
Single-atom alloy catalysts: structural analysis, electronic properties and catalytic activities
Tianjun
Zhang†
ab,
Andrew G.
Walsh†
a,
Jihong
Yu
*b and
Peng
Zhang
 *a
*a
aDepartment of Chemistry, Dalhousie University, 6274 Coburg Road, B3H 4R2, Halifax, Canada. E-mail: peng.zhang@dal.ca
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun, 130012, P. R. China. E-mail: jihong@jlu.edu.cn
First published on 10th November 2020
Abstract
Monometallic catalysts, in particular those containing noble metals, are frequently used in heterogeneous catalysis, but they are expensive, rare and the ability to tailor their structures and properties remains limited. Traditionally, alloy catalysts have been used instead that feature enhanced electronic and chemical properties at a reduced cost. Furthermore, the introduction of single metal atoms anchored onto supports provided another effective strategy to increase both the atomic efficiency and the chance of tailoring the properties. Most recently, single-atom alloy catalysts have been developed in which one metal is atomically dispersed throughout the catalyst via alloy bonding; such catalysts combine the traditional advantages of alloy catalysts with the new feature of tailoring properties achievable with single atom catalysts. This review will first outline the atomic scale structural analysis on single-atom alloys using microscopy and spectroscopy tools, such as high-angle annular dark field imaging-scanning transmission electron microscopy and extended X-ray absorption fine structure spectroscopy. Next, progress in research to understand the electronic properties of single-atom alloys using X-ray spectroscopy techniques and quantum calculations will be presented. The catalytic activities of single-atom alloys in a few representative reactions will be further discussed to demonstrate their structure–property relationships. Finally, future perspectives for single-atom alloy catalysts from the structural, electronic and reactivity aspects will be proposed.
 Tianjun Zhang | Tianjun Zhang received his PhD degree in Jilin University in 2020 under the supervision of Prof. Jihong Yu. From 2018 to 2019, he joined Prof. Peng Zhang's group at Dalhousie University as a visiting student. Currently, he works as a postdoctoral fellow in the group of Prof. Jihong Yu at Jilin University. His research focuses on the synthesis and modification of Ti-containing zeolites in selective oxidation catalysis, and the structural characterization of zeolite supported metal catalysts using X-ray absorption spectroscopy. |
 Andrew G. Walsh | Andrew G. Walsh received his BSc and MSc from Memorial University. After completing his MSc, he joined Dalhousie University in May 2019 as a research support staff member of the Zhang research group and is currently in this position. His research focuses on X-ray absorption spectroscopy studies of Ag nanostructures and their applications in catalysis and biomedicine. |
 Jihong Yu | Jihong Yu received her PhD degree in inorganic chemistry from Jilin University in 1995. From 1996–1998, she worked as a postdoctoral fellow first at the Hong Kong University of Science and Technology and then at Tohoku University in Japan. Since 1999, she has been a full Professor in the Chemistry Department at Jilin University. She was elected as a member of the Chinese Academy of Sciences in 2015, academician of TWAS in 2016 and member of Academiae Europaeae in 2019. Her research interests are focused on the designed synthesis of zeolitic nanoporous materials/zeolite-based composite materials and their applications in energy, environment and other emerging fields. |
 Peng Zhang | Peng Zhang received his BSc and MSc from Jilin University and PhD from the University of Western Ontario. After spending two years at McGill University as an NSERC postdoctoral fellow, he joined Dalhousie University in 2005 as an assistant professor and is now a full professor at the institution. His research focuses on synchrotron X-ray spectroscopy studies of nanomaterials and their applications in catalysis and biomedicine. |
1 Introduction
Metal-containing catalysts, especially those containing noble metals,1–3 are widely used in the field of heterogeneous catalysis. However, they are not abundant and are expensive, which greatly hinders the application of noble metal-containing catalysts.4,5 Hence, developing a new class of cost-effective and efficient metal-containing catalysts is important for the future of heterogeneous catalysis.The field of heterogeneous catalysis, specifically those involving alloy catalysts (which consist of more than one metal) with distinguishable electronic and chemical properties, has seen many advances over the past few decades.6–15 Their outstanding properties can be attributed to the intermetallic synergy between the different metals.16–18 Alloy catalysts offer the opportunity to obtain simple and effective new catalysts. Bimetallic alloy catalysts were the first to gain considerable commercial interest in the 1960s.19,20 Compared with monometallic catalysts, they displayed unique activities in hydrocarbon reforming.21 The findings of these unexpected properties of bimetallic catalysts have inspired many extensive investigations into their possible applications; currently bi- or multi-metallic alloy catalysts are widely utilized in many thermo-catalytic and electro-catalytic applications.17,21–28
Atomically dispersed catalysts consisting of single metal atoms anchored onto supports, which are referred to as single-atom catalysts (SACs), have recently emerged as promising new heterogeneous catalysts.29–35 The most important feature of SACs is the isolated metal species at the single-atom level that are dispersed onto the support. This approach not only maximizes the atomic efficiency of the metal,36,37 but also provides more uniform and well-defined active sites than common nanoparticle (NP) catalysts.38–40 With 100% metal dispersion and maximum metal utilization, SACs provide an ideal strategy for creating cost-effective catalysts and they can be used to form highly efficient catalysts based on noble metals.29,30,41 Since the report by Qiao et al. in 2011 on CO oxidation using Pt1/FeOx as the SAC,42 various types of metal SACs have been successfully designed and prepared, thus opening up a new avenue for the specific and atomic level design of catalysts for target reactions.
For the manipulation of SACs, remarkable progress came with one of the earliest reports of a single-atom alloy (SAA) catalyst.43 To be specific, the concept of SAAs can be described as the material formed by bi- and multi-metallic complexes, where one of these metals is atomically dispersed in such material.43–61 Furthermore, the catalytic properties of the active sites are intrinsically induced by the strong metallic interaction (i.e. alloy bonding) between the isolated single atoms and the metal nanostructure/support, which leads to the formation of new atomic and electronic structures.62–69 In this review, some representative works on SAA catalysts will be presented (see Table 1 which provides a comprehensive list of the properties of various SAA catalysts and corresponding applications). Owing to the structural complexity of such materials, we will focus on the metal nanostructure-based SAAs, including their structural and electronic characterization using state-of-the-art techniques as well as some applications in catalytic reactions. Instead of on a 3D metal nanostructure, literature works about the deposition of single atoms on 2D metal materials, such as the pioneering work done by the Sykes group on the model catalyst synthesized using an ultrahigh vacuum (UHV),44,56,66,70,71 are not covered in this review.
| Metal single-atom | Metal host | Molar ratio (Maximum) | Synthetic method | Application | Ref. |
|---|---|---|---|---|---|
| Pd | Au | 0.15 Pd (0.15) Au (0.85) | Sequential reduction | Electrocatalytic H2O2 production | 43 |
| Pd | Ag | 0.024 Pd (0.025) Ag (1.00) | Incipient wetness co-impregnation | Selective hydrogenation of acetylene | 69 |
| Pd | Cu | 0.021 Pd (0.03 wt%) Cu (0.85 wt%) | Incipient wetness sequential impregnation | Selective hydrogenation of acetylene | 72 |
| Pd | Ni | 0.028 Pd (0.98 wt%) Ni (19 wt%) | Atomic layer deposition | Hydrogenation of nitriles | 54 |
| Pd | Au | 0.020 | Surface decoration | Electrochemical CO2 reduction | 29 |
| Pd | Au | 0.020 | Seed-mediated growth | CO oxidation | 27 |
| Pd | Cu | 0.17 Pd (2.0 wt%) Cu (6.0 wt%) | Impregnation | NO reduction | 73 |
| Pd | Ag | 0.0013 Pd (0.01 wt%) Ag (8 wt%) | Incipient wetness impregnation | Selective hydrogenation of acrolein | 45 |
| Pd | Au | 0.0040 Pd (1) Au (250) | Sequential reduction | Selective hydrogenation of 1-Hexyne | 55 |
| Pd | Au | 0.0014 | Seed-mediated growth | Selective oxidation of 2-propanol | 74 |
| Pd | Au | 0.091 Pd (1) Au (10) | Ion exchange and reduction | Ullmann reaction of aryl chlorides in water | 75 |
| Pd | Cu | 0.006 | Incipient wetness co-impregnation | Semihydrogenation of acetylene | 76 |
| Pd | Ag | 0.20 Pd (1) Ag (4) | Co-reduction | Oxygen reduction in alkaline media | 17 |
| Pd | Cu | 0.0018 Pd (0.0018 at%) Cu (100 at%) | Galvanic replacement | Selective hydrogenation of phenylacetylene | 77 |
| Pd | Au | 0.024 Pd (2.5 at%) Au(100 at%) | Ion exchange and reduction | Selective hydrogenation of acetylene | 36 |
| Pd | Au | 0.0040 | Sequential reduction | Selective hydrogenation of 1-hexyne | 52 |
| Pd | Au | 3 wt% total metal | Colloidal chemistry | Selective Hydrosilylation | 78 |
| Pt | Cu | 0.0048 Pt (0.1 wt%) Cu (6.7 wt%) | Incipient wetness co-impregnation | Propane dehydrogenation | 57 |
| Pt | SbSnO2 (Pt–Sn) | 0.063 Pt (8 wt%) SbSnO2 (92 wt%) | Incipient wetness impregnation | Formic acid oxidation reaction | 59 |
| Pt | Au | 0.070 Pt (7 at%) Au (93 at%) | Colloidal chemistry | Formic acid oxidation reaction | 68 |
| Pt | Cu | 0.029 Pt (0.030) Cu (1.00) | Galvanic replacement | Glycerol hydrogenolysis | 63 |
| Pt | Cu | 0.016 Pt (0.2 at%) Cu (12 at%) | Galvanic replacement | Selective hydrogenation of 1,3-butadiene | 79 |
| Pt | Ni | 0.016 Pt (0.016) Ni (1.00) | Galvanic replacement | Selective Hydrogenation of 3-nitrostyrene | 80 |
| Pt | AlFe | 0.0020 | Czochralski method | Propyne hydrogenation | 51 |
| Pt | Cu | 0.0079 Pt (1) Cu (125) | Sequential reduction | H2 activation and acetylene hydrogenation | 81 |
| Pt | Cu | 0.010 | Galvanic replacement | Non-oxidative dehydrogenation of butane | 70 |
| Pt | Cu | 0.0083 Pt (0.3 wt%) Cu (11.7 wt%) | Incipient wetness impregnation | Hydrogenation of methyl glycolate | 67 |
| Pt | Ni | 0.050 | Co-impregnation | Selective hydrogenation of CO | 62 |
| Ni | Cu | 0.010 | Electroless galvanic deposition | Selective non-oxidative dehydrogenation of ethanol | 49 |
| Ni | Au | 0.0050 | Sequential reduction | Non-oxidative dehydrogenation of ethanol | 46 |
| Ni | Cu | 0.0010 | Electroless galvanic deposition | Selective non-oxidative ethanol dehydrogenation | 61 |
| Ni | Au | 0.0050 | Sequential reduction | Selective oxidation of methacrolein | 82 |
| Au | Pd | 0.082 Au (12 atoms) Pd (135 atoms) | Galvanic replacement | Glucose oxidation | 83 |
| Au | Ru | 0.15 Au (15.35 at%) Ru(84.65 at%) | Laser ablation in liquid | Electrocatalytic hydrogen evolution | 84 |
| Cu | Pt | 0.0020 | Underpotential deposition | CO oxidation | 7 |
| Ru | PtCu (Ru–Pt, Ru–Cu) | 0.082 Ru (8.2 at%) Pt (68.7 at%) Cu (23.1 at%) | Sequential etching and electrochemical leaching | Electrocatalytic oxygen evolution reaction | 58 |
| Ir | Co | 0.0051 Ir (1.7 wt%) Co (100 wt%) | Atomically confined alloying | Dinuclear tandem water oxidation | 41 |
2. Structural analysis of SAAs
A variety of microscopic and spectroscopic methods have been used to study the structure of single-atom catalysts.85 Herein we focus on two of the most widely used experimental techniques, one from the direct imaging perspective and the other on in-depth spectroscopic analysis of the local structure.2.1 Direct imaging of SAAs
Aberration-corrected high-angle annular dark field imaging-scanning transmission electron microscopy (HAADF-STEM), which is a powerful technique in electron microscopy, has been extensively used to directly image single-atom sites on SAAs.43,57–59,63,68,69,72,79–81,86 In ideal cases, the individual atoms can be clearly observed by excellent image contrast due to their different atomic numbers.51,54,63,64,73,84,87 Another imaging technique, scanning tunnelling microscopy (STM), has also been used for SAA structural study, but it is mainly used for 2D surfaces instead of NP-based SAA samples.44,66,70An example of a study done on SAAs using HAADF-STEM is that by Sun et al., who reported the synthesis of Pt/Cu SAAs using incipient wetness co-impregnation.57 The structure of Pt/Cu SAAs was deduced using HAADF-STEM. The occurrence of single, brighter Pt atoms throughout the Cu NPs is shown in Fig. 1. The lattice spacing of Pt/Cu SAAs was determined to be 0.21 nm, which agrees with the lattice spacing of the Cu(111) surface. This value indicates that Pt atoms were dispersed throughout the Cu NPs. Single Pt atoms were consistently seen in different portions of the reduced 0.1Pt10Cu/Al2O3 catalyst.
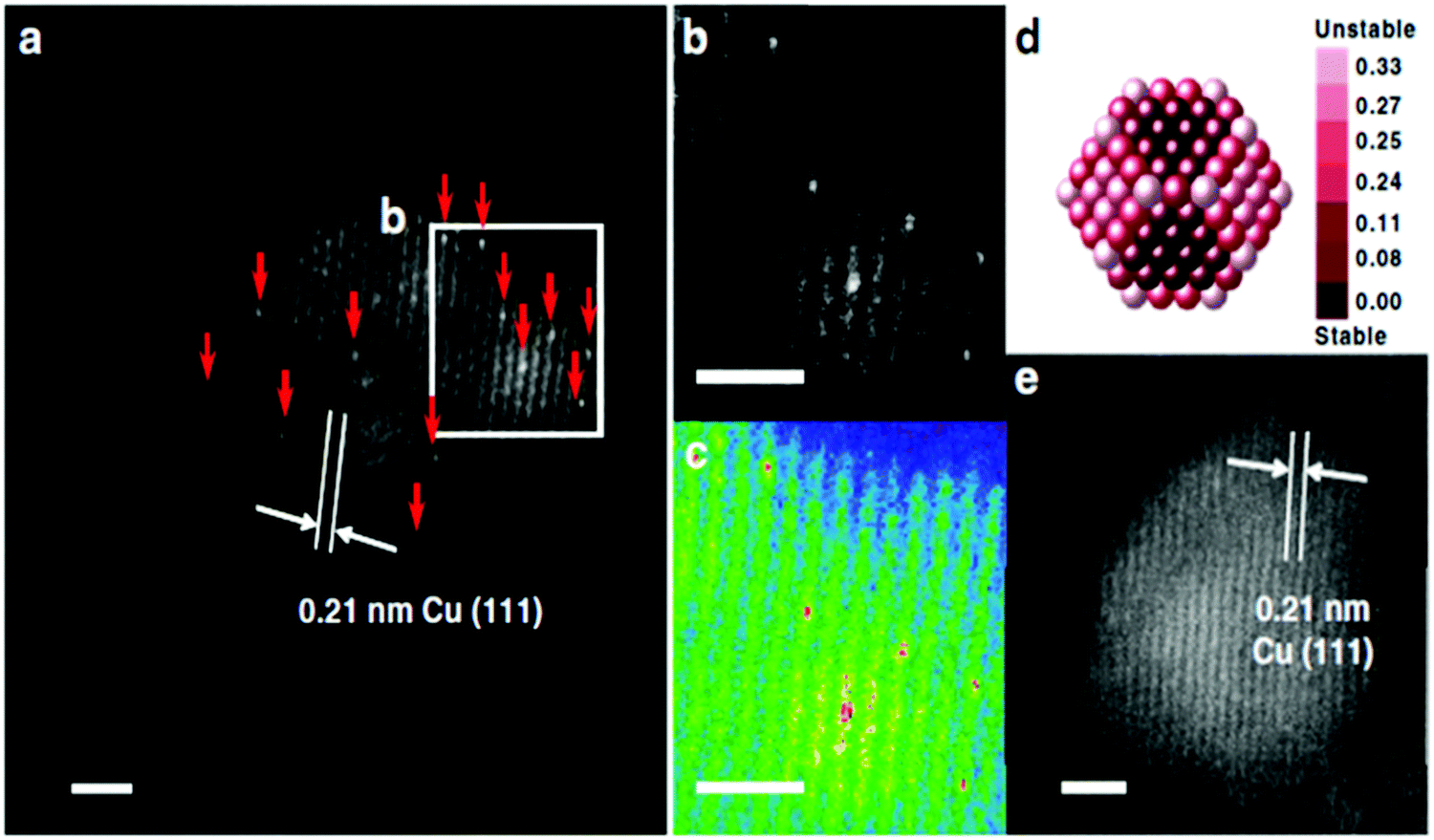 | ||
| Fig. 1 Morphology of γ-Al2O3-supported SAA. (a) HAADF-STEM images with the typical region of the reduced 0.1Pt10Cu/Al2O3 catalyst; Pt atoms are highlighted using red arrows. (b) The enlarged image and (c) the coloured intensity map from the selected region in (a). (d) Relative stability of Pt single atoms over Cu (approximately 2.1 nm) nanoparticles. Dark red indicates stable locations of Pt atoms. The colour bar unit is eV per Pt atom. (e) HAADF-STEM images with the typical region of the reduced 10Cu/Al2O3 catalyst. Reprinted with permission from ref. 57. Copyright 2018 Springer Nature. | ||
Another example of the use of HAADF-STEM in SAA structural analysis is the work done by Lucci et al. in distinguishing isolated Pt atoms from the Pt/Cu SAAs that were synthesized using galvanic replacement.79 In their work, several bright, single atoms were seen within the Cu lattice. Furthermore, the lattice spacing of Pt/Cu SAAs was comparable to the lattice spacing of pure Cu; this correlation supported the dispersion of Pt atoms that was observed.
HAADF-STEM can also be used in combination with other techniques to obtain more detailed imaging. By combining the HAADF-STEM images and energy-dispersive X-ray (EDX) spectroscopy elemental mapping images, the structure of more complex SAAs with three components can also be fully investigated. For example, Yao et al. used HAADF-STEM elemental mapping, as shown in Fig. 2a–f, to demonstrate the formation of core–shell PtCux/Pt-skin structures and the atomic dispersion of Ru1.58 Ordered and straight-edge structures were suggested through Pt-resolved elemental mapping (Fig. 2b). However, when using Cu-resolved elemental mapping (Fig. 2c), these ordered structures were not observed. An ordered arrangement and regular geometry profile were visible upon the overlap of the Pt and Cu elemental maps (Fig. 2e); this is similar to the corresponding HAADF-STEM image in Fig. 2a. As shown by the red dashed lines in Fig. 2e, the lack of Cu in the very top layer provides evidence for Pt-skin shell formation, which was suggested to be due to the separation of the Pt-skin shell located in the inner Cu species from oxygenated species (Fig. 2g). The elemental distribution of Ru in Pt3Cu, which is seen in Fig. 2d, suggested high atomic dispersion of Ru (marked arrows in Fig. 2f) in the Pt3Cu/Pt-skin matrix.
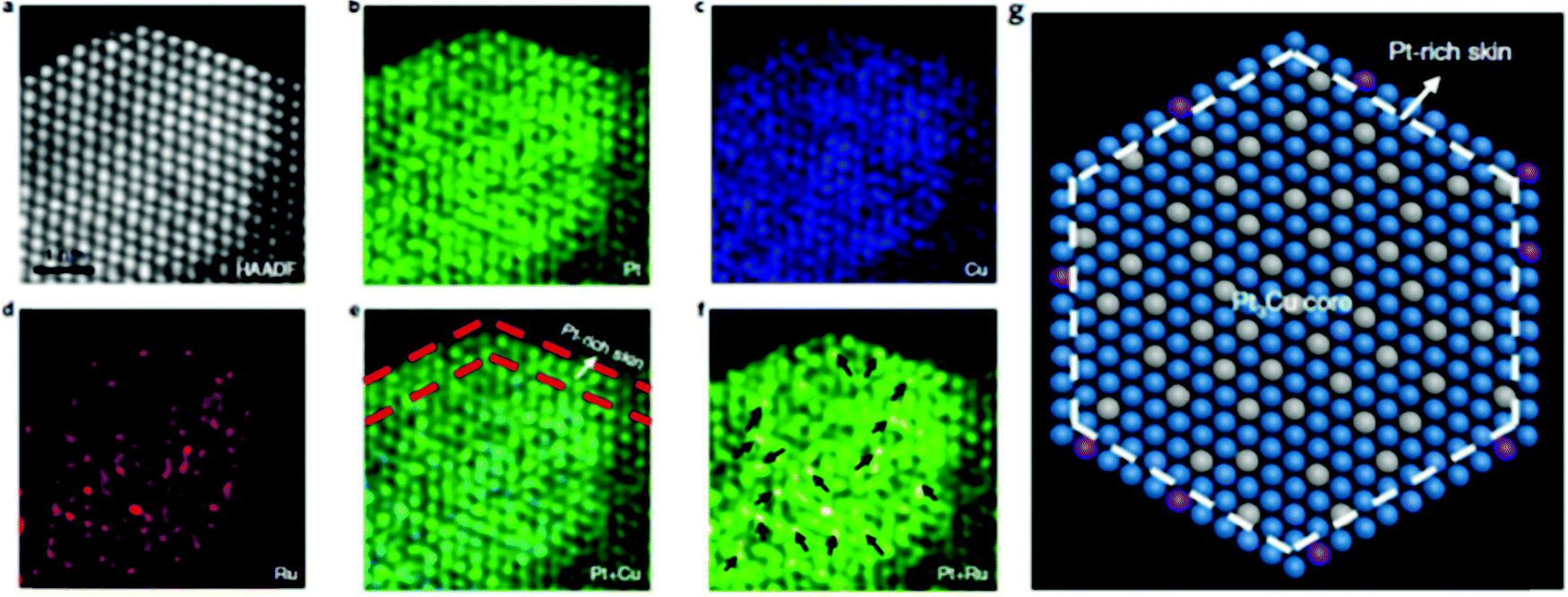 | ||
| Fig. 2 The structure characterizations of Ru1–Pt3Cu. (a)–(f) Atomically resolved elemental mapping of Ru1–Pt3Cu. (g) Atomic model of (a). Blue, grey and purple represent Pt, Cu and Ru, respectively. Adapted with permission from ref. 58. Copyright 2019 Springer Nature. | ||
Another application for the use of HAADF-STEM EDX mapping is when the SAA is composed of two metal elements having similar atomic numbers (e.g. Pt and Au atoms). This technique is useful, not only in determining the existence of single atoms, but in understanding the formation of the SAA. An example of this application is the work done by Duchesne et al., who reported the characterization of PtAu SAAs using HAADF-STEM EDX mapping images.68 As shown in Fig. 3a and b, by overlapping EDX maps of representative particles, it was determined that the signal due to Pt was enhanced and more diffuse at the particle edges while the signal due to Au came mostly from the NP cores. The appearance of a Pt-rich surface related to a surface structure containing single Pt atoms. For an ideal, shortened octahedral NP as shown in Fig. 3d, the maximum surface coverage by single-atom sites having no adjacent Pt atoms was determined to occur with a Pt content of 5.5%. Although the synthesized PtAu SAAs were mostly polycrystalline as shown by the HAADF-STEM EDX elemental mapping, the single-crystal estimate served as an estimated upper concentration limit where only single-atom Pt sites could exist for both Pt4Au96 and Pt7Au93. According to the combined results obtained from HAADF-STEM EDX, extended X-ray absorption fine structure (EXAFS), electrochemical analysis and valence band analysis, the PtAu samples were listed according to their surface structure: Pt4Au96 and Pt7Au93 contained Au with single-atom Pt sites, Pt17Au83 contained Au with both single-atom and few-atom Pt sites, while Pt53Au47 and Pt78Au22 showed an Au-core/Pt-shell configuration (Fig. 3c). Fig. 3e–g show this progression in the SAAs from single-atom to near-complete shell coverage.
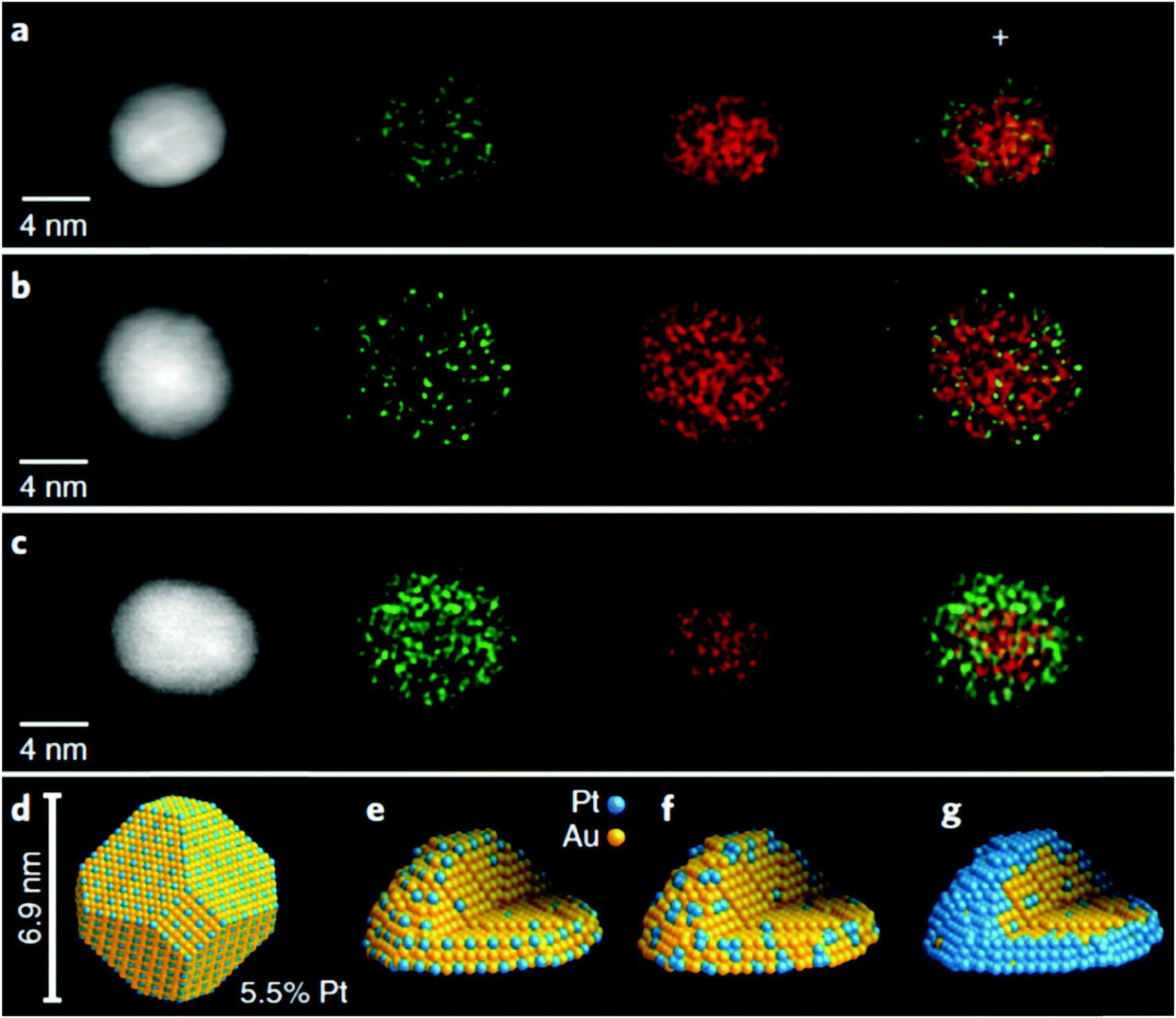 | ||
| Fig. 3 HAADF-STEM images and structural models. (a)–(c). STEM/EDX mapping images of individual particles from (a) Pt4Au96 and (b) Pt7Au93 single-atom Pt samples and from (c) a Pt78Au22 core–shell sample. (d) Structural model of an ideal, truncated octahedral nanoparticle with an optimal single-atomic-site coverage. (e)–(g) Models that depict the proposed evolution of PtAu surface structures from (e) single-atom Pt sites to (f) few-atom Pt clusters to (g) a complete Pt shell as a result of increased Pt content. Reprinted with permission from ref. 68. Copyright 2018 Springer Nature. | ||
2.2 In-depth local structural analysis of SAAs
Although atomic-resolution HAADF-STEM can provide direct visualization of SAAs, it only offers information on the specific local area. In contrast, X-ray absorption spectroscopy (XAS) can be used to shed light on the detailed local information of elements from the perspectives of inter-atomic distance (at approximately 0.01 Å level), type of chemical bonds, coordination number and structural disorder. It is particularly suitable for the analysis of low-concentration, multi-element samples like SAAs due to the element-specific nature of this technique and its high sensitivity originated from the ultra-bright synchrotron light source.45,54,55,57–59,61–63,73–76,79–81,84 Based on the Fourier-transformed (FT) EXAFS spectra and fitting results, a thorough investigation into the atomic structure of SAAs can be achieved.An example of the use of XAS measurements to determine the atomic structure of distinguishable SAAs is the work by Wrasman et al., in which the authors reported the synthesis and characterization of PdAu SAAs having various amounts of Pd.74 The Pd K-edge and Au L3-edge EXAFS spectra are shown in Fig. 4a and b, respectively. The Pd−Pd coordination number (CN) was determined to be approximately zero for all samples within experimental error. This result assured that either a dilute or single-atom alloy had been formed since most of the Pd atoms in the alloy were isolated from each other. The Pd–Au bond length was intermediate with respect to both pure Pd and pure Au; it was also in agreement with the bond lengths of other Pd/Au alloys previously reported in the literature.89,90 For all eight samples analysed, a good comparison of Pd distribution among Au was achieved between the model used for surface alloying and the experimentally determined CNs (Fig. 5). The modelled Pd–Au CNs were less than 9, which indicated that Pd was mainly located on the surface atomic layer. As expected, the Pd–Au CNs decreased with increasing XPd/D.
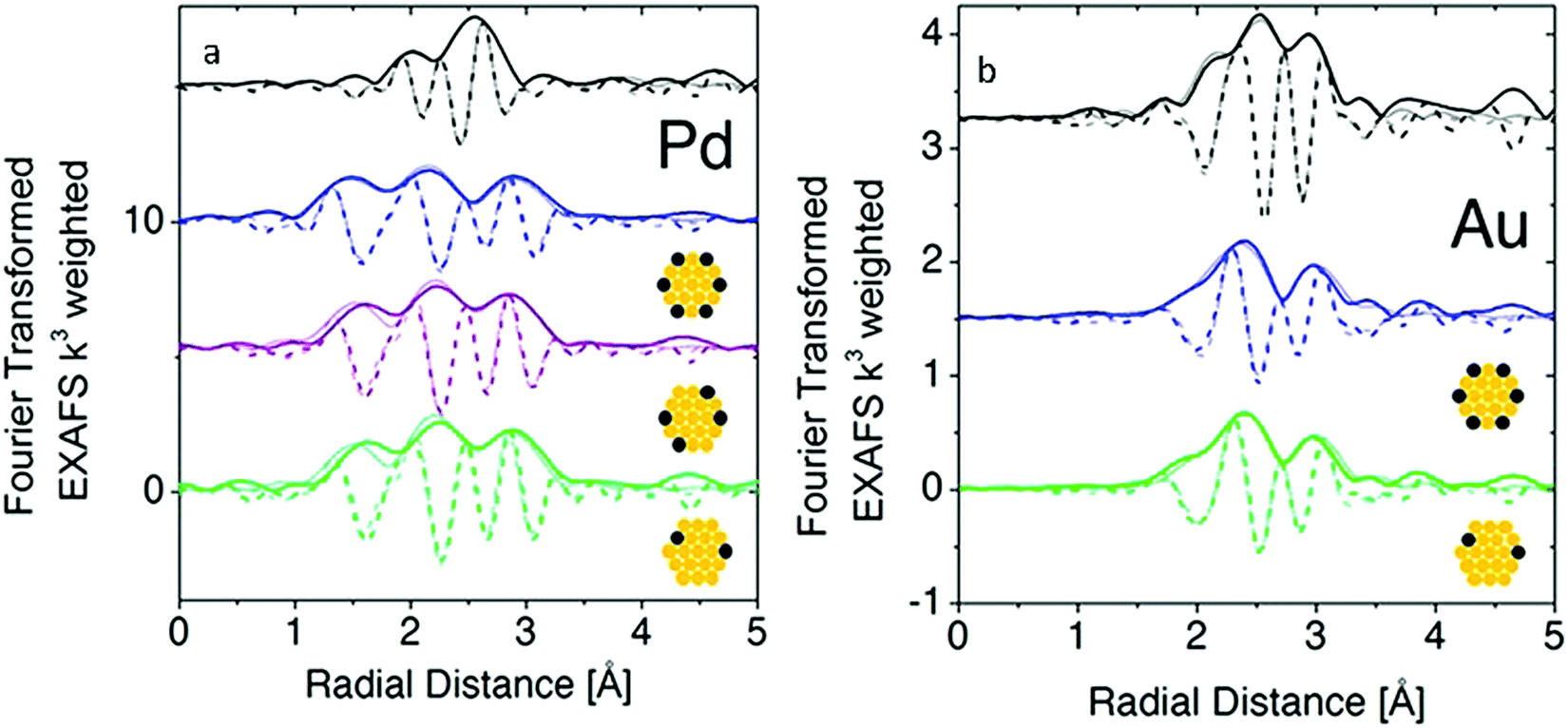 | ||
| Fig. 4 Fourier-transform EXAFS at the Pd K-edge and Au L3-edge with model fits (a and b, respectively) for Pd(1.4%)/Au (green), Pd(4.1%)/Au (purple), and Pd(7.0%)/Au (blue) along with Pd or Au foils (black) where the dark lines represent data, the faded lines represent best-fit models, the solid lines represent FT magnitude and dotted lines represent the imaginary component of the Fourier transform. Adapted with permission from ref. 74. Copyright 2018 American Chemical Society. | ||
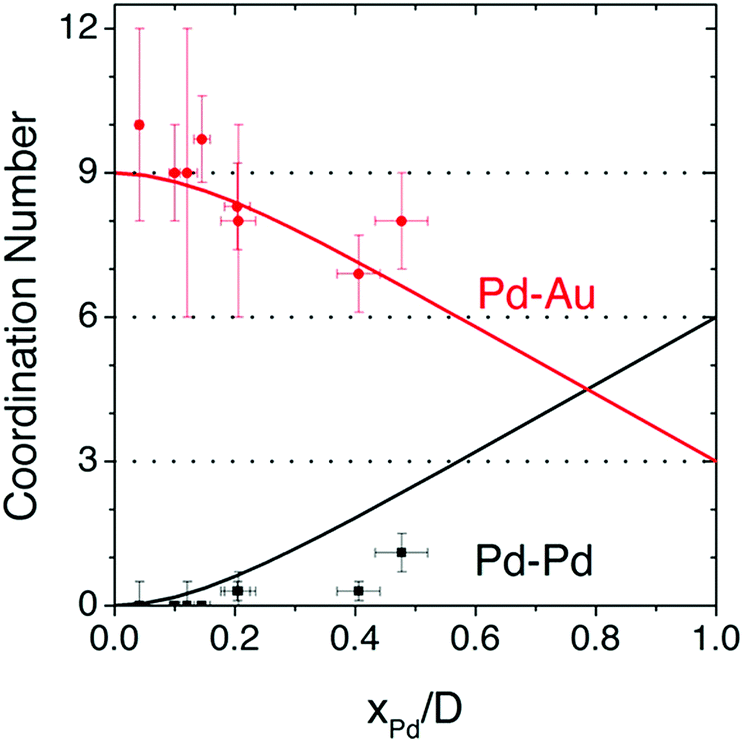 | ||
| Fig. 5 Pd−Pd and Pd−Au coordination numbers predicted by the surface-alloy model (solid lines), overlaid with experimentally determined coordination numbers from all 8 samples analysed using XAS. Reprinted with permission from ref. 74. Copyright 2018 American Chemical Society. | ||
In addition to Au, Pd can also be alloyed with Cu to form distinguishable SAA catalysts as shown by Pei et al.76 The FT- EXAFS spectra were determined for Pd foil (Fig. 6a), Pd0.006/SiO2 (Fig. 6b), CuPd0.015/SiO2 (Fig. 6c) and CuPd0.006/SiO2 (Fig. 6d). The spectra indicated that the first shell distance for the CuPd catalysts was shorter than that of Pd foil. This result confirmed the formation of CuPd alloys. For the bimetallic catalyst containing a higher Pd content (CuPd0.015/SiO2), both Pd–Cu and Pd–Pd bonds were present having CNs of 9.8 and 1.5, respectively. Furthermore, the isolation of Pd atoms from Cu was confirmed since only Pd–Cu bonds appeared in the CuPd0.006/SiO2 catalyst. The FT-EXAFS spectra were in good agreement with the experimental spectra as seen in Fig. 6, and thus the formation of a CuPd SAA for the CuPd0.006/SiO2 catalyst was verified. Neither Pd–O nor Pd–Si bonds were observed for both bimetallic catalysts. This result suggested that there was no Pd attached to the SiO2 support, which was attributed to the simplicity of formation of the CuPd alloys.
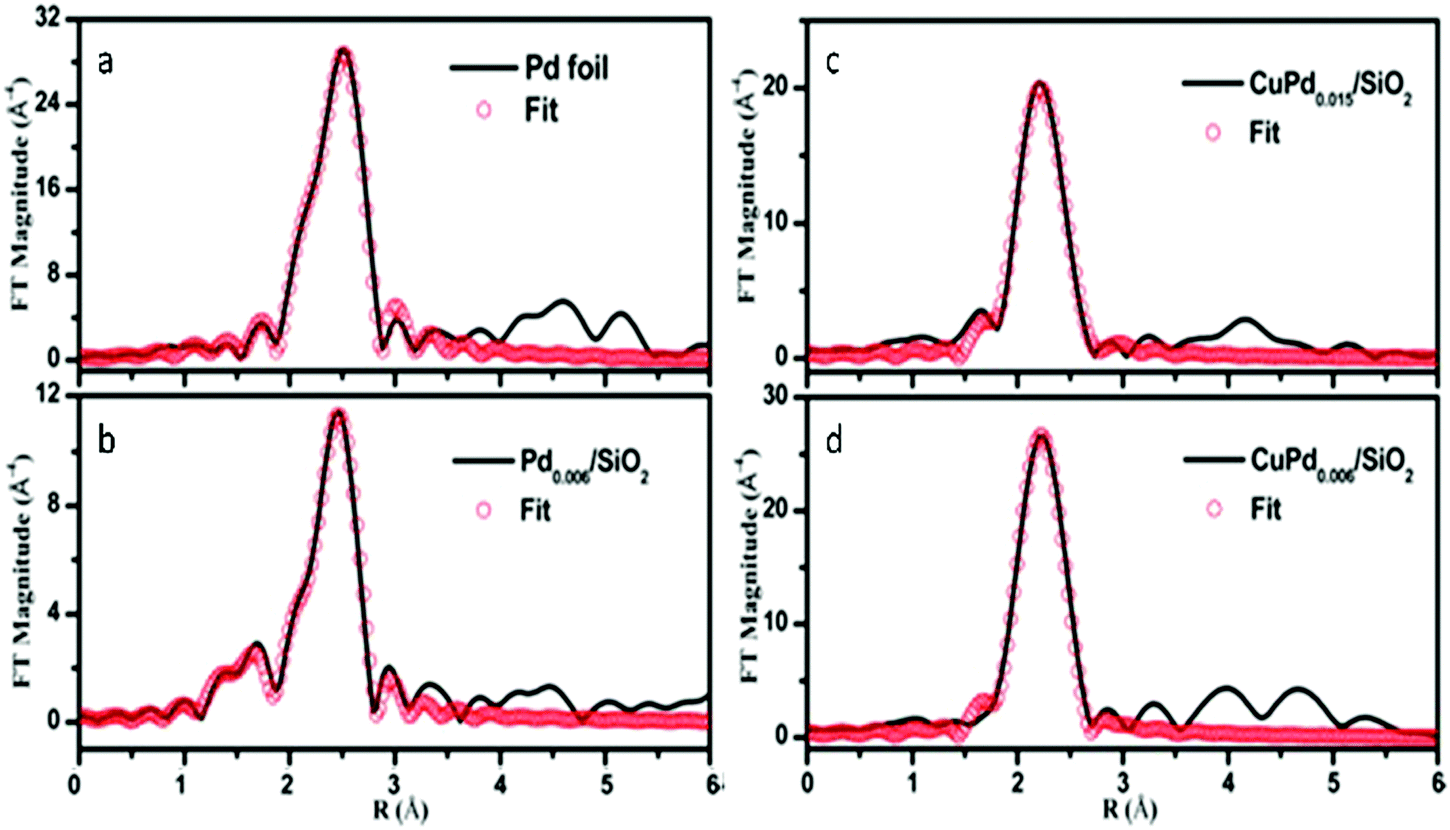 | ||
| Fig. 6 k 3-Weighted FT-EXAFS spectra of (a) Pd foil, (b) Pd0.006/SiO2, (c) CuPd0.015/SiO2 and (d) CuPd0.006/SiO2 catalysts. Adapted with permission from ref. 76. Copyright 2017 American Chemical Society. | ||
Another combination of metals that can be used to form distinguishable SAA catalysts is Pt and Sn. For example, PtSn SAA/antimony-doped tin oxides (ATOs) were synthesized by Kim et al.59 The substrate for this SAA, ATO, is more complex than those of conventional SAAs (which normally is a metal NP). The atomic structure of this SAA catalyst was determined using XAS analysis. Based on the FT-EXAFS spectra in Fig. 7, it was determined that the sample reduced at 400 °C (Pt1/ATO) showed Pt–Sn interactions, while both Pt/C and the sample reduced at 100 °C (Pt NP/ATO) only showed Pt–Pt bonds. Furthermore, the Pt–Sn CN was determined to be 2.61 for the sample reduced at 400 °C and no Pt–Pt bonding was observed. These results confirmed that Pt was atomically dispersed as single atoms throughout the Pt1/ATO catalyst. In addition, no Pt–Cl bonds were observed for the Pt1/ATO catalyst, whereas a weak Pt–Cl interaction having a CN of 0.38 was observed for the Pt NP/ATO. This result was suggested to be due to the Cl− ligands becoming detached during reduction at high temperature.
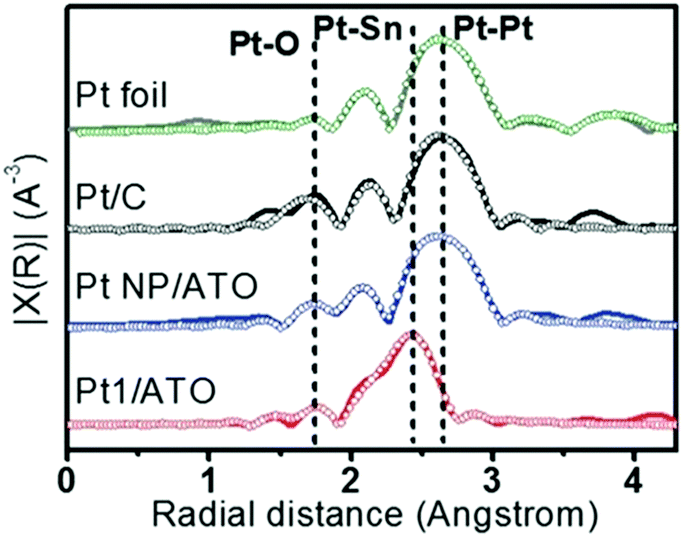 | ||
| Fig. 7 EXAFS (line: measured, symbol: fitted) data for Pt foil, reduced Pt/C, Pt NPs/ATO and Pt1/ATO samples. Commercial Pt/C was reduced at 100 °C for 1 h prior to the measurements. Reprinted with permission from ref. 59. Copyright 2017 Wiley Materials. | ||
One example of indistinguishable SAAs is the synthesis of PtAu SAA catalysts by Duchesne et al.68 As shown in the wavelet transformation (WT) EXAFS spectra, the presence of a single metal–metal (M–M) bonding peak, located at around 7.5 Å−1 and 2.5 Å, further highlighted the fact that neither atomic mass nor bond length (represented by the x-axis and y-axis in Fig. 8a, respectively) can be used to distinguish the M–Pt and M–Au scattering paths. In order to elucidate further structural information about these PtAu samples, an EXAFS analysis was conducted on the PtAu SAAs at both the Au-L3 and Pt-L3 absorption edges. As shown in the FT-EXAFS spectra (Fig. 8b) and the EXAFS fitting results, the scattering from both Au and Pt could be treated as one path (denoted Au–M and Pt–M for the Au-L3 and Pt-L3 edges, respectively) due to the similarity of both the Au and Pt paths. Examination of the Au–M and Pt–M bond lengths indicated that the Pt atoms in the most active PtAu samples (Pt4Au96 and Pt7Au93) were mainly present as single atomic sites surrounded by Au. This observation was based on the close agreement between the measured Pt–M bond lengths (2.82 Å) and the expected bimetallic bond length of 2.822 Å for a homogeneous PtAu alloy, which was calculated from experimental Pt–Pt and Au–Au bond lengths in pure Pt and pure Au NPs. EXAFS analysis of the local CNs revealed that Pt atoms were undercoordinated in the PtAu SAAs, especially those that showed highly active single-atom sites. CNs for the Pt–M scattering path ranged from 9 in Pt78Au22 to 5 in Pt4Au96, which was lower than the CN of 11 observed for pure Pt NPs. Since the Au–M CNs were between 11 and 12, an Au-core/PtAu shell NP structure having mostly surface Pt was suggested. In addition, X-ray absorption near-edge structure (XANES) analysis of the samples indicated the presence of both metallic Pt and metallic Au in these SAAs; a positive binding-energy shift and an increased white-line breadth provided evidence for the presence of Pt single atoms.
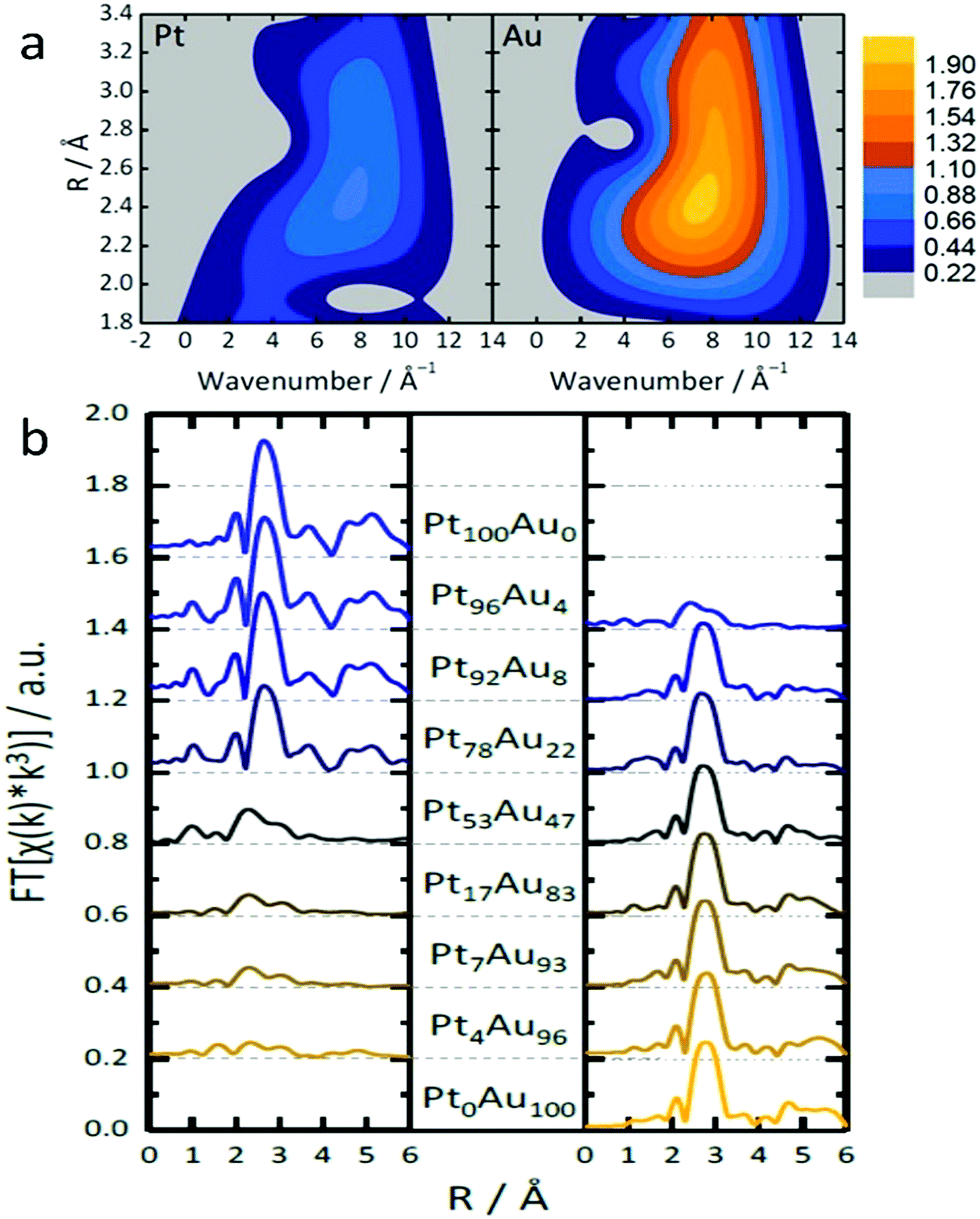 | ||
| Fig. 8 (a) Topographical maps of wavelet transform-EXAFS (WT-EXAFS) spectra obtained from the Pt and Au L3-edges of Pt4Au96. (b) The complete set of FT-EXAFS spectra (left: Pt, right: Au) for the nanoparticles so prepared. Adapted with permission from ref. 68. Copyright 2018 Springer Nature. | ||
An example in this regard is shown in the work by Yao et al., in which they conducted both ex situ and in situ XAS measurements to fully analyse the atomic structure of the Ru1–Pt3Cu SAA.58 Initially, they carried out traditional ex situ XAS analysis. The ex situ Ru K-edge XAS spectra for Ru1–Pt3Cu was shown to be situated between Ru foil and bulk RuO2. This result indicated that excess oxidation was prevented at the surface exposed Ru1 sites. In addition, the FT-EXAFS spectra for Ru1–Pt3Cu showed three peaks: the first peak at 1.58 Å was attributed to Ru–O scattering, while two peaks at 2.30 Å and 2.73 Å were deemed to be due to Ru–Pt and Ru–Cu bonding, respectively. Furthermore, since Ru–Ru bonds were not observed at 2.38 Å, no metallic Ru clusters existed in the sample. Similarly, the lack of Ru–O–Ru coordination at higher distances eliminated the possibility of RuO2 particles in the sample. These results further supported the presence of single-atom Ru species located within the core–shell Pt3Cu/Pt-skin particles.
In order to study the oxidation of Ru1–Pt3Cu as a function of potential during the oxygen evolution reaction (OER), in situ electrochemical Ru K-edge XAS analysis was performed.58 As the potential was increased from 0 V to 1.86 V, the oxidation state of Ru1–Pt3Cu remained fairly constant. This observation was shown by the similar absorption edge positions (Fig. 9a) and coordination (Fig. 9b) for the Ru K-edge throughout the scan. These results proved that, throughout the OER, the Ru single atoms remained stable. In addition, in situ electrochemical Pt L3-edge XAS was performed. As seen in Fig. 9c, an increase in the Pt L3-edge white line intensity suggested an increase in Pt d band vacancies. The increase in d band vacancies was suggested to be due to electron transfer from Pt to Ru1.
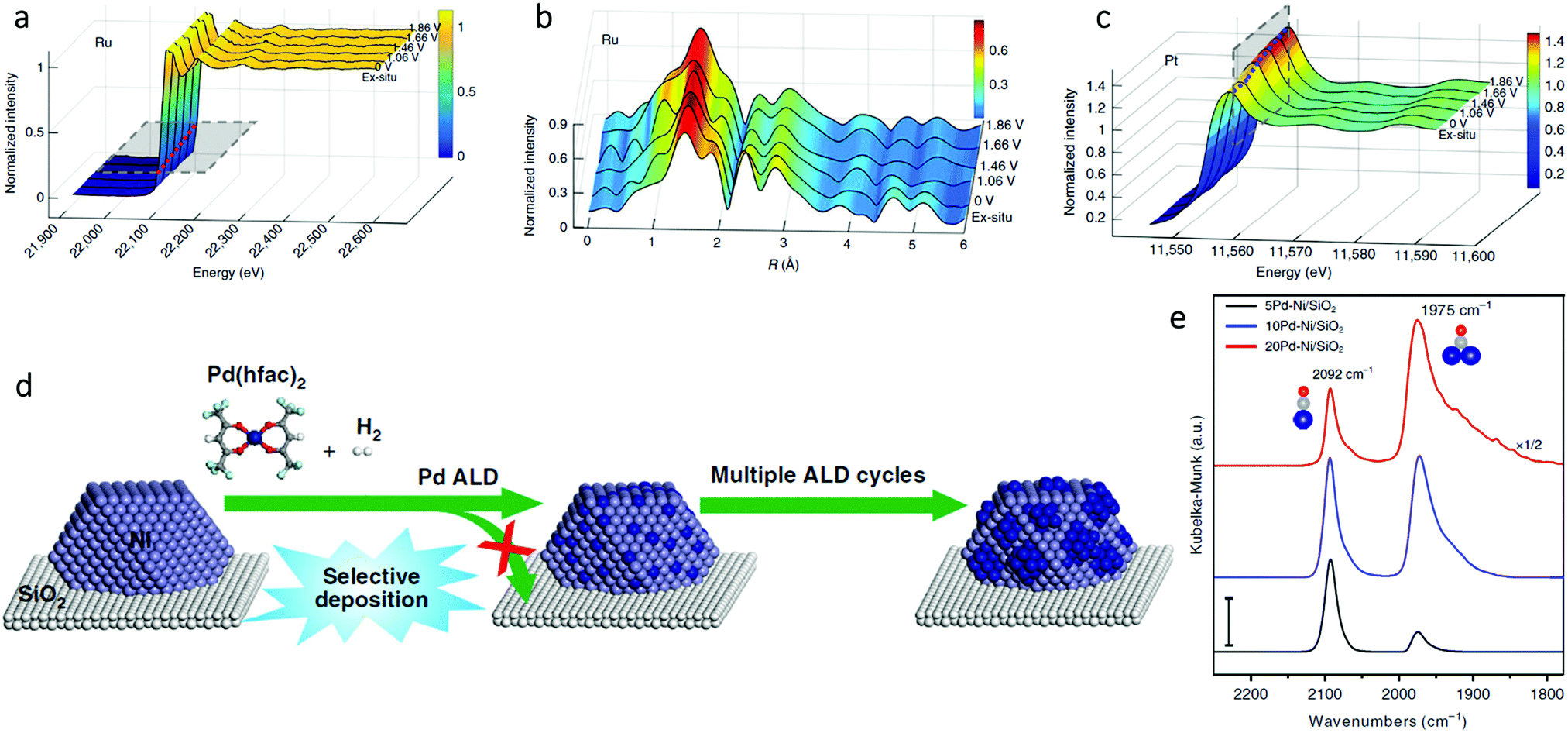 | ||
| Fig. 9 In situ (a) X-ray absorption and (b) FT-EXAFS spectra change from 0 to 1.86 V potential of the Ru K-edge. (c) In situ XAFS spectra change of the Pt L3-edge. Adapted with permission from ref. 58. Copyright 2019 Springer Nature. (d) Schematic illustration of the synthesis of xPd–Ni/SiO2 bimetallic catalysts using selective Pd ALD. (e) DRIFTS CO chemisorption of the xPd–Ni/SiO2 samples (x = 5, 10, and 20) at the CO saturation coverage. The dark blue, gray, and red balls are Pd, C, and O atoms, respectively. Scale bar: 0.01. Adapted with permission from ref. 54. Copyright 2019 Springer Nature. | ||
In addition to quantitative in situ XAS techniques, in situ infrared (IR) spectroscopy also plays a complementary role in the qualitative structural analysis of SAAs. With the appropriate probe molecule (usually CO or NO), IR offers a fast and convenient characterization method to directly identify single atoms from nanoparticles.75 For example, Wang et al. performed in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) CO chemisorption measurements to determine the surface structure of PdNi SAAs (Fig. 9d).54 As shown in Fig. 9e, on 5Pd–Ni/SiO2, the dominant peak at 2092 cm−1 (due to linear CO on Pd), was more than the peak at 1975 cm−1 (due to bridge-bonded CO). Therefore, Pd was present as a single atom within Ni. However, as the number of Pd atomic layer deposition (ALD) cycles increased, the peak associated with bridge-bonded CO considerably strengthened; thus, the existence of large Pd ensembles or a continuous Pd shell on the Ni NP surfaces was proven. The SAA structure of Pd1Ni not only maximized the Pd utilization, but also modified the strong metal-selectivity relations in the benzonitrile hydrogenation reaction.
3. Electronic properties of SAAs
The electronic properties of SAAs are frequently overlooked in their analysis. However, they often play a crucial role in understanding the catalytic performance of SAAs. A remarkable example has been shown in the work by Greiner et al. in which the authors observed that the Cu d states of AgCu SAAs were like the free-atom state and thus resulted in significantly different catalytic activity from the monometallic Cu catalysts.91 While the electronic behaviour of single atoms in SAAs is normally very complicated, convincing conclusions sometimes cannot be obtained solely using experimental characterization methods. Considering this statement, the comprehensive data of XANES,58,59,61–63,68,69,72,73,75,79,83,88,89 X-ray photoelectron spectroscopy (XPS)46,49,51,52,54,58–60,67–69,76,79,82,83,85,91 and theoretical models obtained from computational chemistry48,53,57,58,65,68,75,79,81–83,85,92–101 can help reveal the electronic properties of SAAs.An example of the examination of the electronic properties of SAAs is shown by Kim et al., who reported the electronic properties of Pt atoms in PtSn SAAs supported by antimony-doped tin oxides (Pt1/ATO) using XANES and XPS.59 As shown in Fig. 10a, when compared to other Pt samples, the location of the white line peak of the Pt1/ATO XANES was shifted to higher energy. Meanwhile, the Pt 4f XPS peak of Pt1/ATO was shown to have a higher binding energy than the other Pt samples as seen in Fig. 10b. These results suggested that, as seen for other Pt alloys,102,103 electron transfer occurred between Pt and the ATO support.
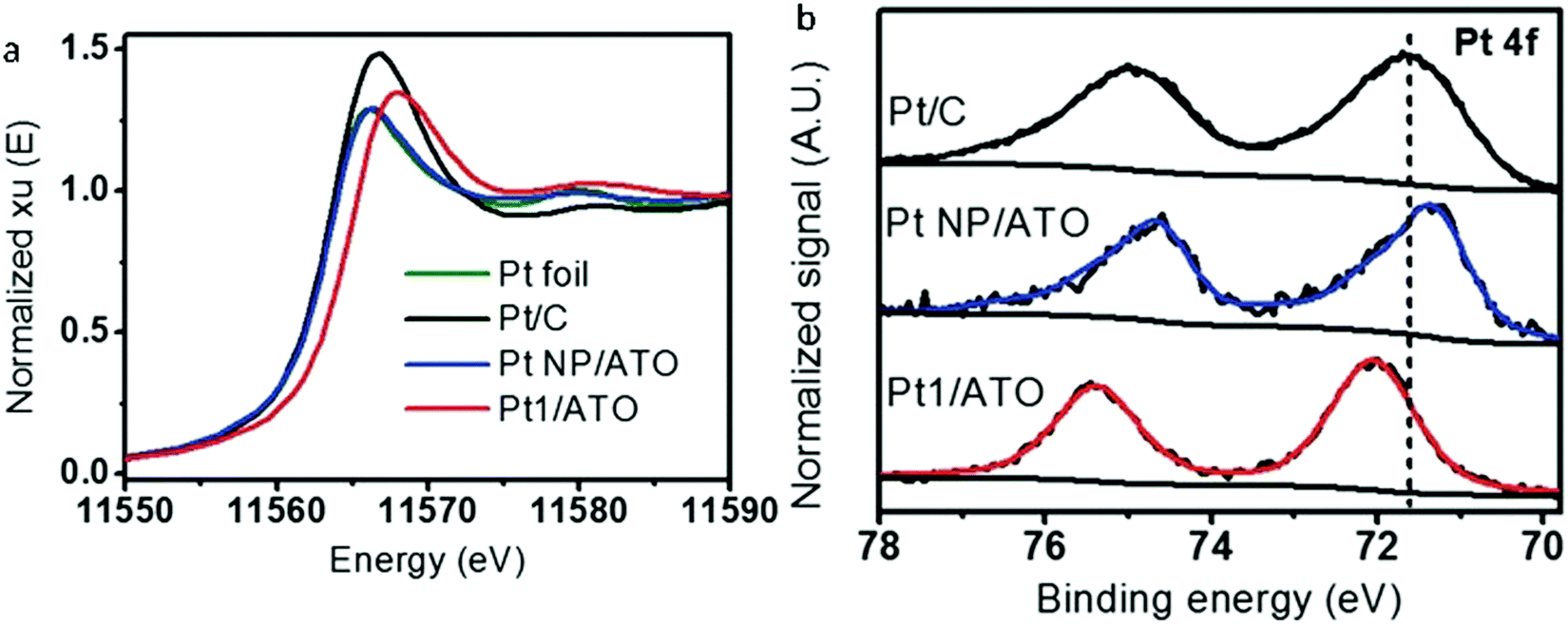 | ||
| Fig. 10 (a) XANES Pt-L3 edge and (b) XPS Pt 4f spectra for Pt foil, reduced Pt/C, Pt NPs/ATO, and Pt1/ATO samples. Commercial Pt/C was reduced at 100 °C for 1 h prior to the measurements. Adapted with permission from ref. 59. Copyright 2017 Wiley Materials. | ||
As with standard structural studies, in situ studies can be used to investigate the electronic properties of SAAs. For instance, Pei et al. used XANES and in situ XPS to investigate the electronic properties of surface Ag and Pd in AgPd SAAs.69 Firstly, as presented in the Pd K-edge XANES spectra in Fig. 11a, palladium was found to be zero valent within both SAAs at 400 °C. However, compared to both Pd foil and Pd0.01/SiO2, AgPd0.01/SiO2 showed an absorption edge shifted to lower energy. The authors suggested that this corresponds to a slightly lower oxidation charge of anionic palladium in the SAA with charge transfer occurring between silver and palladium.104 This observation was also in agreement with in situ Ag 3d5/2 and Pd 3d5/2 XPS measurements for AgPd/SiO2 at various temperatures (Fig. 11b–g), which showed that the binding energy was lowered as the temperature was increased. As a result, the enhancement in ethylene selectivity with pre-reduction temperature could be attributed to charge transfer.105,106
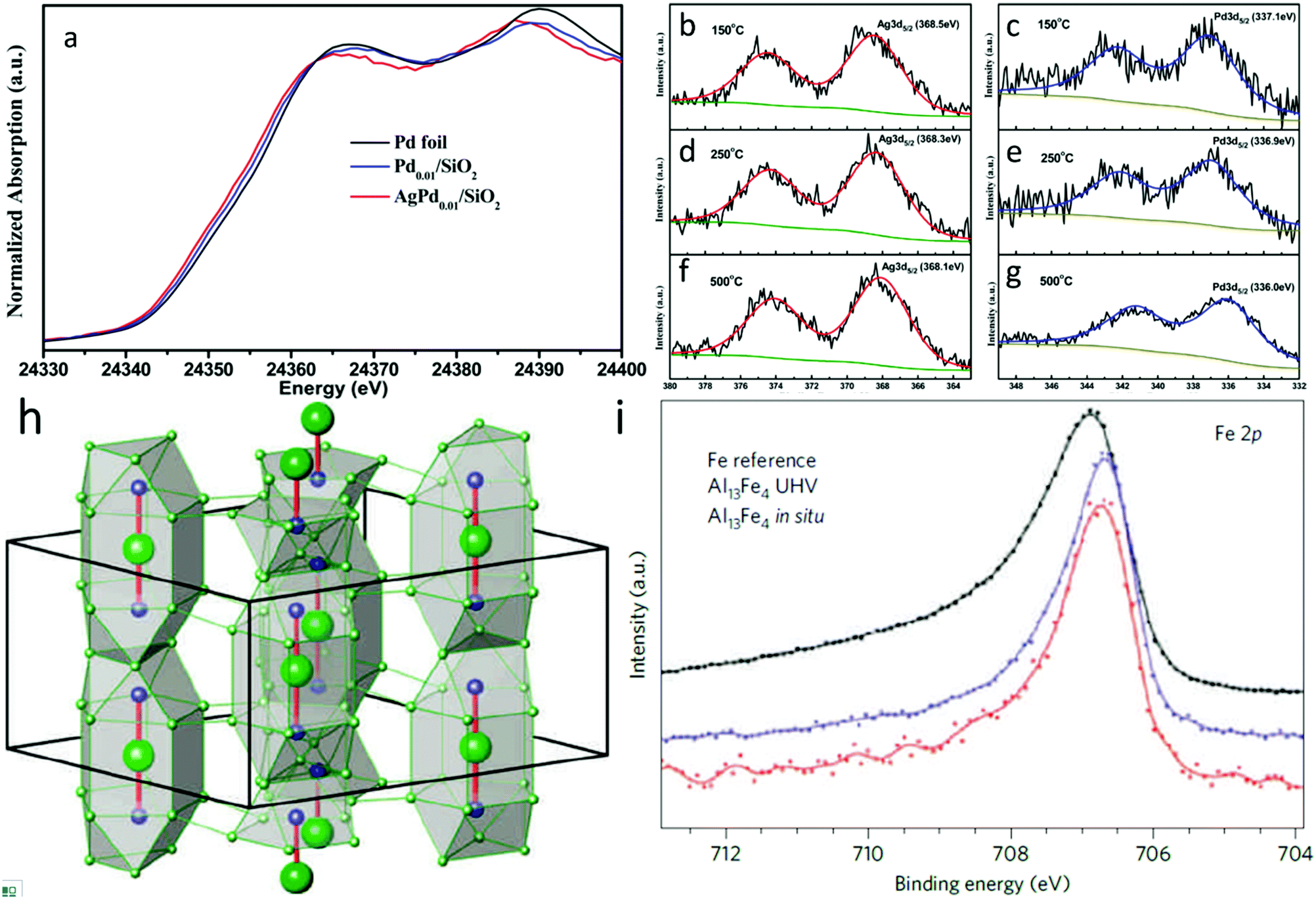 | ||
| Fig. 11 (a) Normalized Pd K-edge XANES spectra of Pd0.01/SiO2 and AgPd0.01/SiO2 after 400 °C prereduction. A Pd foil reference spectrum is shown for comparison. (b), (d), (f) Ag 3d and (c), (e), (g) Pd 3d XPS spectra of Ag1Pd1/SiO2 catalyst reduced in situ at 150 °C, 250 °C, and 500 °C. Adapted with permission from ref. 69. Copyright 2015 American Chemical Society. (h) Unit cell of Al13Fe4 emphasizing the structurally isolated Fe–Al–Fe units (Al, green; Fe, blue). (i) Fe 2p XPS spectra of the single-crystalline (010) surface of Al13Fe4 and elemental Fe foil as a reference with 860 and 900 eV photon energy, respectively. Adapted with permission from ref. 107. Copyright 2012 Springer Nature. | ||
As a special type of SAAs, IMC-based SAA catalysts normally possess a well-defined and uniform local coordination environment that makes their electronic properties significantly different from those of the constituents. Thus, a thorough study into the electronic properties of IMC-based SAAs can help researchers to unambiguously establish the catalytic role of such materials. For example, Armbrüster et al. revealed the electronic properties of the Al13Fe4 (Fig. 11h) SAA through XPS study.107 As shown in Fig. 11i (Fe 2p core-level XPS spectra), the small binding energy shift and smaller half-width at full maximum for the signal of Al13Fe4 is different than elemental iron, which removes the possibility of additional Fe nanoparticles being present (whose spectrum would have had a higher energy and broader Fe 2p peak). These differences are common for intermetallic materials consisting of isolated transition-metal atoms evenly distributed in either other transition metals or main group elements (such as for Pd in PdGa108–110 or PdIn111). They can be related to changes in the electronic properties of the materials, which provides unique core hole screening. Consequently, such low-cost noble-metal-free SAA catalysts exhibit excellent catalytic properties in the semi-hydrogenation of acetylene, which can be assigned to the combination of site-isolation and the alteration of the electronic structure resulting from its chemical bonding.
In addition, XPS can be used as a complement to calculated atomic models. To shed light on the unique electronic properties of RuAu SAAs, Chen et al. conducted XPS measurements on RuAu SAAs and compared these results with their theoretical atomic models.84 As shown in Fig. 12a, the Ru 3d5/2 peak of RuAu-0.2, which is located at 280.7 eV, increased in binding energy by 0.18 eV compared to pure Ru.112,113 In contrast to the Ru 3d5/2 peak, the Au 4f peak (Fig. 12b) shifted to a lower binding energy by 0.2 eV with respect to pure Au.112 The contrast in binding energy shifts was attributed to electron transfer between Au and Ru. This assessment was confirmed by the Bader charge analysis represented by the atomic models in Fig. 12c and d. As seen in Fig. 12e and f, the peak shift of RuAu-0.2 was greater than those of RuAu-0.3 and RuAu-0.5. This result suggested that, in SAAs with higher amounts of Au, phase separation of Au from the Ru host occurred. Changes in d-band filling resulted in changes in the electronic properties for both Au and Ru. For Ru, the d-band filling decreased since electrons were lost. Since electrons were gained from Ru, the d-band filling increased for Au. By applying d-band centre theory, these changes could affect hydrogen absorption.114,115
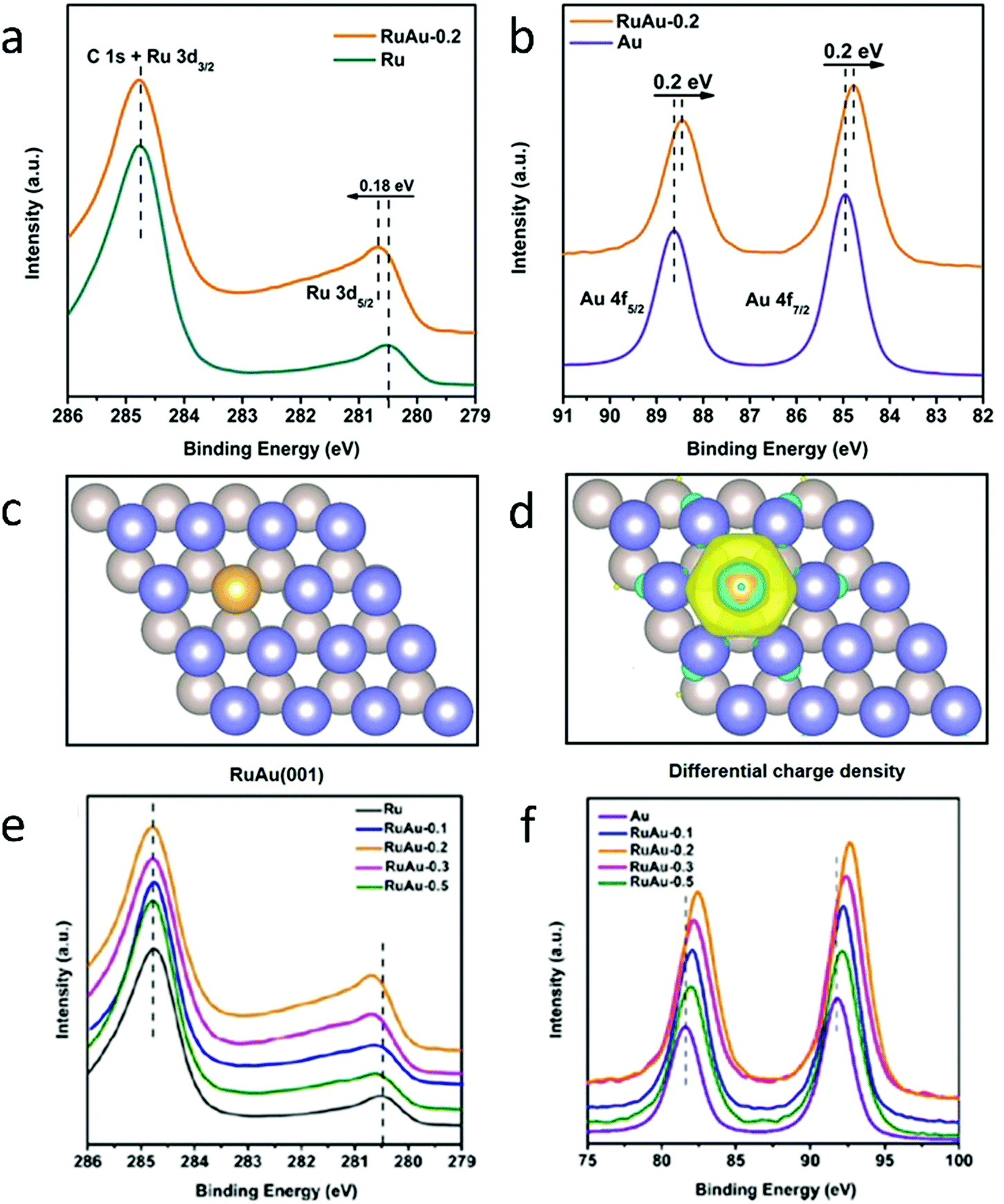 | ||
| Fig. 12 Charge redistribution in RuAu alloy. (a) Ru 3d XPS spectra of Ru and RuAu-0.2. (b) Au 4f XPS spectra of Au and RuAu-0.2. (c) Atomic model of the RuAu (001) surface; the yellow ball represents the Au atom and deep blue and silver grey balls indicate the top and bottom layers of Ru, respectively. (d) Differential charge densities of RuAu (001) surface. Yellow and blue contours represent electron accumulation and depletion, respectively. (e) Ru 3d and (f) Au 4f XPS spectra of the metal reference, RuAu-0.1, RuAu-0.2, RuAu-0.3, and RuAu-0.5. Adapted with permission from ref. 84. Copyright 2019 Wiley Materials. | ||
XPS can also be used in combination with density of states (DOS) calculated using the density functional theory (DFT) method to determine the electronic properties of SAAs. For example, Au-supported Pt SAAs having sensitive surface structures were analysed by Duchesne et al. using the comprehensive data from DOS calculations as well as varied excitation energies.68 As shown by the DFT DOS results in Fig. 13a, the intensity near the Fermi level (0 eV) for the single-atomic-site Pt surface was lower than that of the pure Pt surface. As seen in the XPS valence band measurements (Fig. 13b–e), the near-Fermi level DOS (−2 to 0 eV) was sensitive to the local Pt environment. Both Pt4Au96 and Pt7Au93, the samples having Pt single atoms, showed the lowest DOS intensity between −2 and 0 eV since they were like pure Au. However, the DOS intensity near the Fermi level was higher for Pt17Au83, which contained few-atom Pt clusters. Furthermore, Pt53Au47 showed the highest near-Fermi level DOS with its intensity approaching that of pure Pt, which suggested that a more complete Pt shell was formed at the surface of the Au NP. The catalytic performance of the PtAu SAAs was also affected by their valence band structure. Due to the very low d-electron density for the Pt SAA samples near the Fermi level, the normally strong Pt–CO interaction induced by the so-called donation-and-back-donation bonding mechanism was significantly weakened, as the Pt d-electron density participating in π back-donation to CO molecules was dramatically reduced. Thus, the CO adsorption energies on these surfaces were lowered.
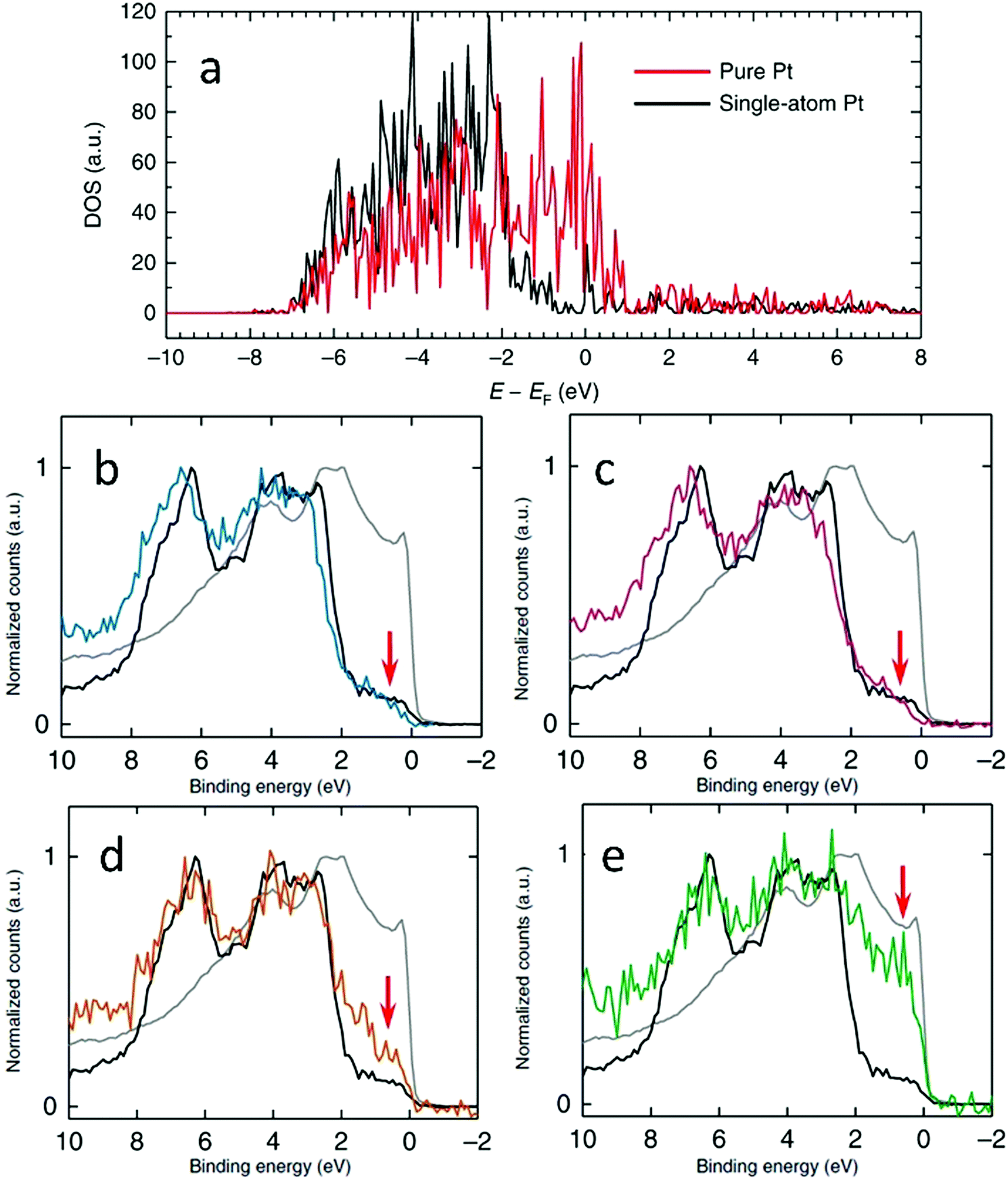 | ||
| Fig. 13 (a) Calculated DFT DOS plots that correspond to the pure and single-atom Pt surfaces used to calculate the CO adsorption energies. (b)–(e) XPS valence band spectra of Pt4Au96 (b), Pt7Au93 (c), Pt17Au83 (d) and Pt53Au47 (e) samples compared with those of Pt (grey) and Au (block) foil references. The red arrows indicate the near-Fermi level region. Reprinted with permission from ref. 68. Copyright 2018 Springer Nature. | ||
Another example of the use of DOS calculations is the work done by Yao et al., who computed the projected density of states (PDOS) of Ru1 (Fig. 14a) in order to determine the electronic origin of the reactivity of atomically dispersed Ru1.58 It was determined that, as the Pt to Cu ratio increased (PtCu3 < PtCu < Pt3Cu < pure Pt), the PDOS of Ru1 approached the Fermi level. As shown in Fig. 14b, the d-band centre for Ru1 (εRu-d) shifted from −3.37 eV to −2.76 eV, which was suggested to be due to the slow release of compressive surface strain. Consequently, the oxygen adsorption energy (EO) was strengthened from −1.63 eV for weakly binding pure Cu to −2.48 eV for strongly binding pure Pt. This strengthening led to an inverse plot for the OER overpotential as a function of lattice constant shown in Fig. 14c. These results provided an explanation for the experimental findings in Fig. 9.
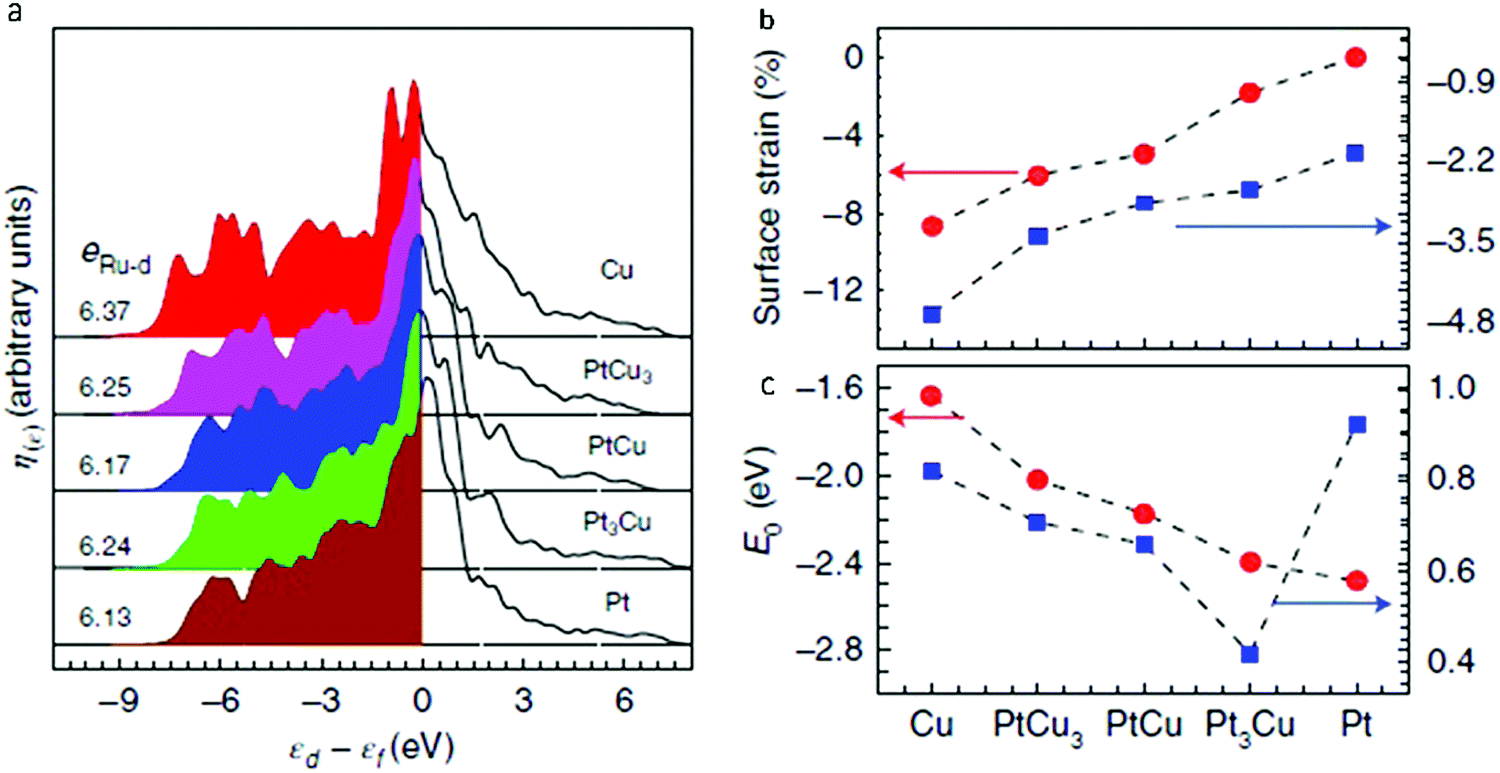 | ||
| Fig. 14 (a) PDOS of surface-embedded Ru1 4d with respect to the Fermi level, where eRu-d is total 4d electrons of the Ru1 atom. (b) In-plane lattice contraction relative to the Pt(111) pristine surface (red circles) and corresponding d-band centre εRu-d of Ru1 (blue squares). (c) Corresponding adsorption energy EO of the oxygen atoms (red circles) and OER overpotential, η (blue squares). Adapted with permission from ref. 58. Copyright 2019 Springer Nature. | ||
4. Catalytic activities of SAAs
Due to their unique structural and electronic properties, SAAs have shown outstanding performance in various catalytic reactions.43–47,49,50,52–70,72–83,85,86,88,89,91,94–97,99–102,116–125 By establishing the structure-performance relationship of SAAs in different reactions, the rational design of more efficient SAAs can be performed to meet specific reaction requirements. In this section, some representative catalytic reactions are selected to illustrate the unique structure–property relationship of SAAs.4.1 Oxidation reactions
The first class of reactions that have commonly been studied with SAAs is oxidation reactions. Among the wide variety of oxidation reactions that have been studied, the formic acid oxidation (FAO) reaction and the glucose oxidation reaction are used as examples for our discussion.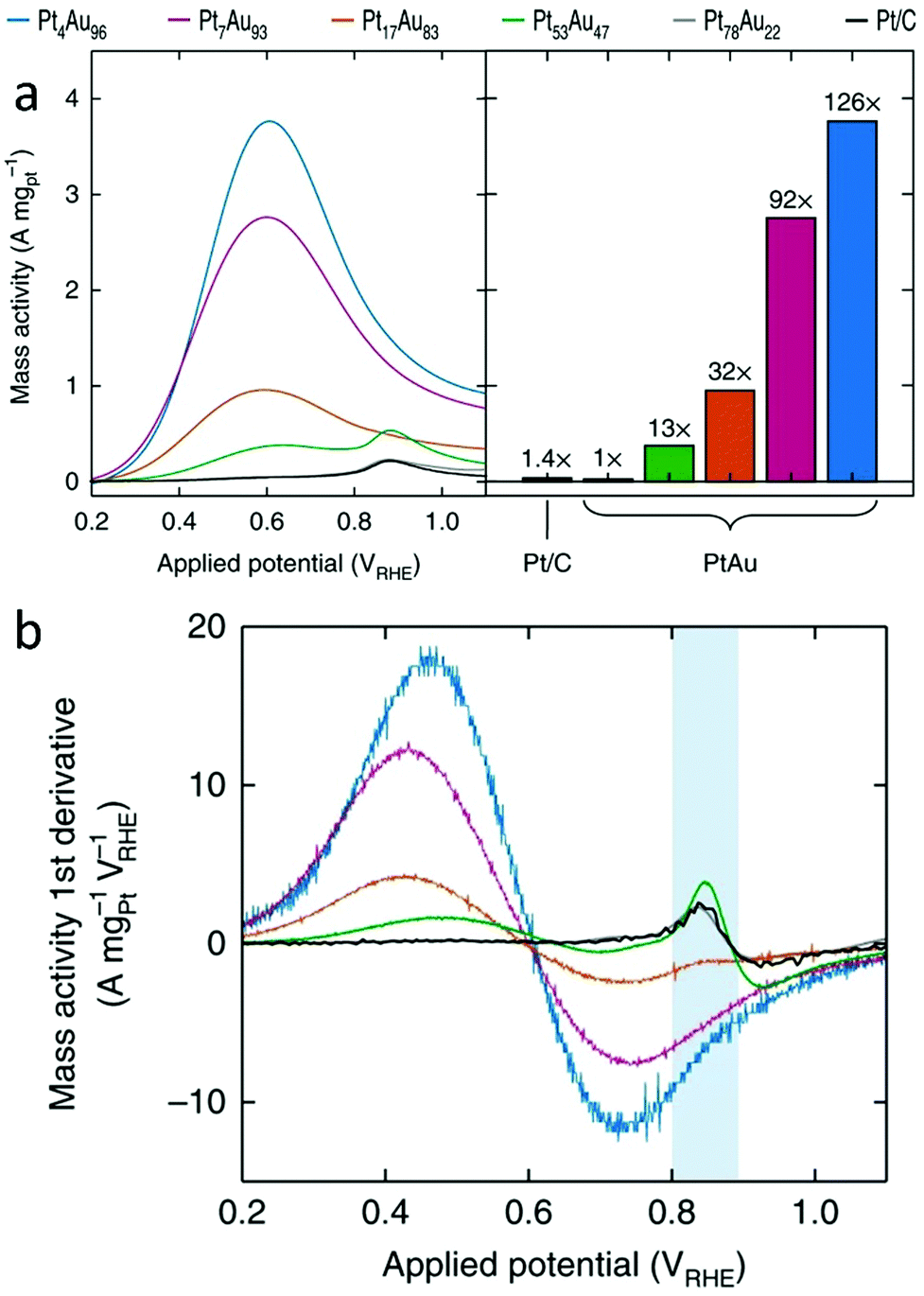 | ||
| Fig. 15 (a) Pt mass-normalized anodic sweeps obtained from PtAu NP catalysts, with the peak currents graphed for comparison (left). (b) The first derivative of the Pt mass-normalized FAO voltammograms. The blue area highlights the sensitivity of the first-derivative peak at ∼0.85 VRHE to the Pt surface structure. Adapted with permission from ref. 68. Copyright 2018 Springer Nature. | ||
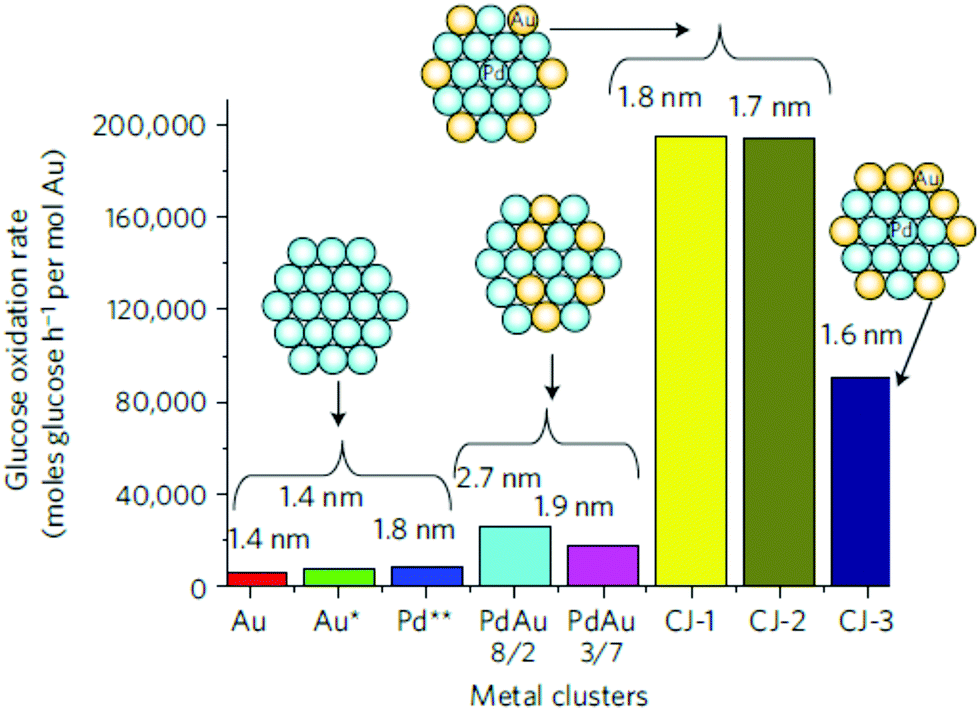 | ||
| Fig. 16 Comparison of the catalytic activity of CJ-Au/Pd, Au, Pd and Pd/Au alloy NCs for aerobic glucose oxidation. Schematic insets and numbers shown at the top of each bar indicate the structure models and the average particle sizes, respectively, of the NCs; Au*, the activity was normalized by the number of surface Au atoms in NCs; Pd**, the activity was normalized by the number of surface Pd atoms. Reprinted with permission from ref. 83. Copyright 2011 Springer Nature. | ||
4.2 Selective hydrogenation reactions
Besides oxidation reactions, another class of frequently studied reactions with SAAs is selective hydrogenation reactions. This type of reaction can involve diluting Pt single atoms in Cu NPs, which offers a practical method for reducing the amount of Pt in SAAs while enhancing catalyst selectivity for partial hydrogenation reactions.47,63,67,79,81,92,100,117 An example of the use of SAAs in selective hydrogenation reactions is the work by Lucci et al., who reported the synthesis of PtCu SAAs by using galvanic replacement.79 It was determined that single Pt atoms could enhance selective hydrogenation of 1,3-butadiene on a less active metal, such as Cu. As shown in Fig. 17a, the Pt0.1Cu14/Al2O3 SAA had a hydrogenation reaction onset at 40 °C. This temperature was 35 °C lower than that for the Cu catalyst without Pt. In addition, the hydrogenation reaction rate over Pt0.1Cu14/Al2O3 was roughly ten times greater than the monometallic Cu catalyst at 60 °C, while the Pt0.2Cu12/Al2O3 SAA showed an even higher hydrogenation rate than Pt0.1Cu14/Al2O3. This difference was attributed to a higher amount of surface Pt in Pt0.2Cu12/Al2O3 at a higher Pt loading. Despite the increase in catalytic activity, the selectivity of Pt0.2Cu12/Al2O3 was retained due to the isolation of Pt atoms. Furthermore, the PtCu SAAs showed high selectivity to butenes; 90% selectivity was achieved towards butene isomers. This high selectivity was comparable to that of Cu15/Al2O3. In contrast, pure Pt catalysts converted butadiene to butane, which was attributed to pure Pt over-hydrogenating dienes and alkynes.126,127 Finally, in order to test the hydrogenation ability of the Pt/Cu SAAs, the selective hydrogenation activity of Pt0.1Cu14/Al2O3 was examined upon the addition of excess propylene. As seen in Fig. 17b, the Pt/Cu SAAs showed both high stability and high selectivity under such a condition. Therefore, this work demonstrated that, by the rational design of the single-atom structure, the Pt/Cu SAAs can offer promising selective hydrogenation properties in addition to a significant reduction in the use of the costly element Pt.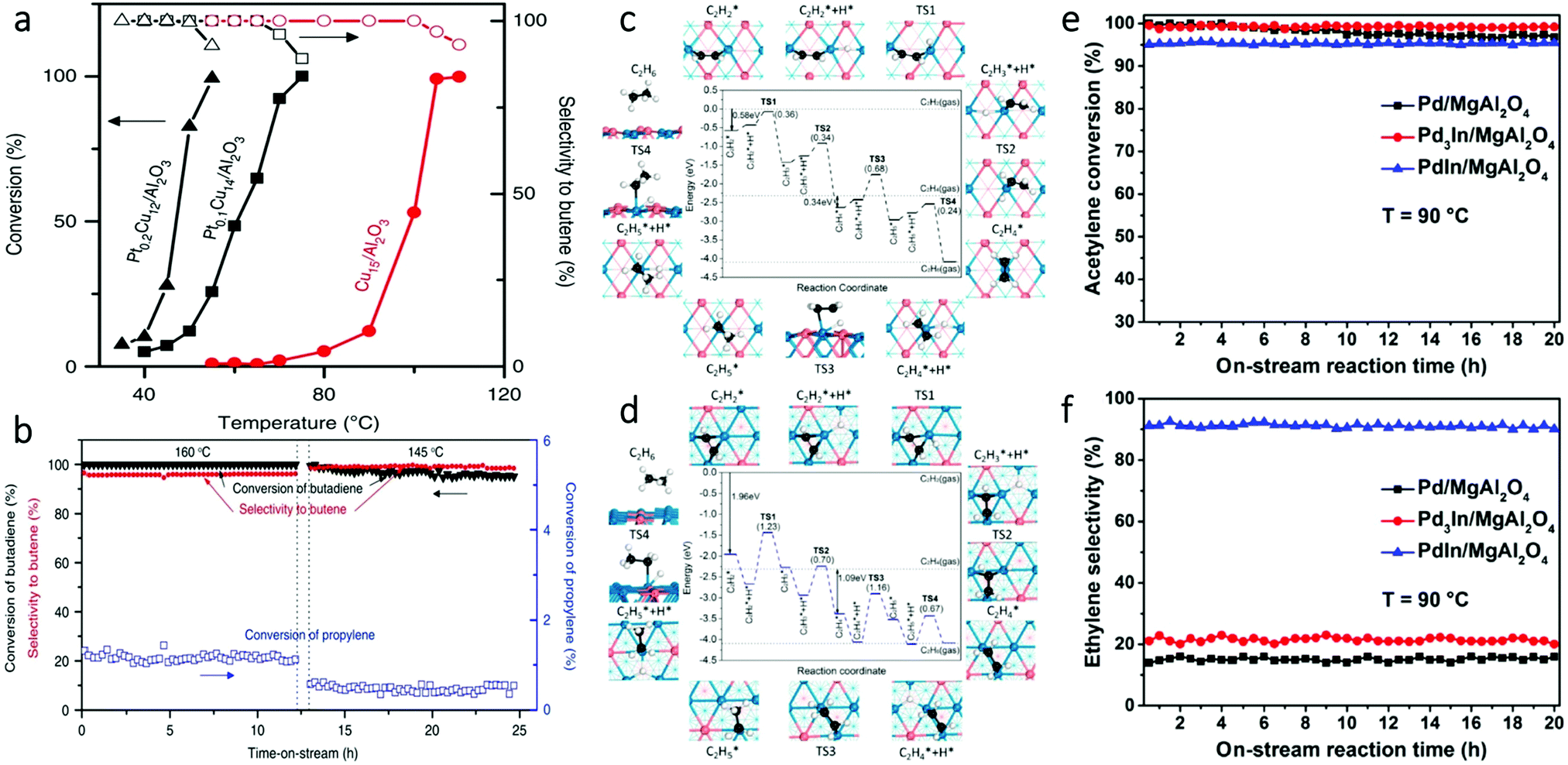 | ||
| Fig. 17 (a) Selective hydrogenation of butadiene. Hydrogenation shown as a function of temperature over Cu15/Al2O3, Pt0.1Cu14/Al2O3 and Pt0.2Cu12/Al2O3 NPs. (b) Butadiene conversion in the presence of excess propylene. Adapted with permission from ref. 79. Copyright 2015 Springer Nature. (c) Hydrogenation mechanism of acetylene to ethane on the PdIn(110) surface. (d) Hydrogenation mechanism of acetylene to ethane on the Pd3In(111) surface. In (c) and (d), numbers in the parentheses indicate the barriers of elementary steps; Pd, blue; In, orange; C, black; H, white. (e) Acetylene conversion and (f) ethylene selectivity in long-term selective hydrogenation of acetylene at 90 °C on the three as-obtained catalysts. Adapted with permission from ref. 111. Copyright 2017 American Chemical Society. | ||
Intermetallic SAAs have also been used in selective hydrogenation reactions. An example is the work done by Feng et al. who used theoretical studies to guide the synthesis and catalysis of PdIn intermetallic SAAs.111 DFT modelling showed that the PdIn (110) surface containing single Pd sites had high selectivity for the semi-hydrogenation of acetylene. In contrast, the Pd3In (111) surface having Pd trimer sites exhibited low selectivity (Fig. 17c and d). Guided by the theoretical predictions, experimental results further demonstrated that intermetallic PdIn NPs with a (110) surface structure (92%) showed greater selectivity for acetylene hydrogenation than Pd3In NPs with a (111) surface structure (21%) as seen in Fig. 17e and f. These findings illustrate an interesting approach for the rational design of SAA catalysts with specific catalytic properties.
4.3 Selective dehydrogenation reactions
SAAs have also been studied as catalysts for selective dehydrogenation reactions. Among various metals tested, Cu-based SAAs have been found to be particularly active in various dehydrogenation reactions, which can be attributed to the presence of isolated metal single atoms such as Ni, Pd and Pt, which serve as entrance and exit routes for hydrogen dissociation and re-combination.46,49,57,61,75,119,120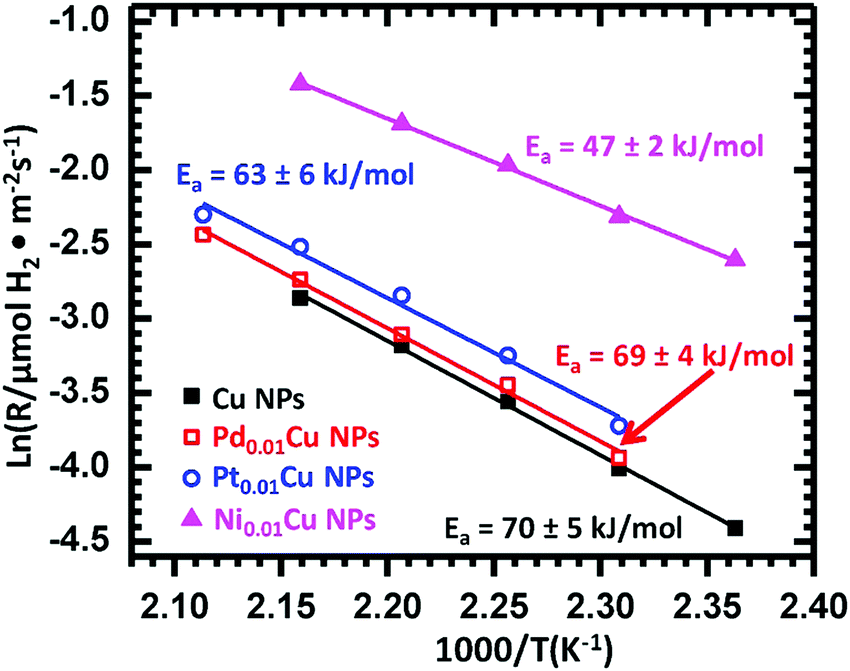 | ||
| Fig. 18 Arrhenius-type plots of the reaction rate in ethanol dehydrogenation, normalized by the surface area of Cu over monometallic Cu NPs, Pd0.01Cu NPs, Pt0.01Cu NPs, and Ni0.01Cu NPs. Reprinted from ref. 61, Copyright (2018), with permission from Elsevier. | ||
To increase both selectivity and catalytic activity during the dehydrogenation of propane, Sun et al. synthesized PtCu SAAs supported on γ-alumina using atomic dilution co-impregation.57 DFT calculations revealed that, during the dehydrogenation of propane, the PtCu scaling relationship was broken. This suggestion was supported by the fact that the reaction had high propylene selectivity while only incurring a slight increase in the activation energy. As shown in Fig. 19a, the initial propylene formation rate was 3.2 times greater for the 0.1Pt10Cu/Al2O3 SAA than for the monometallic 0.1Pt/Al2O3. Meanwhile, propylene selectivity and conversion were both significantly higher for 0.1Pt10Cu/Al2O3 than for 0.1Pt/Al2O3. In addition, DFT calculations were used to determine the deactivation rate for propane dehydrogenation. It was found that using 0.1Pt10Cu/Al2O3 resulted in a far lower deactivation rate than when 0.1Pt/Al2O3 was used. Furthermore, the dehydrogenation reaction temperature was increased from 520 °C to 550 °C. As shown in Fig. 19a, 0.1Pt10Cu/Al2O3 still showed higher activity and a lower deactivation rate than when using 0.1Pt/Al2O3. Finally, the performance of 0.1Pt10Cu/Al2O3 was examined for successive oxidation–reduction cycles. After five cycles, there was no change in either the selectivity or the conversion of the SAA (Fig. 19b). This result proved its good regeneration ability and high stability after propane dehydrogenation. As seen in Fig. 19c, upon regeneration of the SAA, most of its initial activity was restored. This result provided additional evidence for the cycling stability of 0.1Pt10Cu/Al2O3.
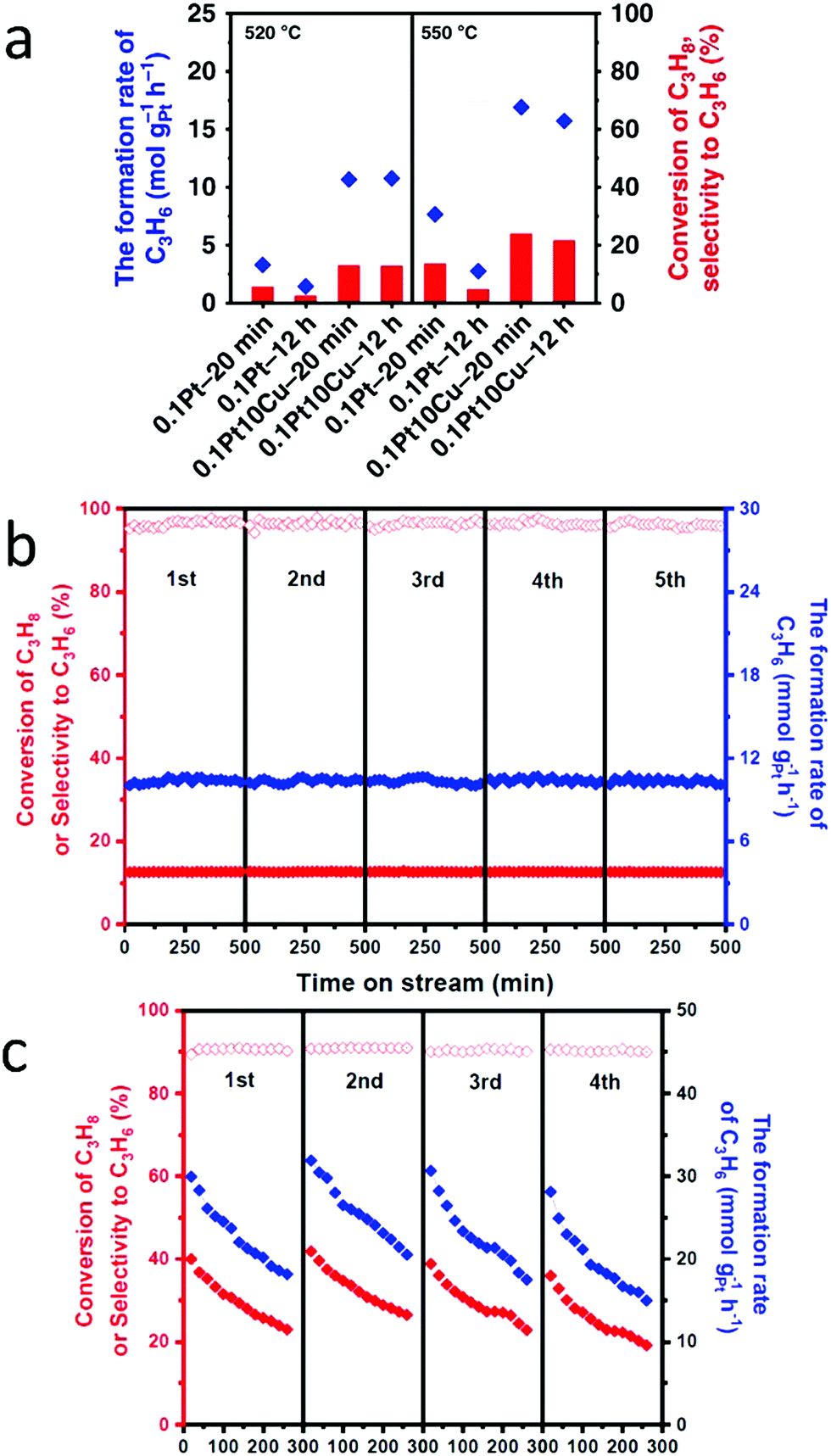 | ||
| Fig. 19 (a) Catalytic performances at the initial and final period at 520 °C and 550 °C during 12 h PDH, showing significantly improved performances of Pt/Cu SAA compared with Pt nanoparticles. (b) Catalytic performances on the 0.1Pt10Cu/Al2O3 catalyst for each of the five successive dehydrogenation cycles. (c) Catalytic performances on the 0.1Pt10Cu/Al2O3 catalyst for each of the four successive dehydrogenation cycles. Adapted with permission from ref. 57. Copyright 2018 Springer Nature. | ||
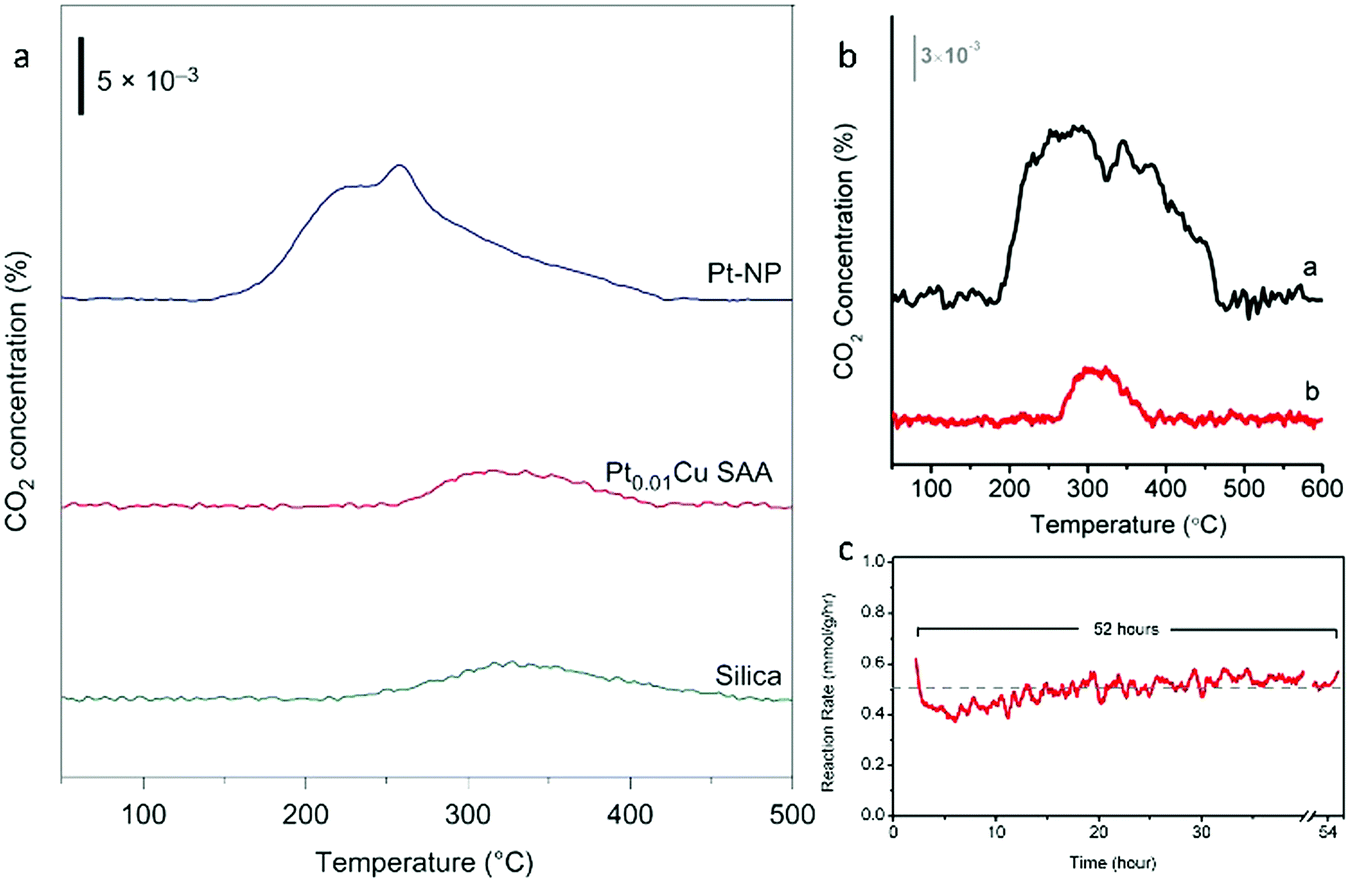 | ||
| Fig. 20 (a) TPO of Pt–NP (blue), Pt0.01Cu SAA (red) and silica (green). (b) Comparing the degree of coking in Pt NP vs. PtCu SAA catalysts. TPO of (a) Pt–NP and (b) Pt0.01Cu-SAA. (a) and (b) are spent catalysts after butane dehydrogenation reaction. (c) Long-term butane dehydrogenation reaction over Pt0.01Cu-SAA catalyst. Adapted from ref. 70. Copyright 2018 Springer Nature. | ||
As a solution to the coking problem, Marcinkowski et al. studied Pt/Cu SAAs in order to maximize C–H activation.70 To determine whether coking and carbon deposition due to butane-deuterium isotope scrambling occurred in the catalysts, temperature-programmed oxidation (TPO) studies were conducted. For the Pt NPs, a broad peak due to CO2 production was observed and peaks corresponding to two different types of carbon were present (Fig. 20a). However, for the Pt0.01Cu SAA, these peaks were not seen, and the amount of CO2 evolved resembled that of the SiO2 support. Therefore, it was concluded that the Pt/Cu SAAs were coke resistant. In addition, the non-oxidative dehydrogenation of butane to butene was performed to determine the coke resistance of the Pt/Cu SAAs. Minimal carbon deposition was observed for the Pt0.01Cu SAA (Fig. 20b), while the activity of the SAAs remained stable over 52 h (Fig. 20c). These results illustrated that the Pt/Cu SAA strategy is an excellent approach for the development of low-cost, high-stability and coke-resistant C–H activation catalysts.
5. Conclusions and outlook
In summary, single-atom alloy (SAA) catalysts containing an atomically dispersed metal in a bi- or multi-metallic complex have emerged as a promising system for heterogeneous catalysis. This review summarizes the recent progress made in the structural analysis, electronic properties and catalytic activities of SAA nanoparticle catalysts.The atomic structure of SAAs has been examined on catalysts such as AuPd, PdCu, PtCu, PtSn, RuAu, AgPd, NiCu and PtAu. High-angle annular dark field imaging-scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray (EDX) spectroscopy have been used to directly image single atoms in SAAs; they are very successful for the analysis of distinguishable SAAs. X-ray absorption spectroscopy (XAS), including multi-element extended X-ray absorption fine structure (EXAFS) fitting analysis and other associated techniques, has been used to obtain detailed local structural information of SAAs, such as bond distances, due to its element-specific feature and high-precision (approximately 0.01 Å scale). It is particularly useful for the analysis of indistinguishable SAAs, where it is difficult to distinguish elements having very similar atomic numbers using HAADF-STEM. Additionally, widely available in situ XAS and infrared (IR) spectroscopy techniques have been used to determine the atomic structure of SAAs during real reactions. Structural analyses of SAAs, particularly in situ techniques, are very helpful in understanding the detailed atomic structure of SAAs during catalytic reactions.
While the electronic properties of SAAs are often overlooked when studying SAAs, the detailed information obtained by combining spectroscopy experiments (for example, X-ray absorption near-edge structure (XANES) and X-ray photoelectron spectroscopy (XPS)) and density functional theory (DFT) modelling has been useful in elucidating the electronic properties of SAAs. Details revealed by XANES and XPS on the valence band and charge transfer in SAAs can be of great assistance in understanding their catalytic properties. In addition to these ex situ tools, a variety of in situ spectroscopy methods (such as XANES, XPS and IR) can provide additional information about the important role of electronic properties in determining the reactivities of SAAs during dynamic catalytic processes. Therefore, these techniques will be very helpful in the rational design and fabrication of high-performance SAA catalysts.
A few representative catalytic reactions involving SAAs have been selected to illustrate the structure–property relationships of SAA catalysts. The role of SAAs in oxidation reactions, such as the formic acid oxidation reaction and the glucose oxidation reaction, has been investigated due to their importance in fuel cells and in technology. In addition, SAAs have been used in selective hydrogenation reactions in order to improve metal efficiency as well as increase their catalytic activity and selectivity. Furthermore, the use of SAAs as catalysts in selective dehydrogenation reactions, such as in the dehydrogenation of ethanol, propane and butane, can help generate products such as acetaldehyde and propylene to be used in industrial applications. A more thorough understanding of the catalytic activities and reaction mechanisms of SAAs can lead to the discovery of further applications of SAAs.
Nevertheless, some major challenges remain in this research field. The detection of the detailed atomic structure of multi-element SAAs, particularly those containing elements with similar atomic numbers, will continue to be a major difficulty in the structural analysis of SAA catalysts. As such, the development of higher resolution imaging and spectroscopy tools, and the joint use of multiple such techniques, will play a more important role in this regard. In addition, more in-depth studies on the electronic properties of SAAs using various experimental methods, in combination with theoretical methods, are needed to understand the catalytic performance of SAAs. This aspect of research on SAAs is particularly under-developed when compared to the structural and catalytic studies of SAAs. Furthermore, the increased use of SAAs in real reactions is needed to help enhance their applications in more practically useful reactions. Toward this end, more powerful in situ and operando research tools, especially those having high resolution in space and time domains, are required to provide a complete understanding of the structural properties of SAAs during real reactions. In addition to the aforementioned in situ X-ray techniques, in situ transmission electron microscopy can also be used to observe nanocrystals in real time, in the liquid phase and with high resolution.133 Future work involving SAAs should also include investigating new combinations of metals, particularly those consisting of many more elements than the currently available bimetallic samples, such as the recently emerged high-entropy alloy catalysts.134
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from NSERC Canada. T. Z. acknowledges the financial support from China Scholarship Council for his visit to Dalhousie University. This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy (DOE) Office of Science by Argonne National Laboratory, and was supported by the US DOE under contract no. DE-AC02-06CH11357, and the Canadian Light Source (CLS) and its funding partners. The CLS is supported by the CFI, NSERC, NRC, CIHR, the University of Saskatchewan, the Government of Saskatchewan and Western Economic Diversification Canada. J. Y. and P. Z. thank the 111 project of China (B17020) for supporting international collaboration.References
- A. Vazquez, A. Kato and L. Schmidt, J. Catal., 1982, 78, 306–318 CrossRef.
- A. Gutierrez, R. Kaila, M. Honkela, R. Slioor and A. Krause, Catal. Today, 2009, 147, 239–246 CrossRef CAS.
- M. Hegde, G. Madras and K. Patil, Acc. Chem. Res., 2009, 42, 704–712 CrossRef CAS.
- Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364–370 CrossRef CAS.
- C.-H. Tu, A.-Q. Wang, M.-Y. Zheng, X.-D. Wang and T. Zhang, Appl. Catal., A, 2006, 297, 40–47 CrossRef CAS.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS.
- L. Luo, L. Zhang, Z. Duan, A. S. Lapp, G. Henkelman and R. M. Crooks, ACS Nano, 2016, 10, 8760–8769 CrossRef CAS.
- A. Holewinski, J.-C. Idrobo and S. Linic, Nat. Chem., 2014, 6, 828–834 CrossRef CAS.
- N. Nilius, T. M. Wallis and W. Ho, J. Phys. Chem. B, 2004, 108, 14616–14619 CrossRef CAS.
- E. V. Carino and R. M. Crooks, Langmuir, 2011, 27, 4227–4235 CrossRef CAS.
- X. Liu, A. Wang, X. Wang, C.-Y. Mou and T. Zhang, Chem. Commun., 2008, 3187–3189 RSC.
- K. Shin, L. Zhang, H. An, H. Ha, M. Yoo, H. M. Lee, G. Henkelman and H. Y. Kim, Nanoscale, 2017, 9, 5244–5253 RSC.
- H. Hsieh, Y. Chang, W. Pong, J. Pieh, P. Tseng, T. Sham, I. Coulthard, S. Naftel, J. Lee and S. Chung, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 15204 CrossRef CAS.
- C. Jiao, Z. Huang, H. Zhang and S. Zhang, Prog. Chem., 2015, 27, 472 CAS.
- L. Luo, J. Timoshenko, A. S. Lapp, A. I. Frenkel and R. M. Crooks, Langmuir, 2017, 33, 12434–12442 CrossRef CAS.
- J. N. Butler, J. Giner and J. M. Parry, Surf. Sci., 1969, 18, 140–158 CrossRef CAS.
- D. A. Slanac, W. G. Hardin, K. P. Johnston and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 9812–9819 CrossRef CAS.
- D. Wang, A. Villa, F. Porta, D. Su and L. Prati, Chem. Commun., 2006, 1956–1958 RSC.
- H. E. Swift and J. E. Bozik, J. Catal., 1968, 12, 5–14 CrossRef CAS.
- D. Cadenhead and N. Masse, J. Phys. Chem., 1966, 70, 3558–3566 CrossRef CAS.
- J. H. Sinfelt, Acc. Chem. Res., 1977, 10, 15–20 CrossRef CAS.
- C. H. Bartholomew and R. J. Farrauto, Can. J. Chem. Eng., 2007, 85, 127 Search PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS.
- X. Liu, A. Wang, X. Yang, T. Zhang, C.-Y. Mou, D.-S. Su and J. Li, Chem. Mater., 2009, 21, 410–418 CrossRef CAS.
- K. Tedsree, T. Li, S. Jones, C. Chan and K. Yu, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS.
- M. Luneau, T. Shirman, A. Filie, J. Timoshenko, W. Chen, A. Trimpalis, M. Flytzani-Stephanopoulos, E. Kaxiras, A. I. Frenkel and J. Aizenberg, Chem. Mater., 2019, 31, 5759–5768 CrossRef CAS.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg and H. Zong, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS.
- M. Yang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2013, 135, 3768–3771 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo and D. Zang, Science, 2016, 352, 797–800 CrossRef CAS.
- Y. Wang, B. Yu, K. Liu, X. Yang, M. Liu, T.-S. Chan, X. Qiu, J. Li and W. Li, J. Mater. Chem. A, 2020, 8, 2131–2139 RSC.
- A. J. Hensley, J. Zhang, I. Vinçon, X. P. Hernandez, D. Tranca, G. Seifert, J.-S. McEwen and Y. Wang, J. Catal., 2018, 361, 414–422 CrossRef CAS.
- T. He, L. Zhang, G. Kour and A. Du, J. CO2 Util., 2020, 37, 272–277 CrossRef CAS.
- Z. Kou, W. Zang, W. Pei, L. Zheng, S. Zhou, S. Zhang, L. Zhang and J. Wang, J. Mater. Chem. A, 2020, 8, 3071–3082 RSC.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. H. Antink, S. Park and K.-S. Lee, Nat. Mater., 2020, 19, 436–442 CrossRef CAS.
- G. X. Pei, X. Y. Liu, A. Wang, L. Li, Y. Huang, T. Zhang, J. W. Lee, B. W. Jang and C.-Y. Mou, New J. Chem., 2014, 38, 2043–2051 RSC.
- X. Ren, J. Li, S. Wang, D. Zhang and Y. Wang, J. Chem. Technol. Biotechnol., 2020, 95, 1675–1682 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- M. S. Hamdy, M. A. Eissa and S. M. Keshk, Green Chem., 2017, 19, 5144–5151 RSC.
- D. D. Babu, Y. Huang, G. Anandhababu, X. Wang, R. Si, M. Wu, Q. Li, Y. Wang and J. Yao, J. Mater. Chem. A, 2019, 7, 8376–8383 RSC.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS.
- P. Aich, H. Wei, B. Basan, A. J. Kropf, N. M. Schweitzer, C. L. Marshall, J. T. Miller and R. Meyer, J. Phys. Chem. C, 2015, 119, 18140–18148 CrossRef CAS.
- G. Giannakakis, A. Trimpalis, J. Shan, Z. Qi, S. Cao, J. Liu, J. Ye, J. Biener and M. Flytzani-Stephanopoulos, Top. Catal., 2018, 61, 475–486 CrossRef CAS.
- F. R. Lucci, M. D. Marcinkowski, T. J. Lawton and E. C. H. Sykes, J. Phys. Chem. C, 2015, 119, 24351–24357 CrossRef CAS.
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Top. Catal., 2018, 61, 428–438 CrossRef CAS.
- J. Shan, N. Janvelyan, H. Li, J. Liu, T. M. Egle, J. Ye, M. M. Biener, J. Biener, C. M. Friend and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2017, 205, 541–550 CrossRef CAS.
- J. Han, J. Lu, M. Wang, Y. Wang and F. Wang, Chin. J. Chem., 2019, 37, 977–988 CrossRef CAS.
- T. Yamada, T. Kojima, E. Abe, S. Kameoka, Y. Murakami, P. Gille and A. P. Tsai, J. Am. Chem. Soc., 2018, 140, 3838–3841 CrossRef CAS.
- J. Liu, J. Shan, F. R. Lucci, S. Cao, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Catal. Sci. Technol., 2017, 7, 4276–4284 RSC.
- M. T. Darby, R. Réocreux, E. C. H. Sykes, A. Michaelides and M. Stamatakis, ACS Catal., 2018, 8, 5038–5050 CrossRef CAS.
- H. Wang, Q. Luo, W. Liu, Y. Lin, Q. Guan, X. Zheng, H. Pan, J. Zhu, Z. Sun and S. Wei, Nat. Commun., 2019, 10, 1–9 CrossRef.
- J. Liu, M. B. Uhlman, M. M. Montemore, A. Trimpalis, G. Giannakakis, J. Shan, S. Cao, R. T. Hannagan, E. C. H. Sykes and M. Flytzani-Stephanopoulos, ACS Catal., 2019, 9, 8757–8765 CrossRef CAS.
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS.
- G. Sun, Z.-J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li and S. C. Purdy, Nat. Commun., 2018, 9, 1–9 CrossRef.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang and G. Wu, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- J. Kim, C. W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1701476 CrossRef.
- S. Yang and H. Lee, ACS Catal., 2013, 3, 437–443 CrossRef CAS.
- J. Shan, J. Liu, M. Li, S. Lustig, S. Lee and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2018, 226, 534–543 CrossRef CAS.
- S. Kikkawa, K. Teramura, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2019, 123, 23446–23454 CrossRef CAS.
- X. Zhang, G. Cui, H. Feng, L. Chen, H. Wang, B. Wang, X. Zhang, L. Zheng, S. Hong and M. Wei, Nat. Commun., 2019, 10, 1–12 CrossRef.
- M. Cargnello, V. V. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS.
- S. Nigam and C. Majumder, Nanoscale, 2018, 10, 20599–20610 RSC.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2018, 52, 237–247 CrossRef.
- C. Yang, Z. Miao, F. Zhang, L. Li, Y. Liu, A. Wang and T. Zhang, Green Chem., 2018, 20, 2142–2150 RSC.
- P. N. Duchesne, Z. Li, C. P. Deming, V. Fung, X. Zhao, J. Yuan, T. Regier, A. Aldalbahi, Z. Almarhoon and S. Chen, Nat. Mater., 2018, 17, 1033–1039 CrossRef CAS.
- G. X. Pei, X. Y. Liu, A. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. Pan, X. Yang, X. Wang and Z. Tai, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS.
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325 CrossRef CAS.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev. DOI:10.1021/acs.chemrev.0c00078.
- X. Cao, A. Mirjalili, J. Wheeler, W. Xie and B. W.-L. Jang, Front. Chem. Sci. Eng., 2015, 9, 442–449 CrossRef CAS.
- F. Xing, J. Jeon, T. Toyao, K.-i. Shimizu and S. Furukawa, Chem. Sci., 2019, 10, 8292–8298 RSC.
- C. J. Wrasman, A. Boubnov, A. R. Riscoe, A. S. Hoffman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 12930–12939 CrossRef CAS.
- L. Zhang, A. Wang, J. T. Miller, X. Liu, X. Yang, W. Wang, L. Li, Y. Huang, C.-Y. Mou and T. Zhang, ACS Catal., 2014, 4, 1546–1553 CrossRef CAS.
- G. X. Pei, X. Y. Liu, X. Yang, L. Zhang, A. Wang, L. Li, H. Wang, X. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- M. B. Boucher, B. Zugic, G. Cladaras, J. Kammert, M. D. Marcinkowski, T. J. Lawton, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Phys. Chem. Chem. Phys., 2013, 15, 12187–12196 RSC.
- H. Miura, K. Endo, R. Ogawa and T. Shishido, ACS Catal., 2017, 7, 1543–1553 CrossRef CAS.
- F. R. Lucci, J. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- Y. Peng, Z. Geng, S. Zhao, L. Wang, H. Li, X. Wang, X. Zheng, J. Zhu, Z. Li and R. Si, Nano Lett., 2018, 18, 3785–3791 CrossRef CAS.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS.
- A. Trimpalis, G. Giannakakis, S. Cao and M. Flytzani-Stephanopoulos, Catal. Today, 2020, 355, 804–814 CrossRef CAS.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49 CrossRef.
- C. H. Chen, D. Wu, Z. Li, R. Zhang, C. G. Kuai, X. R. Zhao, C. K. Dong, S. Z. Qiao, H. Liu and X. W. Du, Adv. Energy Mater., 2019, 9, 1803913 CrossRef.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- W. H. Lai, B. W. Zhang, Z. Hu, X. M. Qu, Y. X. Jiang, Y. X. Wang, J. Z. Wang, H. K. Liu and S. L. Chou, Adv. Funct. Mater., 2019, 29, 1807340 CrossRef.
- W. Lefebvre, V. Kopp and C. Pareige, Appl. Phys. Lett., 2012, 100, 141906 CrossRef.
- Y. Wang, H. Kageyama, T. Mori, H. Yoshikawa and J. Drennan, Solid State Ionics, 2006, 177, 1681–1685 CrossRef CAS.
- Y.-L. Fang, J. T. Miller, N. Guo, K. N. Heck, P. J. Alvarez and M. S. Wong, Catal. Today, 2011, 160, 96–102 CrossRef CAS.
- E. K. Gibson, A. M. Beale, C. R. A. Catlow, A. Chutia, D. Gianolio, A. Gould, A. Kroner, K. M. Mohammed, M. Perdjon and S. M. Rogers, Chem. Mater., 2015, 27, 3714–3720 CrossRef CAS.
- M. T. Greiner, T. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbrüster, A. Knop-Gericke and R. Schlögl, Nat. Chem., 2018, 10, 1008–1015 CrossRef CAS.
- J. P. Simonovis, A. Hunt, S. D. Senanayake and I. Waluyo, Surf. Sci., 2019, 679, 207–213 CrossRef CAS.
- J. P. Simonovis, A. Hunt, R. M. Palomino, S. D. Senanayake and I. Waluyo, J. Phys. Chem. C, 2018, 122, 4488–4495 CrossRef CAS.
- Z.-T. Wang, M. T. Darby, A. J. Therrien, M. El-Soda, A. Michaelides, M. Stamatakis and E. C. H. Sykes, J. Phys. Chem. C, 2016, 120, 13574–13580 CrossRef CAS.
- Z. Zhao and G. Lu, J. Phys. Chem. C, 2019, 123, 4380–4387 CrossRef CAS.
- X. Cao, Q. Fu and Y. Luo, Phys. Chem. Chem. Phys., 2014, 16, 8367–8375 RSC.
- H. Thirumalai and J. R. Kitchin, Top. Catal., 2018, 61, 462–474 CrossRef CAS.
- F. R. Lucci, M. T. Darby, M. F. Mattera, C. J. Ivimey, A. J. Therrien, A. Michaelides, M. Stamatakis and E. C. H. Sykes, J. Phys. Chem. Lett., 2016, 7, 480–485 CrossRef CAS.
- M. Kettner, S. Maisel, C. Stumm, M. Schwarz, C. Schuschke, A. Görling and J. Libuda, J. Catal., 2019, 369, 33–46 CrossRef CAS.
- K. Yang and B. Yang, Phys. Chem. Chem. Phys., 2017, 19, 18010–18017 RSC.
- M. T. Darby, F. R. Lucci, M. D. Marcinkowski, A. J. Therrien, A. Michaelides, M. Stamatakis and E. C. H. Sykes, J. Phys. Chem. C, 2019, 123, 10419–10428 CrossRef CAS.
- J. Zhou, W. An, Z. Wang and X. Jia, Catal. Sci. Technol., 2019, 9, 4314–4326 RSC.
- K. D. Sabnis, M. C. Akatay, Y. Cui, F. G. Sollberger, E. A. Stach, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2015, 330, 442–451 CrossRef CAS.
- B. Y. Xia, H. B. Wu, N. Li, Y. Yan, X. W. Lou and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 3797–3801 CrossRef CAS.
- A. D. Benavidez, P. D. Burton, J. L. Nogales, A. R. Jenkins, S. A. Ivanov, J. T. Miller, A. M. Karim and A. K. Datye, Appl. Catal., A, 2014, 482, 108–115 CrossRef CAS.
- R. N. Lamb, B. Ngamsom, D. L. Trimm, B. Gong, P. L. Silveston and P. Praserthdam, Appl. Catal., A, 2004, 268, 43–50 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Friedrich, D. Teschner, G. Wowsnick, M. Hahne, P. Gille, L. Szentmiklósi, M. Feuerbacher, M. Heggen, F. Girgsdies, D. Rosenthal, R. Schlögl and Y. Grin, Nat. Mater., 2012, 11, 690–693 CrossRef.
- K. Kovnir, M. Armbrüster, D. Teschner, T. V. Venkov, L. Szentmiklósi, F. C. Jentoft, A. Knop-Gericke, Y. Grin and R. Schlögl, Surf. Sci., 2009, 603, 1784–1792 CrossRef CAS.
- J. Osswald, R. Giedigkeit, R. E. Jentoft, M. Armbrüster, F. Girgsdies, K. Kovnir, T. Ressler, Y. Grin and R. Schlögl, J. Catal., 2018, 258, 210–218 CrossRef.
- J. Prinz, R. Gaspari, C. A. Pignedoli, J. Vogt, P. Gille, M. Armbrüster, H. Brune, O. Gröning, D. Passerone and R. Widmer, Angew. Chem., Int. Ed., 2012, 51, 9339–9343 CrossRef CAS.
- Q. Feng, S. Zhao, Y. Wang, J. Dong, W. Chen, D. He, D. Wang, J. Yang, Y. Zhu, H. Zhu, L. Gu, Z. Li, Y. Liu, J. Li and Y. Li, J. Am. Chem. Soc., 2017, 139, 7294–7301 CrossRef CAS.
- J. H. Kang, E. W. Shin, W. J. Kim, J. D. Park and S. H. Moon, J. Catal., 2002, 208, 310–320 CrossRef CAS.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 1–10 Search PubMed.
- J. Liu, Y. Zheng, D. Zhu, A. Vasileff, T. Ling and S.-Z. Qiao, Nanoscale, 2017, 9, 16616–16621 RSC.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- M.-J. Cheng, E. L. Clark, H. H. Pham, A. T. Bell and M. Head-Gordon, ACS Catal., 2016, 6, 7769–7777 CrossRef CAS.
- Q. Fu and Y. Luo, J. Phys. Chem. C, 2013, 117, 14618–14624 CrossRef CAS.
- C. M. Kruppe, J. D. Krooswyk and M. Trenary, ACS Catal., 2017, 7, 8042–8049 CrossRef CAS.
- M. D. Marcinkowski, J. Liu, C. J. Murphy, M. L. Liriano, N. A. Wasio, F. R. Lucci, M. Flytzani-Stephanopoulos and E. C. H. Sykes, ACS Catal., 2017, 7, 413–420 CrossRef CAS.
- Z.-T. Wang, R. A. Hoyt, M. El-Soda, R. J. Madix, E. Kaxiras and E. C. H. Sykes, Top. Catal., 2018, 61, 328–335 CrossRef CAS.
- M. Xing, S. Guo and L. Guo, Theor. Chem. Acc., 2018, 137, 18 Search PubMed.
- N. Gerrits, D. Migliorini and G.-J. Kroes, J. Chem. Phys., 2018, 149, 224701 CrossRef.
- M. Jørgensen and H. Grönbeck, J. Am. Chem. Soc., 2019, 141, 8541–8549 CrossRef.
- F. R. Lucci, L. Zhang, T. Thuening, M. B. Uhlman, A. C. Schilling, G. Henkelman and E. C. H. Sykes, Surf. Sci., 2018, 677, 296–300 CrossRef CAS.
- S. Stevanović, D. Tripković, V. Tripkovic, D. Minić, A. Gavrilović, A. Tripković and V. Jovanović, J. Phys. Chem. C, 2014, 118, 278–289 CrossRef.
- Z. Dobrovolná, P. Kačer and L. Červený, J. Mol. Catal. A: Chem., 1998, 130, 279–284 CrossRef.
- J. Boitiaux, J. Cosyns and E. Robert, Appl. Catal., 1987, 32, 145–168 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sørensen, C. H. Christensen and J. K. Nørskov, Science, 2008, 320, 1320–1322 CrossRef CAS.
- S. Zha, G. Sun, T. Wu, J. Zhao, Z.-J. Zhao and J. Gong, Chem. Sci., 2018, 9, 3925–3931 RSC.
- E. Ruckenstein and Y. H. Hu, Appl. Catal., A, 1995, 133, 149–161 CrossRef CAS.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, J. Catal., 2010, 276, 268–279 CrossRef CAS.
- F. Yang, J. Koeller and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 4759–4762 CrossRef CAS.
- H. Zheng, R. K. Smith, Y.-w. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu and M. Rehwoldt, Science, 2018, 359, 1489–1494 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |