DOI:
10.1039/D0CS01506G
(Review Article)
Chem. Soc. Rev., 2021,
50, 2984-3012
Status and prospects of the decentralised valorisation of natural gas into energy and energy carriers
Received
4th December 2020
First published on 24th February 2021
Abstract
Natural gas is widely considered as the key feedstock to enable the transition from the oil to the renewables era. Despite its vast reserves, the use of this resource to produce energy and chemicals does not match its full potential. The main reason lies in the nature of its wells, which are often found in remote locations around the globe, rendering access and transportation challenging. To aid this development, several technologies for energy and energy carrier production have been developed, all of which have in common the goal of upgrading natural gas directly at the source of extraction. Following this direction, this review firstly analyses the advances in process design towards decentralised generation of electricity and of liquefied natural gas. Subsequently, recent efforts in progress made in catalysed alkane transformations using heterogeneous catalysts are reviewed for small-scale chemicals and fuels production. The presented analysis identifies that techno-economic and life-cycle assessments should be widely performed to enable proper technological benchmarking of these technologies. The integration of these multidisciplinary fields is key to foster synergies between researchers in the areas of decentralised energy and energy carrier generation in view of developing effective and efficient processes for valorising natural gas directly on-site.

Guido Zichittella
| Guido Zichittella (Erice, Italy, 1991) studied Chemical Engineering at Politecnico di Milano (BSc) and at ETH Zurich (MSc). He then joined the group of Prof. Dr J. Pérez-Ramírez at ETH Zurich to pursue his doctoral studies, which he completed in December 2019. He is now continuing his research activities as a scientist in the same group. His work embraces the development of catalytic technologies for the direct functionalization of light alkanes. For his doctoral work, he was awarded the ETH Medal and the ABB Research Award. |

Javier Pérez-Ramírez
| Javier Pérez-Ramírez (Benidorm, Spain, 1974) holds the Chair of Catalysis Engineering at ETH Zurich. His research pursues the design of heterogeneous catalysts and reactor concepts tackling current and future energy, resource and environmental challenges of society. The main topics of interest include the valorisation of renewables, carbon dioxide and natural gas using nanostructured materials. His work has been recognised by several awards, most recently the Paul H. Emmett Award in Fundamental Catalysis of the North American Catalysis Society. He directs a National Competence Centre of Research in Catalysis and is Visiting Professor at the National University of Singapore. |
1. Introduction
1.1. Evolution in primary energy demand
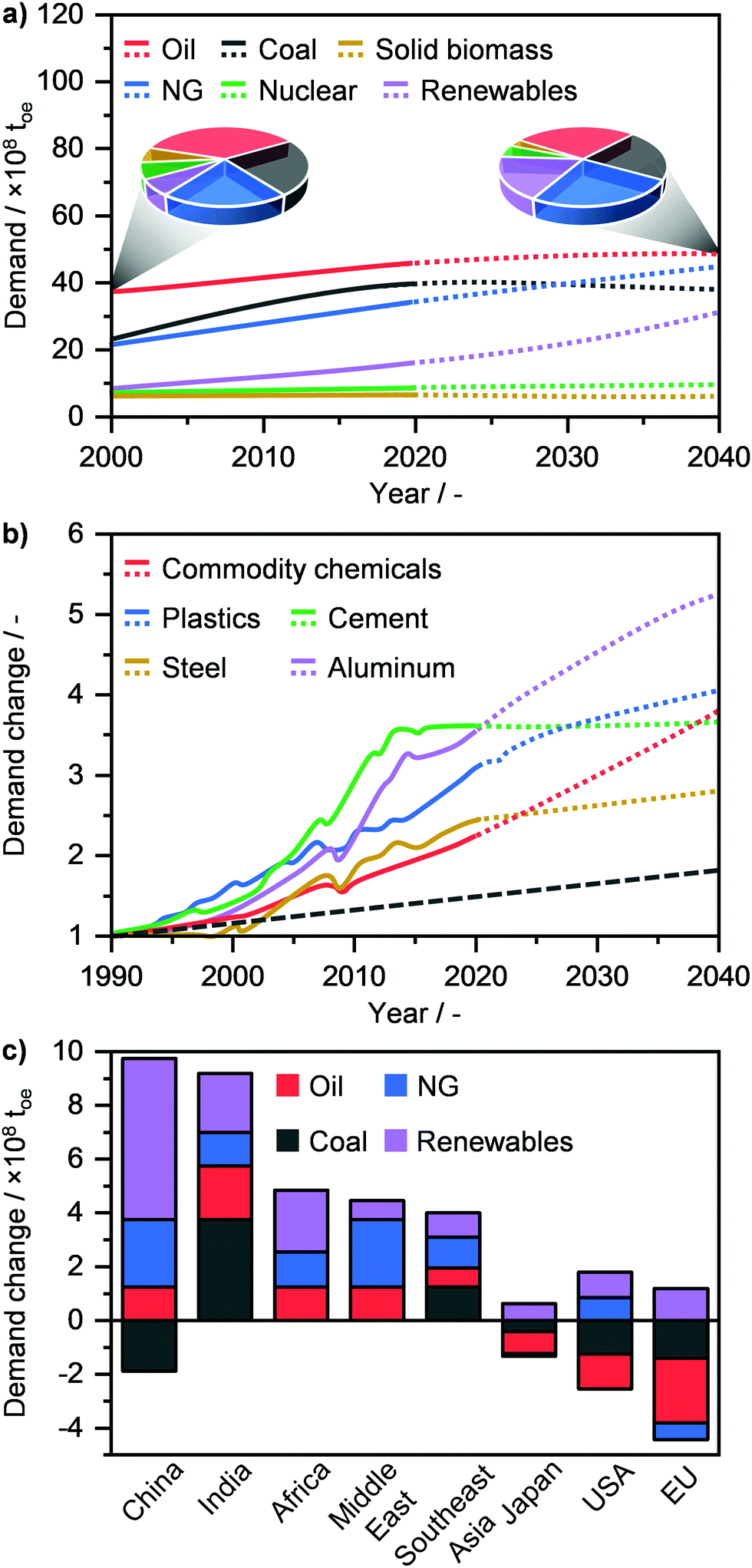
Modern society strongly relies on the availability and efficient utilisation of raw materials to produce energy and energy carriers (E + EC), such as liquid fuels and chemicals. These feedstocks are categorised into fossil fuels, i.e., oil, coal, and natural gas (NG) and into low emitting fuels, such as biomass, nuclear and renewables, which include solar, hydro and wind energies.1 Currently, the demand for primary energy, which in 2019 stood at ca. 14.5 billion tons of oil equivalent (toe), is dominated by fossil fuels and in particular by oil (30.0%) followed by coal (26.5%) and NG (22.8%) (Fig. 1a).2,3 According to the International Energy Agency, due to the continuous increase in world population and living standards, the primary energy demand is forecasted to grow to ca. 17.5 billion toe by 2040.2–5 This translates into a necessary and constantly increasing supply not only of energy, but also of commodities, such as aluminium, cement, chemicals, steel and plastics to enable our modern society to prosper (Fig. 1b).2,6 Given that the world consumption rate of fossil fuels, and especially of oil, greatly exceeds that of their natural production cycle, the use of primary raw materials is expected to change significantly in order to sustain societal growth with an acceptable environmental impact.7 According to the World Energy Outlook, oil demand will level off by 2040, while that of coal will decrease, after reaching a peak in the next 5 years (Fig. 1a). On the other hand, the use of renewables will see the highest rate of increase, going from ca. 6.5% in 2000 to 17.5% of the energy share in 4 decades.2,3 Although these are considered to be the ultimate solution for sustainable energy generation, the use of renewables still faces different challenges today in view of becoming the predominant energy source. Notably, despite the considerable advances in energy storage (e.g. the use of steam and molten salts) and distribution (e.g. smart grids),8,9 the generation of electricity and heat by wind, sun and/or water has typically a discontinuous character. Furthermore, processes that enable the harvesting of solar energy, water (H2O) and carbon dioxide (CO2) for the manufacture of chemicals and fuels have a low TRL (technology readiness level) and will likely require several years or decades to become economically competitive.10 Consequently, the winning strategy to bridge the gap between the incremental societal needs and the depleting resources, as also highlighted by the European Commission in late 2018, is most likely to maximise the efficient utilisation of the current fossil resources in the transition towards the renewables era.11 In this perspective, NG is widely considered as the most suited feedstock for E + EC production, given its vast availability and much lower environmental footprint compared to oil and especially coal.7,11–13 As a matter of fact, NG will see a strong rise in its demand, reaching up to 25.1% of the primary energy share by 2040 (Fig. 1a), thus surpassing coal (21.3%) and achieving a comparable consumption rate to oil (27.7%). This shift in an almost NG-dominated energy industry is forecasted to be primarily led by China and the Middle East, followed by India, Africa, Southeast Asia and the USA, while Japan and the EU are predicted to decrease their NG demand (Fig. 1c).2,3
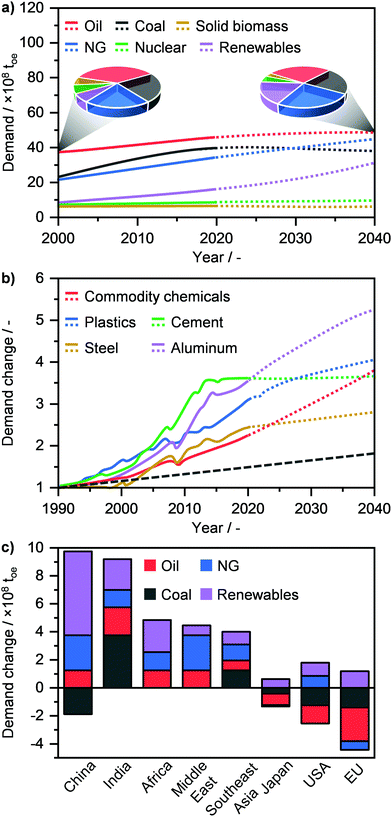 |
| | Fig. 1 (a) Annual global primary energy demand by fuel with projections (dotted line) up to 2040. The pie charts compare the change in resource use between 2000 and 2040. (b) Annual global demand of commodities in relation to their demand in 1990 with projections (dotted line) up to 2040. The dashed grey line denote the concomitant change in the global population. (c) Change in primary energy demand in 2040 by region.2 | |
1.2. Natural gas reserves
Although there is no hard record of when this resource was discovered for the first time, the use of NG has been documented in China as early as 400 BC. The fossil fuel, which was extracted during drilling for brine, was transported using bamboo pipelines and then mixed with air in order to yield a fire that would evaporate the water to recover the salt.14 This is possible thanks mainly to the energy released by the combustion of its primary constituent, methane (CH4), which accounts for 75–99 mol% of the overall molar composition (Fig. 2, left). Ethane (C2H6, 1–15 mol%), propane (C3H8, 1–10 mol%) and higher hydrocarbons (C4+, 0–5 mol%) are typically present in low-to-moderate amounts and they constitute, after separation from methane and liquefaction, the so-called natural gas liquids. Furthermore, H2O, mercury (Hg), hydrogen sulfide (H2S) and other sulfur compounds as well as CO2 are found in variable quantities, ranging from a few ppm to ca. 30 mol%, while other components such as helium (He), hydrogen (H2), nitrogen (N2) and oxygen (O2) are generally present as impurities.15
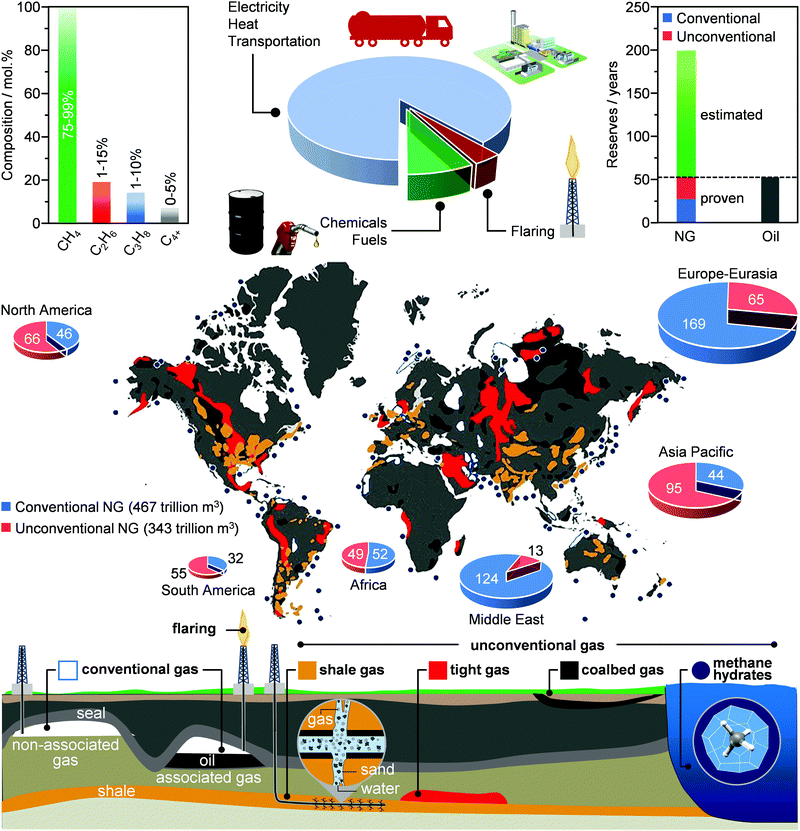 |
| | Fig. 2 NG is an attractive source of light alkanes (left panel) for several industrial sectors. Conventional and unconventional gas wells (bottom panel) hold large reserves, rivalling those of oil (right panel). However, they are often found in remote locations (bottom panel), where the high cost of transportation and the lack of technologies enabling the on-site production of readily transportable products, including liquid chemicals, fuels and LNG, often results into flaring (middle panel), which causes economic losses and negative environmental footprint.16,22,26 | |
Gas wells can be classified as conventional or unconventional (Fig. 2, bottom). The main distinction between these two classes of basins stems from their geology: conventional gas can be found accumulated in easily accessible porous rocks (non-associated gas), or jointly with oil (associated gas). On the other hand, unconventional gas is locked inside deep and low-permeable sandstones (shale and tight gas), in the pores of surface coal deposits (coalbed methane) and in crystalline cage-like structures formed by water molecules at low temperatures and high pressures (methane hydrates), conditions typical of permafrost areas and deep marine sea environments.16–20 Accordingly, different extraction technologies, ranging from conventional vertical drilling to more specialised and unconventional fracking, are required in order to retrieve profit-making quantities of these globally scattered reserves.16,21 The advances in extraction techniques have driven a revolution in the field of NG extraction: an increment of at least 28% and 50% compared to 2009 and 1999, respectively, has been registered in 2019, resulting into a total amount of ca. 200 trillion m3 of proven NG reserves, equivalent to ca. 50 years of use.2,3,22 The current situation is exemplified by the discovery of a vast gas reservoir in the Eastern Mediterranean Sea in 2018, estimated at ca. 3.5 trillion m3.23 Similarly, in the United States, the increasing amounts of shale gas found are driving a revolution in this sector, with this unconventional gas forecasted to become the main game player in the US energy sector by 2040.2,24 In addition, experts predict that if technologies improve enough to be able to extract methane hydrates, whose most conservative assessment foresees ca. 440 trillion m3 of gas, the proven reserves will have the potential to last for at least 200 years (Fig. 2, right).25–27
1.3. Natural gas as feedstock
Today, ca. 90% of the NG is used for the generation of heat and electricity and for transportation (Fig. 2, middle).28,29 In contrast, only up to 8% of this extracted resource is used for the production of chemical commodities, including ammonia (NH3), methanol (CH3OH), light olefins (C2H4, C3H6), H2, aromatics (C6H6, C7H8, C8H10) and long-chain hydrocarbons (e.g., CnH2n+2 and CnH2n, n = 5). The key reason for this unbalanced use of NG as an energy and carbon feedstock derives from its high transportation costs.29–31 Specifically, its much lower mass and energy density generally leads to an increase in shipment expenses by up to 3 orders of magnitude compared to oil.15,32 Accordingly, NG transportation, today predominantly performed using pipelines that can require investments up to 7 billion USD,33 becomes economically attractive only when a considerably high volume of gas is transported over short-to-medium distances. To mitigate this fundamental problem, NG can be condensed into a liquid, also called liquefied natural gas (LNG), which opens possibilities to increase transportation and use of this resource as feedstock for the generation of E + EC.32 Still, the cryogenic temperatures (ca. 110 K) required and the considerable energy losses typically translate into the construction of massive liquefaction plants, in order for these processes to be economically competitive.32,34
Another key reason for the limited use of NG in the chemical industry originates from the fact that today there are few processes that are able to directly convert alkanes into valuable products. The cause for such difficulty relates to their chemical inertness, which arises from the strong and localised C–C and C–H bonds of alkane molecules, thus leading to a lack of empty orbitals of low energy or filled orbitals of high energy that could readily participate in a chemical reaction.30,31 Consequently, aggressive reagents and/or harsh reaction conditions are typically required for alkane activation.12,16 Two commercial technologies, namely steam reforming and steam cracking, are mainly used to convert light alkanes into commodities, such as CH3OH and light olefins.35–37 Still, the harsh conditions of temperatures and pressures, typically needed to operate these processes, translate into high capital and operating costs, which render them the most energy-consuming processes in the chemical industry and globally utilising >8% of the sector's total primary energy use.37–39 Consequently, similarly to liquefaction routes, the only strategy that allows the economic competitiveness of these technologies is the construction of megaplants. However, these trends go against the intrinsic nature of NG wells, since a significant portion (>30%), mainly comprised of unconventional reserves, is constituted by small reservoirs that are scattered around the globe (Fig. 2, bottom).16,29,39 This, in combination with the high transportation costs, often results in flaring practices (Fig. 2, middle), burning around 13 billion USD worth of NG and releasing into the atmosphere more than 350 million tons of CO2 every year.40 Therefore, a strategy to unlock the potential of these reserves and mitigate this negative environmental footprint needs to be developed and implemented.
1.4. Towards a decentralised society
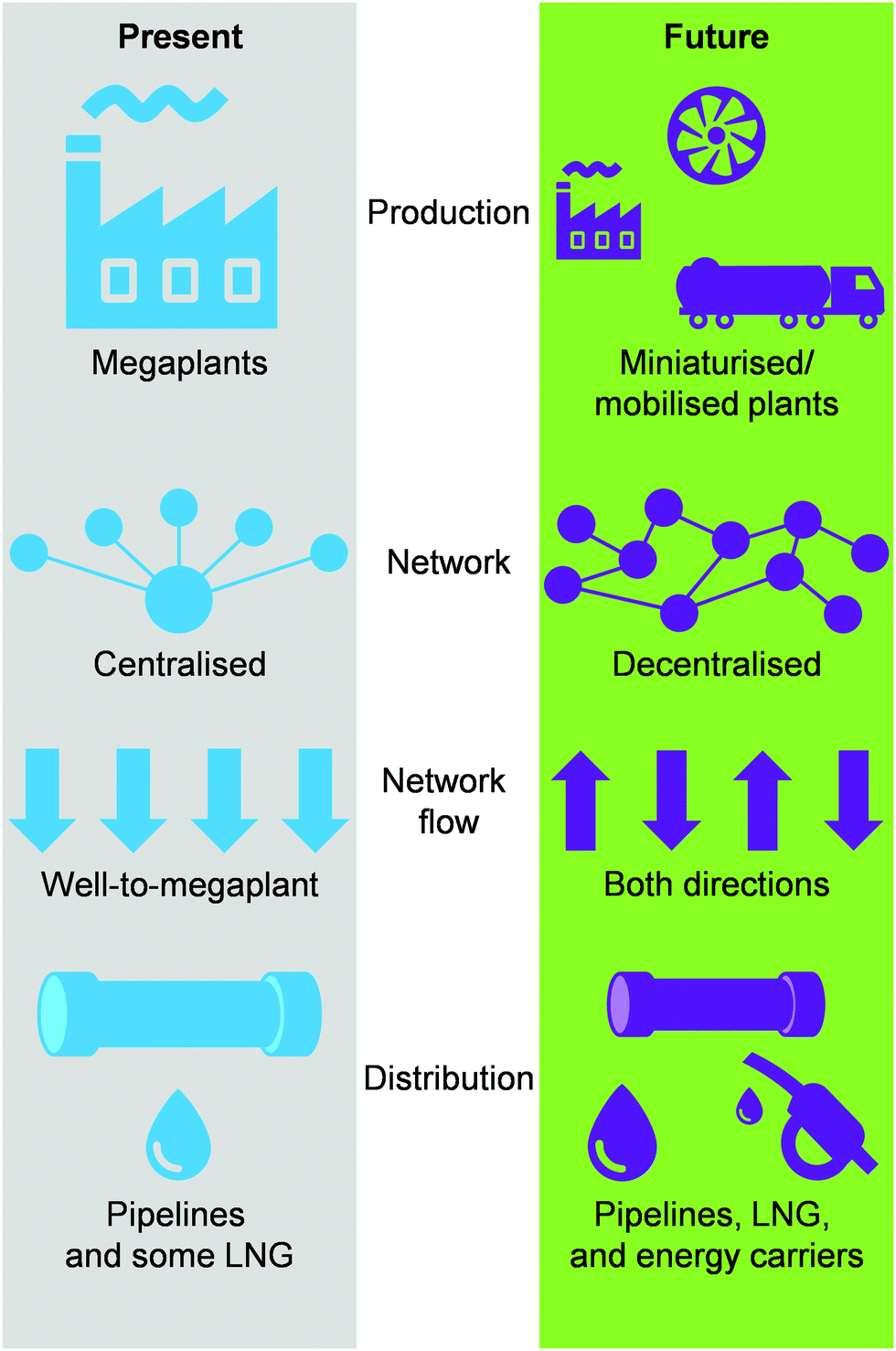
Today, the energy and chemical industries are primarily based on a centralised model, where a few megaplants, which are fed by continuous, unilateral feedstock streams originating from several locations, are responsible for generating the majority of the energy, i.e., electricity, as well as energy carriers, e.g. LNG, chemicals, fuels, needed to sustain a highly populated area (Fig. 3, left).29,37 While this model, mainly propelled by oil and coal, has so far served society well, the advent of a new era with renewables and the transition towards it, led by NG,2,3 necessitates a drastic change in the way E + EC is produced in order to efficiently utilise natural resources and to sustain an ever-growing population and living standards at a minimal-to-null environmental impact. This goal could be accomplished by transitioning to a decentralised system, which entails the generation of E + EC in a highly distributed and multilateral network of miniaturised, modular and/or mobile plants (Fig. 3, right). Accordingly, this would facilitate NG to be upgraded into electricity, LNG and/or chemicals directly at the point of extraction. In this way, small, far-located and stranded NG reserves could be efficiently utilised and the high transportation costs would be abated, ultimately resulting in the reduction or even elimination of undesired flaring practices.
