
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive organic emission profiles, secondary organic aerosol production potential, and OH reactivity of domestic fuel combustion in Delhi, India†
Gareth J.
Stewart
 a,
Beth S.
Nelson
a,
W. Joe F.
Acton‡
b,
Adam R.
Vaughan
a,
James R.
Hopkins
ac,
Siti S. M.
Yunus
d,
C. Nicholas
Hewitt
b,
Eiko
Nemitz
e,
Tuhin K.
Mandal
fg,
Ranu
Gadi
h,
Lokesh. K.
Sahu
i,
Andrew R.
Rickard
ac,
James D.
Lee
ac and
Jacqueline F.
Hamilton
*a
a,
Beth S.
Nelson
a,
W. Joe F.
Acton‡
b,
Adam R.
Vaughan
a,
James R.
Hopkins
ac,
Siti S. M.
Yunus
d,
C. Nicholas
Hewitt
b,
Eiko
Nemitz
e,
Tuhin K.
Mandal
fg,
Ranu
Gadi
h,
Lokesh. K.
Sahu
i,
Andrew R.
Rickard
ac,
James D.
Lee
ac and
Jacqueline F.
Hamilton
*a
aWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, York, YO10 5DD, UK. E-mail: Jacqui.hamilton@york.ac.uk
bLancaster Environment Centre, Lancaster University, Lancaster LA1 4YQ, UK
cNational Centre for Atmospheric Science, University of York, York, YO10 5DD, UK
dSchool of Water, Environment and Energy, Cranfield University, Cranfield, MK43 0AL, UK
eUK Centre for Ecology and Hydrology, Penicuik, EH26 0QB, UK
fCSIR-National Physical Laboratory, Dr. K.S. Krishnan Marg, New Delhi, Delhi 110012, India
gAcademy of Scientific & Innovative Research, Ghaziabad, Uttar Pradesh-201 002, India
hIndira Gandhi Delhi Technical University for Women, Kashmiri Gate, New Delhi, Delhi 110006, India
iPhysical Research Laboratory (PRL), Ahmedabad 380009, India
First published on 21st January 2021
Abstract
Domestic solid fuel combustion is a major source of organic compounds to the atmosphere in gas and aerosol phases; however, large uncertainties exist in the current understanding of the gas-to-particle partitioning and the drivers of the reactivity of these emissions. This study developed comprehensive, model-ready organic emission profiles for domestic solid fuel combustion sources collected from Delhi, India. It also examined the organic species responsible for secondary organic aerosol (SOA) production potential and hydroxyl radical (OH) reactivity of these emissions. The profiles spanned the entire volatility range, including non-methane volatile organic compounds (NMVOCs, effective saturation concentration, C* = 3 × 106 to 1011 μg m−3), intermediate-volatility organic compounds (IVOCs, C* = 300 to 3 × 106 μg m−3), semi-volatile organic compounds (SVOCs, C* = 0.3–300 μg m−3) as well as low- and extremely low-volatility organic compounds (L/ELVOCs, where LVOC C* ≤ 0.3 μg m−3). The profiles predicted that IVOCs would contribute significantly to SOA production and that the combustion of fuel wood and charcoal released some of the smallest proportions of SVOCs. A model was developed to examine SOA production from burning emissions which estimated that phenolics would contribute 10–70% of the SOA. Furanics were the most important reactive species, contributing 9–48% of the OH reactivity and 9–58% of the SOA. Different combustion sources were also compared, with emissions from fuel wood, crop residue, cow dung cake and municipal solid waste (MSW) burning shown to be 30, 90, 120 and 230 times more reactive with the OH radical than emissions from liquefied petroleum gas (LPG) fuel. This study also estimated 3–4 times more SOA from cow dung cake combustion and 6–7 more from MSW combustion than fuel wood under comparable combustion conditions. The results of this study suggest that emissions from the combustion of domestic solid fuel sources in Delhi have the potential to significantly degrade local and regional air quality. As a result, more effective mitigation strategies are required to limit the impacts of solid fuel combustion on human health in countries like India.
Environmental significanceApproximately 3 billion people use solid fuel combustion to meet their daily residential energy requirements, however, little is known about the subsequent impact that these emissions have on the atmosphere. Recent studies have shown that burning releases large amounts of lower volatility material, however, these are not accurately reflected in chemical transport models. This study addresses this, by providing model-ready inputs to better constrain burning related organic emissions. It also provides insight into the species of interest released from solid fuel combustion which require further lab studies and implementation into chemical models to truly understand the global impact of solid fuel combustion. |
1. Introduction
Around 3 billion people globally use solid fuels to meet their daily cooking energy requirements.1 Emissions from residential solid fuel combustion are significant and have been shown to cause indoor air pollution which resulted in 2.8–3.9 million premature deaths globally,1–3 with around 25% of ambient particulate matter (PM) in South Asia related to cooking emissions.4 Approximately a quarter of worldwide residential solid fuel use is in India,5 where cooking domestically over biomass remains popular because biomass fuel is cheaper than liquefied petroleum gas (LPG) and meals cooked with traditional methods perceived to be tastier.6 Recent studies have shown that 16%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 of non-methane hydrocarbons and 27%8 of non-methane volatile organic compounds (NMVOCs) by mixing ratio at different urban sites in Delhi were from solid fuel combustion sources. Furthermore, Aerosol Mass Spectrometer measurements found that crop residue burning and solid fuel combustion jointly accounted for 24% (35.8 μg m−3) of the concentration of PM with a diameter <1 μm (PM1) during the post-monsoon in Delhi, with likely additional contributions to the SOA.9
7 of non-methane hydrocarbons and 27%8 of non-methane volatile organic compounds (NMVOCs) by mixing ratio at different urban sites in Delhi were from solid fuel combustion sources. Furthermore, Aerosol Mass Spectrometer measurements found that crop residue burning and solid fuel combustion jointly accounted for 24% (35.8 μg m−3) of the concentration of PM with a diameter <1 μm (PM1) during the post-monsoon in Delhi, with likely additional contributions to the SOA.9
Studies focussed on organic emissions from both open biomass burning and domestic solid fuel combustion have shown that organic components are released over a range of volatilities.10–14 These include non-methane volatile organic compounds (NMVOCs, effective saturation concentration, C*, 3 × 106 to 1011 μg m−3), intermediate-volatility organic compounds (IVOCs, C* = 300 to 3 × 106 μg m−3), semi-volatile organic compounds (SVOCs, C* = 0.3–300 μg m−3) as well as low- and extremely low-volatility organic compounds (L/ELVOCs, where LVOC C* ≤ 0.3 μg m−3).15 As a result, I/SVOCs from domestic solid fuel combustion potentially represent a large global source of SOA, however, the effect of I/SVOCs on OH reactivity, aging and SOA formation remains poorly understood.16–18
The factors controlling SOA formation are complex. These include the oxidation of NMVOCs to less volatile products which partition into the particle phase, the heterogeneous oxidation of particle-phase SVOCs, and plume dilution with subsequent SVOC evaporation followed by further gas-phase oxidation.19 Of 17 studies examining the enhancement factor of organic aerosol (OA) to CO from the aging of open biomass burning emissions, 10 found no increase in SOA, 4 found an increase and 3 reported a decrease.20 Despite varied results, a recent lab study has shown SOA formation from combustion of fuels relevant to open biomass burning to be significant. Lim et al. (2019) showed a carbon yield of SOA from NMVOCs emitted from the combustion of western U.S. fuels of 24 ± 4% when exposed to atmospheric aging equivalent to 6 hours, which increased to 56 ± 9% after aging equivalent to 6 days.19
Formation of SOA from open biomass burning has been examined as part of several recent studies. Hatch et al. (2015) estimated that 8–15% of SOA from the combustion of black spruce, cut grass, Indonesian peat and ponderosa pine was because of furanic compounds. The contribution of furanic compounds to SOA was estimated to be greater still (28–50%) from rice straw and wiregrass.11 Gilman et al. (2015) examined the relative contributors to SOA from the combustion of U.S. fuels and found the main contributors to be polyunsaturated oxygenated NMVOCs.21 High SOA formation potential was driven by benzene diols, benzaldehyde, and phenols. Ahern et al. (2019) showed that for the combustion of coniferous fuels, which were dominated by the burning of biomass needles, biogenic NMVOCs were the most important class of SOA precursor.22 Akherati et al. (2020) reported that oxygenated aromatic compounds resulted in just under 60% of the SOA from the combustion of western U.S. fuels.23 These studies have also shown that reactive chemical species such as furanics, oxygenated aromatics and aliphatics are important drivers of the OH reactivity of open biomass burning emissions.10,21,24 Recent model simulations by Coggon et al. (2019) focussed on modelling the OH radical chemistry in emissions from the combustion of fuels from the western U.S. showed that up to 10% of O3 in the first 4 h after emission was a result of the oxidation of furanic compounds.25
Few studies have examined SOA formation from fuels used for domestic solid fuel combustion, with little known about the impact of the species released on the reactivity of emissions. Bruns et al. (2016) examined SOA formation from the combustion of beech fuel wood and demonstrated that the main contributors were 22 compounds, and in some cases up to 80% of the SOA produced was estimated to be formed from phenol, naphthalene and benzene.26 A further study suggested that furanic and phenolic compounds were important precursors to SOA as a result of spruce combustion.24
The concentration of primary organic aerosol (POA) is determined by dynamic gas-to-particle partitioning of an extremely complex mixture of organics over a wide range of volatilities. Understanding the gas-to-particle partitioning represents one of the main difficulties in accurately characterising SOA formation, as measurements of organic emissions using multiple measurement techniques are required. As a result, gas-phase emissions are traditionally considered up to C12 (saturation vapour concentration, C*, ∼106 μg m−3) and POA as non-volatile.27–30 Consequently, many models neglect the importance of I/SVOCs as SOA precursors. The effect is a significant underestimation of SOA production and an overestimation of POA in chemical transport models.20,31,32 The concentration of organic aerosol (OA) is determined by the volatility of species and ambient conditions, with many source tests occurring at unrealistically high OA concentrations. Laboratory-based source studies typically enhance the POA emission factor relative to more dilute ambient conditions.28,33 The inclusion of I/SVOCs leads to better agreement between modelled and measured values.32,34–36 A range of studies have been conducted to comprehensively characterise organic emissions from mobile sources30,37–39 and aircraft engines,40,41 however, a need has been highlighted to develop source profiles for both open and domestic biomass burning.30 These have the potential to result in a better understanding of the SOA formed from the I/SVOCs released. Comprehensive source profiles are far better suited to predicting SOA formation than traditional separated gas- and particle-phase emission factors developed at the point of emission. Comprehensive profiles can be adjusted to real-world dilutions, aerosol concentrations and temperatures. These parameters are all likely to have a large influence on the mass of SOA present.
This study develops comprehensive, model-ready organic emission profiles for solid fuels routinely burnt in the Delhi area of India. These profiles account for the full range of volatilities of organic emissions to better constrain the impact of domestic biomass burning on SOA formation. This study also compares the relative impacts of different solid fuel combustion sources to SOA production potential and OH reactivity and examines the most important chemical contributors.
2. Methods
2.1 Datasets
This paper combines previously published data13,14 of organic emissions from a detailed field campaign designed to measure organic emissions from the combustion of solid fuels widely used in Delhi, India. The data has been used to create comprehensive organic emission profiles and examine the largest contributors to secondary organic aerosol production potential and the OH reactivity of emissions.Fuels were collected from across Delhi in a manner designed to reflect the range and variability of solid fuels used across the region. Sample collection included a range of fuel woods (Melia azedarach, Prosopis spp, Eucalyptus spp, Azadirachta indica, Mangifera indica, Morus spp, Pithecellobium spp, Shorea spp, Ficus religosa, Syzgium spp, Ficus spp, Vachellia spp, Dalbergai sissoo, Ricinus spp, Holopetlea spp, Saraca indica and plywood), cow dung cake, municipal solid waste (MSW, collected from 3 landfill sites: Ghazipur, Bhalswa and Okhla), crop residues (Brassica spp, Solanum melongena and Cocos nucifera) and individual samples of charcoal, sawdust and LPG. Samples were stored in a manner akin to local storage practices prior to combustion. Combustion experiments were conducted using expert local judgement to ensure that the experimental setup replicated real-world burning conditions, using a combustion-dilution chamber at the CSIR-National Physical Laboratory (NPL), New Delhi, that has been well described previously (see the ESI1† for a schematic of the combustion chamber used).42–45
Fuel (200 g) was rapidly heated to spontaneous ignition using an electric heater, with emissions convectively driven into a hood and up a flue to allow enough dilution, cooling and residence time to achieve the quenching typical of indoor environments. These conditions have been previously optimised so that the chamber setup did not alter combustion conditions.42 The methodology was designed to replicate the convection-driven conditions of real-world combustion and was adapted from the VITA water-boiling test. Temperature measurements were made directly above the fuel being combusted (see the ESI2† for combustion temperatures by fuel type) and ambient temperatures during experiments ranged from 25–38 °C. All instruments made measurements over a 30 minute period, to ensure that emissions were collected from both high- and low-temperature combustion conditions. All measurements used the same procedures to characterise emissions to create a self-consistent dataset of speciated organic emissions spanning a large range of volatilities.
NMVOCs were sampled from the top of the flue down a ¼′′ PFA sample line, which was subsampled by three separate online gas-phase instruments designed to target a wide range of NMVOCs of different functionality and volatility. A dual-channel gas chromatograph with flame ionisation detection (DC-GC-FID) was used to sample alkanes from ethane to n-hexane and a range of small alkenes from ethene–isoprene (see the ESI3† for DC-GC-FID method). A two-dimensional gas chromatograph with flame ionisation detection (GC×GC-FID) was used to sample alkanes from n-heptane to n-dodecane, aromatic species from benzene to monoaromatics with up to 5 carbon substituents and up to 12 monoterpenes (see the ESI4† for GC×GC-FID method). GC instruments were calibrated using 4 ppbv gas standards containing a range of alkanes, alkenes, and aromatics purchased from the British National Physical Laboratory. Blank measurements were made at the beginning, middle and end of the day, with mean values subtracted from measured emission factors for both GC instruments.
A proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS, PTR 8000; Ionicon Analytik, Innsbruck) was used to sample a range of small oxygenates, oxygenated aromatics, alkenes, furanic species and nitrogen-containing volatile organic compounds (see the ESI5† for PTR-ToF-MS method). Calibrations were performed twice a week with a gas calibration unit (Ionicon Analytik, Innsbruck) using a calibration gas that contained 18 compounds (Apel-Riemer Environmental Inc., Miami). Before each burn, ambient air was sampled to provide a background for the measurement. Measurements were made by the PTR-ToF-MS at 1 second, which were averaged to the 30 minute sample window of the GC instruments to create the self-consistent dataset of gas-phase organic emissions in this study.
Aerosol phase organics were collected onto polytetrafluoroethylene (PTFE) filters and residual low-volatility organic gases were adsorbed to the surface of C18 coated solid phase extraction disks (SPE) placed behind these. SPE disks and PTFE filters were spiked with an internal standard (EPA 8270 Semivolatile Internal Standard Mix, 2000 μg mL−1 in DCM), extracted using accelerated solvent extraction (ASE 350, Dionex, ThermoFisher Scientific) following the methodology of Farren et al. (2015)46 and analysed using GC×GC-ToF-MS (Leco Pegasus BT 4D). Additional details of the accelerated solvent extraction procedure and GC×GC-ToF-MS method are given in the ESI6.†
Emission factors of 192 speciated NMVOCs, which achieved on average 94% speciation of total measured NMVOCs, were combined from n fires sampled by the DC-GC-FID (n = 51), GC×GC-FID (n = 74), PTR-ToF-MS, (n = 75) and SPE-GC×GC-ToF-MS (n = 28), with information on organic aerosol composition given by PTFE-GC×GC-ToF-MS (n = 28). Speciation profiles were based on a subset of tests that included SPE/PTFE samples from fuel wood (n = 16), cow dung cake (n = 3), MSW (n = 3), crop residue (n = 3), LPG (n = 1), charcoal (n = 1), sawdust (n = 1) and blank measurements (n = 8).
2.2 Mapping organics to volatility basis data set
The volatility-basis dataset (VBS) is designed to simulate the emission and evolution of I/SVOCs into the atmosphere and places NMVOCs into logarithmically spaced bins of C* at 298 K.47 Emissions from fuel wood, cow dung cake, MSW and LPG were mapped onto a VBS to visualise and compare emissions across the entire range of volatilities measured using data collected by the DC-GC-FID, GC×GC-FID, PTR-ToF-MS, SPE-GC×GC-ToF-MS (>C12) and PTFE-GC×GC-ToF-MS. C* values were calculated for individual NMVOCs measured online using the DC-GC-FID, GC×GC-FID and PTR-ToF-MS instruments. For SPE-GC×GC-ToF-MS and PTFE-GC×GC-ToF-MS analyses, organics were lumped into groups of unspeciated compounds. These were spaced between n-alkanes, with the volatility assigned as the mean volatility of the alkanes either side of the bin. For NMVOCs where insufficient data was available for a calculation of C*, the volatility was assigned as the C* of the n-alkane with the nearest boiling point. Despite calibrating C* of organic material to n-alkanes potentially leading to an overestimation of the volatilities of polycyclic aromatic hydrocarbons (PAHs) and aromatic oxygenates,48 this unspeciated material represented a small fraction of the total organic matter. The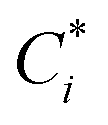 for each NMVOCi was calculated using (1):
for each NMVOCi was calculated using (1):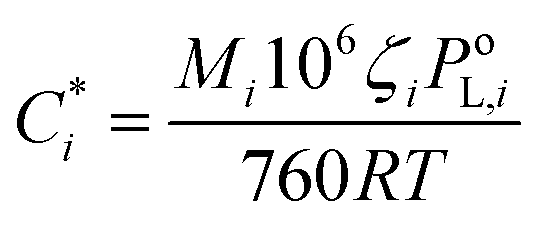 | (1) |
Experimental and/or predicted vapour pressures of species, especially the n-alkanes used for assigning volatility bins, remained uncertain. We adopted a similar approach to Lu et al. (2018), with the factor of 10 spacing of volatility bins to minimise the chance of volatility misassignment.30 IVOCs were in the n-alkane range ∼ C12 to C22, SVOCs from C23 to C32, and L/ELVOCs from C33 to C40. Care was taken to avoid double counting of species measured using multiple techniques (see the ESI7† for the hierarchy of instruments used to measure different species). Gas-phase species, which were possible to measure using either of the GC instruments or the PTR-ToF-MS, were counted once only. In summary C2–C6 non-methane volatile organic compounds (alkanes/alkenes) were measured using the DC-GC-FID. C7–C12 non-methane volatile organic compounds (alkanes and benzene to C3 substituted monoaromatics) were measured using the GC×GC-FID. Remaining NMVOCs and gas-phase I/SVOCs were measured using the PTR-ToF-MS (C4–C5 substituted monoaromatics, phenolics, furanics, oxygenated aromatics, oxygenated aliphatics and nitrogen containing volatile organic compounds). The unidentified gaseous I/SVOC fraction was estimated using SPE-GC×GC-ToF-MS. The organic aerosol fraction was measured using the PTFE-GC×GC-ToF-MS. To allow incorporation of I/SVOCs species from SPE disks, species and their isomers measured using the PTR-ToF-MS were removed from the SPE and PTFE analyses.
2.3 Comparison of EPA and fuel wood source profiles
Fuel wood source profiles were compared to those from the EPA SPECIATE 5.0 (2019) database. Notably profiles from the EPA for burning sources were split into either gas- or particle-phase measurements. All available profiles for residential combustion were considered from sets of experiments including fireplace wood combustion (4640–4642), residential combustion using wood and pellet stoves (95129–95138), residential wood stove combustion (95156–95159) and residential wood combustion (G95467–G95470). EPA profiles 95156–95159 for residential wood stove combustion were not directly compared due to the low number of organic species measured (n = 37). Comparison was made to Pinus ponderosa (G95467), Eucalyptus spp (4640) and a wood stove (95133). This placed into context the VBS developed in this work, because the multiple and complementary techniques used here allowed simultaneous measurement of organics in both gas and aerosol phases.
EPA G95467 was a source profile derived from measurement of 179 organic species from combustion of Pinus ponderosa.50 C2–C12 compounds were collected into canisters and analysed by GC-FID/-MS, C8–C20 compounds were collected onto Tenax tubes and analysed by GC-FID/-MS, carbonyls were collected onto 2,4-dinitrophenylhydrazine cartridges and analysed by HPLC and fine particles and SVOCs were collected onto filter/PUF/XAD/PUF cartridges and analysed by GC-MS. EPA 4640 was a source profile derived from measurement of 85 organic species from Eucalyptus spp. This profile was chosen as Eucalyptus spp was also measured as part of this study. Gas phase semi-volatile species were collected onto PUF cartridges, particles collected onto filters and carbonyls onto C18 cartridges impregnated with dinitrophenylhydrazine. Samples were then extracted and analysed by GC-MS.51 EPA 95133 was developed by sampling VOCs into Tedlar gas sampling bags followed by GC-FID analysis and semi-volatile PAHs were collected onto PUF plugs, extracted and analysed by GC-MS.52
2.4 Estimation of the SOA formation potential
The overall yield of SOA, γSOA, from gas-phase emissions from domestic biomass burning samples (mass of SOA produced/mass of NMOG emissions) was estimated using:30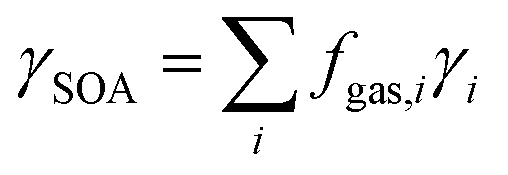 | (2) |
2.5 Estimation of OH reactivity
The OH reactivity of emissions from different fuel types was examined to understand the largest contributors. The mean concentrations of NMVOCs from the DC-GC-FID, GC×GC-FID and PTR-ToF-MS were used to calculate OH reactivity, s−1, using:| s−1 = ([NMVOC](ppbv) × 10−9 × [M]) × kOH(298 K) | (3) |
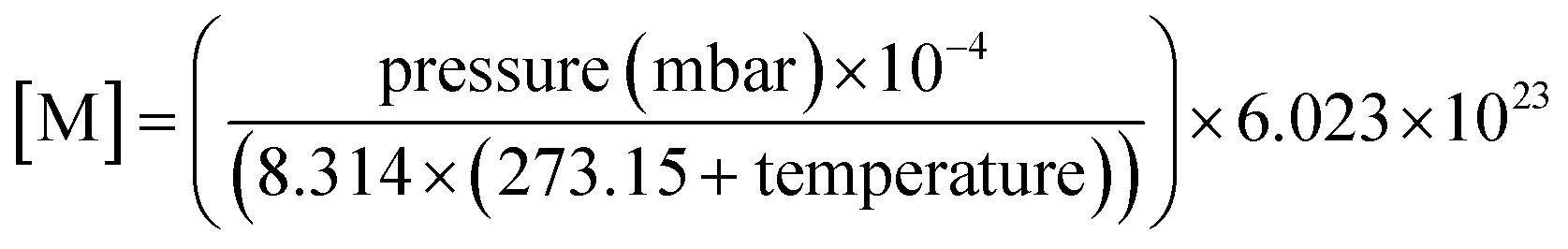 | (4) |
2.6 Estimation of PAH toxicity
Toxicity equivalence factors (TEFs) were used to assess the relative toxicity of PAH emissions per kg of fuel burnt. TEFs indicate the relative toxicity of a PAH to benzo[a]pyrene (BaP), one of the most carcinogenic PAHs.53 The toxicity of a PAH was expressed in BaP equivalents ([BaP]eq), which was calculated in (5) by multiplying the concentration of PAHi, in nanograms per cubic metre (ng m−3), by the corresponding TEF for i, TEFi, given in the ESI10.†54–56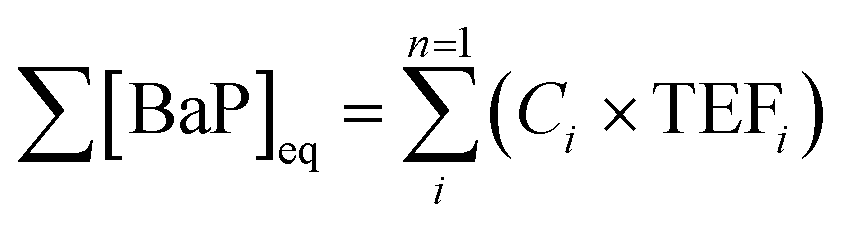 | (5) |
3. Results and discussions
3.1 Volatility distribution
Fig. 1 shows the mean volatility distribution of characterised organic emissions for (A) all fuel wood types studied (n = 16) and (B) cow dung cake (n = 3) classified by measurement technique: PTR-ToF-MS (orange), DC-GC-FID (green), GC×GC-FID (purple), SPE-GC×GC-ToF-MS (blue) and PTFE-GC×GC-ToF-MS (red). Fig. 1 displays a comprehensive characterisation of organic emissions and emphasised the importance of using multiple techniques to measure organic emissions, covering a volatility range of over 13 orders of magnitude. Cow dung cake combustion released more SVOCs and L/ELVOCs than fuel wood.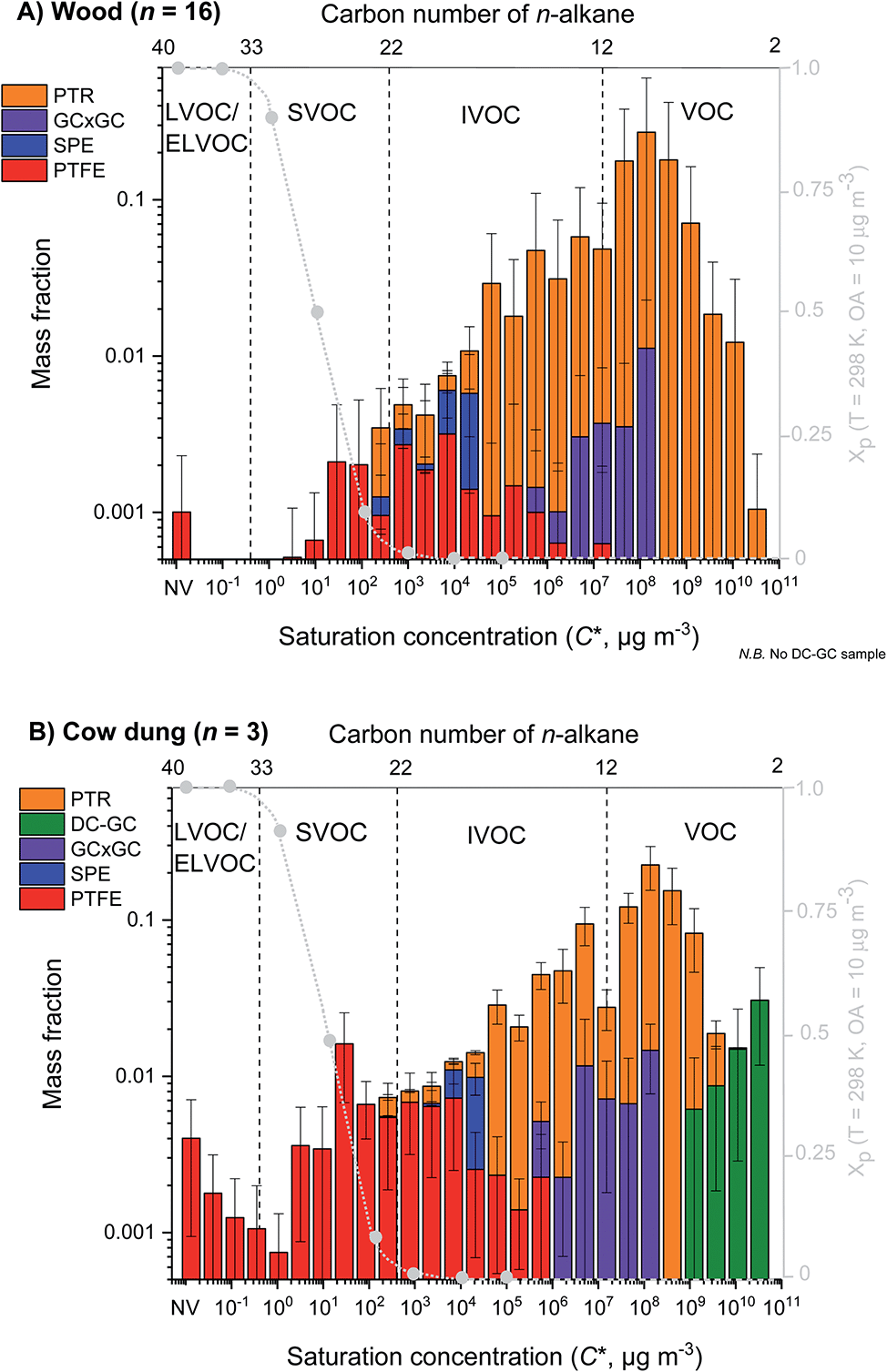 | ||
| Fig. 1 Mean volatility distribution of organics emitted during fuel wood (top) and cow dung cake (bottom) combustion. Emissions are classified by sampling technique with PTR-ToF-MS (orange), DC-GC-FID (green), GC×GC-FID (purple), SPE-GC×GC-ToF-MS (blue) and PTFE-GC×GC-ToF-MS (red). The grey dashed line indicates the particle fraction, assuming the emissions form a quasi-ideal solution when diluted to ambient conditions at organic aerosol concentration = 10 μg m−3 and temperature = 298 K. See the ESI7† for the hierarchy of instruments used to measure fractions of the organic mass. | ||
Fig. 1 illustrates the particle fraction, Xp, which was calculated according to the method in Lu et al. (2018), assuming all the organic emissions formed a quasi-ideal solution when diluted to ambient conditions.30 The particle fraction demonstrated the gas-to-particle partitioning of organics at typical atmospheric conditions (T = 298 K and OA concentration = 10 μg m−3). IVOCs were predominantly found in the gas phase and SVOCs were present in both phases. The predicted particle fraction suggested that there should have been more gas-phase contributions in the I/SVOC range. The amount of organic material in the gas and particle phase is dependent on multiple factors such as temperature and concentration of OA. It is likely that at the high concentrations (OA > 10 μg m−3) during source testing, a larger fraction of I/SVOCs partitioned into the particle phase.
3.2 Chemical composition distribution
Fig. 2 shows the mean volatility distributions of organic emissions from seven different source categories (A) fuel wood, (B) cow dung cake, (C) MSW, (D) crop residue, (E) charcoal, (F) sawdust and (G) LPG. The largest mass fraction at emission for all sources, expect LPG, was in the range C* ∼ 107–109 μg m−3 and a result of small oxygenated species. LPG emission was dominated by fugitive emissions of propane and butane from C* 109–1010 μg m−3. Fig. 2 highlights how changes in the type of source influenced emissions of I/S/L/ELVOCs. All sources, except LPG, had significant emissions of IVOCs.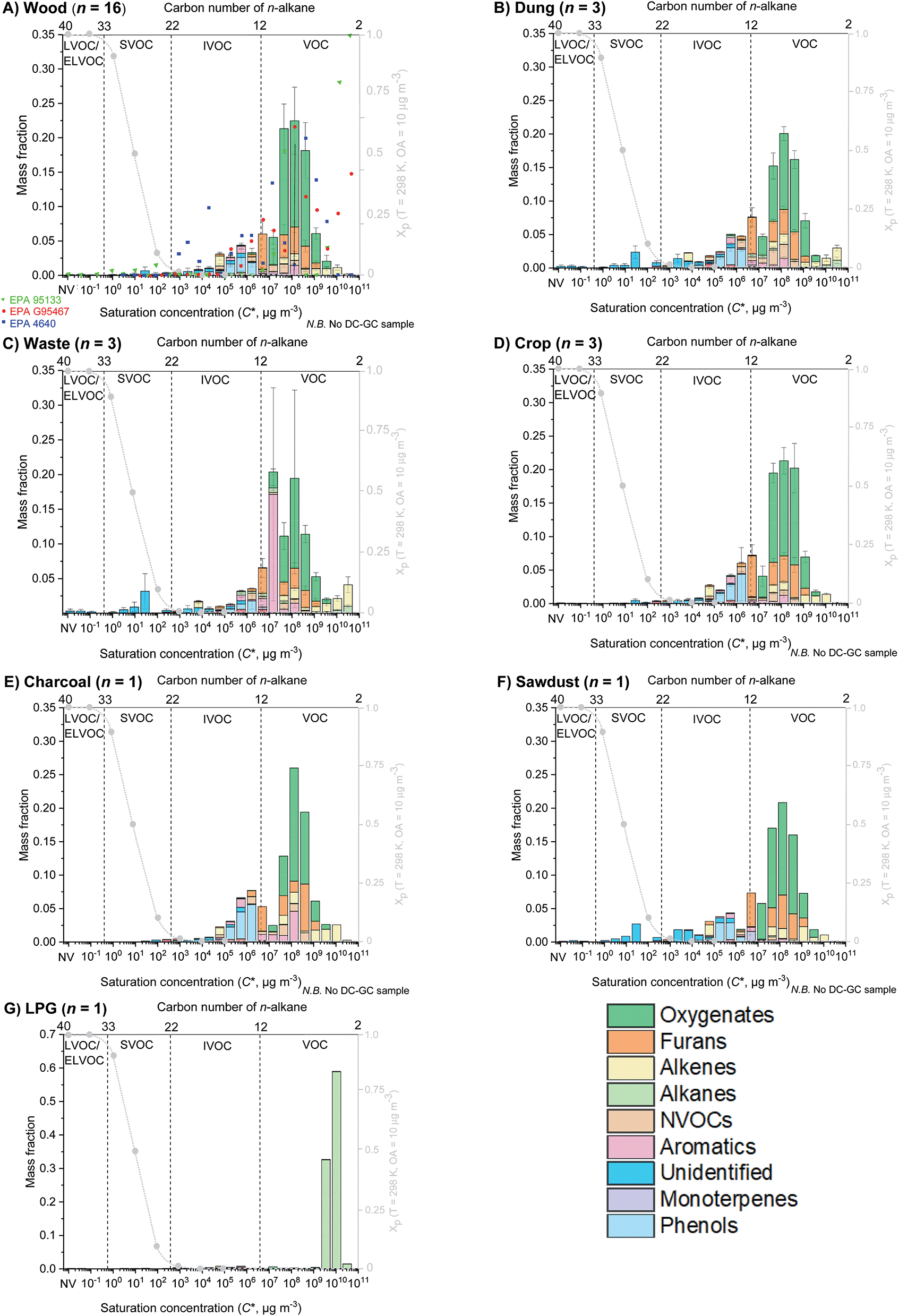 | ||
| Fig. 2 Mean volatility distribution of organic emissions for (A) fuel wood, (B) cow dung cake, (C) municipal solid waste, (D) crop residue, (E) charcoal, (F) sawdust and (G) LPG, with composition indicated by colour. Fuel wood profiles are compared to EPA inventories G95467 for softwood (red circle), EPA 95133 for a wood stove (green triangle) and EPA 4640 for Eucalyptus spp (blue square). | ||
Fig. 2A also shows comparison to EPA source profiles G95467 for Pinus ponderosa (red circles), 95133 for a wood stove (green triangles) and 4640 for Eucalyptus spp (blue squares). These profiles highlighted the difficulties in using current source profiles to predict SOA from domestic biomass burning, due to significantly different predictions in the range C* ∼ 102–106 μg m−3.
EPA 95133 reported essentially no IVOCs, EPA G95467 showed some IVOCs in the range C* ∼ 105–106 μg m−3 and EPA 4640 showed considerably higher IVOC emissions. EPA 95133 did not measure important I/SVOC species released from domestic biomass burning such as phenolics and furanics and therefore no organic matter was represented for C* < 5 × 106 μg m−3. EPA G95467 was one of the best current source profiles, however, no organic matter was present in this profile for C* < 105 μg m−3. This may be due to lack of simultaneous gas- and particle-phase measurements of all organic species present. As a result, gas-phase organic species may have partitioned into the particle phase because of high organic aerosol concentrations during source testing and are therefore were not represented. EPA 4640 measured more phenolic and furanic compounds. Despite this, the measurement of only 85 organic species in EPA 4640 overemphasised the importance of I/SVOCs as a mass fraction. This therefore still posed significant problems when using EPA 4640 to model SOA formation. These issues demonstrated the benefit of the VBS developed here, as simultaneous measurement of organics in both gas and aerosol phases should alleviate these problems. For some sources, such as the combustion of MSW, cow dung cake, crop residues and sawdust a greater mass fraction of I/SVOCs was released. The use of a VBS for these sources is likely even more important due the presence of large amounts of I/SVOC material.
The comparison results suggested that the profile presented for Indian domestic fuel wood from this study was significantly lower than the data in EPA G95467 and 95133 in the two most volatile bins (C* ∼ 1010–1011 μg m−3). However, this was an artefact due to the lack of measurements with the DC-GC-FID, which targeted the most volatile species. However, the underestimation of emissions from C* ∼ 1010–1011 μg m−3 is unlikely to be significant when calculating SOA formation using the measured species and VBS presented.
Table 1 shows the mass fraction of organic material presented in Fig. 2 from the 7 different sources studied here, presented in volatility bins spanning over 13 orders of magnitude. For certain sources, such as LPG and charcoal, only one sample was taken. The lack of repeat measurements significantly increased the uncertainty associated with the VBS presented. Despite this, multiple gas-phase NMVOC measurements were made. These showed similar results and therefore these VBS were included. The results in Table 1 should be used to better characterise SOA formation in chemical-transport models from domestic biomass combustion sources as the volatility distribution of organic emissions presented can be accurately adjusted to atmospheric dilutions, aerosol concentrations and temperatures.
| C* | Range | Wood | Dung | MSW | Sawdust | LPG | Charcoal | Crop |
|---|---|---|---|---|---|---|---|---|
| NV | ELVOC | 0.001 | 0.006 | 0.006 | 0.002 | 0.000 | 0.007 | 0.001 |
| 10−1 | SVOC | 0.000 | 0.002 | 0.002 | 0.001 | 0.000 | 0.000 | 0.000 |
| 100 | L/SVOC | 0.001 | 0.008 | 0.012 | 0.014 | 0.000 | 0.000 | 0.000 |
| 101 | SVOC | 0.004 | 0.023 | 0.025 | 0.027 | 0.001 | 0.001 | 0.004 |
| 102 | S/IVOC | 0.008 | 0.015 | 0.006 | 0.017 | 0.001 | 0.009 | 0.008 |
| 103 | IVOC | 0.012 | 0.021 | 0.014 | 0.033 | 0.000 | 0.011 | 0.010 |
| 104 | IVOC | 0.040 | 0.043 | 0.027 | 0.047 | 0.000 | 0.036 | 0.038 |
| 105 | IVOC | 0.066 | 0.065 | 0.035 | 0.071 | 0.001 | 0.087 | 0.062 |
| 106 | I/VOC | 0.090 | 0.142 | 0.117 | 0.091 | 0.001 | 0.129 | 0.135 |
| 107 | VOC | 0.224 | 0.149 | 0.278 | 0.206 | 0.000 | 0.122 | 0.202 |
| 108 | VOC | 0.449 | 0.379 | 0.348 | 0.387 | 0.001 | 0.471 | 0.434 |
| 109 | VOC | 0.090 | 0.101 | 0.073 | 0.093 | 0.350 | 0.101 | 0.092 |
| 1010 | VOC | 0.013 | 0.046 | 0.058 | 0.011 | 0.646 | 0.027 | 0.014 |
Fig. 3 shows that the mean mass fractions of IVOCs emitted increased from MSW (0.12 ± 0.02) to fuel wood (0.15 ± 0.04) to crop residue (0.16 ± 0.04) to cow dung cake (0.18 ± 0.02). SVOC emissions for fuel wood and crop residue were the lowest mass fraction (0.01 ± 0.01) and larger for cow dung cake (0.04 ± 0.02) and MSW (0.05 ± 0.04). L/ELVOC emissions for crop residue (0.001 ± 0.001) and fuel wood (0.002 ± 0.002) were the lowest and larger for cow dung cake (0.006 ± 0.004) and MSW (0.009 ± 0.008). SVOC and L/ELVOC emissions from crop residue and charcoal were similarly low to fuel wood. These low S/L/ELVOC emissions may be a result of the different fire conditions caused by the difference in composition of samples. Fires which are intense and flaming have been shown to have high black carbon emissions, whilst those which are more towards the smouldering phase have higher OA emissions.57–60 It is likely that the higher emissions of OA from cow dung cake, MSW and sawdust were a result of the lower combustion efficiency of these samples.
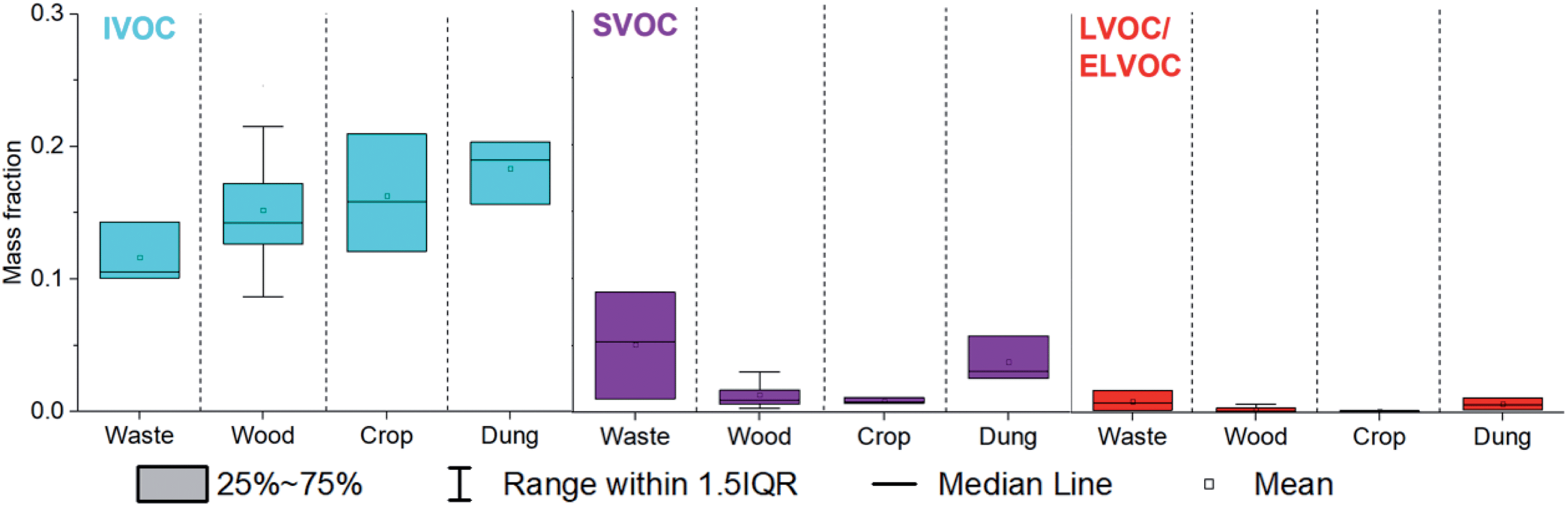 | ||
| Fig. 3 IVOC, SVOC and L/ELVOC mass fractions emitted from combustion of municipal solid waste, fuel wood and cow dung cake. SVOC and L/ELVOC material represented on average a smaller mass fraction from fuel wood and crop residue than from municipal solid waste and cow dung cake. | ||
3.3 SOA formation potential
Fig. 4 shows the sum of the mass fraction of NMVOCs released from domestic fuel burning in this study. Only the species identified as SOA precursors and assigned with SOA yields in the ESI8† were included. The mass fraction of SOA precursors from fuel wood, crop residue and cow dung cake samples were from 0.3–0.5. Compared to sources calculated using the same method,30 the mass fraction which resulted in SOA was less, with the exception of MSW burning, compared to gasoline (∼0.65) and diesel (∼0.7) engines. This was principally due to the large emission of smaller oxygenated species from burning samples. MSW burning samples released the largest mass fraction of SOA precursors (0.4–0.65).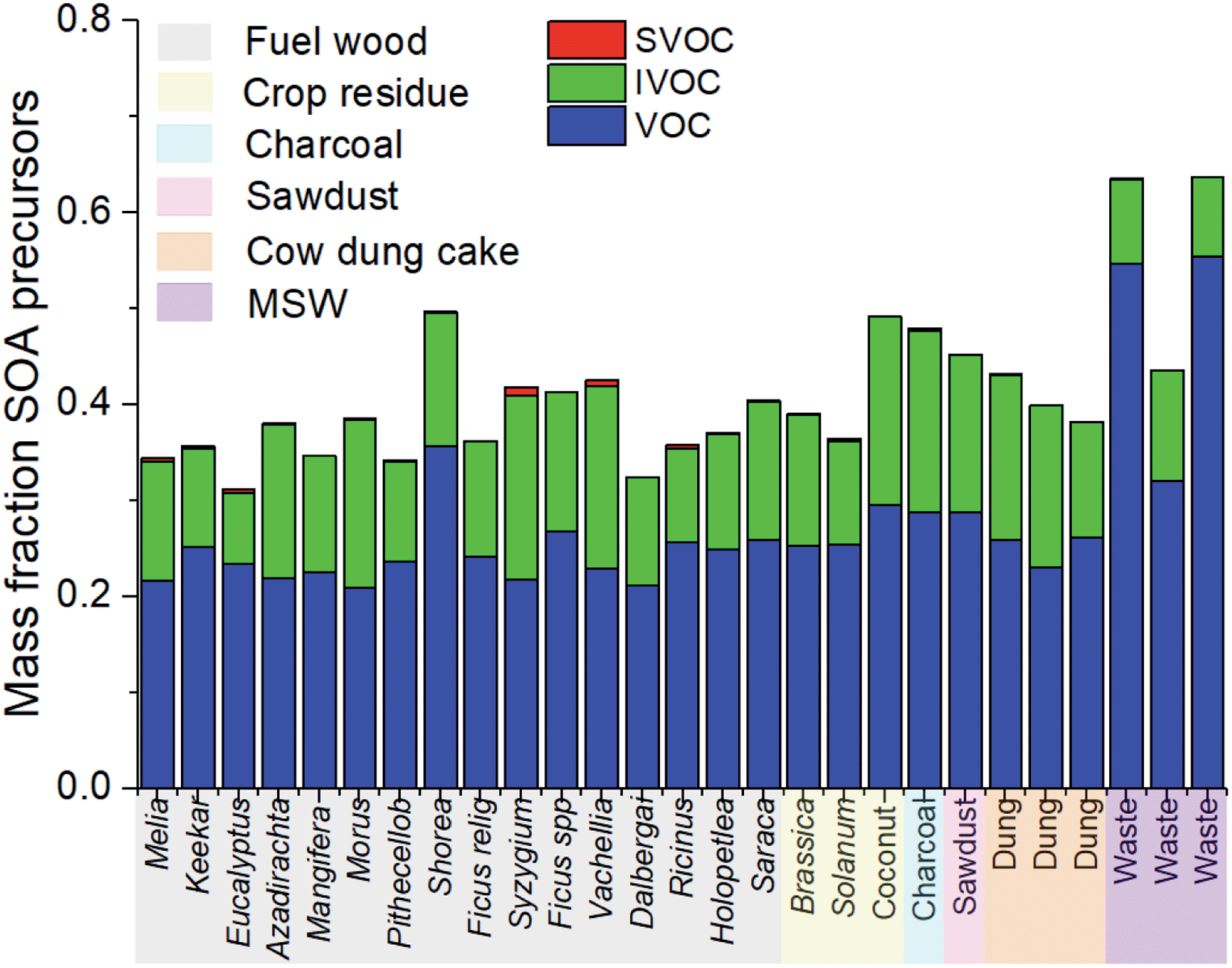 | ||
| Fig. 4 Mass fraction of different NMVOCs from burning which were SOA precursors. | ||
Fig. 5A and B show the estimated SOA yields from burning samples under high and low NOx conditions. These were intended to represent idealised systems for photo-oxidation of SOA precursors. Under high NOx conditions, RO2 radicals react with NOx and under low NOx conditions RO2 radicals react with HO2.61 Consideration was given to both cases since domestic biomass burning in India impacts both urban high NOx regions and rural lower NOx regions. Under high NOx conditions, SOA yields were lower and IVOCs represented a larger proportion of the total SOA produced. Under low NOx conditions, SOA yields were greater, and NMVOCs resulted in a greater proportion of the total SOA due to higher estimated SOA yields from aromatic and furanic species. Other studies examining emissions from burning have traditionally considered yields from only one of these regimes, but greater SOA production under low NOx conditions has been well described previously.62,63
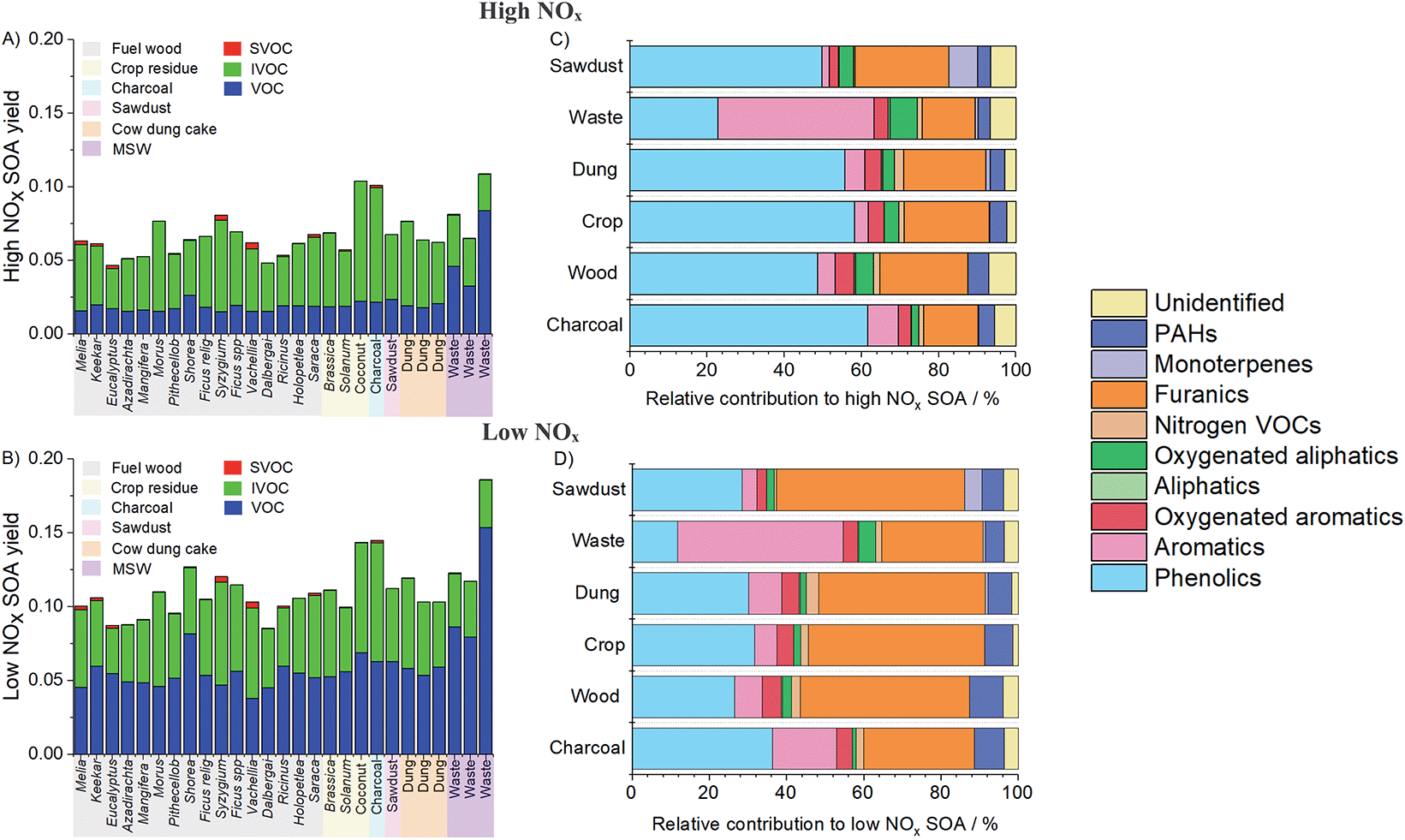 | ||
| Fig. 5 Results of SOA model with (A) SOA yields as mass fraction of NMVOC released from domestic combustion under high NOx conditions, (B) SOA yields as mass fraction of NMVOC released from domestic combustion under low NOx conditions, (C) relative NMVOC contributions to SOA formation under high NOx conditions and (D) relative NMVOC contributions to SOA formation under low NOx conditions. Unidentified corresponds to bulk material from SPE/PTFE filters. | ||
Fig. 5C shows that high NOx SOA yields from sawdust, charcoal, cow dung cake, fuel wood and crop residue were likely dominated by phenolics (light blue, 21–70%), with a significant contribution from furanics (orange, 9–33%) due to high emission factors of these species and high SOA yields. Other important SOA contributions were from aromatics (2–8%), oxygenated aromatics (2–8%), oxygenated aliphatic species (2–9%), monoterpenes (0–7%) and PAHs (2–16%). A larger proportion of SOA (40%) from MSW samples under high NOx conditions was from aromatics due to a high emission factor of styrene from these samples.
Fig. 5D shows that for sawdust, charcoal, cow dung cake and fuel wood samples, furanic species (17–58%) and aromatics (4–16%) were likely to provide a greater proportion of total SOA under low NOx conditions. The contribution of phenolic compounds was less (10–43%) due to larger aromatic and estimated furanic SOA yields under these conditions. Contributions remained small from oxygenated aromatics (3–11%), aliphatic species (0–2%), oxygenated aliphatics (0–2%), nitrogen containing NMVOCs (0.5–3%), monoterpenes (0–2%) and PAHs (5–15%). The contribution of aromatics to SOA from MSW remained high (43%).
Bruns et al. (2016) showed that around 26% of SOA formed from the combustion of beech fuel wood was from phenolics. This was notably higher than the 5–9% contribution of phenol from Picea abies (spruce) reported by Hartikainen et al. (2018), who reported that 12–14% of the total SOA was from phenolic compounds. The results of this study appear more like that of Bruns et al. (2016), with between 10-70% of the total SOA from biomass combustion a result of phenolic compounds.
The yields under high NOx conditions for aromatics in this study (2–8%) were similarly low to those reported by Hartikainen et al. (2018) of 1.9–2.6% of the SOA from benzene and 1.9–3.3% from naphthalene, with low NOx conditions in this study suggesting aromatics could result in greater SOA yields. This study found relatively low SOA yields from monoterpenes from biomass sources (0–7%), which was like Hartikainen et al. (2018) who found that monoterpenes contributed ∼1–3% to SOA.24 This contrasted with Hatch et al. (2015) who showed that monoterpenes could result in 42–58% of SOA from black spruce and Ponderosa pine, however the fuel woods studied by Hatch et al. (2015) were likely larger emitters of monoterpenes.11
It remained difficult to accurately characterise SOA yields from furanic species, as there is a lack of chamber simulation studies. This study suggested that furanic compounds could act as a large SOA precursor source, similar to several other studies.11,24 In this study, the SOA yields of 2-methanol furanone, 2-(3H)-furanone, 5-hydroxymethyl-2[3H]-furanone, furfurals and methyl furfurals were estimated using the toluene yield, as a previous study by Gilman et al. (2015) indicated they had similar secondary organic aerosol formation potentials (SOAP).21 This resulted in two different cases. Under high NOx conditions, the SOA yield in this study of furanics was 0.08, which was similar to that of by Hatch et al. (2015)11 who used 0.10 based on the chemistry of 3-methyl furan measured from a previous study.64 The low NOx yield used in this study was 0.33, which was like Bruns et al. (2016), who used a furfural yield of 0.32 based on the average SOAP of all assigned ≥C6 compounds. The true SOA yields from furanic species from domestic biomass burning samples remained uncertain and requires further chamber studies. This issue was previously highlighted.65 While following a different approach, this study arrived at similar estimated yields of furanic compounds as those used previously. It highlighted that SOA formation from domestic biomass burning smoke from solid fuels collected in India was predominantly driven by phenolic and furanic compounds as well as aromatics.
Table 2 shows the mass fraction of NMVOCs released which were identified as SOA precursors from yield data, and the mass fraction of NMVOCs which resulted in SOA under high and low NOx conditions. These were presented as a mass fraction of total organic compounds measured during this study. Some sources, such as cow dung cake and MSW, released significantly more NMVOCs per kg of fuel burnt than fuel wood (MSW ∼ 88 g kg−1, cow dung cake ∼ 62 g kg−1 and fuel wood ∼ 19 g kg−1).14 Multiplying the emission factor by the mass fraction of NMVOC which would result in SOA highlighted interesting differences in SOA production between different source types. Table 2 shows this result, with the mass of SOA which would result per kg of fuel burnt under high (SOAh, g kg−1) and low (SOAl, g kg−1) NOx conditions. The amount of SOA produced by each source was considered relative to fuel wood, due to difficulties establishing SOA precursor from the chamber background for LPG. Emissions from cow dung cake and MSW resulted in ∼3–4- and ∼6–7-times greater SOA per kg of fuel burnt than fuel wood, respectively. The result for SOA formation from cow dung cake combustion was consistent with that calculated by Fleming et al. (2018), who reported a factor of 3 times higher SOA from dung-chulha stoves than brushwood-chulha for samples collected from Haryana, India.66 It is also noteworthy that SOA estimated from chamber yield data and that observed experimentally during burning experiments have been shown to agree within a factor of 2.22
| Sample | Mass fraction | Mass formed (g kg−1 fuel) | |||
|---|---|---|---|---|---|
| SOA precursors | SOAh | SOAl | SOAh | SOAl | |
| Wood | 0.38 | 0.061 | 0.103 | 1.1 (0.3–5.9) | 1.9 (0.4–10.0) |
| Dung | 0.40 | 0.068 | 0.109 | 4.2 (2.4–5.6) | 6.7 (3.8–9.0) |
| MSW | 0.57 | 0.085 | 0.142 | 7.4 (4.8–10.1) | 12.4 (8.0–16.9) |
| Charcoal | 0.48 | 0.101 | 0.145 | 0.5 (0.2–0.8) | 0.8 (0.3–1.1) |
| Sawdust | 0.45 | 0.068 | 0.112 | 4.9 (1.9–7.7) | 8.1 (3.2–12.8) |
| Crop | 0.41 | 0.076 | 0.121 | 2.9 (0.7–5.6) | 4.5 (1.1–8.9) |
The estimates of SOA formation should be considered relative to the heat output of specific fuels. Energy densities have been reported for LPG (45837 kJ kg−1), charcoal (25715 kJ kg−1), acacia fuel wood (15099 kJ kg−1), Eucalyptus spp fuel wood (15333 kJ kg−1), rice straw (13027 kJ kg−1), Brassica spp (11763 kJ kg−1) and dung cakes (11763 kJ kg−1).67 This highlights that whilst all sources were likely to result in SOA production, the burning of fuels such as cow dung cake is inadvisable due to the low calorific value and high emission factor. The result is that more fuel is required to be burnt to achieve the same heat output, which will lead to greater levels of NMVOC emission. These will subsequently degrade local and regional air quality through the formation of a greater quantity of secondary pollutants.
3.4 OH reactivity
Fig. 6A shows that LPG OH reactivity was principally driven by alkanes (∼75%). The contributions of other species were small and may have arisen from difficulties in background correction for this low emission fuel. For charcoal, the reactivity with OH was principally caused by furanics (33%), phenolics (19%) and oxygenates (15%). The reactivity of fuel wood emissions with OH was driven by furanics (34%), oxygenates (27%), phenolics (13%) and alkenes (12%). Emissions from cow dung cake with OH were due to by furanics (32%), oxygenates (21%), alkenes (16%), phenolics (12%) and nitrogen containing NMVOCs (11%). The OH reactivity from crop residue was from furanics (38%), oxygenates (23%), phenolics (14%) and alkenes (11%). For sawdust, reactivity with OH was a result of furanics (34%), oxygenates (24%), phenolics (15%) and monoterpenes (9%). However, for charcoal and sawdust only 2 samples were measured. The OH reactivity from MSW samples was different and a result of aromatics (30%), followed by oxygenates (22%), furanics (19%) and phenolics (5%).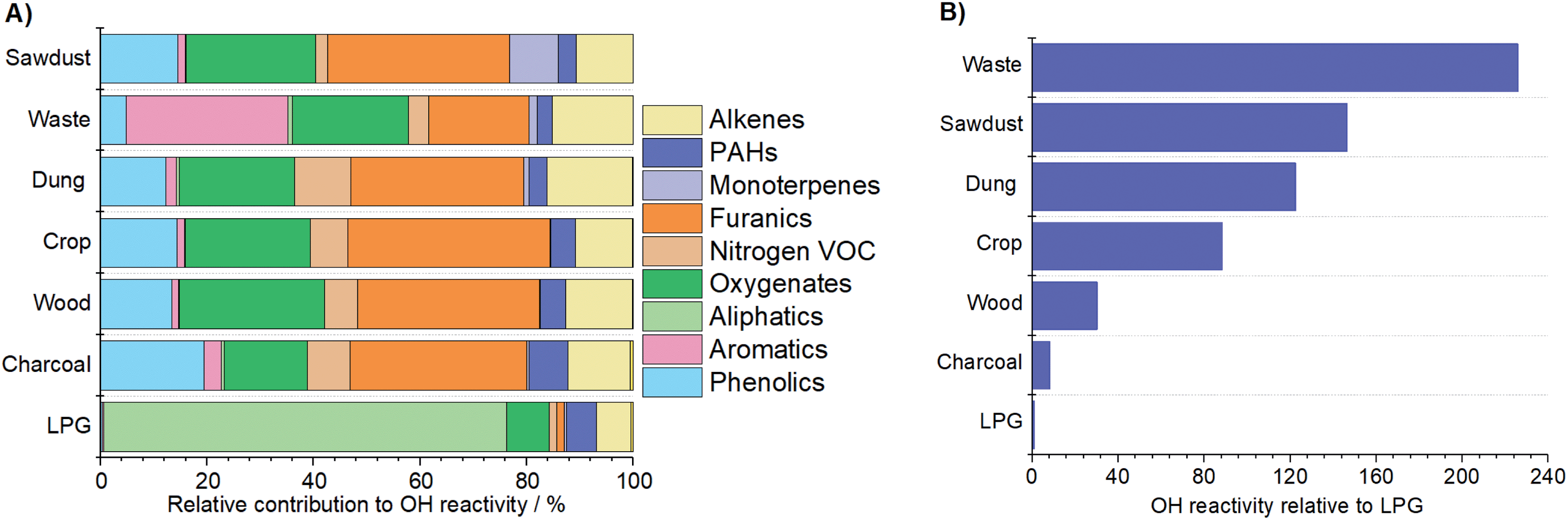 | ||
| Fig. 6 OH reactivity of emissions from different fuel types with (A) relative contribution to OH reactivity and (B) total OH reactivity of fuel types at the top of flue relative to LPG, which is set to 1. | ||
This study identified the species with the largest reactivity with the OH radical from Indian solid fuels. Ozone production from emissions when these fuels are combusted will be more complex and ultimately depend on NMVOC/NOx ratios, meteorology and solar radiation.25 Whilst the phenolic compounds here show relatively large contributions to OH reactivity (5–19%), their emission may both decrease or increase ambient O3. In NMVOC limited environments in India, a reduction in NOx could lead to an increase in O3.68 In NOx limited environments this may result in negative O3 formation due to the formation of nitrophenols, which reduces the amount of NO2 available for NMVOC oxidation.69
Gilman et al. (2015) calculated the relative contribution of different functionalities to the OH reactivity of fuel types from the U.S.21 The fuel types studied by Gilman et al. (2015) showed that alkenes contributed 25–29% of the OH reactivity, which was larger than found in this study (7–16%) for Indian fuels. The contribution to OH reactivity of OVOC for U.S. fuels (41–54%) was less than found in this study (45–76%). The contributions of monoterpenes for fuels from the U.S. were slightly larger (4–14%) than for those from India (0–7%). This was likely due to a greater contribution of monoterpene emitting fuel woods, such as pine, to fuels from the U.S. studied by Gilman et al. (2015). Both studies found a small contribution of aromatics (<5%) and nitrogen containing NMVOCs (<11%) to OH reactivity.
Fig. 6B shows the OH reactivity of each source at the top of the combustion chamber relative to LPG. This was calculated by multiplying the mean OH reactivity of flue gases by the volume of air sampled and normalising to the total reactivity of LPG. The OH reactivity of LPG was the lowest. Emissions from charcoal, fuel wood, crop residue, cow dung cake and sawdust were respectively ∼8, 30, 90, 120 and 150 times more reactive with OH than those from LPG. The OH reactivity of emissions from MSW was the greatest and approximately 230 times greater than from LPG. Fuel wood, cow dung cake and MSW burning are large NMVOC sources in India.70,71 The significantly greater OH reactivity of emissions from these sources is likely to substantially deteriorate local and regional air quality compared to users cooking over LPG.
3.5 PAH toxicity
A need has been identified to better understand the impact of PAHs from combustion sources in cities such as Delhi, where concentrations have been shown to be high and suggested to be enhanced by emissions from burning sources.72Fig. 7 shows that when comparing the toxicity of 21 PAHs released, fuel wood, crop residue, cow dung cake and MSW were respectively 20, 60, 130 and 220 times more toxic than LPG per kg of fuel burnt. Toxic emissions from these 21 PAHs released from LPG were small, and were principally driven by naphthalene (43%), fluoranthene (24%) and methylnaphthalenes (11%). The largest drivers for fuel wood/crop residue toxicity were benzo[a]pyrene (38%/48%), naphthalene (14%/11%) and benzo[b]fluoranthene (8%/8%), respectively. The contribution of naphthalene to the toxicity of cow dung cake and MSW was lower, with their toxicities driven by benzo[a]pyrene (49%/42%), dibenz[a,h]anthracene (13%/16%) and benzo[b]fluoranthene (8%/13%). The real-world effect of this toxicity would be significantly enhanced for fuel wood and cow dung cake, by around a further factor of 10. This is because significantly more fuel wood and cow dung cake fuel is used per user than LPG, due to the higher energy density of LPG and more efficient burning of this fuel.73,74 These results reinforced findings of other studies assessing the health benefits of LPG vs. solid fuels which suggested that to significantly reduce the impacts of combustion, a shift to cleaner cooking technologies was required.75–77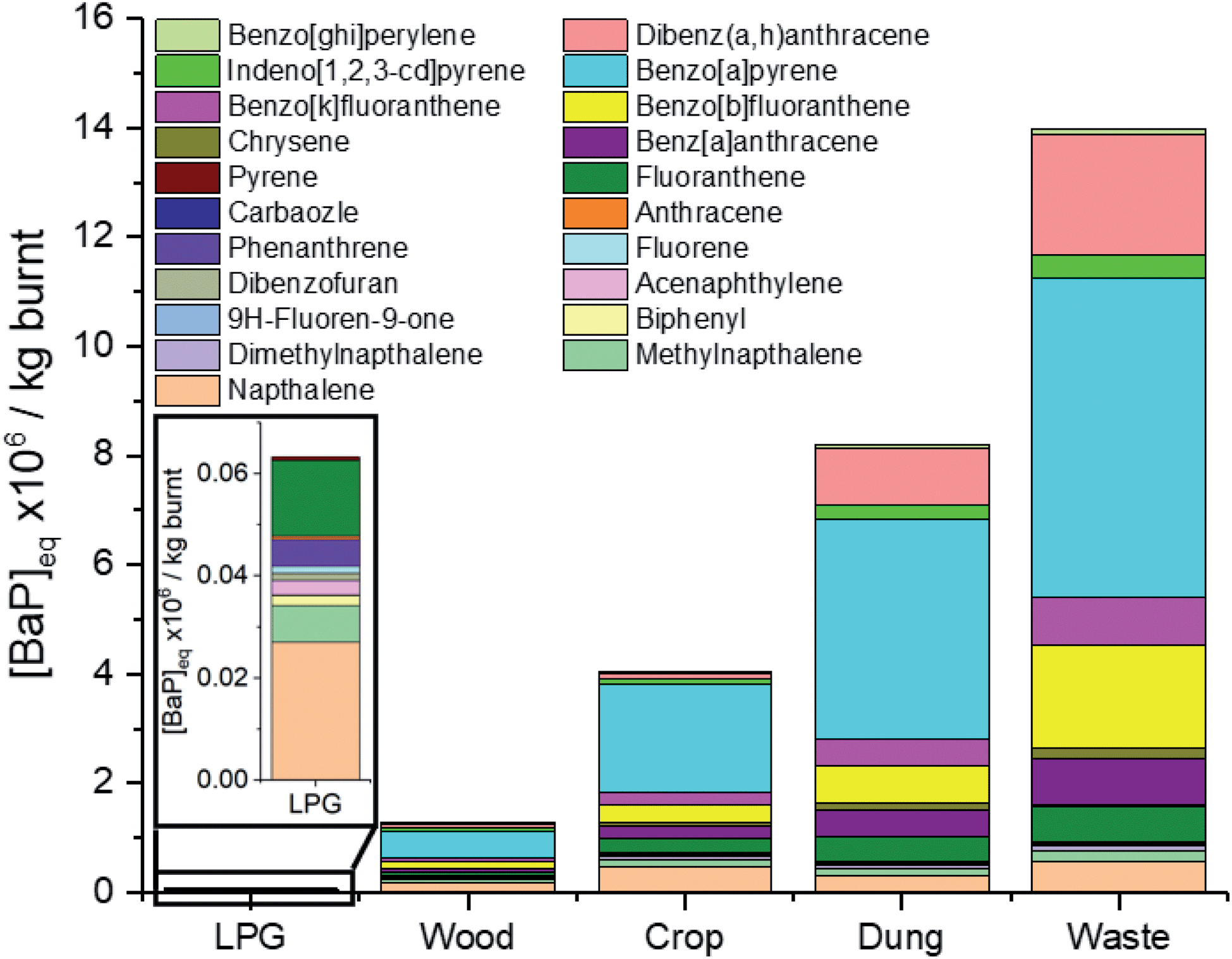 | ||
| Fig. 7 Comparison of PAH equivalent toxicity of fuel types. | ||
4. Conclusions
This study compiled comprehensive measurements of organic emissions from the combustion of a range of domestic fuels common to India. A range of detailed and complementary techniques allowed a VBS to be generated across a wide range of C* values. This highlighted that IVOC emissions should be better represented in models for an improved understanding of SOA production from emissions caused by domestic solid fuel combustion.The results estimated that phenolics and furanics were important to both the SOA production potential and the OH reactivity, respectively accounting for 10–70% and 9–58% of the SOA production potential and 5–22% and 9–48% of the OH reactivity of domestic biomass burning emissions. The contribution of smaller oxygenated species to the OH reactivity was also significant at 15–42%. Different combustion sources were compared, which showed that emissions from fuel wood, crop residue, cow dung cake and MSW burning were 30, 90, 120 and 230 times more reactive with the OH radical and that PAH emissions were 20, 60, 130 and 220 times more toxic than LPG, respectively. This also showed that NMVOCs released from the combustion of cow dung cake and MSW samples in this study resulted in ∼ 3–4 and 6–7 times more SOA production per kg burnt than fuel wood, respectively. This demonstrated that reduction of emissions from these sources is important to improve local and regional air quality across India.
Limited measurements were made from MSW, cow dung cake, crop residue and LPG samples in this study. Future studies are needed to improve the emission profiles of these domestic fuels to better understand the impact of burning emissions. This study also highlights that the C* of many species measured, including alkanes, remain uncertain and future studies are required to better understand the C* of these species. In addition, there have only been a limited number of chamber studies to determine the SOA formed during the oxidation of furanic species under high and low NOx conditions. More studies of the oxidation, and subsequent SOA formation, of these important domestic biomass burning emissions are required to better understand the impact of domestic solid fuel use on the atmosphere.
Author contributions
GJS developed volatility basis dataset, analysed I/SVOC fractions, conducted inventory comparison, developed SOA and OH reactivity models and lead paper. BSN, JRH and WJFA assisted with data analysis. SSMY contributed code to map of organics onto VBS. EN, ARR, JDL, TKM, RG and LKS contributed to data interpretation. JFH provided overall guidance for analysis and writing of paper. All authors contributed to the discussion, writing and editing of the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the Newton-Bhabha fund administered by the UK Natural Environment Research Council, through the DelhiFlux project of the Atmospheric Pollution and Human Health in an Indian Megacity (APHH-India) programme. The authors gratefully acknowledge the financial support provided by the UK Natural Environment Research Council and the Earth System Science Organization, Ministry of Earth Sciences, Government of India under the Indo-UK Joint Collaboration vide grant no. NE/P016502/1, NE/P01643X/1 and MoES/16/19/2017/APHH (DelhiFlux) to conduct this research. The paper does not discuss policy issues and the conclusions drawn in the paper are based on interpretation of results by the authors and in no way reflect the viewpoint of the funding agencies. GJS and BSN acknowledge the NERC SPHERES doctoral training programme for studentships. TKM is thankful to Director, CSIR-National Physical Laboratory, New Delhi for allowing to carry out this work. LKS acknowledges Physical Research Laboratory (PRL), Ahmedabad, India for the support and permission to deploy PTR-ToF-MS during the experimental campaign.References
- World Health Organisation, Household air pollution and health, accessed 05 Aug 2020, https://www.who.int/news-room/fact-sheets/detail/household-air-pollution-and-health Search PubMed.
- J. K. Kodros, E. Carter, M. Brauer, J. Volckens, K. R. Bilsback, C. L'Orange, M. Johnson and J. R. Pierce, Geohealth, 2018, 2, 25–39 CrossRef CAS.
- K. R. Smith, N. Bruce, K. Balakrishnan, H. Adair-Rohani, J. Balmes, Z. Chafe, M. Dherani, H. D. Hosgood, S. Mehta, D. Pope and E. Rehfuess, Annu. Rev. Public Health, 2014, 35, 185–206 CrossRef.
- Z. A. Chafe, M. Brauer, Z. Klimont, R. Van Dingenen, S. Mehta, S. Rao, K. Riahi, F. Dentener and K. R. Smith, Environ. Health Perspect., 2014, 122, 1314–1320 CrossRef CAS.
- World Bank, Tracking SDG 7: The Energy Progress Report 2020. Chapter 2: Access To Clean Fuels And Technologies For Cooking, International Bank for Reconstruction and Development, Washington, DC, 2020 Search PubMed.
- R. Mukhopadhyay, S. Sambandam, A. Pillarisetti, D. Jack, K. Mukhopadhyay, K. Balakrishnan, M. Vaswani, M. N. Bates, P. Kinney, N. Arora and K. Smith, Glob. Health Action, 2012, 5, 19016 CrossRef.
- G. J. Stewart, B. S. Nelson, W. S. Drysdale, W. J. F. Acton, A. R. Vaughan, J. R. Hopkins, R. E. Dunmore, C. N. Hewitt, E. G. Nemitz, N. Mullinger, B. Langford, Shivani, E. R. Villegas, R. Gadi, A. R. Rickard, J. D. Lee and J. F. Hamilton, Faraday Discuss., 2021 10.1039/d0fd00087f.
- L. Wang, J. G. Slowik, N. Tripathi, D. Bhattu, P. Rai, V. Kumar, P. Vats, R. Satish, U. Baltensperger, D. Ganguly, N. Rastogi, L. K. Sahu, S. N. Tripathi and A. S. H. Prévôt, Atmos. Chem. Phys. Discuss., 2020, 2020, 1–27 Search PubMed.
- J. M. Cash, B. Langford, C. D. Marco, N. Mullinger, J. Allan, E. Reyes-Villegas, R. Joshi, M. R. Heal, W. J. F. Acton, N. Hewitt, P. K. Misztal, W. Drysdale, T. K. Mandal, Shivani, R. Gadi and E. Nemitz, Atmos. Chem. Phys. Discuss., 2020 DOI:10.5194/acp-2020-1009.
- C. E. Stockwell, P. R. Veres, J. Williams and R. J. Yokelson, Atmos. Chem. Phys., 2015, 15, 845–865 CrossRef.
- L. E. Hatch, W. Luo, J. F. Pankow, R. J. Yokelson, C. E. Stockwell and K. C. Barsanti, Atmos. Chem. Phys., 2015, 15, 1865–1899 CrossRef CAS.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. de Gouw, Atmos. Chem. Phys., 2018, 18, 3299–3319 CrossRef CAS.
- G. J. Stewart, B. S. Nelson, W. J. F. Acton, A. R. Vaughan, N. J. Farren, J. R. Hopkins, M. W. Ward, S. J. Swift, R. Arya, A. Mondal, R. Jangirh, S. Ahlawat, L. Yadav, S. S. M. Yunus, C. N. Hewitt, E. G. Nemitz, N. Mullinger, R. Gadi, A. R. Rickard, J. D. Lee, T. K. Mandal and J. F. Hamilton, Atmos. Chem. Phys. Discuss., 2020 DOI:10.5194/acp-2020-860.
- G. J. Stewart, W. J. F. Acton, B. S. Nelson, A. R. Vaughan, J. R. Hopkins, R. Arya, A. Mondal, R. Jangirh, S. Ahlawat, L. Yadav, R. E. Dunmore, S. S. M. Yunus, C. N. Hewitt, E. Nemitz, N. Mullinger, R. Gadi, A. R. Rickard, J. D. Lee, T. K. Mandal and J. F. Hamilton, Atmos. Chem. Phys. Discuss., 2020 DOI:10.5194/acp-2020-892.
- N. M. Donahue, J. H. Kroll, S. N. Pandis and A. L. Robinson, Atmos. Chem. Phys., 2012, 12, 615–634 CrossRef CAS.
- X. Liu, L. G. Huey, R. J. Yokelson, V. Selimovic, I. J. Simpson, M. Müller, J. L. Jimenez, P. Campuzano-Jost, A. J. Beyersdorf, D. R. Blake, Z. Butterfield, Y. Choi, J. D. Crounse, D. A. Day, G. S. Diskin, M. K. Dubey, E. Fortner, T. F. Hanisco, W. Hu, L. E. King, L. Kleinman, S. Meinardi, T. Mikoviny, T. B. Onasch, B. B. Palm, J. Peischl, I. B. Pollack, T. B. Ryerson, G. W. Sachse, A. J. Sedlacek, J. E. Shilling, S. Springston, J. M. St. Clair, D. J. Tanner, A. P. Teng, P. O. Wennberg, A. Wisthaler and G. M. Wolfe, J. Geophys. Res.: Atmos., 2017, 122, 6108–6129 CAS.
- Z. C. J. Decker, K. J. Zarzana, M. Coggon, K.-E. Min, I. Pollack, T. B. Ryerson, J. Peischl, P. Edwards, W. P. Dubé, M. Z. Markovic, J. M. Roberts, P. R. Veres, M. Graus, C. Warneke, J. de Gouw, L. E. Hatch, K. C. Barsanti and S. S. Brown, Environ. Sci. Technol., 2019, 53, 2529–2538 CrossRef CAS.
- D. Sengupta, V. Samburova, C. Bhattarai, A. C. Watts, H. Moosmüller and A. Y. Khlystov, Atmos. Chem. Phys. Discuss., 2020, 2020, 1–50 Search PubMed.
- C. Y. Lim, D. H. Hagan, M. M. Coggon, A. R. Koss, K. Sekimoto, J. de Gouw, C. Warneke, C. D. Cappa and J. H. Kroll, Atmos. Chem. Phys., 2019, 19, 12797–12809 CrossRef CAS.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Rev. Geophys., 2017, 55, 509–559 CrossRef.
- J. B. Gilman, B. M. Lerner, W. C. Kuster, P. D. Goldan, C. Warneke, P. R. Veres, J. M. Roberts, J. A. de Gouw, I. R. Burling and R. J. Yokelson, Atmos. Chem. Phys., 2015, 15, 13915–13938 CrossRef CAS.
- A. T. Ahern, E. S. Robinson, D. S. Tkacik, R. Saleh, L. E. Hatch, K. C. Barsanti, C. E. Stockwell, R. J. Yokelson, A. A. Presto, A. L. Robinson, R. C. Sullivan and N. M. Donahue, J. Geophys. Res.: Atmos., 2019, 124, 3583–3606 CAS.
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS.
- A. Hartikainen, P. Yli-Pirilä, P. Tiitta, A. Leskinen, M. Kortelainen, J. Orasche, J. Schnelle-Kreis, K. E. J. Lehtinen, R. Zimmermann, J. Jokiniemi and O. Sippula, Environ. Sci. Technol., 2018, 52, 4979–4988 CrossRef CAS.
- M. M. Coggon, C. Y. Lim, A. R. Koss, K. Sekimoto, B. Yuan, J. B. Gilman, D. H. Hagan, V. Selimovic, K. J. Zarzana, S. S. Brown, J. M. Roberts, M. Müller, R. Yokelson, A. Wisthaler, J. E. Krechmer, J. L. Jimenez, C. Cappa, J. H. Kroll, J. de Gouw and C. Warneke, Atmos. Chem. Phys., 2019, 19, 14875–14899 CrossRef CAS.
- E. A. Bruns, I. El Haddad, J. G. Slowik, D. Kilic, F. Klein, U. Baltensperger and A. S. H. Prévôt, Sci. Rep., 2016, 6, 27881 CrossRef CAS.
- A. L. Robinson, N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce and S. N. Pandis, Science, 2007, 315, 1259–1262 CrossRef CAS.
- Y. Fujitani, K. Saitoh, A. Fushimi, K. Takahashi, S. Hasegawa, K. Tanabe, S. Kobayashi, A. Furuyama, S. Hirano and A. Takami, Atmos. Environ., 2012, 59, 389–397 CrossRef CAS.
- A. A. May, A. A. Presto, C. J. Hennigan, N. T. Nguyen, T. D. Gordon and A. L. Robinson, Atmos. Environ., 2013, 77, 128–139 CrossRef CAS.
- Q. Lu, Y. Zhao and A. L. Robinson, Atmos. Chem. Phys., 2018, 18, 17637–17654 CrossRef CAS.
- A. Hodzic, J. L. Jimenez, S. Madronich, M. R. Canagaratna, P. F. DeCarlo, L. Kleinman and J. Fast, Atmos. Chem. Phys., 2010, 10, 5491–5514 CrossRef CAS.
- M. C. Woody, K. R. Baker, P. L. Hayes, J. L. Jimenez, B. Koo and H. O. T. Pye, Atmos. Chem. Phys., 2016, 16, 4081–4100 CrossRef CAS.
- E. M. Lipsky and A. L. Robinson, Environ. Sci. Technol., 2006, 40, 155–162 CrossRef CAS.
- R. Ots, D. E. Young, M. Vieno, L. Xu, R. E. Dunmore, J. D. Allan, H. Coe, L. R. Williams, S. C. Herndon, N. L. Ng, J. F. Hamilton, R. Bergström, C. Di Marco, E. Nemitz, I. A. Mackenzie, J. J. P. Kuenen, D. C. Green, S. Reis and M. R. Heal, Atmos. Chem. Phys., 2016, 16, 6453–6473 CrossRef CAS.
- B. N. Murphy, M. C. Woody, J. L. Jimenez, A. M. G. Carlton, P. L. Hayes, S. Liu, N. L. Ng, L. M. Russell, A. Setyan, L. Xu, J. Young, R. A. Zaveri, Q. Zhang and H. O. T. Pye, Atmos. Chem. Phys., 2017, 17, 11107–11133 CrossRef CAS.
- S. H. Jathar, M. Woody, H. O. T. Pye, K. R. Baker and A. L. Robinson, Atmos. Chem. Phys., 2017, 17, 4305–4318 CrossRef CAS.
- A. A. May, N. T. Nguyen, A. A. Presto, T. D. Gordon, E. M. Lipsky, M. Karve, A. Gutierrez, W. H. Robertson, M. Zhang, C. Brandow, O. Chang, S. Y. Chen, P. Cicero-Fernandez, L. Dinkins, M. Fuentes, S. M. Huang, R. Ling, J. Long, C. Maddox, J. Massetti, E. McCauley, A. Miguel, K. Na, R. Ong, Y. B. Pang, P. Rieger, T. Sax, T. Truong, T. Vo, S. Chattopadhyay, H. Maldonado, M. M. Maricq and A. L. Robinson, Atmos. Environ., 2014, 88, 247–260 CrossRef CAS.
- Y. L. Zhao, N. T. Nguyen, A. A. Presto, C. J. Hennigan, A. A. May and A. L. Robinson, Environ. Sci. Technol., 2015, 49, 11516–11526 CrossRef CAS.
- Y. Zhao, N. T. Nguyen, A. A. Presto, C. J. Hennigan, A. A. May and A. L. Robinson, Environ. Sci. Technol., 2016, 50, 4554–4563 CrossRef CAS.
- A. A. Presto, N. T. Nguyen, M. Ranjan, A. J. Reeder, E. M. Lipsky, C. J. Hennigan, M. A. Miracolo, D. D. Riemer and A. L. Robinson, Atmos. Environ., 2011, 45, 3603–3612 CrossRef CAS.
- E. S. Cross, J. F. Hunter, A. J. Carrasquillo, J. P. Franklin, S. C. Herndon, J. T. Jayne, D. R. Worsnop, R. C. Miake-Lye and J. H. Kroll, Atmos. Chem. Phys., 2013, 13, 7845–7858 CrossRef.
- C. Venkataraman, G. Negi, S. Brata Sardar and R. Rastogi, J. Aerosol Sci., 2002, 33, 503–518 CrossRef CAS.
- T. Saud, T. K. Mandal, R. Gadi, D. P. Singh, S. K. Sharma, M. Saxena and A. Mukherjee, Atmos. Environ., 2011, 45, 5913–5923 CrossRef CAS.
- T. Saud, R. Gautam, T. K. Mandal, R. Gadi, D. P. Singh, S. K. Sharma, M. Dahiya and M. Saxena, Atmos. Environ., 2012, 61, 212–220 CrossRef CAS.
- D. P. Singh, R. Gadi, T. K. Mandal, T. Saud, M. Saxena and S. K. Sharma, Atmos. Environ., 2013, 68, 120–126 CrossRef CAS.
- N. J. Farren, N. Ramírez, J. D. Lee, E. Finessi, A. C. Lewis and J. F. Hamilton, Environ. Sci. Technol., 2015, 49, 9648–9656 CrossRef CAS.
- N. M. Donahue, A. L. Robinson, C. O. Stanier and S. N. Pandis, Environ. Sci. Technol., 2006, 40, 2635–2643 CrossRef CAS.
- A. A. Presto, C. J. Hennigan, N. T. Nguyen and A. L. Robinson, Aerosol Sci. Technol., 2012, 46, 1129–1139 CrossRef CAS.
- EPA, USEPA: Estimation Programs Interface Suite™ for Microsoft® Windows v 4.11, https://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface Search PubMed.
- J. D. McDonald, B. Zielinska, E. M. Fujita, J. C. Sagebiel, J. C. Chow and J. G. Watson, Environ. Sci. Technol., 2000, 34, 2080–2091 CrossRef CAS.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Environ. Sci. Technol., 2001, 35, 1716–1728 CrossRef CAS.
- E. Pettersson, C. Boman, R. Westerholm, D. Boström and A. Nordin, Energy Fuels, 2011, 25, 315–323 CrossRef CAS.
- OEHHA, Benzo[a]pyrene as a Toxic Air Contaminant, 1994 Search PubMed.
- N. Ramírez, A. Cuadras, E. Rovira, M. Marcé Rosa and F. Borrull, Environ. Health Perspect., 2011, 119, 1110–1116 CrossRef.
- S. Tomaz, P. Shahpoury, J.-L. Jaffrezo, G. Lammel, E. Perraudin, E. Villenave and A. Albinet, Sci. Total Environ., 2016, 565, 1071–1083 CrossRef CAS.
- A. Elzein, R. E. Dunmore, M. W. Ward, J. F. Hamilton and A. C. Lewis, Atmos. Chem. Phys., 2019, 19, 8741–8758 CrossRef CAS.
- L. F. Radke, D. A. Hegg, P. V. Hobbs, D. J. Nance, J. H. Lyons, K. K. Laursen, R. E. Weiss, P. J. Riggan and D. E. Ward, Particulate and trace gas emissions from large biomass fire in North America, in Global Biomass Burning: Atmospheric, Climatic, and Biospheric Implications, ed. J. S. Levine, The MIT Press, Cambridge, Massachusetts, 1991, pp. 209–216 Search PubMed.
- R. J. Yokelson, I. T. Bertschi, T. J. Christian, P. V. Hobbs, D. E. Ward and W. M. Hao, J. Geophys. Res.: Atmos., 2003, 108(D13), 8478 CrossRef.
- G. R. McMeeking, S. M. Kreidenweis, S. Baker, C. M. Carrico, J. C. Chow, J. L. Collett Jr, W. M. Hao, A. S. Holden, T. W. Kirchstetter, W. C. Malm, H. Moosmüller, A. P. Sullivan and C. E. Wold, J. Geophys. Res.: Atmos., 2009, 114, D19210 CrossRef.
- M. Kortelainen, J. Jokiniemi, P. Tiitta, J. Tissari, H. Lamberg, J. Leskinen, J. Grigonyte-Lopez Rodriguez, H. Koponen, S. Antikainen, I. Nuutinen, R. Zimmermann and O. Sippula, Fuel, 2018, 233, 224–236 CrossRef CAS.
- L. D. Yee, K. E. Kautzman, C. L. Loza, K. A. Schilling, M. M. Coggon, P. S. Chhabra, M. N. Chan, A. W. H. Chan, S. P. Hersey, J. D. Crounse, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2013, 13, 8019–8043 CrossRef.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2007, 7, 3909–3922 CrossRef CAS.
- A. W. H. Chan, K. E. Kautzman, P. S. Chhabra, J. D. Surratt, M. N. Chan, J. D. Crounse, A. Kürten, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2009, 9, 3049–3060 CrossRef CAS.
- C. M. Strollo and P. J. Ziemann, Atmos. Environ., 2013, 77, 534–543 CrossRef CAS.
- L. E. Hatch, R. J. Yokelson, C. E. Stockwell, P. R. Veres, I. J. Simpson, D. R. Blake, J. J. Orlando and K. C. Barsanti, Atmos. Chem. Phys., 2017, 17, 1471–1489 CrossRef CAS.
- L. T. Fleming, R. Weltman, A. Yadav, R. D. Edwards, N. K. Arora, A. Pillarisetti, S. Meinardi, K. R. Smith, D. R. Blake and S. A. Nizkorodov, Atmos. Chem. Phys., 2018, 18, 15169–15182 CrossRef CAS.
- EPA, Greenhouse gases from small-scale combustion devices in devloping countries: phase IIA household stoves in India, 2000 Search PubMed.
- Y. Chen, G. Beig, S. Archer-Nicholls, W. Drysdale, W. J. F. Acton, D. Lowe, B. Nelson, J. Lee, L. Ran, Y. Wang, Z. Wu, S. K. Sahu, R. S. Sokhi, V. Singh, R. Gadi, C. N. Hewitt, E. N. Nemitz, A. Archibald, G. McFiggan and O. Wild, Faraday Discuss., 2021 10.1039/D0FD00079E.
- A. Lauraguais, C. Coeur-Tourneur, A. Cassez, K. Deboudt, M. Fourmentin and M. Choël, Atmos. Environ., 2014, 86, 155–163 CrossRef CAS.
- C. Wiedinmyer, R. J. Yokelson and B. K. Gullett, Environ. Sci. Technol., 2014, 48, 9523–9530 CrossRef CAS.
- S. Sharma, A. Goel, D. Gupta, A. Kumar, A. Mishra, S. Kundu, S. Chatani and Z. Klimont, Atmos. Environ., 2015, 102, 209–219 CrossRef CAS.
- A. Elzein, G. J. Stewart, S. J. Swift, B. S. Nelson, L. R. Crilley, M. S. Alam, E. Reyes-Villegas, R. Gadi, R. M. Harrison, J. F. Hamilton and A. C. Lewis, Atmos. Chem. Phys., 2020, 14303–14319, DOI:10.5194/acp-20-14303-2020.
- NSSO, Household Consumption of Various Goods and Services in India 2011-2012, NSS 68th round, 2014 Search PubMed.
- NSSO, Energy Sources of Indian Households for Cooking and Lighting, 2011-12, NSS 68th Round, National Sample Survey Office, Ministry of Statistics and Programme Implementation, Government of India, 2015 Search PubMed.
- G. L. Simon, R. Bailis, J. Baumgartner, J. Hyman and A. Laurent, Energy Sustainable Dev., 2014, 20, 49–57 CrossRef.
- D. Pope, N. Bruce, M. Dherani, K. Jagoe and E. Rehfuess, Environ. Int., 2017, 101, 7–18 CrossRef CAS.
- S. Sambandam, K. Balakrishnan, S. Ghosh, A. Sadasivam, S. Madhav, R. Ramasamy, M. Samanta, K. Mukhopadhyay, H. Rehman and V. Ramanathan, EcoHealth, 2015, 12, 25–41 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ea00009d |
| ‡ Now at: School of Geography, Earth and Environmental Sciences, University of Birmingham, B15 2TT, Birmingham, UK. |
| This journal is © The Royal Society of Chemistry 2021 |