
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcentrated dual-cation electrolyte strategy for aqueous zinc-ion batteries†
Yunpei
Zhu‡
a,
Jun
Yin‡
 b,
Xueli
Zheng‡
c,
Abdul-Hamid
Emwas
d,
Yongjiu
Lei
a,
Omar F.
Mohammed
b,
Yi
Cui
ce and
Husam N.
Alshareef
*a
b,
Xueli
Zheng‡
c,
Abdul-Hamid
Emwas
d,
Yongjiu
Lei
a,
Omar F.
Mohammed
b,
Yi
Cui
ce and
Husam N.
Alshareef
*a
aMaterials Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: husam.alshareef@kaust.edu.sa
bAdvanced Membranes & Porous Materials Center, KAUST Catalysis Center, Division of Physical Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
cDepartment of Materials Science and Engineering, Stanford University, Stanford, California 94305, USA
dCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
eStanford Institute for Materials and Energy Sciences, SLAC National Accelerator Laboratory, Menlo Park, California 94025, USA
First published on 3rd July 2021
Abstract
Rechargeable Zn-ion batteries are highly promising for stationary energy storage because of their low cost and intrinsic safety. However, due to the poor reversibility of Zn anodes and dissolution of oxide cathodes, aqueous Zn-ion batteries encounter rapid performance degradation when operating in conventional low-concentration electrolytes. Herein, we demonstrate that an aqueous Zn2+ electrolyte using a supporting Na salt at a high concentration is efficient to address these issues without sacrificing the power densities, cycling stability, and safety of zinc-ion batteries. We show that the high-concentration solute minimizes the number of free water molecules and the changes in the electronic state of the electrolyte. A combination of experimental and theoretical investigations reveals that a unique interphase, formed on the Zn anode, enables reversible and uniform Zn plating. Utilizing a cathode of sodium vanadate synthesized through a scalable strategy, the Zn–sodium vanadate battery with the concentrated bi-cation electrolyte shows improved cycling stability, decent rate performance, and low self-discharge. This work provides new insights on electrolyte engineering to achieve high-performance aqueous batteries.
Broader contextElectrolytes play a critical role in determining the performance of electrochemical storage devices, such as cycling stability, rate performance, and self-discharge. To date, the promising aqueous Zn-ion batteries still suffer from low cycling performance due to the low reversibility of the Zn anode and serious dissolution of active cathode materials during cycling in conventional aqueous electrolytes. Recently, highly concentrated “water-in-salt” electrolytes have been proposed to decrease the amount of free water molecules for improved cyclability, though the selection is still limited to expensive organic or toxic solutes. Herein, to address the challenges of developing formulations of aqueous electrolytes, we design a dual-cation electrolyte that features high ionic conductivity, low cost, and intrinsic safety. Importantly, the high concentration can not only suppress water activity and decrease the amount of free water molecules, but also induce the change of electronic state of the electrolyte. The former prohibits cathode dissolution, while the latter ensures a high reversibility for the anode. These factors together enable stable aqueous Zn-ion batteries with decent rate performance and improved self-discharge performance. |
Introduction
Compared to the prominent energy storage technologies (e.g., Li-ion batteries and Pb–acid batteries), aqueous Zn-ion batteries (ZIBs) show the advantages of safety, affordability, and environmental friendliness.1 Aqueous ZIBs benefit from the direct use of a Zn metal anode with high capacity (820 mA h g−1 and 5854 mA cm−3) and decent electrochemical potential (−0.76 V vs. standard hydrogen electrode (SHE)). These features make aqueous ZIBs particularly promising for grid-scale storage and other applications (e.g., home batteries) in which energy density is not a major concern.2 Importantly, aqueous ZIBs can be compatible with the existing Li-ion battery manufacturing infrastructure, in which billions of dollars have been invested,3 a significant indicator for potentially commercializing aqueous ZIBs. Their full levelized cost of electricity should be further reduced to promote their application at scale. This will benefit from the cost reduction expected for scale-up fabrication (e.g., preparing cathode materials at scale) and increased battery cycle life.4The working mechanism of an aqueous ZIB depends on the shuttling of Zn2+ between a Zn metal anode and a cathode capable of reversible Zn2+ (de)insertion. Different formats of Zn metals (e.g., foil, powder, and mesh) are commercially available at a low price. However, the conventional aqueous electrolytes lead to fast performance degradation during battery operation because of:2,5–10 (1) the poor reversibility of the Zn metal anode caused by the side reactions (e.g., hydrogen evolution reaction and corrosion) and non-uniform Zn plating, and (2) the dissolution of cathodes into the bulk electrolytes upon cycling, which is more conspicuous when operating at low current densities. These issues are strongly related to the existence of a large number of free water molecules in the conventional aqueous electrolytes (e.g., 2 M ZnSO4). The recent development of “water-in-salt” (WIS) electrolytes and deep-eutectic electrolytes, both of which feature limited or even no free water molecules, renders an unprecedented opportunity to overcome the aforementioned shortcomings of aqueous ZIBs.5,11–18 Based on the eutectic effect, WIS electrolytes with an extremely high concentration of 75 m (m: mol kgH2O−1) could be even achieved.19 On the one hand, the dissolution of cathodes can be impeded, enabling improved cycling stability.13,14 On the other hand, the reorganized solvation-sheath structure of cations (e.g., Zn2+) (typically dominated by binding anions) suppresses the hydrolysis effect efficiently, providing a high possibility for reversible Zn plating/stripping with high coulombic efficiency (CE).11,20,21 Accordingly, engineering electrolytes through using solutes with intrinsically high solubility or additives can enable high-performance aqueous rechargeable batteries.22,23 In addition, the physicochemical properties (i.e., ionic conductivity and electrochemical stability window) and economic and environmental aspects of the aqueous electrolytes should be considered when developing aqueous ZIBs for potential practical applications.22 However, most WIS electrolytes with excess reaction-irrelevant anions (e.g., costly fluorinated metal salts) lead to cost and safety concerns, which compromises the economic and environmental benefits anticipated for aqueous ZIBs.2 For instance, the frequently used lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) (>2.8 euros per g) in the classical WIS electrolytes shows acute dermal and oral toxicity as well as chronic aquatic toxicity.13 Additionally, the classical WIS electrolytes show high viscosity and low ionic conductivity, thereby resulting in poor rate performance and low energy efficiency.13,24,25 Recently, 30 m ZnCl2 WIS electrolyte with high viscosity of ∼1000 mPa s and low ionic conductivity of ∼2 mS cm−1 has been shown as a low-cost electrolyte for ZIBs with decent cyclability and rate capability, though few cathodes are likely workable in this electrolyte.21,26 All these factors make it extremely challenging to apply the current WIS electrolyte chemistry into potential commercial energy storage devices for intermittent renewable energy storage.
Significant progress has been achieved in identifying cathode materials for aqueous ZIBs.2,10,27 The synthesis of these cathode materials typically utilizes hydrothermal methods with prolonged heat treatment, which not only increases the instrumental cost but also leads to low product yield (e.g., ∼100 mg per batch). To achieve the potential application of aqueous ZIBs for future energy storage, a scalable approach to fabricate high-performance cathodes should be developed. Herein, we develop a room-temperature, scalable dissolution–recrystallization method to prepare Na2V6O16·nH2O (hereafter referred to as NVO) nanofibers as an efficient cathode for aqueous ZIBs. To improve the cycling stability of our NVO cathode, we design a low-cost and environmentally benign WIS electrolyte of Zn(ClO4)2 with a highly concentrated supporting salt of NaClO4, specifically, 0.5 m Zn(ClO4)2 with 18 m NaClO4 (see the price comparison of different solutes in Fig. S1, ESI†). The supporting salt of NaClO4 can efficiently adjust the solvation structure and electronic states of the electrolyte upon increasing the concentrations, as discussed below. Therefore, the highly concentrated electrolyte shows the scarcity of free water molecules, low viscosity, and high ionic conductivity, which can efficiently prohibit the dissolution of the NVO cathode to enable remarkable cycling stability and high rate performance.