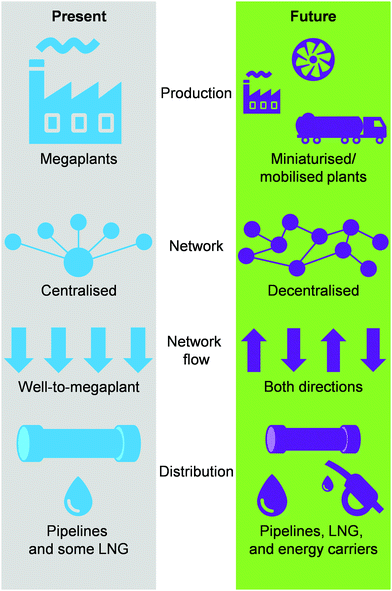 |
| | Fig. 3 Expected shift from a centralised to a decentralised-based production and distribution network for a more efficient utilisation of NG as valuable and low-emitting feedstock for electricity, LNG and chemicals generation. | |
1.5. Scope of the review
The potential of NG as energy and carbon feedstock to sustain the rising demand in primary energy and commodities with decreased environmental impact in the transition towards renewables is unparalleled among other fossil feedstocks.2 Still, as of today, it remains largely untapped. This stems from the fact that, currently, the energy industry primarily works with a centralised model, which has been tailored for the use of coal and especially oil as feedstock. Instead, the much more scattered nature of gas reserves calls for the establishment of a decentralised system for NG upgrading, where the latter can be transformed into valuable energy, i.e., electricity, and/or energy carriers, such as LNG and chemicals, directly at the source of extraction. In this regard, this review examines current developments in the decentralised valorisation of NG. Firstly, we analyse the advancements towards decentralised generation of electricity as well as of LNG. Subsequently, we examine recent efforts in progress made in heterogeneously catalysed selective alkane transformations in view of valorising NG into chemicals and fuels directly at the source of extraction. Particular focus is placed on the advances in the process, reactor and/or catalyst design that allows for process intensification. Furthermore, we provide an overview of the commercial initiatives that have been spent or are undergoing in these fields. This multidisciplinary approach of integrating the fields of decentralised electricity, LNG and chemicals production provides a unique and broad perspective to identify challenges, outline future directions in research and policy making as well as foster potential synergies between researchers in these areas in view of accelerating the development of effective and efficient technologies for on-site natural gas valorisation.
2. Technologies for decentralised electricity production
Electricity is one of the highest forms of energy and constitutes a foundational basis of our modern living standards.41 According to the International Energy Agency, the current global electricity generation stands at ca. 27 million GW h, and it is expected to surpass 40 million GW h by 2040 (Fig. 4).2,3 Coal has been so far the predominantly used resource to satisfy this demand, accounting for almost 40% of the feedstock share in 2000, followed by NG (ca. 18%). Still, its use has been predicted to flatten out by 2040, while NG as well as solar and wind energies will increase substantially, reaching a comparable feedstock share of approximately 22% (Fig. 4).2,3 Despite this significant growth, electricity production remains mainly conducted in centralised power plants. Even if electrical grids have connected billions of people, this system has led to a persistent and widespread energy poverty, where still billions of people worldwide lack even the most basic or reliable sources.42 Instead, a decentralised electrical energy generation system would not only utilize resources efficiently and effectively, but also improve the quality of life and enhance human development across the globe. Accordingly, NG represents an optimal feedstock, thanks to its presence in developing countries as well as given the rather high efficiency in its transformation into electrical energy by means of different scalable technologies, such as gas turbines and fuel cells.43–45 In this section, we overview the recent development in decentralised NG transformation into electricity by means of gas microturbines, following Brayton or inverted Brayton cycles, as well as by solid oxide and protonic ceramic fuels cells. In addition, we outline current efforts by companies and start-ups to implement on-site NG-to-electricity microplants. For more details and broader discussion on these topics, we refer the reader to other studies.43–48
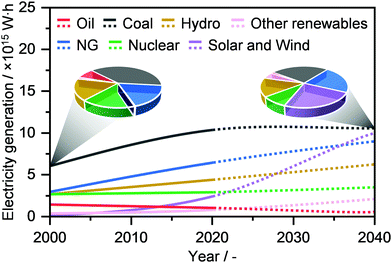 |
| | Fig. 4 Annual global electricity generation by fuel with projections (dotted line) up to 2040. The pie charts compare the change in resource use between 2000 and 2040.2 | |
2.1. Gas-to-power
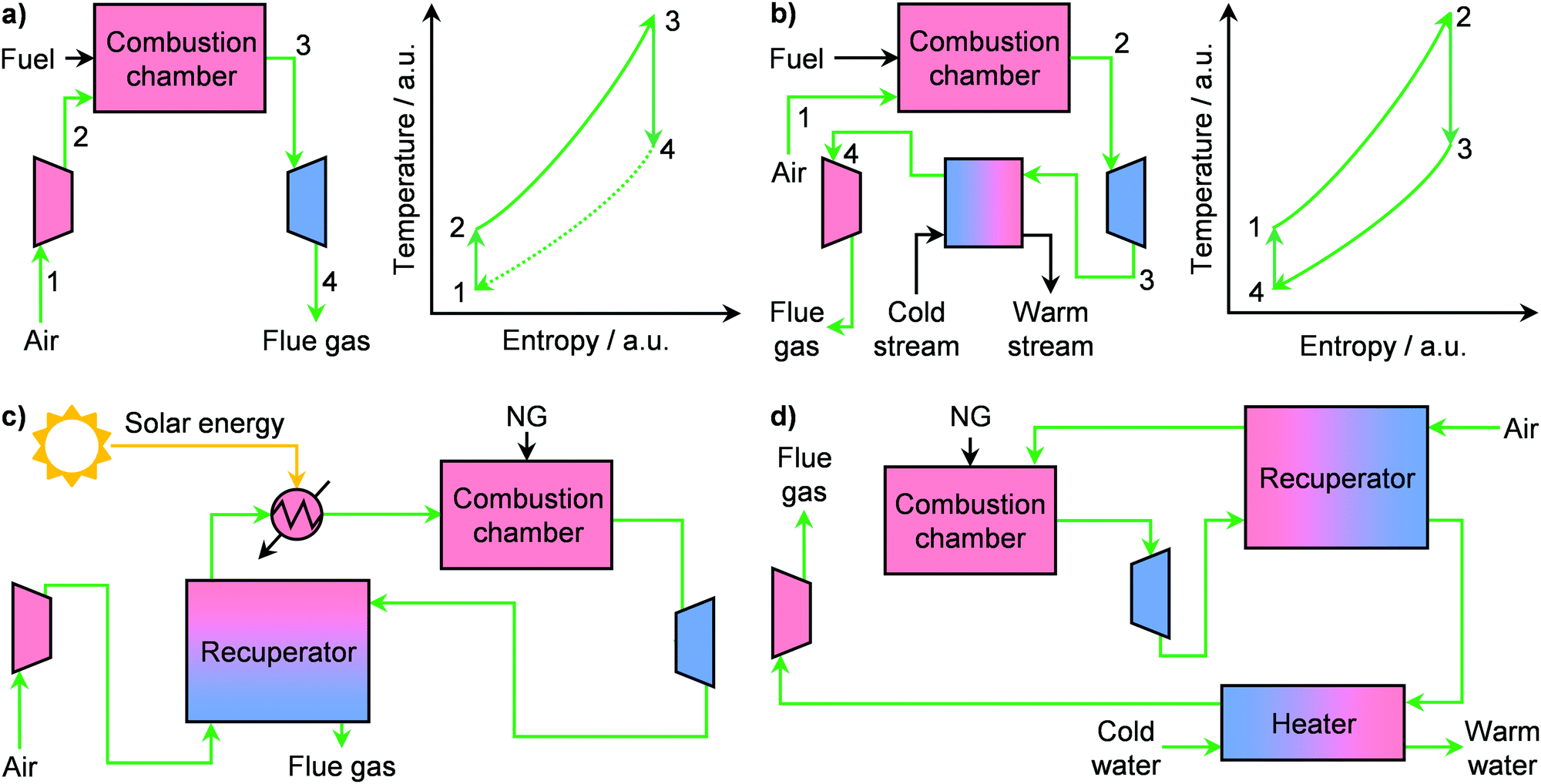
During the 20th century, economies of scale dominated the electricity sector, leading to an ever-increasing size of power plants. However, in the 1980s, technological progress as well as a shift in energy policy were the first seeds of change.49 This is exemplified by the case of the United States, where in 1978 the Power Utility Regulatory Act broke the monopoly of the electricity sector and fostered the expansion of distributed energy generation in the country.43,49 Accordingly, some companies, such as The Advanced Energy System Corporation, strode towards independent power production by targeting the use of small-scale and standardised NG plants.49 This led to extensive research in order to develop commercially viable small gas turbines, or microturbines.43 These are energy generators whose capacity ranges typically from 15 to 300 kW and they possess key advantages, such as higher load variability, simplicity and compactness, compared to large systems.43 The working principle of microturbines relies on different versions of the thermodynamic Brayton cycle (Fig. 5a and b). Notably, these systems, which consist of a compressor, a combustion chamber, and a turbine, typically follow an open Brayton cycle (Fig. 5a, left). There, air is compressed, mixed with a fuel, such as NG, and fed to the combustion chamber where both temperature and entropy of the working fluid increase considerably (Fig. 5a, right). The latter is finally expanded in a turbine, which provides the work necessary to power the compressor as well as additional energy that is converted into electricity by a generator. However, this cycle as such generally provides an energy efficiency of only 17%, which is too low for viable operation. The key factor that affects the performance of this microsystem is the inlet temperature at the turbine.43,50 By using a recuperator, a gas–gas heat exchanger that recovers the energy of the turbine exhaust, the inlet air at the combustion chamber can be preheated, thus ultimately increasing the temperature of the working fluid at the turbine entrance. This alone boosts the efficiency up to 30%.43 Still, commercially viable operation at small scales requires efficiencies ≥40%, especially under fluctuating fuel prices.49,50 Accordingly, a major approach to solve this problem, currently investigated in the literature, is the introduction of a renewable energy source, typically solar, in addition to the recuperator.51–53 Aichmayer et al. have demonstrated that this hybrid design, schematised in Fig. 5c, allows preheating the air before the combustion chamber up to 1053 K. In this way, an inlet turbine temperature of 1173 K can be reached (ca. 31% efficiency), which translates into (i) lower fuel consumption, (ii) ≤43% lower electricity costs and (iii) ≤35% less CO2 emission compared to competitor diesel generators.51 Despite the promising results, a key challenge remains the intermittent nature of solar energy. Although the introduction of energy storage devices have been shown to be marginally beneficial,52 efforts are still needed to reach the target of ≥40%. In this context, we anticipate that, with the increasingly stronger role of renewables in the energy mix, the development of hybrid systems, which can include solar, wind, battery storage and microturbines,54 will become pivotal in the establishment of small-scale NG-to-power plants.
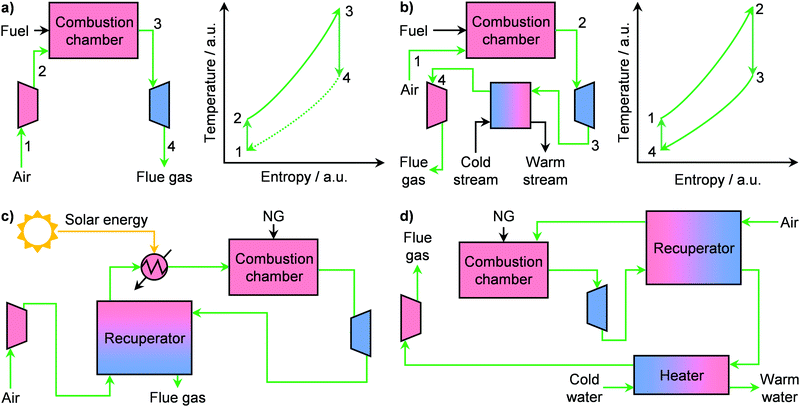 |
| | Fig. 5 Schematic flowsheet (left panel) and corresponding temperature–entropy diagrams (right panel) of (a) a Brayton cycle-based process for production of electricity and (b) an inverted Brayton cycle-based process for the cogeneration of electricity and heat. (c) Flowsheet of a hybrid solar micro gas-turbine operating by an internally fired recuperated cycle for the small-scale production of electricity from NG. (d) Flowsheet of a small-scale process for the cogeneration of electricity and steam from NG based on an inverted Brayton cycle. | |
In addition to electricity, gas microturbines can also be used to provide heat on-site, in the so-called combined heat and power (CHP) system.49 Such microplants can provide a wide range of electrical (1–150 kW) as well as thermal (≤200 kW) energy.55–57 By using NG as fuel, which could be supplied as LNG (vide infra), CHP systems can (i) produce low-price electricity and heat, (ii) guarantee energy security since they are independent of the electricity grid and (iii) lower greenhouse gas emissions.56 Accordingly, these microplants could find application in households and, in particular, in hospitals, research institutions, food processing plants and other small manufacturing operations where electric and thermal power outages are especially disruptive and costly.49,56 Still, gas turbines for micro CHP plants typically require an electricity outlet of ≥30 kW in order to maintain energy efficiency ≥30%. The major challenges for the development of efficient microturbines with only a few kW of power, which are suited for the above-discussed applications, are the higher relative weights of thermal and mechanical losses as well as high specific costs at small scales.56,58 To overcome these limitations, different strategies have been proposed in the literature. The first targets the use of ceramic-based materials in the turbine, combustion chamber, as well as the recuperator, which can reach a 7.5 kW electrical output at an efficiency of 30%.50 Alternatively, the compressor and turbine pair can be replaced with a reciprocating engine, which has the advantage of higher single cycle efficiencies as well as lower capital intensiveness although it suffers from a higher footprint.59 Accordingly, a micro CHP plant with 5 kW and 8 kW of electrical and thermal output can be achieved at 33% electrical efficiency.60 Another approach was presented by Aigner and co-workers, who have shown that a CHP microplant can be operated in an inverted Brayton cycle configuration.61 In this thermodynamic cycle, the combustion occurs at atmospheric pressure (Fig. 5b). Then, the working fluid is expanded in a turbine to below 1 bar, and its energy is recovered in a heat exchanger by warming a water stream. Finally, a compressor returns the flue gas to atmospheric pressure before discharge.49 Remarkably, they demonstrated that in this configuration, a recuperated micro CHP plant can be operated at similar fuel capacity to non-inverted systems, reaching a design electrical and thermal power of 1 kW and 3.5 kW, respectively (Fig. 5d). However, the mechanical losses in their system were too high to allow for any practical operation.61 Still, the idea of using an inverted Brayton cycle opens interesting possibilities in the field of CHP microplants and calls for further developments to improve efficiencies.
In particular, in the last decade, different commercialisation attempts have been reported for small-scale NG-to-power, which are overviewed in Table 1.62,63 In particular, microturbines are found among the most targeted technologies for practical implementation. Companies, including Aggreko PLC, Capstone Turbine Corporation, Unicorn Power Solutions Pvt Ltd, and LPP Combustion LLC, have launched microturbine systems with a power outlet ranging between 30 kW and at least 1 MW, which are in operation or under installation in several countries across the world. These systems allow for highly flexible operations, since they can be loaded in modular, mobile, and/or containerised installations. In addition, they tolerate not only fluctuating gas feeds but they are also able to process gas that would otherwise be flared (Table 1).62 Still, no considerable applications of microturbines below 30 kW and of CHP systems relevant for households, hospitals and small industrial sites have been announced.
