
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous alkane dehydrogenation catalysts investigated via a surface organometallic chemistry approach
Scott R.
Docherty
 ,
Lukas
Rochlitz
,
Pierre-Adrien
Payard
and
Christophe
Copéret
*
,
Lukas
Rochlitz
,
Pierre-Adrien
Payard
and
Christophe
Copéret
*
Department of Chemistry and Applied Biosciences – ETH Zürich, Vladimir Prelog 2, CH8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
First published on 24th March 2021
Abstract
The selective conversion of light alkanes (C2–C6 saturated hydrocarbons) to the corresponding alkene is an appealing strategy for the petrochemical industry in view of the availability of these feedstocks, in particular with the emergence of Shale gas. Here, we present a review of model dehydrogenation catalysts of light alkanes prepared via surface organometallic chemistry (SOMC). A specific focus of this review is the use of molecular strategies for the deconvolution of complex heterogeneous materials that are proficient in enabling dehydrogenation reactions. The challenges associated with the proposed reactions are highlighted, as well as overriding themes that can be ascertained from the systematic study of these challenging reactions using model SOMC catalysts.
 Scott R. Docherty | Scott R. Docherty obtained a Masters in Chemistry with a Year Abroad from the University of Edinburgh in 2018, spending a year in the lab of Prof. Christophe Copéret before completing projects in the groups of Dr Stephen Thomas and Prof. Polly Arnold. Since September 2018 Scott has been a doctoral candidate in the lab of Prof. Christophe Copéret at ETH Zürich. His work focuses on the role of dopants in heterogeneous catalytic processes. |
 Lukas Rochlitz | Lukas Rochlitz received his Bachelor's and Master's degree in Chemistry from ETH Zürich. He carried out his Master thesis project in the laboratory of Prof. T. Don Tilley at the UC Berkeley in 2017, working on Fe-catalysed hydrosilylation. In January 2018, he joined the group of Prof. Christophe Copéret for his PhD studies focusing on the development of supported, Pt-metal based catalysts for propane dehydrogenation. |
 Pierre-Adrien Payard | Pierre-Adrien Payard completed his Bachelor's and Master's degree in Chemistry at Ecole Normale Supérieure Paris-Saclay, and carried out his PhD at Ecole Normale Supérieure under the joint supervision of Dr Laurence Grimaud and Dr Marc Pera-Titus. In 2019, he joined the team of Prof. Christophe Copéret at ETH Zürich as a postdoctoral researcher before starting his independent career as associate professor at the University of Lyon 1 (Oct. 2020). His current research is dedicated to experimental and computational studies of reaction mechanisms targeting to overcome the limitations encountered in catalytic systems. |
 Christophe Copéret | Christophe Copéret (CCH) obtained a PhD in 1996 with E.-i. Negishi (Purdue University) and was a postdoctoral fellow with K. B. Sharpless (Scripps Research Institute). CCH joined CNRS in 1998, became Research director in 2008 and moved to ETH Zürich in 2010, as Chair of Surface and Interfacial Chemistry. His research interest lies at the interface between molecular, surface and material chemistries. The objective of his laboratory is to bring a molecular-level understanding of complex inorganic systems such as heterogeneous catalysts, hence the development of Surface organometallic chemistry methodology as well as spectroscopic techniques such as solid-state NMR. |
1. Introduction
In recent years, the shift away from traditional Naptha-based cracking processes, due to the appearance of abundant source of light alkanes with the emergence of Shale gas, has resulted in a dramatic reduction in the production of short chain alkenes. In parallel, the abundance of light alkanes, coupled with an increasing global demand of light alkenes,1–4 has pushed the use of alternative technologies for the production of light alkenes, such as the direction conversion of alkanes to alkenes via dehydrogenation (Scheme 1a).3,5,6 This reaction is however highly endothermic (standard enthalpy: +124.3 kJ mol−1), hence requiring high operating temperatures.6 For instance, equilibrium conversion (Xe) for propane is only circa 30% for working pressures of 1 bar(a) at 550 °C.6 Such harsh conditions results in increased side reactions such as (thermal) cracking and coke formation; these conditions also result in increased sintering of supported nanoparticles, leading to limited catalyst lifetimes. To overcome coke formation and catalyst deactivation, constant regeneration through oxidative de-coking and re-reduction cycles is required in industrial settings.7 These aforementioned constraints highlight the challenge to develop selective and stable catalysts for the non-oxidative dehydrogenation of light alkanes. Several processes have however been developed and implemented in industry for on-purpose production of short chain alkenes. Today three catalytic systems are currently employed commercially: the established ABB Lummus CATOFIN (CrOx/Al2O3) (Scheme 1b) and UOP OLEFLEX (Pt/Sn/Al2O3) processes (Scheme 1c),3,8 as well as the most recent process developed by Dow based on Pt/Ga/Al2O3.9 One may note that the analogous oxidative dehydrogenation processes have been more difficult to implement due to the high exothermicity of the reaction, and the difficulty to obtain high alkene selectivity at high conversion. However, recent findings may lead to further development in this field.10,11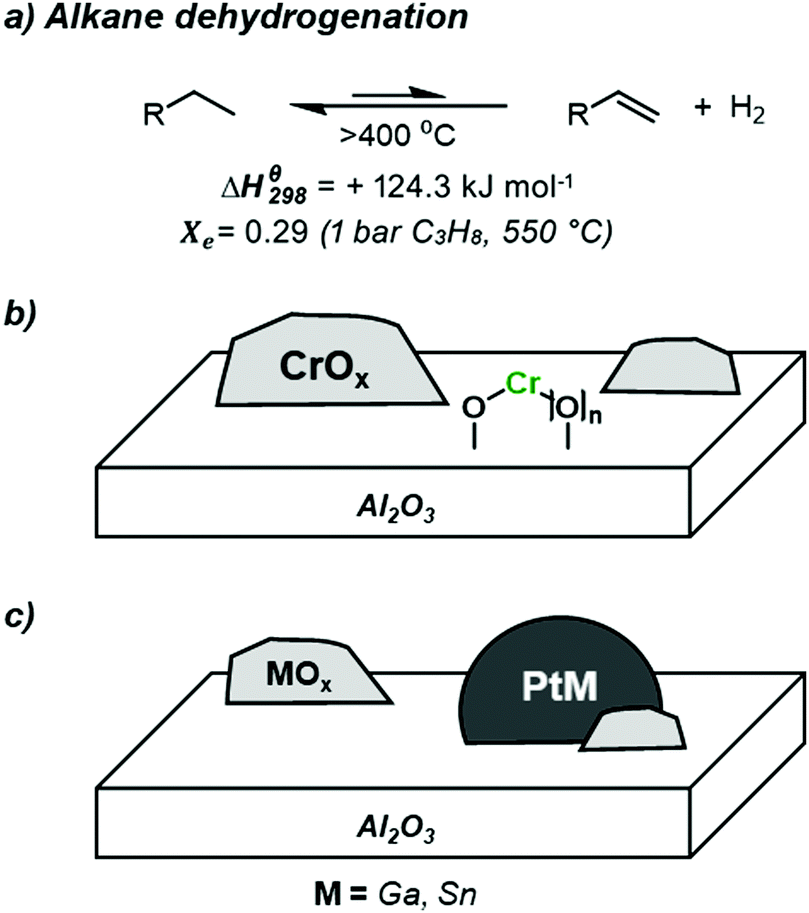 | ||
| Scheme 1 (a) Dehydrogenation of light alkanes (thermodynamic constraints, equilibrium conversion of propane at 1 bar and 550 °C; (b) general structure of CATOFIN®-type catalysts (CrOx/Al2O3), and (c) general structure of platinum-based catalysts for the dehydrogenation of light alkanes (PtM/MOx/Al2O3). | ||
Coming back to the non-oxidative alkane dehydrogenation process, while these aforementioned catalysts have been widely studied, the complex nature of the catalytic systems leaves many unanswered questions regarding the nature of the active site, strategies for the suppression of side reactions (cracking, coking), and how catalyst structure can influence selectivity in alkane dehydrogenation processes.
Thus, the development of realistic model systems is highly desired, with the goal of unravelling the nature of the active sites, the mechanism of this reaction, and to better understand deactivation processes. The increased demand of these processes and the emergence of strategies for the synthesis of well-defined sites and the control of interfaces between metal particles on metal oxide supports has recently triggered interest in this field of research. In this review, we limit our discussion to the development of molecular-level understanding of light alkane dehydrogenation using model catalysts synthesized via Surface Organometallic Chemistry (SOMC).12 SOMC, a precise molecular approach which exploits the formation of covalent bonds between molecular species and supports (through grafting), has emerged as a powerful technique to understand heterogeneous catalysis when used in conjunction with in situ or operando spectroscopy combined with computational approaches. This approach is particularly powerful to interrogate the structure of active sites and reaction mechanism (vide infra).13–15 The review is divided into three main sections: (i) a section describing the SOMC methodology, and two additional sections related to the use of this methodology for the development of alkane dehydrogenation catalysts based on (ii) isolated metal sites on oxide supports that parallel the industrial Cr-based catalysts, and (iii) supported nanoparticles on doped oxide supports as the second important class of catalysts. This review will end with a view on the possible role of SOMC in this field and future research directions.
2. SOMC methodology
Surface organometallic chemistry has emerged as an effective tool for the generation of materials with well-defined surface sites.13–20 Through the design of molecular precursors, such as thermolytic molecular precursors (TMP),21,22 and rigorous characterization at every stage of synthesis, one is now able to selectively generate uniform supported, isolated species (so-called single sites) and supported metallic nanoparticles with a narrow particle size distribution, where the average particle size is largely independent of the dopant(s) and support – enabling direct comparison of physicochemical properties and catalytic performance.14 The precise molecular control conferred by this approach enables explicit study of the interplay between structure and activity, complementing, and deconvoluting, experimental observations from more complex materials synthesized using traditional synthetic approaches.This approach has been exploited for the synthesis of isolated Mn+ sites dispersed on oxide supports. Through the selective reaction of homoleptic metal siloxides, hydrocarbyls, alkoxides and aluminates with hydroxyl groups found on the surface of oxides (henceforth described as grafting), it has been possible to generate isolated surface species which can be subsequently thermally treated, under reduced pressure, inert or even reactive conditions (e.g. synthetic air), to generate well-defined surface sites of known oxidation state, geometry and nuclearity (Scheme 2a).13,19 The materials generated can be considered surface-doped oxide materials, with effective metal utilization (Msurface = 100%). This approach has been widely exploited for an array of transition metals (Ti, Zr, Hf, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni) as well as Cu, Zn, main group elements (Al, Ga, etc.) and lanthanides, where the strong M–O bonds allow grafting and retention of highly dispersed metal sites. In some cases, like Ni and Cu, oxidative treatment is needed to avoid reduction of the metal sites and to retain isolated metal sites.23,24
 | ||
| Scheme 2 SOMC/TMP as a strategy for the generation of model catalysts. (a) Generation of isolated metal sites at the surface of metal oxides; (b) synthesis of supported metal nanoparticles via SOMC; (c) combined approach for the synthesis of bimetallic materials using and SOMC approach, (i) monometallic particles on doped supports for study of the role of interface, (ii) bimetallic nanoparticles for the study of alloying in supported catalysts. (d) Hydride functionalization strategies for the formation of adatom surface species and the synthesis of bimetallic nanoparticles; and (e) representative analytical tools used in conjunction with the SOMC/TMP approach. | ||
Secondly, SOMC can also be used for the synthesis of supported metal nanoparticles. Following the grafting of late-transition metal precursors (siloxides, hydrocarbyls, alkoxides, amides etc.) through the surface hydroxyl groups of oxide supports, that enables the generation of materials with a uniform initial distribution of metal across the support, the treatment under hydrogen (typically 1 bar(a), in flow) at elevated temperatures yields small, uniform nanoparticles, free of organic ligands/capping agents (Scheme 2b). This approach has been exploited for metals which possess greater ‘noble’ character (Ir, Rh, Pt, Cu, Ag, and Au), though the lower M–O bond strength reduces the thermodynamic impetus for grafting, and renders the range of relevant precursors more limited.25–29
These two approaches can also be combined, exploiting the stability of doped oxide materials generated by SOMC (Scheme 2a), and further functionalizing these materials through the grafting of a second molecular precursor, through remaining/regenerated surface hydroxyls.14 Upon treatment under H2, one of two outcomes occurs: (i) the reduction of the second grafted species to form monometallic nanoparticles, of uniform size, atop silica containing isolated metal ions (Scheme 2c); or (ii) the reduction of the second grafted species to form metallic nanoparticles, of uniform size, atop oxide supports containing isolated metal ions, with gradual reduction and intercalation of the second metal to the nanoparticle, forming an alloy (Scheme 2c).30–32 The use of this approach, in the absence of alloying enables the disambiguation of bulk/surface effects often described in heterogeneous catalysis, as well as explicit study of particle–support interfaces. Where alloying occurs, it is possible to study, in the absence of large volumes of spectator species (typically found in the bulk of pure oxides), the role of alloying in determining the reactivity for reactions catalysed by supported nanoparticles.
In addition, SOMC has also been shown to generate so-called single atom alloys as well as bimetallic bulk alloyed materials. In this approach, supported metal (M) nanoparticles, prepared via classical or SOMC approach, are reacted, typically under H2, with organometallic compounds or molecular hydrides (E = Sn, Si, Ge, Pb, As).33–38 This approach involves (Scheme 2d) the selective reaction of surface hydrides present on the metallic nanoparticles under H2 with the organometallic reagent to generate usually organometallic fragment covalently bound to the surface of the metallic particles. Further treatment under H2 at elevated temperatures leads to the removal of the organic fragments via hydrogenolysis leaving a bare surface adatom. Increasing the temperature allows migration of the adatom into the bulk yielding alloys or solid solutions (MEx).33–37,39
While the materials generated using these molecular approaches vary greatly in terms of composition and reactivity, the uniformity of these materials renders all of them amenable to microscopy and spectroscopic study, in particular XAS, XPS as well as IR and NMR, in combination with probe molecules. Furthermore, the effective metal utilization associated with these systems, as well as the absence of complex dissolution/precipitation processes associated with conventional catalyst synthesis approaches, e.g. impregnation techniques, and the absence of complex ionic ligands and capping agents, which are known to have a significant influence on reactivity, enables deconvolution of the role of specific catalyst components. As all dopant atoms are either at or on the surface, spectroscopic study, and in particular nucleus specific spectroscopic study (e.g. X-ray absorption spectroscopy (XAS), or solid state nuclear magnetic resonance spectroscopy (SS-NMR)) focuses exclusively on exposed (and thus the most likely to be catalytically relevant) species – a clear advantage over materials synthesized using traditional approaches. Materials can be exhaustively characterized by an array of spectroscopic techniques (IR, SS-NMR, XAS), electron microscopy, and traditional reactivity studies (catalyst evaluation, chemisorption, kinetics), where the numbers of active sites are known, and rates can be ascertained with a greater degree of accuracy. Furthermore, these uniform materials, as model catalysts, are appealing as reference systems for ab initio modelling studies due to their well-defined nature, and the low nuclearity of the nanoparticles formed (in the case of late transition metals).
Note that henceforth, materials denoted Mn+@M′Ox represent materials prepared by SOMC that have been fully characterized after post-treatment (i.e. in the state that they would be in upon exposure to reaction gas), while the ‘/’ notation is used where either (a) the grafted species, prepared by SOMC, was only characterized prior to pretreatment, or (b) the material was prepared by conventional impregnation methods (further denoted (IWI)).
3 Modelling supported metal oxides via SOMC
The CATOFIN process is based on chromium oxide dispersed on alumina, Cr2O3/Al2O3, that falls into a broader class of alkane dehydrogenation catalysts composed of transition-metal or main group metal oxides dispersed on redox-inert supports. This section will first start by examining SOMC models developed to understand the Cr-based CATOFIN catalysts, before looking at alternative systems based on other supported metal oxides.3.1 Models for supported CrOx
Regarding Cr-based catalysts, the identity of the active sites remains unknown, in part due to the range of Cr oxidation states detected in CrOx/Al2O3 under reaction conditions.40–48 In addition, the presence of mononuclear and polynuclear sites has been discussed. Both CrII and CrIII have been proposed to be catalytically active for alkane dehydrogenation with a large consensus pointing to CrIII being the most relevant active species.45–56Therefore, there has been a strong interest in generating isolated Cr sites of known oxidation state and nuclearity at the surface of tailored oxide supports, to help the identification of active sites in supported metal oxides. One approach is the combined SOMC/TMP approach (see previous section), which provides access to dispersed low coordinated metal sites on oxide supports with controlled oxidation state and nuclearity.13,19 For instance, SOMC/TMP was used to generate a series of isolated CrIII sites at the surface SiO2, SiO2–Al2O3 and Al2O3. More specifically, these materials were obtained by grafting CrIII molecular precursors followed by a subsequent thermal treatment under vacuum: Cr(OSi(OtBu)3)3(THF)2 on dehydroxylated SiO2–700,57,58 and Al2O3–700 as well as Cr(Al(OiPr)4)3 on dehydroxylated Al2O3–700 (Scheme 3).43 This approach enables the formation of CrIII isolated mononuclear sites on silica, silica-alumina and alumina.
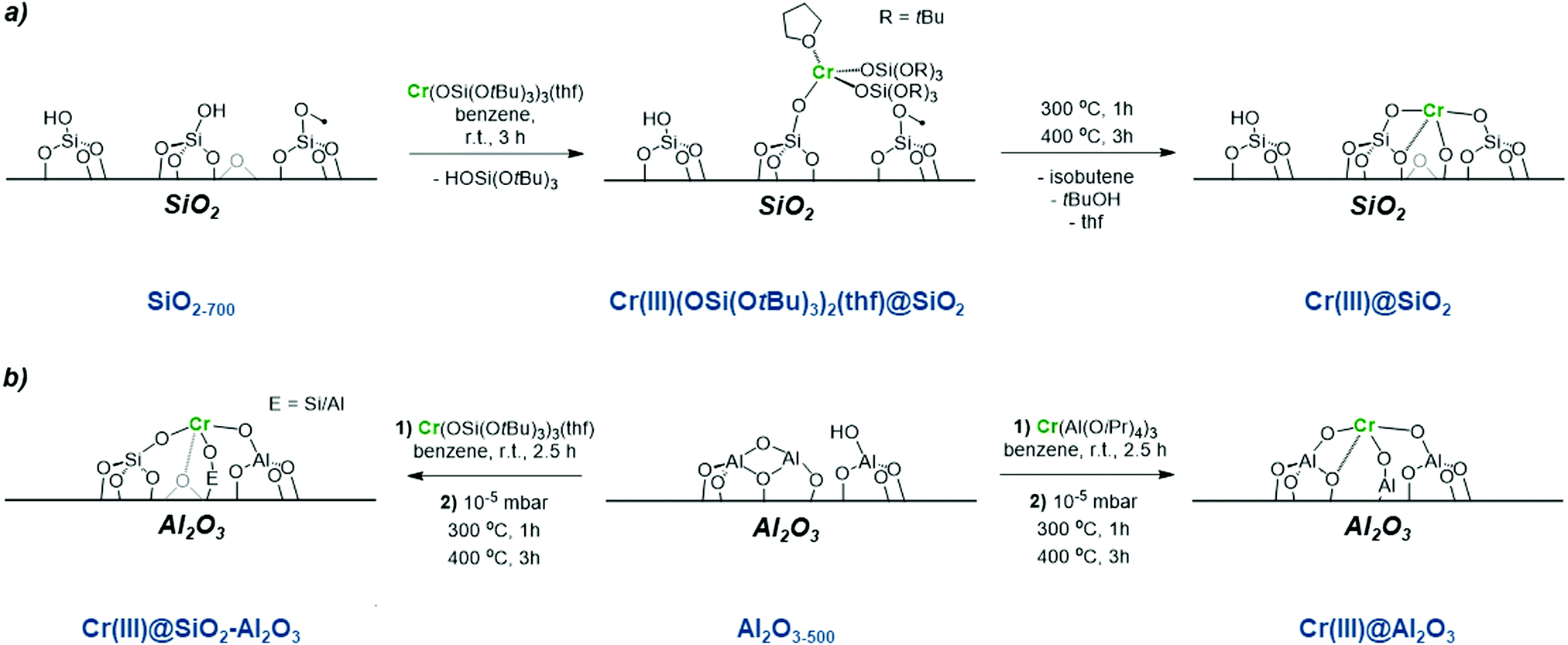 | ||
| Scheme 3 Isolated CrIII sites prepared using an SOMC/TMP approach (a) synthesis of CrIII single sites on SiO2 partially dehydroxylated at 700 °C; and (b) SiO2–Al2O3 and Al2O3. | ||
In the specific case of silica, CO adsorption monitored by FT-IR, in combination with DFT, evidenced the presence of two types of CrIII sites: tri-coordinated sites containing three Si–O groups coordinated to a CrIII center as well as tetra-coordinated sites featuring an additional siloxane bridge coordinated to the metal center (Scheme 4).58 The oxidation state of Cr after treatment was confirmed to be CrIII by EPR and XANES. Such CrIII sites are able to catalyse PDH, albeit with low activity (Table 1).
 | ||
| Scheme 4 (a) Proposed non-redox mechanism for the dehydrogenation of light alkanes over MIII isolated sites (M = Cr & E = Si or Al; or M = Ga & E = Si); and (b) structure of model tri- and tetra-coordinated CrIII sites on Al2O3. Reproduced from ref. 43 with permission from American Chemical Society, copyright 2016. | ||
| Material | Feed | Time (h) | T (°C) | Conversion (%) | Selectivitya (%) | Productivity (gprod gGa−1 h−1) | wt% Cr | Ref. |
|---|---|---|---|---|---|---|---|---|
| CrIII@SiO2 | 20% propane/Ar | 0 | 550 | 10 | 72 | 16 | 0.95 | 43 and 58 |
| 2 | 3 | — | 4.2 | |||||
| CrIII@SiO2–Al2O3 | 20% propane/Ar | 0 | 550 | 5 | 70 | 43 | 0.21 | 43 |
| 2 | 3 | — | 24 | |||||
| CrIII@Al2O3 | 20% propane/Ar | 0 | 550 | 4 | 66 | 51 | 0.18 | 43 |
| 2 | 3 | — | 29 | |||||
| CrOx/Al2O3 (IWI) | 20% propane/Ar | 0 | 550 | 16 | 88 | 22 | 1.5a | 43 |
| 2 | 17 | — | 26 |
In contrast, CrIII sites dispersed on alumina, independently of the molecular precursors (Table 1), show significantly higher activity in the dehydrogenation of propane. Their activity even exceeds that of catalysts prepared by classical impregnation methods (Table 1, entry 4), probably because of the increased concentration of active sites in the former. On the other hand, these single sites catalysts are significantly less selective, presumably due to the presence of Brønsted sites (e.g. Si–OH or Al–OH interacting with adjacent Lewis sites), favouring cracking. Further surface chemistry can be foreseen to tune the acidity of the surface towards the development of highly active and selective PDH catalysts.
To further understand the reactivity of these sites, the hydrogenation of propene, that has the advantage to take place at much lower temperatures than the reverse PDH reaction, was studied in detail. Kinetic investigation on CrIII single-site catalysts show low activation energies (0–40 kJ mol−1) while activation entropies are very negative (−145 to −194 J K−1 mol−1). Under reaction pressure (2 bar(g)), estimation of reaction order revealed first order kinetics law with respect to H2 for all catalysts and first or zero order rate law with respect to propene for catalysts CrIII@SiO2–Al2O3 and CrOx/Al2O3 and for catalysts CrIII/Al2O3 and CrIII@Al2O3, respectively. At lower pressures, the rate law always depends on the concentration of propene, following a Langmuir-like behaviour. This observation is likely due to a strong adsorption of propene on the sites (site-saturation). Furthermore, ParaHydrogen-Induced Polarization (PHIP),59–62 was used to probe the mechanistic pathway for hydrogenation – and thereby the microscopic reverse reaction (dehydrogenation) – on CrIII supported single sites. PHIP shows that hydrogenation likely takes place via pairwise heterolytic H2 activation, as evidenced by the partial induced polarization transfer, in agreement with earlier proposed mechanism.44,58 Modelling the CrIII sites dispersed on alumina by periodic DFT calculations further support the proposed mechanism, which involves the non-redox heterolytic cleavage of a terminal C–H bond on a Cr–O bond (Scheme 4a) leading to the formation of Cr-alkyl and OH group followed by β-hydrogen transfer, olefin de-coordination and desorption of H2 to regenerate the initial Cr–O reactive sites.43 This mechanism is overall more favoured on tetra- than tri-coordinated CrIII sites (Scheme 4b), and is favoured on alumina compared to silica, likely because of the greater polarization of the Cr–O bonds in the more ionic alumina support.
3.2 Supported GaIII and related systems
In parallel to CrIII-based systems employed industrially, main group oxides dispersed on oxide supports have also been shown to selectively dehydrogenate alkanes.63 In fact, the Cyclar® process (BP/UOP) that is used industrially to produce BTX aromatics is based on a Ga-exchanged (ZSM-5) zeolite, where isolated GaIII sites and Brønsted acid sites are thought to dehydrogenate propane and aromatize the olefins, respectively.64–66 A wide variety of Ga- as well as In-exchanged zeolites show similar reactivity.67–71 Intriguingly, it has been observed that when the Ga is introduced into the zeolite using an organometallic precursor (e.g. GaMe3), the resulting material is selective for the dehydrogenation of propane to propene, likely because GaMe3 reacts with the Brønsted acid sites – quenching the Brønsted acidity, and preventing subsequent reaction steps.72 Furthermore, numerous Ga-containing systems, that are not supported on hierarchical materials, have been shown to be proficient in light alkane dehydrogenation. For example, numerous Ga2O3-based systems have been reported to selectively catalyse propane dehydrogenation, though these systems typically suffer from rapid deactivation due to Ga2O3 reduction.63,73–75To suppress reduction of GaIII in reaction conditions, isolated GaIII sites dispersed on silica, a non-reducible support, have been developed and are highly active, selective and stable PDH catalysts. These GaIII isolated sites are prepared by the SOMC/TMP approach, similarly the CrIII sites discussed above (Scheme 5a). Here, (Ga(OSi(OtBu)3)(THF) is grafted on silica (Aerosil® 200) partially dehydroxylated at 700 °C, followed by a thermal treatment under reduced pressure (10−3 Pa, 500 °C) that removes the organic ligands leaving GaIII isolated sites.76 Using a combination of Ga K-edge EXAFS, wavelet transform analysis (WT) of the EXAFS and analysis of Ga K-edge XANES, the thermally treated material (Ga@SiO2) was shown to be composed of isolated tetracoordinate Ga sites dispersed on the surface of silica. The material shows a high activity per Ga, which is attributed to effective metal utilization associated with SOMC. In addition, a high selectivity (93%) is observed (Table 2), which is likely related to the absence of Brønsted acid sites that are often invoked in cracking and dehydroaromatization reactions.77 Recent computational studies demonstrate that GaIII-catalyzed propane dehydrogenation most likely proceeds through an analogous mechanism to that described for CrIII (Scheme 4a), and highlight that the presence of strain and specific configuration are critical for highly active isolated GaIII sites.78 While isolated GaIII sites are thought to be formed via impregnation method using (Ga(NO3)3.nH2O/citric acid/Davisil 646®), their activity and selectivity in PDH are significantly lower (Table 2) than the materials prepared via SOMC, indicating that the nature of the surface must be different.79 This may be attributed to the complexity of the material prepared by impregnation in water, where dissolution/precipitation can readily take place, thus leading to more complex materials.
 | ||
| Scheme 5 (a) Depiction of synthetic procedure employed for the generation of isolated Ga(III) sites supported on silica using an SOMC/TMP approach; and (b) reaction scheme and proposed structures for Ga(iBu)3 grafted on partially dehydroxylated silica and alumina. | ||
| Material | Feed | Time (h) | T (°C) | Conv. (%) | Select.a (%) | Productivitya (gprod gGa−1 h−1) | wt% Ga | Ref. |
|---|---|---|---|---|---|---|---|---|
| a 20 h on-stream. b Not disclosed. | ||||||||
| Ga@SiO2 | 3.3% propane in Ar (2 bar) | 0.1 | 550 | 9.3 | 94 | 13 | 1.6 | 76 |
| 20 | 6.5 | 93 | 9 | |||||
| Ga/SiO2 | 2.3% propane in Ar (1.4 bar) | 1 | 550 | 9.8 | 97 | 0.9 | 2.64 | 79 |
| Ga(iBu)3/SiO2–700 | 20% propane in Ar (1 bar) | 1 | 550 | 8 | 90 | 4.7 | 3.34 | 80 |
| 25 | 2.5 | 72 | 2.3a | |||||
| Ga(iBu)3/Al2O3–500 | 20% propane in Ar (1 bar) | 1 | 550 | 24 | —b | 13 | 1.97 | 80 |
| 25 | 8 | 90 | 8a | |||||
Using an approach akin to the aforementioned work with GaMe3 on zeolites, the SOMC approach has also been used with silica and alumina support materials, using the homoleptic precursor tri-isobutylgallane – Ga(iBu)3 (Scheme 5b).80 A dimeric surface species is proposed to be formed upon grafting of Ga(iBu)3 on silica partially dehydroxylated at 700 °C (Ga(iBu)3/SiO2–700, Degussa Aerosil 200), as evidenced by WT-EXAFS and NMR. Conversely, monomeric Ga sites are formed on Al2O3 partially dehydroxylated at 500 °C (Ga(iBu)3/Al2O3–500), γ-alumina) as evidenced by EXAFS and NMR. The alumina supported catalyst is significantly more active in PDH (550 °C, 20% propane in Ar) than the silica-supported material (Table 2); the difference of reactivity is attributed to the greater reactivity towards C–H bonds of Ga–O bonds in [–Ga–O–Al–] vs. [–Ga–O–Si–] systems, which parallels what is found for Cr (vide supra).58,80 Note however that Al2O3 also displays a considerable PDH activity and selectivity (ca. 4% conversion, ≈40% total activity after 20 hours on-stream).81 In this case, the high background activity of alumina precludes more direct comparison of site requirements (such as nuclearity).
3.3 1st row transition metal single-site PDH catalysts
While supported vanadium oxides are key catalysts for the oxidative dehydrogenation of propane, they have also been studied in non-oxidative PDH discussed in this review. These catalysts are thought to require partial reduction of V2O5 to VOx sites. Pre-treatment by H2 has been shown to influence the activity,82 and both VIII and VIV isolated sites have been proposed to catalyse this reaction.82–91To gain insight into the active structures for the vanadium catalysed dehydrogenation of propane, silica-supported VIII and VV single sites have thus been prepared via SOMC (Scheme 6) using the well-defined VIII and VV molecular precursors VIII(Mes)3(THF) and VV(O)(Mes)3.83,89,92 Characterization by FT-IR, UV-vis, solid-state 1H and 51V NMR and XAS shows that these precursors can generate the corresponding mono [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)VV(O)Mes2] or bis-grafted [(SiO)2VV(O)Mes] sites depending on the temperature at which the silica support is dehydroxylated (Scheme 6). Activity for these catalysts is reported in Table 3. Following an induction period, [(SiO)VV(O)Mes2] shows high PDH activity (Table 3) compared to V2O5/SiO2 prepared by impregnation, likely due to the absence of inactive polynuclear VOx species.83 [(SiO)VIIIMes2(THF)] shows comparable TOF to [(SiO)VV(O)Mes2], albeit with a significantly reduced induction period, suggesting that VIII species are the active species (Table 3). Pre-treatment of (OSi)VIIIMes2(THF) under H2 allows the total suppression of this induction period. This has been attributed to the formation of surface hydride species.89 Furthermore, a more recent study indicates that a VIII species is likely formed upon hydrogen treatment of [(SiO)2VV(O)Mes] at elevated temperatures (250–500 °C),93 suggesting that in spite of the different precursors used, the active site structures and ‘activated’ materials are very similar. While various plausible pathways have been proposed, initiation likely starts with the C–H activation of propane to form mesitylene and a propyl vanadium species, followed by β-H elimination and formation of a propylene π-complex. An alternative pathway invokes the cleavage of a V–O bond in a fashion akin to that described for CrIII (vide supra, Scheme 4a).83 However, to obtain further insight into the mechanism additional in situ or operando studies would be beneficial. Studies using PHIP show that pairwise replacement of two hydrogens from propane with the ones originating from para-H2 is not an efficient process over [(SiO)VV(O)Mes2], further highlighting the ambiguity in interpretation of the propane dehydrogenation mechanism in reaction conditions.93 Detailed mechanistic studies of the microscopic reverse reaction (i.e. propene hydrogenation) show that by switching between hydrogenation and dehydrogenation conditions, the hydrogenation capacity of the catalyst is reduced, suggesting that the hydrogenation is enabled by a site that is not stable in relevant conditions – precluding mechanistic insights being obtained using this strategy.89
SiO)VV(O)Mes2] or bis-grafted [(SiO)2VV(O)Mes] sites depending on the temperature at which the silica support is dehydroxylated (Scheme 6). Activity for these catalysts is reported in Table 3. Following an induction period, [(SiO)VV(O)Mes2] shows high PDH activity (Table 3) compared to V2O5/SiO2 prepared by impregnation, likely due to the absence of inactive polynuclear VOx species.83 [(SiO)VIIIMes2(THF)] shows comparable TOF to [(SiO)VV(O)Mes2], albeit with a significantly reduced induction period, suggesting that VIII species are the active species (Table 3). Pre-treatment of (OSi)VIIIMes2(THF) under H2 allows the total suppression of this induction period. This has been attributed to the formation of surface hydride species.89 Furthermore, a more recent study indicates that a VIII species is likely formed upon hydrogen treatment of [(SiO)2VV(O)Mes] at elevated temperatures (250–500 °C),93 suggesting that in spite of the different precursors used, the active site structures and ‘activated’ materials are very similar. While various plausible pathways have been proposed, initiation likely starts with the C–H activation of propane to form mesitylene and a propyl vanadium species, followed by β-H elimination and formation of a propylene π-complex. An alternative pathway invokes the cleavage of a V–O bond in a fashion akin to that described for CrIII (vide supra, Scheme 4a).83 However, to obtain further insight into the mechanism additional in situ or operando studies would be beneficial. Studies using PHIP show that pairwise replacement of two hydrogens from propane with the ones originating from para-H2 is not an efficient process over [(SiO)VV(O)Mes2], further highlighting the ambiguity in interpretation of the propane dehydrogenation mechanism in reaction conditions.93 Detailed mechanistic studies of the microscopic reverse reaction (i.e. propene hydrogenation) show that by switching between hydrogenation and dehydrogenation conditions, the hydrogenation capacity of the catalyst is reduced, suggesting that the hydrogenation is enabled by a site that is not stable in relevant conditions – precluding mechanistic insights being obtained using this strategy.89
 | ||
| Scheme 6 Preparation of silica supported VIII and VV aryl complexes for PDH, (a) SOMC approach for the preparation of monopodal and bipodal supported VV single sites employed in vanadium catalysed propane dehydrogenation; and (b) preparation of VIII single sites for propane dehydrogenation prepared using an SOMC approach. | ||
| Material | Feed | Time (h) | T (°C) | Conv. (%) | Select. (%) | Productivity (gprodgM−1 h−1) | wt% M | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The reported corrected selectivity is 99% upon subtraction of cracking products – the observed selectivity is below 70%. b Calculated based on metal loading in the material prior to reduction in H2. c Corrected selectivity by subtracting the amount of thermal cracking attributed to silica. | ||||||||
| [(SiO)VV(O)Mes2] |
20% propane/Ar | 0.1 | 500 | 17 | 83 | — | V: 1.40 | 83 |
| 168 | 9 | 83 | — | |||||
| [(SiO)2VV(O)Mes] |
20% propane/Ar | 0.1 | 500 | 17 | 90 | — | V: 1.64 | 83 |
| 168 | 12 | 90 | — | |||||
| [(SiO)2VIIIMes] |
2.3% propene/Ar | 0.3 | 550 | 8 | 94 | 0.19 | V: 2.8 | 89 |
| 200 | 4.5 | 92 | 0.11 | |||||
| FeII@SiO2 | 3% propane/Ar | 0 | 650 | 4.1 | 69a | 0.83 | Fe: 1.11 | 97 |
| 18 | 6.3 | 69a | 1.05 | |||||
| Fe0/SiO2–700 | 3% propane/Ar | 0 | 650 | 27 | 14 | 32.3 | Fe: 1.83b | 97 |
| Fe2O3/SiO2 | 3% propane/Ar | 0 | 650 | — | — | — | Fe: 4.4 | 97 |
| CoII@SiO2 (SOMC) | 20% propane/Ar | 0.1 | 550 | 9.5 | 73 | — | Co: 1.77 | 98 |
| 1.5 | 550 | 6.5 | 92 | 8.9 | ||||
| 10 | 550 | 4 | 89 | 3.7 | ||||
| CoII/SiO2 (IWI) | 3% propane/Ar | 0 | 550 | 4 | 91c | 0.52 | Co: 2.1 | 96 |
| 20 | 10 | 95c | 1.34 | |||||
3.4 Late-transition metal PDH catalysts based on single-sites
Later 3d transition-metals dispersed on oxide supports such as Fe and Co have also shown PDH activity.6,94–96 For example, silica-supported FeII single sites have been prepared by grafting bis(η5-2,4-dimethylpentadienyl)iron(II) on silica (Scheme 7a). As evidenced NMR, FTIR and analysis of washings, the grafting takes place by reaction of one Si–OH group with bis(η5-2,4-dimethylpentadienyl)iron(II) leading to the release of 1 equiv. of 2,4-dimethylpentadiene (Scheme 7). Reduction of this material under H2 at 400 °C leads to the formation of small iron oxide clusters that can be re-dispersed by thermal treatment at 650 °C. The associated FeII isolated sites show PDH activity (Table 3), while the corresponding iron nanoparticles show poor selectivity (coke formation is the dominant reaction), and iron oxide nanoparticles supported on silica show significantly lower activity and selectivity for PDH, suggesting that FeII isolated sites are catalytically competent in PDH.97 | ||
| Scheme 7 Preparation of silica supported FeII and CoII isolated sites for PDH, (a) SOMC approach for the preparation of supported FeII single sites using bis-2,4-dimethylpentadienide iron(II) as molecular precursor; and (b) preparation of CoII single sites for propane dehydrogenation prepared using a combined SOMC/TMP approach. | ||
Using the SOMC/TMP approach, mononuclear silica-supported CoII PDH catalysts have been prepared starting from the dinuclear CoII precursor [Co2(OSi(OtBu)3]2 (Scheme 7b).98 EXAFS data, consistent with the presence of a Co–O path of degeneracy 4 (1.94 Å), attributed to anionic oxygen directly bound to Co centre and adjacent siloxane oxygens interacting in a dative fashion, was used to establish the proposed surface structure. Worth of note, the activity toward PDH of such SOMC-TMP prepared CoII single sites is significantly higher than the one of single sites prepared via impregnation approach (Table 3).96,98 However the stability of this catalyst is poor, due to the formation of nanoparticles under reaction conditions. A mechanism similar to the one proposed for CrIII (vide supra, Scheme 4a) has been calculated on model clusters and is consistent with kinetic and PHIP experiments on SOMC prepared single sites in the reverse hydrogenation reaction.96 Both experiment and calculations indicate that PDH likely involves the heterolytic C–H activation of propane, similarly to what has been proposed for CrIII.98
4 Modelling alkane dehydrogenation catalysts using supported metal nanoparticles and alloys prepared via SOMC
Early studies have shown that PtM (M = Pd, Rh, Ir, Sn) nanoparticles can catalyze cyclohexane dehydrogenation as well as other important hydrocarbon conversion reactions.99–105 The addition of a second metal component has been shown to result in enhanced selectivity and stability. The superior catalytic performances of bimetallic catalysts in alkane dehydrogenation, when compared to their monometallic counterparts, has raised a considerable interest in bimetallic systems, and has led to the development of industrial systems for the dehydrogenation of light alkanes.33,101,102 First commercialized in the 1990s, the UOP Oleflex process consists of alloyed Pt–Sn nanoparticles supported on alumina, and is used for the selective dehydrogenation of propane. In addition to the licensed Pt–Sn systems, there are an array of related systems that have garnered attention in both academia and industry.6 This broad class of catalysts, of general structure PtM/M′Ox, have been widely studied as model systems for the Oleflex catalyst.106–112In spite of the complexity of these systems and the presence of multiple metal species, SOMC has also been used to study supported metal nanoparticles as models for the Oleflex system. Building upon pioneering work on the Pt–Sn system by Yermakov in the late 1970's,113,114 SOMC research in the 1980's and 1990's focused on modifying supported metal nanoparticles and generating alloys for alkane dehydrogenation through the selective grafting of organometallic compounds on the surface hydrides present at the surface of platinum particles. In the specific case of Pt–Sn, this approach could be successfully carried out by reacting organotin species selectively with supported Pt nanoparticles at ambient temperature (ca. 25 °C), followed by a high temperature treatment under H2 (Scheme 2d). The first step leads to the formation of R3Sn–Pt moieties at the nanoparticle surface, which, upon treatment under H2 at elevated temperatures, lose their alkyl ligands and form a Pt–Sn alloy by migration of the Sn inside of the particle.115–118
This approach was used more recently, to prepare a trimetallic Pt–Sn–Li material active in the dehydrogenation of 2,3-dimethylbutane (Scheme 8a).119 The treatment of alumina with BuLi and Pt(acac)2, followed by a calcination under air at 400 °C and a reduction at 550 °C under H2 yielded a material containing dispersed Li+ aluminate sites (quenching of Brønsted acidity for the purpose of suppressing side reactions) and small Pt particles. Subsequent reaction of this material with Bu4Sn, in the presence of H2 (550 mbar), followed by a treatment under H2 at 550 °C did not lead to a change in particle size. The addition of both Li and Sn by SOMC leads to an increased productivity and stability compared to the monometallic Pt catalyst (Table 4). Addition of Sn leads to an increased selectivity, and is manifested in H2 chemisorption measurements. The beneficial role of Li has been attributed to decreasing the acidity of the Al2O3 support and increasing Pt dispersion, leading to an overall improved catalytic performance.
 | ||
| Scheme 8 (a) Depiction of synthetic procedure employed for the generation of PtSn alloyed nanoparticles supported on Gi-doped alumina using an SOMC metal functionalisation approach; and (b) depiction of synthetic procedure employed for the generation of PtSn alloyed nanoparticles on tin-doped θ-alumina using an SOMC/TMP approach (n = 1, 3). | ||
| Material (particle size, nm) | Feed | Time (h) | T (°C) | Conv. (%) | Select.a (%) | Productivitya (gprod/gPt h) | wt% M | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Only desired products are included. C6 olefins in the case of 2,3-dimethylbutane, propene in the case of propane, butenes in the case of n-butane. | ||||||||
| Pt/SiO2 (2.2 ± 0.8) | 20% propane/argon | 0.1 | 550 | 2.5 | 75 | 14.5 | 3.96 Pt | 112 |
| 2 | 1.5 | 47 | 8.7 | |||||
| Pt–Sn–Li/Al2O3 (n.d.) | 35% 2,3-dimethylbutane/H2 | 0.1 | 500 | 40 | 77 | 55 | 2.0 Pt, 1.32 Sn, 2.8 Li | 119 |
| 30 | 34 | 80 | 47 | |||||
| Pt–Sn3/Al2O3 (0.8 ± 0.2) | 10% propane/10% H2/Helium | 0.1 | 550 | 43 | 99 | 860 | 0.5 Pt, 1.5 Sn | 120 |
| 72 | 38 | >99.8 | 766 | |||||
| Pt–Ga/SiO2 (1.0 ± 0.2) | 20% propane/argon | 0.3 | 550 | 32 | >99 | 661 | 4.82 Pt, 1.48 Ga | 111 |
| 20 | 17 | >99 | 357 | |||||
| Pt–Zn/SiO2 (0.8 ± 0.2) | 20% propane/argon | 0.3 | 550 | 30 | 98 | 703 | 3.05 Pt, 1.54 Zn | 112 |
| 30 | 16 | 95 | 375 | |||||
| Pt–Zn/SiO2 (ALD) (1.2 ± 0.3) | 5% n-butane/argon | 0.1 | 500 | 79 | 92.5 | 165 | 0.6 Pt, 9.9 Zn | 121 |
| 20 | 52 | 99 | 116 | |||||
| Pt–B/SiO2 (2.6. ± 0.6) | 1% n-butane/argon | 0.1 | 550 | 1.6 | 70 | 8.7 | 0.021 Pt, 0.60 B | 122 |
| 25 | 1.5 | 70 | 8.2 | |||||
Alternatively, the consecutive grafting of organometallic precursors via the OH groups of metal oxide supports was implemented for the synthesis of bimetallic materials (Scheme 8b). For instance, PtSn supported on θ-Al2O3 have been recently prepared via SOMC through the consecutive grafting of [Pt(COD)Me2] and HSnPh3 onto the partially dehydroxylated support via Pt–Me and Sn–H cleavage as evidenced by GC analysis (with the respective formation of CH4 and H2).120 Powder XRD measurements only show contributions of the θ-Al2O3 structure which indicates no significant amounts of crystalline PtSn, Pt or Sn domains in the material prior to a reduction step under H2 at 150 °C that yields supported sub-nanometric (0.8 nm), highly dispersed and size-homogeneous Pt–Sn particles as evidenced by HAADF-STEM. CO adsorption IR indicates formation of a Pt–Sn alloy as the bridge-type CO vibrational band (νCO < 2000 cm−1) is absent in these spectra. XPS analysis indicates the coexistence of SnIV, SnII and Sn0 species with increasing volumes of oxidized species with increased Sn/Pt ratio. While Sn0 enhances the selectivity due to alloy formation, the presence of SnII/SnIV is proposed to prevent sintering, hence the formation of small particles. In this study, a PtSn material containing a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Sn:Pt shows highest PDH performance with 40% conversion over 72 h at >99.5% selectivity for propene – stable in a WHSV range from 11.8 h−1 to 3300 h−1 – at 550 °C (Table 4).
:1 Sn:Pt shows highest PDH performance with 40% conversion over 72 h at >99.5% selectivity for propene – stable in a WHSV range from 11.8 h−1 to 3300 h−1 – at 550 °C (Table 4).
Besides Sn, Ga has garnered attention in alkane dehydrogenation processes, and has recently been implemented industrially, with the Fluidized Catalytic Dehydrogenation process introduced by Dow Chemical in 2017.9,110
To gain insight into this family of catalysts, which contain both Pt and Ga, a PtGa bimetallic material was prepared via SOMC by grafting of [Pt(COD)(OSi(OtBu)3)2] onto the silica-supported GaIII single-sites discussed above (Ga@SiO2, Section 3.2) followed by a thermal treatment under H2 (Scheme 9a).111 This approach builds on the presence of isolated GaIII sites in close proximity of –OH groups (ca. 0.6 Ga and 1.3 –OH nm−2) that enables introduction of a controlled quantity of Pt through grafting onto the –OH groups. After H2 treatment, highly dispersed and size-homogeneous 1 nm particles are generated, even at a high Pt loading of 4.37 wt%.111 Detailed CO adsorption IR, XPS and XAS studies have shown that a Pt–Ga alloy is formed along a 0.1 to 0.5 fraction of the overall Ga remaining as GaIII on the support surface. EXAFS fitting indicates that bulk Ga2O3 domains are not present. The material shows high initial productivity (Table 4) and selectivity (>99%) in the PDH reaction at 550 °C. The productivity of this catalyst, per g, exceeds that of the GaIII single-site PDH catalysts, described in Section 3.2 by ca. two orders of magnitude. In addition to the high selectivity that is observed, this material also shows an unusually high long-term stability over 20 h during which the average structure of the catalyst does not change, according to in situ XAS. Finally, this material has been shown to be partially regenerated upon consecutive O2/H2 treatments, with a recovery of 75% of the initial activity with a high propene selectivity (>99%) retained (Table 4).
 | ||
| Scheme 9 (a) Depiction of synthetic procedure employed for the generation of PtGa alloyed nanoparticles supported on Ga-doped silica using an SOMC/TMP approach; and (b) depiction of synthetic procedure employed for the generation of sub-nanometric PtZn alloyed nanoparticles on zinc-doped silica using an SOMC/TMP approach. | ||
In an analogous approach to the synthesis of bimetallic Pt–Ga materials prepared via SOMC described above, the corresponding silica-supported Pt–Zn bimetallic system was prepared, starting from isolated ZnII sites supported on silica.112 This approach yielded silica-supported subnanometric Pt–Zn clusters (0.8 nm). CO adsorption IR, XPS and XAS studies confirm the formation of a Pt–Zn alloy with an approximate 1:1 ratio of Zn:Pt in the alloyed particles while an overall 3:2 ratio of Zn:Pt is evidenced by EA. This indicates that 33% of ZnII remains on the support surface, in line with observations from XPS and XANES linear combination fits (LCF) indicating the presence of 20–45% of ZnII (Scheme 9b). The material shows excellent performance in the PDH reaction with a higher initial productivity (Table 4) than most other Pt–Zn based systems. Propene selectivity (>98%) drops slightly after 30 h on stream despite the relatively high stability over 30 h when compared to other Pt-based propane dehydrogenation catalysts (Table 4).
The use of one-cycle atomic layer deposition (ALD) has also recently been applied to prepare Pt–Zn nanoclusters on a SiO2 support, that are shown to be active in the dehydrogenation of n-butane to butenes and 1,3-butadiene.121 Consecutive ALD of Zn(C2H5)2 followed by (MeCp)PtMe3 yields a material containing 10 wt% Zn and 0.5 wt% Pt. After reduction in an H2 stream at 550 °C for 2 h, the material consists of 1.2 nm alloyed PtZn particles as shown by HAADF-STEM, XAS and XPS and CO adsorption studies. The material exhibits high selectivity to butenes (>90%, of which <20% 1,3-butadiene) at high conversion levels at 550 °C. (Table 4) A long-term stability test over 7 days shows a 60% deactivation of the catalyst while the 1,3-butadiene selectivity increases from 12% to 28%.
Besides the combination of late and post-transition metals a recent study by Teplyakov et al. utilizes the SOMC approach to combine Pt and B supported on SiO2 for butane dehydrogenation.122 The material was synthesized via sequential grafting of B(OiPr)3 and [(MeCp)PtMe3] onto partially dehydroxylated SiO2 with an intermediate calcination step at 400 °C. A treatment under H2 yielded 2.6 nm monometallic Pt particles supported on B doped SiO2 as evidenced by HAADF-STEM and HR-TEM. XPS revealed similar trigonal and tetrahedral B sites in Pt–B and B materials as well as mainly PtII/PtIV and Pt0 in the material before and after H2 treatment, respectively. 11B MQMAS NMR additionally showed significant differences of the B sites in Pt–B and B materials post-catalysis. In the B–Pt material a new feature consistent with a tetrahedral coordination environment appears in the post-catalysis sample which is consistent with calculations that invoke the formation of B–C bonds. Dehydrogenation performance of the B–Pt material shows a two-fold increase in activity compared to Pt, a selectivity of around 70% for butenes and minor deactivation over 25 h (Table 4). Redispersion of Pt species can be observed during catalysis as evidenced by post-catalysis HAADF-STEM, being more significant for the B–Pt sample compared to a Pt only material. In addition, Raman and TGA studies show that less coke is formed on the bimetallic material over the course of the reaction.
We have shown that bimetallic systems can be prepared via SOMC and that they show high activity towards propane dehydrogenation. Because it is possible to introduce dopants selectively at the support interface, these systems can be characterized exhaustively to provide molecular-level information about the structure of multi-metallic systems. This opens the possibility for more detailed operando and computational studies to address the question of the role of dopants, their effect on catalyst activity and stability as well as the structure and dynamics of active sites. Such insights could then inform the design of better performing catalysts.
5. Conclusions
SOMC, and the combination of SOMC & TMP, have been shown to provide molecular level insight into the structure and nature of active centres for the dehydrogenation of light alkanes for the two classes of dehydrogenation catalysts based on isolated sites or supported Pt nanoparticles. Through the systematic use of tailored molecular precursors, and rigorous characterization of materials at every step of the synthesis, it has been possible to obtain valuable insight into dehydrogenation processes using model catalysts. Furthermore, the ability to systematically change a single component of a system (i.e. identity of dopant), while fixing all other variables other metals, surface area, metal loading etc.) enables direct comparison of otherwise identical materials – something which is extremely challenging using traditional synthetic approaches such as incipient wetness impregnation. This has enabled rigorous study of general classes of catalysts employed in light alkane dehydrogenation.Specifically, in the case of isolated metal sites (Mn+) on non-reducible supports, it has been possible to develop CrIII/MOx type catalysts, containing isolated metal ions which catalysethe dehydrogenation of light alkanes. This insight indicates that isolated metal ions are proficient in the dehydrogenation of light alkanes, and that there is most likely a non-redox mechanism that enables this reactivity. Broader study of a range of metal ions (GaIII, VIII/V, FeII and CoII) shows that this is a more general approach for the synthesis of oxide-based catalysts for the dehydrogenation of light alkanes. In the case of Ga, it has also been shown that through the dilution of GaIII sites, and the formation of strong Ga–O–E (E = Si, Al) linkages, reduction (and thus deactivation) of GaIII can be suppressed.
For multimetallic systems based on Pt-nanoparticles, the SOMC approach has been able to show, by using detailed spectroscopic study, that the presence of an alloyed PtM phase is critical to the stability of Pt-based systems. More generally, it has been possible to demonstrate that alloying, as a general strategy possible with an array of metals (Sn, Ga, Zn), enables the synthesis of more active, selective and stable catalysts for PDH. It has been shown, by using this molecular approach, that alloying serves as an effective strategy for the dilution of Pt centres at the surface of the supported nanoparticles – suppressing cracking and coke formation as well as augmenting selectivity. Interestingly, the materials synthesized using this approach possess a stability which exceeds that of existing catalysts. This can most likely be attributed to the existence of remaining dispersed Lewis acidic sites on the surface of these materials which serve as anchoring sites for the alloyed nanoparticles, hence reducing sintering. The molecular approach employed, and the ability to selectively introduce metal functions in the absence of complex anionic species typical of traditional synthetic approaches enables explicit study of the role of the metal. In future these well-defined model systems could be used to explore the possibility of further optimized ternary systems, similar to those described in the recent literature.123
In summary, SOMC provides unprecedented insight into the molecular-level structure of heterogeneous catalysts for light alkane dehydrogenation, informing strategies for the synthesis of improved catalytic systems, and providing a basis for understanding of the subtle interplay between structure and reactivity. As spectroscopic, computational and data-driven high-throughput experimentation approaches improve, we envisage that the insights provided using this approach will serve as a platform for the systematic study of new materials for the dehydrogenation of light alkanes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swiss National Science Foundation (SNSF fond numbers: 200020B_192050 and 200021_169134) for funding. Prof. C. Copéret would like to thank numerous undergraduate and doctoral students as well as postdoctoral fellows that have participated in developing this research area; their names appear within the cited references.Notes and references
- D. Malakoff, The gas surge, Science, 2014, 344, 1464–1467 CrossRef CAS PubMed.
- D. F. Rodríguez-Vallejo, G. Guillén-Gosálbez and B. Chachuat, What Is the True Cost of Producing Propylene from Methanol? The Role of Externalities, ACS Sustainable Chem. Eng., 2020, 8, 3072–3081 CrossRef.
- Z. Nawaz, Light alkane dehydrogenation to light olefin technologies: a comprehensive review, Rev. Chem. Eng., 2015, 31, 413–436 CAS.
- T. Ridha, Y. Li, E. Gençer, J. J. Siirola, J. T. Miller, F. H. Ribeiro and R. Agrawal, Valorization of shale gas condensate to liquid hydrocarbons through catalytic dehydrogenation and oligomerization, Processes, 2016, 6, 139 CrossRef.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Shale Gas Revolution: An Opportunity for the Production of Biobased Chemicals?, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, New Trends in Olefin Production, Engineering, 2017, 3, 171–178 CrossRef CAS.
- B. V. Vora, Development of Dehydrogenation Catalysts and Processes, Top. Catal., 2012, 55, 1297–1308 CrossRef CAS.
- M. T. Pretz and M. W. Stewart, Catalytic dehydrogenation process, US Pat., 9725382 B2, Dow Global Technologies LLC, 2016 Search PubMed.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Selective oxidative dehydrogenation of propane to propene using boron nitride catalysts, Science, 2016, 354, 1570–1573 CrossRef CAS PubMed.
- J. M. Venegas, Z. Zhang, T. O. Agbi, W. P. McDermott, A. Alexandrova and I. Hermans, Why Boron Nitride is such a Selective Catalyst for the Oxidative Dehydrogenation of Propane, Angew. Chem., Int. Ed., 2020, 59, 16527–16535 CrossRef CAS PubMed.
- C. Coperet, C–H Bond Activation and Organometallic Intermediates on Isolated Metal Centers on Oxide Surfaces, Chem. Rev., 2010, 110, 656–680 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Surface Organometallic and Coordination Chemistry toward Single-Site Heterogeneous Catalysts: Strategies, Methods, Structures, and Activities, Chem. Rev., 2016, 116, 323–421 CrossRef.
- C. Copéret, Single-Sites and Nanoparticles at Tailored Interfaces Prepared via Surface Organometallic Chemistry from Thermolytic Molecular Precursors, Acc. Chem. Res., 2019, 52, 1697–1702 CrossRef PubMed.
- C. Copéret, Fuels and energy carriers from single-site catalysts prepared via surface organometallic chemistry, Nat. Energy, 2019, 4, 1018–1024 CrossRef.
- T. J. Marks, Surface-bound metal hydrocarbyls. Organometallic connections between heterogeneous and homogeneous catalysis, Acc. Chem. Res., 1992, 25, 57–65 CrossRef CAS.
- S. L. Wegener, T. J. Marks and P. C. Stair, Design Strategies for the Molecular Level Synthesis of Supported Catalysts, Acc. Chem. Res., 2012, 45, 206–214 CrossRef CAS PubMed.
- M. M. Stalzer, M. Delferro and T. J. Marks, Supported Single-Site Organometallic Catalysts for the Synthesis of High-Performance Polyolefins, Catal. Lett., 2015, 145, 3–14 CrossRef CAS.
- M. K. Samantaray, V. D’Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, The Comparison between Single Atom Catalysis and Surface Organometallic Catalysis, Chem. Rev., 2020, 120, 734–813 CrossRef CAS.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Bridging the Gap between Industrial and Well-Defined Supported Catalysts, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed.
- K. L. Fujdala, R. L. Brutchey and T. D. Tilley, in Surface and Interfacial Organometallic Chemistry and Catalysis, ed. C. Copéret and B. Chaudret, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 69–115 Search PubMed.
- K. L. Fujdala and T. D. Tilley, Design and synthesis of heterogeneous catalysts: the thermolytic molecular precursor approach, J. Catal., 2003, 216, 265–275 CrossRef CAS.
- J. Meyet, K. Searles, M. A. Newton, M. Wörle, A. P. van Bavel, A. D. Horton, J. A. van Bokhoven and C. Copéret, Monomeric Copper(II) Sites Supported on Alumina Selectively Convert Methane to Methanol, Angew. Chem., Int. Ed., 2019, 131, 9841–9845 CrossRef PubMed.
- I. B. Moroz, A. Lund, M. Kaushik, L. Severy, D. Gajan, A. Fedorov, A. Lesage and C. Copéret, Specific Localization of Aluminum Sites Favors Ethene-to-Propene Conversion on (Al)MCM-41-Supported Ni(II) Single Sites, ACS Catal., 2019, 7476–7485 CrossRef CAS.
- P. Laurent, L. Veyre, C. Thieuleux, S. Donet and C. Copéret, From well-defined Pt(II) surface species to the controlled growth of silica supported Pt nanoparticles, Dalton Trans., 2013, 42, 238–248 RSC.
- G. Noh, S. R. Docherty, E. Lam, X. Huang, D. Mance, J. L. Alfke and C. Copéret, CO2 Hydrogenation to CH3OH on Supported Cu Nanoparticles: Nature and Role of Ti in Bulk Oxides vs Isolated Surface Sites, J. Phys. Chem. C, 2019, 123, 31082–31093 CrossRef CAS.
- K. Boukebbous, N. Merle, C. Larabi, A. Garron, W. Darwich, E. A. Laifa, K. Szeto, A. De Mallmann and M. Taoufik, Silica supported copper nanoparticles prepared via surface organometallic chemistry: active catalysts for the selective hydrogenation of 2,3-dimethylbutadiene, New J. Chem., 2017, 41, 427–431 RSC.
- C. Copéret, E. Lam, J. J. Corral-Pérez, K. Larmier, G. Noh, P. Wolf, A. Comas-Vives and A. Urakawa, CO2 Hydrogenation on Cu/Al2O3: Role of Metal/Support Interface in Driving Activity and Selectivity of a Bifunctional Catalyst, Angew. Chem., Int. Ed., 2019, 58, 13989–13996 CrossRef PubMed.
- F. Heroguel, Controlled growth and interfaces of supported metal nanoparticles from late transition metal siloxides, PhD thesis, ETH Zurich, 2014 Search PubMed.
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, Isolated Zr Surface Sites on Silica Promote Hydrogenation of CO2 to CH3OH in Supported Cu Catalysts, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS PubMed.
- E. Lam, G. Noh, K. W. Chan, K. Larmier, D. Lebedev, K. Searles, P. Wolf, O. V. Safonova and C. Copéret, Enhanced CH3OH selectivity in CO2 hydrogenation using Cu-based catalysts generated via SOMC from GaIII single-sites, Chem. Sci., 2020, 11, 7593–7598 RSC.
- G. Noh, E. Lam, J. L. Alfke, K. Larmier, K. Searles, P. Wolf and C. Copéret, Selective Hydrogenation of CO2 to CH3OH on Supported Cu Nanoparticles Promoted by Isolated TiIV Surface Sites on SiO2, ChemSusChem, 2019, 12, 968–972 CrossRef CAS PubMed.
- J. Margitfalvi, S. Szabó and F. Nagy, Supported bimetallic catalysts prepared by controlled surface reactions, Stud. Surf. Sci. Catal., 1986, 27, 373–409 CrossRef CAS.
- J. Margitfalvi, S. Szabó and F. Nagy, in Catalytic Hydrogenation, LBT-SSS and C. Cerveny, Elsevier, 1986, vol. 27, pp. 373–409 Search PubMed.
- J. L. Margitfalvi, M. Hegedüs and E. Tálas, Reactions of methylcyclopentane on Pt/Al2O3 and Sn-Pt/Al2O3 catalysts, J. Mol. Catal., 1989, 51, 279–284 CrossRef CAS.
- F. Humblot, F. Lepeltier, J. P. Candy, J. Corker, O. Clause, F. Bayard and J. M. Basset, Surface Organometallic Chemistry on Metals: Formation of a Stable Sn(n-C4H9) Fragment as a Precursor of Surface Alloy Obtained by Stepwise Hydrogenolysis of Sn(n-C4H9)4 on a Platinum Particle Supported on Silica, J. Am. Chem. Soc., 1998, 120, 137–146 CrossRef CAS.
- J. L. Margitfalvi, I. Borbáth, M. Hegedûs, S. Gõbölös, A. Tompos and F. Lònyi, in 12th International Congress on Catalysis, ed. A. Corma, F. V. Melo, S. Mendioroz and C. Fierro, Elsevier, 2000, vol. 130, pp. 1025–1030 Search PubMed.
- J.-P. Candy, C. Copéret and J.-M. Basset, in Surface and Interfacial Organometallic Chemistry and Catalysis, ed. C. Copéret and B. Chaudret, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 151–210 Search PubMed.
- B. Didillon, C. Houtman, T. Shay, J. P. Candy and J. M. Basset, Surface organometallic chemistry on metals. Evidence for a new surface organometallic material, Rh[Sn(n-C4H9)x]y/SiO2, obtained by controlled hydrogenolysis of tetra-n-butylstannane on a rhodium/silica catalyst, J. Am. Chem. Soc., 1993, 115, 9380–9388 CrossRef CAS.
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Surface Chemistry and Spectroscopy of Chromium in Inorganic Oxides, Chem. Rev., 1996, 96, 3327–3350 CrossRef CAS PubMed.
- S. De Rossi, G. Ferraris, S. Fremiotti, A. Cimino and V. Indovina, Propane dehydrogenation on chromia/zirconia catalysts, Appl. Catal., A, 1992, 81, 113–132 CrossRef CAS.
- D. Shee and A. Sayari, Light alkane dehydrogenation over mesoporous Cr2O3/Al2O3 catalysts, Appl. Catal., A, 2010, 389, 155–164 CrossRef CAS.
- M. F. Delley, M.-C. Silaghi, F. Nuñez-Zarur, K. V. Kovtunov, O. G. Salnikov, D. P. Estes, I. V. Koptyug, A. Comas-Vives and C. Copéret, X–H Bond Activation on Cr(III),O Sites (X = R, H): Key Steps in Dehydrogenation and Hydrogenation Processes, Organometallics, 2017, 36, 234–244 CrossRef CAS.
- S. Lillehaug, K. J. Børve, M. Sierka and J. Sauer, Catalytic dehydrogenation of ethane over mononuclear Cr(III) surface sites on silica. part I. C–H activation by σ-bond metathesis, J. Phys. Org. Chem., 2004, 17, 990–1006 CrossRef CAS.
- S. De Rossi, G. Ferraris, S. Fremiotti, V. Indovina and A. Cimino, Isobutane dehydrogenation on chromia/zirconia catalysts, Appl. Catal., A, 1993, 106, 125–141 CrossRef.
- S. Derossi, G. Ferraris, S. Fremiotti, E. Garrone, G. Ghiotti, M. C. Campa and V. Indovina, Propane Dehydrogenation on Chromia/Silica and Chromia/Alumina Catalysts, J. Catal., 1994, 148, 36–46 CrossRef CAS.
- U. Olsbye, A. Virnovskaia, Ø. Prytz, S. J. Tinnemans and B. M. Weckhuysen, Mechanistic Insight in the Ethane Dehydrogenation Reaction over Cr/Al2O3 Catalysts, Catal. Lett., 2005, 103, 143–148 CrossRef CAS.
- A. Kytökivi, J.-P. Jacobs, A. Hakuli, J. Meriläinen and H. H. Brongersma, Surface Characteristics and Activity of Chromia/Alumina Catalysts Prepared by Atomic Layer Epitaxy, J. Catal., 1996, 162, 190–197 CrossRef.
- H. J. Lugo and J. H. Lunsford, The dehydrogenation of ethane over chromium catalysts, J. Catal., 1985, 91, 155–166 CrossRef CAS.
- F. M. Ashmawy, Surface composition and catalytic activity of chromia–alumina catalysts, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 2096–2101 RSC.
- A. Hakuli, A. Kytökivi, A. O. I. Krause and T. Suntola, Initial Activity of Reduced Chromia/Alumina Catalyst in n-Butane Dehydrogenation Monitored by On-Line FT–IR Gas Analysis, J. Catal., 1996, 161, 393–400 CrossRef CAS.
- B. M. Weckhuysen, A. Bensalem and R. A. Schoonheydt, Insitu UV–VIS diffuse reflectance spectroscopy–on-line activity measurements Significance of Cr+ species (n = 2, 3 and 6) in n-butane dehydrogenation catalyzed by supported chromium oxide catalysts, J. Chem. Soc., Faraday Trans., 1998, 94, 2011–2014 RSC.
- C. Marcilly and B. Delmon, The activity of true Cr2O3-Al2O3 solid solutions in dehydrogenation, J. Catal., 1972, 24, 336–347 CrossRef CAS.
- F. Cavani, M. Koutyrev, F. Trifirò, A. Bartolini, D. Ghisletti, R. Iezzi, A. Santucci and G. Del Piero, Chemical and Physical Characterization of Alumina-Supported Chromia-Based Catalysts and Their Activity in Dehydrogenation of Isobutane, J. Catal., 1996, 158, 236–250 CrossRef CAS.
- A. Hakuli, M. E. Harlin, L. B. Backman and A. O. I. Krause, Dehydrogenation of i-Butane on CrOx/SiO2 Catalysts, J. Catal., 1999, 184, 349–356 CrossRef CAS.
- B. M. Weckhuysen, A. A. Verberckmoes, J. Debaere, K. Ooms, I. Langhans and R. A. Schoonheydt, In situ UV–Vis diffuse reflectance spectroscopy—on line activity measurements of supported chromium oxide catalysts: relating isobutane dehydrogenation activity with Cr-speciation via experimental design, J. Mol. Catal. A: Chem., 2000, 151, 115–131 CrossRef CAS.
- M. F. Delley, F. Núñez-Zarur, M. P. Conley, A. Comas-Vives, G. Siddiqi, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Proton transfers are key elementary steps in ethylene polymerization on isolated chromium(III) silicates, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11624–11629 CrossRef CAS PubMed.
- M. P. Conley, M. F. Delley, F. Núñez-Zarur, A. Comas-Vives and C. Copéret, Heterolytic Activation of C–H Bonds on CrIII–O Surface Sites Is a Key Step in Catalytic Polymerization of Ethylene and Dehydrogenation of Propane, Inorg. Chem., 2015, 54, 5065–5078 CrossRef CAS PubMed.
- S. B. Duckett and R. E. Mewis, Application of Parahydrogen Induced Polarization Techniques in NMR Spectroscopy and Imaging, Acc. Chem. Res., 2012, 45, 1247–1257 CrossRef CAS PubMed.
- J. Natterer and J. Bargon, Parahydrogen induced polarization, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 31, 293–315 CrossRef.
- K. V. Kovtunov, V. V. Zhivonitko, I. V. Skovpin, D. A. Barskiy and I. V. Koptyug, in Hyperpolarization Methods in NMR Spectroscopy, ed. L. T. Kuhn, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 123–180 Search PubMed.
- P. J. Carson, C. R. Bowers and D. P. Weitekamp, The PASADENA Effect at a Solid Surface: High-Sensitivity Nuclear Magnetic Resonance of Hydrogen Chemisorption, J. Am. Chem. Soc., 2001, 123, 11821–11822 CrossRef CAS PubMed.
- B. Zheng, W. Hua, Y. Yue and Z. Gao, Dehydrogenation of propane to propene over different polymorphs of gallium oxide, J. Catal., 2005, 232, 143–151 CrossRef CAS.
- A. Bhan and W. N. Delgass, Propane Aromatization over HZSM-5 and Ga/HZSM-5 Catalysts, Catal. Rev., 2008, 50, 19–151 CrossRef CAS.
- J. H. Gregor, C. D. Gosling and H. E. Fullerton, Upgrading Fischer–Tropsch LPG (liquefied petroleum gas) with the Cyclar process, United States, 1989 Search PubMed.
- P. Schulz and M. Baerns, Aromatization of ethane over gallium-promoted H-ZSM-5 catalysts, Appl. Catal., 1991, 78, 15–29 CrossRef CAS.
- N. M. Phadke, J. Van der Mynsbrugge, E. Mansoor, A. B. Getsoian, M. Head-Gordon and A. T. Bell, Characterization of Isolated Ga3+ Cations in Ga/H-MFI Prepared by Vapor-Phase Exchange of H-MFI Zeolite with GaCl3, ACS Catal., 2018, 8, 6106–6126 CrossRef CAS.
- E. Mansoor, M. Head-Gordon and A. T. Bell, Computational Modeling of the Nature and Role of Ga Species for Light Alkane Dehydrogenation Catalyzed by Ga/H-MFI, ACS Catal., 2018, 8, 6146–6162 CrossRef CAS.
- E. A. Pidko, V. B. Kazansky, E. J. M. Hensen and R. A. van Santen, A comprehensive density functional theory study of ethane dehydrogenation over reduced extra-framework gallium species in ZSM-5 zeolite, J. Catal., 2006, 240, 73–84 CrossRef CAS.
- M. S. Pereira and M. A. C. Nascimento, Theoretical study of the dehydrogenation reaction of ethane catalyzed by zeolites containing non-framework gallium species: The 3-step mechanism × the 1-step concerted mechanism, Chem. Phys. Lett., 2005, 406, 446–451 CrossRef CAS.
- Z. Maeno, S. Yasumura, X. Wu, M. Huang, C. Liu, T. Toyao and K. Shimizu, Isolated Indium Hydrides in CHA Zeolites: Speciation and Catalysis for Nonoxidative Dehydrogenation of Ethane, J. Am. Chem. Soc., 2020, 142, 4820–4832 CrossRef CAS PubMed.
- N. Rane, A. R. Overweg, V. B. Kazansky, R. A. van Santen and E. J. M. Hensen, Characterization and reactivity of Ga+ and GaO+ cations in zeolite ZSM-5, J. Catal., 2006, 239, 478–485 CrossRef CAS.
- M. Chen, J. Xu, F.-Z. Su, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Dehydrogenation of propane over spinel-type gallia–alumina solid solution catalysts, J. Catal., 2008, 256, 293–300 CrossRef CAS.
- C.-T. Shao, W.-Z. Lang, X. Yan and Y.-J. Guo, Catalytic performance of gallium oxide based-catalysts for the propane dehydrogenation reaction: effects of support and loading amount, RSC Adv., 2017, 7, 4710–4723 RSC.
- P. Castro-Fernández, D. Mance, C. Liu, I. B. Moroz, P. M. Abdala, E. A. Pidko, C. Copéret, A. Fedorov and C. R. Müller, Propane Dehydrogenation on Ga2O3-Based Catalysts: Contrasting Performance with Coordination Environment and Acidity of Surface Sites, ACS Catal., 2021, 907–924 CrossRef.
- K. Searles, G. Siddiqi, O. V. Safonova and C. Copéret, Silica-supported isolated gallium sites as highly active, selective and stable propane dehydrogenation catalysts, Chem. Sci., 2017, 8, 2661–2666 RSC.
- N. Rane, M. Kersbulck, R. A. van Santen and E. J. M. Hensen, Cracking of n-heptane over Brønsted acid sites and Lewis acid Ga sites in ZSM-5 zeolite, Microporous Mesoporous Mater., 2008, 110, 279–291 CrossRef CAS.
- C. S. Praveen, A. P. Borosy, C. Copéret and A. C. Vives, Strain in Silica-Supported Ga(III) Sites: Neither Too Much nor Too Little for Propane Dehydrogenation Catalytic Activity, Inorg. Chem., 2021 DOI:10.1021/acs.inorgchem.0c03135.
- A. “Bean” Getsoian, U. Das, J. Camacho-Bunquin, G. Zhang, J. R. Gallagher, B. Hu, S. Cheah, J. A. Schaidle, D. A. Ruddy, J. E. Hensley, T. R. Krause, L. A. Curtiss, J. T. Miller and A. S. Hock, Organometallic model complexes elucidate the active gallium species in alkane dehydrogenation catalysts based on ligand effects in Ga K-edge XANES, Catal. Sci. Technol., 2016, 6, 6339–6353 RSC.
- K. C. Szeto, Z. R. Jones, N. Merle, C. Rios, A. Gallo, F. Le Quemener, L. Delevoye, R. M. Gauvin, S. L. Scott and M. Taoufik, A Strong Support Effect in Selective Propane Dehydrogenation Catalyzed by Ga(i-Bu)3 Grafted onto γ-Alumina and Silica, ACS Catal., 2018, 8, 7566–7577 CrossRef CAS.
- P. Wang, Z. Xu, T. Wang, Y. Yue, X. Bao and H. Zhu, Unmodified bulk alumina as an efficient catalyst for propane dehydrogenation, Catal. Sci. Technol., 2020, 10, 3537–3541 RSC.
- Z.-J. Zhao, T. Wu, C. Xiong, G. Sun, R. Mu, L. Zeng and J. Gong, Hydroxyl-Mediated Non-oxidative Propane Dehydrogenation over VOx/γ-Al2O3 Catalysts with Improved Stability, Angew. Chem., Int. Ed., 2018, 57, 6791–6795 CrossRef CAS PubMed.
- K. C. Szeto, B. Loges, N. Merle, N. Popoff, A. Quadrelli, H. Jia, E. Berrier, A. De Mallmann, L. Delevoye, R. M. Gauvin and M. Taoufik, Vanadium Oxo Organometallic Species Supported on Silica for the Selective Non-oxidative Dehydrogenation of Propane, Organometallics, 2013, 32, 6452–6460 CrossRef CAS.
- Z. Wu and P. C. Stair, UV Raman spectroscopic studies of V/θ-Al2O3 catalysts in butane dehydrogenation, J. Catal., 2006, 237, 220–229 CrossRef CAS.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, Effect of support on selectivity and on-stream stability of surface VOx species in non-oxidative propane dehydrogenation, Catal. Sci. Technol., 2014, 4, 1323–1332 RSC.
- U. Rodemerck, S. Sokolov, M. Stoyanova, U. Bentrup, D. Linke and E. V. Kondratenko, Influence of support and kind of VOx species on isobutene selectivity and coke deposition in non-oxidative dehydrogenation of isobutane, J. Catal., 2016, 338, 174–183 CrossRef CAS.
- U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, Influence of the kind of VOx structures in VOx/MCM-41 on activity, selectivity and stability in dehydrogenation of propane and isobutane, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- P. Hu, W.-Z. Lang, X. Yan, X.-F. Chen and Y.-J. Guo, Vanadium-doped porous silica materials with high catalytic activity and stability for propane dehydrogenation reaction, Appl. Catal., A, 2018, 553, 65–73 CrossRef CAS.
- D. M. Kaphan, M. S. Ferrandon, R. R. Langeslay, G. Celik, E. C. Wegener, C. Liu, J. Niklas, O. G. Poluektov and M. Delferro, Mechanistic Aspects of a Surface Organovanadium(III) Catalyst for Hydrocarbon Hydrogenation and Dehydrogenation, ACS Catal., 2019, 9, 11055–11066 CrossRef CAS.
- P. Hu, Y. Chen, X. Yan, W.-Z. Lang and Y.-J. Guo, Correlation of the Vanadium Precursor and Structure Performance of Porous VOX-SiO2 Solids for Catalytic Dehydrogenation of Propane, Ind. Eng. Chem. Res., 2019, 58, 4065–4073 CrossRef CAS.
- Y. Xie, R. Luo, G. Sun, S. Chen, Z.-J. Zhao, R. Mu and J. Gong, Facilitating the reduction of V–O bonds on VOx/ZrO2 catalysts for non-oxidative propane dehydrogenation, Chem. Sci., 2020, 11, 3845–3851 RSC.
- H. Sohn, J. Camacho-Bunquin, R. R. Langeslay, P. A. Ignacio-de Leon, J. Niklas, O. G. Poluektov, C. Liu, J. G. Connell, D. Yang, J. Kropf, H. Kim, P. C. Stair, M. Ferrandon and M. Delferro, Isolated, well-defined organovanadium(III) on silica: single-site catalyst for hydrogenation of alkenes and alkynes, Chem. Commun., 2017, 53, 7325–7328 RSC.
- V. V. Zhivonitko, I. V. Skovpin, K. C. Szeto, M. Taoufik and I. V. Koptyug, Parahydrogen-Induced Polarization Study of the Silica-Supported Vanadium Oxo Organometallic Catalyst, J. Phys. Chem. C, 2018, 122, 4891–4900 CrossRef CAS PubMed.
- Y. Sun, L. Tao, T. You, C. Li and H. Shan, Effect of sulfation on the performance of Fe2O3/Al2O3 catalyst in catalytic dehydrogenation of propane to propylene, Chem. Eng. J., 2014, 244, 145–151 CrossRef CAS.
- Y. Sun, Y. Wu, H. Shan and C. Li, Studies on the Nature of Active Cobalt Species for the Production of Methane and Propylene in Catalytic Dehydrogenation of Propane, Catal. Lett., 2015, 145, 1413–1419 CrossRef CAS.
- B. Hu and A. Bean, Getsoian, N. M. Schweitzer, U. Das, H. Kim, J. Niklas, O. Poluektov, L. A. Curtiss, P. C. Stair, J. T. Miller and A. S. Hock, Selective propane dehydrogenation with single-site CoII on SiO2 by a non-redox mechanism, J. Catal., 2015, 322, 24–37 CrossRef CAS.
- B. Hu, N. M. Schweitzer, G. Zhang, S. J. Kraft, D. J. Childers, M. P. Lanci, J. T. Miller and A. S. Hock, Isolated FeII on Silica As a Selective Propane Dehydrogenation Catalyst, ACS Catal., 2015, 5, 3494–3503 CrossRef CAS.
- D. P. Estes, G. Siddiqi, F. Allouche, K. V. Kovtunov, O. V. Safonova, A. L. Trigub, I. V. Koptyug and C. Copéret, C–H Activation on Co,O Sites: Isolated Surface Sites versus Molecular Analogs, J. Am. Chem. Soc., 2016, 138, 14987–14997 CrossRef CAS.
- R. A. Ross and J. H. Valentine, The dehydrogenation of cyclohexane vapor on silica-supported nickel catalysts, J. Catal., 1963, 2, 39–44 CrossRef CAS.
- J. L. Gland, K. Baron and G. A. Somorjai, Low-energy electron diffraction, work function charge and mass spectrometric studies of chemisorption and dehydrogenation of cyclohexane, cyclohexene and 1,3-cyclohexadiene on the Pt(111) surface, J. Catal., 1975, 36, 305–312 CrossRef CAS.
- J. H. Sinfelt, Catalysis by Alloys and Bimetallic Clusters, Acc. Chem. Res., 1977, 10, 15–20 CrossRef CAS.
- J. Völter, Supported mono- and bimetallic catalysts in hydrocarbon conversions, Stud. Surf. Sci. Catal., 1986, 27, 337–372 CrossRef.
- J. Völter, G. Lietz, M. Uhlemann and M. Hermann, Conversion of cyclohexane and n-heptane on Pt-Pb/Al2O3 and Pt-Sn/Al2O3 bimetallic catalysts, J. Catal., 1981, 68, 42–50 CrossRef.
- H. Dang Lanh, H. Si Thoang, H. Lieske and J. Vőlter, Conversion of cyclohexane on bimetallic Pt-Sb/Al2O3 catalysts, Appl. Catal., 1984, 11, 195–200 CrossRef.
- J. H. Sinfelt, Polymetallic cluster compositions useful as hydrocarbon conversion catalysts, US Pat., 3953368, 1974 Search PubMed.
- L. G. Cesar, C. Yang, Z. Lu, Y. Ren, G. Zhang and J. T. Miller, Identification of a Pt3Co Surface Intermetallic Alloy in Pt–Co Propane Dehydrogenation Catalysts, ACS Catal., 2019, 9, 5231–5244 CrossRef CAS.
- V. J. Cybulskis, B. C. Bukowski, H.-T. Tseng, J. R. Gallagher, Z. Wu, E. Wegener, A. J. Kropf, B. Ravel, F. H. Ribeiro, J. Greeley and J. T. Miller, Zinc Promotion of Platinum for Catalytic Light Alkane Dehydrogenation: Insights into Geometric and Electronic Effects, ACS Catal., 2017, 7, 4173–4181 CrossRef CAS.
- E. C. Wegener, Z. Wu, H.-T. Tseng, J. R. Gallagher, Y. Ren, R. E. Diaz, F. H. Ribeiro and J. T. Miller, Structure and reactivity of Pt–In intermetallic alloy nanoparticles: Highly selective catalysts for ethane dehydrogenation, Catal. Today, 2018, 299, 146–153 CrossRef CAS.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, A combined in situ time-resolved UV–Vis, Raman and high-energy resolution X-ray absorption spectroscopy study on the deactivation behavior of Pt and PtSn propane dehydrogenation catalysts under industrial reaction conditions, J. Catal., 2010, 276, 268–279 CrossRef CAS.
- J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Platinum-Promoted Ga/Al2O3 as Highly Active, Selective, and Stable Catalyst for the Dehydrogenation of Propane, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS PubMed.
- K. Searles, K. Wing Chan, J. Augusto Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, Highly Productive Propane Dehydrogenation Catalyst Using Silica-Supported Ga–Pt Nanoparticles Generated from Single-Sites, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS PubMed.
- L. Rochlitz, K. Searles, J. Alfke, D. Zemlyanov, O. V. Safonova and C. Copéret, Silica-supported, narrowly distributed{,} subnanometric Pt–Zn particles from single sites with high propane dehydrogenation performance, Chem. Sci., 2020, 11, 1549–1555 RSC.
- Y. I. Yermakov and B. N. Kuznetsov, Supported metallic catalysts prepared by decomposition of surface ornagometallic complexes, J. Mol. Catal., 1980, 9, 13–40 CrossRef.
- Y. I. Yermakov, Supported Catalysts Obtained by Interaction of Organometallic Compounds of Transition Elements with Oxide Supports, Catal. Rev., 1976, 13, 77–120 CrossRef.
- J. L. Margitfalvi, E. Tálas and S. Gőbölös, Preparation of alumina supported tin-platinum catalysts by reaction of tin tetraethyl with hydrogen adsorbed on platinum. Study of the formation and decomposition of the primary surface complex, Catal. Today, 1989, 6, 73–80 CrossRef.
- J. L. Margitfalvi, I. Borbáth, E. Tfirst and A. Tompos, Formation of multilayered tin organometallic surface species. Preparation of new type of supported Sn–Pt catalysts, Catal. Today, 1998, 43, 29–49 CrossRef CAS.
- J.-P. Candy, B. Didillon, E. L. Smith, T. B. Shay and J.-M. Basset, Surface organometallic chemistry on metals: a novel and effective route to custom-designed bimetallic catalysts, J. Mol. Catal., 1994, 86, 179–204 CrossRef CAS.
- J. P. Candy, F. Humblot, B. Didillon, F. Le Peltier and J. M. Basset, in Catalyst Deactivation 1999, ed. G. F. Froment and B. Delmon, Elsevier, 1999, vol. 126, pp. 237–247 Search PubMed.
- P. Rouge, A. Garron, S. Norsic, C. Larabi, N. Merle, L. Delevoye, R. M. Gauvin, K. C. Szeto and M. Taoufik, A smarter approach to catalysts by design: Combining surface organometallic chemistry on oxide and metal gives selective catalysts for dehydrogenation of 2,3-dimethylbutane, Mol. Catal., 2019, 471, 21–26 CrossRef.
- Z. Xu, R. Xu, Y. Yue, P. Yuan, X. Bao, E. Abou-Hamad, J. M. Basset and H. Zhu, Bimetallic Pt-Sn nanocluster from the hydrogenolysis of a well-defined surface compound consisting of [(≡AlO–)Pt(COD)Me] and [(≡AlO–)SnPh3] fragments for propane dehydrogenation, J. Catal., 2019, 374, 391–400 CrossRef CAS.
- J. Camacho-Bunquin, M. S. Ferrandon, H. Sohn, A. J. Kropf, C. Yang, J. Wen, R. A. Hackler, C. Liu, G. Celik, C. L. Marshall, P. C. Stair and M. Delferro, Atomically Precise Strategy to a PtZn Alloy Nanocluster Catalyst for the Deep Dehydrogenation of n-Butane to 1,3-Butadiene, ACS Catal., 2018, 10058–10063 CrossRef CAS.
- C. Byron, S. Bai, G. Celik, M. S. Ferrandon, C. Liu, C. Ni, A. Mehdad, M. Delferro, R. F. Lobo and A. V. Teplyakov, Role of Boron in Enhancing the Catalytic Performance of Supported Platinum Catalysts for the Nonoxidative Dehydrogenation of n-Butane, ACS Catal., 2020, 10, 1500–1510 CrossRef CAS.
- Y. Nakaya, J. Hirayama, S. Yamazoe, K. Shimizu and S. Furukawa, Single-atom Pt in intermetallics as an ultrastable and selective catalyst for propane dehydrogenation, Nat. Commun., 2020, 11, 2838 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |