
Emerging perovskite quantum dot solar cells: feasible approaches to boost performance
Jingxuan
Chen
a,
Donglin
Jia
a,
Erik M. J.
Johansson
 b,
Anders
Hagfeldt
c and
Xiaoliang
Zhang
*a
b,
Anders
Hagfeldt
c and
Xiaoliang
Zhang
*a
aSchool of Materials Science and Engineering, Beihang University, Beijing 100191, China. E-mail: xiaoliang.zhang@buaa.edu.cn
bDepartment of Chemistry-Ångström, Physical Chemistry, Uppsala University, Uppsala 75120, Sweden
cLaboratory of Photomolecular Science, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne CH-1015, Switzerland
First published on 9th November 2020
Abstract
Lead halide perovskite quantum dots (PQDs), also called perovskite nanocrystals, are considered as one of the most promising classes of photovoltaic materials for solar cells due to their prominent optoelectronic properties and simple preparation techniques. Remarkable achievements in PQD solar cells (PQDSCs) have been made. In particular, the power conversion efficiency of PQDSCs has been largely pushed from 10.77% to 17.39% (certified 16.6%) by finely controlling the surface chemistry of PQDs and the device physics of PQDSCs. In this review, we summarize the latest advances of emerging PQDSCs and discuss various strategies applied to improve the device performance of PQDSCs, including the synthesis methods, compositional engineering and surface chemistry of PQDs. Moreover, the device operation of PQDSCs is discussed to highlight the effect of device architecture on the photovoltaic performance of PQDSCs. Facing the practical applications of the PQDSCs under ambient conditions, device stability is also highlighted. Finally, conclusions and perspectives are presented along with the possible challenges and opportunities to promote development steps of PQDSCs with higher photovoltaic performance and robust stability.
 Jingxuan Chen | Jingxuan Chen is a PhD student at Beihang University (BUAA). She received her Bachelor's degree in materials science and engineering from Nanjing University of Aeronautics and Astronautics in 2018. Currently, she is pursuing her PhD degree under the guidance of Prof. Xiaoliang Zhang. Her current research interest mainly focuses on utilizing surface management to enhance the performance of perovskite quantum dot solar cells. |
 Donglin Jia | Donglin Jia received his Bachelor's degree in 2017 from Beijing University of Chemical Technology (BUCT), Republic of China. He is currently pursuing his PhD degree from Beihang University (BUAA), Republic of China, under the supervision of Prof. Xiaoliang Zhang. His research focuses on regulating the surface chemistry of quantum dots for application in photovoltaic devices. |
 Erik M. J. Johansson | Erik M. J. Johansson is a professor in Physical Chemistry at Uppsala University. He received his PhD degree in Physics from Uppsala University in 2006. After postdoctoral work at Lund University, he was an assistant professor at Uppsala University. His research field is nanostructured solar cells including perovskite, quantum dot and dye-sensitized solar cells. His research spans understanding the fundamental processes and the atomic-scale structures in these solar cells to the function of the full devices. |
 Anders Hagfeldt | Anders Hagfeldt has been a professor at the Institute of Chemical Sciences and Engineering, Ēcole Polytechnique Fédérale de Lausanne (EPFL), and the Head of the Laboratory of Photomolecular Science since 2014. He obtained his PhD in 1993 from Uppsala University and was a postdoctoral fellow with Prof. Michael Grätzel (1993–1994) at EPFL, Switzerland. Before 2014 he was a professor in Physical Chemistry and the Director of the Center for Molecular Devices at Uppsala University, Sweden. He is a member of the Royal Swedish Academy of Sciences, the European Academy of Sciences, the Royal Society of Sciences in Uppsala, and the Royal Swedish Academy of Engineering Sciences in Stockholm. He is an Honorary Doctor of the University of Paris-Diderot. |
 Xiaoliang Zhang | Xiaoliang Zhang is a professor at Beihang University, China, and directs a research group working on optoelectronic materials and devices. He received his PhD degree in Materials Physics and Chemistry from Beihang University. Then, he joined Uppsala University, Sweden, as a postdoctoral researcher and subsequently was promoted as a Senior Researcher at Uppsala University. He joined Beihang University as a full professor in 2018. He has particular interest in photovoltaic device physics, such as electro-optics and interfacial engineering in photovoltaic and optoelectronic devices, with a current emphasis on semiconducting nanocrystals. |
Broader contextTo address the issues of fossil energy shortage and environmental pollution, efficient utilization of solar energy has been becoming important since solar energy is a kind of inexhaustible energy and can be directly converted into electric power using photovoltaic devices. Metal-halide perovskites as a new class of photovoltaic materials have been springing up rapidly in the last few years, bringing new opportunities to efficiently use solar energy. However, perovskite solar cells (PSCs) may suffer from long-term operation under harsh conditions. Therefore, by retaining the superb optoelectronic features of perovskites, the materials are prepared into low-dimensional perovskite quantum dots (PQDs) to improve their stability by finely controlling the surface chemistry of PQDs. Remarkable progress has been made in PQD solar cells (PQDSCs), and a PQDSC with a power conversion efficiency of over 16% was obtained. Although the efficiency of PQDSCs was largely improved, it is still far behind the theoretical efficiency of PQDSCs, likely due to serious recombination in PQDSC devices. This review provides a comprehensive summary of various approaches to improve the performance of PQDSCs, and the possible challenges and opportunities of emerging PQDSCs are discussed. |
1. Introduction
Burgeoning metal-halide perovskites have received tremendous attention for many applications, such as solar cells, light-emitting diodes (LEDs) and photodetectors, due to their outstanding optoelectronic properties with low exciton binding energy and long carrier lifetimes, simple preparation techniques and low material cost.1–9 Metal-halide perovskites have a formula of ABX3 in the crystal lattice, in which A represents an organic (formamidine cation (FA+) or methylammonium cation (MA+)) or inorganic (Cs+) cation, B is the Pb2+ or Sn2+ cation, and X is a halide anion (Cl−, Br− and I−).10–12 Therefore, the designable composition of ABX3 perovskites provides feasible strategies for fine-tuning the optoelectronic properties and bandgap energy (Eg) of perovskites by mixing different cations or anions on the sites of the crystal lattice toward different applications. In the past few years, with breakthroughs in the fundamental studies on the compositional engineering, interfacial engineering and device architecture optimization of perovskite solar cells (PSCs), extraordinary progress has been made.13–18 In particular, single-junction PSCs with a power conversion efficiency (PCE) of over 25% have been obtained, which are comparable to commercial photovoltaic devices, such as crystalline silicon solar cells, showing great potential for photovoltaic applications.19 However, due to the severe degradation issues of hybrid PSCs, they may suffer from long-term high-power operation under harsh conditions, such as high temperature, high humidity and strong illumination.20–23 Therefore, in addition to improving device efficiency, more and more studies focused on improving device stability to meet application requirements.24,25Replacing organic A-site cations with inorganic Cs+ cations was believed to be a feasible avenue to improve the thermal stability of PSCs.26–28 However, due to the low Goldschmidt tolerance factor caused by the smaller inorganic Cs+ cations, all-inorganic CsPbI3 perovskites exhibit poor phase stability at room temperature.29 Wang et al. used organic cations to regulate the crystallization of CsPbI3 perovskites, and the β-CsPbI3 perovskite with better phase stability was obtained under mild conditions. Meanwhile, by using choline iodide to passivate surface defects, the efficiency of all-inorganic PSCs has exceeded 18%.30 However, the desired α-CsPbI3 perovskite films could only be stable at high temperatures (>200 °C).31 The emergence of all-inorganic perovskite quantum dots (PQDs), also called nanocrystals (NCs), provided an implemented solution for this dilemma.32 After preparing all-inorganic perovskites into low-dimensional quantum dots or nanocrystals, the phase stability of perovskites was largely improved due to the reduced surface strain caused by reducing the size of perovskites.33,34 Furthermore, the spectroscopic properties of PQDs can be widely adjusted by controlling the halide composition, dimensional size and shape of PQDs.32,35,36 More delightfully, multiple exciton effects in PQDs may provide a possibility to make full use of high-energy photons and thus overcome the Shockley–Queisser theoretical efficiency limitation for single-junction solar cells.37 As a new research field that is just beginning to flourish, the PCE of perovskite quantum dot solar cells (PQDSCs) has been largely improved from 10.77% to 17.39% (certified 16.6%) in a short time, showing great potential for solution-processed high-performance solar cells.34,38
PQDs are generally synthesized using wet-chemistry methods, and long-chain organic ligands, such as oleic acid (OA) and oleylamine (OAm), are used to cap dot surfaces and make them uniformly dispersed in the colloidal systems.36 However, long-chain insulating ligands seriously affect interdot coupling and carrier transport within the PQD solid films. Therefore, molecules with a low steric hindrance, such as FAI and GASCN, are usually applied for ligand exchange to lower the energy barrier of carrier transport and thus improve carrier mobility in the PQD solid films.39–41 Besides, nano-sized PQDs always present a large number of surface defects, which can act as non-radiative recombination centers of photoinduced carriers, significantly affecting the charge extraction of solar cell devices.42,43 Coupled with the purification and ligand exchange process using anti-solvent, more surface defects on the PQDs are produced, leading to energy loss in PQDSC devices.44 Efficient surface passivation of PQDs can reduce the surface defects of PQDs and thus lower recombination in the PQD solid films.45,46 Moreover, since PQDSCs are generally constructed by stacking different functional layers, such as charge (electron and hole) transport layers and multiple photoactive PQD layers, finely controlling interfacial properties is critical to decreasing interfacial recombination and thus improving device photovoltaic performance.47–50 Part of the progress of PQDSCs has been summarized in previous reviews.51–54 However, overviews focusing on the latest advances and systematic improvement of the photovoltaic performance and stability of PQDSCs through different strategies are still scarce, which would provide a deep understanding of the feasible approaches to realize high-performance PQDSCs. In this review, we comprehensively summarize the latest advances of emerging PQDSCs and discuss various strategies applied to improve the photovoltaic performance and stability of PQDSCs toward the development steps of commercialization.
2. State-of-the-art of PQDSCs
Previous studies on highly-efficient quantum dot solar cells (QDSCs) focused on traditional chalcogenide colloidal quantum dots (CQDs), such as lead sulfide (PbS) CQDs.55–58 In the past decade, significant progress in the device operation and device physics has been made in PbS-CQDSCs and the efficiency of PbS-CQDSCs has largely boosted from 2.9% in 2010 to recently over 13%.59 PQDs as a new class of solution-processable semiconductors are promising for solar cells due to their tunable light absorption spectrum with strong light absorption and high quantum yields.32 As shown in Fig. 1a, an exponentially increasing number of publications of PQDs with application in solar cells has been published, suggesting a good trend in the development of PQDSCs. Fig. 1b presents the certified efficiencies of QDSCs by the National Renewable Energy Laboratory (NREL). In 2016, a PQDSC with an efficiency of 10.77% was reported by Swarnkar et al., who used all-inorganic CsPbI3 PQDs as the photoactive material in the solar cell.34 Meanwhile, thanks to the negative surface energy and size-induced lattice strain of PQDs, the CsPbI3 PQD solid films exhibited better stability than bulk CsPbI3 perovskite films, showing advantages in solar cell applications.33 With fundamental studies on the surface chemistry and compositional engineering of PQDs, the efficiency of PQDSCs was largely improved to 16.6%,38 which is much higher than that of conventional PbS-CQDSCs, showing great potential for next-generation QDSCs.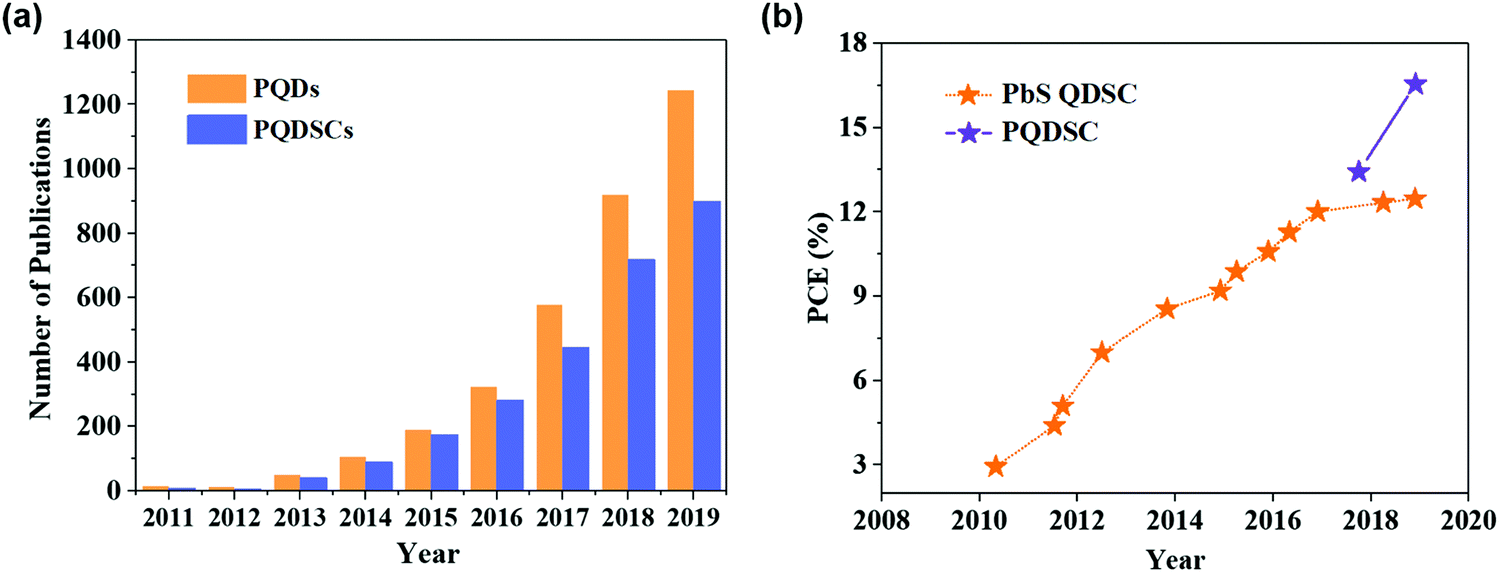 | ||
| Fig. 1 State-of-the-art of PQDSCs. (a) The number of annual publications (from 2011 to 2019) of PQDs (searched using the keyword: perovskite quantum dot) and PQDSCs (searched using the keyword: perovskite quantum dots and solar cells). The data were retrieved from Scopus. (b) Evolution of the certified PCEs of QDSCs by the NREL.19 | ||
For an efficient solar cell, efficient extraction of photoinduced charge carriers is required and obtained by minimizing charge recombination in the solar cell device. Thus, the device efficiency of PQDSCs was increased rapidly by diminishing charge recombination by means of various methods, such as surface and compositional engineering of PQDs, as well as the optimization of device architecture and operation. Although the efficiency of PQDSCs has been largely improved in the past years, their efficiency has largely lagged behind that of bulk perovskite solar cells.28 Firstly, the photovoltaic performance and stability of PQDSCs are determined by the surface chemistry of PQDs that is determined by surface ligands, which seriously affects the carrier mobility of PQD solid films. The carrier mobility of PQD solid films can be improved through ligand regulation or post-treatment of PQD surfaces to further promote device efficiency.39–41 However, a large number of defects appeared on the PQD surface during the ligand exchange process, which has a serious impact on charge recombination in the PQD solid films. The photogenerated charge carriers in the PQDs are likely preferably captured by these defect states rather than transported to charge collection layers.60 Meanwhile, due to the ionic crystal nature of the perovskite lattice, the surface ligands will be removed from the PQD surfaces during the purification process of PQDs, which will cause amounts of surface dangling bonds that capture photons as defect states.44 Furthermore, during the preparation of PQDSC devices, the ligand exchange process also leads to the production of surface defects of PQDs. Therefore, reducing the paths of non-radiative recombination in PQDs is critical to increasing charge extraction of PQDSCs.
In addition, PQD solid films are generally prepared using a layer-by-layer spin-coating method and finally several PQD layers are stacked to form a PQD solid film with enough thickness to harvest more photons. The PQD solid film is sandwiched between an electron transport layer (ETL) and a hole transport layer (HTL), and therefore the interfacial properties within the PQD solid films, as well as at the PQD/ETL and PQD/HTL interfaces, also need to be finely controlled to reduce the interfacial recombination of charge carriers. Thus, charge transport materials with high carrier mobility and suitable energy levels are very important to obtain high-performance PQDSCs.
Therefore, in this review, we comprehensively summarize the latest advances of emerging PQDSCs from the viewpoint of improving device photovoltaic performance and stability through various methods, as shown in Fig. 2. In Section 3, we make a systematic retrospect on the latest research situation of PQDSCs, as demonstrated above. In Sections 4 and 5, we summarize the major properties and synthesis methods of PQDs to emphasize the importance of synthesis methods on non-radiative combination in the PQDs. In Section 6, we emphatically introduce several feasible strategies with great influence on boosting device performance by lowering charge recombination, including the compositional engineering of PQDs, surface chemistry regulation of PQDs, and device operations of PQDSCs. In Section 7, the stability of PQDs and PQDSCs affected by hydrothermal, storage and illumination conditions is discussed in-depth to identify the feasible methods for further enhancing the device stability of PQDSCs. Finally, we present conclusions, and the possible challenges and prospects for future developing high-performance PQDSCs are discussed.
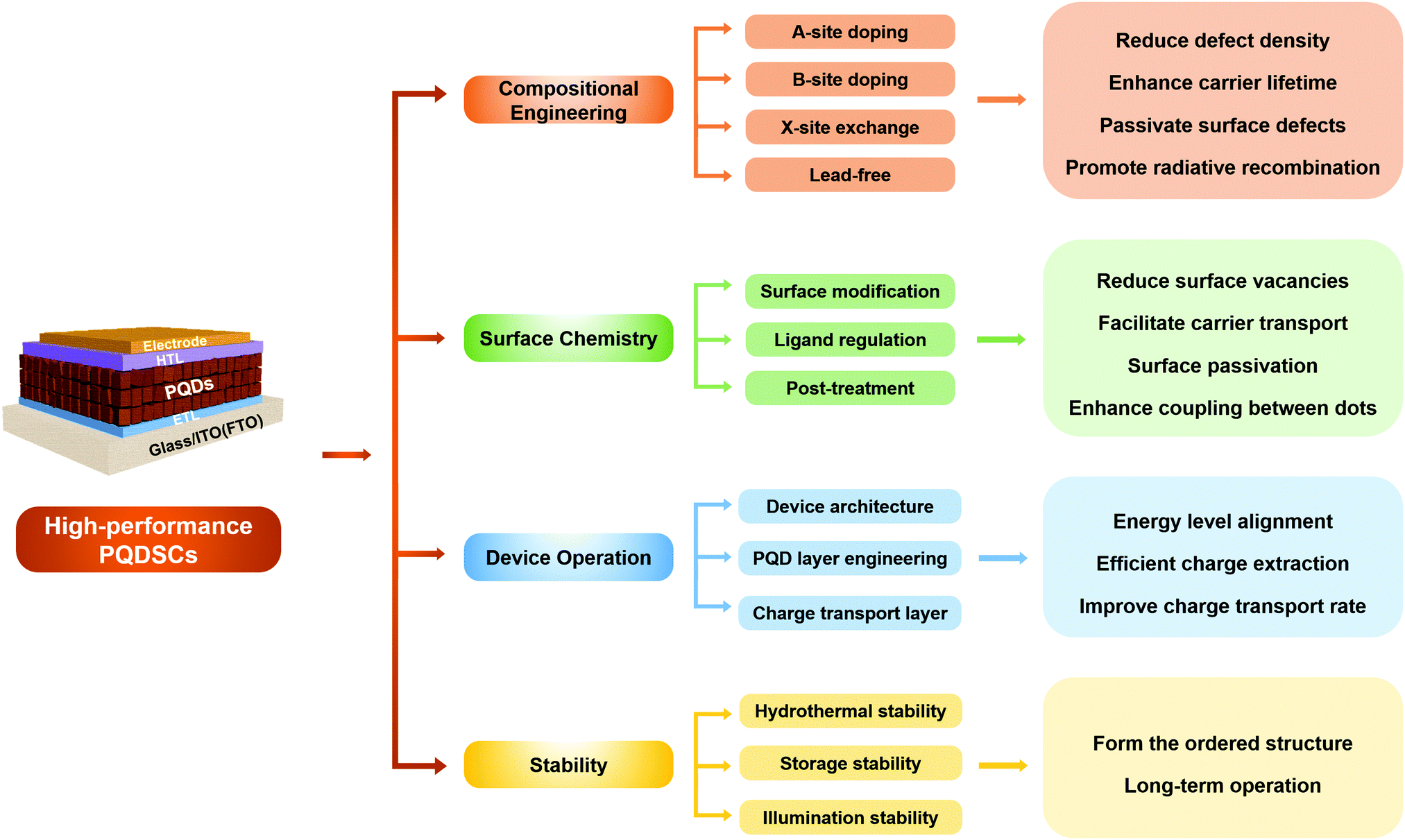 | ||
| Fig. 2 The overall framework of the review. Different approaches applied to improve photovoltaic performance and device stability are presented. | ||
3. PQDs
Among the various optoelectronic nanomaterials, CQDs (also called colloidal nanocrystals) are one of the most important branches, which have attracted extensive attention in recent years.32,61–63 CQDs are a kind of nano-dimensional semiconductor material, whose excitons are restricted to three spatial dimensions.64 Compared with bulk optoelectronic materials, the most important advantage of CQDs is the quantum confinement effect that appears as the size of semiconducting materials is reduced to a certain extent (2–20 nm), and exceptional optical and electrical properties were observed.65 PQDs, as an up-rising star of optoelectronic nanomaterials, exhibit unique properties, such as tunable light absorption spectra, high light absorption coefficients and high defect tolerance, making them ideal light-absorbing materials for next-generation solar cells.66 In this section, we will discuss the characteristics of PQDs and synthesis methods of PQDs in detail.3.1. Crystal structure and unique properties of PQDs
The typical ABX3 perovskite lattice is displayed in Fig. 3a, where the A-site is usually a Cs+, MA+ or FA+ cation, the B-site is the Pb2+ or Sn2+ cation and the X-site is Cl−, Br− or I− (or mixed halide ions), as demonstrated in the Introduction. The perovskite lattice is supported by the corner-sharing [BX6] octahedral frameworks and the A-site cations locate between the cavities, which is also considered as the photoactive cubic phase.67 However, the ideal cubic phase is easily converted into an edge-sharing orthorhombic phase because of the ionic nature of the perovskite lattice, especially in inorganic I-based perovskites.34,68 In 2015, Protesescu et al., for the first time, applied the hot-injection method to synthesize visible color-tunable inorganic PQDs by controlling the composition of halide ions in the PQDs (Fig. 3b).32 Due to the high surface energy and size-induced lattice strain of PQDs, phase-stable CsPbX3 PQDs with a cubic phase could be formed at room temperature.33 Moreover, the CsPbX3 PQDs with bright and narrow PL emission presented a faster radiative lifetime than the bulk counterparts (Fig. 3c). Therefore, compared with the bulk counterparts, PQDs are regarded as one of the most promising optoelectronic materials owing to their good phase stability, narrow PL emission, tunable light absorption spectrum and high photoluminescence quantum yield (PLQY), which have shown great potential for application in optoelectronic devices, such as solar cells,38,40,45 electroluminescent diodes,69–71 lasers and photodetectors.72,73 | ||
| Fig. 3 Crystal structure and optical properties of PQDs. (a) Schematic diagram of the PQD structure. (b) Photograph of CsPbX3 (X = Cl, Br, I) PQD solutions under a UV lamp (λ = 365 nm) and representative PL spectra. (c) Time-resolved PL decays of CsPbX3 PQDs (except for CsPbCl3). (b and c) Reprinted with permission from ref. 32. Copyright 2015 American Chemical Society. (d) Temperature-dependent absorption spectra of CsPbI3 PQDs. Reprinted with permission from ref. 34. Copyright 2016 American Association for the Advancement of Science. (e) Band-structure of two types of semiconductors: Conventional defect-intolerant semiconductors (left) and defect-tolerant PQDs (right). Reprinted with permission from ref. 80. Copyright 2018 Nature Publishing Group. (f) Schematic of the MEG process in CQDs. Reprinted with permission from ref. 84. Copyright 2020 The Royal Society of Chemistry. | ||
Compared with bulk semiconductor materials, quantum dots have distinctive electronic structures. Due to the quantum confinement effect, when the geometric radius of semiconductors decreases to be comparable with (or below) the exciton Bohr radius of semiconductors, the continuous band structure of semiconductors starts to form several quasi-discrete levels and the Eg increases with reducing size.74 Thus, the Eg values of some semiconductors that satisfy dimensional requirements could be tuned by altering particle size.33,34 PQDs synthesized using the hot-injection method generally have a cube shape with an edge length of 4–15 nm.32 Therefore, the Eg of PQDs can be easily adjusted by controlling PQD size, which can be achieved by controlling the reaction temperature during PQD synthesis. With the size of PQDs increasing, the light absorption edge becomes red-shifted in the spectrum (Fig. 3d). Meanwhile, the optical properties of PQDs can also be adjusted by changing the chain length and ratio of surface ligands during the synthesis process.35,36,75,76 It is notable that compared with traditional metal chalcogenide CQDs, the weak quantum size effect of PQDs makes PQDs relatively easy to precisely control spectrum regulation.
The undercoordinated atoms on the QD surface and disorder distribution of ions in the QDs usually result in forming sub-bandgap states (trap states), which act as non-radiative recombination centers of photoinduced carriers, thus significantly affecting the optoelectronic properties of QDs. Therefore, surface defect passivation through surface ligand formulation or core/shell structure formation is necessary for traditional metal chalcogenide QDs.77–79 In contrast, owing to the low defect formation energy in the PQDs, the localized electronic states associated with the point defects produced during the purification process are within the valence and conduction bands or near the edge of the electronic band (Fig. 3e).80 Therefore, due to the unique band structure of PQDs, the interstitial and anti-site defects in the PQDs hardly exist.81 Benefiting from the high defect tolerance of PQDs (appears as shallow defect-related energy levels), the impeding effect of surface defects on charge recombination and transport is not as aggravated as that of chalcogenide QDs.
Generally, when semiconducting materials absorb one high-energy photon, the absorbed photon energy will excite one photoinduced electron–hole pair, while the surplus energy that exceeds the Eg will be wasted as heat.82 Researchers have observed multiple exciton generation (MEG) in quantum-confined semiconducting materials, which could effectively use dissipated energy.83 The specific process of MEG is that the CQDs can absorb one high-energy photon to produce multiple excitons, as shown in Fig. 3f.84 Therefore, additional electrons and holes formed by the surplus energy can be separated, transported and collected to produce a higher photocurrent in CQDSCs.37 Also, CQDs have higher exciton binding energy than bulk materials due to the quantum confinement effect, which was conducive to the reduction of non-radiative recombination.33 However, less attention was paid to the MEG of PQDs, which may need more fundamental studies to figure out the possibility to improve photovoltaic performance by using MEG.
3.2. PQD synthesis
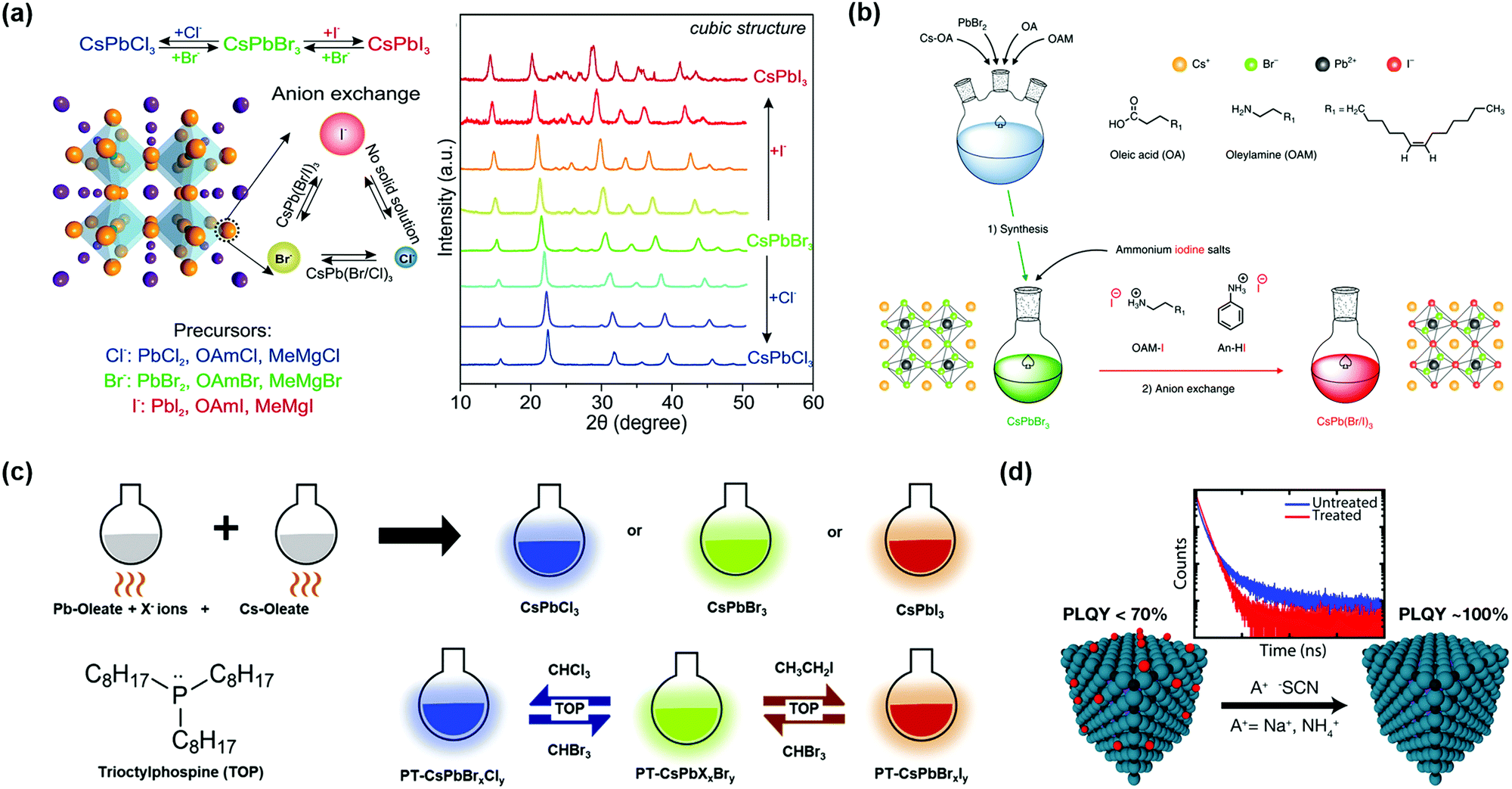
The synthesis methods of high-quality CsPbX3 PQDs with less non-radiative recombination (as a burgeoning kind of optoelectronic material) have attracted significant attention. Protesescu et al. applied the hot-injection method to synthesize monodispersed CsPbX3 PQDs with sizes of 4–15 nm.32 A colloidal PQD solution was obtained by injecting Cs-oleate precursor solution into an organic solvent (octadecene, ODE) containing lead(II)-halide (PbX2) and ligands (OAm and OA) at a temperature of 140–200 °C (Fig. 4a). After quickly cooling the PQD solution, the PQDs needed to be purified using tert-butanol (BuOH) and finally stored in hexane. Subsequently, Swarnkar et al. developed a new method to purify PQDs using methyl acetate (MeOAc) as an anti-solvent to remove the excess unreacted precursors and wash PQDs, as well as prevent the agglomeration and phase transformation of PQDs.34 This improved synthesis pathway and purification method can make the α-CsPbI3 PQDs maintain the cubic phase under ambient conditions for several months. However, it is notable that the washing step will affect the synthetic yield of PQDs, and some of the OAm and OA surface ligands will be washed away during the washing step, which could lower the PLQY of PQDs. The tunable photoluminescence of CsPbX3 PQDs in the wavelength region 410–700 nm was obtained by adjusting the halide composition in the PQDs and the injection temperature of Cs-oleate precursor solution. However, due to the fact that the PbX2 salt was used as both lead and halide sources for the synthesis of CsPbX3 PQDs, the precisely controlled amounts of reactant in the PQDs remain challenging. Imran et al. proposed an optimized three-precursor synthesis route to achieve high-quality CsPbX3 PQDs, in which benzoyl halides were used as the sources of halide precursors, and thus the composition of CsPbX3 PQDs could be precisely controlled to finely regulate the optical properties of PQDs.85 Owing to the halide-rich environment, CsPbX3 PQDs with lead halide-terminated surfaces showed impressive phase stability and excellent optical properties under ambient conditions.86 Meanwhile, the halogen vacancies on the surfaces of the PQDs were largely reduced to avoid non-radiative recombination.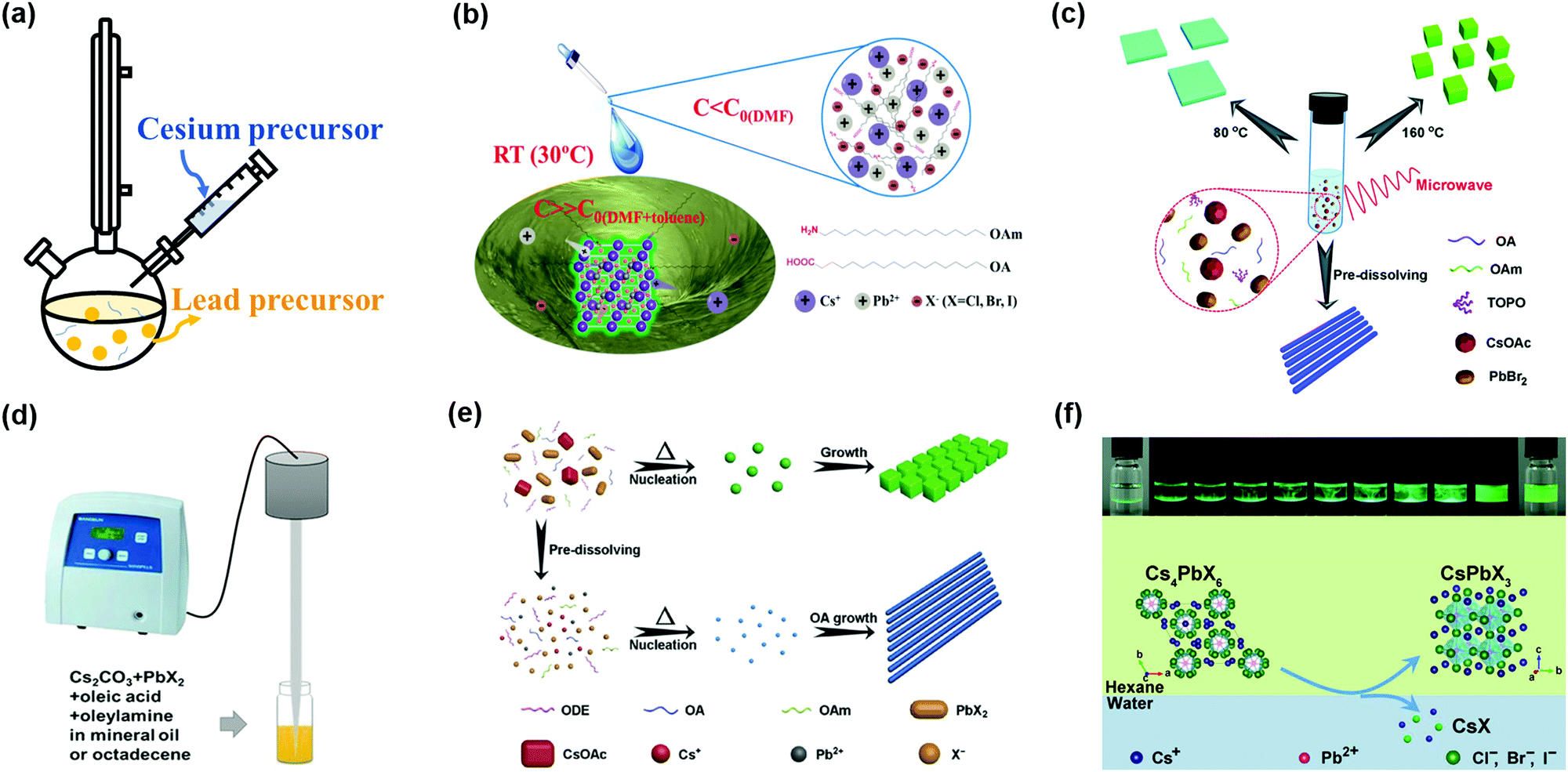 | ||
| Fig. 4 Synthesis methods of PQDs with less non-radiative recombination. (a) Hot-injection synthesis method of PQDs. (b) Room temperature supersaturated recrystallization synthesis method of PQDs. Reprinted with permission from ref. 71. Copyright 2016 Wiley-VCH. (c) Microwave-assisted synthesis method of PQDs. Reprinted with permission from ref. 89. Copyright 2017 The Royal Society of Chemistry. (d) Ultrasonication synthesis method of PQDs. Reprinted with permission from ref. 92. Copyright 2016 Wiley-VCH. (e) Solvothermal synthesis method of PQDs. Reprinted with permission from ref. 93. Copyright 2017 Wiley-VCH. (f) Water-triggered transformation synthesis of PQDs through CsX-stripping. Reprinted with permission from ref. 94. Copyright 2017 American Chemical Society. | ||
The hot-injection method is still one of the most widely used approaches, so far, to synthesize PQDs, and could yield high-quality PQDs with uniform size distribution. However, such a method needs to be carried out at a high temperature and under the protection of an inert atmosphere. The synthesis efficiency is low and it is difficult for large-scale production of PQDs toward commercialization. The room temperature (RT) reprecipitation method proposed by Li et al. can afford CsPbX3 PQDs with excellent optical properties, obviating complex requirements during the PQD synthesis.71 As shown in Fig. 4b, superior PQDs were fabricated by transferring perovskite precursor solution from a polar solvent (dimethyl formamide, DMF, or dimethyl sulfoxide, DMSO) into a nonpolar solvent (toluene) with the aid of surfactants (OA and OAm). PQDs were obtained by a supersaturated recrystallization process, which takes advantage of the different solubilities of ion sources between these two solvents. More interestingly, imperfect PQDs’ surfaces can be self-passivated due to the reversible process of dissolution and recrystallization.87 However, with the RT reprecipitation method it was difficult to precisely control the growth process of PQDs because of an intense and rapid process in supersaturated recrystallization reaction.88
The size and morphology of PQDs significantly affect the physical and chemical properties of PQDs. To understand the formation mechanism of PQDs, Pan et al. put forward a novel microwave-assisted method to synthesize PQDs with a controllable morphology.89 During the microwave-assisted synthesis of PQDs, the powders of two precursors (PbBr2 and Cs2CO3) and a mixture of organic solvents (ODE, OA, OAm and TOPO) were loaded into a quartz boat and then heated under the microwave irradiation (Fig. 4c). It has been revealed that the reaction temperature, time, and capping ligand all contributed to control of the morphology of PQDs.32,35,90 Compared with conventional methods, such as hot-injection and RT reprecipitation methods, the microwave-assisted method was beneficial for uniform heating and nucleation to form well-dispersed PQDs due to the high heating rate of microwave irradiation. PQDs with a narrow size distribution can reduce the bandtail state, resulting in efficient charge transport in PQD solid films.91
Another disadvantage of the traditional hot-injection method is that when scaling up the synthesis, it is really difficult to produce high-quality PQDs. Thus, Tong et al. reported a one-step ultrasonication method to fabricate PQDs, which was suitable for industrial production.92 To achieve CsPbX3 PQDs with high luminescence, a mixture of precursor salts (Cs2CO3 and PbX2) and capping ligands (OAm, OA) was directly ultrasonicated for some minutes under ambient conditions (Fig. 4d). By using the ultrasonication method, the scalable synthesis of PQDs with uniform nucleation and consistent optical properties can be obtained. Recently, Chen et al. also produced shape controllable CsPbX3 PQDs by a facile solvothermal method, which can also ensure the maximum batch to batch consistency.93 Mixing the precursor solutions with desired ligands in a stainless-steel autoclave for a certain period can yield monodisperse CsPbX3 PQDs (Fig. 4e). Similar to the microwave-assisted method, adjusting the morphology of PQDs by controlling the temperature and pre-reaction time of the above mentioned synthesis methods was practicable.
Even though different methods have been developed to directly synthesize PQDs, little attention has been paid to the chemical transformation route. Recently, a novel CsX-stripping mechanism was reported to convert non-luminescent Cs4PbX6 (no emission in the visible range) NCs into highly luminescent CsPbX3 NCs.94 This process utilized the high solubility of CsX in water to synthesize monodisperse and stable CsPbX3 NCs. During the transformation, Cs4PbX6 NCs (as a CsX-rich structure) can be stripped of excess CsX by the interfacial reaction between water and a nonpolar solvent (Fig. 4f). Akkerman et al. also proposed a simple method to transform insulator Cs4PbX6 NCs into photoactive CsPbX3 NCs by further reaction with some extra lead halide salts.95
Besides the commonly used synthesis strategies mentioned above (Table 1), some modified approaches have also been proposed to improve the optical properties of PQDs. The near-unity PLQY of all kinds of PQDs (especially Cl-based PQDs) was achieved by injecting alkylammonium halide salts into equimolar Cs and Pb (non-halide) precursor solution at an optimized temperature (220–260 °C).96 During the purification process, the highly dynamic proton exchange between OA and OAm molecules made it easy to lose surface ligands.44 The amine-free synthesis method reported by Yassitepe et al. showed a good effect on improving the stability of PQD solution, which used tetraoctylammonium halide (TOAX) salts to synthesize OA-capped PQDs.97 Luo et al. designed a novel cooling strategy called ultrafast thermodynamic control (UTC) to enhance crystal quality and finally achieve an ultrahigh PLQY of PQDs.98 The ultrafast cooling rate could avoid the generation of undesired crystals and impelled a qualitative leap in the spectral stability of mixed halide PQDs. Lignos et al. controlled and detected the formation process of CsPbX3 PQDs using a microfluidic platform.99 The rapid and controllable synthesis technology was beneficial to future standard industrial production.
| Hot injection | Room-temperature reprecipitation | Microwave-assisted synthesis | Ultrasonication synthesis | Solvothermal synthesis | |
|---|---|---|---|---|---|
| Ligand | Oleic acid (OA) and oleylamine (OAm) | ||||
| Temperature | 140–200 °C | Room-temperature | ∼160 °C | — | ∼160 °C |
| Morphology | Nanocube | Nanocube | Nanocube, nanoplate and nanorod | Nanocube and nanoplate | Nanocube and nanowire |
| PLQY | ∼90% | ∼80% | ∼75% | ∼90% | ∼80% |
| Environmental requirement | N2 | Free from inert gas | Microwave irradiation | Sonication | Solvothermal |
4. Compositional engineering of PQDs
During the construction of a PQDSC device, a PQD solid film is applied as a photoactive layer in the device. Therefore, a PQD solid film with minimized defects is preferred in the devices due to the fact that the defects in the PQD solid film will work as recombination centers for photoinduced charge carriers, leading to lowered charge extraction and thus degraded device photovoltaic performance. To improve the photovoltaic performance of PQDSCs by minimizing charge recombination, the PQDs are generally doped with some cations or anions to tune the optical and electrical properties of PQDs, as well as stabilize the crystal structure of PQDs. In this section, we will comprehensively discuss the doping principle of PQDs, and doping at different sites, such as A, B and X sites, in the perovskite crystal of PQDs to decrease charge recombination in the PQDSCs.4.1. Doping principle of PQDs
The crystal stability of PQDs is of significant importance for their application in solar cell devices. If the perovskite crystal of PQDs is not stable enough, phase transition (transformation from the black phase to a yellow phase) will occur, and thus a large number of defects will be formed in PQD solid films. These defects generally act as non-radiative recombination centers for photoinduced charger carriers, resulting in energy losses in PQDSCs.100 Eliminating vacancy defects caused by the instability of the lattice structure is an urgent issue to be solved. The compositional engineering of PQDs not only offered a doable way to stabilize the perovskite crystal structure of PQDs,101 but could also modulate the optical and electrical properties of PQDs.102 Therefore, considerable efforts have been made towards improving the crystal stability of PQDs and meanwhile lowering energy losses in PQDSCs through compositional engineering of PQDs. Fig. 5 schematically presents the crystal structure and compositional engineering of PQDs by doping PQDs with different cations (A and B site) or anions (X site) in the perovskite lattice.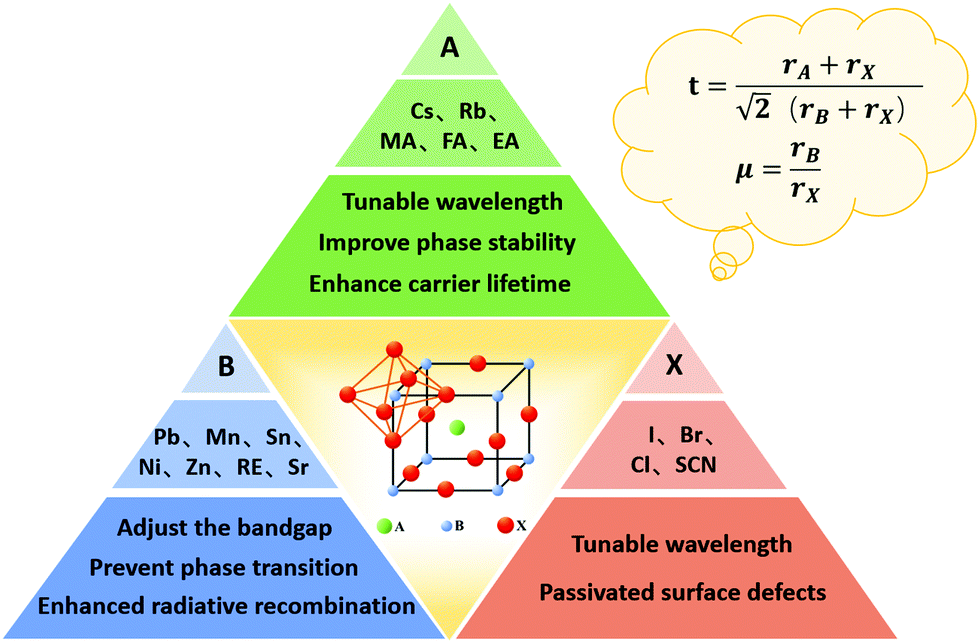 | ||
| Fig. 5 Doping of PQDs. Doping PQDs with different cations (A and B sites) or anions (X site) in the perovskite lattice. When doping PQDs with different ions, both the Goldschmidt tolerance factor and octahedral factor should be taken into account. | ||
The versatile properties of perovskite materials are primarily owing to their tunable composition, as by adjusting the composition of PQDs, their physical properties, such as optical and electrical properties, can be largely tuned. Contributing to the ionic crystalline structure of PQDs, doping PQDs with some cations (at A and B sites) or anions (at X sites) offered a relatively simpler approach than doping traditional semiconductors with covalent bond structures.103 A typical structure of perovskite ABX3 comprises a corner-shared [PbX6] octahedral framework and a large cation A, such as MA+, FA+ or Cs+, locates between the interstitials. During the doping/alloying process of PQDs, the Goldschmidt tolerance factor (GTF), t, is firstly considered, which can be expressed as
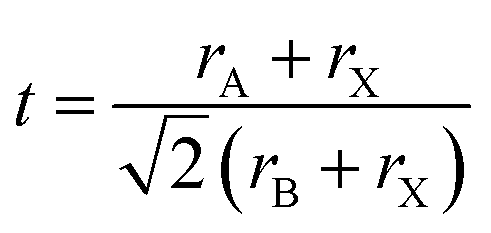
Besides regulating the perovskite lattice of PQDs according to the GTF within a specific range, the octahedral factor, μ, is also a considerable evaluation criterion for compositional engineering.108 The formula of μ is given by
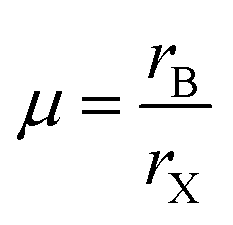
In addition to stabilizing the crystal structure of PQDs by doping ions at different sites in the crystal lattice, other purposes can also be realized for their application in solar cell devices. Firstly, the number of defects of PQDs or in PQD solids can be largely decreased using doping approaches.104,110 During the operation of PQDSC devices, the vacancies located on the PQD surface or inner PQDs will work as trap states that significantly affect the charge extraction of PQDSCs. Therefore, applicable doping could provide a valid avenue to minimize non-radiative recombination in the PQDs, resulting in an improved photovoltaic performance of PQDSCs. Secondly, a PQD solid film with enhanced optoelectronic properties can be achieved through the doping method.111 The high carrier mobility in a PQD solid film will reduce the possibility of charge carriers being captured by defects. Therefore, the influence of doping at different sites in the perovskite lattice on the defects and optoelectronic properties of PQDs will be discussed in detail in the following section.
4.2. A-Site doping of PQDs
In a typical ABX3 perovskite crystal structure, the function of the cation at the A-site is to maintain the balance of the perovskite lattice. From the perspective of the stable cubic phase structure of perovskites, the cation with too large or too small size at the A-site in the perovskite lattice will cause lattice distortion and phase transitions. Thus, an appropriate tolerance factor between 0.9 and 1 is preferred to stabilize the perovskite crystal structure. Moreover, the cation at the A-site in the perovskite lattice could also slightly change the Eg of PQDs. Upon increasing the radius of the cation at the A-site, the perovskite lattice will slightly expand and therefore result in a lower Eg with a redshift of the optical absorption band edge.112 Thus, the A-site doping of the perovskite lattice could result in a higher short-circuit current density (Jsc), leading to a higher PCE in PQDSCs.Generally, the MA+ cation has a more appropriate tolerance factor for an ideal cubic phase of PQDs. However, the hygroscopicity, volatility and thermal instability of organic cations in the perovskite lattice hindered the further photovoltaic application of organic–inorganic PQDs.20 MA-Based PQDs will be easily decomposed and the black phase of MAPbI3 PQDs turns into a yellow phase with a high Eg under heat, moisture, oxygen or light conditions. To replace volatile organic molecules, recent studies revealed that the Cs+ cation is one of the ideal substitutes that can make PQDs exhibit better thermal stability and maintain excellent photoelectronic properties.113 It has been discovered that all-inorganic halide perovskite materials generally have a higher thermal-decomposition temperature than organic–inorganic hybrid counterparts.26 However, Cs-based PQDs also face phase stability issues caused by their small GTF value (t ≈ 0.81), which affects lattice stability.114 It is worth noting that by decreasing the size of perovskite materials to the nanoscale, such as PQDs or nanocrystals, the stability of the perovskite lattice will be largely improved due to the size-induced lattice strain and enhanced contribution from surface energy, showing advantages of PQDs over perovskite bulk materials.115,116
To further increase the GTF of Cs-based perovskite materials, Li et al. presented a universal chemical composition design protocol. Specifically, introducing cations with a large radius into the CsPbI3 (small GTF value) perovskite lattice resulted in better stability of materials and devices.117 However, for a bulk organic–inorganic hybrid perovskite, a higher percentage of Cs+ cations in the perovskite lattice will induce phase segregation of the mixed systems.118 Solution-processed PQDs provide a convenient platform for doping of arbitrary component contents and it is possible to achieve desired PQDs just by simply mixing additional ions within the colloidal PQD solution. Hazarika et al. reported a cation exchange post-treatment method to tune cations at the A-site in PQDs, which exhibited tunable PL emission within a range of 650–800 nm (Fig. 6a).119 The A-site cations (FA+ and Cs+) could cross-exchange to achieve a desired alloy composition structure. Therefore, simply mixing two types of pure-component PQD solutions with different proportions could continuously adjust the PL emission peak positions. However, the occurrence of A-site cation exchange seems not as easy as the halide anion exchange in the PQDs,103 and thus a driving force for A-site cation exchange of PQDs may be needed. Fig. 6b presents a temperature-dependent cation exchange between CsPbI3 and FAPbI3 PQDs, which finally formed Cs1−xFAxPbI3 PQDs, which indicated that the thermally driven approach is one of the practical ways for A-site cation exchange of PQDs. Resulting from the quantum confinement effect,34 Cs1−xFAxPbI3 PQDs were applied for the fabrication of PQDSC devices, and the devices had a higher photovoltage than the bulk competitors.
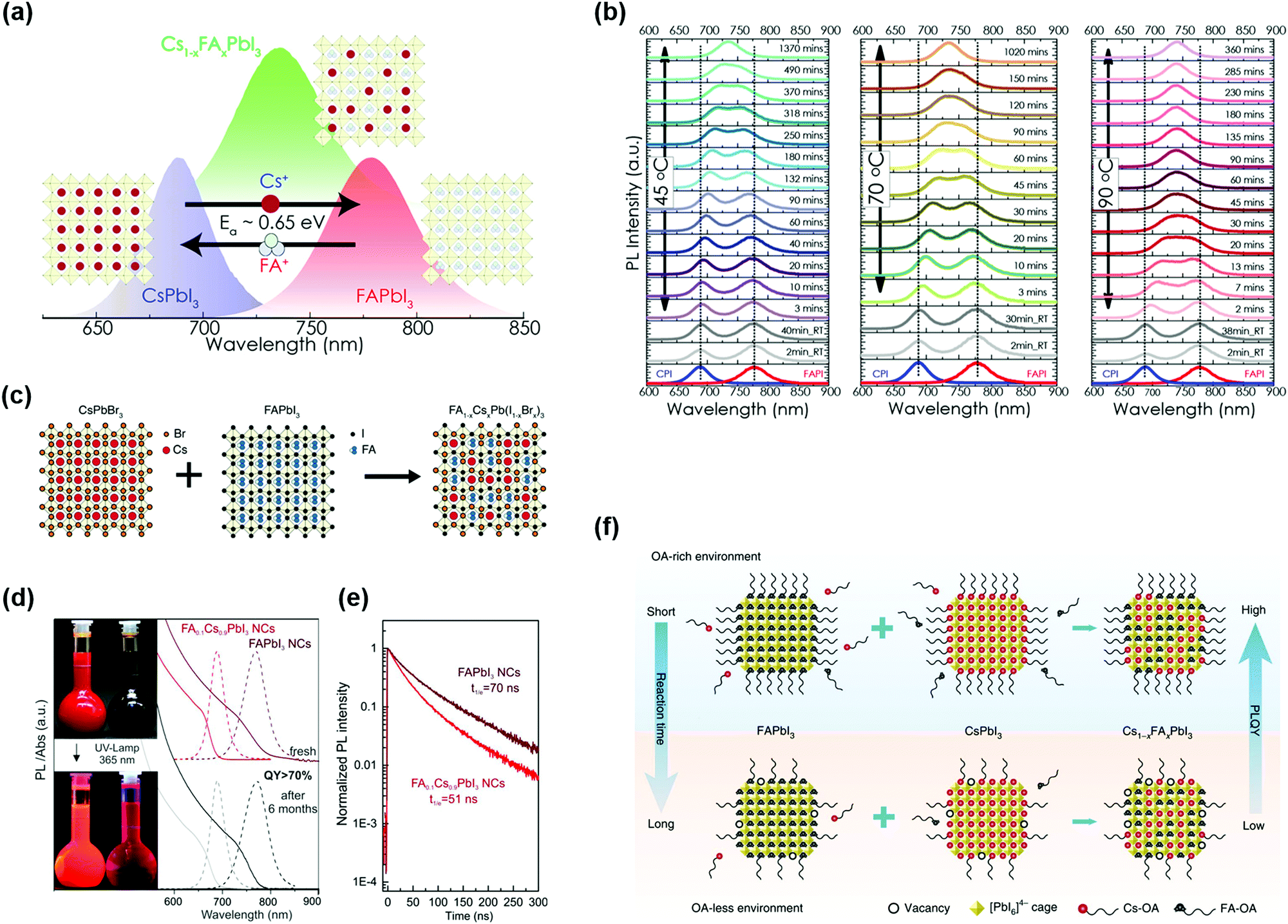 | ||
| Fig. 6 B-Site doping in the perovskite crystal structure of PQDs. (a) Illustration of the A-site cation exchange process and the corresponding PL spectra of PQDs. (b) Temperature-dependent kinetics of A-site cation exchange of PQDs. (a and b) Reprinted with permission from ref. 119. Copyright 2018 American Chemical Society. (c) Ion-exchange mechanism of FA1−xCsxPb(I1−xBrx)3 PQDs. Reprinted with permission from ref. 125. Copyright 2019 American Chemical Society. (d) Storage stability of FA0.1Cs0.9PbI3 NCs (left) and FAPbI3 NCs (right), and the corresponding absorption and PL spectra. (e) PL decay for FA0.1Cs0.9PbI3 NCs and FAPbI3 NCs. (d and e) Reprinted with permission from ref. 126. Copyright 2017 American Chemical Society. (f) Schematic diagram of ligand-assisted A-site cation exchange of PQDs. Reprinted with permission from ref. 38. Copyright 2020 Nature Publishing Group. | ||
According to the GTF formula, in order to further improve the crystal stability of the perovskite lattice, a smaller X-site anion should be introduced into the CsPbI3 perovskite lattice. But such a procedure induced serious halide segregation in the mixed halide bulk perovskites, which produced two different Eg regions under illumination. The lower Eg region served as the recombination center and impedes charge extraction in solar cell devices.120–122 Fortunately, previous studies reported that reducing charge carrier diffusion lengths was conducive to inhibiting phase separation.123 Thus, QD-based perovskite materials provided a potential way to mitigate phase separation of mixed halide perovskites. Resulting from the increased incorporation of Br− anions, mixed halide CsPb(I1−xBrx)3 PQDs presented an obvious decrease in their carrier lifetime and particularly showed a low open-circuit voltage (Voc) contrary to the expected results.124 Recently, by incorporation of additional cations and anions into PQDs, called the co-alloying method (combining A- and X-sites), the hybrid systems could be optimized, which resulted in a stable perovskite lattice (Fig. 6c).125 Suri et al. fabricated PQDSCs with wide bandgap FA1−xCsxPb(I1−xBrx)3 PQDs as a light absorber and the devices yielded a high Voc of up to 1.29 V, which shows potential as a top sub-cell for the construction of tandem solar cells.125 The device with a long carrier lifetime and wide-bandgap was obtained by means of balancing the ratio of FA+ to Cs+ and Br− to I− ions, respectively. Particularly attention needs to be paid that the alloying process also required mixing two PQD solutions at 70 °C, as described above.
