
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon-based catalysts for Fischer–Tropsch synthesis†
Yanping
Chen
 *a,
Jiatong
Wei
ad,
Melis S.
Duyar
c,
Vitaly V.
Ordomsky
*d,
Andrei Y.
Khodakov
b and
Jian
Liu
*ac
*a,
Jiatong
Wei
ad,
Melis S.
Duyar
c,
Vitaly V.
Ordomsky
*d,
Andrei Y.
Khodakov
b and
Jian
Liu
*ac
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: chenyp@dicp.ac.cn; jianliu@dicp.ac.cn
bUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: vitaly.ordomsky@univ-lille.fr
cDICP-Surrey Joint Centre for Future Materials, Department of Chemical and Process Engineering, and Advanced Technology Institute, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: jian.liu@surrey.ac.uk
dInstitute of Chemistry for Functionalized Materials, School of Chemistry and Chemical Engineering, Liaoning Normal University, 850 Huanghe Road, Dalian 116029, China
First published on 4th January 2021
Abstract
Fischer–Tropsch synthesis (FTS) is an essential approach to convert coal, biomass, and shale gas into fuels and chemicals, such as lower olefins, gasoline, diesel, and so on. In recent years, there has been increasing motivation to deploy FTS at commercial scales which has been boosting the discovery of high performance catalysts. In particular, the importance of support in modulating the activity of metals has been recognized and carbonaceous materials have attracted attention as supports for FTS. In this review, we summarised the substantial progress in the preparation of carbon-based catalysts for FTS by applying activated carbon (AC), carbon nanotubes (CNTs), carbon nanofibers (CNFs), carbon spheres (CSs), and metal–organic frameworks (MOFs) derived carbonaceous materials as supports. A general assessment of carbon-based catalysts for FTS, concerning the support and metal properties, activity and products selectivity, and their interactions is systematically discussed. Finally, current challenges and future trends in the development of carbon-based catalysts for commercial utilization in FTS are proposed.
 Yanping Chen | Yanping Chen received her PhD from Zhejiang University, China, in 2014. Now she is an Assistant Professor at the State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences. She experienced two years of postdoctoral research focusing on iron-catalyzed Fischer–Tropsch synthesis in DICP and two years of postdoctoral research focusing on cobalt/ruthenium-catalyzed Fischer–Tropsch synthesis in Unite de Catalyse et Chimie du Solide UMR 8181 (UCCS, CNRS) in France. Now she works mainly on carbon-based metal catalysts for Fischer Tropsch synthesis. |
 Jiatong Wei | Jiatong Wei received her bachelor's degree from Shenyang Normal University in 2018. She is now a joint master's student under the supervision of Prof. Jian Liu at Dalian Institute of Chemical Physics, Chinese Academy of Sciences. Her research is focused on the design and synthesis of nanoreactor for Fischer–Tropsch synthesis. |
 Melis S. Duyar | Melis S. Duyar is Lecturer of Chemical and Process Engineering at the University of Surrey. She obtained her MS (2013) and PhD (2015) from Columbia University and conducted post-doctoral research (2015–2017) at Stanford University. Prior to her academic appointment at the University of Surrey, Dr Duyar worked at the US Department of Energy's SLAC National Accelerator Laboratory as Associate Staff Scientist and was also Lecturer of Chemical Engineering at Stanford University (2017–2019). Her research interests include CO2 capture and catalyst development for the production of sustainable fuels and chemicals. |
 Vitaly V. Ordomsky | Vitaly V. Ordomsky received his MSc (2006) and PhD (2009) degree from Moscow State University in the field of zeolite catalysis. He was a postdoctoral researcher in the Department of Chemical Engineering at the Eindhoven University in the Netherlands, working on biomass conversion. Afterwards, he received a permanent position as a researcher in 2013 at the CNRS (France) in the Energy group of Lille University working in C1 chemistry mainly focusing in Fischer–Tropsch synthesis. From 2016 to 2019 he was CNRS researcher in E2P2L laboratory in Shanghai. His main research field is development of new nano-materials for sustainable catalysis. |
 Andrei Y. Khodakov | Andrei Y. Khodakov obtained his PhD degree in 1991 from Zelinsky Institute of Organic Chemistry of USSR Academy of Sciences. In 1992–1999, he worked as a postdoctoral researcher in academic and industrial laboratories in France, UK and USA. From 1999, he is a researcher of the National Centre for Scientific Research in France. Since 2017, he is a CNRS Senior Research Director in the UCCS-CNRS Laboratory in the University of Lille and Centrale Lille Institute. One of the major research topics addresses synthesis of fuels and chemicals from syngas via Fischer–Tropsch synthesis. |
 Jian Liu | Jian Liu received his PhD degree in physical chemistry from the Dalian Institute of Chemical Physics, Chinese Academy of Science, China, in 2008, and worked at the Australian Institute for Bioengineering and Nanotechnology (AIBN), University of Queensland (UQ) as a research fellow. After working at Curtin University as a senior lecturer from 2013 to 2017, he then joined DICP as a full professor and group leader of micro–nanoreactor and reaction engineering. He also held an adjunct Reader position in the Department of Chemical and Process Engineering, University of Surrey, UK. His research focuses on the design of micro–nanoreactors, porous carbon spheres, and industrialization of homogeneous catalysis in heterogeneous systems. His h-index is 58 (Web of Science). |
1. Introduction
Because of the depletion of crude oil and environmental concerns, utilisation of non-petroleum carbon resources such as natural gas, coal, and biomass is currently becoming more an attractive strategy to obtain fuels and chemicals. Natural gas, coal, and biomass can be converted into syngas through gasification and reforming, and then syngas may be transformed into lower olefins, gasoline, diesel, wax, oxygenates, α-olefins, and other chemicals via Fischer–Tropsch synthesis (FTS). FTS products are raw chemicals for the synthesis of fibers, rubbers, and plastics, lubricating oil, surfactants, detergents, and so on (Fig. 1). Syngas (CO + H2) is the most important platform that bridges various carbon resources with fuels and chemicals. FTS is a typical catalytic process that has been thoroughly investigated for the conversion of syngas, and the commercial FTS catalysts with precipitated bulk Fe and supported Co are widely applied in the industry.1–5 However, the precipitated bulk Fe suffers from low activity and stability due to its low iron dispersion, fragmentation during FTS, and low catalytic activity induced by severe metal–support interactions.6 FTS is structure sensitive reaction, hence metal nanoparticle size and distribution are some of the significant parameters affecting the activity of supported nanoparticles.7–10 Typical supports used for FTS include SiO2, Al2O3, TiO2, MgO, mesoporous materials and zeolite supports which achieve metal high surface areas.11–15 However, strong interaction between metal and these supports can lead to the formation of inert mixture compounds. For instance, the strong metal–support interaction between cobalt and Al2O3, SiO2, and TiO2 leads to the formation of Co2SiO2, Co2AlO4, or CoTiO4 respectively, which are reducible only at high temperatures.16–21 Carbonaceous supports might overcome these drawbacks because of their weak interaction with metal precursors. Activated carbon (AC), carbon nanotubes (CNTs), carbon nanofibers (CNFs), carbon spheres (CSs), metal–organic frameworks (MOFs) derived carbonaceous materials, and other carbonaceous materials have been employed in FTS (Fig. 2).22–28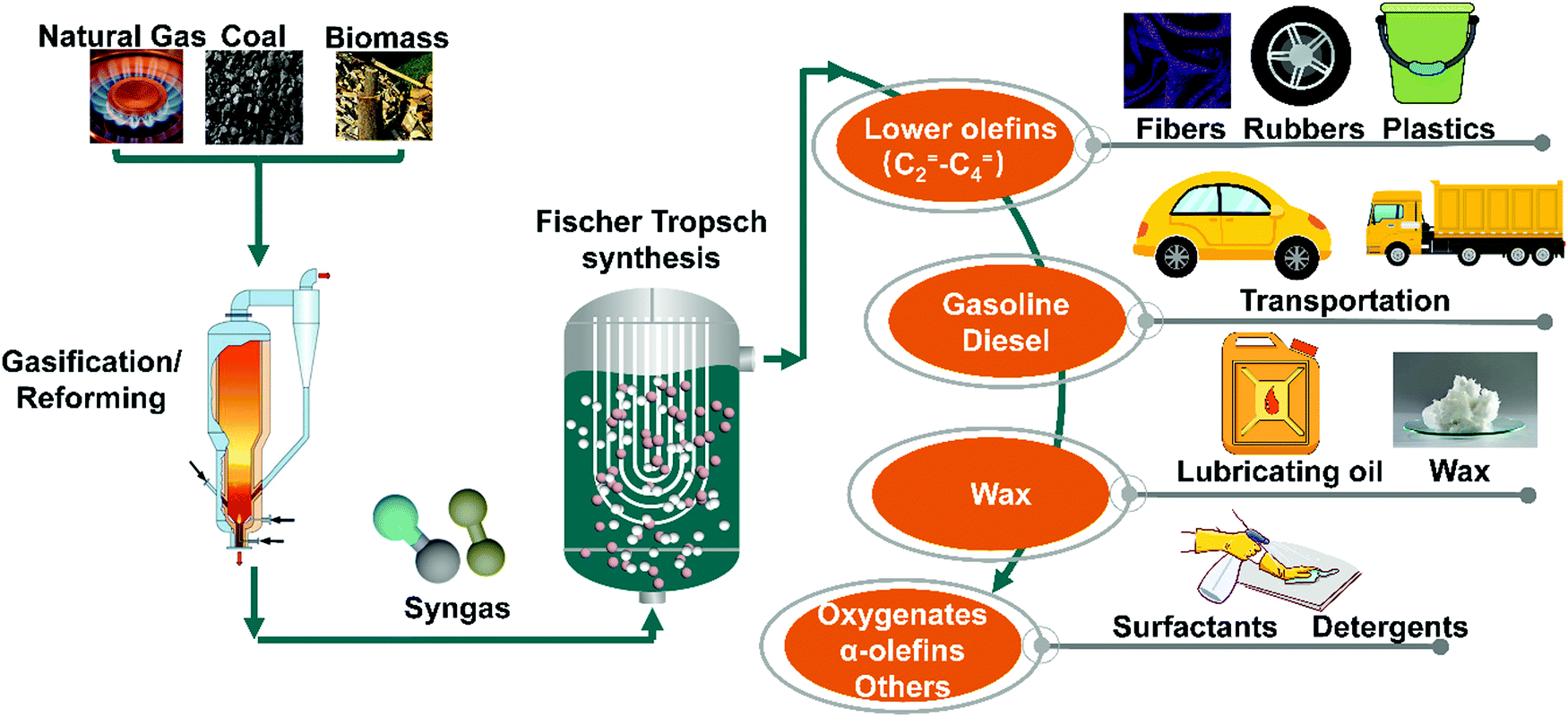 | ||
| Fig. 1 Schematic illustration for the overall transformation process of Fischer–Tropsch synthesis. | ||
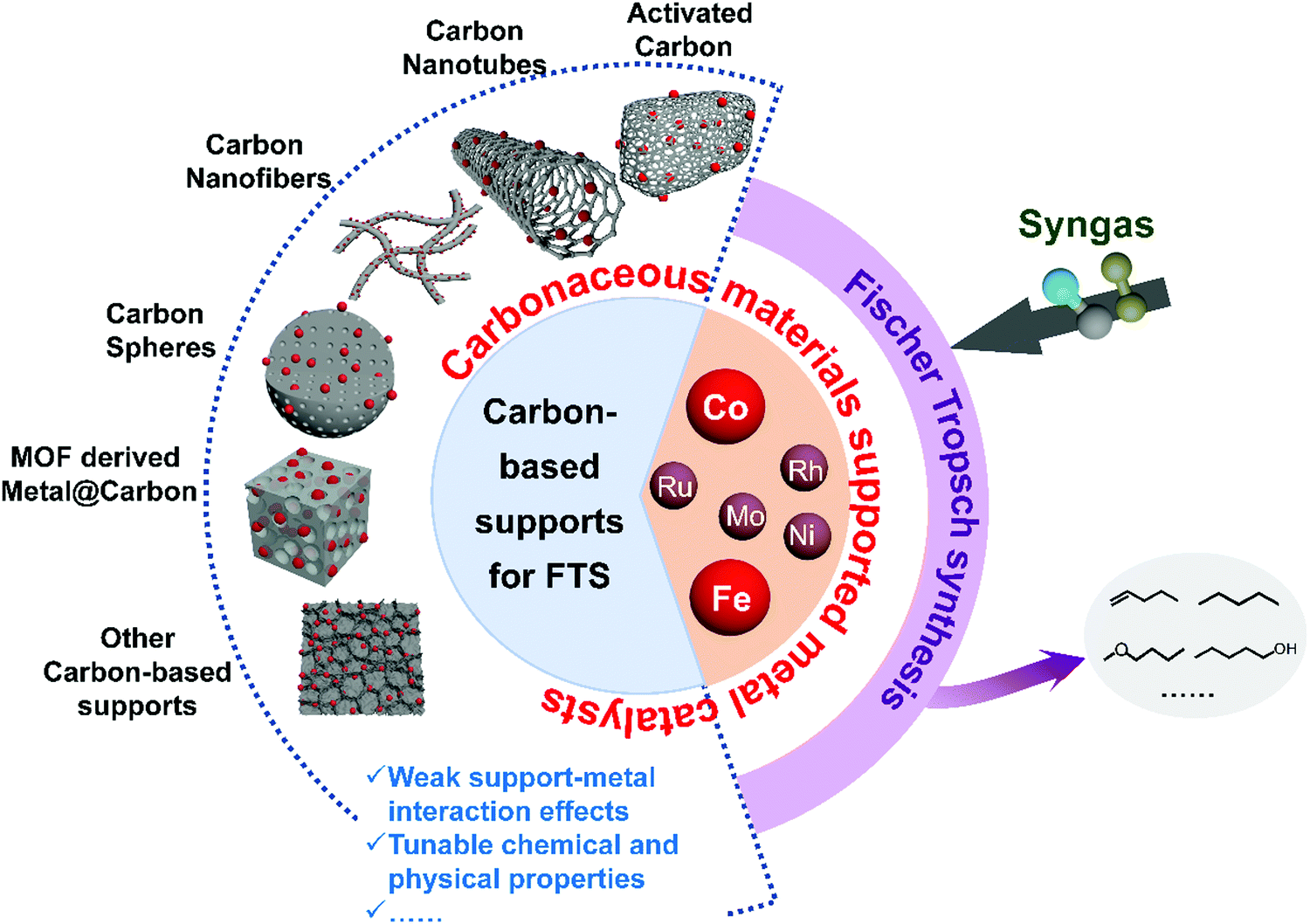 | ||
| Fig. 2 Carbonaceous materials supported metal catalysts for FTS. | ||
Generally, these carbonaceous materials can adjust the interaction between the active phase and support. Meanwhile, those carbons are chemically and thermally stable under harsh conditions in an inert atmosphere. Their unique properties, such as tunable surface area, tailorable surface chemistry, and favorable recycling characteristics, are suitable for developing them as catalytic support materials for FTS. The use of carbonaceous materials as supports is an effective approach to enhance the reducibility of cobalt and iron oxides in FTS catalysts.1,7,8 The surface of carbon-based supports can be easily modified leading to the tunable interaction between iron oxides and supports, which induces a facile reduction of iron oxides and facilitates the formation of the active iron carbide phases. Iron and cobalt catalysts supported on carbonaceous materials have shown higher activity in FTS compared to the oxide supported counterparts, which could be attributed to the possible electronic transfer between carbon and metal species. This review addresses use of several carbonaceous materials for the design of efficient FTS catalysts.
The advantages of AC as supports include low toxicity, high stability towards chemicals, radiation, and heat, as well as the rigid porous structure and mechanical strength. AC supports have an intricate porous structure with large amounts of micropores. The porous structure and high surface area of AC facilitates good dispersion of metals at high loadings resulting in high FTS activity.29,30 AC can be prepared from residual biomass and waste materials which is attractive from both economics and sustainability. The removal of residual impurities in the biomass-derived AC is however an important issue for their utilization in catalysis. The presence of various oxygenated functional groups on the AC surface enhances the dispersion of metal particles.31,32 The inert surface of AC facilitates the reduction or carburization of the metal precursor. Besides, it was reported that hydrocarbons produced on Co/AC were mainly distributed in C10–C20 and almost no wax was generated in the products.33,34 AC supported metal catalysts also have advantages of good resistance to high water partial pressure and easy treatment of deactivated catalysts to retrieve the metal components.
CNTs can be envisioned as a tubular structure formed by rolling up graphene layers.35 According to the number of graphene layers, there are two types of CNTs which are distinguished as single-walled CNTs and multiwalled CNTs (MWCNTs).36–38 The curvature of CNTs walls alters the hybridization of electronic orbitals, which deforms the sp2 hybridization of graphene with a sp3 character, inducing different chemical environments inside the channels and on the outer surface of CNTs. The curvature of CNTs walls shifts the π electron density of the graphene layers from the concave inner to the convex outer surface and leads to an electric potential difference and further to various redox properties of metal particles, because of unique tubular morphology, CNTs are expected to utilize as nanoreactors with guest metal particles encapsulated inside their well-defined channels.39–47 The encapsulated metal particles are restricted to the nanometer and even the sub-nanometer scales by the rigid nanotubes. In terms of FTS, interactions between the encapsulated metal particles and the CNTs surfaces may be further adjusted to improve the catalytic performances. Consequently, properties and chemical reactivities are different for molecules and metal particles on the exterior walls of CNTs and those confined within CNTs.38,44 These inside and outside activity differences vary as functions of metals used and the reactions catalyzed. All the properties above have evoked wide interest for the catalytic applications of CNTs in FTS.
The CNFs consist of interwoven graphitic carbon fibers with high chemical inertness, high purity, and high mechanical strength, which make them a very promising novel graphitic support material for applications in FTS. CNFs have been widely used as excellent supports to investigate the intrinsic cobalt or iron particle size effects on the FTS catalytic performance.48–50 FTS is a structure sensitive reaction and the catalytic performance is strongly associated with the particle size of the metal, with particle sizes larger than 6–8 nm leading to higher intrinsic activity for cobalt catalysts.51 However, on oxide-based supports such as SiO2 and Al2O3, smaller cobalt particles are more inclined to form irreducible mixed oxides such as cobalt aluminate or cobalt silicate.51–53 This implies that oxidic supports may mask the cobalt particle size effects on the catalytic performance for FTS. However, CNFs exhibit weak metal–support interaction and uniformly distributed metal particles which are more suitable for investigating the intrinsic metal properties.
The CSs supports provide advantages of regular geometry, tunable porosity, and controllable size distribution of metal particles.23,54,55 CSs ranging in size from nanometers to micrometers have been applied as supports in FTS. Recent preliminary studies on the Co/CS catalysts exhibited that the cobalt oxide could be auto-reduced by the CSs which results in better FTS performance in comparison with cobalt catalysts reduced in hydrogen.54,56 More significantly, it was observed that the tunable chemical and physical properties of CSs make it an ideal model catalyst support to investigate the effect of metal particle size, promoters, and deactivation on FTS.
MOFs have attracted considerable attention in catalysis due to their special structures and functional properties. Unlike zeolites, most MOFs suffer from low thermal and hydrothermal stability under the high-temperature and/or high-pressure conditions, which prevents their use as FTS catalysts. However, MOFs can serve as a sacrificial template to prepare carbon-based catalysts. Upon annealing in an inert atmosphere at high temperatures, the framework collapses and the organic linker is transformed into a carbon matrix, leading to the formation of a highly dispersed encapsulated metal phase. The resulting catalysts are referred to as nanoparticles@carbon (NP@C).57–60 The confinement of metal nanoparticles inside carbon matrix has been shown to inhibit aggregation of metal nanoparticles and also can tune the selectivity of FTS.3,61 The spatial restriction of metal nanoparticles encapsulated in the carbon matrix minimizes the sintering of metal nanoparticles. The catalysts display properties of “nanoreactors”, enabling tunability of catalytic activity via confinement effects. Meanwhile, MOF-derived NP@C also exhibits high surface area, high pore volume, and uniformly dispersed metal nanoparticles, which can further improve the catalytic performance. Fe- or Co-containing MOFs have been exploited in the synthesis of MOF-derived catalysts for FTS and exhibited unique characteristics in catalytic performances.
Other carbon-based supports include reduced graphene oxide (rGO), N-doped graphene (NG), carbon–silica composite materials (SiC), graphitic carbon nitride (g-C3N4), carbonaceous supports prepared from polymers as carbon precursors.
The present review focuses on the carbon-based catalysts for FTS, and carbon-based supports include AC, CNTs, CNFs, CSs, MOFs derived-materials. The unique characteristics of carbon-based catalysts are emphasized. The current developments, future challenges, and potential applications of carbon-based catalysts for FTS are discussed through the review.
2. Cobalt supported on carbonaceous materials for Fischer–Tropsch synthesis
2.1 Activated carbon supported cobalt catalysts
Researchers have investigated Co/AC for FTS since 1981.62–65 The sources of AC play an important role in the physical and chemical surface properties which may affect the catalytic performance of supported metal catalysts. Lahti et al. have developed a novel AC produced by carburization and steam activation of lignin, a waste fraction from the process of Kraft pulping.66 Ash content could be reduced by acid (HNO3) treatment which also has a positive effect on the reduction of cobalt particles. Wang et al. have confirmed that the AC supports promote Co2C formation, whereas traditional supports such as SiO2 and Al2O3 inhibit it.67 Small Co particles enhance the thermodynamic driving force toward Co2C formation. Ying et al. have investigated the product distributions of FTS over Co/AC catalyst under different reaction conditions.68 High temperature, space velocity, H2/CO ratio in feed gas, and low pressures leads to the production of light hydrocarbons.69,70 The product distributions deviate from the Anderson–Schulz–Flory (ASF) distribution due to the re-adsorption of alkenes and alkene secondary reactions. Ying et al. have developed a comprehensive kinetic model for the Co/AC catalyst under a wide range of operating conditions which provides a new insight into the activation energies of FTS.71 Cheng et al. have developed a novel CoZr/AC@ZSM-5 core–shell catalyst by coating ZSM-5 shell on the core of CoZr/AC.72 The CoZr/AC@ZSM-5 catalysts showed high selectivity to short chain hydrocarbons while long chain hydrocarbons are suppressed due to the ZSM-5 shell.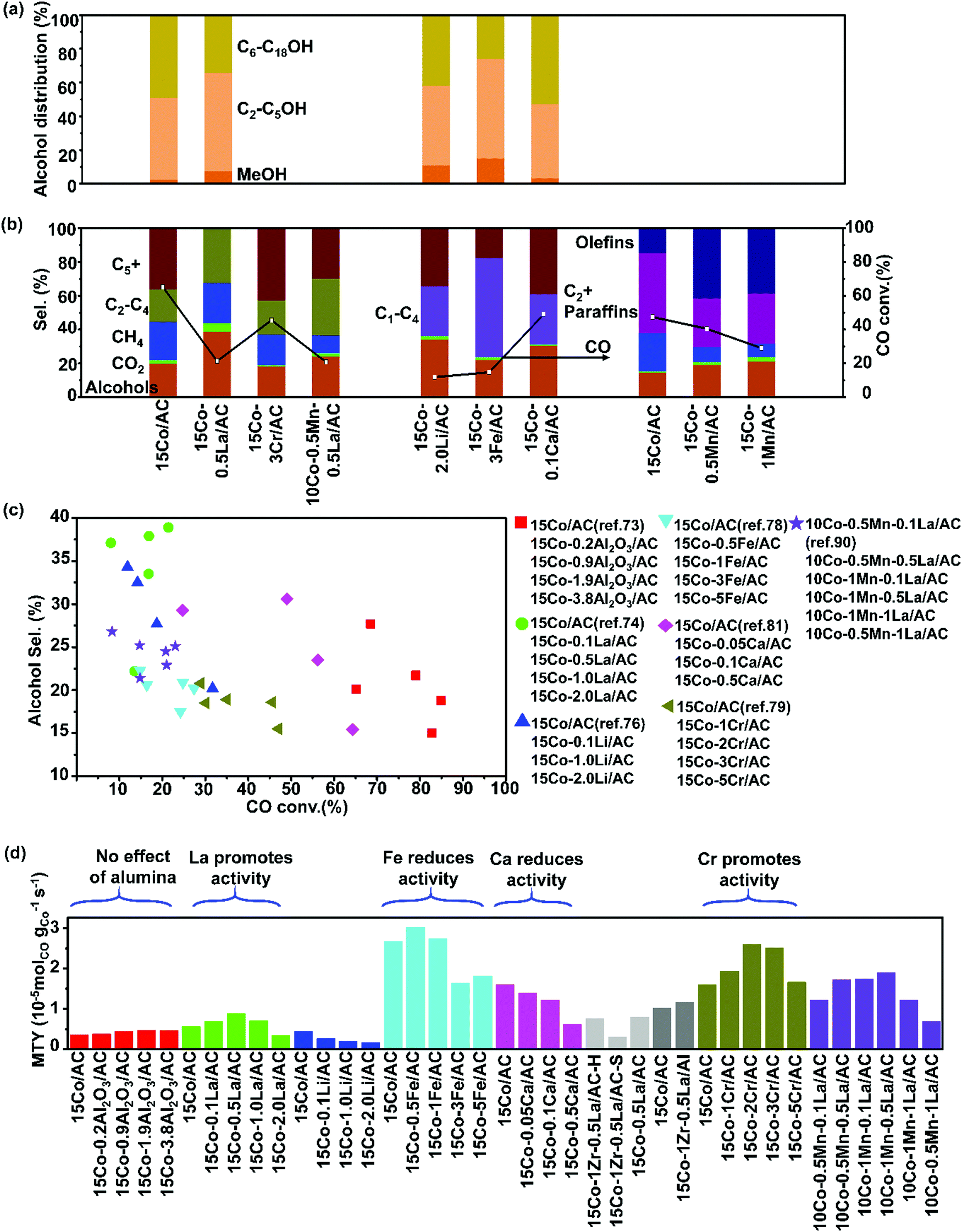 | ||
| Fig. 3 Catalytic performances of activated carbon supported cobalt catalysts with various promoters. Alcohol distribution (a), C selectivity (b), CO conversion vs. C5+ selectivity (c) and MTY (d). (Reaction conditions: 220 °C, H2/CO = 2, P = 3 MPa, gas hourly space velocity (GHSV) = 0.2–4 L gcat−1 h−1 and time on stream (TOS) = 24–60 h.) (a) Adapted from ref. 73, 74, 76, 78 and 81; (b) from ref. 73, 74, 76, 78, 79, 81, 88 and 99; (c) and (d) from ref. 73, 74, 76, 78, 79, 81 and 88. | ||
The Co/AC catalysts were found to achieve the CO conversions of up to 87.4%, while exhibiting low methane selectivity and high C5+ selectivity, when 4 wt% V was added as a promoter.75 The addition of vanadium enhances the CO dissociation and leads to a higher concentration of surface-active carbon species, improving the formation of long-chain hydrocarbons (Fig. 3). Lithium has also been used as a promoter in Co/AC and significantly suppressed the formation of gaseous hydrocarbons and increased the selectivity to higher hydrocarbons (C5+) and mixed linear α-alcohols (C1–C18).76 The 15 wt% Co/AC (15Co–2.0Li/AC) catalysts with 2.0 wt% amounts of Li exhibits selectivity to alcohols of 34.3%, compared to 20.2% over Co/AC (Fig. 3). The promotion with Li decreases the reducibility of Co2+ resulting in suppressed CO dissociation and lower activity (Fig. 3). The addition of Li may tune the relative content of surface Co and Co2C species.85 Co2C has been shown to exist in some Co based catalysts for the selective synthesis of alcohols. It shifts selectivity through the molecular binding of CO on its surface.67,86–88
Cobalt supported on almond AC shows lower alcohol selectivity than supported on AC made from coconut shells.33 Syngas conversion and CH4 selectivity increase and the content of the C12–C20 hydrocarbons in the liquid organic product decreases as Co loading amount rises from 7 to 20 wt%. The addition of K to the Co/AC catalyst decreased the activity and CH4 selectivity, but increased the selectivity to CO2. The Ce promoted Co/AC catalyst shows high activity and CH4 selectivity which is due to the improved Co dispersion and interaction between Co oxide and AC surface. The 5 wt% Fe doped 15Co/AC catalyst (15Co–5Fe/AC) demonstrates an alcohol selectivity of 20.6 with the main production of the C2–C5 alcohols.78 The metal impregnation sequence plays an important role in the activity, selectivity, and product distributions of the Fe/AC catalysts.89 The impregnation of Fe followed by Co on a 15Co–5Fe/AC catalyst was found to result in a more active catalyst than the reversed order of impregnation. The Co–Fe alloy was observed in bimetallic 15Co–5Fe/AC which exhibits high CO uptake and more cobalt active sites on the surface of the catalyst.
The Cr promotion of the Co/AC catalysts was revealed as an alternative method of tuning the selectivity in FTS by enhancing the hydrogenation step, rather than CO dissociation. 2 wt% Cr promoted 15Co/AC (15Co–2Cr/AC) shows much higher activity than 15Co/AC (Fig. 3).79 The CO conversion was elevated from 28.9% to 47.0% and the C5+ selectivity increased from 32.0% to 41.4% with the addition of Cr. The high selectivity to paraffins was attributed to the relative H-rich surface environment, which was caused by the enhancement of H2 uptake by the Cr promotion. The α-hydrogen addition step was facilitated, while the β-hydride elimination and CO insertion steps were prohibited simultaneously with the Cr addition. The Cr addition inhibits the formation of Co2C because of the enhancement of H2 adsorption. The Cr2O3 particles are easy to aggregate on the surfaces of AC supports and gradually cover Co nanoparticles when the loading of Cr is above 2 wt%.
0.2 wt% La was added into 10Co–4Zr/AC. The promoted catalyst showed high CO conversion and low methane selectivity (Fig. 3).80 The high activity comes from the higher reducibility of cobalt promoted by La. Excess of La (0.3–1.0 wt%) exhibits higher methane selectivity due to the decrease in the degree of reduction and the high concentration of Co2+ sites. The addition of 1 wt% La into 15Co–0.5Mn/AC improved the selectivity towards alcohols.90 The hydrogenation of alkyl chains was favored in La promoted CoxMn/AC due to the enhancement of H2 chemisorption, resulting in a shift of the product selectivity toward short-chain length hydrocarbons. A relatively low loading of La provides a strong promotion effect on the alcohol synthesis. An excess amount of La tends to aggregate on the surface of Co nanoparticles leading to an opposite effect. The addition of La into Co–Zr/AC leads also to the alcohol selectivity as high as 42% with a contribution of C6–C18 alcohols reaching 50.4% in total alcohols (Fig. 3).91 Co2C plays an important role in the synthesis of alcohols over the Co–Zr–La/AC catalyst. The addition of 0.1% CaO to the 15Co/AC catalyst improved the selectivity to alcohols accompanied by a drop of CO conversion (Fig. 3).81 The doping with CaO increases the amount of the Co2C phase in the catalyst, which is considered active for the synthesis of alcohols. CaO tends to interact with metallic Co during the reduction process which is more likely to transform into Co2C phase rather than face-centered cubic (fcc) Co. The synergistic effect of the Co and Co2C phases is responsible for the selective synthesis of alcohols over the 15Co–0.1CaO/AC catalysts in FTS. The Co2C species were also found in the Co/AC catalysts after reduction at 350 °C.92 The activity of the reduced Co/AC was, however, lower than for other Co-based catalysts due to the formation of less active Co2C species (Fig. 3). The doping with Mn and Cr promoters could restrain the formation of Co2C species, thus improving the activity in the CO hydrogenation.93,94
In short, the main goal of the promotion of Co/AC is enhancement of the formation of the Co2C phases, increase in reducibility of cobalt oxides, and enhancement of CO dissociation. This results in high alcohol selectivity, high activity of FTS, and low methane selectivity. Representative catalytic data of promoter effects on Co/AC for FTS have been listed in Table S1 in ESI.†
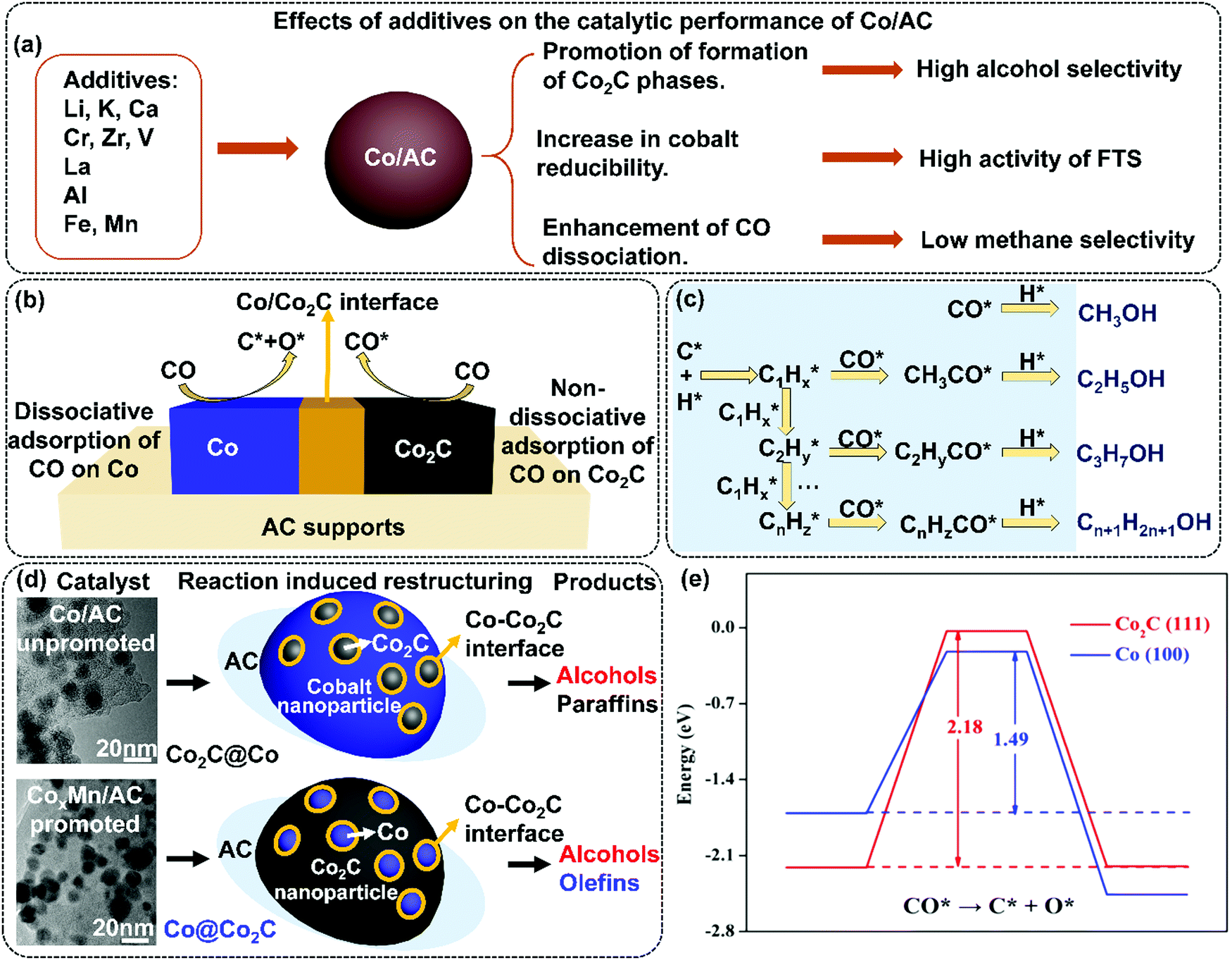 | ||
| Fig. 4 Effects of additives on the catalytic performance of Co/AC (a); dissociation and non-dissociation of CO on Co/Co2C interface (b); the alcohol formation mechanism (c); TEM images, conceptual frameworks, and products of Co/AC and CoxMn/AC catalysts (d), and energetic and geometric information for direct CO activation on Co2C(111) (red) and fcc Co(100) (blue) surfaces: potential energy diagram for direct CO dissociation (activation barriers and reaction energies in eV are indicated) (e). (a) Adapted from ref. 67, 73–83, 85, 86 and 89–92; (b) and (c) from ref. 74, 81, 82 and 92; (d) Reprinted with permission from ref. 99. Copyright (2017) American Chemical Society. (e) Reprinted with permission from ref. 82. Copyright (2015) American Chemical Society. | ||
The mechanism of Co2C formation was investigated in the Mn promoted Co/AC catalysts. Mn was found to promote the transformation of metallic Co to Co2C.99 As shown in Fig. 4(d), the spent 15Co1Mn/AC (15 wt% Co and 1 wt% Mn) and 15Co/AC catalysts exhibited different microstructures. It can be proposed that the active site of 15Co/AC can be related to the Co2C phase on the surface of Co nanoparticles (∼11 nm), named Co2C@Co, and that in 15Co1Mn/AC, the residual small Co nanoparticles (<6 nm) are distributed on the surface of large Co2C nanoparticles (∼20 nm), named Co@Co2C. The 15Co1Mn/AC catalysts demonstrate lower CO conversion (29.1%) than that of 15Co/AC (47.5%), yet the lower methane selectivity (8.1%) and higher olefin selectivity (38.5%) was observed than those of 15Co/AC (22.9% for methane and 14.7% for olefins). The selectivity to alcohol over both catalysts is similar (∼20%), as shown in Fig. 3(b). The lower content of the Co sites compared to Co2C sites is responsible for the lower activity of the 15Co1Mn/AC catalyst, while its C-rich and H-lean surface leads to high selectivity to olefins. The synergistic effect between Co and Co2C phases is responsible for the formation of alcohols. The authors conclude that the Mn promoter facilitates the dissociation and disproportionation of CO on the surface of Co/AC catalyst resulting in the C-rich and H-lean surface chemical environment. Density functional theory (DFT) calculations show that the calculated barrier of CO dissociation is 2.18 eV on Co2C(111), which is indeed much higher than that of 1.49 eV on Co(100), as shown in Fig. 4(e). Hence, it can be concluded that Co2C is highly efficient for the CO non-dissociative adsorption, whereas the Co metal is highly active for the CO dissociative adsorption and the subsequent carbon-chain growth. The interface between the cobalt metal and its carbide phase could be used to rationalize the catalyst design for the synthesis of oxygenates.
2.2 Carbon nanotubes supported cobalt catalysts
The main drawback associated with traditional supports like SiO2, Al2O3, and TiO2 is their strong interaction with metal particles. CNTs can serve as inert supports to overcome this drawback. Tavasoli et al. compared the effect of CNTs and γ-Al2O3 on the size, dispersion and reducibility of cobalt particles and further on the activity and selectivity of FTS.100 Co/CNT showed lower reduction temperature than Co/γ-Al2O3 which resulted from the weaker metal–support interactions between cobalt particles and CNTs and smaller size of cobalt particles. The consequent FTS activity of Co/CNT was better than that of Co/γ-Al2O3. Co/CNT favours the production of lower hydrocarbons. From the industrial scale, the higher activity of Co/CNT may decrease on the one hand, the reactor volume requirements and then decrease the capital cost of the plant as compared to the Co/Al2O3 catalyst. On the other hand, the lower amount of C20+ wax products of Co/CNT may decrease the size of hydro-cracker reactor for further product upgrading. However, Co/CNT showed a more rapid FTS rate drop with the reaction time than that for Co/Al2O3 due to the faster sintering of Co particles. CNTs were prepared and grown on MgO and Al2O3 to get CNT-MgO and CNT-Al2O3. Then, the elaborated materials were used as supports for cobalt catalysts.101 AC, MgO, and conventional Al2O3 were considered as reference supports. The deposition of CNTs on MgO and Al2O3 significantly suppressed the interaction of cobalt particles with both MgO and Al2O3 supports, which in turn led to good reducibility of cobalt species. 15Co/CNT-MgO significantly enhances the selectivity to C5+ hydrocarbons, with about ten-fold enhancement in the olefin to paraffin (O/P) ratio. The observed effects result from the synergy effect between CNTs and MgO.CNT was also compared with other carbon supports for the preparation of cobalt catalyzed FTS catalysts.102 Coville's group prepared a series of the Co/CNT and Co/CS catalysts with different cobalt particle sizes (3–45 nm) by different methods and using different cobalt precursors.103 The Co/CNT and Co/CS catalysts can be auto-reduced by the supports by calcination at 480 °C in N2. They demonstrated better FTS activity than those reduced above 400 °C in H2. The catalytic activity depends only on the particle size no matter what kinds of precursors and preparation methods were used. The turnover frequency (TOF) was constant for cobalt particles above 10 nm and decreased sharply for catalysts with cobalt particles below 10 nm (Fig. 5). Cobalt catalysts were prepared using three kinds of carbon supports of CNTs, CNFs, and fibrous material (FM).104 The FM support demonstrated a relatively large surface area with more surface oxygen groups and defects, which favored the formation of small cobalt particles with more significant hydrogen uptake. Hydrogen spillover was promoted by the surface oxygen groups and defects on the support.105,106 As a result of hydrogen spillover, the 15Co/FM catalyst showed the highest selectivity to methane and alkanes, as well as the lowest O/P ratio, compared with 15Co/CNT and 15Co/CNF.
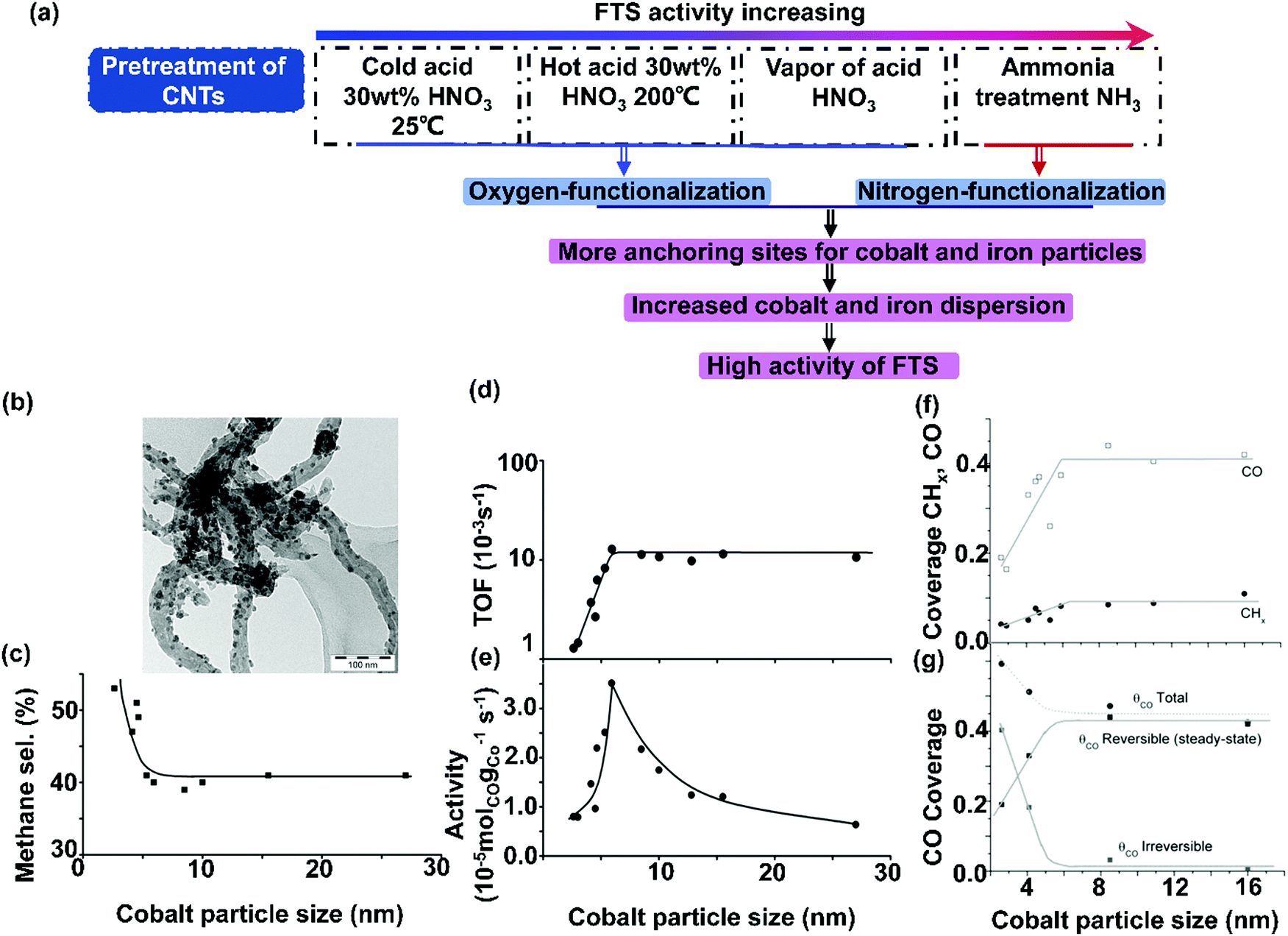 | ||
| Fig. 5 Effects of pretreatment of CNTs on catalytic performances of FTS (a); TEM image of Co/CNF-HDP9 with cobalt particle sizes of around 14 nm distributed over CNFs (b); the influence of cobalt particle size on methane selectivity (220 °C, H2/CO = 2, 1 bar) (c); the influence of cobalt particle size on the TOF (220 °C, H2/CO = 2, 1 bar) (d); the influence of cobalt particle size of Co/CNF on activity (220 °C, H2/CO = 2, 1 bar) (e); coverage of reversibly bonded CHx and CO as function of cobalt particle size (210 °C, 1.85 bar, H2/CO = 10) (f) and CO coverage from CO introduction, reversibly (from steady-state measurements) and irreversibly bonded CO (210 °C, 1.85 bar, H2/CO = 10) (g) (Θco total = the total CO coverage; Θco reversible(steady-state) = the coverage of the reversibly bonded CO during steady-state isotopic switches; Θco irreversible = the coverage of the irreversibly bonded CO). (a) Adapted from ref. 115–118; (b)–(e) Reprinted with permission from ref. 51. Copyright (2006) American Chemical Society. (f) and (g) Reprinted with permission from ref. 53. Copyright (2009) American Chemical Society. | ||
CNTs can be functionalized before being used as a support material for metal catalysts, to improve the wetting properties for aqueous solutions, as well as enhancing metal precursor deposition and creating anchoring sites for the metal nanoparticles.107,108 Many approaches have been used to functionalize the surface of CNTs, such as acid treatment, base treatment, oxidation and plasma treatment, vacuum-ultraviolet photochemical reactions, and microwave procedures.109–112 The most efficient and lowest cost process of liquid-phase oxidation is acid treatment by using nitric acid, sulfuric acid, or acid mixtures (Fig. 5(a)).113–115 Dalai et al. has investigated the influence of acid (30 wt% HNO3) treatment on CNTs supported cobalt catalysts.116 The as-prepared CNTs were used for the preparation of 10Co/CNT (10 wt% Co). The 10Co/CNT-cold acid catalyst and 10Co/CNT-hot acid catalyst were prepared by treatment of the as-prepared CNTs with 30 wt% HNO3 at 25 °C and 100 °C respectively. The BET surface area of acid-treated CNTs increased by 18 and 25% for the 10Co/CNT-cold acid and 10Co/CNT-hot acid catalyst, respectively, in comparison with 10Co/CNT. Simultaneously, the cobalt particle size was decreased and the cobalt dispersion was increased. The CO conversion increased from 35 to 50% for the 10Co/CNT-cold acid and 10Co/CNT-hot acid catalyst respectively due to the improved reducibility (Fig. 6). The product selectivity of the acid-treated catalyst shifted towards methane. Zhang et al. found that acid pretreatment of CNTs removed impurities and oxidized the surface. This can enhance to some extent the catalytic activity of FTS, yet it does not affect cobalt dispersion.117 CNTs with different diameters exhibited similar cobalt particle sizes and reducibility, and therefore they also demonstrate similar FTS catalytic performance. It appears that the cobalt particle size of Co/CNT was influenced by chemical interactions between cobalt particles and CNTs likely originating from the decomposition of cobalt precursor, nucleation, and growth of cobalt oxide crystallites.
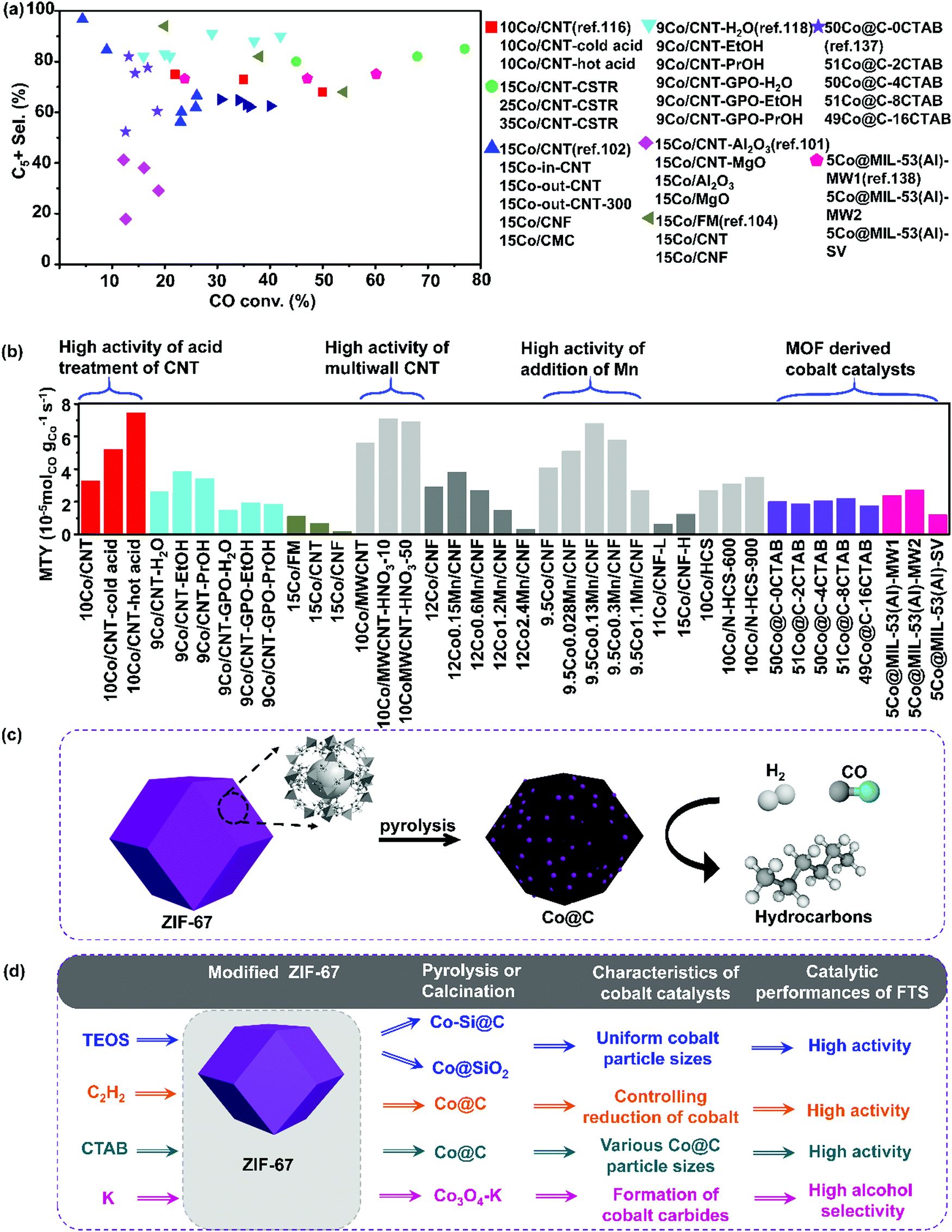 | ||
| Fig. 6 Catalytic performances of carbon-based cobalt catalysts with various carbon supports. CO conversion vs. C5+ selectivity (a) and MTY (b). (Reaction conditions: 220–240 °C, H2/CO = 2, P = 1–2 MPa, GHSV = 0.7–6.8 L gcat−1 h−1 and TOS = 10–72 h.) ZIF-67 derived Co@C catalysts for hydrocarbons production from syngas (c) and modified ZIF-67 derived cobalt catalysts for FTS (d). (a) Adapted from ref. 34, 101, 102, 104, 116, 118, 137 and 138; (b) from ref. 107, 108, 116–118, 134, 137 and 138; (c) from ref. 144 and (d) from ref. 136, 139 and 143–145. | ||
Dalai et al. investigated the influence of Co loading (15, 25, 35, and 40 wt%) on CNTs on their catalytic performance for FTS using a continuous stirred tank reactor.34 The activity of Co/CNT was improved largely with cobalt loading increased from 15 to 40 wt% due to the smaller size of Co nanoparticles even at higher cobalt loadings, good reducibility and also the reduced metal–support interactions between Co nanoparticles and CNTs support. 40 wt% cobalt loading demonstrated the highest FTS activity. De Jong et al. also prepared 9Co/CNT–H2O, 9Co/CNT–EtOH, and 9Co/CNT–PrOH through incipient wetness impregnation of CNTs with 1.5 M water, ethanol, and 1-propanol solutions of cobalt nitrate hexahydrate respectively.118 Also, 9Co/CNT–GPO–H2O, 9Co/CNT–GPO–EtOH, and 9Co/CNT–GPO–PrOH were prepared by using CNTs treated with vapors of nitric acid (65% nitric acid was heated to reflux) as supports. Co3O4 nanoparticles manifest better reducibility when ethanol and 1-propanol are used as solvents for impregnation compared to water. The catalysts prepared with alcohol solutions showed a superior cobalt-weight based activity than those prepared from water solution, as they had higher specific metal surface areas (Fig. 6). The activity of untreated 9Co/CNT was higher than that of the acid-treated 9Co/CNT owing to the formation of hexagonal closest packed Co phases. However, 9Co/CNT with acid treatment shows higher stability than those without acid treatment due to aggregation of cobalt particles induced by the lack of anchoring sites in the non-treated samples. Multiwall carbon nanotubes (MWNTs) were used as supports for preparing 10Co/MWNTs and Fe/MWNTs by two methods of impregnation with acetate precursors and deposition of pre-prepared metal oxide nanoparticles.119 The 10Co/MWNTs and 10Fe/MWNTs catalysts showed lower degrees of reducibility of the metal particles, resulting in lower activity. Fig. 6(b) demonstrates metal-time-yield (MTY) of carbon-based cobalt catalysts with various carbon supports. Multiwall CNT and acid pretreated CNT supported cobalt catalysts exhibit high activity. In a different study, CoCu/CNT was prepared by constant pH co-precipitation of metallic components on CNTs, the impregnated catalyst showed higher selectivity to oxygenates such as butanol and dimethyl ether than CNT-free conventional co-precipitated Co–Cu counterpart.120
2.3 Carbon nanofibers supported cobalt catalysts
Fischer–Tropsch reaction is a structure sensitive reaction, which indicates that the catalytic performance is strongly associated with the particle size of the active phase. The effect of cobalt particle size on the catalytic performance might be obscured by the effect of strong metal–support interactions.121–123 To study the intrinsic particle size effect and to eliminate the support effects, CNFs can be used as an inert carrier material. The research group of Krijn P. De Jong of Utrecht University has done numerous studies on the cobalt size effects in FTS by using CNFs as supports. They found that CNFs were promising supports for FTS. Co/CNF showed stable activity at a running time of 400 h.124 The Co particle size effect was investigated further where a series of Co/CNF catalysts with cobalt particle sizes varying from 2.6 to 27 nm were prepared by using different synthesis methods and various weight loadings. As shown in Fig. 5(b), representative Co/CNF-HDP9 was prepared by homogeneous deposition precipitation (HDP) with cobalt loading of 9%. The sample exhibited rather long fibers decorated with 14 nm cobalt particles.51 The TOF and hydrocarbon selectivity were independent of the cobalt particle size for the catalysts with the Co particle sizes larger than 6 nm, but both activity and selectivity to the C5+ decreased for the catalysts with Co particle sizes below 6 nm. Fig. 5(e) shows that the cobalt-specific activity achieves the maximum value for Co/CNF with a cobalt particle size of 6 nm. The TOF was calculated using the dispersion obtained from X-ray photoelectron spectroscopy. The TOF data are plotted in Fig. 5(d). It can be concluded that TOF is rather constant with a value of around 10−2 s−1 for the cobalt particles ranging from 6 to 27 nm. For the cobalt particles with particle sizes below 6 nm, TOF steeply decreases to a value close to 10−3 s−1. In Fig. 5(c), the methane selectivity is plotted as a function of cobalt particle size. The catalysts with the cobalt nanoparticle sizes smaller than 5 nm displayed progressively higher methane selectivity. The formation of methane indicates that the catalyst surface is enriched with dissociated hydrogen. The higher selectivity to methane also indicates a lower abundance of the sites active for chain growth. Therefore, it is confirmed that the minimum desired cobalt particle size for FTS is 6 nm. Extended X-ray absorption fine structure (EXAFS) evidenced that the cobalt coordination number decreased under reaction conditions, which resulted from the reconstruction of the cobalt particles. Therefore, it was argued that a combination of CO-induced surface reconstruction and non-classical structure sensitivity can be considered as a reason for the observed cobalt particle size effect.Steady-state isotopic transient kinetic analysis (SSITKA) was used to investigate the cobalt particle size effects.53 It was concluded from SSITKA that the lower intrinsic activity of smaller cobalt particles (<6 nm) was the result of a significant increase in the CHx residence time and a decrease in the CO residence time, together with a decrease in the surface coverage of the CHx, OHx and CO intermediates (Fig. 5(f)). The higher methane selectivity stems from the higher coverage of hydrogen. The residence times and surface coverages of CHx, OHx, and CO appeared to be constant for Co/CNF with the Co particle size above 6 nm, therefore, the activity of Co/CNF is independent of the size for larger Co particles (>6 nm).52 The total CO coverage was calculated as a function of Co particle size and compared with the coverage of the reversibly bonded CO from SSITKA (Fig. 5(g)). This graph shows that the total CO coverage increased with decreasing Co particle size (<6 nm). The amount of reversibly bonded CO decreased for small cobalt particles during steady-state isotopic switches. The difference between those two coverages reveals an increase in the irreversible CO coverage with decreasing particle size. This means that the surface of smaller particles was partly blocked with unreactive CO.
Other researchers have also investigated the promoter effects on Co/CNF for FTS. CNFs provide suitable supports, which enable the study of a promoter effect without interference between metal particles and support.125 Manganese was deposited onto the Co/CNF sample and the addition of 0.15 wt% Mn brought about a 25% increase in the C5+ hydrocarbon selectivity.107 It was considered that manganese oxides were closely associated with cobalt nanoparticles on CNFs thereby improving the selectivity in FTS. The addition of manganese to Co/CNF retarded cobalt reduction.126 CNFs support materials are also ideal supports to investigate the deposition–precipitation technique of cobalt catalysts for FTS.108 Co/CNF-L and Co/CNF-H were prepared through the conventional deposition from an acidic solution using urea hydrolysis and basic solution using ammonia evaporation respectively. The cobalt particle size of Co/CNF-L is 25 nm and Co/CNF-H is 8 nm. The smaller size of Co/CNF-H comes from the basic solution, which enhances the ion adsorption and further improves the cobalt dispersion on CNFs. Therefore, Co/CNF-H shows 2–4 times higher cobalt-specific activity in FTS than the Co/CNF-L catalyst (Fig. 6(b)).
CNFs with fishbone and platelet structure (CNF-P) were used as supports for cobalt catalysts.127 Co/CNF-P achieved high cobalt dispersion due to a high number of edge sites and surface oxygen groups of platelet structure. Hence the activity and C5+ selectivity were enhanced. The effect of water on the activity and selectivity of FTS has been investigated by using Co/CNF.128 Addition of 20 and 33 mol% water to the inlet syngas increased the reaction rates, but the deactivation rates also increased, due to an irreversible deactivation because of cobalt oxidation and sintering. A uniform SiO2 layer was deposited on CNFs to get a better catalyst.129 The Co/CNF prepared with a coating of SiO2 shows higher activity and C5+ selectivity than the uncoated catalysts, because the SiO2 layer hinders the oxidation of the cobalt species and thus enhances the stability of the Co catalyst.
2.4 Carbon spheres supported cobalt catalysts
CSs have many advantages including regular geometry, good mechanical strength, tunable porosity, surface functional groups, and controllable distribution of metal particles, which make them ideal model catalyst supports to investigate the effect of varying structural properties along with metal particle size and promoters on FTS.23,54,55 Co@C core–shell catalysts were synthesized through a direct carbonization method using the industrially available and renewable lignin as carbon sources.56 Co@C catalysts with carbonization temperatures of 600 °C showed remarkably high activity due to the core–shell structure facilitating electronic conductivity between cobalt and CO molecules and suppressing aggregation of cobalt nanoparticles. Nitrogen-doped carbons displayed superior activity when compared to oxygen-functionalized carbons for FTS catalysts.130,131 The mesoporous carbon-supported cobalt catalyst (15Co/MC) was more active and selective to the C5+ hydrocarbon production than 15Co/AC.132 The FTS activity was related to the ratio of adsorption heats of CO and H2. The best ratio was found of 1.2. The addition of 1 wt% K inhibited the FTS activity due to the increased surface basicity, which induced increasing adsorption heat of CO and decreasing the adsorption heat of H2. In contrast, the addition of ZrO2 increased the surface acidity which resulted in increasing heat adsorption of H2.132Carbon Molecular Sieves-3 (CMK-3) was used as supports for cobalt catalysts in FTS. CMK-3 was prepared using SBA-15 as a template and sucrose as a carbon source.133 Co/CNT, Co/CMK-3 and Co/AC were prepared by incipient wetness. Co3O4 particles of Co/CNT and Co/CMK-3 were mainly dispersed inside the pores yet those of Co/AC were outside the pores. Co/CNT exhibited the best FTS performances among three kinds of catalysts. Good crystallized graphitic structure of CNTs facilitated the electron transfer between the cobalt and CO molecules (Fig. 6). Porous hollow carbon spheres (HCSs) materials were functionalized by N doping through a post-synthesis method with melamine as the nitrogen source.134 Various levels of N doping and different bonding configurations of HCSs were obtained through performing carbonization at different temperatures. Marginal defects of the carbon framework of HCSs were found. Both the activity and stability were improved on the N-doped HCSs supported cobalt catalyst (10Co/N-HCS). The cobalt nanoparticle size of spent 10Co/N-HCS was below 15 nm which was much smaller than that of 10Co/HCS of 30 nm, demonstrating the inhibition of sintering of cobalt particles over the N-doped HCSs supports. Representative catalytic data of Co/CNT, Co/CNF, Co/CS, and Co-MOF derived Co@C catalysts has been presented in Table S2 in ESI.†
2.5 MOF-derived carbon supported cobalt catalysts
High metal loading is essential for high catalytic activity. However, during FTS, larger nanoparticles and/or clusters may form, leading to lower metal dispersion and lower reaction rate. Therefore, the maximum metal loading that can be achieved via the widely-used method of impregnation remains below 30 wt%.4,135 In this respect, it is important to develop alternative preparation methods and obtain FTS catalysts with both high metal loading and optimal particle size distribution. The MOFs derived Co@carbon nanoreactors have been exploited for this purpose. Zeolitic imidazolate frameworks-67 (ZIF-67) is a typical kind of Co-MOF material and the preparation process of cobalt catalysts derived from ZIF-67 is shown in (Fig. 6(c)). The preparation process of modified ZIF-67 derived cobalt catalysts has been summarized in Fig. 6(d).136–143Kapteijn et al. developed Co@SiO2 catalyst by a stepwise methodology of making use of a cobalt-containing MOFs as a hard template.144 The first step is the impregnation and hydrolysis of TEOS molecules in the pores of ZIF-67. The second step is the pyrolysis of ZIF-67@SiO2 in N2 resulting in the Co@C–SiO2 catalyst. The final step is the calcination of Co@C–SiO2 in the air to remove carbon. This preparation method results in well-dispersed cobalt nanoparticles with sizes of 5–15 nm. The cobalt loading was as high as ∼50 wt% with cobalt oxide reducibility of the order of 80%. Most importantly, the obtained Co@SiO2 catalyst showed higher activity than the traditional impregnated Co/SiO2 counterpart (Fig. 6(d)). The CO conversion of Co@SiO2 is twice higher that of Co/SiO2 and three times higher that of Co@C derived from the pyrolysis of cobalt-containing MOFs. The CH4 selectivity of Co@SiO2 is similar with that of Co/SiO2, but with improved stability.
The FTS activity of MOF-derived Co-based catalysts is heavily influenced by the carbon pore structure and pore size distribution in the final catalyst.4,145,146 Lower porosity has been shown to lead to lower activity in FTS. Nitrogen species in the carbon matrix of the MOFs derived cobalt catalysts can also affect the FTS activity. Ma's group investigated both the effect of porosity and nitrogen species in the MOF-derived cobalt-based catalysts by using two different kinds of MOFs.145 One kind of MOFs used was Co-MOF-74 and after pyrolysis, the Co@C nanoreactors without N atom in the carbon matrix were obtained. Another kind of MOFs used was ZIF-67, and after pyrolysis, Co@NC nanoreactors with N atom in the carbon matrix were produced. It was observed that Co@C had a broader mesopore distribution with a larger average pore size of 7 nm, whereas Co@NC had a smaller average pore size of 3 nm. Larger mesopores in the MOF-derived nanoreactors could facilitate gas diffusion in the carbon matrix. Hence Co-MOF-74 derived Co@C demonstrated higher catalytic activity than ZIF-67 derived Co@NC nanoreactors. N species in the carbon matrix are viewed as efficient electron donors to accelerate the CO adsorption–dissociation process and tune the product selectivity towards short-chain hydrocarbons in FTS. Co@NC exhibited lower CO conversion of 10%, lower selectivity to C5+ products of 31%, and higher selectivity to C2–C4 of 37%, while Co@C showed higher CO conversion of 30%, higher selectivity to the C5+ products of 65%, and lower selectivity to the C2–C4 hydrocarbons of 10%.
Liu's group investigated the size effect of cobalt metallic nanoparticles in the Co/C catalysts derived from ZIF-67 on the catalytic performance of FTS.144 Co/C catalysts with Co particle sizes ranging from 8.4 to 74.8 nm were obtained by pyrolysis of ZIF-67 under temperatures from 450 to 900 °C. The decomposition of ZIF-67 started at about 350 °C and metal cobalt nanoparticles entrapped in partially graphitized carbon matrix were produced after pyrolysis of ZIF-67 above 450 °C. The carbon matrix was polished by some pyridinic and graphitic N and a small amount of adsorbed oxygen. There was nearly no occurrence of sintering and re-oxidation during FTS reaction. This suggests that the uniformly distributed Co particles confined in porous carbon matrix exhibited good stability. The TOF value increased from 1.8 × 10−2 to 4.0 × 10−2 s−1 when the size of cobalt nanoparticles rose from 8.4 to 10.5 nm, yet staying stable in the range of Co nanoparticle sizes from 10.5 to 74.8 nm. The product selectivity is independent of cobalt particle size between 8.4 and 47.8 nm. Notably, they studied the effect of N species inside the ZIF-67 derived Co/C on the catalytic performances. EXAFS fitting revealed that there were no Co–N bonds in the Co/C catalyst and the nitrogen species were well distributed in the carbon matrix with pyridinic and graphitic structure. This confirms there were no electronic effects between N and Co atoms and there was no enhancement of CO adsorption–dissociation effect by N species. In short, they concluded that the pyrolysis of ZIFs might offer an alternative method for the preparation of uniformly distributed cobalt nanoparticles on carbon support with excellent stability. The cobalt nanoparticles of 10 nm demonstrated the optimum performance in FTS.
Li's group focused on the Si-doped Co@C species impregnated on Co-MOF-71 for FTS.141 The mean size of the Si-doped Co@C catalysts was tailored by varying the amount of Si species. High Co site density and good cobalt dispersion were achieved. The Si-doped Co@C catalysts showed good FTS activity and unprecedented high diesel selectivity. The C5+ hydrocarbon space time yield was 1.45 g gcat−1 h−1, which was much higher than that of traditional supported catalysts.
Isaeva and Kustov have demonstrated that MIL-53(Al) can be utilized as a carrier for Co nanoparticle deposition. The obtained Co@MIL-53(Al) catalysts exhibited higher selectivity to the C5+ hydrocarbons and lower selectivity to methane than conventional Co/Al2O3.138 The Co nanoparticles were immobilized on MIL-53(Al) under H2 flow at 400 °C without any degradation of the porous host matrix. In a follow-up study, they attempted to elucidate the influence of the morphology and particle size of the MIL-53(Al) on the catalytic performance of the subsequent Co-catalyst in FTS. The enhanced activity and selectivity in FTS can be obtained on the Co-catalyst derived from the nano-scaled MIL-53(Al) support. This effect can be due to the decreased diffusion limitations of nanoscale MIL-53(Al) support and more homogeneous distribution of cobalt nanoparticles in the metal–organic matrix. Also, the nanocrystalline form of MIL-53(Al) support demonstrated a pronounced confinement effect, which prevented cobalt nanoparticles from aggregation during FTS. It can be seen from Fig. 6(a) that the C5+ selectivity is significantly higher over the CNT and CNF supported cobalt catalysts than MOF derived cobalt catalysts. This could be explained by defective cobalt in the MOF derived cobalt catalysts providing high methane selectivity and low C5+ selectivity.
The Co@C catalysts derived directly from Co-MOF have a broad size distribution with cobalt nanoparticle sizes varying from ten to hundreds of nanometres. Moreover, cobalt catalyzes graphitization of carbon during pyrolysis under high temperatures. The graphitized carbon shell around cobalt nanoparticles may not be porous, which hinders the transport of chemical reactants to the active sites. In order to overcome these drawbacks of MOF-derived cobalt-based catalysts, Li's group developed a new preparation approach for the MOF-derived Co@C catalysts.139 Co-MOF are established for the fabrication of cobalt nanoparticles in porous carbon shells and chemical vapor deposition (CVD) of ethyne over MOFs is utilized for the manufacture of ultrasmall cobalt species. The cobalt nanoparticles in the Co-MOF when were then reduced by the hydrogen released from ethyne during pyrolysis (Fig. 6). The reduced cobalt nanoparticles were encapsulated in the carbon matrix, which came from ethyne decomposition. The resulting carbon shells are porous and accessible for the reactants and products. The obtained Co@C nanoparticles exhibited high catalytic activity and selectively converted syngas (CTY = 254.1–312.1 μmolCO gCo−1 s−1) into hydrocarbons (4.0–5.2 gHC gcat−1 h−1) at 260 °C and 3.0 MPa. The excellent catalytic performance in FTS reactions were linked to the permeability of the porous carbon shell of Co@C. Note that Fig. 6(a) shows significantly higher C5+ selectivity over CNT and CNF supported cobalt catalysts than over the MOF derived cobalt catalysts. This could be explained by defective cobalt of MOF derived cobalt catalysts providing higher methane selectivity and lower C5+ hydrocarbon selectivity. Fig. 6(b) demonstrates MTY of carbon-based cobalt catalysts with various carbon supports. Multiwall CNT and acid pretreated CNT supported cobalt catalysts exhibit higher activity, whereas the MOF derived cobalt catalysts manifest moderate activity. The addition of Mn promotes the activity of Co/CNF catalysts.
Representative catalytic data of Co/CNT, Co/CNF, Co/CS, and Co-MOF derived Co@C catalysts are presented in Table S2 in ESI.†
3. Iron supported on carbonaceous materials for Fischer–Tropsch synthesis
3.1 Activated carbon supported iron catalysts
Early studies have shown that the C1–C6 hydrocarbons are produced over a 3–5 wt% Fe/AC catalyst at 1 atm of pressure.63,147,148 More recently, it was reported that the C1–C20 hydrocarbons were the main product of FTS over Fe/AC.33,149 The pore structure of AC plays a role in tailoring the chain length of the hydrocarbons. Ding's group has found that Fe supported on AC catalyst favors during FTS the formation of hydrocarbons with fewer than 20 carbon atoms.150Potassium promoter also is influential in controlling activity and product distribution during FTS on Fe/AC catalyst.151 0.9 wt% K promoted Fe/AC showed high FTS and water-gas shift activities, whereas an opposite trend is observed on the 2 wt% K promoted catalyst. Potassium content of 0.9 wt% K in Fe/AC greatly decrease the amount of n-paraffins and internal olefins and dramatically increased branched paraffins and α-olefins, with the product selectivity shifting towards C5+ hydrocarbons and C2–C5 alcohols. Oxygenate selectivity of Fe/AC decreased with temperature, yet that of K promoted Fe/AC is almost independent of the temperature. The addition of Cu to the Fe–K/AC catalyst promotes oxygenates and internal olefins formation, without change of overall HC distribution.152 Increasing the Cu loading from 0.8 to 2.0 wt% results in the decreased activity due to the suppressed carburization of Fe by Cu. It is confirmed that Cu promotes hydrogen and CO adsorption and inhibits CO dissociation on the surface of Fe–K/AC catalyst. The addition of Mn to Fe–Cu/AC leads to higher yield and selectivity towards the lower olefins. Mn distributes uniformly on the AC surface and simultaneously forms mixed species with iron oxide.153 The addition of 6 wt% Mo to 15.7Fe–0.8Cu–0.9K/AC results in the improved catalyst stability without sacrificing activity. The effect is due, inhibition of agglomeration of iron by Mo.154 The reduction of 15.7Fe–0.8Cu–0.9K–6Mo/AC was suppressed by strong interaction between the Fe and Mo oxides.
AC can also be treated to improve anchoring of the catalytically active phase. In a study,155 focused on the FT to olefins (FTO) process, different concentrations of KMnO4 solutions were used to do the pretreatment of AC. The KMnO4 pre-treatment resulted in a uniform distribution of K-doped MnO2, oxygen-containing groups and defects on the support surface. The enrichment with defects and oxygen-containing groups anchors α-Fe2O3 particles with small average particle sizes. Manganese in AC serves as H2 adsorption competitor which may decrease the H2/CO ratio over active sites and further reduce the hydrogenation of olefins. Residual potassium favors the formation of iron carbides leading to high activity. The KMnO4 pretreated AC (10MnK-AC) was used as support for preparing N-doped Fe catalysts by using ammonium iron citrate as an iron precursor.156 The lower olefin selectivity of 10FeN–27Mn–5K/AC accounts for 44.7%. An improvement in O/P ratio was simultaneously observed. The promotion effect comes from the nitrogen atoms doped into iron lattice leading to the electron donor effect and suppression of the secondary hydrogenation by 10MnK-AC.
Preparation method of AC supported Fe–K catalysts play an important role in the FTS catalytic performances.157 The preliminary alkalized AC support exhibits small iron particles (average diameter 5–7 nm) with narrow size distribution. Yet the reverse order of the Fe and K deposition over the catalyst results in larger iron particles (average diameter 15–16 nm) with a broad size distribution. The former one manifests higher activity due to higher concentration of active sites of γ-Fe5C2.
3.2 Carbon nanotubes supported iron catalysts
The CNTs with unique tubular morphology can be used as nanoreactors to create novel composites through encapsulating iron carbide nanoparticles within their well-defined channels.158Firstly, they have developed strategies for the preparation of homogeneously dispersed nanoparticles inside CNTs, focusing mainly on MWCNTs with an inner diameter smaller than 10 nm and double-walled CNTs with 1.0 nm. CNTs with well-defined morphology and unique electronic structure provide a unique confinement environment for metal particles. They have demonstrated that catalytic performances are different for metal or metal oxide nanoparticles confined inside CNTs and the same species deposited on the exterior surface of CNTs. Furthermore, these differences of catalytic performances vary based on the used metals and catalytic reactions.
Secondly, they presented the first example of the CNT-confined iron metal particles obtained through direct reduction of incorporated iron oxide nanoparticles by the CNTs host.38 Multi-walled CNTs with an inner diameter of 4–8 nm and an outer diameter of 10–20 nm were loaded with Fe2O3 (8 wt%) and the reduction of Fe2O3 particles was monitored in situ by High Resolution Transmission Electron Microscope with the temperature heating from 20 to 600 °C. They demonstrated the direct experimental evidence of facile reduction of Fe2O3 nanoparticles deposited inside the inner surface of CNTs at 600 °C with respect to the reduction of those on the outer surface of CNTs at 800 °C.
Thirdly, following their previous findings that confinement within CNTs can modify the redox properties of encapsulated iron oxides, they later demonstrated how the resultant redox properties can affect the catalytic performances of iron catalysts in FTS.44 Fe-in-CNT (10 wt% Fe) with iron particles encapsulated inside CNTs shows higher FTS activity than Fe-out-CNT with iron particles deposited outside CNTs. This effect is due to the iron species of Fe-in-CNT preferentially existing in a reduced state. These reduced iron species form more active species of iron carbides under reaction conditions. The yield of the C5+ hydrocarbons of FTS over Fe-in-CNT catalyst is twice higher that over the Fe-out-CNT and more than 6 times higher that over Fe/AC. The enhancement of catalytic activity is attributed to the confinement of iron particles within the CNTs channels. The improvement of the C5+ hydrocarbon selectivity benefits from trapping the reaction intermediates inside channels of CNTs. The trapping prolongs their contact time with iron catalysts, leading to the growth of longer chain hydrocarbons. Furthermore, the spatial restriction of the channels of CNTs also retards the aggregation of the iron species during the reaction.
Finally, DFT calculations were performed to further understand the physical and chemical differences between Fe-in-CNT and Fe-out-CNT and the interactions between the iron species and walls of CNTs.164 A series of one-dimensional Fe and FeO nanowires, Fe atom and dimers were chosen as probes to study the interactions of iron with the interior and exterior CNTs surfaces. The Fe species bind stronger with the outer CNTs wall than with the inner one, which comes from the more electron-enriched frontier orbitals that are mostly located on the outer CNTs surface. Analysis of the electronic structure indicates that more electrons are distributed on the exterior surface of the CNTs. A more in-depth research was focused on the concept of “confinement energy” of CNTs, which enables the prediction of the catalytic performances in different reactions.165 A variety of transition metal clusters of Fe, Re, Ru, FeCo, and RhMn confined inside CNTs and deposited outside CNTs were investigated by DFT calculations. The findings revealed that the confinement was an intrinsic property of CNTs. The nano space was formed by the concomitant electronic structures of the curved graphene wall of CNTs. The weakened binding of molecules of CO, N2, and O2 over the encapsulated metal clusters was observed due to the downshifted d-band states of encapsulated metal clusters with respect to the metal clusters sitting on the CNTs exterior walls. Thus, the electronic effect of the confined space of CNTs shifts the volcano curve of the catalytic activities toward the metals with higher binding energies. This concept well described the catalytic activities of Fe/CNT in FTS.
The deformation of π bonding results in the electron transfer from the crowded concave side to the convex side, when a tube is formed through the rolling up of graphene, inducing a different electron distribution on the inner and outer surface of the CNTs. The electronic structure of the CNTs was studied by looking at the distribution of electrons on the occupied molecular orbital of CNTs. The inner surface of CNTs demonstrates an electron-poor environment and the outer surface of an electron-rich environment, as shown in Fig. 7(a) and (d). It is confirmed that the frontier orbitals of the CNTs are mainly located outside the CNTs. Therefore, combining all findings from the aforementioned studies (ref. 38, 44, 164 and 165) the respectively higher and lower FTS activity of Fe-in-CNT and Fe-out-CNT can be attributed to the evolution of iron species during reduction and reaction induced by the different electron distribution on the inner and outer surface of the CNTs, as shown in Fig. 7(b), (c), (e), and (f).
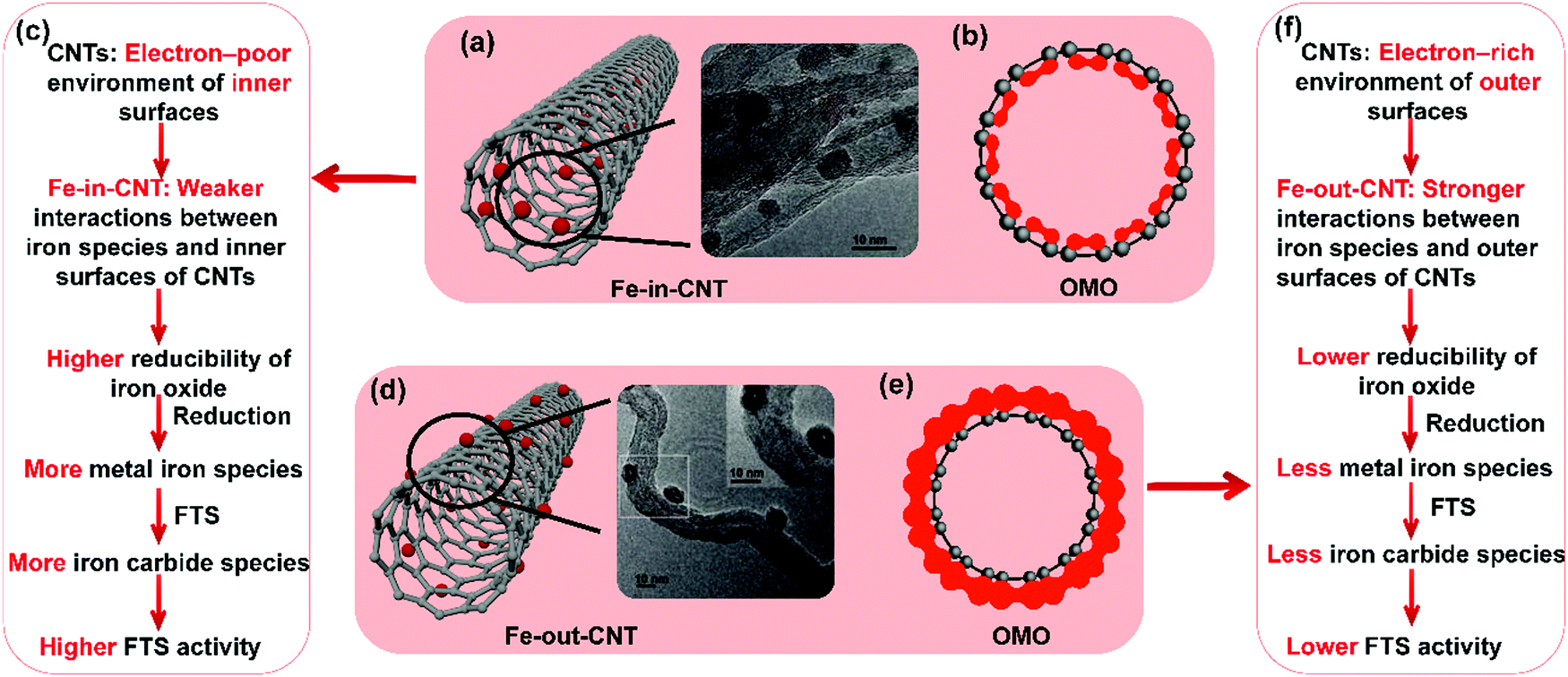 | ||
| Fig. 7 Summary of Fe–CNT catalyst properties. Scheme and TEM image (a), electronic density distribution of the occupied molecular orbital (OMO) of inner surface of CNTs (b), and confinement effects on the catalytic activity (c) of iron particles confined inside carbon nanotube (Fe-in-CNT). Scheme and TEM image (d), electronic density distribution of OMO of outer surface of CNTs (e), and interaction between iron particles and outer surfaces of CNTs on the catalytic activity (f) of iron particles supported on the outside carbon nanotube (Fe-out-CNT). (a) and (d) Reprinted with permission from ref. 44. Copyright (2008) American Chemical Society. (b), (c), (e), and (f) Summarized from ref. 38, 44, 164 and 165. | ||
The confinement effect of CNTs was also confirmed by several researchers. Fe-in-CNT and Fe-out-CNT with 70–80% of iron oxide particles deposited inside the inner and outside of the outer surface of CNTs respectively, were successfully obtained.166 Fe-in-CNT shows an easier reduction of iron oxide at 381 °C, which is lower than 418 °C for Fe-out-CNT. 12Fe-in-CNT exhibited higher selectivity to heavier hydrocarbons and more stable activity, while Fe-out-CNT experienced deactivation after testing for 125 h due to iron sintering.
The results of Khodakov's group167,168 also suggest that iron carbonization proceeds much easier for iron species confined inside CNTs and promoted with Bi and Pb. Iron nanoconfinement inside CNTs results in much higher iron dispersion. Nanoconfinement combined with the promotion with Bi or Pb resulted in a 10-fold higher yield of light olefins. The selective synthesis of light olefins from syngas can even occur at atmospheric pressure over confined iron catalysts promoted with Bi and Pb.
FexN–CNT has also been investigated as a catalyst for the FTS reaction.170 It has previously been shown that doping nitrogen atoms into the matrix of carbon can change the electronic environment and increase olefin selectivity.22,171,172 Nitrogen atoms act as an electron donor and suppress the secondary hydrogenation of lower olefins, resulting in higher olefins selectivity. 5.6Fe-in-CNT catalysts are prone to deactivation owing to oxidation of iron species by water under the reaction conditions. Nitrogen atoms can be incorporated into the Fe lattice to form iron nitride which overcomes this issue by enhancing resistance to oxidation of iron species. Iron can be nitrided with varying contents of nitrogen. For the first time, cubic FexN nanoparticles were synthesized through incorporation in CNTs channels to form FexN/CNT catalysts. 5.6FexN-in-CNT with FexN confined inside CNT channels exhibits higher activity than 5.2FexN-out-CNT with FexN particles located on the CNTs exterior walls in FTS. This might be attributed to the enhanced formation of iron carbides and nitrides in 5.6FexN-in-CNT along with their smaller particle size. The addition of 1 wt% Mn to FeN/CNT reduces CO conversion by almost half, yet the selectivity of lower olefins increases. Mn is thought to enhance the formation of iron nitride with an orthorhombic structure rather than cubic FeN.173 Heteroatom doping of carbonaceous materials with N, P, S, and other atoms can be developed. The co-doping may further enrich the applications of these materials.
Fe3−xMnxO4/CNT (x = 0–0.5) was used as model catalysts to study the promotion with Mn on the iron-based catalysts for FTS.174 It is found that the incorporation of Mn into iron-based catalysts could enhance the reduction of Fe3O4 to FeO, but hinders the further reduction of FeO to metallic Fe. The addition of Mn leads to an increase in C5+ hydrocarbon yield and C2–C4 olefin selectivity without any loss of FTS activity, yet excessive addition of Mn (Mn/Fe > 0.024) may cause a significant decrease of the reaction rate.
10Fe0.25Ru/CNT (10 wt% Fe and 0.25 wt% Ru) was prepared by co-impregnation of Fe nitrate and Ru acetate salts on CNTs. The Fe–Ru metal particles with sizes of 2.1 nm were obtained in the CNTs supports.175 The promoting effects of Cu and K were investigated. It was found that Cu and K promoted 10Fe0.25Ru/CNT demonstrated similar trends of product selectivity and FTS activity compared to the un-promoted catalysts. 10Fe0.25Ru/CNT was found to be remarkably stable because of higher resistance to the sintering of metal particles induced by the interaction of CNTs and metals. The potassium promoted Fe/CNT catalysts exhibited higher activity, selectivity to CO2 and C2 olefins, and lower methane selectivity compared to the un-promoted Fe/CNT catalysts.176 The addition of copper to Fe/CNT enhanced the catalyst activity but it did not affect the product selectivity of FTS.
Recently, Khodakov's group uncovered a new type of liquid metal promoters (Bi, Pb, Sb and Sn) for iron catalysts supported over CNT.177 The promoting effects of bismuth and lead result in a better reducibility and easier carbonization of iron nanoparticles. Slowing down secondary hydrogenation of olefins and a decrease in the chain growth probability were major reasons of better yield of light olefins over these catalysts. The extensive in-depth characterization of the bismuth-promoted iron catalysts supported by CNT under the reacting gas and reaction temperatures was conducted by a combination of cutting-edge in situ techniques: in situ scanning transmission electron microscopy, near-atmospheric pressure X-ray photoelectron spectroscopy and in situ X-ray adsorption near edge structure.179 We uncovered a significant mobility of bismuth species during the activation and reaction. The Bi migration leads to the formation of larger spherical bismuth droplets and iron–bismuth core–shell structures. In the working catalysts, metallic bismuth located at the interface of iron species undergoes continuous oxidation and reduction cycles, which facilitate carbon monoxide dissociation and result in a substantial increase in the reaction rate.
3.3 Carbon nanofibers supported iron catalysts
12Fe/CNF catalysts (12 wt% Fe) for FTS can display remarkably high selectivity to lower olefins (52 wt%), as shown by Krijn P. de Jong's group in 2012.178 This higher selectivity to lower olefins stems from the weak interaction between homogeneously dispersed iron nanoparticles and CNFs supports (Fig. 8(a–c)). Fe/CNF demonstrated suppression of methane production and maximization of the C2–C4 olefin fraction. The addition of trace amounts of Na and S to 12Fe/CNF further enhanced the C2–C4 olefin selectivity. Several other supports were also studied. The catalysts displaying weak metal support interaction, such as 12Fe/CNF, 6Fe/α-Al2O3, and 8Fe/β-SiC, achieved high activity-FTY compared to 13Fe/γ-Al2O3, and bulk iron catalysts. Dissimilar dispersion of Fe was achieved on γ-Al2O3 and CNFs supports despite the comparable Fe loading amounts. The addition of trace amounts of Na and S to 12Fe/CNF further enhanced the C2–C4 olefins selectivity. The surface carbide mechanism, shown in Fig. 8(d) is thought to dominate over Fe catalysts during FTS.178,180 In this mechanism, CO dissociation and carbon hydrogenation occur on the surface of the catalyst to form a CH3 fragment, which acts as a chain initiator. Methylene monomer units (CH2) are then added to this group to achieve carbon chain growth. α-Olefins are formed through the termination of chain growth by β-hydride abstraction, while the paraffins are produced by hydrogenation. The addition of Na and S to an iron catalyst is proposed to suppress methane formation by promoting the termination step via β-hydride abstraction. As shown in Fig. 8(e), a significant enhancement of lower olefins production was only observed on the iron catalysts promoted with Na and S over the CNT inert support. In contrast, a high selectivity to methane was noticed on the 13Fe/γ-Al2O3 catalyst. It is evidenced that the inert support of CNFs promotes activity through weak interaction with iron particles and the resulting catalyst can further be promoted with Na and S.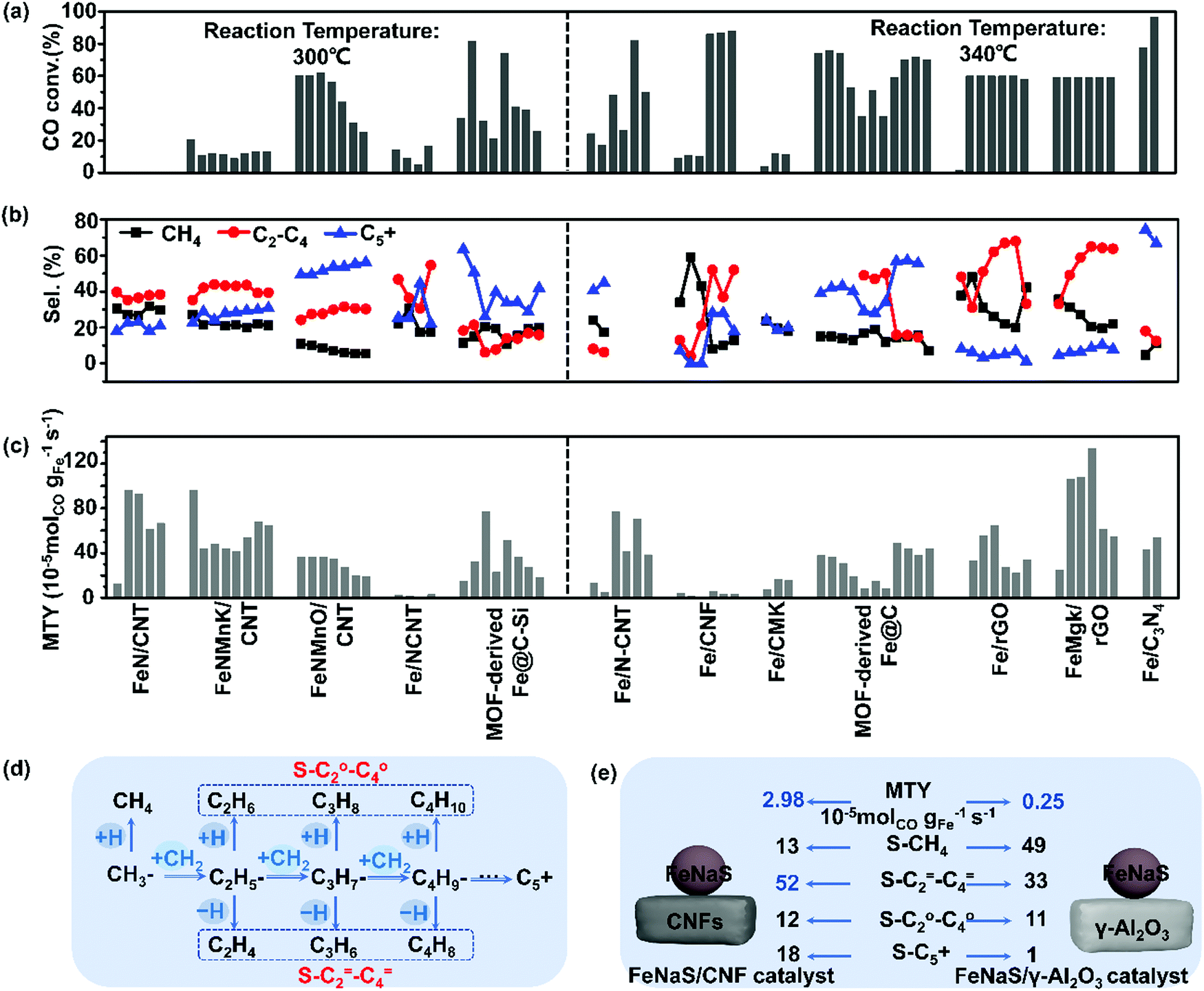 | ||
| Fig. 8 Catalytic performance of carbon-based iron catalysts with various carbon-based supports. CO conversion (a), selectivity of CH4, C2–C4 and C5+ (b); MTY (c). (Reaction conditions: 300 and 340 °C, H2/CO = 1–2, P = 0.1–2 MPa, GHSV = 4–20 L gcat−1 h−1 and TOS = 1–140 h.) The surface carbide mechanism of FTS (d) and the catalytic performance of FeNaS/CNF and FeNaS/γ-Al2O3 (e). (Reaction condition: 340 °C, 2.0 MPa, H2/CO = 1.0, TOS (h) = 64. S-CH4 denotes CH4 selectivity.) (a)–(c) Summarized from ref. 22, 170, 173, 174, 181, 186, 188, 189, 191, 194, 208, 209, 212 and 213 (d) and (e) adapted from ref. 178. | ||
Meanwhile, a series of Fe/CNF catalysts with different iron loadings (1, 2, 5, 10, and 20 wt% Fe) were prepared by using incipient wetness impregnation with aqueous solutions of ammonium iron citrate and a colloidal synthesis based on the thermal decomposition of iron oleate.181 Fe/CNF with iron particle sizes varying from 2 to 27 nm were obtained. Simultaneously, FeNaS/CNF was prepared from iron precursors containing traces of sodium and sulfur. Smaller iron carbide particles display higher surface-specific activities mainly due to higher methane production. The surface-specific activity based on the initial activity of Fe/CNF at 1 bar increased 6–8 folds when the iron carbide size decreased from 7 to 2 nm, yet methane and lower olefins selectivity were not affected. FeNaS/CNF achieved a 2-fold increase in the surface-specific activity, which mainly resulted from the smallest iron particles responsible for a high yield of methane. There are abundant highly active low coordination sites at corners and edges of small iron carbide particles which favour methane formation. However, the available terrace sites of iron particles promoted with Na and S enhance the production of lower olefins. It is inferred that the iron carbide particle size plays an important role in the design of active and selective FTO catalysts.
3.4 Carbon spheres supported iron catalysts
The fabrication of FexOy@C spheres with highly dispersed iron oxide nanoparticles embedded in carbon matrix was achieved through a facile and efficient one pot route by hydrothermal treatment of a glucose solution containing iron nitrate at a mild temperature.28 The surrounding carbonaceous matter promoted the formation of iron carbides during H2 activation, facilitated the formation of C5+ hydrocarbons and restricted the aggregation of iron carbide nanoparticles during the reaction processes. This preparation route is applicable to fabricating other core–shell catalysts by using a wide range of other saccharides and metal nitrates as sources.Well-dispersed Fe3C nanoparticles in the porous carbon matrix with the Fe3C@C core–shell structure were also obtained through a pyrolysis process employing a nitrate of iron mixed with urea and glucose as a precursor.182 The intimate contact with Fe and C from this preparation method led to the formation of iron carbides. The high activity of Fe3C@C was related to large amounts of iron carbides and a small size of iron species with a good distribution. Moreover, the addition of the alkali metal promoters such as Na, Mg, Ca, and K, decreased the methane, increased the lower olefin selectivity, and shifted the product distribution toward higher molecular weight hydrocarbons.
The porous Fe-based catalysts supported on polystyrene mesoporous carbon were studied in FTS.183 The pre-existing carbides phases obtained upon using elevated calcination temperatures were not completely re-oxidized during the reaction, and the formation of carbides through the polymeric carbon reaction with magnetite and/or metallic iron led to high activity for FTS.
The CMK-3 with a well-defined pore structure was applied as a carrier material to prepare FeSNa/CMK-3 catalysts and to study the relationship between the calcination/activation conditions and catalytic performance. At the calcination temperature of 800 or 1000 °C, the carrier material performed carbothermal reduction of carbon oxides resulting in a higher proportion of catalytically active iron carbides. The low catalytic activity was due to the particle growth and blockage of catalytic active sites with dense graphite layers. CMK-3 with different surface modifications was applied as a carrier for iron-based catalysts in FTS.184 The iron (carbide) supported on the nitrogen-rich CMK-3 and the CMK-3 support with a lower concentration of functional groups showed higher catalytic activity than the counterpart with an oxygen-rich surface. It was found that even iron nanoparticles confined in CMK-3 mesopores continued to grow leading to lower activity under FTS conditions.185 The CMK-3 carbon material with a hexagonal mesopore arrangement used as a support for iron-based catalysts showed a high selectivity to lower olefins under industrially relevant FTO conditions, low methane production, and stable operation for up to 140 hours.186 It was confirmed that the weak metal support interaction between CMK-3 carbon material and iron produced highly active FTS catalysts, compared with iron catalysts supported by SBA-15 ordered mesoporous silica. Somewhat lower activity of Fe/CMK-3 compared to the CNT-supported iron catalysts might be also explained by a lower extent of carburisation.20
Conventional carbon supports are generally treated with acid or base solutions to produce functionalized groups on the surface which benefit anchoring active metal or metal oxide species. The nitrogen-doped carbon materials with N atoms in the carbon materials, where the N atoms act as anchoring sites can be prepared. However, this should be achieved without the environmentally unfriendly step of acid and base treatment. The 5Fe/NCS-ver, 5Fe/NCS-hor, and 5Fe/NCS-hyd catalysts (5 wt% Fe) were obtained through three different strategies of CVD in a horizontal furnace, CVD in a vertical furnace, and using a hydrothermal approach respectively.187 5Fe/NCS-ver presented the highest N content and the most defect sites, with the smallest diameter and highest surface areas, among three catalysts. 5Fe/NCS-hyd demonstrated the lowest quantity of defect sites and lowest N content, with the largest diameter and lowest surface areas. Pyrrolic and pyridinic N atoms play a key role in binding the Fe atoms of Fe/NCS and that quaternary N atoms play a minor role. 5Fe/NCS-ver with well-dispersed Fe oxide particles on carbon matrix induced by the highest N content exhibited higher activity in FTS than 5Fe/NCS-hor and 5Fe/NCS-hyd catalysts.
3.5 MOF-derived iron catalysts
Recently, the Fe-MOF derived Fe@C catalysts demonstrated high activity and stability in FTS.189,191–193 The obtained MOFs derived Fe@C catalysts showed a high iron dispersion and extremely small Fe nanoparticles confined within a porous carbon matrix.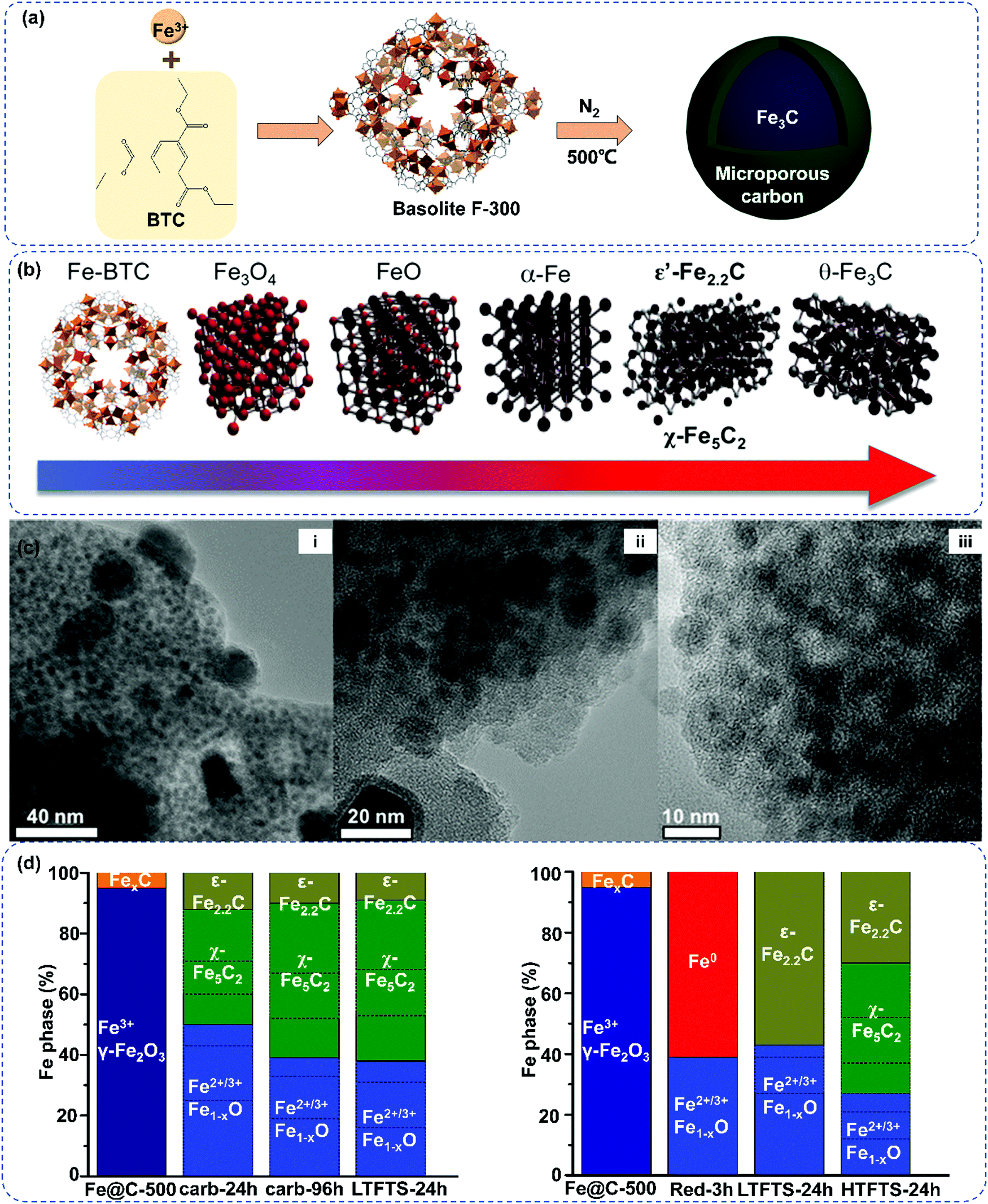 | ||
| Fig. 9 MOF derived carbon-based iron catalysts for FTS: direct pyrolysis of basolite F-300 (a); evolution of iron phases during pyrolysis of Fe-BTC, reduction and Fischer–Tropsch reaction (b); micrographs of fresh Fe@C-500 (c(i)) catalysts, and images of spent catalysts after 140 h carburization (c(ii)) and after 3 h reduction (c(iii)) under different magnification and the small iron nanoparticles embedded in the carbon matrix are visible as low-contrast dark circles, in comparison to the lighter carbon matrix. Mössbauer data summary of Fe@C-500 measured at 4.2 K after carburization and subsequent LTFT conditions. Fe@C-500 after reduction, subsequent LTFT and additional HTFT. The bar graphs represent the spectral contributions of iron phases (d). (a) Reprinted with permission from ref. 188. Copyright 2015, Springer Nature. (b) Reprinted with permission from ref. 189. Copyright (2008) American Chemical Society. (c) and (d) Reprinted from ref. 190, Copyright (2018), with permission from Elsevier. | ||
Potassium serves as an excellent electronic donor for iron in FTS. It plays an important role in increasing activity and tuning the selectivity of hydrocarbons.2 Nitrogen species have been also known to act as electronic donors in carbon-supported iron materials, which increase the activity of FTS.196 For the confirmation of the electronic effects of nitrogen species in iron catalysts, Wang's group prepared a Fe-MIL-88B-NH2 derived Fe-based catalyst, which exhibited uniformly dispersed iron nanoparticles encapsulated in a N-doped carbon matrix.191 The obtained iron catalysts demonstrated higher activity than most of the iron catalysts reported in the literature. The high activity was found to correlate with the special Fe3O4@χ-Fe5C2 core–shell structure of the MOF-derived iron catalysts. Their work has provided new insights into the relationship between catalytic performance and iron phase transformation in FTS.
To investigate the relationship between the initial MOFs topology of the Fe@C precursors, catalyst activity and selectivity during FTS, Kapteijn and Gascon selected Fe-MOF (MIL-68, MIL-88A, MIL-100, MIL-101, MIL-127, and Fe-BTC) as precursors to prepare the Fe@C catalyst.192 The carboxylate linkers of these precursors differ both in size and connectivity. The use of commercial MOFs to mediate the Fe@C catalyst in this study is also helpful in assessing the potential industrial applications. The pyrolysis of the most porous MOFs produces the smallest Fe nanoparticles, which confirms that the structure and porosity of the original MOFs precursor affects the final metal dispersion. The surface areas of Fe@C catalyst are independent of the topology of MOFs precursor, indicating a similar degree of carbonization of the framework by decarboxylation and thus a similar C matrix. The Fe@C catalysts with smaller Fe particle sizes demonstrate higher activity in FTS, and this effect is even more pronounced in the K-promoted samples. This work also highlights the influence of impurities and other heteroatoms on the FTS activity and selectivity. In general, the catalytic results are correlated to many factors, such as the Fe nanoparticle size, porosity, crystallite phase, and impurities. Especially, the type and content of impurities vary for each MOF-derived Fe@C catalyst. More research efforts into the influence of each factor on the catalytic performance would be beneficial for the further development of the Fe@C catalysts. The direct correlation between the FTS catalyst performance and iron carbide phase was also investigated by Kapteijn and Gascon (Fig. 9).190 MOFs derived Fe@C exhibits good dispersion of iron nanoparticles (Fig. 9c(i)) and excellent anti-sintering properties of iron nanoparticles after FTS (Fig. 9c(ii and iii)). Fe@C has been analyzed by the in situ Mössbauer spectroscopy in order to investigate the carburization of iron. The active phase of ε′-Fe2.2C phase has been monitored under low temperature Fischer–Tropsch (LTFT) conditions, yet χ-Fe5C2 phase was monitored under HTFT (Fig. 9(d)). Inspired by the Fe/γ-Al2O3 catalysts, with strong mechanical strength, they also prepared MOF-derived iron-based catalyst with enhanced dispersion of the active phase and excellent mechanical strength for producing lower olefins from HTFT.193 The Fe@C/Al catalyst was obtained by wet extrusion of different amounts of Basolite F300 and AlOOH followed by subsequent pyrolysis, which showed enhanced C2–C4 selectivity and mechanical stability. There are various MOF derived carbonaceous materials, such as nanoporous hybrid carbon being worthy of further study.199,200
3.6 Other carbon materials supported iron catalysts
Polyaniline (PANI) is a kind of conducting polymer which is used as a carbon precursor to prepare iron catalysts. A 11Fe/PANI catalyst (11 wt% Fe) was synthesized by a simple sonochemical method and displayed an excellent catalytic performance in FTS with the selectivity to lower olefins as high as 50%.201 The CO conversion and selectivity of lower olefins of the 11Fe/PANI catalyst were much higher than that of the 10Fe/AC and 11Fe/N-AC catalysts. The support of PANI results in uniform and higher dispersion of Fe particles, which further facilitate the reduction of iron oxide and the formation of iron carbides. Carbon nanosheets (CNS) supported iron catalysts showed superior iron time yield and a stable activity of more than 100 hours, with a light olefin selectivity of 41%.94 K-Promoted CNS can stabilize the metallic iron nanoparticles during the reduction by hydrogen and enhances the formation of iron carbide during FTS reaction conditions. Yolk–shell nanoparticles with metal particles confined in carbonaceous materials may be developed to enhance the catalytic stability in FTS.202,203Iron catalysts supported by reduced graphene oxide (rGO) and promoted with K were also investigated for the FTO reaction.204 With an increment of the K content from 0 to 2.0 wt%, the surface areas, basicity, adsorption capacities of CO and H2, and particle sizes of Hägg carbide increased accordingly. The MTY exhibited a volcanic evolution with the increasing content of K, which can be attributed to the promotion effect of K on the formation of iron carbide with suppression of aggregation of iron carbide. The addition of K to Fe/rGO effectively suppressed the production of CH4 and the secondary hydrogenation of lower olefins, resulting in higher selectivity to lower olefins. The addition of Mg and K to the Fe/rGO catalysts was investigated.205 The addition of K promoted the carburization of iron and the double addition of Mg and K enhanced the dissociation of CO. These phenomena were proposed responsible for the high activity of the Mg and K dual-decorated Fe/rGO catalysts. A series of model catalysts were prepared by using monodisperse iron oxide nanoparticles as the metal precursor and pyrolytic GOs as catalyst support.206 The Fe/GO catalysts have been employed in FTS to study the carbon support effect on the iron active phase. Thermal treatment of support materials resulted in the decrease in oxygen and sulfur species and in the enhancement of the graphitization degree. The decrease in the amount of surface oxygen groups changed the surface properties of the active iron phase, leading to higher catalytic activities.
N-Doped graphene (NG) is an efficient electron donor for iron catalysts, which was found to enhance the selectivity to lower olefins.196 Fe/NG demonstrated a good reducibility which was the key factor to promote the selectivity to lower olefins. The Fe–K-nanoparticle catalyst supported on high surface area graphene nanosheets showed high activity and excellent stability.207 Compared with CNTs, graphene supported iron catalysts showed a lower selectivity to methane and carbon dioxide. The presence of defects in the graphene lattice can serve as favorable nucleation sites for anchoring iron nanoparticles and provide tuneable metal–support interactions. Similarly, carbon-based supports, such as nitrogen-doped mesoporous CSs,208 g-C3N4,209 and C3N4210,211 may exhibit the same electron donor properties for iron catalysts resulting in high catalytic performances.
A series of iron catalysts supported on SiC were prepared by mild hydrothermal method using fructose.212 The higher carbon content in the carbon–silica composite catalyst was beneficial for the direct interaction between iron carbide material and carbon support. The iron nanoparticles on the carbon–silica support exhibited a lower oxidation state and hence higher catalytic activity compared to similar catalysts supported on silica and pure carbon. An interesting strategy was employed with g-C3N4 when it was used as a sacrificial support.213 The chemical state of iron in the resulting Fe/g-C3N4 catalysts was found to be a more reduced state (FeO) on this support, compared to Fe/CNT which contained primarily Fe2O3. After pretreatment in hydrogen, the fully reduced metal iron phase was dominant in Fe/g-C3N4, while incomplete reduction of iron oxide was observed in Fe/CNT. This facilitated the conversion of Fe/g-C3N4 to crystalline Hägg carbide (χ-Fe5C2) during the FTS reaction, leading to improved CO conversion and high selectivity to C5+ products.
Representative catalytic data of carbon-based iron catalysts has been presented in Table S3 in ESI.† The CO conversion, selectivity and MTY of carbon-based iron catalysts with various carbon supports are compared in Fig. 8(a–c). It can be concluded that FeN/CNT shows higher activity than MOF-derived Fe@C–Si catalysts under reaction temperatures of 300 °C. Graphene oxide supported iron catalysts and carbon nanotube supported FeN catalysts exhibit higher activity than other carbon-based iron catalysts. Graphene oxide supported iron catalysts demonstrate selectivity to lower olefins as high as 60% with about 30% of methane selectivity.
4. Other metals (Mo, Ni, Rh, Ru) supported on carbonaceous materials for Fischer–Tropsch synthesis
Besides cobalt and iron-based catalysts, some other metals, such as ruthenium and molybdenum-based catalysts, are also good for FTS. Hence, the catalysts with other metals supported on structured porous carbon have also been investigated.4.1 Activated carbon supported metal catalysts
Mesoporous AC which has a larger surface area, was used as a support for the K–Co–Mo/AC catalyst synthesized by an improved sol–gel method.214 Compared with the unsupported catalysts, K–Co–Mo/AC has higher selectivity to mixed alcohols, especially, the content of C2 + OH increased significantly. It can be explained that the supported catalyst has a high active surface area, and the use of a mesoporous structure is proposed to prolong the residence time of alcohol-forming intermediates in the pores to the benefit of the formation of higher alcohols. The surface Moδ+ (1 < δ < 4) species formed on the reduced catalyst are favourable for the synthesis of alcohol. At the reduction temperature of 798 K, the Mo/AC weight ratio of 40% catalyst showed high activity, which may be due to the high content of Moδ+ (1 < δ < 4) surface species.The K-promoted NiMo catalysts supported on AC also show enhanced catalytic performances.215 Compared with unsupported material, the K-promoted NiMo/AC catalyst has a three-fold increase in the selectivity to higher alcohols, which may be due to the reduced crystallinity of the active phase NiMoO4 and its partial transition from α to β form. Compared with K NiMo/AC1a, the conversion over the K NiMo/AC2a catalyst is as high as 45% when using acid-treated support with a higher specific surface area. The increase in the activity can be attributed to better dispersion of active phase on the AC support with high surface area, therefore the exposure degree of active Ni–O–Mo sites is higher. In summary, for all K-promoted bimetallic NiMo catalysts, the reduction of acidity was beneficial for the increase in alcohol production.
The K/MoS2–MMO-AC catalyst was studied for the synthesis of higher alcohols using a mixture of mesoporous AC and mixed MgAl oxide (MMO) as supports.216 It is found that a large amount of Mo migrates from AC to MMO during the reaction and the high C3 + OH selectivity is related to the percentage of double [002] MoS2 layers on both supports.
4.2 Carbon nanotubes and nanofibers supported metal catalysts
Other metals supported on CNTs were applied in FTS such as Rh,217,218 Ru,219–221 NiMo.222 The Rh particles confined within nanotubes have a significantly enhanced catalytic activity for the conversion of CO and H2 to ethanol. The total ethanol formation rate inside the nanotubes (30.0 mol molRh−1 h−1) is an order of magnitude higher than the total generation rate outside the nanotubes, although the latter is easier to obtain. 5 wt% Ru nanoparticles confined in channels or dispersed on the outer surface of CNTs were synthesized using capillary action or deposition method. The 5 wt% Ru/CNTs-in catalyst showed excellent activity under the condition of complete CO oxidation. The conclusion was that nanochannels of CNTs could selectively increase the density of reactants, and the encapsulation of Ru nanoparticles in CNTs channels could provide a microenvironment for the enhancement of the reactivity of catalytic sites. The Ru nanoparticles supported on CNTs have excellent selectivity to the C10–C20 hydrocarbons. Both the unique hydrogen-absorbing species and acidic functional groups on the surface of CNTs may play roles in the mild hydrocracking of C10–C20 from heavier hydrocarbons. C10–C20 selectivity and TOF for CO conversion depended on the average size of Ru particles. The Ru/CNT catalysts with an average Ru size of about 7 nm showed the best C10–C20 selectivity and relatively higher TOF.The Ni–Mo–K sulfide catalysts doped with CNTs were synthesized and showed high activity and selectivity to alcohols, especially to ethanol.222 A Ni0.5Mo1K0.5–15% CNTs catalyst exhibited ethanol selectivity of 33.1% (CO2-free) under the reaction conditions of 8.0 MPa and 593 K. CNTs can adsorb and activate hydrogen to generate a surface microenvironment and a higher stationary-state concentration of the adsorbed hydrogen species resulting in a sharp increase in the concentration of the catalytically active Mo4+/Mo5+ species on the surface of catalysts.
4.3 Other carbon-based FTS catalysts
Recently, the K promoted mesoporous carbon (Starbon 800) has also been used in FTS and was found to promote the activity and higher alcohol selectivity of molybdenum phosphide (MoP) catalysts.223 It was shown that when supported on mesoporous carbon, and promoted with K, MoP produced mainly higher alcohols, with ethanol forming the highest weight fraction among products. A similar effect was also observed on the K/MoS2 catalysts supported on mesoporous carbon, which were found to produce higher amounts of ethanol compared to those supported on MMO.216 Carbon coated alumina (CCA) has also been used as a support for sulfide catalysts for syngas conversion.224 In a study focusing on the K promoted CoMoS catalysts, CCA was found to increase the alcohol/hydrocarbons ratio in the products.The MoP catalysts have attracted attention because of their powerful methanol synthesis activity.223 The mesoporous carbon supported KMoP catalysts were used to synthesize higher alcohols with the idea of the promotion with alkali metals such as K for CO-dissociation and with MoP for hindering the CO-dissociation. Carbon modified alumina was used as supports for molybdenum-based catalysts and the obtained KCoMoS/C/Al2O3 catalysts were prepared to produce alcohols.224 It was noted that the carbon support modified alumina promoted alcohol synthesis catalyst through enhancing the CO conversion and the alcohols/hydrocarbons ratio compared with pure alumina.
Ru supported on porous carbon materials has been investigated in FTS.225 A series of 3 wt% Ru catalysts was prepared. Ru nanoparticles were embedded on the carbon walls of ordered mesoporous carbon (OMC) catalysts with different pore sizes.226 The 3 wt% Ru–OMC catalyst was found to exhibit a highly ordered mesoporous structure and a large surface area. During the FTS reaction, 3 wt% Ru–OMC catalysts embedded in the carbon walls were more stable than 3 wt% Ru/AC counterparts, because the aggregation, movement, and oxidation of the particles were suppressed. It was found that catalytic activity and C5+ selectivity increased with increasing pore size, but CH4 selectivity showed the opposite trend. These changes could be explained by the special environment of the active Ru sites and the diffusion of products in the catalyst pores. The activity and hydrocarbon selectivity were related to the pore size of the OMC, but not to the size of Ru particles. For comparison, three types of Ru catalysts supported on OMC, AC, and CNTs were synthesized by conventional impregnation methods, and exposed to FTS under the same reaction conditions.227 The Ru–OMC catalyst exhibited a highly ordered mesoporous structure and large surface area, which was similar with the parent OMC material. On this catalyst, the Ru nanoparticles were indeed embedded in the carbon wall with close contact with the carbon support. It was suggested that this feature might generate some electron-deficient pieces at the interface, which promote the transfer of spilled hydrogen and improve hydrogen dissociation on the catalyst surface.
The graphite with high surface areas was applied as support to investigate the local chemical environment and electronic structure of active Ru and Cs promoter.225 It is revealed that the Ru reduction proceeds through a two-step mechanism with an intermediate oxidation state and partial reduction of Cs occurs simultaneously.
5. Conclusions and perspectives
5.1 Summaries and conclusions
FTS reaction is an important process that can be used to convert coal, shale gas, biomass and captured carbon dioxide into fuels and chemicals. FTS has been industrialized for decades, yet the development of new catalysts is still advancing. The commercial cobalt and iron-based catalysts, which have been used widely in the industry can be bulk or supported on metal oxide supports, such as SiO2, Al2O3, TiO2. The strong interaction between metals and these metal oxide supports and the formation of metal–support mixed compounds (aluminate or silicate) result in low activity for FTS. However, carbon-based materials as supports for FTS allow a tunable interaction between iron/cobalt and carbon and reduce in-desirable strong metal–support interactions. The pore structure and surface chemistry of carbon-based materials can be easily controlled by different activation and treatment procedures. The pores and functional groups of carbon materials act as anchoring sites for metal precursors and therefore result in better dispersion of active phase and particle size control. Recently, numerous reports have addressed carbon-based materials as supports for FTS catalysts. This review summarized literature on FTS catalysts supported by AC, CNTs, CNFs, CSs, MOFs derived materials, and other carbonaceous materials and it particularly focuses on research since the early 2000s.Extensive research on Co/AC for FTS revealed that the synergistic effect of Co and Co2C was responsible for the generation of alcohols. The active sites of Co/AC are located at the Co/Co2C interface, in which the metallic Co species are active for the dissociative adsorption of CO and the subsequent carbon-chain growth, and the Co2C species act as active sites for non-dissociative adsorption of CO and promotes the CO insertion into the adsorbed alkyl chain. Electronic additives such as Li, K, and Ca are essential to tune the electronic environment of Co/AC, resulting in enhanced chain growth capabilities. Electronic and structural additives of Cr and La promotes the activity of Co/AC, and additives of Fe and Ca promotes the formation of Co2C phases resulting in high alcohol selectivity.
The electronic interaction of the confined iron particles with the CNTs walls can facilitate activation of syngas and enhance the catalytic activity of FTS. In addition, the CNTs surface influences the diffusion behavior and results in enriched reactant concentrations inside the CNTs channels, which further promotes the catalytic activity. There are electronic differences between the inner channel and the outer surface of CNTs, which come from the curvature of CNTs walls induced shifting the π electron density of the graphene layers from the concave inner to the convex outer surface. Hence, the auto-reduction of iron oxide nanoparticles is facilitated within the CNTs channels compared to the particles located on the out surface of CNTs. The reduction temperature decreases with the inner diameter of CNTs. The oxidation of metal iron particles inside CNTs is suppressed compared with that of particles located on the outer CNTs surface. Pretreatment of CNTs plays an important role in the preparation of the CNTs supported metal catalysts. High temperatures of the acid pretreatment enhance oxygen functionalization and produce more anchoring sites for metal particles leading to increased dispersion.
CNFs are ideal supports for investigating the intrinsic cobalt particle size effects. The catalytic performance was independent on the cobalt particle size for the catalysts with Co particle sizes larger than 6 nm, but both activity and selectivity to the C5+ hydrocarbons decreased for the catalysts with Co particle sizes smaller 6 nm. Therefore, it is confirmed that the cobalt particle size in the FTS catalysis should be larger than 6 nm. The Fe/CNF catalysts demonstrate high selectivity to lower olefins due to the weak interaction between homogeneously dispersed iron nanoparticles and CNFs supports. The addition of Na and S to Fe/CNF catalyst promotes negative deviations from the ASF distribution by decreasing methane selectivity through limiting the hydrogenation reactions and enhancing the termination step via β-hydride abstraction.
There are many advantages of CSs as supports for FTS catalysts including regular geometry, good mechanical strength, tunable porosity and surface functional groups, easy doping of uniformly distributed heteroatom, and controllable distribution of metal particles. The tunable porosity of CSs exhibits controllable distribution of metal particles. The MOF-derived iron or cobalt catalysts exhibit uniform dispersion with iron or cobalt nanoparticles encapsulated by a porous carbon matrix. The preparation of AC, CNTs, and CNFs supported metal catalysts usually involves impregnation methods and the metal loadings are normally lower than 30 wt% in order to achieve well-distributed metal particles. However, the iron or cobalt catalysts derived from MOFs prepared by pyrolysis can achieve metal loadings as high as 50 wt% with uniformly dispersed metal nanoparticles within the porous carbon. It greatly increases both the number and efficiency of iron or cobalt active sites. The special structure of MOF-derived iron or cobalt catalysts inhibits the aggregation of metal nanoparticles during FTS.
AC offers the advantage of low cost but its microporous structure can impose additional transport limitations. The main differences between CNFs and CNTs is the absence of hollow cavities for the former one. The meso-structures of CNTs facilitate transport processes. CSs and MOF-derived carbonaceous materials are generally made from the pyrolysis of organic frameworks which impose cost burdens on the preparation. In addition to the unique structural and electronic advantages offered by each carbon supports, there are also supplementary appealing properties they share that make them suitable for FTS. For instance, retrieving metals from the spent catalysts is possible by burning off the carbon. Simultaneously, the carbon surfaces can be functionalized by simple procedures of acidic or basic treatments, which can make the surfaces hydrophilic to be dispersed in polar solvents during the catalyst loading process or serves as anchoring sites for metal particles. Additionally, doping heteroatoms into carbon supports provides an alternative approach to tune its catalytic properties.
The most distinctive feature of the AC supported cobalt catalysts is the production of alcohols. The La promoted 15 wt% Co/AC catalysts demonstrate a high alcohol selectivity of 38.9% with the alcohol distribution of methanol of 7.7%, C2–C5OH of 58.1%, and C6–C18OH of 34.2%. Yet the cobalt time yield is just 0.88 × 10−5 (molCO gCo−1 s−1). Despite the fact that the surface area of AC is almost above 700 m2 g−1, the cobalt and iron time yields over the AC supported catalysts are lower than those of the CNTs/CNFs supported catalysts under the same reaction conditions. This may come from the complicated channel structure, which induces uneven metal particle size distribution.
The Na and S promoted Fe/CNF catalyst demonstrates a lower olefins selectivity of 52.0% at temperatures above 300 °C. The Mn and K promoted FeN/CNT catalyst exhibits a lower olefins selectivity of 43.6% with a high MTY of 54.0 × 10−5 (molCO gFe−1 s−1). The MOF-derived cobalt catalysts show a relatively low activity and high methane selectivity under reaction temperatures of 220 °C possibly due to the carbon layer formed on the surface of cobalt particles, which slows down and influences the diffusion of reactants and products.
5.2 Challenges and recommendations
Some challenges are associated with using carbon-based supports for FTS. The main challenge is still the high cost of carbon-based materials. The carbon-based materials should be manufactured using available raw material at a low preparation cost. The high price makes it not suitable for Fischer–Tropsch's large-scale production process. The second challenge comes from the insufficient stability of carbon-based materials in oxidizing atmosphere. Catalyst regeneration may require oxidative posttreatments, which can be problematic for the carbon-based catalysts. Another challenge arises from the possible deactivation of metal catalysts supported over carbon materials. Weak metal–support interaction may lead to aggregation of the metal particles during reaction resulting in deactivation by loss of active surface area. Finally, it is necessary to pelletize the nano-sized carbon-supported catalysts to an appropriate size and shape to satisfy the industrial application. Although the mechanical strength of some carbon-based supports is high, the mechanical strength of pelletized carbon-based catalysts is usually weak and requires further investigation for industrial applications. The weak mechanical strength of pelletized catalysts may cause fragmentation with an increase of the pressure in packed bed reactors and plugging of separation equipment in slurry bed reactors and recirculating fluidized bed. Additionally, the density of carbon-based supports is much lower than that of traditional supports such as SiO2, Al2O3, and TiO2. The low density and non-adhesive properties of carbon-based materials may further aggravate the fragmentation of catalysts during the reaction.5.3 New trends and prospects
Based on the extensive research summarized herein, few promising research paths emerge for future investigations.First, carbon-based materials with structured carbons containing tunable pore sizes and hierarchical porous structures should be developed as supports for FTS along with the development of advanced porous carbon fabrication technologies. It is important to prepare carbon-based supports with pore structures tunable in the micro- and meso-ranges and construct hierarchical structures by connecting the micropores to mesopores and macropores. Ordered porous structures are essential to serve as model catalysts for the investigation of the effects of pore structures. Micropores can assist in dispersing the metal particles and forming nano-sized particles, meanwhile, hierarchical structures can facilitate mass transfer, enhance reaction activity, and tune the selectivity of FTS. The hierarchical structures also would enhance the thermal conductivity of carbon-based materials and inhibit the hotspots in the reactors since FTS reaction is exothermic. Currently, substantial progresses have been made in the methodology to tune the texture structures of carbon-based supports to improve the performance of supported metal catalysts.228–234 The extension of Stöber method for the preparation of carbon-based supports opens up new areas of carbon spheres.235,236 Carbon-based supports impregnated with highly monodispersed metal nanoparticles have been successfully made and applied in catalysis.237 It is essential to explore new synthesis methods to maximize interparticle spacing and to achieve narrower particle size distributions and a more homogeneous distribution of iron nanoparticles on the carbon-based supports in the future.
Second, the drawbacks of carbon-based materials such as weak mechanical strength, weak stability and inability to regenerate them in the oxidizing atmosphere can be overcome by using hybrid structures, composed by both oxide and carbon. For example, deposition of a thin layer of carbonaceous materials over an oxide support can be considered in order to enhance the metal dispersion, intrinsic catalytic activity of iron species over the carbonaceous shell, but the same time maintain high mechanical and chemical stabilities of the resulting hybrid catalysts due to the oxide core.238,239
Third, metal loading techniques of metal-supported catalysts should be improved to control metal particle size, dispersion and catalytic performances of FTS. Except for those conventional preparation methods, such as impregnation, co-precipitation, sol–gel technology, and precipitation–deposition methods, novel techniques concerning introducing metal particles into carbon-based supports need to be developed to achieve uniformly dispersed crystalline nanoparticles. Since various carbon-based supports have been discovered, some in situ metal loading techniques were developed accordingly. Hydrothermal methods, evaporative methods, CVD methods, and organic complex methods can be applied in the future. MOFs serve as important precursors to get well-formed metal@shell structures. It is meaningful to develop cheap precursors for MOFs for their potential commercial application in FTS.
Fourth, carbon-based materials can be designed to serve as model supports for various heterogeneous catalysis applications, where intrinsic catalytic properties of metal particles (particle sizes, promoter effects, formation of active phases) should be investigated. For example, CNTs supported iron catalysts bring a breakthrough discovery of different properties of iron particles inside and outside the channel of CNTs which provides a novel approach to modify the redox properties of the confined metal oxides. The obtained information about the intrinsic activity of metal particles is especially significant for the rational design of catalysts based on carbonaceous materials.
Finally, the effect of carbon-based materials on the formation of iron and/or cobalt carbides should be investigated since their formation heavily influence FTS activity. Suggestion for academic research includes the investigation of the iron particle size effects on carbon lay-down during reaction using the Tapered Element Oscillating Microbalance and the effects on surface coverage and residence times on reaction intermediates using SSITKA. The use of in situ and operando techniques such as High Resolution Transmission Electron Microscope, Infrared Spectroscopy, Mössbauer Spectroscopy, X-ray Absorption Spectroscopy, and the rigorous analysis of catalyst structure and chemical states during reaction will be essential for further probing metal–support interactions at reaction conditions relevant for industrial application. Advances in the in situ microscopy techniques would provide a powerful tool in the future to study the aggregation and fragmentation of iron-containing particles and promoters under the reaction conditions. Additionally, the relationship between carbon-based supports and cobalt/iron carbides formation should be investigated systematically together with the role of cobalt/iron carbides in the formation of high value-added products of oxygenates, lower olefins, and α-olefins.
Abbreviations
| AC | Activated carbon |
| ASF | Anderson–Schulz–Flory |
| CNTs | Carbon nanotubes |
| CNFs | Carbon nanofibers |
| CSs | Carbon spheres |
| CNF-P | Carbon nanofibers with fishbone structure and platelet structure |
| CMK-3 | Carbon Molecular Sieves-3 |
| Co/N-HCS | N-Doped hollow carbon spheres supported cobalt catalyst |
| CVD | Chemical vapor deposition |
| CTY | Converting syngas |
| CCA | Carbon coated alumina |
| CNS | Carbon nanosheets |
| DFT | Density functional theory |
| EXAFS | Extended X-ray absorption fine structure |
| FTS | Fischer–Tropsch synthesis |
| FM | Fibrous material |
| FTO | Fischer–Tropsch to olefins |
| Fe-in-CNT | Iron particles confined inside carbon nanotube |
| Fe-out-CNT | Iron particles supported on the outside carbon nanotube |
| GPO | Gas phase oxidation |
| GHSV | Gas hourly space velocity |
| g-C3N4 | Graphitic carbon nitride |
| HDP | Homogeneous deposition precipitation |
| HCSs | Hollow carbon spheres |
| HTFT | High-temperature Fischer–Tropsch |
| LTFT | Low temperature Fischer–Tropsch |
| MOFs | Metal–organic frameworks |
| MWCNTs | Multiwalled carbon nanotubes |
| MTY | Metal time yield |
| MWNTs | Multiwall carbon nanotubes |
| MMO | Mixed MgAl oxide |
| MoP | Molybdenum phosphide |
| NG | N-Doped graphene |
| N-CNTs | Nitrogen doping carbon nanotubes |
| NP@C | Nanoparticles@carbon |
| OMO | Occupied molecular orbital |
| OMC | Ordered mesoporous carbon |
| O/P | Olefin to paraffin |
| PANI | Polyaniline |
| rGO | Reduced graphene oxide |
| SSITKA | Steady-state isotopic transient kinetic analysis |
| SiC | Carbon–silica composite materials |
| TOS | Time on stream |
| TOF | Turnover frequency |
| ZIF-67 | Zeolitic imidazolate frameworks-67 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Liaoning Natural Science Foundation (2020-MS-021). Supported by LiaoNing Revitalization Talents Program (XLYC1807077). Dalian National Laboratory for Clean Energy (DNL), DNL Cooperation Fund, CAS (DNL180402). The authors acknowledge financial support of the French National Research Agency (NANO4-FUT, Ref. ANR-16-CE06-0013) and from European Union (Interreg V project PSYCHE).References
- H. Xiong, L. L. Jewell and N. J. Coville, ACS Catal., 2015, 5, 2640–2658 CrossRef CAS.
- Y. Chen, Y. Ni, Y. Liu, H. Liu, X. Ma, S. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2018, 8, 5943–5954 RSC.
- Y. Chen, N. Batalha, M. Marinova, M. Impéror-Clerc, C. Ma, O. Ersen, W. Baaziz, J. A. Stewart, D. Curulla-Ferré and A. Y. Khodakov, J. Catal., 2018, 365, 429–439 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- Y. Yang, K. Chiang and N. Burke, Catal. Today, 2011, 178, 197–205 CrossRef CAS.
- B. Sun, K. Xu, L. Nguyen, M. Qiao and F. Tao, ChemCatChem, 2012, 4, 1498–1511 CrossRef CAS.
- Y. Chen, Y. Xu, D. G. Cheng, Y. Chen, F. Chen, X. Lu, Y. Huang and S. Ni, J. Chem. Technol. Biotechnol., 2015, 90, 415–422 CrossRef CAS.
- E. Rytter and A. Holmen, Catal. Today, 2016, 275, 11–19 CrossRef CAS.
- W. Hou, B. Wu, X. An, T. Li, Z. Tao, H. Zheng, H. Xiang and Y. Li, Catal. Lett., 2007, 119, 353–360 CrossRef CAS.
- H. M. T. Galvis, A. C. Koeken, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Catal., 2013, 303, 22–30 CrossRef.
- K. Jothimurugesan and S. Gangwal, Ind. Eng. Chem. Res., 1998, 37, 1181–1188 CrossRef CAS.
- S.-H. Kang, J. W. Bae, K.-J. Woo, P. S. Prasad and K.-W. Jun, Fuel Process. Technol., 2010, 91, 399–403 CrossRef CAS.
- S.-H. Kang, J. W. Bae, P. S. Prasad and K.-W. Jun, Catal. Lett., 2008, 125, 264–270 CrossRef CAS.
- G. Jacobs, T. K. Das, Y. Zhang, J. Li, G. Racoillet and B. H. Davis, Appl. Catal., A, 2002, 233, 263–281 CrossRef CAS.
- A. Tavasoli, Y. E. Mortazavi, A. A. Khodadadi, S. M. A. Mousavian, K. Sadagiani and A. Karimi, Iran. J. Chem. Chem. Eng., 2005, 24, 9–17 CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- H. M. Torres Galvis and K. P. de Jong, ACS Catal., 2013, 3, 2130–2149 CrossRef CAS.
- K. Cheng, V. V. Ordomsky, M. Virginie, B. Legras, P. A. Chernavskii, V. Kazak, C. Cordier, S. Paul, Y. Wang and A. Y. Khodakov, Appl. Catal., A, 2014, 488, 66–77 CrossRef CAS.
- Q. Zhang, K. Cheng, J. Kang, W. Deng and Y. Wang, ChemSusChem, 2014, 7, 1251–1264 CrossRef CAS.
- J. Lu, L. Yang, B. Xu, Q. Wu, D. Zhang, S. Yuan, Y. Zhai, X. Wang, Y. Fan and Z. Hu, ACS Catal., 2014, 4, 613–621 CrossRef CAS.
- M. Moyo, M. A. Motchelaho, H. Xiong, L. L. Jewell and N. J. Coville, Appl. Catal., A, 2012, 413, 223–229 CrossRef.
- K. P. De Jong and J. W. Geus, Catal. Rev., 2000, 42, 481–510 CrossRef CAS.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS.
- C. Zhu, M. Zhang, C. Huang, L. Zhong and K. Fang, New J. Chem., 2018, 42, 2413–2421 RSC.
- G. Yu, B. Sun, Y. Pei, S. Xie, S. Yan, M. Qiao, K. Fan, X. Zhang and B. Zong, J. Am. Chem. Soc., 2010, 132, 935–937 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- J. Zhu, A. Holmen and D. Chen, ChemCatChem, 2013, 5, 378–401 CrossRef CAS.
- M. Jeguirim, M. Belhachemi, L. Limousy and S. Bennici, Chem. Eng. J., 2018, 347, 493–504 CrossRef CAS.
- J. Figueiredo, M. Pereira, M. Freitas and J. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- W.-P. Ma, Y.-J. Ding and L.-W. Lin, Ind. Eng. Chem. Res., 2004, 43, 2391–2398 CrossRef CAS.
- A. Tavasoli, R. M. M. Abbaslou, M. Trepanier and A. K. Dalai, Appl. Catal., A, 2008, 345, 134–142 CrossRef CAS.
- P. M. Ajayan, Chem. Rev., 1999, 99, 1787–1800 CrossRef CAS.
- M. May, Science, 2013, 339, 858–860 CrossRef.
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef CAS.
- W. Chen, X. Pan, M.-G. Willinger, D. S. Su and X. Bao, J. Am. Chem. Soc., 2006, 128, 3136–3137 CrossRef CAS.
- E. Dujardin, T. Ebbesen, H. Hiura and K. Tanigaki, Science, 1994, 265, 1850–1852 CrossRef CAS.
- J.-M. Nhut, L. Pesant, J.-P. Tessonnier, G. Winé, J. Guille, C. Pham-Huu and M.-J. Ledoux, Appl. Catal., A, 2003, 254, 345–363 CrossRef CAS.
- Y. Zhang, H.-B. Zhang, G.-D. Lin, P. Chen, Y.-Z. Yuan and K. Tsai, Appl. Catal., A, 1999, 187, 213–224 CrossRef CAS.
- M. Menon, A. N. Andriotis and G. E. Froudakis, Chem. Phys. Lett., 2000, 320, 425–434 CrossRef CAS.
- S. Westerberg, C. Wang, K. Chou and G. A. Somorjai, J. Phys. Chem. B, 2004, 108, 6374–6380 CrossRef CAS.
- W. Chen, Z. Fan, X. Pan and X. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS.
- D. Ugarte, A. Chatelain and W. De Heer, Science, 1996, 274, 1897–1899 CrossRef CAS.
- R. Haddon, Science, 1993, 261, 1545–1550 CrossRef CAS.
- B. Shan and K. Cho, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 081401 CrossRef.
- J. Li, G. Jacobs, T. Das, Y. Zhang and B. Davis, Appl. Catal., A, 2002, 236, 67–76 CrossRef CAS.
- P. Van Berge, J. Van de Loosdrecht, S. Barradas and A. Van Der Kraan, Catal. Today, 2000, 58, 321–334 CrossRef CAS.
- G. Jacobs, P. M. Patterson, Y. Zhang, T. Das, J. Li and B. H. Davis, Appl. Catal., A, 2002, 233, 215–226 CrossRef CAS.
- G. L. Bezemer, J. H. Bitter, H. P. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS.
- J. P. den Breejen, J. R. Sietsma, H. Friedrich, J. H. Bitter and K. P. de Jong, J. Catal., 2010, 270, 146–152 CrossRef CAS.
- J. Den Breejen, P. Radstake, G. Bezemer, J. Bitter, V. Frøseth, A. Holmen and K. P. de Jong, J. Am. Chem. Soc., 2009, 131, 7197–7203 CrossRef CAS.
- H. Xiong, M. Moyo, M. A. Motchelaho, L. L. Jewell and N. J. Coville, Appl. Catal., A, 2010, 388, 168–178 CrossRef CAS.
- Z. Wang and Z. Kang, Carbon, 1997, 35, 419–426 CrossRef CAS.
- H. Qin, S. Kang, Y. Wang, H. Liu, Z. Ni, Y. Huang, Y. Li and X. Li, ACS Sustainable Chem. Eng., 2016, 4, 1240–1247 CrossRef CAS.
- W. Zhang, X. Jiang, X. Wang, Y. V. Kaneti, Y. Chen, J. Liu, J.-S. Jiang, Y. Yamauchi and M. Hu, Angew. Chem., Int. Ed., 2017, 56, 8435–8440 CrossRef CAS.
- W. Zhang, X. Jiang, Y. Zhao, A. Carné-Sánchez, V. Malgras, J. Kim, J. H. Kim, S. Wang, J. Liu, J.-S. Jiang, Y. Yamauchi and M. Hu, Chem. Sci., 2017, 8, 3538–3546 RSC.
- C.-A. H. Price, L. Pastor-Pérez, T. Ramirez Reina and J. Liu, React. Chem. Eng., 2018, 3, 433–436 RSC.
- H. Tian, C. Zhang, P. Su, Z. Shen, H. Liu, G. Wang, S. Liu and J. Liu, J. Energy Chem., 2020, 40, 137–143 CrossRef.
- V. Subramanian, K. Cheng, C. Lancelot, S. Heyte, S. Paul, S. Moldovan, O. Ersen, M. Marinova, V. V. Ordomsky and A. Y. Khodakov, ACS Catal., 2016, 6, 1785–1792 CrossRef CAS.
- J. J. Venter and M. A. Vannice, Catal. Lett., 1990, 7, 219–240 CrossRef CAS.
- J. Venter, M. Kaminsky, G. L. Geoffroy and M. A. Vannice, J. Catal., 1987, 103, 450–465 CrossRef CAS.
- M. Vannice, P. Walker Jr, H. Jung, C. Moreno-Castillat and O. Mahajan, Stud. Surf. Sci. Catal., 1981, 7, 460–474 CrossRef CAS.
- A. Chen, M. Kaminsky, G. Geoffroy and M. Vannice, J. Phys. Chem., 1986, 90, 4810–4819 CrossRef CAS.
- R. Lahti, D. Bergna, H. Romar, T. Hu, A. Comazzi, C. Pirola, C. L. Bianchi and U. Lassi, Top. Catal., 2017, 60, 1415–1428 CrossRef CAS.
- V. M. Lebarbier, D. Mei, D. H. Kim, A. Andersen, J. L. Male, J. E. Holladay, R. Rousseau and Y. Wang, J. Phys. Chem. C, 2011, 115, 17440–17451 CrossRef CAS.
- W. Qian, H. Zhang, W. Ying and D. Fang, J. Nat. Gas Chem., 2011, 20, 389–396 CrossRef CAS.
- R. Luque, A. R. de la Osa, J. M. Campelo, A. A. Romero, J. L. Valverde and P. Sanchez, Energy Environ. Sci., 2012, 5, 5186–5202 RSC.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- W. Qian, H. Zhang, W. Ying and D. Fang, Chem. Eng. J., 2013, 228, 526–534 CrossRef CAS.
- F. Chen, W. Jin, D.-G. Cheng, X. Zhan and Y. S. Lin, J. Chem. Technol. Biotechnol., 2013, 88, 2133–2140 CrossRef CAS.
- Y. Pei, Y. Ding, H. Zhu, J. Zang, X. Song, W. Dong, T. Wang and Y. Lu, Catal. Lett., 2014, 144, 1433–1442 CrossRef CAS.
- G. Jiao, Y. Ding, H. Zhu, X. Li, J. Li, R. Lin, W. Dong, L. Gong, Y. Pei and Y. Lu, Appl. Catal., A, 2009, 364, 137–142 CrossRef CAS.
- T. Wang, Y. Ding, J. Xiong, L. Yan, H. Zhu, Y. Lu and L. Lin, Catal. Lett., 2006, 107, 47–52 CrossRef CAS.
- Y. Pei, Y. Ding, J. Zang, X. Song, W. Dong, H. Zhu, T. Wang and W. Chen, Chin. J. Catal., 2013, 33, 808–812 Search PubMed.
- T. Wang, Y.-J. Ding, J.-M. Xiong, W.-M. Chen, Z.-D. Pan, Y. Lu and L.-W. Lin, Stud. Surf. Sci. Catal., 2004, 147, 349–354 CrossRef CAS.
- H. Du, H. Zhu, T. Liu, Z. Zhao, X. Chen, W. Dong, W. Lu, W. Luo and Y. Ding, Catal. Today, 2017, 281, 549–558 CrossRef CAS.
- Z. Zhao, W. Lu, C. Feng, X. Chen, H. Zhu, R. Yang, W. Dong, M. Zhao, Y. Lyu, T. Liu, Z. Jiang and Y. Ding, J. Catal., 2019, 370, 251–264 CrossRef CAS.
- T. Wang, Y. Ding, Y. Lü, H. Zhu and L. Lin, J. Nat. Gas Chem., 2008, 17, 153–158 CrossRef CAS.
- H. Du, H. Zhu, X. Chen, W. Dong, W. Lu, W. Luo, M. Jiang, T. Liu and Y. Ding, Fuel, 2016, 182, 42–49 CrossRef CAS.
- Y.-P. Pei, J.-X. Liu, Y.-H. Zhao, Y.-J. Ding, T. Liu, W.-D. Dong, H.-J. Zhu, H.-Y. Su, L. Yan and J.-L. Li, ACS Catal., 2015, 5, 3620–3624 CrossRef CAS.
- K. Fang, D. Li, M. Lin, M. Xiang, W. Wei and Y. Sun, Catal. Today, 2009, 147, 133–138 CrossRef CAS.
- M. Ao, G. H. Pham, J. Sunarso, M. O. Tade and S. Liu, ACS Catal., 2018, 8, 7025–7050 CrossRef CAS.
- Y. Pei, Y. Ding, H. Zhu, J. Zang, X. Song, W. Dong, T. Wang, L. Yan and Y. Lu, React. Kinet., Mech. Catal., 2013, 111, 505–520 CrossRef.
- G. Volkova, T. Yurieva, L. Plyasova, M. Naumova and V. Zaikovskii, J. Mol. Catal. A: Chem., 2000, 158, 389–393 CrossRef CAS.
- J. A. Singh, A. S. Hoffman, J. Schumann, A. Boubnov, A. S. Asundi, S. S. Nathan, J. Nørskov, S. R. Bare and S. F. Bent, ChemCatChem, 2019, 11, 799–809 CrossRef CAS.
- C. Göbel, S. Schmidt, C. Froese, Q. Fu, Y.-T. Chen, Q. Pan and M. Muhler, J. Catal., 2020, 383, 33–41 CrossRef.
- H. Du, H. Zhu, Z. Zhao, W. Dong, W. Luo, W. Lu, M. Jiang, T. Liu and Y. Ding, Appl. Catal., A, 2016, 523, 263–271 CrossRef CAS.
- Z. Zhao, W. Lu, H. Zhu, W. Dong, Y. Lyu, T. Liu, X. Chen, Y. Wang and Y. Ding, J. Catal., 2018, 361, 156–167 CrossRef CAS.
- G. Jiao, Y. Ding, H. Zhu, X. Li, W. Dong, J. Li and Y. Lü, Chin. J. Catal., 2009, 30, 92–94 CrossRef CAS.
- J. Xiong, Y. Ding, T. Wang, L. Yan, W. Chen, H. Zhu and Y. Lu, Catal. Lett., 2005, 102, 265–269 CrossRef CAS.
- Y. Zhou, S. Natesakhawat, T. D. Nguyen-Phan, D. R. Kauffman, C. M. Marin, K. Kisslinger, R. Lin, H. L. Xin, E. Stavitski and K. Attenkofer, ChemCatChem, 2019, 11, 1625–1632 CrossRef CAS.
- S. Iqbal, T. E. Davies, D. J. Morgan, K. Karim, J. S. Hayward, J. K. Bartley, S. H. Taylor and G. J. Hutchings, Catal. Today, 2016, 275, 35–39 CrossRef CAS.
- M. Claeys, M. Dry, E. Van Steen, E. Du Plessis, P. Van Berge, A. Saib and D. Moodley, J. Catal., 2014, 318, 193–202 CrossRef CAS.
- B. Wang, D. Liang, Z. Guan, D. Li and R. Zhang, J. Phys. Chem. C, 2020, 124, 5749–5758 CrossRef CAS.
- Y. An, T. Lin, K. Gong, X. Wang, L. Zhong, H. Wang and Y. Sun, ChemCatChem, 2020, 12, 1630–1638 CrossRef CAS.
- Y. Pei, Y. Ding, J. Zang, X. Song, W. Dong, H. Zhu, T. Wang and W. Chen, Chin. J. Catal., 2013, 34, 1570–1575 CrossRef CAS.
- Z. Zhao, W. Lu, R. Yang, H. Zhu, W. Dong, F. Sun, Z. Jiang, Y. Lyu, T. Liu, H. Du and Y. Ding, ACS Catal., 2017, 8, 228–241 CrossRef.
- A. Tavasoli, K. Sadagiani, F. Khorashe, A. A. Seifkordi, A. A. Rohani and A. Nakhaeipour, Fuel Process. Technol., 2008, 89, 491–498 CrossRef CAS.
- M. Zaman, A. Khodadi and Y. Mortazavi, Fuel Process. Technol., 2009, 90, 1214–1219 CrossRef CAS.
- H. Xiong, M. A. M. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, Catal. Today, 2013, 214, 50–60 CrossRef CAS.
- H. Xiong, M. A. M. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, J. Catal., 2011, 278, 26–40 CrossRef CAS.
- A. C. Ghogia, S. Cayez, B. F. Machado, A. Nzihou, P. Serp, K. Soulantica and D. Pham Minh, ChemCatChem, 2019, 12, 1117–1128 CrossRef.
- S. Chen, C. Pei and J. Gong, Energy Environ. Sci., 2019, 12, 3473–3495 RSC.
- A. d. Klerk, Energy Environ. Sci., 2011, 4, 1177–1205 RSC.
- G. L. Bezemer, U. Falke, A. J. van Dillen and K. P. de Jong, Chem. Commun., 2005, 731–733 RSC.
- G. L. Bezemer, P. Radstake, V. Koot, A. Van Dillen, J. Geus and K. De Jong, J. Catal., 2006, 237, 291–302 CrossRef CAS.
- M. Omraei, S. Sheibani, S. Sadrameli and J. Towfighi, Ind. Eng. Chem. Res., 2013, 52, 1829–1835 CrossRef CAS.
- J. Y. Yook, J. Jun and S. Kwak, Appl. Surf. Sci., 2010, 256, 6941–6944 CrossRef CAS.
- P.-L. Girard-Lauriault, R. Illgen, J.-C. Ruiz, M. R. Wertheimer and W. E. Unger, Appl. Surf. Sci., 2012, 258, 8448–8454 CrossRef CAS.
- A. Amiri, M. Maghrebi, M. Baniadam and S. Z. Heris, Appl. Surf. Sci., 2011, 257, 10261–10266 CrossRef CAS.
- J.-P. Tessonnier, D. Rosenthal, T. W. Hansen, C. Hess, M. E. Schuster, R. Blume, F. Girgsdies, N. Pfänder, O. Timpe and D. S. Su, Carbon, 2009, 47, 1779–1798 CrossRef CAS.
- T. G. Ros, A. J. Van Dillen, J. W. Geus and D. C. Koningsberger, Chem. – Eur. J., 2002, 8, 1151–1162 CrossRef CAS.
- H. J. Schulte, B. Graf, W. Xia and M. Muhler, ChemCatChem, 2012, 4, 350–355 CrossRef CAS.
- M. Trépanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2009, 90, 367–374 CrossRef.
- H. Zhang, C. Lancelot, W. Chu, J. Hong, A. Y. Khodakov, P. A. Chernavskii, J. Zheng and D. Tong, J. Mater. Chem., 2009, 19, 9241–9249 RSC.
- T. O. Eschemann, W. S. Lamme, R. L. Manchester, T. E. Parmentier, A. Cognigni, M. Rønning and K. P. de Jong, J. Catal., 2015, 328, 130–138 CrossRef CAS.
- L. Guczi, G. Stefler, O. Geszti, Z. Koppany, Z. Konya, E. Molnar, M. Urban and I. Kiricsi, J. Catal., 2006, 244, 24–32 CrossRef CAS.
- H. B. Zhang, X. Dong, G. D. Lin, X. L. Liang and H. Y. Li, Chem. Commun., 2005, 5094–5096 RSC.
- M. Boudart and M. A. McDonald, J. Phys. Chem., 1984, 88, 2185–2195 CrossRef CAS.
- R. A. Van Santen, Acc. Chem. Res., 2009, 42, 57–66 CrossRef CAS.
- A. Wielers, A. Kock, C. Hop, J. Geus and A. van Der Kraan, J. Catal., 1989, 117, 1–18 CrossRef CAS.
- G. Bezemer, A. Van Laak, A. Van Dillen and K. De Jong, Stud. Surf. Sci. Catal., 2004, 147, 259–264 CrossRef CAS.
- N. E. Tsakoumis, R. Dehghan, R. E. Johnsen, A. Voronov, W. van Beek, J. C. Walmsley, Ø. Borg, E. Rytter, D. Chen, M. Rønning and A. Holmen, Catal. Today, 2013, 205, 86–93 CrossRef CAS.
- G. Bezemer, P. Radstake, U. Falke, H. Oosterbeek, H. Kuipers, A. Vandillen and K. Dejong, J. Catal., 2006, 237, 152–161 CrossRef CAS.
- Z. Yu, Ø. Borg, D. Chen, B. C. Enger, V. Frøseth, E. Rytter, H. Wigum and A. Holmen, Catal. Lett., 2006, 109, 43–47 CrossRef CAS.
- Ø. Borg, Z. Yu, D. Chen, E. A. Blekkan, E. Rytter and A. Holmen, Top. Catal., 2013, 57, 491–499 CrossRef.
- J. Zhu, J. Yang, A. H. Lillebø, Y. Zhu, Y. Yu, A. Holmen and D. Chen, Catal. Today, 2013, 215, 121–130 CrossRef CAS.
- S. Campisi, C. E. Chan-Thaw and A. Villa, Appl. Sci., 2018, 8, 1159 CrossRef.
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 30, 1706836 CrossRef.
- L. Chen, G. Song, Y. Fu and J. Shen, J. Colloid Interface Sci., 2012, 368, 456–461 CrossRef CAS.
- T. Fu, Y. Jiang, J. Lv and Z. Li, Fuel Process. Technol., 2013, 110, 141–149 CrossRef CAS.
- M. W. Dlamini, T. N. Phaahlamohlaka, D. O. Kumi, R. Forbes, L. L. Jewell and N. J. Coville, Catal. Today, 2020, 342, 99–110 CrossRef CAS.
- K. Cheng, J. Kang, D. L. King, V. Subramanian, C. Zhou, Q. Zhang and Y. Wang, Adv. Catal., 2017, 60, 125–208 CAS.
- Z. Wang, G. Laddha, S. Kanitkar and J. J. Spivey, Catal. Today, 2017, 298, 209–215 CrossRef CAS.
- Y. Chen, X. Li, M. U. Nisa, J. Lv and Z. Li, Fuel, 2019, 241, 802–812 CrossRef CAS.
- V. I. Isaeva, O. L. Eliseev, R. V. Kazantsev, V. V. Chernyshev, A. L. Tarasov, P. E. Davydov, A. L. Lapidus and L. M. Kustov, Polyhedron, 2019, 157, 389–395 CrossRef CAS.
- C. Zhang, X. Guo, Q. Yuan, R. Zhang, Q. Chang, K. Li, B. Xiao, S. Liu, C. Ma, X. Liu, Y. Xu, X. Wen, Y. Yang and Y. Li, ACS Catal., 2018, 8, 7120–7130 CrossRef CAS.
- V. I. Isaeva, O. L. Eliseev, R. V. Kazantsev, V. V. Chernyshev, P. E. Davydov, B. R. Saifutdinov, A. L. Lapidus and L. M. Kustov, Dalton Trans., 2016, 45, 12006–12014 RSC.
- Y. Pei, Z. Li and Y. Li, AlChE J., 2017, 63, 2935–2944 CrossRef CAS.
- N. Li, C.-P. Ma, C.-H. Zhang, Y. Yang and Y.-W. Li, J. Fuel Chem. Technol., 2019, 47, 428–437 CrossRef CAS.
- X. Sun, A. I. O. Suarez, M. Meijerink, T. van Deelen, S. Ould-Chikh, J. Zecevic, K. P. de Jong, F. Kapteijn and J. Gascon, Nat. Commun., 2017, 8, 1680 CrossRef.
- Q.-X. Luo, L.-P. Guo, S.-Y. Yao, J. Bao, Z.-T. Liu and Z.-W. Liu, J. Catal., 2019, 369, 143–156 CrossRef CAS.
- B. Qiu, C. Yang, W. Guo, Y. Xu, Z. Liang, D. Ma and R. Zou, J. Mater. Chem. A, 2017, 5, 8081–8086 RSC.
- K. Holt, L. Jewell, H. Niemantsverdriet, L. Macheli, M. Shozi, G. Hutchings, T. Wezendonk, M. Bowker, R. Catlow and S. Adam, Faraday Discuss., 2017, 197, 353–388 RSC.
- H. Jung, M. Vannice, L. Mulay, R. Stanfield and W. Delgass, J. Catal., 1982, 76, 208–224 CrossRef CAS.
- A. Sepulveda-Escribano and F. Rodriguez-Reinoso, J. Mol. Catal., 1994, 90, 291–301 CrossRef CAS.
- E. van Steen and F. F. Prinsloo, Catal. Today, 2002, 71, 327–334 CrossRef CAS.
- W. Ma, Y. Ding, J. Yang, X. Liu and L. Lin, React. Kinet. Catal. Lett., 2005, 84, 11–19 CrossRef CAS.
- W. Ma, E. L. Kugler and D. B. Dadyburjor, Energy Fuels, 2007, 21, 1832–1842 CrossRef CAS.
- W. Ma, E. L. Kugler and D. B. Dadyburjor, Energy Fuels, 2011, 25, 1931–1938 CrossRef CAS.
- K. Asami, K. Komiyama, K. Yoshida and H. Miyahara, Catal. Today, 2018, 303, 117–122 CrossRef CAS.
- W. Ma, E. L. Kugler, J. Wright and D. B. Dadyburjor, Energy Fuels, 2006, 20, 2299–2307 CrossRef CAS.
- Z. Tian, C. Wang, Z. Si, L. Ma, L. Chen, Q. Liu, Q. Zhang and H. Huang, Appl. Catal., A, 2017, 541, 50–59 CrossRef CAS.
- Z. Tian, C. Wang, Z. Si, C. Wen, Y. Xu, W. Lv, L. Chen, X. Zhang and L. Ma, Catalysts, 2019, 9, 505–514 CrossRef CAS.
- P. A. Chernavskii, G. V. Pankina, R. V. Kazantsev and O. L. Eliseev, ChemCatChem, 2018, 10, 1313–1320 CrossRef CAS.
- X. Pan and X. Bao, Acc. Chem. Res., 2011, 44, 553–562 CrossRef CAS.
- R. M. Malek Abbaslou, A. Tavasoli and A. K. Dalai, Appl. Catal., A, 2009, 355, 33–41 CrossRef CAS.
- T. Vo, Y.-D. Wu, R. Car and M. Robert, J. Phys. Chem. C, 2008, 112, 8400–8407 CrossRef CAS.
- Y. Yagi, T. M. Briere, M. H. Sluiter, V. Kumar, A. A. Farajian and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075414 CrossRef.
- S. B. Fagan, R. Mota, A. J. da Silva and A. Fazzio, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205414 CrossRef.
- U. M. Graham, A. Dozier, R. A. Khatri, M. C. Bahome, L. L. Jewell, S. D. Mhlanga, N. J. Coville and B. H. Davis, Catal. Lett., 2009, 129, 39–45 CrossRef CAS.
- L. Yu, W.-X. Li, X. Pan and X. Bao, J. Phys. Chem. C, 2012, 116, 16461–16466 CrossRef CAS.
- J. Xiao, X. Pan, S. Guo, P. Ren and X. Bao, J. Am. Chem. Soc., 2015, 137, 477–482 CrossRef CAS.
- R. M. M. Abbaslou, A. Tavassoli, J. Soltan and A. K. Dalai, Appl. Catal., A, 2009, 367, 47–52 CrossRef CAS.
- B. Gu, S. He, D. V. Peron, D. R. Strossi Pedrolo, S. Moldovan, M. C. Ribeiro, B. Lobato, P. A. Chernavskii, V. V. Ordomsky and A. Y. Khodakov, J. Catal., 2019, 376, 1–16 CrossRef CAS.
- B. Gu, C. Zhou, S. He, S. Moldovan, P. A. Chernavskii, V. V. Ordomsky and A. Y. Khodakov, Catal. Today, 2019, 05, 054 Search PubMed.
- H. Xiong, M. A. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, Appl. Catal., A, 2014, 482, 377–386 CrossRef CAS.
- Z. Yang, S. Guo, X. Pan, J. Wang and X. Bao, Energy Environ. Sci., 2011, 4, 4500–4503 RSC.
- C. Galeano, J. C. Meier, M. Soorholtz, H. Bongard, C. Baldizzone, K. J. Mayrhofer and F. Schüth, ACS Catal., 2014, 4, 3856–3868 CrossRef CAS.
- J. Xie, J. Yang, A. I. Dugulan, A. Holmen, D. Chen, K. P. de Jong and M. J. Louwerse, ACS Catal., 2016, 6, 3147–3157 CrossRef CAS.
- Z. Yang, X. Pan, J. Wang and X. Bao, Catal. Today, 2012, 186, 121–127 CrossRef CAS.
- J.-D. Xu, K.-T. Zhu, X.-F. Weng, W.-Z. Weng, C.-J. Huang and H.-L. Wan, Catal. Today, 2013, 215, 86–94 CrossRef CAS.
- M. C. Bahome, L. L. Jewell, K. Padayachy, D. Hildebrandt, D. Glasser, A. K. Datye and N. J. Coville, Appl. Catal., A, 2007, 328, 243–251 CrossRef CAS.
- M. C. Bahome, L. L. Jewell, D. Hildebrandt, D. Glasser and N. J. Coville, Appl. Catal., A, 2005, 287, 60–67 CrossRef CAS.
- B. Gu, V. V. Ordomsky, M. Bahri, O. Ersen, P. A. Chernavskii, D. Filimonov and A. Y. Khodakov, Appl. Catal., B, 2018, 234, 153–166 CrossRef CAS.
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef CAS.
- B. Gu, D. V. Peron, A. J. Barrios, M. Bahri, O. Ersen, M. Vorokhta, B. Šmíd, D. Banerjee, M. Virginie, E. Marceau, R. Wojcieszak, V. V. Ordomsky and A. Y. Khodakov, Chem. Sci., 2020, 11, 6167–6182 RSC.
- A. Steynberg, Stud. Surf. Sci. Catal., 2004, 152, 1–63 CrossRef CAS.
- H. M. Torres Galvis, J. H. Bitter, T. Davidian, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, J. Am. Chem. Soc., 2012, 134, 16207–16215 CrossRef CAS.
- X. Zhao, S. Lv, L. Wang, L. Li, G. Wang, Y. Zhang and J. Li, Mol. Catal., 2018, 449, 99–105 CrossRef CAS.
- M. G. A. Cruz, F. A. N. Fernandes, A. C. Oliveira, J. M. Filho, A. C. Oliveira, A. F. Campos, E. Padron-Hernandez and E. Rodríguez-Castellón, Catal. Today, 2017, 282, 174–184 CrossRef CAS.
- M. Oschatz, J. P. Hofmann, T. W. van Deelen, W. S. Lamme, N. A. Krans, E. J. Hensen and K. P. de Jong, ChemCatChem, 2017, 9, 620–628 CrossRef CAS.
- M. Oschatz, T. W. van Deelen, J. L. Weber, W. S. Lamme, G. Wang, B. Goderis, O. Verkinderen, A. I. Dugulan and K. P. de Jong, Catal. Sci. Technol., 2016, 6, 8464–8473 RSC.
- M. Oschatz, W. S. Lamme, J. Xie, A. I. Dugulan and K. P. de Jong, ChemCatChem, 2016, 8, 2846–2852 CrossRef CAS.
- H. Xiong, M. Moyo, M. A. Motchelaho, Z. N. Tetana, S. M. A. Dube, L. L. Jewell and N. J. Coville, J. Catal., 2014, 311, 80–87 CrossRef CAS.
- V. P. Santos, T. A. Wezendonk, J. J. Jaen, A. I. Dugulan, M. A. Nasalevich, H. U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451–6458 CrossRef CAS.
- T. A. Wezendonk, V. P. Santos, M. A. Nasalevich, Q. S. Warringa, A. I. Dugulan, A. Chojecki, A. C. Koeken, M. Ruitenbeek, G. Meima and H.-U. Islam, ACS Catal., 2016, 6, 3236–3247 CrossRef CAS.
- T. A. Wezendonk, X. Sun, A. I. Dugulan, A. J. F. van Hoof, E. J. M. Hensen, F. Kapteijn and J. Gascon, J. Catal., 2018, 362, 106–117 CrossRef CAS.
- B. An, K. Cheng, C. Wang, Y. Wang and W. Lin, ACS Catal., 2016, 6, 3610–3618 CrossRef CAS.
- T. A. Wezendonk, Q. S. Warringa, V. P. Santos, A. Chojecki, M. Ruitenbeek, G. Meima, M. Makkee, F. Kapteijn and J. Gascon, Faraday Discuss., 2017, 197, 225–242 RSC.
- L. Oar-Arteta, M. J. Valero-Romero, T. Wezendonk, F. Kapteijn and J. Gascon, Catal. Sci. Technol., 2018, 8, 210–220 RSC.
- M. Oschatz, S. Krause, N. A. Krans, C. Hernandez Mejia, S. Kaskel and K. P. de Jong, Chem. Commun., 2017, 53, 10204–10207 RSC.
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC.
- X. Chen, D. Deng, X. Pan, Y. Hu and X. Bao, Chem. Commun., 2015, 51, 217–220 RSC.
- D. Peña, A. Cognigni, T. Neumayer, W. van Beek, D. S. Jones, M. Quijada and M. Rønning, Appl. Catal., A, 2018, 554, 10–23 CrossRef.
- V. V. Ordomsky, B. Legras, K. Cheng, S. Paul and A. Y. Khodakov, Catal. Sci. Technol., 2015, 5, 1433–1437 RSC.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS.
- J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS.
- B. Gu, S. He, W. Zhou, J. Kang, K. Cheng, Q. Zhang and Y. Wang, J. Energy Chem., 2017, 26, 608–615 CrossRef.
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. Lou, X. Xing and G. Q. Lu, Chem. Commun., 2011, 47, 12578–12591 RSC.
- J. Liu, S. Z. Qiao, S. Budi Hartono and G. Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 4981–4985 CrossRef CAS.
- Y. Cheng, J. Lin, K. Xu, H. Wang, X. Yao, Y. Pei, S. Yan, M. Qiao and B. Zong, ACS Catal., 2015, 6, 389–399 CrossRef.
- Y. Cheng, J. Lin, T. Wu, H. Wang, S. Xie, Y. Pei, S. Yan, M. Qiao and B. Zong, Appl. Catal., B, 2017, 204, 475–485 CrossRef CAS.
- H. Zhao, Q. Zhu, Y. Gao, P. Zhai and D. Ma, Appl. Catal., A, 2013, 456, 233–239 CrossRef CAS.
- S. O. Moussa, L. S. Panchakarla, M. Q. Ho and M. S. El-Shall, ACS Catal., 2014, 4, 535–545 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 54, 588–593 CAS.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892–3896 CrossRef CAS.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith, M. Jaroniec, G. Q. Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS.
- V. Subramanian, V. V. Ordomsky, B. Legras, K. Cheng, C. Cordier, P. A. Chernavskii and A. Y. Khodakov, Catal. Sci. Technol., 2016, 6, 4953–4961 RSC.
- H. Park, D. H. Youn, J. Y. Kim, W. Y. Kim, Y. H. Choi, Y. H. Lee, S. H. Choi and J. S. Lee, ChemCatChem, 2015, 7, 3488–3494 CrossRef CAS.
- M. Lv, W. Xie, S. Sun, G. Wu, L. Zheng, S. Chu, C. Gao and J. Bao, Catal. Sci. Technol., 2015, 5, 2925–2934 RSC.
- E. T. Liakakou, E. Heracleous, K. S. Triantafyllidis and A. A. Lemonidou, Appl. Catal., B, 2015, 165, 296–305 CrossRef CAS.
- M. Taborga Claure, S.-H. Chai, S. Dai, K. A. Unocic, F. M. Alamgir, P. K. Agrawal and C. W. Jones, J. Catal., 2015, 324, 88–97 CrossRef CAS.
- L. Chen, Y. Zhu, H. Zheng, C. Zhang, B. Zhang and Y. Li, J. Chem. Technol. Biotechnol., 2012, 87, 112–122 CrossRef CAS.
- X. Pan, Z. Fan, W. Chen, Y. Ding, H. Luo and X. Bao, Nat. Mater., 2007, 6, 507–511 CrossRef CAS.
- B. Li, C. Wang, G. Yi, H. Lin and Y. Yuan, Catal. Today, 2011, 164, 74–79 CrossRef CAS.
- K. Murata, K. Okabe, M. Inaba, I. Takahara and Y. Liu, J. Jpn. Pet. Inst., 2009, 52, 16–20 CrossRef CAS.
- J. Kang, S. Zhang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2009, 48, 2565–2568 CrossRef CAS.
- J.-J. Wang, J.-R. Xie, Y.-H. Huang, B.-H. Chen, G.-D. Lin and H.-B. Zhang, Appl. Catal., A, 2013, 468, 44–51 CrossRef CAS.
- I. C. ten Have, E. Valle, A. Gallo, J. L. Snider, M. S. Duyar and T. F. Jaramillo, Energy Technol., 2019, 7, 1801102 CrossRef.
- V. S. Dorokhov, E. A. Permyakov, P. A. Nikulshin, V. V. Maximov and V. M. Kogan, J. Catal., 2016, 344, 841–853 CrossRef CAS.
- J. L. Eslava, A. Iglesias-Juez, G. Agostini, M. Fernández-García, A. Guerrero-Ruiz and I. Rodríguez-Ramos, ACS Catal., 2016, 6, 1437–1445 CrossRef CAS.
- K. Xiong, Y. Zhang, J. Li and K. Liew, J. Energy Chem., 2013, 22, 560–566 CrossRef CAS.
- K. Xiong, J. Li, K. Liew and X. Zhan, Appl. Catal., A, 2010, 389, 173–178 CrossRef CAS.
- M. Perovic, Q. Qin and M. Oschatz, Adv. Funct. Mater., 2020, 1908317 Search PubMed.
- Y. Boyjoo, M. Wang, V. K. Pareek, J. Liu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 6013–6047 RSC.
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS.
- J. Tang, J. Liu, N. L. Torad, T. Kimura and Y. Yamauchi, Nano Today, 2014, 9, 305–323 CrossRef CAS.
- H. Tian, J. Liang and J. Liu, Adv. Mater., 2019, 31, 1903886 CrossRef CAS.
- J. Liu, T. Yang, D.-W. Wang, G. Q. Lu, D. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 CrossRef.
- T. Yang, R. Zhou, D. W. Wang, S. P. Jiang, Y. Yamauchi, S. Z. Qiao, M. J. Monteiro and J. Liu, Chem. Commun., 2015, 51, 2518–2521 RSC.
- J. Liu, S. Z. Qiao, H. Liu, J. Chen, A. Orpe, D. Zhao and G. Q. M. Lu, Angew. Chem., Int. Ed., 2011, 50, 5947–5951 CrossRef CAS.
- H. Tian, J. Liu, K. O'Donnell, T. Liu, X. Liu, Z. Yan, S. Liu and M. Jaroniec, J. Colloid Interface Sci., 2016, 476, 55–61 CrossRef CAS.
- T. Yang, H. Ling, J.-F. Lamonier, M. Jaroniec, J. Huang, M. J. Monteiro and J. Liu, NPG Asia Mater., 2016, 8, 240 CrossRef.
- K. Cheng, V. Subramanian, A. Carvalho, V. V. Ordomsky, Y. Wang and A. Y. Khodakov, J. Catal., 2016, 337, 260–271 CrossRef CAS.
- A. Carvalho, M. Marinova, N. Batalha, N. R. Marcilio, A. Y. Khodakov and V. V. Ordomsky, Catal. Sci. Technol., 2017, 7, 5019–5027 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cs00905a |
| This journal is © The Royal Society of Chemistry 2021 |