
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAre rate and selectivity correlated in iridium-catalysed hydrogen isotope exchange reactions?†‡
Daria S. Timofeeva ,
David M. Lindsay*,
William J. Kerr* and
David J. Nelson*
,
David M. Lindsay*,
William J. Kerr* and
David J. Nelson*
WestCHEM Department of Pure and Applied Chemistry, Thomas Graham Building, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: david.nelson@strath.ac.uk
First published on 9th July 2021
Abstract
Herein we qualitatively examine the relationship between reaction rate and reaction selectivity in iridium-catalysed hydrogen isotope exchange (HIE) reactions directed by Lewis basic functional groups. We have recently developed a directing group scale that allows semi-quantitative predictions of Lewis base directed selectivity in HIE, formally ranking ‘relative rates’ determined from a structured set of competition experiments. Here, we show that selectivity and rate are in fact not correlated, but that different types of behaviour emerge in competition experiments and that the observed behaviour can be predicted from our established selectivity scale.
Introduction
Achieving useful levels of site-selectivity in a robust and predictable manner is an enduring challenge of C–H activation chemistry. While most directing group approaches lead to ortho-selectivity,1–5 the factors at play in determining selectivity can vary between different catalyst systems and can be a function of the mechanism of the C–H activation step.6,7 We seek a detailed understanding of C–H activation selectivity, and have elected to deploy iridium-catalysed HIE reactions as a model system as part of this research programme.Hydrogen isotope exchange (HIE) is a crucial tool in many areas of chemistry, including in drug discovery, where it is critical in enabling the selective incorporation of 2H (D) and 3H (T) into drug molecules for pharmacokinetic studies, and in mechanistic studies, via measurement of kinetic isotope effects (KIE).8,9 An ideal HIE process should be general, operationally simple, and lead to high and predictable incorporation of the isotopic atom. Reactions catalysed by iridium complexes, which rely on a C–H activation step directed by a Lewis base, have emerged as reliable methods that fulfil these criteria (Fig. 1).10–12 These processes are therefore ideal vehicles for this study of C–H activation selectivity, where the regioselectivity of the reaction of a complex substrate with multiple Lewis basic groups may be difficult to predict a priori. Besides the commercial Crabtree's catalyst [Ir(COD)(PCy3)(py)][PF6] and Kerr's catalysts, such as [Ir(COD)(IMes)(PPh3)][X] (X = PF6 or BArF24), which remain the most widely used for isotope labelling in industry (COD = 1,5-cyclooctadiene; IMes = 2,6-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene; Cy = cyclohexyl; py = pyridine; BArF24 = tetrakis(3,5-bis(trifluoromethyl)phenyl) borate), numerous complexes of iridium have been developed to overcome the limitations and improve the scope of the ortho-directed H/D exchange; these include neutral [IrCl(COD)(NHC)] catalysts,13 new generations of iridium catalysts bearing bidentate P,N-ligands14,15 or NHC,N ligand (Burgess's catalyst)16 and [Ir(COD)(L)(NHC)] complexes with anionic N-heterocyclic carbenes that contain weakly coordinating anionic borate moieties to promote H/D exchange in nonpolar media.17 Homogeneous iridium-catalysed HIE reactions using D2 (or T2) gas have proved to be highly efficient for ortho-selective labelling of compounds next to directing groups such as ketones, amides, esters, nitroarenes, and sulfonamides, as well as various heterocycles such as pyridines, pyrimidines, pyrazoles, imidazole(in)es, thiazole(in)es, oxazole(in)es and their benzo-fused analogues.13,18–23
 | ||
| Fig. 1 Rate studies on iridium catalysed HIE reactions. | ||
We have recently constructed an empirical directing group scale that can be used to predict the selectivity of iridium catalysed HIE reactions in substrates with multiple directing groups.24 This scale was obtained by conducting a structured series of competition experiments using representative HIE catalysts [Ir(COD)(IMes)(PPh3)][BArF24] and [IrCl(COD)(IMes)]. This quantitative reactivity scale is formally ordered according to 'relative rate constants' krel and ranks nineteen commonly used directing groups, including various pharmaceutically relevant, nitrogen-containing heterocycles, and can be used for semi-quantitative regioselectivity predictions in hydrogen isotope exchange reactions of complex molecules. Derdau and co-workers recently reported DFT calculations to reveal the reactivity order of directing groups based on the relative free energies ΔGrel for the formation of their coordination complex.25 The proposed catalytic cycle for these reactions proceeds via binding of a substrate via its directing group, followed by steps that break and form C–H/D and H–H/D–D bonds (Scheme 1).18,26 All of the steps in the catalytic cycle are formally reversible. With competing directing groups, two possible reasons for selectivity differences are: (i) that there is a simple rate difference between the two parallel reactions; or (ii) that an equilibrium between the two substrate-bound intermediates (II) dictates the observed reactivity.
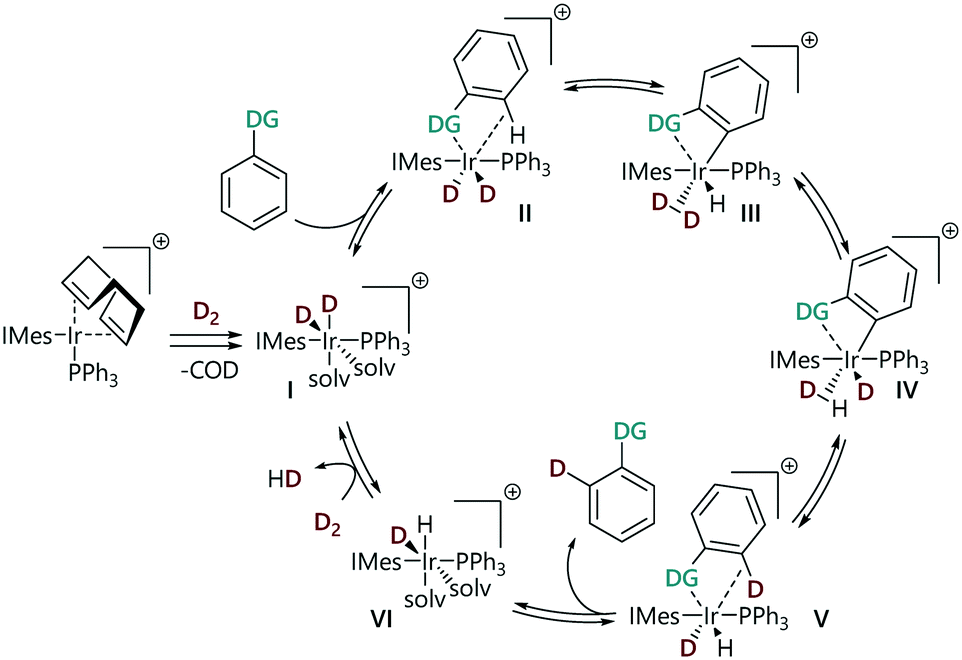 | ||
| Scheme 1 Mechanism of HIE catalysed by iridium complexes. | ||
In order to examine the relationship between the rate and selectivity of HIE reactions, we have elected to collect time-resolved data for a series of representative reactions. No kinetic investigation has been carried out previously to determine the rates of iridium-catalysed hydrogen isotope exchange. Absolute rates for the deuteration (kD) of acetophenone,18 an arylsulfonamide,13 and methyl phenylacetate,27 and for the hydrogenation of corresponding dideuterated species (kH), were determined in the course of the kinetic isotope effect (KIE) measurements for these reactions. In other examples, reactions were monitored by sampling at time points to build reaction profiles, but in such cases no rate constants were determined.28
In this work, we present a series of experiments in which the rates of HIE reactions catalysed by cationic iridium complex [Ir(COD)(IMes)(PPh3)][BArF24] are measured, and compared to data gathered previously for reaction selectivity. Kinetic studies on a single substrate were carried out first, in order to compare the observed rates of HIE. Selected intra- and intermolecular competition kinetic experiments were then performed to identify if reactions occur in parallel with different rates, or if one substrate inhibits the reaction of the other via competitive binding.
Results and discussion
The progress of each a series of representative HIE reactions catalysed by [Ir(COD)(IMes)(PPh3)][BArF24] (Scheme 2) was monitored by the withdrawal of aliquots from reaction mixtures at time intervals; these aliquots were analysed by 1H NMR spectroscopy and the degree of deuteration was quantified by integration of the peak of interest versus a site at which deuteration does not occur (see the ESI‡).29 Reactions were carried out in chloroform-d to enable the direct analysis of samples after quenching with a few drops of acetonitrile. We collected real-time quantitative information about the extent of isotopic labelling under representative laboratory conditions of D2 pressure (using a balloon), temperature, and stirring.§ Using this method we were able to monitor the HIE reactions of representative substrates chosen from the spectrum of established directing group strengths.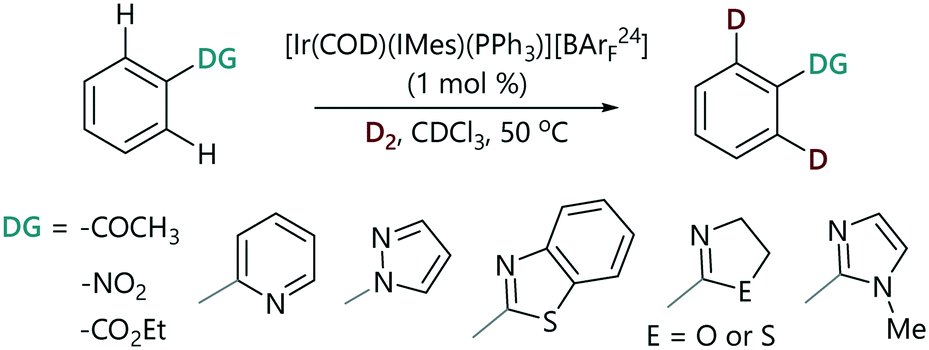 | ||
| Scheme 2 HIE reaction kinetic monitoring. | ||
The HIE reactions of acetophenone performed in CDCl3 showed no significant solvent effect when compared to data obtained in DCM.18 The use of CDCl3 as a reaction solvent allows for a wider temperature range and minimises the number of operations required after the withdrawal of the aliquot from the reaction.
Initial attempts to obtain the rate expression for the overall reaction revealed complex kinetic behaviour due to multiple factors, including gas solubility and mass transfer between phases.30 In this work, substrates with different directing groups were studied under otherwise identical conditions to allow an empirical comparison of the reaction rates. We assume that the same reaction mechanism is in operation with all the substrates, based on a number of computational studies of this class of reaction,13,18 therefore any difference in reactivity should be a function of the substrate. Under the conditions deployed here, reactions did not go to completion and approached equilibrium position, but labelling reactions proceeded further towards completion when they were conducted in larger vessel; there is a correlation between the approximate reaction solution surface area and the reaction rate (see the ESI‡).
A plot of the concentration of unlabelled substrate and deuterium incorporation (% D) of the substrate versus time shows clear mono-exponential kinetic behaviour for the representative examples of acetophenone, nitrobenzene, and 2-phenyloxazoline (Fig. 2(a)). The expected linearity of the ln [substrate] versus time plot was observed with a gradient of −kobs (Fig. 2(b)). Rate constants (kobs) are quoted in Table 1 and showed a high degree of reproducibility in duplicate experiments. These rate constants cover a very small range (ca. four-fold) and show very little correlation with the reactivity scale obtained from competition experiments (krel). The differences between the reaction rates observed for ketone, nitro, and ester groups (up to ca. 1.5-fold) were found to be much smaller compared to those observed for heterocyclic directing groups (ca. 5-fold).31 Pyridine is at least 15-fold more strongly directing than acetophenone according to competition experiments, and yet there is an apparent inverted difference in reactivity (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.6) when kobs are compared.
:2.6) when kobs are compared.
 | ||
| Fig. 2 (a) Representative kinetic profiles for disappearance of Ph-DG (filled symbols ●■▲; [Ph-DG] versus time; primary y-axis), and formation of labelled product (empty symbols ○□△; % D versus time; secondary y-axis) of the HIE reaction in CDCl3 at 50 °C. (b) Fitting a first-order kinetic model to the data. Product concentrations were determined by 1H NMR. | ||
| Directing group | Kinetic study | Competition study | |
|---|---|---|---|
| k obs (s−1)a | k rel | k rel | |
| a The indicated uncertainties are the observed standard deviations for replicates of the experiment performed over time, using fresh materials. b From reference.24 | |||
| N-Methylimidazole | (4.90 ± 0.21) × 10−4 | 4.2 | 10.1 |
| Oxazoline | (5.87 ± 0.71) × 10−4 | 5.1 | 2.80 |
| Thiazoline | (2.22 ± 0.26) × 10−4 | 1.9 | 2.26 |
| Pyrazole | (4.41 ± 0.44) × 10−4 | 3.8 | 1.77 |
| Pyridine | (1.16 ± 0.17) × 10−4 | 1.0 | 1.00 |
| Benzothiazole | (2.22 ± 0.02) × 10−4 | 1.9 | 0.27 |
| Acyl (C(O)Me) | (3.93 ± 0.05) × 10−4 | 3.4 | 0.06 |
| Nitro (NO2) | (2.53 ± 0.17) × 10−4 | 2.2 | 0.03 |
| Ethyl ester (CO2Et) | (2.63 ± 0.02) × 10−4 | 2.3 | 0.01 |
These data were gathered by considering the total degree of deuteration from the integration of the 1H NMR spectrum, but this analytical technique cannot distinguish between the d0, d1, and d2 isotopomers of the symmetrical substrates used here. To overcome this limitation, and to provide additional insight into the progress of these reactions, the HIE reactions of acetophenone and 2-phenylpyridine were monitored by mass spectrometry. This allows the distribution of the d0, d1 and d2 isotopomers to be determined, and how these vary over time (Fig. 3).
 | ||
| Fig. 3 The distribution of non-deuterated (d0), mono-deuterated (d1), and di-deuterated (d2) Ph-DG substrate over time for the HIE reaction in CDCl3 at 50 °C. | ||
These data show that the labelling of acetophenone proceeds via significant amounts of the d1-isotopomer, which peak at ca. 50% of the total acetophenone charge; similar observations were made by Heys et al. using Raman monitoring of the reaction of 2-phenylimidazole using a similar catalyst system.32 However, in stark contrast to this, 2-phenylpyridine is essentially converted directly from 2-phenylpyridine-d0 to 2-phenylpyridine-d2, with less than 20% of the initial 2-phenylpyridine charge being present as 2-phenylpyridine-d1 at any given time point. Further investigations are underway in our laboratories at present, but these initial results are consistent with a more strongly binding pyridine directing group remaining bound to the iridium centre for more than one cycle of C–H activation/H–D exchange/C–D reductive elimination.
We then studied selected two-substrate kinetic experiments (Scheme 3(top)). The reactions were conducted under the same conditions to those for single substrate kinetics, but using 1 mol% of the catalyst in total to allow reaction rates and selectivity to be monitored for both substrates in these intermolecular competition systems. In each case, the rate of HIE for the more reactive directing group in each pair (Ph-DG1) was similar to that from the corresponding single substrate reaction. The rate of the reaction of the second (less strongly directing) substrate (Ph-DG2) decreased.
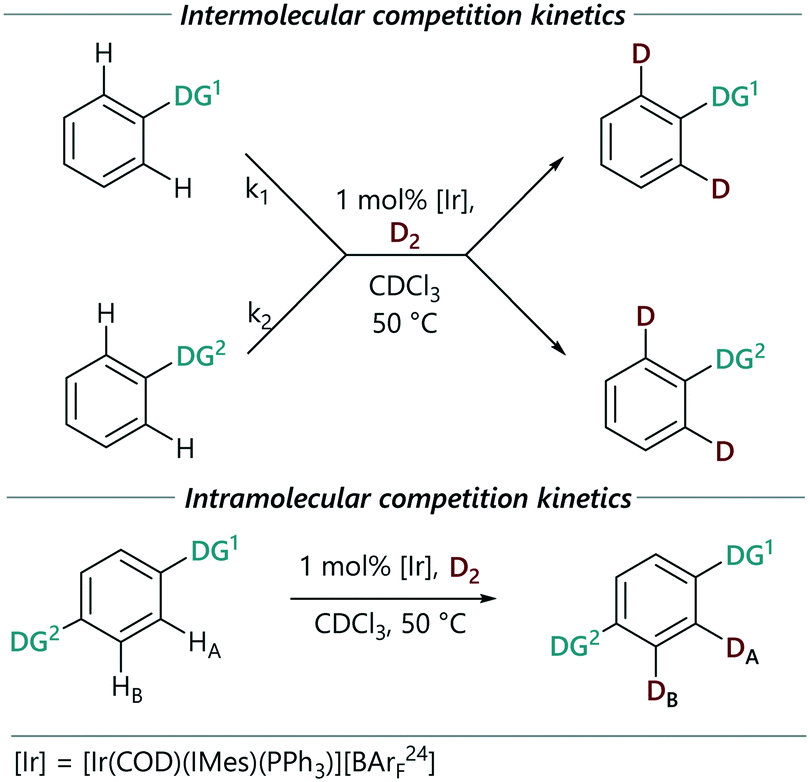 | ||
| Scheme 3 Competition reaction kinetic monitoring. | ||
Additionally, we monitored the reactions of a selection of substrates with multiple directing groups (Scheme 3(bottom)) to understand whether the same behaviour is observed in intramolecular competition experiments. Three general scenarios were observed: i) the two sites react in parallel, but at different rates; ii) the two sites react in series, with the second site undergoing labelling once a significant proportion of the first site has labelled; iii) one site reacts and the other does not. Fig. 4 shows the representative examples for each scenario, including kinetic profiles for intra- and intermolecular competition kinetics and single substrate kinetics.
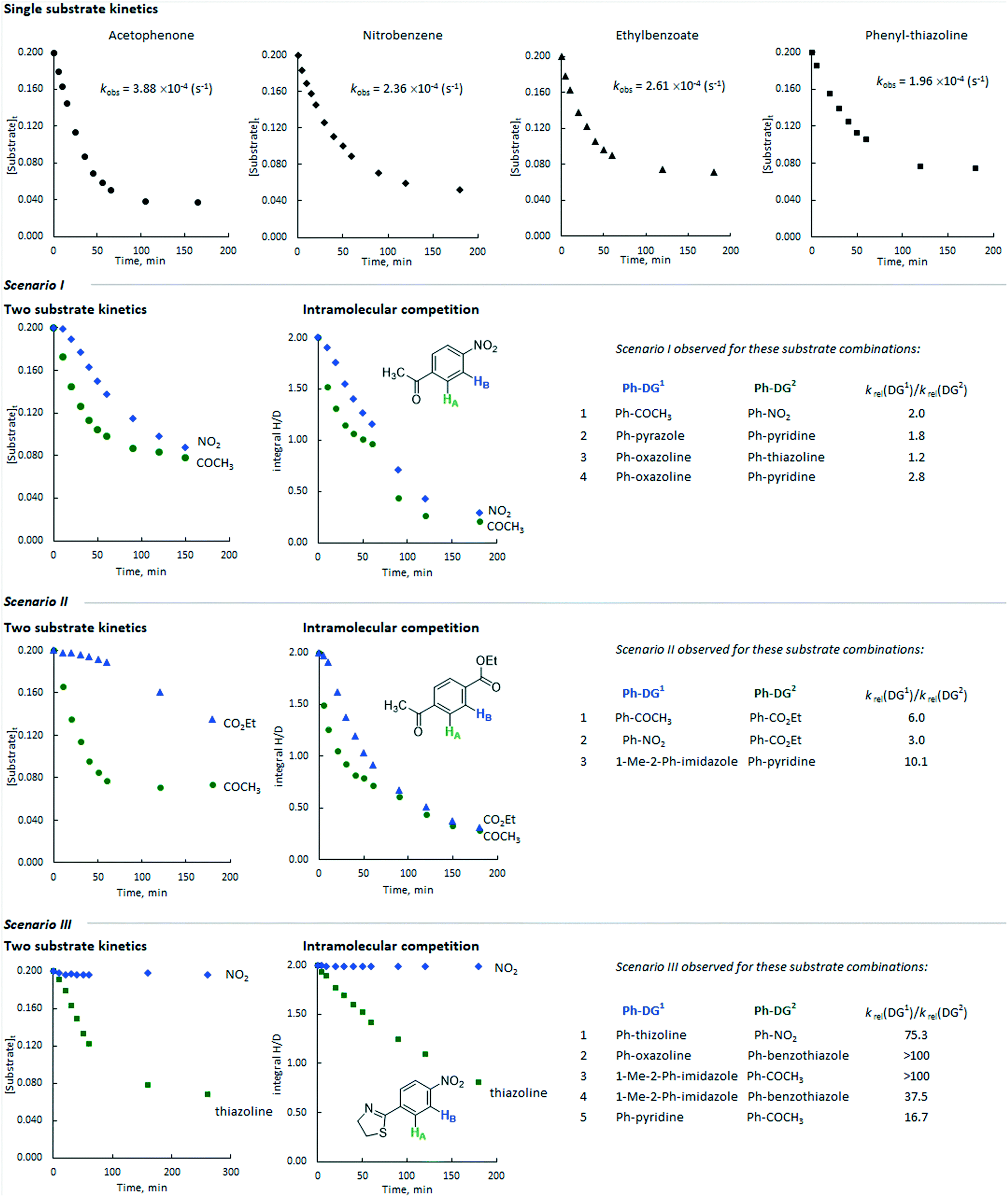 | ||
| Fig. 4 Representative profiles for inter- and intramolecular competition kinetics compared to single substrate kinetics. The values of krel taken from reference.24 | ||
Scenario 1
This is represented by the HIE reaction with competing ketone and nitro groups. As discussed above, the single substrate kinetic experiments gave comparable rates from these substrates. When the labelling reaction of acetophenone and nitrobenzene was carried out in the same flask, the rate of deuterium incorporation for both substrates decreased by ca. 2 and 3 times, respectively, with their reactions proceeding in parallel with approximately constant selectivity at each time point. The same scenario was observed for the following directing group combinations: pyrazole versus pyridine, oxazoline versus thiazoline, and oxazoline versus pyridine.Scenario 2
This is represented by the competition between ketone and ester directing groups. As can be deduced from the intramolecular competition kinetics, the ketone group inhibits the isotopic labelling ortho to the ester group in the initial phases of the reaction. When the reaction reaches relatively high conversion (labelling) ortho to the ketone group, ester directed labelling begins to occur, albeit with a significantly lower rate (kobs ∼10−5 s−1) compared to the corresponding single substrate experiment (kobs ∼10−4 s−1). The combinations of nitro versus ester and N-methyl-imidazole versus pyridine demonstrated similar behaviour.Scenario 3
This is obtained for pairs of directing groups with large differences in reactivity. The representative example of the competition between thiazoline and nitro showed that the latter is fully inhibited by the more reactive thiazoline group and almost no deuterium labelling was observed ortho to the nitro group. Similar behaviour was obtained for competition reactions between oxazoline and ketone, pyridine and ketone, N-methylimidazole and ketone, N-methylimidazole and benzothiazole, and oxazoline and benzothiazole.For a given substrate pair, the same qualitative behaviour is obtained in both intermolecular competition experiments with two different substrates and intramolecular competition experiments with a single bifunctional substrate. However, in some cases the profiles from these experiments are somewhat different in quantitative terms. For example, in the competition between the ethyl ester and the methyl ketone (Fig. 4, scenario II), the sequential nature of labelling is far more pronounced for the intermolecular competition experiment than it is for the labelling of ethyl 4-acetylbenzoate. There may be a pathway for the exchange of binding site without substrate dissociation, and computational studies to explore this are currently underway.
As shown for various directing group combinations (Fig. 4, right), the ratios of krel for the corresponding directing groups from our directing group power scale can predict which coordinating groups can inhibit otherwise productive catalytic reactions:
Scenario 1
This is observed when the ratio of krel from our directing group scale is less than ca. 3. The directing groups are of broadly similar strength and the reactions occur in parallel at similar rates.Scenario 2
This is observed when the ratio of krel of the two groups lies between ca. 3 and 10. In these situations, one of the directing groups is clearly stronger than the other. The substrate bearing the more strongly directing Lewis basic group inhibits the reaction of that with the less strongly directing group in the initial phases of the reaction. The second substrate undergoes labelling subsequently.Scenario 3
This is observed when the ratio of krel for the two directing groups is greater than ca. 10, and so one directing group is overwhelmingly more strongly directing than the other. The substrate bearing more strongly directing Lewis basic group fully inhibits the reaction of the other substrate.To further probe whether the reactions are guided by the binding abilities of the directing group, we conducted the HIE reaction of acetophenone in the presence of pyridine (0.50 mmol). No isotope labelling ortho to the ketone was observed, because the binding of the more Lewis basic pyridine to the iridium centre completely inhibits acetophenone binding. This is consistent with our observations in systems where acetophenone and 2-phenylpyridine compete (ratio of krel = 16.7 in favour of pyridine).
Conclusions
In summary, we have systematically studied the kinetic behaviour of iridium-catalysed hydrogen isotope exchange reactions using a sampling method. The reactions fit a mono-exponential treatment quite well, allowing us to obtain the rate constants (kobs) for a range of directing groups that are both heterocyclic and non-heterocyclic. Surprisingly, the obtained rates do not show significant differences and do not correlate with previously determined krel values from competition experiments. Instead, the two-substrate kinetic profiles showed that reactions occur in parallel with different rates for the substrates with very close directing group abilities, and one substrate inhibits the reaction of the other via competitive binding for the directing groups with bigger differences in reactivity. The ratio of the krel of the directing groups can be used to predict the behaviour of reactions in which there are multiple directing groups, whether these be on discrete molecules or on one bifunctional substrate. This allows us to predict scenarios where, despite the presence of two directing groups, labelling will only occur at one site. Further studies are underway within our laboratories to understand this process in more detail.Author contributions
DST: data curation; formal analysis; investigation; methodology; writing - original draft. DML: conceptualisation; funding acquisition; project administration; supervision; writing – review & editing. WJK: conceptualisation; funding acquisition; resources; supervision. DJN: conceptualisation; funding acquisition; methodology; project administration; resources; supervision; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leverhulme Trust for a Research Project Grant (RPG-2018-207). We thank Mr Frank McGeoch, Mr Craig Irving, Ms Patricia Keating, and Dr John Parkinson for assistance with technical and analytical facilities.Notes and references
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS.
- A. Tomberg, M. É. Muratore, M. J. Johansson, I. Terstiege, C. Sköld and P.-O. Norrby, iScience, 2019, 20, 373–391 CrossRef CAS PubMed.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- J. McIntyre, I. Mayoral-Soler, P. Salvador, A. Poater and D. J. Nelson, Catal. Sci. Technol., 2018, 8, 3174–3182 RSC.
- R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, Faraday Discuss., 2019, 220, 386–403 RSC.
- R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, J. Am. Chem. Soc., 2019, 141, 8896–8906 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- W. J. Kerr, G. J. Knox and L. C. Paterson, J. Labelled Compd. Radiopharm., 2020, 63, 281–295 CrossRef CAS PubMed.
- M. Reid, Iridium Catalysts for Hydrogen Isotope Exchange, in Iridium Catalysts for Organic Reactions, Topics in Organometallic Chemistry, ed. L. A. Oro and C. Claver, Springer, Cham, vol. 69, 2020 Search PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- W. J. Kerr, M. Reid and T. Tuttle, ACS Catal., 2015, 5, 402–410 CrossRef CAS.
- M. Parmentier, T. Hartung, A. Pfaltz and D. Muri, Chem. – Eur. J., 2014, 20, 11496–11504 CrossRef CAS PubMed.
- K. Jess, V. Derdau, R. Weck, J. Atzrodt, M. Freytag, P. G. Jones and M. Tamm, Adv. Synth. Catal., 2017, 359, 629–638 CrossRef CAS.
- A. Burhop, R. Prohaska, R. Weck, J. Atzrodt and V. Derdau, J. Labelled Compd. Radiopharm., 2017, 60, 343–348 CrossRef CAS PubMed.
- M. Koneczny, L. Phong Ho, A. Nasr, M. Freytag, P. G. Jones and M. Tamm, Adv. Synth. Catal., 2020, 362, 3857–3863 CrossRef CAS.
- J. A. Brown, A. R. Cochrane, S. Irvine, W. J. Kerr, B. Mondal, J. A. Parkinson, L. C. Paterson, M. Reid, T. Tuttle, S. Andersson and G. N. Nilsson, Adv. Synth. Catal., 2014, 356, 3551–3562 CrossRef CAS.
- J. A. Brown, S. Irvine, A. R. Kennedy, W. J. Kerr, S. Andersson and G. N. Nilsson, Chem. Commun., 2008, 1115–1117 RSC.
- J. Atzrodt, V. Derdau, W. J. Kerr, M. Reid, P. Rojahn and R. Weck, Tetrahedron, 2015, 71, 1924–1929 CrossRef CAS.
- A. R. Kennedy, W. J. Kerr, R. Moir and M. Reid, Org. Biomol. Chem., 2014, 12, 7927–7931 RSC.
- W. J. Kerr, R. J. Mudd, P. K. Owens, M. Reid, J. A. Brown and S. Campos, J. Labelled Compd. Radiopharm., 2016, 59, 601–603 CrossRef CAS PubMed.
- A. R. Cochrane, S. Irvine, W. J. Kerr, M. Reid, S. Andersson and G. N. Nilsson, J. Labelled Compd. Radiopharm., 2013, 56, 451–454 CrossRef CAS PubMed.
- D. S. Timofeeva, D. M. Lindsay, W. J. Kerr and D. J. Nelson, Catal. Sci. Technol., 2020, 10, 7249–7255 RSC.
- M. Valero, T. Kruissink, J. Blass, R. Weck, S. Güssregen, A. T. Plowright and V. Derdau, Angew. Chem., Int. Ed., 2020, 59, 5626–5631 CrossRef CAS PubMed.
- A. Y. L. Shu, W. Chen and J. R. Heys, J. Organomet. Chem., 1996, 524, 87–93 CrossRef CAS.
- M. Valero, D. Becker, K. Jess, R. Weck, J. Atzrodt, T. Bannenberg, V. Derdau and M. Tamm, Chem. – Eur. J., 2019, 25, 6517–6522 CrossRef CAS PubMed.
- J. Devlin, W. Kerr, D. Lindsay, T. McCabe, M. Reid and T. Tuttle, Molecules, 2015, 20, 11676 CrossRef CAS PubMed.
- Initial attempts to monitor reactions in situ using an NMR tube with a J. Young valve were unsuccessful. These reactions are very slow, presumably due to the very small interfacial area between the deuterium gas and the reaction solution. See the supporting information for full details..
- K. Shirono, T. Morimatsu and F. Takemura, J. Chem. Eng. Data, 2008, 53, 1867–1871 CrossRef CAS.
- The reactions of a series of para-substituted acetophenone substrates were monitored with the aim of examining a Hammett correlation. Unfortunately, the very small differences between the observed rates precluded such an analysis (see the ESI‡)..
- J. R. Heys, M. E. Powell and D. E. Pivonka, J. Labelled Compd. Radiopharm., 2004, 47, 983–995 CrossRef CAS.
Footnotes |
| † All raw data underpinning this publication are openly available from the University of Strathclyde Knowledge Base at https://doi.org/10.15129/cc0cb14f-9329-4e42-bb99-36ca1fbbddd9. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy00708d |
| § Representative procedure: reactions were carried out in 30 mL microwave vials with septum-fitted caps. The vessel was kept at a constant temperature (thermostat/Drysyn block), and a balloon containing D2 was connected to the flask to maintain a constant supply (and excess) of D2. To calculate the concentration of the unlabelled substrate, the residual proton signal from the site of incorporation (ortho to the directing group) was compared against that of a site where deuterium incorporation did not occur. |
| This journal is © The Royal Society of Chemistry 2021 |