
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation state variation in bis-calix[4]arene supported decametallic Mn clusters†
Lucinda R. B.
Wilson
 a,
Marco
Coletta
a,
Reshma
Jose
b,
Gopalan
Rajaraman
*b,
Scott J.
Dalgarno
*c and
Euan K.
Brechin
*a
a,
Marco
Coletta
a,
Reshma
Jose
b,
Gopalan
Rajaraman
*b,
Scott J.
Dalgarno
*c and
Euan K.
Brechin
*a
aEastCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, Edinburgh, Scotland EH9 3FJ, UK. E-mail: ebrechin@ed.ac.uk
bDepartment of Chemistry, Indian Institute of Technology Bombay, Mumbai, 400076, India. E-mail: rajaraman@chem.iitb.ac.in
cInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh, Scotland EH14 4AS, UK. E-mail: S.J.Dalgarno@hw.ac.uk
First published on 19th November 2021
Abstract
The reaction of MnCl2·4H2O, H8L (2,2′-bis-p-tBu-calix[4]arene) and NEt3 in a dmf/MeOH solvent mixture results in the formation of a mixed valent decametallic cluster of formula [MnII6MnIII4(L)2(μ3-OH)4(μ-OH)4(MeOH)4(dmf)4(MeCN)2]·MeCN (3). Complex 3 crystallises in the monoclinic space group P21/n with the asymmetric unit comprising half of the compound. Structure solution reveals that the bis-calix[4]arene ligands are arranged such that one TBC[4] moiety in each has undergone inversion in order to accommodate a [MnIII4MnII6] metallic skeleton that describes three vertex-sharing [MnIII2MnII2] butterflies. The structure is closely related to the species [MnIII6MnII4(L)2(μ3-O)2(μ3-OH)2(μ-OMe)4(H2O)4(dmf)8]·4dmf (4), the major difference being the oxidation level of the Mn ions in the core of the compound. DFT calculations on the full structures reveal that replacing the MnIII ions in 4 for MnII ions in 3 results in a significant decrease in the magnitude of some antiferromagnetic exchange contributions, a switch from ferromagnetic to antiferromagnetic in others, and the loss of significant spin frustration.
Introduction
Coordination compounds of Mn maintain long standing interest across a breadth of topics from bioinorganic chemistry1 to molecular magnetism.2 Key to manipulating and enhancing physicochemical behaviour for any application is structural control, and for polymetallic compounds the self-assembly process and identity of the final product is largely dominated by choice of bridging ligand.3We, and others,4–7 have employed p-tBu-calix[4]arene (H4TBC[4], Fig. 1A) for the synthesis of a range of transition metal (TM) and lanthanide metal (LnM) species in which the TM/LnM-TBC[4] moiety acts as a metalloligand that encapsulates an oxo/hydroxo-bridged polymetallic core. Structurally, this means it acts as a capping vertex in the resulting metallic skeleton.8 The calix[4]arene tetraphenolic pocket is particularly well suited to bonding Jahn–Teller (JT) distorted ions such as CuII and MnIII because it will preferentially coordinate metals possessing four short equatorial and two long axial bonds.9,10 Illustrative examples include the complexes [CuII9(OH)3(TBC[4])3Cl2(DMSO)5.5(EtOH)0.5][CuICl2] (1, Fig. 1B) and [MnIII2MnII2(OH)2(TBC[4])2(dmf)6] (2, Fig. 1C). In the former the TM-TBC[4] metalloligand encapsulates a [CuII6(OH)3] trigonal prism, and in the latter a [MnII2(OH)2] dimer. This general bonding motif has been observed in the vast majority of TM/LnM complexes we have isolated under ambient conditions and this has allowed us to develop a specific set of empirical metal ion binding rules for TBC[4].11 (A) TBC[4] preferentially binds TMIII ions; (B) TBC[4] will bind TMII ions in the absence of TMIII ions; (C) TBC[4] will bind LnMIII ions in the absence of TMII or TMIII ions.12
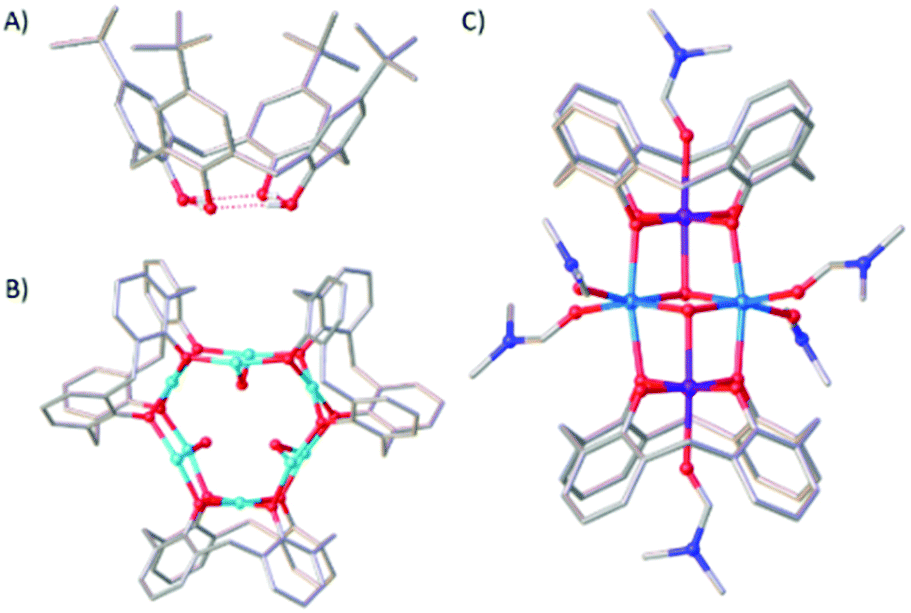 | ||
| Fig. 1 Single crystal X-ray structures of H4TBC[4] (A), 1 (B) and 2 (C). Colour code C – grey, O – red, N – dark blue, H – white, CuII – turquoise, MnII – pale blue, MnIII – purple. H atoms, tBu groups of TBC[4], ligated solvent molecules and co-crystallised solvent/anions omitted for clarity. | ||
As an extension to this science we have recently begun investigating the coordination chemistry of 2,2′-bis-p-tBu-calix[4]arene (H8L), which contains two TBC[4] units linked at the methylene bridge (Fig. 2). The conformational flexibility (ring inversion) of TBC[4] in solution is well understood13,14 and when applied to H8L upon coordination-driven self-assembly it proffers a ligand with eight phenolic O-atoms in close proximity and oriented in the same direction.15 Initial studies16 have shown that H8L displays a systematic extension of the empirical metal ion binding rules established for TBC[4], rather than, for example, mimicking the behaviour of p-tBu-calix[8]arene.17 Herein, we outline the synthesis, structure and magnetic behaviour of a new bis-TBC[4]-supported complex, [MnIII4MnII6(L)2(μ3-OH)4(μ-OH)4(MeOH)4(dmf)4(MeCN)2]·MeCN (3), together with an experimental and theoretical magneto-structural comparison to the structurally related (and previously published) species [MnIII6MnII4(L)2(μ3-O)2(μ3-OH)2(μ-OMe)4(H2O)4(dmf)8]·4dmf (4).18
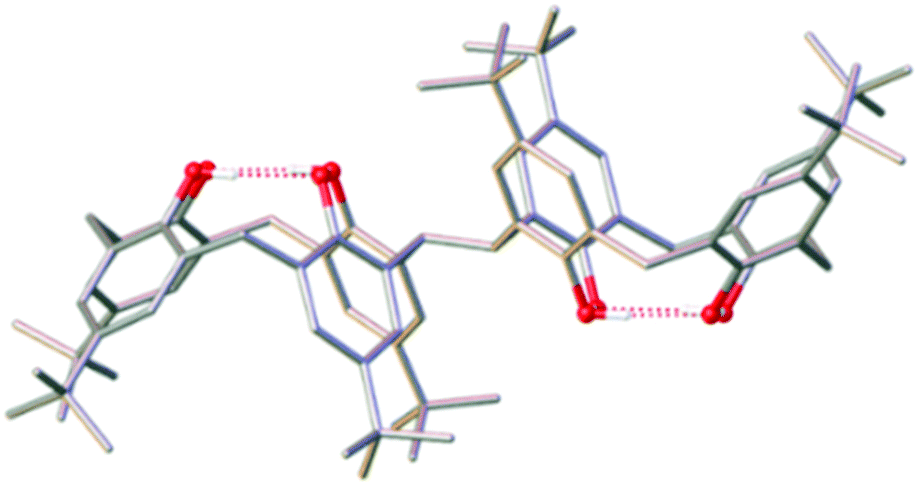 | ||
| Fig. 2 Single crystal X-ray structure of H8L. Colour code C – grey, O – red, H – white. | ||
Experimental
MnCl2·4H2O was purchased from Fluorochem and was used without further purification. 2,2′-Bis-p-tBu-calix[4]arene (H8L) was prepared as previously described.19Synthesis of [MnII6MnIII4(L)2(μ3-OH)4(μ-OH)4(MeOH)4(dmf)4(MeCN)2]·MeCN (3): MnCl2·4H2O (0.86 mmol, 170 mg), H8L (0.154 mmol, 200 mg) and NEt3 (2.87 mmol, 0.4 mL) were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (dmf/MeOH) mixture (24 ml) and stirred for 2 hours. After filtration, MeCN was diffused into the mother liquor, affording crystals of 3 in 31% yield after 7 days. Elemental analysis (%) calculated for 3: C, 62.64%; H, 6.82%; N, 2.58%. Found: C, 62.18%; H, 6.30%; N, 2.05%. Yield 180 mg (30.7%).
:1 (dmf/MeOH) mixture (24 ml) and stirred for 2 hours. After filtration, MeCN was diffused into the mother liquor, affording crystals of 3 in 31% yield after 7 days. Elemental analysis (%) calculated for 3: C, 62.64%; H, 6.82%; N, 2.58%. Found: C, 62.18%; H, 6.30%; N, 2.05%. Yield 180 mg (30.7%).
Single crystal X-ray data for 3 (CCDC 2099440†): C200H270Mn10N8O32 (M = 3847.62 g mol−1): monoclinic, space group P21/n (no. 14), a = 22.322(6) Å, b = 18.903(5) Å, c = 27.501(7) Å, β = 106.949(16)°, V = 11101(5) Å3, Z = 2, T = 100.0 K, Bruker D8 Venture Diffractometer operating with a Photon III detector, μ(CuKα) = 4.960 mm−1, Dcalc = 1.151 g cm−3, 157579 reflections measured (4.506° ≤ 2θ ≤ 146.326°), 21932 unique (Rint = 0.1384, Rsigma = 0.0760) which were used in all calculations. The final R1 was 0.1271 (I > 2σ(I)) and wR2 was 0.3950 (all data).