To overcome the challenge of the “perovskite red wall”, Protesescu et al. synthesized highly monodisperse cubic phase FAPbI3 and FA0.1Cs0.9PbI3 PQDs, which exhibited better stability than Cs-based or MA-based PQDs.126 As shown in Fig. 6d, incorporating FA+ cations into CsPbI3 PQDs resulted in the retention of ∼70% PLQY after storage for six months. Meanwhile, a number of studies have demonstrated that FA+ rotates faster than other A-site organic cations.127 The faster rotation of A-site organic cations could lead to a better orbital overlap, which can more effectively reduce recombination and increase the carrier lifetime.128 As previously reported, FAPbI3 and FA0.1Cs0.9PbI3 PQDs exhibited a much longer carrier lifetime than all-inorganic PQDs (Fig. 6e). Additionally, they also provided a distinctive method by using FA-oleate or Cs-oleate as a precursor for A-site cation exchange. Previous studies generally promoted this cation exchange process by increasing the temperature and it was time-consuming due to the high kinetic barrier. Distinguishing from the thermally driven approach, utilization of FA-oleate or Cs-oleate as a precursor of A-site cations was a process of rearranging the corresponding atoms by virtue of vacancies. Recently, Hao et al. demonstrated a feasible ligand-assisted cation exchange strategy of PQDs, in which no heating was needed and high-quality Cs1−xFAxPbI3 PQDs with good stability were obtained.38 The oleic acid-rich (OA-rich) environment could promote A-site ion migration and diffusion in the CsPbI3 and FAPbI3 matrices (Fig. 6f). Encouragingly, the PQDSC device fabricated with Cs0.5FA0.5PbI3 PQDs delivered a certified efficiency of 16.6%, which is the highest PCE of PQDSCs reported so far. In general, A-site regulation in the perovskite lattice of PQDs produces a positive effect on the phase stability of PQDs and meanwhile offers an avenue to adjust the spectrum of PQDs.
4.3. B-Site doping of PQDs
The metal cation at the B-site in a typical perovskite lattice is generally a Pb2+ cation, together with halogen X− anions forming a [PbX6] octahedron. According to the tolerance factor and octahedral factor, the B-site cation also plays a vital role in stabilizing the perovskite crystal structure. Changing the size of B-site cations can shrink or expand the perovskite lattice and subsequently influence the GTF of the perovskite lattice, affecting the stability of PQDs. The ecotoxicity of lead-based perovskite materials is progressively becoming an obstacle to optoelectronic applications, and therefore considerable efforts have been made towards exploring non-toxic or less hazardous elements, such as Sn2+, Ge2+ and Mn2+ cations, which could replace (or partly replace) Pb2+ cations in the perovskite lattice. Thus, the desired properties of PQDs can be obtained by altering B-site cations in the perovskite lattice.With respect to the B-site substitution in the perovskite lattice of PQDs, the doping or alloying process is much more difficult than that of A- and X-site substitution owing to the high formation energy.129 Firstly, a suitable solvent that is able to dissolve additional metal salt acting as a source of B-site cations as much as possible without any damage to the PQD host is required. Secondly, the lattice strain and defects were sometimes induced by the incorporation of B-site cations with different sizes and charge numbers.130 Therefore, appropriate B-site doping in the perovskite lattice of PQDs is more arduous than that in bulk perovskite materials.131 To obtain doped PQDs with preferable properties, the most commonly used strategy is to mix metal chloride salts into the PQD precursor solutions at a high temperature.132–134 An alternative method for B-site doping in the perovskite lattice of PQDs is called the halide exchange-driven cation exchange strategy, which focused on the post-synthetic doping of PQDs.135 During the halide exchange of PQDs, the rigid [PbX6] octahedra were opened that facilitated replacement of the Pb2+ cation in the center.136
Recently, researchers found that partly replacing Pb2+ cations with Mn2+ cations in PQDs could result in better structural and optical stability of PQDs.101,135,137–141 Akkerman et al. demonstrated that the stability of CsPbI3 PQDs was largely improved after alloying CsPbI3 PQDs with Mn2+ cations.141 Partial substitution of Pb2+ cations with smaller radius Mn2+ cations will be accompanied by a lattice contraction, which is beneficial to obtaining a suitable GTF value of PQDs. As verified by density functional theory (DFT) calculations, this substitution strategy indicated that the existence of Mn2+ cations could enhance the cohesive energy of the perovskite crystal. Thus, such an optimized tactic could be capable of effectively ameliorating the thermal stability of CsPbI3 PQDs, in which the α-phase of PQDs with a high photoactivity could easily degrade to non-emitting δ-phase materials at room temperature. For Cl-based PQDs, through substitution of Pb2+ cations with Mn2+ cations, the photoluminescence quantum yield (PLQY) was significantly increased from 5% to 54%, and meanwhile, an extra PL emission at ∼580 nm was observed.142 But in I-based PQDs, due to the negligible contributions of Mn orbitals to the band edges, the optical properties of CsPbxMn1−xI3 PQDs had little change after alloying (Fig. 7a). The NCs generally exhibit high surface energy as the size of the material is becoming smaller, suggesting that it is a challenge to obtain stable PQDs with a few nanometer sizes. Yao et al. reported a strontium-substitution strategy that was beneficial to reducing the structural distortion of the perovskite lattice and hence improving the stability of α-CsPbI3 PQDs with a few nanometer sizes.143 By incorporation of Sr2+ cations into the CsPbI3 PQDs, forming CsPbxSr1−xI3 PQDs, the stability of PQDs can be improved, especially their optical stability (Fig. 7b). Meanwhile, the CsPbxSr1−xI3 PQDs had a higher PLQY value compared with CsPbI3 PQDs due to the improved radiative recombination of excitons, which is necessary for the construction of efficient solar cell devices.
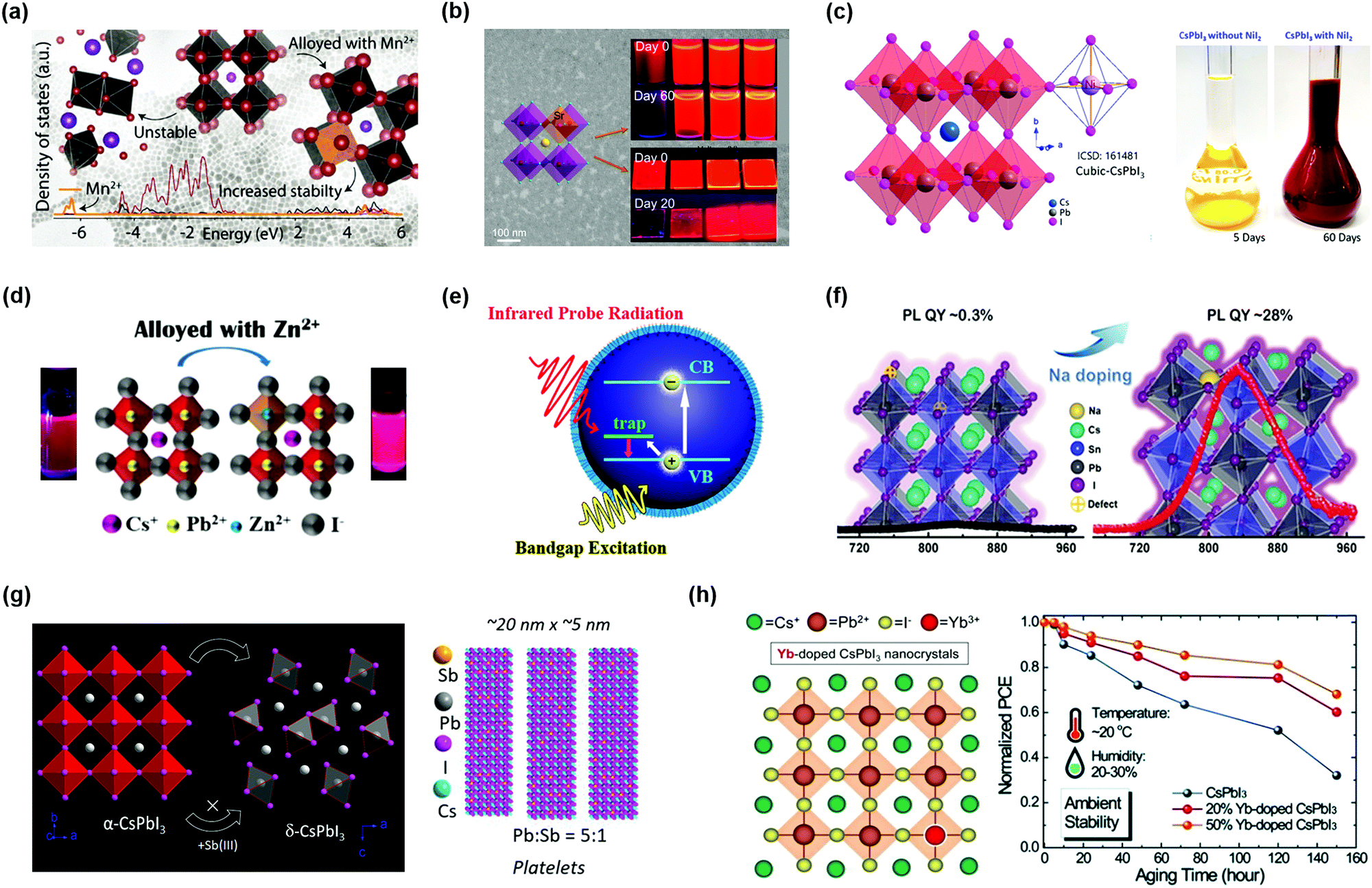 | ||
| Fig. 7 B-Site doping in the perovskite crystal structure of PQDs. (a) The density of states (DOS) of CsPbxMn1−xI3 PQDs. Reprinted with permission from ref. 141. Copyright 2017 American Chemical Society. (b) Strontium doped CsPbxSr1−xI3 PQDs and the stability of PQDs. Reprinted with permission from ref. 143. Copyright 2019 American Chemical Society. (c) The crystal structure of CsPbxNi1−xI3 PQDs and the stability of PQD solution. Reprinted with permission from ref. 144. Copyright 2019 American Chemical Society. (d) Atomic model and photographs of Zn-alloyed CsPbI3 PQDs. Reprinted with permission from ref. 114. Copyright 2019 American Chemical Society. (e) The trap-to-band transitions in the CsSn1−xPbxI3 PQDs. Reprinted with permission from ref. 147. Copyright 2017 American Chemical Society. (f) Sodium doping for reviving the near-infrared emission of CsSn1−xPbxI3 PQDs. Reprinted with permission from ref. 148. Copyright 2020 Wiley-VCH. (g) The B-site doping induced shape-changing of CsPbxSb1−xI3 NCs. Reprinted with permission from ref. 133. Copyright 2019 American Chemical Society. (h) Schematic illustration of Yb doped α-CsPbI3 PQDs and the device stability of PQDSCs. Reprinted with permission from ref. 110. Copyright 2019 The Royal Society of Chemistry. | ||
According to the GTF formula, the preferable radius of the doped cation at the B-site should be as small as possible. A few metal cations could somewhat stabilize PQDs, inhibiting phase transition. Behera et al. successfully synthesized stable Ni(II) doped α-CsPbI3 PQDs without bringing any extra PL emission features.144 Using the Ni2+ cation with a smaller Shannon radius to substitute the Pb2+ cation could suppress the octahedral tilting of the perovskite lattice and thus consolidate the perovskite structure of PQDs (Fig. 7c). In the case of violet-emitting CsPbCl3 PQDs that had a large number of intrinsic structural defects, the phase stability also showed a large improvement after implementing the Ni(II) doping strategy. Based on the experiments and theoretical calculations, Yong et al. confirmed that the presence of Ni2+ cations can increase the defect formation energy and enhance the lattice order of PQDs, thus lowering the non-radiative recombination rate.132 A similar phenomenon was also found in Zn-alloyed CsPbI3 PQDs in that the alloyed PQD-based device showed a noticeable increase in PL (Fig. 7d).114 These results indicated that the non-radiative recombination in the PQDs determined by the defects was significantly mitigated due to the increased exciton binding energy through incorporation of Zn2+ into the PQDs. Bi et al. used ZnCl2 to synthesize CsPb1−xZnxI3 PQDs and fabricated PQDSCs that gave an efficiency as high as 14.8%. The combination of Zn alloying and Cl passivation promotes PQDs with ultralow defect density.145
Another promising substitute cation to stabilize the perovskite structure of PQDs is Sn2+, which is in the same group as Pb on the periodic table that shows similar chemical properties to Pb. The Sn-based PQDs or partial replacement Pb2+ with Sn2+ provides an implemented pathway for near-infrared optoelectronic devices. However, the development of Sn-based PQDs is hindered by their poor stability.146 In order to overcome such issues, Pb2+ and Sn2+ were combined in a preferred ratio to decrease the instability of CsSn1−xPbxI3 PQDs.147 By using such a way, the light absorption of PQDs was enhanced, but the low defect formation energy in the PQDs caused by the incorporation of Sn2+ provided the possibility to form Sn vacancies in the alloyed CsSn1−xPbxI3 PQDs. Fig. 7e schematically illustrates the trap-to-band transitions in the CsSn1−xPbxI3 PQDs, indicating that the Sn-rich PQDs had some charge trapping and non-radiative recombination centers within the bandgap. The undesirable internal defects impede the stability of the PQDs and the carrier transport in the PQD solid films. Liu et al. used ultra-low Na+ cations to revive the near-infrared emission of the alloyed CsSn1−xPbxI3 PQDs (Fig. 7f).148 The Na+ doping strategy had a positive effect on enhancing the chemical interaction between Sn2+ and I−, hence suppressing the appearance of vacancy defects. Meanwhile, the Na+ dopants also increased the PL lifetime of the alloyed PQD system. However, due to the small radius of Na+ cations, they cannot remain stable in the CsSn1−xPbxI3 PQD lattice and thus the stability of the alloyed PQDs needs to be improved in future studies.
Except for alloying with divalent metal cations, heterovalent cations were also taken into account. Bera et al. prepared heterovalent Sb3+ doped CsPbxSb1−xI3 PQDs with a stable cubic phase.133 Similar to the stabilization mechanism mentioned above, doped PQDs with Sb3+also presented lattice contraction and increased cohesive energy. However, it is notable that the successful replacement of the Pb2+ cation at the B-site in the perovskite lattice is relatively limited due to the heterovalent doping. Excessively doping with Sb3+ cations will convert PQDs from nanocubes into nanoplatelets (Fig. 7g), which lowered the phase stability of PQDs under ambient exposure and deteriorated the emission efficiency. After adjusting the effective doping amount of Sb3+ cations in the PQDs, the device efficiency was increased to 9.4% and showed good stability. Likewise, lanthanide cations could be regarded as other potential candidates for doping PQDs, and by incorporation of lanthanide cations, it is possible to widely tune the spectral properties of PQDs with multicolor emission.102 DFT calculations prove that lanthanide cations can enter the perovskite structure by substituting the Pb2+ site. As the radius of the doped cations decreases, the spectrum shows a blue-shift due to the lattice contraction. Compared with undoped PQDs, the presence of impurity cations could enhance the overall PLQY of CsPbCl3 PQDs owing to their bright intrinsic emission. It is notable that some lanthanide cations, for instance, Eu3+ cations, have the capacity to remove Cl− anion vacancies in the perovskite lattice, which acted as recombination centers, as reported by Wang et al.104 A further study proved that in situ doped PQDs with ytterbium (Yb) brought an alternative route for the development of high-efficiency and stable optoelectronic devices.110 With regard to I-based PQDs, Yb3+ doping hardly changes the optical properties and original crystal structure of PQDs, whereas the Yb3+ doping could promote the thermal stability and effectively passivate the surface defects of PQDs. Therefore, the devices fabricated with Yb-doped PQDs maintained high efficiency under ambient conditions (Fig. 7h).
Overall, most of the B-site cation doping approaches had a negligible impact on the morphology of PQDs, and the PQDs maintained a cubic shape with a nearly monodisperse system, but the optoelectronic properties of PQDs could be well-tuned by using different dopants at the B-site. Moreover, the doped cations with a smaller size than Pb2+ will lead to lattice contraction and therefore improve the intrinsic stability of PQDs. Equally important, the formation of deep-level defect states can be evidently suppressed by the incorporation of dopants with increased radiative recombination in the PQDs.
4.4. X-Site exchange of PQDs
The widely tunable optical properties of PQDs can be manipulated by controlling the experimental conditions of PQD synthesis, such as the hot-injection temperature, composition and post-treatment of PQDs. The Eg of PQDs is temperature-dependent during the PQD synthesis such that with increasing hot-injection temperature, the size of the synthesized PQDs is increasing, leading to a decrease in Eg and a red-shift in the light absorption spectrum. But too high or too low hot-injection temperature will adversely affect the formation of uniform-sized PQDs. Full-spectrum luminescence in the visible region can be expeditely achieved by altering their halide composition in the PQDs, which significantly affects the Eg of PQDs. Besides altering the synthesis temperature and the kind of halide salt precursor, anion exchange processes also provide an avenue to regulate their optical properties.Due to the unstable surface properties and highly ionic crystal structure of PQDs, the anion exchange assisted by halide-ion vacancies is a convenient and efficient approach to tune the physical properties of PQDs. Nedelcu et al. reported a halide exchange process using a post-treatment strategy for the formation of homogeneous solutions (Fig. 8a).103 A halide-containing precursor, such as PbX2, oleylammonium halides (OAmX) and organometallic Grignard reagents (MeMgX), was introduced into the synthesized PQD solution and the anion exchange process can be accomplished in a relatively short time at 40 °C. The parent lattice did not change during the anion exchange process, and therefore after the anion exchange, the I-based PQDs showed better stability than directly synthesized I-based PQDs. Akkerman et al. also demonstrated that by anion-exchange reactions it is possible to adjust the spectra of CsPbX3 PQDs without changing their shape, size and crystal structure.149 Chiba et al. used ammonium iodine salts, such as oleylammonium iodide (OAM-I) and aniline hydroiodide (An-HI), to conduct anion exchange in the original CsPbBr3 PQD solution and the corresponding LED device with an external quantum efficiency (EQE) of 21.3% was successfully fabricated (Fig. 8b).70 Resulting from different surface ligand densities after anion exchange, the I-based PQDs presented longer thermal stability. Halogenated alkanes applied as feasible sources can release halide ions, which could precisely control the PQD composition.85 However, the poor solubility of organic alkyl halide salts in a nonpolar solvent restricted the anion exchange process. Yoon et al. reported a reversible post-treatment method to exchange halide ions in PQDs.150 Haloalkane solvents were used as halide sources and amalgamated with nucleophilic phosphine ligands, which were applied to accelerate the substitution reaction (Fig. 8c).151 It is proved by DFT calculations that in the presence of trioctylphosphine (TOP) the dissociation free energy of halide ions was greatly reduced. Moreover, the anion exchange of PQDs can be easily adjusted by changing the concentration of TOP in the PQD solutions and the time for anion exchange. Instead of using organic reagents, Zhang et al. reported a facile and low costly anion exchange process in that zinc halogenide salts were used as halide sources for anion exchange of PQDs.152 The PQDs obtained using anion exchange with metal halide salts could remain stable for several weeks, which was due to the fact that the ligands were still anchored onto the surfaces of the X-site exchanged PQDs.
 | ||
| Fig. 8 X-Site changes in the perovskite crystal structure of PQDs. (a) Anion exchange in CsPbX3 (X = Cl, Br, I) PQDs achieved by a series of precursors and XRD patterns of the corresponding PQDs. Reprinted with permission from ref. 103. Copyright 2015 American Chemical Society. (b) Using halide-containing long alkyl ammonium and aryl ammonium salts for anion exchange. Reprinted with permission from ref. 70. Copyright 2018 Nature Publishing Group. (c) Schematic Illustration of CsPbX3(X = Cl, Br, I) PQD synthesis and haloalkane post-treatment for anion exchange. Reprinted with permission from ref. 150. Copyright 2018 Elsevier. (d) Schematic illustrations of thiocyanate post-treatment for essentially trap-free PQDs. Reprinted with permission from ref. 43. Copyright 2017 American Chemical Society. | ||
X-Site regulation in the perovskite lattice of PQDs also provides a strategy to passivate surface defects. During the purification process of PQDs using anti-solvents (such as methyl acetate and ethyl acetate), the dissociation of surface ligands results in high-density surface defects and therefore forms shallow traps, which is detrimental to charge extraction in PQDSC devices.44,153 To address the surface defect passivation of PQDs, Koscher et al. largely improved the PLQY of aged CsPbBr3 PQDs by using thiocyanate salt (NaSCN, NH4SCN) to treat PQD surfaces, as shown in Fig. 8d.43 The introduced SCN− anion has similar size and properties to the halide anion, which can remove excess Pb2+ cations from the PQD surface. The surface state of PQDs could be completely restored after the treatment with thiocyanate salt, and thus essentially trap-free CsPbBr3 PQDs were obtained, leading to a near-unity PLQY of PQDs. Bian et al. further proved that thiocyanate anions played an important role in increasing carrier transport between PQDs.111 After capping PQDs with SCN− anions, the PCE of CsPbI3 based PQDSCs was increased by 10% compared with that of the controlled device. In addition to regulating their spectrum and passivating surface defects, X-site variation can also improve the stability of PQDs, especially for I-based PQDs. The decomposition energy of PQDs was improved by appropriate halide anion substitution and thus the stability of PQDs was improved.154 Due to the relatively low solubility of CsBr, it is difficult to prepare CsPbBr3 films for bulk materials. Fortunately, CsPbBr3 PQD ink technologies provide a possibility to prepare CsPbBr3 perovskite films and the CsPbBr3 PQDSCs show good stability with a high Voc.155
4.5. Lead-free PQDs
Up to now, most of the high-efficiency perovskite devices contain lead, a soluble heavy metal, which is harmful to the environment and human health. To solve the issues of the “lead dependence” in PQDs, it is urgent to develop low toxicity PQDs that show similar optical and electrical properties to lead-based PQDs. The alternative element that is in the same group as lead, such as tin (Sn) and germanium (Ge), provides an option to substitute lead for lead-free perovskites. Jellicoe et al. reported the synthesis and optical properties of lead-free CsSnX3 (X = Cl, Br, I) PQDs.156 By changing the halide composition in the CsSnX3 PQDs, the optical emission in the whole visible region and even in the near-infrared region can be obtained (Fig. 9a). The Sn-based PQDs had a redshift compared to Pb-based PQDs in the light absorption spectrum, which was attributed to the formation of [SnX6] octahedra that altered the Eg of PQDs.157 Due to the low defect formation energy in the Sn-based PQDs, the PLQY was seriously decreased, resulting from defect-assisted non-radiative recombination. Besides using Sn2+ as B-site cations, Zhao et al. successfully proposed the design criterion of lead-free A2M+M3+X6 (M+ = Na+, Ag+, In+, and so on, M3+= Bi3+, Sb3+) double-perovskite structures.158 Through DFT calculations and photovoltaic performance studies, some suitable structures were reported to replace Pb-based perovskites in the solar cells (Fig. 9b). Zhang et al. found that Sb-based PQDs could exhibit high stability and good optical performance as the Br-rich surfaces of Cs3Sb2Br9 PQDs prepared using a ligand-assisted reprecipitation method could remove surface defects and dangling bonds. Notably, the derived quantum-well band structure and large exciton binding energy could improve radiative recombination in the PQDs.159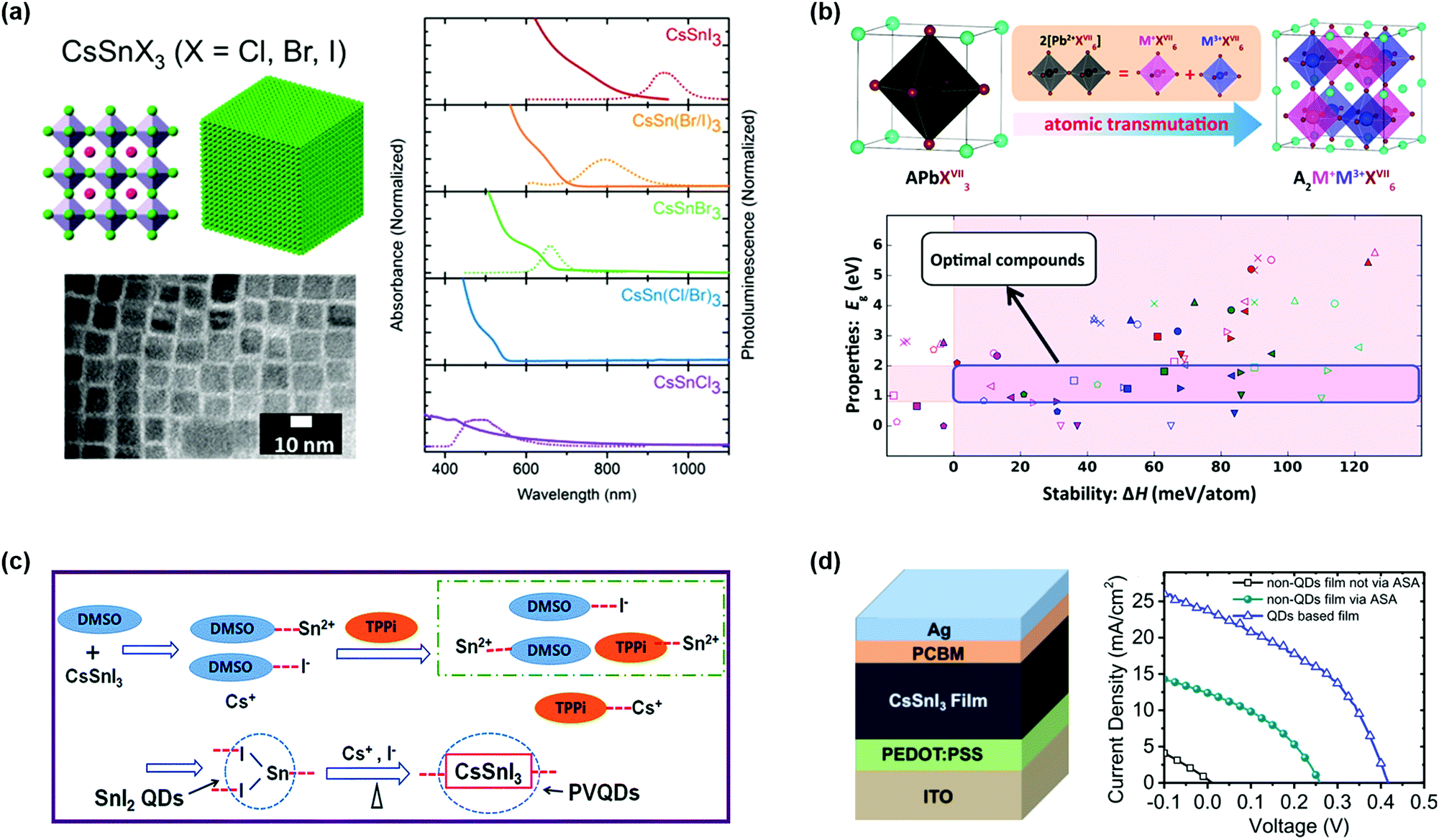 | ||
| Fig. 9 Lead-free PQDs and PQDSCs. (a) The atomic models of CsSnX3 (X = Cl, Br, I) PQDs and the corresponding absorbance and steady-state PL spectra. Reprinted with permission from ref. 156. Copyright 2016 American Chemical Society. (b) The schematic design strategy of cation-transmutation of lead-free perovskites and energy design criteria. Reprinted with permission from ref. 158. Copyright 2017 American Chemical Society. (c) Schematic diagram of the synthesis process of CsSnI3 PQDs. (d) The structure and performance of CsSnI3-based PQDSCs. (c and d) Reprinted with permission from ref. 161. Copyright 2019 The Royal Society of Chemistry. | ||
However, the oxidative degradation issues of lead-free PQDs for photovoltaic applications need to be considered. The oxidation of Sn-based perovskite, the most promising alternative to APbX3, is particularly serious. Several studies have shown that using SnF2 or excess SnI2 in bulk ASnX3 perovskites can reduce Sn vacancies and thus improve device performance.157,160 Wang et al. reported a facile synthesis method of CsSnI3 PQDs, which could inhibit oxidation from Sn2+ to Sn4+ by adding triphenyl phosphite (TPPi) used as an antioxidant solvent additive (ASA), as shown in Fig. 9c.161 Further analysis with electrical impedance spectroscopy (EIS) verified that the TPPi in the precursor solution could lower charge recombination resistance. However, the photovoltaic performance of the lead-free PQDSCs needs to be largely improved toward commercial development (Fig. 9d).
5. Surface properties of PQDs
The surface properties of PQDs play a crucial role in their optoelectronic properties, since with decreasing dot size, the specific surface area of PQDs largely increases, resulting in the distribution of a large number of atoms on the PQD surface. Thus, the optoelectronic properties and stability of PQDs are determined by the surface properties of PQDs.79 In this section, we comprehensively discuss the surface properties of PQDs from the perspectives of surface passivation using ligands and surface treatment of PQDs.5.1. Surface modification of PQDs
During the synthesis of PQDs using wet chemistry approaches, the PQD surface is generally capped with long-chain organic ligands, such as OA and OAm, which enable their uniform dispersion in nonpolar solvents, forming colloidal systems. Meanwhile, the long-chain organic ligands can efficiently passivate the surface defects of PQDs and thus eliminate shallow traps, leading to a high PLQY of PQD colloidal systems. However, the soft and ionic crystal nature of PQDs makes them quite sensitive to a polar solvent and the surface ligands do not tightly bind with surface atoms. During the purification process of PQDs, a large number of surface ligands are washed away from the PQD surface and thus create enormous amounts of undercoordinated atoms, which induces surface defect formation, impeding charge collection and transport in PQDSC devices.79,162 Moreover, the solvent with a high polarity could even cause a phase transition of PQDs from the black phase to yellow phase.32,68,163 Although PQDs have better defect tolerance than metal chalcogenide QDs, the presence of non-negligible surface defects still causes a serious energy loss in photovoltaic devices,81,164 which indicates that finely controlling the surface properties of PQDs is critical for the development of highly efficient and stable PQDSCs.Considering the binding behavior between PQD surfaces and ligands, the interaction between PQDs and surface ligands can be described according to the classification of covalent bonds between surface ligands and PQDs. Three types of ligands are generally applied to passivate PQD surfaces, which are called L-, X- and Z-type ligands that contribute two electrons, one electron, and zero electron, respectively, to bind to the dot surface (Fig. 10a).165 For instance, protonated oleylammonium can combine with X-site halide ions by weak hydrogen bonding or interact with the deprotonated OA to form X-type ligand pairs, which are located on the PQD surface. Recent computational studies revealed that the protonated OAm could fill Cs+ vacancies on the PQD surface.153,166 Due to the ionic crystal nature of PQDs, highly dynamic binding between the PQD surface and organic ligands often occurred, and the native ligand on the PQD surface would gradually be removed after purification of PQDs and deposition of PQD solid films (Fig. 10b).44,69,167,168 The imbalance of surface ligands (such as OA and OAm) of PQDs promoted that the CsPbX3 perovskite structure showed potential to transform into other non-luminescent structures, such as Cs4PbX6.169 Moreover, the atoms on the PQD surface, such as Cs+ and halide ions, were easily separated from the perovskite crystal under the influence of anti-solvents, leading to the formation of surface vacancies and agglomeration of PQDs.170 As shown in Fig. 10c, for the PQDs with a CsX terminated facet, the imperfect PQD crystal generally presents two types of surface defects, named A-site and X-site vacancies, respectively. Nenon et al. studied the relationship between the surface chemistry and properties of PQDs by combining experimental studies and theoretical calculations.153 Compared with the Br- and I-based PQDs, the concentration of surface halide vacancies in the Cl-based PQDs had a more negative impact on the PLQY of PQDs because of deep midgap states in the Cl-based PQDs.171 The undercoordinated Pb atoms on the PQD surface (also known as halide vacancies) generally act as charge-trapping centers, which can inhibit charge extraction in the photovoltaic devices. Therefore, surface defect passivation of PQDs plays a pivotal role in achieving high-performance photovoltaic devices.
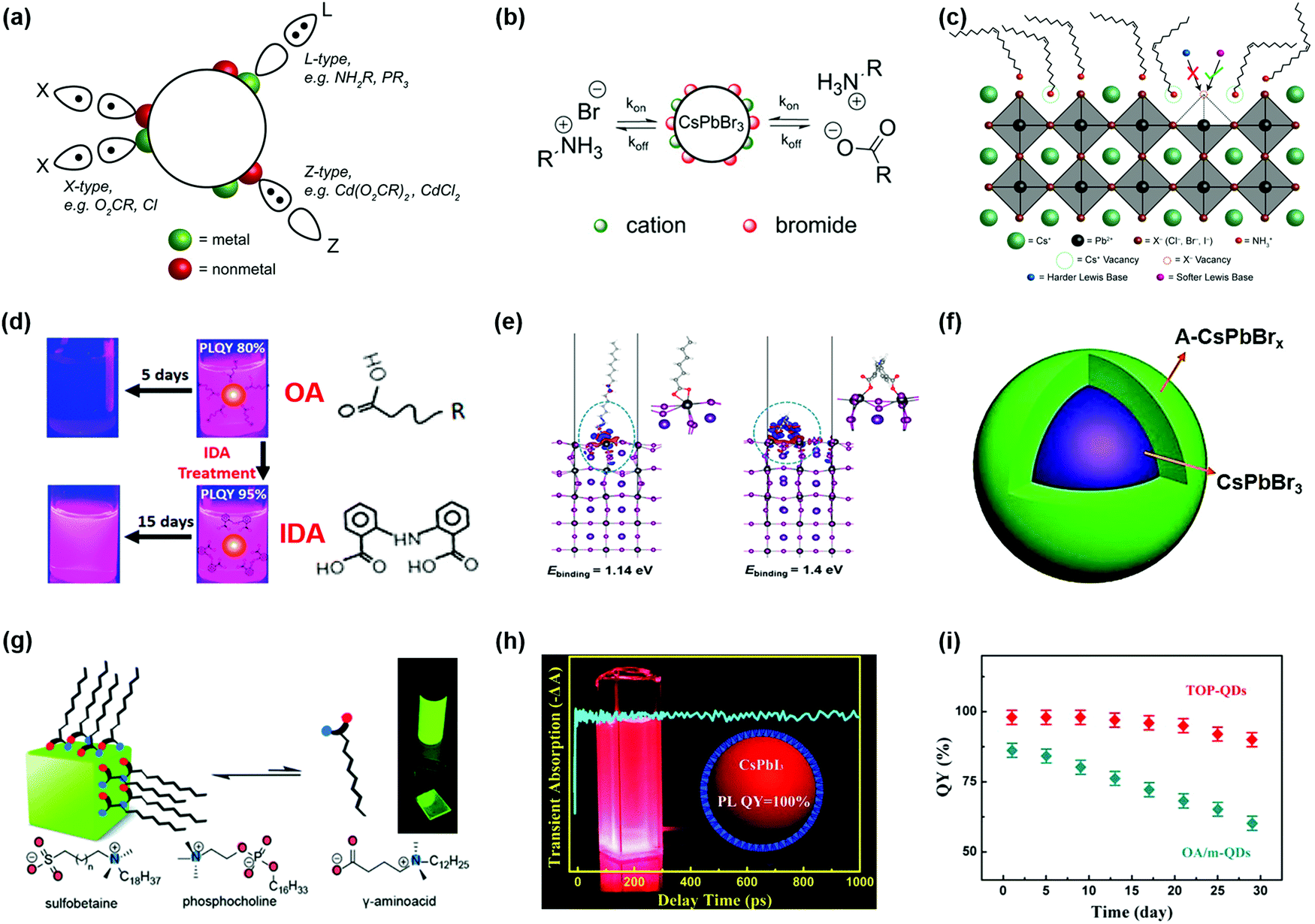 | ||
| Fig. 10 Surface modification of PQDs with different surface ligands. (a) The covalent bond classification scheme of PQDs. Reprinted with permission from ref. 153. Copyright 2018 American Chemical Society. (b) Schematic diagram of the dynamic change of surface ligands in CsPbBr3 PQDs. Reprinted with permission from ref. 44. Copyright 2016 American Chemical Society. (c) Schematic diagram of a CsX-terminated PQD structure. Reprinted with permission from ref. 153. Copyright 2018 American Chemical Society. (d) Photographs of untreated and bidentate ligand treated CsPbI3 PQDs and molecular structures of OA and IDA ligands. (e) DFT calculations of charge redistributions for OA and IDA passivated PQD surfaces. (d and e) Reprinted with permission from ref. 173. Copyright 2018 American Chemical Society. (f) Schematic diagram of CsPbBr3 PQDs with an amorphous CsPbBrx(A-CsPbBrx) shell. Reprinted with permission from ref. 186. Copyright 2017 American Chemical Society. (g) Several zwitterionic capping ligands for improving the chemical durability of PQDs. Reprinted with permission from ref. 188. Copyright 2018 American Chemical Society. (h) Photos of TOP-prepared CsPbI3 QDs with high PLQY. (i) The QY stability of TOP-PQDs and OA/m-PQDs. (h and i) Reprinted with permission from ref. 199. Copyright 2017 American Chemical Society. | ||
The highly dynamic binding between PQD surfaces and ligands provides a possibility of ligand exchange in the PQDs. Recent studies have shown that softer X-type Lewis bases (such as organic carboxylates and thiophene) were available to combine with undercoordinated Pb atoms on the PQD surface.153,172–174 Pan et al. used a bidentate ligand, called 2,2′-iminodibenzoic acid (IDA), to fully passivate the surfaces of CsPbI3 PQDs.173 The post-treatment of CsPbI3 PQDs not only promoted carrier transport between PQDs but also enhanced the stability of PQD solution due to the chelate effect of the bidentate ligand (Fig. 10d).175 In contrast to long-chain OA ligands, the double carboxylic acid groups can simultaneously passivate two exposed Pb atoms on the PQD surface, resulting in a much higher binding energy between PQDs and ligands (Fig. 10e). Besides the carboxylic acid groups, the halide salts were also beneficial to passivating the surface defects of PQDs, which released halide ions to decrease halide vacancies on the PQD surface.86,176–181 Dai et al. used 1,8-octyldiamine bromide salt (BOABr2) to alleviate surface bromine vacancies in CsPbBr3 PQDs, which were synthesized by a room temperature recrystallization method.181 The double-terminal diamine bromide salts can efficiently passivate the surfaces of PQDs and at the same time also replace part of insulated OAm, which sharply improve charge transport in the PQDs. Wang et al. introduced benzyl iodide (BI) salts to improve the PLQY of CsPb(Br/I)3 PQDs by reducing iodine vacancies in the PQDs, while changing the energy level of PQDs through an aromatic ring made charge extraction more efficient.182 As the above demonstrates, the Cl-based PQDs generally suffer from extremely low PLQYs due to the mid-gap traps of Cl-deficient surfaces.153 To overcome such issues, Ahmed et al. used YCl3, a trivalent metal chloride salt, to passivate the surfaces of CsPbCl3 PQDs and enhanced the PLQY up to 60%.183
As mentioned above, during separation and purification of PQDs, the undercoordinated Pb atoms are produced as halide ions on the dot surface were dislodged from the perovskite crystal. Another strategy used to eliminate surface defects, resulting from undercoordinated Pb atoms was to remove supernumerary Pb atoms from the PQD surface. Koscher et al. modified Pb-rich PQD surfaces with thiocyanate salts to achieve near-unity PLQYs of CsPbBr3 PQDs.43 The stoichiometric ratio of Pb to Br atoms reached 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 after removing supernumerary Pb atoms from the PQD surface. To fully remove excessive Pb atoms from the PQD surface, Ahmed et al. applied tetrafluoroborate salts to treat PQD surfaces, which could improve the PLQY of CsPbX3 PQDs, especially blue-emitting Cl-based PQDs.184 Similarly, Wang et al. used hydrated nitrates to bind uncoordinated cations on the surface, and finally a complete surface and almost no defects in PQD were obtained.185
:3 after removing supernumerary Pb atoms from the PQD surface. To fully remove excessive Pb atoms from the PQD surface, Ahmed et al. applied tetrafluoroborate salts to treat PQD surfaces, which could improve the PLQY of CsPbX3 PQDs, especially blue-emitting Cl-based PQDs.184 Similarly, Wang et al. used hydrated nitrates to bind uncoordinated cations on the surface, and finally a complete surface and almost no defects in PQD were obtained.185
Inspired by the studies on metal chalcogenide QDs,78 Wang et al. designed and synthesized cubic CsPbBr3@amorphousCsPbBrx PQDs with a core/shell structure using a hot-injection method (Fig. 10f).186 Compared with the crystal phase of CsPbBr3 PQDs, the amorphous shell could enhance the absorption ability of core CsPbBr3 PQDs and increase the PLQY up to 84% with blue emission. Furthermore, the special core/shell structure also exhibited superior chemical stability owing to an amorphous shell on the crystal phase of CsPbBr3 PQDs. Tang et al. constructed core/shell QDs by coating CsPbBr3 nanocrystals with a CdS shell, which exhibited a higher PLQY due to their reduced surface defects.187 The core/shell structure can limit the migration of carriers in the PQDs through a protective shell with a high Eg. Therefore, it is necessary to use materials with suitable energy band structures as shells of QDs. Moreover, it is notable that when the core/shell structure of PQDs was used to improve device performance, the degree of lattice mismatch between core and shell materials should be well considered to minimize interfacial defects at the core/shell interface.78,187
Though OA/OAm ligands are widely used as native ligands for the synthesis of PQDs, the weak binding energy between PQD surfaces and ligands generally induces desorption of ligands from the PQD surfaces, resulting in a dramatic decrease in the PLQY and stability of PQDs, and even phase transition.36,97 Therefore, it is feasible to introduce functional molecules as substitutes for OA/OAm ligands. Krieg et al. reported a novel ligand capping strategy that replaced conventional OA/OAm ligands with zwitterionic molecules.188 Long-chain zwitterionic ligands, such as sulfobetaine containing both anion and cation groups, could coordinate with the corresponding atoms on the PQD surface (Fig. 10g), which showed a much better chemical durability compared with conventional OA/OAm ligands.189 Yang et al. applied dodecylbenzene sulfonic acid (DBSA) as a single ligand to passivate exposed Pb atoms by filling bromide vacancies (VBr), also called the “Br-equivalent” ligand strategy.170 Imran et al. used quaternary ammonium bromides (R4NBr) to simultaneously passivate anion and cation vacancies, while ensuring high PLQYs and greatly improving the colloidal stability of quantum dots.190 In addition to replacing organic acid ligands, studies also suggested that incorporation of didodecyl dimethyl ammonium bromide (DDAB) into PQDs for partial replacement of amine ligands can improve the optical properties of PQDs.191–193 Li et al. applied π-conjugation ligands, such as phenethylamine (PEA) molecules, to enhance charge carrier transportation within PQDs.194 Dai et al. used an aromatic amine molecule, such as 3-phenyl-2-propen-1-amine (PPA), to replace long-chain hydrocarbon amine ligands, which not only improved the stability of PQDs but also enhanced carrier mobility owing to the higher conductivity of conjugated PPA ligands.195
Phosphorus-containing organic molecules were also regarded as functional ligands for the synthesis of metal chalcogenide QDs. The lone pair electrons of phosphorus atoms make it possible to dissolve insoluble precursor salts, leading to a strong interaction between the phosphorus atom and Pb atom.196–198 Liu et al. used the TOP as a coordinating solvent to prepare CsPbI3 PQDs, which showed nearly 100% PLQY and good stability (Fig. 10h).199 Although no phosphorus signal was detected on the PQD surface, the PQDs synthesized using TOP-PbI2 precursor (namely TOP-PQDs) had a better crystal quality and fewer interior defects compared to the PQDs synthesized with a traditional OA/OAm-based route. As shown in Fig. 10i, the TOP based PQDs can retain more than 85% quantum yield (QY) after storage for one month. Moreover, PQDs with branched capping ligands always have better chemical stability due to the steric effect of their surface ligands, such as trioctylphosphine oxide (TOPO) and (3-aminopropyl)triethoxysilane (APTES).198,200 We could conclude here that the surface defects of PQDs are effectively passivated by using post-treatment or improving the synthesis strategies of PQDs to enhance the optoelectronic properties and stability of PQDs toward efficient and stable photovoltaic devices.
5.2. Surface ligand regulation of PQDs
Through extensive studies on the nanocrystal nucleation and growth processes of PQDs, the interaction between perovskite precursors and surface ligands was fundamentally understood and it was found that the long hydrocarbon chain ligands played an important role in synthesizing PQDs and stabilizing dot surfaces.201 The carboxylic acids and amines with different chain lengths were used to regulate the size and morphology of PQDs.35,36 The native ligands significantly affected the reactivity of perovskite precursors, allowing fine control over the growth rate of PQDs. During the initial nucleation stage of PQDs, the ligands had a positive effect on the reactivity of precursors. Whereas during the grain growth stage of PQDs, the anchoring headgroup is attached to the surface atoms of PQDs and thus restricted the growth of nanocrystals.79 However, the long-chain insulating ligands on the dot surface usually obstructed carrier transportation within PQD solid films and thus induced charge recombination, affecting their photovoltaic performance. Therefore, to improve the conductivity of PQD solid films, the original long-chain ligands were always replaced with shorter ones through solid-state or liquid-state ligand exchange. Currently, most PQDSCs with a high-performance rely on solid-state ligand exchange.Two kinds of surfactants (OA and OAm) are generally applied to the synthesis of PQDs, which makes ligand exchange of PQDs more complicated than that of metal chalcogenide QDs. During the construction of PQDSC devices, a solid-state ligand exchange method with a two-step ligand exchange process is generally carried out to thoroughly remove long-chain OA and OAm ligands, as presented by Luther and coworkers.202 Generally, the original OA ligands are partially replaced with acetic acid by immersing the as-deposited PQD solid films in methyl acetate (MeOAc) solution. It is worth noting that the participation of adventitious water molecules is required to hydrolyze MeOAc to acetate (COO−). During the second step ligand exchange, when the PQD films reach the required thickness, the remaining OAm ligands need to be removed by introducing some short-chain ligands, such as MAI and FAI, which should be regulated within a narrow dynamic range (Fig. 11a). After solid-state ligand exchange, it is necessary to ensure the stability of CsPbI3 PQD solid films and simultaneously promote charge carrier extraction and transportation within PQDSCs.
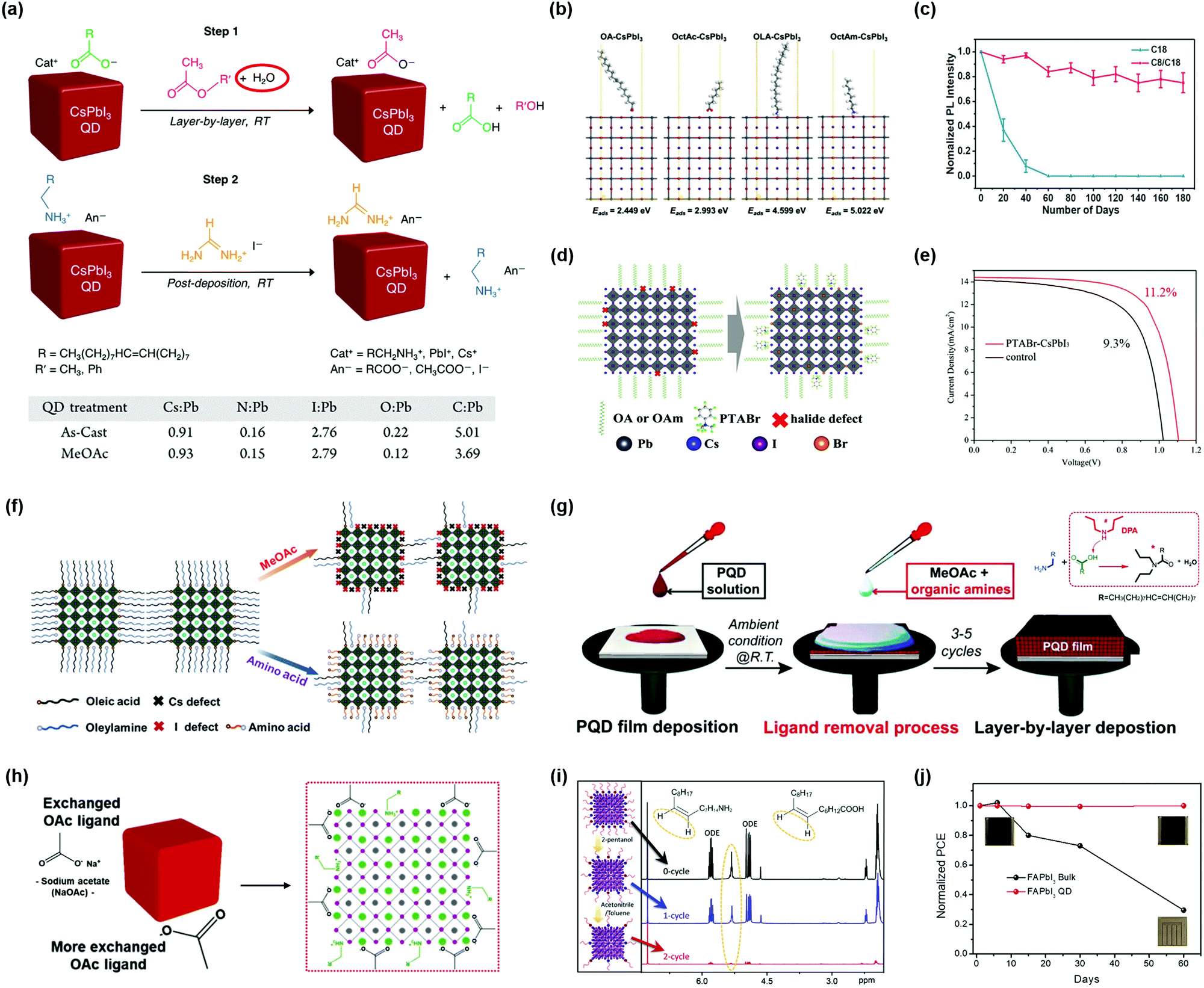 | ||
| Fig. 11 Surface ligand regulation of PQDs. (a) Ligand exchange process and element analysis of CsPbI3 PQD solid films. Reprinted with permission from ref. 202. Copyright 2018 American Chemical Society. (b) Adsorption energies of C18 capped and C8/C18 co-capped CsPbI3 PQDs (c) PL stability of PQDs capped with different ligands. (b and c) Reprinted with permission from ref. 41. Copyright 2019 Wiley-VCH. (d) Schematic diagram of CsPbI3 PQDs passivated with PTABr. (e) Device performances of PTABr-CsPbI3 PQDSCs and control devices. (d and e) Reprinted with permission from ref. 203. Copyright 2019 Wiley-VCH. (f) Schematic illustration of typical MeOAc rinse and dual-passivation with amino acids. Reprinted with permission from ref. 46. Copyright 2020 Wiley-VCH. (g) Illustration of the ligand removal process using organic amines. Reprinted with permission from ref. 204. Copyright 2020 Wiley-VCH. (h) Schematic of NaOAc-assisted ligand exchange in CsPbI3 PQDs. Reprinted with permission from ref. 208. Copyright 2019 Elsevier. (i) NMR spectra of FAPbI3 PQD solid films with different cycles of surface treatment. (j) Device stability of FAPbI3 PQDSCs and the corresponding bulk PSCs. (i and j) Reprinted with permission from ref. 167. Copyright 2018 Elsevier. | ||
Chen et al. introduced short-chain capping ligands, such as octanoic acid and octylamine (OctAc and OctAm, C8), into the synthesis of CsPbI3 PQDs to partly substitute original insulating ligands (OA and OAm, C18) and further enhance the charge carrier extraction of PQDSCs.41 The DFT calculation indicated that the adsorption energy (Eads) of the C8-capped surface on the PQD surface was stronger than that of native C18-capped ligands (Fig. 11b). Therefore, the surface defects produced due to the weak binding energy of primitive ligands were largely avoided, and both the phase stability of α-CsPbI3 PQDs and the corresponding device photovoltaic performance were improved (Fig. 11c). The short-chain ligand substitution strategy may provide an avenue to alleviate the adverse effects of insulated capping ligands on charge transport within PQD solid films. To optimize the quality of PQD solid films toward scalable fabrication of CsPbI3 PQDSCs, Yuan et al. reported a spray-coating technology with a continuous automatic spraying and purification function for a large area deposition.203 The phenyltrimethylammonium bromide (PTABr) molecule was introduced to passivate the surfaces of CsPbI3 PQDs and further improve the device efficiency, in which the PTA group served the same function as amine ligands, and Br− ions could fill the iodine vacancies of the perovskite crystal (Fig. 11d). Finally, the device efficiency was improved to 11.2% through the combined effect of spray-coating and ligand engineering, providing a feasible strategy for future large-scale preparation of PQDSC devices (Fig. 11e).
During the layer-by-layer deposition of PQD films, Jia et al. proposed a simple one-step ligand exchange approach to regulate the surface characteristics of CsPbI3 PQDs, which provided a workable strategy to reduce surface defects during the spin-coating of each layer.46 Using amino acid as a dual passivation ligand could contemporaneously passivate cationic (Cs+ vacancies) and anionic (I− vacancies) surface defects caused by an antisolvent immersion process (Fig. 11f). The device photovoltaic performance was significantly improved due to the combination of reduced defects on the PQD surface and the improved carrier mobility of PQD solid films. Moreover, this ligand exchange strategy also avoided using time-sensitive FAI post-treatment. Recently, Wang et al. proposed a different ligand removal strategy, which resulted in the improved coupling between dots by using di-N-propylamine (DPA) to control the surface ligand density.204 The acylation reaction between the secondary amine and OA was used to promote removal of the original ligands in PQDs (Fig. 11g). Thanks to the improved electronic coupling and reduced recombination of PQDs, the device efficiency was increased to nearly 15%. In the conventional solid-state ligand exchange of PQDs, the production of COO− was accompanied by a series of side reaction products (such as methanol and protons), which had negative impacts on the perovskite crystal. To address such issues, the ionic compound NaOAc was used to directly generate COO− anions for more efficient exchange with OA ligands, as shown in Fig. 11h. Meanwhile, during such a ligand exchange process, fewer metal hydroxide (Pb-OH) species were produced, which have been proved as trap states in PbS CQDs.205
The weak interaction between polar organic FA+ cations and inorganic [PbX6] frameworks makes it more challenging to maintain colloidal structural integrity during the separation and purification processes of FAPbI3 PQDs.11 Xue et al. used gradient solvents with appropriate polarity to regulate surface ligand density and achieved a good balance between dispersion and charge carrier transport (Fig. 11i).167 FAPbI3 PQDSCs were successfully prepared, and an efficiency of 8.38% was achieved, which also showed better stability compared to bulk competitors (Fig. 11j). This surface ligand manipulation method was also applied to CsPbBrI2 PQDSCs by Liu et al.206 Therefore, during the fabrication of PQDSC devices with solid-state ligand exchange techniques, thorough removal of long-chain ligands to ensure unhindered charge transport and dot coupling is largely required. Meanwhile, introducing specific functional ligands could reduce surface defects and enhance electronic coupling between dots.39 Due to the unstable ionic crystal structure of perovskites, liquid-phase ligand exchange for PQDs is very difficult. Very recently, Dong et al. successfully realized the ligand exchange process of PQDs in the liquid-phase with a bipolar-shell, which can produce PQD ink with high mobility and low defect density through electrostatic protection and resurfacing processes.207
Hence, surface ligand regulation engineering provides an important way to improve the photovoltaic performance and stability of PQDSC devices. However, new ligands or detailed processes of ligand exchange in both the liquid-state and solid-state may need more fundamental studies to thoroughly remove long-chain ligands on the PQD surface and meanwhile maintain the high stability of PQD solid films.
5.3. Post-treatment of PQD solid films
Surface ligand engineering of PQDs provides a feasible strategy to improve the photovoltaic performance of PQDSCs by decreasing charge recombination in the PQD solid film. Additionally, to obtain high-efficiency solar cell devices, various studies have focused on surface defect passivation of PQDs by using post-treatment of PQDs or PQD solid films and thus improve electronic coupling between dots.As described above, during the layer-by-layer deposition of PQD solid films using the solid-state ligand exchange method, the original long-chain amine ligands are still retained on the PQD surface after soaking PQD solid films in MeOAc solution, hindering charge carrier transport in the PQD solid films.202 To thoroughly remove long-chain amine ligands, Sanehira et al. reduced the density of original surface ligands through post-treatment of PQDs with A-site cation halide (AX) salt, which showed that the A-site cations could substantially replace the surface amine ligands of PQDs (Fig. 12a). It is worth noting that the post-treatment of PQDs with optimized AX salt only had effect on the dot surface and did not cause the alloying, shell coating or Ostwald ripening phenomenon of PQDs. The results of the charge carrier dynamics of PQD solid films suggested that the carrier mobility of CsPbI3 PQD solid films with FAI post-treatment was nearly double that of the original CsPbI3 PQD solid film without any post-treatment (Fig. 12b). Meanwhile, the post-treatment of CsPbI3 PQDs had a negligible effect on the carrier lifetime, whereas the electronic coupling between PQDs was significantly improved and the PQDSC gave a recorded efficiency of 13.43% in 2017 (Fig. 12c).39
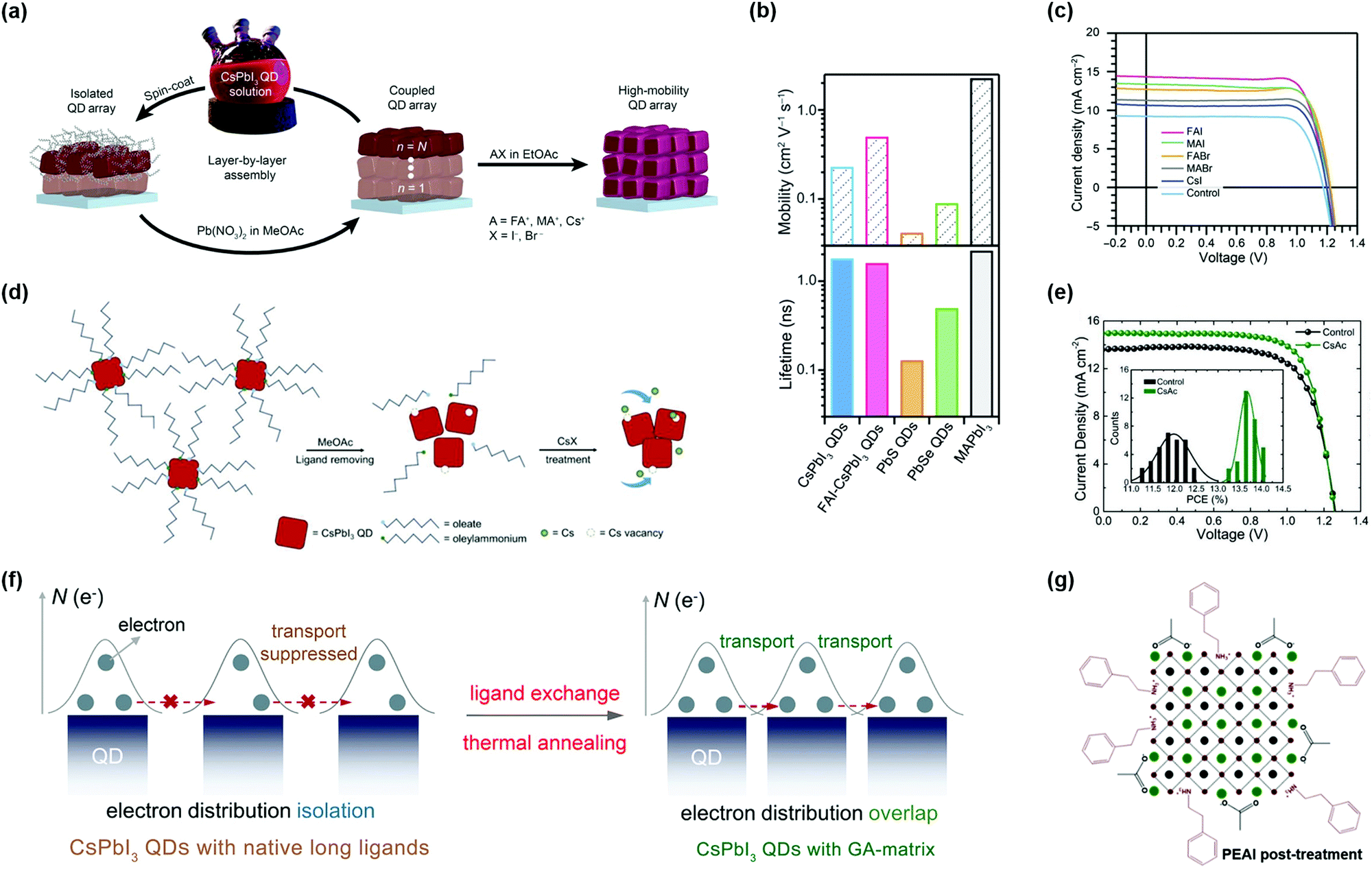 | ||
| Fig. 12 Post-treatment of PQDs or PQD solid films. (a) Deposition of CsPbI3 PQD solid films with AX salt post-treatment. (b) The mobilities and lifetimes of PQD solid films. (c) J–V curves of PQDSCs with different AX salt post-treatments. (a–c) Reprinted with permission from ref. 39. Copyright 2017 American Association for the Advancement of Science. (d) Schematic diagram for the CsX post-treatment process. (e) J–V curves and PCE distribution histograms of CsPbI3 PQDSCs with and without post-treatment. (d and e) Reprinted with permission from ref. 45. Copyright 2019 Wiley-VCH. (f) Illustration of carrier transport in a CsPbI3 solid film and a GA-matrix CsPbI3 PQD solid film. Reprinted with permission from ref. 40. Copyright 2020 Wiley-VCH. (g) Schematic illustration of CsPbI3-PQD with PEAI post-treatment. Reprinted with permission from ref. 215. Copyright 2020 Elsevier. | ||
The purification of the as-synthesized PQDs and deposition of PQD solid films assisted by anti-solvent also generated amounts of surface vacancies, resulting in defect-assisted non-radiative recombination in the PQDs. To decrease the surface defects of PQDs, different cesium salts were applied for the post-treatment of PQD solid films by filling Cs vacancies on the PQD surface to improve charge extraction of PQDSCs (Fig. 12d).45 The absence of vacancies (defects) could reduce undesired non-radiative recombination in the PQDs, leading to improved photocurrent density. A slight redshift in the absorption and PL spectra of PQD solid films revealed that the electronic coupling between individual dots was largely enhanced after post-treatment of PQD solid films with cesium acetate (CsAc).209 Thanks to the improved defect passivation and electronic coupling between dots, the performance of PQDSCs was largely enhanced and an efficiency of up to 14.1% was obtained (Fig. 12e).
Compared with bulk inorganic PSCs, PQDSCs still had a lower photocurrent density likely due to carrier recombination in PQD solid films, which indicated that there is still room for improving the photovoltaic performance of PQDSCs.28 Therefore, further increasing charge extraction is a crucial point to enhance the efficiency of PQDSCs. The guanidine (GA+) group molecule has been widely used in two-dimensional perovskite materials, which could significantly improve the stability of perovskites films and meanwhile eliminate interfacial defects within perovskite films.210–212 Ling et al. achieved a device efficiency of 15.21% by combining post-treatment of PQDs with guanidinium thiocyanate (GASCN) and a mild thermal annealing process (called an “LE-TA” strategy).40 The GA+ cations can adsorb on the PQD surface to replace the original amine ligands and fill surface defects during the post-treatment of PQDs. Meanwhile, the reduced dot-to-dot distance after the GASCN treatment also enhanced electronic coupling between PQDs, thereby improving the carrier mobility of PQD solid films (Fig. 12f). The further thermal annealing process of PQD solid films was beneficial to forming smooth and closely-packed PQD solid films, reducing the distance between dots. Applying such a post-treatment method for Br-based PQDs could also produce stable solar cells with a high photovoltage, which were expected to be used for semitransparent devices.213
Overall, the post-treatment of PQDs with AX salt offered a good method to manage the surface chemistry of PQDs. However, the post-treatment of PQDs with AX salt sometimes had issues. For instance, the PQDs with FA+ cations on the dot surface that came from post-treatment of PQDs showed poor humidity resistivity, lowering device stability under ambient conditions.202 Compared with three-dimensional (3D) perovskite materials, low-dimensional perovskite materials have better moisture stability and thus more studies have begun to combine these two types of perovskite materials.214 Based on previous studies on the NaOAc exchange (called Ac-exchange) strategy, Kim et al. developed a new post-treatment method of PQDs to replace OAm ligands with the PEA group (Fig. 12g).215 The PEA group, an aromatic organic cation with a larger size than that of Cs+ cations, can effectively prevent the phase decomposition of perovskite materials caused by water molecules.194,216 The PEA groups can successfully replace OAm ligands on the PQD surface and thus the electronic coupling between dots was largely improved. Finally, the device efficiency of PEA-based PQDSCs was improved to 14.1% and maintained excellent stability within 15 days.
6. Device operation
Among various PQDs, iodine-based PQDs were believed to be the most promising photoactive materials for the construction of PQDSCs, and the device efficiency has improved dramatically in the past years, as summarized in Fig. 13. A typical PQDSC device generally consists of five functional parts to minimize charge recombination that occurred in the PQD solid films or at the interfaces within devices. The first functional part is a transparent electrode, such as indium tin oxide (ITO) or fluorine-doped tin oxide (FTO) on a glass or flexible substrate, that allows incoming light to reach the photoactive layer. Secondly, on both sides of the photoactive layer are an electron transport layer (ETL) and a hole transport layer (HTL), respectively, which could efficiently extract photoinduced electrons or holes from the photoactive layer. The most important layer in the PQDSC device is the PQD solid film that produces photocarriers after harvesting photons. Under the built-in electric field or by diffusion, the photocarriers transport to the ETL or HTL. A metal film, such as Ag, Au, Al or Cu, prepared by a thermal evaporation method is generally applied as a back-contact electrode. Therefore, finely regulating each functional layer and interface within the device is critical to suppressing unexpected carrier recombination in solar cells. | ||
| Fig. 13 Device efficiencies of PQDSCs. The PCE evolution of iodine-based PQDSCs with different cations (the square frames in red represent the certified efficiencies by the NREL).34,38–41,45–50,110,119,133,167,203,204,208,215,217–221 | ||
In the above sections, we discussed in detail the feasible ways to reduce the non-radiative recombination and increase the carrier mobility of PQD solid films through compositional engineering and surface chemistry regulation of PQDs. However, the device performance not only largely depends on the PQD solid films, and the device operation also significantly affects the photovoltaic parameters (such as Voc, Jsc, and fill factor (FF)) of solar cell devices. Compared to bulk PSCs, the original long-chain ligands of PQDs largely hinder the charge extraction of PQDSCs, which affects the Jsc of PQDSCs. Currently, the device Jsc is still largely lagging behind the theoretical values, which suggests that it is possible to improve the device Jsc by increasing the thickness of PQD solid films and decreasing charge recombination. Meanwhile, the solid-state ligand exchange of PQDs and the layer-by-layer spin-coating method of PQD layers are generally combined to achieve a PQD solid film with an enough thickness (300–500 nm). The multi-interfaces within the PQD solid films may lead to increased series resistance and charge recombination, which could lower the device FF and Voc. Therefore, finely regulating the device structure could decrease the energy loss in the device and thus improve the photovoltaic performance of PQDSCs.
In this section, we comprehensively discuss the effect of functional layers on photovoltaic performance by decreasing charge recombination.
6.1. Device architectures and working mechanisms
So far, different device architectures of PQDSCs have been developed to take advantage of the unique properties of PQDs. Generally, the device architectures of PQDSCs have originated from other types of solar cells. In this section, we summarize the device architectures of PQDSCs, including PQD-sensitized solar cells, and conventional structure PQDSCs and inverted PQDSCs, and device working mechanisms.Dye-sensitized solar cells (DSSCs) have been widely studied due to their high photovoltaic performance, low-cost and low power consumption for device fabrication.222 However, due to the limited light collection ability of organic sensitizers, extensive studies were carried out to replace organic dyes with metal chalcogenide QDs to enhance the light absorption of solar cells.223 Inspired by DSSCs, Kojima et al., for the first time, fabricated liquid-state perovskite-sensitized solar cells using a nanocrystalline MAPbI3 perovskite-sensitized mesoporous TiO2 (m-TiO2) layer as a light absorber and combining with iodide/iodine electrolyte, and the solar cell gave an efficiency of 3.8% that opened the research field of perovskite solar cells.224 The stoichiometric perovskite precursors were directly deposited on the surface of m-TiO2 by a wet chemistry method, which finally formed perovskite nanocrystalline particles. The iodide/iodine electrolyte was applied as a hole transporter, which was used for the reduction and regeneration of sensitizers. Park and co-workers further improved the efficiency of MAPbI3 PQD sensitized solar cells (PQDSSCs) to 6.54% by adjusting the thickness of m-TiO2 layers and the annealing temperature of photoactive layers.225 The perovskite nanocrystalline particles could efficiently fill the interspaces of m-TiO2 layers to increase photoelectron injection and thus improve the electron transport rate. Since Kovalenko and co-workers successfully synthesized all-inorganic CsPbX3 PQDs for the first time, Liu et al. used CsPbI3 PQDs as sensitizers to prepare PQDSSCs.226Fig. 14a shows the device architecture of such liquid-state PQDSSCs. However, the perovskite structure of PQDs was degraded in a short period of time due to the high solvent-sensitivity of MAPbI3 perovskite materials, leading to a significant reduction in device efficiency.
 | ||
| Fig. 14 Different device structures of PQDSCs. (a) Schematic illustration of liquid-state PQDSSCs. (b) Schematic illustration of PQDSSC. Reprinted with permission from ref. 50. Copyright 2020 American Chemical Society. (c) Schematic illustration of conventional planar PQDSCs. Reprinted with permission from ref. 34. Copyright 2016 American Association for the Advancement of Science. (d) Schematic illustration of inverted PQDSCs. Reprinted with permission from ref. 221. Copyright 2019 Wiley-VCH. | ||
In 2012, solid-state PSCs with 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-MeOTAD) as a hole-conductor was reported by Kim et al. to avoid the degradation issues of perovskite materials.227 After that time the PSCs were developed very rapidly and the device efficiency was recently improved to 25.2%.1,19,228 Very recently, Chen et al. used m-TiO2 as an ETL to prepare solid-state PQDSCs, which had a high photocurrent density of 17.77 mA cm−2 due to increased electron injection from PQDs into m-TiO2 layers (Fig. 14b).50 Detailed information will be further provided in the following section.
The successful synthesis of all-inorganic CsPbX3 PQDs provided a possible approach for the fabrication of efficient and stable PQDSCs.32 Luther and co-workers proposed to use MeOAc as a gentle anti-solvent to purify CsPbI3 PQDs and remove long-chain ligands on the PQD surface to avoid agglomeration of PQDs. Through layer-by-layer deposition of PQDs, the first all-inorganic PQDSC with a conventional device structure (n–i–p) of FTO/TiO2/CsPbI3 PQDs/spiro-OMeTAD/MoOx/Al was successfully prepared (Fig. 14c),34 which had a similar structure to planar PSCs. Compared with the complex formation requirements of bulk inorganic perovskites, CsPbI3 PQD solid films can be prepared at room temperature and showed good phase stability due to the controlled surface properties of PQDs. Thanks to good electronic coupling between dots and energy band alignment within the device, a device efficiency of up to 10.77% was achieved and it could be stable for more than 60 days under dry conditions.
In a PQDSC with a conventional device structure, a compact electron transport layer of TiO2 is generally prepared at a high temperature (450–500 °C), which is not compatible with ITO electrodes and flexible substrates. Therefore, accompanied by diversified development requirements, the device with an inverted architecture (p–i–n) has received increasing attention due to the relatively simple preparation process at a low-temperature, which has been widely studied in flexible and tandem solar cells.210,229 For instance, Tavakoli et al. successfully prepared semitransparent PQDSCs with an inverted device structure of ITO/PTAA/PQDs/C60/BCP/graphene, as shown in Fig. 14d.221 An average visible transmittance (AVT) of up to 53% and device efficiency of 6.8% were achieved. The HTL in the inverted PSCs generally used polymers (such as PEDOT:PSS and PTAA) or p-type inorganic semiconductors (such as NiOx and Cu2O), which should have a high light transmittance and good energy level alignment with the photoactive layer. Similarly, the ETL (based on fullerenes and their derivatives, etc.) also needs to have suitable energy levels to match well with the photoactive layer, minimizing the interfacial recombination of photoinduced carriers.230
Therefore, the efficient operation of PQDSCs requires the following points:
(i) Photoactive materials with strong light absorption are required to generate electron–hole pairs. PQDs have suitable bandgap energy and can absorb light over a wide wavelength range, as well as can, in addition, convert photons into electron–hole pairs effectively.
(ii) The photoinduced electron–hole pairs should be efficiently separated and transferred to the charge transport layers, which need a high carrier mobility and defect-less photon absorbing material.
(iii) Excellent charge extraction and charge transport properties are always needed to achieve a better transformation of the opposite types of charge carriers into an external circuit, which is enabled by the suitable charge transport layers.
6.2. Interfacial engineering of PQD solid layers
A series of studies focused on considering heterojunction or interfacial modification of PQD solid films to improve device photovoltaic performance. The adjustment of the A-site component of bulk perovskite materials is limited by the multi-component film formation process. Specifically, due to the different formation temperatures of bulk perovskites with different A-site components, hybrid perovskites with adjustable compositions are not prepared easily.121 However, PQDs with continuously adjustable A-site cations can be used to widen the light absorption range.119 Zhao et al. used a layer-by-layer deposition method to prepare heterostructured PQD solid films with different A-site compositions in the PQDs, which largely improved charge separation and collection in the CQD solid films (Fig. 15a).220 The internally heterostructured PQD solid films were designed according to the energy levels of PQDs with different A-site compositions. It can be revealed that the photoinduced electrons were easily driven to PQD solid films with more FA+ cations in the PQDs, while holes tended to be transferred to PQDs with a higher Cs+ content in the PQDs, resulting in decreased charge carrier recombination within the CQD solid films. As a result, the optimized PQDSCs had an efficiency of 17.39% by using internally heterostructured PQD solid films in the device and that efficiency was one of the highest efficiencies of PQDSCs at that time (Fig. 15b). | ||
| Fig. 15 Interfacial engineering of PQD solid layers. (a) Layer-by-layer deposition and band structure of PQDs. (b) Device performances of Cs0.25FA0.75PbI3 and CsPbI3 PQDSCs. (a and b) Reprinted with permission from ref. 220. Copyright 2019 Nature Publishing Group. (c) Device structure of α-CsPbI3/FAPbI3 PQDSCs. (d) Device performances of PQDSCs based on different PQDs (CsPbI3, FAPbI3, CsPbI3 + FAPbI3). (c and d) Reprinted with permission from ref. 218. Copyright 2019 American Chemical Society. (e) Device structure of a PQDSC with a polymer-PQD bulk heterojunction connecting layer. Reprinted with permission from ref. 217. Copyright 2020 The Royal Society of Chemistry. (f) Deposition process of an FAPbI3 PQD/ITIC film. Reprinted with permission from ref. 48. Copyright 2019 Wiley-VCH. (g) Three factors to improve the performance of PQDSCs with μ-graphene crosslinked PQDs. Reprinted with permission from ref. 47. Copyright 2018 Wiley-VCH. | ||
The Eg value of CsPbI3 PQDs with a value of 1.73 eV is larger than that of ideal photoactive layers of a single-junction solar cell, which is not conducive to fully use solar energy. To expand the light absorption range of PQDs, the composition of PQDs can be engineered by combining CsPbX3 PQDs with other PQDs, such as FAPbI3 PQDs with a narrow bandgap (Eg = 1.5 eV).76 Li et al. took advantage of these two types of PQDs to construct an α-CsPbI3/FAPbI3 bilayer structure and obtained a gradient composition within PQD solid films through a thermal annealing process, which largely increased electronic coupling at the bilayer interface (Fig. 15c).218 PQDSCs constructed with a bilayer structured PQD solid film had a wide light absorption and thus the photocurrent density was largely improved (from 13.83 to 17.26 mA cm−2), leading to a device efficiency of ∼15.6% (Fig. 15d). Meanwhile, compared with Cs-based PQDs, the FAPbX3 perovskite structure had better stability because of the larger ionic radius of the FA+ cation in the perovskite lattice.108 Therefore, the devices with top layered FAPbI3 PQDs not only satisfied energy level alignment within devices to facilitate charge transfer but also improved device stability.
In addition to building graded PQD solid films to increase charge extraction by reducing charge recombination within the CQD solid films, interface optimization between the charge transport layer and PQDs is also critical to obtaining high-performance solar cells. The typical HTL for a conventional PQDSC is organic molecules or polymers (such as Spiro-OMeTAD and PTAA). However, the photoinduced holes could not always be efficiently injected from PQDs to the HTL due to the large difference of surface energy between the PQD solid layer and organic HTL.231 Ji et al. reported a method of bridging PQDs and the HTL by incorporating a polymer–PQD bulk heterojunction (BHJ) layer to improve charge transfer, mitigating the interfacial recombination of photoinduced carriers.217 As shown in Fig. 15e, the well-blended polymer–PQD layer could improve light collection efficiency and accelerate charge extraction at the interface.
As mentioned above, the unique surface chemistry of PQDs and the layer-by-layer deposition method of PQD solid films offer a chance to sequentially deposit PQD solid films with different components in the PQDs or heterojunction connecting layers. Several studies have proven that the use of functional molecular layers within PQD solid films can provide a feasible way to fabricate efficient and stable devices.47,48,232 Xue et al. used conjugated molecules (ITIC) as charge drivers in FAPbI3 PQD solid films to promote electron transport from PQDs to the SnO2 layer and then reduce charge recombination.48 The fabrication process of PQD solid films is shown in Fig. 15f. As the original OA/OAm ligands on the dot surface were removed, the conjugated organic molecules were introduced to enhance dot coupling.233 By adjusting the distribution of small molecules among PQD solid layers, an FAPbI3-based PQDSC with an efficiency of 12.7% was obtained. Moreover, Ashley et al. found that the carrier type and density of CsPbI3 PQDs can be adjusted by treating PQD solid films with molecular dopants.232
As the long-chain ligands capped on the PQD surface were difficult to thoroughly remove,69 PQD solid films showed poor conductivity, thus affecting the charge carrier extraction of PQDSCs. Wang et al. successfully crosslinked high mobility micrometer-sized graphene (μGR) with PQDs, which greatly improved the conductivity of PQD solid films as the μGR acted as an electron transporting channel in the PQD solid films.47 In addition, the μGR coordinated on the PQD surface also acted as a moisture barrier that could protect PQDs from the external environment and prevent PQD aggregation (Fig. 15g). The crosslinked μGR-PQD films presented a faster charge extraction and the device efficiency was increased by 12%. Therefore, the PQD layer regulation engineering provides a feasible solution to obtain a highly efficient and stable PQDSC.
6.3. Charge transport layers
After the harvesting of photons by PQD solid films in PQDSC devices, photoinduced electrons/holes are produced and subsequently transported to the corresponding charge transport layer. Thus, charge transport layers play an important role in efficiently extracting charge carriers, thus determining device photovoltaic performance. In order to reduce interfacial recombination at the ETL/PQD or PQD/HTL interface, the energy levels of the charge transport layers should be matched well with PQD solid films, and meanwhile the charge mobility of the ETL and HTL should be high enough for charge transport.234 In this section, we discuss the regulation of the ETL and HTL in PQDSCs to lower interfacial recombination.The c-TiO2 layer is widely applied as an ETL in PQDSCs since it has suitable energy levels with PQDs, and also the flat and smooth surface of c-TiO2 layers is beneficial to depositing PQD solid films using a spin-coating method.34 As studied in DSSCs and perovskite solar cells, the m-TiO2 layer with a large surface area may benefit charge injection from light absorbers into the m-TiO2 layer.223 To explore the implementation of m-TiO2 layers in PQDSCs, Chen et al. modified the surfaces of m-TiO2 layers with CsAc solution and successfully used m-TiO2 to improve electron separation and transport from PQDs to the m-TiO2 layers, in order to construct high-performance mesoscopic CsPbI3 PQDSCs (Fig. 16a).50 According to the results of element detection, direct deposition of PQD solid films on the m-TiO2 layer cannot make PQDs thoroughly fill the mesoporous structure and thus hinder electron injection and transport to m-TiO2 layers, resulting in low efficiency. After the CsAc-treatment, the PQDs could more easily migrate into the m-TiO2 layer and the Cs+ can also passivate the surface defects of PQDs (Fig. 16b). As a result, a device efficiency of 14.32% was attained in mesoscopic CsPbI3 PQDSCs (Fig. 16c). The improved efficiency was attributed to the enhanced electron injection rate by three times after the CsAc-treatment of the m-TiO2 layer (calculated according to the TA kinetics shown in Fig. 16d).
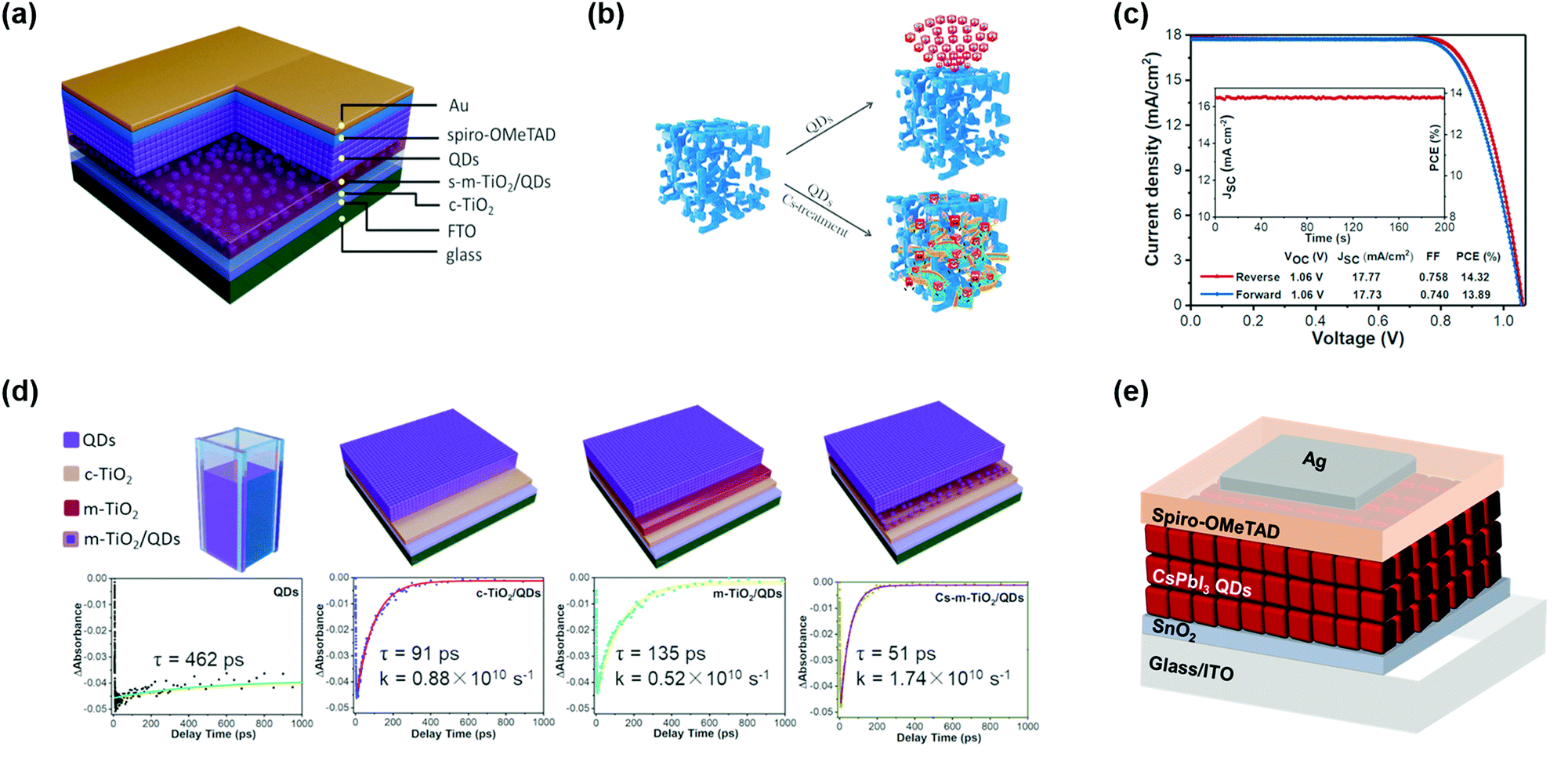 | ||
| Fig. 16 Electron transport materials in PQDSCs. (a) Device architecture of mesoscopic PQDSCs. (b) Schematic illustration of Cs-treatment on the m-TiO2 layer. (c) Device performance of CsPbI3 PQDSCs with a mesoporous structure. (d) Schematic and TA kinetics of CsPbI3 PQD solution and a series of TiO2/CsPbI3 PQD films. (a–d) Reprinted with permission from ref. 50. Copyright 2020 American Chemical Society. (e) The device structure of SnO2-based CsPbI3 PQDSCs. Reprinted with permission from ref. 46. Copyright 2020 Wiley-VCH. | ||
Even though TiO2 has been successfully applied for construction of high-efficiency PQDSCs, it also has drawbacks, such as high sintering temperature (>450 °C) and instability under ultraviolet light illumination, which may limit its application in new generational flexible devices and may be not compatible with ITO electrodes. Jiang et al. used SnO2 layers that had much higher electron mobility than TiO2 as the ETL material for the preparation of planar-structured PSCs.236 Since SnO2 can be prepared at a low temperature (∼150 °C) and has good electron mobility and light transmittance, it can also be used in PQDSCs and flexible devices. Jia et al. used the low-temperature processed SnO2 layer as an efficient ETL in PQDSCs, and the device gave a high efficiency of 13.66% (Fig. 16e).237 However, the ETL/PQD interface in the planar-structured device has no large contact area, which may need further studies to promote charge transfer at such an interface through interfacial engineering.238
It is worth noting that compared with conventional planar-structured devices the ETL material in the inverted devices needs to be prepared at low temperatures to avoid the influence of processing of the ETL on PQD solid films. Benefitting from their excellent electrical properties and thermal stability, fullerenes and their derivatives can be used as ETL materials for inverted PQDSCs.221 However, compared with the PQDSCs with the conventional n–i–p device structure, the efficiency of the inverted devices is still low, likely due to the interfacial recombination of photoinduced charge carriers.
Thus, further studies need to be carried out to improve the photovoltaic performance of inverted devices by overcoming charge transfer between PQDs and the ETL. More ETL materials may need to be explored, such as ZnO, phenyl-C61-butyric acid methyl ester (PCBM) and fullerene (C60), which were widely applied in PSCs.239,240
6.3.2. Hole transport layer
Correspondingly, HTL materials should also have suitable energy levels and high hole mobility, which could effectively extract the holes and block the electrons from reaching the contact.241 In addition, the HTL could also act as a protective barrier for the photoactive layer to prevent the degradation phenomenon, thereby improving device stability.242 So far, most efficient PQDSCs have used small organic molecule Spiro-OMeTAD, an aromatic polyamine compound with a wide distribution of electron clouds, as a hole conductor in the device. However, the deficient hole-transporting ability of Spiro-OMeTAD significantly needs to be improved using some dopants, such as lithium salt (LiTFSI) and cobalt complexes (FK209), which may exacerbate the degradation of perovskite materials under ambient conditions.243 Therefore, from the perspective of improving device stability, it is urgent to find a material with good hole transportability and without any doping treatment.In order to obtain high-efficiency and stable CsPbI3 PQDSCs, Yuan et al. studied a series of organic conjugated polymers, such as poly-3-hexylthiophene (P3HT) and poly[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl-alt-3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophene-4,6-diyl] (PTB7), to replace Spiro-OMeTAD as a HTL in PQDSCs (Fig. 17a).49 It is worth noting that when PQD solid films were covered with various HTL materials, the PL intensity and lifetime were significantly reduced, revealing that the internal dipole of the polymers could enhance charge extraction and separation (Fig. 17b).244 The uniform surface PL quenching of PQD solid films also confirmed the excellent film quality of undoped polymers. Although P3HT has better hole mobility, the energy levels of polymer PTB7 could match better with PQDs, which facilitated extraction of charge carriers at the interface (Fig. 17c). Since the Spiro-OMeTAD needs to be oxidized and easily absorb moisture after being doped with a lithium salt, the reproducibility of devices was poor and exhibited an obvious hysteresis phenomenon in the J–V measurements (Fig. 17d). In contrast, the CsPbI3 PQDSCs based on PTB7 as a hole conductor reached the highest efficiency of 12.55% among these devices, with a Voc of 1.27 V (Fig. 17e). Meanwhile, the stability of PQDSCs was also significantly improved due to the absence of dopants in the HTL. With respect to obtaining a higher photovoltaic performance of PQDSCs, we believed that the surface/interfacial modification between the charge transport layer and PQD solid layer may be still insufficient. The chemical engineering of charge transport layers, such as doping, could be used to improve the electron/hole mobility of ETL/HTL materials. Furthermore, we should pay more attention to interfacial engineering to improve charge transport at the interfaces, thereby reducing interfacial recombination losses.
 | ||
| Fig. 17 Hole transport materials in PQDSCs. (a) The device structure of α-CsPbI3 PQDSCs based on the PTB7 hole conductor. (b) PL intensities and lifetimes of an α-CsPbI3 PQD solid film and with different conjugated polymers. (c) The band structures of the α-CsPbI3 PQD solid film and with different conjugated polymers. (d) Device performance of α-CsPbI3 PQDSCs based on the Spiro-OMeTAD hole conductor. (e) Champion device performance of α-CsPbI3 PQDSCs based on the PTB7 hole conductor. (a–e) Reprinted with permission from ref. 49. Copyright 2018 Elsevier. | ||
The device architectures and photovoltaic performances of some highly efficient PQDSCs are summarized in Table 2. The studies on PQDs in photovoltaic applications are relatively late (starting from 2016); the device physics and device operation are still not very clear. Thus, the device efficiency is still largely behind those of bulk PSCs, which means that the photovoltaic efficiencies of PQDSCs are much lower than theoretical efficiencies (Fig. 18) and indicates that there is large room for further improving device efficiency. We believe that with a more fundamental understanding of and breakthrough in the surface chemistry of PQDs and the device operation of PQDSCs, the device efficiency and stability of PQDSCs will be largely improved in the near future.
| Architecture (n–i–p/p–i–n) | PQD type | Cell stack | Size of PQDs (nm) | PCE (%) | V oc (V) | J sc (mA cm−2) | FF | Ref. |
|---|---|---|---|---|---|---|---|---|
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/MoOx/Al | 9 | 10.77 | 1.23 | 13.47 | 0.65 | 34 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/MoOx/Al | ∼9 | 13.43 | 1.16 | 15.24 | 0.76 | 39 | |
| CsPbI3 PQDs | FTO/TiO2/μGR-PQDs/PTAA/Au | 10 | 11.64 | 1.18 | 13.59 | 0.72 | 47 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/PTB7/MoOx/Ag | 8 | 12.55 | 1.27 | 12.89 | 0.80 | 49 | |
| FAPbI3 PQDs | ITO/SnO2/PQDs/spiro-OMeTAD/Au | 17.7 | 8.38 | 1.10 | 11.84 | 0.64 | 167 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/MoOx/Al | ∼12 | 13.47 | 1.18 | 15.50 | 0.73 | 119 | |
| FAPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/MoOx/Al | ∼12 | 8.52 | 1.12 | 11.81 | 0.64 | 119 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/Au | 12 | 12.15 | 1.11 | 14.80 | 0.74 | 219 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/PTB7/MoOx/Ag | 10 | 13.12 | 1.25 | 14.18 | 0.74 | 110 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/Au | 9.86 | 11.87 | 1.04 | 16.98 | 0.67 | 41 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/Au | 14 | 9.4 | 1.04 | 13.15 | 0.69 | 133 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/PTAA/MoOx/Ag | ∼9 | 14.1 | 1.25 | 14.96 | 0.76 | 45 | |
| Heterostructured PQDs | ITO/TiO2/PQDs/spiro-OMeTAD/MoOx/Al | ∼12 | 17.39 | 1.20 | 18.91 | 0.76 | 220 | |
| n–i–p | FAPbI3 PQDs | ITO/SnO2/PQDs/spiro-OMeTAD/Ag | 14 | 12.7 | 1.10 | 15.40 | 0.75 | 48 |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/Au | 10 | 11.2 | 1.11 | 14.40 | 0.70 | 203 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/MoOx/Ag | 10 | 13.3 | 1.18 | 15.21 | 0.74 | 208 | |
| Bilayer structured PQDs | FTO/TiO2/PQDs/PTAA/MoOx/Ag | ∼12 | 15.6 | 1.22 | 17.26 | 0.74 | 218 | |
| CsPbI3 PQDs | FTO/c-TiO2/m-TiO2/PQDs/spiro-OMeTAD/Au | 9.1 | 14.32 | 1.06 | 17.77 | 0.75 | 50 | |
| Cs0.25FA0.75PbI3 PQDs | ITO/SnO2/PQDs/spiro-OMeTAD/Au | 14 | 16.6 | 1.17 | 18.30 | 0.78 | 38 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/PTAA/MoOx/Ag | ∼10 | 13.8 | 1.22 | 15.10 | 0.75 | 217 | |
| FAPbI3 PQDs | FTO/TiO2/PQDs/PTAA/MoOx/Ag | ∼10 | 13.2 | 1.12 | 16.7 | 0.71 | 217 | |
| CsPbI3 PQDs | ITO/SnO2/PQDs/spiro-OMeTAD/Ag | 15 | 13.66 | 1.22 | 17.66 | 0.63 | 46 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/PTAA/MoOx/Ag | 9 | 15.21 | 1.25 | 15.85 | 0.77 | 40 | |
| CsPbI3 PQDs | ITO/TiO2/PQDs/spiro-OMeTAD/MoOx/Ag | 10 | 14.1 | 1.23 | 15.30 | 0.75 | 215 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/spiro-OMeTAD/Ag | ∼15 | 14.8 | 1.19 | 16.4 | 0.76 | 145 | |
| CsPbI3 PQDs | FTO/TiO2/PQDs/PTAA/MoOx/Ag | 9 | 14.9 | 1.24 | 15.84 | 0.75 | 204 | |
| p–i–n | CsPbI3 PQDs | ITO/PTAA/PQDs/C60/BCP/graphene | 9 | 6.8 | 1.09 | 10.90 | 0.57 | 221 |
 | ||
| Fig. 18 The highest PCEs of different QDSCs. The Shockley–Queisser theoretical efficiency limitation curve of singe-junction solar cells is included for comparison.38,40,48,59,245 | ||
In the above section, we have introduced feasible methods to improve the photovoltaic performance of PQDSC devices, such as compositional engineering, surface regulation, and device operation. We believe that these methods may also work for other optoelectronic devices to improve device performance. For instance, in PQD light-emitting diode (LED) devices, radiative recombination is preferred in the PQDs and therefore we believe that these methods, such as compositional engineering and surface chemistry regulation of PQDs, might also be suitable for the preparation of high-performance LED devices by lowering the energy losses in the devices.
7. Stability of PQDSCs
It can be seen that the device efficiency of PQDSCs has significantly boosted in the past few years,19 but one of the pivotal issues toward future commercial applications is the device stability of PQDSCs. The volatile organic component in organic–inorganic hybrid PSCs was easily decomposed under high temperature or high humidity conditions, leading to the deterioration of device performance.106,246 Therefore, an increasing effort has focused on replacing sensitive organic groups with inorganic cesium. Although high surface energy and long-chain protecting ligands can stabilize a colloidal system of PQDs, the PQDs will also degrade when exposed to high humidity, high temperature or continuous illumination conditions. Therefore, the stability of PQDs, such as hydrothermal stability and light stability, is of great importance for PQDSC devices. As such, the stability of PQDs and PQDSCs is discussed in-depth in the following section to identify possible approaches to improve device stability.7.1. Hydrothermal stability
PQDs are sensitive to polar solvents due to the ionic nature of the perovskite crystal structure. The surface atoms of PQDs easily dissociate under the influence of polar solvents, causing surface defects and agglomeration of PQDs.247 In addition, the surface ligand interaction with PQDs was believed as a highly dynamic and constantly changing system.44 When PQDs were exposed to moist air or anti-solvent solution during the preparation of CQD solid films using a layer-by-layer deposition process, H2O molecules would permeate into PQDs from surface defects and thus destroy the perovskite crystal structure of PQDs. Therefore, using functional molecules to passivate the surface defects of PQDs is a promising strategy to improve material stability. As demonstrated in the above section, most of the post-treatment of PQD solid films can reduce the defect density, and thus somehow improve the stability of PQDSCs.45Photoactive PQD solid films are generally post-treated with FAI, but the hygroscopic FA cations make devices prone to degradation under ambient conditions.106 Kim et al. treated PQD solid films with PEAI to improve the hydrophobic stability of the films and meanwhile replace original OAm ligands, which resulted in the PEAI-based PQD solid film having better stability than the FAI-based PQD film (Fig. 19a).215 Thanks to the hydrophobic phenyl-terminated molecules, unencapsulated PQDSCs maintained 90% of the initial efficiency after 15 days. Similarly, Jia et al. passivated the surface defects of PQDs with amino acid molecules, as a result of which the increased surface ligand density could protect the PQDs from moisture.46 Wang et al. crosslinked hydrophobic μ-GR with PQDs and the resulting PQDSCs could work stably at a high humidity of 60%.47 In addition, polymers or organic molecules were also introduced into PQDs to cap the PQD surface as an inert protective shell, which improved the resistance of PQDs to the external environment.248–252
 | ||
| Fig. 19 Stability of PQDs and PQDSCs. (a) Photographs of PQD solid films with different post-treatments (under 30% RH). Reprinted with permission from ref. 215. Copyright 2020 American Chemical Society. (b) Thermal stability of Yb-doped CsPbI3 PQD solid films. Reprinted with permission from ref. 110. Copyright 2019 The Royal Society of Chemistry. (c) Photographs of C18-CsPbI3 PQD and C8/C18-CsPbI3 PQD solutions after storage for 180 days. Reprinted with permission from ref. 41. Copyright 2019 Wiley-VCH. (d) Long-term stability of CsxFA1−xPbI3 PQD solid films and Cs0.25FA0.75PbI3 bulk perovskite films. Reprinted with permission from ref. 38. Copyright 2020 Nature Publishing Group. (e) PL spectral shifts of KBr-passivated CsPbI3−xBrx PQDs and pristine CsPbI3−xBrx PQDs. Reprinted with permission from ref. 176. Copyright 2020 American Chemical Society. | ||
Due to the heat-induced reassembly process of PQDs, the quality of PQD solid films can be enhanced through a thermal annealing process of PQD solid films.40,50,218 However, at excessively high temperatures, the surface defect states formed by detached ligands will capture charge carriers and reduce radiative recombination, which in turn causes PL quenching.253 Therefore, the studies on the thermal stability of PQDs are equally important with the improvement of the photovoltaic efficiency of PQDSCs. Shi et al. strengthened lattice stability using Yb-doped PQDs and achieved PQDSCs with better thermal stability (Fig. 19b).110 Improving the formation energy of PQDs by a doping strategy also provided an avenue to stabilize the lattice structure of perovskite materials.254
7.2. Storage stability
CsPbI3 PQDs possess a low structural tolerance factor that is prone to undergo undesirable phase transitions. Therefore, adjusting the composition of perovskites to obtain appropriate tolerance factors can fundamentally improve the stability of materials.133,141,144,145,254,255 B-site doping or alloying can increase the cohesive energy of perovskite crystals, which is more conducive to resisting the adverse effects of the external environment. In addition, it is well known that surface ligands play an important role in stabilizing the perovskite lattice of PQDs. As mentioned above, the dynamic proton exchange between OA and OAm ligands could easily lead to agglomeration of PQDs, resulting in poor stability of colloidal systems.97 Yassitepe et al. improved the colloidal stability of PQDs by using OA alone as capping ligands, which largely eliminated proton exchange between OA and OAm ligands.97 The results revealed that ligands that have stronger binding energy with PQD surfaces could be applied to substitute OA/OAm ligands and that the purpose of improving the stability of colloidal systems was achieved.41,173 C8/C18-CsPbI3 PQDs obtained by partially replacing OA/OAm ligands with short-chain ligands (OctAc/OctAm) exhibited excellent stability within 180 days (Fig. 19c). The short-chain ligands with higher binding energy not only protected the perovskite crystal but also avoided adverse effects on charge transport. Bi et al. used 2-aminoethanethiol (AET), a short-chain ligand with the –SH group, to partially replace long-chain OA/OAm ligands, and subsequently, a dense ligand protective shell was formed around PQDs.73 Unencapsulated devices based on the AET-CsPbI3 PQDs maintained 95% of the initial efficiencies when the device was placed in ambient air for 40 hours. Khan et al. used the surface-passivating ligand anchoring strategy to treat PQD solid films, and the corresponding devices can maintain good efficiency after ∼70 days of storage under ambient conditions (a low RH of <5%). After the surface ligand exchange, the short Lewis base ligands, which contain the thiol group (–SH), can improve the electronic coupling of the PQD solid films and reduce the surface defect states. Therefore, such a strategy not only improved device efficiency but also greatly improved device stability.256 Some studies revealed that precursor engineering for the synthesis of PQDs could also improve PQD stability.85,86,219,257 Liu et al. used GeI2 as an additive halide source to avoid the formation of lead-rich surfaces in PQDs and thus improve the PQDs’ stability due to the fact that the reduction of halide vacancies could decrease the surface defect density.219 Finally, GeI2-based PQDSCs presented excellent stability that maintained 85% of the device efficiency after storage for 90 days. Therefore, precursor regulation offers a feasible method to control the composition of PQDs, resulting in improved stability of PQDSCs.An interesting emission recovery phenomenon was proposed by Wang et al. – by adding TOP into aged PQD solutions could obtain long-term storage PQD solutions.258,259 During the liquid-phase post-treatment of PQDs, the participation of TOP reduced surface defects and perfect surface conditions were achieved. Notably, the TOP did not change the perovskite crystal structure, which was in good agreement with previous studies reported by Liu et al.199 In addition, the polymer has excellent light stability, and low water and oxygen transmission rates,260 which could be applied to protect PQDs against degradation.261 However, compared with embedding PQDs in polymers, the construction of core/shell structured PQDs also improved the stability of PQDs and did not affect charge transport between dots.186,187,262,263 Compared with the wide range of applications of core/shell structures in chalcogenide QDs, there is a relatively little exploration of core/shell structured PQDs. Under the precondition of ensuring efficient charge transfer between PQDs, finding a suitable shell structure to improve the stability of PQDSCs still remains a challenge.
7.3. Illumination stability
Light-induced degradation of PQDs may play a key role in the performance deterioration of PQDSCs. The capping ligands that can inhibit PQD aggregation under continuous illumination conditions can therefore avoid the formation of non-radiative recombination centers that capture charge carriers at the surface defects of PQDs.264 Meanwhile, the ionic defects in perovskites will migrate under the synergistic effect of illumination and heat, which causes device hysteresis and lowered device stability.129,265,266 Unlike resistance to the external environment, the degradation of perovskites caused by ion migration might not be fully addressed by coating or encapsulation methods. Therefore, it is necessary to reduce defects and increase vacancy formation energy to suppress the migration phenomenon, hence improving device stability.For a bulk PSC, the mixed cations and anions generally suffer from phase segregation under illumination or electric field conditions, and the defect recombination centers formed by segregation regions generally lead to a decrease in the device Voc.267 Previous studies proved that phase segregation can be suppressed by controlling the grain size of perovskite materials.120 As the size of the perovskite crystal is decreased, an almost segregation-free perovskite film is obtained. The capping ligands of PQDs also act as a hindrance to ion migration. Hao et al. demonstrated that under illumination Cs1−xFAxPbI3 PQD solid films presented higher stability than bulk films with the same composition.38 Since there was almost no change in the energy barrier of ion migration in the PQD solid films before and after illumination, the Cs1−xFAxPbI3 PQDSCs maintained more than 90% of the initial efficiency after 900 hours’ illumination (Fig. 19d).
Compared with the migration of A-site cations in PQDs, the halide anions are more common to migrate due to their lower activation energy for ion migration.129,268 Yang et al. passivated the surface vacancies of CsPbI3−xBrx PQDs with KBr molecules to obtain a high PLQY and meanwhile prevent ion segregation.176 An appropriate amount of K+ ions combined with Br atoms located on the PQD surface could stabilize the perovskite lattice and reduce surface defect density, which resulted in a stable PL spectrum (Fig. 19e). Luo et al. reported an ultrafast thermodynamic cooling strategy that directly added liquid nitrogen into a high-temperature reaction system to obtain a high cooling rate of the reaction system.98 Cooperating with the surface passivation with Pb(BrCl)2 molecules, the CsPbBrxCl3−x PQD solid films had excellent spectral stability.
Even though significant efforts had been made towards improving the device efficiency of PQDSCs, the studies on device stability are relatively unenlightened. Since the light-absorbing layer and the functional layer materials in the PQDSCs are relatively sensitive to water, oxygen and light, the lifetime of the devices is relatively short in ambient air. How to improve device stability under the premise of ensuring the high efficiency of PQDSCs is becoming increasingly important. Encapsulation technology can effectively isolate the working area of the devices from the external environment,269 which is a promising method for PQDSCs. Furthermore, it is also a feasible approach to explore some suitable surface passivation molecules to provide a hydrophobic surface without sacrificing their electrical properties.270 We believe that with more fundamental studies on the thermodynamics and dynamics of the degradation of PQDs, by combining comprehensive theoretical calculations and experimental studies, the stability of PQDs will be significantly enhanced to face future commercial applications of PQDSCs.
8. Summary and perspectives
In this review, we comprehensively summarized the recent advances of emerging PQDSCs from the viewpoints of improving photovoltaic performance and device stability through different strategies, from reducing non-radiative recombination in the PQDs to device operation with decreased interfacial recombination within the devices. The outstanding optoelectronic properties of PQDs make them emerging candidates for the development of next-generation solar cells, and in a surprisingly short time, the device efficiency has skyrocketed from ∼10% to over 16% by lowering energy losses in PQDSCs. Starting off from improving the optoelectronic properties of PQDs, PQDs with a stable structure and fewer internal defects were successfully prepared through compositional engineering (doping strategy, anion and cation exchange, etc.) to lower non-radiative recombination, which provides a strong basis for constructing high-performance PQDSCs. Secondly, substantial surface chemical regulation methods were undertaken to reduce the surface defects of PQDs, which seriously affect charge transport in PQD solid films, minimizing defect-assisted non-radiative recombination and thus increasing charge extraction of PQDSCs. Thirdly, a series of device operations were discussed in-depth to decrease interfacial energy losses induced by interfacial recombination in the devices: for instance, constructing a graded junction within devices, and interfacial and charge transport layer engineering, which provided feasible strategies to further improve device photovoltaic performance. Finally, the device stability of PQDSCs was discussed to figure out the possibility of PQDSCs toward future commercial applications.Even though the efficiency of PQDSCs has been largely improved by increasing carrier mobility and lowering energy losses in the devices, there are still some important issues pertaining to the improvement of device performance, which need to be fully addressed toward commercial application in the future, as presented in Fig. 20.
 | ||
| Fig. 20 Possible challenges and opportunities of PQDSCs toward future development. | ||
(i) Considering the detrimental effect of abundant defects on device performance, it is of great significance to decrease and understand the defect-assisted recombination behavior in PQDs through a combination of experimental studies and theoretical calculations. PQDs have been found to have higher defect density than bulk perovskite materials.46,271 The highly dynamic binding and desorption of surface ligands during the purification and film deposition processes will produce a large number of surface defects,44 which results in the trapping of charge carriers and thus leads to a lowered photovoltage caused by defect-assisted recombination. Besides, due to the existence of surface defects, the incomplete surface is easily dissolved by anti-solvent during the deposition of PQD solid films. Hence, the limited thickness of PQD solid films is insufficient for absorbing enough photons to achieve an ideal photocurrent.272 Moreover, the imperfect surface defect passivation of PQDs will act as the intrusion point for the external environment to destroy perovskite crystals. Therefore, in order to achieve a higher efficiency of PQDSCs, more attention should be paid to passivation of the surface defects of PQDs to minimize the non-radiative recombination and simultaneously improve the stability of PQDSC devices. In addition, several studies have shown that the optoelectronic properties (PL lifetime, PLQY, etc.) of PQDs could be greatly improved using liquid-phase treatment approaches, such as additive engineering and constructing core/shell structures.173,186,203 However, very few studies of PQDSC devices focused on defect passivation in the liquid-state, which may reduce the defect density of PQDs before deposition of PQD solid films. Therefore, further investigation should be centered on exploring suitable liquid-phase treatments to modulate the carrier transport and defect density of PQDs.
(ii) Further exploring the methods to enhance charge transfer between interfaces within PQDSCs is considered to be a promising route to improve the performance of PQDSCs. Since the complex ligand dynamic balance on the surfaces of PQDs makes it more sensitive and difficult to be controlled than that of traditional chalcogenide QDs, it is more challenging to perform liquid-state ligand exchange. Compared with bulk PSCs, the photoactive active layer in PQDSCs is generally prepared by stacking multiple PQD layers and some of the original ligands still bonded onto the PQD surface, which hinders charge transport, leading to a lowered photocurrent density. Moreover, the disordered and inhomogeneous system caused by anti-solvent immersion also produces bandtail states in the PQD solid films, resulting in a decrease in photovoltage and impeding carrier transport between dots.233,273,274 Thus, during solid-state ligand exchange, the solvent type and ligand exchange time should be well controlled to improve carrier mobility and meanwhile avoid PQD fusion caused by inconsistent ligand exchanges. Additionally, layer-by-layer deposition of PQD solid films makes PQDSCs have more interfaces than bulk devices, which induces undesired interfacial recombination. In-depth research of solid film modification should be implemented to suppress energy losses between multilayers within devices. Meanwhile, non-negligible carrier recombination at the interfaces between the PQD solid films and charge transport layers need to be finely suppressed using effective interface regulation. For instance, double-layer electron transport materials may be used to promote charge injection between interfaces by establishing a better energy level alignment; using a self-assembled molecular layer with specific functional groups can bridge the ETL and PQD layers and meanwhile passivate surface defects on the metal oxide (TiO2, SnO2).275
(iii) In order to solve toxicity restrictions on the commercial application of PSCs, tin-based perovskites applied as environment-friendly absorbers with narrow bandgaps may provide a good choice for the development of high-efficiency lead-free solar cells. Quite recently, the efficiency of tin-based PSCs reached a value of 12.4% through ETL design,276 but these PSCs still did not achieve the desired efficiencies due to their high Voc losses. Similar to bulk perovskites, tin-based PQDs also suffer from high defect density and strong non-radiative recombination issues.148,156 So far, none of the lead-free devices have shown comparable photovoltaic performance to lead-based competitors. Alleviating high concentration vacancies (defects) in Sn-based PQDs might be still a troublesome challenge that needs to be solved in lead-free PQDSCs. Therefore, additive engineering or doping of PQDs may suppress the internal defects and keep short-range regular arrangement in the PQDs, which may alleviate recombination in lead-free PQDs. Besides tin-based perovskites, there are still a large number of compounds having perovskite-type structures, and thus it is necessary to use theoretical calculations to filter the potential candidates that are thermodynamically stable and have suitable bandgaps for solar cell preparation.
(iv) With continuous breakthroughs in the device efficiency, PQDSCs need to advance towards the scalable and stable industrialization requirements. Under the normal synthesis routes, PQDs need to be purified with anti-solvent and repeatedly centrifuged to obtain a colloidal solution that is suitable for the deposition of PQD solid films, but the relatively low synthetic yield of PQDs is not conducive to realizing large-scale production.204 Therefore, controllable preparation of large-scale uniformly dispersed PQD solutions without using complex preparation conditions remains a significant challenge. Meanwhile, most of the extraordinary efficiencies are achieved on small devices with an area of less than 1 cm2, and the device performance degrades rapidly due to the inferior PQD solid film quality as the cell area increases. Moreover, making full use of low-cost and solution-processable PQDs to deposit uniform and dense films could be achieved using spray-coating, blade-coating or ink-jet printing methods. Therefore, it is necessary to explore viable fabrication methods, such as spray-coating, blade-coating and ink-jet printing, which can fully utilize low-cost, solution-processable PQDs to deposit uniform and dense films. By improving deposition techniques, the non-radiative recombination caused by uneven films may be suppressed to prepare large-area efficient and stable PQDSCs. In addition, due to the ionic nature of PQDs, the stability of materials and devices is still an intractable problem to be solved for future commercial exploitation. As mentioned earlier, various studies have shown that the stability of PQDs could be improved by surface coating, doping, and ligand engineering of PQDs. Thus, the microscopic degradation mechanism of PQDs should be explored through fundamental research to further understand the unique surface chemistry in PQDs.
Overall, even though there remains much scope for improvement of the photovoltaic performance of PQDSCs, we believe that PQDs show great potential for future photovoltaic applications. Solution-processable PQDs can be synthesized at a low temperature, making their large-scale preparation cost-effective and their application in flexible and lightweight devices possible. Uniformly dispersed PQDs allow for depositing a solid film with a uniform thickness and low stress (without thermal stress due to the high-temperature annealing), which may help the devices maintain good stability in stress alternation. Transparent/semi-transparent devices based on PQDs may be used as windows for buildings and vehicles, generating electricity while maintaining transparency.213,221 Moreover, PQDs may be an ideal candidate for construction of tandem solar cells due to their excellent optoelectronic properties and tunable Eg as PQDs with appropriate Eg (1.6–1.75 eV) absorb high-energy photons to ensure a high voltage, while low-energy photons are fully utilized by the complementary bottom cell.277 To fully utilize the advantages of PQDs and improve device performance lay a good foundation for PQDSCs to step from the laboratory research toward mass production.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51872014), the Recruitment Program of Global Experts, the Fundamental Research Funds for the Central Universities, the “111” project (B17002), and the Swedish Energy Agency.References
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Gratzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS.
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T.-W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582–587 Search PubMed.
- N. Pellet, P. Gao, G. Gregori, T.-Y. Yang, M. K. Nazeeruddin, J. Maier and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS.
- Y. Chen, S. Tan, N. Li, B. Huang, X. Niu, L. Li, M. Sun, Y. Zhang, X. Zhang, C. Zhu, N. Yang, H. Zai, Y. Wu, S. Ma, Y. Bai, Q. Chen, F. Xiao, K. Sun and H. Zhou, Joule, 2020, 4, 1961–1976 CrossRef CAS.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. I. Seok, Science, 2019, 366, 749–753 CrossRef CAS.
- W. Pan, H. Wu, J. Luo, Z. Deng, C. Ge, C. Chen, X. Jiang, W.-J. Yin, G. Niu, L. Zhu, L. Yin, Y. Zhou, Q. Xie, X. Ke, M. Sui and J. Tang, Nat. Photonics, 2017, 11, 726–732 CrossRef CAS.
- S. A. Veldhuis, P. P. Boix, N. Yantara, M. Li, T. C. Sum, N. Mathews and S. G. Mhaisalkar, Adv. Mater., 2016, 28, 6804–6834 CrossRef CAS.
- Z. Fang, W. Chen, Y. Shi, J. Zhao, S. Chu, J. Zhang and Z. Xiao, Adv. Funct. Mater., 2020, 30, 1909754 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS.
- M. Kulbak, S. Gupta, N. Kedem, I. Levine, T. Bendikov, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2016, 7, 167–172 CrossRef CAS.
- G. E. Eperon and D. S. Ginger, ACS Energy Lett., 2017, 2, 1190–1196 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS.
- W. Li, W. Zhang, S. Van Reenen, R. J. Sutton, J. Fan, A. A. Haghighirad, M. B. Johnston, L. Wang and H. J. Snaith, Energy Environ. Sci., 2016, 9, 490–498 RSC.
- W. Chen, Y. Wu, J. Liu, C. Qin, X. Yang, A. Islam, Y.-B. Cheng and L. Han, Energy Environ. Sci., 2015, 8, 629–640 RSC.
- J. Chen, Y. Rong, A. Mei, Y. Xiong, T. Liu, Y. Sheng, P. Jiang, L. Hong, Y. Guan, X. Zhu, X. Hou, M. Duan, J. Zhao, X. Li and H. Han, Adv. Energy Mater., 2016, 6, 1502009 CrossRef.
- Best-research-cell-efficiencies. NREL, 2019, https://www.nrel.gov/pv/cell-efficiency.html.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. D. Angelis and H.-G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef.
- Y. H. Lin, N. Sakai, P. Da, J. Wu, H. C. Sansom, A. J. Ramadan, S. Mahesh, J. Liu, R. D. J. Oliver, J. Lim, L. Aspitarte, K. Sharma, P. K. Madhu, A. B. Morales-Vilches, P. K. Nayak, S. Bai, F. Gao, C. R. M. Grovenor, M. B. Johnston, J. G. Labram, J. R. Durrant, J. M. Ball, B. Wenger, B. Stannowski and H. J. Snaith, Science, 2020, 369, 96–102 CrossRef CAS.
- Z. Liu, L. Qiu, L. K. Ono, S. He, Z. Hu, M. Jiang, G. Tong, Z. Wu, Y. Jiang, D.-Y. Son, Y. Dang, S. Kazaoui and Y. Qi, Nat. Energy, 2020, 5, 596–604 CrossRef CAS.
- G. E. Eperon, G. M. Paternò, R. J. Sutton, A. Zampetti, A. A. Haghighirad, F. Cacialli and H. J. Snaith, J. Mater. Chem. A, 2015, 3, 19688–19695 RSC.
- Q. Ye, Y. Zhao, S. Mu, F. Ma, F. Gao, Z. Chu, Z. Yin, P. Gao, X. Zhang and J. You, Adv. Mater., 2019, 31, 1905143 CrossRef CAS.
- Y. Wang, X. Liu, T. Zhang, X. Wang, M. Kan, J. Shi and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16691–16696 CrossRef CAS.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712–4715 RSC.
- Y. Wang, M. I. Dar, L. K. Ono, T. Zhang, M. Kan, Y. Li, L. Zhang, X. Wang, Y. Yang, X. Gao, Y. Qi, M. Gratzel and Y. Zhao, Science, 2019, 365, 591–595 CrossRef CAS.
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J.-M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- Q. Zhao, A. Hazarika, L. T. Schelhas, J. Liu, E. A. Gaulding, G. Li, M. Zhang, M. F. Toney, P. C. Sercel and J. M. Luther, ACS Energy Lett., 2019, 5, 238–247 CrossRef.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS.
- C. de Weerd, L. Gomez, A. Capretti, D. M. Lebrun, E. Matsubara, J. Lin, M. Ashida, F. C. M. Spoor, L. D. A. Siebbeles, A. J. Houtepen, K. Suenaga, Y. Fujiwara and T. Gregorkiewicz, Nat. Commun., 2018, 9, 4199 CrossRef.
- M. Hao, Y. Bai, S. Zeiske, L. Ren, J. Liu, Y. Yuan, N. Zarrabi, N. Cheng, M. Ghasemi, P. Chen, M. Lyu, D. He, J.-H. Yun, Y. Du, Y. Wang, S. Ding, A. Armin, P. Meredith, G. Liu, H.-M. Cheng and L. Wang, Nat. Energy, 2020, 5, 79–88 CrossRef CAS.
- E. M. Sanehira, A. R. Marshall, J. A. Christians, S. P. Harvey, P. N. Ciesielski, L. M. Wheeler, P. Schulz, L. Y. Lin, M. C. Beard and J. M. Luther, Sci. Adv., 2017, 3, eaao4204 CrossRef.
- X. Ling, J. Yuan, X. Zhang, Y. Qian, S. M. Zakeeruddin, B. W. Larson, Q. Zhao, J. Shi, J. Yang, K. Ji, Y. Zhang, Y. Wang, C. Zhang, S. Duhm, J. M. Luther, M. Grätzel and W. Ma, Adv. Mater., 2020, 32, 2001906 CrossRef CAS.
- K. Chen, Q. Zhong, W. Chen, B. Sang, Y. Wang, T. Yang, Y. Liu, Y. Zhang and H. Zhang, Adv. Funct. Mater., 2019, 29, 1900991 CrossRef.
- G.-J. A. H. Wetzelaer, M. Scheepers, A. M. Sempere, C. Momblona, J. Ávila and H. J. Bolink, Adv. Mater., 2015, 27, 1837–1841 CrossRef CAS.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS.
- X. Ling, S. Zhou, J. Yuan, J. Shi, Y. Qian, B. W. Larson, Q. Zhao, C. Qin, F. Li, G. Shi, C. Stewart, J. Hu, X. Zhang, J. M. Luther, S. Duhm and W. Ma, Adv. Energy Mater., 2019, 9, 1900721 CrossRef.
- D. Jia, J. Chen, M. Yu, J. Liu, E. M. J. Johansson, A. Hagfeldt and X. Zhang, Small, 2020, 16, 2001772 CrossRef CAS.
- Q. Wang, Z. Jin, D. Chen, D. Bai, H. Bian, J. Sun, G. Zhu, G. Wang and S. F. Liu, Adv. Energy Mater., 2018, 8, 1800007 CrossRef.
- J. Xue, R. Wang, L. Chen, S. Nuryyeva, T. H. Han, T. Huang, S. Tan, J. Zhu, M. Wang, Z. K. Wang, C. Zhang, J. W. Lee and Y. Yang, Adv. Mater., 2019, 31, 1900111 CrossRef.
- J. Yuan, X. Ling, D. Yang, F. Li, S. Zhou, J. Shi, Y. Qian, J. Hu, Y. Sun, Y. Yang, X. Gao, S. Duhm, Q. Zhang and W. Ma, Joule, 2018, 2, 2450–2463 CrossRef CAS.
- K. Chen, W. Jin, Y. Zhang, T. Yang, P. Reiss, Q. Zhong, U. Bach, Q. Li, Y. Wang, H. Zhang, Q. Bao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 3775–3783 CrossRef CAS.
- J. Gan, J. He, R. L. Z. Hoye, A. Mavlonov, F. Raziq, J. L. MacManus-Driscoll, X. Wu, S. Li, X. Zu, Y. Zhan, X. Zhang and L. Qiao, ACS Energy Lett., 2019, 4, 1308–1320 CrossRef CAS.
- F. Zhou, Z. Li, H. Chen, Q. Wang, L. Ding and Z. Jin, Nano Energy, 2020, 73, 104757 CrossRef CAS.
- A. a. O. El-Ballouli, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2019, 31, 6387–6411 CrossRef CAS.
- J. Yuan, A. Hazarika, Q. Zhao, X. Ling, T. Moot, W. Ma and J. M. Luther, Joule, 2020, 4, 1160–1185 CrossRef CAS.
- J. Xu, O. Voznyy, M. Liu, A. R. Kirmani, G. Walters, R. Munir, M. Abdelsamie, A. H. Proppe, A. Sarkar, F. P. Garcia de Arquer, M. Wei, B. Sun, M. Liu, O. Ouellette, R. Quintero-Bermudez, J. Li, J. Fan, L. Quan, P. Todorovic, H. Tan, S. Hoogland, S. O. Kelley, M. Stefik, A. Amassian and E. H. Sargent, Nat. Nanotechnol., 2018, 13, 456–462 CrossRef CAS.
- M. Liu, Y. Chen, C.-S. Tan, R. Quintero-Bermudez, A. H. Proppe, R. Munir, H. Tan, O. Voznyy, B. Scheffel, G. Walters, A. P. T. Kam, B. Sun, M.-J. Choi, S. Hoogland, A. Amassian, S. O. Kelley, F. P. García de Arquer and E. H. Sargent, Nature, 2019, 570, 96–101 CrossRef CAS.
- D. Jia, J. Chen, S. Zheng, D. Phuyal, M. Yu, L. Tian, J. Liu, O. Karis, H. Rensmo, E. M. J. Johansson and X. Zhang, Adv. Energy Mater., 2019, 9, 1902809 CrossRef CAS.
- X. Zhang, J. Zhang, D. Phuyal, J. Du, L. Tian, V. A. Öberg, M. B. Johansson, U. B. Cappel, O. Karis, J. Liu, H. Rensmo, G. Boschloo and E. M. J. Johansson, Adv. Energy Mater., 2018, 8, 1702049 CrossRef.
- B. Sun, A. Johnston, C. Xu, M. Wei, Z. Huang, Z. Jiang, H. Zhou, Y. Gao, Y. Dong, O. Ouellette, X. Zheng, J. Liu, M.-J. Choi, Y. Gao, S.-W. Baek, F. Laquai, O. M. Bakr, D. Ban, O. Voznyy, F. P. García de Arquer and E. H. Sargent, Joule, 2020, 4, 1542–1556 CrossRef CAS.
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2019, 5, 44–60 CrossRef.
- K. Kalyanasundaram, E. Borgarello, D. Duonghong and M. Grätzel, Angew. Chem., Int. Ed. Engl., 1981, 20, 987–988 CrossRef.
- R. Rossetti, S. Nakahara and L. E. Brus, J. Chem. Phys., 1983, 79, 1086–1088 CrossRef CAS.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS.
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Science, 2016, 353, aac5523 CrossRef.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS.
- C. K. Møller, Nature, 1958, 182, 1436 CrossRef.
- J.-K. Sun, S. Huang, X.-Z. Liu, Q. Xu, Q.-H. Zhang, W.-J. Jiang, D.-J. Xue, J.-C. Xu, J.-Y. Ma, J. Ding, Q.-Q. Ge, L. Gu, X.-H. Fang, H.-Z. Zhong, J.-S. Hu and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 11705–11715 CrossRef CAS.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef.
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y.-J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- J. Chen, W. Du, J. Shi, M. Li, Y. Wang, Q. Zhang and X. Liu, InfoMat, 2019, 2, 170–183 CrossRef.
- C. Bi, S. V. Kershaw, A. L. Rogach and J. Tian, Adv. Funct. Mater., 2019, 29, 1902446 CrossRef.
- L. Brus, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. Garcia Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS.
- R. Wang, Y. Shang, P. Kanjanaboos, W. Zhou, Z. Ning and E. H. Sargent, Energy Environ. Sci., 2016, 9, 1130–1143 RSC.
- G. S. Selopal, H. Zhao, Z. M. Wang and F. Rosei, Adv. Funct. Mater., 2020, 30, 1908762 CrossRef CAS.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS.
- Q. A. Akkerman, G. Raino, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS.
- J. Kang and L.-W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS.
- A. J. Nozik, Chem. Phys. Lett., 2008, 457, 3–11 CrossRef CAS.
- X. Zhang, J. Liu and E. M. J. Johansson, Nanoscale, 2015, 7, 1454–1462 RSC.
- Y. Zhang, G. Wu, F. Liu, C. Ding, Z. Zou and Q. Shen, Chem. Soc. Rev., 2020, 49, 49–84 RSC.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- X. Li, D. Yu, F. Cao, Y. Gu, Y. Wei, Y. Wu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 5903–5912 CrossRef CAS.
- G. Li, H. Wang, T. Zhang, L. Mi, Y. Zhang, Z. Zhang, W. Zhang and Y. Jiang, Adv. Funct. Mater., 2016, 26, 8478–8486 CrossRef CAS.
- Q. Pan, H. Hu, Y. Zou, M. Chen, L. Wu, D. Yang, X. Yuan, J. Fan, B. Sun and Q. Zhang, J. Mater. Chem. C, 2017, 5, 10947–10954 RSC.
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D’Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS.
- J. Werner and M. Peisl, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 6881–6883 CrossRef CAS.
- Y. Tong, E. Bladt, M. F. Ayguler, A. Manzi, K. Z. Milowska, V. A. Hintermayr, P. Docampo, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2016, 55, 13887–13892 CrossRef CAS.
- M. Chen, Y. Zou, L. Wu, Q. Pan, D. Yang, H. Hu, Y. Tan, Q. Zhong, Y. Xu, H. Liu, B. Sun and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1701121 CrossRef.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS.
- A. Dutta, R. K. Behera, P. Pal, S. Baitalik and N. Pradhan, Angew. Chem., Int. Ed., 2019, 58, 5552–5556 CrossRef CAS.
- E. Yassitepe, Z. Yang, O. Voznyy, Y. Kim, G. Walters, J. A. Castañeda, P. Kanjanaboos, M. Yuan, X. Gong, F. Fan, J. Pan, S. Hoogland, R. Comin, O. M. Bakr, L. A. Padilha, A. F. Nogueira and E. H. Sargent, Adv. Funct. Mater., 2016, 26, 8757–8763 CrossRef CAS.
- C. Luo, C. Yan, W. Li, F. Chun, M. Xie, Z. Zhu, Y. Gao, B. Guo and W. Yang, Adv. Funct. Mater., 2020, 30, 2000026 CrossRef CAS.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. deMello and M. V. Kovalenko, Nano Lett., 2016, 16, 1869–1877 CrossRef CAS.
- C. Liu, Q. Zeng and B. Yang, Adv. Mater. Interfaces, 2019, 6, 1901136 CrossRef CAS.
- S. Zou, Y. Liu, J. Li, C. Liu, R. Feng, F. Jiang, Y. Li, J. Song, H. Zeng, M. Hong and X. Chen, J. Am. Chem. Soc., 2017, 139, 11443–11450 CrossRef CAS.
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005–8011 CrossRef CAS.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS.
- L. Wang, H. Zhou, J. Hu, B. Huang, M. Sun, B. Dong, G. Zheng, Y. Huang, Y. Chen, L. Li, Z. Xu, N. Li, Z. Liu, Q. Chen, L. D. Sun and C. H. Yan, Science, 2019, 363, 265–270 CrossRef CAS.
- T. Duong, H. K. Mulmudi, H. Shen, Y. Wu, C. Barugkin, Y. O. Mayon, H. T. Nguyen, D. Macdonald, J. Peng, M. Lockrey, W. Li, Y.-B. Cheng, T. P. White, K. Weber and K. Catchpole, Nano Energy, 2016, 30, 330–340 CrossRef CAS.
- Y. Hu, T. Qiu, F. Bai, X. Miao and S. Zhang, J. Mater. Chem. A, 2017, 5, 25258–25265 RSC.
- W. Zhao, Z. Yao, F. Yu, D. Yang and S. Liu, Adv. Sci., 2018, 5, 1700131 CrossRef.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702–707 CrossRef CAS.
- J. Shi, F. Li, J. Yuan, X. Ling, S. Zhou, Y. Qian and W. Ma, J. Mater. Chem. A, 2019, 7, 20936–20944 RSC.
- H. Bian, D. Bai, Z. Jin, K. Wang, L. Liang, H. Wang, J. Zhang, Q. Wang and S. Liu, Joule, 2018, 2, 1500–1510 CrossRef CAS.
- I. Lignos, V. Morad, Y. Shynkarenko, C. Bernasconi, R. M. Maceiczyk, L. Protesescu, F. Bertolotti, S. Kumar, S. T. Ochsenbein, N. Masciocchi, A. Guagliardi, C.-J. Shih, M. I. Bodnarchuk, A. J. deMello and M. V. Kovalenko, ACS Nano, 2018, 12, 5504–5517 CrossRef CAS.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- X. Shen, Y. Zhang, S. V. Kershaw, T. Li, C. Wang, X. Zhang, W. Wang, D. Li, Y. Wang, M. Lu, L. Zhang, C. Sun, D. Zhao, G. Qin, X. Bai, W. W. Yu and A. L. Rogach, Nano Lett., 2019, 19, 1552–1559 CrossRef CAS.
- Y. Jiang, J. Yuan, Y. Ni, J. Yang, Y. Wang, T. Jiu, M. Yuan and J. Chen, Joule, 2018, 2, 1356–1368 CrossRef.
- Z. Li, F. Zhou, Q. Wang, L. Ding and Z. Jin, Nano Energy, 2020, 71, 104634 CrossRef CAS.
- Z. Li, M. Yang, J.-S. Park, S.-H. Wei, J. J. Berry and K. Zhu, Chem. Mater., 2015, 28, 284–292 CrossRef.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Gratzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 7232–7238 CrossRef CAS.
- A. Hazarika, Q. Zhao, E. A. Gaulding, J. A. Christians, B. Dou, A. R. Marshall, T. Moot, J. J. Berry, J. C. Johnson and J. M. Luther, ACS Nano, 2018, 12, 10327–10337 CrossRef CAS.
- A. F. Gualdrón-Reyes, S. J. Yoon, E. M. Barea, S. Agouram, V. Muñoz-Sanjosé, Á. M. Meléndez, M. E. Niño-Gómez and I. Mora-Seró, ACS Energy Lett., 2018, 4, 54–62 CrossRef.
- W. Li, M. U. Rothmann, A. Liu, Z. Wang, Y. Zhang, A. R. Pascoe, J. Lu, L. Jiang, Y. Chen, F. Huang, Y. Peng, Q. Bao, J. Etheridge, U. Bach and Y.-B. Cheng, Adv. Energy Mater., 2017, 7, 1700946 CrossRef.
- S. Seth, T. Ahmed, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 1610–1618 CrossRef CAS.
- S. Draguta, O. Sharia, S. J. Yoon, M. C. Brennan, Y. V. Morozov, J. S. Manser, P. V. Kamat, W. F. Schneider and M. Kuno, Nat. Commun., 2017, 8, 200 CrossRef.
- D. P. McMeekin, G. Sadoughi, W. Rehman, G. E. Eperon, M. Saliba, M. T. Horantner, A. Haghighirad, N. Sakai, L. Korte, B. Rech, M. B. Johnston, L. M. Herz and H. J. Snaith, Science, 2016, 351, 151–155 CrossRef CAS.
- M. Suri, A. Hazarika, B. W. Larson, Q. Zhao, M. Vallés-Pelarda, T. D. Siegler, M. K. Abney, A. J. Ferguson, B. A. Korgel and J. M. Luther, ACS Energy Lett., 2019, 4, 1954–1960 CrossRef CAS.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bar, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C. J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Pechy, S. M. Zakeeruddin, M. Gratzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef CAS.
- J. Gong, M. Yang, X. Ma, R. D. Schaller, G. Liu, L. Kong, Y. Yang, M. C. Beard, M. Lesslie, Y. Dai, B. Huang, K. Zhu and T. Xu, J. Phys. Chem. Lett., 2016, 7, 2879–2887 CrossRef CAS.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- J. Zhang, Q. Di, J. Liu, B. Bai, J. Liu, M. Xu and J. Liu, J. Phys. Chem. Lett., 2017, 8, 4943–4953 CrossRef CAS.
- G. M. Dalpian and J. R. Chelikowsky, Phys. Rev. Lett., 2006, 96, 226802 CrossRef.
- Z. J. Yong, S. Q. Guo, J. P. Ma, J. Y. Zhang, Z. Y. Li, Y. M. Chen, B. B. Zhang, Y. Zhou, J. Shu, J. L. Gu, L. R. Zheng, O. M. Bakr and H. T. Sun, J. Am. Chem. Soc., 2018, 140, 9942–9951 CrossRef CAS.
- S. Bera, D. Ghosh, A. Dutta, S. Bhattacharyya, S. Chakraborty and N. Pradhan, ACS Energy Lett., 2019, 4, 1364–1369 CrossRef CAS.
- A. Shrestha, M. Batmunkh, A. Tricoli, S. Z. Qiao and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 5202–5224 CrossRef CAS.
- G. Huang, C. Wang, S. Xu, S. Zong, J. Lu, Z. Wang, C. Lu and Y. Cui, Adv. Mater., 2017, 29, 1700095 CrossRef.
- D. Chen, S. Zhou, G. Fang, X. Chen and J. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 39872–39878 CrossRef CAS.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS.
- D. Gao, B. Qiao, Z. Xu, D. Song, P. Song, Z. Liang, Z. Shen, J. Cao, J. Zhang and S. Zhao, J. Phys. Chem. C, 2017, 121, 20387–20395 CrossRef CAS.
- W. van der Stam, J. J. Geuchies, T. Altantzis, K. H. van den Bos, J. D. Meeldijk, S. Van Aert, S. Bals, D. Vanmaekelbergh and C. de Mello Donega, J. Am. Chem. Soc., 2017, 139, 4087–4097 CrossRef CAS.
- Q. Wang, X. Zhang, Z. Jin, J. Zhang, Z. Gao, Y. Li and S. F. Liu, ACS Energy Lett., 2017, 2, 1479–1486 CrossRef CAS.
- Q. A. Akkerman, D. Meggiolaro, Z. Dang, F. De Angelis and L. Manna, ACS Energy Lett., 2017, 2, 2183–2186 CrossRef CAS.
- H. Liu, Z. Wu, J. Shao, D. Yao, H. Gao, Y. Liu, W. Yu, H. Zhang and B. Yang, ACS Nano, 2017, 11, 2239–2247 CrossRef CAS.
- J. S. Yao, J. Ge, K. H. Wang, G. Zhang, B. S. Zhu, C. Chen, Q. Zhang, Y. Luo, S. H. Yu and H. B. Yao, J. Am. Chem. Soc., 2019, 141, 2069–2079 CrossRef CAS.
- R. K. Behera, A. Dutta, D. Ghosh, S. Bera, S. Bhattacharyya and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7916–7921 CrossRef CAS.
- C. Bi, X. Sun, X. Huang, S. Wang, J. Yuan, J. X. Wang, T. Pullerits and J. Tian, Chem. Mater., 2020, 32, 6105–6113 CrossRef CAS.
- N. K. Noel, S. D. Stranks, A. Abate, C. Wehrenfennig, S. Guarnera, A.-A. Haghighirad, A. Sadhanala, G. E. Eperon, S. K. Pathak, M. B. Johnston, A. Petrozza, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2014, 7, 3061–3068 RSC.
- F. Liu, C. Ding, Y. Zhang, T. S. Ripolles, T. Kamisaka, T. Toyoda, S. Hayase, T. Minemoto, K. Yoshino, S. Dai, M. Yanagida, H. Noguchi and Q. Shen, J. Am. Chem. Soc., 2017, 139, 16708–16719 CrossRef CAS.
- F. Liu, J. Jiang, Y. Zhang, C. Ding, T. Toyoda, S. Hayase, R. Wang, S. Tao and Q. Shen, Angew. Chem., Int. Ed., 2020, 59, 8421–8424 CrossRef CAS.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS.
- Y. J. Yoon, K. T. Lee, T. K. Lee, S. H. Kim, Y. S. Shin, B. Walker, S. Y. Park, J. Heo, J. Lee, S. K. Kwak, G.-H. Kim and J. Y. Kim, Joule, 2018, 2, 2105–2116 CrossRef CAS.
- S. G. Charles, H. DePuy, A. Mullin and V. M. Bierbaum, J. Am. Chem. Soc., 1990, 112, 8650–8655 CrossRef.
- T. Zhang, G. Li, Y. Chang, X. Wang, B. Zhang, H. Mou and Y. Jiang, CrystEngComm, 2017, 19, 1165–1171 RSC.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L. W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS.
- J. Hieulle, X. Wang, C. Stecker, D. Y. Son, L. Qiu, R. Ohmann, L. K. Ono, A. Mugarza, Y. Yan and Y. Qi, J. Am. Chem. Soc., 2019, 141, 3515–3523 CrossRef CAS.
- Q. A. Akkerman, M. Gandini, F. Di Stasio, P. Rastogi, F. Palazon, G. Bertoni, J. M. Ball, M. Prato, A. Petrozza and L. Manna, Nat. Energy, 2016, 2, 16194 CrossRef.
- T. C. Jellicoe, J. M. Richter, H. F. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Bohm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS.
- M. H. Kumar, S. Dharani, W. L. Leong, P. P. Boix, R. R. Prabhakar, T. Baikie, C. Shi, H. Ding, R. Ramesh, M. Asta, M. Graetzel, S. G. Mhaisalkar and N. Mathews, Adv. Mater., 2014, 26, 7122–7127 CrossRef CAS.
- X. G. Zhao, J. H. Yang, Y. Fu, D. Yang, Q. Xu, L. Yu, S. H. Wei and L. Zhang, J. Am. Chem. Soc., 2017, 139, 2630–2638 CrossRef CAS.
- J. Zhang, Y. Yang, H. Deng, U. Farooq, X. Yang, J. Khan, J. Tang and H. Song, ACS Nano, 2017, 11, 9294–9302 CrossRef CAS.
- T.-B. Song, T. Yokoyama, S. Aramaki and M. G. Kanatzidis, ACS Energy Lett., 2017, 2, 897–903 CrossRef CAS.
- Y. Wang, J. Tu, T. Li, C. Tao, X. Deng and Z. Li, J. Mater. Chem. A, 2019, 7, 7683–7690 RSC.
- A. J. Houtepen, Z. Hens, J. S. Owen and I. Infante, Chem. Mater., 2017, 29, 752–761 CrossRef CAS.
- F. Zhang, S. Huang, P. Wang, X. Chen, S. Zhao, Y. Dong and H. Zhong, Chem. Mater., 2017, 29, 3793–3799 CrossRef CAS.
- S. ten Brinck and I. Infante, ACS Energy Lett., 2016, 1, 1266–1272 CrossRef CAS.
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127–148 CrossRef CAS.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS.
- J. Xue, J.-W. Lee, Z. Dai, R. Wang, S. Nuryyeva, M. E. Liao, S.-Y. Chang, L. Meng, D. Meng, P. Sun, O. Lin, M. S. Goorsky and Y. Yang, Joule, 2018, 2, 1866–1878 CrossRef CAS.
- T. Chiba, K. Hoshi, Y.-J. Pu, Y. Takeda, Y. Hayashi, S. Ohisa, S. Kawata and J. Kido, ACS Appl. Mater. Interfaces, 2017, 9, 18054–18060 CrossRef CAS.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S. T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS.
- D. Yang, X. Li, W. Zhou, S. Zhang, C. Meng, Y. Wu, Y. Wang and H. Zeng, Adv. Mater., 2019, 31, 1900767 CrossRef.
- X. Zheng, S. Yuan, J. Liu, J. Yin, F. Yuan, W.-S. Shen, K. Yao, M. Wei, C. Zhou, K. Song, B.-B. Zhang, Y. Lin, M. N. Hedhili, N. Wehbe, Y. Han, H.-T. Sun, Z.-H. Lu, T. D. Anthopoulos, O. F. Mohammed, E. H. Sargent, L.-S. Liao and O. M. Bakr, ACS Energy Lett., 2020, 5, 793–798 CrossRef CAS.
- N. K. Noel, A. Abate, S. D. Stranks, E. S. Parrott, V. M. Burlakov, A. Goriely and H. J. Snaith, ACS Nano, 2014, 8, 9815–9821 CrossRef CAS.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A. H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS.
- S. M. Park, A. Abtahi, A. M. Boehm and K. R. Graham, ACS Energy Lett., 2020, 5, 799–806 CrossRef CAS.
- C. Querner, P. Reiss, J. Bleuse and A. Pron, J. Am. Chem. Soc., 2004, 126, 11574–11582 CrossRef CAS.
- J. N. Yang, Y. Song, J. S. Yao, K. H. Wang, J. J. Wang, B. S. Zhu, M. M. Yao, S. U. Rahman, Y. F. Lan, F. J. Fan and H. B. Yao, J. Am. Chem. Soc., 2020, 142, 2956–2967 CrossRef CAS.
- M. I. Bodnarchuk, S. C. Boehme, S. Ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Gunther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63–74 CrossRef CAS.
- F. Li, Y. Liu, H. Wang, Q. Zhan, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 8546–8554 CrossRef CAS.
- F. Di Stasio, S. Christodoulou, N. Huo and G. Konstantatos, Chem. Mater., 2017, 29, 7663–7667 CrossRef.
- B. B. Zhang, S. Yuan, J. P. Ma, Y. Zhou, J. Hou, X. Chen, W. Zheng, H. Shen, X. C. Wang, B. Sun, O. M. Bakr, L. S. Liao and H. T. Sun, J. Am. Chem. Soc., 2019, 141, 15423–15432 CrossRef CAS.
- J. Dai, J. Xi, Y. Zu, L. Li, J. Xu, Y. Shi, X. Liu, Q. Fan, J. Zhang, S. Wang, F. Yuan, H. Dong, B. Jiao, X. Hou and Z. Wu, Nano Energy, 2020, 70, 104467 CrossRef CAS.
- H. Wang, Y. Dou, P. Shen, L. Kong, H. Yuan, Y. Luo, X. Zhang and X. Yang, Small, 2020, 16, 2001062 CrossRef CAS.
- G. H. Ahmed, J. K. El-Demellawi, J. Yin, J. Pan, D. B. Velusamy, M. N. Hedhili, E. Alarousu, O. M. Bakr, H. N. Alshareef and O. F. Mohammed, ACS Energy Lett., 2018, 3, 2301–2307 CrossRef CAS.
- T. Ahmed, S. Seth and A. Samanta, Chem. Mater., 2018, 30, 3633–3637 CrossRef CAS.
- S. Wang, Y. Wang, Y. Zhang, X. Zhang, X. Shen, X. Zhuang, P. Lu, W. W. Yu, S. V. Kershaw and A. L. Rogach, J. Phys. Chem. Lett., 2019, 10, 90–96 CrossRef CAS.
- S. Wang, C. Bi, J. Yuan, L. Zhang and J. Tian, ACS Energy Lett., 2017, 3, 245–251 CrossRef.
- X. Tang, J. Yang, S. Li, Z. Liu, Z. Hu, J. Hao, J. Du, Y. Leng, H. Qin, X. Lin, Y. Lin, Y. Tian, M. Zhou and Q. Xiong, Adv. Sci., 2019, 6, 1900412 CrossRef CAS.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. Ten Brinck, P. Aellen, A. Suess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C. J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS.
- Q. Wang, X. Zheng, Y. Deng, J. Zhao, Z. Chen and J. Huang, Joule, 2017, 1, 371–382 CrossRef CAS.
- M. Imran, P. Ijaz, L. Goldoni, D. Maggioni, U. Petralanda, M. Prato, G. Almeida, I. Infante and L. Manna, ACS Energy Lett., 2019, 4, 819–824 CrossRef CAS.
- H. Wu, Y. Zhang, M. Lu, X. Zhang, C. Sun, T. Zhang, V. L. Colvin and W. W. Yu, Nanoscale, 2018, 10, 4173–4178 RSC.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS.
- J. Pan, S. P. Sarmah, B. Murali, I. Dursun, W. Peng, M. R. Parida, J. Liu, L. Sinatra, N. Alyami, C. Zhao, E. Alarousu, T. K. Ng, B. S. Ooi, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2015, 6, 5027–5033 CrossRef CAS.
- G. Li, J. Huang, H. Zhu, Y. Li, J.-X. Tang and Y. Jiang, Chem. Mater., 2018, 30, 6099–6107 CrossRef CAS.
- J. Dai, J. Xi, L. Li, J. Zhao, Y. Shi, W. Zhang, C. Ran, B. Jiao, X. Hou, X. Duan and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 5754–5758 CrossRef CAS.
- Y. Tan, Y. Zou, L. Wu, Q. Huang, D. Yang, M. Chen, M. Ban, C. Wu, T. Wu, S. Bai, T. Song, Q. Zhang and B. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 3784–3792 CrossRef CAS.
- C. Wang, A. S. Chesman and J. J. Jasieniak, Chem. Commun., 2016, 53, 232–235 RSC.
- L. Wu, Q. Zhong, D. Yang, M. Chen, H. Hu, Q. Pan, H. Liu, M. Cao, Y. Xu, B. Sun and Q. Zhang, Langmuir, 2017, 33, 12689–12696 CrossRef CAS.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS.
- B. Luo, Y. C. Pu, S. A. Lindley, Y. Yang, L. Lu, Y. Li, X. Li and J. Z. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8864–8868 CrossRef CAS.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS.
- L. M. Wheeler, E. M. Sanehira, A. R. Marshall, P. Schulz, M. Suri, N. C. Anderson, J. A. Christians, D. Nordlund, D. Sokaras, T. Kroll, S. P. Harvey, J. J. Berry, L. Y. Lin and J. M. Luther, J. Am. Chem. Soc., 2018, 140, 10504–10513 CrossRef CAS.
- J. Yuan, C. Bi, S. Wang, R. Guo, T. Shen, L. Zhang and J. Tian, Adv. Funct. Mater., 2019, 29, 1906615 CrossRef CAS.
- Y. Wang, J. Yuan, X. Zhang, X. Ling, B. W. Larson, Q. Zhao, Y. Yang, Y. Shi, J. M. Luther and W. Ma, Adv. Mater., 2020, 32, 2000449 CrossRef CAS.
- M. Yuan, M. Liu and E. H. Sargent, Nat. Energy, 2016, 1, 16016 CrossRef CAS.
- C. Liu, Q. Zeng, Y. Zhao, Y. Yu, M. Yang, H. Gao, H. Wei and B. Yang, Sol. RRL, 2020, 4, 2000102 CrossRef CAS.
- Y. Dong, Y.-K. Wang, F. Yuan, A. Johnston, Y. Liu, D. Ma, M.-J. Choi, B. Chen, M. Chekini, S.-W. Baek, L. K. Sagar, J. Fan, Y. Hou, M. Wu, S. Lee, B. Sun, S. Hoogland, R. Quintero-Bermudez, H. Ebe, P. Todorovic, F. Dinic, P. Li, H. T. Kung, M. I. Saidaminov, E. Kumacheva, E. Spiecker, L.-S. Liao, O. Voznyy, Z.-H. Lu and E. H. Sargent, Nat. Nanotechnol., 2020, 15, 668–674 CrossRef CAS.
- J. Kim, B. Koo, W. H. Kim, J. Choi, C. Choi, S. J. Lim, J.-S. Lee, D.-H. Kim, M. J. Ko and Y. Kim, Nano Energy, 2019, 66, 104130 CrossRef CAS.
- R. Koole, P. Liljeroth, C. de Mello Donega, D. Vanmaekelbergh and A. Meijerink, J. Am. Chem. Soc., 2006, 128, 10436–10441 CrossRef CAS.
- J. Tong, Z. Song, D. H. Kim, X. Chen, C. Chen, A. F. Palmstrom, P. F. Ndione, M. O. Reese, S. P. Dunfield, O. G. Reid, J. Liu, F. Zhang, S. P. Harvey, Z. Li, S. T. Christensen, G. Teeter, D. Zhao, M. M. Al-Jassim, M. van Hest, M. C. Beard, S. E. Shaheen, J. J. Berry, Y. Yan and K. Zhu, Science, 2019, 364, 475–479 CrossRef CAS.
- Y. Zheng, X. Yang, R. Su, P. Wu, Q. Gong and R. Zhu, Adv. Funct. Mater., 2020, 30, 2000457 CrossRef CAS.
- W. Li, Z. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 16099 CrossRef.
- X. Zhang, Y. Qian, X. Ling, Y. Wang, Y. Zhang, J. Shi, Y. Shi, J. Yuan and W. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 27307–27315 CrossRef CAS.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2018, 4, 4–22 CrossRef.
- J. Kim, S. Cho, F. Dinic, J. Choi, C. Choi, S. M. Jeong, J.-S. Lee, O. Voznyy, M. J. Ko and Y. Kim, Nano Energy, 2020, 75, 104985 CrossRef CAS.
- L. N. Quan, M. Yuan, R. Comin, O. Voznyy, E. M. Beauregard, S. Hoogland, A. Buin, A. R. Kirmani, K. Zhao, A. Amassian, D. H. Kim and E. H. Sargent, J. Am. Chem. Soc., 2016, 138, 2649–2655 CrossRef CAS.
- K. Ji, J. Yuan, F. Li, Y. Shi, X. Ling, X. Zhang, Y. Zhang, H. Lu, J. Yuan and W. Ma, J. Mater. Chem. A, 2020, 8, 8104–8112 RSC.
- F. Li, S. Zhou, J. Yuan, C. Qin, Y. Yang, J. Shi, X. Ling, Y. Li and W. Ma, ACS Energy Lett., 2019, 4, 2571–2578 CrossRef CAS.
- F. Liu, C. Ding, Y. Zhang, T. Kamisaka, Q. Zhao, J. M. Luther, T. Toyoda, S. Hayase, T. Minemoto, K. Yoshino, B. Zhang, S. Dai, J. Jiang, S. Tao and Q. Shen, Chem. Mater., 2019, 31, 798–807 CrossRef CAS.
- Q. Zhao, A. Hazarika, X. Chen, S. P. Harvey, B. W. Larson, G. R. Teeter, J. Liu, T. Song, C. Xiao, L. Shaw, M. Zhang, G. Li, M. C. Beard and J. M. Luther, Nat. Commun., 2019, 10, 2842 CrossRef.
- M. M. Tavakoli, M. Nasilowski, J. Zhao, M. G. Bawendi and J. Kong, Small Methods, 2019, 3, 1900449 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- Z. Pan, H. Rao, I. Mora-Sero, J. Bisquert and X. Zhong, Chem. Soc. Rev., 2018, 47, 7659–7702 RSC.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- J. H. Im, C. R. Lee, J. W. Lee, S. W. Park and N. G. Park, Nanoscale, 2011, 3, 4088–4093 RSC.
- F. Liu, Y. Zhang, C. Ding, T. Toyoda, Y. Ogomi, T. S. Ripolles, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, J. Phys. Chem. Lett., 2018, 9, 294–297 CrossRef CAS.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS.
- Z. Yang, Z. Yu, H. Wei, X. Xiao, Z. Ni, B. Chen, Y. Deng, S. N. Habisreutinger, X. Chen, K. Wang, J. Zhao, P. N. Rudd, J. J. Berry, M. C. Beard and J. Huang, Nat. Commun., 2019, 10, 4498 CrossRef.
- L. Meng, J. You, T.-F. Guo and Y. Yang, Acc. Chem. Res., 2016, 49, 155–165 CrossRef CAS.
- J. Yuan, A. Gallagher, Z. Liu, Y. Sun and W. Ma, J. Mater. Chem. A, 2015, 3, 2572–2579 RSC.
- E. A. Gaulding, J. Hao, H. S. Kang, E. M. Miller, S. N. Habisreutinger, Q. Zhao, A. Hazarika, P. C. Sercel, J. M. Luther and J. L. Blackburn, Adv. Mater., 2019, 31, 1902250 CrossRef.
- C. R. Kagan and C. B. Murray, Nat. Nanotechnol., 2015, 10, 1013–1026 CrossRef CAS.
- J. Chen and N. G. Park, Adv. Mater., 2018, 31, 1803019 CrossRef.
- P. Wang, J. Xie, K. Xiao, H. Hu, C. Cui, Y. Qiang, P. Lin, V. Arivazhagan, L. Xu, Z. Yang, Y. Yao, T. Lu, Z. Wang, X. Yu and D. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 22320–22328 CrossRef CAS.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2016, 2, 16177 CrossRef.
- L. Xiong, Y. Guo, J. Wen, H. Liu, G. Yang, P. Qin and G. Fang, Adv. Funct. Mater., 2018, 28, 1802757 CrossRef.
- X. Zhang and E. M. J. Johansson, J. Mater. Chem. A, 2017, 5, 303–310 RSC.
- M. Zhang, Q. Chen, R. Xue, Y. Zhan, C. Wang, J. Lai, J. Yang, H. Lin, J. Yao, Y. Li, L. Chen and Y. Li, Nat. Commun., 2019, 10, 4593 CrossRef.
- D. Luo, W. Yang, Z. Wang, A. Sadhanala, Q. Hu, R. Su, R. Shivanna, G. F. Trindade, J. F. Watts, Z. Xu, T. Liu, K. Chen, F. Ye, P. Wu, L. Zhao, J. Wu, Y. Tu, Y. Zhang, X. Yang, W. Zhang, R. H. Friend, Q. Gong, H. J. Snaith and R. Zhu, Science, 2018, 360, 1442–1446 CrossRef CAS.
- L. Calio, S. Kazim, M. Gratzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef CAS.
- Z. H. Bakr, Q. Wali, A. Fakharuddin, L. Schmidt-Mende, T. M. Brown and R. Jose, Nano Energy, 2017, 34, 271–305 CrossRef CAS.
- L. Zhang, C. Liu, J. Zhang, X. Li, C. Cheng, Y. Tian, A. K. Y. Jen and B. Xu, Adv. Mater., 2018, 30, 1804028 CrossRef.
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS.
- S.-W. Baek, S. Jun, B. Kim, A. H. Proppe, O. Ouellette, O. Voznyy, C. Kim, J. Kim, G. Walters, J. H. Song, S. Jeong, H. R. Byun, M. S. Jeong, S. Hoogland, F. P. García de Arquer, S. O. Kelley, J.-Y. Lee and E. H. Sargent, Nat. Energy, 2019, 4, 969–976 CrossRef CAS.
- G. Divitini, S. Cacovich, F. Matteocci, L. Cinà, A. Di Carlo and C. Ducati, Nat. Energy, 2016, 1, 15012 CrossRef CAS.
- G. Almeida, I. Infante and L. Manna, Science, 2019, 364, 833–834 CrossRef CAS.
- H. Wu, S. Wang, F. Cao, J. Zhou, Q. Wu, H. Wang, X. Li, L. Yin and X. Yang, Chem. Mater., 2019, 31, 1936–1940 CrossRef CAS.
- H. Zhang, X. Wang, Q. Liao, Z. Xu, H. Li, L. Zheng and H. Fu, Adv. Funct. Mater., 2017, 27, 1604382 CrossRef.
- S. N. Raja, Y. Bekenstein, M. A. Koc, S. Fischer, D. Zhang, L. Lin, R. O. Ritchie, P. Yang and A. P. Alivisatos, ACS Appl. Mater. Interfaces, 2016, 8, 35523–35533 CrossRef CAS.
- V. González-Pedro, S. A. Veldhuis, R. Begum, M. J. Bañuls, A. Bruno, N. Mathews, S. Mhaisalkar and Á. Maquieira, ACS Energy Lett., 2018, 3, 1409–1414 CrossRef.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. a. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- J. B. Hoffman, G. Zaiats, I. Wappes and P. V. Kamat, Chem. Mater., 2017, 29, 9767–9774 CrossRef CAS.
- W. J. Mir, A. Swarnkar and A. Nag, Nanoscale, 2019, 11, 4278–4286 RSC.
- J. Liang, Z. Liu, L. Qiu, Z. Hawash, L. Meng, Z. Wu, Y. Jiang, L. K. Ono and Y. Qi, Adv. Energy Mater., 2018, 8, 1800504 CrossRef.
- J. Khan, X. Zhang, J. Yuan, Y. Wang, G. Shi, R. Patterson, J. Shi, X. Ling, L. Hu, T. Wu, S. Dai and W. Ma, ACS Energy Lett., 2020, 5, 3322–3329 CrossRef CAS.
- P. Liu, W. Chen, W. Wang, B. Xu, D. Wu, J. Hao, W. Cao, F. Fang, Y. Li, Y. Zeng, R. Pan, S. Chen, W. Cao, X. W. Sun and K. Wang, Chem. Mater., 2017, 29, 5168–5173 CrossRef CAS.
- H. Wang, N. Sui, X. Bai, Y. Zhang, Q. Rice, F. J. Seo, Q. Zhang, V. L. Colvin and W. W. Yu, J. Phys. Chem. Lett., 2018, 9, 4166–4173 CrossRef CAS.
- H. Wang, X. Zhang, N. Sui, Y. Hu, V. L. Colvin, W. W. Yu and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 11769–11777 CrossRef CAS.
- H. Huang, B. Chen, Z. Wang, T. F. Hung, A. S. Susha, H. Zhong and A. L. Rogach, Chem. Sci., 2016, 7, 5699–5703 RSC.
- M. Meyns, M. Perálvarez, A. Heuer-Jungemann, W. Hertog, M. Ibáñez, R. Nafria, A. Genç, J. Arbiol, M. V. Kovalenko, J. Carreras, A. Cabot and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2016, 8, 19579–19586 CrossRef CAS.
- A. Loiudice, M. Strach, S. Saris, D. Chernyshov and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 8254–8263 CAS.
- S. Bhaumik, S. A. Veldhuis, Y. F. Ng, M. Li, S. K. Muduli, T. C. Sum, B. Damodaran, S. Mhaisalkar and N. Mathews, Chem. Commun., 2016, 52, 7118–7121 RSC.
- R. An, F. Zhang, X. Zou, Y. Tang, M. Liang, I. Oshchapovskyy, Y. Liu, A. Honarfar, Y. Zhong, C. Li, H. Geng, J. Chen, S. E. Canton, T. Pullerits and K. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 39222–39227 CrossRef CAS.
- K. Domanski, B. Roose, T. Matsui, M. Saliba, S.-H. Turren-Cruz, J.-P. Correa-Baena, C. R. Carmona, G. Richardson, J. M. Foster, F. De Angelis, J. M. Ball, A. Petrozza, N. Mine, M. K. Nazeeruddin, W. Tress, M. Grätzel, U. Steiner, A. Hagfeldt and A. Abate, Energy Environ. Sci., 2017, 10, 604–613 RSC.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS.
- W. Zhou, S. Chen, Y. Zhao, Q. Li, Y. Zhao, R. Fu, D. Yu, P. Gao and Q. Zhao, Adv. Funct. Mater., 2019, 29, 1809180 CrossRef.
- F. Yang, H. Chen, R. Zhang, X. Liu, W. Zhang, J. Zhang, F. Gao and L. Wang, Adv. Funct. Mater., 2020, 30, 1908760 CrossRef CAS.
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 368 Search PubMed.
- A. N. Singh, S. Kajal, J. Kim, A. Jana, J. Y. Kim and K. S. Kim, Adv. Energy Mater., 2020, 10, 2000768 CrossRef CAS.
- A. A. Zhumekenov, M. I. Saidaminov, M. A. Haque, E. Alarousu, S. P. Sarmah, B. Murali, I. Dursun, X.-H. Miao, A. L. Abdelhady, T. Wu, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 32–37 CrossRef CAS.
- X. Lan, O. Voznyy, A. Kiani, F. P. Garcia de Arquer, A. S. Abbas, G. H. Kim, M. Liu, Z. Yang, G. Walters, J. Xu, M. Yuan, Z. Ning, F. Fan, P. Kanjanaboos, I. Kramer, D. Zhitomirsky, P. Lee, A. Perelgut, S. Hoogland and E. H. Sargent, Adv. Mater., 2016, 28, 299–304 CrossRef CAS.
- M. Liu, F. Che, B. Sun, O. Voznyy, A. Proppe, R. Munir, M. Wei, R. Quintero-Bermudez, L. Hu, S. Hoogland, A. Mandelis, A. Amassian, S. O. Kelley, F. P. García de Arquer and E. H. Sargent, ACS Energy Lett., 2019, 4, 1225–1230 CrossRef CAS.
- M. Liu, O. Voznyy, R. Sabatini, F. P. Garcia de Arquer, R. Munir, A. H. Balawi, X. Lan, F. Fan, G. Walters, A. R. Kirmani, S. Hoogland, F. Laquai, A. Amassian and E. H. Sargent, Nat. Mater., 2017, 16, 258–263 CrossRef CAS.
- G. H. Kim, F. P. Garcia de Arquer, Y. J. Yoon, X. Lan, M. Liu, O. Voznyy, L. Krishnan Jagadamma, A. Saud Abbas, Z. Yang, F. Fan, A. H. Ip, P. Kanjanaboos, S. Hoogland, A. Amassian, J. Y. Kim and E. H. Sargent, Nano Lett., 2016, 15, 7691–7696 CrossRef.
- X. Jiang, F. Wang, Q. Wei, H. Li, Y. Shang, W. Zhou, C. Wang, P. Cheng, Q. Chen, L. Chen and Z. Ning, Nat. Commun., 2020, 11, 1245 CrossRef CAS.
- T. Leijtens, K. A. Bush, R. Prasanna and M. D. McGehee, Nat. Energy, 2018, 3, 828–838 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |