLCAS99,100 suite of programs. For this purpose, an ANO-RCC-MB basis set was used, as well as the scalar relativistic Douglas–Kroll–Hess correction87 to the one-electron Hamiltonian and Cholesky decomposition for the computation of the two-electron integrals.101 For these calculations, gas phase was assumed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr Sebastian Schott and Jarno Riefer for their help with preliminary measurements during the early phase of this project, and to Prof. Dr Tobias Brixner for his support. We thank the Center of Solvation Science (ZEMOS) at Ruhr-University Bochum for providing equipment and the Vienna Scientific Cluster for generous computational resources. We further acknowledge the Deutsche Forschungsgemeinschaft (DFG) [Priority Program SPP 2102 “Light-controlled reactivity of metal complexes” (Grants GO 1059/8-1, NU 263/4-1, and SCHA 962/13-1)].References
- A. K. Mustafa, M. M. Gadalla and S. H. Snyder, Signaling by gasotransmitters, Sci. Signaling, 2009, 2, re2 Search PubMed.
- M. Kajimura, R. Fukuda, R. M. Bateman, T. Yamamoto and M. Suematsu, Interactions of Multiple Gas-Transducing Systems: Hallmarks and Uncertainties of CO, NO, and H2S Gas Biology, Antioxid. Redox Signaling, 2010, 13, 157–192 CrossRef CAS PubMed.
- T. L. Poulos, Heme Enzyme Structure and Function, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- M. D. Maines, The heme oxygenase system: a regulator of second messenger gases, Annu. Rev. Pharmacol. Toxicol., 1997, 37, 517–554 CrossRef CAS PubMed.
- T. Matsui, M. Unno and M. Ikeda-Saito, Heme Oxygenase Reveals Its Strategy for Catalyzing Three Successive Oxygenation Reactions, Acc. Chem. Res., 2010, 43, 240–247 CrossRef CAS PubMed.
- B. R. Crane, J. Sudhamsu and B. A. Patel, Bacterial Nitric Oxide Synthases, Annu. Rev. Biochem., 2010, 79, 445–470 CrossRef CAS PubMed.
- C. Szabo, Gaseotransmitters: New Frontiers for Translational Science, Sci. Transl. Med., 2010, 2, 59ps54 CAS.
- B. Wegiel, D. W. Hanto and L. E. Otterbein, The social network of carbon monoxide in medicine, Trends Mol. Med., 2013, 19, 3–11 CrossRef CAS PubMed.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Nitric Oxide Donors:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Chemical Activities and Biological Applications, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
Chemical Activities and Biological Applications, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed. - C. Napoli and L. J. Ignarro, Nitric oxide-releasing drugs, Annu. Rev. Pharmacol. Toxicol., 2003, 43, 97–123 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, The therapeutic potential of carbon monoxide, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Developing drug molecules for therapy with carbon monoxide, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- U. Schatzschneider, Novel lead structures and activation mechanisms for CO-releasing molecules (CORMs), Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS PubMed.
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Carbon monoxide – physiology, detection and controlled release, Chem. Commun., 2014, 50, 3644–3660 RSC.
- U. Schatzschneider, Photoactivated Biological Activity of Transition-Metal Complexes, Eur. J. Inorg. Chem., 2010, 1451–1467 CrossRef CAS.
- U. Schatzschneider, PhotoCORMs: Light-triggered release of carbon monoxide from the coordination sphere of transition metal complexes for biological applications, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- R. D. Rimmer, A. E. Pierri and P. C. Ford, Photochemically activated carbon monoxide release for biological targets. Toward developing air-stable photoCORMs labilized by visible light, Coord. Chem. Rev., 2012, 256, 1509–1519 CrossRef CAS.
- I. Chakraborty, S. J. Carrington and P. K. Mascharak, Design Strategies to Improve the Sensitivity of Photoactive Metal Carbonyl Complexes (photoCORMs) to Visible Light and Their Potential as CO-Donors to Biological Targets, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed.
- S. Romanski, B. Kraus, U. Schatzschneider, J.-M. Neudörfl, S. Amslinger and H.-G. Schmalz, Acyloxybutadiene iron tricarbonyl complexes as enzyme-triggered CO-releasing molecules (ET-CORMs), Angew. Chem., Int. Ed., 2011, 50, 2392–2396 CrossRef CAS PubMed.
- N. S. Sitnikov, Y. Li, D. Zhang, B. Yard and H.-G. Schmalz, Design, Synthesis, and Functional Evaluation of CO-Releasing Molecules Triggered by Penicillin G Amidase as a Model Protease, Angew. Chem., Int. Ed., 2015, 54, 12314–12318 CrossRef CAS PubMed.
- J. Niesel, A. Pinto, H. W. P. N’Dongo, K. Merz, I. Ott, R. Gust and U. Schatzschneider, Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane (tpm) manganese tricarbonyl complex, Chem. Commun., 2008, 1798–1800 RSC.
- J. D. Simon and X. Xie, Photodissociation of chromium hexacarbonyl in solution: direct observation of the formation of pentacarbonyl(methanol)chromium, J. Phys. Chem., 1986, 90, 6751–6753 CrossRef CAS.
- J. Z. Zhang and C. B. Harris, Photodissociation dynamics of Mn2(CO)10 in solution on ultrafast time scales, J. Chem. Phys., 1991, 95, 4024–4032 CrossRef CAS.
- T. P. Dougherty and E. J. Heilweil, Ultrafast transient infrared absorption studies of M(CO)6 (M = Cr, Mo or W) photoproducts in n-hexane solution, Chem. Phys. Lett., 1994, 227, 19–25 CrossRef CAS.
- J. C. Owrutsky and A. P. Baronavski, Ultrafast infrared study of the ultraviolet photodissociation of Mn2(CO)10, J. Chem. Phys., 1996, 105, 9864–9873 CrossRef CAS.
- T. Jiao, Z. Pang, T. J. Burkey, R. F. Johnston, T. A. Heimer, V. D. Kleiman and E. J. Heilweil, Ultrafast Ring Closure Energetics and Dynamics of Cyclopentadienyl Manganese Tricarbonyl Derivatives, J. Am. Chem. Soc., 1999, 121, 4618–4624 CrossRef CAS.
- L. M. Kiefer, J. T. King and K. J. Kubarych, Dynamics of Rhenium Photocatalysts Revealed through Ultrafast Multidimensional Spectroscopy, Acc. Chem. Res., 2015, 48, 1123–1130 CrossRef CAS PubMed.
- P. Rudolf, F. Kanal, J. Knorr, C. Nagel, J. Niesel, T. Brixner, U. Schatzschneider and P. Nuernberger, Ultrafast Photochemistry of a Manganese-Tricarbonyl CO-Releasing Molecule (CORM) in Aqueous Solution, J. Phys. Chem. Lett., 2013, 4, 596–602 CrossRef CAS PubMed.
- P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, F. M. F. de Groot, K. J. Gaffney, S. Techert, M. Odelius and A. Föhlisch, Orbital-specific mapping of the ligand exchange dynamics of Fe(CO)5 in solution, Nature, 2015, 520, 78–81 CrossRef CAS PubMed.
- K. R. Sawyer, R. P. Steele, E. A. Glascoe, J. F. Cahoon, J. P. Schlegel, M. Head-Gordon and C. B. Harris, Direct Observation of Photoinduced Bent Nitrosyl Excited-State Complexes, J. Phys. Chem. A, 2008, 112, 8505–8514 CrossRef CAS PubMed.
- M. S. Lynch, M. Cheng, B. E. Van Kuiken and M. Khalil, Probing the Photoinduced Metal−Nitrosyl Linkage Isomerism of Sodium Nitroprusside in Solution Using Transient Infrared Spectroscopy, J. Am. Chem. Soc., 2011, 133, 5255–5262 CrossRef CAS PubMed.
- H. J. B. Marroux, B. F. E. Curchod, C. A. Faradji, T. A. Shuttleworth, H. A. Sparkes, P. G. Pringle and A. J. Orr-Ewing, Spin Changes Accompany Ultrafast Structural Interconversion in the Ground State of a Cobalt Nitrosyl Complex, Angew. Chem., Int. Ed., 2017, 56, 13713–13716 CrossRef CAS PubMed.
- A. A. Raheem, M. Wilke, M. Borgwardt, N. Engel, S. I. Bokarev, G. Grell, S. G. Aziz, O. Kühn, I. Y. Kiyan, C. Merschjann and E. F. Aziz, Ultrafast kinetics of linkage isomerism in Na2[Fe(CN)5NO] aqueous solution revealed by time-resolved photoelectron spectroscopy, Struct. Dyn., 2017, 4, 044031 CrossRef PubMed.
- A. Gavriluta, G. E. Büchel, L. Freitag, G. Novitchi, J. B. Tommasino, E. Jeanneau, P.-S. Kuhn, L. González, V. B. Arion and D. Luneau, Mechanism Elucidation of the cis–trans Isomerization of an Azole Ruthenium–Nitrosyl Complex and Its Osmium Counterpart, Inorg. Chem., 2013, 52, 6260–6272 CrossRef CAS PubMed.
- L. Freitag and L. González, Theoretical Spectroscopy and Photodynamics of a Ruthenium Nitrosyl Complex, Inorg. Chem., 2014, 53, 6415–6426 CrossRef CAS PubMed.
- J. Sanz García, F. Alary, M. Boggio-Pasqua, I. M. Dixon, I. Malfant and J.-L. Heully, Establishing the Two-Photon Linkage Isomerization Mechanism in the Nitrosyl Complex trans-[RuCl(NO)(py)4]2+ by DFT and TDDFT, Inorg. Chem., 2015, 54, 8310–8318 CrossRef PubMed.
- F. Talotta, J.-L. Heully, F. Alary, I. M. Dixon, L. González and M. Boggio-Pasqua, Linkage Photoisomerization Mechanism in a Photochromic Ruthenium Nitrosyl Complex: New Insights from an MS-CASPT2 Study, J. Chem. Theory Comput., 2017, 13, 6120–6130 CrossRef CAS PubMed.
- F. Talotta, L. González and M. Boggio-Pasqua, CASPT2 Potential Energy Curves for NO Dissociation in a Ruthenium Nitrosyl Complex, Molecules, 2020, 25, 2613 CrossRef CAS PubMed.
- F. Talotta, M. Boggio-Pasqua and L. González, Early Relaxation Dynamics in the Photoswitchable Complex trans-[RuCl(NO)(py)4]2+, Chem. – Eur. J., 2020, 26, 11522–11528 CrossRef CAS PubMed.
- W. Wang, F. Chen, J. Lin and Y. She, Time-resolved IR studies of gas-phase photochemistry of Co(CO)3NO, J. Chem. Soc., Faraday Trans., 1995, 91, 847–853 RSC.
- H. Nakata, K. Nagamori, M. Haze, K. Yamasaki and H. Kohguchi, Primary and Secondary Loss of CO and NO Ligands in the Ultraviolet Photodissociation of the Heteroleptic Co(CO)3NO Complex, J. Phys. Chem. A, 2020, 124, 10694–10704 CrossRef CAS PubMed.
- M. Kubota, M. K. Chan, D. C. Boyd and K. R. Mann, Thermal and photolytic reactions of nitrosyl-carbonyl complexes of rhodium and iridium with triphenylphosphine, Inorg. Chem., 1987, 26, 3261–3264 CrossRef CAS.
- A. T. McPhail, G. R. Knox, C. G. Robertson and G. A. Sim, Metal–carbonyl and metal–nitrosyl complexes. Part X. Photochemical generation of organometallic nitrenes and the crystal structure of a carbonylation product, isocyanatocarbonyl-π-cyclopentadienylbis(triphenylphosphine)molybdenum, J. Chem. Soc. A, 1971, 205–214 RSC.
- J. Böhmer, G. Haselhorst, K. Wieghardt and B. Nuber, The First Mononuclear Nitrosyl(oxo)molybdenum Complex: Side-On Bonded and μ3-Bridging NO Ligands in [{MoL(NO)(O)(OH)}2]NaPF6·H2O, Angew. Chem., Int. Ed. Engl., 1994, 33, 1473–1476 CrossRef.
- G. Haselhorst, S. Stoetzel, A. Strassburger, W. Walz, K. Wieghardt and B. Nuber, Synthesis and co-ordination chemistry of the macrocycle 1,4,7-triisopropyl-1,4,7-triazacyclononane, J. Chem. Soc., Dalton Trans., 1993, 83–90 RSC.
- J. A. McCleverty, Chemistry of Nitric Oxide Relevant to Biology, Chem. Rev., 2004, 104, 403–418 CrossRef CAS PubMed.
- R. A. Love, H. B. Chin, T. F. Koetzle, S. W. Kirtley, B. R. Whittlesey and R. Bau, X-ray and neutron diffraction studies on HW2(CO)8(P(OCH3)3). A compound with a slightly asymmetric hydrogen bridge bond, J. Am. Chem. Soc., 1976, 98, 4491–4498 CrossRef CAS.
- L. Ling-Kang, L. Jiann-T'suen and F. Duencheng, On the non-isostructures of M(CO)2(CH3CN)2(NO)(Sn(C6H5)3) (M = W, Cr), Inorg. Chim. Acta, 1989, 161, 239–245 CrossRef.
- K.-B. Shiu, S.-T. Lin, D.-W. Fung, T.-J. Chan, S.-M. Peng, M.-C. Cheng and J. L. Chou, Steric and Electronic Effects on Ligand Substitution of Cationic Carbonyl Nitrosyl Complexes of Molybdenum(0) Containing Bis(3,5-dimethylpyrazol-1-yl)methane-N,N’, Inorg. Chem., 1995, 34, 854–863 CrossRef CAS.
- A. Włodarczyk, R. M. Richardson, M. D. Ward, J. A. McCleverty, M. H. B. Hursthouse and S. J. Coles, Complexes of tris(3,5-dimethylpyrazolyl)borates alkylated on the 4-position of the pyrazolyl rings. X-ray crystal structure of molybdenum dicarbonyl nitrosyl tris(3,5-dimethyl-4-n-butylpyrazolyl)borate, Polyhedron, 1996, 15, 27–35 CrossRef.
- E. M. Holt, S. L. Holt, F. Cavalito and K. J. Watson, Crystal and Molecular Structure of Dicarbonylnitrosyl[hydrotris(pyrazol-l-yl)borato]molybdenum, HB(C3N2H3)3Mo(CO)2NO, Acta Chem. Scand., Ser. A, 1976, 30, 225–228 CrossRef.
- M. Cano, J. V. Heras, A. Monge, E. Gutierrez, C. J. Jones, S. L. W. McWhinnie and J. A. McCleverty, Synthesis and electrochemistry of [Mo(NO){HB(mppz)3}X2](mppz = 3-p-methoxyphenylpyrazolyl, X = CO or Cl); crystal structure of [Mo(NO){HB(mppz)3}(CO)2], J. Chem. Soc., Dalton Trans., 1992, 2435–2438 RSC.
- J. C. Jeffery, S. S. Kurek, J. A. McCleverty, E. Psillakis, R. M. Richardson, M. D. Ward and A. Wlodarczyk, Syntheses of 4-benzyl-3,5-dimethylpyrazolylborato complexes of molybdenum and tungsten nitrosyls: molecular structure of [Mo(CO)2(NO){HB(3,5-Me2-4-PhCH2C3N2)3}], a complex with an ‘inverted’ bowl-like structure, J. Chem. Soc., Dalton Trans., 1994, 2559–2564 RSC.
- N. C. Harden, J. C. Jeffery, J. A. McCleverty, L. H. Rees and M. D. Ward, Dinuclear molybdenum and gadolinium complexes of new ‘back-to-back’ B–B linked bis[tris(pyrazolyl)borate] ligands, New J. Chem., 1998, 22, 661–663 RSC.
- J. A. Campo, M. Cano, J. V. Heras, E. Pinilla, A. Monge and J. A. McCleverty, Chemistry of bulky tetrakis(pyrazolyl)borate ligands [B(pzR)4]− (R = p-CH3OC6H4 or C6H11), J. Chem. Soc., Dalton Trans., 1998, 3065–3070 RSC.
- A. L. Rheingold, L. N. Zakharov and S. Trofimenko, The Effect of a 3-Benzyl Group on the Coordination Chemistry of Homoscorpionate Ligands, Inorg. Chem., 2003, 42, 827–833 CrossRef CAS PubMed.
- C. Nagel, PhD thesis, Universität Würzburg, 2015.
- G. L. Geoffroy and M. S. Wrighton, Organometallic Photochemistry, Academic Press, New York, 1979 Search PubMed.
- H.-M. Berends and P. Kurz, Investigation of light-triggered carbon monoxide release from two manganese photoCORMs by IR, UV–Vis and EPR spectroscopy, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS.
- U. Sachs, G. Schaper, D. Winkler, D. Kratzert and P. Kurz, Light- or oxidation-triggered CO release from [MnI(CO)3(κ3-L)] complexes: reaction intermediates and a new synthetic route to [Mn2III/IV(μ-O)2(L)2] compounds, Dalton Trans., 2016, 45, 17464–17473 RSC.
- P. Hamm, Coherent effects in femtosecond infrared spectroscopy, Chem. Phys., 1995, 200, 415–429 CrossRef CAS.
- P. Nuernberger, K. F. Lee, A. Bonvalet, T. Polack, M. H. Vos, A. Alexandrou and M. Joffre, Suppression of perturbed free-induction decay and noise in experimental ultrafast pump-probe data, Opt. Lett., 2009, 34, 3226–3228 CrossRef CAS PubMed.
- K. M. Mullen and I. H. M. van Stokkum, TIMP: An R Package for Modeling Multi-way Spectroscopic Measurements, J. Stat. Softw., 2007, 18, 1–46 CrossRef.
- S. Mai, F. Plasser, J. Dorn, M. Fumanal, C. Daniel and L. González, Quantitative wave function analysis for excited states of transition metal complexes, Coord. Chem. Rev., 2018, 361, 74–97 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- U. Megerle, I. Pugliesi, C. Schriever, C. F. Sailer and E. Riedle, Sub-50 fs broadband absorption spectroscopy with tunable excitation: putting the analysis of ultrafast molecular dynamics on solid ground, Appl. Phys. B: Lasers Opt., 2009, 96, 215–231 CrossRef CAS.
- S. Schott, A. Steinbacher, J. Buback, P. Nuernberger and T. Brixner, Generalized magic angle for time-resolved spectroscopy with laser pulses of arbitrary ellipticity, J. Phys. B: At., Mol. Opt. Phys., 2014, 47, 124014 CrossRef.
- K. J. Kubarych, M. Joffre, A. Moore, N. Belabas and D. M. Jonas, Mid-infrared electric field characterization using a visible charge-coupled-device-based spectrometer, Opt. Lett., 2005, 30, 1228–1230 CrossRef PubMed.
- K. F. Lee, P. Nuernberger, A. Bonvalet and M. Joffre, Removing cross-phase modulation from midinfrared chirped-pulse upconversion spectra, Opt. Express, 2009, 17, 18738–18744 CrossRef CAS PubMed.
- J. Knorr, P. Rudolf and P. Nuernberger, A comparative study on chirped-pulse upconversion and direct multichannel MCT detection, Opt. Express, 2013, 21, 30693–30706 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, ıt OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- olex2.solve, https://www.olexsys.org/tags/olex2.solve/, (accessed 22 March 2021).
- olex2.refine, https://www.olexsys.org/tags/olex2.refine/, (accessed 22 March 2021).
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Dolg, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the rare earth elements, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09 Revision A.02, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- M. Lundberg and P. E. M. Siegbahn, Agreement between experiment and hybrid DFT calculations for O–H bond dissociation enthalpies in manganese complexes, J. Comput. Chem., 2005, 26, 661–667 CrossRef CAS PubMed.
- K. P. Jensen, B. O. Roos and U. Ryde, Performance of density functionals for first row transition metal systems, J. Chem. Phys., 2007, 126, 014103 CrossRef PubMed.
- K. A. Moltved and K. P. Kepp, Performance of Density Functional Theory for Transition Metal Oxygen Bonds, ChemPhysChem, 2019, 20, 3210–3220 CrossRef CAS PubMed.
- B. A. Hess, Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3742–3748 CrossRef CAS PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- D. Peng and M. Reiher, Exact decoupling of the relativistic Fock operator, Theor. Chem. Acc., 2012, 131, 1081 Search PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Barbatti and K. Sen, Effects of different initial condition samplings on photodynamics and spectrum of pyrrole, Int. J. Quantum Chem., 2016, 116, 762–771 CrossRef CAS.
- M. Richter, P. Marquetand, J. González-Vázquez, I. Sola and L. González, SHARC: ab Initio Molecular Dynamics with Surface Hopping in the Adiabatic Representation Including Arbitrary Couplings, J. Chem. Theory Comput., 2011, 7, 1253–1258 CrossRef CAS PubMed.
- S. Mai, P. Marquetand and L. González, Nonadiabatic dynamics: The SHARC approach, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1370 Search PubMed.
- S. Mai, M. Richter, M. Heindl, M. F. S. J. Menger, A. Atkins, M. Ruckenbauer, F. Plasser, L. M. Ibele, S. Kropf, M. Oppel, P. Marquetand and L. González, SHARC2.1: Surface Hopping Including Arbitrary Couplings – Program Package for Non-Adiabatic Dynamics, http://sharc-md.org, 2019 Search PubMed.
- F. Plasser and H. Lischka, Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- F. Plasser, M. Wormit and A. Dreuw, New tools for the systematic analysis and visualization of electronic excitations. I. Formalism, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- F. Plasser, TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- F. Aquilante, J. Autschbach, A. Baiardi, S. Battaglia, V. A. Borin, L. F. Chibotaru, I. Conti, L. De Vico, M. Delcey, I. F. Galván, N. Ferré, L. Freitag, M. Garavelli, X. Gong, S. Knecht, E. D. Larsson, R. Lindh, M. Lundberg, P. Å. Malmqvist, A. Nenov, J. Norell, M. Odelius, M. Olivucci, T. B. Pedersen, L. Pedraza-González, Q. M. Phung, K. Pierloot, M. Reiher, I. Schapiro, J. Segarra-Martí, F. Segatta, L. Seijo, S. Sen, D.-C. Sergentu, C. J. Stein, L. Ungur, M. Vacher, A. Valentini and V. Veryazov, Modern quantum chemistry with [Open]Molcas, J. Chem. Phys., 2020, 152, 214117 Search PubMed.
- V. Veryazov, P.-O. Widmark, L. Serrano-Andrés, R. Lindh and B. O. Roos, 2MOLCAS as a development platform for quantum chemistry software, Int. J. Quantum Chem., 2004, 100, 626–635 CrossRef CAS.
- F. Aquilante, R. Lindh and T. B. Pedersen, Analytic derivatives for the Cholesky representation of the two-electron integrals, J. Chem. Phys., 2008, 129, 034106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1414615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cp03514b |
| This journal is © the Owner Societies 2021 |