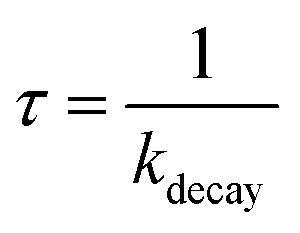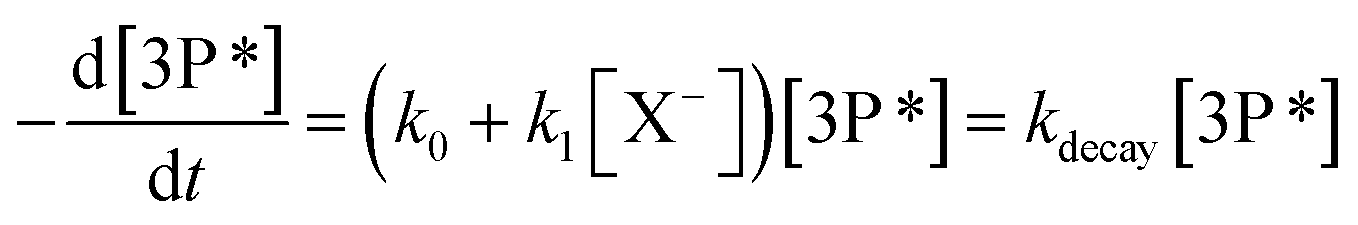Quenching of ketone triplet excited states by atmospheric halides
Received
26th October 2020
, Accepted 30th November 2020
First published on 23rd December 2020
Abstract
The photosensitized chemistry of three aromatic ketones (xanthone, flavone, and acetophenone) and also of secondary organic aerosol (SOA) arising from the photo-oxidation of naphthalene was investigated by means of transient absorption spectroscopy. Halide ions were selected to probe the reactivity of the generated triplet states. The quenching rate constants ranged from 109 M−1 s−1 with iodide ions to less than 105 M−1 s−1 with chloride ions. The halide-triplet state interactions produced the corresponding radical anion (X2˙−) along with halogenated and more oxidized organic compounds as identified by liquid chromatography and mass spectrometry. Deoxygenated naphthalene SOA solutions showed strong transient absorption at 420 nm when excited at 355 nm, and were also quenched by iodide ions similar to the single compound experiments indicating that compounds in naphthalene SOA can act as photosensitizers. Combining the study of these individual and known photosensitizers with those formed in the atmosphere (in this case through the oxidation of naphthalene) demonstrates that tropospheric photosensitization may involve a large variety of compounds of primary or secondary nature and will introduce new, unconsidered chemical pathways that impact atmospheric multiphase chemistry.
Environmental significance
Understanding the processing chain of tropospheric aerosols and radicals from sources to sinks is key for our ability to understand climate change. Numerous indicators, however, show that our knowledge is far from complete. Photosensitized reactions have been suggested to be an important atmospheric oxidation pathway; we therefore investigated the quenching rates of several triplet states of selected aromatic ketones relevant to the troposphere using halide ions as quenchers. We show here that this chemistry proceeds with fast rates. In addition, we also demonstrate, for the first time, that secondary organic aerosol produced from the oxidation of naphthalene also carry photosensitizing properties. This study provides knowledge for a quantitative assessment of these oxidation routes in air.
|
1. Introduction
The hydroxyl radical (˙OH) is certainly one of the dominant tropospheric oxidants in both gas and aqueous phases.1 However, besides the ˙OH radicals, a number of other oxidative pathways including nitrate radicals, ozone, halogenated radicals, singlet molecular oxygen, peroxyl radicals, peroxides, and triplet excited states of organic compounds (3C*) have also been shown to play a significant role in the troposphere.2,3
Very recently, the potential importance of triplet state chemistry has been unraveled for the atmospheric aqueous phase by novel ambient measurements of triplet state concentrations in fog and aerosol water, and by reactivity measurements under atmospheric conditions of various photosensitizers known to be present in the particulate matter.3–11 For instance, triplet excited states of aromatic ketones have been shown to oxidize phenols commonly found in biomass burning aerosol,12,13 and halide ions commonly found in sea spray aerosol.14 In the latter case, the halide ion oxidation induces the formation of halogen atoms (X˙) or radical anions (X2˙−),10 which are important reactive species for the oxidation of organic and inorganic compounds throughout the troposphere. Halide ions such as iodide, bromide, and chloride represent compounds of interest because of the high reactivity of their associated atomic radicals and because of their ubiquity.15–18
Here, we report the reactivity of common halide ions toward a subset of organic triplet states from three aromatic ketones: xanthone, acetophenone, and flavone. These compounds are present in different environmental compartments and were previously considered as aqueous phase photosensitizers.19 They are also present in the gaseous and particulate atmospheric phases. Of these three compounds, xanthone is more abundant in the particulate phase having a concentration of hundreds of pg m−3 in urban areas.20,21 Different primary and secondary sources of xanthone have been identified such as wood and agricultural waste burning, and combustion of diesel, gasoline, and city gas.22–26 Acetophenone has been detected in aerosols but it mainly partitions into the gas phase.22 Primary and secondary sources of acetophenone have also been identified, including wood combustion and diesel emissions.23,24 Flavone has been identified in biogenic emissions as flavonoids are known as plant-protective antioxidants.25 In a recent study, flavone has also been identified in biomass burning emissions.26
To answer the question whether direct emissions are the unique way of introduction of primary photosensitizing compounds into the troposphere, we also investigated the possibility of producing such compounds in situ in air. We previously showed that particulate phase chemistry induces the production of such secondary photosensitizers.27 Here, we extended this approach to secondary organic aerosol (SOA). Specifically, we have examined the triplet-state reactivity towards I− for a complex mixture of organic compounds in SOA produced by the photooxidation of naphthalene under high-NOx conditions. This naphthalene SOA has been shown to strongly absorb near-UV and visible radiation.28 UV irradiation of naphthalene SOA has been shown to result in an efficient photosensitized production of singlet oxygen,29 and reactivity towards limonene.30 Our working hypothesis was that a subset of compounds in naphthalene SOA has sufficiently high triplet state energies to oxidize halide ions.
This work aims to explore the effect of the triplet state chemistry of these compounds on the formation yield of reactive halogen species (X˙, X2˙−) and their kinetics. The identification of products from these aqueous photosensitized reactions also provides insights into the potential chemical mechanisms. It is noteworthy to point out that the chemistry investigated here is not only of importance for liquid particles, but also for the description of photochemical processes at the air/sea interface. As recently highlighted by our previous work, there is rich photochemistry taking place in the sea surface microlayer,31 which has now been demonstrated to involve PAHs and organic compounds of marine interest.32,33 Therefore, PAHs can also be efficient photosensitizers at the sea surface and in aerosols as well.
2. Experimental
2.1. Materials and solution preparation for single-component experiments
The chemicals used in this study – xanthone (97% Sigma-Aldrich), acetophenone (96% Sigma-Aldrich), flavone (97% Sigma-Aldrich), sodium chloride (>99% Sigma-Aldrich), sodium iodide (>99% Sigma-Aldrich), and sodium bromide (>99% Sigma-Aldrich) – were all used without further purification. All solutions were freshly prepared using 18 MΩ ultra-pure water. The photosensitizer was dissolved in water using magnetic agitation in the dark for two hours. Aqueous solutions containing ≈2 μM of photosensitizer were deoxygenated with a stream of argon for 20 min to avoid any interference or electron transfer with singlet oxygen. All the experiments are performed at a temperature maintained at 23 °C.
2.2. SOA preparation
Naphthalene SOA was prepared under high NOx conditions and 40% relative humidity (RH) in a 5 m3 Teflon batch chamber surrounded by 42 UV-B lamps. The chamber was equipped with a proton-transfer-reaction mass spectrometer to monitor naphthalene concentrations and a scanning mobility particle sizer to monitor particle concentrations and size distributions in the chamber. The NOx concentration and RH of the chamber were monitored using a Thermo Scientific 42i-Y and Vaisala HMT333 probe, respectively. The starting naphthalene, H2O2, and NOx mixing ratios were 0.4, 2, and 0.4 parts per million by volume (ppmv), respectively. After the naphthalene was added to the chamber and the concentration stabilized, H2O2 and NO were added and the UV-B lamps were turned on to initiate the photo-oxidation process (with a steady state OH concentration of ∼106 molecules per cm3). The OH steady-state concentration was determined from the rate at which the starting organic compound is removed from the chamber as measured by PTR-MS. Once the particle mass concentration peaked (∼3 h), the SOA was collected onto a poly(tetrafluoroethylene) (PTFE) filter (FGLP04700, Millipore, 47 mm diameter, 0.2 μm pore size). The filter was then extracted with HPLC-grade water to produce a 0.170 g ml−1 SOA solution. Aliquots of an aqueous KI solution were added to the SOA solution for quenching studies. All the samples were deoxygenated for 20 minutes before the transient absorption experiments, as mentioned for single compound photosensitizer experiments.
2.3. Laser transient absorption
The transient absorption spectra of the excited photosensitizers (xanthone, flavone, and acetophenone) were followed with a classical laser transient absorption apparatus.34 The third harmonic of an Nd:YAG laser (Surelite II 10, Continuum) was used as the excitation source. Based on the photosensitizers' absorption spectra (Fig. 1), the laser wavelength was set at 266 nm where the three photosensitizers absorb. The laser was operated in a single-shot mode, and its energy was set to ∼10 mJ per pulse. Using moderate energy helps to limit as much as possible the direct photolysis of the photosensitizer and therefore avoid possible interferents from its products. To get closer to the atmosphere conditions, another set of experiments was carried out at a laser wavelength of 355 nm. At this wavelength, only xanthone absorbs efficiently, but 1.7 times less than at 266 nm. Therefore, in this case a slightly higher laser energy was used (Fig. 1).
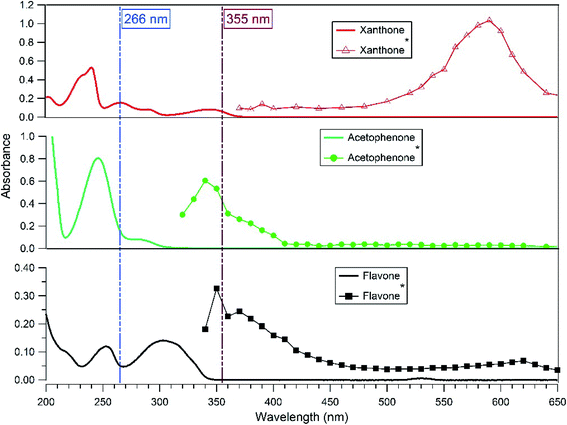 |
| | Fig. 1 Absorption and transient absorption spectra of solutions of xanthone, acetophenone and flavone. The solid line (—) shows the (UV-Vis) absorption spectra of solutions before the laser excitation. Absorption spectra recorded 330 ns after the 266 nm laser pulse of an aqueous solution of xanthone (-Δ-), acetophenone (-●-) and flavone (-■-) (10−2 mM). | |
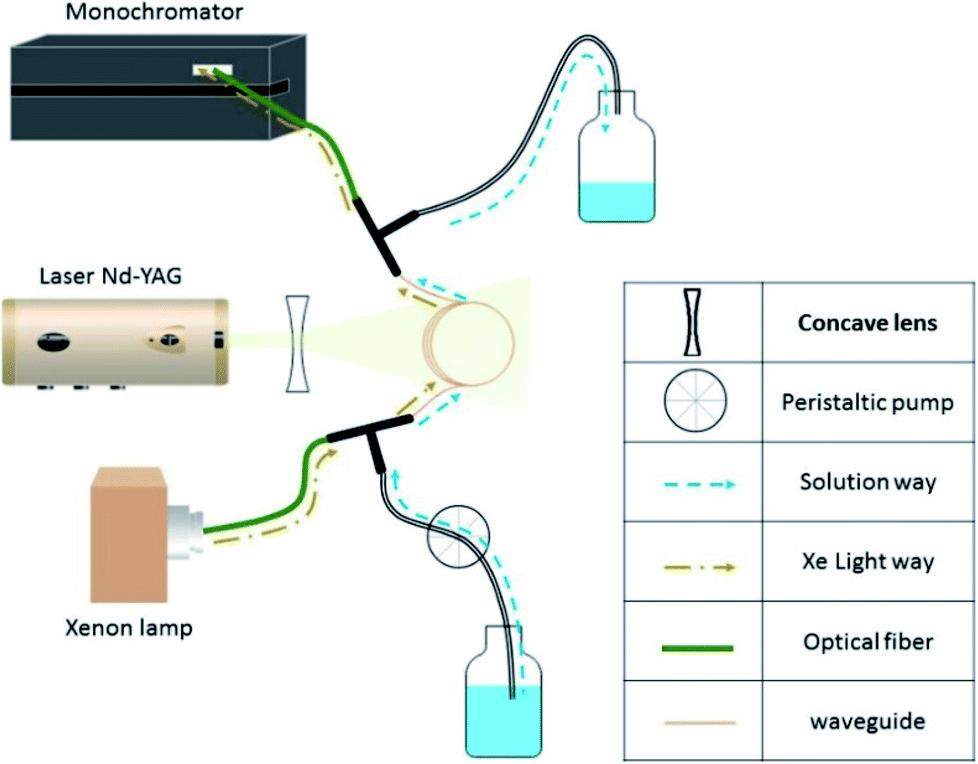
Fig. 2 illustrates the experimental setup. It consists of a standard laser transient absorption system but the reaction cell is replaced with a Teflon AF liquid waveguide. The laser output was shaped by a dispersing lens to irradiate homogeneously the outer surface of this 1 m long transparent waveguide wounded in a reel. Using a peristaltic pump, the solutions containing all reactants were flowing through the waveguide to constantly replenish their content between each laser shot (the residence time of the sample in the cell being less than one second). All connections were made from either glass or PTFE tubing. This is important to limit the exposure of the solution to repeated laser pulses to maintain a constant temperature in the flow cell and to reduce the degradation/photolysis of the solution. Transient species produced by the pulsed laser beam were monitored employing time-resolved absorption spectroscopy. A 150 W high-pressure xenon arc lamp was used as a broadband radiation source. The light from the lamp passed along the axis of the waveguide and the optical fibers and was collected then by a 1/4 m monochromator (Spectral Products DK240) equipped with a 2400 grooves per mm grating and coupled with a photomultiplier (PMT, Hamamatsu H7732-01). The output signal of the PMT passed through a high-speed current amplifier/discriminator (Femto) and the AC component recorded on a 300 MHz oscilloscope (Tektronix TDS3032c). Typically, signals from 512 laser pulses were averaged for each measurement. To construct the transient absorption spectrum, measurements were repeated every 10–15 nm between 300 and 650 nm. The full transient absorption spectrum was reconstructed from the steady and transient signals (330 ns after the laser pulse), and reported in a wavelength region free of spectral interferents from the ground state solution.
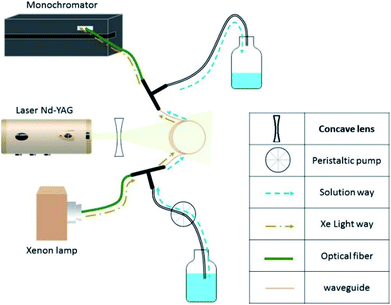 |
| | Fig. 2 Experimental setup for the laser flash photolysis. | |
The transient absorption experiments on naphthalene SOA solutions were conducted using a different laser transient absorption apparatus at the University of California, Irvine. The sample was placed in a standard 10 mm fused silica cuvette. Circulation was not used due to the limited amount of available sample (see below). The solution was excited at 355 nm (40 mJ per pulse) and the transients were probed using a similar xenon arc lamp, monochromator, and oscilloscope setup (averaging 40 pulses). Exciting the molecules at 355 nm ensured that the transients formed would be tropospherically relevant and would lead to photochemical aging of organic aerosols in the lower atmosphere.
2.4. Chemical characterization
Chemical analysis of the reaction products was performed in the case of xanthone. For this purpose, solutions were prepared, containing the same concentration of xanthone, without halide ions, with sodium iodide (0.89 mM) and finally with sodium bromide (0.1 M). Half of each solution was kept in the dark for reference, and the remaining half was exposed to laser pulses at 355 nm in the waveguide. These six solutions were then desalinated and concentrated by solid phase extraction (SPE) (Waters, Oasis MAX 6 cc Vac Cartridge, copolymer, 30 μm). The extracts were dried under a nitrogen flow and reconstituted in 1 ml of acetonitrile. The samples were analyzed by ultra-high performance liquid chromatography (UPLC, Dionex 3000, Thermo Scientific) coupled with a Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific) with an electrospray ionization source. The separation relied on a Water Acquity HSS C18 column (1.8 μL, 100 × 2.1 mm) at a flow rate of 0.3 ml min−1 with a mobile phase gradient using acidified water (eluent A: 0.1%, v/v, formic acid; Optima liquid chromatography (LC/MS), Fisher Scientific) and acidified acetonitrile (eluent B: 0.1%, v/v, formic acid; Optima LC/MS, Fisher Scientific).
In addition, we have examined the distribution of compounds found in naphthalene SOA using the same UPLC-Q-Exactive system equipped with a UV photo-detector array (PDA). The instrument was operated in the negative polarity mode. The most relevant compounds in SOA are the ones that strongly absorb near-UV radiation, and they were assigned by examining the correlation between chromatographic peaks detected by both PDA and mass spectroscopy detectors. In addition, we analyzed the chromatograms using Compound Discoverer to locate compounds with high abundance in the mass spectrum regardless of their light-absorption properties. All peaks were assigned to the formulae of [CcHhOoNn–H]−, where c, h, o, and n represent the corresponding number of atoms in the ions. Identified peaks met a sample![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :blank ratio greater than 5 and the formula assignments were limited to 30 C, 50 H, 20 O, and 10 N atoms with a maximum error of 5 ppm. When multiple formula possibilities existed, we picked compounds that could be conceivably derived from naphthalene. Assignments were not verified with standards, so the formulae cited below should be regarded as tentative.
:blank ratio greater than 5 and the formula assignments were limited to 30 C, 50 H, 20 O, and 10 N atoms with a maximum error of 5 ppm. When multiple formula possibilities existed, we picked compounds that could be conceivably derived from naphthalene. Assignments were not verified with standards, so the formulae cited below should be regarded as tentative.
3. Results
3.1. Triplet state transient absorption spectrum
The transient absorption spectrum recorded after the 266 nm laser pulse is characterized by a strong absorption peak with a maximum absorption wavelength of around 590 nm for xanthone, 340 nm for acetophenone, and 350 nm for flavone (Fig. 1). These wavelengths are in reasonably good agreement with previously published spectra as discussed below. The maximum absorption wavelength for excited xanthone and acetophenone depends on the nature of the solvent. In non-polar solvents, the n–π* transition triplet state is favored and the maximum wavelength absorption is at 600 nm for xanthone and 350 nm for acetophenone.35–39 As shown in Fig. 1, the triplet flavone transient absorption spectrum presents three peaks at 350, 370 (appearing as shoulders due to broad absorption and limited spectral resolution), and 620 nm. The highest one is at 350 nm and the weakest one is at 620 nm. These results are in good agreement with the work published by Avila and Previtali,40 who observed two absorption maxima at 370 nm (the most intense) and 620 nm, and these maxima were practically independent of the solvent used (benzene, ethanol, and acetonitrile). For the determination of lifetimes and quenching rate coefficients, the triplet state decay was monitored at the wavelengths reported above.
3.2. Characteristics of the photosensitizer triplet state
Laser flash photolysis of deoxygenated solutions containing the selected photosensitizers showed strong transient absorptions as shown in Fig. 3A for flavone. Over the 0–7 μs range, the absorption decay was fitted with a single exponential decay with an offset (eqn (1)) (Fig. 3B). The triplet state lifetime without quenchers was thus calculated following eqn (2) where kdecay is the observed rate constant.| | 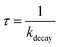 | (2) |
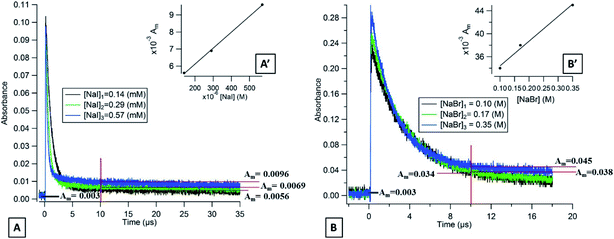 |
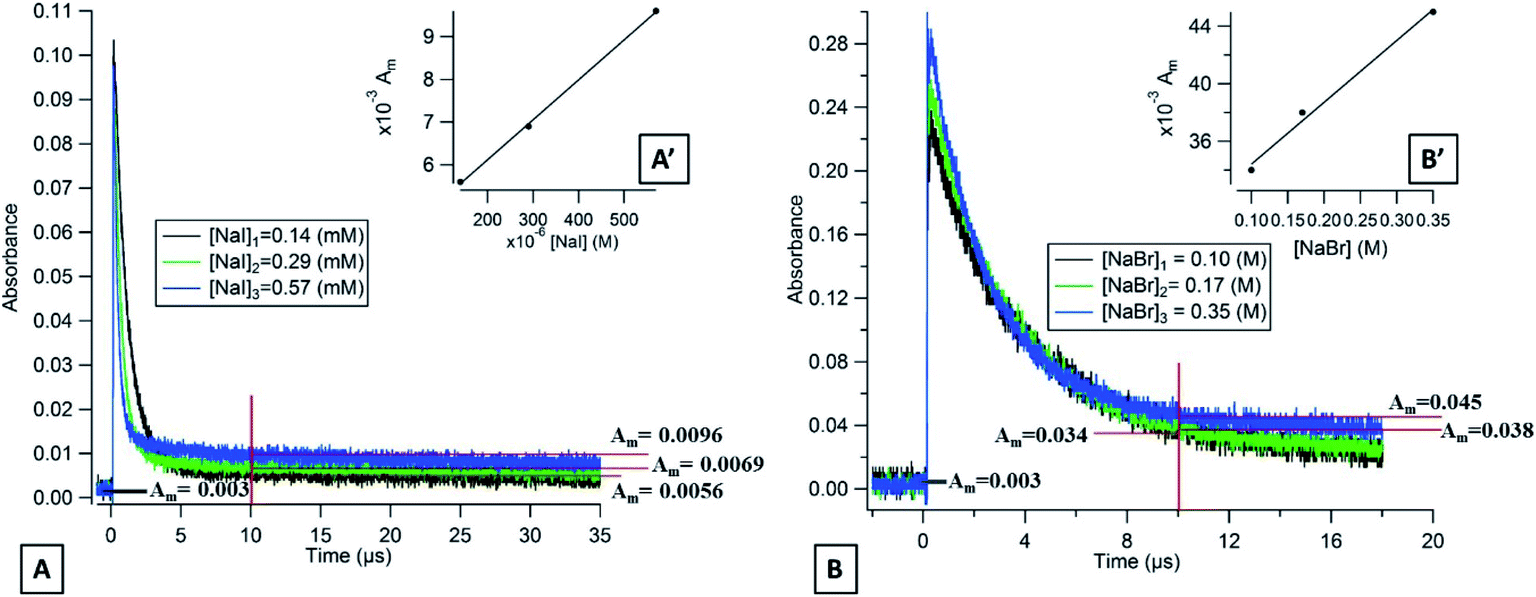
| | Fig. 3 (A) Transient absorption decays of flavone triplet states in deoxygenated aqueous solutions containing different concentrations of NaI observed at 390 nm and (B) with different concentrations of NaBr observed at 350 nm. | |
These constants in deoxygenated solutions are presented in Table 1. For the three compounds, the lifetime of the triplet state ranged from 1.85 μs for acetophenone to 5.5 μs for flavone. These values are in agreement, with triplet state excitation lifetimes reported in the literature for a large variety of compounds, which are in the range of 10−7 to 1 s.41 The triplet lifetimes of cyclic ketones vary with the ring size and with the degree of substitution; for cyclohexanone it varies between 0.6 and 40 ns.42 In the presence of oxygen, the decay became faster indicating a quenching by oxygen, probably by energy transfer producing reactive oxygen species such as singlet oxygen.43
Table 1 First-order rate constants and triplet lifetimes in the absence of quenchers. The ground state reduction potential E°, the triplet state energy ET, and the triplet state reduction potentials E°* of the three photosensitizers are also given61–66
|
|
k
decay (μs−1) 266 nm |
τ (μs) 266 nm |
k
decay (μs−1) 355 nm |
E° (V) |
E
T (V) |
E°* (V) |
| Xanthone |
0.44 ± 0.02 |
2.27 |
0.43 ± 0.03 |
−1.21 (63) |
3.17 (64) |
1.96 |
| Acetophenone |
0.54 ± 0.04 |
1.85 |
|
−1.42 (65) |
3.19 (66) |
1.77 |
| Flavone |
0.18 ± 0.02 |
5.5 |
|
−1.18 (67) |
2.69 (68) |
1.51 |
3.3. Triplet state quenching rate constant
We investigated the quenching of the selected photosensitizers by halide ions. Different concentrations of NaI, NaBr, and NaCl were added into the deoxygenated aqueous solutions of the different photosensitizers. Their decay became faster with increasing concentration. This chemistry is expected to produce atomic halogens according to (R1) in the next mechanism:14| | | 3P* → quenching by solvent | (R-0) |
| | | X2˙− + X2˙− → X2 + 2X− | (R-5) |
Under pseudo-first-order conditions, the quenchers are added in excess compared to the triplet state, and the quenching rate coefficients can be determined by the following Stern–Volmer equation (eqn (3)):
| | 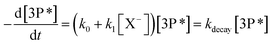 | (3) |
In
eqn (3),
k0 corresponds to the rate constant of the triplet state decay in the absence of oxygen and other quenchers in pure water, while
k1 corresponds to the quenching rate constant by the halide X
− in reaction
(1).
41 For a constant concentration of photosensitizer,
k1 corresponds to the slope of the curve obtained from the plot of the observed rate coefficient as a function of quencher concentration. The different quenching rate coefficients are summarized in
Table 2.
Table 2 Reaction (1) rate constants k1 for quenching of ketone triplet states by different halide ions following excitation at 266 nm and 355 nm, redox potentials (E(X−)/X) for the anions and the free energy of the electron transfer ΔGET. Values were obtained with 3 experimental repetitions61–66
|
|
I− |
Br− |
Cl− |
|
E(X−/X) (V) |
0.535 |
1.087 |
1.36 |
|
|
ΔGET (V) |
k
I (M−1 s−1) |
ΔGET (V) |
k
Br (M−1 s−1) |
ΔGET (V) |
k
Cl (M−1 s−1) |
| Xanthone (266 nm) |
−1.425 |
(8.34 ± 0.43) × 109 |
−0.873 |
(6.46 ± 0.01) × 108 |
−0.6 |
≤105 |
| Xanthone (355 nm) |
(1.86 ± 0.82) × 109 |
(1.06 ± 0.23) × 107 |
≤05 |
| Acetophenone (266 nm) |
−1.235 |
(3.47 ± 0.28) × 109 |
−0.683 |
(9.89 ± 0.6) × 106 |
−0.41 |
≤105 |
| Flavone (266 nm) |
−0.975 |
(4.85 ± 0.35) × 109 |
−0.423 |
(1.28 ± 0.28) × 105 |
−0.15 |
≤105 |
The ground state reduction potential E° and the triplet state energy ET for the different photosensitizers studied are presented in Table 1. The triplet state reduction potential E°* is the sum between ET and E°. Using these values, we can calculate the free energy (ΔG) of one electron transfer between the excited photosensitizer and the quencher (eqn (4)) presented in Table 2. The deactivation of the excited photosensitizer by reaction (R1) will take place spontaneously only if ΔGET is negative. This quantity may be calculated using the Rehm–Weller equation:44
| | | ΔGET = nF(E(X−/X) − E° − ET) + C | (4) |
nF is the total charge transferred during the reaction. The entropic variation and the coulombic term (
C) are considered here as negligible. If we consider each photosensitizer individually and compare Δ
GET with different halides, we note that thermodynamically, the electron transfer reactions are the most favorable, as expected, with iodine having a Δ
GET ranging from −0.975 to −1.425 (eV). In the presence of bromide and chloride, the electron transfer is thermodynamically less favorable than for iodide with Δ
GET values ranging from −0.873 to −0.423 (eV) and −0.6 to −0.15 (eV), respectively. These results may explain the different measured values of
k1 for the same photosensitizer with the different halide ions. And indeed, efficient quenching assumed to correspond to a rapid electron transfer, has been measured for iodide, but with somewhat lower rates for the bromide, and a much slower rate for chloride.
If we compare the measured values of k1 for the different photosensitizers with the same halide ion, we observed that in all cases, xanthone presents the highest values for k1. This can be explained by considering the triplet state reduction potential, E°*, which is a critical value for the kinetics of electron transfer reactions.45 The E°* for xanthone is the highest, compared to those of acetophenone or flavone, and corresponds to 1.96, 1.77, and 1.51 V, respectively.44 This observation has been proven in the literature where two studies show that the E°* influences and controls the kinetics of the quenching oxidation reaction.46,47 Based on the same concept and comparing the measured values obtained for acetophenone to those obtained for flavone, we note that E°* can explain the differences observed with bromide where acetophenone k1 is almost a 100 times higher than k1 of flavone. In the case of iodide, a different trend is observed with flavone k1 being 1.4 times higher than acetophenone k1. This difference, a factor 10 to 100 with bromide while a factor of 1.4 for iodide, can be due to the diffusion effect in the solution which plays a limiting role in this case. Fig. 3A shows the transient absorption spectrum of acetophenone in the presence of I− at 330 ns after the laser shot. The maximum absorption corresponds to 350 nm, which is slightly higher than the value obtained for acetophenone in the absence of any quencher (340 nm). This shift may correspond to the formation of an absorbing transient compound (see below) produced rapidly after the laser discharge. Consequently, the underlying decay kinetics are affected and more uncertain.
These measurements have also been carried out, for xanthone, at an excitation wavelength of 355 nm. At this wavelength, there is still a pronounced reactivity with the halide ions, which is in the same order of magnitude with iodide whereas with bromide the kinetics is more than 50 times slower than at 266 nm (Table 2).
3.4. Transient absorption spectrum of transient species and products
Based on reactions (R1) and (R2), iodine and bromine atoms are expected to be produced after the quenching. While no transient compounds were observed in the absence of photosensitizers, the situation appeared more complex with added iodide or bromide ions in the solution. Indeed, after the laser discharge, the transient absorption should return to its initial background absorption (A0) due to the relaxation of the triplet state. The time needed to return to the initial background absorption depends on the lifetime of the triplet and the presence of other quenchers. When the absorption does not return to the A0 level, or does so over much longer timescales that are expected for triplet state reactions, it indicates the formation of a transient or new product.
3.4.1. Flavone.
In the presence of NaI and NaBr, the absorption decay of the flavone triplet does indeed not return to its background value, indicating the presence of transient species. Fig. 3 shows the flavone absorption decays at different concentrations of NaI at 390 nm (Fig. 3A) and at different concentrations of NaBr at 350 nm (Fig. 3B). We observe that the long-lived absorption values at this wavelength are a function of the halide concentrations. It is important to note that the maximum intensity of this long-lived absorption is observed at 390 nm for NaI and at 350 nm for NaBr and that lower values are observed at wavelengths of 20 nm around these wavelengths. Based on the literature, for NaI solutions, this maximum absorption wavelength could be assigned to the radical I2˙− exhibiting absorption bands at 385 and 725 nm with an extinction coefficient of 9500 and 3000 l mol−1 cm−3, respectively.48 Our system did not allow us to detect the second peak at 725 nm because of its lower intensity. For the NaBr solutions, the maximum absorption wavelength at 350 nm would correspond to Br2˙− having an absorption band at 354 nm with an extinction coefficient of 9900 l mol−1 cm−3.49
If we consider the extinction coefficient of I2˙− at 390 nm (9000 l mol−1 cm−3) and for Br2˙− at 350 nm (9900 l mol−1 cm−3), we can calculate that the yield of I2˙− is 239 times higher than the one for Br2˙− at 10 μs after the laser pulse, which is consistent with our results presented above.
Flavone presents the lowest values of triplet state energy and triplet state reduction potentials compared to acetophenone and xanthone. In consequence, it should lead to the lowest halide ion oxidation rates.
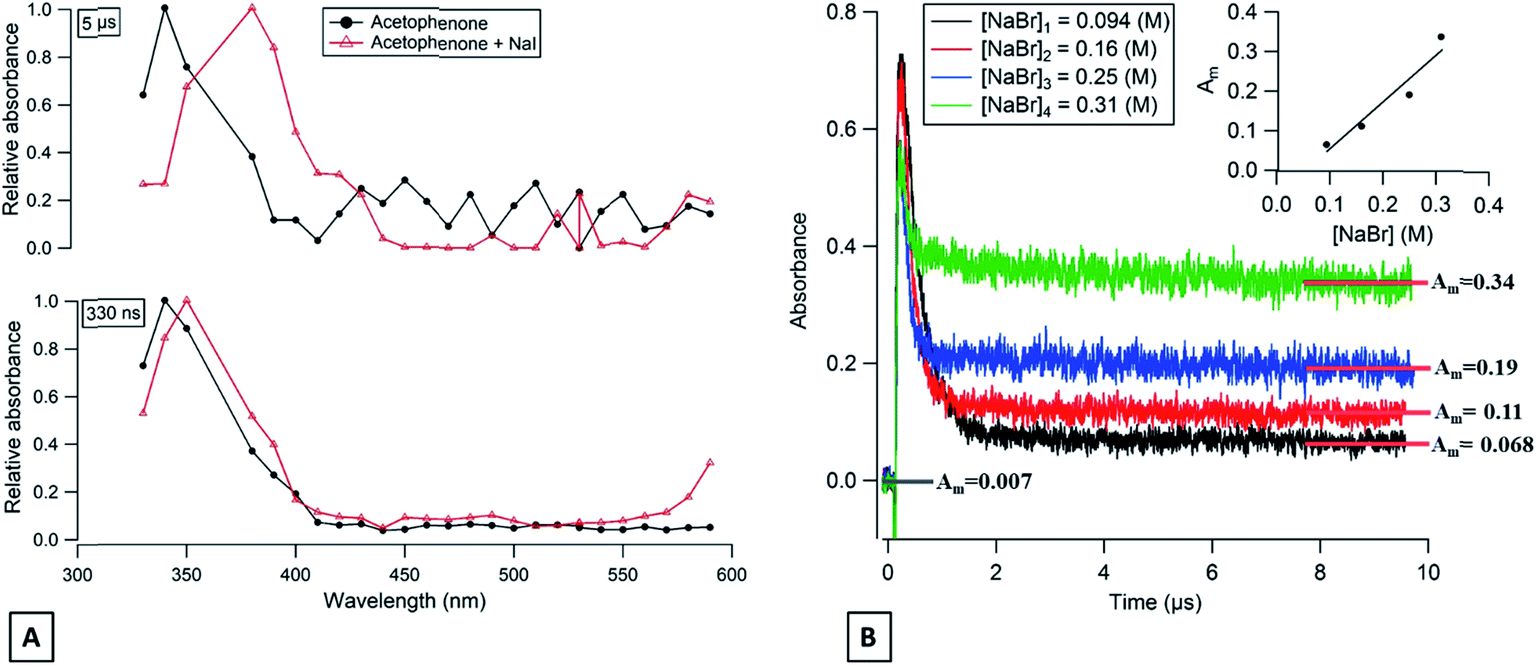
3.4.2. Acetophenone.
For acetophenone solutions in the presence of halide ions (Br− and I−), transient products have also been detected. A comparison of the transient absorption spectra, between acetophenone in pure water or in the presence of NaI, at 330 ns and 5 μs after the laser discharge is shown in Fig. 4A. It can be seen that a new absorption band at 390 nm appears a few μs after the laser discharge for the solution containing NaI. The acetophenone triplet lifetime is expected to last for 1.85 μs and therefore the observed absorption band at 5 μs is unlikely to be explained by the acetophenone triplet state but could be attributed to I2˙−.
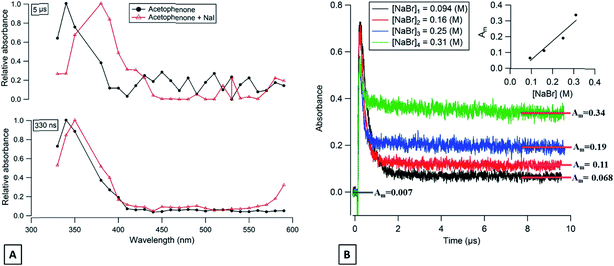 |
| | Fig. 4 (A) Transient absorption spectra of deoxygenated acetophenone solution (red triangles) and deoxygenated acetophenone with NaI (black circles) at 330 ns (bottom) and at 5 μs (top) after the laser shot. (B) Transient absorbance decays of acetophenone triplet states in aqueous solutions containing different concentrations of NaBr observed at 350 nm. | |
The long-lived absorption at 350 nm for acetophenone solutions in the presence of a different concentration of NaBr is presented in Fig. 4B. In this case, the long-lived absorption is more pronounced than for flavone for the same NaBr concentration range. To quantify the yield of Br2˙− formed and to compare with the results obtained with flavone, the correlation between the absorption at 10 μs and the different concentrations of NaBr is presented in Fig. 4B′. A simple first-order fit for these data gives a slope of about 1.2, i.e., corresponding to 6 times more Br2˙− production with acetophenone than with flavone. This observation could be explained by the different values of triplet state energies. The acetophenone has ET higher than that for flavone and slightly higher than ET for xanthone, while a value of triplet state reduction potential is in between the two values of other photosensitizers.
3.4.3. Xanthone.
Because of technical limitations, the transient absorption spectra of xanthone alone, xanthone with NaI and xanthone with NaBr, at 330 ns and at 5 μs after the laser pulse, start at 370 nm in Fig. 5A and A′ (no signal is detected for wavelength < 370 nm). Approximately 5 μs after the laser pulse, half of the absorbance at 590 nm decreases in the absence of any quencher, while a new absorbance appears with a maximum at 390 nm for a solution containing NaI. Another weaker peak appears around 700 nm. The ratio of the absorbance (0.56/0.19) of these two peaks (390/700) equals the ratio of the extinction coefficients (6/1.8) of these two peaks from the I2˙− spectrum48 confirming the formation of these transient radicals. For the solutions containing NaBr, the new absorbance band appears around 370 nm but we cannot confirm the wavelength of the maximum absorbance.
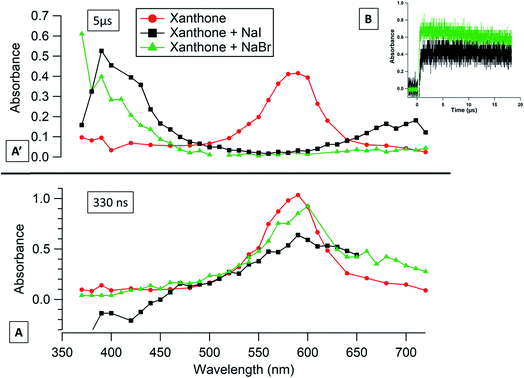 |
| | Fig. 5 Transient absorption spectra of deoxygenated xanthone solution (red circles), deoxygenated xanthone with NaI solution (black squares) and deoxygenated xanthone with NaBr solution (green triangles) (A) at 330 ns (bottom) and (A′) at 5 μs (top) after the laser shot. (B) Transient absorption decays of xanthone triplet states in a deoxygenated aqueous solution, in black containing NaI observed at 390 nm, and in green containing NaBr observed at 370 nm. | |
Fig. 5B shows the absorbance as a function of time at 370 nm for xanthone in a 0.21 M NaBr solution and at 390 nm for xanthone in a 0.56 M NaI solution. An absorbance at 370 nm and at 390 nm was seen in the literature, showing the formation of these anion radicals.49,50 Taking into account the extinction coefficient of Br2˙− at 370 nm (9200 l mol−1 cm−3), the concentration of NaBr in the solution and the values of long-lived absorbance, we can conclude that photoexcited xanthone can oxidize Br− to Br˙, followed by the formation of Br2˙− 4 times more efficiently than acetophenone and 24 times more efficiently than flavone. Likewise, xanthone shows a high capacity to oxidize iodide, up to 42 times larger than for flavone. Compared to the other photosensitizers, xanthone has the highest E°*, the highest ΔGET, and the fastest kinetics with halides, which can explain the behavior of this photosensitizer.
3.5. Chemical characterization
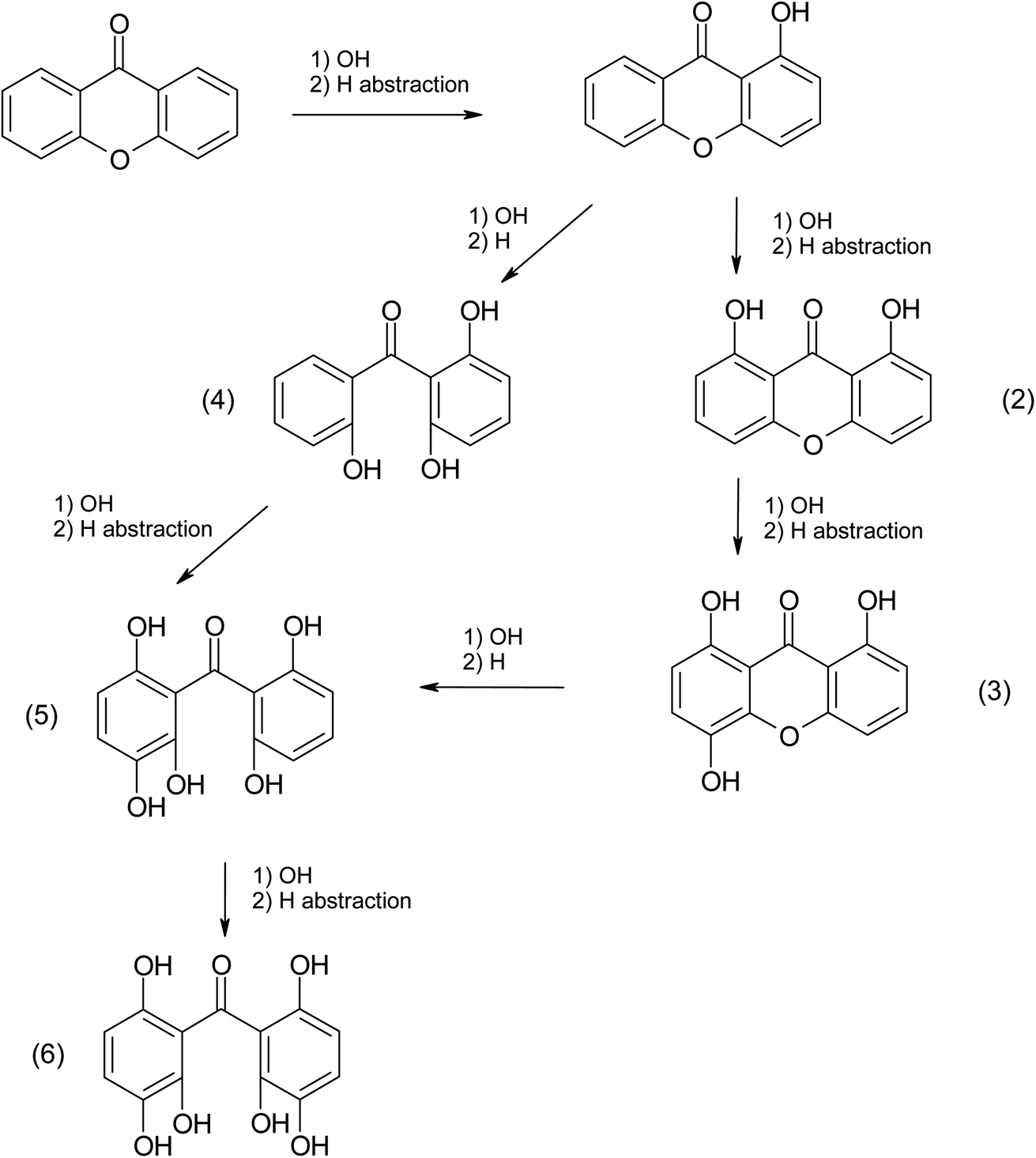
Based on (R-3) and (R-4), electron transfer and H abstraction reactions are expected due to the reaction between the radicals X˙, X2˙− and the surrounding organic compounds. As a result, halogenated products should be detected in the aqueous phase and some of them may escape to the gas phase.51 Previous studies have detected the formation of halogenated products and X2 in the gas phase from photosensitized reactions.14,52 In this study, we could only examine the products present in the aqueous phase.
As the solutions were not degassed, in the absence of halide ions, the products after the laser discharge correspond to chemistry involving oxygen (and peroxy radicals).53,54 Using the UPLC-MS analyses, the molecular formulae were identified based on their molecular masses and isotopic abundance. Without tandem analysis (MS-MS) only the empiric formula could be confirmed (Table 3).
Table 3 Compounds identified (D, detected; ND, not detected) in the presence of xanthone and halide ions upon irradiation (I) or in darkness (NI) by UHPLC-MS: theoretical m/z, measured m/z, empirical formula, and normalized peak abundances
|
|
Normalized peak abundances |
| Theoretical mass-to-charge ratio (m/z) |
Measured mass-to-charge ratio (m/z) |
Empirical formula |
Xanthone (NI) |
Xanthone (I) |
Xanthone + NaI (I) |
Xanthone + NaBr (I) |
| 197.0603 |
197.19 |
C13H8O2 |
1 |
1 |
1 |
1 |
| 223.0759 |
223.08 |
C15H10O2 |
ND |
2.1 × 10−1 |
1.8 × 10−1 |
1.9 × 10−1 |
| 229.0501 |
229.0511 |
C13H8O4 |
ND |
4.3 × 10−2 |
7.2 × 10−3 |
3.8 × 10−4 |
| 245.0450 |
245.0462 |
C13H8O5 |
ND |
1.7 × 10−2 |
1.4 × 10−3 |
1.3 × 10−4 |
| 247.0606 |
247.05 |
C13H10O4 |
ND |
7.1 × 10−3 |
3.0 × 10−3 |
5.0 × 10−5 |
| 263.0556 |
263.0569 |
C13H10O6 |
ND |
1.5 × 10−2 |
2.5 × 10−3 |
1.4 × 10−4 |
| 279.0504 |
301.03 |
C13H10O7 |
ND |
1.4 × 10−2 |
4.3 × 10−3 |
1.5 × 10−4 |
| 341.9389 |
341.93 |
C12H7O4I |
ND |
ND |
7.9 × 10−4 |
ND |
| 372.9573 |
372.95 |
C13H9O5I |
ND |
ND |
1.5 × 10−3 |
ND |
| 421.2331 |
421.2353 |
C21H41I |
ND |
ND |
5.6 × 10−4 |
ND |
Only xanthone (m/z 197.19) was identified in the non-irradiated sample, however, six new products were identified in the irradiated samples in the presence or absence of halide ions. The most abundant product was C15H10O2, and it represents the only product that has not gained additional oxygen atoms. This molecule could be simply the combination of an excited xanthone with the C2H2 fragment produced from the photolysis. Five molecules gaining two or more oxygens have been identified in the three irradiated samples. These molecules are the most abundant in the samples without halide ions. This could be due to the concentration of oxygen in the solution, by increasing the concentration of salt in a solution the solubility of the oxygen decreases, which explains the decrease in the abundance of these products with increasing salt concentration ([NaBr] > [NaI]).55 This observation can also show that oxygen is the limiting reagent in this photosensitized reaction. Organo-iodide molecules were detected only in the irradiated sample containing NaI. While no molecule containing bromine could be identified, it can be due to an insufficient concentration of NaBr to produce a detectable concentration by the instrument. Some of the observations presented above can be explained by the mechanism depicted in Scheme 1, which only shows the first oxidation steps. In fact, the production of halogenated and organic radicals induces a rich and complex chemistry where a multitude of radical recombination reactions may take place. These reactions would correspond to the halogen–halogen or organic radical recombination reactions as shown by Roveretto et al.,52 explaining some of the products listed in Table 3.
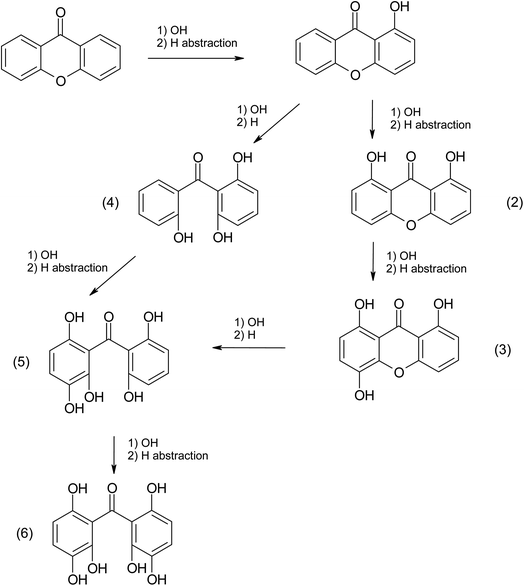 |
| | Scheme 1 Xanthone photosensitized oxidation mechanism. | |
3.6. Naphthalene SOA composition
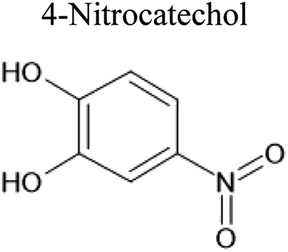
An aqueous solution of naphthalene SOA was analyzed using a UPLC-PDA high resolution mass spectrometer to help identify potential photosensitizers. The PDA chromatogram (Fig. 6) showed several absorbing species in the 300–500 nm spectral region, and these peaks correlated well with peaks appearing in the total ion chromatogram (TIC), which allowed us to assign the absorbing species to specific ions. The neutral formulae of the chromophores are listed beside each of their mass spectrometry chromatograms (Fig. 6) and include multiple nitrogen containing species such as nitro-catechol (C6H5O4N), nitro-naphthol (C10H7O3N), and dinitro-naphthol, one isomer of which is known as Martius yellow (C10H6O5N2). Most of these compounds were previously observed in naphthalene photooxidation by Kautzman et al.56 The ten most prominent compounds found by the Compound Discoverer (regardless of their absorbing characteristics) are shown in Table 4, and are also dominated by nitrogen containing compounds. Although much lower in intensity, we also found smaller amounts of compounds with the same formula as acetophenone (0.3% of the sum of all assigned compound intensities).
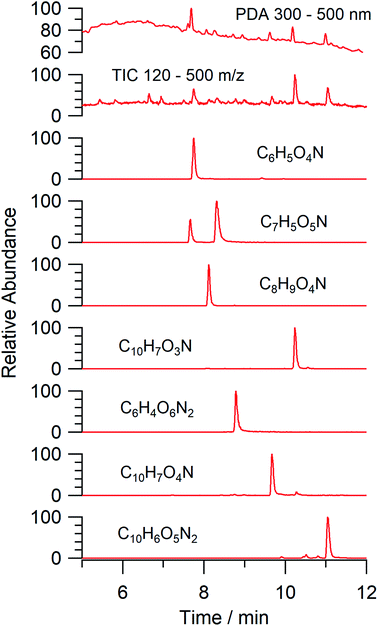 |
| | Fig. 6 A PDA chromatogram of naphthalene SOA integrated between 300 and 500 nm shows multiple absorbing species. TIC of molecular species between m/z 120–500 overlap with peaks in the PDA chromatogram affording molecular formula assignment to major chromophores. The neutral formulae of the chromophores are listed beside each of their single ion chromatogram. | |
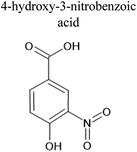
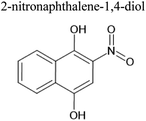
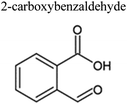
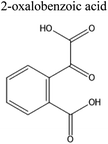
Table 4 Ten most prominent NAPH/OH/NOx compounds (in descending order of abundance) from UPLC-PDA-MS analysis of the SOA solution. Proposed structures are also given but are not confirmed as no standards or MS-MS were conducted on the sample
| MW |
Compound formula |
Representative structure with this formula |
Retention time/min |
Measured m/z |
Delta mass/ppm |
| 189 |
C10H7O3N |
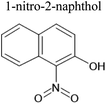
|
10.24 |
188.03491 |
2.14 |
| 234 |
C10H6O5N2 |
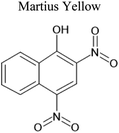
|
11.05 |
233.01987 |
2.25 |
| 155 |
C6H5O4N |
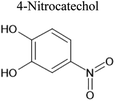
|
7.74 |
154.01424 |
2.23 |
| 166 |
C8H6O4 |
|
6.64 |
165.01891 |
2.57 |
| 183 |
C7H5O5N |
|
8.31 |
182.00903 |
2.53 |
| 205 |
C10H7O4N |
|
9.67 |
204.02977 |
2.25 |
| 150 |
C8H6O3 |
|
6.95 |
149.02402 |
2.67 |
| 194 |
C9H6O5 |
|
5.43 |
193.01375 |
2.57 |
| 183 |
C8H9O4N |
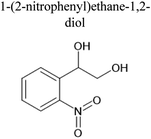
|
8.11 |
182.04544 |
2.41 |
| 139 |
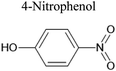
C6H5O3N |
|
8.59 |
138.01936 |
2.29 |
3.7. Naphthalene SOA transient reactivity
Laser flash photolysis of deoxygenated naphthalene SOA solutions showed strong transient absorption at 420 nm (Fig. 7A) when excited at 355 nm. A strong fluorescence was also observed immediately after the laser excitation. This was expected, as naphthalene SOA is known to fluoresce with an effective quantum yield of a few percent.28,57 After the fluorescence, there was a buildup of transient absorption, followed by a slow exponential decay (∼20 μs). The decay portion was fitted with a single exponential (eqn (1)) as done for single photosensitizer molecule solutions (Fig. 7B). As expected, the transient lifetimes from the exponential fits decreased with increasing KI concentrations and were then used to conduct a Stern–Volmer analysis to obtain the reaction rate constant (k1) of 3.0 × 108 M−1 s−1. This is similar to the single compound experiments but is an order of magnitude lower, which is reasonable considering that not every absorber in naphthalene SOA can act as a photosensitizer.
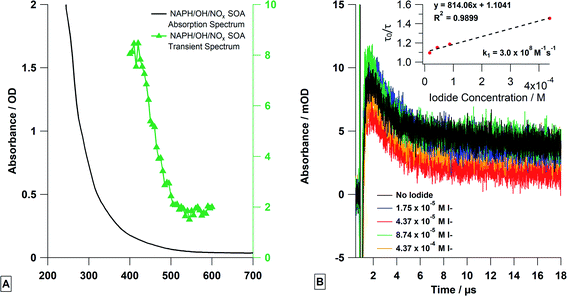 |
| | Fig. 7 (A) Steady state (black line) and transient absorption spectra (green triangles) of deoxygenated naphthalene SOA solution. The transient spectrum was the average signal from 1.2–2 μs. Please note the transient spectrum is measured in mOD. (B) The transient absorption decays at 420 nm were measured and fitted to produce a Stern–Volmer analysis at varying concentrations of KI. All solutions were deoxygenated. | |
Comparing the SOA solution experiments to the single photosensitizer experiments indicate some similarities as the absorption values at later times were not equal to the absorption values before the laser pulse, indicating the formation of a long-lived transient or a stable product (Fig. 7B). Unlike the single compound experiments where the long-time absorption increased linearly with quencher concentration, the long-time absorption was independent of quencher concentration for SOA solution experiments. The reason for this is not entirely clear but is not entirely surprising either, as the SOA solution is a mixture of thousands of molecular species including quinones, and other aromatic ketones, many of which can exhibit transient absorption ∼420 nm. Although speculative, we expect that the transient absorption at 420 nm is most likely a composite absorption of multiple triplets and free radicals, some of which react with iodide and/or other SOA species. Similar behavior has been observed in the chromophoric dissolved organic matter (CDOM) community.58–60 Thus, the formation of new transients and products at long-times is expected to be complex and will include atomic iodine as well as multiple cross-reaction products between atomic iodine, radical iodide, and SOA radical species—the amounts of which depend on the initial amount of iodide present. Increasing the amount of iodide present may increase the yield of one photoproduct but may decrease it for another, resulting in a similar absorption signal. This is further demonstrated by the y-intercept of the Stern–Volmer plot. The y-intercept of the plot is larger than the theoretical value of 1, where the y-axis is the ratio of the lifetime without iodide (τ0) and the lifetime with a certain concentration of iodide (τ). At the y-intercept, the ratio should equal one, but the larger value here indicates chemical reactivity even when the iodide concentration is zero, likely due to the aforementioned chemical complexity and cross reactions in the solution.
The SOA system is complex, with multiple reactive species undergoing multiple generations of cross reactions with each other, and cannot be well-explained by a classical triplet-quencher mechanism. As such, we consider our results as a study of the “effective” reactivity of the system based on the triplet/radical pool at 420 nm.
4. Conclusion
These experimental results indicate that aromatic ketones contribute towards photosensitized reactivity in atmospheric particles as a source of anion radical formation in the atmosphere. We show that compounds that come from primary emissions (xanthone, flavone, and acetophenone) as well as from secondary oxidation of aromatic compounds (such as naphthalene) can act as potential photosensitizers that are capable of abstracting electrons from halide ions. The rate of the resulting radical anion formation depends on the free energy of the electron transfer of the photosensitizer, and the oxidation potential of the halide ions. Furthermore, oxidized compounds and halogenated organic compounds are also formed by irradiation at 355 nm in the presence of oxygen, which is atmospherically relevant. These products are highly oxygenated, and their presence in the atmosphere can be a marker of the photosensitization process from xanthone in the atmosphere. This study demonstrates that tropospheric photosensitization may involve a large variety of compounds of primary or secondary nature and will introduce new, unconsidered chemical pathways that impact atmospheric multiphase chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Agence National de la Recherche (ANR) under the grant Photosoa (grant ANR-16-CE92-0017). SAN, DAF and VB acknowledge support by the US NSF grant AGS-1853639.
References
- H. Herrmann, D. Hoffmann, T. Schaefer, P. Bräuer and A. Tilgner, Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools, ChemPhysChem, 2010, 11, 3796–3822 CrossRef CAS.
- C. Anastasio, B. C. Faust and C. J. Rao, Aromatic Carbonyl Compounds as Aqueous-Phase Photochemical Sources of Hydrogen Peroxide in Acidic Sulfate Aerosols, Fogs, and Clouds. 1. Non-Phenolic Methoxybenzaldehydes and Methoxyacetophenones with Reductants (Phenols), Environ. Sci. Technol., 1997, 31, 218–232 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. Harrison, R. I. Olariu and C. Arsene, Photochemical reactions in the tropospheric aqueous phase and on particulate matter, Chem. Soc. Rev., 2006, 35, 441–453 CAS.
- Y. Chen, N. Li, X. Li, Y. Tao, S. Luo, Z. Zhao, S. Ma, H. Huang, Y. Chen, Z. Ye and X. Ge, Secondary organic aerosol formation from 3C*-initiated oxidation of 4-ethylguaiacol in atmospheric aqueous-phase, Sci. Total Environ., 2020, 723, 137953 CrossRef CAS.
- M. Jang and S. R. McDow, Products of Benz[a]anthracene Photodegradation in the Presence of Known Organic Constituents of Atmospheric Aerosols, Environ. Sci. Technol., 1997, 31, 1046–1053 CrossRef CAS.
- R. Kaur, B. M. Hudson, J. Draper, D. J. Tantillo and C. Anastasio, Aqueous reactions of organic triplet excited states with atmospheric alkenes, Atmos. Chem. Phys., 2019, 19, 5021–5032 CrossRef CAS.
- N. K. Richards-Henderson, A. T. Pham, B. B. Kirk and C. Anastasio, Secondary Organic Aerosol from Aqueous Reactions of Green Leaf Volatiles with Organic Triplet Excited States and Singlet Molecular Oxygen, Environ. Sci. Technol., 2015, 49, 268–276 CrossRef CAS.
- S. Rossignol, K. Z. Aregahegn, L. Tinel, L. Fine, B. Nozière and C. George, Glyoxal Induced Atmospheric Photosensitized Chemistry Leading to Organic Aerosol Growth, Environ. Sci. Technol., 2014, 48, 3218–3227 CrossRef CAS.
- J. D. Smith, V. Sio, L. Yu, Q. Zhang and C. Anastasio, Secondary Organic Aerosol Production from Aqueous Reactions of Atmospheric Phenols with an Organic Triplet Excited State, Environ. Sci. Technol., 2014, 48, 1049–1057 CrossRef CAS.
- L. Tinel, S. Dumas and C. George, A time-resolved study of the multiphase chemistry of excited carbonyls: Imidazole-2-carboxaldehyde and halides, C. R. Chim., 2014, 17, 801–807 CrossRef CAS.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 2014, 14, 13801–13816 CrossRef.
- S. Canonica, B. Hellrung, P. Müller and J. Wirz, Aqueous Oxidation of Phenylurea Herbicides by Triplet Aromatic Ketones, Environ. Sci. Technol., 2006, 40, 6636–6641 CrossRef CAS.
- S. Canonica, B. Hellrung and J. Wirz, Oxidation of Phenols by Triplet Aromatic Ketones in Aqueous Solution, J. Phys. Chem. A, 2000, 104, 1226–1232 CrossRef CAS.
- A. Jammoul, S. Dumas, B. D'Anna and C. George, Photoinduced oxidation of sea salt halides by aromatic ketones: a source of halogenated radicals, Atmos. Chem. Phys., 2009, 9, 4229–4237 CrossRef CAS.
- G. Honninger, C. von Friedeburg and U. Platt, Multi axis differential optical absorption spectroscopy (MAX-DOAS), Atmos. Chem. Phys., 2004, 4, 231–254 CrossRef.
- U. Platt and G. K. Moortgat, Heterogeneous and Homogeneous Chemistry of Reactive Halogen Compounds in the Lower Troposphere, J. Atmos. Chem., 1999, 34, 1–8 CrossRef CAS.
- T. Sherwen, M. J. Evans, R. Sommariva, L. D. J. Hollis, S. M. Ball, P. S. Monks, C. Reed, L. J. Carpenter, J. D. Lee, G. Forster, B. Bandy, C. E. Reeves and W. J. Bloss, Effects of halogens on European air-quality, Faraday Discuss., 2017, 200, 75–100 RSC.
- W. R. Simpson, S. S. Brown, A. Saiz-Lopez, J. A. Thornton and R. von Glasow, Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 2015, 115, 4035–4062 CrossRef CAS.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
-
S. Tomaz, Study of polycyclic aromatic compounds in the atmosphere: molecular characterization and chemical processes related to organic aerosols, PhD thesis, 2015.
- S. Tomaz, P. Shahpoury, J.-L. Jaffrezo, G. Lammel, E. Perraudin, E. Villenave and A. Albinet, One-year study of polycyclic aromatic compounds at an urban site in Grenoble (France): seasonal variations, gas/particle partitioning and cancer risk estimation, Sci. Total Environ., 2016, 565, 1071–1083 CrossRef CAS.
- W. Cautreels and K. Van Cauwenberghe, Experiments on the distribution of organic pollutants between airborne particulate matter and the corresponding gas phase, Atmos. Environ., 1978, 12, 1133–1141 CrossRef CAS.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks, Environ. Sci. Technol., 1999, 33, 1578–1587 CrossRef CAS.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Measurement of Emissions from Air Pollution Sources. 3. C1–C29 Organic Compounds from Fireplace Combustion of Wood, Environ. Sci. Technol., 2001, 35, 1716–1728 CrossRef CAS.
- G. Ketseridis, J. Hahn, R. Jaenicke and C. Junge, The organic constituents of atmospheric particulate matter, Atmos. Environ., 1976, 10, 603–610 CrossRef CAS.
- P. Lin, P. K. Aiona, Y. Li, M. Shiraiwa, J. Laskin, S. A. Nizkorodov and A. Laskin, Molecular Characterization of Brown Carbon in Biomass Burning Aerosol Particles, Environ. Sci. Technol., 2016, 50, 11815–11824 CrossRef CAS.
- K. Z. Aregahegn, B. Noziere and C. George, Organic aerosol formation photo-enhanced by the formation of secondary photosensitizers in aerosols, Faraday Discuss., 2013, 165, 123–134 RSC.
- P. K. Aiona, J. L. Luek, S. A. Timko, L. C. Powers, M. Gonsior and S. A. Nizkorodov, Effect of Photolysis on Absorption and Fluorescence Spectra of Light-Absorbing Secondary Organic Aerosols, ACS Earth Space Chem., 2018, 2, 235–245 CrossRef CAS.
- A. Manfrin, S. A. Nizkorodov, K. T. Malecha, G. J. Getzinger, K. McNeill and N. Borduas-Dedekind, Reactive Oxygen Species Production from Secondary Organic Aerosols: The Importance of Singlet Oxygen, Environ. Sci. Technol., 2019, 53, 8553–8562 CrossRef CAS.
- K. T. Malecha and S. A. Nizkorodov, Feasibility of Photosensitized Reactions with Secondary Organic Aerosol Particles in the Presence of Volatile Organic Compounds, J. Phys. Chem. A, 2017, 121, 4961–4967 CrossRef CAS.
- R. Ciuraru, L. Fine, M. van Pinxteren, B. D'Anna, H. Herrmann and C. George, Unravelling New Processes at Interfaces: Photochemical Isoprene Production at the Sea Surface, Environ. Sci. Technol., 2015, 49, 13199–13205 CrossRef CAS.
- M. Mekic, J. Zeng, B. Jiang, X. Li, Y. G. Lazarou, M. Brigante, H. Herrmann and S. Gligorovski, Formation of Toxic Unsaturated Multifunctional and Organosulfur Compounds From the Photosensitized Processing of Fluorene and DMSO at the Air-Water Interface, J. Geophys. Res.: Atmos., 2020, 125, e2019JD031839 CAS.
- M. Brigante, T. Charbouillot, D. Vione and G. Mailhot, Photochemistry of 1-Nitronaphthalene: A Potential Source of Singlet Oxygen and Radical Species in Atmospheric Waters, J. Phys. Chem. A, 2010, 114, 2830–2836 CrossRef CAS.
- D. I. Reeser, A. Jammoul, D. Clifford, M. Brigante, B. D'Anna, C. George and D. J. Donaldson, Photoenhanced Reaction of Ozone with Chlorophyll at the Seawater Surface, J. Phys. Chem. C, 2009, 113, 2071–2077 CrossRef CAS.
- M. Goez and B. H. M. Hussein, Photoionization of xanthone via its triplet state or via its radical anion, Phys. Chem. Chem. Phys., 2004, 6, 5490–5497 RSC.
- H. Lutz, E. Breheret and L. Lindqvist, Effects of solvent and substituents on the absorption spectra of triplet acetophenone and the acetophenone ketyl radical studied by nanosecond laser photolysis, J. Phys. Chem. C, 1973, 77, 1758–1762 CrossRef CAS.
- H. Lutz, M. C. Duval, E. Breheret and L. Lindqvist, Solvent effects on acetophenone photoreduction studied by laser photolysis, J. Phys. Chem. C, 1972, 76, 821–822 CrossRef CAS.
-
M. Montalti, A. Credi, L. Prodi and M. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 2006 Search PubMed.
- R. Tang, P. Zhang, H. Li, Y. Liu and W. Wang, Photosensitized xanthone-based oxidation of guanine and its repair: a laser flash photolysis study, J. Photochem. Photobiol., B, 2011, 105, 157–161 CrossRef CAS.
- V. Avila and C. M. Previtali, Triplet state properties of flavone in homogeneous and micellar solutions. A laser flash photolysis study, J. Chem. Soc., Perkin Trans. 2, 1995, 2281–2285, 10.1039/P29950002281.
-
A. M. Braun, Technologie Photochimique, Presses Polytechniques Romandes, Lausanne, 1986 Search PubMed.
- P. J. Wagner and R. W. Spoerke, Triplet lifetimes of cyclic ketones, J. Am. Chem. Soc., 1969, 91, 4437–4440 CrossRef CAS.
- C. Grewer and H.-D. Brauer, Mechanism of the Triplet-State Quenching by Molecular Oxygen in Solution, J. Phys. Chem. C, 1994, 98, 4230–4235 CrossRef CAS.
- S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, Reexamination of the Rehm–Weller Data Set Reveals Electron Transfer Quenching That Follows a Sandros–Boltzmann Dependence on Free Energy, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS.
-
L. Eberson, in Advances in Physical Organic Chemistry, ed. V. Gold and D. Bethell, Academic Press, 1982, vol. 18, pp. 79–185 Search PubMed.
- S. Canonica, U. Jans, K. Stemmler and J. Hoigne, Transformation Kinetics of Phenols in Water: Photosensitization by Dissolved Natural Organic Material and Aromatic Ketones, Environ. Sci. Technol., 1995, 29, 1822–1831 CrossRef CAS.
- K. M. Parker and W. A. Mitch, Halogen radicals contribute to photooxidation in coastal and estuarine waters, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5868–5873 CrossRef CAS.
- R. Devonshire and J. J. Weiss, Nature of the transient species in the photochemistry of negative ions in aqueous solution, J. Phys. Chem. C, 1968, 72, 3815–3820 CrossRef CAS.
- I. Lampre, J.-L. Marignier, M. Mirdamadi-Esfahani, P. Pernot, P. Archirel and M. Mostafavi, Oxidation of Bromide Ions by Hydroxyl Radicals: Spectral Characterization of the Intermediate BrOH˙−, J. Phys. Chem. A, 2013, 117, 877–887 CrossRef CAS.
- B. G. Ershov and E. Janata, The one electron reduction of Hg2+ by 1-hydroxyalkyl radicals in aqueous solution: a pulse radiolysis study, Radiat. Phys. Chem., 2004, 69, 55–57 CrossRef CAS.
- D. O. Mártire, J. A. Rosso, S. Bertolotti, G. C. Le Roux, A. M. Braun and M. C. Gonzalez, Kinetic Study of the Reactions of Chlorine Atoms and Cl2˙− Radical Anions in Aqueous Solutions. II. Toluene, Benzoic Acid, and Chlorobenzene, J. Phys. Chem. A, 2001, 105, 5385–5392 CrossRef.
- M. Roveretto, M. Li, N. Hayeck, M. Brüggemann, C. Emmelin, S. Perrier and C. George, Real-Time Detection of Gas-Phase Organohalogens from Aqueous Photochemistry Using Orbitrap Mass Spectrometry, ACS Earth Space Chem., 2019, 3, 329–334 CrossRef CAS.
- Z. Mehrdad, A. Noll, E.-W. Grabner and R. Schmidt, Sensitization of singlet oxygen via encounter complexes and via exciplexes of ππ* triplet excited sensitizers and oxygen, Photochem. Photobiol. Sci., 2002, 1, 263–269 RSC.
- Z. Mehrdad, C. Schweitzer and R. Schmidt, Formation of O2(1Σg+), O2(1Δg), and O2(3Σg-) during Oxygen Quenching of nπ* Triplet Phenyl Ketones: The Role of Charge Transfer and Sensitizer-Oxygen Complex Structure, J. Phys. Chem. A, 2002, 106, 228–235 CrossRef CAS.
-
W. Xing, M. Yin, Q. Lv, Y. Hu, C. Liu and J. Zhang, in Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, ed. W. Xing, G. Yin and J. Zhang, Elsevier, Amsterdam, 2014, pp. 1–31, DOI:10.1016/B978-0-444-63278-4.00001-X.
- K. E. Kautzman, J. D. Surratt, M. N. Chan, A. W. H. Chan, S. P. Hersey, P. S. Chhabra, N. F. Dalleska, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Chemical Composition of Gas- and Aerosol-Phase Products from the Photooxidation of Naphthalene, J. Phys. Chem. A, 2010, 114, 913–934 CrossRef CAS.
- H. J. Lee, P. K. Aiona, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon, Environ. Sci. Technol., 2014, 48, 10217–10226 CrossRef CAS.
-
A. M. Fischer, J. S. Winterle and T. Mill, in Photochemistry of Environmental Aquatic Systems, American Chemical Society, 1987, ch. 11, vol. 327, pp. 141–156 Search PubMed.
- M. V. Martin, R. A. Mignone, J. A. Rosso, P. David Gara, R. Pis Diez, C. D. Borsarelli and D. O. Mártire, Transient spectroscopic characterization and theoretical modeling of fulvic acid radicals formed by UV-A radiation, J. Photochem. Photobiol., A, 2017, 332, 571–579 CrossRef CAS.
-
J. F. Power, D. K. Sharma, C. H. Langford, R. Bonneau and J. Joussot-Dubien, in Photochemistry of Environmental Aquatic Systems, American Chemical Society, 1987, ch. 12, vol. 327, pp. 157–173 Search PubMed.
- T. A. Geissman and S. L. Friess, Flavanones and Related Compounds. VI. The Polarographic Reduction of Some Substituted Chalcones, Flavones and Flavanones, J. Am. Chem. Soc., 1949, 71, 3893–3902 CrossRef CAS.
- R. A. Day and R. E. Biggers, Polarography of p-Chlorobenzophenone and Xanthone, J. Am. Chem. Soc., 1953, 75, 738–739 CrossRef.
- G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. O. Cowan, R. C. Counsell, V. Vogt and C. Dalton, Mechanisms of Photochemical Reactions in Solution. XXII.1 Photochemical cis-trans Isomerization, J. Am. Chem. Soc., 1964, 86, 3197–3217 CrossRef CAS.
- A. J. G. Barwise, A. A. Gorman, R. L. Leyland, P. G. Smith and M. A. J. Rodgers, A pulse radiolysis study of the quenching of aromatic carbonyl triplets by norbornadienes and quadricyclenes. The mechanism of interconversion, J. Am. Chem. Soc., 1978, 100, 1814–1820 CrossRef CAS.
- P. J. Wagner, R. J. Truman, A. E. Puchalski and R. Wake, Extent of charge transfer in the photoreduction of phenyl ketones by alkylbenzenes, J. Am. Chem. Soc., 1986, 108, 7727–7738 CrossRef CAS.
- P. B. Merkel and J. P. Dinnocenzo, Thermodynamic energies of donor and acceptor triplet states, J. Photochem. Photobiol., A, 2008, 193, 110–121 CrossRef CAS.
Footnotes |
| † Now at: TPSH, 4 Rue Jean Mermoz, Courcouronnes, France. |
| ‡
Now at: INRS, Nancy, France.
|
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
D. A.
Fishman
b,
S. A.
Nizkorodov
b,
D. A.
Fishman
b,
S. A.
Nizkorodov