
Sodium transition metal oxides: the preferred cathode choice for future sodium-ion batteries?
Qiannan
Liu
 ab,
Zhe
Hu
b,
Weijie
Li
b,
Chao
Zou
a,
Huile
Jin
a,
Shun
Wang
*a,
Shulei
Chou
*b and
Shi-Xue
Dou
b
ab,
Zhe
Hu
b,
Weijie
Li
b,
Chao
Zou
a,
Huile
Jin
a,
Shun
Wang
*a,
Shulei
Chou
*b and
Shi-Xue
Dou
b
aKey Laboratory of Carbon Materials of Zhejiang Province, Institute of New Materials and Industrial Technologies, Wenzhou University, Wenzhou, Zhejiang 325027, China. E-mail: shunwang@wzu.edu.cn
bInstitute for Superconducting and Electronic Materials, Australian Institute for Innovative Materials, University of Wollongong, Innovation Campus, Squires Way, North Wollongong, NSW 2522, Australia. E-mail: shulei@uow.edu.au
First published on 28th October 2020
Abstract
The exploration of next-generation sodium-ion batteries (SIBs) is a worldwide concern to replace the current commercial lithium-ion batteries, mitigating the increasing exhaustion of Li resources. Sodium transition metal oxides are considered to be one of the most promising cathode materials for SIBs. The anionic redox reaction in Li-rich transition metal oxides is capable of providing extra capacity in addition to the cationic redox activities in lithium-ion batteries. A similar phenomenon exists in SIBs, which even applies to Na-deficient transition metal oxides. Moreover, transition metal oxides with mixed phase also demonstrate great potential. In this review, studies on anionic redox are first systematically introduced. The up-to-date advances on high-capacity transition metal oxide cathode materials for SIBs are then classified and summarized in different groups associated with or without anionic redox. The existing challenges as well as available solutions and strategies are discussed, and proposals with new insights are made at the end. It is expected that this work can provide new perspectives on controlling the anionic redox activity and finding novel high-capacity oxide cathode materials for SIBs.
 Qiannan Liu | Qiannan Liu received her Bachelor's degree (2011) and Master's degree (2014) in material science from the Central South University, China. She obtained her PhD from the Institute for Superconducting and Electronic Materials, University of Wollongong in 2018. Her research focuses on the synthesis and characterization of electrode materials for sustainable energy storage and conversion applications. |
 Zhe Hu | Zhe Hu received his Bachelor's degree (2011) in chemistry and Master's degree (2013) in material engineering from the Nankai University, China. He obtained his PhD from the Institute for Superconducting and Electronic Materials, University of Wollongong in 2019. His research focuses on lithium-ion batteries and sodium-ion batteries. |
 Huile Jin | Huile Jin is a Professor at college of Chemistry and Materials Engineering, Wenzhou University. His research interests are in the development of advanced energy materials for supercapacitors, lithium-ion batteries and fuel cells. He has published more than 90 journal articles including Adv. Mater., Adv. Energy Mater., J. Am. Chem. Soc., Angew. Chem., Int. Ed., etc. He is also listed as an inventor on over 30 patents. He is a Managing editor of Carbon Energy. |
 Shun Wang | Shun Wang is a Distinguished Professor at the College of Chemistry and Materials Engineering, Wenzhou University. His research is focused on nanostructured functional materials, including carbon-based nanocomposites, functional Te nanocrystals, hierarchically structured and assembled materials for electrochemical energy storage and conversion technology. He has published over 100 papers including Nat. Commun., Adv. Mater., Adv. Energy Mater., J. Am. Chem. Soc., etc. He also filed over 30 patents and patent applications. He is an editor-in-chief of Carbon Energy and has been named a Fellow of the Royal Society of Chemistry. |
 Shulei Chou | Shulei Chou is a Professor at the ISEM, University of Wollongong. He obtained his Bachelor's degree (1999) and Master's degree (2014) from the Nankai University, China. He received his PhD degree from the University of Wollongong in 2010. His research interests include energy storage materials for battery applications, especially novel composite materials, new binders, and new electrolytes for Li/Na batteries. |
 Shi-Xue Dou | Shi-Xue Dou is a Distingiushed Professor at University of Wollongong, the founding director of ISEM. He received his PhD in chemistry at Dalhousie University, Canada in 1984 and DSc at the University of New South Wales in 1998. He was elected as a Fellow of the Australian Academy of Technological Science and Engineering in 1994, and awarded the Australian Government's Centenary Medal in 2003 and Australian Order of Member in 2019. He is named as a highly cited researcher in materials science by Thomson Reuters. He is also a program leader in the Auto CRC 2020. |
1. Introduction
With the increasing consumption of fossil fuels, the development of clean and sustainable energy sources is of more and more significance to society. Secondary rechargeable batteries have proved their value, which has been greatly acknowledged by the Nobel Prize awarded for lithium ion batteries (LIBs) in 2019.1 The continuous depletion of reserves and increasing cost of lithium, however, make finding alternatives a necessity.2,3 Sodium-ion batteries (SIBs) are regarded as one of the most promising candidates to replace the current commercial LIBs due to the abundant reserves and low cost of sodium and their similar characteristics to LIBs.4,5 It is essential to find suitable electrode materials with high capacity for practical applications. Among the various types of cathodes investigated for rechargeable SIBs,6,7 namely transition metal oxides, polyanionic compounds, hexacyanoferrates (HCF) or Prussian blue and its analogues (PBAs), and organic compounds, Na transition metal oxide compounds are considered to be the most promising, owing to their compact crystal structure and similarities to the successful Li transition metal oxides in LIB systems.8Generally, charge compensation of Li/Na oxide cathodes during Li+/Na+ de-intercalation/intercalation is balanced by the oxidation/reduction of transition metals (such as Ni and Mn), and the Li/Na storage capacities depend solely on the cationic redox reactions. The discovery of the Li-rich layered transition metal oxides (Li1+xTM1−xO2 or Li1+xM1−xO2, TM/M = 3d, 4d, or 5d transition metals, 0 < x ≤ 1/3) for use in LIBs, however, breaks this rule and has attracted researchers’ attention around the world.9–11 For example, Li1.2Ni0.13Mn0.54Co0.13O2 displays an abnormally high capacity of 290 mA h g−1 with a long plateau at 4.5 V in the initial charge process, which is impossible to attribute merely to cationic redox reactions.12 Extensive efforts have been performed to investigate the underlying mechanisms, and it is widely acknowledged that oxygen anions participate in the charge compensation process in addition to the cationic metals, providing extra capacity.13 Unfortunately, the anionic capacity achieved during the charge process is partly irreversible during discharging. The study of this anionic oxygen redox is of great interest for the SIB system as well, which can be identified from the explosive growth in the number of published reports in the literature on oxygen redox related SIB oxide cathodes. It is critical to make the best of additional capacities provided by anionic redox reactions. The periodic table of elements with ionic radii and highlighted elements reported in SIB oxide cathodes is summarized in Fig. 1. A great many oxide materials have been synthesized or predicted. Active electrochemical redox couples are mainly found in 3d transition metals. Transition metals in the 4d and 5d groups are mostly related to Na-rich oxides, such as Na2RuO314 and Na2IrO3.15 In addition, unlike the case in Li-based systems, Na-deficient oxides, such as Na2/3Ni1/3Mn2/3O2,16 can also exhibit anionic oxygen redox activity with additional capacity. The voltage activating oxygen redox activities in SIBs is normally lower than that needed in LIBs. Differences in specific characteristics between SIBs and LIBs demand new discoveries and comprehensive analysis in SIBs rather than simple duplication from LIB systems.
 | ||
| Fig. 1 Periodic table of elements with ionic radius.17 Elemental status for Na transition metal oxide cathodes are shown in the shadowed entries. The unit of ionic radius is Angstrom. | ||
Oxygen redox activities are generally triggered at high voltages, which imposes strict requirements on the electrolyte and cycling stability of the electrode materials. Many challenges, such as the voltage decay and gas release issues that possibly accompany it, still exist, resulting in performance deterioration.18 Modification strategies need to be carried out. There are several review papers discussing the anionic redox behaviour in LIBs with some involvement of SIBs.19–26 Besides Na-rich and Na-deficient oxide cathodes that could deliver extra capacity from anionic redox at high voltages, it is also important to develop new types of oxide cathodes with satisfactory capacities under moderate voltage ranges, such as those with mixed P-/O-/T-phase. In this review, studies of anionic redox are first systematically introduced. Current advances on high-capacity transition metal oxide cathodes for SIBs are then classified and summarized, which are associated with or without anionic redox in different groups. The existing challenges as well as available solutions and strategies are discussed, and proposals with new insights are made at the end. It is expected that this work can provide new perspectives on controlling the oxygen redox activity and finding novel high-capacity oxide cathode materials for SIBs.
2. Anionic redox
The anionic redox reaction was proposed and has been intensively studied in Li-rich transition metal oxides for LIBs. Typically, as shown in Fig. 2a–c, the Li-rich layered oxide Li1.2Ni0.13Mn0.54Co0.13O2 contains extra Li in the transition metal layers and could deliver a high discharge capacity of 290 mA h g−1 and energy density of 1000 W h kg−1 in the first cycle, nearly double those of the traditional layered oxide LiCoO2.12 The excess capacity is attributed to the oxygen redox activities at potentials above 4.5 V. During the charging process, Li1.2Ni0.13Mn0.54Co0.13O2 experiences transition metal oxidation below 4.5 V and then oxygen oxidation at higher potentials. Oxygen reduction occurs before the reduction of transition metals when discharging. In Fig. 2d, for the Li1.17Ni0.21Co0.08Mn0.54O2 reported by Gent et al., however, the transition metals are reduced first, followed by oxygen reduction below 3.65 V during discharging.27 They proposed that the coupling between the O2−/O− redox couple and TM migration altered the local oxygen coordination environment and modulated the electrochemical potential, inverting the TM–O/O redox sequence after the first charge.27 | ||
| Fig. 2 (a) The structure of layered oxides compared with that of Li-rich layered oxides, and voltage profiles of (b) LiCoO2 and (c) Li1.2Ni0.13Mn0.54Co0.13O2. Reproduced with permission.12 Copyright 2018, Nature Publishing Group. (d) dQ/dV curve of the first cycle voltage profile of Li1.17Ni0.21Co0.08Mn0.54O2. Reproduced with permission.27 Copyright 2017, Nature Publishing Group. | ||
The process and reversibility of oxygen redox reactions in Li-rich transition metal oxides was interpreted by Doublet's group.28 In their report, Li2Ru4+O3 as an example first undergoes a classical cationic (Ru4+/Ru5+) oxidation to form a LiRu5+O3 phase with no structural modification, which further experiences an anionic oxidation that creates unstable O-holes in the oxygen network, inducing its structural reorganisation (Fig. 3a). Three types of oxygen may be involved in these reactions, namely the oxo (O2−) species, the peroxo-like (O2)n− species, and the peroxo (O2)− species (Fig. 3b). The O2− and (O2)n− species occur during reversible cationic and anionic redox reactions, respectively. The 2O2−/(O2)2− transformation is catalysed by the TM, which allows for TM(d)–O2(σ) (Ru(4d)–O2(σ) in the Li2RuO3 case) covalent interactions and may be stabilized through a reductive coupling mechanism. As long as the (O2)n− species are covalently bonded to the TM, the anionic redox reaction should be reversible, leading to increased capacity for the material. In the case of a complete 2O2−/(O2)2− transformation, the (O2)− species de-coordinate the TM prior to being fully oxidized to O2, leading to irreversibility in discharge (Fig. 3b).28
 | ||
| Fig. 3 (a) Schematic illustration of the band structure for cationic vs. anionic redox for Li2Ru4+O3 and LiRu5+O3, and (b) illustration of oxygen participation in the redox activity of a transition metal oxide. Reproduced with permission.28 Copyright 2016, Royal Society of Chemistry. (c) Illustration of 2D ordered cation layer with coplanar unhybridized O 2p orbitals and 3D disordered cation framework with randomly distributed unhybridized O 2p orbitals for Li-rich transition metal oxides. Reproduced with permission.30 Copyright 2019, Wiley-VCH. (d) Schematic illustration of galvanostatic curves for different scenarios of the cationic and anionic processes in alkali-metal-rich transition metal oxides. Reproduced with permission.31 Copyright 2019, Nature Publishing Group. | ||
The source of oxygen redox reactions in Li-rich transition metal oxides was theoretically explained by Ceder's group.29 In conventional transition metal oxide cathodes, O anions are coordinated by three TM cations. The O 2p orbitals mainly participate in σ-type bonding states that are situated far below the Fermi level, and the occurrence of oxygen oxidation is unlikely. In Li-rich transition metal oxides, O anions are coordinated by two or less TM cations, leaving nonbonding (orphaned) oxygen 2p orbitals located on top of the TM–O bonding orbitals. In contrast to the Li-rich transition metal oxides with a two-dimensional (2D) ordered cation structure, the Li-rich transition metal oxides with a three-dimensional (3D) disordered cation framework shows a relatively stable oxygen lattice structure with randomly distributed nonbonding O 2p orbitals (Fig. 3c).30 During the processes of Li extraction and transition metal oxidation, orphaned oxygen levels rise to the Fermi level and are capable of donating electrons, acting as reversible redox centres. This theory was accepted for a long time in the early stage of research, but was challenged recently, which will be discussed later.
Different scenarios for the cationic and anionic processes in alkali-metal-rich transition metal oxides are summarized as shown in Fig. 3d.31 There is no O2 gas release and no cation migration during charging in the fully reversible cationic and anionic processes (i in Fig. 3d). For the ii–iv types, a hysteresis between charge and discharge occurs: (ii) O2 release occurs in the first charge due to irreversible cationic migration, leading to a persistent hysteresis and an S-shaped curve due to the accumulation of transition metal and (O–O) redox centres; (iii) the charge plateaus are recovered if no O2 release occurs in the high charge stage and if cationic TM migrations are fully reversible in discharge; and (iv) the capacity loss caused by O2 release in charge is partially compensated in discharge by the activation of a novel TM redox couple. It is critical to achieve reversible oxygen redox reactions for satisfactory cyclability.
3. Transition metal oxide cathodes for SIBs
3.1. Sodium-rich oxide cathodes
Triggered by Li-rich transition metal oxides, the attention on high-capacity cathode materials for SIBs has expanded to sodium-rich transition metal oxides (Na1+xM1−xO2), which may present reversible capacities exceeding 1 Na per formula unit and deliver additional capacity through combining cationic and anionic redox activities. It is improper and not rational, however, to duplicate the theory directly from Li systems to Na1+xM1−xO2 due to their unique chemistry. For example, detrimental TM migration would be suppressed if the ionic radii of Li/Na and TM differ greatly. In this regard, Na1+xM1−xO2 shows more potential with larger contrast in ionic radii with 3d transition metals than its Li counterparts.32 Contrary to the popular ordered layered AMO2 and A1+xM1−xO2 structures (A = alkali metal), disordered rock-salt structures would be formed if the A and M ions are randomly distributed and the two layers are not obviously distinguished in A1+xM1−xO2 (Fig. 4a).33 A highly reversible insertion reaction would take place in rock-salt materials despite the increased disorder of the crystal structure, which could be attributed to the presence of a percolating 0-TM network.34 The synthesis of and report on Na1+xM1−xO2 have been far less than for its Li analogues. In the following part, Na1+xM1−xO2 with different formulas, namely, Na2MO3, NaMO2, Na2MO3·NaM′O2 compound, Na3MO4, and Na2Mn3O7 with TM vacancy, are summarized and reviewed in detail. | ||
| Fig. 4 (a) Schematic illustration and descriptions of layered and disordered rock-salt structures of alkali metal oxides. Reproduced with permission.33 Copyright 2020, Royal Society of Chemistry. (b) Structures of Na-rich O3-Na2MO3, O1-Na2MO3 and desodiated O1′-Na1MO3, (c) phase stability (ΔHstb: relative energy of a given phase against all other stable materials in the phase diagram) of O3-Na2MO3, formation energy convex hull for (d) Na2−xIrO3 and (e) Na2−xMnO3, and (f) crystal structures of fully desodiated MO3 phases. Reproduced with permission.35 Copyright 2020, Elsevier. | ||
Widespread attention on Na2MO3 started from the compound Na2Ru1−ySnyO3, reported by Tarascon’ group.36 Its charge–discharge cycle was based on a solid solution–biphasic–solid solution process, in contrast to the report on its Li analogue.37 This material delivered a high capacity of 140 mA h g−1 from the combined cationic (Ru4+/Ru5+) process at 2.8 V and anionic (O2−/O2n−) redox process at 3.8 V in the first cycle. The capacity decayed to 100 mA h g−1, however, after 50 cycles, which was attributed to the irreversibility of the high voltage oxygen redox reaction.
Na2RuO3 was subsequently reported and widely studied by Yamada's group, both experimentally and theoretically. Based on density functional theory calculation, they claimed that the average voltage for R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m phase NaxRuO3 with 1 < x < 2 is ∼2.28 V and that the average voltage for x < 1 driven by the desodiation of ilmenite type Na1RuO3 is ∼3.85 eV (Fig. 5a).38 The synthesized ordered Na2RuO3 (O-Na2RuO3) with honeycomb-ordered [Na1/3Ru2/3]O2 slabs was able to deliver a capacity of 180 mA h g−1, corresponding to a 1.3-electron reaction, whereas disordered Na2RuO3 only delivered 135 mA h g−1 (Fig. 5b).39 The extra 30% capacity of ordered Na2RuO3 was enabled by the formation of intermediate O1-type Na1RuO3 (Fig. 5c), which demonstrated honeycomb-type cation ordering in its [Na1/3M2/3]O2 slabs and induced frontier orbital reorganization to trigger the oxygen redox reaction.40 Recently, the group further demonstrated a self-repairing phenomenon in the stacking faults of Na2RuO3 upon desodiation through synchrotron X-ray diffraction coupled with planar-defect refinement analysis. The ordered vacancies mediating long-range cooperative Coulombic interactions between RuO3 slabs not only resulted in the generation of nonbonding O 2p orbitals, but also stabilized the phase transformation for highly reversible oxygen redox reactions.14 Later reports on Ru-based Na2MO3 materials including Na2Ru0.95Zr0.05O3 (Zr doping)41 and Na2Ru0.8Mn0.2O3 (Mn doping)42 demonstrated enhanced cyclability.
m phase NaxRuO3 with 1 < x < 2 is ∼2.28 V and that the average voltage for x < 1 driven by the desodiation of ilmenite type Na1RuO3 is ∼3.85 eV (Fig. 5a).38 The synthesized ordered Na2RuO3 (O-Na2RuO3) with honeycomb-ordered [Na1/3Ru2/3]O2 slabs was able to deliver a capacity of 180 mA h g−1, corresponding to a 1.3-electron reaction, whereas disordered Na2RuO3 only delivered 135 mA h g−1 (Fig. 5b).39 The extra 30% capacity of ordered Na2RuO3 was enabled by the formation of intermediate O1-type Na1RuO3 (Fig. 5c), which demonstrated honeycomb-type cation ordering in its [Na1/3M2/3]O2 slabs and induced frontier orbital reorganization to trigger the oxygen redox reaction.40 Recently, the group further demonstrated a self-repairing phenomenon in the stacking faults of Na2RuO3 upon desodiation through synchrotron X-ray diffraction coupled with planar-defect refinement analysis. The ordered vacancies mediating long-range cooperative Coulombic interactions between RuO3 slabs not only resulted in the generation of nonbonding O 2p orbitals, but also stabilized the phase transformation for highly reversible oxygen redox reactions.14 Later reports on Ru-based Na2MO3 materials including Na2Ru0.95Zr0.05O3 (Zr doping)41 and Na2Ru0.8Mn0.2O3 (Mn doping)42 demonstrated enhanced cyclability.
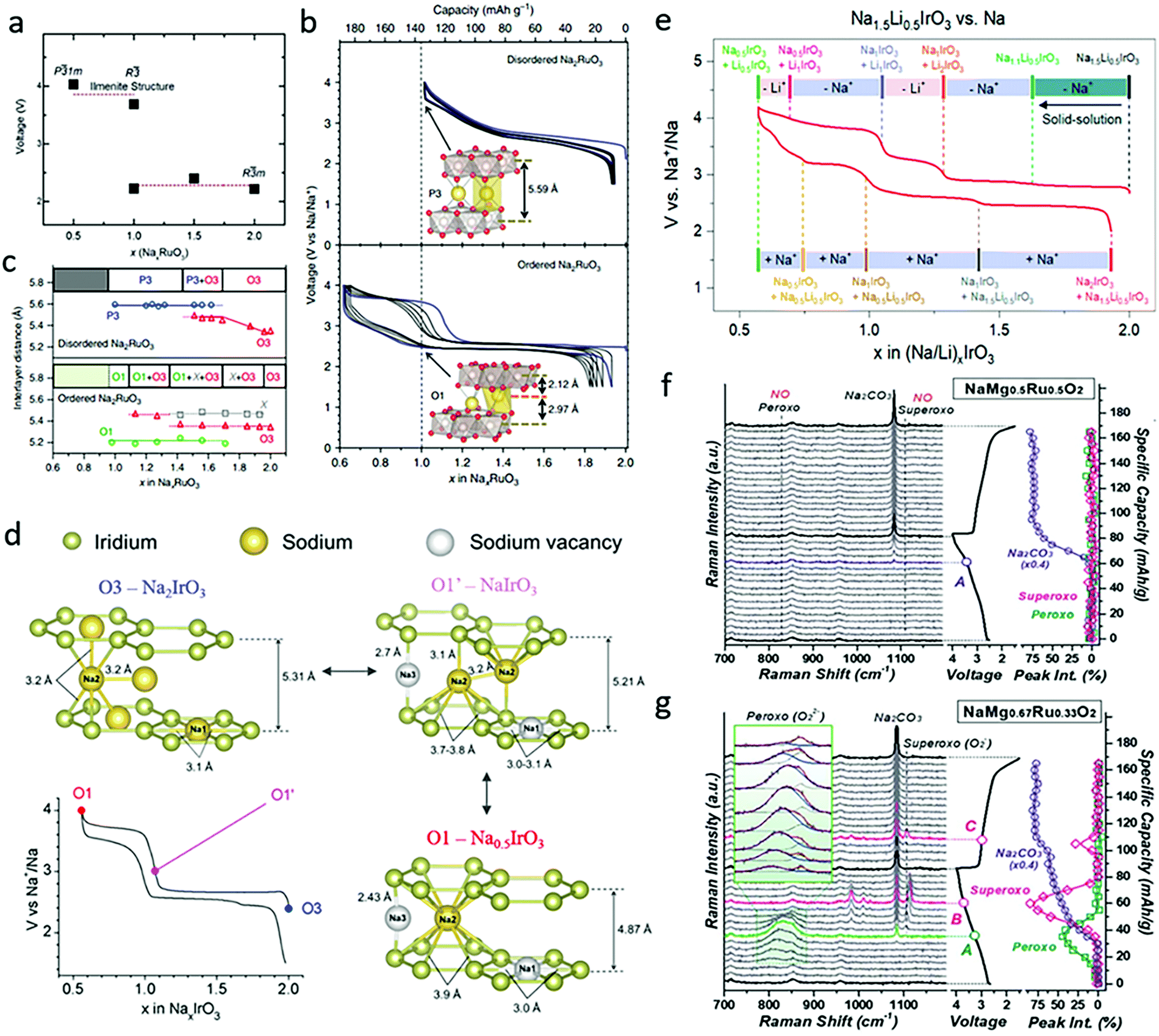 | ||
| Fig. 5 (a) Calculated voltage versus Na content (x) in NaxRuO3 compounds. Reproduced with permission.38 Copyright 2018, Royal Society of Chemistry. (b) Galvanostatic charge/discharge curves with the first cycle shown in blue (with the insets showing crystal structure at x = 1.0), and (c) corresponding interlayer distances and phase transformations as a function of x in disordered and ordered NaxRuO3. Reproduced with permission.39 Copyright 2016, Nature Publishing Group. (d) First cycle galvanostatic charge/discharge curve and corresponding illustration of the phase transformation in Na2IrO3. Reproduced with permission.15 Copyright 2016, American Chemical Society. (e) Galvanostatic charge/discharge curves and phase transformation of Na1.5Li0.5IrO3 in SIBs. Reproduced with permission.43 Copyright 2019, American Chemical Society. Capacity-dependent in situ Raman spectra collected during the first cycle for (f) NaMg0.5Ru0.5O2 and (g) NaMg0.67Ru0.33O2. Reproduced with permission.44 Copyright 2019, Royal Society of Chemistry. | ||
As a typical oxide with a 5d metal, Na2IrO3 was widely studied as another Na-rich cathode material. It has stable intermediate phases, Na1IrO3 and Na0.5IrO3, which exhibits O1′- and O1-type structures respectively (Fig. 5d), consistent with the results shown in Fig. 4d. Na2IrO3 was reported by Tarascon's group to feature the reversible extraction/insertion 1.5 Na+ per formula unit, while not experiencing oxygen release nor cationic migrations.15 Its large capacity was attributed to simultaneous cationic and anionic redox reactions occurring at 2.7 V, as demonstrated by complementary X-ray photoelectron spectroscopy (XPS), X-ray/neutron diffraction (XRD/ND), and transmission electron microscopy (TEM) measurements. Further Na+ removal was limited since the Na sites were stabilized by the O1′-phase formation. The first cycle voltage hysteresis was inhibited, since the large delocalization of 5d orbitals enables strong covalent Ir–O bonds, which block Ir migration from the TM layers to the interlayer sites. Other Na2MO3 compounds, including Na2Ti0.94Cr0.06O2.97 (Cr-doping)45 and Y-doped Na2ZrO3,46 have also been reported. Since Ti, Cr and Zr are all electrochemically inert in these materials, they only exhibit oxygen redox activities without participation of cationic redox reactions.
NaMO2 with only traditional 3d metals can also present anionic redox activity. Yusuke et al. have clarified the oxygen contribution to the redox reaction of O3-NaFe0.5Ni0.5O2 by a combination of X-ray absorption spectroscopy, Mössbauer spectroscopy, and density functional theory calculations.49 The material retains 91% capacity after 10 cycles in the voltage range of 2.0–3.8 V, slightly higher than the 82% capacity retention of NaTi0.5Ni0.5O2. This difference was attributed to the different oxygen orbital contributions to the redox reaction for NaFe0.5Ni0.5O2 (80%) and NaTi0.5Ni0.5O2 (40%).
An Ir-based Na-rich Na1.2Mn0.4Ir0.4O2 was reported by Zhang et al., and presented both cationic and anionic redox reactions during cycling in the voltage range of 1.5–4.4 V.51 The cation redox step relies on the Mn3+/Mn4+ couple, whereas Ir atom functions to build a strong Ir–O covalency and effectively suppress the O2 release, confirmed by the combination of in situ Raman, ex situ XPS, soft-X-ray absorption spectroscopy (XAS) and gas chromatography-mass spectrometry (GC-MS) measurements. CO2 release was detected, however, and is believed to come from the decomposition of carbonate electrolyte solvents.
Nb5+ ions also could effectively stabilize the redox reaction of O2− when coupled with Mn.52 A cation-disordered rocksalt oxide, Na1.3Nb0.3Mn0.4O2, was prepared by mechanical milling method.53 While the sample synthesized by traditional calcination delivered a capacity of 95 mA h g−1 based on the Mn3+/Mn4+ redox, the sample obtained by the mechanical milling delivered a large reversible capacity of ∼200 mA h g−1 at 50 °C based on both the Mn2+/Mn4+ redox and oxygen redox reactions. First-principle calculation, however, indicated that the oxide ion redox was stable in the system and contributed little to the capacity. The authors attributed this contradictory conclusion with the experimental observation to the nano-size of particles prepared by mechanical milling. Moreover, the cyclability could be improved by the addition of sodium bis(fluorosulfonyl)amide (NaFSA) to the electrolyte.
 | ||
| Fig. 6 (a) Proposed anionic redox activity, (b) first-cycle voltage profile, and (c) in situ Raman spectral contour plot of Na3RuO4. Reproduced with permission.54 Copyright 2018, Royal Society of Chemistry. (d) Quantification of the oxygen redox cyclability at the 2nd, 10th, and 30th cycles of Na3RuO4. Reproduced with permission.55 Copyright 2019, American Chemical Society. (e) The average voltage profile of NaxMn3O7 in the range of 0.5 < x < 2.0. Reproduced with permission.57 Copyright 2017, Royal Society of Chemistry. (f) Band structure of Na transition metal oxides with vacancy, and (g) voltage profile with corresponding structural evolution of Na4/7[□1/7Mn6/7]O2 (Na2Mn3O7), where □ represents vacancy. Reproduced with permission.58 Copyright 2019, American Chemical Society. | ||
Both Ru cation and oxygen anion activities, however, were reported to be involved in the charge compensation process of Na3RuO4 in another study, in which the initial charge capacity of Na3RuO4 was 115 mA h g−1 in the voltage range of 1.5–4.0 V.55 A quantitative investigation of the cationic and anionic redox reactions was performed by combining full-range mapping of resonant inelastic X-ray scattering and bulk-sensitive XAS. It was found that the Ru redox reaction was highly reversible over extended electrochemical cycling, while the lattice oxygen redox gradually deteriorated, with only 36% retention after 30 cycles (Fig. 6d), which was mainly responsible for the capacity fading of Na3RuO4. In addition, it was strikingly reported that 3 Na can be extracted from Na3RuO4 along with the oxidation of Ru5+ to Ru6+, leading to the formation of Na2RuO4 and then the oxidation of oxygen during the rest of the charge.56 This conclusion also highlights the difference between Li and Na materials regarding anionic redox, since Ru never reaches the +6 oxidation state during lithium removal.
It is generally believed that one (of three) O 2p orbitals is nearly unhybridized in the Na–O–Na configuration in the layered (or rocksalt) sodium transition metal oxides due to the strong ionic property of the Na–O bonds. Thus, if Na+ ions on the TM layers are replaced by vacancies, the unhybridized oxygen 2p bands (purple-colored) are expected to be much narrower in the □–O–Na or □–O–□ configuration relative to the Li–O–Li (Na) configuration in the conventional layered Li/Na-excess transition metal oxides (Fig. 6f).58 Thus, the vacancy in Na4/7(□1/7Mn6/7)O2 is capable of stabilizing the voltage profile of its oxygen-redox reactions while generating nonbonding O 2p orbitals along the □–O–Na and □–O–□ axes in charge compensation process. Song et al. demonstrated exceptional small-voltage hysteresis (<50 mV) in Na2Mn3O7, and attributed it to the well-maintained oxygen stacking sequence and the absence of irreversible gliding of O layers and cation migration from the TM layers.58 They further argued that the 4.2 V charge/discharge plateau was associated with a zero-strain de-intercalation/intercalation process of Na+ ions from distorted octahedral sites, while the 4.5 V plateau was linked to a reversible shrinkage/expansion process of the Mn vacancy at distorted prismatic sites (Fig. 6g). In another report, Mortemard de Boisse et al. demonstrated that Na2Mn3O7 exhibited the Mn3+/Mn4+ redox reaction and a highly reversible 4.1 V oxygen redox reaction, which leaded to an extra reversible capacity of ∼75 mA h g−1, corresponding to the (de)intercalation of 1.0 Na+ per formula unit (Na2Mn3O7 ↔ NaMn3O7).32 The active redox couples and electrochemical properties of various Na-rich transition metal oxide cathodes are summarized in Table 1.
| Material | Structure | Active redox couples | Voltage range (V) | Current density (mA g−1) | First charge/discharge capacity (mA h g−1) | Capacity (mA h g−1) (number of cycles) | Ref. |
|---|---|---|---|---|---|---|---|
| Na2RuO3 | Ordered O3 | Ru5+/Ru4+; O2−/O2n− | 1.5–4.0 | 30 | ∼180/180 | 160 (50) | 39 |
| Disordered O3 | Ru5+/Ru4+ | 1.5–4.0 | 30 | 135/∼130 | 130 (50) | 39 | |
| Na2IrO3 | O3 | O2−/O2n− | 1.5–4.0 | C/5 | 130/130 | ∼55 (50) | 15 |
| Na2ZrO3 | Monoclinic | O2−/O22− | 1.5–4.5 | 14.45 | 95/20 | — | 46 |
| Y-Doped Na2ZrO3 | Monoclinic | O2−/O22− | 1.5–4.5 | 14.45 | 382/158 | 180 (50) | 46 |
| Na2Ru0.75Sn0.25O3 | Hexagonal | Ru4+/Ru5+; O2−/O2n− | 1.5–4.2 | C/20 | 140/60.3 | 100 (50) | 36 |
| Na2Ru0.95Zr0.05O3 | Ordered monoclinic + disordered hexagonal | Ru4+/Ru5+ | 1.5–3.5 | 137 | ∼100/128 | 105 (200) | 41 |
| Na2Ru0.8Mn0.2O3 | O3 | Mn4+/Mn3+; Ru4+/Ru5+; O2−/O2n− | 1.5–4.0 | 27 | 163/178 | 120 (100) | 42 |
| Na2TiO3 | Monoclinic | O2−/O22− | 1.5–4.5 | 18.9 | ∼490/217 | 229 (15) | 45 |
| Na2Ti0.94Cr0.06O2.97 | Monoclinic | O2−/O22− | 1.5–4.5 | 18.9 | ∼960/336 | 182 (50) | 45 |
| NaMg0.67Ru0.33O2 | O3 | O2−/O−/O2−; Ru5+/Ru4+ | 1.5–4.0 | 10 | 87/81 | ∼70 (50) | 44 |
| NaMg0.5Ru0.5O2 | Ru4+/Ru5+ | ∼86/86 | ∼63 (50) | 44 | |||
| Na1.3Nb0.3Mn0.4O2 | Disordered rocksalt | Mn2+/Mn4+; O2−/O22− | 1.0–4.0 | 10 at 50 °C | ∼200 | ∼100 (30) | 53 |
| Na2.3Cu1.1Mn2O7−δ | Triclinic | Cu2+/Cu3+; Mn3+/Mn4+ | 2.1–4.05 | 10 | ∼107/107 | — | 65 |
| 2.1–4.05 | 2000 | — | ∼100 (1000) | ||||
| Na1.2Mn0.4Ir0.4O2 | Hexagonal O3 | Mn3+/Mn4+; O2−/O2n− | 1.5–4.4 | 20.3 | ∼170/140 | — | 51 |
| Na1.2Ni0.2Mn0.2Ru0.4O2 | Hexagonal O3 | — | 1.5–3.8 | 52.4 | 140/128 | 122 (60) | 50 |
| Na3RuO4 | Monoclinic | O2−/O2− | 1.5–4.0 | 50 | 321/∼130 | — | 54 |
| Na3RuO4 | Monoclinic | Ru5+/Ru6+; O2−/O2− | 1.5–4.0 | — | 115/92 | 33 (30) | 55 |
| Na2Mn3+0.3Mn4+2.7O6.85 | Hexagonal P2 + triclinic | Mn4+/Mn3+; O2−/(O2)n− | 1.5–4.5 | 30 | 75/213 | ∼105 (50) | 59 |
| Na4/7[□1/7Mn6/7]O2 (Na2Mn3O7) | Triclinic | Mn4+/Mn3+; O2−/(O2)n− | 1.5–4.7 | C/20 | ∼80/∼200 | — | 32 |
| 3.0–4.7 | C/20 | ∼110/75 | 57 (20) | ||||
| Na4/7[□1/7Mn6/7]O2 | Triclinic | Mn3+/Mn4+; O2−/(O2)n− | 1.5–4.4 | 20 | 96/220 | ∼100 (50) | 66 |
3.2. Sodium-deficient oxide cathodes
In Na-deficient oxides, since there are potential Na vacancies in NaO2 slabs and fully occupied TMO2 slabs, the local components around O are normally Na–O–TM and/or V–O–TM configurations in contrast to the Li–O–Li configurations in Li-rich compounds, leading to their special structural and chemical features which can increase O 2p orbitals near the Fermi level, triggering oxygen oxidation prior to TM oxidation towards a high-valence state. Therefore, it is possible to trigger oxygen redox chemistry in P-type Na-deficient electrode materials, which is different from the case in LIBs. High-Na content oxides are always preferred with high capacity and good structural stability. For low-Na P2-type materials, phase transformation from P2 to O2 or OP4/Z′ occur at voltages lower than ∼4.2 V when the Na content in the host drops below 1/3 mol per unit (Fig. 7a).60 The larger amount of Na in high-Na P2-type materials could reduce the average oxidation state of the TM ions below that of low-Na P2 materials, which would induce a larger contribution of TM cations in the valence band (Fig. 7b) and allow more Na+ ions to be maintained in the interlayers during charging, ensuring better structural and phase stability of the materials. Fig. 7c compares the phase transformations for different P2-type metal oxide cathodes along with Na+ de-insertion.61 The O redox reaction could be involved through the introduction of vacancy, Li, Mg, or Zn into the framework of classical P2 materials, the same as in Na-rich oxides. In most cases, the structural transition from P2 to O2 or OP4 could be inhibited in those materials through those cation or vacancy substitution. Other elements, such as Sn, Sb, Nb, Ti, Al, Fe and Zr, could also be doped into P2-type oxides to stabilize their structures.62–64 In the following part, different Na-deficient Nax(XyM1−y)O2 (M = Ni, Mn; X = Li, Mg, Zn, Ti, Fe, Cu, vacancy etc.; x, y < 1) cathodes are discussed based on the types of transition metal or substitution elements.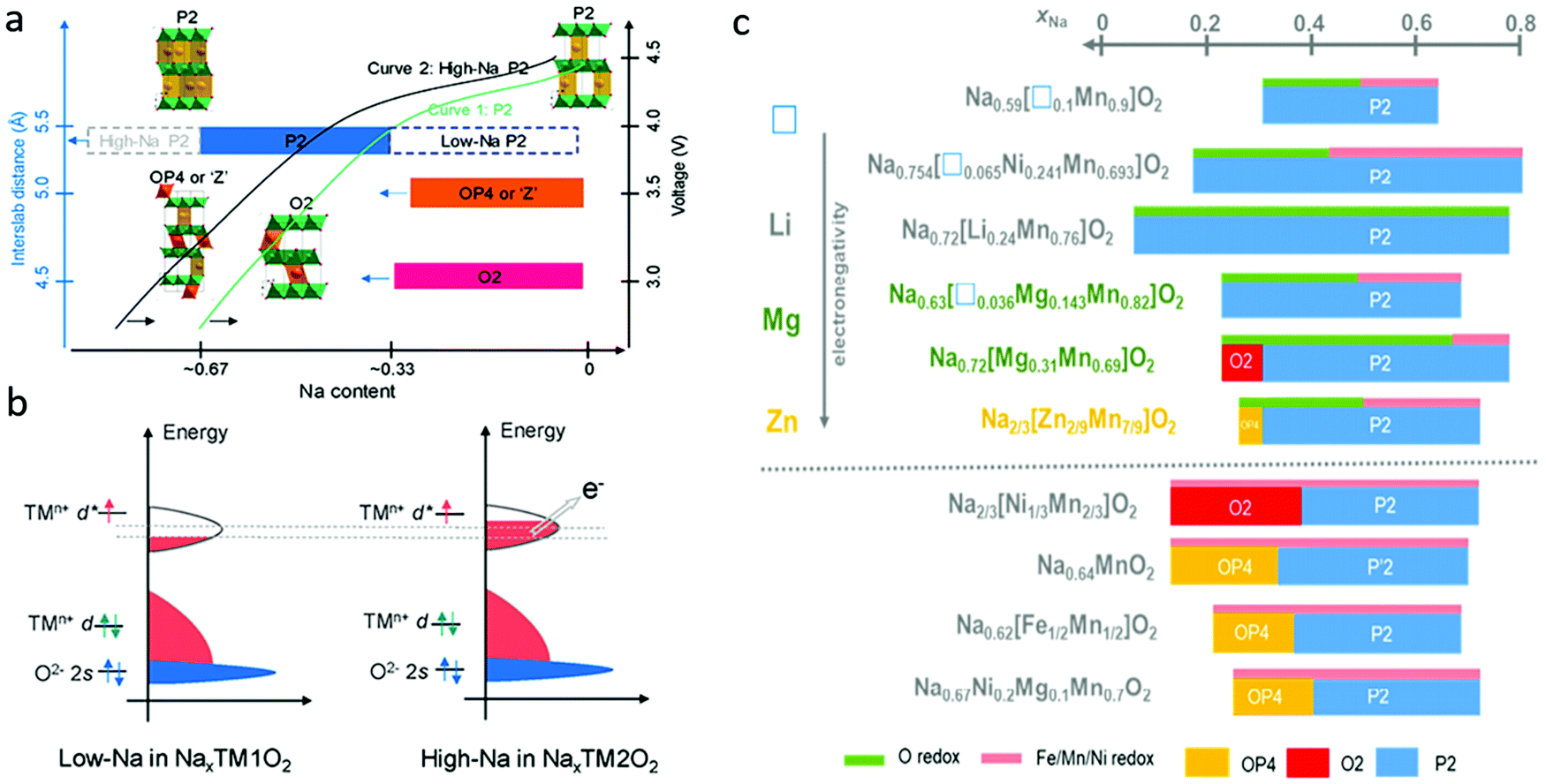 | ||
| Fig. 7 (a) Structural evolution and phase transformation of different types of P2 Na-deficient oxides during desodiation, and (b) electronic structure of low-Na and high-Na P2 oxides. Reproduced with permission.60 Copyright 2020, American Chemical Society. (c) Summaries of phase evolution for different P2-type metal oxide cathodes during Na+ extraction. Reproduced with permission.61 Copyright 2020, Elsevier. | ||
 | ||
| Fig. 8 (a) Galvanostatic charge/discharge profiles and charge compensation mechanism of Na0.67Ni0.2Mn0.8O2. Reproduced with permission.69 Copyright 2019, American Chemical Society. (b) Schematic illustration of in-plane Mn migrations based on honeycomb, ribbon, and mesh superstructures, respectively, and first-cycle voltage curves for (c) honeycomb-ordered Na0.75[Li0.25Mn0.75]O2 and (d) ribbon-ordered Na0.6[Li0.2Mn0.8]O2. Reproduced with permission.71 Copyright 2020, Nature Publishing Group. (e) Structural evolution and illustration of P3-type Na2/3Mg1/3Mn2/3O2 during the first cycle. Reproduced with permission.72 Copyright 2019, Royal Society of Chemistry. | ||
In particular, by comparing honeycomb-ordered Na0.75[Li0.25Mn0.75]O2 with ribbon-ordered Na0.6[Li0.2Mn0.8]O2, House et al. concluded that the first cycle voltage hysteresis was determined by the superstructure, namely the local ordering of Li and TM ions in the TM layers.71 In-plane Mn migrations, indicated by the arrows in Fig. 8b, are thought to be required to form O2− molecules (orange ellipses) in the TM layers of the charged honeycomb, ribbon, and mesh structures (Fig. 8b). More Mn migrations are required to form O2 in the ribbon and mesh structures, making O2 formation less likely to occur compared with that in the honeycomb structure. The honeycomb superstructure of Na0.75[Li0.25Mn0.75]O2 is lost on charging due to the formation of molecular O2, which reforms O2− during discharging. In the meantime, the Mn migration taking place within the plane changes the O2− coordination and lowers the discharge voltage (Fig. 8c). The ribbon superstructure of Na0.6[Li0.2Mn0.8]O2 inhibits Mn disorder and hence O2 formation, suppressing voltage hysteresis (Fig. 8d) and facilitating stable electron holes on O2−.
With Mg incorporation, P3-type Na2/3Mg1/3Mn2/3O2 was reported to deliver charge capacity larger than 190 mA h g−1 from lattice oxygen redox alone with a cut-off voltage of 4.65 V.72 High discharge capacity of 220 mA h g−1 could even be achieved when Mn3+/Mn4+ redox was partially involved in addition to the oxygen redox reaction. The voltage hysteresis here was attributed to a P3–O3 phase transition along with Mg2+ migration as shown in Fig. 8e.72 Through density functional theory analysis and modelling of both O2- and P2-Na2/3Mg1/3Mn2/3O2, Vergnet et al. argued that the oxygen network could be stabilized through a disproportion ration of oxygen pairs in O stacking or a highly reversible collective distortion in P stacking.77 P2-Na2/3[Mg0.28Mn0.72]O2 was reported to exhibit additional capacity caused by the oxygen redox with no O2 loss, since Mg2+ remained in the lattice and interacted with O 2p orbitals during cycling.78
By introducing Zn into the framework, P2-Na2/3[Mn0.7Zn0.3]O2 was able to deliver a specific discharge capacity of approximately 190 mA h g−1, based on the cumulative O2−/O1− and Mn4+/Mn3+ redox reactions between 1.5 and 4.6 V.79 A high capacity retention of 80% was achieved after 200 cycles. Similarly, the Zn-doped Na0.833[Li0.25Mn0.75]O2 (Na0.833Zn0.0375[Li0.25Mn0.7125]O2) achieved charge and discharge capacities of 2410 mA h g−1 and 1660 mA h g−1 respectively, and maintained a discharge capacity of 1620 mA h g−1 in the 100th cycle.80 In both cases, the excellent cyclability was attributed to the electrochemically inert Zn2+, which effectively mitigated the Jahn–Teller Mn3+ distortion and stabilized the structure.
Fig. 9a illustrates the structural evolution of typical alkali metal oxide cathodes as a function of the TM/O ratio. The TM/O ratio can be regulated via increasing the alkali metal content to more than 1 mol per formula unit in A-rich materials and decreasing the TM content less than 1 mol per formula unit in A-deficient materials (purple part).81 Decreasing TM content to form TM vacancy was proposed as a potential strategy to enable oxygen redox activity in A-deficient materials.81 This statement is supported by Na0.653Mn0.929O2, which possesses a layered P2 structure with disordered Mn vacancies distributed in MnO2 slabs in octahedral sites and Na in trigonal prismatic sites. Although the voltage plateau at ∼4.2 V associated with the O2−/(O2)n− oxygen redox showed a more severe capacity fade in the voltage range of 1.5–4.3 V than 2.5–4.3 V, Na0.653Mn0.929O2 maintained a capacity of ∼182 mA h g−1 after 60 cycles in a voltage range of 1.5–4.3 V at the rate of 0.1C (Fig. 9b and c). Different doping components may play different roles with different proportions during anionic redox activation. A combination of vacancies and Mg doping was utilized for P2-Na0.63[□0.036Mg0.143Mn0.820]O2.61 It was found that the vacancies and Mg doping triggered independent anionic redox processes at different redox voltage (4.1 and 4.35 V for vacancy and Mg doping activated anionic redox, respectively). The one associated with vacancies occurs at lower potential and is irreversible while the anionic redox process is reversible when the Mg2+ distribution is ordered during the charging process.
 | ||
| Fig. 9 (a) Structural evolution of typical reported oxide cathodes utilizing transition metal and/or oxygen redox activity as a function of the TM/O ratio, (b) XPS spectra of O 1s and Mn 2p peaks at different charge–discharge states of Na0.653Mn0.929O2, and (c) galvanostatic charge/discharge profiles of the 1st and 2nd cycles of Na0.653Mn0.929O2 in the voltage range of 1.5–4.3 V at 0.1C. Reproduced with permission.81 Copyright 2019, Elsevier. | ||
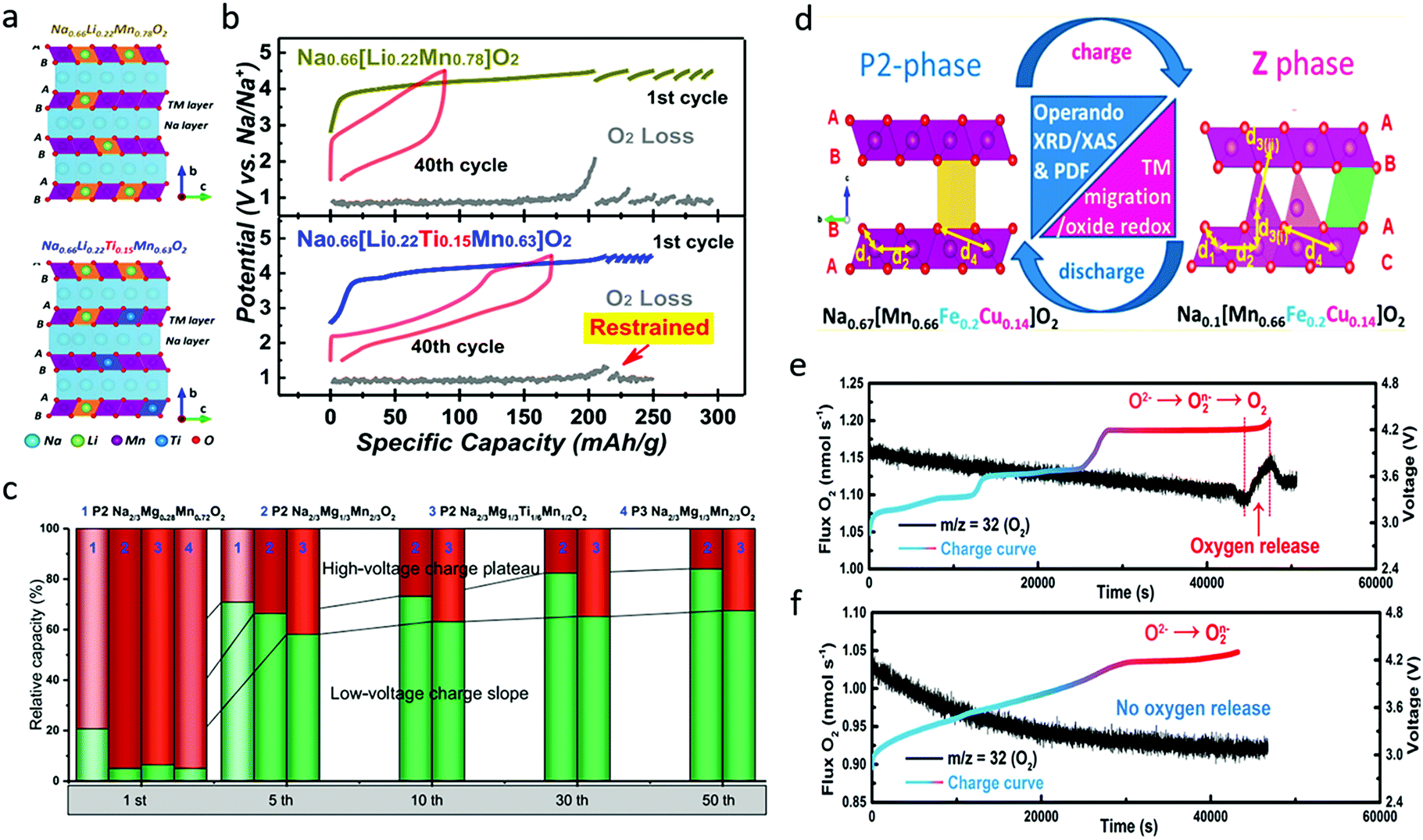 | ||
| Fig. 10 (a) Crystal structure illustration and (b) galvanostatic charge/discharge profiles of the 1st and 40th cycles and in situ DEMS results for O2 gas evolution during initial charging and subsequent galvanostatic intermittent charging processes of Na0.66Li0.22Mn0.78O2 and Na0.66Li0.22Ti0.15Mn0.63O2. Reproduced with permission.83 Copyright 2019, American Chemical Society. (c) Relative capacity of different Na–Mg–Mn oxides in the high-voltage charge plateau and low-voltage charge slope areas during cycling. Reproduced with permission.84 Copyright 2019, Elsevier. (d) Schematic illustration of crystal structure and phase of Na0.67[Mn0.66Fe0.20Cu0.14]O2 before and after desodiation. Reproduced with permission.85 Copyright 2017, American Chemical Society. In situ DEMS analysis of oxygen release during the first charge for (e) Na2/3Ni1/3Mn2/3O2 and (f) Na2/3Fe2/9Ni2/9Mn5/9O2. Reproduced with permission.16 Copyright 2020, American Chemical Society. | ||
Cu has also been incorporated to provide improved performance.63 P2-type Na0.67[Mn0.66Fe0.20Cu0.14]O2 delivered a high specific capacity of ∼176 mA h g−1 in the voltage range of 1.5–4.3 V at the rate of 13 mA g−1.85 Through operando XRD, the ex situ pair distribution function (PDF), and operando XPS analysis, it was found that the material was converted from P2 phase to a new high-voltage Z phase during first charge and discharge processes, as presented in Fig. 10d. The local structural evolution included Mn3+/Mn4+ redox below ∼3.4 V, followed by Cu and Fe ion redox activities at higher voltages. No transition metal evolution occurred above 4.1 V where the Z phase growth started, implying the reversible contribution of the oxygen redox couple to the capacity, coincident with metal migration. The effects of Cu doping were systematically investigated by Li et al. in Cu-doped P2-Na0.67Mn0.8Fe0.1Co0.1O2, which showed much larger O22−/O2− peak area ratio than the un-doped sample.86 The Cu-doped sample delivered a discharge specific capacity of 178 mA h g−1 in the first cycle and could maintain 162 mA h g−1 at the 50th cycle with a capacity retention of 91%. Through differential scanning calorimetry, ex situ XPS, and ND analysis, it was concluded that Cu doping increased both the a and the c lattice parameters, enlarged the interslab spacing and Na–O bond length, and decreased the Mn3+/Mn4+ ratio, resulting in alleviated Jahn–Teller distortion, and an enhanced Na+ diffusion coefficient and rate capability. Moreover, Cu doping effectively mitigated lattice volume change and phase transitions during cycling processes, which suppressed O2 release and improved the reversibility of oxygen redox. All these effects could contribute to improved structural stability and cycling stability.
O2−/O2n−/O2 evolution was also found in the traditional Na2/3Ni1/3Mn2/3O2, and the oxygen release problem was solved through Fe substitution (Fig. 10e and f).16 Irreversible O2−/O2n−/O2 evolution occurs at the 4.2 V plateau, which is due to the lack of TM–O hybridization and causes densification of Na2/3Ni1/3Mn2/3O2. Through Fe substitution, the oxygen release can be greatly suppressed due to the formation of the Fe–(O–O) species in Na2/3Fe2/9Ni2/9Mn5/9O2, which guarantees the reversibility of the O2−/O2n− redox reaction at a high operating voltage. This is similar to the effect of Ru–O–O covalent bonds in Li-rich Li2Ru1−ySnyO3 cathode, which is capable of minimizing oxygen release at a high operating voltage.37 As a result, the irreversible capacity loss in the first cycle is reduced from 25% in Na2/3Ni1/3Mn2/3O2 to 4% in Na2/3Fe2/9Ni2/9Mn5/9O2.
Co-doping of more than one component has been conducted and proved to be synergistically effective.87 Cu and Mg co-doping of Na0.67Mn0.75Ni0.25O2 endows Na0.67Mn0.71Cu0.02Mg0.02Ni0.25O2 with enhanced reversibility of both cationic and anionic redox activities.88 It was found that Cu/Mg co-doping effectively shortened the TM–O bonds, enhanced the TM–O bonding energy, improved the thermal decomposition temperature and the reversible P2–O2 transformation, and reduced the Mn3+/Mn4+ ratio, leading to improved oxygen redox reversibility, enhanced structural stability, and alleviation of the Jahn–Teller effect. More Na-deficient transition metal oxide cathodes exhibiting oxygen redox activities are listed in Table 2.
| Material | Structure | Active redox couples | Voltage range (V) | Current density (mA g−1) | First charge/discharge capacity (mA h g−1) | Capacity (mA h g−1) (number of cycles) | Ref. |
|---|---|---|---|---|---|---|---|
| Na0.5Ni0.25Mn0.75O2 | P3 | Ni2+/Ni4+; O2−/O− | 3.75–4.25 | 20 | ∼180/180 | — | 70 |
| 50 | ∼100/100 | ∼85 (100) | |||||
| Na0.78Ni0.23Mn0.69O2 | P2 | Ni2+/Ni4+; O2−/O− | 2.0–4.5 | 12.1 | 180/138 | 125 (10) | 68 |
| Na0.67Ni0.2Mn0.8O2 | P3 | Ni2+/Ni4+; O2−/O− | 1.8–4.4 | 10 | —/204 | 100 (25) | 69 |
| Ni2+/Ni4+ | 1.8–3.8 | 10 | —/150 | 139.5 (25) | 69 | ||
| Na2/3Ni1/3Mn2/3O2 | P2 | Ni2+/Ni3+/Ni4+; O2−/O2n− | 2.6–4.3 | 10 | 158/119 | — | 16 |
| 80 | — | 58.4 (100) | |||||
| Na0.6(Li0.2Mn0.8)O2 | P3 | Mn3+/Mn4+; O2−/(O2)n− | 2.0–4.5 | 15 | ∼130/120 | ∼148 (50) | 74 |
| Na0.6(Li0.2Mn0.8)O2 | P2 | Mn3+/Mn4+; O2−/(O2)n− | 2.0–4.6 | C/15 | ∼162/162 | 190 (100) | 76 |
| Na0.72Li0.24Mn0.76O2 | P2 | Mn3+/Mn4+; O2−/O− | 1.5–4.5 | 10 | 212/271 | 140 (30) | 103 |
| Na2/3Mg1/3Mn2/3O2 | P3 | Mn3+/Mn4+; O2−/O− | 1.6–4.4 | 15 | >150/220 | 153 (30) | 72 |
| Na2/3Mg0.28Mn0.72O2 | P2 | Mn3+/Mn4+; O2−/O− | 1.5–4.4 | 10 | >200 | >150 (30) | 104 |
| Na0.6Mg0.2Mn0.6Co0.2O2 | P2 | Co3+/Co4+; Mn3.75+/Mn4+; O2−/O− | 1.5–4.6 | 26 | 150/214 | 186 (100) | 105 |
| Na2/3Mn0.7Zn0.3O2 | P2 | Mn3+/Mn4+; O2−/O− | 1.5–4.6 | 26 | 115/190 | 152 (200) | 79 |
| Na0.63[□0.036Mg0.143Mn0.820]O2 | P2 | Mn3+/Mn4+; O2−/O− | 1.5–4.5 | ∼14 | 127/198 | — | 61 |
| Na45/54Li4/54Ni16/54Mn34/54O2 | P2 | Ni2+/Ni4+ | 2.0–4.0 | 22 | ∼103/103 | — | 60 |
| Ni2+/Ni4+; O2−/(O2)n− | 2.0–4.6 | 22 | 150/130 | — | 60 | ||
| Na0.85Li0.12Ni0.22Mn0.66O2 | P2 | Ni2+/Ni4+ | 2.0–4.3 | 22.4 | ∼136/123 | 115 (100) | 106 |
| Ni2+/Ni4+; O2−/(O2)n− | 2.0–4.5 | 22.4 | 173/120 | — | |||
| Na0.66Li0.22Ti0.15Mn0.63O2 | P2 | Mn3+/Mn4+; O2−/(O2)n− | 1.5–4.5 | 10 | 195/228 | 150 (50) | 83 |
| Na2/3Mg1/3Ti1/6Mn1/2O2 | P2 | Mn3+/Mn4+; O2−/O− | 1.5–4.5 | 20 | 175/250 | >160 (50) | 84 |
| Na0.67Cu0.28Mn0.72O2 | P2 | Mn3+/Mn4+; Cu2+/Cu3+; O2−/(O2)n− | 2.0–4.5 | 10 | ∼102/109 | 109 (50) | 107 |
| Na0.67Mn0.66Fe0.20Cu0.14O2 | P2 | Mn3+/Mn4+; Cu2+/Cu3+; Fe3+/Fe4+; O2−/(O2)n− | 1.5–4.3 | 13 | ∼176/176 | 70 (100) | 85 |
| 2.1–4.1 | 13 | ∼94/94 | 79 (100) | ||||
| Na2/3Fe2/9Ni2/9Mn5/9O2 | P2 | Ni2+/Ni3+/Ni4+; Fe3+/Fe4+; O2−/O2n− | 2.6–4.3 | 80 | —/130.9 | 102.1 (100) | 16 |
| Na0.67Mn0.77Fe0.1Co0.1Cu0.03O2 | P2 | Mn3+/Mn4+; O2−/O22− | 1.5–4.2 | 20 | —/178 | 162 (50) | 86 |
3.3. Mixed phase oxide cathodes
Transition metal oxides with mixed phase could provide enhanced electrochemical performance and phase stability in ambient atmosphere,8,89 combining the advantages of different structures. P2/P3-, P2/O3-, P2/tunnel-, and even materials with three phases, such as P2/O3/O1-type materials, have been reported. No oxygen redox has been reported to be involved in those materials.Based on the content of Li, Na1−xLixNi0.5Mn0.5O2+d demonstrates different phases, as shown in Fig. 11a.90 A synergistic effect from the P2/O3 intergrowth structure and Li incorporation beneficially endows Na0.7Li0.3Ni0.5Mn0.5O2+d with an unexpected enhancement of both specific capacity and rate capability as compared to single phase O3-NaNi0.5Mn0.5O2.90 It maintains almost 95% of its capacity even at the high current density of 150 mA g−1. Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+d integrating minor O3 structure into the Li-substituted P2-majority was also prepared.91 The redox peaks at ∼2.0 V, 2.0–3.9 V, and above 4 V are attributed to the redox reactions of manganese, cobalt and low valence state nickel, and high valence state nickel, respectively. This material delivered a large discharge capacity of 200 mA h g−1 at 0.1C with high energy density of 640 W h kg−1.
 | ||
| Fig. 11 (a) XRD patterns of Na1−xLixNi0.5Mn0.5O2+d with corresponding major phases evolution shown in the right panel. Reproduced with permission.90 Copyright 2014, Wiley-VCH. (b) High resolution TEM image of layered-tunnel intergrowth Na0.6MnO2, and (c) first cycle galvanostatic charge/discharge curves versus specific capacity (left) and specific energy (right) of layered-tunnel intergrowth, layered, and tunnel electrodes. Reproduced with permission.92 Copyright 2018, Wiley-VCH. (d) In operando synchrotron XRD patterns of NaxNi1/3Co1/3Mn1/3O2 cathode during the first cycle. Reproduced with permission.94 Copyright 2017, Royal Society of Chemistry. (e) Comparison among capacities and average voltages of mixed phase metal oxide cathodes with energy density curves superimposed.90–102 | ||
Tunnelled structures can also be included in this group. P2-tunnelled Na0.6MnO2 composite (Fig. 11b) electrode combined the high capacity advantage of layered structures with the excellent cycling stability and rate performance of tunnel structures, and delivered a reversible discharge capacity of 198.2 mA h g−1 at 0.2C together with a high energy density of 520.4 W h kg−1, superior to the performance of materials with only the layered or tunnel structures (Fig. 11c).92 Another mixed P2 + T NaxCo0.1Mn0.9O2 (0.44 < x < 0.7) sample also demonstrated an excellent discharge capacity of 219 mA h g−1 at 0.1C and retained 117 mA h g−1 even at 5C.93 The Na+ diffusion coefficient of the P2 + T phase was drastically improved, reaching 6 and 200 times that of pure T and P phases, respectively.
P2/O3/O1-intergrowth NaxNi1/3Co1/3Mn1/3O2 demonstrated better thermal stability and electrochemical performance than both P2/O3-structured and P-phase dominated NaxNi1/3Co1/3Mn1/3O2.94 It delivered a high initial reversible capacity of 142.8 mA h g−1 with a high Coulombic efficiency of 95% as well as a 93% capacity retention up to 50 cycles at 0.1C. The P2/O1/O3 intergrowth effectively inhibited the irreversible P2–O2 phase transition and improved the structural stability of the O3 and O1 phases (Fig. 11d). The average voltages and capacities of different mixed phase oxide cathodes are summarized in Fig. 11e. It could be found that Li and Mg substitutions also play an important role in this group.
4. Challenges and strategies
4.1. With anionic redox activity
Fig. 12 compares the high-voltage charge plateau and discharge capacity in the first cycle for different Na oxide cathodes. Normally, the anionic redox reaction is activated in the deep charge state represented by the high-voltage charge plateau that provides extra capacity. For Na-rich oxides, high voltage of >4.5 V is needed for Na2TiO3 and Y-doped Na2ZrO3 (in grey shadow) that do not have active cationic redox. With vacancy introduced, the voltage needed is reduced for Na2Mn3O7 (in yellow shadow). Anionic redox could be activated at lower voltage for Na-rich Ru- or Ir-based oxides (in green shadow), but the obtained capacity is relatively lower. On the other hand, Na-deficient oxides, which mainly feature the P2 phase with few featuring the P3 phase, normally present anionic redox activities at ∼4.0 V. More details are presented in Tables 1 and 2. Their long-term cyclability, however, may not be satisfactory. Although the oxygen redox reaction can contribute to additional capacity, it may cause voltage hysteresis in the first cycle and structural degradation during cycling due to the TM migration and oxygen loss, which would lead to progressive voltage decay and capacity loss. In Na-rich oxides, the extraction of Na+ ions from the Na and TM layers would result in severe underbonded oxygen, which promotes oxygen loss and could trigger further parasitic electrolyte degradation, oxygen-blocking surface reconstruction, and TM reduction.28 While oxygen evolution may occur merely at high voltage in near-surface regions, the TM migration occur throughout the bulk of the particles as well as the surface area, greatly influencing the electrochemical behaviour of materials.27 Although Na-deficient oxides suffer less from these issues, the progressively growing oxygen oxidation may still induce deterioration of both the bulk diffusion coefficient and the interfacial charge-transfer kinetics among them.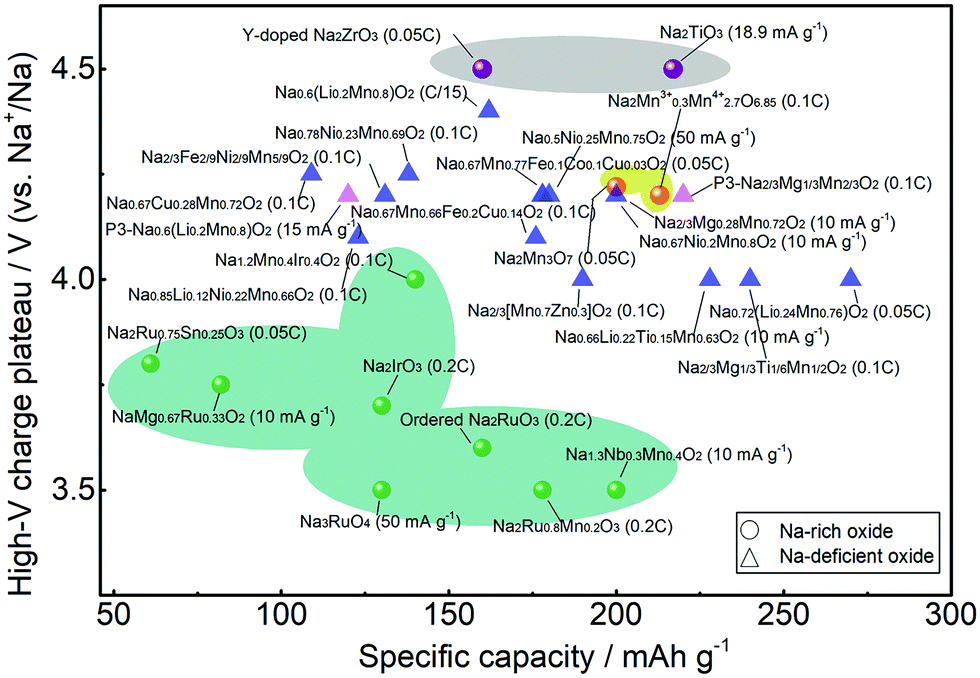 | ||
| Fig. 12 High-voltage charge plateau and discharge capacity in the first cycle for different Na-rich and Na-deficient oxide cathodes from Tables 1 and 2 with Na-rich oxides presented in shadow. | ||
Through designing a unified picture of anionic redox and introduction of an electron localization function in alkali-metal-rich transition metal oxides, Doublet's group concluded that the oxygen redox chemistry was tuned through structural modifications rather than the commonly supposed TM–O covalency modifications.31 This conclusion could explain the fact that systems containing no obvious nonbonding O 2p states (systems having neither alkaline earth/d10 TM cations, nor TM vacancies in the TMO2 layers), such as Na0.67Ni0.2Mn0.8O2,69 could perform oxygen redox reactions. They also suggested that O holes (ho) of 1/3 is the upper limit to avoid O2 release and achieve reversible anionic capacity in Li/Na-rich oxide cathode materials.31 The same emphasis on the effect of localized electron holes on O atoms was proposed for Li1.2[Ni0.13Co0.13Mn0.54]O2 cathode.108 Very recently, Chueh's group proposed the importance of the defect formation energy, which they believe can be used to control the nature of the oxidized species while minimizing structural disorder of high-valent redox couples in interaction cathodes.109
Although there is no widely accepted conclusion about the mechanism of oxygen redox, it could be accepted that advantageous structures that could reduce the structural transition induced TM migration, such as ribbon superstructures71 and disordered rocksalt structures,34,53 and appropriate chemical substitution/doping that increases the TM–O covalency or lowers ho through the activation of a cationic redox pair are efficient strategies to favour structural stability and reversible anionic activities. Sodium transition metal oxide cathodes that could operate with high structural stability and suppression of TM migration and O2 release are highly expected. The partial substitution of 3d metal ions in the TMO2 layer, by alkali metals such as Li+/Na+, alkaline-earth metals (Mg2+, etc.), d0 metals (Ti4+, Zr4+, Nb5+, etc.), d10 metals (Zn2+, Sb5+, Sn4+, etc.) or TM vacancies, is able to stabilize the oxidized oxygen species, and is the most effective way to enhance the reversibility of oxygen redox activity. In addition, anionic doping/substitution (F−; Cl−; S2−, etc.) has also been widely studied in Li-rich oxides.110–113 Compared with S and Cl, F has been demonstrated to be the best doping candidate because it can promote increased average voltage, improved ionic conductivity, reduced cation mixing, and minimized oxygen release, which provides superior stability for the Li-rich Li1.2Mn0.60Ni0.20O2 during high temperature cycling (Fig. 13a).112 Similar effects could be expected in SIBs. For layered transition metal oxide, F-doped P2-Na2/3Ni1/3Mn2/3O2,156 O3-NaNi1/3Fe1/3Mn1/3O2,114 and P3-type Na0.65Mn0.75Ni0.25O2115 were reported. It was claimed that F-doping could stabilize the structure and bring higher capacity for doped materials. The effect of F-doping on tunnel-phase Na0.44MnO2116 and Na0.66[Mn0.66Ti0.34]O2117 was also reported. A layer-tunnel hybrid structure was found in Na0.44MnO1.93F0.07, which displayed a superb capacity retention of ∼97% over 400 cycles at 5C. Al and F co-doping was further reported in Na0.44MnO2 and a 2D structure was formed in Na0.46Mn0.93Al0.07O1.79F0.21 (Fig. 13b),118 demonstrating much better structural stability and enhanced performance comparing with the undoped sample.
 | ||
| Fig. 13 (a) CV curves of first cycle (solid lines) and second cycle (dotted lines) for F-, S-, and Cl-doped Li1.2Mn0.60Ni0.20O2 in comparison with a pristine sample. Reproduced with permission.112 Copyright 2020, American Chemical Society. (b) Illustration of crystal structure, cyclic voltammograms at 0.1 mV s−1, and galvanostatic charge–discharge profiles at 0.3C of Na0.44MnO2 (NMO) and Al, F co-doped Na0.44MnO2 (AFNMO). Reproduced with permission.118 Copyright 2020, Wiley-VCH. (c) Schematic illustration of the structure and morphology evolution (left) and the mechanism for suppression of the O3–P3 phase transition (right) for NaCrO2 with enriched grain boundaries. Reproduced with permission.119 Copyright 2017, American Chemical Society. (d) Schematic illustration showing the transition from the initial material composed of Rm, C2/m, and nanocomposite intergrowth of Rm and C2/m to the spinel structure, which is partially inhibited by an AlF3 coating. Reproduced with permission.120 Copyright 2013, American Chemical Society. (e) Illustration of mechanism for the pristine and gas–solid interface reaction (GSIR) Li-rich NCM before charging and after full initial charge. Reproduced with permission.121 Copyright 2016, Nature Publishing Group. | ||
4.2. Besides anionic redox activity
A high charge voltage is normally needed to activate the anionic oxygen reaction, which would impose a higher requirement on both the electrode material and the electrolyte to perform over a wider voltage window. Irrespective of the oxygen redox reactions, Na transition metal oxides may suffer from the drawbacks of insufficient electrochemical performance, complex phase transitions and air sensitivity in SIBs.122 In this regard, besides triggering anionic redox reactions, composition design, cation doping, anion doping, and vacancy doping can also act as stabilizing tactics to provide enhanced structural stability. In addition, rational materials design (morphology control, nanostructure engineering, etc.) has demonstrated advantageous effects.119,123–128 For example, cation-disordered rock-salt and nanosized NaCrO2 were prepared by mechanical milling and further transformed to layered O3-type and nanosized NaCrO2 with enriched grain boundaries after heat-treatment (Fig. 13c).119 The gliding of CrO2 layers and O3–P3 phase transition were suppressed due to the presence of enriched grain boundaries and randomly oriented crystalline structures in individual particles, leading to better capacity retention compared with bulk O3-type NaCrO2. Moreover, surface coating modification, including protective layer coating and surface defect incorporation, has also been widely performed and proved its potential to mitigate phase transformation and capacity decay.129–133 AlF3 coating was proven to partially relieve the layer-to-spinel structural transformation during cycling, as illustrated in Fig. 13d, resulting in a slower capacity decay.120 As confirmed by both experimental characterization and theoretical calculations, Li[Li0.144Ni0.136Co0.136Mn0.544]O2 was found to experience less electrode/electrolyte corrosion and structural transformation after surface modification by oxygen vacancies created on the surface (Fig. 13e), delivering a high discharge capacity of 300 mA h g−1 with little voltage degradation after 100 cycles.121 Improvements due to the effects of protective carbon, polypyrrole, NaPO3, AlPO4, Mg3(PO4)2, TiO2, ZrO2, Al2O3, MgO, CuO and AlF3 layers have been verified for various oxide cathodes for SIBs as well.134–144Limiting the voltage range to suppress unfavourable phase transitions could also be useful but at the expense of energy density.145 The development of functional electrolyte, including effective additives, is necessary to withstand decomposition during long-term cycling.146 There is still a lack of sufficient experimental evidence to make clear of how the electrolyte influences the cathode,147 and how TM dissolution and structural changes proceed during the cycling process. The usage of advanced characterization techniques, such as GC-MS and DEMS, to detect possible gas formation from electrolyte decomposition or from oxygen evolution and quantize the contribution of anionic chemistry during cycling, is highly recommended to elucidate how much each mechanism contributes to the capacity.148 Further in-depth investigation and advanced theoretical modelling/predictions on the electrochemical mechanism could act as a powerful tool to help understand and design materials with desired properties.149 Valuable information on the structural evolution, migration energy barriers and optimal sodium migration path of electrode materials could be obtained as indicated in above examples, and have been used to help design, analysis and predict novel electrode materials.150 Combining experimental evidence and computational analysis needs to be rationally carried out to provide evidence and guidance for the development of high-performance oxide cathodes for SIBs.
5. Conclusion and perspectives
The exploration of next-generation SIBs is a worldwide concern. High capacity is always desirable for electrode materials. Anionic redox activities have been widely utilized and studied in Li-rich oxide cathodes for LIBs to provide extra capacity. No consensus on the sources of the anionic redox reaction, however, has been reached. Further and deeper studies are still needed for in-depth clarification. The possible induced voltage hysteresis, TM migration and O2 release during this process need to be suppressed to guarantee good performance. Studies on anionic redox in SIBs is still at an early stage. There is controversy about it in the literature, such as in the case of Na3RuO4.54,55 Besides Na-rich oxides with an emphasis on 4d and 5d TM elements, many Na-deficient oxides only containing 3d transition metals are capable of exhibiting oxygen redox activities, unlike the situation in Li-based systems. Different types of high-capacity Na transition metal oxide cathodes are summarized in Fig. 14a, which include Na-rich oxides, Na-deficient oxides mainly with P-phase, and mixed phase oxides. The former two families can exhibit anionic redox at high charge voltages. Most elements related to Na-rich oxides are 4d (Ru) or 5d (Ir) elements with minor 3d elements. Na-deficient oxides are dominated by 3d TM elements. Substitution of TMs with Li+, Mg2+, Zn2+, Ti4+, Zr4+, Nb5+, Sn4+, etc. or vacancy, is effectively performed to activate and stabilize oxygen redox activity in those materials. Moreover, Na oxides with mixed P-/O-/T-phase have also demonstrated their potential to provide high capacity and enhanced air stability, and are promising high-performance SIB cathodes as well. | ||
| Fig. 14 (a) Summary of high-capacity sodium transition metal oxide cathode materials; (b) available strategies and future research directions from the aspects of material development with those related to anionic redox shown in blue and mechanism studies. | ||
It is important to master the kinetic and thermodynamic mechanism of anionic redox to find approaches for suppressing TM migration and O2 release, mitigating voltage hysteresis and capacity decay to make the best of extra capacity that could be obtained from anionic redox chemistry.151 To achieve high capacity, effective strategies exploited for Na transition metal oxides could include structure engineering (composition design; morphology control; nanostructure engineering; etc.), cation/anion/vacancy doping/substitution that could motivate oxygen redox reactions and stabilize material structures, surface engineering (protective coating; defect surface coating) that could provide enhanced surface stability, and the development of high-performance mixed phase materials. Complementary developments of advanced characterization techniques and theoretical computational analysis can help provide a comprehensive understanding of the underlying electrochemical theory. Available and effective strategies for material development associated with or without anionic redox and mechanism studies are summarized in Fig. 14b.
When it comes to practical application of SIBs, in regard to polyanionic compound cathode, it is hard to get uniform and homogeneous composition from scale-up production. Inert gas protection and compounding with conductive materials are essentially needed to ensure material stability and electrical conductivity.152 Prussian blue and its analogues normally suffer from low capacity and intrinsic water and vacancies, leading to unsatisfactory energy density.153 Organic compounds show low operating voltages and are easy to dissolve in organic electrolytes.154 The introduction of solid-state electrolyte may be helpful to some extent but with extensive challenges that need to be tackled. Although low-speed electric vehicles powered by SIBs using transition metal oxide cathodes have been reported,155 many unknown and new materials with high performance are waiting to be found and developed. The voltage activating oxygen redox activities in SIBs is normally lower than that in LIBs. Future studies cannot completely rely on existing theories. On the one hand, it is not reasonable to completely transport the electrochemical mechanism, relating to anionic redox or otherwise, from Li-based systems to Na-based systems. More in-depth investigation of the mechanism and improvement strategies needs to be carried out. On the other hand, the development of novel materials, such as multi-phase oxides, which do not necessarily suffer from detrimentally irreversible capacity fade, is a promising direction. Moreover, the exploration of anode materials, high-voltage electrolyte (salt, solvent, additive), binder, separator, and even current collector needs to be cooperatively conducted at the same time. From the preliminary success of low-speed electric vehicles, with more and more sophisticated characterization techniques and advanced theoretical calculation approaches, it can be expected that existing challenges and difficulties will be finally conquered and that the commercialization of SIBs could be realized in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by an Australian Renewable Energy Agency (ARENA S4 project) grant and funds from the National Natural Science Foundation of China (Grant No. 51772219 and 51872209), the Zhejiang Provincial Special Support Program for High-level Talents (2019R52042) and the Zhejiang Provincial Natural Science Foundation of China (LZ21E010001). The authors are grateful to Dr Tania Silver for her critical revision of the manuscript. All the authors have given approval to the final version of the manuscript.References
- Y.-S. Hu and Y. Lu, ACS Energy Lett., 2019, 4, 2689–2690 CrossRef CAS.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- T. Or, S. W. Gourley, K. Kaliyappan, A. Yu and Z. Chen, Carbon Energy, 2020, 2, 6–43 CrossRef CAS.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 RSC.
- X. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, S. L. Chou and S. X. Dou, Small, 2019, 15, 1805381 CrossRef.
- J. Hong, H. Gwon, S.-K. Jung, K. Ku and K. Kang, J. Electrochem. Soc., 2015, 162, A2447 CrossRef CAS.
- P. Oh, S. Myeong, W. Cho, M.-J. Lee, M. Ko, H. Y. Jeong and J. Cho, Nano Lett., 2014, 14, 5965–5972 CrossRef CAS.
- B. Xiao, H. Liu, N. Chen, M. N. Banis, H. Yu, J. Liang, Q. Sun, T.-K. Sham, R. Li, M. Cai, G. A. Botton and X. Sun, Angew. Chem., Int. Ed., 2020, 59, 14313–14320 CrossRef CAS.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- W. Lee, S. Muhammad, C. Sergey, H. Lee, J. Yoon, Y. M. Kang and W. S. Yoon, Angew. Chem., Int. Ed., 2020, 59, 2578–2605 CrossRef CAS.
- B. M. de Boisse, M. Reynaud, J. Ma, J. Kikkawa, S.-i. Nishimura, M. Casas-Cabanas, C. Delmas, M. Okubo and A. Yamada, Nat. Commun., 2019, 10, 2185 CrossRef.
- A. J. Perez, D. Batuk, M. Saubanère, G. Rousse, D. Foix, E. Mccalla, E. J. Berg, R. Dugas, K. H. W. van den Bos, M.-L. Doublet, D. Gonbeau, A. M. Abakumov, G. V. Tendeloo and J.-M. Tarascon, Chem. Mater., 2016, 28, 8278–8288 CrossRef CAS.
- Y. Zhang, M. Wu, J. Ma, G. Wei, Y. Ling, R. Zhang and Y. Huang, ACS Cent. Sci., 2020, 6, 232–240 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2015, 162, A2490 CrossRef CAS.
- B. Li and D. Xia, Adv. Mater., 2017, 29, 1701054 CrossRef.
- C. Li, F. Geng, B. Hu and B. Hu, Mater. Today Energy, 2020, 17, 100474 CrossRef.
- C. Zhao, Q. Wang, Y. Lu, Y.-S. Hu, B. Li and L. Chen, J. Phys. D: Appl. Phys., 2017, 50, 183001 CrossRef.
- A. Manthiram, J. C. Knight, S. T. Myung, S. M. Oh and Y. K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef.
- M. Li, T. Liu, X. Bi, Z. Chen, K. Amine, C. Zhong and J. Lu, Chem. Soc. Rev., 2020, 49, 1688–1705 RSC.
- K. Zhang, B. Li, Y. Zuo, J. Song, H. Shang, F. Ning and D. Xia, Electrochem. Energy Rev., 2019, 2, 606–623 CrossRef CAS.
- E. Zhao, X. Yu, F. Wang and H. Li, Sci. China: Chem., 2017, 60, 1483–1493 CrossRef CAS.
- Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1701785 CrossRef.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. McIntire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef.
- M. Saubanère, E. McCalla, J.-M. Tarascon and M.-L. Doublet, Energy Environ. Sci., 2016, 9, 984–991 RSC.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692 CrossRef CAS.
- E. Zhao, Q. Li, F. Meng, J. Liu, J. Wang, L. He, Z. Jiang, Q. Zhang, X. Yu, L. Gu, W. Yang, H. Li, F. Wang and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 4323–4327 CrossRef CAS.
- M. B. Yahia, J. Vergnet, M. Saubanère and M.-L. Doublet, Nat. Mater., 2019, 18, 496–502 CrossRef.
- B. Mortemard de Boisse, S. i. Nishimura, E. Watanabe, L. Lander, A. Tsuchimoto, J. Kikkawa, E. Kobayashi, D. Asakura, M. Okubo and A. Yamada, Adv. Energy Mater., 2018, 8, 1800409 CrossRef.
- W. Lee, J. Kim, S. Yun, W. Choi, H. Kim and W.-S. Yoon, Energy Environ. Sci., 2020 10.1039/d0ee01277g.
- R. Clément, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345–373 RSC.
- J. Do, I. Kim, H. Kim and Y. Jung, Energy Storage Mater., 2020, 25, 62–69 CrossRef.
- P. Rozier, M. Sathiya, A.-R. Paulraj, D. Foix, T. Desaunay, P.-L. Taberna, P. Simon and J.-M. Tarascon, Electrochem. Commun., 2015, 53, 29–32 CrossRef CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. Laisa, H. Vezin, M. T. Sougrati, M.-L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. B. Hassine, L. Dupont and J.-M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS.
- M. Assadi, M. Okubo, A. Yamada and Y. Tateyama, J. Mater. Chem. A, 2018, 6, 3747–3753 RSC.
- B. M. De Boisse, G. Liu, J. Ma, S.-i. Nishimura, S.-C. Chung, H. Kiuchi, Y. Harada, J. Kikkawa, Y. Kobayashi, M. Okubo and A. Yamada, Nat. Commun., 2016, 7, 11397 CrossRef.
- M. Assadi, M. Okubo, A. Yamada and Y. Tateyama, J. Electrochem. Soc., 2019, 166, A5343 CrossRef CAS.
- S. Song, M. Kotobuki, F. Zheng, Q. Li, C. Xu, Y. Wang, W. D. Z. Li, N. Hu and L. Lu, J. Power Sources, 2017, 342, 685–689 CrossRef CAS.
- S. Liu, F. Lu, H. Li, X. Tian, H. Wang, J. Li and Y. Yang, J. Power Sources, 2019, 421, 14–22 CrossRef CAS.
- A. J. Perez, G. l. Rousse and J.-M. Tarascon, Inorg. Chem., 2019, 58, 15644–15651 CrossRef CAS.
- M. Jia, Y. Qiao, X. Li, K. Jiang and H. Zhou, J. Mater. Chem. A, 2019, 7, 20405–20413 RSC.
- S. Song, M. Kotobuki, Y. Chen, S. Manzhos, C. Xu, N. Hu and L. Lu, Sci. Rep., 2017, 7, 373 CrossRef.
- S. Song, M. Kotobuki, F. Zheng, C. Xu, N. Hu, L. Lu, Y. Wang and W.-D. Li, ACS Sustainable Chem. Eng., 2017, 5, 4785–4792 CrossRef CAS.
- J. Xu, D. H. Lee, R. l. J. Clément, X. Yu, M. Leskes, A. J. Pell, G. Pintacuda, X.-Q. Yang, C. P. Grey and Y. S. Meng, Chem. Mater., 2014, 26, 1260–1269 CrossRef CAS.
- D. Kim, M. Cho and K. Cho, Adv. Mater., 2017, 29, 1701788 CrossRef.
- Y. Nanba, T. Iwao, B. M. d. Boisse, W. Zhao, E. Hosono, D. Asakura, H. Niwa, H. Kiuchi, J. Miyawaki, Y. Harada, M. Okubo and A. Yamada, Chem. Mater., 2016, 28, 1058–1065 CrossRef CAS.
- N. Su, Y. Lyu and B. Guo, Electrochem. Commun., 2018, 87, 71–75 CrossRef CAS.
- X. Zhang, Y. Qiao, S. Guo, K. Jiang, S. Xu, H. Xu, P. Wang, P. He and H. Zhou, Adv. Mater., 2019, 31, 1807770 CrossRef.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS.
- K. Sato, M. Nakayama, A. M. Glushenkov, T. Mukai, Y. Hashimoto, K. Yamanaka, M. Yoshimura, T. Ohta and N. Yabuuchi, Chem. Mater., 2017, 29, 5043–5047 CrossRef CAS.
- Y. Qiao, S. Guo, K. Zhu, P. Liu, X. Li, K. Jiang, C.-J. Sun, M. Chen and H. Zhou, Energy Environ. Sci., 2018, 11, 299–305 RSC.
- Y. Hu, T. Liu, C. Cheng, Y. Yan, M. Ding, T.-S. Chan, J. Guo and L. Zhang, ACS Appl. Mater. Interfaces, 2019, 12, 3617–3623 CrossRef.
- M. Otoyama, Q. Jacquet, A. Iadecola, M. Saubanère, G. Rousse and J. M. Tarascon, Adv. Energy Mater., 2019, 9, 1803674 CrossRef.
- Z. Zhang, D. Wu, X. Zhang, X. Zhao, H. Zhang, F. Ding, Z. Xie and Z. Zhou, J. Mater. Chem. A, 2017, 5, 12752–12756 RSC.
- B. Song, M. Tang, E. Hu, O. J. Borkiewicz, K. M. Wiaderek, Y. Zhang, N. D. Phillip, X. Liu, Z. Shadike, C. Li, L. Song, Y.-Y. Hu, M. Chi, G. M. Veith, X.-Q. Yang, J. Liu, J. Nanda, K. Page and A. Huq, Chem. Mater., 2019, 31, 3756–3765 CrossRef CAS.
- Q. Wang, W. Yang, F. Kang and B. Li, Energy Storage Mater., 2018, 14, 361–366 CrossRef.
- C. Zhao, Z. Yao, Q. Wang, H. Li, J. Wang, M. Liu, S. Ganapathy, Y. Lu, J. Cabana, B. Li, X. Bai, A. Aspuru-Guzik, M. Wagemaker, L. Chen and Y.-S. Hu, J. Am. Chem. Soc., 2020, 142, 5742–5750 CrossRef CAS.
- X. Bai, A. Iadecola, J.-M. Tarascon and P. Rozier, Energy Storage Mater., 2020, 31, 146–155 CrossRef.
- X. Rong, F. Gao, F. Ding, Y. Lu, K. Yang, H. Li, X. Huang, L. Chen and Y.-S. Hu, J. Mater. Sci. Technol., 2019, 35, 1250–1254 CrossRef.
- S. Kumakura, Y. Tahara, S. Sato, K. Kubota and S. Komaba, Chem. Mater., 2017, 29, 8958–8962 CrossRef CAS.
- R. W. Smaha, J. H. Roudebush, J. T. Herb, E. M. Seibel, J. W. Krizan, G. M. Fox, Q. Huang, C. B. Arnold and R. J. Cava, Inorg. Chem., 2015, 54, 7985–7991 CrossRef CAS.
- K. Jiang, S. Xu, S. Guo, X. Zhang, X. Zhang, Y. Qiao, T. Fang, P. Wang, P. He and H. Zhou, Nano Energy, 2018, 52, 88–94 CrossRef CAS.
- Y. Li, X. Wang, Y. Gao, Q. Zhang, G. Tan, Q. Kong, S. Bak, G. Lu, X. Q. Yang, L. Gu, J. Lu, K. Amine, Z. Wang and L. Chen, Adv. Energy Mater., 2019, 9, 1803087 CrossRef.
- E. Talaie, V. Duffort, H. L. Smith, B. Fultz and L. F. Nazar, Energy Environ. Sci., 2015, 8, 2512–2523 RSC.
- C. Ma, J. Alvarado, J. Xu, R. l. J. Clément, M. Kodur, W. Tong, C. P. Grey and Y. S. Meng, J. Am. Chem. Soc., 2017, 139, 4835–4845 CrossRef CAS.
- E. J. Kim, L. A. Ma, L. C. Duda, D. M. Pickup, A. V. Chadwick, R. Younesi, J. T. Irvine and A. R. Armstrong, ACS Appl. Energy Mater., 2019, 3, 184–191 CrossRef.
- Q. Li, Y. Qiao, S. Guo, K. Jiang, Q. Li, J. Wu and H. Zhou, Joule, 2018, 2, 1134–1145 CrossRef CAS.
- R. A. House, U. Maitra, M. A. Pérez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502–508 CrossRef CAS.
- B. Song, E. Hu, J. Liu, Y. Zhang, X.-Q. Yang, J. Nanda, A. Huq and K. Page, J. Mater. Chem. A, 2019, 7, 1491–1498 RSC.
- N. Yabuuchi, R. Hara, M. Kajiyama, K. Kubota, T. Ishigaki, A. Hoshikawa and S. Komaba, Adv. Energy Mater., 2014, 4, 1301453 CrossRef.
- K. Du, J. Zhu, G. Hu, H. Gao, Y. Li and J. B. Goodenough, Energy Environ. Sci., 2016, 9, 2575–2577 RSC.
- X. Rong, J. Liu, E. Hu, Y. Liu, Y. Wang, J. Wu, X. Yu, K. Page, Y.-S. Hu, W. Yang, H. Li, X.-Q. Yang, L. Chen and X. Huang, Joule, 2018, 2, 125–140 CrossRef CAS.
- E. de la Llave, E. Talaie, E. Levi, P. K. Nayak, M. Dixit, P. T. Rao, P. Hartmann, F. Chesneau, D. T. Major, M. Greenstein, D. Aurbach and L. F. Nazar, Chem. Mater., 2016, 28, 9064–9076 CrossRef CAS.
- J. Vergnet, M. Saubanère, M.-L. Doublet and J.-M. Tarascon, Joule, 2020, 4, 420–434 CrossRef CAS.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS.
- A. Konarov, J. H. Jo, J. U. Choi, Z. Bakenov, H. Yashiro, J. Kim and S.-T. Myung, Nano Energy, 2019, 59, 197–206 CrossRef CAS.
- K. Zhang, D. Kim, Z. Hu, M. Park, G. Noh, Y. Yang, J. Zhang, V. W.-h. Lau, S.-L. Chou, M. Cho, S.-Y. Choi and Y.-M. Kang, Nat. Commun., 2019, 10, 5203 CrossRef CAS.
- C. Zhao, Q. Wang, Y. Lu, L. Jiang, L. Liu, X. Yu, L. Chen, B. Li and Y.-S. Hu, Energy Storage Mater., 2019, 20, 395–400 CrossRef.
- C. Li, C. Zhao, B. Hu, W. Tong, M. Shen and B. Hu, Chem. Mater., 2020, 32, 1054–1063 CrossRef CAS.
- X. Cao, X. Li, Y. Qiao, M. Jia, F. Qiu, Y. He, P. He and H. Zhou, ACS Energy Lett., 2019, 4, 2409–2417 CrossRef CAS.
- C. Zhao, Z. Yao, J. Wang, Y. Lu, X. Bai, A. Aspuru-Guzik, L. Chen and Y.-S. Hu, Chem, 2019, 5, 2913–2925 CAS.
- E. Talaie, S. Y. Kim, N. Chen and L. F. Nazar, Chem. Mater., 2017, 29, 6684–6697 CrossRef CAS.
- L. Li, H. Wang, W. Han, H. Guo, A. Hoser, Y. Chai and X. Liu, J. Electrochem. Soc., 2018, 165, A3854 CrossRef CAS.
- H.-R. Yao, P.-F. Wang, Y. Gong, J. Zhang, X. Yu, L. Gu, C. OuYang, Y.-X. Yin, E. Hu, X.-Q. Yang, E. Stavitski, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2017, 139, 8440–8443 CrossRef CAS.
- W. Kong, R. Gao, Q. Li, W. Yang, J. Yang, L. Sun and X. Liu, J. Mater. Chem. A, 2019, 7, 9099–9109 RSC.
- J. Liu, Z. Liu, X. Xu, S. Ji, L. Zeng and D. Zhang, Chem. – Eur. J., 2020, 26, 7747–7766 CrossRef.
- E. Lee, J. Lu, Y. Ren, X. Luo, X. Zhang, J. Wen, D. Miller, A. DeWahl, S. Hackney, B. Key, D. Kim, M. D. Slater and C. S. Johnson, Adv. Energy Mater., 2014, 4, 1400458 CrossRef.
- S. Guo, P. Liu, H. Yu, Y. Zhu, M. Chen, M. Ishida and H. Zhou, Angew. Chem. Int. Ed., 2015, 54, 5894–5899 CrossRef CAS.
- Y. Xiao, P. F. Wang, Y. X. Yin, Y. F. Zhu, X. Yang, X. D. Zhang, Y. Wang, X. D. Guo, B. H. Zhong and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1800492 CrossRef.
- G. Gao, D. Tie, H. Ma, H. Yu, S. Shi, B. Wang, S. Xu, L. Wang and Y. Zhao, J. Mater. Chem. A, 2018, 6, 6675–6684 RSC.
- G.-L. Xu, R. Amine, Y.-F. Xu, J. Liu, J. Gim, T. Ma, Y. Ren, C.-J. Sun, Y. Liu, X. Zhang, S. M. Heald, A. Solhy, I. Saadoune, W. L. Mattis, S.-G. Sun, Z. Chen and K. Amine, Energy Environ. Sci., 2017, 10, 1677–1693 RSC.
- Y. F. Zhu, Y. Xiao, W. B. Hua, S. Indris, S. X. Dou, Y. G. Guo and S. L. Chou, Angew. Chem., 2020, 132, 9385–9390 CrossRef.
- Y.-N. Zhou, P.-F. Wang, Y.-B. Niu, Q. Li, X. Yu, Y.-X. Yin, S. Xu and Y.-G. Guo, Nano Energy, 2019, 55, 143–150 CrossRef CAS.
- X. Chen, X. Zhou, M. Hu, J. Liang, D. Wu, J. Wei and Z. Zhou, J. Mater. Chem. A, 2015, 3, 20708–20714 RSC.
- X. Qi, L. Liu, N. Song, F. Gao, K. Yang, Y. Lu, H. Yang, Y.-S. Hu, Z.-H. Cheng and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 40215–40223 CrossRef CAS.
- Z.-Y. Li, J. Zhang, R. Gao, H. Zhang, L. Zheng, Z. Hu and X. Liu, J. Phys. Chem. C, 2016, 120, 9007–9016 CrossRef CAS.
- Y. Xiao, Y. F. Zhu, W. Xiang, Z. G. Wu, Y. C. Li, J. Lai, S. Li, E. Wang, Z. G. Yang, C. L. Xu, B.-H. Zhong and X.-D. Guo, Angew. Chem., Int. Ed., 2020, 59, 1491–1495 CrossRef CAS.
- M. Keller, D. Buchholz and S. Passerini, Adv. Energy Mater., 2016, 6, 1501555 CrossRef.
- Q. Huang, J. Liu, S. Xu, P. Wang, D. G. Ivey, B. Huang and W. Wei, Chem. Mater., 2018, 30, 4728–4737 CrossRef CAS.
- X. Rong, E. Hu, Y. Lu, F. Meng, C. Zhao, X. Wang, Q. Zhang, X. Yu, L. Gu and Y.-S. Hu, Joule, 2019, 3, 503–517 CrossRef CAS.
- N. Yabuuchi, R. Hara, K. Kubota, J. Paulsen, S. Kumakura and S. Komaba, J. Mater. Chem. A, 2014, 2, 16851–16855 RSC.
- H. J. Kim, A. Konarov, J. H. Jo, J. U. Choi, K. Ihm, H. K. Lee, J. Kim and S. T. Myung, Adv. Energy Mater., 2019, 9, 1901181 CrossRef.
- T. Jin, P.-F. Wang, Q.-C. Wang, K. Zhu, T. Deng, J. Zhang, W. Zhang, X.-Q. Yang, L. Jiao and C. Wang, Angew. Chem., 2020, 132, 14619–14624 CrossRef.
- W. Zheng, Q. Liu, Z. Wang, Z. Wu, S. Gu, L. Cao, K. Zhang, J. Fransaer and Z. Lu, Energy Storage Mater., 2020, 28, 300–306 CrossRef.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684 CrossRef CAS.
- W. E. Gent, I. I. Abate, W. Yang, L. F. Nazar and W. C. Chueh, Joule, 2020, 4, 1369–1397 CrossRef CAS.
- H. Yan, B. Li, Z. Yu, W. Chu and D. Xia, J. Phys. Chem. C, 2017, 121, 7155–7163 CrossRef CAS.
- Z. Shadike, Y.-N. Zhou, L.-L. Chen, Q. Wu, J.-L. Yue, N. Zhang, X.-Q. Yang, L. Gu, X.-S. Liu, S.-Q. Shi and Z.-W. Fu, Nat. Commun., 2017, 8, 566 CrossRef.
- P. Vanaphuti, S. Bong, L. Ma, S. Ehrlich and Y. Wang, ACS Appl. Energy Mater., 2020, 3, 4852–4859 CrossRef CAS.
- W. D. Richards, S. T. Dacek, D. A. Kitchaev and G. Ceder, Adv. Energy Mater., 2018, 8, 1701533 CrossRef.
- Q. Zhang, Y. Huang, Y. Liu, S. Sun, K. Wang, Y. Li, X. Li, J. Han and Y. Huang, Sci. China Mater., 2017, 60, 629–636 CrossRef CAS.
- Y. Wang, X. Wang, X. Li, R. Yu, M. Chen, K. Tang and X. Zhang, Chem. Eng. J., 2019, 360, 139–147 CrossRef CAS.
- W.-J. Shi, Y.-W. Yan, C. Chi, X.-T. Ma, D. Zhang, S.-D. Xu, L. Chen, X.-M. Wang and S.-B. Liu, J. Power Sources, 2019, 427, 129–137 CrossRef CAS.
- Q.-C. Wang, Q.-Q. Qiu, N. Xiao, Z.-W. Fu, X.-J. Wu, X.-Q. Yang and Y.-N. Zhou, Energy Storage Mater., 2018, 15, 1–7 CrossRef.
- M. S. Chae, H. J. Kim, J. Lyoo, R. Attias, Y. Gofer, S. T. Hong and D. Aurbach, Adv. Energy Mater., 2020, 2002205 CrossRef CAS.
- Y. Tsuchiya, A. M. Glushenkov and N. Yabuuchi, ACS Appl. Nano Mater., 2017, 1, 364–370 CrossRef.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, ACS Nano, 2013, 7, 760–767 CrossRef CAS.
- B. Qiu, M. Zhang, L. Wu, J. Wang, Y. Xia, D. Qian, H. Liu, S. Hy, Y. Chen, K. An, Y. Zhu, Z. Liu and Y. S. Meng, Nat. Commun., 2016, 7, 12108 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, S. L. Chou, Y. Liu and S. X. Dou, Adv. Funct. Mater., 2020, 30, 1909530 CrossRef CAS.
- B. Peng, Z. Sun, S. Jiao, G. Wang and G. Zhang, Batteries Supercaps, 2020, 3, 147–154 CrossRef CAS.
- J. Xiao, F. Zhang, K. Tang, X. Li, D. Wang, Y. Wang, H. Liu, M. Wu and G. Wang, ACS Cent. Sci., 2019, 5, 1937–1945 CrossRef CAS.
- H. He, X. Zeng, H. Wang, N. Chen, D. Sun, Y. Tang, X. Huang and Y. Pan, J. Electrochem. Soc., 2014, 162, A39 CrossRef.
- D. Su, C. Wang, H. j. Ahn and G. Wang, Chem. – Eur. J., 2013, 19, 10884–10889 CrossRef CAS.
- J. Lamb and A. Manthiram, Chem. Mater., 2020, 32, 8431–8441 CrossRef CAS.
- V. Soundharrajan, B. Sambandam, M. H. Alfaruqi, S. Kim, J. Jo, S. Kim, V. Mathew, Y.-k. Sun and J. Kim, J. Mater. Chem. A, 2020, 8, 770–778 RSC.
- Y. Liu, X. Fang, A. Zhang, C. Shen, Q. Liu, H. A. Enaya and C. Zhou, Nano Energy, 2016, 27, 27–34 CrossRef CAS.
- Y. Yu, W. Kong, Q. Li, D. Ning, G. Schuck, G. Schumacher, C. Su and X. Liu, ACS Appl. Energy Mater., 2020, 3, 933–942 CrossRef CAS.
- J. Ahn, J. H. Kim, B. W. Cho, K. Y. Chung, S. Kim, J. W. Choi and S. H. Oh, Nano Lett., 2017, 17, 7869–7877 CrossRef CAS.
- Y. Pei, Q. Chen, M. Wang, B. Li, P. Wang, G. Henkelman, L. Zhen, G. Cao and C.-Y. Xu, Nano Energy, 2020, 71, 104644 CrossRef CAS.
- Y. Xiao, N. M. Abbasi, Y. F. Zhu, S. Li, S. J. Tan, W. Ling, L. Peng, T. Yang, L. Wang, X. D. Guo, Y.-X. Yin, H. Zhang and Y.-G. Guo, Adv. Funct. Mater., 2020, 30, 2001334 CrossRef CAS.
- S. Guo, Q. Li, P. Liu, M. Chen and H. Zhou, Nat. Commun., 2017, 8, 135 CrossRef.
- X. Ke, Z. Zhao, J. Liu, Z. Shi, Y. Li, L. Zhang, H. Zhang, Y. Chen, Z. Guo, Q. Wu and L. Liu, Appl. Energy, 2017, 194, 540–548 CrossRef CAS.
- J. H. Jo, J. U. Choi, A. Konarov, H. Yashiro, S. Yuan, L. Shi, Y. K. Sun and S. T. Myung, Adv. Funct. Mater., 2018, 28, 1705968 CrossRef.
- J.-J. Ding, Y.-N. Zhou, Q. Sun and Z.-W. Fu, Electrochem. Commun., 2012, 22, 85–88 CrossRef CAS.
- H.-H. Sun, J.-Y. Hwang, C. S. Yoon, A. Heller and C. B. Mullins, ACS Nano, 2018, 12, 12912–12922 CrossRef CAS.
- J.-Y. Hwang, T.-Y. Yu and Y.-K. Sun, J. Mater. Chem. A, 2018, 6, 16854–16862 RSC.
- C.-Y. Yu, J.-S. Park, H.-G. Jung, K.-Y. Chung, D. Aurbach, Y.-K. Sun and S.-T. Myung, Energy Environ. Sci., 2015, 8, 2019–2026 RSC.
- K. Kaliyappan, J. Liu, B. Xiao, A. Lushington, R. Li, T. K. Sham and X. Sun, Adv. Funct. Mater., 2017, 27, 1701870 CrossRef.
- R. Dang, Q. Li, M. Chen, Z. Hu and X. Xiao, Phys. Chem. Chem. Phys., 2019, 21, 314–321 RSC.
- Y. Wang, K. Tang, X. Li, R. Yu, X. Zhang, Y. Huang, G. Chen, S. Jamil, S. Cao, X. Xie, Z. Luo and X. Wang, Chem. Eng. J., 2019, 372, 1066–1076 CrossRef CAS.
- D. Lu, Z. Yao, Y. Zhong, X. Wang, X. Xia, C. Gu, J. Wu and J. Tu, ACS Appl. Mater. Interfaces, 2019, 11, 15630–15637 CrossRef CAS.
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, Q. Gu and S. Chou, J. Mater. Chem. A, 2019, 7, 9215–9221 RSC.
- J. G. Han, K. Kim, Y. Lee and N. S. Choi, Adv. Mater., 2019, 31, 1804822 CrossRef.
- H. Y. Asl and A. Manthiram, Science, 2020, 369, 140–141 CrossRef CAS.
- C. Zhao, Y. Lu, Y. Li, L. Jiang, X. Rong, Y. S. Hu, H. Li and L. Chen, Small Methods, 2017, 1, 1600063 CrossRef.
- J. Lamb, L. Stokes and A. Manthiram, Chem. Mater., 2020, 32, 7389–7396 CrossRef CAS.
- Q. Bai, L. Yang, H. Chen and Y. Mo, Adv. Energy Mater., 2018, 8, 1702998 CrossRef.
- Y.-X. Huang, F. Wu and R.-J. Chen, Natl. Sci. Rev., 2020, 7, 1367–1386 CrossRef.
- M. Chen, L. Chen, Z. Hu, Q. Liu, B. Zhang, Y. Hu, Q. Gu, J. L. Wang, L. Z. Wang, X. Guo, S.-L. Chou and S.-X. Dou, Adv. Mater., 2017, 29, 1605535 CrossRef.
- W. J. Li, C. Han, G. Cheng, S. L. Chou, H. K. Liu and S. X. Dou, Small, 2019, 15, 1900470 CrossRef.
- Q. Zhao, Y. Lu and J. Chen, Adv. Energy Mater., 2017, 7, 1601792 CrossRef.
- X. Rong, L. Lu, X. Qi, Q. Zhou, W. Kong, K. Tang, L. Chen and Y. Hu, Energy Storage Sci. Technol., 2020, 9, 515 Search PubMed.
- K. Liu, S. Tan, J. Moon, C. J. Jafta, C. Li, T. Kobayashi, H. Lyu, C. A. Bridges, S. Men, W. Guo, Y. Sun, J. Zhang, M. P. Paranthaman, X.-G. Sun and S. Dai, Adv. Energy Mater., 2020, 10, 2000135 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |