
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissociations of free radicals to generate protons, electrophiles or nucleophiles: role in DNA strand breaks
John C.
Walton

EaStCHEM School of Chemistry, University of St. Andrews, St. Andrews, Fife KY16 9ST, UK. E-mail: jcw@st-andrews.ac.uk
First published on 21st May 2021
Abstract
The concept behind the research described in this article was that of marrying the ‘soft’ methods of radical generation with the effectiveness and flexibility of nucleophile/electrophile synthetic procedures. Classic studies with pulse radiolysis and laser flash photolysis had shown that free radicals could be more acidic than their closed shell counterparts. QM computations harmonised with this and helped to define which radical centres and which structural types were most effective. Radicals based on the sulfonic acid moiety and on the Meldrum's acid moiety (2,2-dimethyl-1,3-dioxane-4,6-dione) were found to be extreme examples in the superacid class. The ethyne unit could be used as a very effective spacer between the radical centre and the site of proton donation. The key factor in promoting acidity was understood to be the thermodynamic stabilisation of the conjugate anion-radicals released on deprotonation. Solvation played a key part in promoting this and theoretical microhydration studies provided notable support. A corollary was that heterolytic dissociations of free radicals to yield either electrophiles or nucleophiles were also enhanced relative to non-radical models. The most effective radical types for spontaneous releases of both these types of reagents were identified. Ethyne units were again effective as spacers. The enhancement of release of phosphate anions by adjacent radical centres was an important special case. Reactive oxygen species and also diradicals from endiyne antibiotics generate C4′-deoxyribose radicals from nucleotides. Radicals of these types spontaneously release phosphate and triphosphate and this is a contributor to DNA and RNA strand breaks.
 John C. Walton | John C. Walton studied chemistry at Sheffield University and then joined the faculty of Dundee University in 1967 to work with John Tedder. He moved to St. Andrews University in 1970 and became research professor in 2007. He is enthusiastic about research and has worked on the application of physical organic methods, particularly EPR spectroscopy, to organic mechanisms. Recently he has been developing radical-mediated synthetic protocols including those based around: “pro-aromatic” cyclohexadienyl reagents, oxime derivatives, photoredox methods employing titanium dioxide and on radical enhancement of heterolysis. He was elected Fellow of the Royal Society of Edinburgh in 1995. |
1. Introduction
Most mainline organic synthetic procedures rely on the availability of nucleophiles and/or electrophiles. In fact, these species are the workhorses of retrosynthetic analytical methods. Accounts of these methods in the literature and in University level courses generally only make passing reference to free radical mediated protocols.1 Powerful bases or acids that are corrosive and may be pyrophoric and toxic are often required to get such procedures underway. Techniques for generating radicals are much ‘softer,’ particularly now that visible light photoredox catalytic methods have been developed;2 some even avoiding transition metals.3 Thermolytic radical generation can also be accomplished at temperatures only slightly above ambient by means of azo-initiators such as 1,1′-azobis(cyclohexane-1-carbonitrile) (V-40), dimethyl 2,2′-azobis(2-methylpropionate) (V-601) and even in water with 2,2′-azobis[2-methyl-N-(2-hydroxyethyl)propionamide] (VA-086) or 2,2′-azobis(2-methylpropionamidine)dihydrochloride (V-50). Minimal energy consumption and environmental impact are achievable by these means.It would be synthetically advantageous if soft radical generation could be married to the power and flexibility of nucleophile/electrophile strategies. Furthermore, this would enable the convenience and effectiveness of conventional retrosynthetic analysis to simultaneously be made use of. The concept then is to generate an appropriate radical by a soft method and have it spontaneously convert to a desired nucleophile or electrophile (Scheme 1). Formation of the radical must be immediately followed by heterolysis either to yield an electrophile together with an anion-radical (1a, b) or a nucleophile together with a cation-radical (eqn (2), Scheme 1).
 | ||
| Scheme 1 Radical mediated generation of electrophiles and nucleophiles. | ||
Clues about how to achieve the concepts in Scheme 1 were obtained from a consideration of the chemical behaviour of acid radicals. The proton is a special archetype electrophile that is itself extremely useful as an acid catalyst and also for the generation of other electrophiles (eqn (1a), Scheme 1). In the 1960s and 70s several research groups started to measure the pKa values of reactive free radicals. They observed that free radicals were often stronger acids than their neutral precursor molecules. Instances of free radicals that were very strong acids and, on generation, ionised to protons essentially instantaneously were noted. This research remained dormant in the literature but eventually re-emerged. The idea that, with appropriate design, the phenomenon could be generalised so as to provide ranges of electrophiles and possibly nucleophiles was conceived. This article reviews the research that has been carried out to examine the scope and underlying causes of acidity in radicals and then to generalise the concept for release of more complex cations and anions.
2. Experimental determinations of ionization constants of transient radicals
From the 1960s onwards there was a huge flowering of research in radiation chemistry and photochemistry as the fast techniques of Pulse Radiolysis (PR) and Laser Flash Photolysis (LFP) were developed. These methods were applied to the study of acid/base equilibria of transient species. Much of this chemistry was reviewed in 1974 by Hayon and Simic in an important article.4 It was observed that the optical absorptions of acidic free radicals were usually blue shifted compared to those of their conjugate bases. The ionisation constants (pKa) of free radicals could then be determined on the basis of the spectral differences. The EPR (ESR) spectra of acidic radicals and their conjugate base anion radicals differed and this also enabled pKa values to be determined for several such ionizations.5 Conductivity and polarography measurements were also utilised for determining pKa values of transient radicals.6 Nibbering and coworkers measured the acidities of halo-, cyano-, and isocyano-substituted free radicals using a bracketing method and FT-ICR mass spectrometry.7α-Hydroxylalkyl radicals (ketyl radicals) 1 (Scheme 2) received the most attention. The pKa values observed for these types were in the narrow range of 10.7 to 12.1 units depending on R1 and R2. That is, they were about 5 pH units more acidic than those of corresponding non-radical alcohols.8
 | ||
| Scheme 2 Transient acid radicals studied by PR and LFP. | ||
Alcohols with unpaired electrons (upe) on C(sp3)-atoms β or γ to their OH groups had pKa values close to those of corresponding closed shell alcohols; that is their acidities were not enhanced. However when the radical C˙–OH group was conjugated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds or aromatic rings the acidity of the OH group was increased further. The strongest inductive effect was observed when a carbonyl group occupied an α-position to the C˙–OH radical. Thus, pKa values of 4.4 and 5.2 were obtained for biacetyl and hexenedione units respectively. On the other hand, the pKa values measured for α-carboxyalkyl radicals 2 (Scheme 2) of about 4.9 were close to those of parent carboxylic acids.9 Peptide radicals 3 (Scheme 2) were observed to ionize by proton loss from their NH groups with pKa values much smaller than those of parent non-radical peptides.10 Another PR study found pKa values in the range 5.2–5.8 for peroxyl radicals of type 4 indicating substantial increases in acidity in comparison with models.11
C bonds or aromatic rings the acidity of the OH group was increased further. The strongest inductive effect was observed when a carbonyl group occupied an α-position to the C˙–OH radical. Thus, pKa values of 4.4 and 5.2 were obtained for biacetyl and hexenedione units respectively. On the other hand, the pKa values measured for α-carboxyalkyl radicals 2 (Scheme 2) of about 4.9 were close to those of parent carboxylic acids.9 Peptide radicals 3 (Scheme 2) were observed to ionize by proton loss from their NH groups with pKa values much smaller than those of parent non-radical peptides.10 Another PR study found pKa values in the range 5.2–5.8 for peroxyl radicals of type 4 indicating substantial increases in acidity in comparison with models.11
The conclusions from these experimental studies were that: (i) ionization constants of free radicals can differ considerably from those of parent compounds, (ii) the pKa values of free radicals are significantly lowered only when the ionising group occupies an α-position relative to the carbon atom carrying the upe, (iii) the pKa values of α-hydroxylalkyl radicals correlate linearly with the redox potentials of parent compounds.12
3. Computational studies of proton release from functionalised radicals
3.1 Development of dependable QM methods
This experimental research raised a number of intriguing questions about the phenomenon of deprotonation of radicals. What structural features were required for radicals to deprotonate readily? How would the ease of proton loss vary with the nature of the radical centre? What kinds of acid and proton donor groups would participate? Was it essential that the proton donor group be α to the atom bearing the upe? Could spacer groups that transmitted the effect to greater distances be found? What was the limit of acidity that could be reached? What were the underlying causes of the increased acidity?Computational methods were needed to find answers for these questions because of the specialised and complex resources required for experimental studies. Radom and co-workers made a computational study of deprotonation of radicals of type ˙CH2X for donor groups X = NH2, OH, OCH3, PH2, SH, F, Cl, and Br and for acceptor groups X = BH2, AlH2, CHO, NO2, CN, and NC, with G2 ab initio theory, albeit in the gas phase only.13 They predicted the gas phase acidities of ˙CH2X radicals would exceed those of parent molecules H–CH2X for π-donor groups X but be less for X = π-acceptor groups (except CN and NC). They associated the higher acidities of the radicals with the greater stabilities of their conjugate anions.
A follow-up study on similar radicals in the gas phase using the high level W1w ab initio method came to similar conclusions.14 The results were rationalised with reference to resonance and orbital interactions in the conjugate radical anions ˙CH2X−. Radom and co-workers also examined the effect of a connector group W on the interaction between a substituent X and the radical centre in ˙CH2WX radicals.15 They found that W = CH2 as connector turned off π-electron effects but allowed transmission of σ-effects although at reduced levels. For connector groups W = –CHCH– and –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– the only effects transmitted were just those of the connectors themselves.
C– the only effects transmitted were just those of the connectors themselves.
My interest in the phenomenon resulted from research in my group into the chemistry of bicarbonate radicals 6 (Scheme 3). Dissolution of CO2 in water yields carbonic acid which ionizes to the bicarbonate anion 5 (pKa = 3.6) and then the carbonate dianion. In blood serum and intercellular media these equilibria constitute the ‘bicarbonate buffer system.’ Various enzymes16 oxidise the bicarbonate anion to the neutral bicarbonate radical 6 (Scheme 3). Bicarbonate is ubiquitous in living systems so the bicarbonate radical and its conjugate base, the carbonate radical anion 7, are very important. The chemistries of the two species 6 and 7 will be very different because 6 is neutral whereas 7 is negatively charged. The extent of delocalisation of the upe is different in the two species as well. It was therefore important to know the pKa of this ionisation. Attempts to measure this pKa experimentally had shown 6 was a strong acid but the pKa could not be quantified.17 A computational study suggested this pKa might be as low as −4 units.18 This acidity led to the expectation that the carbonate radical anion 7 would be the dominant partner in aqueous media and this has been assumed in biochemical work.19
 | ||
| Scheme 3 Deprotonations of bicarbonate, carboxyl and hydroperoxyl radicals. | ||
In comparison with carbonic acid [HOC(O)OH], as a model in which the upe is replaced by an H-atom, the bicarbonate radical is orders of magnitude more acidic. Experimental pKa values had been measured for the carboxyl radical 8 (−0.2)20 and the hydroperoxyl radical (perhydroxyl) 10 (4.88)21 and these were 4 and 7 log units lower than the pKas of model formic acid and hydrogen peroxide respectively.
These, and the experimental findings referred to above, were intriguing results that sparked a comprehensive computational study of the phenomenon by my group. For convenience the phenomenon of Radical-Enhanced Dissociation was dubbed a molecular “RED-shift” from the initials, but also because increased acidity generally corresponds to the development of red colour in indicators. A DFT computational method for free radicals was developed by means of a benchmarking study.22 Twenty-three DFT functionals were tested with nine radical reaction types and compared with the high-level composite ab initio G4 method.23 The PBE0 (PBE1PBE),24 and CAM-B3LYP25 functionals both gave very satisfactory results.
To obtain pKa data for transient radicals, linear correlations were first obtained of DFT computed free energies of deprotonation (ΔGA-HA) with experimentally known pKas for five classes of model proton donors. The CAM-B3LYP functional was employed with the 6-311+G(2d,p) basis set together with the CPCM continuum model26 with water as solvent. The experimental free energy of solvation for the proton (−264.2 kcal mol−1)27 was included.
Good linear regression plots were obtained for sets of carboxylic, sulfonic and sulfinic acids, for imides, and for carbonyl compounds.28 These relationships enabled pKas to be reproduced, in most instances to within 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log units. This method then enabled realistic estimates to be made of the pKas of transient radicals of each proton donor class. Support for this conclusion came from the good correspondence of the computed pKas with the experimental values for the radicals of Scheme 3.29,30 A useful measure of the enhanced acidity conferred by the presence of a free radical centre in a proton donor (i.e. the RED-shift) was defined as:
log units. This method then enabled realistic estimates to be made of the pKas of transient radicals of each proton donor class. Support for this conclusion came from the good correspondence of the computed pKas with the experimental values for the radicals of Scheme 3.29,30 A useful measure of the enhanced acidity conferred by the presence of a free radical centre in a proton donor (i.e. the RED-shift) was defined as:
| RED-shift = ΔpKa = pKa(model) − pKa(radical) |
3.2 Influence of different radical centres on the acidities of carboxylic, sulfinic and sulfonic acids
The free energies of dissociation and pKa values were computed for series of radicals containing carboxylic acid, sulfinic acid and sulfonic acid groups as proton donors. The analogous data was obtained for corresponding model acids. The enhancements of acidity ΔpKa then followed from the difference. For deprotonations of carboxylic acids ˙Z–CO2H it was found that for Z = CH2–, CH2CH2–, CHCH–, C6H4– and CH2C6H4– the pKas were all in the range 4.3 to 5.2, similar to those of their models. This was in good agreement with the experimental (PR/LFP) results. Consequently, their RED-shifts (ΔpKa) were all close to zero. In their conjugate bases, ˙Z–CO2−, the upe and negative charge were well separated such that no extra resonance was possible.
Two conformations of the carboxyl radical (8 = 12a, H–O–C˙O) are possible in which the O–H group is either trans- or cis-to the CO group. The computed pKas were fairly similar (see Scheme 4) and both showed substantial RED-shifts. For the acids 12b–d, in which the upe was situated on CH2-, NH-groups, and O-atoms respectively, the pKas decreased substantially with the electronegativity of the radical centre. The RED-shifts also increased with increasing electronegativity of the radical centre (see Scheme 4). The ethynyl group was exceptionally effective as an enhancer of acidity. Thus, the carboxyethynyl radical 12e was as acidic as a strong mineral acid and a large RED-shift was observed.
 | ||
| Scheme 4 Deprotonation data for carboxylic, sulfinic and sulfonic acid radicals. [*For 12a(trans), pKa = 0.6, ΔpKa = 3.2, nd = not determined, u = unstable]. | ||
It was apparent that stabilization of the conjugate radical anion bases was a key factor. In support of this, a plot of the computed relative electronic charges [Δq(O), (model − radical)] on the carboxylate O-atoms against ΔpKa showed a monotonic trend for the series of carboxylic acid radicals. For the same series of carboxylate radical anions a plot of the computed spin densities on oxygen ρ(O) against ΔpKa also showed a monotonic trend. The observed rule was that the radical anions derived from radicals showing greater acidity had charge delocalized away from their anionic O-atoms, together with spin density transferred toward these O-atoms. When the structures permitted both spin and charge to be delocalised, in the conjugate bases, enhanced acidity resulted.
The small or negative pKas of selected sulfinic acid radicals revealed that they were all strong acids and furthermore, sulfonic acid radicals were significantly stronger (Scheme 4). Just as with the carboxymethyl acid radical (12b) essentially no RED-shift was present in the sulfinylmethyl acid radical 13b. By way of contrast, a significant RED-shift of 2.21 was obtained for the sulfonylmethyl radical 14b. A possible cause of this was the pyramidality of the SO3 group which ensured that the p-orbital of the CH2˙ group necessarily eclipsed one of the SO bonds with consequent spin delocalisation. No such eclipsing occurred with the planar CO2 of the non-RED-shifted carboxymethyl radical 12b or the SO2 group of the sulfinylmethyl radical 13b.
As with carboxylic series member (12e) the ethynyl group gave enhanced acidity to the ethynylsulfinic acid radical 13e and ethynylsulfonic acid radical 14e. In the ethynyl type radicals the upe is (formally) associated with an sp hybridised C-atom so that it is in a σ-type orbital, which facilitated thermodynamic stabilization in the conjugate bases.
The upes of cage bridgehead radicals also reside in σ-orbitals. Furthermore, spin density is transmitted through the centres of the smaller cages,31 so the effect of such spacers seemed worth investigating. That the bicyclo[1.1.1]pentane cage would be the most suitable for spin transmission was suggested by the exceptionally large γ-H-atom hyperfine coupling in the EPR spectrum of its radical.32 The sulfinic acid radical 13f containing this cage spontaneously dissociated to [1.1.1]propellane and the HSO2˙ radical. However, the carboxylic acid radical 12f and the sulfonic acid radical 14f, with bicyclo[1.1.1]pentane spacers between their upes and acid groups, both showed significant RED-shifts (Scheme 4).
3.3 Influence of different radical centres on the acidities of imides
The effect of introducing radical centres to proton donors with their H-atoms situated on N-atoms was also examined.28 Some imides were known to be good proton donors. Of these, the azetidine-2,4-dione unit (15) has a planar symmetrical structure permitting comparatively close through space approach of an upe on C-3 to the incipient anionic centre on the N-atom in the conjugate base (Scheme 5). | ||
| Scheme 5 Deprotonation data for radicals with imide and related structures. | ||
Data was computed for structures 15a–f containing electron-withdrawing and electron-releasing substituents on C-3 (see Scheme 5). RED-shifts were modest (ΔpKa = 0.9 to 1.8) or close to zero for all substituents and no trend with the electronic nature of the substituent was revealed. Similarly, the 1,3,5-dioxazinane-4,6-dion-4-yl radical 16 and the dihydro-1,3,5-triazinyl radical 17 had only small RED-shifts (Scheme 5). It was evident that charge and/or spin was not appreciably delocalised by either the C(O) units of azetidine-2,4-diones 15, or the C(O)O units of 16, or the CN double bonds of 17.
3.4 Influence of different radical centres on the acidities of carbon acids
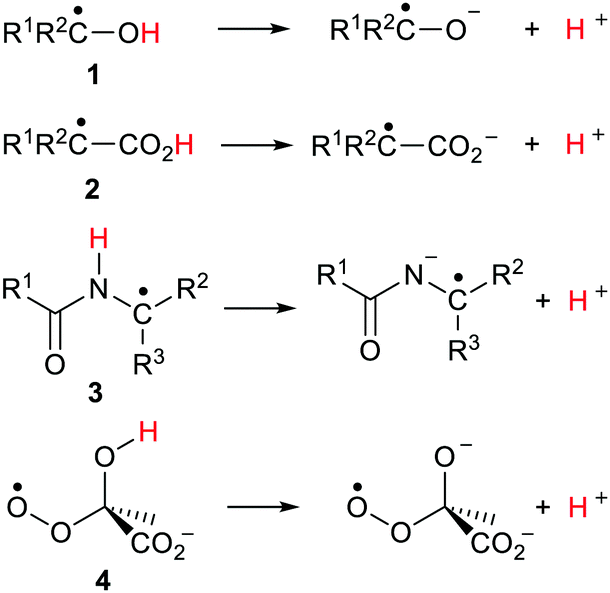
The effect of neighbouring radicals on deprotonation of C–H bonds is of special interest because the resultant carbanions are extremely important in numerous organic preparative sequences. Two types of 1,3-dicarbonyl type compounds, useful for enolate generation, were therefore studied.28,29 Model closed shell 1,3-diketones are weak acids (pKas 9–11 units) whereas Meldrum's acid (2,2-dimethyl-1,3-dioxane-4,6-dione)33 and derivatives are stronger (pKas about 5 units). Radicals based on various centres were examined within the pentan-2,4-dione set 18a–f (Scheme 6). Large ΔpKa values were obtained for radicals 18a, b and 18c. The enhancements appeared to increase with increasing electronegativity of the radical centre, although the O-centred radical 18c was unstable and dissociated to 2-oxopropanal and acetyl radicals. As with other acid types, the ethynyl unit was exceptionally effective and in radical 18d gave rise to a huge RED-shift of 16.1 units making it almost a superacid. The formyl radical 18e had only a small RED-shift, probably because delocalisation of the upe was ineffective. A significant RED-shift of 2.9 was computed for nitroxide radical 18f. For more persistent nitroxides (18f; R = Ph, But) essentially zero RED-shifts were obtained; probably because steric factors blocked delocalisation of the upe.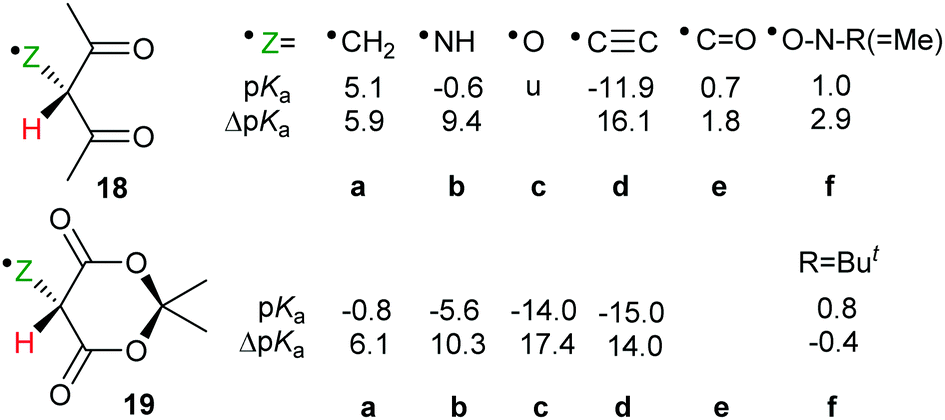 | ||
| Scheme 6 Deprotonation of carbon acid radicals. | ||
A radical substituent on C-5 in the quasi-chair Meldrum's acid structure may be quasi-axial or quasi-equatorial. The computed pKas and RED-shifts of the two conformers differed by only small amounts so only the quasi-equatorial values are shown in Scheme 6. This structure stood out as exceptionally effective and gave rise to enormous RED-shifts for certain radicals. Even the ˙CH2 type 19a had a substantial ΔpKa (6.1 units) and the ˙NH radical 19b had a huge RED-shift of 10.3 units. The oxyl radical 19c, with an even more electronegative centre, had a remarkable computed pKa of −14 units. The acidity of this radical had been increased by 17.4 units in comparison with the closed shell model Meldrum's alcohol (Z = OH). The superacids fluorosulfuric and triflic acid have pKas of −10 and −13 units respectively so Meldrum's radical 19c belongs in the same category although, of course, it is a transient species. The radical 19d, based on an ethynyl unit, had a pKa of −15.0 which placed it also in the superacid class. By way of contrast, the Meldrum's nitroxide 19f and the Meldrum's bicyclo[1.1.1]pentanyl radical (not shown) displayed essentially zero RED-shifts. Examination of the charge and spin distribution in the conjugate bases of the Meldrum's series 19 revealed the same pattern as with the carboxylic series 12. When the structures of the conjugate bases permitted both spin and charge to be delocalised, enhanced acidity resulted.
3.5 Effect of spacer groups on deprotonation of radicals
Large enhancements of deprotonation had been obtained for several acid types having their radical centres on ethynyl units, see structures 12e–14e in Scheme 4 and 18d, 19d in Scheme 6. It seemed, therefore, that other spacers between the formal radical centre and the charge centre might also be effective. However, for the carboxylic acid series of radicals, saturated chain spacers (CH2, CH2CH2) were ineffective as were CC linkages and aromatic rings. The effect of increasing the number of ethyne units in the spacers of carboxylic, sulfinic, sulfonic and 1,3-dicarbonyl species was also tested (Scheme 7).28,29
 | ||
| Scheme 7 Influence of spacers on pKas of various radical types. | ||
For the carboxylic, sulfinic and sulfonic acid series, 20, 22 and 23 the computed pKas increased (or became less negative) as the number of ethyne units in the spacers increased. Significant RED-shifts were, never-the-less, obtained on inclusion of up to four ethyne spacer units. Remarkably, the polyethynyl carboxylic acid radicals remained much more acidic than the carboxymethyl radical 12b even with six ethyne spacers, as in species 20f.
The radicals with sulfonic (23), pentandione (24), and Meldrum's (25) units, even those with 10 ethyne spacers, were all very strong acids. The pKas of the monoethynyl members 23a, 24a, and 25a were highly negative, comparable with those of the well-known sulfonic superacids (see above). The dicarbonyl-based radicals 24a and 25a are members, albeit transient ones, of the uncommon class of carbon superacids.34 The acidity of the radicals slowly tended towards those of the corresponding closed shell models as the number of spacer alkyne units increased. Remarkably, for both the pentandione 24 and Meldrum's series 25, a sizeable RED-shift was observed even beyond 10 ethyne units.
With the carboxylic acid and sulfonic acid series, modest enhancements of deprotonation were observed with the bicyclo[1.1.1]pentane spacer; see Scheme 4, structures 12f and 14f. The effect of increasing the number of bicyclo[1.1.1]pentane spacer units was also investigated for the diverse acids of Scheme 7. In the carboxylic acid series 21 a small enhancement (ΔpKa = 0.93) was obtained with just one of these spacers. However, with two or more the enhancement plummeted to essentially zero (21b, c). With the Meldrum's structure 26a even one bicyclo[1.1.1]pentane spacer effectively suppressed any enhancement i.e. ΔpKa = 0.5.
The distributions of spin and charge in the occupied orbitals of the pentandione and Meldrum's series of conjugate radical anions (from 24 and 25) were carefully examined. This demonstrated that the first members of the series 24a and 25a, having the largest ΔpKa values, were characterized by very significant delocalisation of both spin density and charge.
For each member of the polyethynyl series having up to 10 ethyne units, dispersion of both spin density and charge occurred efficiently along their polyethyne chains. Computed SOMO and spin density distributions extended throughout the length of their polyethyne chains to include the 1,3-dicarbonyl unit. This is illustrated in Fig. 1 which shows these functions for Meldrum's radical anion 25f−. Consequently, although ΔpKa values diminished, they remained appreciable even when the dissociating proton was separated from the formal radical centre by chains of up to 20-C-atoms.
 | ||
| Fig. 1 HOMO amplitudes and spin density distribution for the hexaethynyl-Meldrum's radical anion 25f−. | ||
The enhancement of acidity of the alkyne-containing radicals is very intriguing. In these systems the upe is formally contained in sp hybridized acetylenic type orbitals. The enhancement may be a manifestation of Bent's rule which states that: “s-character concentrates in orbitals directed toward electropositive substituents” or, alternatively, that “atoms direct hybrid orbitals with more p-character towards more electronegative elements.”35,36 In all the series 20 and 22–25 the acid end groups of the radicals are electronegative moieties so Bent's rule predicts a shift of electron density towards the other acetylenic end. The effect would be to confer some acetylide character with concomitant positive hole character delocalised throughout the molecule. Weakening of the O–H bonds and greater acidity would then be expected.37
4. Microhydration and the acidity of radicals
Both experimental and DFT studies had revealed that an appropriately placed radical centre could impart greatly enhanced acidity. That this enhancement was associated with the thermodynamic stabilization of the conjugate radical anions was also apparent. The latter species are open shell and charged so solvation was thought likely to play a key part. In the DFT computations solvation was accounted for by an electrostatic continuum model that effectively smears out and averages solute–solvent interactions. In reality, charge and spin are distributed in particular patterns in the conjugate radical anions, depending on the details of their molecular structures. It seemed likely therefore that specific interactions between the radical anions and individual solvent molecules would play a part. The continuum solvent models might well not be reliable for such small and strong acids.Experiment and theory had shown that strong mineral acids spontaneously ionized on association with just a few microsolvating water molecules. For instance, HCl dissociated into Cl−(H2O)3 + (H3O+) upon association with just four water molecules.38 Similar behaviour by other strong mineral acids has been reviewed.39 It was established that as the acidity of an acid increased, the number of water molecules required to induce ionization tended to decrease.40,41 This trend afforded means of corroborating the remarkably high acidities computed for radicals.
The number of possible 3D arrangements of water molecules around a solute increases steeply as the number of water molecules increases. The potential energy surfaces become complex, with many shallow minima, making it difficult and very time-consuming to pinpoint global minima. Never-the-less we also investigated the effect of microhydration on a small selection of radicals with and without RED-shifts. Radicals associated with ˙Z–CO2 units were the carboxyl 12a, bicarbonate 12d, and carboxyethynyl 12e all showing appreciable RED-shifts, contrasted with carboxymethyl, 12b, for which DFT found no RED-shift. To extend the scope, the hydroperoxyl radical 10, in which the upe is associated with two adjacent O-atoms, was also studied.41
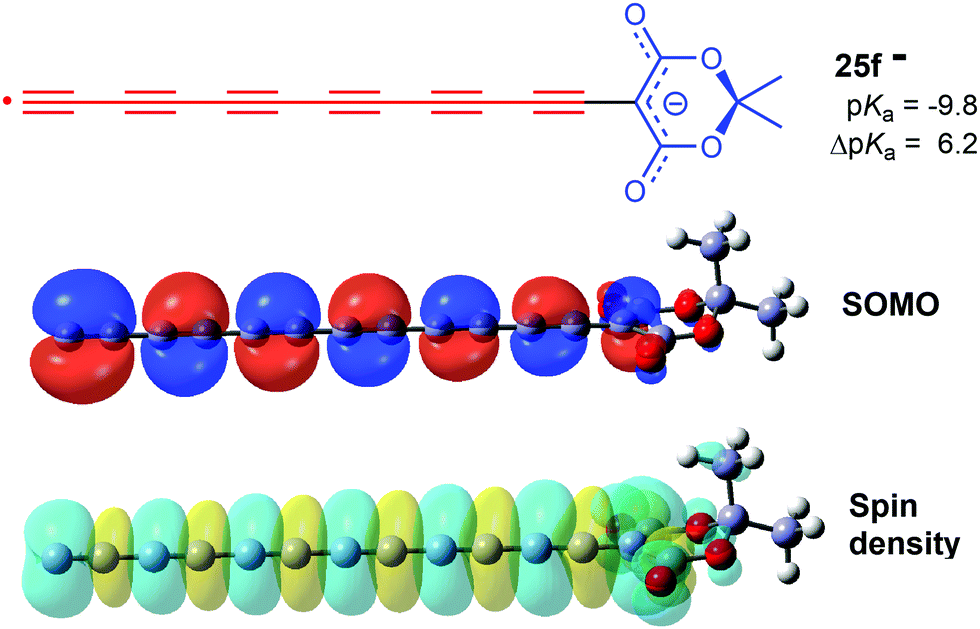
The structures and energies of individual complexes of these radicals were investigated starting with one water and gradually incrementing by one additional water at a time. Some surprising results were obtained. The carboxyl radical was known to exist as a pair of cis- and trans-conformers 12ac and 12at (= 8ac & 8at) with the latter about 1.7 kcal mol−1 lower in energy.42 From a stereoelectronic point of view, 12ac where the OH group is antiperiplanar with a radical would be anticipated to be more acidic than the trans conformer 12at. Scheme 4 shows that 12ac was indeed more acidic, although the difference was small. Microhydration of clusters of the cis-conformer [12ac·nH2O] were examined by DFT for n = 1 to 8 and for trans-conformer clusters [12at·nH2O] for n = 1 to 12. For each value of n several structures, sometimes many structures, with different arrangements of the water molecules, which were local minima were obtained. For clusters of the cis-conformer 12ac, partly ionised local minima first appeared with 4 × H2O ligands, but the global minimum remained unionized. For clusters with 5 × H2O, ten local minimum cluster structures were obtained and the global minimum was found to be an ion pair in which the carboxyl proton transferred to a water (see Fig. 2). An ion pair [12ac·5H2O] was obtained as a global minimum by five hydrating water molecules (NiG = 5). Surprisingly, many more water molecules were required to induce ionisation of trans-carboxyl 12at. Deprotonation did not take place until 11 water molecules were included in the cluster (NiG = 11). The cis arrangement of the HO group with the adjacent C–O bond in 12ac enabled two molecules to H-bond between the OH and carboxyl C–O, thus forming a 5-O-atom ring. Additional waters could then add whilst keeping ring structures intact. An ionized 3D cage of rings was obtained with 5 × H2O in which the carboxyl moiety sensed two H-bonds and each water sensed at least two (Fig. 2).
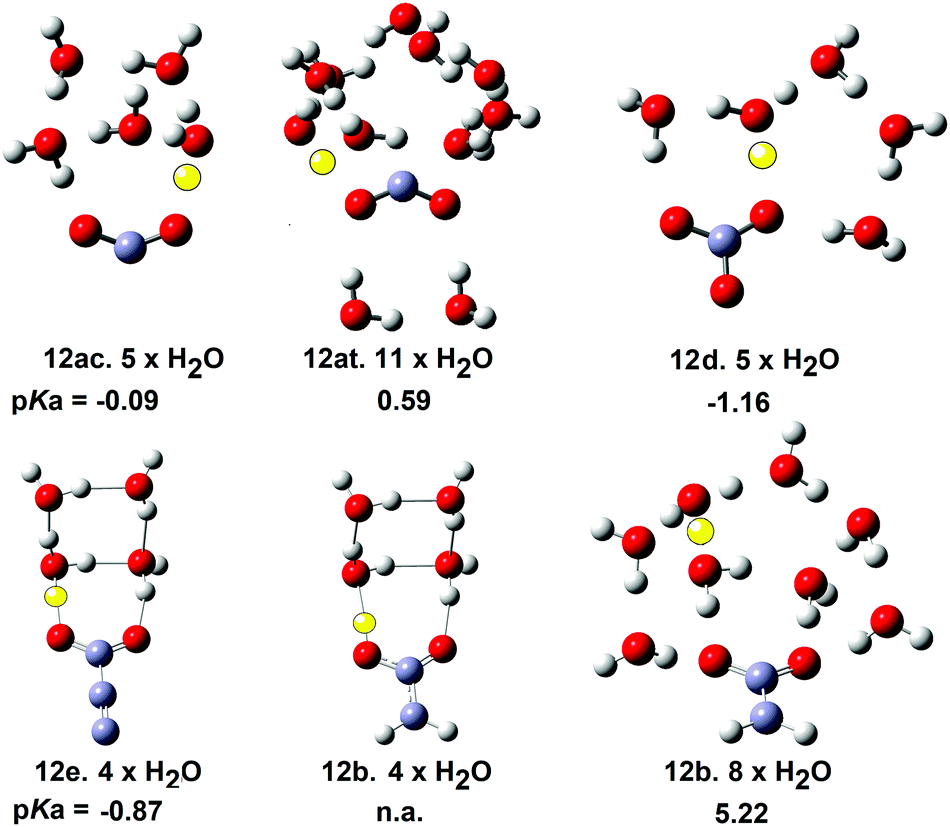 | ||
| Fig. 2 Microsolvated structures for radicals with carboxylate structures. The ionising proton is shown in yellow (n.a. = not applicable). | ||
With trans-conformer 12at, however, the trans-arrangement of the H–O group with the adjacent C–O meant that only a looser and larger ring of waters could stretch across the carboxyl group. The resulting H-bonds were weaker so that four water H-bonds to the carboxyl were required before ionization could take place (see Fig. 2).
In the gas phase the trans-conformer 12at is lower in energy than cis12ac. In aqueous solution conformer 12at remained lower in energy only when associated with 1 or 2 water molecules. For larger clusters (n > 2) cis12ac·nH2O were lower in energy than 12t·nH2O. In aqueous solution, therefore, thermodynamic control should ensure the cis-conformer predominates and deprotonation is completed by five waters. Ionization by only 5 waters is an indication of a strong mineral type acid so the microhydration data supports the experimental and DFT pKas (−0.2 and −0.1 respectively).
DFT study of microhydrated clusters for the bicarbonate radical 12d indicated that 5 × H2O were needed for spontaneous deprotonation (Fig. 2). Similarly, 4 × H2O were required for deprotonation of the carboxyethynyl radical 12e. The mineral acids HCl and H2SO4 require 4 and 5 microsolvating waters respectively for spontaneous ionisation. Thus radicals 21e and 12d have similar acidities to these mineral acids and this is in good accord with their computed pKa values.
Comparison with the microhydration data for the carboxymethyl radical 12b was very interesting. The structure of the global minimum cluster for 12b.4 × H2O was very similar to that of the carboxyethynyl radical 12e·4 × H2O (see Fig. 2). Note the much longer bond from the CO to the yellow proton in the 12e·4 × H2O structure signifying full ionization. Compare this with the shorter bond from the CO to the yellow proton in the 12b·4 × H2O structure signifying non-ionic character. Eight microhydrating H2O were needed for spontaneous ionization of 12b. This was in very satisfactory agreement with its much less acidic character computed by DFT (pKa = 5.22) and practically zero RED-shift. Thus the microhydration studies provided good evidence corroborating the magnitudes of RED-shifts derived from the DFT computations.
In the carboxylate-containing radicals the hydration clusters did not form round either the protruding CH2 or CC moieties but primarily round the CO2 units. The carboxyethynyl radical 12e had significant negative charge associated with its CC unit. A local minimum exhibiting H-bonding to the terminal C-atom of the ethyne unit in 12e·5 × H2O was found. However, this cluster was 10 kcal mol−1 higher in energy than the global minimum shown in Fig. 2. It was evident that H-bonding of H2O molecules to ethyne units contributed nothing to the stabilization the conjugate radical anion. It is likely that in general, for most carboxylic acids, water molecules cluster around the CO2 units in structures similar to those shown in Fig. 2.
The hydroperoxyl radical/superoxide radical anion conjugate pair (10/11) plays important roles in lipid peroxidations and in atmospheric chemistry.43 Different hydration behaviour was expected because HOO˙ contains two adjacent O-atoms rather than a CO2 unit. Many more H2O ligands were needed to induce deprotonation. The first cage containing ionized O2−˙ and H+ was obtained with 12 × H2O ligands, but was only a local minimum. Complete exploration of the entire potential energy landscapes for larger clusters was not possible. Both ionized and un-ionized forms were obtained for 12 up to 16 H2O ligands. For the 10·17 × H2O cluster the majority of structures were ionized. Remarkably, only a small structural difference was required to bring about spontaneous ionisation. A single water molecule moving from one peripheral site to another in the cluster was sufficient (“a” coloured blue in Fig. 3). It is probable that these larger hydration clusters existed as equilibria of ionized and un-ionized cages. The ‘number of water molecules needed to induce ionization’ became a fuzzy concept of little value for estimating acidity.
 | ||
| Fig. 3 Microsolvated structures of the hydroperoxyl radical 10 with 17 × H2O ligands. Left: Ionised proton in yellow, migrating water “a” in blue. Right: Unionised proton in yellow, migrating water “a” in blue. | ||
5. Release of electrophiles from free radicals
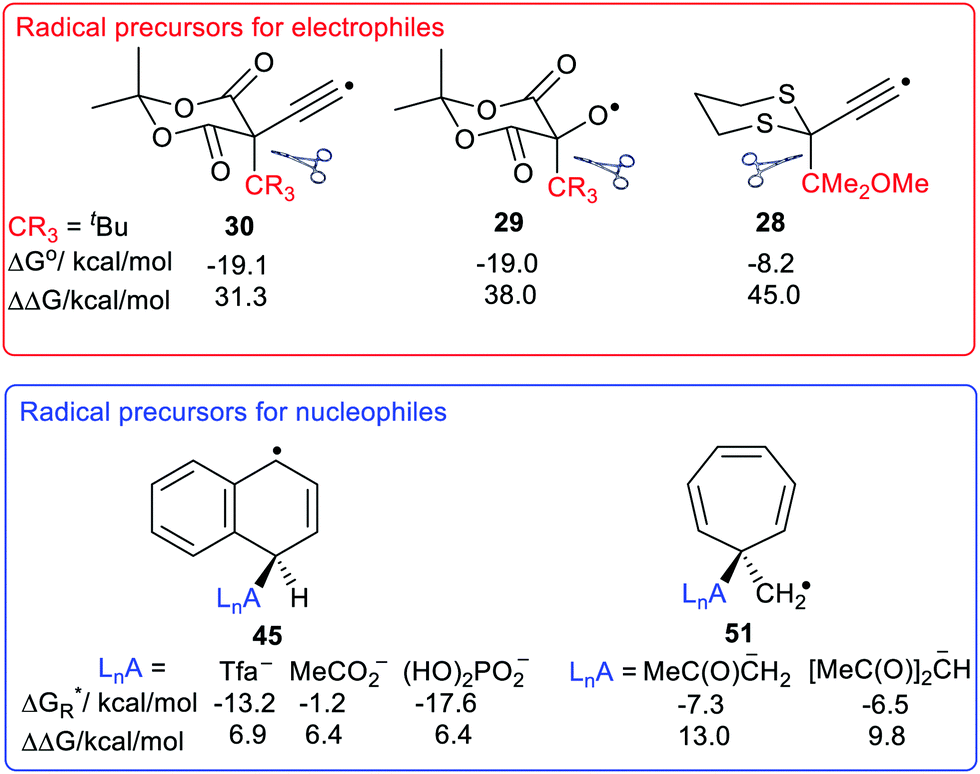
The large enhancements of deprotonations found for radicals suggested the intriguing possibility that suitable radicals RmZ˙–XRn could spontaneously undergo heterolytic dissociation with release of carbocations, i.e. electrophiles RnX+. Of course, electrophiles play crucial roles in a multiplicity of contemporary organic syntheses,44 as well as in cationic polymerisations. Homoallylic/cyclopropyl rearrangements, pinacol and other rearrangements are important in many natural product syntheses. Friedel–Crafts alkylations and acylations are amongst the most important routes to functionalized aromatic compounds. There were good practical reasons therefore for seeking soft ways, via free radicals, of producing these intermediates.We used a similar computational approach to test this scenario, starting with RmZ units that had shown the largest enhancements for proton release.45 We found that carbocation release was indeed significantly enhanced for species having certain radical centres adjacent to potential cations. For spontaneous carbocation release, heterolysis needed to be exothermic, or more specifically exergonic, as well as enhanced. Sulfate esters 27 and dithianes 28 furnished exergonic heterolyses with enhanced cation formation. However, the 5-oxyl-Meldrum's 29 and 5-ethynyl-Meldrum's 30 radical types were the most successful platforms that gave enhanced exergonic heterolytic release of the widest variety of cations. Probably, other 1,3-dicarbonyl containing radicals would work nearly as well.
The Meldrum's-based radicals 29 and 30 underwent exergonic dissociations with release of tertiary cations 32 and 33. For most other cation types, including primary 31, allylic 34 and benzylic 35, an electron releasing group such as MeO had to be incorporated to ensure exergonicity. Acyl type cations, 36 were released from the corresponding radicals of type 29 or 30 in exergonic processes (Chart 1). All these cation releases took place most readily in water as solvent. However exergonic heterolyses were also predicted for methanol, DMSO and acetonitrile as solvents as well as in mixed solvents with water.
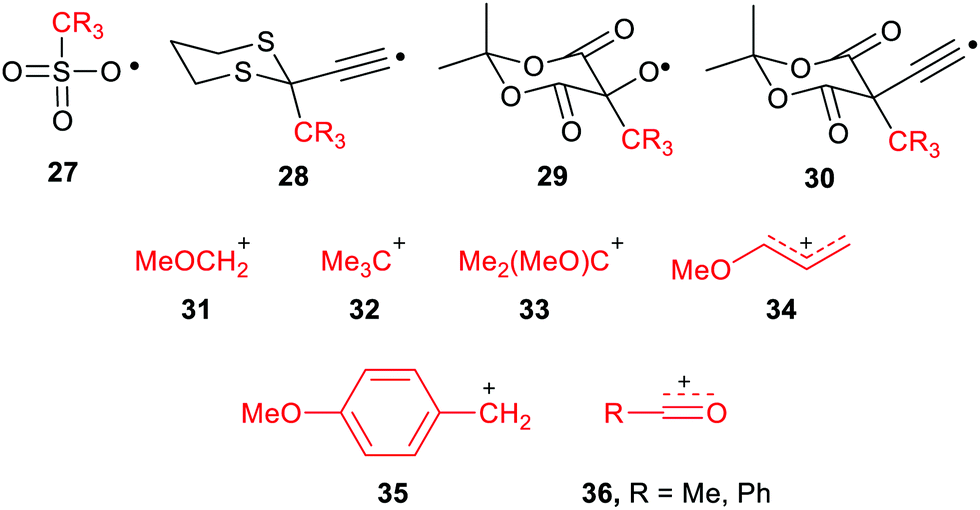 | ||
| Chart 1 Electrophiles and the radicals that spontaneously release them. | ||
Because of its importance in battery technology, radical enhancement of Li cation release was also examined. However, enhancements of Li cation release were small for all radical precursors, and all the heterolyses were endergonic. This was probably because the Li-substituted radical precursors were already rather salt like and at least partly ionic in character.
The ethyne spacer had proved very effective in enhancing deprotonations, so its effect was also examined in heterolyses forming carbocations. In the heterolytic release of t-butyl cations from Meldrum's precursor radicals 38 (CR3 = Me3C) the process remained exergonic, even when the formal radical centre was more than 6 ethyne units away from the departing cation (Scheme 8). The  values ranged from −19.9 for n = 1 to −7.5 kcal mol−1 for n = 6 and the enhancements reduced from ΔΔG = 33.4 to 12.2 kcal mol−1.46 For release of the 2-methoxyprop-2-yl cation from Meldrum's type radicals 38 (CR3 = Me2(MeO)C) the heterolyses were even more exergonic ranging from −42.1 to −45.6 with enhancements ΔΔG of 33.3 to 30.0 kcal mol−1 as ‘n’ increased from 1 to 6.
values ranged from −19.9 for n = 1 to −7.5 kcal mol−1 for n = 6 and the enhancements reduced from ΔΔG = 33.4 to 12.2 kcal mol−1.46 For release of the 2-methoxyprop-2-yl cation from Meldrum's type radicals 38 (CR3 = Me2(MeO)C) the heterolyses were even more exergonic ranging from −42.1 to −45.6 with enhancements ΔΔG of 33.3 to 30.0 kcal mol−1 as ‘n’ increased from 1 to 6.
 | ||
| Scheme 8 Influence of spacers on carbocation release from various radicals. | ||
The polyethynyl-dithiane radicals 41 also underwent exergonic heterolyses for all values of ‘n’ and the enhancements ΔΔG remained high at 38.5 kcal mol−1 even with 6 ethyne spacers (Scheme 8). Release of lithium ions from the polyethynyl-lithium precursors 43, having up to 7 ethynyl spacers, was investigated in tetrahydrofuran solvent. These dissociations were all endergonic with  for n = 1 to n = 6. In every case, however, modest enhancements relative to the non-radical species were obtained i.e. ΔΔG reduced from 10.4 to 4.1 kcal mol−1 for n = 1 to n = 6.
for n = 1 to n = 6. In every case, however, modest enhancements relative to the non-radical species were obtained i.e. ΔΔG reduced from 10.4 to 4.1 kcal mol−1 for n = 1 to n = 6.
The same trend as with deprotonations was observed. That is of more facile carbocation release when the conjugate radical anions were stabilised by charge delocalized away from their anionic centres, coupled with spin density distributed toward these centres.
6. Radical-stimulated nucleophile release
6.1 Competition between heterolysis and 1,2-ester migration
The success of radicals in enhancing heterolytic dissociations with release of protons and carbocations raised the question of whether the inverse heterolytic dissociations that released nucleophiles could also be facilitated. Thus, model compounds HRmZ–ALn [Scheme 1, eqn (2)] contain a potential radical centre RmZ bonded to the potential nucleophile −ALn. The radical species generated by loss of the H atom could heterolytically dissociate to the cation-radical RmZ+˙, together with nucleophile −ALn. Could this be significantly easier than with the closed-shell precursor? Clearly the structural features required for electron delocalization/stabilization of radical-cations RmZ˙+ would be rather different than for the radical-anions RmZ˙− that accompanied deprotonations.Nucleophiles are key reagents for a huge variety of organic synthetic applications. Those with one or more stabilizing substituents such as enolates and azaenolates are workhorses for preparations of carbon–carbon and carbon–heteroatom bonds. For carboxylate anion nucleophiles experimental evidence already exists that they can be released by heterolytic fragmentation of β-(acyloxy)alkyl radicals (37, A = C; Scheme 9).
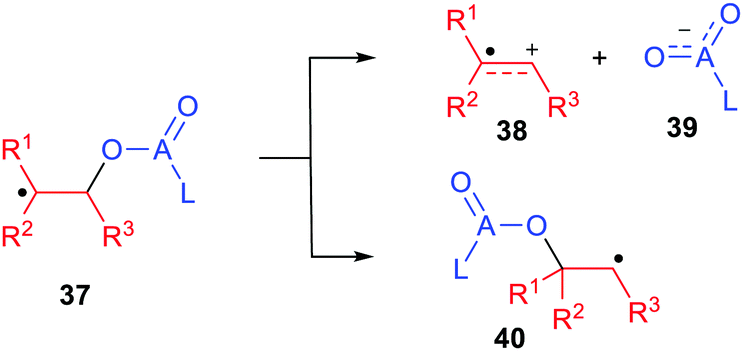 | ||
| Scheme 9 Heterolytic dissociations and 1,2-shifts of β-(acyloxy)alkyl and β-(phosphatoxy)alkyl radicals. | ||
Product analyses provided good evidence of heterolytic fragmentation to radical cations 38 together with carboxylate anions 39 (A = C) that was backed up by EPR spectroscopic and electrochemical data.47,48
This dissociation was found to be in competition with the 1,2-migration of the ester moiety yielding the rearranged radical 40 (Scheme 9). Research from the groups of Surzur,49 Tanner,50 Beckwith,51 Giese52 and others demonstrated this.53 Crich and co-workers,54 and Giese and co-workers,55 discovered that β-(phosphatoxy)alkyl radicals 37 (A = P) undergo an analogous but even faster 1,2-migration. These migrations occur in competition with heterolytic dissociations that release the phosphate ester nucleophile as demonstrated by research from the groups of Schulte-Frohlinde,53 Giese,56,57 Crich,58 and others.
These reactions are biologically significant because the nucleotide C4′ radicals, formed by radical damage of nucleic acids, contain structural units similar to that of 37 (A = P). Their heterolytic fragmentation is implicated in RNA and DNA strand breaks. Model nucleotide radicals fragment to radical cations plus phosphate anions as was demonstrated in several important studies.57,58 Beckwith, Crich and co-workers published a comprehensive and illuminating review bringing into focus this whole elaborate field.59 For two types of ester nucleophiles there was, therefore, good evidence that they were released from appropriate radicals.
6.2 Influence of radical type on nucleophile release
We used our DFT computational method to explore the types of radicals for which release of nucleophiles would be spontaneous.60 For useful releases of nucleophiles, that would be concerted with radical generation, heterolyses that were enhanced (ΔΔG positive) and also exergonic were sought. Strongly stabilised nucleophiles would be needed to achieve this so the ‘Meldrumate’ anion (Mm) was chosen for C-centred nucleophiles and the 2,2,2-trifluoroacetate anion (Tfa) was selected to represent O-centred nucleophiles (see Scheme 10). For the former nucleophile it was found that considerable resonance stabilisation of the accompanying radical-cation was required. A good range of structures were tested and structures containing the 1,4-dihydronaphthalene, 45, 9,10-dihydroanthacene, 42, 46 or the fluorenyl, 43, 44, units were generally useful (Scheme 10).
were sought. Strongly stabilised nucleophiles would be needed to achieve this so the ‘Meldrumate’ anion (Mm) was chosen for C-centred nucleophiles and the 2,2,2-trifluoroacetate anion (Tfa) was selected to represent O-centred nucleophiles (see Scheme 10). For the former nucleophile it was found that considerable resonance stabilisation of the accompanying radical-cation was required. A good range of structures were tested and structures containing the 1,4-dihydronaphthalene, 45, 9,10-dihydroanthacene, 42, 46 or the fluorenyl, 43, 44, units were generally useful (Scheme 10).
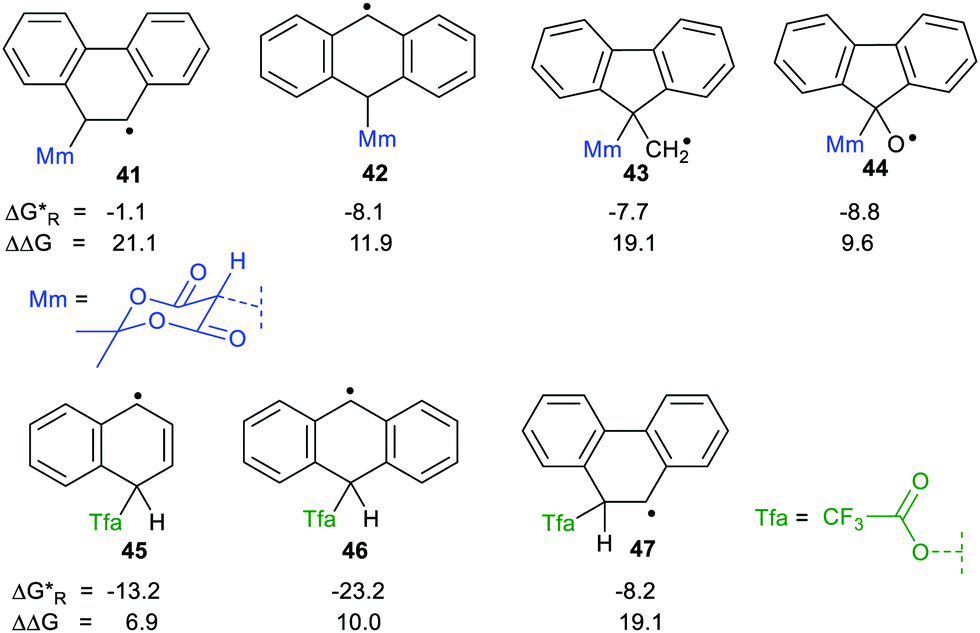 | ||
| Scheme 10 Energetics for release of ‘Meldrumate’ and trifluoroacetate nucleophiles from selected radicals. | ||
The Gibbs free energies were computed for both heterolyses and 1,2-migrations for the trifluoroacetate-containing radicals. The 1,2-migrations were found to be the lower energy pathways in most cases. Successful release of trifluoroacetate, and other carboxylate nucleophiles could, however, be achieved with radicals 45 and 46 where the radical centre is not vicinal and with radical 47 where 1,2-migration is degenerate, i.e. gives back the same precursor radical.
Spontaneous release of the C-centred ‘Meldrumate’ nucleophile was more difficult than release of O-centred trifluoroacetate; as might be expected from the greater electronegativity of the O-atom. The computations showed that the enhancements of the heterolyses were greater for C-centred radical precursors compared to O-centred radicals. This is the reverse of what was observed for heterolyses of radicals yielding protons or carbocations. This makes good sense because delocalisation of the positive charges in the radical-cations formed on nucleophile release would not be favoured by nearby O-atoms. On the other hand, delocalisation of the negative charges in the radical-anions released from heterolyses giving cations would be favoured by neighbouring electronegative O-atoms.
6.3 Scope for nucleophile release
Scheme 11 shows the considerable range of nucleophile types that could be spontaneously obtained from dihydronaphthalenyl carrier radicals (as in 45). | ||
| Scheme 11 Energetics of heterolytic dissociations of dihydronaphthalenyl-linked nucleophiles. | ||
The dissociations gave rise to enhancements in the range 6.2–6.7 kcal mol−1, for each of the nucleophiles. Exergonic heterolyses were found for carboxylate, bicarbonate, sulfonate and phosphate esters; only hydroxide was endergonic. The implication of the data for RCO2− type nucleophiles is that all types of carboxylates would be spontaneously released from this platform.
The radical carriers 49, 50 and 51 (Chart 2) were tested for release of chosen enolates (a, b) and 1,3-dicarbonyl nucleophiles (c, d and e). Although substantial enhancements were computed for all three carriers, only the cyclohepta-2,4,6-trienylmethyl carrier 51 gave exergonic releases of the enolates a and b. A similar picture emerged with the 1,3-dicarbonyl nucleophiles; only 51 afforded enhancement plus exergonicity (Chart 2).
 | ||
| Chart 2 Energetics for release of enolate and 1,3-dicarbonyl nucleophiles. | ||
6.4 Radical enhanced delivery of phosphate nucleophiles and cleavage of nucleic acids
Release of phosphate [(HO)2P(O)O−], when linked to the dihydronaphthalenyl carrier 45, was predicted to be exergonic and enhanced even though it was three bonds distant from the radical centre (see Scheme 11). The fluorenylmethyl (43 & 50) and cycloheptatrienyl (51) carriers for phosphate would probably yield heterolyses that were even more exergonic. However, in agreement with experimental results mentioned above, DFT computations indicated that 1,2-phosphate migration would be preferred for these and related precursors where the radical centre and leaving nucleophile are vicinal.The exergonic release of phosphate from 45 was of special interest because phosphate is a key constituent of nucleotides, DNA and RNA. It is well established that Reactive Oxygen Species (ROS) including hydroxyl radicals,61 and hydroperoxyl radicals62 abstract H-atoms from nucleotides to yield, amongst other intermediates, C4′ sugar radicals. These C4′-nucleotide radicals (type 52) are generated by enediyne antitumor agents,63 including calicheamicin, esperamicin, and neocarzinostatin, which abstract H atoms from C4′-sugar sites of DNA and RNA.49,64 (Scheme 12) These radical types are reckoned to be important players in DNA single and double strand breaks by heterolytic dissociation of their phosphate linker groups.65 To investigate the role of radical enhancement of these strand breaks the energetics of dissociation of the phosphate-substituted deoxyribose radicals 52, depicted in Scheme 12, were computed.60
 | ||
| Scheme 12 DFT Computed energetics of heterolytic dissociation of model C4′ nucleotide radicals. | ||
In mode (a), dissociation of the C5′-phosphate linkage of the C4′-radical (52) yields a phosphate anion [LnP5−], together with the exocyclic radical-cation 53. In mode (b), dissociation of the C3′-phosphate linkage yields a phosphate anion [LnP3−] together with cyclic radical-cation 54. 1,2-Migration of phosphate in C4′-nucleotide radicals like 52 is disfavoured because the rearranged radicals would be thermodynamically less stable. The computed Gibbs free energies66 ΔG* and enhancement factors ΔΔG, relative to models with the upe replaced by H-atoms, are in Scheme 12.
The initial cations released on heterolyses of the closed-shell deoxyribose models (analogous to 53 and 54 but with H in place of the upes) were unstable and rearranged to lower energy cations. Consequently the ΔΔG enhancements in Scheme 12 are less than the true enhancements. Never-the-less the data showed significant enhancements for breaking all the C5′-phosphate linkages and all the C3′-phosphate linkages. Similar energetics were also obtained for analogous phosphate-ribose radicals (not shown). It can be concluded that vicinal radical centres enhance heterolytic dissociation of nucleotides based on both types of sugar.
The heterolyses of the C3′-phosphates (b) were found to be more exoergonic than dissociations of the C5′-phosphates (a) (Scheme 12). C1′-Substitution by cytosine (and also OH, not shown) had little effect on the free energies or enhancements of C5′-phosphate release (a) from 52. Interestingly, the release of the triphosphate group [(HO)4P3O6−] was more energetically favourable for both C5′- and C3′-linkages than release of the dihydrogen phosphate group. The dihydrogen phosphate and triphosphate groups were released more readily in the deoxyribose series than in the ribose series (not shown). This is in good accord with experimental results showing diminished reactivity of RNA compared to DNA.59,67
Predictably, both C3′ and C5′ cleavages were computed to be endergonic for doubly charged HOPO32− ions and even more endergonic for triply negative PO43− ions. These results harmonise with the pH dependence found experimentally for various oligonucleotide fragmentations.59 The large rate decreases (∼3 orders of magnitude), reported by Schulte-Frohlinde and co-workers53 for fragmentations of model phosphatoxy-substituted alkyl radicals, as the charge on the phosphate groups increased, are also in agreement. These DFT results are fully supportive of the afore-mentioned experimental findings that generation of C4′-nucleotide radicals will lead to strand breaks in DNA and RNA.
Release of triphosphate or phosphate from ATP and AMP and related nucleotides is also predicted to occur on generation of their C4′-radicals. The phosphate and triphosphate linkages in these species are deprotonated at normal physiological pH. As Scheme 12 shows, heterolytic dissociations with release of the ‘bare’ doubly charged HOPO22− or triply charged PO43− ions are endergonic. However, in inter- and intra-cellular fluids the charged phosphate O-atoms are associated with Mg2+ cations which act as counter-ions neutralising the negative charges.68 Consequently, the heterolyses are expected to resemble those of the protonated species and to readily occur.
6.5 Solvent effects on release of nucleophiles
DFT studies on release of Meldrumate and on release of trifluoroacetate ions from a benzylmethyl radical precursor were carried out with Truhlar's SMD continuum description of the solvent.26,69 Both heterolyses became less favourable as the solvent polarity decreased from water through methanol, acetonitrile and THF to n-heptane.60 The free energies of heterolysis became less favourable fairly smoothly as the Hildebrand solvent parameters δ decreased. Trifluoroacetate release was exergonic in methanol and acetonitrile as well as in water. The enhancement factors (ΔΔG) were practically independent of the solvent. Likely this is because the solvation extents were similar for the reactants and products for the neutral models and their corresponding radicals. Water was always the best solvent for heterolyses, but mixed solvents of water with methanol, acetonitrile, DMF, or DMSO could be used with little diminution in enhancement.6.6 Effect of spacers on nucleophile release from radicals
Heterolytic dissociations of radical precursors were computed to be inhibited by introduction of the CH2 unit, or longer hydrocarbon chains, between the formal site of an upe and the nucleophile.60 The same was true for the bicyclo[1.1.1]pentane spacer. Unlike the cases of proton or cation release, the simple ethynyl spacer was usually ineffective too. The key factor was, of course, the stabilisation of the radical-cation released along with the nucleophile. The ethynyl radical is a σ-radical with its upe in an sp-orbital orthogonal to the adjacent π-system, and this prevents delocalisation of the upe. To circumvent this problem the ˙CH2CC–, ˙CH2CHCH– and ˙CH2C6H6– units, where the upe resides in a p-orbital available for conjugation with its neighbour bonds, were examined as spacers (Scheme 13).60
 | ||
| Scheme 13 Influence of spacers on radical-promoted release of trifluoroacetate. | ||
With trifluoroacetate as the attached nucleophile heterolyses were endergonic or only marginally exergonic, irrespective of chain length, for these three spacers. Interestingly, however, release of trifluoroacetate was enhanced, relative to the corresponding models, for all three types of spacer. The enhancement reduced to essentially zero for two or more ethenyl spacers 56. Remarkably, for the aryl series 57, enhancement (ΔΔG +ve) was observed for up to 3 phenyl ring spacers. Surprisingly, therefore, an upe separated by 3 aryl rings, that is by 13 C–C bonds, from the incipient positive charge had a detectible effect in the cation radicals. The ethynyl spacers 55 were the most effective in that modest ΔΔG values remained measurable out to at least six ethyne spacers.
Several lines of evidence supported the conclusion that the key factor favouring heterolytic dissociation to a nucleophile is the resonance stabilization of the released radical-cations (RmZ+˙). The much greater π-conjugation in the frontier SOMOs of the radical-cations RmZ+˙ compared to the HOMOs of the cations HRmZ+, released in the analogous heterolyses of the model compounds, gave an apt illustration of this.
The greater extent of π-conjugation in the SOMO of the fluorenylmethyl radical cation (Fig. 4a, right) compared to the cation from the model compound (Fig. 4a, left) is quite clear. The frontier orbital of the model tetradeca-2,4,6,8,10,12-hexayn-1-ylium cation (Fig. 4b, left) takes the form of an orthodox π-system. This contrasts with the remarkable π-double helix of the resultant radical-cation (Fig. 4b, right). This SOMO extends over all 14 C-atoms and has only one helically disposed node. The additional conjugation in the SOMOs of the radical cations results in greater thermodynamic stabilization than for the cations of the model compounds.
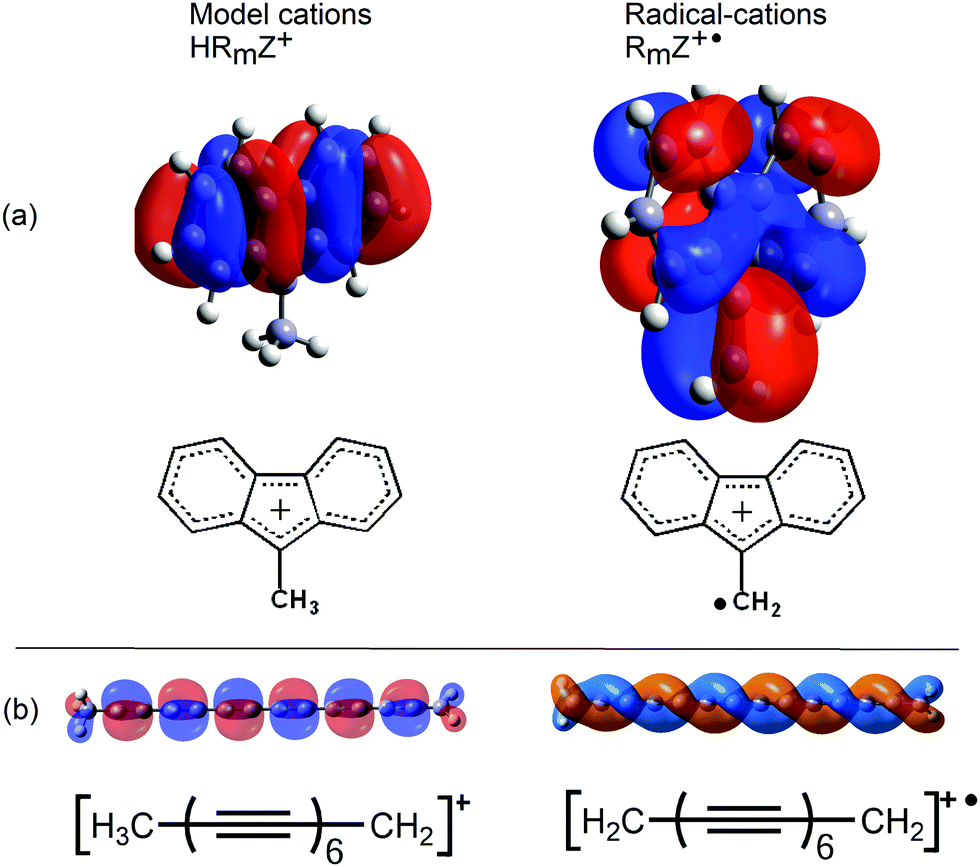 | ||
| Fig. 4 Frontier HOMOs and SOMOs of fluorenylmethyl and hexaynylmethyl model cations and radical-cations. | ||
7. Significance of enhanced heterolysis in biological systems
Radicals are produced in biological systems in a variety of ways. ROS, especially hydroxyl radicals, abstract H-atoms with production of neutral free radicals from lipids and other components. Electron transfer either by enzymes or by exogenous ionising radiation can generate radical anions and radical cations. Single electron transfer by reductases to carbonyl-containing substrates generates radical-anions such that nearby H-atoms possess enhanced acidity. Buckel and co-workers studied the dehydratase promoted reaction of (R)-2-hydroxyglutaryl-CoA 55a.70 Ketyl type radical 56a was generated by enzymatic one electron transfer and subsequently eliminated its 2-hydroxy group to produce radical 57a (Scheme 14). The acidity of the γ-H-atom of 57a was enhanced by the adjacent radical centre and so 57a readily deprotonated. The resulting allyl type radical 58a was then oxidised to afford (E)-glutaconyl-CoA. | ||
| Scheme 14 Radical enhanced enzymatic dehydration. | ||
A pKa of ca. 14 units was computed for radical 57a which corresponded to an enhancement of 7 units compared to the non-radical model.71 A similar mechanism was proposed for the enzyme-catalysed dehydration of (R)-2-hydroxy-4-methylpentanoyl-CoA 55b. In this instance the allyl-type radical 58b was characterised by EPR spectroscopy.72 This sequence could be adapted for the functionalisation of γ-H-atoms in ketones containing β-hydroxy, or other, leaving groups.
Wessig and Muehling described another type of system where the increased acidity of the proton plays an important role. In α-hydroxy- or α-alkoxy-radicals 59, with an adjacent leaving group X, a rapid elimination takes place to afford either a ketyl radical 60 or radical cation 61.73 The spin centre shifts one atom during this process (Scheme 15). They showed that this process was important in the deoxygenation of ribonucleotides to deoxyribonucleotides and in the inactivation of ribonucleotide reductase. Furthermore the scope of the process in several synthetic methods was also explored.
 | ||
| Scheme 15 Eliminations from α-hydroxy- or α-alkoxy radicals. | ||
Radical centres are produced in DNA and other polynucleotides by impacting ionising radiation, by ROS, by enediyne antitumor agents, and by redox enzymes. Several types of oxidising agents remove single electrons from DNA and the most easily oxidised site is the guanine base.74 The proximity of the bases one to another along the helical axis of DNA promotes efficient inter-base electron transfer. Thus oxidations of nucleic acids tend to generate radical-cations of type 63 with their positive charges associated with the guanine unit (Scheme 16).
 | ||
| Scheme 16 Facilitation of inter-base proton transfers in DNA. | ||
The DNA double helical structure places the bases together in pairs and this has a marked effect on the chemistry.75 Steenken reviewed reports of how proton transfers from base to base can be facilitated with consequent effects on reactivity.76 The deoxyguanosine in polynucleotide 62 is a weak acid with a pKa of 9.4. The radical-cation 63, formed on oxidation, is a much stronger acid with a pKa of 3.9. In the DNA double strand the structure is perfectly aligned for proton transfer between bases. For radical-cation 63 proton transfer to the complementary cytosine unit readily takes place with production of species 64 having a different distribution of charge and unpaired spin and hence different properties and reactivity.
As mentioned above, the H-atoms of the sugar units of DNA and RNA can readily be abstracted by ROS or the diradicals from enediyne antibiotics, so C4′-nucleotide radicals 65 of the type shown in Scheme 17, are formed by these means together with other radicals. The C4′-radical centre promotes spontaneous heterolysis with elimination of phosphate. The result for DNA or RNA is a strand break as in structure 66. Subsequent radical attack on a sugar of the complementary polynucleotide strand will lead to a second heterolysis and complete break of the double helical structure. The radical enhancement of heterolysis with phosphate nucleophile release thus rationalises the initiation this type of DNA degradation.
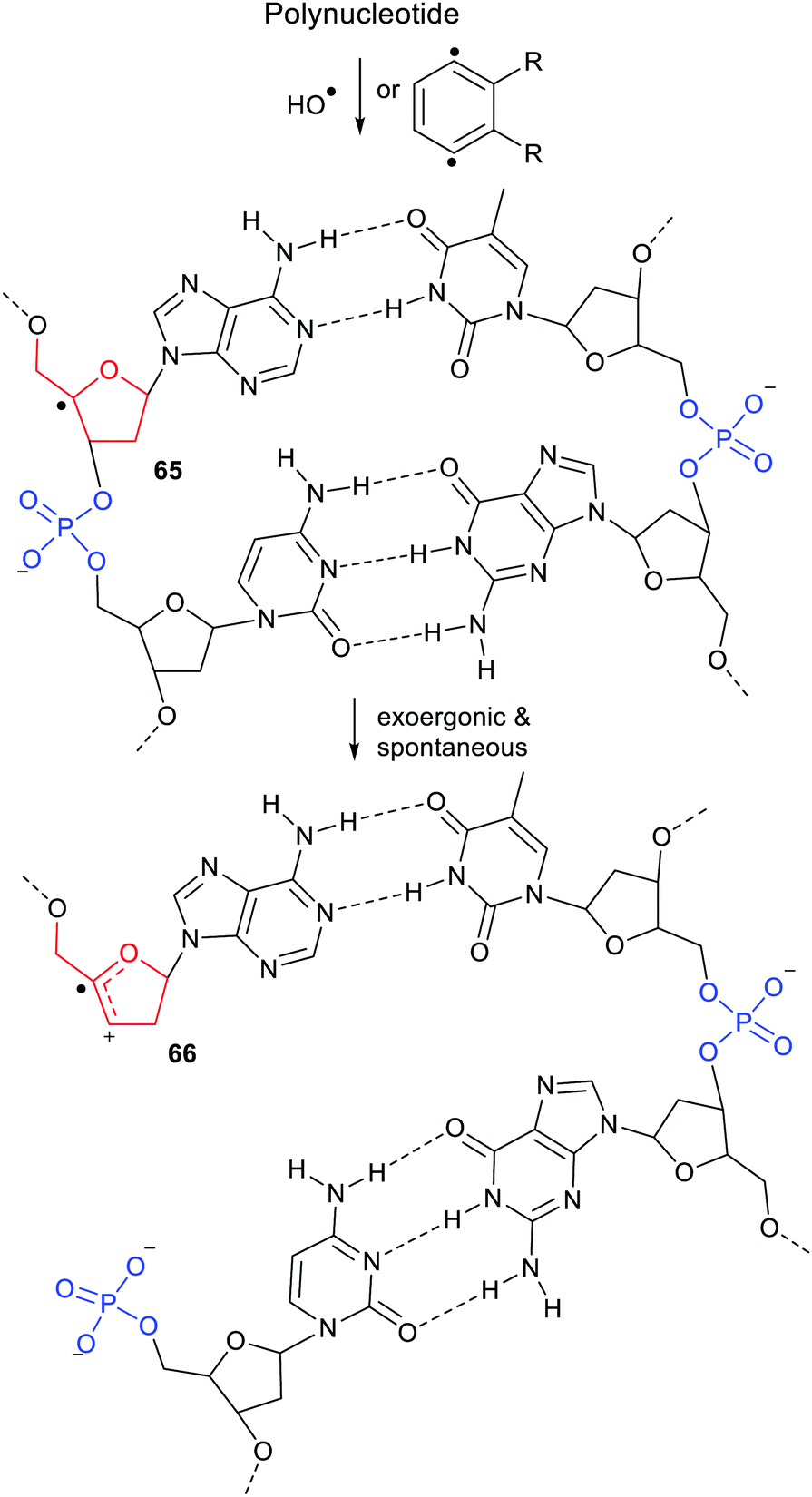 | ||
| Scheme 17 DNA strand break by radical enhanced heterolysis. | ||
8. Miscellaneous actual and potential applications
Inter- and intra-molecular homolytic aromatic substitutions are important processes for functionalising arenes and heteroarenes. Intramolecular versions are particularly popular and numerous preparations of quinolines, phenanthridines and related heterocycles have been reported. For example the natural product trispheridine 70 has been accessed by a variety of such approaches (Scheme 18). Iminyl radical 68 generated from an oxime derivative 67 (X = Ar, Ph, C(O)ON = CHAr, etc.) ring closes to produce cyclohexadienyl type radical 69 that is oxidised to trispheridine.77 Many variants of this strategy exist with C-centred or N-centred radicals adding or ring closing to different types of aromatic acceptors to yield functionalised arenes and heteroarenes.78 The exo-cyclic H-atom of the ring-closed radical 69 is rendered acidic because of the proximate radical centre. Only weak base is needed to complete the oxidative aromatisation step (Scheme 18).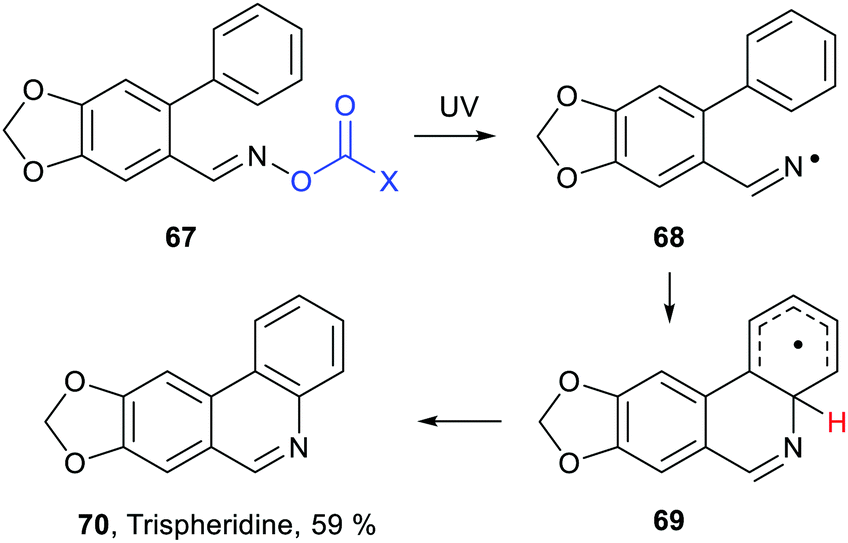 | ||
| Scheme 18 Example of intramolecular homolytic aromatic substitution. | ||
With some bases B− electron transfer to an adduct radical 71 accompanies H-abstraction (Scheme 19). The anion-radical 72 may then transfer an electron to the radical source R–L so providing the aromatised substitution product 73 and initiating another cycle in a chain process. In these specific instances the strong reductants 72 that are formed provide examples of electron ‘upconversion’ that mediates the chain process.79 These processes are Base Promoted Homolytic Aromatic Substitutions (BHAS). Many examples of intermolecular and intramolecular BHAS reactions have been reported,80 and alternative electron-catalysed mechanisms have been proposed.81
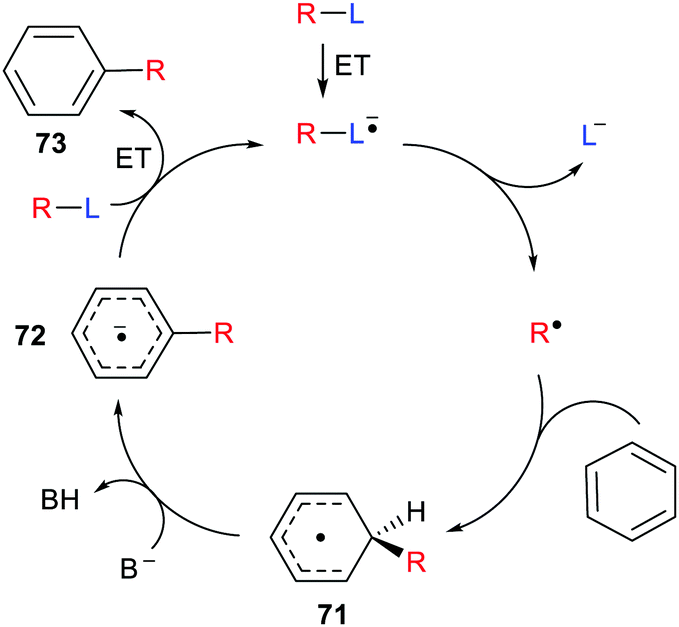 | ||
| Scheme 19 Mechanism of base promoted homolytic aromatic substitutions. | ||
Transient acidic radicals can be generated either with steady or pulsed irradiation or by heat. Several different devices based on this property appear possible. Control of pH simply by applying light could be achieved. With a pulsed light source, intermittent or programmed pH variation could be obtained. By means of pH sensitive dyes, light addressed display devices could be constructed. Because conductivity usually varies with pH, light operated switches also appear feasible. Several radical types, for example 12e, 14e, 19c and 19d (Scheme 20) exhibited extreme acidity of the superacid class. These are Brønsted acids and so might be milder and more user-friendly reagents than the corrosive Lewis superacid mixtures usually employed for generating carbocations from hydrocarbons.
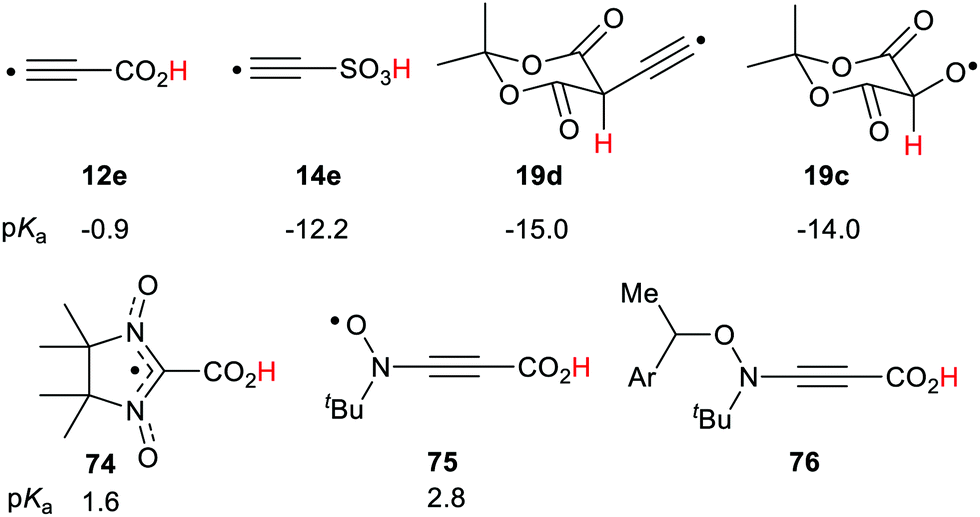 | ||
| Scheme 20 Selected transient and persistent acid radicals. | ||
4,4,5,5-Tetramethyl-imidazolyl-3-oxyl-2-carboxylic acid 74 is an isolable compound,82 and a strong acid (pKa = 1.6). It had a small ΔΔG of 0.9 units making it a stable, persistent acid radical. The t-butyl-2-carboxyethynyl nitroxide 75, with the alkyne spacer, was found to be a strong acid with an appreciable enhancement (ΔΔG = 1.34). A library of persistent acidic radicals could be obtained based on these compound types. Nitroxide radicals can exist in equilibrium with suitable alkoxyamines so that radical 75 could be generated by mild thermal dissociation of a precursor such as 76. This type of acidic nitroxide radical could find useful applications in nitroxide mediated radical polymerizations (NMP). These examples show possible applications of the RED-shift phenomenon. Prospects for novel chemical transformations incorporating radical promoted –CH(R)– activation appear very promising.
9. Conclusions
Experiment and theory support one another in showing that nearby radical centres enhance the ease of heterolytic dissociations of C–H, C–C and C–X bonds. The enhancements can be very large so that, for example, radicals of types 14e, 19d and 19c belong to the class of superacids. The main factor causing this phenomenon was found to be the additional thermodynamic stabilisation of the radical anions or radical cations released during the heterolyses. This stabilization resulted when their structures permitted both spin and charge to be delocalised from their formal sites of origin. Radical centres immediately adjacent to the bond undergoing heterolysis were the most effective. For release of protons or electrophiles, radical precursors with electrophilic radical centres were more effective the order being: C˙ < N˙ < O˙. For heterolyses releasing nucleophiles this order was reversed. Hydrocarbon and other spacers between the upe and the breaking bond usually abolished any enhancement. The main exception was ethyne spacers. Introduction of ethyne units dampened the enhancement but this could still be appreciable even with long polyethyne chains between the upe and breaking bond.The objective of marrying soft radical generation with the ease and power of nucleophile/electrophile synthetic strategies is clearly achievable. As sources of protons a host of carboxylic, sulfonic and carbon acid radicals are available including 12d, 12e, 18b, d, 19a–d. For use as sources of electrophiles, radicals that spontaneously dissociate on generation are required. Very effective radical platforms for this purpose were the Meldrum's types 29 and 30 (Chart 3). These spontaneously released tertiary-cations and also primary, allylic, benzylic and acyl cations, provided an electron releasing group was incorporated. The ethynyl-1,3-dithiane radical types 28 were also potentially useful, particularly for release of electrophiles stabilised with electron-releasing groups.
 | ||
| Chart 3 Radical precursors of choice for spontaneous release of electrophiles and nucleophiles. | ||
As precursors for spontaneous release of ester and phosphate nucleophiles the 1,4-dihydronaphthalen-4-yl radical types 45 were good choices. The QM computations indicated that for release of enolates or 1,3-dicarbonyl type nucleophiles radicals of the cyclohepta-2,4-6-trien-1-ylmethyl 51 would be effective (Chart 3).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I’m very grateful to Dr Herbert Fruchtl and Professor Michael Bühl for their unsparing help with the computations. I thank several generations of undergraduate and postgraduate students including Rory McCaughan, Tom Francis and Tallulah Hutson for their able and enthusiastic help. Thanks to the University of St. Andrews and the EaStCHEM Research Computing Facility for financial and computational support.Notes and references
- See for example:
E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, Wiley, New York, 1995 CrossRef CAS PubMed
; S. Warren and P. Wyatt, Organic Synthesis: The Disconnection Approach, Wiley, 2008 CrossRef CAS PubMed
- See for example: M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed
- E. Hayon and M. Simic, Acc. Chem. Res., 1974, 7, 114 CrossRef CAS
-
(a) H. Zeldes and R. Livingston, J. Chem. Phys., 1967, 47, 1465 CrossRef CAS
-
(a) J. Lilie, G. Beck and A. Henglein, Ber. Bunsen-Ges., 1971, 75, 458 CrossRef CAS
-
(a) H. E. K. Matimba, A. M. Crabbendam, S. Ingemann and N. M. M. Nibbering, Int. J. Mass Spectrom. Ion Processes, 1992, 114, 85 CrossRef CAS
-
(a) M. Simic, P. Neta and E. Hayon, J. Phys. Chem., 1969, 73, 3794 CrossRef CAS
-
(a) A. Fojtik, G. Czapski and A. Henglein, J. Phys. Chem., 1970, 74, 3204 CrossRef CAS
-
(a) M. Simic, P. Neta and E. Hayon, J. Am. Chem. Soc., 1970, 92, 4763 CrossRef CAS PubMed
- E. Hayon and M. Simic, J. Am. Chem. Soc., 1973, 95, 6681 CrossRef CAS
- P. S. Rao and E. Hayon, Anal. Chem., 1976, 48, 564 CrossRef CAS
-
(a) P. M. Mayer, M. N. Glukhovtsev, J. W. Gauld and L. Radom, J. Am. Chem. Soc., 1997, 119, 12889 CrossRef
- M. Morris, B. Chan and L. Radom, J. Phys. Chem. A, 2012, 116, 12381 CrossRef CAS PubMed
- A. S. Menon, T. Bally and L. Radom, J. Phys. Chem. A, 2012, 116, 10203 CrossRef CAS PubMed
- See for example:
(a) S. V. Lymar and J. K. Hurst, J. Am. Chem. Soc., 1995, 117, 8867 CrossRef CAS
-
(a) O. P. Chawla and R. W. Fessenden, J. Phys. Chem., 1975, 79, 2693 CrossRef CAS
- D. A. Armstrong, W. L. Waltz and A. Rauk, Can. J. Chem., 2006, 84, 1614 CrossRef CAS
-
(a) D. Lapenna, G. Ciofani, S. D. Pierdomenico, M. A. Giamberardino and F. Cuccurullo, FEBS Lett., 2005, 579, 245 CrossRef CAS PubMed
- A. S. Jeevarajan, I. Carmichael and R. W. Fessenden, J. Phys. Chem., 1990, 94, 1372 CrossRef CAS
-
(a) D. Behar, G. Czapski, J. Rabani, L. M. Dorfman and H. Schwarz, J. Phys. Chem., 1970, 74, 3209 CrossRef CAS
-
T. Francis, Bisulfite radical – what is its lifecycle in living systems?, MChem dissertation, University of St. Andrews, 2018 Search PubMed
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed
-
(a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS
- M. D. Tissandier, K. A. Cowen, W. Y. Feng, E. Gundlach, M. H. Cohen, A. D. Earhart, J. V. Coe and T. R. Tuttle, J. Phys. Chem. A, 1998, 102, 7787 CrossRef CAS
- J. C. Walton, J. Phys. Chem. A, 2018, 122, 1422 CrossRef CAS PubMed
- J. C. Walton, J. Phys. Chem. A, 2017, 121, 7761 CrossRef CAS PubMed
- M. Bühl, P. DaBell, D. W. Manley, R. P. McCaughan and J. C. Walton, J. Am. Chem. Soc., 2015, 137, 16153 CrossRef PubMed
-
(a) J. C. Walton, Chem. Soc. Rev., 1992, 21, 105 RSC
-
(a) B. Maillard and J. C. Walton, J. Chem. Soc., Chem. Commun., 1983, 900 RSC
-
(a) H. McNab, Chem. Soc. Rev., 1978, 7, 345 RSC
- C. A. Reed, K.-C. Kim, R. D. Bolskar and L. J. Mueller, Science, 2000, 289, 101 CrossRef CAS PubMed
-
(a) H. A. Bent, J. Chem. Phys., 1959, 33, 1258 CrossRef
- I. V. Alabugin, S. Bresch and G. dos Passos Gomes, J. Phys. Org. Chem., 2015, 28, 147 CrossRef CAS
- I thank a reviewer for drawing my attention to the Bent's rule connection .
- A. Gutberlet, G. Schwaab, O. Birer, M. Masia, A. Kaczmarek, H. Forbert, M. Havenith and D. Marx, Science, 2009, 324, 1545 CrossRef CAS PubMed
- K. R. Leopold, Annu. Rev. Phys. Chem., 2011, 62, 327 CrossRef CAS PubMed
- K. H. Weber and F.-M. Tao, J. Phys. Chem. A, 2001, 105, 1208 CrossRef CAS
- J. C. Walton, Molecules, 2018, 23, 423 CrossRef PubMed
-
(a) S. V. Ryazantsev, V. I. Feldman and L. Khriachtchev, J. Am. Chem. Soc., 2017, 139, 9551 CrossRef CAS PubMed
-
(a) B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041 CrossRef CAS
- R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef CAS PubMed
- J. C. Walton, ACS Omega, 2018, 3, 13290 CrossRef CAS PubMed
- Gas phase enthalpies and Gibbs free energies ΔG° were adjusted for zero point and thermal corrections to 1 atm and 298 K. For solution processes the standard state of an ideal gas at a concentration of 1 mol L−1 is more appropriate; Gibbs free energies are then denoted by ΔG*. The relationship of these standard states is: ΔG* = ΔG° − RTln(24.46) = ΔG° − 1.89 kcal mol−1.
-
(a) R. Lenz and B. Giese, J. Am. Chem. Soc., 1997, 119, 2784 CrossRef CAS
-
(a) B. C. Gilbert, J. P. Larkin and R. O. C. Norman, J. Chem. Soc., Perkin Trans. 2, 1972, 794 RSC
- J. M. Surzur and P. Teissier, Bull. Soc. Chim. Fr., 1970, 3060 CAS
- D. D. Tanner and F. C. P. Law, J. Am. Chem. Soc., 1969, 91, 7535 CrossRef CAS
-
(a) A. L. J. Beckwith and P. K. Tindal, Aust. J. Chem., 1971, 24, 2099 CrossRef CAS
-
(a) B. Giese, K. S. Groeninger, T. Witzel, H.-G. Korth and R. Sustmann, Angew. Chem., Int. Ed. Engl., 1987, 26, 233 (
Angew. Chem.
, 1987
, 99
, 246
) CrossRef
-
(a) G. Behrens, G. Koltzenburg, A. Ritter and D. Schulte-Frohlinde, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1978, 33, 163 CrossRef CAS PubMed
- D. Crich and Q. Yao, J. Am. Chem. Soc., 1993, 115, 1165 CrossRef CAS
- A. Koch, C. Lamberth, F. Wetterich and B. Giese, J. Org. Chem., 1993, 58, 1083 CrossRef CAS
- B. Giese, P. Erdmann, L. Giraud, T. Gobel, M. Petretta, T. Schaefer and M. von Raumer, Tetrahedron Lett., 1994, 35, 2683 CrossRef CAS
-
(a) B. Giese, J. Burger, T. W. Kang, C. Kesselheim and T. Wittmer, J. Am. Chem. Soc., 1992, 114, 7322 CrossRef CAS
- D. Crich and X.-S. Mo, J. Am. Chem. Soc., 1997, 119, 249 CrossRef CAS
- A. L. J. Beckwith, D. Crich, P. J. Duggan and Q. Yao, Chem. Rev., 1997, 97, 3273 CrossRef CAS PubMed
- J. C. Walton, J. Org. Chem., 2019, 84, 12606 CrossRef CAS PubMed
-
(a) M. M. Greenberg, M. R. Barvian, G. P. Cook, B. K. Goodman, T. J. Matray, C. Tronche and H. Venkatesan, J. Am. Chem. Soc., 1997, 119, 1828 CrossRef CAS
-
(a) M. L. Taverna Porro and M. M. Greenberg, J. Am. Chem. Soc., 2013, 135, 16368 CrossRef CAS PubMed
- R. K. Mohamed, P. W. Peterson and I. V. Alabugin, Chem. Rev., 2013, 113, 7089 CrossRef CAS PubMed
-
(a) P. C. Dedon and I. H. Goldberg, Chem. Res. Toxicol., 1992, 5, 311 Search PubMed
-
(a) M. Dizdaroglu, C. Von Sonntag and D. Schulte-Frohlinde, J. Am. Chem. Soc., 1975, 97, 2277 CrossRef CAS PubMed
- Gibbs free energies (denoted by ΔG*) referring to the standard state of an ideal gas at a concentration of 1 mol L−1 were computed.
-
(a) S. M. Hecht, Bioconjugate Chem., 1994, 5, 513 CrossRef CAS PubMed
-
(a) G. S. Manning, Q. Rev. Biophys., 1978, 11, 179 CrossRef CAS PubMed
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed
- W. Buckel and R. Keese, Angew. Chem., Int. Ed. Engl., 1995, 34, 1502 CrossRef CAS
- D. M. Smith, W. Buckel and H. Zipse, Angew. Chem., Int. Ed., 2003, 42, 1867 CrossRef CAS PubMed
- J. Kim, D. J. Darley, W. Buckel and A. J. Pierik, Nature, 2008, 452, 239 CrossRef CAS PubMed
- P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2219–2232 CrossRef CAS
-
(a)
G. Scholes, in Photochemistry and Photobiology of Nucleic Acids, ed. S. Y. Wang, Academic, New York, 1976, vol. 1, p. 521 Search PubMed
- L. Kar and W. A. Bernhard, Radiat. Res., 1983, 93, 232 CrossRef CAS
- S. Steenken, Chem. Rev., 1989, 89, 503 CrossRef CAS
-
(a) F. Portela-Cubillo, J. Lymer, E. M. Scanlan, J. S. Scott and J. C. Walton, Tetrahedron, 2008, 64, 11908 CrossRef CAS
- See for example:
(a)
D. Spagnolo and D. Nanni, in Encyclopedia of Radicals in Chemistry, Biology, and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, 2012, vol. 2, pp. 1019–1057 Search PubMed
- M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem., Int. Ed., 2019, 58, 5532 CrossRef CAS PubMed
-
(a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673 CrossRef CAS PubMed
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed
- D. G. B. Boocock, R. Darcy and E. F. Ullman, J. Am. Chem. Soc., 1968, 90, 5945 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2021 |