Table 1 Companies and start-ups active in the field of decentralised NG-to-power technologies62,63
| Company |
Technology |
Feed range/×103 m3 STP d−1 |
Flexibility |
Status |
Country of origin |
| Aggreko |
Gas microturbine |
0.3–20 |
• Tolerates flare gas |
Plants in operation in Russia, Romania, and South Africa |
UK |
| • Applicable offshore |
| BINGO Interests |
Radial gas turbines |
2.5–310 |
• Multi-fuel operation |
— |
USA |
| • Applicable offshore |
| Capstone Turbine Corporation |
Gas microturbine |
0.3–8.0 |
• Multi-fuel operation |
Plants in operation in USA, Germany, and Russia |
USA |
| • Modular plant |
| • Applicable offshore |
| • Tolerates flare gas |
| General Electric |
Reciprocating gas engines |
1–100 |
• Tolerates variations in gas feed |
Plants in operation in the Middle East |
USA |
| • Multi-fuel operation |
| • Applicable offshore |
| LPP Combustion |
Gas (micro)turbines |
0.2–2000 |
• Modular, mobile and containerised process |
Plants in operation in Canada |
USA |
| • Tolerates flare gas |
| • Applicable offshore |
| Moser Energy System |
Generator |
0.3–7.5 |
• Tolerates flare gas |
Plants in operation |
USA |
| • Multi-fuel operation |
| OPRA Turbines |
Radial gas turbines |
9.5–95 |
• Modular, mobile and containerised process |
Plants in operation in Russia, Brazil and the North Sea |
The Netherlands |
| • Tolerates flare gas |
| • Applicable offshore |
| Turboden |
Thermal boiler |
8.5–500 |
• Modular, mobile and containerised process |
Plants in operation in Russia |
Italy |
| • Tolerates flare gas |
| • Applicable offshore |
| Unicorn Power Solutions |
Gas microturbines |
1.8–8.5 |
• Tolerates flare gas |
Plants under installation |
India |
| • Multi-fuel operation |
| • Applicable offshore |
| Wärtsilä |
Gas turbines |
100–4000 |
• Modular, mobile process |
Plants in operation in several countries |
Finland |
| • Multi-fuel operation |
| • Applicable offshore |
| Mitsubishi Hitachi Power Systems |
Hybrid microturbine with solid oxide fuel cell |
1.5–6 |
• Modular process |
Plants in demonstration in Japan |
Japan |
| Ceres Power |
Solid oxide fuel cell |
0.01–10 |
• Modular, mobile process |
Plants in operation in UK |
UK |
| Fuel Cell Energy |
Solid oxide fuel cell |
15–35 |
• Modular process |
Plants in operation |
USA |
2.2. Fuel cells
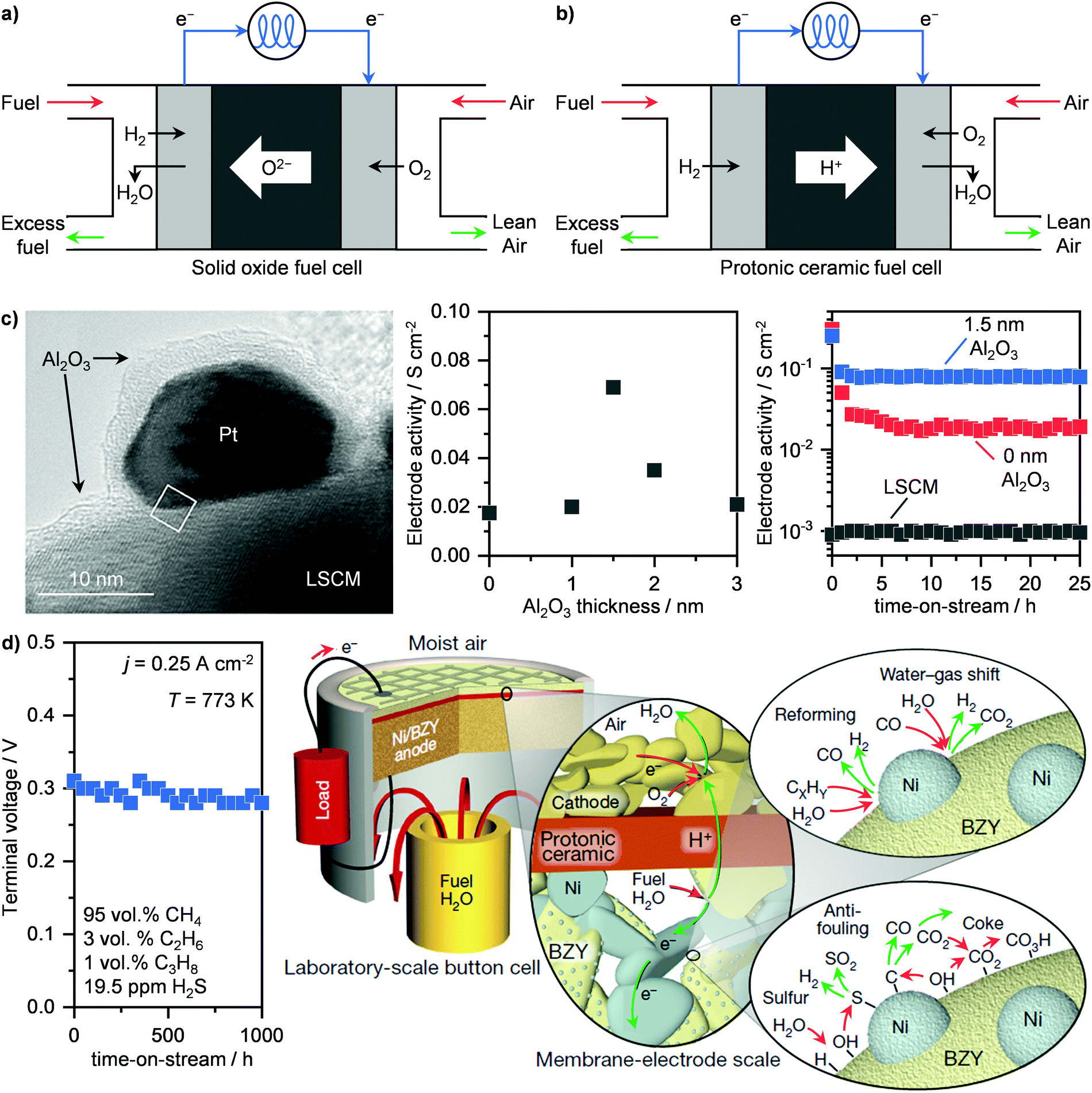
Fuel cells represent an alternative and newer technology compared to turbines for the electrochemical transformation of a feedstock into electricity. While several configurations exist, we focus our attention on solid oxide fuel cells (SOFCs) and protonic ceramic fuel cells (PCFCs) (Fig. 6a and b), which operate at 773–1273 K. Specifically, SOFCs, which generate electrical power in the range of 100 kW to 2 MW, can reach up to 65% energy efficiency, with very low emissions of greenhouse gases and long lifetime (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h).45,64 Their working principle is based on the separation of the oxidating and reducing semi-reactions, (Fig. 6a). In this way, the two electrodes are separated by an electrolyte, which allows for oxygen ions to diffuse towards the anode so that the reaction can occur and electricity is thus generated. Particularly, this energy is obtained by the combustion of H2 and CO that are obtained in situ from the reforming of CH4. Due to the chemical inertness of the simplest of hydrocarbons, high temperatures (873–1273 K), are typically required as well as the use of an electrocatalyst to aid the reaction.64 Consequently, a good electrode/electrolyte pair should be able to have (i) high thermal stability, (ii) good oxide-ion conductivity under relevant conditions and (iii) high activity for CH4 reforming and syngas combustion. Materials that possess such properties are yttria-stabilised zirconia and especially nickel-supported zirconia, due to the high reactivity of this late transition metal in CH4 reforming.65,66 However, due to the presence of contaminants in NG like H2S as well as the temperature-induced formation of carbonaceous species, these systems typically deactivate and require the development of sulphur- and carbon-resistant electrodes for SOFCs to be viable.66–68 In this field, important advances have been made by Nobel Laureate John Goodenough, who demonstrated that double perovskites Sr2Mg1−xMnxMoO6−δ (SMMO) can meet the requirement of long-term stability with tolerance to sulphur and carbon and show good electrocatalytic performance.67 Alternatively, perovskite La0.75Sr0.25Cr0.5Mn0.5O3 (LSCM) was shown to achieve stable operation under a sulphur-containing feed.66 Despite these remarkable achievements, the main disadvantage of oxide-based electrode remains the too low reactivity in CH4 activation.64 To solve this problem, Seo et al. have deposited platinum nanoparticles over LSCM, which boost the catalytic activity by 100-fold and still retain the high chemical stability, without incurring temperature-induced particle sintering.69 This was possible by encapsulating the platinum nanoparticle in a gas-permeable Al2O3 shell (Fig. 6c, left), which was obtained via atomic layer deposition over the platinum-supported LSCM. Despite being an insulator, the optimal thickness of 1.5 nm of the Al2O3 film reduces the impedance of the bare nanoparticles, thus promoting the catalytic activity (Fig. 6c, centre). In addition, the layer enhances the resistance to sulphur and coke and hinders particle sintering, allowing for stable operation (Fig. 6c, right).69 These results have motivated companies, including Mitsubishi Hitachi Power Systems Ltd, Ceres Power Plc, and FuelCell Energy Inc., to promote implementation of SOFCs in the energy production industry (Table 1).62,63 Still, there is a serious lack of internationally accepted codes and standards, which regulate the uniform and safe performance of these systems across countries. This situation hampers the widespread commercial application of fuel cells, and calls for policymakers to take action in order to propel the evolution of this energy sector.64
000 h).45,64 Their working principle is based on the separation of the oxidating and reducing semi-reactions, (Fig. 6a). In this way, the two electrodes are separated by an electrolyte, which allows for oxygen ions to diffuse towards the anode so that the reaction can occur and electricity is thus generated. Particularly, this energy is obtained by the combustion of H2 and CO that are obtained in situ from the reforming of CH4. Due to the chemical inertness of the simplest of hydrocarbons, high temperatures (873–1273 K), are typically required as well as the use of an electrocatalyst to aid the reaction.64 Consequently, a good electrode/electrolyte pair should be able to have (i) high thermal stability, (ii) good oxide-ion conductivity under relevant conditions and (iii) high activity for CH4 reforming and syngas combustion. Materials that possess such properties are yttria-stabilised zirconia and especially nickel-supported zirconia, due to the high reactivity of this late transition metal in CH4 reforming.65,66 However, due to the presence of contaminants in NG like H2S as well as the temperature-induced formation of carbonaceous species, these systems typically deactivate and require the development of sulphur- and carbon-resistant electrodes for SOFCs to be viable.66–68 In this field, important advances have been made by Nobel Laureate John Goodenough, who demonstrated that double perovskites Sr2Mg1−xMnxMoO6−δ (SMMO) can meet the requirement of long-term stability with tolerance to sulphur and carbon and show good electrocatalytic performance.67 Alternatively, perovskite La0.75Sr0.25Cr0.5Mn0.5O3 (LSCM) was shown to achieve stable operation under a sulphur-containing feed.66 Despite these remarkable achievements, the main disadvantage of oxide-based electrode remains the too low reactivity in CH4 activation.64 To solve this problem, Seo et al. have deposited platinum nanoparticles over LSCM, which boost the catalytic activity by 100-fold and still retain the high chemical stability, without incurring temperature-induced particle sintering.69 This was possible by encapsulating the platinum nanoparticle in a gas-permeable Al2O3 shell (Fig. 6c, left), which was obtained via atomic layer deposition over the platinum-supported LSCM. Despite being an insulator, the optimal thickness of 1.5 nm of the Al2O3 film reduces the impedance of the bare nanoparticles, thus promoting the catalytic activity (Fig. 6c, centre). In addition, the layer enhances the resistance to sulphur and coke and hinders particle sintering, allowing for stable operation (Fig. 6c, right).69 These results have motivated companies, including Mitsubishi Hitachi Power Systems Ltd, Ceres Power Plc, and FuelCell Energy Inc., to promote implementation of SOFCs in the energy production industry (Table 1).62,63 Still, there is a serious lack of internationally accepted codes and standards, which regulate the uniform and safe performance of these systems across countries. This situation hampers the widespread commercial application of fuel cells, and calls for policymakers to take action in order to propel the evolution of this energy sector.64
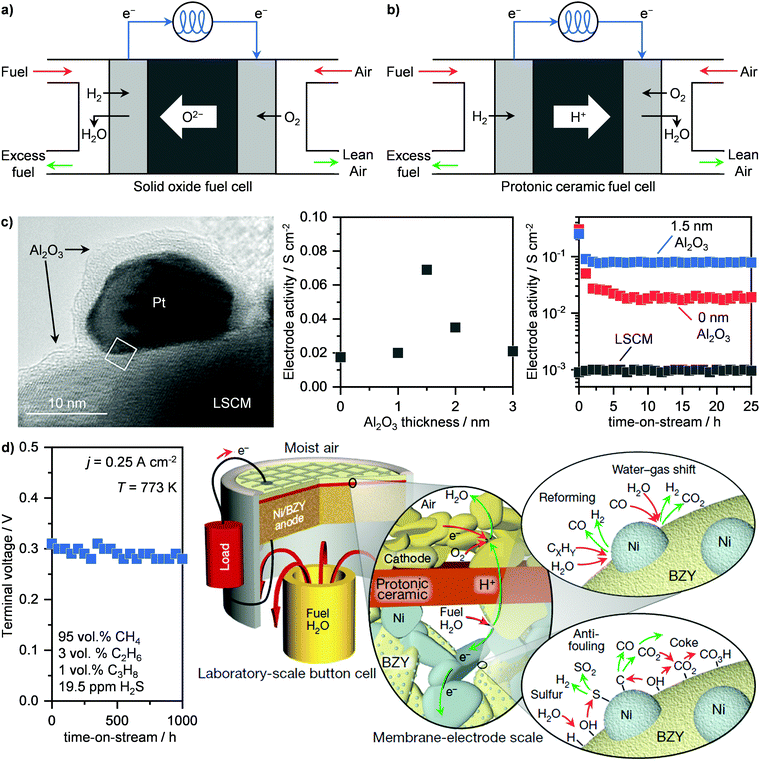 |
| | Fig. 6 Schematic representation of the working principles of (a) SOFCs and (b) PCFCs. (c) Transmission electron microscopy (left panel), activity as a function of Al2O3 thickness (middle panel) and time-on-stream (right panel) of an Al2O3-encapsulated platinum nanoparticle-based system supported on a lanthanum-based electrode (LSCM) for the electrocatalytic oxidation of methane via SOFC technology.69 Transmission electron micrograph reproduced with permission from ref. 69. Copyright (2020) ACS Publications. (d) Voltage as a function of time-on-stream of a nickel supported on an yttrium-doped barium zirconate (Ni/BZY) anode for the electrocatalytic oxidation of simulated sour NG (left panel) via PCFC technology. On the right panel, a schematic representation of the mechanism of hydrocarbon reforming, water–gas-shift reaction, and sulphur and carbon removal pathways, giving Ni/BZY anodes their unparalleled performance, is presented.71 Right scheme reproduced with permission from ref. 71. Copyright (2018) Springer Nature. | |
An intrinsic challenge of SOFCs is represented by the high temperature required to convert CH4. In addition to the thermal stress on the materials, the start-up and shutdown operations typically require weeks, even if high energy efficiency and stable performance are possible.64 Accordingly, this makes SOFCs highly valuable for steady-state applications, such as in power plants or for continuous electricity generation at a moderate-to-big gas well, but not for more discontinuous operations, including households or in containerised microplants that converts small gas volumes where and when it is needed. A potential solution might come from PCFCs. The main difference compared to SOFCs is in the electrolyte, which, instead of transferring oxygen anions towards the anode, it conducts protons to the cathode (Fig. 6b). This avoids water production at the anode and, consequently, fuel dilution. Accordingly, higher equilibrium potentials can be achieved, reducing considerably the operating temperature (≤823 K) compared to SOFCs.70–73 The pioneering work of Duan et al. has demonstrated that PCFCs could become assets in the energy production industry. Notably, they developed highperformance cathode– and anode–electrolyte pairs based on BaCo0.4Fe0.4Zr0.4Y0.1O3−δ and nickel supported on yttrium-doped barium zirconate (Ni/BZY), respectively, which allows PCFCs to convert CH4 as well as simulated NG at high currents (≤1.2 A cm−2) and power (≤0.35 W cm−2) at ≤773 K.70,71 They further demonstrated the stable operation of these systems, which in the case of Ni/BZY, could be maintained for 1000 h on stream in the presence of H2S (Fig. 6d, left).71 They ascribed this high sulphur and coke tolerance to the particular structure of Ni/BZY systems. As schematised in Fig. 6d, right, Ni nanoparticles are highly dispersed onto the BZY support/electrolyte. Such a high contact area allows combining two key properties: the catalytic reforming ability of Ni and the high capacity of BZY to dissociate water and provide surface hydroxyl and hydrogen species. This bifunctionality allows the carbon and H2S that adsorbs over Ni nanoparticles to be oxidised to CO2 and SO2, respectively, thus preventing poisoning.71 Although PCFCs show great promise for NG-to-power operations, especially at small scales, no commercial applications have been announced yet.
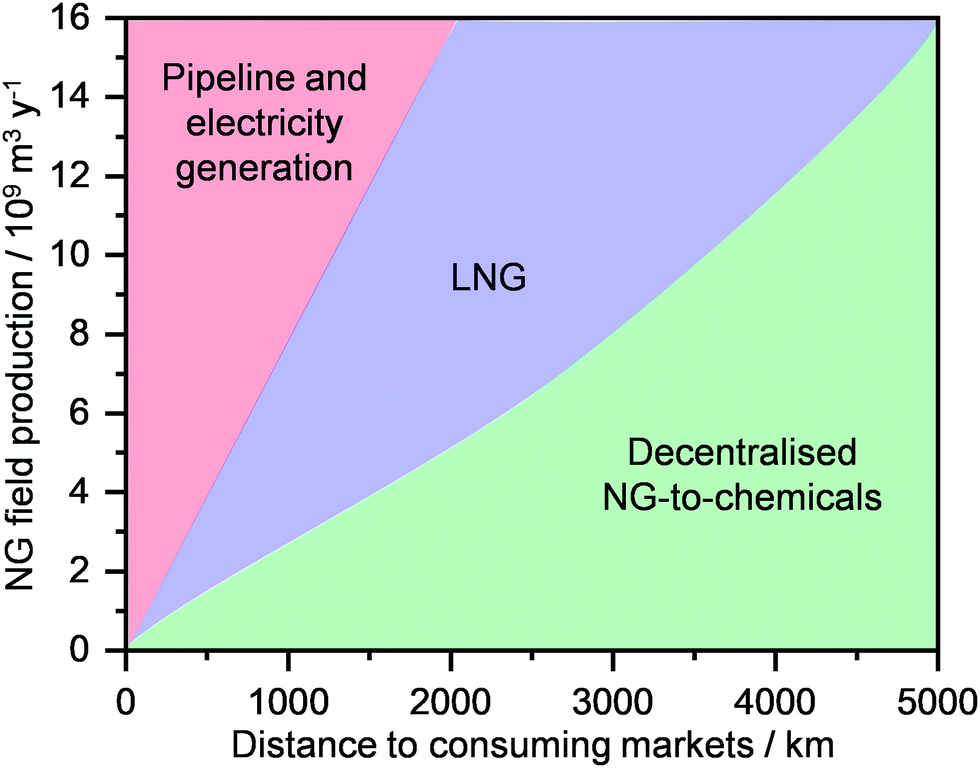
3. Technologies for decentralised LNG production
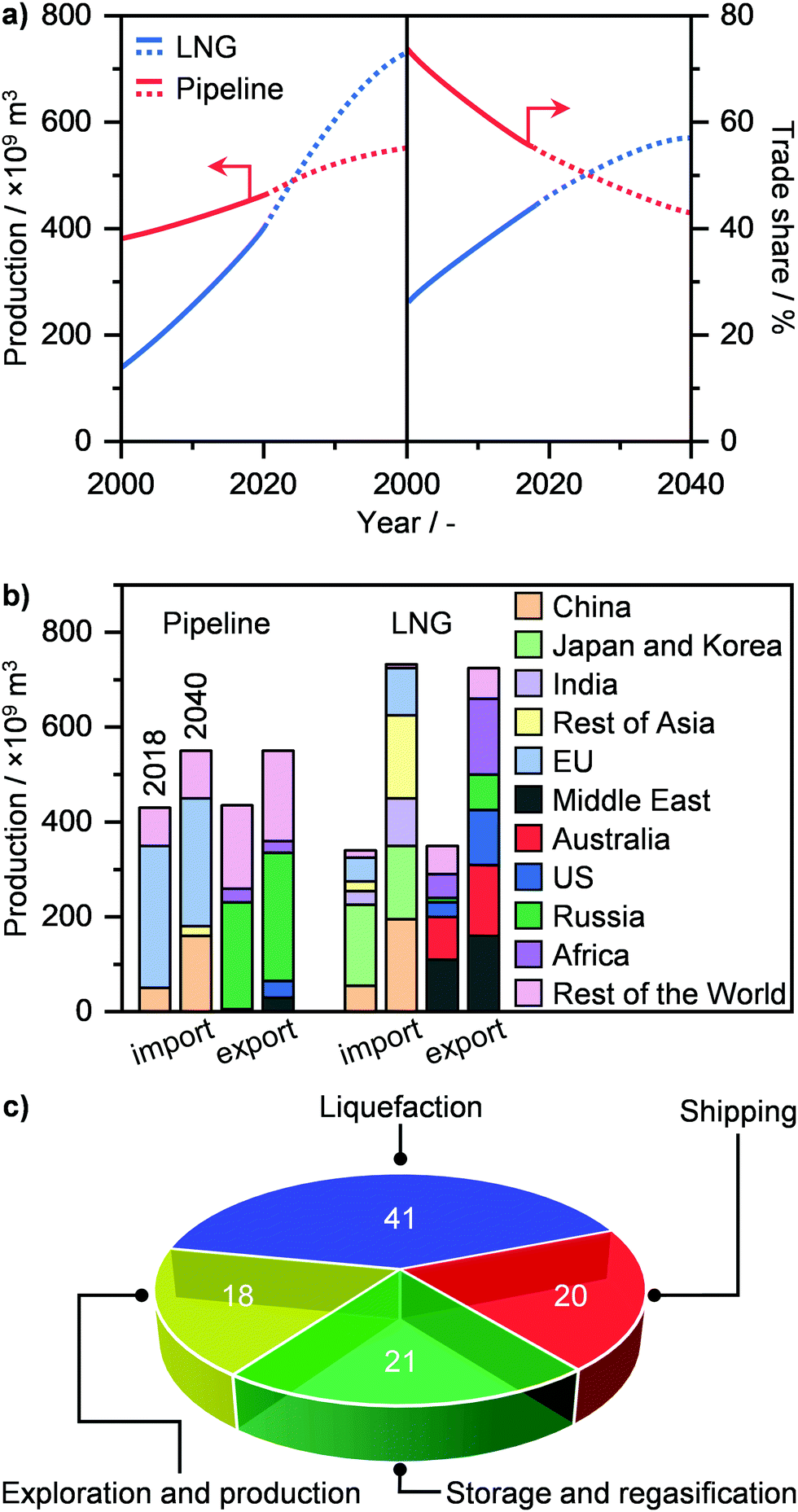
NG transport today mainly occurs via a pipeline, where the gas, pressurised at up to 80 bar, can travel as far as 4000 km from its point of extraction. Currently, the global gas pipeline network, which in 2019 transported approximately 450 trillion m3 of gas (Fig. 7a),2 is estimated to cumulatively extend to 900000 km around the globe.74 Still, this approach is extremely costly. While a typical onshore pipeline costs on average ≤3 million USD per km, offshore installations can reach up to 160 million USD per km.33 Accordingly, only a high volume of gas, shipped from a medium-to-big size well to a centralised plant, can justify such an investment. Furthermore, the lack of flexibility in the transport route, the supply dependence on long-term contracts and the fixed gas capacity due to the design pressure differential, render pipelines not suited for exploiting small-to-medium gas reserves in remote areas as well as serving dynamic consuming markets.34 Alternatively, this resource could be transported as LNG, which, given its liquid state, has the advantage of considerably reducing specific transportation costs, allowing much longer distances than typical pipelines by land and sea to be reached.32,34 As a matter of fact, the production of LNG has seen a strong growth in the last 20 years, and it is expected to surpass pipeline shipments and become a major means of gas transportation and trade within the next decade (Fig. 7a).2,3,22 By 2040, Middle East, Australia, Africa, and USA will be the main producers and exporters of LNG, while EU and predominantly Asia, i.e., China, India, Japan and Korea, will become major importers (Fig. 7b). Pipeline transportation will still see a moderate increase in the next 20 years, although it will be circumscribed to mainly Russia as the exporter and China and especially the EU as importers.2
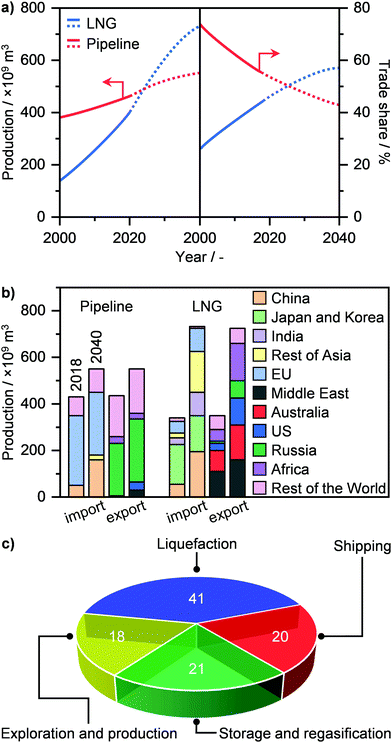 |
| | Fig. 7 (a) Annual global production (left panel) and trade share (right panel) of LNG and pipeline gas with projections (dotted line) up to 2040.2 (b) Total import and export production of LNG and pipeline gas in 2018 and in 2040 by region.2 (c) Cost breakdown of the LNG value chain.77 | |
Still, the liquefaction process as well as the storage and regasification of LNG represent the major source of costs in its value chain (Fig. 7c). This is due to the need for using cryogenic temperatures, reaching down to 110 K. Consequently, the considerable energy input needed as well as potential energy losses typically results in the need for large liquefaction facilities, with a production of over 1 million tons per year, in order to be economically competitive.34 In the last decade however, advances in process design, cryogenic liquefaction and storage, as well as in regasification operations and utilisation of cold energy have substantially increased the energy efficiency of production, storage, and employment of LNG. These breakthroughs represent the basis of the ongoing revolution in gas transportation (Fig. 7a), and are key to the development and widespread implementation of small-scale (≤100000 tons per year) plants for LNG generation and utilisation.34,75–77 These would not only support the flexible capacity to sustain increasingly dynamic markets but it would especially enable exploitation of small, scattered and stranded gas reserves on land and sea as well as to service end-users in remote and pipeline-scarce areas around the world.34 In this section, we cover an overview of the recent advances in the design of gas liquefaction and cold utilisation processes that pave the way for the establishment of small-scale decentralised LNG plants. In addition, we outline current efforts by companies and start-ups to implement on-site LNG production. For more details and broader discussion on these topics, we refer the reader to other studies in the literature.34,76–82
3.1. Gas liquefaction
Prior to liquefaction, NG has to be treated by separating propane and butane, which will be used as Liquefied Petroleum Gas (LPG) as well as water, CO2, H2S and C5+ hydrocarbons. Consequently, the remaining components are C2H6 and especially CH4, forming the bulk constituents of LNG, which require low temperatures (≈110 K) to be condensed.77 An ideal liquefaction process is composed of four main components: a heater/evaporator, a cooler/condenser, an expander or an expansion valve and a compressor to cycle the refrigerant (Fig. 8a, right). Accordingly, two refrigeration systems are possible: the reverse Rankine and the reverse Brayton cycles (Fig. 8a, left). While the former involves a change of state of the cooling medium, the second cycle utilises a gas as the refrigerant. In both cases, the working principle is to extract heat from NG by evaporating or raising the temperature of a coolant, which will then be compressed, cooled or condensed and finally expanded to continue the cycle (Fig. 8a). The two major technologies following the reverse Ranking and Brayton cycles are the single-mixed refrigerant (SMR) and the N2 expander (Fig. 8b), respectively.34,77 While the latter uses N2 as the refrigerant, the former typically implements a mixture of hydrocarbons, including CH4, C2H6, C3H8 and/or C4H10 often diluted in N2, as a cooling medium that can be at least in part condensed.77 The SMR technology as well as its variations, comprising different refrigerants as mixtures or as pure phases, finds wide implementation in LNG production at all scales.34,77 Although the simplicity of its configuration is an advantage at small scales due to low capital costs, this comes at the expense of low energy efficiency.83 This is due to the high compression power needed in this refrigeration cycle, which depends on the temperature gradient in the main LNG cryogenic heat exchanger.84,85 Accordingly, process optimisation is required in order to promote small-scale on- and offshore applications. To achieve this goal, several strategies have been proposed in the literature. While the replacement of expansion valves can achieve power savings of around 6%,86 structural alterations alone are not enough to significantly improve the liquefaction performance.87 Instead, optimization of the plant design, including compositional and/or flow changes of the refrigerant, have been identified as key factors for process improvement.84,87 In this regard, Lee et al. have shown that splitting the cooling medium in different phases across the process improves energy efficiency. In particular, after the partial evaporation of the refrigerant, the latter is divided into its gaseous and liquid phases (Fig. 9a, left).83 These are compressed separately by compressors and pumps, respectively, and are finally re-combined to serve as a single-mixed refrigerant in the main heat exchanger. This reduces the energy input needed from 7.5 to 5 kW h kmol−1 of LNG produced, which is comparable to the energy requirement of a propane-mixed refrigerant (C3MR) process, considered to be one of the most efficient liquefaction approaches.77,83 This corresponds to an energy saving of ca. 40% compared to a not optimised SMR process (Fig. 9a, right).83 An alternative method is the use of non-hydrocarbon-based condensable refrigerants, such as CO2 and hydrofluorocarbons. Among them the use of nitrous oxide (N2O) is highly promising, as it reaches specific energy input in between those of SMR and C3MR configurations, which renders this technology possibly suitable for offshore floating and floating, production, storage and offloading units.88,89 Still, mixed-refrigerant technologies have an intrinsic limitation. Since cooling medium losses are present, such leakages must be replaced by a makeup system. However, if the refrigerant is a hydrocarbon, CO2, or N2O that cannot be generated on-site, an external makeup import needs to be provided, which would make operations in remote areas logistically challenging.90 This could be approached by using the N2 expander process, which employs N2 as the cooling medium that can be extracted directly from air. However, this technology requires ≤4.5 times the energy input of a standard SMR unit.89,90 Therefore, process optimization is required in order for this configuration to be viable at the small-scale. Several approaches have been proposed to enhance the energy efficiency. The first strategy, constituting the so-called N2 double expander process, involves the introduction of a second expander, which can be configured in series or in parallel.91,92 This allows a 3.75-fold reduction in energy requirement compared to the simpler route, reaching an energetic input of ca. 8.75 kW h kmol−1 of LNG produced.92 However, this is still considerably higher than the energy needed for C3MR or optimised SMR processes.77,83 Another approach consists in the addition of a pre-cooling step with an alternative refrigerant. Accordingly, different pre-coolants, including CO2, C3H8 and difluoromethane (CH2F2; R410a), have been shown to obtain up to 12% energy savings (Fig. 9b, right).93,94 Nevertheless, despite these improvements, the major advantage of N2-based processes of not requiring an external makeup system would be lost, due to the need for replacing the pre-refrigerant. A solution to this problem has been proposed by Qyyum et al. that has demonstrated a highly efficient route based only on N2 as the cooling medium.92 The process employs 4 intermediately cooled compressors in series and a recuperator, which utilises a cold N2 stream to pre-cool a warmer N2 flow in a closed cycle (Fig. 9b, left). This strategy enables up to 24% energy savings compared to the double expander process (Fig. 9b, right) without the need for using additional cooling agent,92 showing great promise for potential implementation to generate LNG directly at remote and stranded gas reserves.
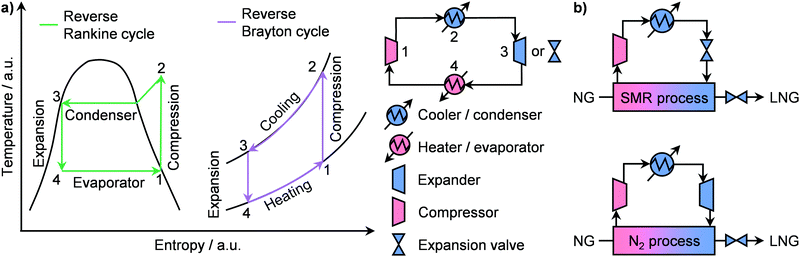 |
| | Fig. 8 (a) Temperature–entropy diagram of an ideal reverse Rankine and a reverse Brayton cycles (left panel). The corresponding flowsheet is presented on the right section of panel (a). (b) Schematic representation of a N2-expander and a single-mixed refrigerant (SMR)-based process for LNG production. | |
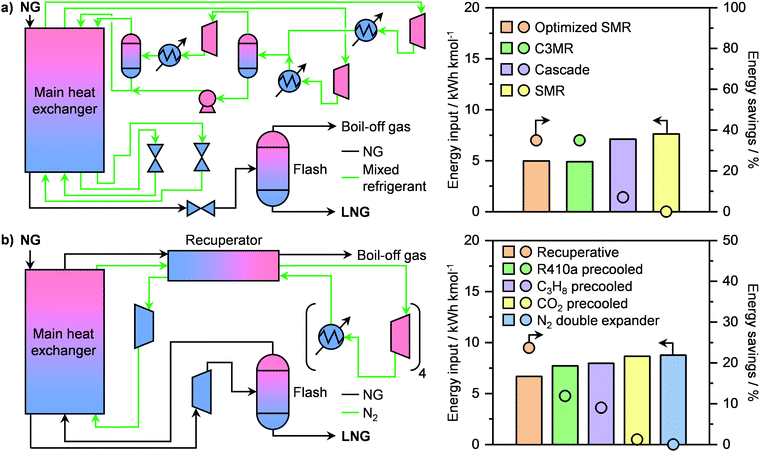 |
| | Fig. 9 (a) Flowsheet of an optimised single-mixed refrigerant (SMR) process for small-scale LNG liquefaction (left panel) and its energy input and savings in comparison to other double-phase refrigerant-based technologies (right panel).83 (b) Flowsheet of a closed-loop self-cooling recuperative N2 expander process for small-scale LNG liquefaction (left panel) and its energy input and savings in comparison to other N2 expander-based technologies (right panel).92 | |
These advancements have prompted several companies around the world to embark on different commercialisation projects (Table 2).62,95 Specifically, Expansion Energy LLC and Galileo Technologies S.A. have constructed several liquefaction microplants across the American continent based on hydrocarbon refrigerants. In particular, such processes can reach down to 10000 m3 per day of gas, equivalent to ca. 2500 tons per year of LNG, and can be containerised and used offshores.62 Baker Hughes plc, Chart Industries Inc. and Calvert Energy Group LLC have adopted mixed N2 and hydrocarbon refrigerant cycles for their small-scale plants, which can reach down to similar productivities to those above described (Table 2). Finally, Linde Cryostar SAS and Air Products Inc. have implemented modular and offshore applicable processes based on a N2 expansion cycle, which can reach down to 10000 tons per year of LNG.62,95 This shows great promise for small-scale LNG production facilities and calls for continued efforts to further expand this important sector. In addition, we anticipate that the hybridisation of these microplants will be pivotal. This will entail the integration of technologies, such as microturbines, fuels cells and/or solar power that have already shown great promise at medium-to-large scales,96–99 in order to further improve energy efficiency in view of boosting decentralised liquefaction applications.
Table 2 Companies and start-ups active in the field of decentralised NG-to-LNG technologies62,95
| Company |
Technology |
Feed range/×103 m3 STP d−1 |
Flexibility |
Status |
Country of origin |
| Expansion Energy |
Methane expansion cycle |
10–1200 |
• Tolerates variations in gas feed and contaminates |
Plants in operation |
USA |
| • Applicable offshore |
| Galileo Technologies |
Closed propane cycle |
20 |
• Tolerates variations in gas feed and contaminates |
Plants in operation in USA, Argentina, Australia, and Colombia |
Argentina |
| • Modular and containerised process |
| Baker Hughes |
Open N2 expansion and methane cycle, pre-cooled mixed refrigerant |
800–3000 |
• Tolerates variations in gas feed |
Plants in operation |
USA |
| Chart Industries |
Closed N2 expansion and mixed refrigerants cycles |
100–4000 |
• Tolerates variations in gas feed |
Plants in operation in USA and Canada |
USA |
| • Applicable offshore |
| Calvert Energy Group |
Closed N2 expansion and mixed refrigerants cycles |
10–1200 |
• Tolerates variations in gas feed |
Plants in operation in USA, Mexico, and Nigeria |
Belgium |
| • Applicable offshore |
| Linde Cryostar |
Closed N2 expansion cycle |
40–300 |
• Tolerates variations in gas feed |
Plants in operation |
France |
| • Applicable offshore |
| Air Products |
Closed N2 expansion cycle |
140–850 |
• Modular process |
Plants in operation in USA, Japan, and England |
USA |
3.2. Cold utilisation
Regasification is the last step before supplying NG to a consumer and represents the second major source of costs in the LNG value chain (Fig. 7c).77,81 Currently, around 100 plants are in operation around the world for large-scale LNG regasification.100 In view of the increasing efficiency, this step is typically combined with another thermal process that can provide a warm stream to vaporise LNG, which acts as a phase-changing refrigerant. Such configuration, which is schematised in Fig. 10a, is called a cold utilisation system.81,82,101 This strategy becomes even more critical for small-scale applications, which require high energy efficiencies in order to be economically viable.102 In this direction, two main strategies have been proposed in the literature. The first combines LNG regasification units with microturbines.82 Notably, the working fluid of an open Brayton cycle can be used, after the recuperator, to provide the heat necessary to evaporate LNG and reach an overall exergetic efficiency of ca. 27.5%.103 Alternatively, the second strategy envisages the combination with a micro CHP plant to generate both power and heat, which can be further complemented by cryogenic CO2 capture (Fig. 10b).102–106 In this way, the cold energy of LNG can be used not only to increase energy efficiency but also to reduce environmental impact. Through life-cycle-integrated thermo- and enviroeconomic analysis, it was shown that a small-scale LNG regasification-CHP combined process decreases costs by 19% compared to a cold utilisation-power generation system and reaches a 32% energy efficiency.106 Although the integration of a cryogenic CO2 capture step increased the costs by 7.5%, this configuration has a high estimated sustainability index.106 Despite these results, no commercial applications of LNG cold utilisation microplants have been announced yet. This calls for not only further technological developments in this area, but also the introduction of policies that can favour small-scale implementation. Besides, we anticipate that renewables will have a strong role in this sector. Specifically, we forecast that the integration of solar energy, which has already been successfully simulated at large scales,107–109 will become pivotal in the decentralised cold utilisation field. In a similar trend, novel kinds of applications could also boost implementation. Given the exponentially increasing need for information storage and processing in our society, data centres could make use of LNG cold energy for refrigeration of (super)computers, which can be installed on- as well as offshore, making them especially attractive for technologically expanding regions with a limited land area, such as Singapore.82
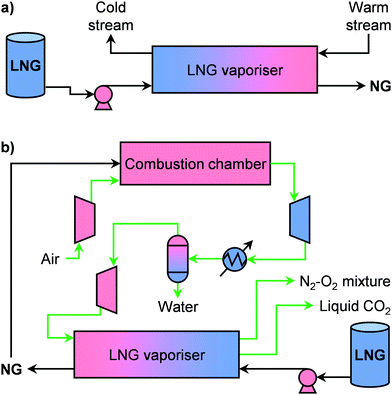 |
| | Fig. 10 (a) Scheme of a cold utilization plant that utilizes the heat of vaporisation of LNG to cool down a desired stream. (b) Flowsheet of a small-scale process for the generation of electricity and for cryogenic CO2 capture that uses LNG as fuel and cold source. | |
4. Technologies for decentralised chemicals and fuels production
Primary chemicals, including NH3, CH3OH, light olefins (C2H4, C3H6) and aromatics (C6H6, C7H8, C8H10; BTX), as well as fuels constitute the backbone of our society. However, their importance and growing role is one of the key “blind spots” in the global energy debate, likely due to the diversity and complexity of this sector in comparison to others, such as electricity and LNG.110 The worldwide manufacture of these commodities stood at almost 650 million tons in 2017 and their demand is expected to rise substantially in the next decades (Fig. 11a). Currently, around 76.5 million toe of NG are used to satisfy this production, which is equivalent to ca. 14% of the resource share. Still, the role of NG as feedstock is forecasted to grow considerably by 2050 (Fig. 11b), which will be primarily driven by the rise in NH3 and CH3OH demand.2,110 In 2017, the Southeast Asia and Oceania area was the world-leading region in primary chemicals production (Fig. 11c), and it is forecasted to maintain its dominant role in the next decades.110 On the other hand, Africa, the Middle East and North America are expected to undergo a strong local increase in the manufacture of NH3 and particularly CH3OH by 2050, which is likely to develop jointly with the predicted rise in these regions in the use of NG as the primary energy source (vide supraFig. 1c).2,110
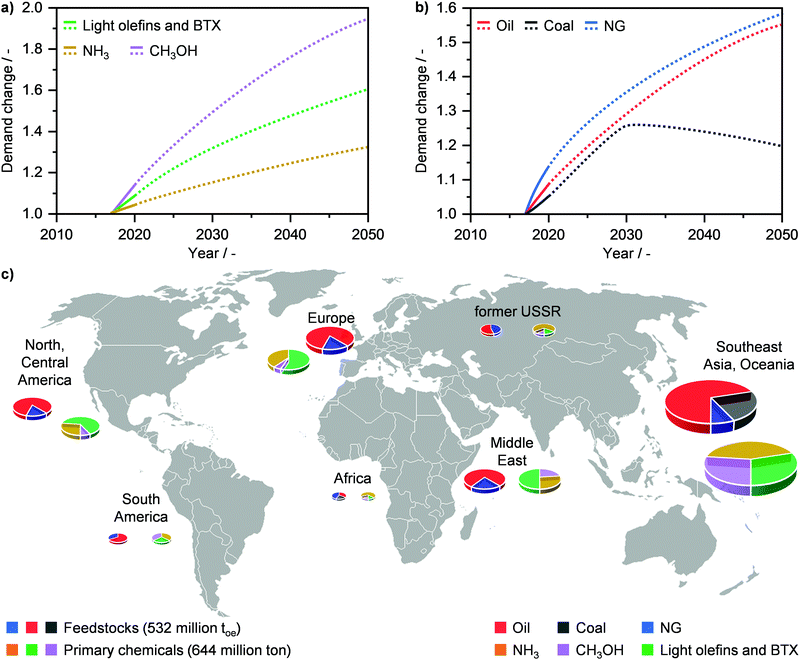 |
| | Fig. 11 Annual global demand of (a) primary chemicals, such as methanol (CH3OH), ammonia (NH3), and light olefins as well as benzene, toluene, xylenes (BTX), and (b) feedstocks in relation to their demand in 2017 with projections (dotted line) up to 2050. (c) Feedstock use and primary energy production by region in 2017.110 | |
In order to generate primary chemicals from NG, two main commercial processes are currently used: steam reforming and steam cracking (Fig. 12). The first converts CH4 in the presence of steam over a Ni-based catalyst into a mixture of carbon monoxide (CO) and H2, also known as syngas.28,30 This product can be further transformed over a Cu/ZnO/Al2O3 catalyst into CH3OH, a key precursor of light olefins and BTX, or into long-chain hydrocarbons, including fuels and waxes, via Fischer–Tropsch synthesis over iron- or cobalt-based systems.28 Furthermore, H2 from steam reforming can be further used in the Haber–Bosch process to generate NH3, and thus fertilizers, over an iron-based catalyst.31,36 On the other hand, steam cracking is a non-catalytic process that converts higher alkanes, i.e. ethane, liquid petroleum gas (e.g. C3H8 and/or C4H10), or light naphtha (C5+), into light olefins in the presence of steam.37,38 Still, their high-energy intensiveness makes these technologies economical only at large scales.38 An alternative strategy is the use of the direct catalytic process for alkane upgrading. This would boost efficiency, thus opening the possibility of NG valorisation into chemicals and fuels at small scales.7,36–38 In this direction, several routes have been intensively studied over the last decades (Fig. 12). These can be divided into two categories: non-oxidative and oxidative. The first targets the direct catalytic functionalisation of light alkanes into primary chemicals as light olefins and BTX in the absence of O2, which includes methane dehydroaromatisation and catalytic dehydrogenation of light alkanes (Fig. 12). These have the advantage of generating H2 as a valuable by-product, but they typically suffer from thermodynamic limitations.28,37 The second category, instead, utilises O2 to convert alkanes over a solid catalyst into olefins, oxygenates and other intermediates, and comprises oxidative dehydrogenation, partial oxidation, oxidative coupling and (oxy)halogenation routes (Fig. 12). This eases thermodynamic limitations by driving the reaction towards the formation of by-product H2O. However a major challenge is the generation of carbon oxides (COx; CO + CO2), which requires precise tuning of the properties of the catalytic material in order to hamper it.16,28,111 In this section, we cover an overview of the recent advances in catalyst and reactor design for efficient and direct alkane functionalisation via non-oxidative and oxidative pathways, which paves the way towards decentralised NG-to-chemicals microplants. In addition, we outline current efforts by companies and start-ups to implement on-site chemicals production. For more details and broader discussion on these topics, we refer the reader to other studies in the literature.16,28,37,111–117
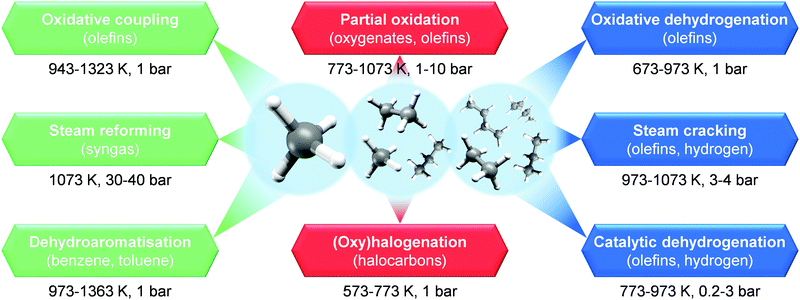 |
| | Fig. 12 Widely investigated routes in the last decades for the functionalization of methane (orange), C1–C3 (green), and C2–C5 (grey) alkanes. Target products of each upgrading approach are mentioned in brackets, while the typical operating conditions of these processes are shown on the bottom of each route. | |
4.1. Non-oxidative alkane upgrading routes
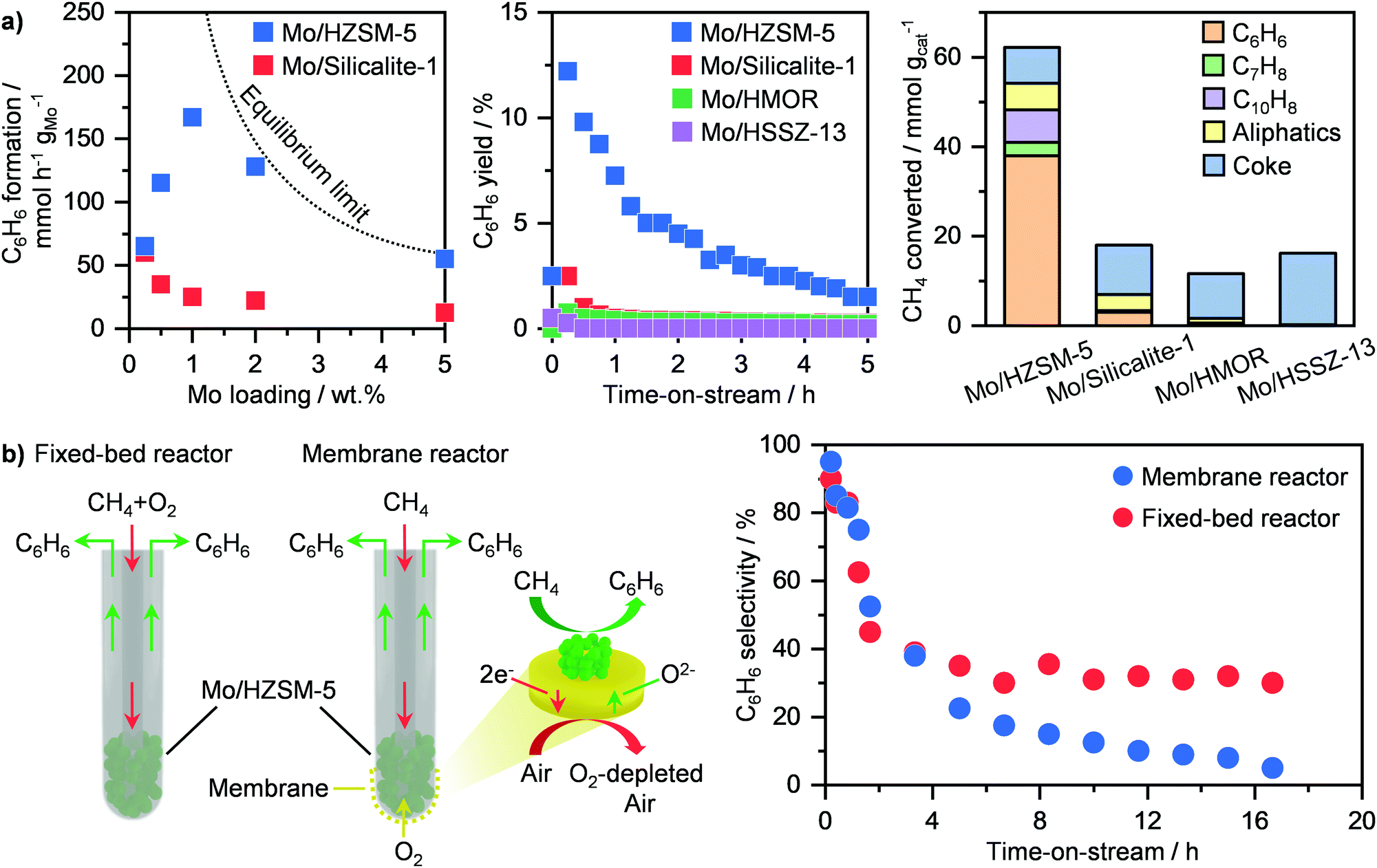
Methane dehydroaromatisation (MDA) represents one of the mainly investigated routes for the direct non-oxidative transformation of the lightest of hydrocarbons into valuable chemicals.31,117 This route comprises the reaction of CH4 over a solid catalyst to yield C6H6 and H2 at ≥973 K and atmospheric pressure. Among the several systems reported, molybdenum-based catalysts represent the most studied.116,117 Although different carriers have been investigated including Al2O3, SiO2, zeolites, HZSM-5 has been shown as the best support, achieving high C6H6 production rates (Fig. 13a, left).116,118 This stems from the bifunctional nature of this system. The zeolite, aside from anchoring the metal-based sites, provides a shape-selective environment that favours C6H6 formation, while molybdenum is responsible for CH4 activation.118 Still, there is a strong debate on the exact role and structure of the active Mo sites. In particular, earlier studies proposed that MoC3 clusters were the main active phase, which transforms CH4 into C6H6via generation of light hydrocarbon intermediates, such as C2H4.118–121 On the other hand, Hensen and co-workers have suggested partially reduced single-atom Mo sites stabilized within the zeolite framework to be responsible for the activation of CH4. Accordingly, radicals and primary coupled C2Hx fragments are formed and further react with polyaromatic species confined in the pores of the zeolite to generate C6H6, thus following a hydrocarbon-pool mechanism.122 Recently, Gascon et al. have provided evidence by means of advanced nuclear magnetic resonance spectroscopy that both MoC3- and hydrocarbon pool-driven CH4 conversion pathways co-exist during MDA.123 Consequently, these results have shed light on the induction period typically observed in this reaction (Fig. 13a, middle),118 which originates from the in situ CH4-driven carburisation of dispersed Mo–oxo species into partially or fully reduced Mo sites as well as from the building up of hydrocarbon species inside the zeolite pores for the auto-catalytic C6H6 generation route.122,123 In addition to the simplest of aromatics, other products, including toluene and aliphatic hydrocarbons can be generated (Fig. 13a, right). Still, a key challenge of this route is the severe catalyst deactivation (Fig. 13a, middle),118 which primarily hampers potential implementation. While sintering of Mo-based phases has been shown to contribute to activity losses,119,120 the accumulation of carbonaceous species over the surface and inside the pores of the zeolite is predominantly responsible for performance degradation.116–120,124,125 To improve catalyst lifetime, several strategies have been adopted, involving synthesis, optimization of reaction conditions and reactor design. The first tackles tuning of metal loading and bottom-up synthetic routes. While the use of a 2 wt% Mo/HZSM-5 compared to state-of-the-art 5 wt% Mo/HZSM-5 has been shown to improve regenerability,126 the use of a solvothermal approach with supercritical water–ethanol mixture as solvent enables the generation of a Mo-based system that can sustain a CH4 conversion of ca. 12% for 15 h on stream.127 In an alternative to synthesis, Hensen and co-workers demonstrated that rising the reaction pressure to 15 bar led not only to a tenfold increase in aromatics productivity, but especially to higher stability of Mo/HZSM-5 due to an improved coke hydrogenation under these conditions.128 Furthermore, they showed that another potential strategy is to co-feed O2 during MDA, which allows partial burning of the coke species, improving catalyst lifetime by a factor of 4.129 While the benefit in stability is apparent, this approach might result in the undesired combustion of aromatic products. To mitigate this possibility without compromising on the beneficial lifetime improvement, Cao et al. have proposed a reactor design in which a Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSFC) perovskite membrane allows for controlled oxygen transport (Fig. 13b, left). Accordingly, after initial equilibration, the Mo/HZSM-5 catalyst can reach a more stable performance for up to ca. 17 h on stream (Fig. 13b, right).130 Following a similar approach, electrochemical BaZrO3-based membrane can be used to achieve a high (ca. 11%) aromatic yield and a prolonged lifetime of 45 h over a Mo/HMCM-22 catalyst.131 This was possible thanks to the proton and oxide ion conductivity properties of the membrane that enables separation of H2, thus shifting the equilibrium towards products, and at the same time introduction of O2 to decrease coke generation. By performing process simulation analyses, this design was shown to reach a carbon efficiency of 80%, which is comparable to that obtained in Fischer–Tropsch plants.131 Accordingly, this MDA process would allow for high modularity and flexibility to adapt production to the size of the NG field, thus representing a major step in small scale gas-to-chemicals technologies.
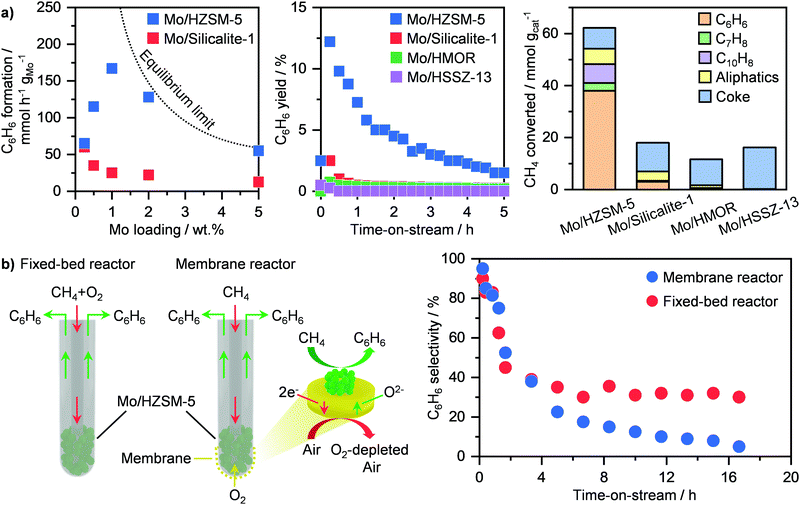 |
| | Fig. 13 (a) Rate of C6H6 formation (left), C6H6 yield as a function of time-on-stream (middle) and product distribution (right) over different Mo-based catalysts in methane dehydroaromatisation.118 (b) Schematic representation of a fixed-bed and a membrane reactor for oxygen-assisted methane dehydroaromatisation (left). The obtained C6H6 selectivity as a function of time-on-stream is reported on the right panel.130 | |
Catalytic dehydrogenation represents an alternative non-oxidative approach that targets the conversion of higher alkanes, including C2H6, C3H8, C4H10, to light olefins over a solid catalyst. Similarly to MDA, this route suffers from thermodynamic limitations, which become more stringent at decreasing the carbon chain length (Fig. 14a), and requires high temperatures (≥873 K) to achieve moderate conversions.37 Accordingly, major efforts have been directed towards catalytic dehydrogenation of butane and especially of propane. These represent highly relevant on-purpose routes for butylenes and propylene production, which found commercial application at medium-to-large scales as the Catofin and the Oleflex processes.37 While the former utilizes a CrOx/Al2O3-based system, the latter employs a Pt–Sn/Al2O3 catalyst to generate the desired olefin. Still, due to extensive coking as well as particle sintering, catalyst regeneration is required as often as every 12 minutes of reaction.37 Although this has been approached by using several reactors in parallel going through reaction-regeneration cycles in the commercial processes, such configuration would hinder application at small scales. Accordingly, considerable efforts have been spent in improving catalyst lifetime, especially of Pt-based systems. Given the structure sensitivity of this reaction,132,133 two main strategies that target the engineering of single atoms and nanoclusters of active platinum, respectively, have been investigated. Since single platinum atoms supported on metal oxides, such as cerium oxide (CeO2), undergo sintering during reaction,132 the first approach focuses on single-atom alloys (SAAs) to stabilize these highly active centres. Specifically, by dispersing platinum single atoms into copper, the olefin adsorption energy can be reduced, which favours product desorption, thus allowing high olefin selectivity, and prevents further dehydrogenation, leading to coking.134 Accordingly, bulk and supported Pt/Cu SAA catalysts were shown to reach high selectivity and maintain moderate olefin yields (≤11%) up to 120 h on stream in the dehydrogenation of propane and butane.134,135 Still, the most prominent example is represented by a modified SiO2-supported PtGa intermetallic system. This metal combination generates isolated (Pt1) and trimeric (Pt3) platinum sites (Fig. 14b, top left).136 Since the latter catalyses C–H and C–C bond scission, Nakaya et al. modified the catalyst with lead (Pb) in order to selectively poison Pt3 and only expose Pt1 centres (Fig. 14b, bottom left). Accordingly, they demonstrated that this system reaches full C3H6 selectivity at ca. 30% C3H8 conversion for at least 50 h on stream (Fig. 14b, right).136 The second strategy to improve performance and lifetime of platinum-based catalysts envisage the modification of Pt clusters with late or post-transition metals. Notably, Weckhuysen and co-workers have shown that by introducing Ga in Pt/Al2O3 and Sn in Pt/CeO2, a high (ca. 90%) C3H6 selectivity could be achieved at around 35% alkane conversion, which could be maintained even after several regeneration cycles.137,138 Recently, Liu et al. have shown that low-nuclearity PtSn clusters can be precisely dispersed within the 10MR pores of MFI zeolites by introducing potassium as the dopant prior to a multi-hydrogenation step (Fig. 14c, left).139,140 They demonstrated that by optimizing the H2 treatment time at 873 K to ≥12 h, low nuclearity PtSn clusters with Pt–O–Sn bridges are formed. By hydrogenating the system for 22 h, the obtained catalyst reached full C3H6 selectivity at around 35% C3H8 conversion and quasi stable behaviour (deactivation constant ca. 0.01 h−1; Fig. 14c, right).140 Following a comparable approach, Wang and co-workers developed a ligand-protected hydrogen reduction method for dispersing PtZn nanoclusters within silicalite-1 zeolites.141 In particular, they showed that the introduction of zinc promotes activity and stability, allowing a quasi stable 45% C3H8 conversion at 95% C3H6 for over 200 h on stream to be reached, with a deactivation constant of only 0.001 h−1. They argue that this represents the highest catalytic lifetime reported among any state-of-the-art Pt-based systems for propane dehydrogenation.141 Other catalyst families, including metal oxides and metal free systems, have been reported for propane dehydrogenation (Fig. 14d).37 Among these, chromium oxide-based materials represent the most studied, followed by vanadium and gallium oxide as well as nitrogen-doped and nitrogen-free carbons. Although Cr2O3 finds application in the Catofin process, these families are typically considerably less active, requiring around 3 times lower space velocity to reach comparable C3H6 yields (Fig. 14d), as well as less stable than Pt-based systems, with a deactivation constant ranging between 0.1 and 10 h−1.37
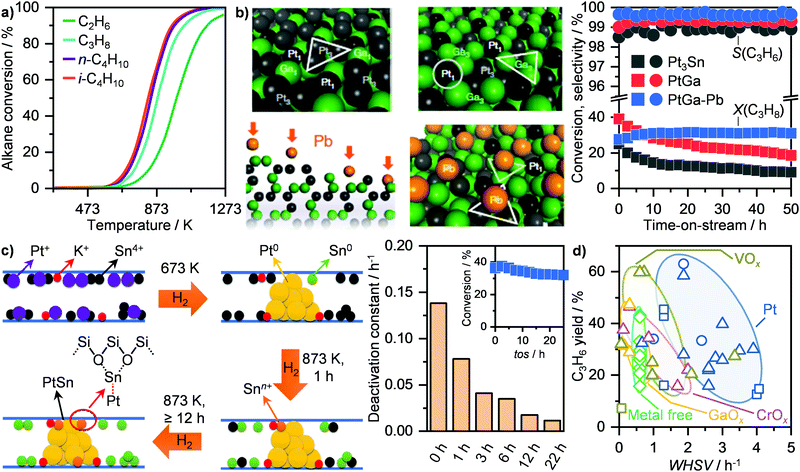 |
| | Fig. 14 (a) Equilibrium alkane conversion as a function of temperature in the dehydrogenation of light alkanes at atmospheric pressure.37 (b) Schematic representation of PtGa (top, left) and Pb-modified PtGa (bottom, left) intermetallic catalysts. Colour code: single Pt atoms (Pt1; grey), Pt trimers (Pt3; black), Ga (green), Pb (orange).136 Left scheme reproduced with permission from ref. 136. Copyright (2020) Springer Nature. Right panel, the corresponding C3H6 selectivity and C3H8 conversion, together with the performance of benchmark Pt3Sn and PtGa, are shown as a function of time.136 (c) Scheme of the structural evolution of K-PtSn/MFI during different reduction steps (left).140 Left scheme reproduced with permission from ref. 140. Copyright (2020) Springer Nature. The corresponding deactivation constant in propane dehydrogenation as a function of the pre-reduction time is shown on the left panel. The inset represents the alkane conversion as a function of time on stream (tos) over K-PtSn/MFI pre-reduced for 22 h.140 (d) C3H6 yield as a function of the weight hourly space velocity (WHSV) over different catalyst families in propane dehydrogenation.37 | |
Despite the important progress made in the field of catalytic methane dehydroaromatization and alkane dehydrogenation, currently disclosed commercial implementations of non-oxidative routes primarily focus on Fischer–Tropsch-based technologies for NG-to-chemical applications (Table 3).62,142 Accordingly, companies, such as Calvert Energy Group LLC, CompactGTL Limited, INFRA Technology Group, Emerging Fuels Technology Inc. and Greyrock Energy Inc., as well as the US Department of Energy have embarked in different projects that target the production of synthetic crude or diesel at small scales with modular, containerised, and/or offshore applicable processes (Table 3).62,143 Although steam reforming and Fischer–Tropsch processes do not scale down favourably, process intensification, in the form of catalyst and reactor design, has likely played a key role in making these small-scale applications economically viable.144 In particular, minimizing the CH4 selectivity by optimizing the nuclearity and structure of the Fischer–Tropsch catalyst as well as designing reactors capable to concentrate and utilise solar energy to sustain the endothermic steam reforming reaction have been shown as effective strategies to boost the efficiency of these technologies.145–147 Furthermore, another approach to foster implementation of these processes at small scales is the construction of a series of modular units. Accordingly, Boffito and co-workers have simulated that investment costs can be reduced by a factor of 2 for the case of Fischer–Tropsch microplants.148 Still, besides these advances, we envisage that implementation of efficient methane dehydroaromatisation and alkane dehydrogenation catalytic technologies could further boost the application of highly flexible microplants for on-site transformation of NG to valuable aromatics and olefins.
Table 3 Companies and start-ups active in the field of decentralised NG-to-chemicals technologies62,142
| Company |
Technology |
Product |
Feed range/×103 m3 STP d−1 |
Flexibility |
Status |
Country of origin |
| Calvert Energy group |
Catalytic plasma reformer |
Synthetic diesel |
5–3000 |
• Tolerates H2S, N2, CO2 impurities |
Operating plants in Argentina and North Dakota, USA |
Belgium |
| • Applicable offshore |
| CompactGTL |
Two-stage Fischer–Tropsch |
Synthetic crude and diesel |
400–4000 |
• Modular and containerised process |
Commercial plant in construction in Kazakhstan and Brazil |
UK |
| • Applicable offshore |
| INFRA Technology |
Two-stage Fischer–Tropsch |
Synthetic diesel |
30–3000 |
• Modular and containerised process |
Plant under demonstration in Texas, USA and Russia |
USA |
| • Applicable offshore |
| Emerging Fuels Technology |
Fischer–Tropsch |
Synthetic crude |
10–200 |
• Modular process |
Licensed technology to Juniper GTL and Fulcrum Bioenergy |
USA |
| Greyrock Energy |
Fischer–Tropsch |
Synthetic diesel |
10 |
• Modular process |
Demonstrated technology in Ohio, USA |
USA |
| Primus Green Energy |
Two-stage methanol synthesis |
Methanol and gasoline |
150–500 |
• Tolerates variations in gas composition and volume and sour gas |
Plant under demonstration in West Virginia, USA |
USA |
| GasTechno Energy & Fuels |
Partial oxidation |
Methanol, ethanol and formaldehyde |
8.5–300 |
• Modular and containerised process |
Operating plant in Michigan, USA |
USA |
| • Applicable offshore |
| Siluria Technologies |
Oxidative coupling |
Ethylene and propylene |
200–4000 |
• Tolerates lean, rich, and/or contaminated gas |
Plant under demonstration in Texas, USA |
USA |
| Sulzer-GTC Technology |
Bromination |
Aromatics |
300 |
• Adaptable to efficient synthesis of other petrochemicals |
Under demonstration |
USA |
| Grillo AG |
Sulfonation |
Methanesulfonic acid |
— |
— |
Licenced technology |
Germany |
4.2. Oxidative alkane upgrading routes
Oxidative routes use O2 in the feed to ease thermodynamic limitation. However, their major challenge is the formation of COx, which can only be kinetically hindered by catalyst and/or reactor design. In this perspective, decades of research have been conducted in the field of partial oxidation of methane, ethane, propane, oxidative coupling of methane, as well as oxidative dehydrogenation of ethane till pentane. The first route targets the formation of oxygenates such as alcohols, aldehydes, ethers, esters, and carboxylic acids.115 Although attractive yields (>40%) and selectivities (>80%) in the first set of reactions have been achieved using homogeneous systems and enzymes, the low space-time yields and high costs of these materials together with the significantly poorer performance of any reported heterogeneous catalyst (yields <1.5%) have hampered so far the potential industrialization of this route,115,149–154 and will, thus, not be analysed in detail in this review. On the other hand, methane oxidative coupling (OCM) has shown much greater promise for the efficient production of C2H4 over solid catalysts.28 Studies have observed that basicity represents a key catalyst property for selective CH4 acitvation.31,115 In particular, it is widely accepted that the OCM reaction proceeds via transformation of CH4 to methyl radicals over the catalyst surface. Once radicals desorb, they couple in the gas phase to generate C2H6, which undergoes catalytic oxidative dehydrogenation to form C2H4.116,155 Accordingly, three main catalyst families have been disclosed in the literature: (i) alkali metal-doped alkaline earth metal oxides, (ii) alkali or alkaline earth metal-doped lanthanide oxides, (iii) alkali and transition metal-doped manganese-based oxides.28,115 The first class is represented by Li-doped magnesium oxide (MgO), which, despite has a much simpler formulation compared to other systems, suffers from limited (<20%) C2H4 + C2H6 combined yields and poor stability.156,157 Prominent examples of the second family are Sr-doped lanthanum oxide (La2O3) and Na-promoted praseodymium oxide (Pr6O11), which can reach up to 20% combined yield and moderately stable operation.156 The third category comprises of more complex formulations, such as SiO2-supported sodium tungstate (Na2WO4)-doped manganese oxide (Mn2O3) catalysts. This represents the most active and stable family of catalytic systems reported so far, which can obtain close to 30% C2H4 + C2H6 combined yields (Fig. 15a).115,158–160 Despite the complexity of these multi-metallic materials, some studies have attempted to rationalize their superior performance. Interestingly, by evaluating SiO2-supported catalysts with Na, W, and Mn and combination thereof as active elements in OCM, it was shown that the blending of Na, W and Mn was necessary to achieve the highest performance (Fig. 15a). While the role of each metal is hard to clarify exactly due to their structural evolution under reaction conditions,160 some evidence and hypotheses have been put forward. Specifically, sodium has been proposed to have different key functions. Studies have shown that this alkali metal favours the transformation from amorphous to α-cristobalite SiO2, the latter being inert towards CH4 combustion.161 In addition, in situ X-ray diffraction (XRD) analyses have shown that this element converts into sodium peroxide (Na2O2) during reaction, which generates hydroxyl radicals that have been suggested as responsible for H abstraction from CH4.162,163 On the contrary, Trunschke and co-workers have recently shown by means of in situ XRD and operando Raman spectroscopy that the oxide of sodium (Na2O) is responsible for the OCM activity of the SiO2-supported Na2WO4-doped Mn2O3 catalyst. In addition, they shed light on the role of Mn and W, which act as oxygen donor and stabilising agents for the Na2O transient species, resulting in the observed high activity, selectivity and stability of this system.164 Still, despite these results, decades of research have so far failed to produce a catalyst that can reach the estimated commercial threshold of 30% C2H4 + C2H6 single-pass yield.28,115 A possible solution might come from reactor design. In particular, Cruellas et al. have simulated different configurations and found that by distributing the O2 feed along the reactor axial length a 3-fold increase in the C2H4 + C2H6 yield could be achieved compared to a fixed-bed design over a La2O3/CaO catalyst. The simulations have shown that this was possible by using a BSFC perovskite membrane, which has been demonstrated as highly relevant in MDA,130 reaching up to 60% C2H4 + C2H6 yield (Fig. 15b).165 Chemical looping represents an alternative approach, which consists in separating the methane activation and the catalyst re-oxidation semi-reactions (Fig. 15c). Accordingly, CH4 is converted into products over a catalyst that acts as oxygen-donor. The thus reduced system is then reoxidized in a separated step using a moving bed, closing the catalytic cycle (Fig. 15c, left).166 This strategy reaches higher selectivities at comparable conversion with respect to a fixed-bed reactor, as exemplified for the case of Na-doped Pr6O11 (Fig. 15c, right).156 Still, despite several experimental studies have been conducted over state-of-the art Mn-based systems, no reports of >30% C2H4 + C2H6 yields have been disclosed yet.166–168
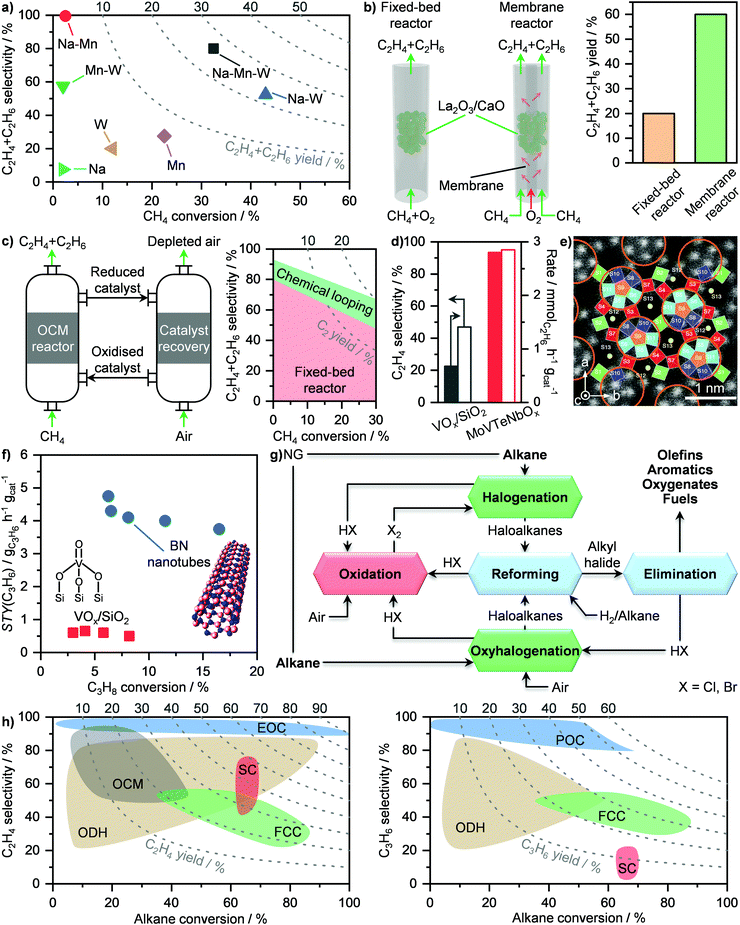 |
| | Fig. 15 (a) C2H4+C2H6 selectivity as a function of CH4 conversion over SiO2-supported Na, Mn, W-based catalysts in OCM.160 (b) Schematic representation of a fixed-bed and membrane reactor (left) and corresponding achievable C2H4+C2H6 yield (right) in catalytic OCM.165 (c) Scheme of a chemical loop-operating OCM reactor (left) and corresponding achievable C2H4+C2H6 selectivities as a function of CH4 conversion using as Na-doped Pr6O11 catalyst (right).156 (d) C2H4 selectivity and rate of C2H6 consumption in the ODH of ethane over VOx/SiO2 and MoVTeNbOx catalysts.170 (e) High-angle annular dark-field scanning transmission electron microscopy of a [001] oriented MoVTeNbOx crystal. Blue: Mo6+; orange: Nb5+; yellow: Te4+; cyan: Mo6+/V5+; green: Mo5+/V4+; red: Mo5+/6+/V4+/5+; orange circle: Nb-centred M6O11 clusters.171 Reproduced with permission from ref. 171. Copyright (2019) Springer Nature. (f) C3H6 space-time-yield (STY) as a function of C3H8 conversion over VOx/SiO2 and BN nanotubes.182 (g) Flowsheet of a halogen-based process for the upgrading of NG into chemicals and fuels. (h) Relative comparison of state-of-the-art performance (coloured areas) achieved across different routes for ethylene (left) and propylene (right) production. EOC: ethane oxychlorination; POC: propane oxychlorination; OCM: oxidative coupling of methane; ODH: oxidative dehydrogenation of light alkanes; SC: steam cracking; FCC: fluid catalytic cracking.215,219,225 | |
The catalytic oxidative dehydrogenation (ODH) of light alkanes, such as C2H6, C3H8 and C4H10, represents an interesting route for on-purpose generation of light olefins. Similar to OCM, a key challenge of this reaction is the formation of COx, which requires precise catalyst design to curb it.111–113 Historically, bulk and supported oxides of transition metals, including nickel, molybdenum and especially vanadium, as well as of rare earths, such as lanthanum, samarium and cerium, have been the most studied systems in the ODH of ethane.111 Notably, VOx/Al2O3 has shown a comparably high activity among this class of materials, thanks to the facile reducibility of this transition metal. Despite the efforts spent in doping strategies to improve performance, vanadium-based systems still suffer from low C2H4 selectivity (Fig. 15d, left), due to the low energy of vanadium-oxygen bonds that facilitates COx formation.111,115 Accordingly, two main approaches were investigated in the literature to tackle this challenge. The first comprises the use of halogens, such as fluorine, chlorine and bromine. Specifically, by introducing halogenating agents, including alkali and alkali earth halides as well as hydrochloric acid, the formation of metal (oxy)halides was detected, which was associated to an increase in C2H4 selectivity.111 A notable example is represented by a dysprosium–magnesium oxide-supported alkali metal chloride-based catalyst, which exhibited a high (ca. 95%) C2H4 selectivity at considerable C2H6 conversions (≤65%).169 Still, the stability of this material remains unknown, while the mechanism over this system has been only speculated to involve the formation of a transient molten hypochlorite phase that activates C–H bonds.111,169 Another strategy is the use of mixed metal oxides. In particular, bulk and supported systems based on the orthorhombic crystalline form of the oxide of molybdenum, vanadium, tellurium and niobium (MoVTeNbOx), also known as M1 phase, have been reported as among the best catalysts for ethane ODH, reaching considerably higher reactivity and C2H4 selectivity compared to archetypical supported vanadium oxide (Fig. 15d).115,170–173 The reason for the superior performance of these materials stems from their structure, which consists of Nb-pillars surrounded by edge and corner-sharing MoO6 and VO6 octahedra. Such units are connected by further metal–oxygen clusters forming hexagonal and heptagonal channels, which are partially occupied by tellurium ions (Fig. 15e).171,174 Thanks to the tight confinement, these micropores stabilise C–H activation transition states through van der Waals interactions and hinder O-insertion, limiting COx formation.170,175 While these systems are highly effective for ethylene generation, they show poorer olefin selectivity in the ODH of propane and butane.176 The higher reactivity of these substrates dictates different design criteria for optimal systems. Notably, metal carbides, such as multielement MAX phases, as well as metal-free materials, including carbon nanotubes and boron nitrides (BN), have been demonstrated as effective for the catalytic production of C3H6 and C4H8.177–182 In particular, Hermans and co-workers have reported BN nanotubes as a superior system in the ODH of propane. This catalyst reached around 80% C3H6 selectivity at relevant (ca. 15%) alkane conversion as well as up to 5 gC3H6 h−1 gcat−1, which is an order of magnitude higher C3H6 space-time-yield than conventional supported vanadium-based catalysts (Fig. 15f).182 Although surface passivation of BN is key in the ODH reaction,183,184 the mechanistic reasons for the observed superior performance are still debated. Specifically, Hermans and co-workers have hypothesised based on theoretical calculations that the high C3H6 selectivity stems from its formation in the gas phase. Thus, the role of the surface is to activate C3H8 into propyl radicals, which upon desorption propagate the reaction and further convert into the olefin.185 However, recent work has detected only methyl radicals over BN during ODH of propane by means of synchrotron vacuum ultraviolet photoioniasation mass spectroscopy, indicating that only formation of cracking products occurs via a gas-phase radical pathway while generation of C3H6 is surface bound.186 Similarly to OCM, chemical looping strategies has been investigated in these routes. While major efforts have been focused on ethane ODH, disclosing bulk as well as alkali- and transition metal-doped manganese-based and lanthanum–strontium–iron perovskite systems reaching up to 60% C2H4 yield at >90% olefin selectivity, recent work has also demonstrated that similarly selective behaviour could be obtained over archetypical VOx-based catalysts in propane ODH.166,187–189 Although studies in this sector have been so far limited, we anticipate that the combination of chemical looping with advanced catalyst design, could open further opportunities to improve performance.
An alternative approach consists in the functionalisation of alkanes with halogens, such as chlorine and bromine, which selectively converts these hydrocarbons under mild conditions of temperature (<800 K) and atmospheric pressure into alkyl halides, important building blocks from which chemicals and fuels can be obtained via hydrogen halide (HX, X = Cl, Br) elimination.7,16 Accordingly, the backbone of such process should comprise: (i) alkane halogenation into haloalkanes and (ii) elimination of HX to higher hydrocarbons or oxygenates (Fig. 15g). While the former step can be conducted non-catalytically as well as over solid catalysts, including metal oxyhalides, supported precious metals, sulphated metal oxides, and zeolites, halogenation inevitably results into the formation of polyhalogenated species.190,191 These compounds often act as catalyst poisons in the alkyl halide upgrading step, thus significantly contributing to any carbon and especially halogen loss.16,192 Consequently, technologies that selectively reform polyhalocarbons to alkyl halides are required for a process to be viable (Fig. 15g). Although few studies have been reported, two main reforming agents were investigated: H2 (hydrodehalogenation) and alkanes (reproportionation). While the latter reaction occurs non-catalytically,193,194 several catalysts, including SiO2-supported metal nanoparticles and epitaxially-directed iridium nanostructure over TiO2, have been shown to selectively (≤96%) reform dibromo- (CH2Br2) and dichloromethane (CH2Cl2) into the corresponding methyl halide at relevant conversions (≤25%).195–197 Still, a key challenge is represented by the low stability of these systems due to coking, sintering and/or halogenation,196,197 which calls for further developments in this field. The thus reformed polyhalocarbons and the alkyl halides from the halogenation step can be further upgraded via several catalytic routes, including carbonylation into acetic acid, hydrolysis into alcohols and ethers, and dehydrohalogenation into olefins (Fig. 15g).198–201 Among them, the coupling of methyl chloride (CH3Cl) and methyl bromide (CH3Br) into olefins and aromatics represent the most relevant.16 Catalysts for this reaction are predominantly based on ZSM-5 zeolites and molecular sieve SAPO-34, which have been reported in both protonic and metal-exchanged form.16,202–205 Although its mechanism is still contested, methyl halide coupling has been strongly linked to the methanol-to-olefin (MTO) pathway, since both routes have been proposed to follow a hydrocarbon-pool mechanism in which polyaromatic species confined in the pores of the zeolite generates the products.203 Despite these similarities, considerable distinctions have been observed. Notably, CH3OH generally reaches at least three times higher rates compared to CH3Cl and especially CH3Br coupling under comparable conditions, which has been assigned to differences in proton affinity.16,203 Furthermore, methyl halide transformation generates HX as by-product, which in addition to coking typically observed also in MTO, can further results into zeolite dealumination and thus more severe catalyst deactivation.203,206,207 Accordingly, no considerably active and stable catalyst has been reported today for these reactions. Notably, all these backbone steps releases HX, which needs to be recycled for any process to be viable. Two catalytic strategies have been investigated in the last decades. The first comprises the oxidation of HX into molecular halogen (X2) over a solid catalyst (Fig. 15g). Notably, several systems, including those based on the oxides of ruthenium (RuO2), cerium (CeO2), iridium (IrO2) as well as uranium (U3O8), have been disclosed as highly active and stable catalyst for the oxidation of HCl to Cl2, despite the strong thermodynamic limitations of the Deacon reaction.208–211 In spite of the apparent chemical similarities, the oxidation of HBr to Br2, which is much more exothermic and much less thermodynamically constrained, requires significantly different catalyst design strategies. As a matter of fact, Deacon catalysts are much more active in HBr oxidation, as testified by the 120–330 K drop in operating temperature compared to the chlorine-based reaction, while they suffered strong deactivation due to much more pronounced bulk halogenation of the material.16 Still, extensive analysis from Pérez-Ramírez and co-workers led to the discovery of novel materials, such as titanium oxide (TiO2) and europium oxybromide (EuOBr), which were found active and stable in HBr oxidation, thanks to their ability to limit bulk bromination.212,213 An alternative strategy for halogen recovery is alkane oxyhalogenation (Fig. 15g). The latter comprises the reaction of the hydrocarbon with O2 and HX over a solid catalyst. Several classes have been disclosed for the oxyhalogenation of CH4 and of higher alkanes, which includes metal oxides, oxyhalides, carbides, phosphates as well as supported metal nanoparticles.213–220 Still, depending on the alkane and halogen of choice, different catalyst design criteria must be considered. Specifically, alkane activation in oxybromination and in CH4 oxychlorination, which predominantly generate the corresponding alkyl halides, has been shown to follow a gas-phase mechanism. By combining operando prompt gamma activation analysis (PGAA) and photoelectron photoion coincidence (PEPICO) spectroscopies, it was demonstrated that the role of the catalyst is to generate in situ Cl2 or Br2, which then evolve in the gas phase to activate the hydrocarbon via a radical-driven pathway.220–223 Although formation of alkyl halide and concomitant polyhalogenation steps are thus material-independent, tuning of oxidizing properties remains key in order to tailor undesired formation of COx especially in the case of CH4 oxyhalogenation. Accordingly, systems, such as SiO2-supported palladium nanoparticles (Pd/SiO2), EuOBr, MgO-supported and bulk CeO2 as well as vanadyl pyrophosphate ((VO)2P2O7) allows to reach virtually up to full CH3X + CH2X2 (X = Cl, Br) combined selectivity at relevant (≤25%) alkane conversion in the oxychlorination and oxybromination of CH4.213,214,218,220,224 On the other hand, the activation of ethane and propane in oxychlorination, has been shown to occur exclusively over the catalyst surface, which generates selectively (≤98%) C2H4 and C3H6.221,222 In particular, it was shown, by combining operando PGAA and PEPICO spectroscopies with kinetics analyses and theoretical calculations, that the alkane is catalytically converted to a surface-bound alkyl chloride, which is further transformed into the corresponding olefin. Therefore, optimal catalysts should possess not only balanced redox properties to allow for COx suppression but also good ability to dehydrohalogenate the obtained alkyl chloride for efficient olefin generation. Accordingly, this understanding led to the discovery of catalysts such as europium oxychloride (EuOCl) as well as chromium phosphate (CrPO4) and nickel oxide-doped CeO2 (NiO–CeO2) that combined stable operation for up to 150 h on stream and high single-pass yields of C2H4 (≤90%) and C3H6 (≤55%), respectively, which are unparalleled by any existing olefin production technology (Fig. 15h).215,219,225
Different attempts have been disclosed for the commercialisation of direct oxidative routes for NG upgrading (Table 3).62,142 A prominent example is represented by Siluria Technologies Inc., which is the first company to bring catalytic oxidative coupling of methane towards implementation, with a plant currently under demonstration in Texas, USA.62,115 Although no details have been disclosed, GasTechno Energy & Fuels LLC has developed a modular and containerised process for the non-catalytic partial oxidation of light alkanes to alcohols and aldehydes, currently operating in Michigan, USA.62 Similarly, Sulzer-GTC Technology and Grillo AG have licensed and are demonstrating radical-based technologies for selective alkane transformation via bromine and sulphur chemistry to aromatics and platform molecule methanesulfonic acid, respectively.7,142 These can be considered as highly relevant model cases that should further boost the implementation of highly flexible microplants for on-site transformation of NG to valuable chemicals.
5. Discussion
The transition to a decentralised system for the efficient valorisation of scattered and stranded NG reserves is critical in order to secure the generation of E + EC, needed to sustain an ever-growing population and living standards at minimal-to-null environmental impact, as well as to boost the progressive shift towards renewable resources. This can be achieved by converting NG to electricity, LNG and chemicals in plants at small scales. Accordingly, we identified four key characteristics that a process should have for effective decentralised E + EC generation. The first is flexibility in terms of both feedstock tolerance and product variability. This allows not only conversion of a wide variety of feed compositions, but also rapid reaction to changes in product demand. Secondly, modularity of operations is another key feature that can provide with (i) faster time-to-market, (ii) scalable capacity, (iii) up to 40% lower capital costs compared to conventional processes, (iv) simplified manufacture and (v) improved safety.144 Last but not least, mobilisation as well as compactness represent other important properties, since they permit individual units, potentially installed within containers, to react to alterations in both feedstock availability and product demand across a wide geographical area. A process with these characteristics can be defined as a “plant-on-wheels” (Fig. 16). The latter could be successfully shipped, installed and operated directly at NG extraction sites (Fig. 16a), such as small, remote, on- and offshore gas reserves, which typically presents high logistic costs and insufficient feedstock supply for conventional processes.144,226 Furthermore, such units could be coupled with cold utilisation approaches at LNG regasification stations to further upgrade the obtained gas. In addition to the NG upgrading part, the plant-on-wheels could accommodate upstream and downstream processing sections (Fig. 16b). While upstream would involve NG sweetening and dehydration for which highly efficient absorbers have been disclosed,227,228 the downstream section would tackle heat exchange as well as separation and purification of products especially in chemicals producing plants-on-wheels. While applications at small scales have been shown for fine chemistry and pharmaceutical production,229 more efforts are required for developing miniaturised and effective unit operations for on-site NG-to-chemicals. Nevertheless, the core of any plant-on-wheels is represented by the technology that transforms NG to electricity, heat, LNG or chemical commodities. Based on the work of Mokhatab et al.32 and the herein presented analysis, we anticipate that the reserve size and distance between gas well and product consuming markets are key in selecting the most suitable NG upgrading process for commercially viable decentralised applications (Fig. 17). In particular, given the comparatively low energy efficiency of microturbines compared to fuel cells, these technologies could find implementation at medium-to-small and small-to-micro scales, respectively. In addition, due to the lack of electrical infrastructure in certain remote areas,42 decentralised gas-to-power routes are most likely to have important impact where electricity could be readily used. This can include NG extraction sites where electricity can be utilised to generate locally H2 and NH3 for fuels and fertilisers, respectively, as well as at LNG regasification units. Given that transportation of LNG compared to electricity is much more efficient over long distances,32,77 a plant-on-wheels for LNG production could target medium-to-small and comparably faraway NG wells (Fig. 17). While on-site cryogenic storage might present some logistic challenges at distant locations, the decentralised generation of easily liquefiable or liquid chemicals, such as olefins, BTX and/or fuels, represent a valid alternative. Such a plant-on-wheels could allow to unlock small, highly remote and stranded NG reserves (Fig. 17). Furthermore, potential synergies might arise from the combination of different plants-on-wheels. A valid example would be the coupling of LNG and chemicals producing microplants, which could utilize the energy produced by an exothermic catalytic reaction to extract heat from NG in order to efficiently produce both chemicals and LNG on-site. Still to confirm these predictions, life-cycle assessments at plant and planetary level would be required, taking into account the logistics of storage and/or distribution of the generated energy or energy carrier.
 |
| | Fig. 16 Schematic representation of (a) a plant-on-wheels installed at the NG extraction site and of (b) its interior for decentralised NG upgrading. | |
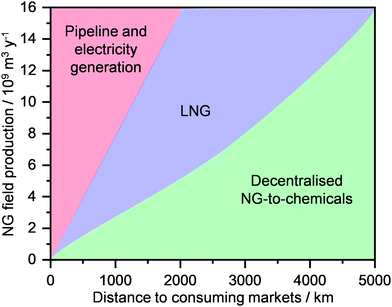 |
| | Fig. 17 Natural gas field production as a function of the distance to gas-consuming markets. Three areas are identified, indicating the optimal scenario for gas transportation and/or valorisation based on the production/distance-to-market ratio: pipeline and electricity generation (high production, small distance; red), liquefaction (medium-to-high production, medium distance; blue), on-site transformation into energy vectors and transportation thereof (small-to-medium production, large distance; green).32 | |
Currently, a natural gas grid is defined as the distribution of NG pipelines across a defined area.230 However, with pipeline transportation becoming less predominant (vide supraFig. 7) in an increasingly less centralised energy industry, the concept of NG grid should be redefined. In a decentralised world, we envision that a NG grid would consist of a highly dispersed and multilateral network of plants-on-wheels, which distributes NG-derived power, LNG and chemicals to urban and industrial areas (Fig. 18). Thanks to their modularity, the number of these microplants could be scaled in order to meet the size- and population-dependent demand of different regions. If widespread implemented, according to the International Energy Agency,231 such kind of energy solution could not only improve the use of NG resources but also help to accelerate the transition towards renewables and to reduce environmental impact. In addition, we anticipate that the proposed NG grid design could boost the economies of second and third world countries, where often stranded NG wells are located. This is of critical importance in order to improve quality of life and decrease energy poverty.42,232 Although applications have been demonstrated for distributed E + EC production (vide supra), policies at regional and national level are critical in order to promote their extensive implementation. This would entail the introduction of subsidies and grants to support commercial enterprises,232 as well as the establishment of new laws that target the improved utilisation of natural resources and the reduction of environmental impact. A valid example is represented by the Waste Prevention Rule of 2016 in the USA that incentivized operators to capture and use previously wasted NG, reducing methane emissions by over 35%.233 Furthermore, these domestic efforts should be complemented with international negotiations,234 in order to promote a cooperative response and give significant momentum to the implementation of a decentralised society at a global level.
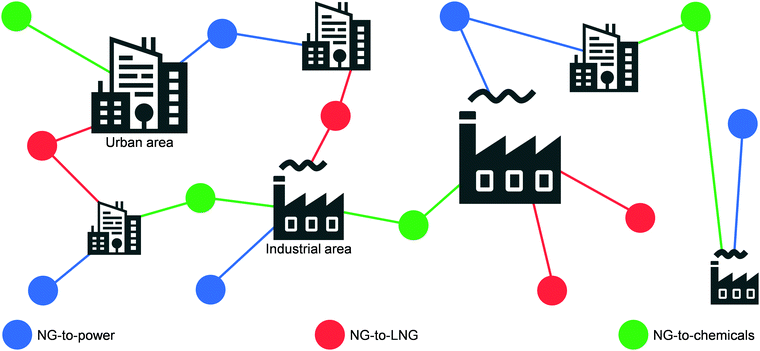 |
| | Fig. 18 Schematic representation of a natural gas grid that is based on a decentralised network to supply electricity, LNG, and chemicals to small, medium, and large-sized urban and industrial areas. | |
6. Conclusions and outlook
The implementation of a decentralised industry is key in order to secure the generation of energy and energy carriers that are required to sustain an ever-growing population and living standards at a minimal-to-null environmental impact. Furthermore, such a system would allow scattered and stranded NG reserves to be efficiently valorised as well as ultimately boosting the progressive shift towards renewable resources. Accordingly, in this review we have analysed the recent progress in view of upgrading NG into E + EC, such as electricity, LNG and chemicals, directly at the source of extraction. This integrative approach of combining these multidisciplinary fields is necessary to provide a unique and broad perspective that can foster potential synergies between researchers in these areas in view of developing effective and efficient technologies for on-site natural gas valorisation. Such vision could be achieved by using flexible, modular, compact and mobile microplants, which we have defined as plants-on-wheels, that can meet and rapidly adapt to the market- and region-dependent demand of desired E + EC. In this direction, important technologies for on-site NG-to-power are microturbines as well as solid oxide and protonic ceramic fuel cells. While the limited energy efficiency of the former represent the major barrier for a more widespread application, fuel cell-based routes allow electricity to be effectively generated from NG, but the limited flexibility of SOFCs and understanding of PCFCs have so far hindered implementation. On the other hand, important efforts have been made in gas liquefaction and LNG cold utilisation. While the former could be conducted at NG wells with a recuperated N2-based process, the second step could be performed both at the source of NG utilisation and extraction. Notably, the latter could be combined with electricity- and/or chemical-producing plant-on-wheels in order to use the cold LNG energy to boost efficiency as well as allow for on-site CO2 capture. Finally, the upgrading of NG to commodities for decentralised applications is at a far less developed stage compared to electricity and LNG production, likely due to the complexity of this sector. Still, important progress has been made to develop different catalytic routes, including methane dehydroaromatization, propane dehydrogenation, methane oxidative coupling and halogen-based alkane functionalization, for the direct conversion of light alkanes into valuable liquid and easily liquefiable chemicals, such olefins and aromatics. Still, besides increased efforts required to develop more selective and stable catalysts for these routes, key advances in the field of decentralised NG upgrading would come from life-cycle assessments at plant and planetary level. In particular, we anticipate that detailed techno-economic and life-cycle analyses, which can accurately compare the capital and operating costs as well as the logistics of different technologies for the on-site upgrading of natural gas into E + EC, will be essential in order to promote application-oriented advances and boost potential cooperations across fields. Accordingly, this would be a keystone for the implementation of a NG grid, which we envision would consist of a highly dispersed and multilateral network of plants-on-wheels, distributing NG-derived power, LNG and chemicals to urban and industrial areas. If widespread implemented, such kind of energy solution could not only improve the use of NG resources and accelerate the transition towards renewables but could also boost the economies of second and third world countries in order to improve quality of life and decrease energy poverty. Still, to become reality, we anticipate that this vision, in addition to technological advancements, would require the combined efforts of national policies as well as international negotiations. This is essential in order to promote a cooperative response and give significant momentum at both regional and global level in view of implementing a decentralised-based energy industry for a less environmentally impactful society.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by ETH Research Grant ETH-04 16-1. This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation.
References
- W. Steffen, K. Richardson, J. Rockstrom, S. E. Cornell, I. Fetzer, E. M. Bennett, R. Biggs, S. R. Carpenter, W. de Vries, C. A. de Wit, C. Folke, D. Gerten, J. Heinke, G. M. Mace, L. M. Persson, V. Ramanathan, B. Reyers and S. Sorlin, Science, 2015, 347, 1259855 CrossRef
 .
.
- World Energy Outlook, 2019, available at http://www.iea.org.
- BP Statistical Review of World Energy, 2020, available at: http://www.bp.com.
- P. Gerland, A. E. Raftery, H. Sevcikova, N. Li, D. Gu, T. Spoorenberg, L. Alkema, B. K. Fosdick, J. Chunn, N. Lalic, G. Bay, T. Buettner, G. K. Heilig and J. Wilmoth, Science, 2014, 346, 234–237 CrossRef CAS .
- Q. Schiermeier, Nature, 2014 DOI:10.1038/nature.2014.15956 .
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS .
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS .
- E. González-Roubaud, D. Pérez-Osorio and C. Prieto, Renewable Sustainable Energy Rev., 2017, 80, 133–148 CrossRef .
- G. Carpinelli, G. Celli, S. Mocci, F. Mottola, F. Pilo and D. Proto, IEEE Trans. Smart Grid, 2013, 4, 985–995 Search PubMed .
- P. N. Vennestrom, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef CAS .
- In-depth analysis in support of the commision communication COM(2018) 773: a clean planet for all: a European long-term strategic vision for a prosperous, modern, competitive and climate neutral economy, 2018, available at: https://ec.europa.eu.
- H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 10096–10115 CrossRef CAS .
- The future of natural gas, 2011, available at: http://energy.mit.edu.
-
R. Temple, The Genius of China: 3,000 Years of Science, Discovery, and Invention, Prion, 2007, pp. 1–288 Search PubMed .
-
G. Hammer, T. Lübcke, R. Kettner, M. R. Pillarella, H. Recknagel, A. Commichau, H.-J. Neumann and B. Paczynska-Lahme, Natural Gas, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Weinheim, 2012, pp. 740–792 Search PubMed .
- R. Lin, A. P. Amrute and J. Pérez-Ramírez, Chem. Rev., 2017, 117, 4182–4247 CrossRef CAS .
- D. Saha, H. A. Grappe, A. Chakraborty and G. Orkoulas, Chem. Rev., 2016, 116, 11436–11499 CrossRef CAS .
- R. D. Vidic, S. L. Brantley, J. M. Vandenbossche, D. Yoxtheimer and J. D. Abad, Science, 2013, 340, 1235009 CrossRef CAS .
- R. A. Kerr, Science, 2010, 328, 1624–1626 CrossRef CAS .
- X.-S. Li, C.-G. Xu, Y. Zhang, X.-K. Ruan, G. Li and Y. Wang, Appl. Energy, 2016, 172, 286–322 CrossRef CAS .
- B. K. Sovacool, Renewable Sustainable Energy Rev., 2014, 37, 249–264 CrossRef .
- ENI World Gas and Renewables Review 2019, available at: https://www.eni.com.
- A. C. Goldthau, J. Richert and S. Stetter, Rev. Policy Res., 2020 DOI:10.1111/ropr.12387 .
- M. Melikoglu, Renewable Sustainable Energy Rev., 2014, 37, 460–468 CrossRef .
- Ş. Merey, J. Nat. Gas Sci. Eng., 2016, 35, 1167–1179 CrossRef .
- K. C. Hester and P. G. Brewer, Annu. Rev. Mar. Sci., 2009, 1, 303–327 CrossRef .
- J. B. Klauda and S. I. Sandler, Energy Fuels, 2005, 19, 459–470 CrossRef CAS .
- R. Horn and R. Schlögl, Catal. Lett., 2014, 145, 23–39 CrossRef .
- C. Hammond, S. Conrad and I. Hermans, ChemSusChem, 2012, 5, 1668–1686 CrossRef CAS .
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS .
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS .
-
S. Mokhatab, J. Y. Mak, J. V. Valappil and D. A. Wood, Handbook of Liquefied Natural Gas, Elsevier, Oxford, 2014, pp. 1–591 Search PubMed .
- Global Energy Mornitor, Oil and Gas Pipeline Construction Costs https://www.gem.wiki/Oil_and_Gas_Pipeline_Construction_Costs, accessed November 01st, 2020, 10:00 GMT.
- J. Zhang, H. Meerman, R. Benders and A. Faaij, Appl. Therm. Eng., 2020, 166, 114736 CrossRef .
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS .
- E. V. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Catal. Sci. Technol., 2017, 7, 366–381 RSC .
- J. J. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS .
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS .
- R. Khalilpour and I. A. Karimi, Energy, 2012, 40, 317–328 CrossRef CAS .
- J. Tollefson, Nature, 2016 DOI:10.1038/nature.2016.19141 .
-
J. Jensen, Energy Storage, 1980, pp. 5–9 DOI:10.1016/b978-0-408-01225-6.50006-7 .
- P. Alstone, D. Gershenson and D. M. Kammen, Nat. Clim. Change, 2015, 5, 305–314 CrossRef .
-
M. A. R. do Nascimento, L. de E. C. dos Santos, E. E. Batista-Gomes, F. L. Goulart, E. I. Gutirrez Velsques and R. Alexis-Miranda, Progress in Gas Turbine Performance, 2013, ch. 5 Search PubMed .
- A. Choudhury, H. Chandra and A. Arora, Renewable Sustainable Energy Rev., 2013, 20, 430–442 CrossRef CAS .
- H. Wasajja, R. E. F. Lindeboom, J. B. van Lier and P. V. Aravind, Fuel Process. Technol., 2020, 197, 106215 CrossRef CAS .
- G. Xiao, T. Yang, H. Liu, D. Ni, M. L. Ferrari, M. Li, Z. Luo, K. Cen and M. Ni, Appl. Energy, 2017, 197, 83–99 CrossRef .
- K. A. Al-attab and Z. A. Zainal, Appl. Energy, 2015, 138, 474–487 CrossRef CAS .
- H.-I. Ji, J.-H. Lee, J.-W. Son, K. J. Yoon, S. Yang and B.-K. Kim, J. Korean Ceram. Soc., 2020, 57, 480–494 CrossRef CAS .
-
C. Burger and J. Weinmann, The Decentralized Energy Revolution, Palgrave MacMillan, London, 2013, pp. 1–221 Search PubMed .
- C. F. McDonald and C. Rodgers, Appl. Therm. Eng., 2008, 28, 60–74 CrossRef CAS .
- L. Aichmayer, J. Spelling, B. Laumert and T. Fransson, J. Eng. Gas Turbines Power, 2013, 135, 113001 CrossRef .
- J. Chen, G. Xiao, M. L. Ferrari, T. Yang, M. Ni and K. Cen, Renewable Energy, 2020, 154, 187–200 CrossRef .
- A. Mohammadi and M. Mehrpooya, J. Cleaner Prod., 2016, 139, 372–383 CrossRef .
- S. Saib, A. Gherbi, R. Bayindir and A. Kaabeche, Arabian J. Sci. Eng., 2019, 45, 1553–1566 CrossRef .
- U.S. Department of Energy, Combined Heat and Power Report, 2016, available at: https://www.energy.gov.
- M. M. Maghanki, B. Ghobadian, G. Najafi and R. J. Galogah, Renewable Sustainable Energy Rev., 2013, 28, 510–524 CrossRef .
- K. Thu, B. B. Saha, K. J. Chua and T. D. Bui, Appl. Energy, 2016, 178, 600–608 CrossRef .
- W. P. J. Visser, S. A. Shakariyants and M. Oostveen, J. Eng. Gas Turbines Power, 2011, 133, 042301 CrossRef .
- J. M. Clark and E. J. Wolfe, Nav. Eng. J., 1967, 79, 103–106 CrossRef .
- R. W. Moss, A. P. Roskilly and S. K. Nanda, Appl. Energy, 2005, 80, 169–185 CrossRef .
-
E. Agelidou, T. Monz, A. Huber and M. Aigner, Proceedings of ASME Turbo Expo 2017: Turbomachinery Technical Conference and Exposition, 2017, p. 64490 DOI:10.1115/GT2017-64490.
- The World Bank, Global Gas Flaring Reduction Partnership, Technology Overview Report, 2018, available at: http://pubdocs.worldbank.org.
- Fortune Business Insights, https://www.globenewswire.com/news-release/2019/12/16/1960725/0/en/Solid-Oxide-Fuel-Cell-Market-to-reach-USD-2-00-Billion-by-2026-Growing-Need-to-Reduce-GHG-Emissions-to-Boost-the-Market-Fortune-Business-Insights.html, accessed November 01st, 2020, 10:00 GMT.
- K. Al-Khori, Y. Bicer and M. Koç, J. Cleaner Prod., 2020, 245, 118924 CrossRef .
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828–1847 CrossRef CAS .
- S. Tao and J. T. Irvine, Nat. Mater., 2003, 2, 320–323 CrossRef CAS .
- Y. H. Huang, R. I. Dass, Z. L. Xing and J. B. Goodenough, Science, 2006, 312, 254–257 CrossRef CAS .
- P. Boldrin, E. Ruiz-Trejo, J. Mermelstein, J. M. Bermudez Menendez, T. Rami Rez Reina and N. P. Brandon, Chem. Rev., 2016, 116, 13633–13684 CrossRef CAS .
- J. Seo, N. Tsvetkov, S. J. Jeong, Y. Yoo, S. Ji, J. H. Kim, J. K. Kang and W. Jung, ACS Appl. Mater. Interfaces, 2020, 12, 4405–4413 CrossRef CAS .
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS .
- C. Duan, R. J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. J. Crumlin, D. Hook, R. Braun, N. P. Sullivan and R. O'Hayre, Nature, 2018, 557, 217–222 CrossRef CAS .
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O’Hayre, Nat. Energy, 2019, 4, 230–240 CrossRef CAS .
- S. Choi, T. C. Davenport and S. M. Haile, Energy Environ. Sci., 2019, 12, 206–215 RSC .
- Gaznat Report on Natural Gas, 2017, available at: https://www.gaznat.ch.
- Air Products, LNG Technology Report, 2018, available at: https://www.gastechevent.com.
- L. Castillo and C. A. Dorao, J. Nat. Gas Sci. Eng., 2010, 2, 302–309 CrossRef .
- W. Lim, K. Choi and I. Moon, Ind. Eng. Chem. Res., 2013, 52, 3065–3088 CrossRef CAS .
- M. S. Khan, I. A. Karimi and D. A. Wood, J. Nat. Gas Sci. Eng., 2017, 45, 165–188 CrossRef .
- M. A. Qyyum, K. Qadeer and M. Lee, Ind. Eng. Chem. Res., 2017, 57, 5819–5844 CrossRef .
- T. He, I. A. Karimi and Y. Ju, Chem. Eng. Res. Des., 2018, 132, 89–114 CrossRef CAS .
- B. B. Kanbur, L. Xiang, S. Dubey, F. H. Choo and F. Duan, Renewable Sustainable Energy Rev., 2017, 79, 1171–1188 CrossRef .
- T. He, Z. R. Chong, J. Zheng, Y. Ju and P. Linga, Energy, 2019, 170, 557–568 CrossRef .
- S. Lee, N. V. D. Long and M. Lee, Ind. Eng. Chem. Res., 2012, 51, 10021–10030 CrossRef CAS .
- M. A. Qyyum, N. V. D. Long, L. Q. Minh and M. Lee, Cryogenics, 2018, 89, 131–140 CrossRef .
- M. Wang, R. Khalilpour and A. Abbas, Energy Convers. Manage., 2014, 88, 947–961 CrossRef CAS .
- M. F. M. Fahmy, H. I. Nabih and M. El-Nigeily, Energy Convers. Manage., 2016, 112, 308–318 CrossRef CAS .
- A. Vatani, M. Mehrpooya and A. Palizdar, Energy Convers. Manage., 2014, 78, 720–737 CrossRef .
-
A. O. Fredheim and P. Paurola, US Pat., US7386996B2, 2008 Search PubMed .
- C.-J. Lee, K. Song, S. Shin, Y. Lim and C. Han, Ind. Eng. Chem. Res., 2015, 54, 11106–11112 CrossRef CAS .
-
T. Kohler, M. Bruentrup, D. R. Key and T. Edvardsson, Choose the best refrigeration technology for small-scale LNG production, 2014, available at: https://www.linde-engineering.com/en/images/41469_ePrint.pdf_tcm19-129829.pdf.
- T. He and Y. Ju, Energy, 2014, 75, 349–359 CrossRef .
- M. Abdul Qyyum, K. Qadeer and M. Lee, ACS Sustainable Chem. Eng., 2018, 6, 5021–5033 CrossRef CAS .
- M. S. Khan, S. Lee, M. Hasan and M. Lee, J. Nat. Gas Sci. Eng., 2014, 18, 263–273 CrossRef CAS .
- T. B. He and Y. L. Ju, Cryogenics, 2014, 61, 111–119 CrossRef CAS .
- Air Products, Technology Report for Small-Scale LNG Applications, 2019, available at: https://www.airproducts.com/.
- M. Mehrpooya, B. Ghorbani and F. K. Bahnamiri, J. Cleaner Prod., 2020, 262, 121401 CrossRef CAS .
- F. A. Boyaghchi and A. Sohbatloo, Energy Convers. Manage., 2018, 162, 77–89 CrossRef .
- B. Ghorbani, M. Mehrpooya and E. Omid, Energy Convers. Manage., 2020, 207, 112512 CrossRef CAS .
- M. Mehrpooya, Energy, 2016, 111, 468–483 CrossRef CAS .
- A. Atienza-Márquez, J. C. Bruno and A. Coronas, Appl. Therm. Eng., 2018, 132, 463–478 CrossRef .
-
T. Sung and K. C. Kim, Energy
Solutions to Combat Global Warming, 2017, ch. 3, vol. 33, pp. 47–66 Search PubMed .
- B. B. Kanbur, L. Xiang, S. Dubey, F. H. Choo and F. Duan, Energy Convers. Manage., 2019, 181, 507–518 CrossRef CAS .
- B. B. Kanbur, X. Liming, S. Dubey, C. F. Hoong and F. Duan, Energy Procedia, 2017, 143, 680–685 CrossRef .
- B. B. Kanbur, L. Xiang, S. Dubey, F. H. Choo and F. Duan, Energy, 2017, 129, 171–184 CrossRef .
- B. B. Kanbur, L. Xiang, S. Dubey, F. H. Choo and F. Duan, Appl. Energy, 2017, 204, 1148–1162 CrossRef CAS .
- B. B. Kanbur, L. Xiang, S. Dubey, F. H. Choo and F. Duan, Int, J. Energy Res., 2019, 43, 4104–4126 CrossRef CAS .
- M. Mehrpooya and M. M. M. Sharifzadeh, Appl. Therm. Eng., 2017, 114, 1090–1104 CrossRef CAS .
- M. Mehrpooya, M. M. Moftakhari Sharifzadeh and M. A. Rosen, Energy, 2016, 95, 324–345 CrossRef CAS .
- B. Ghorbani, K. B. Mahyari, M. Mehrpooya and M.-H. Hamedi, Renewable Energy, 2020, 148, 1227–1243 CrossRef .
- International Energy Agency, The Future of Petrochemicals Report, 2018, available at: https://www.iea.org.
- C. A. Gärtner, A. C. van Veen and J. A. Lercher, ChemCatChem, 2013, 5, 3196–3217 CrossRef .
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS .
- C. A. Carrero, R. Schlögl, I. E. Wachs and R. Schomäcker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS .
- F. Cavani, Catal. Today, 2010, 157, 8–15 CrossRef CAS .
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS .
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS .
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC .
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, ACS Catal., 2017, 7, 520–529 CrossRef CAS .
- I. Lezcano-Gonzalez, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215–5219 CrossRef CAS .
- J. Gao, Y. Zheng, J. M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS .
- W. Ding, S. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 506–513 CrossRef CAS .
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016–1020 CrossRef CAS .
- M. Caglayan, A. Lucini Paioni, E. Abou-Hamad, G. Shterk, A. Pustovarenko, M. Baldus, A. D. Chowdhury and J. Gascon, Angew. Chem., Int. Ed., 2020, 59, 2–8 CrossRef .
- C. H. L. Tempelman and E. J. M. Hensen, Appl. Catal., B, 2015, 176-177, 731–739 CrossRef CAS .
- X. Jiao, X. Huang and K. Wang, Catal. Sci. Technol., 2019, 9, 6552–6555 RSC .
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, J. Catal., 2017, 346, 125–133 CrossRef CAS .
- I. Julian, M. B. Roedern, J. L. Hueso, S. Irusta, A. K. Baden, R. Mallada, Z. Davis and J. Santamaria, Appl. Catal., B, 2020, 263, 118360 CrossRef CAS .
- N. Kosinov, E. A. Uslamin, L. Meng, A. Parastaev, Y. Liu and E. J. M. Hensen, Angew. Chem., Int. Ed., 2019, 58, 7068–7072 CrossRef CAS .
- N. Kosinov, F. J. Coumans, E. Uslamin, F. Kapteijn and E. J. Hensen, Angew. Chem., Int. Ed., 2016, 55, 15086–15090 CrossRef CAS .
- Z. Cao, H. Jiang, H. Luo, S. Baumann, W. A. Meulenberg, J. Assmann, L. Mleczko, Y. Liu and J. Caro, Angew. Chem., Int. Ed., 2013, 52, 13794–13797 CrossRef CAS .
- S. H. Morejudo, R. Zanon, S. Escolastico, I. Yuste-Tirados, H. Malerod-Fjeld, P. K. Vestre, W. G. Coors, A. Martinez, T. Norby, J. M. Serra and C. Kjolseth, Science, 2016, 353, 563–566 CrossRef CAS .
- L. Liu, D. M. Meira, R. Arenal, P. Concepcion, A. V. Puga and A. Corma, ACS Catal., 2019, 9, 10626–10639 CrossRef CAS .
- W. Zhang, H. Wang, J. Jiang, Z. Sui, Y. Zhu, D. Chen and X. Zhou, ACS Catal., 2020, 10, 12932–12942 CrossRef CAS .
- G. Sun, Z. J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef .
- M. D. Marcinkowski, M. T. Darby, J. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325–332 CrossRef CAS .
- Y. Nakaya, J. Hirayama, S. Yamazoe, K. I. Shimizu and S. Furukawa, Nat. Commun., 2020, 11, 2838 CrossRef CAS .
- J. J. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2014, 53, 9251–9256 CrossRef CAS .
- H. Xiong, S. Lin, J. Goetze, P. Pletcher, H. Guo, L. Kovarik, K. Artyushkova, B. M. Weckhuysen and A. K. Datye, Angew. Chem., Int. Ed., 2017, 56, 8986–8991 CrossRef CAS .
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Nat. Mater., 2019, 18, 866–873 CrossRef CAS .
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Nat. Catal., 2020, 3, 628–638 CrossRef CAS .
- Q. Sun, N. Wang, Q. Fan, L. Zeng, A. Mayoral, S. Miao, R. Yang, Z. Jiang, W. Zhou, J. Zhang, T. Zhang, J. Xu, P. Zhang, J. Cheng, D. C. Yang, R. Jia, L. Li, Q. Zhang, Y. Wang, O. Terasaki and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS .
- F. Schüth, Science, 2019, 363, 1282–1283 CrossRef .
- US Department of Energy, Ultra-Clean Fischer-Tropsch Fuels Production and Demonstration Project Report, 2005, available at: https://www.osti.gov/biblio/860805.
- M. Baldea, T. F. Edgar, B. L. Stanley and A. A. Kiss, AIChE J., 2017, 63, 4262–4272 CrossRef CAS .
- E. J. Sheu, E. M. A. Mokheimer and A. F. Ghoniem, Int. J. Hydrogen Energy, 2015, 40, 12929–12955 CrossRef CAS .
- B. Todić, V. V. Ordomsky, N. M. Nikačević, A. Y. Khodakov and D. B. Bukur, Catal. Sci. Technol., 2015, 5, 1400–1411 RSC .
- J. Jin, X. Wei, M. Liu, Y. Yu, W. Li, H. Kong and Y. Hao, Appl. Energy, 2018, 226, 797–807 CrossRef CAS .
- G. S. Patience and D. C. Boffito, J. Adv. Manuf. Process., 2020, 2, e10039 Search PubMed .
- B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232–1237 CrossRef CAS .
- M. M. Konnick, B. G. Hashiguchi, D. Devarajan, N. C. Boaz, T. B. Gunnoe, J. T. Groves, N. Gunsalus, D. H. Ess and R. A. Periana, Angew. Chem., Int. Ed., 2014, 53, 10490–10494 CrossRef CAS .
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS .
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocella, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS .
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS .
- F. Xu, S. G. Bell, J. Lednik, A. Insley, Z. Rao and L. L. Wong, Angew. Chem., Int. Ed., 2005, 44, 4029–4032 CrossRef CAS .
- L. Luo, X. Tang, W. Wang, Y. Wang, S. Sun, F. Qi and W. Huang, Sci. Rep., 2013, 3, 1625 CrossRef .
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 9, 8592–8621 CrossRef CAS .
- K. Kwapien, J. Paier, J. Sauer, M. Geske, U. Zavyalova, R. Horn, P. Schwach, A. Trunschke and R. Schlögl, Angew. Chem., Int. Ed., 2014, 53, 8774–8778 CrossRef CAS .
- A. Palermo, J. P. Holgado Vazquez and R. M. Lambert, Catal. Lett., 2000, 68, 191–196 CrossRef CAS .
- S.-f. Ji, T.-c. Xiao, S.-b. Li, C.-z. Xu, R.-l. Hou, K. S. Coleman and M. L. H. Green, Appl. Catal., A, 2002, 225, 271–284 CrossRef CAS .
- D. Kiani, S. Sourav, J. Baltrusaitis and I. E. Wachs, ACS Catal., 2019, 9, 5912–5928 CrossRef CAS .
- A. Palermo, J. Holgadovazquez, A. Lee, M. Tikhov and R. Lambert, J. Catal., 1998, 177, 259–266 CrossRef CAS .
- K. Takanabe and E. Iglesia, J. Phys. Chem. C, 2009, 113, 10131–10145 CrossRef CAS .
- K. Takanabe, A. M. Khan, Y. Tang, L. Nguyen, A. Ziani, B. W. Jacobs, A. M. Elbaz, S. M. Sarathy and F. F. Tao, Angew. Chem., Int. Ed., 2017, 56, 10403–10407 CrossRef CAS .
- M. J. Werny, Y. Wang, F. Girgsdies, R. Schlögl and A. Trunschke, Angew. Chem., Int. Ed., 2020, 59, 14921–14926 CrossRef CAS .
- A. Cruellas, T. Melchiori, F. Gallucci and M. van Sint Annaland, Energy Technol., 2019, 8, 1900148 CrossRef .
- X. Zhu, Q. Imtiaz, F. Donat, C. R. Müller and F. Li, Energy Environ. Sci., 2020, 13, 772–804 RSC .
- V. Fleischer, U. Simon, S. Parishan, M. G. Colmenares, O. Görke, A. Gurlo, W. Riedel, L. Thum, J. Schmidt, T. Risse, K.-P. Dinse and R. Schomäcker, J. Catal., 2018, 360, 102–117 CrossRef CAS .
- Z. Cheng, D. S. Baser, S. G. Nadgouda, L. Qin, J. A. Fan and L.-S. Fan, ACS Energ. Lett., 2018, 3, 1730–1736 CrossRef CAS .
- C. P. Kumar, S. Gaab, T. E. Müller and J. A. Lercher, Top. Catal., 2008, 50, 156–167 CrossRef CAS .
- L. Annamalai, Y. Liu, S. Ezenwa, Y. Dang, S. L. Suib and P. Deshlahra, ACS Catal., 2018, 8, 7051–7067 CrossRef CAS .
- D. Melzer, G. Mestl, K. Wanninger, Y. Zhu, N. D. Browning, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 4012 CrossRef .
- B. Chu, H. An, T. A. Nijhuis, J. C. Schouten and Y. Cheng, J. Catal., 2015, 329, 471–478 CrossRef CAS .
- D. Dang, X. Chen, B. Yan, Y. Li and Y. Cheng, J. Catal., 2018, 365, 238–248 CrossRef CAS .
- D. Melzer, P. Xu, D. Hartmann, Y. Zhu, N. D. Browning, M. Sanchez-Sanchez and J. A. Lercher, Angew. Chem., Int. Ed., 2016, 55, 8873–8877 CrossRef CAS .
- Y. Liu, L. Annamalai and P. Deshlahra, J. Phys. Chem. C, 2019, 123, 28168–28191 CrossRef CAS .
- J. M. López Nieto, B. Solsona, P. Concepción, F. Ivars, A. Dejoz and M. I. Vázquez, Catal. Today, 2010, 157, 291–296 CrossRef .
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 73–77 CrossRef CAS .
- X. Liu, B. Frank, W. Zhang, T. P. Cotter, R. Schlogl and D. S. Su, Angew. Chem., Int. Ed., 2011, 50, 3318–3322 CrossRef CAS .
- J. Li, P. Yu, J. Xie, J. Liu, Z. Wang, C. Wu, J. Rong, H. Liu and D. Su, ACS Catal., 2017, 7, 7305–7311 CrossRef CAS .
- W. H. K. Ng, E. S. Gnanakumar, E. Batyrev, S. K. Sharma, P. K. Pujari, H. F. Greer, W. Zhou, R. Sakidja, G. Rothenberg, M. W. Barsoum and N. R. Shiju, Angew. Chem., Int. Ed., 2018, 57, 1485–1490 CrossRef CAS .
- C. Chen, J. Zhang, B. Zhang, C. Yu, F. Peng and D. Su, Chem. Commun., 2013, 49, 8151–8153 RSC .
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS .
- H. Li, J. Zhang, P. Wu, S. Xun, W. Jiang, M. Zhang, W. Zhu and H. Li, J. Phys. Chem. C, 2019, 123, 2256–2266 CrossRef CAS .
- Z. Zhang, E. Jimenez-Izal, I. Hermans and A. N. Alexandrova, J. Phys. Chem. Lett., 2019, 10, 20–25 CrossRef CAS .
- J. M. Venegas, Z. Zhang, T. O. Agbi, W. P. McDermott, A. Alexandrova and I. Hermans, Angew. Chem., Int. Ed., 2020, 59, 16527–16535 CrossRef CAS .
- X. Zhang, R. You, Z. Wei, X. Jiang, J. Yang, Y. Pan, P. Wu, Q. Jia, Z. Bao, L. Bai, M. Jin, B. Sumpter, V. Fung, W. Huang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 8042–8046 CrossRef CAS .
- L. M. Neal, S. Yusuf, J. A. Sofranko and F. Li, Energy Technol., 2016, 4, 1200–1208 CrossRef CAS .
- Y. Gao, X. Wang, J. Liu, C. Huang, K. Zhao, Z. Zhao, X. Wang and F. Li, Sci. Adv., 2020, 6, eaaz9339 CrossRef CAS .
- S. Chen, C. Pei, X. Chang, Z. J. Zhao, R. Mu, Y. Xu and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 22072–22079 CrossRef CAS .
- V. Paunović and J. Pérez-Ramírez, Catal. Sci. Technol., 2019, 9, 4515–4530 RSC .
- P. Batamack, I. Bucsi, A. Molnar and G. A. Olah, Catal. Lett., 1994, 25, 11–19 CrossRef CAS .
- M. H. Nilsen, S. Svelle, S. Aravinthan and U. Olsbye, Appl. Catal., A, 2009, 367, 23–31 CrossRef CAS .
- K. Ding, A. Zhang and G. D. Stucky, ACS Catal., 2012, 2, 1049–1056 CrossRef CAS .
- I. M. Lorkovic, S. Sun, S. Gadewar, A. Breed, G. S. Macala, A. Sardar, S. E. Cross, J. H. Sherman, G. D. Stucky and P. C. Ford, J. Phys. Chem. A, 2006, 110, 8695–8700 CrossRef CAS .
- K. Ding, A. R. Derk, A. Zhang, Z. Hu, P. Stoimenov, G. D. Stucky, H. Metiu and E. W. McFarland, ACS Catal., 2012, 2, 479–486 CrossRef CAS .
- A. J. Saadun, G. Zichittella, V. Paunović, B. A. Markaide-Aiastui, S. Mitchell and J. Pérez-Ramírez, ACS Catal., 2019, 10, 528–542 CrossRef .
- A. J. Saadun, S. Pablo-García, V. Paunović, Q. Li, A. Sabadell-Rendón, K. Kleemann, F. Krumeich, N. López and J. Pérez-Ramírez, ACS Catal., 2020, 10, 6129–6143 CrossRef CAS .
- K. X. Wang, H. F. Xu, W. S. Li, C. T. Au and X. P. Zhou, Appl. Catal., A, 2006, 304, 168–177 CrossRef CAS .
- Y. Fan, D. Ma and X. Bao, Catal. Lett., 2009, 130, 286–290 CrossRef CAS .
- G. K. S. Prakash, J. C. Colmenares, P. T. Batamack, T. Mathew and G. A. Olah, J. Mol. Catal. A: Chem., 2009, 310, 180–183 CrossRef CAS .
- G. Zichittella, B. Puértolas, V. Paunović, T. Block, R. Pöttgen and J. Pérez-Ramírez, Catal. Sci. Technol., 2018, 8, 2231–2243 RSC .
- S. Svelle, S. Aravinthan, M. Bjorgen, K. Lillerud, S. Kolboe, I. Dahl and U. Olsbye, J. Catal., 2006, 241, 243–254 CrossRef CAS .
- U. Olsbye, O. V. Saure, N. B. Muddada, S. Bordiga, C. Lamberti, M. H. Nilsen, K. P. Lillerud and S. Svelle, Catal. Today, 2011, 171, 211–220 CrossRef CAS .
- D. Jaumain and B.-L. Su, J. Mol. Catal. A: Chem., 2003, 197, 263–273 CrossRef CAS .
- Z. Jiang, B.-x. Shen, J.-g. Zhao, L. Wang, L.-t. Kong and W.-g. Xiao, Ind. Eng. Chem. Res., 2015, 54, 12293–12302 CrossRef CAS .
- M. Ibáñez, M. Gamero, J. Ruiz-Martínez, B. M. Weckhuysen, A. T. Aguayo, J. Bilbao and P. Castaño, Catal. Sci. Technol., 2016, 6, 296–306 RSC .
- V. Paunović, S. Mitchell, R. Verel, S. S. Lee and J. Pérez-Ramírez, J. Phys. Chem. C, 2019, 124, 722–733 CrossRef .
- A. P. Amrute, C. Mondelli, M. Moser, G. Novell-Leruth, N. López, D. Rosenthal, R. Farra, M. E. Schuster, D. Teschner, T. Schmidt and J. Pérez-Ramírez, J. Catal., 2012, 286, 287–297 CrossRef CAS .
- A. P. Amrute, F. Krumeich, C. Mondelli and J. Pérez-Ramírez, Chem. Sci., 2013, 4, 2209 RSC .
- A. P. Amrute, C. Mondelli, T. Schmidt, R. Hauert and J. Pérez-Ramírez, ChemCatChem, 2013, 5, 748–756 CrossRef CAS .
- M. Moser, C. Mondelli, A. P. Amrute, A. Tazawa, D. Teschner, M. E. Schuster, A. Klein-Hoffman, N. López, T. Schmidt and J. Pérez-Ramírez, ACS Catal., 2013, 3, 2813–2822 CrossRef CAS .
- M. Moser, I. Czekaj, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2014, 53, 8628–8633 CrossRef CAS .
- V. Paunovic, R. Lin, M. Scharfe, A. P. Amrute, S. Mitchell, R. Hauert and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2017, 56, 9791–9795 CrossRef CAS .
- G. Zichittella, V. Paunović, A. P. Amrute and J. Pérez-Ramírez, ACS Catal., 2017, 7, 1805–1817 CrossRef CAS .
- G. Zichittella, N. Aellen, V. Paunović, A. P. Amrute and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2017, 56, 13670–13674 CrossRef CAS .
- J. He, T. Xu, Z. Wang, Q. Zhang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2012, 51, 2438–2442 CrossRef CAS .
- G. Zichittella, B. Puértolas, S. Siol, V. Paunovic, S. Mitchell and J. Perez-Ramirez, ChemCatChem, 2018, 10, 1282–1290 CrossRef CAS .
- V. Paunović, G. Zichittella, M. Moser, A. P. Amrute and J. Pérez-Ramírez, Nat. Chem., 2016, 8, 803–809 CrossRef .
- G. Zichittella, S. Stähelin, F. M. Goedicke and J. Pérez-Ramírez, ACS Catal., 2019, 9, 5772–5782 CrossRef CAS .
- V. Paunović, G. Zichittella, P. Hemberger, A. Bodi and J. Pérez-Ramírez, ACS Catal., 2019, 9, 1710–1725 CrossRef .
- G. Zichittella, M. Scharfe, B. Puértolas, V. Paunović, P. Hemberger, A. Bodi, L. Szentmiklosi, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2019, 58, 5877–5881 CrossRef CAS .
- G. Zichittella, P. Hemberger, F. Holzmeier, A. Bodi and J. Perez-Ramirez, J. Phys. Chem. Lett., 2020, 11, 856–863 CrossRef CAS .
- V. Paunović, P. Hemberger, A. Bodi, N. López and J. Pérez-Ramírez, Nat. Catal., 2018, 1, 363–370 CrossRef .
- V. Paunovic, G. Zichittella, S. Mitchell, R. Hauert and J. Perez-Ramirez, ACS Catal., 2018, 8, 291–303 CrossRef CAS .
- Q. Xie, H. Zhang, J. Kang, J. Cheng, Q. Zhang and Y. Wang, ACS Catal., 2018, 8, 4902–4916 CrossRef CAS .
- C. L. Lara, D. E. Bernal, C. Li and I. E. Grossmann, Comput. Chem. Eng., 2019, 127, 295–310 CrossRef CAS .
- G. Liu, A. Cadiau, Y. Liu, K. Adil, V. Chernikova, I. D. Carja, Y. Belmabkhout, M. Karunakaran, O. Shekhah, C. Zhang, A. K. Itta, S. Yi, M. Eddaoudi and W. J. Koros, Angew. Chem., Int. Ed., 2018, 57, 14811–14816 CrossRef CAS .
- Y. Belmabkhout, P. M. Bhatt, K. Adil, R. S. Pillai, A. Cadiau, A. Shkurenko, G. Maurin, G. Liu, W. J. Koros and M. Eddaoudi, Nat. Energy, 2018, 3, 1059–1066 CrossRef CAS .
- L. Bittorf, F. Reichmann, M. Schmalenberg, S. Soboll and N. Kockmann, Chem. Eng. Technol., 2019, 42, 1985–1995 CrossRef CAS .
- M. Chaczykowski and P. Zarodkiewicz, Energy, 2017, 134, 681–698 CrossRef CAS .
- Scaling-up renewables through decentralised energy solutions, 2017, available at: http://www.iea.org.
- The World Bank, Decentralized Energy Services to Fight Poverty: Outcome Driven Engagement of Small and Medium Size Enterprises in the Provision of Energy Services in IDA Country, 2009, available at: https://openknowledge.worldbank.org.
- The State of Methane Report, 2018, available at: https://www.taxpayer.net.
- A. Galán-Martín, C. Pozo, A. Azapagic, I. E. Grossmann, N. Mac Dowell and G. Guillén-Gosálbez, Energy Environ. Sci., 2018, 11, 572–581 RSC .
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Javier
Pérez-Ramírez
* and
Javier
Pérez-Ramírez