Magnetic data were collected on a Quantum Design MPMS magnetometer operating in the temperature/field ranges 1.8–300 K/0–7 T. Diamagnetic corrections were applied using Pascal's constants. Elemental analyses were performed on a Thermo Fisher Scientific Flash SMART instrument. See the ESI† for full computational details.
Results and discussion
Reaction of H8L with MnCl2·4H2O in a basic dmf/MeOH mixture affords single crystals of [MnIII4MnII6(L)2(μ3-OH)4(μ-OH)4(MeOH)4(dmf)4(MeCN)2]·MeCN (3, Fig. 3), following vapour diffusion of MeCN into the mother liquor. The crystals were found to be in a monoclinic cell and structure solution was carried out in the space group P21/n. The asymmetric unit (ASU) comprises half of the cluster, with an inversion centre located in the middle of the Mn5–O14–Mn5′–O14′ rhomb. Pertinent bond lengths and angles (Table S1†) and BVS calculations (Table S2) are given in the ESI.†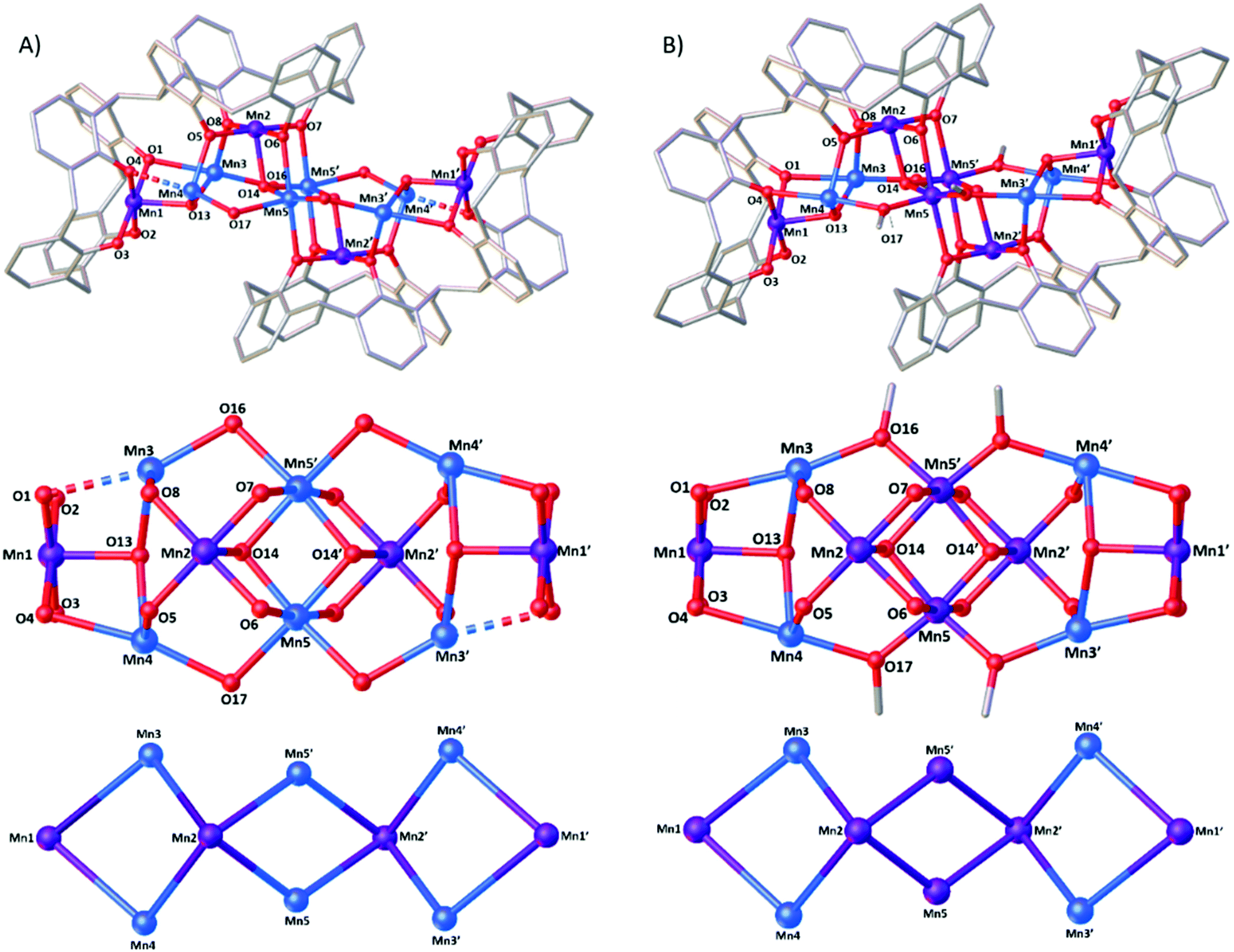 | ||
| Fig. 3 Single crystal X-ray structures (top), cluster cores (middle) and metallic skeletons (bottom) of 3 (A) and 4 (B). Colour code C – grey, O – red, MnII – pale blue, MnIII – purple. H atoms, tBu groups of TBC[4], ligated solvent molecules and co-crystallised solvent/anions omitted for clarity. Dashed lines represent longer bond lengths (as discussed in text). | ||
Both L ligands are arranged such that one TBC[4] moiety in each has undergone inversion in order to accommodate a [MnIII4MnII6] metallic skeleton that describes three vertex-sharing [MnIII2MnII2] butterflies (Fig. 3). These are of two types. The central butterfly (MnIII2, MnII5, O6 (ORL), O7 (ORL), O14 (OH) and symmetry equivalent) is of the form [Mn4(μ3-OH)2(μ-ORL)4] and is reminiscent of the structure of 2 in that the MnIII ions occupy the wings and the MnII ions occupy the body positions of the butterfly (Fig. 1). The peripheral butterflies (MnIII1, MnIII2, MnII3, MnII4, O1 (ORL), O4 (ORL), O5 (ORL), O8 (ORL), O13 (OH)) appear rather asymmetric and are of the form [Mn4(μ3-OH)(μ-ORL)4] with just one μ3-bridging OH anion (O13) linking Mn1, Mn3 and Mn4. O13 H-bonds to the neighbouring μ3-OH ion in the central butterfly (O13⋯O14, 2.831 Å). The metal–oxygen core is completed by the presence of four “external” μ-OH ions that link the MnII ions in the body positions of the central butterfly to the MnII ions in the body positions of peripheral butterflies (Mn5′–O16–Mn3; Mn5–O17–Mn4). Thus, as expected, the L ligands bind the MnIII ions within their polyphenolic pockets with the MnII ions encapsulated between them, linked through multiple OH anions.
The MnIII ions (Mn1, Mn2 and symmetry equivalent) are six coordinate and in JT distorted octahedral geometries, with the JT axes defined by the HO–Mn–NCMe vector in each case. The acetonitrile molecules are disordered over two positions within the calix[4]arene cavities, with Mn–N distances of ∼2.34 and ∼2.58 Å and N–Mn–O angles of ∼169 and ∼172°. The MnII ions are also six coordinate, but with rather different geometries. The two central Mn ions (Mn5) have [Mn(OH)4(ORL)2] coordination spheres with Mn–O bond lengths in the range ∼1.99–2.29 Å, and cis/trans angles of ∼78–98°/163–172°. Mn3 and Mn4 have their coordination spheres completed by one MeOH and one dmf, affording [Mn(OH)2(ORL)2(Osolvent)2] with (highly) distorted octahedral geometries. For both, five of the six Mn–O bond lengths fall in the range ∼2.1–2.2 Å (Mn3) and ∼2.1–2.3 Å (Mn4). The sixth bond, to the L O-atom, is much longer (Mn3–O1 = 2.577(7) Å and Mn4–O4 = 2.424(7) Å). A search of the Cambridge Structural Database reveals that of over 93000 Mn–O bond distance entries, only 325 are found to be between 2.5–2.6 Å, with 98.8% of the entries lying within the 1.8–2.5 Å range (Fig. S1†). cis/trans angles are ∼70–122°/154–176° (Mn3), and ∼79–116°/154–175° (Mn4). The O-atoms of the terminally bonded MeOH molecules on Mn3 and Mn4 are H-bonded to the two terminally bonded ORL atoms at distances of O15⋯O2, ∼2.56 Å, and O18⋯O3, ∼2.54 Å.
Examination of the extended structure (Fig. S2†) reveals that the closest intermolecular interactions are between coordinated dmf molecules and the tBu C-atoms at C⋯C distances ≥ 3.2 Å, and between neighbouring tBu groups, C⋯C ≥ 3.8 Å. The closest M⋯M distance is ∼12.3 Å between the Mn1 ions of distinct molecules, meaning they are structurally isolated thanks to the framework of the L ligands and overall shape of the assembly.
A comparison of the structures of 3 and 4 (Fig. 3) shows them to be very closely related. Inspection reveals three significant differences: (a) the oxidation states of the central Mn5/Mn5′ ions are different – MnII in 3 and MnIII in 4. (b) The bridging groups connecting Mn3/Mn4 to Mn5 are μ-OH in 3 but μ-OCH3 in 4. (c) The O1⋯Mn3 distance in 3 is 2.577(7) Å compared to 2.436(4) Å in 4. The former would be expected to have significant impact upon magnetic behaviour (vide infra). Note that 3 and 4 result from the methodical screening of reaction conditions, where changes in reagent stoichiometry and crystallisation conditions leads to the selective formation of either 3 or 4. Indeed the only major differences in the preparation of 3 and 4 comes in the ligand:metal ratio (1:6 (3), 1:8 (4)) and the method of crystallisation – MeCN diffusion into the DMF/MeOH mother liquor for 3 and slow evaporation of the DMF/MeOH solution for 4.
Fig. 4 shows that the structure of 3 is also related to the compounds [MnIII6MnII2GdIII2(L)2(μ4-O)2(μ3-OH)2(μ-OMe)2(μ-OH)2(MeOH)4(dmf)8(NO3)2(H2O)2] (5)18 and [MnIII4MnII4(L)2(μ3-OH)2(μ-OH)(μ-Cl)(H2O)(MeOH)(dmf)4] (6),16 which was the very first compound isolated with H8L. The metallic skeleton of 5 describes three vertex-sharing [MnIII2MnIILnIII] butterflies, and that of 6 two vertex-sharing [MnIII2MnII2] butterflies. Indeed, from an inspection of the metallic skeletons of 3–6 (Fig. 3 and 4) it is clear to see that it is the MnIIIL metalloligand that directs structure formation, with the additional MnII/LnIII ions encapsulated within this framework, connected via bridging hydroxides/alkoxides. What is also clear is that these encapsulated metal ions can be replaced, whilst maintaining the general structure. This is illustrated by complex 6 undergoing selective MnII/LnIII substitution to form [MnIII4MnII2GdIII2(L)2(Cl)2(μ3-OH)4(MeOH)2(dmf)8], 7 (Fig. 4).16 This mirrors the behaviour observed for TBC[4]. For example, the MnII ions in 2 can be replaced in a stepwise fashion with LnIII ions, from [MnIII2MnII2] to [MnIII2MnIILnIII] and [MnIII2LnIII2].20 The isolation of multiple, structurally-related compounds is reflective of the versatility of the bis-calix[4]arene ligand for the construction of polymetallic clusters where subtle changes in reactants/conditions can be exploited to direct the nature of the metallic core and associated physical properties.
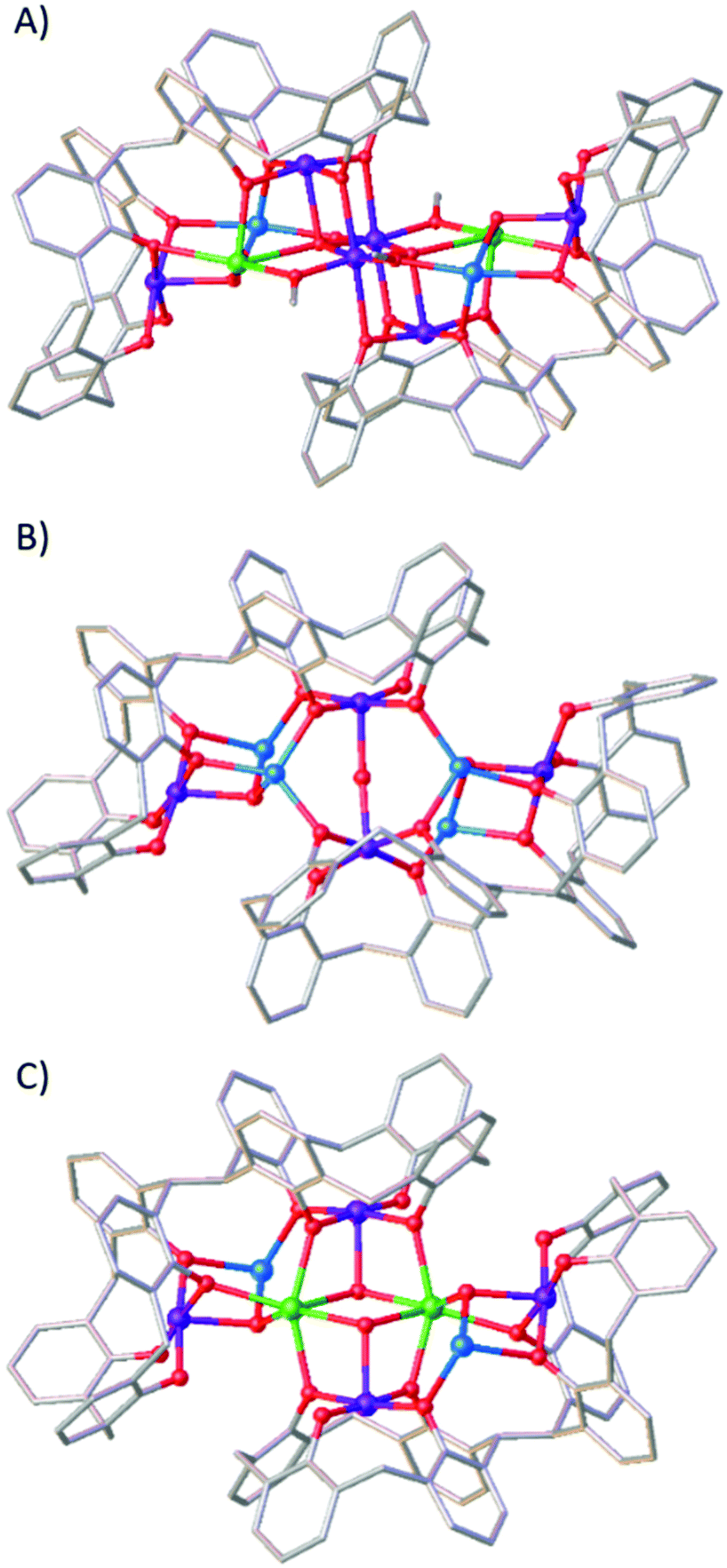 | ||
| Fig. 4 Single crystal X-ray structures of 5 (A), 6 (B) and 7 (C). Colour code C – grey, O – red, MnII – pale blue, MnIII – purple, GdIII – green. H atoms, tBu groups of TBC[4], ligated solvent molecules and co-crystallised solvent/anions omitted for clarity. | ||
Magnetic measurements
Direct current (dc) magnetic susceptibility studies were performed on a polycrystalline sample of 3 over the temperature range T = 2–298 K, in an applied magnetic field B = 0.1 T (Fig. 5), where χ = M/B and M is the magnetisation. At 298 K, the χMT value of 37.4 cm3 mol−1 K is in agreement with the expected value for spin-only contributions to the susceptibility for a [MnIII4MnII6] unit (38.25 cm3 mol−1 K, g = 2.0). Upon cooling, the χMT product decreases slowly until approximately T = 100 K, wherefrom it decreases more rapidly, reaching a value of 8.6 cm3 mol−1 K at 2 K. Variable-temperature-variable-field (VTVB) magnetisation data (inset of Fig. 5, Fig. S3†) shows M rising slowly with increasing B, reaching a maximum value of M = 17.6 μB at 7 T but without saturating. Both are suggestive of the presence of weak, competing exchange interactions dominated by the antiferromagnetic contributions. The large nuclearity of 3 and the presence of six different exchange interactions prevents a quantitative analysis of the susceptibility and magnetisation data. Previously published Mn complexes of TBC[4] and H8L show that exchange interactions tend to be relatively weak, with JMn(III)–Mn(II) being weakly ferromagnetic, JMn(II)–Mn(II) being weakly antiferromagnetic and JMn(III)–Mn(III) being borderline anti/ferromagnetic.16,20 In order to investigate this in more detail, we now turn to theory.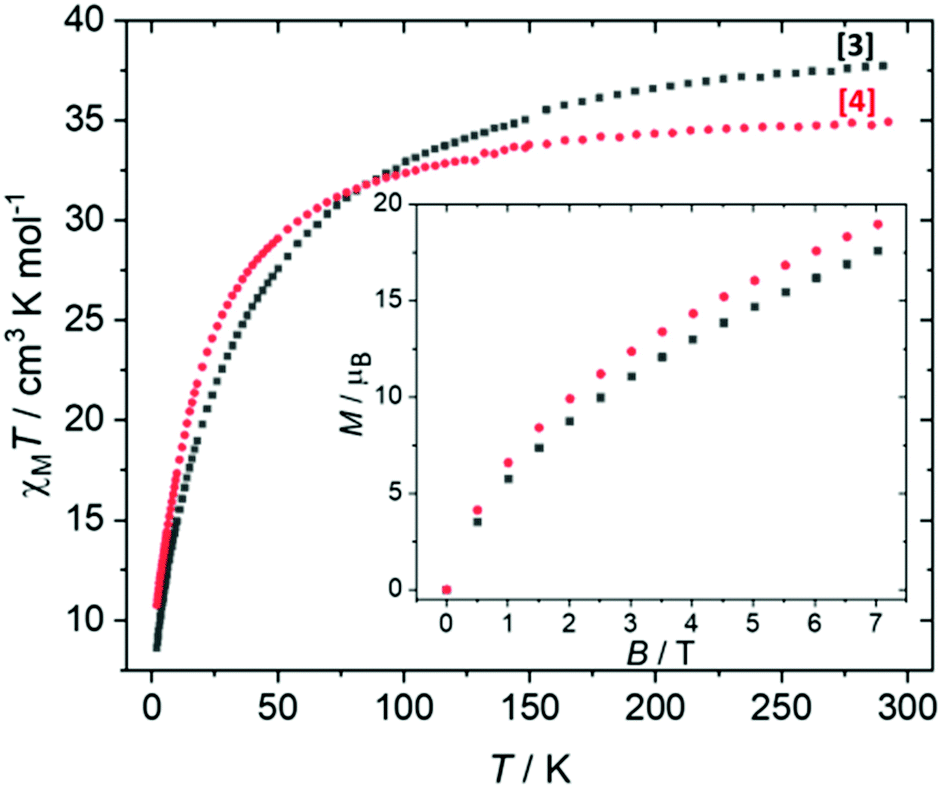 | ||
| Fig. 5 Experimental χMT versus T data for 3 measured in the T = 2–298 K temperature range in an applied field of 0.1 T. Inset: Variable-temperature-variable-field (VTVB) magnetisation data of 3 at T = 2 K and B = 0–7 T. The equivalent data for complex 4 is added for comparison. | ||
Estimation of magnetic exchange interactions using DFT
For full details of the computational methodology, see the ESI.† Analysis of the structures of 3 and 4 reveals a total of six different exchange interactions (J1–6), as shown in Fig. 6. The magnitude and sign of the DFT calculated exchange constants is provided in Table 1, alongside pertinent structural information describing the bond distances and angles in each pairwise interaction. In each case the sign and magnitude of the J values obtained can be easily explained via magneto-structural correlations previously published for O-bridged MnII/III complexes,21 and are in close agreement with those calculated for a mixed-valent [MnIII2MnII4] cage published by Milios and co-workers.22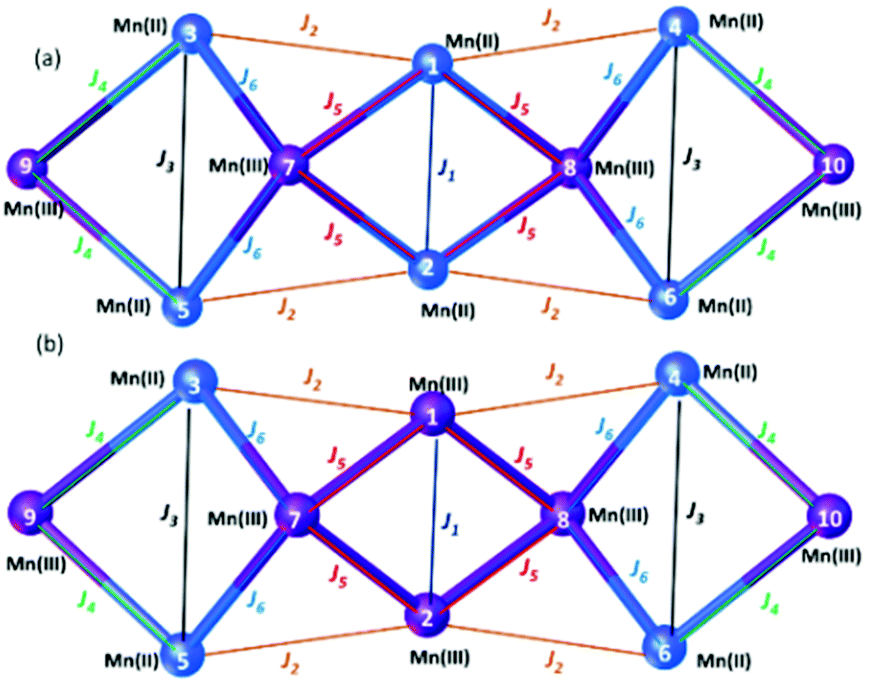 | ||
| Fig. 6 Schematic representation of the six different exchange interactions present in 3 (a) and 4 (b). The corresponding spin-Hamiltonian is given in the ESI.† | ||

|
For complex 3, the J values are all found to be weakly antiferromagnetic (−0.2 ≤ J ≤ −4.8 cm−1), with J1 the strongest and J5 the weakest. J1 is mediated between the two central MnII ions via two μ3-OH bridges, with (short) Mn–O/Mn–Mn distances of 1.99/3.00 Å and (relatively acute) Mn–O–Mn of angles of 97.7°. The J2–J5 interactions are more weakly antiferromagnetic than J1 due to the larger Mn–O–Mn angles and larger Mn–O/Mn–Mn distances present. The value of J6, bridged by a single MnIII–μOR–MnII unit is comparable to that of J1 due to the large Mn–O–Mn angle (116.0°). Magneto-structural correlations of such units predict antiferromagnetic exchange in the range of ∼5 cm−1.23 Based on these J values complex 3 has an S = 2 ground state (Fig. S4†).
For complex 4, the J values are found to be in a much larger range, with both ferro- and antiferromagnetic exchange being observed (+4.1 ≤ J ≤ −40.4 cm−1). J1, between two central MnIII ions connected by two μ3-OH bridges, is the largest antiferromagnetic exchange present due to the small Mn–O–Mn angles (97.6°) and short Mn–O (1.92 Å) and Mn–Mn (2.88 Å) distances. The large J value has been confirmed through calculations on a dinuclear model complex adapted from the X-ray structure (Fig. S5†). We note that the JT axes of the two MnIII ions are oriented parallel to the bridging ligands, which can be classified as a type I structure based on detailed studies of the dinuclear {MnIII2(OR)2} motif. In this class of interactions, a large JAF contribution and a negligible JF contribution are expected, leading to strong antiferromagnetic coupling, as observed here.21,24
J 2 is ferromagnetic in nature, mediated between MnII and MnIII ions via a single μ-OR bridge with a short Mn–O distance (2.03 Å) and a large Mn–O–Mn angle (122.8°). Interestingly, although J3 (MnII–OH–MnII) is mediated via similar bridging angles and distances as that seen for J2, it is weakly antiferromagnetic in nature, highlighting the important role played by the oxidation state of the Mn ion in controlling the sign and magnitude of the exchange.
J 3, J4 and J6 are much more weakly antiferromagnetic in comparison to J1 due to the larger Mn–O/Mn distances and larger Mn–O–Mn angles present, just as they are in 3. J5, between two MnIII ions bridged by a μ3-O ion and a μ-OR group, is weakly ferromagnetic in nature. J5 can be classified as a type II {MnIII2(OR)2} unit in which the JT axes of the MnIII ions are oriented parallel to each other and lying along μ-oxo bridge. This leads to borderline ferro/antiferromagnetic coupling dictated by Mn–O–Mn angle and Mn–O distances. The highly frustrated nature of the interactions in complex 4 precludes the identification of an isolated spin ground state.
From these calculations we can conclude that a change in oxidation state levels from [MnIII6MnII4] in compound 4 to [MnIII4MnII6] in compound 3 results in a significant decrease in the magnitude of some antiferromagnetic exchange contributions (J1 = −4.8 cm−1 (3), −40.4 cm−1 (4)), a switch from ferromagnetic to antiferromagnetic in others (J2 = −1.1 cm−1 (3), +3.1 cm−1 (4); J5 = −0.2 cm−1 (3), +4.1 cm−1 (4)), and the loss of significant spin frustration. The computed spin density plots for the high spin state of complexes 3 and 4 are given in Fig. S6,† with the spin density plots for all the broken symmetries computed given in Fig. S7 and 8.†
Conclusions
Altering reaction stoichiometry results in a change in oxidation state distribution in bis-calix[4]arene supported [Mn10] cages, without significantly altering structural topology. DFT calculations reveal that the shift in oxidation state levels from [MnIII6MnII4] in compound 4 to [MnIII4MnII6] in compound 3 results in a significant decrease in the magnitude of some antiferromagnetic exchange contributions, a switch from ferromagnetic to antiferromagnetic in others, and the loss of significant spin frustration. Indeed, the change in oxidation state from MnIII–MnIII to MnII–MnII in the central J1 interaction causes a 10-fold change in the magnitude of the exchange interaction.These results, alongside those we and others have already reported in the chemistry of p-tBu-calix[4]arene and 2,2′-bis-p-tBu-calix[4]arene, suggest that the MnIIIL metalloligand can be employed as a structure-directing unit capable of encapsulating additional TM (and LnM) moieties in its core. The ability to construct families of structurally analogous species, but whose oxidation state distribution, or metal identity, differs allows for more detailed investigation of the magneto-structural relationship. This ability underpins the design of magnetic molecules whose properties can be tuned toward a specific application. It also has wider design implications for scientists interested in molecules that can robustly cycle through multiple oxidation levels in, for example, bioinorganic chemistry and catalysis.
Author contributions
LRBW and MC performed the synthetic chemistry and collected the magnetic data. SJD collected the single crystal X-ray data and performed structure solution. RJ and GR performed the theoretical calculations. EKB and SJD conceived the concept. All authors contributed to the writing/editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EKB thanks the EPSRC for funding a studentship (LRWB) and grant EP/N01331X/1 (MC). GR would like to thank DST/SERB (CRG/2018/000430, DST/SJF/CSA03/2018-10; SB/SJF/2019-20/12) for funding.Notes and references
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- S. Hill, R. S. Edwards, N. Aliaga-Alcade and G. Christou, Science, 2003, 302, 1015–1018 CrossRef CAS PubMed.
- A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC.
- M. M. Olmstead, G. Sigel, H. Hope, X. Xu and P. P. Power, J. Am. Chem. Soc., 1985, 107, 8087–8091 CrossRef CAS.
- C. Aronica, G. Chastanet, E. Zueva, S. A. Borshch, J. M. Clemente-Juan and D. Luneau, J. Am. Chem. Soc., 2008, 130, 2365–2371 CrossRef CAS PubMed.
- F. Corazza, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Chem. Commun., 1990, 1083–1084 RSC.
- C. Redshaw, D. Homden, D. L. Huges, J. A. Wright and M. R. J. Elsegood, Dalton Trans., 2008, 1231–1242 Search PubMed.
- S. M. Taylor, R. D. McIntosh, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2012, 48, 11190–11192 RSC; S. M. Taylor, R. D. McIntosh, C. M. Beavers, S. J. Teat, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 1440–1442 RSC; S. Sanz, K. Fereira, R. D. McIntosh, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 9042–9044 RSC.
- G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285–8288 CrossRef CAS PubMed.
- G. Karotsis, S. Kennedy and S. J. Dalgarno, Chem. Commun., 2010, 46, 3884–3886 RSC.
- M. Coletta, E. K. Brechin and S. J. Dalgarno, Calixarenes and Beyond, Springer International Publishing, Switzerland, 2016 Search PubMed.
- S. Sanz, R. D. McIntosh, C. M. Beavers, S. J. Teat, M. Evangelisti, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2012, 48, 1449–1451 RSC.
- C. D. Gutsche, B. Dhawan, J. A. Levine, K. H. No and L. Bauer, Tetrahedron, 1983, 39, 409–426 CrossRef CAS.
- S. Fischer, P. D. J. Grootenhuis, L. C. Groenen, W. P. van Hoorn, F. C. J. M. van Veggel, D. N. Reinhoudt and M. Karplus, J. Am. Chem. Soc., 1995, 117, 1611–1620 CrossRef CAS.
- P. Murphy, S. J. Dalgarno and M. J. Paterson, J. Phys. Chem. A, 2014, 118, 7986–8001 CrossRef CAS PubMed.
- R. McLellan, M. A. Palacios, C. M. Beavers, S. J. Teat, S. Piligkos, E. K. Brechin and S. J. Dalgarno, Chem. – Eur. J., 2015, 21, 2804–2812 CrossRef CAS PubMed.
- S. M. Taylor, S. Sanz, R. D. McIntosh, C. M. Beavers, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Chem. – Eur. J., 2012, 18, 16014–16022 CrossRef CAS PubMed.
- M. Coletta, R. McLellan, A. Waddington, S. Sanz, K. J. Gagnon, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2016, 52, 14246–14249 RSC.
- L. T. Carroll, P. A. Hill, C. Q. Ngo, K. P. Klatt and J. L. Fantini, Tetrahedron, 2013, 69, 5002–5007 CrossRef CAS.
- M. A. Palacios, R. McLellan, C. M. Beavers, S. J. Teat, H. Weihe, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. – Eur. J., 2015, 21, 11212–11218 CrossRef CAS PubMed.
- N. Berg, T. Rajeshkumar, S. M. Taylor, E. K. Brechin, G. Rajaraman and L. F. Jones, Chem. – Eur. J., 2012, 18, 5906–5918 CrossRef CAS PubMed; K. R. Vignesh, S. K. Langley, C. J. Gartshore, B. Moubaraki, K. S. Murray and G. Rajaraman, Inorg. Chem., 2017, 56, 1932–1949 CrossRef PubMed; C. J. Milios, R. Inglis, A. Vinslava, R. Bagai, W. Wernsdorfer, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 12505–12511 CrossRef PubMed.
- C. J. Milios, M. Manoli, G. Rajaraman, A. Mishra, L. E. Budd, F. White, S. Parsons, W. Wernsdorfer, G. Christou and E. K. Brechin, Inorg. Chem., 2006, 45, 6782–6793 CrossRef CAS PubMed.
- K. R. Vignesh, S. K. Langley, K. S. Murray and G. Rajaraman, Chem. – Eur. J., 2015, 21, 2881–2892 CrossRef CAS PubMed.
- T. Gupta and G. Rajaraman, Chem. Commun., 2016, 52, 8972–9008 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of bond lengths and angles, additional figures and computational details. CCDC 2099440. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03410c |
| This journal is © The Royal Society of Chemistry 2021 |