Results and discussion
In conventional aqueous electrolytes, vanadium-oxide-based cathodes experience rapid capacity degradation because of the dissolution of cathodes during cycling (Fig. S2, ESI†).1,2,8 One strategy to address this issue is to add salt additives into the electrolytes, wherein the cation of the additive is the same as the metal cation in the cathode (e.g., Na+ for NaV3O8·nH2O and Mn2+ for MnO2).28,29 This may shift the dissolution equilibrium of active cathodes, thereby suppressing their continuous dissolution during battery operation. However, this strategy is still inefficient in retaining the capacity when operating at low current densities (e.g., 0.1 A g−1). This problem correlates with: (1) the structural disintegration of vanadium oxides at a high degree of Zn2+ intercalation (high capacities), and (2) the slow cathode dissolution and low Zn plating/stripping efficiency (increased side reaction of hydrogen evolution at low current densities) because of the existence of a large number of free water molecules. The recent development of superconcentrated WIS electrolytes enables the stable operation of aqueous ZIBs even at low current densities through minimizing or even removing free water molecules, which, however, comes at the cost of the rate capability because of the significantly increased viscosity and decreased ionic conductivity.11,26 Additionally, the rate performance of aqueous ZIBs is intuitively associated with the resistance of Zn2+ transport in the electrode. Our NVO electrode has ample vacancies in the layered structure, showing structural stability and flexibility to accommodate fast Zn2+ intercalation at high capacities. Inspired by the WIS concept using an additional supporting salt,11–13,25 we here present a high-concentration Zn-ion electrolyte (hereafter denoted as HCZE) using highly soluble NaClO4 as the supporting salt (0.5 m Zn(ClO4)2 + 18 m NaClO4), which enables stable operation of the ZIB using an NVO cathode with a remarkable rate capability.HCZE possesses a low viscosity of 8.16 mPa s and a high ionic conductivity of 98.5 mS cm−1, which are superior to those of the classical WIS electrolytes14,22,30–32 and state-of-the-art eutectic electrolytes featuring no free water5,12,13 (Fig. 1a and Fig. S3, ESI†). The superior properties of our HCZE electrolyte play an essential role in enabling high-rate, stable Zn2+ storage using the NVO cathode, as discussed later. Even at a high concentration (nwater![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ncation = 3), the low viscosity and high ionic conductivity of HCZE is related to the absence of bulky organic ligands (e.g., acetate and trifluoromethanesulfonate) and additives (e.g., urea, acetamide, and succinonitrile) that are typically utilized to prepare WIS and eutectic electrolytes, respectively. Additionally, the presence of a minor amount of “free” water molecules in HCZE, as confirmed in the following section, induces fast exchange between ClO4−/water and highly concentrated Na+ to ensure high ionic conductivity. Therefore, our HCZE electrolyte shows a pronounced improvement in the trade-off between ionic conductivity and the amount of free water molecules in aqueous electrolytes.
:ncation = 3), the low viscosity and high ionic conductivity of HCZE is related to the absence of bulky organic ligands (e.g., acetate and trifluoromethanesulfonate) and additives (e.g., urea, acetamide, and succinonitrile) that are typically utilized to prepare WIS and eutectic electrolytes, respectively. Additionally, the presence of a minor amount of “free” water molecules in HCZE, as confirmed in the following section, induces fast exchange between ClO4−/water and highly concentrated Na+ to ensure high ionic conductivity. Therefore, our HCZE electrolyte shows a pronounced improvement in the trade-off between ionic conductivity and the amount of free water molecules in aqueous electrolytes.
 | ||
| Fig. 1 Physicochemical properties and electrochemical performance of the HCZE electrolyte. (a) Comparison of ionic conductivity and viscosity with the reported WIS and deep-eutectic electrolytes. (b) Zn plating/striping efficiency and stability on a Ti working electrode at 0.4 mA cm−2. (c) CV plots of Zn deposition/dissolution using two electrolytes at a scan rate of 1 mV s−1. (d) Galvanostatic Zn stripping/plating plots of a Zn/Zn symmetrical cell at 0.2 mA cm−2. (e) SEM images of the Zn anode after cycling tests. (e1): Zn in 0.5 m Zn(ClO4)2 after 20 cycles. Zn in HCZE after 1 cycle (e2), 20 cycles (e3), and 50 cycles (e4). XRD patterns (f) and Cl 2p core level (g) of the Zn metal in HCZE after different cycles. | ||
As an important parameter to evaluate the rechargeability of Zn metal anodes, CE was determined using an asymmetric Ti/Zn half cell at a capacity of 0.4 mA h cm−2, in which a Ti foil was used as the working electrode and Zn metal foil as the counter and reference electrodes. Smooth and stable voltage profiles can be realized in HCZE (Fig. S4, ESI†), along with a high average CE of 98.2% calculated from cycle 1 to 100 at 0.4 mA cm−2 (Fig. 1b). The electrochemical performance of Zn plating/stripping achieved in HCZE outperforms that of the conventional low-concentration electrolytes, including 0.5 m Zn(ClO4)2 and 2 m ZnSO4 (Fig. S5, ESI†). The improved performance indicates that the plated Zn on the Ti substrate is efficiently recovered in the following stripping process, and parasitic reactions (e.g., hydrogen evolution) can be significantly prohibited.5,11,33 In dilute electrolytes (e.g., 0.5 m Zn(ClO4)2), Zn2+ ions are fully hydrated by dipolar water molecules to form [Zn(OH2)6]2+.34 Such Zn2+–H2O interaction would greatly weaken the O–H bond of water molecules,35 leading to the deprotonation of H2O to generate an acidic electrolyte.
The deprotonation process is confirmed by the mildly acidic nature of 0.5 m Zn(ClO4)2 (pH ∼ 5). In contrast, HCZE shows a high concentration of solute, which can efficiently mitigate the parasitic reactions like hydrogen evolution, corrosion, and passivation that can compete with Zn plating/stripping. Accordingly, HCZE enables a remarkable stability of Zn/Zn2+ redox processes with high efficiency, superior to those of the dilute control electrolytes (Fig. S5 and S6, ESI†). Additionally, cyclic voltammetry (CV) tests of Zn plating/stripping (Fig. 1c) reveal that compared to the low-concentration electrolyte, HCZE enables decreased voltage offset and onset potential for Zn deposition/dissolution, suggesting improved reversibility and kinetics in the concentrated electrolyte.
The stability and reversibility of the Zn metal anode in our HCZE electrolyte were further evaluated in a Zn/Zn symmetric cell under galvanostatic conditions. As shown in Fig. 1d, the symmetric cell exhibits a long-term stability at a low current density of 0.2 mA cm−2 without signs of potential fluctuation or cell short-circuiting. This is expected given the enhanced CE of Zn plating/stripping and the scarcity of free water molecules in HCZE. The overpotential is even smaller than that in dilute aqueous electrolytes of ZIBs (Fig. S7, ESI†), suggesting the improved Zn plating/stripping kinetics in our HCZE electrolyte. These results reveal the capability of HCZE in enabling stable and reversible Zn chemistry.
To investigate the Zn plating/stripping mechanism in more detail, Zn was plated with a capacity of 1 mA h cm−2 onto the Zn electrode (0.5 mA cm−2) and then stripped away, followed by multiple plating/stripping cycles with a capacity of 0.5 mA h cm−2 at every half cycle. The scanning electron microscopy (SEM) image shows the corrosion and dendrite formation on the Zn electrode in 0.5 m Zn(ClO4)2 (Fig. 1e), which is the reason for cell short-circuiting and premature failure. In sharp contrast, Zn foil shows a relatively flat surface without obvious dendrites during cycling tests in HCZE (Fig. 1e and Fig. S8, ESI†). X-ray diffraction (XRD) patterns of the Zn electrodes at different cycles show the typical diffractions from the hexagonal phase of zinc (Fig. 1f) when using HCZE, which is different from the formation of undesired byproducts (e.g., Zn4ClO4(OH)7, ZnO) upon Zn plating/stripping in 0.5 m Zn(ClO4)2 (Fig. S9, ESI†).
