
CO2 reduction and ethane dehydrogenation on transition metal catalysts: mechanistic insights, reactivity trends and rational design of bimetallic alloys†
Fatima
Jalid
ab,
Tuhin Suvra
Khan
 *c and
M. Ali
Haider
*a
*c and
M. Ali
Haider
*a
aRenewable Energy and Chemicals Laboratory, Department of Chemical Engineering, Indian Institute of Technology Delhi, Hauz Khas, Delhi, 110016, India. E-mail: haider@iitd.ac.in; Fax: +91 11 2658 2037; Tel: +91 11 26591016
bDepartment of Chemical Engineering, National Institute of Technology Srinagar, Srinagar, Jammu and Kashmir 190006, India
cLight Stock Processing Division, CSIR-Indian Institute of Petroleum, Dehradun, 24805, India. E-mail: tuhins.khan@iip.res.in
First published on 5th October 2020
Abstract
Reactivity trends of transition metal catalysts are studied for the ethane dehydrogenation reaction using CO2 as a mild oxidant. An ab initio microkinetic model (MKM) is constructed to gain insights about the dominant route for CO2 reduction and simultaneous ethylene formation over the terrace (111) and step (211) surfaces of the catalysts. At the terrace sites, Rh and Pt are observed to show high ethane consumption with maximum turnovers to produce ethylene. For CO2 consumption, Rh, Ru, Ni and Co are calculated to exhibit significant activity (TOF ∼1 s−1). CO2 on the (111) surface is predominantly reduced through the reverse water gas shift (RWGS) reaction, since the production rates of H2O and CO are comparable to the consumption rates of CO2. At the step sites, the hydrogenolysis reaction is more pronounced leading to coke formation. Hydrogenolysis at the step surface also led to significant activity for the reforming reaction. Over the (211) surface, the direct dehydrogenation of ethane to produce ethylene is observed to be predominant. For oxygen assisted ethane dehydrogenation, Co, Ru, Ni and Rh are calculated to show appreciable activity (>1 s−1). The same four metals also show significant CO2 consumption at the step surface. The MKM is further utilised to design bimetallic alloys of Ni and Pt to achieve greater CO2 consumption activity and reduced coke formation with significant activity for the dehydrogenation reaction. While most of the alloys undergo reforming and RWGS reactions, three potential bimetallic combinations (NiFe, NiCo and PtCo) are selected, exhibiting appreciable activity for CO2 assisted dehydrogenation of ethane with some reduction in coke formation, compared to their monometallic counterparts.
1. Introduction
The recent discovery of shale gas is promulgating efforts for efficient transformation of lighter alkanes into value added products.1 For example, olefinic (ethylene, propylene, etc.) products which are generally produced from cracking reactions of naphtha2 are now obtained from the dehydrogenation reaction of alkanes (ethane, propane, butane, etc.). In this alternative route, few commercial setups (e.g. UOP's Oleflex process3) have already been developed, which employ transition metal catalysts (Pt-based) for non-oxidative dehydrogenation of propane to produce propylene.4 Depending upon the source of shale gas, the amount of C2–C4 alkanes may go up to 12% for ethane and 3% for propane,5 which can be separated from the natural gas and each of these constituents offers exciting prospects for developing an alternative olefin technology.6 Towards fulfilling this goal, thermodynamics is a constraint (ΔH298° = 137 kJ mol−1 for ethane, ΔH298° = 124.3 kJ mol−1 for propane and ΔH298° = 125.9 kJ mol7 for butane dehydrogenation) and the reaction requires high temperatures (∼823–1023 K) to achieve appreciable equilibrium conversion (18% for ethane, 50% propane and 63% butane at 1 bar and 873 K).6 At such high temperatures, alkanes are likely to undergo a hydrogenolysis (C–C cleavage) reaction to produce coke. In order to overcome thermodynamic constraints, an oxidative route is thought of, in which hydrogen produced in the reaction is removed using oxygen to shift the equilibrium for achieving higher alkane conversion.8 This also facilitates some coke removal.9 However, direct use of oxygen is likely to make the process hazardous leading to potential explosion under uncontrolled conditions.10In oxidative dehydrogenation of alkanes (C2–C4) over transition metal catalysts, recent work by Chen and co-workers11–14 has applied CO2 as a mild oxidant. This is suggested to serve the dual purpose of producing olefins and reducing CO2 to address global warming concerns.14–16 The authors have reported reasonable alkane (up to 24% ethane,11 11.6% propane13 and 30.9% butane12) and CO2 (>40%) conversions with some olefin selectivity on studying an array of transition metal (Ni, Mo, and Pt) and bimetallic alloy (NiFe, CoPt, Fe3Ni, and Fe3Pt) catalysts tested at 873 K. Interestingly in this approach, whenever the olefin product is measured with appreciable selectivity (e.g. 31% ethylene on NiFe,11 24% ethylene on Mo,11 58.2% propylene on Fe3Ni (ref. 13) and 37.9% butylene on Fe3Ni (ref. 12)), the corresponding alkane conversions (9.1% and 5.1% ethane on NiFe and Mo, respectively,11 2.7% propane on Fe3Ni (ref. 13) and 13.9% butane on Fe3Ni (ref. 12)) are observed to be significantly reduced. A plausible reason for this lies in the activity of the reducible support. In almost all of the catalysts tested, CeO2 is used as the support.11–13 It is therefore suggested that CO2 is reduced at the metal–ceria interface via the Mars–van Krevelen mechanism and oxygen from the ceria lattice is made available on the metal surface to carry out oxidative dehydrogenation. Furthermore, metal supported ceria is also known as an active catalyst for reforming reactions resulting in higher alkane and CO2 conversions, with significantly reduced olefin selectivity.11–13
The aim is, therefore, to study the reactivity of transition metal catalysts for the dehydrogenation reaction of ethane in the presence of CO2 as a mild oxidant. Utilising density functional theory (DFT) simulations, an ab initio microkinetic model (MKM) is constructed to understand reactant (CO2 and ethane) conversion and product (CO, H2, C2H4, and H2O) formation trends over the (111) and (211) surfaces of an array of transition metal catalysts (Pt, Pd, Co, Ni, Rh, Ru, Re, Cu, Au and Ag). On analysing the MKM, mechanistic insights are drawn to elucidate the dominant reaction route for CO2 conversion. In general, CO2 is suggested to convert via oxidative dehydrogenation (eqn (1)), reverse water gas shift (RWGS) reaction (eqn (2)), dry reforming (eqn (3)) or reverse Boudouard reaction (eqn (4)).11 All of these pathways are analysed in the ab initio MKM.
| C2H6 + CO2 → C2H4 + CO + H2O | (1) |
| H2 + CO2 → CO + H2O | (2) |
| C2H6 + 2CO2 → 4CO + 3H2O | (3) |
| CO2 + C → 2CO | (4) |
2. Methodology
For CO2 assisted ethane dehydrogenation, a MKM is developed to analyze the reactivity of the terrace (111) and step sites (211) of transition metal catalysts using a descriptor based analysis tool – CatMAP.26 Governing differential equations defining the kinetics of the reactions are solved to obtain steady-state solutions, using the multi-dimensional Newton's root finding method as implemented in the Python mpmath library.27 The MKM follows a mean-field approach, wherein the reactant and the product turnovers are calculated using the required inputs of reaction conditions, gas-phase energetics, binding energies of the adsorbed species and activation barriers of the elementary reaction steps. The reaction energetics of the elementary steps are described by the binding energies of the adsorbed species, transition states and gas phase species.In developing the MKM, the energies of adsorbed species and transition states are obtained from the reported values in CatApp28,29 and other DFT studies.30–33 In order to determine the adsorption energies of the rest of the adsorbed species over the transition metal catalysts, DFT simulations are carried out using the periodic plane wave DFT code of the Vienna ab initio simulation package (VASP).34 The binding energies of the adsorbates over the (111) facets of Cu, Pd, Pt and Rh catalysts are calculated using the slab model, in which the transition metal catalyst surface is modelled with a four layer slab by creating a 4 × 4 supercell and a vacuum of length of 20 Å, perpendicular to the surface. Revised Perdew–Burke–Ernzerhof (RPBE) parameterization of the generalized gradient approximation (GGA) is utilised to account for the exchange and correlation energies of the electrons.35 Metal atoms in the bottom two layers are kept fixed and the top two layers are allowed to relax, to mimic the surface and sub-surface restructuring process during simulations. In order to account for the dispersion effects, vdW corrections are incorporated.36,37 Kohn–Sham one-electron valence states are expanded using a plane wave basis set truncated to a cut-off energy of 396 eV. The iterative diagonalization of the Kohn–Sham Hamiltonian is employed to determine the self-consistent electron density. Core electrons are defined using the projected augmented wave (PAW) potential.34 The surface Brillouin zone is sampled using a 3 × 3 × 1 Monkhorst–Pack grid. The Methfessel–Paxton method for integrations in the irreducible Brillouin zone is implemented.38 For structure optimizations, the energy and force convergence criteria are set to 1 × 10−6 eV and 0.05 eV Å−1, respectively. The adsorbed state configurations of CH3CH2 and CH2CH2, obtained on different metal surfaces, are shown in Fig. SI-1.†
The CatApp data used in this study are obtained using the DACAPO39 plane wave DFT method along with the revised Perdew–Burke–Ernzerhof (RPBE) GGA exchange correlation functional.28,29 The core electrons are defined using Vanderbilt ultrasoft pseudopotentials (USPP). Computational details for CatMAP data collected from different sources are available in Table SI-1.† The DFT calculations that were not available in the literature are performed using the plane wave DFT method available in the VASP, using the same RPBE functional. As both the VASP and DACAPO are plane-wave based DFT methods, the systematic error in the calculated adsorption energies is expected to be low. The carbon and oxygen binding energies as calculated by the VASP and DACAPO are given in Tables SI-2 and SI-3,† respectively.
In addition, it has to be emphasized that the MKM results can be susceptible to error propagation as the DFT data were taken from different sources. The propagation of error in MKM studies is discussed by Medford et al.,40 where the authors have elaborated upon the errors in DFT calculated energetics and underlying correlation between the errors and their propagation in MKM calculated catalytic rates. From this analysis, the authors have concluded that the ab initio MKM describes the catalytic trends better than absolute rates for metal and bimetallic catalysts. Similar suggestions were made by Falsig et al. in their study of direct NO decomposition over transition metal surfaces.41 For the energies which are correlated, error propagation in calculated rates was considerably lower than that of uncorrelated energies.40 Diverse DFT databases were also used by Medford et al. to study the conversion of synthesis gas to higher alcohols. The authors have estimated an error bar of 0.2 eV in estimating the TOFs with respect to the binding energies of descriptors, EC and EO.30 As can be seen from the scaling relationship plots in Fig. SI-2,† the energies used in this study are highly correlated and are likely to be less susceptible to error propagation.
In order to model the graphitic and amorphous forms of coke formed as the undesired product in the MKM, the rate of formation of C2(g) species is calculated. Formation of C2(g) from C–C coupling reactions of adsorbed C species is considered a necessary step for coke formation.42–44 The relative propensity of the catalyst surface for C–C vs. C–O coupling reactions is accounted in the MKM, so as to represent the competing processes of coke formation and carbon removal (eqn (4), reverse Boudouard reaction).45 The energy of C2(g) or ‘model-coke’ is obtained from the formation energy of the carbon cluster on the Ni(111) surface as suggested by Gao et al.46 DFT simulations are further employed to calculate the formation of C2(g) species over the stepped surfaces of Ag, Cu, Pd, Ni and Rh. For the Ni surface, spin polarized calculations are undertaken. To model the stepped (211) surface, a 2 × 4 supercell is employed and the Brillouin zone is sampled using a 2 × 2 × 1 Monkhorst–Pack grid. Optimized configurations of C2 species adsorbed over the catalyst surfaces are shown in Fig. SI-3.† The energetics of the transition state for the C–C coupling reaction are obtained using the universal BEP scaling relationship.41,47
The energies of all the intermediates and transition states are referenced with respect to the gas phase energies of hydrogen, water and methane. The binding energies of carbon (EC) and oxygen (EO) over the terrace (111) and stepped (211) sites of transition metal catalysts are chosen as descriptors.48–50 The adsorbed intermediates involved are known to bind with the metal surface predominantly through the oxygen and/or the carbon atoms.30,51,52 A number of studies based on the MKM have previously employed the binding energies of oxygen and carbon as descriptors to evaluate the reaction network, with reliable scaling of the energetics of the adsorbed species.30,53–55 The transition state energies for the elementary reaction steps where no previous data is available are calculated using the transition state scaling relations,41,47,56 mapping the energy of the transition state with the energy of the corresponding dissociated state. The binding energies of the surface intermediates are scaled with the descriptors. This procedure reduces the dimensionality of the model and parameter space required to get a qualitative description of the reaction energetics involved, thereby allowing the mapping of the entire reaction kinetics onto a limited set of parameters.
The MKM is solved using a fixed entropy assumption57 for defining the gas phase thermochemistry. Adsorbed intermediates are assumed to show a negligible entropy change in the surface reactions, following the frozen adsorbate approximation.26 Since the coke formed during the reaction is likely to remain adsorbed on the catalyst surface, the entropy of C2(g) is considered to have a negligible value of 0.001 eV mol−1 K−1. Owing to the small size of the hydrogen atom, it is unlikely to exert a dominant effect on the adsorption with the other intermediates, thus a separate hydrogen reservoir site is designated.30 Following which, the MKM has two separate adsorption sites, wherein the number of hydrogen sites is kept equal to the rest of the sites. A similar model for H adsorption is assumed previously by Lausche et al.,52 Medford et al.,30 and our group.33,48,58,59 Steady state solutions for the MKM are obtained at a reaction temperature of 873 K, starting with an equal proportion (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of ethane and carbon dioxide, and an initial carbon conversion of 0.1% towards the oxidative dehydrogenation of ethane viaeqn (1). A similar conversion has been considered for MKM analysis in a previous work on non-oxidative dehydrogenation of ethane by Hansen et al.60 The obtained steady state rates are plotted as 2-dimensional volcano plots with respect to the descriptors – EC and EO.
:1) of ethane and carbon dioxide, and an initial carbon conversion of 0.1% towards the oxidative dehydrogenation of ethane viaeqn (1). A similar conversion has been considered for MKM analysis in a previous work on non-oxidative dehydrogenation of ethane by Hansen et al.60 The obtained steady state rates are plotted as 2-dimensional volcano plots with respect to the descriptors – EC and EO.
In constructing the MKM for CO2 assisted dehydrogenations of ethane, adsorbate–adsorbate interactions are considered to have a negligible effect on the overall reactivity.52,55,61 This follows from the assertion made by Lausche et al. in the CO methanation reaction52 and Xu et al. in the steam reforming reaction,55 wherein the authors have observed that these interactions affect the reactivity only above a threshold coverage of 0.5 ML. In addition, Grabow and co-workers demonstrated that the activity trends remain the same qualitatively upon the inclusion of the interactions between adsorbates.61 Similarly, in an ab initio MKM study of non-oxidative ethane dehydrogenation, Nørskov and co-workers have ignored lateral interactions of adsorbates, presuming a negligible effect in determining the reactivity trend.60 Taking a clue from these observations, lateral interactions of adsorbed species are ignored in this study on ethane dehydrogenation.
For in silico bimetallic alloy screening, the method as proposed previously30,55,62 is used to screen alloys with promising reactivity and selectivity towards CO2 assisted ethane dehydrogenation. The bimetallic alloys with A3B (L12 type structure) composition are considered in the alloy screening. The A3B (L12) type alloy has an FCC crystal structure, with 75:25 composition of A and B metals, respectively. The A3B alloys of Ni with Rh, Pt, Pd, Ru, Cu, Fe, Co, and Sn; and Pt with Cu, Zn, Rh, Co, Pd, Ag, Au and Sn, are evaluated in the MKM. In terms of thermodynamic stability, these A3B bimetallic alloys are considered to be potentially stable as their formation energy is less than +0.2 eV per unit cell.55 The AA and AB surface terminations of the (211) facets of Ni and Pt-based A3B alloys are shown in Fig. SI-4.† For the AB termination, the step sites have both A and B metals in 50:50 composition, whereas, for the AA termination, the step sites only have metal A as depicted in Fig. SI-4(a) and (b),† respectively. The model utilises the descriptor binding energies of the alloys to calculate their activity towards CO2 assisted ethane dehydrogenation.55 An error bar of 0.2 eV is considered for the descriptor energies of the transition metals.63
3. Results and discussion
Reactions forming a range of products: ethylene, methane, carbon monoxide, water, and hydrogen via the decomposition of ethane and carbon dioxide over the metal surfaces are depicted in Fig. 1. A comprehensive list of 45 elementary reaction steps, solved in the MKM, is given in section SI-1 of the ESI.† Using the MKM, the reactivity of the transition metal catalysts is analyzed for direct ethane dehydrogenation, CO2 assisted dehydrogenation, RWGS and reforming reactions (eqn (1)–(4)).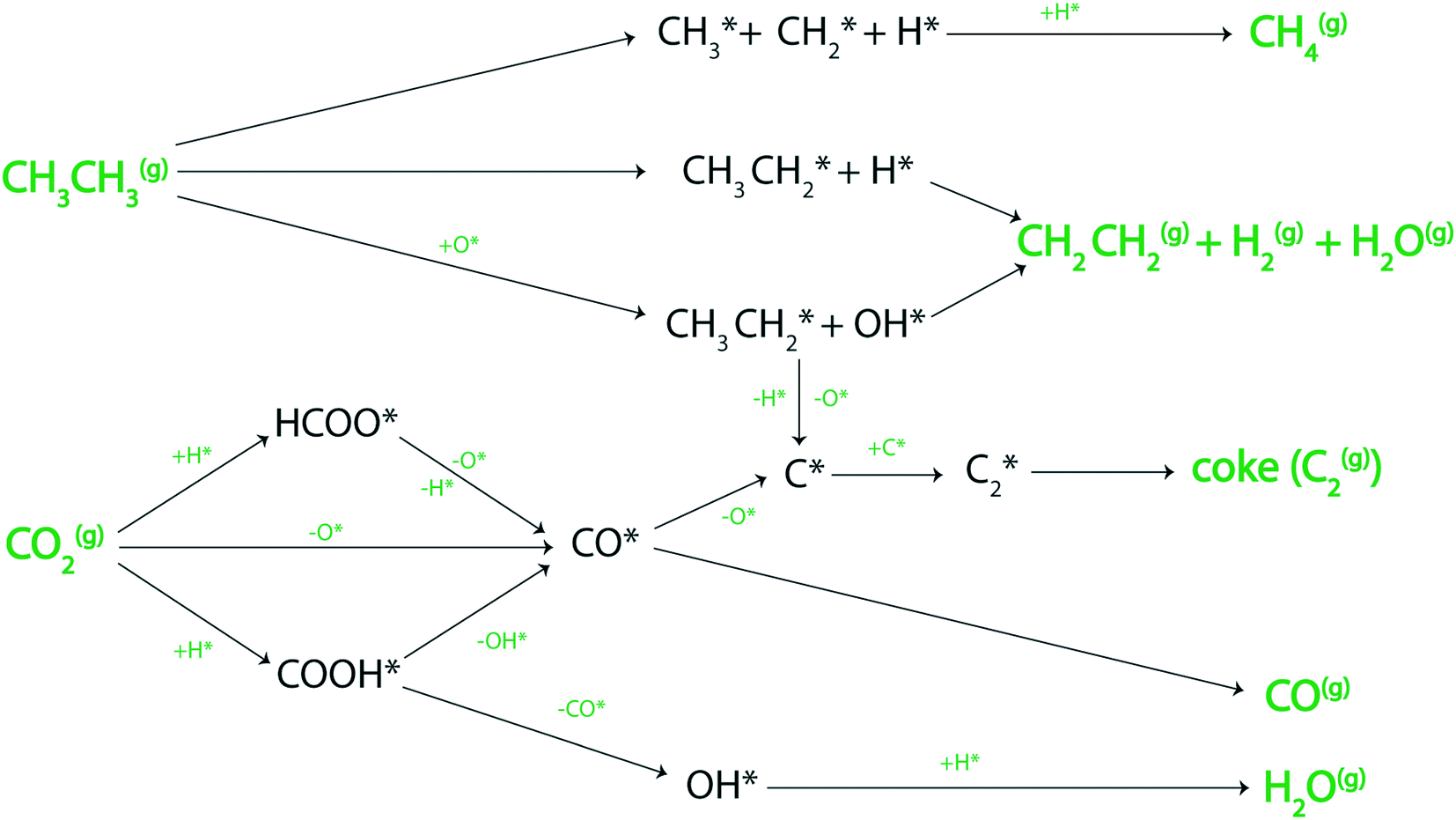 | ||
| Fig. 1 Routes for C2H6 and CO2 decomposition over the catalyst surface to produce CH4, C2H4, H2, H2O, coke (C2(g)) and CO. Surface adsorbed species are marked with an *. | ||
3.1 Reactivity trend of the terrace sites
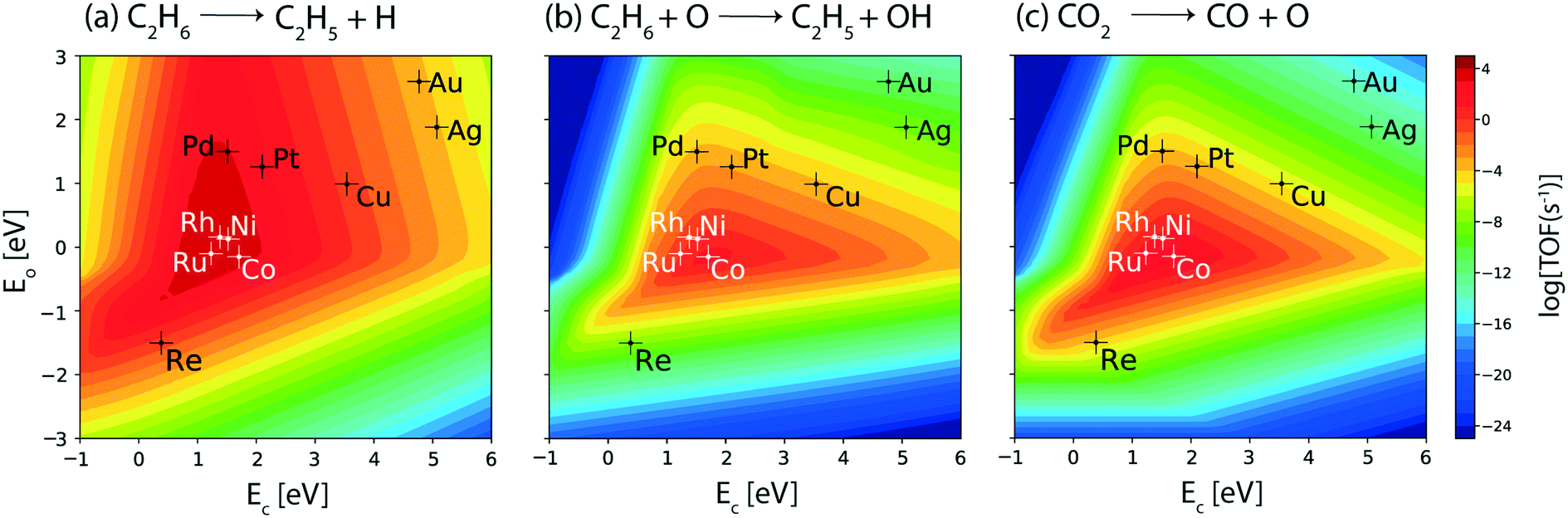
The reaction rates calculated for the consumption of ethane and CO2 over the terrace (111) sites of transition metal catalysts under the reaction conditions studied are depicted in Fig. 2(a) and (b), respectively. The maximum of the consumption volcano plot for ethane corresponds to TOFC2H6 ∼ 105 s−1 which is centered at EC = 1 eV and EO = 0.5 eV, where none of the transition metals screened are lying. Pt and Rh show the maximum activity towards the consumption of ethane, with TOFC2H6 ∼ 104 s−1. In general, Pt is the most widely studied catalyst for this reaction.24,64,65 Haensel demonstrated that Pt-based catalysts are highly active for the dehydrogenation of paraffins to olefins at large.66 Herein, Pd, Ni, Co and Ru display an order of magnitude smaller rates than Rh and Pt (TOFC2H6 ∼ 103 s−1), while Cu shows ethane consumption on the order of 1 s−1. The overall activity trend for the ethane consumption rate is observed to be: Rh > Pt > Pd ∼ Ni ∼ Co > Ru > Cu. While the TOFs calculated using the MKM may differ in value as compared to the experiments, the trends are likely to remain the same.60,64 For example, a similar trend in the reactivity of transition metals is reported in experiments on ethane dehydrogenation, where the ethane conversion trend follows: Rh > Pt > Pd.64 The catalyst reactivity trend studied by Bligaard and co-workers using an ab initio MKM for non-oxidative dehydrogenation of ethane has shown Pt to be more active than Co, Ni and Ru.60 Ethane consumption rates are calculated to be very low (TOFC2H6 < 10−3 s−1) on the (111) surface of Re and noble metals (Ag, Au). In general, the trend in ethane consumption over the transition metal catalysts studied corresponds to the reported trend in C–H bond activation energies of alkanes,67 which indicates that ethane likely undergoes a dehydrogenation reaction over the metal catalysts to produce ethylene. For example, using DFT calculations, Xing et al. have calculated the C–H bond activation of methane to be the lowest on Rh (42.5 kJ mol−1), following the activity trend: Rh > Pt > Ni > Ru.67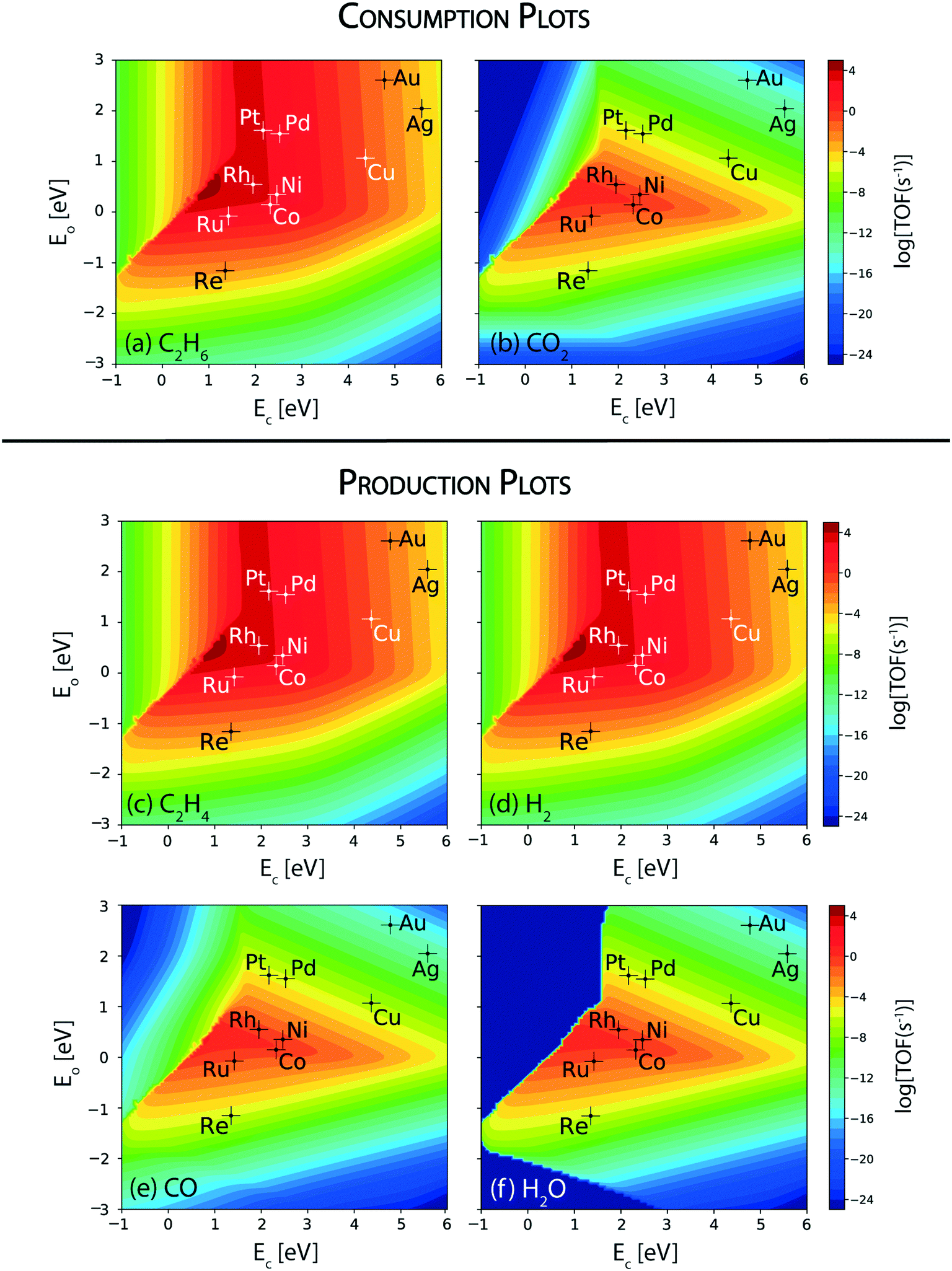 | ||
| Fig. 2 Volcano plots for the consumption rates of (a) C2H6 and (b) CO2; production rates of (c) C2H4, (d) H2, (e) CO and (f) H2O over the (111) sites of transition metal catalysts. Error bar = 0.2 eV. | ||
Since the target reaction is CO2 assisted ethane dehydrogenation, CO2 consumption plots are analyzed next. Fig. 2(b) shows the volcano plot for CO2 consumption. Compared to the ethane consumption plot (Fig. 2(a)), the CO2 consumption plot (Fig. 2(b)) shows lower turnovers on the respective metals and the volcano maximum is shifted to weaker carbon (EC = 1.5 eV) and similar oxygen (EO = 0.5 eV) binding energies. Co, Rh, Ru and Ni catalysts are lying closest to the maximum with TOFCO2 ∼ 1 s−1, which is 3–4 orders of magnitude smaller than the rates calculated for ethane consumption on the same catalysts. This suggests that the direct ethane dehydrogenation (Fig. 2(a)) route is prominent as compared to CO2 assisted ethane dehydrogenation on the (111) surface. The rest of the metals: Pd, Pt, Re, Cu, Ag and Au exhibit negligible rates for CO2 consumption (TOFCO2 < 10−5 s−1, Fig. 2(b)) and predominantly catalyse direct ethane dehydrogenation over the (111) sites.
In order to understand direct ethane activation to produce ethylene, the product formation rates of ethylene (Fig. 2(c)) are analyzed. Interestingly, the volcano plot of ethylene production (Fig. 2(c)) looks similar to that of ethane consumption (Fig. 2(a)), wherein the volcano maximum lies at the same position (EC = 1 eV and EO = 0.5 eV) and individual elements show the same reactivity. Thus, most of the ethane is converted to produce ethylene in this reaction. This is further confirmed on analyzing the hydrogen production plot, Fig. 2(d). The reactivity trends and TOFs calculated for H2 production, over the transition metals screened, are the same as the ethane consumption activity trend. This indicates that ethane predominantly undergoes direct dehydrogenation to produce ethylene and hydrogen. However, a better understanding of this may be developed upon analyzing the CO production plot, Fig. 2(e). This plot exhibits the same reactivity trend and turnovers as those observed for CO2 consumption for all of the metals, in which the four specific metals (Rh, Ru, Ni and Co) show appreciable turnovers for water (TOFH2O ∼ 1 s−1). In fact, the corresponding water production plot (Fig. 2(f)) also looks similar in reactivity to that of CO2 reduction. Interestingly, at very strong carbon and weak oxygen binding energies (dark blue region in the upper left corner; EC < 2 eV, EO > −1 eV) of the water production plot, no product turnover is calculated and in this region water is calculated to be consumed, which is likely due to the activity of the catalyst surface for the steam reforming reaction (Fig. SI-5(a)†). Nevertheless, none of the transition metal catalysts studied lie in this region. Thus, on observing CO2 consumption, H2O and CO production plots, we conclude that most of the CO2 is likely reduced through the RWGS reaction on the (111) surface of the four active metals (Rh, Ru, Ni and Co) to produce CO and H2O. This observation simultaneously excludes the likelihood of the reforming reactions (steam or dry) over the (111) surfaces of the catalysts studied. Since the rates of RWGS are significantly lower than the direct ethane dehydrogenation rates, the hydrogen consumed through RWGS does not influence the H2 production plots (Fig. 2(d)). Among the transition metals, Ni has been experimentally employed as a widely used catalyst for the RWGS reaction.68,69 Dietz et al. conducted a mechanistic study on the RWGS reaction on transition metal catalysts wherein they observed Rh and Ni to exhibit the lowest activation barriers towards CO2 dissociation.70 These metals are therefore suggested to undergo direct CO2 dissociation owing to their higher oxygen affinity.70 Besides Rh and Ni, Ru and Co are also reported to catalyze the RWGS reaction.71,72
To further probe ethylene formation routes, the elementary reaction rates for ethane dehydrogenation, via metal catalyzed C–H bond activation, are computed from the MKM developed for the terrace sites of the catalysts. Results are plotted with respect to the reaction descriptors. Fig. 3(a) shows the volcano plot for the turnovers of the ethane dehydrogenation step over the metal surfaces. The C–H bond activation rate in ethane (Fig. 3(a)) exhibits a similar volcano plot to that of the overall ethane consumption turnovers (Fig. 2(a)). This is also comparable to the production plots of ethylene (Fig. 2(c)) and hydrogen (Fig. 2(d)) alluding to the prevalence of direct ethane dehydrogenation. Interestingly, C–H bond activation in alkanes is known to be affected by surface oxygen species.73 Previously, a number of experimental74–76 as well as theoretical67,77,78 studies have indicated the role played by surface oxygen species in C–H bond activation reactions. Hibbitts and co-workers have performed extended DFT calculations on transition metal surfaces to demonstrate the reactivity of the catalyst surface for C–H activation, assisted by surface oxygen and surface hydroxyl species.73 Specifically, for the coinage metals (Cu, Ag and Au), the authors have observed C–H activation assisted by surface oxygen species, with a lowering of activation energies by about 40 kJ mol−1 as compared to direct metal catalyzed C–H activation. In contrast, for C–H activation in methane on some of the other transition metal catalysts (group 8–10), surface oxygen species are observed to increase the activation energies. Thus, surface oxygen species are expected to play a complex role in the reaction kinetics of the dehydrogenation reaction of ethane. In order to probe this mechanistic route for ethane dehydrogenation, the turnover rates for oxygen assisted C–H bond activation in ethane are plotted in the form of a volcano plot in Fig. 3(b). In the CO2 assisted ethane dehydrogenation, CO2 may undergo dissociation to form carbon monoxide and surface oxygen species. The O* formed on the catalyst surface is likely to facilitate the oxidative route of dehydrogenation. Interestingly, the plot where surface oxygen is applied to assist ethane dehydrogenation shows negligible turnovers (TOF < 10−7 s−1, Fig. 3(b)) over most of the metals except for the four metals (Rh, Ru, Ni and Co) where appreciable turnovers (TOF ∼ 10−1 s−1 on Rh, Ru, and Co and 10−2 s−1 on Ni) are calculated. On these four metals, oxygen assisted C–H bond activation of ethane occurs with one or two orders of magnitude lower rates as compared to CO2 consumption (Fig. 2(b)) and CO formation rates (Fig. 2(e)). This suggests that on these four metals, while most of the CO2 is reduced via the RWGS reaction, some of it also assists the oxidative dehydrogenation of ethane. Nevertheless, direct ethane dehydrogenation rates prevail over all other reactions.
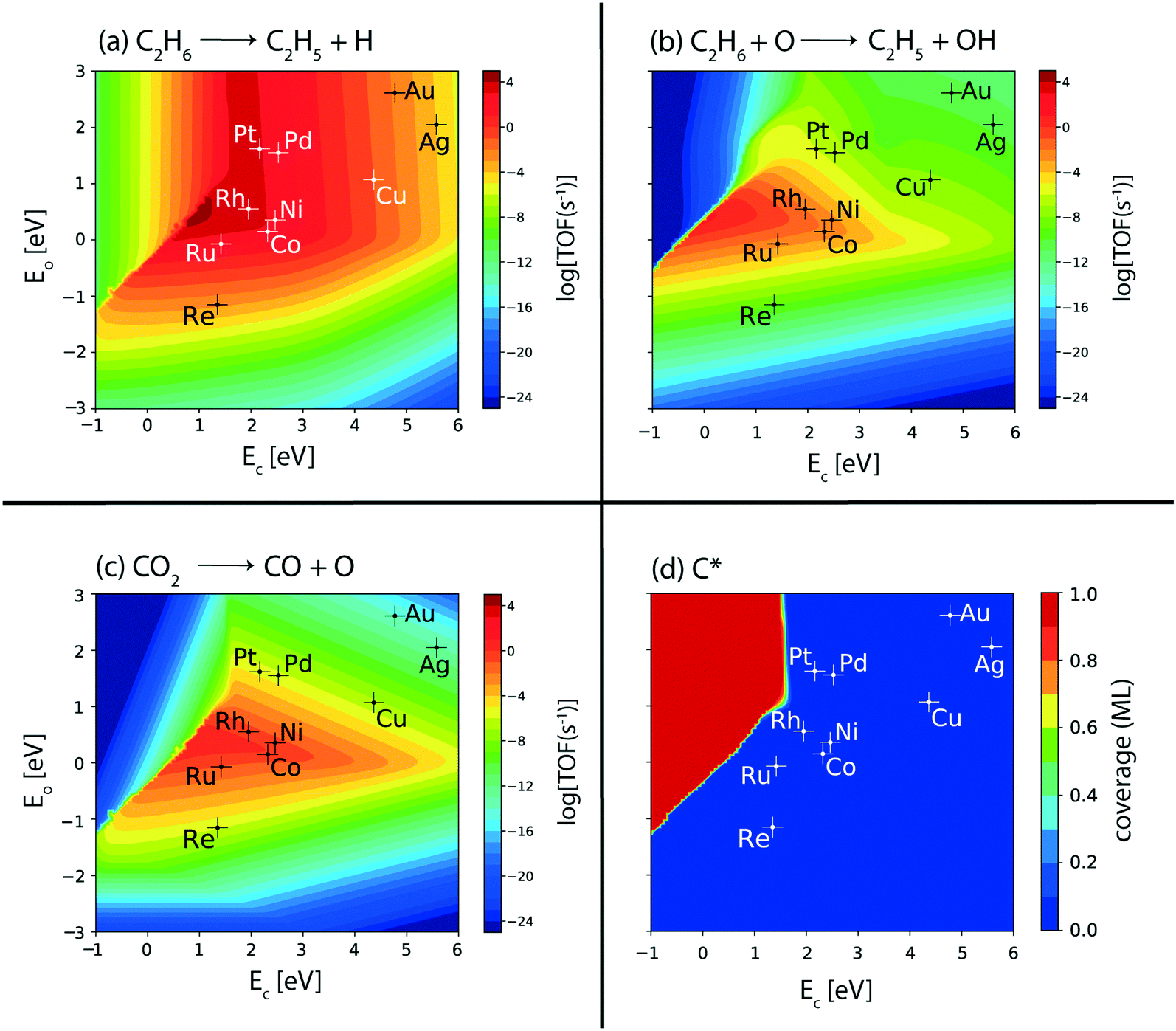 | ||
| Fig. 3 Elementary reaction rates of (a) metal catalyzed ethane dehydrogenation, (b) oxygen assisted ethane dehydrogenation and (c) CO2 dissociation; (d) coverage of C* species over the (111) sites of transition metal catalysts. Error bar = 0.2 eV. | ||
The metals (Rh, Ru, Ni and Co) showing appreciable turnovers for the oxidative route (Fig. 3(b)) are also expected to dissociate CO2 on the surface. In order to confirm this, metal-catalyzed direct CO2 dissociation rates are analyzed as the volcano plot in Fig. 3(c). Indeed, the reaction rates of the direct CO2 dissociation step (Fig. 3(c)) are calculated to be the same as CO2 consumption (Fig. 2(b)) and CO formation (Fig. 2(e)) rates on the respective metals (Rh, Ru, Co, and Ni). Besides direct CO2 dissociation, the transition metal catalysts may catalyze hydrogen assisted CO2 activation via the carboxyl (COOH) and formate (HCOO) mediated pathways.79–81 Interestingly, CO2 dissociation via the carboxyl and formate mediated mechanisms is calculated to be lower (<10−6 s−1, Fig. SI-5(b) and (c)†) than direct CO2 dissociation (>1 s−1, Fig. 3(c)). Therefore, CO2 dissociation is thought to occur via direct dissociation over the transition metal catalysts screened. Experimental studies over Ru, Ni and Co catalysts have alluded towards this assertion that CO2 is dissociated directly on the metal surface.82–84 In a DFT study, Rh and Ni are calculated to dissociate CO2 on the metal surface directly with reduced activation energies as compared to Pt, Cu, Ag and Pd.70 Furthermore, in the same study, Ni, Rh and Cu are shown to favour direct CO2 dissociation as compared to CO2 activation via the carboxyl route.70
CO produced from CO2 dissociation may further dissociate to form adsorbed carbon (C*) species on the metal surface. Carbon is also formed from the C–C cleavage reaction of ethane in reforming or thermal dissociation. For the thermally activated hydrogenolysis reaction, a relatively higher pressure is required, so this reaction is unlikely.6 Over Pt and Pd, some production of CH4 is observed (Fig. SI-5(d)†), which is likely due to the thermal hydrogenolysis reaction of ethane. However, the rates for methane production are calculated to be low (10−4 s−1 and 10−5 s−1, on Pt and Pd, respectively; Fig. SI-5(d)†). In general, reforming of ethane appears to be a dominant route in ethane conversion on the ceria supported metal catalysts tested by Jinguang Chen and co-workers.11–13,85 In order to ascertain any carbon produced on the (111) surface of the metal catalysts, the coverage plot of C* (Fig. 3(d)) is analyzed. None of the metal catalysts screened exhibit any coverage of carbon (θ < 0.1). This indicates reduced likelihood of the (111) metal surface for CO dissociation or the hydrogenolysis reaction, which is also evident in many experiments and theoretical studies.20,31,86–89 A DFT study on ethane dehydrogenation and hydrogenolysis reactions over the surfaces of the Pt catalyst calculated higher barriers for C–C scission at the terrace sites as compared to the step sites.86 Moreover, detailed experimental investigations by Rovik et al. on ethane dehydrogenation over Ni, Ru, Rh and Pd catalysts indicated the step sites to be the active centres for hydrogenolysis, whereas the dehydrogenation reaction is suggested to occur primarily on the terrace surface.20 Liu and Hu have studied the effect of surface structure on C–O bond scission for Rh and Pd, observing that these reactions show greatly reduced barriers on steps sites as compared to terrace sites.87 On increasing the step site density of the Pt catalyst, Somorjai and co-workers have measured a significant increase in the rate of the hydrogenolysis reaction of cyclohexane to produce n-hexane, leading to carbonaceous deposits on the step sites.18
3.2 Reactivity trend of the step sites
In order to develop a comprehensive alloying rationale to carry out the ethane dehydrogenation reaction along with CO2 reduction, the reactivity of the step sites of the transition metal catalysts is analyzed. Fig. 4 shows the corresponding consumption and production plots of the reactants and products, respectively, which are calculated at the (211) surface. The TOF of reactant consumption and product formation rates are computed from the MKM at a reaction temperature of 873 K. Fig. 4(a) and (b) show the rate of consumption of the reactants (viz. ethane and carbon dioxide, respectively). The maximum of the volcano plot for ethane consumption is centered at EC = 1.3 eV and EO = 0.2 eV, Fig. 4(a). Under the reaction conditions, Ru, Rh, Ni, and Co lie at the top of the volcano, exhibiting the highest consumption turnover of 104 s−1 for ethane. Compared to the terrace surface, the step sites of Ru, Ni and Co specifically show increased activity towards ethane consumption. Following these metals, Pd and Pt are calculated to catalyze the consumption of ethane at a rate of 103 s−1. Overall, at the (211) surfaces, the consumption rate of ethane follows the order: Rh ∼ Ni > Ru > Co > Pd > Pt > Re ∼ Cu (Fig. 4(a)). The noble metals, Ag and Au, display the lowest consumption rate of ethane, TOFC2H6 ∼ 10−3 s−1.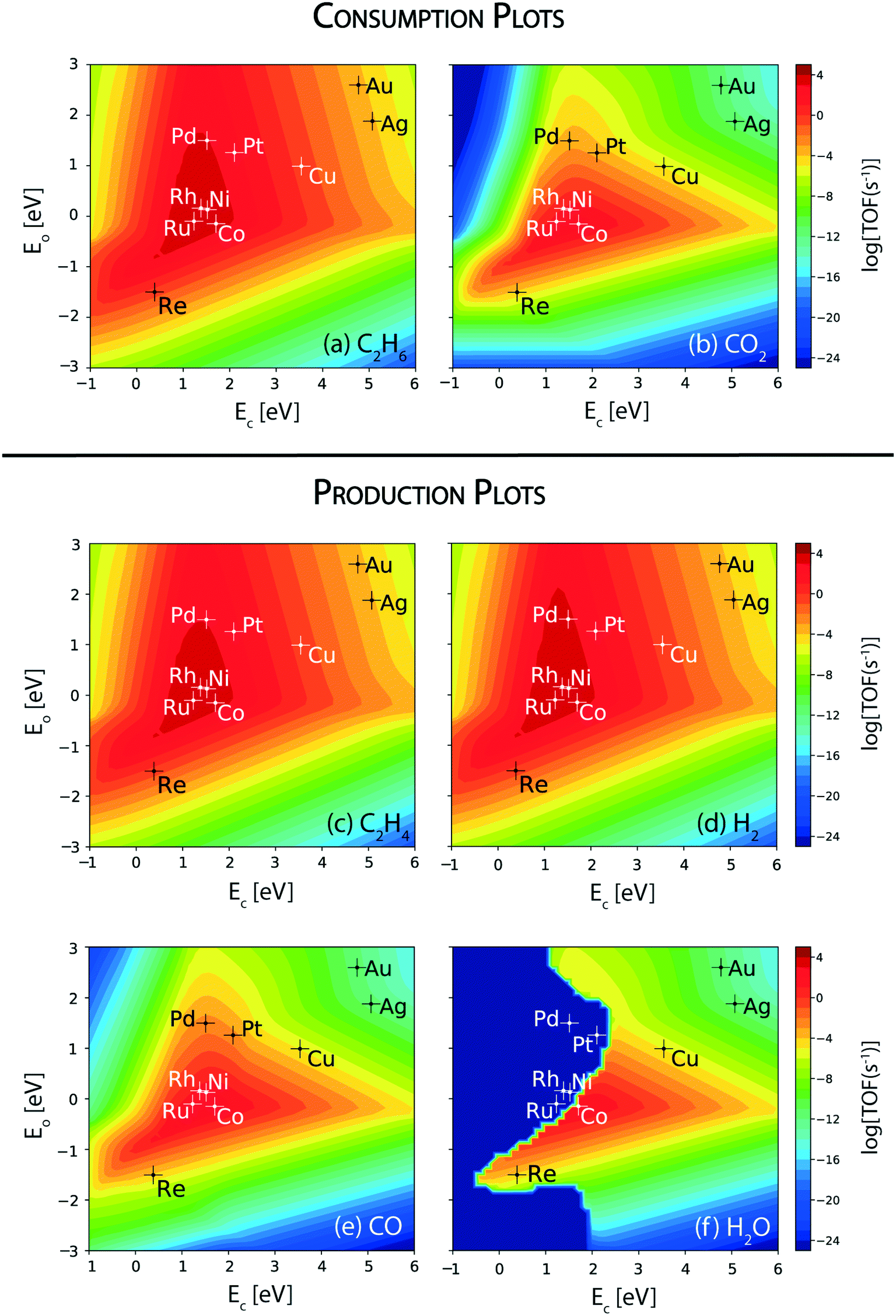 | ||
| Fig. 4 Volcano plots for the consumption rates of (a) C2H6 and (b) CO2; production rates of (c) C2H4, (d) H2, (e) CO and (f) H2O over the (211) sites of transition metal catalysts. Error bar = 0.2 eV. | ||
The trends in the reactivity of the (211) surface for ethane and CO2 consumption are comparable to the reported experimental trends. In experiments, ethane conversion on a ceria supported Ni catalyst (13.3%) for CO2 assisted oxidative dehydrogenation is measured to be higher than that on a ceria supported Co catalyst (4.7%).11 Similarly, for propane oxidative dehydrogenation with CO2, propane conversion over Ni (9.6%) is measured to be significantly higher than that over Pt (1.6%) catalysts.13 In general, Ni is known to show a high activity for C–H bond activation in hydrocarbons (for example, in methane,90,91 ethane91 and propane92). However, ethane conversion alone does not signify the reactivity towards CO2 assisted dehydrogenation of ethane, since ethane may also undergo direct dehydrogenation and reforming reactions.
CO2 consumption rates are therefore analyzed to understand the catalyst activity for CO2 assisted ethane dehydrogenation, RWGS and dry reforming reactions at the step sites. Fig. 4(b) shows the corresponding volcano plot. For CO2 consumption, the maximum of the plot is located at EC = 1.5 eV and EO = −0.2 eV (Fig. 4(b)). Compared to the ethane consumption plot (Fig. 4(a)), the maximum for CO2 consumption lies at a higher oxygen binding energy, which suggests a greater role of oxygen binding over the stepped catalyst surface in activating the CO2 molecule. In general, CO2 consumption rates are calculated to be lower than those of C2H6 on respective metal surfaces (Fig. 4(b)versus (a)). For CO2 consumption, Co is located at the top of the volcano plot, with a turnover of 103 s−1. The (211) sites of Co show a three order of magnitude greater turnover of CO2 than that calculated over the (111) sites. Ru, Rh and Ni also lie close to the volcano maximum with TOFCO2 ∼ 102 s−1. The TOFCO2 calculated over the step sites of these transition metals are observed to be higher than those over the terrace sites, by two orders of magnitude. Pt and Pd (TOFCO2 ∼ 10−3 s−1) are the next active metals for CO2 consumption. This trend in activity is confirmed in the experiments by Chen and co-workers, where the authors have reported a high conversion of CO2 in dehydrogenation of propane over Ni/CeO2 (32.8%) as compared to Pt/CeO2 catalysts (4.2%).13 In this work, the CeO2 support plays a vital role in the activation of CO2; however, the difference in the activity of the catalysts having the same ceria support is attributed to the transition metal.
Overall, the reactivity order for CO2 consumption turnovers from Fig. 4(b) follows: Co > Ru > Ni > Rh > Pt > Pd > Re > Cu. As expected, CO2 consumption rates are calculated to be negligible on the noble metals (Au and Ag), since they are unlikely to activate CO2.67,93,94 By comparing the CO2 consumption plots over the terrace (Fig. 2(b)) and step sites (Fig. 4(b)), it is evident that the step sites catalyze CO2 conversion at a much higher rate. From the consumption rate plots shown in Fig. 4(a) and (b), it can be discerned that Co, Ru, Rh, and Ni are the only transition metal catalysts screened that show appreciable turnovers (TOF > 102 s−1) of both reactants (CO2 and ethane) and thus show potential for CO2 assisted oxidative dehydrogenation of ethane. These are the same four metals which show appreciable CO2 turnovers over the (111) surface (Fig. 2(b)).
Ethylene production in CO2 assisted oxidative dehydrogenation of ethane is analyzed from the volcano plot shown in Fig. 4(c). The shape of the volcano plot for ethylene production (Fig. 4(c)) is similar to that observed for ethane consumption (Fig. 4(a)), wherein the maximum lies at EC = 1.3 eV and EO = 0.2 eV. In fact, the production rates of ethylene on the respective transition metals are similar to ethane consumption rates. The trend in the ethylene (TOFC2H4) production rate follows – Rh ∼ Ni > Ru > Co > Pd > Pt > Re ∼ Cu, Fig. 4(c). Interestingly, the corresponding H2 production plot (Fig. 4(d)) also shows a similar turnover for the metal catalysts, with a slight difference in Pd shifting towards the top of the volcano plot. Thus, most of the ethane is directly dehydrogenated on the metal catalyst surface to produce hydrogen and ethylene. The production rate curves for ethylene (Fig. 4(c)) and hydrogen (Fig. 4(d)) also show an increased TOF, by an order of magnitude over the (211) surface of Ni, Co, Ru, Pd and Re as compared to the (111) sites (Fig. 2). Interestingly, for Pt, the TOF for ethylene decreases by an order of magnitude on the (211) surface (Fig. 2(c)versus4(c)). On the contrary, Rh and Cu show similar TOFs of ethylene and hydrogen over the (211) sites as calculated on the (111) sites.
As the next step, the rate of formation of CO is analyzed from the MKM, which is shown in Fig. 4(e). In comparison to the (111) sites (Fig. 2(e)), the reaction rates of CO formation are observed to be significantly higher on the (211) sites (two orders of magnitude or higher) on all the metal catalysts screened, and Co is calculated to show the highest production of CO, of the order 103 s−1. In fact, the CO production rate on Co is only an order of magnitude lower than the ethylene production rate on the (211) surface. On the contrary, Ru, Rh and Ni show two orders of magnitude lower formation rates for CO (102 s−1) than those for ethylene (104 s−1). The production rates for CO (10−2 s−1) are significantly reduced on the (211) surface of Pd and Pt as compared to ethylene formation (103 s−1). This implies that the dominant reaction route is direct dehydrogenation of ethane, resulting in the production of ethylene and hydrogen on these two metals. Overall, the activity trend for CO formation (Fig. 4(e)) is calculated to be similar to that of CO2 consumption rates (Fig. 4(b)) on all of the metals. This indicates that RWGS is the dominant route for CO2 conversion.
The TOF of H2O production is calculated further to understand the catalyst reactivity trends for the RWGS reaction. Fig. 4(f) shows the corresponding volcano plot. Interestingly, some transition metal catalysts (Rh, Ru, Pd, Pt, Ni) do not show any production rate of H2O. This may be attributed to the steam reforming of ethane, which consumes water to form CO and H2 as products. Fig. SI-6(a)† shows the water consumption rates over these five metals confirming that the water produced in the RWGS reaction over these metals is consumed through steam reforming of ethane. Since the ethylene dehydrogenation rates are dominating with significantly high TOFs for H2 production, the hydrogen obtained from steam reforming does not clearly show up in the trends of the H2 production plot (Fig. 4(d)). On the (211) surface of Co, the TOF of water production is calculated to be 102 s−1 (Fig. 4(f)) which is an order of magnitude smaller than the rate computed for CO formation (TOF ∼ 103 s−1), indicating that some other reactions occur over Co besides RWGS. We hypothesize that Co possibly shows higher activity for dry reforming which leads to less water formation and more CO production.
In order to understand the propensity of the (211) surface of the metal catalysts for oxidative dehydrogenation, elementary reaction rates for direct metal catalyzed and oxygen assisted C–H bond activation of ethane are analyzed. The corresponding volcano plots are shown in Fig. 5(a) and (b), respectively. The calculated rates for the metal catalyzed dehydrogenation (Fig. 5(a)) are similar to those observed for ethane consumption (Fig. 4(a)) and ethylene formation (Fig. 4(c)), indicating the dominance of direct ethane dehydrogenation on the (211) surface. This is similarly observed for the reactivity of the (111) facets (Fig. 3(a)). For the oxidative route to open up, CO2 must dissociate to produce surface oxygen. Direct CO2 dissociation rates (Fig. 5(c)) are calculated to be similar to CO2 consumption rates (Fig. 4(b)), which are appreciable (TOF > 102 s−1) for the four active metals (Rh, Ru, Ni and Co), suggesting that most of the CO2 is converted by direct dissociation on these metals. The TOFs for oxygen assisted dehydrogenation over the (111) surface (Fig. 3(b)) are calculated to be higher than those over the (211) surface (Fig. 5(b)). In general, higher CO2 consumption on the step sites is observed as compared to the terrace sites (Fig. 4(b)versus2(b)). Since, direct CO2 dissociation is the predominant route, hydrogen assisted CO2 dissociation through the carboxyl (Fig. SI-6(b)†) and formate (Fig. SI-6(c)†) routes shows significantly low rates (<10−2 s−1) at the (211) surface for all the metals screened.
 | ||
| Fig. 5 Elementary reaction rates of (a) direct ethane dehydrogenation, (b) oxygen assisted ethane dehydrogenation and (c) CO2 dissociation over the (211) sites of transition metal catalysts. Error bar = 0.2 eV. | ||
The TOFs for oxygen assisted ethylene dehydrogenation are shown in Fig. 5(b). Co exhibits the highest rate for oxygen assisted C–H activation on the metals. Further, the TOF for this reaction step is observed to be an order of magnitude lower than the CO2 dissociation rate on the (211) facets of Co (103 s−1, Fig. 4(b)). Consequently, this indicates that besides oxidative dehydrogenation, Co likely catalyzes the dry reforming and RWGS reactions, which is also inferred in the aforementioned discussions on the water consumption plot (Fig. SI-6(a)†). Rh, Ru and Ni show a TOF of 10 s−1 for the oxygen assisted ethane dehydrogenation step (Fig. 5(b)). The rest of the metals screened show very low turnovers (<10−3 s−1), which rules out the possibility of the oxidative route. From this plot, we screened the four transition metal catalysts (Co, Ni, Rh and Ru) which hold potential for the CO2 assisted ethane dehydrogenation reaction. The same catalysts are also identified from the reactivity trends for the (111) sites.
For the (211) surface, the hydrogenolysis reaction of ethane undergoing C–C cleavage forms a major concern related to catalyst deactivation from coke deposition. The hydrogenolysis activity of the catalyst surface is evaluated from the carbon (C*) coverages, Fig. 6(a). Ru shows the maximum coverage (θ ∼ 1 ML), followed by Rh (θ ∼ 0.8 ML), Ni, Pd (θ ∼ 0.6 ML) and Co (θ ∼ 0.2 ML), Fig. 6(a). In general, the reverse Boudouard reaction does not facilitate the removal of C* species, since the four metals (Co, Ni, Rh and Ru) which show significant turnovers for CO2 consumption rates also show appreciable C* coverage (>0.2 ML, Fig. 6(a)). Thus, the reverse Boudouard reaction rates on these metals are considered significantly lower than the RWGS, reforming and oxygen assisted dehydrogenation rates. Among the active metals (Ni, Co, Rh, Ru, Pd, and Pt), which show significant dehydrogenation activity, only Pt is calculated to exhibit negligible coverage of C* (θ < 0.1 ML). The rest of the inactive metals (Re, Cu, Au, Ag) show no C* coverage. Interestingly, over the (111) facets of all the transition metal catalysts studied, no C* coverage is observed (Fig. 3(d)). This suggests that hydrogenolysis primarily occurs at the (211) surface of the active metals (Ru, Rh, Ni, Co, Pd). The hydrogenolysis activity of the step sites is further evident in CH4 production rates. The volcano plot for CH4 formation from the thermal hydrogenolysis reaction of ethane is presented in Fig. SI-6(d).† Co and Ni exhibit the highest turnover for methane production (10 s−1) followed by Pt and Rh (1 s−1). Pd also shows substantial CH4 formation (10−1 s−1). Overall, the production of CH4 is observed to be significantly enhanced on the (211) facets as compared to the (111) sites, as depicted in Fig. SI-6(d) and SI-5(d),† respectively.
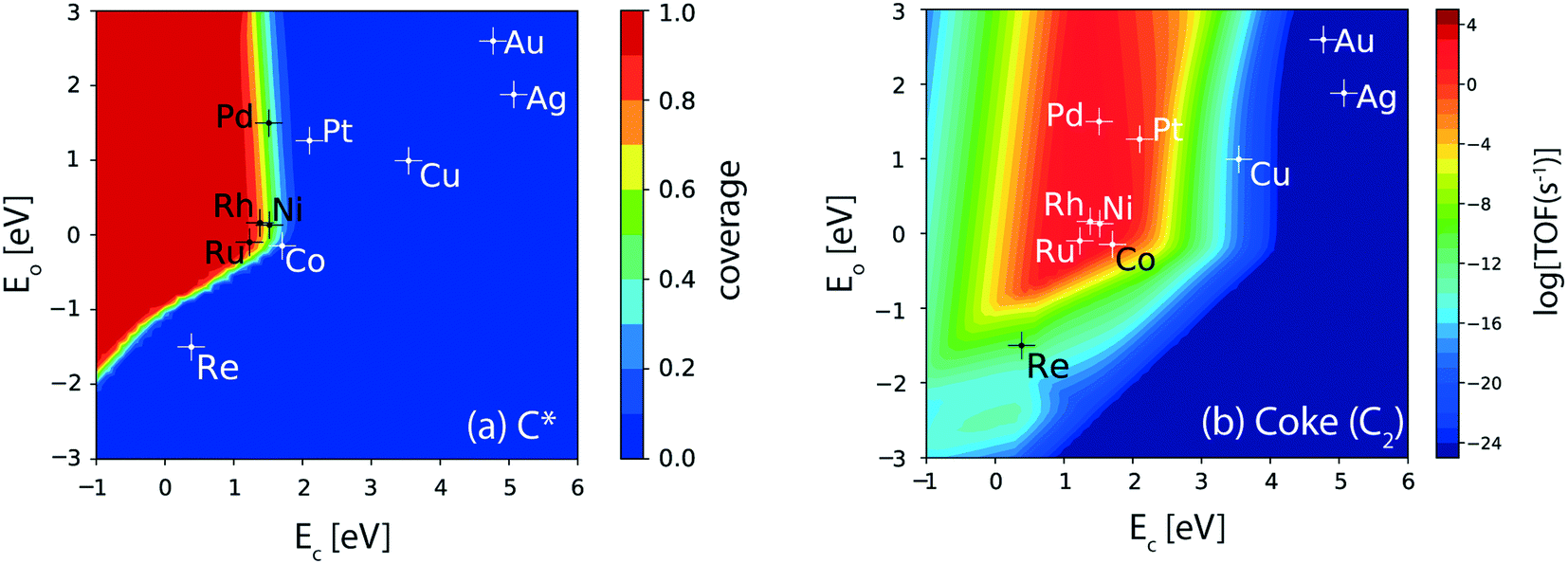 | ||
| Fig. 6 (a) Coverage of C* and (b) production rate of coke (C2) over the (211) facets of transition metal catalysts. Error bar = 0.2 eV. | ||
In order to assess the activity of step sites for coke formation, a model incorporating the C–C coupling reaction of C* species is assumed for the (211) surface, resulting in the formation of product C2(g). C2* formation via the C–C coupling reaction has been shown to be a prerequisite towards coke formation, for both graphitic and amorphous forms.95,96 In an endeavour to design carbon tolerant reforming catalysts, Nikolla et al. have concluded that the long term stability of catalysts can only be ensured by preventing the C–C bond formation reaction.95 Therefore, in this study, the formation of the C2(g) product from C–C coupling is considered as a model for coke deposition. Fig. 6(b) shows the corresponding volcano plot for coke formation at the step surface of the catalysts. Rh, Ru, Ni and Pd lie at the top of the volcano, exhibiting a turnover of the order 103 s−1. Co shows an order of magnitude lower production rate of coke than Ru, Rh, Ni and Pd. Among all the active dehydrogenation catalysts, Pt exhibits two orders of magnitude lower TOF for coke than Ru, Rh, Ni and Pd, which is confirmed by the prevalent application of Pt as an alkane dehydrogenation catalyst. Re and Cu show very low TOFs for coke formation (<10−9 s−1). However, these metals also show reduced dehydrogenation rates (TOF ∼ 1 s−1). Noble metals do not exhibit production of coke and thus may be selected for alloying with the active transition metal catalysts (e.g. Pt, Pd, Rh) to reduce coke formation.
3.3 Rational design of bimetallic alloys
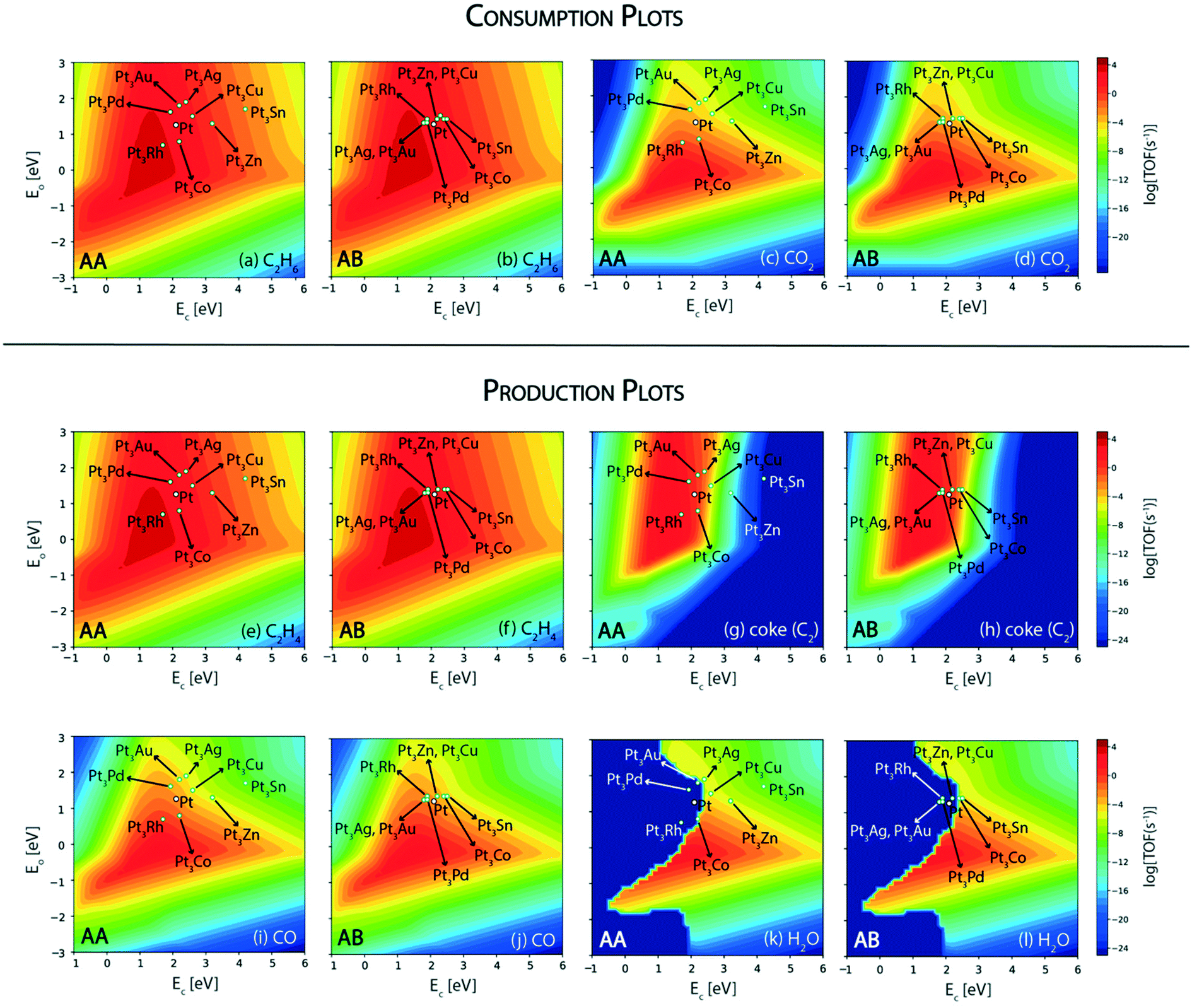
In this reaction, a rationale for designing bimetallic alloys for reduced coke deposition on the (211) surface may be developed from the MKM plots using the interpolation principle.97 This can be done with an aim to achieve high turnover for CO2 consumption and maintain high product (ethylene) selectivity. We have identified two metals (Ni and Pt) for designing alloys in the form of an A3B bimetallic alloy. While Ni is identified as a potential candidate for CO2 assisted ethane dehydrogenation in the aforementioned MKM analysis, Pt is a well-known alkane dehydrogenation catalyst.18,24 Both of these metals have been explored in experiments to synthesize bimetallic alloys for CO2 assisted ethane dehydrogenation.11 For oxidative dehydrogenation of ethane on monometallic and bimetallic catalysts, Myint et al. have observed a decrease in catalyst deactivation on bimetallic compositions as compared to the monometallic catalysts supported on ceria.11 For example, catalyst deactivation is observed to decrease from 48.7% on Pt/CeO2 to 10.4% on CoPt/CeO2.11 Similarly, the deactivation on Fe/CeO2 is found to be 52.8%, whereas on FeNi/CeO2, it is significantly improved to 28.9%.11Fig. 7 shows the screening of Ni-based bimetallic alloy catalysts. Ni is alloyed with Rh, Pt, Pd, Ru, Cu, Fe, Co, and Sn to form two types of (211) surfaces – AA and AB terminations (Fig. SI-4†). The AA and AB terminations of the (211) surface are consistently used as representative model surfaces of A3B bimetallic alloys by us33,48,49,58 and others.30,55,62,98 In a combined experimental and theoretical study, Studt et al. have utilised this model surface to understand the reactivity trends for CO hydrogenation to methanol, wherein calculations on the model surface showed a strong correlation with the experiments, predicting a high product yield on the Cu3Ni alloy.98 On both surface terminations (AA and AB) of the step sites, the reactant (C2H6 and CO2) and product (C2H4, coke, CO and H2O) turnovers are plotted in the form of volcano plots as shown in Fig. 7. These plots are further analyzed to screen suitable bimetallic candidates based on four metrics: (i) the same or higher ethane turnovers, (ii) higher CO2 turnovers, (iii) the same or higher ethylene turnovers and (iv) less coke production on any one of the two surface terminations (AA or AB) as compared to the respective monometallic (Ni) activity. This analysis is presented in the form of an abridged periodic table shown in Fig. 8, wherein the effect of Ni (the A metal) alloying with the other B metals on the reactant (CO2 and C2H6) consumption and product (C2H4, coke, CO and H2O) formation is shown as the colour of the B metal box, corresponding to the TOF. Alloy candidates passing this screening test are shown in colour in Fig. 8. In this representative placing on the periodic table, the alloy showing similar or higher turnovers for CO2 is only shown in C2H4 turnover plots (Fig. 8(c)), while applying the screening criterion of higher or similar turnovers of ethylene. Thus, alloys shown in colour on the C2H4 production plot (Fig. 8(c)) are a subset of the overall alloys showing the same or higher ethylene production rates in Fig. 7(e) and (f). These suitable candidates are finally selected on the basis of reduced coke formation as compared to Ni (Fig. 7(g) and (h)). Plots for H2 production, in general, are similar to ethylene production and are separately shown in Fig. SI-7.†
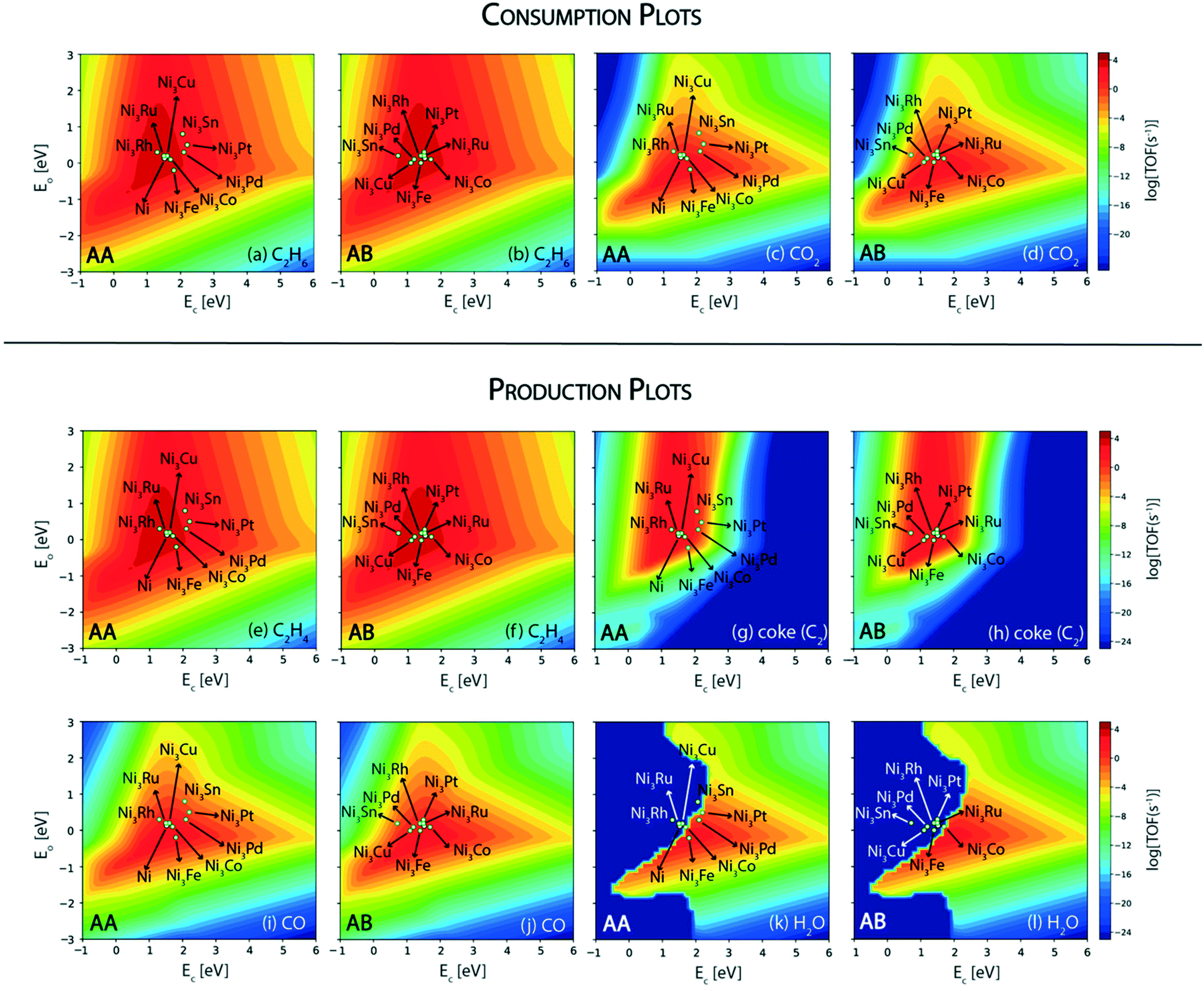 | ||
| Fig. 7 Volcano plots for consumption rates of (a) C2H6 (AA termination), (b) C2H6 (AB termination), (c) CO2 (AA termination), and (d) CO2 (AB termination); production rates of (e) C2H4 (AA termination), (f) C2H4 (AB termination), (g) coke (AA termination), (h) coke (AB termination), (i) CO (AA termination), (j) CO (AB termination), (k) H2O (AA termination) and (l) H2O (AB termination) over Ni-based A3B alloys. | ||
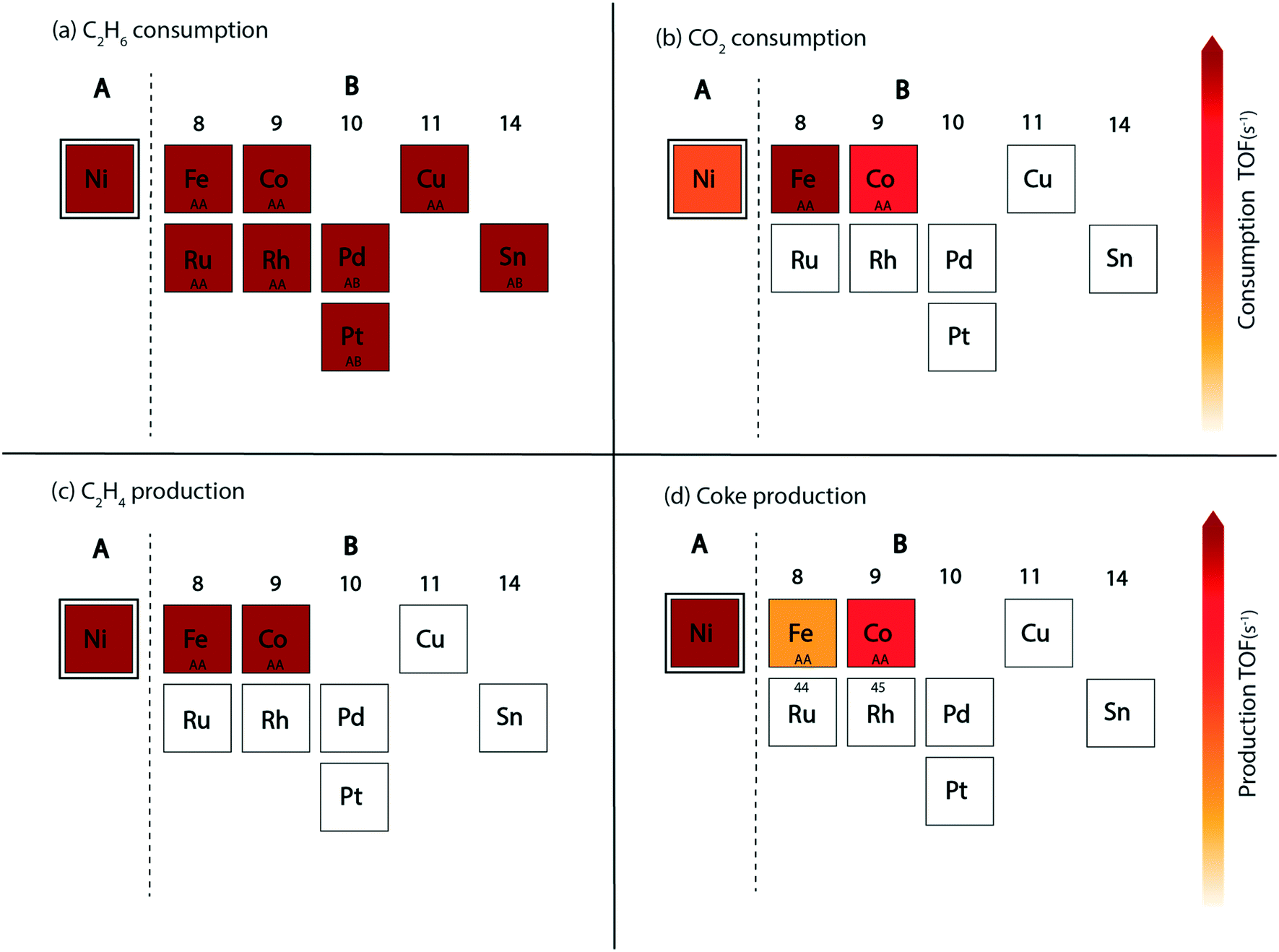 | ||
| Fig. 8 Selection of potential A3B alloy candidates of element A (Ni) for CO2 assisted ethane dehydrogenation. The placement of B-metals alloyed with Ni is shown in the form of an abridged periodic table with their colour corresponding to the TOFs calculated for A3B alloys. Candidates passing the selection criteria: (a) higher or similar C2H6 consumption, (b) higher CO2 consumption, (c) higher or similar C2H4 production (with higher CO2 consumption rate) and (d) reduced coke production are the ones shown in colour (corresponding to their TOFs). Surface termination of the alloy passing the screening criteria is marked as AA or AB in the B-metal boxes. | ||
Since Ni is already lying on the top of the volcano for the ethane consumption plot (Fig. 7(a) and (b)), alloying Ni with most of the metals produces similar turnovers of ethane, with the exception of Sn (on AA termination, Fig. 7(a)), Pt (on AA termination, Fig. 7(a)) and Pd (on AA termination, Fig. 7(a)), where ethane consumption is reduced by an order of magnitude. The effect of alloying in Ni is more visible on CO2 turnovers where Ni3Fe shows an order of magnitude higher rate (TOFCO2 ∼ 103 s−1) on the AA termination (Fig. 7(c)) and a similar rate (102 s−1) on the AB termination (Fig. 7(d)) as compared to pure Ni (TOFCO2 ∼ 102 s−1, Fig. 4(b)). Besides Ni3Fe, Ni3Co shows similar turnovers (TOFCO2 ∼ 102 s−1) on both the AA and AB terminations (Fig. 7(c) and (d)). On the production front, the ethylene production rates (Fig. 7(e) and (f)) are similar to the ethane consumption rates (Fig. 7(a) and (b)) on the (211) surface of the respective bimetallics. Selected bimetallic alloy candidates show higher or similar turnovers of ethylene production (Fig. 7(e) and (f)), while also maintaining a higher turnover of CO2 (Fig. 7(c) and (d)) as compared to Ni, which are highlighted in the periodic table form shown in Fig. 8(c). Both the bimetallic alloys (Ni3Fe and Ni3Co) passing the criteria for CO2 consumption also show similar or higher ethylene consumption as compared to Ni (Fig. 8(c)). The final test for the selection of the two bimetallics lies in the reduction of coke formation at the (211) surface. From the volcano plot (Fig. 7(g) and (h)) of coke formation, it can be observed that Ni3Fe reduces coke formation rates by two orders of magnitude on the AA termination of the (211) surface and Ni3Co reduces coke turnovers by an order of magnitude on both the AA and AB terminations. Thus, both bimetallic candidates (Ni3Fe and Ni3Co) are found suitable for CO2 assisted ethane dehydrogenation with reduced coking compared to Ni. In fact, experiments by Chen and group have identified NiFe as the most suitable bimetallic for CO2 assisted dehydrogenation of ethane,11,85 propane13 and butane.12 There are few other bimetallics (Ni3Sn, Ni3Pd, and Ni3Pt) that show significantly reduced coke formation (Fig. 7(g)) and exhibit potential for direct ethane dehydrogenation. Among them, NiSn is a known bimetallic alloy used for alkane dehydrogenation.99 However, for CO2 assisted dehydrogenation, these bimetallic alloys show reduced CO2 turnovers as compared to Ni and thus are not selected.
Next, we screen the bimetallic alloys of Pt with the same goal of obtaining higher or similar turnovers of ethane and ethylene while reducing more CO2 and producing less coke compared to the pure Pt catalyst. Volcano plots for ethane and CO2 consumption on both surface terminations (AA and AB) of Pt bimetallics are shown in Fig. 9. From the ethane consumption plots (Fig. 9(a) and (b)), the bimetallics showing similar or higher ethane consumption activity compared to Pt are selected and represented in the form of the abridged periodic table shown in Fig. 10. Combining the trends on both surface terminations, Pt3Pd, Pt3Rh, Pt3Ag, Pt3Au and Pt3Co show similar ethane turnovers to those of pure Pt. Interestingly, on the AA surface termination, Pt3Rh lies in the volcano maximum (Fig. 9(a)) with the ethane consumption rate (TOFC2H6 ∼ 104 s−1) calculated to be an order of magnitude higher than Pt. Thus, on applying the selection criteria for ethane consumption, five alloy candidates (Pt3Pd, Pt3Rh, Pt3Ag, Pt3Au and Pt3Co) are selected which are represented in Fig. 10(a). The selection criteria of bimetallics for CO2 consumption require a higher CO2 consumption rate as compared to pure Pt. This screens only two potential bimetallic alloys: Pt3Co (TOFCO2 ∼ 10−1 s−1, Fig. 9(c)) and Pt3Rh (TOFCO2 ∼ 1 s−1, Fig. 9(c)), which show a multifold increase in CO2 turnovers (Fig. 10(b)) as compared to pure Pt (TOFCO2 ∼ 10−3 s−1, Fig. 4(b)). These two bimetallic alloys are further tested for their ethylene formation rates. Indeed, both of them show higher or similar ethylene formation (Fig. 10(a)), since the overall volcano plot and reactivity trend for ethylene formation (Fig. 7(e) and (f)) are the same as those for ethane consumption (Fig. 7(a) and (b)). The final test indeed lies in the coke formation rates on the (211) surface of the bimetallic alloys (Fig. 9(g) and (h)). Here, we expect to reduce coke formation. Of the two bimetallics, Pt3Co reduces coke formation rates by two orders of magnitude on the AA termination (Fig. 9(g)) and one order of magnitude on the AB termination (Fig. 9(h)) while Pt3Rh increases coke formation on both surface terminations as compared to pure Pt. There are other bimetallic combinations which reduce coke (Pt3Au, Pt3Ag, Pt3Cu, Pt3Sn, and Pt3Zn). However, CO2 consumption on these bimetallic alloys is observed to be very low (<10−4 s−1). Previous experimental studies on PtSn have established higher stability of the bimetallic catalyst as compared to the pure metal catalyst for reforming and dehydrogenation reactions of hydrocarbons.24 However, in this case for CO2 assisted ethane dehydrogenation, only one Pt-alloy candidate (Pt3Co) is selected which may reduce overall coke formation with some increase in CO2 consumption compared to pure Pt catalysts. This is shown as the final selection in the periodic table (Fig. 10(d)).
 | ||
| Fig. 9 Volcano plots for consumption rates of (a) C2H6 (AA termination), (b) C2H6 (AB termination), (c) CO2 (AA termination), and (d) CO2 (AB termination); production rates of (e) C2H4 (AA termination), (f) C2H4 (AB termination), (g) coke (AA termination), (h) coke (AB termination), (i) CO (AA termination), (j) CO (AB termination), (k) H2O (AA termination) and (l) H2O (AB termination) over Pt-based A3B alloys. | ||
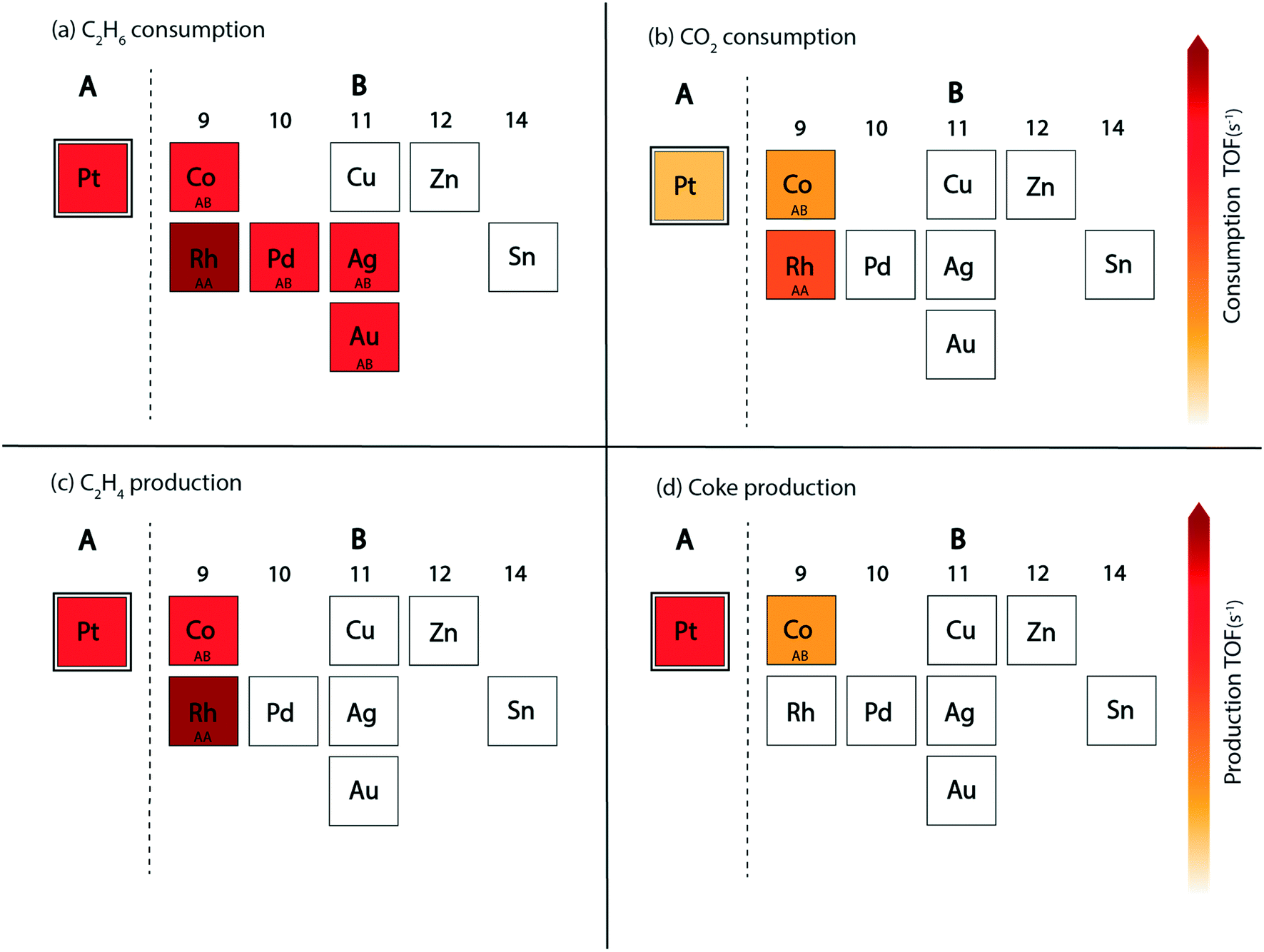 | ||
| Fig. 10 Selection of potential A3B alloy candidates of element A (Pt) for CO2 assisted ethane dehydrogenation. The placement of B-metals alloyed with Pt is shown in the form of an abridged periodic table with their colour corresponding to the TOFs calculated for A3B alloys. Candidates passing the selection criteria: (a) higher or similar C2H6 consumption, (b) higher CO2 consumption, (c) higher or similar C2H4 production (with higher CO2 consumption activity) and (d) reduced coke production are the ones shown in colour (corresponding to their TOFs). Surface termination of the alloy passing the screening criteria is marked as AA or AB in the B-metal boxes. | ||
Among the three bimetallics (Ni3Co, Ni3Fe and Pt3Co) identified as potential alloy compositions for oxygen assisted ethane dehydrogenation, two are tested experimentally.11–13,85 Interestingly, only one of them (NiFe) is suggested as the favored bimetallic for alkane dehydrogenation, while PtCo is observed to produce mostly CO (98.7%, carbon selectivity) with high ethane (24%) and CO2 conversions (44.7%), thus acting as a reforming catalyst.11 On the contrary, on the NiFe bimetallic catalyst, ethylene is produced with significant selectivity (31%).11 However, the corresponding ethane (9.1%) and CO2 conversions (10.8%) are observed to be significantly reduced on the NiFe alloy as compared to the PtCo alloy.11 In order to understand this contrast in reported experimental trends, the reaction rates for oxygen assisted C–H bond activation in ethane are analyzed on the three bimetallic alloys. The corresponding volcano plot for the elementary step is shown in Fig. 11. Pt3Co (AA termination), Ni3Co (AA as well AB termination) and Ni3Fe (AA termination) exhibit elementary rates of 10−1 s−1, 10 s−1 and 102 s−1, respectively, for oxygen assisted ethane dehydrogenation. Indeed, all of the bimetallics show a significant multi-fold increase in oxygen assisted dehydrogenation rates with respect to their monometallic counterparts (Ni ∼ 10 s−1, Pt ∼ 10−1 s−1). Thus, these bimetallic alloys, in general, should be potential candidates for oxidative dehydrogenation of alkanes. Here we also observe that only Ni3Fe lies in the volcano maximum, alluding to the fact that it is the only bimetallic reported so far, showing potential for oxidative dehydrogenation of ethane.11–14 Myint et al. identified the FeNi catalyst supported on CeO2 to be a potential candidate for oxidative dehydrogenation of ethane to produce ethylene.11 However, CO2 and ethane conversions over FeNi were observed to be lower (10.8% and 9.1%, respectively) compared to those over the bimetallic CoPt catalyst (44.7% and 24.0%, respectively); however, CoPt favours reforming reactions to form syngas.
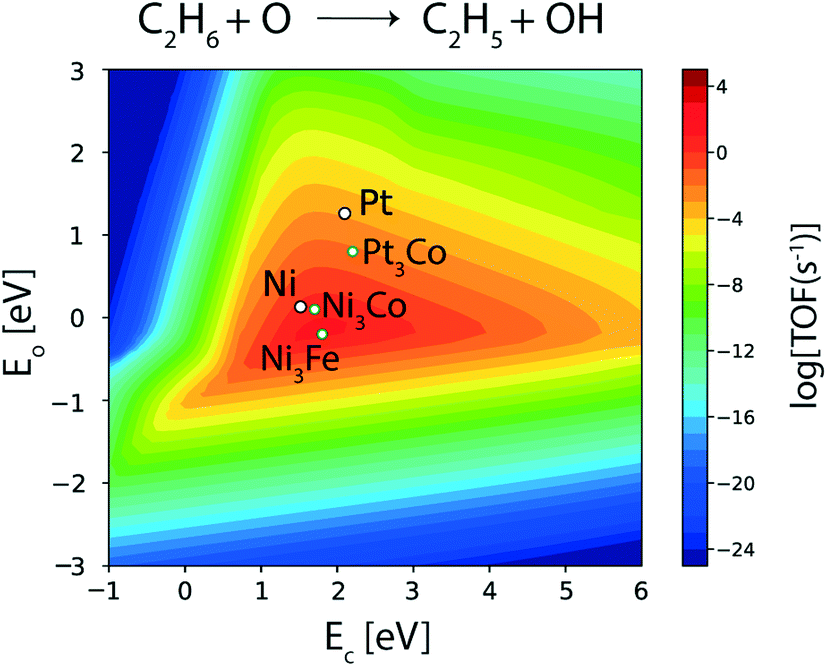 | ||
| Fig. 11 Elementary reaction rate for oxygen assisted ethane dehydrogenation over screened alloy candidates. | ||
The reason for high reforming rates on transition metal catalysts lies in the activity of the metal–ceria interface.100 In all of the metal and bimetallic catalysts tested by Chen and co-workers, ceria is thought to play a prominent role in activating CO2 and providing surface oxygen species for oxidative dehydrogenation.11–13,85 CeO2 supported bimetallic alloys catalyse oxidative dehydrogenation at a high rate as it is a reducible support and the presence of oxygen further prevents coke formation via removal through oxidation.21 For example, on increasing the Fe content in the NiFe bimetallic alloy, an amorphous layer of FeOx is formed at the metal–ceria interface which is observed to inhibit the availability of surface oxygen species on the metal, leading to significantly reduced oxidative ethane conversions.85 For the possibility of oxidative dehydrogenation of ethane, availability of surface oxygen species is a necessity.16,81 Interestingly, the metal–ceria interface is also known for high reforming activity, which possibly leads to ethane hydrogenolysis and significantly reduced ethylene selectivity as observed in experiments.11–13
Since the MKM developed in this study is focused on modeling the trends in transition metal activity by ignoring the support effect, the role of the support in activating CO2 and providing surface oxygen species is not incorporated in the model. At the metal–ceria interface, CO2 is activated on oxygen vacancies in the vicinity of Ce3+ cations.14,81,100 Oxygen species thus available at the interface may be used for diverse reactions which may include reforming to produce CO.100 For reforming reactions of higher alkanes (C2–C4), the hydrogenolysis reaction occurring at the step sites to produce C* species constitutes an essential step. The transition metal and bimetallic alloys studied in this work show a high degree of hydrogenolysis activity, which is evident in their activity for coke formation at the step surface. Thus, whenever the metal–ceria interface becomes active for CO2 activation, the oxygen species produced are utilised for the reforming reaction with a higher rate. For CO2 assisted alkane dehydrogenation, oxygen species are required to assist in C–H bond activation or to facilitate the removal of hydrogen produced (to shift the equilibrium). Thus, reducible supports such as ceria are essential in this reaction.81 However, oxygen assisted dehydrogenation could only be achieved by controlling the hydrogenolysis route which facilitates the reforming activity and coke formation. The hydrogenolysis reaction at the step sites could be moderated by using a relatively noble metal (Ag, Au, Cu, Sn) to block the step sites.19,20,101,102 In addition, an effort could be made to synthesize small metal-clusters103 or apply single metal atoms dispersed on the ceria support for partial or complete removal of step sites, so as to reduce the hydrogenolysis activity, facilitating the oxygen assisted dehydrogenation route. Studies on single-atom catalysts supported on ceria for the water-gas shift reaction104 and CO oxidation105 show a way to synthesize such catalysts which may also be tested for this reaction.
While the MKM is able to predict the activity and selectivity trends of the catalyst surfaces, the morphology of the catalyst plays a vital role in determining the overall catalytic activity.106 The MKM constructed herein doesn't consider the dynamic nature of the catalyst and the effect of morphological changes that the catalyst may undergo under the reaction conditions. Coupling the MKM with ab initio thermodynamics and Wulff–Kaishew construction may facilitate the estimation of changes in active sites of the catalyst in reactive environments, leading to a more realistic estimation of catalytic rates.107
4. Conclusions
An ab initio, mean field MKM is developed to understand the interplay of reactions that take place in CO2 assisted ethane dehydrogenation over the terrace (111) and step (211) sites of transition metal catalysts. The MKM is analyzed at a reaction temperature of 873 K, to understand the trend in reactant and product turnovers on the catalyst surface described by the carbon and oxygen binding energies of the metal catalysts. On the (111) sites, similar rates are calculated for the consumption of ethane and the production of ethylene and hydrogen on the respective metal catalysts, indicating that ethane is predominantly converted to ethylene via direct dehydrogenation. Rh and Pt are screened to be the most active catalysts for the consumption of ethane on the terrace sites, followed by Pd, Ni, Ru and Co. In general, the CO2 consumption rates on the (111) sites are observed to be significantly reduced compared to ethane consumption TOFs, on all the metals. Nevertheless, four metal catalysts (Rh, Ru, Ni and Co) are calculated to show appreciable activity for CO2 conversion. Interestingly on these four metals, the CO and H2O production rates are of the same order as the CO2 consumption rates, indicating the likelihood of CO2 conversion via the RWGS reaction on their (111) surface. On analysing the elementary rates of oxygen assisted dehydrogenation of ethane, the same four catalysts showed appreciable turnovers for oxygen assisted C–H activation; however, the rates are a bit lower than direct dehydrogenation rates. Since none of the metal catalysts calculated any water consumption rates, steam reforming on the (111) surface is unlikely. Moreover, none of the transition metals screened showed coverages of C*, indicating lower rates of the C–C hydrogenolysis reaction over the (111) sites. This is further confirmed in negligible turnovers calculated for methane production on the terrace surface from the thermal hydrogenolysis reaction of ethane.For the (211) surface, a similar observation is made – the ethane consumption plot is comparable to the production plots of ethylene and hydrogen, suggesting the prevalence of metal catalyzed direct dehydrogenation. For CO2 consumption, the same four (Co, Rh, Ru and Ni) metals are calculated to show significantly high turnovers of CO2 on their (211) surface, higher than the respective (111) surface. Among the four metals, the reactivity of Co for consumption of CO2 is observed to be at the maximum on the step sites, followed by Rh, Ru and Ni. In contrast to the (111) surface, the water formation plots on the (211) surface show that only Co produces water. The other three metals (Rh, Ni and Ru) followed by Pd and Pt indicate steam reforming activity by calculating significant turnovers for water consumption. Interestingly, on Co, water is produced at a lower rate than that of CO, implying dry reforming along with RWGS as the route for CO2 reduction. In addition, elementary reaction rates calculated for oxygen assisted C–H activation of ethane point towards significantly high activity on the step sites of Co, Rh, Ru and Ni, illustrating potential for CO2 assisted dehydrogenation. Furthermore, C* coverages and subsequent coke production plots on the (211) surface of all the transition metals underline the prevalence of the C–C hydrogenolysis reaction on the step sites. Co, Ni, Pt and Rh calculated high turnovers on the step sites for thermal hydrogenolysis to produce methane. In general, bimetallic alloys experimented before for CO2 assisted alkane dehydrogenation are observed to produce CO via reforming reactions. Moreover, reforming tends to reduce the dehydrogenation activity of the catalyst. The reason for this lies in the high hydrogenolysis activity of the step surfaces, which leads to both reforming and coke formation. In an attempt to explore bimetallic alloys to reduce coke formation coupled with increased CO2 consumption, three alloys are proposed: Ni3Fe, Ni3Co and Pt3Co, out of which two are tested experimentally and the NiFe catalyst supported on ceria has shown some promise for alkane dehydrogenation along with CO2 reduction. However, MKM results illustrate the rationale for bimetallic design where side reactions enabled by the hydrogenolysis activity of the catalyst lead to undesired product formation. A thoughtful strategy in catalyst synthesis is therefore required to turn off the hydrogenolysis path at the stepped surface, which may be attempted by blocking the step sites or synthesizing single atom catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are appreciative of Prof. Divesh Bhatia for discussions and proof-reading of the manuscript. Authors would like to acknowledge and appreciate the design for cover illustration by Ankur Tomar. Computational resources provided at the high performance computing (HPC) facility of IIT Delhi are acknowledged for all calculations performed in this manuscript. Financial support from the Department of Science and Technology, Government of India Grant DST/TMD/MECSP/2KI7/07 and MTR/20l9/000314 are acknowledged.References
- E. McFarland, Science, 2012, 338, 341–342 CrossRef.
- C. Radcliffe, The FCC unit as a propylene source, (accessed 17 February 2020), https://www.digitalrefining.com/article/1000312#.XkpakigzaUm Search PubMed.
- P. Chaiyavech, Journal of the Royal Institute of Thailand, 2002, 27, 664–673 Search PubMed.
- Z. Nawaz, Rev. Chem. Eng., 2015, 31, 413–436 CAS.
- K. Bullin and P. Krouskop, Oil Gas J., 2009, 107, 50–55 CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- G. B. Kistiakowsky, J. R. Ruhoff, H. A. Smith and W. E. Vaughan, J. Am. Chem. Soc., 1935, 57, 876–882 CrossRef CAS.
- H. H. Kung, Adv. Catal., 1994, 40, 1–38 CAS.
- S. C. Pereira, M. F. Ribeiro, N. Batalha and M. M. Pereira, Greenhouse Gases: Sci. Technol., 2017, 7, 843–851 CrossRef CAS.
- M. M. Bhasin, J. H. McCain, B. V. Vora, T. Imai and P. R. Pujadó, Appl. Catal., A, 2001, 221, 397–419 CrossRef CAS.
- M. N. Z. Myint, B. Yan, J. Wan, S. Zhao and J. G. Chen, J. Catal., 2016, 343, 168–177 CrossRef CAS.
- X. Li, B. Yan, S. Yao, S. Kattel, J. G. Chen and T. Wang, Appl. Catal., B, 2018, 231, 213–223 CrossRef CAS.
- E. Gomez, S. Kattel, B. Yan, S. Yao, P. Liu and J. G. Chen, Nat. Commun., 2018, 9, 1–6 CrossRef CAS.
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem., 2019, 3, 638–649 CrossRef CAS.
- M. D. Porosoff, M. N. Z. Myint, S. Kattel, Z. Xie, E. Gomez, P. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2015, 54, 15501–15505 CrossRef CAS.
- D. Mukherjee, S. E. Park and B. M. Reddy, J. CO2 Util., 2016, 16, 301–312 CrossRef CAS.
- J. H. Sinfelt, Science, 1977, 195, 641–646 CrossRef CAS.
- D. W. Blakely and G. A. Somorjai, J. Catal., 1976, 42, 181–196 CrossRef CAS.
- M. W. Smale and T. S. King, J. Catal., 1989, 119, 441–450 CrossRef CAS.
- A. K. Rovik, S. K. Klitgaard, S. Dahl, C. H. Christensen and I. Chorkendorff, Appl. Catal., A, 2009, 358, 269–278 CrossRef CAS.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. S. Clausen, L. P. Nielsen, A. M. Molenbroek and J. R. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- K. Nagaoka, K. Seshan, K. I. Aika and J. A. Lercherz, J. Catal., 2001, 197, 34–42 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Catal., 1974, 33, 184–201 CrossRef.
- R. Burch and L. C. Garla, J. Catal., 1981, 71, 360–372 CrossRef CAS.
- R. T. Vang, K. Honkala, S. Dahl, E. K. Vestergaard, J. Schnadt, E. Lægsgaard, B. S. Clausen, J. K. Nørskov and F. Besenbacher, Nat. Mater., 2005, 4, 160–162 CrossRef CAS.
- A. J. Medford, C. Shi, M. J. Hoffmann, T. Bligaard and J. K. Nørskov, Catal. Lett., 2015, 145, 794–807 CrossRef CAS.
- F. Johansson, https://code.google.com/archive/p/mpmath/.
- CatApp, http://www.suncat.slac.stanford.edu/catapp Search PubMed.
- J. S. Hummelshøj, F. Abild-Pedersen, F. Studt, T. Bligaard and J. K. Nørskov, Angew. Chem., Int. Ed., 2012, 51, 272–274 CrossRef.
- A. J. Medford, A. C. Lausche, B. Temel, N. C. Schjødt, J. K. Nørskov and F. Studt, Top. Catal., 2014, 57, 135–142 CrossRef CAS.
- T. Jiang, D. J. Mowbray, S. Dobrin, H. Falsig, B. Hvolbœk, T. Bligaard and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, 10548–10553 CrossRef CAS.
- A. A. Peterson, F. Abild-pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- F. Jalid, T. S. Khan, F. Q. Mir and M. A. Haider, J. Catal., 2017, 353, 265–273 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B, 1999, 59, 7413–7421 CrossRef.
- A. Bilic, J. R. Reimers, N. S. Hush, R. C. Hoft and M. J. Ford, J. Chem. Theory Comput., 2007, 2, 1093–1105 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS.
- DACAPO, https://wiki.fysik.dtu.dk/dacapo Search PubMed.
- A. J. Medford, J. Wellendorff, A. Vojvodic, F. Studt, F. Abild-Pedersen, K. W. Jacobsen, T. Bligaard and J. K. Nørskov, Science, 2014, 345, 197–200 CrossRef CAS.
- H. Falsig, J. Shen, T. S. Khan, W. Guo, G. Jones, S. Dahl and T. Bligaard, Top. Catal., 2014, 57, 80–88 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- S. G. Wang, X. Y. Liao, D. B. Cao, Y. W. Li, J. Wang and H. Jiao, J. Phys. Chem. C, 2007, 111, 10894–10903 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, J. Catal., 2007, 250, 85–93 CrossRef CAS.
- Z. Wang, X. M. Cao, J. Zhu and P. Hu, J. Catal., 2014, 311, 469–480 CrossRef CAS.
- J. Gao, J. Yip, J. Zhao, B. I. Yakobson and F. Ding, J. Am. Chem. Soc., 2011, 133, 5009–5015 CrossRef CAS.
- S. Wang, B. Temel, J. Shen, G. Jones, L. C. Grabow, F. Studt, T. Bligaard, F. Abild-Pedersen, C. H. Christensen and J. K. Nørskov, Catal. Lett., 2011, 141, 370–373 CrossRef CAS.
- T. S. Khan, F. Jalid and M. A. Haider, Top. Catal., 2018, 61, 1820–1831 CrossRef CAS.
- T. S. Khan, S. Hussain, U. Anjum and M. A. Haider, Electrochim. Acta, 2018, 281, 654–664 CrossRef CAS.
- S. Saxena, T. S. Khan, F. Jalid, M. Ramteke and M. A. Haider, J. Mater. Chem. A, 2020, 8, 107–123 RSC.
- J. K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef CAS.
- A. C. Lausche, A. J. Medford, T. Suvra, Y. Xu, T. Bligaard, F. Abild-pedersen, J. K. Nørskov and F. Studt, J. Catal., 2013, 307, 275–282 CrossRef CAS.
- A. C. Lausche, H. Falsig, A. D. Jensen and F. Studt, Catal. Lett., 2014, 144, 1968–1972 CrossRef CAS.
- A. C. Lausche, J. S. Hummelshoj, F. Abild-Pedersen, F. Studt and J. K. Norskov, J. Catal., 2012, 291, 133–137 CrossRef CAS.
- Y. Xu, A. C. Lausche, S. Wang, T. S. Khan, F. Abild-pedersen and F. Studt, New J. Phys., 2013, 15, 125021 CrossRef.
- S. Wang, V. Petzold, V. Tripkovic, J. Kleis, J. G. Howalt, E. Skúlason, E. M. Fernández, B. Hvolbæk, G. Jones, A. Toftelund, H. Falsig, M. Björketun, F. Studt, F. Abild-Pedersen, J. Rossmeisl, J. K. Nørskov and T. Bligaard, Phys. Chem. Chem. Phys., 2011, 13, 20760–20765 RSC.
- L. C. Grabow, F. Studt, F. Abild-Pedersen, V. Petzold, J. Kleis, T. Bligaard and J. K. Nørskov, Angew. Chem., Int. Ed., 2011, 50, 4601–4605 CrossRef CAS.
- F. Jalid, T. S. Khan and M. A. Haider, MRS Commun., 2019, 9, 107–113 CrossRef CAS.
- M. I. Alam, T. S. Khan and M. A. Haider, ACS Sustainable Chem. Eng., 2018, 7, 2894–2898 CrossRef.
- M. H. Hansen, J. K. Nørskov and T. Bligaard, J. Catal., 2019, 374, 161–170 CrossRef CAS.
- L. C. Grabow, B. Hvolbæk and J. K. Nørskov, Top. Catal., 2010, 53, 298–310 CrossRef CAS.
- H. Li, A. C. Lausche, A. A. Peterson, H. A. Hansen, F. Studt and T. Bligaard, Surf. Sci., 2015, 641, 105–111 CrossRef CAS.
- A. J. Medford, J. Wellendorff, A. Vojvodic, F. Studt, F. Abild-pedersen, K. W. Jacobsen, T. Bligaard and J. K. Nørskov, Science, 2014, 345, 197–200 CrossRef CAS.
- M. Huff and L. D. Schmidt, J. Phys. Chem., 1993, 97, 11815–11822 CrossRef CAS.
- R. D. Cortright, R. M. Watwe and J. A. Dumesic, J. Mol. Catal. A: Chem., 2000, 163, 91–103 CrossRef CAS.
- V. Haensel, US Pat., 2602772, 1952 Search PubMed.
- B. Xing, X. Y. Pang and G. C. Wang, J. Catal., 2011, 282, 74–82 CrossRef CAS.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Catal. Sci. Technol., 2015, 5, 4154–4163 RSC.
- B. Lu and K. Kawamoto, J. Environ. Chem. Eng., 2013, 1, 300–309 CrossRef CAS.
- L. Dietz, S. Piccinin and M. Maestri, J. Phys. Chem. C, 2015, 119, 4959–4966 CrossRef CAS.
- K. Tsuchiya, J.-D. Huang and K. Tominaga, ACS Catal., 2013, 3, 2865–2868 CrossRef CAS.
- L. Wang, H. Liu, Y. Chen and S. Yang, Int. J. Hydrogen Energy, 2017, 42, 3682–3689 CrossRef CAS.
- D. Hibbitts and M. Neurock, Surf. Sci., 2016, 650, 210–220 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS.
- B. Xu, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2010, 132, 16571–16580 CrossRef CAS.
- J. Gong, D. W. Flaherty, T. Yan and C. B. Mullins, ChemPhysChem, 2008, 9, 2461–2466 CrossRef CAS.
- B. Xu, J. Haubrich, T. A. Baker, E. Kaxiras and C. M. Friend, J. Phys. Chem. C, 2011, 115, 3703–3708 CrossRef CAS.
- J. S. Yoo, T. S. Khan, F. Abild-Pedersen, J. K. Nørskov and F. Studt, Chem. Commun., 2015, 51, 2621–2624 RSC.
- M. Huš, D. Kopač, N. S. Štefančič, D. L. Jurković, V. D. B. C. Dasireddy and B. Likozar, Catal. Sci. Technol., 2017, 7, 5900–5913 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, ChemPhysChem, 2017, 18, 3135–3141 CrossRef.
- D. E. Peebles, D. W. Goodman and J. M. White, J. Phys. Chem., 1983, 87, 4378–4387 CrossRef CAS.
- C. Liu, T. R. Cundari and A. K. Wilson, J. Phys. Chem. C, 2012, 116, 5681–5688 CrossRef CAS.
- T. Avanesian, G. S. Gusmão and P. Christopher, J. Catal., 2016, 343, 86–96 CrossRef CAS.
- B. Yan, S. Yao, S. Kattel, Q. Wu, Z. Xie, E. Gomez, P. Liu, D. Su and J. G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8278–8283 CrossRef CAS.
- Y. Chen and D. G. Vlachos, J. Phys. Chem. C, 2010, 114, 4973–4982 CrossRef CAS.
- Z. P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS.
- S. Gupta, T. S. Khan, B. Saha and M. A. Haider, Ind. Eng. Chem. Res., 2019, 58, 16153–16163 CrossRef CAS.
- S. Gupta, M. I. Alam, T. S. Khan and M. A. Haider, ACS Sustainable Chem. Eng., 2019, 7, 10165–10181 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Sehested and J. K. Nørskov, Adv. Catal., 2002, 47, 65–139 CAS.
- J. R. Rostrup-Nielsen, J. Catal., 1973, 31, 173–199 CrossRef CAS.
- L. B. Råberg, M. B. Jensen, U. Olsbye, C. Daniel, S. Haag, C. Mirodatos and A. O. Sjåstad, J. Catal., 2007, 249, 250–260 CrossRef.
- J. Ko, B.-K. Kim and J. W. Han, J. Phys. Chem. C, 2016, 120, 3438–3447 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- E. Nikolla, A. Holewinski, J. Schwank and S. Linic, J. Am. Chem. Soc., 2006, 128, 11354–11355 CrossRef CAS.
- M. Argyle and C. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- M. P. Andersson, T. Bligaard, A. Kustov, K. E. Larsen, J. Greeley, T. Johannessen, C. H. Christensen and J. K. Nørskov, J. Catal., 2006, 239, 501–506 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, Q. Wu, A. D. Jensen, B. Temel, J.-D. Grunwaldt and J. K. Nørskov, J. Catal., 2012, 293, 51–60 CrossRef CAS.
- G. Wang, H. Wang, H. Zhang, Q. Zhu, C. Li and H. Shan, ChemCatChem, 2016, 8, 3137–3145 CrossRef CAS.
- B. Yan, X. Yang, S. Yao, J. Wan, M. N. Z. Myint, E. Gomez, Z. Xie, S. Kattel, W. Xu and J. G. Chen, ACS Catal., 2016, 6, 7283–7292 CrossRef CAS.
- A. Andreasen, H. Lynggaard, C. Stegelmann and P. Stoltze, Surf. Sci., 2003, 544, 5–23 CrossRef CAS.
- F. Besenbacher, F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Nørskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS.
- H. Wang, J. X. Liu, L. F. Allard, S. Lee, J. Liu, H. Li, J. Wang, J. Wang, S. H. Oh, W. Li, M. Flytzani-Stephanopoulos, M. Shen, B. R. Goldsmith and M. Yang, Nat. Commun., 2019, 10, 1–12 CrossRef.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS.
- F. Dong, Y. Meng, W. Han, H. Zhao and Z. Tang, Sci. Rep., 2019, 9, 12056 CrossRef.
- R. Cheula, A. Soon and M. Maestri, Catal. Sci. Technol., 2018, 8, 3493–3503 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cy01290d |
| This journal is © The Royal Society of Chemistry 2021 |