X-ray photoelectron spectroscopy (XPS) reveals the generation of a characteristic 2p1/2 and 2p3/2 doublet that is attributed to Cl− of ZnCl2 (Fig. 1g). This indicates the efficient reduction of ClO4− to form a Cl−-containing layer on the surface of Zn,36 which could ensure a potential gradient across this surface layer to allow Zn plating underneath. This is in a much similar manner to the LiCl-containing solid–electrolyte interface (SEI) layer on Li metal towards stable Li plating/stripping.37 Although ClO4− reduction could happen in 0.5 m Zn(ClO4)2, other competitive reactions like corrosion and hydrogen evolution1,2 led to the formation of a low-quality protection layer (Fig. S10 and S11, ESI†). This results in low coulombic efficiency and fast cell degradation upon using the low-concentration electrolyte of 0.5 m Zn(ClO4)2. Such findings demonstrate the synergistic effect from high-concentration ClO4− and limited “free” water in HCZE to achieve stable and reversible Zn deposition. Given the high-entropy state of ClO4− and a minimal amount of free water molecules in HCZE, the insoluble Cl−-containing layers most likely exist in the form of stable and coordination-saturated ZnCl2-based species.5,37 As a consequence, the solvation structure of HZCE on uniform Zn deposition is dependent on Zn2+ desolvation from the active ZnCl2-associated Zn solvates and prohibited side reactions at the interface (see the theoretical calculations later).
The unusual interface formed on the Zn metal anode is closely related to the state changes of electrolytes in HCZE. We analyzed the interaction among Zn2+, Na+, ClO4−, and H2O molecules using Raman, Fourier-transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), and nuclear magnetic resonance (NMR) spectroscopy. The Raman spectra show strong O–H stretching vibration modes of water molecules in dilute solutions (Fig. 2a), which is attributed to various hydrogen-bonding environments in water clusters.38 Increasing the concentration of solutions causes a pronounced blue shift and peak intensity weakening of the typical O–H vibrations at 3250 and 3400 cm−1 (Fig. 2a and Fig. S12, ESI†). A similar phenomenon has been found in the FT-IR spectra of the electrolytes upon increasing the concentrations (Fig. 2b and Fig. S11, ESI†). A new sharp peak at 3550 cm−1 in the Raman spectra can be assigned to the signature of crystalline hydrates.5,12 These changes suggest that the minimized quantity of free water molecules has been stabilized in the concentrated electrolytes, namely, the extensive disruption of the water network connected through hydrogen bonding.
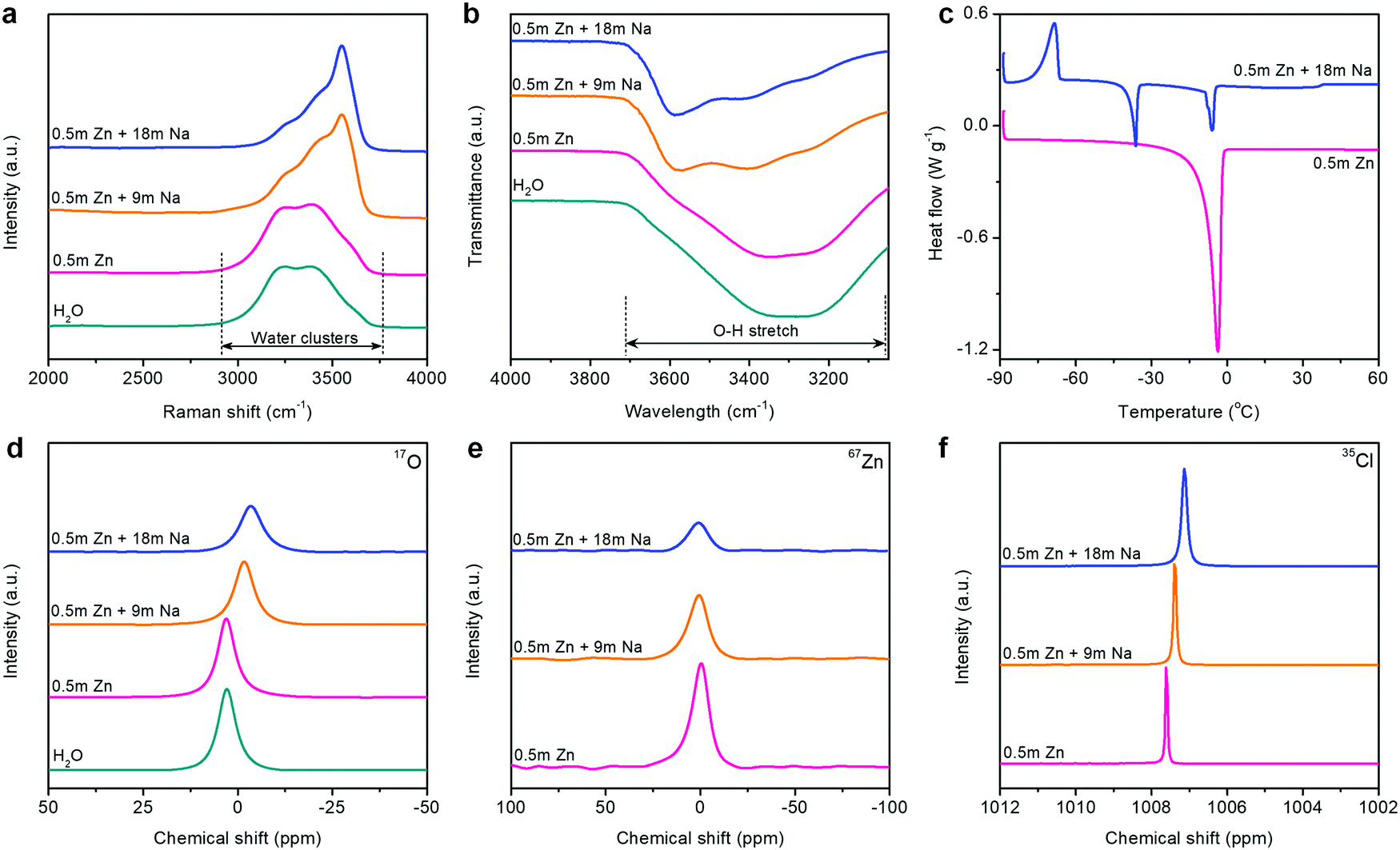 | ||
| Fig. 2 Spectroscopic characterizations of the electrolytes. (a) Raman spectra, (b) FT-IR spectra, (c) DSC profiles, (d) 17O (water) NMR spectra, (e) 67Zn NMR spectra, and (f) 35Cl NMR spectra. | ||
Thermodynamically, HCZE is a stable liquid at room temperature (Fig. S13, ESI†). The dilute electrolyte of 0.5 m Zn(ClO4)2 shows clear melting transitions at −3.9 °C (Fig. 2c), characteristics of low-concentration solutions with a completely dissociated salt.39 Differently, HCZE exhibits an exothermic peak at −68.5 °C, which is attributed to the crystal formation of a salt–water eutectic mixture. Further heating ceases the continuous rearrangement of the eutectic mixture, thereby leading to the melting process after crystal formation. The two endothermic peaks are similar to the features of eutectic electrolytes with a minimum amount of free water molecules.24,25,30
The aforementioned observations reveal that the solvation shells of different ions interpenetrate when reaching the limit of high concentration. We further conducted liquid-state NMR to study the aggregation behaviors of the two salts in HCZE (Fig. 2d, e, f and Fig. S13, ESI†). Compared to the water reference and diluted solutions, the 17O (water) response of HCZE shows a pronounced upfield shift (Fig. 2d). An insignificant peak broadening can be assigned to a slightly decreased T2 relaxation time,13 which in turn confirms the low viscosity of HCZE. This is consistent with our previous analysis and in sharp contrast to those of the classical WIS electrolytes using expensive fluorine-containing anions (Fig. 1a). Notably, the 17O (ClO4−) signal of HCZE shows a slight shift compared to the diluted reference electrolyte (Fig. S14, ESI†). These results reflect that the solvation structures of different ions strongly interpenetrate in the concentrated electrolyte.
As evidenced by Raman and FT-IR, HCZE shows a very limited amount of free water molecules, wherein most of the water molecules would be a part of cation solvation shells. Due to the higher charge density of Zn2+,11,13 Zn2+ possesses a stronger binding ability to water and ClO4− compared to Na+. In the most concentrated electrolyte, the Zn2+ solvation shells would be primarily occupied by ClO4−,11 while the interaction between the lone-pair electrons on water oxygen and Zn2+ could be efficiently suppressed, leading to the shielding effect of 17O (water) and 67Zn resonance signals (Fig. 2d and e). For the concentrated electrolytes, the line width of the 67Zn signal increases marginally, further suggesting the enhanced interaction and complexation between Zn2+ and the abundant anions in the electrolytes. On the other hand, the presence of water and ClO4− in the coordination shell of Na+ results in increased electron density around Na+, thus leading to the upfield peak shift of 23Na for HCZE (Fig. S14, ESI†). The relatively narrow linewidth of the 23Na NMR signal is associated with the fast exchange between Na+ and ClO4−/water in the solvation shell,40 which ensures the high ionic conductivity of HCZE.
The Hofmeister series indicates that ClO4− salts feature a strong capability to destabilize the bulk water structure, implying the probability of adjusting solvation-sheath structures of ion aggregations. At a high concentration for HCZE (0.5 m Zn(ClO4)2 + 18 m NaClO4), the water-breaking ClO4− ions can form complex ion networks with water molecules and cations, giving rise to unique solvation structures and disrupting the formation of hydrogen bonding networks. Therefore, we conducted 35Cl NMR to study the role of ClO4− in changing the solvation-sheath structure of cations (Fig. 2f). The upfield peak shift for highly concentrated electrolytes is indicative of ion shielding. Compared to Na+, the stronger binding capability of Zn2+ to ClO4− could deliver an enhanced ion shielding effect.
Our findings are in good agreement with previous molecular dynamics (MD) simulations,11–13,24,25 which demonstrated that a significant increase in the number of ion aggregates could prevent the formation of an extended hydrogen-bonding network of water molecules in concentrated electrolytes. To further obtain insights into the solvation structures, we performed ab initio MD simulations on our electrolytes with three representative concentrations at different temperatures. Compared to Na+, the stronger binding ability of Zn2+ towards H2O and ClO4− results in a longer residence time of ClO4− in the vicinity of Zn2+. Therefore, a series of MD simulations were conducted from 300 K (room temperature) to 350 K and 400 K (overheated system), aiming at accelerating the dynamics and ensuring the equilibrium of the Zn2+ solvation-sheath structure (Fig. 3 and Fig. S15, S16, ESI†). In a typical dilute solution of 0.5 m Zn(ClO4)2 (Fig. 3a), Zn2+ is expected to be solvated by six water molecules with insignificant contribution from ClO4−. The remaining water molecules generate a network through hydrogen bonding. Upon increasing NaClO4 to the intermediate concentration (i.e., 0.5 m Zn(ClO4)2 + 9 m NaClO4) (Fig. 3b), the ClO4− ions penetrate into the first solvation shell of Zn2+. The formation of cation–anion aggregates has been confirmed to be beneficial for anion reduction on the anode rather than water,24,41 which is vital for prohibiting the side reaction on the Zn anode. For the electrolyte with the highest concentration (0.5 m Zn(ClO4)2 + 18 m NaClO4) (Fig. 3c), the cationic hydration sheaths demonstrate a high degree of interpretation with a significantly decreased amount of free water molecules. The first hydration shell of Zn2+ is mainly occupied by the ClO4− ions with four oxygen atoms from ClO4−. Radial distribution functions (RDFs) were further performed to illustrate the distributions of neighboring molecules of solvated Zn2+ ions (Fig. 3d). RDFs of 0.5 m Zn(ClO4)2 indicate that water molecules dominate both the first and second solvation shells of Zn2+, as revealed by two sharp peaks at 2.05 Å and 4.25 Å. In contrast, two main peaks at 2.05 Å and 3.04 Å appear in the most concentrated electrolyte. This suggests that the water molecules replaced by ClO4− enter the second hydration shell, wherein the ClO4− ions still contribute significantly to the second shell. In combination with Raman and FT-IR spectra (Fig. 2a and b), we confirm that the high concentration of cations in HCZE can efficiently decrease the number of water molecules at a free state. This is an important indication to prevent cathode dissolution when using HCZE in aqueous Zn-ion batteries.
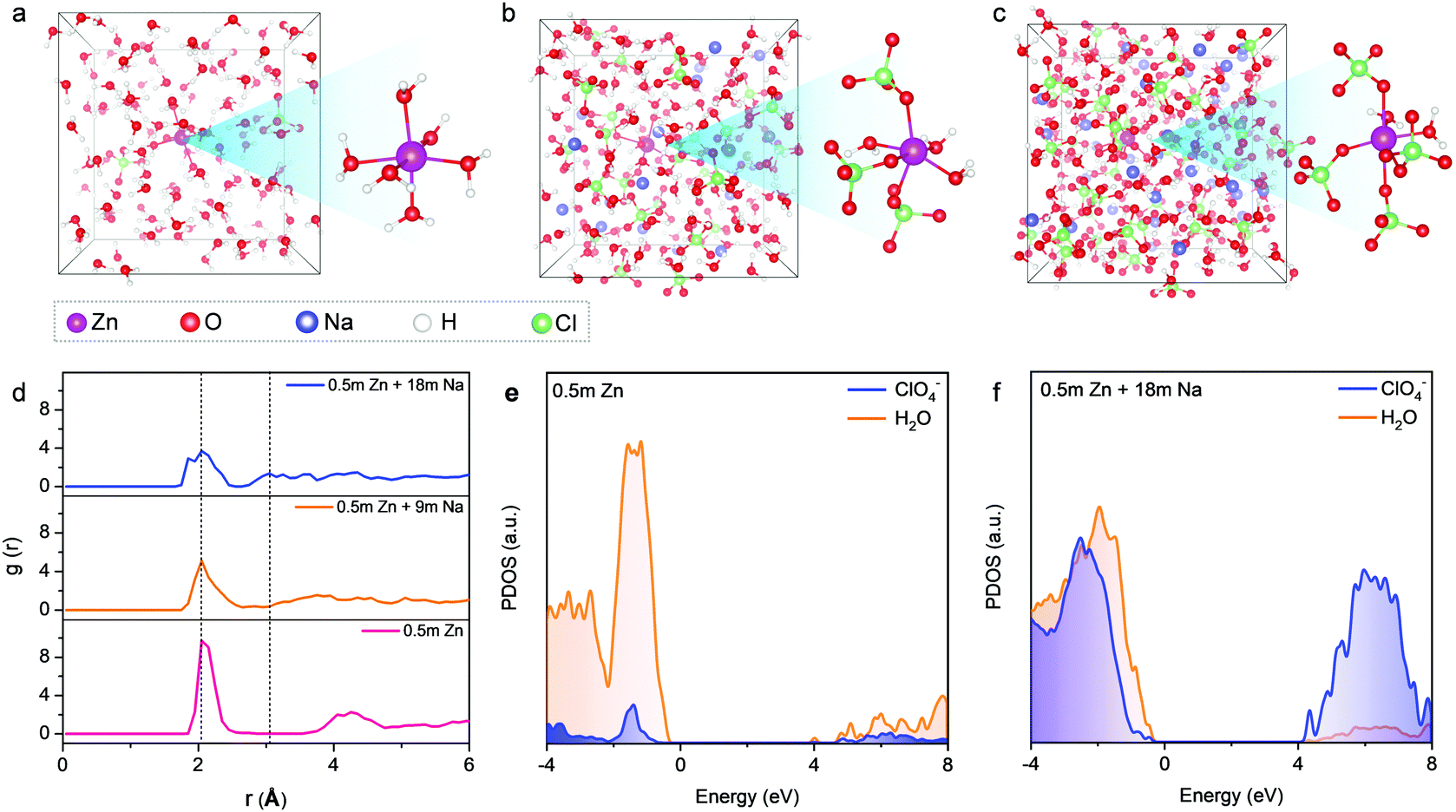 | ||
| Fig. 3 MD simulations and DFT calculations of the electrolytes. Snapshots of equilibrium trajectories from ab initio MD simulations of 0.5 m Zn(ClO4)2 (a), 0.5 m Zn(ClO4)2 + 9 m NaClO4 (b), and 0.5 m Zn(ClO4)2 + 18 m NaClO4 (c). Magenta ball: Zn, red ball: O, blue ball: Na, white ball: H, and green ball: Cl. (d) RDF plots of these three representative electrolytes. Projected density of states (PDOS) plots of 0.5 m Zn(ClO4)2 (e) and 0.5 m Zn(ClO4)2 + 18 m NaClO4 (f). | ||
We further conducted density functional theory (DFT) calculations to investigate the influence of solute concentration on the electronic structures of our electrolytes. For the dilute electrolyte of 0.5 m Zn(ClO4)2 (Fig. 3e), the lowest unoccupied molecular orbital (LUMO) is primarily contributed by water. Accordingly, water molecules would be predominantly reduced on the Zn anode, leading to fast capacity degradation of the battery. At the highest concentration, the ratio of contact ion pairs and aggregates increases, along with a drastically decreased amount of free water molecules. The extensive coordination of water to cations leads to a significant downshift of the orbital levels of ClO4−, delivering a dominant contribution of ClO4− in both the LUMO and the highest occupied molecular orbital (HOMO) levels (Fig. 3f). As a result, ClO4− is predominantly reduced to yield an anion-derived protection layer. Similar features have also been found for different WIS and eutectic electrolytes such as ZnF216 and NaF.25 The anion-derived passivation layer has been proven to show a low interfacial resistance,12,16,24 thus ensuring fast kinetics of Zn plating/stripping. Noticeably, the passivation layer of electrically insulating ZnCl2 could potentially establish a potential gradient across the layer to drive Zn2+ diffusion through the passivation layer, which allows for uniform Zn deposition. The function of the ZnCl2 layer is similar to that of the LiCl-containing SEI layer on the Li metals.37,42
The passivation layer of ZnCl2 could efficiently prohibit parasitic reaction, as evidenced by our DFT calculations (Fig. 4). The adsorption energy of H2O on ZnCl2 (−0.87 eV) is lower than that on Zn (−0.04 eV) (Fig. 4a). The H2O molecule could interact with both Zn and ZnCl2 surfaces via the Zn–O bond, which can be characterized by the electron accumulation (green area) located above the ZnCl2 surface and the electron depletion (purple area) around the O atom. The intensity of the charge redistribution further reveals that the Zn–O bond in the case of ZnCl2 is considerably stronger than that on the Zn surface. Accordingly, the passivation layer induces a higher energy barrier for water dissociation towards hydrogen evolution.43,44 Moreover, the insulating feature of ZnCl2 makes it difficult to obtain electrons from the Zn anode, thus making it kinetically sluggish to proceed hydrogen evolution and the corresponding formation of OH− on the Zn anode. This would significantly mitigate the generation of OH−-involved detrimental byproducts (e.g., Zn4ClO4(OH)7 and ZnO) on the Zn anode. Compared to the Zn2+ adsorption on the Zn metal surface (Fig. 4b), the lower adsorption energy of Zn2+ on ZnCl2 could promote the in-plane diffusion of Zn ad-atoms with a low diffusion barrier, similar to the roles of metal chlorides or fluorides in the SEI of different metal anodes (e.g., Li, Zn, and Al).16,37,45,46 This is also confirmed by the larger charge density difference at the Zn2+/ZnCl2 interfacial region, where the electron accumulation appears above the ZnCl2 surface and the electron depletion is localized in the Zn2+ ion. These metal chlorides or fluorides have been acknowledged as the primary rigid-frame materials to protect metal anodes, wherein they can induce the homogeneous metal nucleation and thus inhibit dendrite growth efficiently.
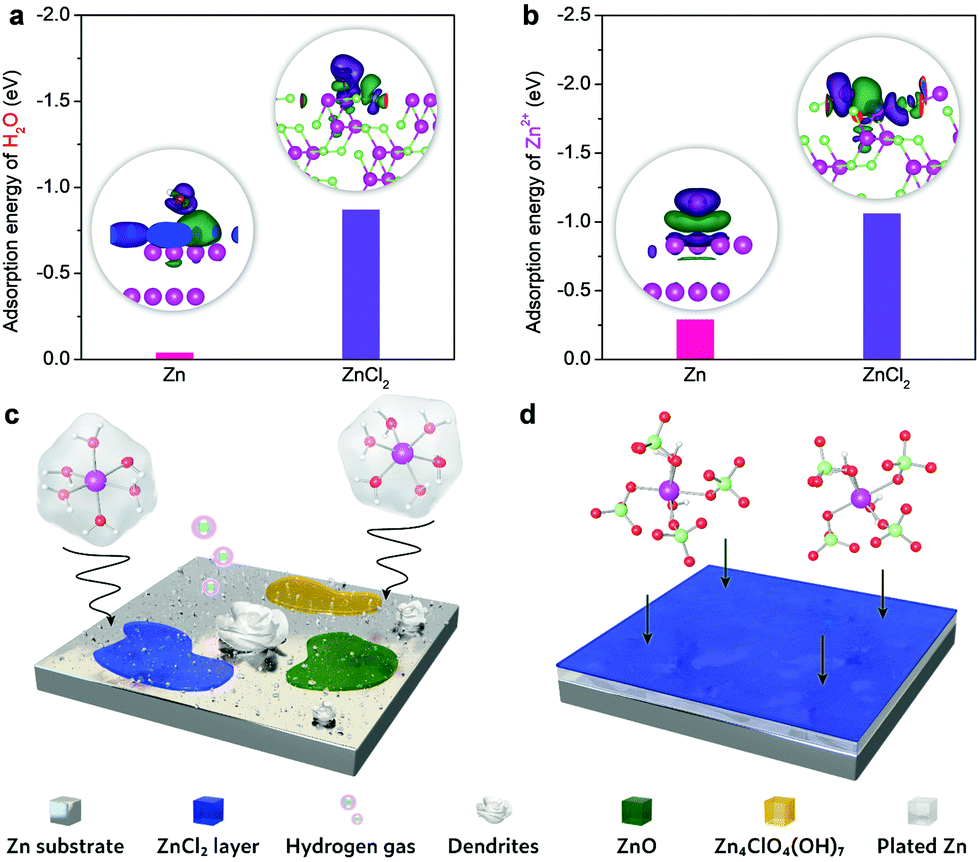 | ||
| Fig. 4 DFT adsorption energy calculations and deposition mechanisms involving the ZnCl2 interface and the pristine Zn. (a) H2O and (b) Zn2+ adsorption. Insets of (a) and (b) show the adsorption with interfacial electron transfers included: green and purple iso-surfaces represent electron accumulation and electron depletion, respectively. Schematics of Zn2+ solvation structures and the corresponding interfacial reactions in (c) 0.5 m (ZnClO4)2 and (d) 0.5 m Zn(ClO4)2 + 18 m NaClO4. | ||
The schematics of interfacial reactions between the Zn anode and electrolytes are shown in Fig. 4c and d. The Zn2+ ions in the most concentrated electrolyte show much higher desolvation energy compared to that of [Zn(H2O)6]2+ in the dilute electrolyte (Fig. S17, ESI†). Therefore, the presence of fragile [Zn(H2O)6]2+ in the dilute electrolyte causes different irreversible parasitic reactions like hydrogen evolution and corrosion. These reactions could compete with Zn plating/stripping at low potentials, thereby promoting Zn dendrite formation and decreasing the coulombic efficiency and cycling stability. However, the primary solvation shell of Zn2+ in the most concentrated electrolyte is mainly occupied by the ClO4− ions. The reduced affinities between Zn2+ and H2O, as a result of the strong coordination of ClO4− to Zn2+, could efficiently mitigate the parasitic reactions on the Zn anode. As such, the synergistic effect of the ZnCl2 passivation layer from perchlorate anion reduction and the intimate Zn2+–ClO4− interaction through increasing Zn2+ desolvation energy could enable stable Zn plating/stripping with high efficiency. Even if the substitution of water by perchlorate ions occurs for Zn2+ solvation, water molecules would be released from the first solvation shell but most like stay in the second solvation shell through a persistent but relatively weak attraction with the Zn2+ center. Accordingly, the improvement of anodic stability can be rationalized. Of note is that the high-concentration feature of HCZE provides abundant internally induced dipole interaction between H2O and ClO4−. This effect could saturate the bipolar sites of water molecules,47,48 further depressing their activity. Given the aforementioned features of our HCZE electrolyte, it is promising to suppress the dissolution of oxide cathodes (e.g., vanadate nanostructures) in conventional low-concentration electrolytes of aqueous ZIBs.
To demonstrate the feasibility and efficiency of HCZE in aqueous ZIBs, we developed NVO nanofibers as the cathodes to pair with Zn metal anodes in HCZE. NVO nanofibers were fabricated via an aqueous processing technique, which is inexpensive, green, and easy for mass production (see synthesis details in ESI†). Here we demonstrate a batch capability of 100 g NVO using a 1 L reactor (Fig. S18, ESI†). Our technique can be potentially scaled up using larger reactors, which shows the first example to produce cathodes of aqueous ZIBs at scale and low cost. NVO shows nanofiber morphology with high purity, aspect ratio, and homogeneity (Fig. S19, ESI†). The monoclinic structure of NVO is constructed from the V3O8 layers featuring two different zigzag chains (chains of edge-sharing V3O8 square pyramids and chains of distorted VO6 octahedra) (Fig. S19, ESI†), while hydrated Na ions are located among the layers. The layered structure and structural flexibility of NVO can enable a good electrochemical performance towards reversible cation (e.g., Zn2+) intercalation. As the crystalline structures of monoclinic NVO and orthorhombic V2O5 have no structural relationship, it is unlikely that a solid-state transformation led to the formation of NVO in aqueous solutions. A dissolution–recrystallization process for the structural transformation has been confirmed (Fig. S20–S25, see Supplementary Note 1 for a detailed analysis, ESI†).
HCZE shows high ionic conductivity, scarcity of free water molecules, and capability to enable stable Zn plating/stripping with high efficiency. The cyclic voltammetry (CV) curve of the NVO electrode shows two pairs of peaks (Fig. 5a), which can be assigned to the redox reactions between V5+ and lower oxidation states during charge/discharge. The electrochemical redox behaviors of our NVO cathode in HCZE resemble those of NVO in 0.5 m Zn(ClO4)2 (Fig. S26, ESI†) and the reported Na2V6O16·nH2O nanostructures in standard dilute aqueous Zn-ion electrolytes.49,50 During the charge/discharge process, no features related to Na+ insertion/removal could be observed even though a supporting salt of NaClO4 at a high concentration was used, as evidenced by the ex situ Na 1s core level (Fig. S27, ESI†). Additionally, the Na 1s core levels remain unchanged at fully discharged and charged states, suggesting the stable and immobile Na+ pillars in the layered structures of NVO upon charge/discharge.51 Similar results have been found in the literature using “water-in-bisalt” electrolytes.11,13 The absence of Na+ intercalation in our case can be explained by the larger cation size of Na+ (1.02 Å) than that of Zn2+ (0.74 Å) (Fig. S28, ESI†).13,20
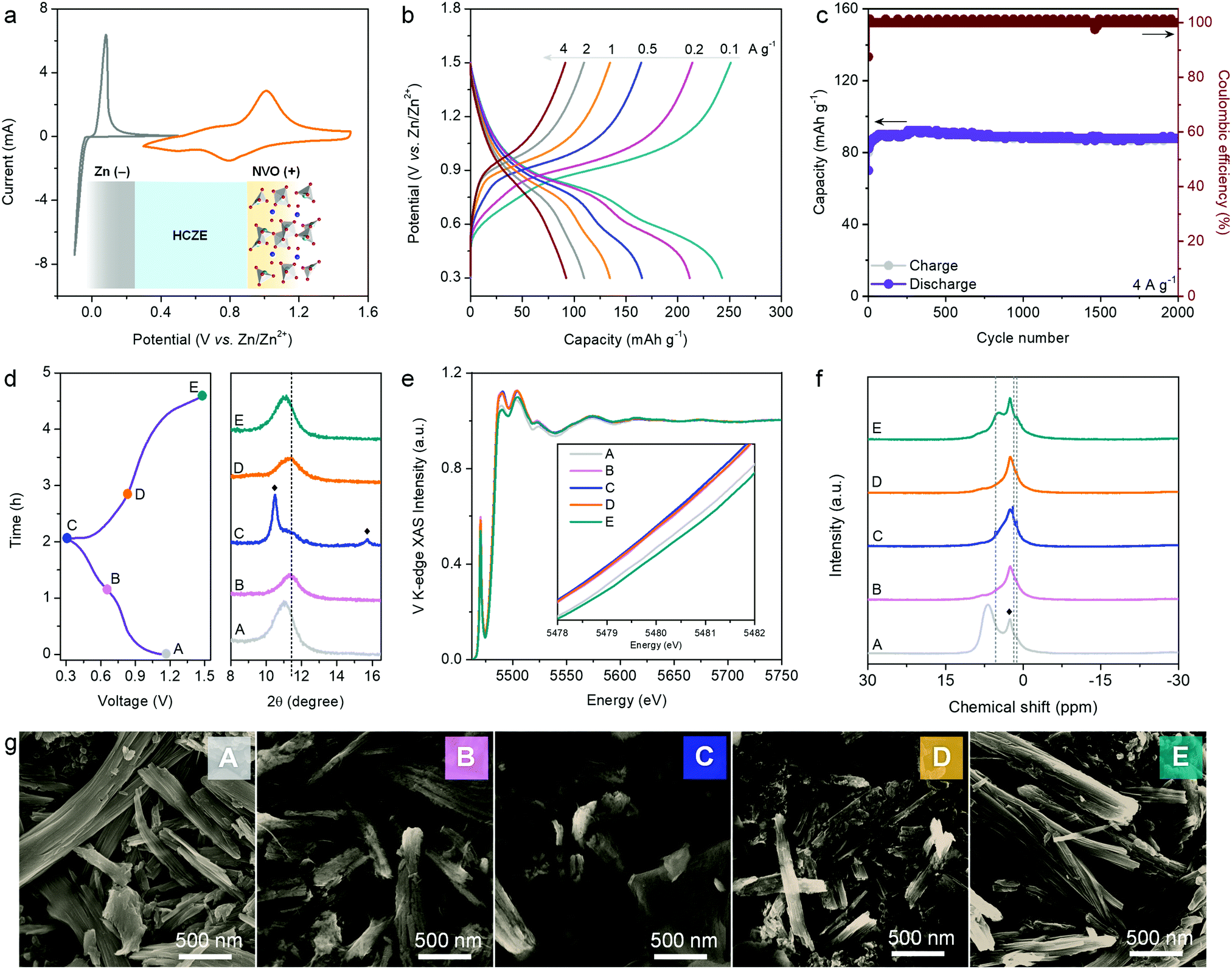 | ||
| Fig. 5 Electrochemical performance and ex situ characterizations of the NVO electrode in HCZE. (a) CV plots of the NVO cathode (orange) against the Zn anode (gray) at 1 mV s−1. Inset shows the battery configuration. (b) Galvanostatic charge–discharge curves at different current densities. (c) Stability test at 4 A g−1. (d) Galvanostatic charge–discharge curves (left) and ex situ XRD patterns (right). The solid rhombus marks in the XRD patterns show the diffractions of Zn4ClO4(OH)7. (e) XANES spectra, (f) 1H NMR spectra, and (g) SEM images of the NVO electrode at selected states. The unchanged signals marked with a solid rhombus in the 1H NMR spectra correspond to the hydrogen from polyvinylidene difluoride (PVDF) binder. The Zn4ClO4(OH)7 nanosheets can be observed in the SEM images at states B, C, and D. | ||
Galvanostatic tests were further carried out to evaluate the rate capability of the NVO electrode (Fig. 5b). The predominantly sloping charge–discharge plots signify a solid-solution type process of cation (de)intercalation. At 0.1 A g−1, a high capacity of 253 mA h g−1 can be obtained. Increasing the rate to 4 A g−1 delivers a decent capacity of 94 mA h g−1 (Fig. 5b). The rate capability of our NVO cathode in HCZE outperforms the intercalation cathodes in conventional WIS and eutectic electrolytes.11–13,16,26 This points to the synergistic effect between the high ionic conductivity of HCZE and the flexible cation-conducting structural framework of the NVO electrode.
As another critical parameter to evaluate the self-discharge of a battery,52,53 the voltage decay of the NVO electrode on the shelf was recorded upon fully charging to 1.5 V at 0.1 A g−1. After resting for 15 days, 80.9% of the original capacity can be maintained, apparently superior to the performance using conventional dilute electrolytes (Fig. S29, ESI†). This low self-discharge rate suggests that the parasitic reactions (e.g., Zn corrosion and water dissociation) and dissolution of the active cathode are efficiently suppressed in our battery (Fig. S1, ESI†). The evaluation of cycling performance is another critical approach to study the effectiveness of HCZE in prohibiting the side reactions and cathode dissolution. As expected, nearly no capacity decay can be observed over 2000 cycles at 4 A g−1 (Fig. 5c), featuring high coulombic efficiency of >99%. Coincident with the electrochemical performance, the nanofiber morphology of our NVO electrode can be maintained after cycling tests (Fig. S30, ESI†). As such, the robust stability of the NVO cathode is strongly associated with the minimized amount of free water molecules to inhibit irreversible parasitic reactions and the flexible, stable layered framework of NVO.
We further conducted different ex situ techniques, including XRD, X-ray absorption spectroscopy (XAS), XPS, solid-state NMR, FT-IR, SEM, and transmission electron microscopy (TEM), to illustrate the electrochemical energy storage mechanism of NVO in HCZE. Ex situ XRD patterns of the NVO cathode demonstrate a slight contraction of the gallery spacing of the (001) plane (2θ = 11.1°) upon discharge to 0.65 V (Fig. 5d and Fig. S31, ESI†), indicating the decrease of interlayer spacing. This is related to an improvement of structural coordination because of the strong electrostatic interaction between the intercalated cations and the V3O8 bilayers of NVO. In the subsequent discharge region of 0.65–0.3 V, we observe the continuous shrinkage of interlayer spacing, which is accompanied by the evolution of a set of new peaks. These diffractions can be assigned to Zn4ClO4(OH)7 (Fig. S31, ESI†),5,29,54 implying a proton intercalation process. The diffractions of Zn4ClO4(OH)7 disappear upon the potential reversal to 1.5 V, along with the recovery of the NVO phase. To reveal the electronic structure of vanadium, we collected V L-edge and V K-edge X-ray absorption spectra for the NVO cathode at certain charging/discharging voltages (Fig. 5e and Fig. S32, ESI†). The line shape and intensity of the V L3-edge at 510–520 eV are used to determine the oxidation state of vanadium. The increasing intensity ratio of 516.8 eV/514.5 eV peaks for NVO electrodes upon charging to 1.5 V indicates higher oxidation of vanadium at charged states (Fig. S31a, ESI†). V K-edge X-ray absorption near edge structure (XANES) spectra show negative shifts of the adsorption edge upon discharge, which reflects the reduction of vanadium (inset of Fig. 5e). During the recharge process, the absorption edge shifts back to higher energy indicating the oxidation of vanadium. Both V L-edge and K-edge spectra reveal the reversible changes of V oxidation states during charge and discharge, which is consistent with ex situ XPS analysis (Fig. S33, ESI†). Wavelet transform analysis shows that the V3O8 bilayers of NVO are well maintained during charge and discharge (Fig. S32, ESI†). The framework stability of NVO is associated with the stable Na+ pillars featuring highly negative formation energy in the presence of structural water.51,55 Accordingly, the reversible contraction/expansion of the gallery spacing of NVO during discharge/charge plays a vital role in ensuring its long-term cycling stability.
Typically, H+ co-intercalation with Zn2+ occurs in conventional electrolytes of aqueous ZIBs. The consumption of H+ from water dissociation leads to the formation of electrolyte-dependent products on the cathodes, e.g., Zn4SO4(OH)6·xH2O in ZnSO4,29 Zn5(OH)8Cl2·xH2O in ZnCl2,26 and triflate-containing Zn-based layered double hydroxides (LDHs) in Zn(CF3SO3)2.56 During discharge, the H+-intercalation byproduct of Zn4ClO4(OH)7 sheets could be formed on the surface of NVO in HCZE (Fig. 5g and Fig. S34–S37, ESI†). Ex situ1H NMR spectra show noticeable yet reversible changes in intensity, peak position, and line width of the 1H signal that is attributed to structural water (6.5 ppm for the pristine NVO) (Fig. 5f). This indicates the participation of structural water in assisting H+ and Zn2+ intercalations through providing an electrostatic shielding effect.55,57 The peaks at 1.1 and 5.4 ppm correspond to the intercalated protons. Another reversible peak at 1.8 ppm is assigned to the proton in Zn4ClO4(OH)7.28,29 Combining the reversible changes of Zn and Cl of energy-dispersive X-ray spectroscopy (EDS) with SEM and TEM at different states (Fig. S36, S37 and Table S1, ESI†), the simultaneous insertion/extraction of H+ and Zn2+ can be confirmed. Such observation is different from previous aqueous ZIBs, such as the sole storage of Zn2+ or H+ in the cathodes26,58 and stepwise intercalation of H+ and Zn2+.59
During discharge, the H+-intercalation byproduct (i.e., Zn4ClO4(OH)7) can also be generated on the NVO cathode in the dilute electrolyte of 0.5 m Zn(ClO4)2. This phenomenon, together with the overlapping of galvanostatic charge/discharge curves to that in HCZE, indicates nearly the same energy storage mechanism in these two electrolytes (Fig. S38, ESI†). The difference between low-concentration aqueous electrolyte and HCZE lies in the changes in solvation structures induced by the high concentration of solutes. In the dilute aqueous electrolyte featuring enough available water molecules, all the Zn2+ ions can be solvated by dipolar water molecules to generate octahedral Zn(H2O)62+. Such cation–water interaction can induce electron departure from the 3a1 orbital of the coordinated water to the empty Zn2+ orbitals, thus significantly weakening the O–H bond of the coordinated water.60 For the dilute electrolyte, such effect could induce deprotonation of water to generate a mildly acidic solution (pH ∼ 5 for 0.5 m Zn(ClO4)2). This can be confirmed by a negative dehydrogenation energy of −3.83 eV for the H2O in octahedral [Zn(H2O)6]2+, as calculated by DFT (Fig. S39, ESI†). The water deprotonation can be efficiently suppressed in concentrated electrolytes because of the unique inter-molecular interaction (e.g., Zn2+–ClO4− and ClO4−–H2O). However, a minor amount of free water molecules still exists in the most concentrated electrolytes (Fig. 2 and 3), which shows three advantages of:30,61 (1) ensuring the high ionic conductivity for high rate performance through decreasing the electrolyte viscosity, (2) minimizing cathode dissolution to ensure cycling stability, and (3) featuring hydrogen bonding networks and providing sources for proton co-intercalation into NVO, which is confirmed to be thermodynamically feasible (Fig. S40, ESI†) (see the detailed energy storage mechanism in Supplementary Note 2, ESI†).
Compared to the previously reported highly concentrated dual-cation aqueous electrolytes (e.g., Zn–Li hybrid battery11 and Li-ion battery13), our battery chemistry shows two advantages: (1) potential scalability for large-scale energy storage, which is enabled by the low-cost dual-cation electrolyte and scalable production of cathode materials and (2) remarkable cycling stability and decent rate capability because of the high ionic conductivity of the electrolyte and the Zn2+/H+ cointercalation mechanism.
Conclusions
We have demonstrated the use of a high-concentration zinc–sodium perchlorate mixture in water, which resembles the classical water-in-salt electrolyte but shows lower cost, higher ionic conductivity, and decreased toxicity. Extensive spectroscopic studies and molecular dynamics simulations reveal that perchlorate anions dominate the cation solvation shells, resulting in suppressed water activity and electronic structure changes of the electrolyte. An anion-derived interphase on the Zn anode formed in situ by perchlorate reduction enables improved stability and efficiency of Zn plating/stripping. Leveraging this high reversibility and the scarcity of free water, a zinc–sodium vanadate battery has been developed to show long cycling stability, enhanced rate capability, and suppressed self-discharge. This battery follows an electrochemical energy storage mechanism of simultaneous H+ and Zn2+ (de)intercalation. The fundamental understanding and the experimental demonstrations of reversible battery chemistry using a bi-cation electrolyte strategy open a viable route to developing aqueous batteries for emerging electrochemical energy storage applications.Author contributions
Y. Z. and H. N. A. conceived the idea and designed the experiments. Y. Z. conducted the experiments of material syntheses, electrochemical tests, and material characterizations including SEM, XRD, XPS, FT-IR, Raman, and DSC. J. Y. and O. F. M. conducted theoretical calculations. X. Z. conducted X-ray synchrotron spectroscopy. A.-H. E. performed the NMR characterization. Y. L. conducted the TEM and EDS characterizations. The manuscript was written by Y. Z. and was revised by the other co-authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this work was supported by King Abdullah University of Science Technology (KAUST).Notes and references
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- S.-B. Wang, Q. Ran, R.-Q. Yao, H. Shi, Z. Wen, M. Zhao, X. Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef PubMed.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3(10), 2620–2640 CrossRef CAS.
- Z. Cao, P. Zhuang, X. Zhang, M. Ye, J. Shen and P. M. Ajayan, Adv. Energy Mater., 2020, 10, 2001599 CrossRef CAS.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- C. Zhang, L. Zhang and G. Yu, Acc. Chem. Rev., 2020, 53, 1648–1659 CrossRef CAS PubMed.
- L. Zhang, C. Zhang, Y. Ding, K. Ramirez-Meyers and G. Yu, Joule, 2017, 1, 623–633 CrossRef CAS.
- S. Cai, X. Chu, C. Liu, H. Lai, H. Chen, Y. Jiang, F. Guo, Z. Xu, C. Wang and C. Gao, Adv. Mater., 2021, 33, 2007470 CrossRef CAS PubMed.
- C.-Y. Chen, K. Matsumoto, K. Kubota, R. Hagiwara and Q. Xu, Adv. Energy Mater., 2019, 9, 1900196 CrossRef.
- Z. Yi, G. Chen, F. Hou, L. Wang and J. Liang, Adv. Energy Mater., 2020, 10, 2003065 Search PubMed.
- T. Zhang, Y. Tang, S. Guo, X. Cao, A. Pan, G. Fang, J. Zhou and S. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 RSC.
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- L. Jiang, L. Liu, J. Yue, Q. Zhang, A. Zhou, O. Borodin, L. Suo, H. Li, L. Chen, K. Xu and Y.-S. Hu, Adv. Mater., 2020, 32, 1904427 CrossRef CAS PubMed.
- L. Zhang, I. A. Rodríguez-Pérez, H. Jiang, C. Zhang, D. P. Leonard, Q. Guo, W. Wang, S. Han, L. Wang and X. Ji, Adv. Funct. Mater., 2019, 29, 1902653 CrossRef.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- L. Chen, J. Zhang, Q. Li, J. Vatamanu, X. Ji, T. P. Pollard, C. Cui, S. Hou, J. Chen, C. Yang, L. Ma, M. S. Ding, M. Garaga, S. Greenbaum, H.-S. Lee, O. Borodin, K. Xu and C. Wang, ACS Energy Lett., 2020, 5, 968–974 CrossRef CAS.
- S. Ko, Y. Yamada, K. Miyazakia, T. Shimada, E. Watanabe, Y. Tateyama, T. Kamiya, T. Honda, J. Akikusa and A. Yamada, Electrochem. Commun., 2019, 104, 106488 CrossRef CAS.
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC.
- M. H. Lee, S. J. Kim, D. Chang, J. Kim, S. Moon, K. Oh, K.-Y. Park, W. M. Seong, H. Park, G. Kwon, B. Lee and K. Kang, Mater. Today, 2019, 29, 26–36 CrossRef CAS.
- H. Liang, Z. Cao, F. Ming, W. Zhang, D. H. Anjum, Y. Cui, L. Cavallo and H. N. Alshareef, Nano Lett., 2019, 19, 3199–3206 CrossRef CAS PubMed.
- Y. K. Sze and D. E. Irish, J. Solution Chem., 1978, 7, 395–415 CrossRef CAS.
- L. Wang, Y. Zhang, H. Hu, H.-Y. Shi, Y. Song, D. Guo, X.-X. Liu and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 42000–42005 CrossRef CAS PubMed.
- X. Liang, Q. Pang, I. R. Kochetkov, M. S. Sempere, H. Huang, X. Sun and L. F. Nazar, Nat. Energy, 2017, 2, 17119 CrossRef CAS.
- B. M. Auer and J. L. Skinner, J. Chem. Phys., 2008, 128, 224511 CrossRef CAS PubMed.
- D. A. McQuarrie, P. A. Rock and E. B. Gallogly, General chemistry, WH Freeman, 1984 Search PubMed.
- E. G. Bloor and R. G. Kidd, Can. J. Chem., 1968, 46, 3425–3430 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- Q. Zhao, Z. Tu, S. Wei, K. Zhang, S. Choudhury, X. Liu and L. A. Archer, Angew. Chem., Int. Ed., 2018, 57, 992–996 CrossRef CAS PubMed.
- B. You, X. Liu, G. Hu, S. Gul, J. Yano, D.-E. Jiang and Y. Sun, J. Am. Chem. Soc., 2017, 139, 12283–12290 CrossRef CAS PubMed.
- C.-T. Dinh, A. Jain, F. P. García de Arquer, P. D. Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- Q. Zhao, M. J. Zachman, W. I. Al Sadat, J. Zheng, L. F. Kourkoutis and L. Archer, Sci. Adv., 2018, 4, eaau8131 CrossRef CAS PubMed.
- Q. Zhang, Y. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed.
- S. Kaur, A. Gupta and H. K. Kashyap, J. Phys. Chem. B, 2020, 124(11), 2230–2237 CrossRef CAS PubMed.
- P. Hu, T. Zhu, X. Wang, X. Wei, M. Yan, J. Li, W. Luo, W. Yang, W. Zhang, L. Zhou, Z. Zhou and L. Mai, Nano Lett., 2018, 18, 1758–1763 CrossRef CAS PubMed.
- V. Soundharrajan, B. Sambandam, S. Kim, M. H. Alfaruqi, D. Y. Putro, J. Jo, S. Kim, V. Mathew, Y.-K. Sun and J. Kim, Nano Lett., 2018, 18, 2402–2410 CrossRef CAS PubMed.
- K. Zhu, T. Wu and K. Huang, Adv. Energy Mater., 2019, 9, 1901968 CrossRef CAS.
- C. Liu, Z. Neale, J. Zheng, X. Jia, J. Huang, M. Yan, M. Tian, M. Wang, J. Yang and G. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC.
- D. Bin, W. Huo, Y. Yuan, J. Huang, Y. Liu, Y. Zhang, F. Dong, Y. Wang and Y. Xia, Chem, 2020, 6, 968–984 CAS.
- T. Wei, Q. Li, G. Yang and C. Wang, Adv. Energy Mater., 2019, 9, 1901480 CrossRef.
- Y. Zhu, G. Huang, J. Yin, Y. Lei, A.-H. Emwas, X. Yu, O. F. Mohammed and H. N. Alshareef, Adv. Energy Mater., 2020, 10, 2002128 CrossRef CAS.
- P. Oberholzer, E. Tervoort, A. Bouzid, A. Pasquarello and D. Kundu, ACS Appl. Mater. Interfaces, 2019, 11, 674–682 CrossRef CAS PubMed.
- J. Shin, D. S. Choi, H. J. Lee, Y. Jung and J. W. Choi, Adv. Energy Mater., 2019, 9, 1900083 CrossRef.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- D. W. Barnum, Inorg. Chem., 1983, 22, 2297–2305 CrossRef CAS.
- R. Bouchal, Z. Li, C. Bongu, S. L. Vot, R. Berthelot, B. Rotenberg, F. Favier, S. A. Freunberger, M. Salanne and O. Fontaine, Angew. Chem., Int. Ed., 2020, 59, 15913–15917 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee01472b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |