
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption of double-stranded ribonucleic acids (dsRNA) to iron (oxyhydr-)oxide surfaces: comparative analysis of model dsRNA molecules and deoxyribonucleic acids (DNA)†
Katharina
Sodnikar
 a,
Kimberly M.
Parker
b,
Simona R.
Stump
a,
Laurel K.
ThomasArrigo
a and
Michael
Sander
*a
a,
Kimberly M.
Parker
b,
Simona R.
Stump
a,
Laurel K.
ThomasArrigo
a and
Michael
Sander
*a
aInstitute of Biogeochemistry and Pollutant Dynamics, ETH Zurich, DUSYS, IBP, Universitätsstrasse 16, CHN H50.3, 8092 Zurich, Switzerland. E-mail: michael.sander@env.ethz.ch; Tel: +41 (0) 44 632 8314
bDepartment of Energy, Environmental & Chemical Engineering, Washington University in St. Louis, St. Louis, Missouri 63130, USA
First published on 3rd March 2021
Abstract
Double-stranded ribonucleic acid (dsRNA) molecules are novel plant-incorporated protectants expressed in genetically modified RNA interference (RNAi) crops. Ecological risk assessment (ERA) of RNAi crops requires a heretofore-missing detailed understanding of dsRNA adsorption in soils, a key fate process. Herein, we systematically study the adsorption of a model dsRNA molecule and of two double-stranded deoxyribonucleic acid (DNA) molecules of varying lengths to three soil iron (oxyhydr-)oxides – goethite, lepidocrocite, and hematite – over a range of solution pH (4.5–10), ionic strength (I = 10–100 mM NaCl) and composition (0.5, 1, and 3 mM MgCl2) and in the absence and presence of phosphate (0.05–5 mM) as co-adsorbate. We hypothesized comparable adsorption characteristics of dsRNA and DNA based on their structural similarities. Consistently, the three nucleic acids (NAs) showed high adsorption affinities to the iron (oxyhydr-)oxides with decreasing adsorption in the order goethite, lepidocrocite, and hematite, likely reflecting a decrease in the hydroxyl group density and positive charges of the oxide surfaces in the same order. NA adsorption also decreased with increasing solution pH, consistent with weakening of NA electrostatic attraction to and inner–sphere complex formation with the iron (oxyhydr-)oxides surfaces as pH increased. Adsorbed NA concentrations increased with increasing I and in the presence of Mg2+, consistent with adsorbed NA molecules adopting more compact conformations. Strong NA–phosphate adsorption competition demonstrates that co-adsorbates need consideration in assessing dsRNA fate in soils. Comparable adsorption characteristics of dsRNA and DNA molecules to iron (oxyhydr-)oxides imply that information on DNA adsorption to soil particle surfaces can inform dsRNA ERA.
Environmental significanceInsecticidal double-stranded RNA (dsRNA) molecules are novel plant-incorporated protectants (PIPs) expressed in genetically modified RNA interference crops. Ecological risk assessment (ERA) of this technology demands information on adsorption of dsRNA molecules in agricultural soils, a key process affecting dsRNA fate. Through systematic studies of dsRNA and DNA adsorption to three iron (oxyhydr-)oxides commonly found in soils, we demonstrate that these NAs have comparable adsorption characteristics across a range of solution pH, ionic strengths and compositions and that their adsorption is competitively suppressed by phosphate as co-solute. Beyond providing insights into the dsRNA adsorption mechanism, this work shows that existing information on DNA adsorption in soils can inform the ERA of dsRNA in the agro-environment. |
1. Introduction
Genetically modified crops expressing plant-incorporated protectants (PIPs) against specific pests play a central role in agricultural pest insect management.1 A new generation of such PIPs are insecticidal double-stranded ribonucleic acid (dsRNA) molecules expressed in so-called RNA interference (RNAi) crops.2–4 Targeted pest insects take up the dsRNA when feeding on the RNAi crops. In the pest organisms, the dsRNA molecules are enzymatically cleaved into small interfering RNA molecules which sequence-specifically bind to messenger RNA (mRNA) that codes for specific, essential proteins for the pest insect. The binding triggers the breakdown of this mRNA5 resulting in retarded development or death of the pest insect.2,6–9The agricultural use of RNAi crops will result in the release of dsRNA molecules to the agro-environment, with soils as major receiving environments.10 An assessment of potential ecological risks associated with this release (e.g. exposure of non-target soil-dwelling organisms to the dsRNA) demands detailed information on dsRNA fate in soils. A key fate process is the adsorption of dsRNA molecules to soil particle surfaces, based on previous studies on the fate of extracellular deoxyribonucleic acids (DNA) in soils.11–17 On the one hand, dsRNA adsorption is expected to decrease its transport and its bioavailability in soils. On the other hand, akin to adsorbed DNA, adsorbed dsRNA molecules may persist for longer in soils if adsorption protects these molecules from (bio)degradation pathways that occur in the soil pore water.18–22 Such adsorptive stabilization may potentially lead to elevated dsRNA solution concentrations if changes in the soil solution chemistry trigger extensive dsRNA desorption from mineral surfaces.
While the importance of dsRNA adsorption to its fate in soils is widely recognized,23 there remains little information on this process. A few studies have demonstrated rapid dissipation of dsRNA in soils (i.e., rapid decreases in detectable dsRNA concentrations in soil and sediment pore waters), which was ascribed to dsRNA breakdown.24–29 However, these studies did not systematically assess the relative contribution of dsRNA adsorption to soil and sediment particle surfaces to overall dissipation, and therefore may have overestimated dsRNA breakdown in soils. In fact, a recent study using 32P-labeled dsRNA demonstrated that dissipation of dissolved dsRNA in soils resulted from both dsRNA adsorption to soil particle surfaces and dsRNA degradation.19 However, adsorption characteristics were not studied in detail and a systematic assessment of dsRNA adsorption to soil particles remains missing.
While systematic studies on dsRNA adsorption to particle surfaces are lacking, numerous studies have assessed adsorption of DNA to soils20,30–33 as well as to mineral and organic soil constitutes including clay minerals,18,20,31,34–48 natural organic matter,49,50 quartz sand,49,51–57 and iron (oxyhydr-)oxides.11,12,35,36,48,58 These studies highlighted the importance of DNA–sorbent electrostatic interactions in driving adsorption.11,34–36,46,49,53,55–60 For variable-charged sorbents, electrostatic interactions between the negatively charged DNA molecules and the sorbent depend on solution pH: attractive electrostatic interactions at pH below the point of zero net charge (PZC) of the sorbent—and thus a net positive sorbent surface charge—may switch to repulsive electrostatic interactions at a solution pH above the PZC—at which the sorbent is net negatively charged.34,36,37,47,61 Furthermore, several studies provided evidence for inner–sphere complex formation between the phosphodiester groups in the backbone of DNAs and the surface hydroxyl groups of iron oxides.11–17,62 While dsRNA and DNA have structural differences (i.e., ribose and base uracil in dsRNA but deoxyribose and base thymine in DNA), these two NAs also share substantial structural similarities: both are double-stranded molecules with a negatively charged phosphodiester backbone chain. As a consequence, we hypothesize that dsRNA adsorption to soil minerals is also driven by electrostatic interactions as well as inner–sphere complex formation.
Another similarity of dsRNA and DNA is that both are semiflexible, charged linear polyelectrolytes.63,64 Their interactions at solid–water interfaces are thus expected to follow polyelectrolyte adsorption theory.64–72 As compared to rigid colloids, adsorption of polyelectrolytes has energetic contributions not only from ‘classical’ sorbate–particle interaction forces (i.e., for NAs dominated by electrostatics and inner–sphere complex formation, see above) but also from (changes in) entropic states that are related to the conformations adopted by adsorbed NAs.69,71,73 Adsorbed segments of the NA backbones (so called ‘trains’) can be interspersed with free dangling internal and end segments of the NA backbone, so called ‘loops’ and ‘tails’.74,75 The NA conformations that form on the sorbent are strongly dependent on solution chemistry: under conditions with NA–sorbent electrostatic attraction, low solution ionic strength leads to electrostatic repulsion between negative charges in the NA backbone as well as between adjacent NA molecules on the sorbent surface, thereby favoring NA adsorption in more elongated, linear conformations, leading to comparatively lower maximum adsorbed concentrations at sorbent surface saturation. Conversely, higher ionic strength screens intra- and intermolecular NA electrostatic repulsion,76–81 allowing for more compact NA conformations in adsorbed states and thus smaller molecular footprints on the sorbent, leading to higher maximum adsorbed concentrations at sorbent surface saturation.70,73,75 Similarly, the presence of dissolved divalent cations, such as Mg2+, is expected to result in more compact NA conformations by forming intramolecular cation bridges between negatively charged phosphodiester groups in the NA backbone.64,82–86 Such cation bridges may also form between negatively charged sorbent surfaces and the negatively charged NA backbone, thereby possibly allowing for adsorption of NAs even to like-charged sorbents.87,88 With decreasing size of NA molecules, their footprint in adsorbed states also decreases. It has previously been shown that smaller NA molecules have higher adsorbed concentrations at sorbent surface saturation as compared to larger NA molecules.32,43
In this study, we aimed to provide the first systematic assessment of dsRNA adsorption over a range of solution conditions to iron (oxyhydr-)oxides that are ubiquitous in agricultural soils. Furthermore, by complementing dsRNA with DNA adsorption experiments, this study is the first to allow for a direct comparison of the adsorption characteristics of dsRNA and DNA. While our work focused on adsorption to iron (oxyhydr-)oxides, comparable adsorption characteristics of dsRNA and DNA to these minerals would likely imply similarity in adsorption also to other soil mineral surfaces not tested herein. Based on the existing DNA adsorption literature and the similarities of the two NAs, our working hypothesis was that adsorption of both NAs follows polyelectrolyte adsorption theory with electrostatic interactions and inner–sphere complex formation playing a key role in driving adsorption. We chose to investigate NA adsorption to the three iron (oxyhydr-)oxides (hereafter referred to as ‘iron oxides’) goethite, lepidocrocite, and hematite. We selected these iron oxides not only because of their different mineralogies but also because they are important in agricultural soils and they carry a pH-dependent surface charge. To test our hypothesis, we assessed adsorption of two types of DNA (i.e., a genomic DNA molecule of ≥17.7 kbp, and a sheared genomic DNA molecule of 425 bp) and one model commercially available dsRNA molecule (polyadenylic–polyuridylic acid molecule (poly(AU), 1287 bp) over a wide range of solution chemistry (i.e., pH, ionic strength and ionic composition) using batch solution depletion experiments. The same experimental approach has been used in numerous past studies on DNA adsorption to isolated soil minerals34,37,38,43,47,82,89 as well as to entire soils.32,33,90 The two differently sized DNA molecules served to assess potential dependencies of NA molecular size on adsorption. In addition to single-solute NA adsorption experiment, we conducted NA adsorption experiments in the presence of phosphate as a potential adsorptive competitor to the NAs. Phosphate is common in agricultural soils under fertilization. The NA adsorption experiments in the presence of phosphate not only served to provide additional information on the adsorption mechanism of the NAs but also to inform on potential competitive effects arising from phosphate on dsRNA adsorption in natural soils. We note that we constrained competition experiments to phosphate but expect that other negatively charged (macro)molecules, such as dissolved organic matter (DOM), may also compete with dsRNA for positively charged adsorption sites.
2. Materials and methods
2.1. Chemicals
All chemicals were of high purity (>98%) and used as received. Detailed information on chemicals are listed in Section S1 in the ESI.†2.2. Solutions
All solutions were prepared with Milli-Q (MQ) water (resistivity >18 MΩ cm; Barnstead NANOpure Water Purification System), contained 10 mM NaCl as background electrolyte and 3 mM of a pH buffering species (i.e., acetate for pH 4.5–5.5, bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane (BisTris) for pH 6–7, 4-(2-hydroxyethyl)piperauzine-1-ethanesulfonic acid (HEPES) for pH 7.5–8, and 2-(cyclohexylamino)ethanesulfonic acid (CHES) for pH 8.6–10). The solution pH was adjusted using 0.5 M NaOH or HCl. As shown in the results and discussion section, there were no buffer-specific offsets in the extent to which NA adsorbed to the iron oxides in pH-dependent adsorption experiments. We therefore concluded that none of the used buffer species showed strong interactions with the iron oxide surfaces that would NA adsorption. To test for the effect of higher ionic strength on NA adsorption at pH 7, we conducted adsorption experiments also in solutions with 50 or 100 mM NaCl while maintaining the same pH buffer concentration (3 mM BisTris). We used NaCl as a model for monovalent cations. To investigate the effect of divalent cations on NA adsorption at pH 7, we assessed NA adsorption in the presence of 0.5, 1 or 3 mM MgCl2 while maintaining a constant total solution ionic strength of 10 mM (i.e., we supplemented the MgCl2 with 8.5, 7 or 1 mM NaCl, respectively) and the same pH buffer concentration (3 mM BisTris). The solutions were either sterile filtered (nominal cutoff of 0.2 μm, sterile polyethersulfone membrane, Pall Life Sciences) or autoclaved (Zirbus Technology, LTA 2 × 3 × 4). Prior to use, all glassware was washed three times each with soapy water followed by MQ water and subsequently baked at 220 °C for at least 24 hours.2.3. Sorbates
All NA concentrations were quantified by measuring the absorbance at a wavelength of 260 nm with a NanoDrop™ 1000 spectrophotometer according to the Beer–Lambert equation with an extinction coefficient (ε) of 0.02 mL (μg cm)−1.91 The size distribution of nucleic acid molecules was determined using the Agilent Tapestation and agarose gel electrophoresis (for details see Section S2, ESI†).Deoxyribonucleic acid sodium salt from salmon testes (herein named genomic DNA (gDNA)) was purchased from Sigma-Aldrich (USA). gDNA stock solutions (concentration 1 mg mL−1) were prepared by dissolving the gDNA in buffer (3 mM of respective pH buffer) containing 10 mM NaCl needed to stabilize the gDNA and were stored at 4 °C. Note that NAs denatured when dissolved in pure MQ water without background electrolyte (Section S3, ESI†). Experimental gDNA solutions were prepared by diluting gDNA stock solutions in the respective buffer solution to a concentration of 100 μg gDNA mL−1, unless indicated otherwise. The median size of the gDNA was 17.7 kbp as determined by Tapestation (Section S2, ESI†) (approximately equal to a length of 6 μm, estimated assuming that 1 bp = 0.34 nm).92
To obtain shorter, sheared DNA (sDNA), we treated the gDNA with a QSonica Q800R3 Sonicator. Briefly, 1 mL of gDNA solution (100 μg gDNA mL−1) in 2 mL tubes (Eppendorf Protein LoBind) was sheared continuously for 8 minutes at a sonicator amplitude setting of 20%, leading to a median size of 425 bp (∼0.14 μm length) as determined by Tapestation (Section S2, ESI†).
The synthetic double-stranded ribonucleic acid (dsRNA) molecule, polyadenylic–polyuridylic acid (polyAU), was supplied by InvivoGen. The median size of the dsRNA was 1287 bp (∼0.44 μm length) as determined by Tapestation (Section S2, ESI†). The lyophilized polyAU was dissolved as described by the manufacturer and then diluted in the respective buffer to the desired concentration.
Phosphate stock solutions for NA–phosphate co-adsorbate experiments were prepared by dissolving NaH2PO4 in 3 mM BisTris – 10 mM NaCl buffer. The pH was adjusted to pH 7 with 10 M sodium hydroxide followed by filtering the solution through 0.2 μm cutoff sterile membrane filters (see above).
2.4. Iron oxides
The iron oxides goethite (α-FeOOH, Bayferrox 910), lepidocrocite (γ-FeOOH, Bayferrox 943), and hematite (α-Fe2O3, Bayferrox 105M) were from Bayer (Germany) and used without any pretreatment. The mineralogy of the iron oxides was confirmed by X-ray diffraction analysis (XRD) and particle morphology was characterized by scanning electron microscopy (SEM) (details in Section S4, ESI†). The specific surface area of the iron oxides was determined by N2-BET analysis on a Nova 3200e Quantachrome BET analyzer. We calculated the pH-dependent speciation of hydroxyl groups on the surface of the iron oxides using published acid dissociation constants (Section S5, ESI†). The key physicochemical characteristics of the iron oxides are listed in Table 1.| Mineral (formula) | SSAa [m2 g−1] | [![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) FeOH]tb [nm−2] FeOH]tb [nm−2] |
PZCd | Acid dissociation constants | Longest particle dimension [μm] |
|---|---|---|---|---|---|
| a SSA = specific surface area, as determined by N2-BET analysis. b Total number of surface hydroxyl groups per nm2. c Used the same material. d pH at which the iron oxide has a zero net charge (PZC). e Used J. T. Baker hematite with a similar SSA as our material. | |||||
| Goethite (α-FeOOH) | 13.0 | 5.5c (ref. 93 and 94) | 7.85c (ref. 93) | pKa1 = 6.7c (ref. 93), pKa2 = 9c (ref. 93) | 0.3–1.5c (ref. 95) |
| Lepidocrocite (γ-FeOOH) | 14.4 | 1.67c (ref. 96) | 7.3c (ref. 96) | pKa1 = 6.45c (ref. 96), pKa2 = 8.1c (ref. 96) | 0.2–0.4c (ref. 97) |
| Hematite (α-Fe2O3) | 11.0 | 0.7c (ref. 94) | 6.95e (ref. 98) | pKa1 = 4.9e (ref. 98), pKa2 = 9e (ref. 98) | 0.09c (ref. 99) |
2.5. Batch adsorption experiments: NAs to iron oxides
We systematically assessed potential effects on NA adsorption resulting from variations in the experimental protocol (i.e., the order of NA and iron oxide addition to the batch reactors, the manner and conditions under which they were added, as well as the total iron oxide loading at constant NA to iron oxide ratio). All tested variations had at most small effects on NA adsorption to the iron oxides, as detailed in Section S6, ESI.† Furthermore, for all variations tested, replicate batch NA adsorption experiments yielded highly reproducible results: variations in the adsorbed NA concentrations were less than 10% of the mean adsorbed NA concentration across all tested variations except for experiments at low goethite and NA concentrations close to the limit of quantification (see Section S6, ESI†). Based on these results, we selected the experimental protocol described in the following.We conducted all adsorption experiments in triplicates in 2 mL tubes (Eppendorf Protein LoBind), unless stated differently. The tubes were placed in a rack on a multi-position stir plate (600 rpm, Variomag Telemodul 20 P, Sterico AG, Switzerland). First, an aliquot of a NA stock solution was added to each 2 mL tube containing a magnetic, Teflon-coated stir bar. Second, during continuous stirring (600 rpm), we dropwise added an aliquot of an iron oxide stock suspension (4 mg iron oxide mL−1 buffer solution (3 mM buffer substance, 10 mM NaCl as background electrolyte)) to the NA solution. Prior to use, this suspension was allowed to equilibrate to the solution for at least 2 hours under continuous stirring (Hytrel, Faust, Switzerland) followed by >12 hours without stirring. Immediately before being transferred to the tubes, we sonicated the iron oxide suspension to disperse all particles (i.e., sonication at 9 Hz for 30 minutes, bath sonicator, Ultrasonic cleaner USC600D, VWR, Switzerland). After mixing of the NA solution and iron oxide suspension in the batch reactors, the experimental iron oxide suspension concentration was 2 mg mL−1 and the nominal ‘initial’ NA concentration was 50 μg NA mL−1. After 4 hours of NA-adsorptive equilibration under continuous stirring, we centrifuged the tubes (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 minutes for goethite and lepidocrocite, and 20 minutes for hematite, Microstar 17, VWR, Switzerland) and quantified the final NA concentration (c(NA)with iron oxide in [μg mL−1]) in the supernatant by measuring its absorbance at a wavelength of 260 nm using a NanoDrop™ 1000 Spectrophotometer. We tested for potential NA adsorption to the tube walls by running triplicate iron-oxide free controls, which were handled in the same manner as the tubes with iron oxides, followed by quantifying the remaining solution concentration after 4 hours, c(NA)without iron oxide (in [μg mL−1]).
000g, 10 minutes for goethite and lepidocrocite, and 20 minutes for hematite, Microstar 17, VWR, Switzerland) and quantified the final NA concentration (c(NA)with iron oxide in [μg mL−1]) in the supernatant by measuring its absorbance at a wavelength of 260 nm using a NanoDrop™ 1000 Spectrophotometer. We tested for potential NA adsorption to the tube walls by running triplicate iron-oxide free controls, which were handled in the same manner as the tubes with iron oxides, followed by quantifying the remaining solution concentration after 4 hours, c(NA)without iron oxide (in [μg mL−1]).
The adsorbed NA concentration (specific surface area-normalized adsorbed NA mass, Q [ng cm−2]) at the end of the adsorptive equilibration was calculated according to eqn (1):
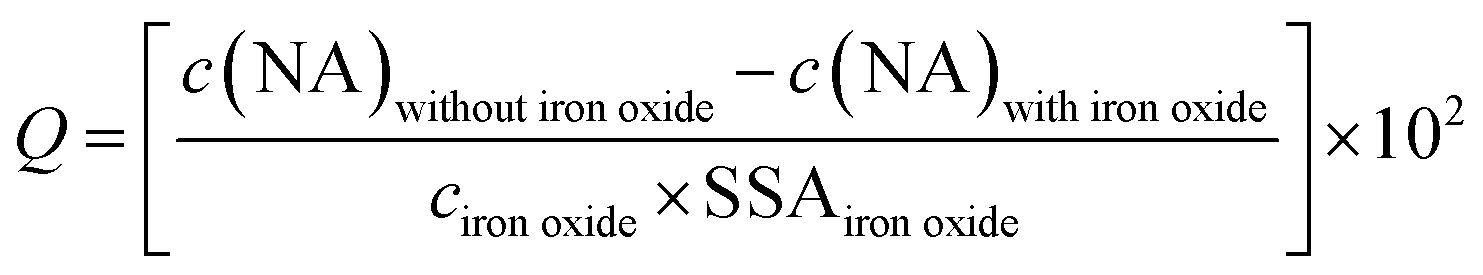 | (1) |
We ran three different sets of batch adsorption experiments. The first set served to determine the kinetics of NA adsorption to the three iron oxides. To be able to repetitively sample each tube at different timepoints up to a total equilibration time of 11 h, we increased the experimental volume from 2 to 5 mL tubes (Eppendorf Protein LoBind) but otherwise followed the same steps as in the experimental protocol described above. At each sampling time, we transferred an aliquot from the respective tubes including controls into 0.5 mL microcentrifuge tubes (Eppendorf Protein LoBind). We centrifuged these aliquots, followed by quantifying NA supernatant concentrations as described above. There was no NA adsorption to the tube walls in controls (i.e., the NA concentrations in all controls were in good agreement with the nominally expected NA concentrations with a standard deviation of only 2 μg mL−1 relative to a starting concentration of 50 μg NA mL−1). The second set of experiments served to determine NA adsorption to the three iron oxides at different initial NA solution concentration (i.e., 5, 10, 20, 30, 40, 50, 60, and 70 μg mL−1, 10 mM NaCl, equilibration time = 4 hours), all at pH 7 and, only for goethite, also at pH 5 and pH 9. Finally, the third set assessed NA adsorption to the three iron oxides over a broad range of solution pH (pH 4.5–10, by using the respective buffer substance described in Section 2.2; the lowest tested pH was set to 4.5 because it corresponds approximately to the reported isoelectric point of DNA,100,101 such that NAs had a net negative charge over the entire tested pH range), solution ionic strengths (10, 50, and 100 mM, adjusted by NaCl) and compositions (0.5, 1, and 3 mM MgCl2 at constant total ionic strength of 10 mM), all at an initial NA concentration of 50 μg NA mL−1 and with an equilibration time of 4 hours.
2.6. Competitive adsorption of NAs and phosphate to goethite
We assessed NA–phosphate co-adsorption to goethite by combining an aliquot of a NA stock solution in a 5 mL tubes (Eppendorf Protein LoBind) with an aliquot of a phosphate stock solution. The initial phosphate concentrations were 0.05, 0.1, 0.2, 0.5, 1, 2, 3, 4, and 5 mM at a constant initial NA concentration of 50 μg NA mL−1. While stirring (600 rpm, Variomag Telemodul 20 P, Sterico AG, Switzerland), sonicated goethite stock suspension was added dropwise to reach a final goethite concentration of 2 mg mL−1. The tubes were stirred for 4 hours, centrifuged (17000g, 10 minutes, Microstar 17, VWR, Switzerland), and the NA concentration in the supernatant of each tube was determined by solution absorbance measurements (see Section 2.5).
In a second set of NA–phosphate co-solute experiments, we tested for effects of the addition order of NA (initial concentration 50 μg mL−1) and phosphate (initial concentration 0.5 mM) on NA adsorption. We either added the phosphate solution at the same time as the NA, prior to, or after the NA. While stirring the first solution (either only NA, only phosphate, or NA and phosphate) (600 rpm, Variomag Telemodul 20 P, Sterico AG, Switzerland), we dropwise added the goethite suspension, followed by adsorptive equilibration for 2 hours. To tubes which contained either only NA or phosphate, we subsequently added the solution with the second missing co-adsorbate and again equilibrated for 2 hours. Therefore, all experiments had a total equilibration time of 4 hours. After centrifugation (17000g, 10 minutes, Microstar 17, VWR, Switzerland), we determined the NA concentration in the supernatant as described in Section 2.5.
3. Results and discussion
3.1. Adsorption kinetics of NAs to iron oxides
We determined the adsorption kinetics of gDNA, sDNA, polyAU to goethite (Fig. 1a) and of gDNA to all three tested iron oxides at an initial NA concentration of 50 μg mL−1 and pH 7 (Fig. 1b). All three NAs rapidly adsorbed to the goethite surface within the first minutes of adsorptive equilibration (Fig. 1a), consistent with kinetic data from a recent study.29 Fast NA adsorption kinetics indicate high affinities of all NAs for the goethite surface, consistent with electrostatic attraction between the negatively charged NAs and the net positively charged goethite surface at pH 7 (PZCgoethite = 7.85, Table 1 and details in Section S5, ESI†). Based on spectroscopic studies we hypothesize that – in addition to electrostatic attraction – inner–sphere complex formation through ligand–exchange reactions between the phosphodiester groups in the NA backbone and the surface hydroxyl groups on the iron oxides surfaces contributed to NA adsorption.11–15,62 Strong evidence in support of inner–sphere complex formation also stems from pH-dependent NA adsorption experiments discussed further below (see Section 3.3). The kinetic data further shows that apparent NA adsorption equilibrium was attained within 4 hours of equilibration: increasing the equilibration time from 4 hours to 10 hours did not lead to a significant increase in adsorbed NA concentrations (one-way ANOVA, p < 0.05). Furthermore, at apparent adsorption equilibrium after four hours, the NA concentration in solution was non-zero, indicating that adsorption ceased when all NA adsorption sites on the goethite surface were occupied by NAs and not because the solution was completely depleted of NAs. We also fitted the kinetic data using a Lagergren's pseudo-first-order kinetic model102 (details in Section S7, ESI†). Fitted adsorption rate constants were in the range of 5.07–10.91 h−1 (depending on the type of NA and the iron oxide), in agreement with our assessment that apparent adsorption equilibrium was attained within 4 hours. We further note that fitted adsorption capacities (qmax) were in good agreement with our calculated maximum adsorbed concentrations (as shown in Section S8, Fig. S9, ESI†).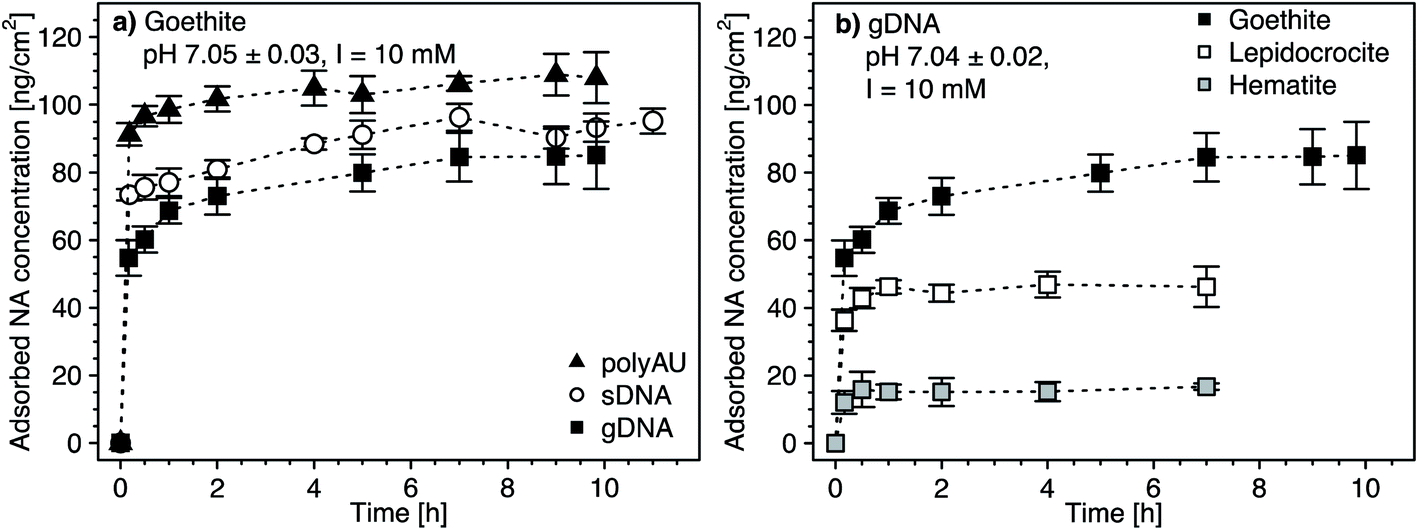 | ||
| Fig. 1 (a) Adsorption kinetics of the three nucleic acid (NA) molecules (i.e., the model dsRNA (polyAU), sheared genomic DNA (sDNA), and the genomic (gDNA)) to the surface of goethite, determined by solution depletion of NAs in batch equilibration reactors. (b) Adsorption kinetics of gDNA to the three different iron oxides (i.e., goethite, lepidocrocite, and hematite). The data of gDNA adsorption to goethite in panel (b) is replotted from panel (a) for comparison. Adsorption kinetics were determined in pH 7 buffer solutions with 3 mM BisTris and 10 mM NaCl background electrolyte and at an initial NA concentration of 50 μg NA mL−1. Data points and error bars represent the mean and the standard deviation of triplicate reactors run in parallel. | ||
The maximum adsorbed concentration at apparent equilibrium (i.e., determined for all timepoints >4 h) of the shorter polyAU (i.e., 106.2 ± 4.0 ng polyAU cm−2) was significantly higher than that of the similarly sized sDNA molecules (92.4 ± 3.7 ng sDNA cm−2) and of the larger gDNA molecules (83.5 ± 5.9 ng gDNA cm−2) (one-way ANOVA, p < 0.05). However, the overall size-dependence of adsorption was small, consistent with previous work on DNA adsorption to clay minerals.43 We ascribe this size-dependence to slightly more efficient packing of smaller than larger NAs on sorbent surfaces.
Initial gDNA adsorption was fast also to lepidocrocite and hematite (Fig. 1b). As discussed above, adsorbed gDNA concentrations on lepidocrocite and hematite also did not significantly increase beyond 4 hours of equilibration (one-way ANOVA, p < 0.05). The maximum adsorbed gDNA concentrations at apparent equilibrium were sorbent-dependent with 83.5 ± 5.9, 46.5 ± 3.8 and 16 ± 1.9 ng gDNA cm−2 for goethite, lepidocrocite and hematite, respectively. We ascribe the substantially lower adsorbed gDNA concentrations on hematite to a combination of factors. First, gDNA adsorption to hematite was likely smaller because its particles are expected to have carried a net neutral charge at the experimental pH 7 (PZChematite = 6.95, Table 1 and details in Section S5, ESI†). By comparison, we expect that goethite and lepidocrocite were net positively charged. Second, we expect that the low net surface charge of hematite in combination with its comparatively small particle size (Table 1) likely led to more aggregation103 than was the case for the other two iron oxides. More pronounced hematite particle aggregation would decrease the available surface area for gDNA adsorption. Aggregation of hematite is indirectly supported by SEM images which, however, were collected after drying the iron oxide suspension (Section S4, ESI†). Finally, since NAs form inner-sphere complexes with surface hydroxyl groups – as shown in numerous past studies (e.g., ref. 11–17) – and are electrostatically attracted to protonated surface hydroxyl groups on the iron oxides, we expected increased NA adsorption with increasing surface densities of these groups. Smaller surface hydroxyl group densities have been reported for hematite (for details see Table 1). The assumption that surface hydroxyl groups are important for NA adsorption is supported by the finding that normalization of the maximum adsorbed NA concentrations on the three iron oxides by their reported maximum surface site densities resulted in more comparable adsorbed NA concentrations (in units of [ng NA (surface OH group)−1)]) for all three iron oxides (Section S7, ESI†).
3.2. Concentration-dependence of NA adsorption
We determined the concentration-dependence of gDNA, sDNA, and polyAU adsorption to goethite, lepidocrocite, and hematite at initial NA solution concentrations from 5 to 70 μg mL−1 and at pH 7. Based on the kinetic data (Fig. 1), we selected a 4 hour equilibration time for adsorption and expected that the NAs adsorbed to the iron oxides at high affinity. Fig. 2 shows the concentration dependence of NA adsorption to goethite (panel a), lepidocrocite (panel b), and hematite (panel c). Fig. 2d compiles the maximum adsorbed concentrations (i.e., adsorption capacities) of the NAs for the three iron oxides, which we calculated by averaging the adsorbed concentrations for solution concentrations at which adsorption plateaued (i.e., solution concentrations above which there was no significant difference in the adsorbed NA concentration; post hoc Tukey test, p < 0.05). In the following section, we first discuss similar trends in the concentration-dependent adsorption of all three NAs to the iron oxides. We subsequently address adsorption characteristics that are more specific to individual NAs. Fitting of the concentration-dependent NA adsorption data with the Langmuir isotherm model104 resulted in fitted maximum adsorption capacities that were in good agreement with the capacities that we determined (as shown in Section S8, ESI†). However, we decided against showing the Langmuir model fits in the manuscript figures as this model makes a number of assumptions that do not fully apply to NA adsorption, as detailed in the ESI.†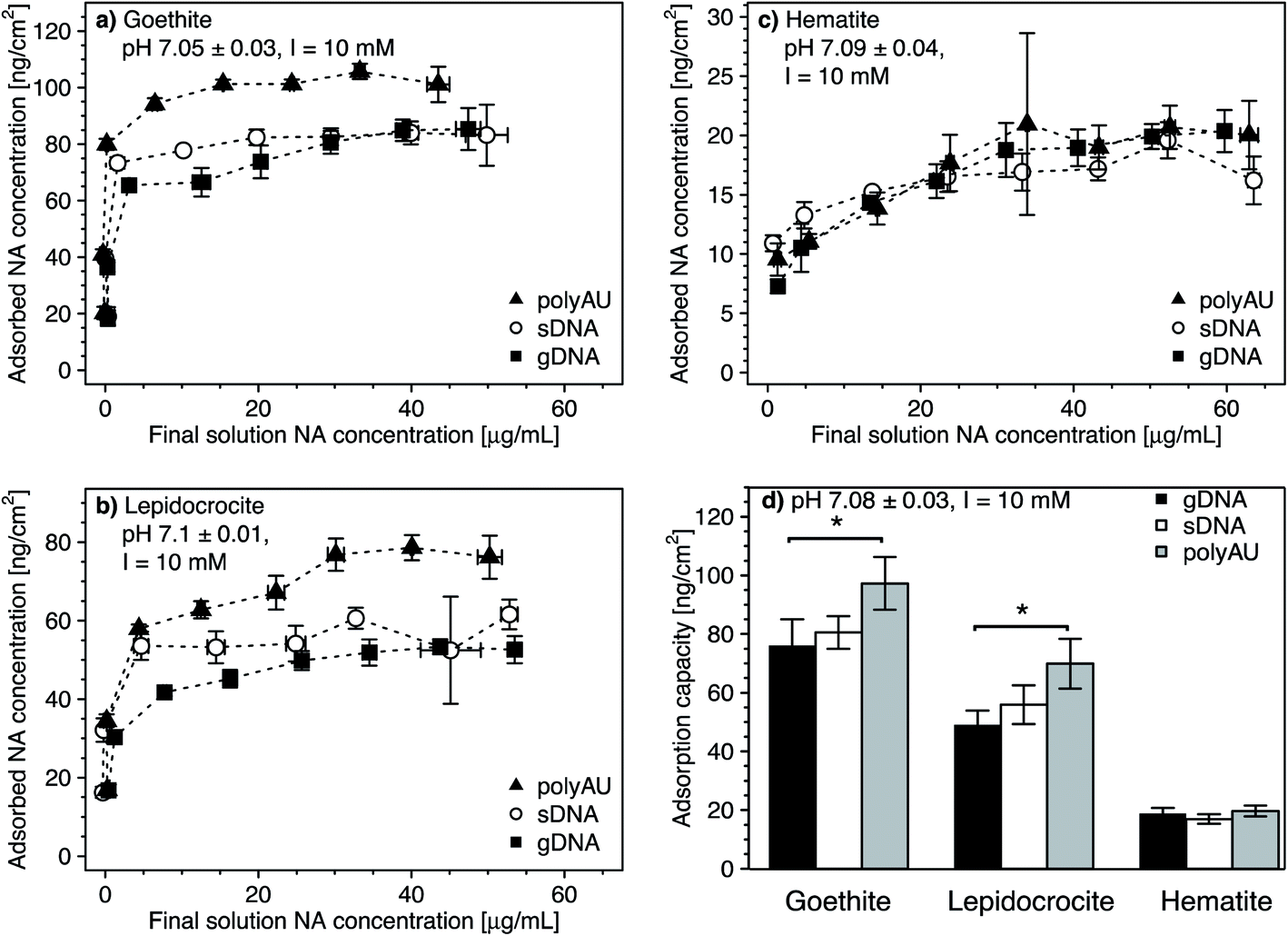 | ||
| Fig. 2 (a–c) Concentration-dependent adsorption of the three nucleic acids (NAs) (i.e., model dsRNA (polyAU), sheared genomic DNA (sDNA), and genomic DNA (gDNA)) to three iron oxides (a) goethite, (b) lepidocrocite, (c) hematite at pH 7 with 3 mM BisTris and 10 mM NaCl background electrolyte, determined by solution depletion of NAs in batch equilibration reactors. (d) Comparison of the maximum adsorbed concentrations (i.e., adsorption capacities) of the three NAs to the three iron oxides tested. We calculated the adsorption capacities by averaging the adsorbed concentrations for solution concentrations at which adsorbed concentrations plateaued. Comparisons between the adsorption capacity of different NAs to the same sorbent were performed using a one-way ANOVA (p < 0.05). Asterisks indicate significant differences between the adsorption capacities of the respective NAs on a given sorbent. Data points and error bars represent the mean and the standard deviation of triplicate reactors run in parallel. | ||
In the cases of goethite and lepidocrocite, adsorption at low initial NA solution concentrations (i.e., 5–20 μg mL−1 for goethite and 5–10 μL mL−1 for lepidocrocite) resulted in complete solution depletion of these NAs at the end of the equilibration time, with goethite exhibiting higher NA adsorption capacities than lepidocrocite. The finding that all added NA molecules adsorbed implies that NA adsorption sites on the iron oxide had not become saturated (Fig. 2a and b). At only slightly higher initial NA concentrations, the adsorbed NA concentrations transitioned into well-defined plateaus. We consider these plateaus to correspond to the adsorption capacities at which all adsorption sites were occupied and no additional NA molecules could adsorb. Complete NA solution depletion at low initial NA concentration and surface saturation at only slightly higher initial NA concentration imply that all three NAs had a high affinity for goethite and lepidocrocite surfaces. High affinity adsorption isotherms likely reflected both NA–iron oxide electrostatic attraction as well as inner–sphere complex formation of the NAs with surface hydroxyl groups on the two iron oxides at pH 7.
As compared to goethite and lepidocrocite, the NA adsorbed concentrations to hematite plateaued at lower values and at higher dissolved NA concentrations. Complete solution depletion of the added NAs was observed only at the lowest tested initial concentration of 5 μg NA mL−1 (Fig. 2c) and thus constrained to lower NA concentrations than observed for goethite and lepidocrocite. Moreover, increasing the initial NA concentrations above 5 μg NA mL−1 resulted in a more gradual increase in the adsorbed NA concentrations on hematite as compared to the other two iron oxides. For hematite, adsorbed NA concentrations only plateaued when initial NA concentrations were higher than 30 μg NA mL−1. As compared to goethite and lepidocrocite, the NAs thus had a lower affinity for the hematite surface which we ascribe to factors discussed in Section 3.1.
Consistent with the size-dependence of NA adsorption to goethite discussed above, the adsorption capacities of all NAs to goethite and lepidocrocite decreased with increasing size of the NA molecules (Fig. 2a and b). When comparing polyAU and gDNA adsorption on a given iron oxide, adsorption capacities of polyAU were significantly higher than for gDNA on both goethite and lepidocrocite (one-way ANOVA, p < 0.05) (Fig. 2d). Conversely, there were no significant differences in the adsorption capacities of the three NAs on hematite (one-way ANOVA, p > 0.05). The absence of size-dependence in NA adsorption to hematite as compared to the other iron oxides likely reflects the lower net surface charge (Section S5, ESI†) and density of surface hydroxyl groups on the hematite particles (Table 1). Furthermore, the absence of an effect of molecular size on NA adsorption may have also been linked to the smaller size of hematite as compared to goethite and lepidocrocite particles. In this case, however, the underlying cause remains unidentified.
3.3. pH-dependence of NA adsorption
We assessed the effect of solution pH on NA adsorption in two sets of experiments. In the first set, we studied NA adsorption to goethite at pH 5 and 9 over a range of initial NA concentrations (Fig. 3a–c, with pH 7 data replotted from Fig. 2), akin to NA concentration-dependent adsorption experiments at pH 7 discussed above. In the second set, we determined gDNA, sDNA, and polyAU adsorption to all three iron oxides (Fig. 3d–f) at one initial NA concentration of 50 μg NA mL−1 over a range of solution pH from pH 4.5–10. In both types of experiments, we expected decreasing NA adsorption with increasing solution pH as the latter leads to increasing deprotonation of surface hydroxyl groups and thus decreasing net positive surface charge on the iron oxide surfaces, thereby weakening NA-oxide electrostatic attraction. Also, inner–sphere complex formation between NAs and iron oxide surfaces through ligand exchange is less favorable at higher solution pH, reflecting that this reaction results in the release of hydroxyl anions from the surface.15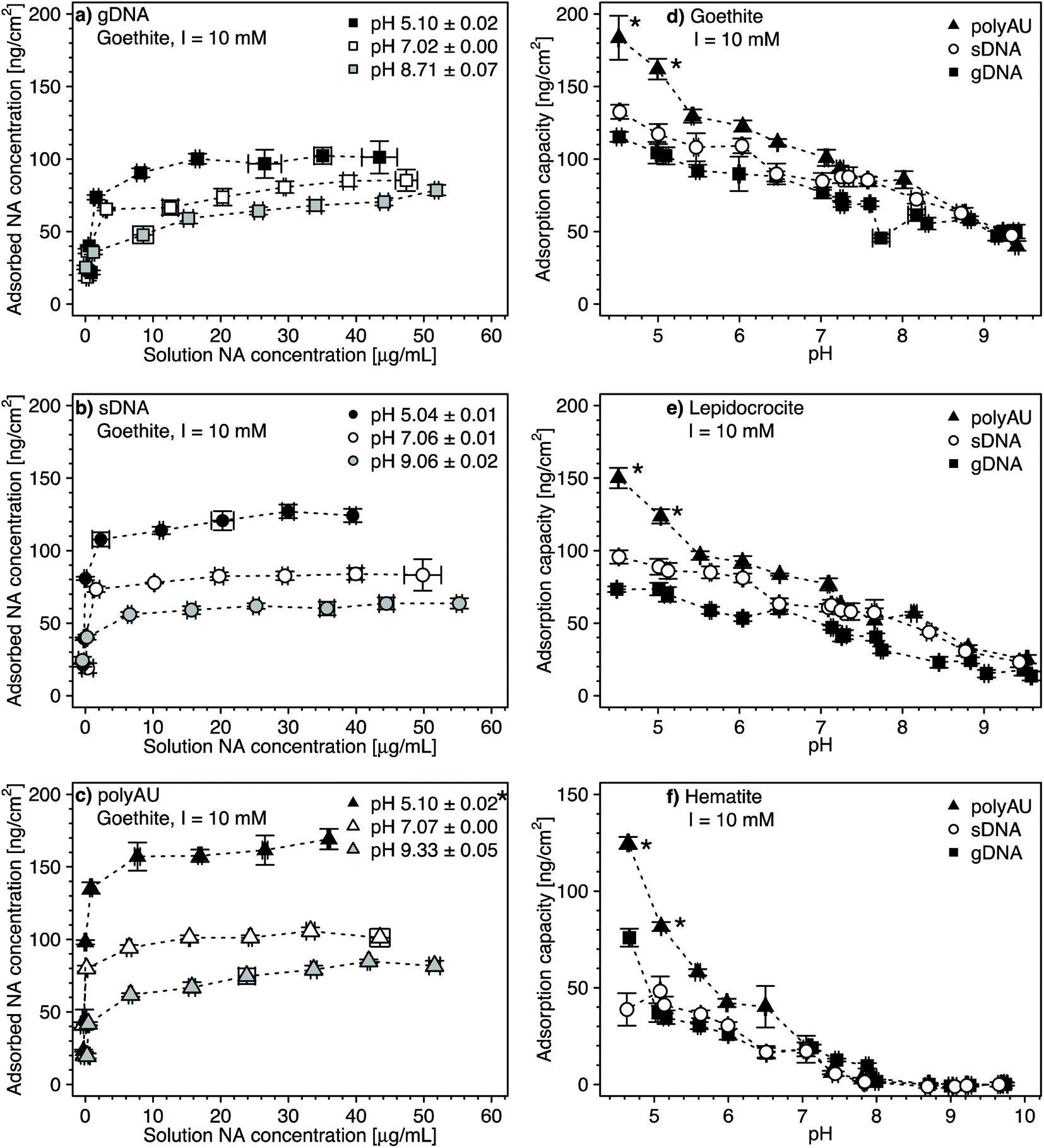 | ||
| Fig. 3 (a–c) Concentration-dependent adsorption of the three nucleic acids (NAs) (a) genomic DNA (gDNA); (b) sheared genomic DNA (sDNA); (c) model dsRNA (polyAU) to goethite at solution pH 5, 7, and 9 (with 10 mM NaCl background electrolyte) determined by solution depletion of NAs in batch equilibration reactors. The pH 7 NA adsorption data in panels a–c were replotted from Fig. 2a for comparison. (d–f) pH-dependent adsorption for polyAU, sDNA, and gDNA (all at an initial NA concentration of 50 μg NA mL−1) to three iron oxides (d) goethite, (e) lepidocrocite, and (f) hematite (with 10 mM NaCl background electrolyte) determined by solution depletion of NAs in batch equilibration reactors. The pH 7 data in panel f was replotted from Fig. 2c at an initial NA concentration of 50 μg NA mL−1. Data points and error bars represent the mean and the standard deviation of the results of triplicate adsorption experiments run in parallel. *At these low pH values, the absorbance of polyAU in iron oxide-free controls slightly increased over the equilibration time, suggesting partial polyAU denaturation. As a consequence, it is possible that the presented data points slightly underestimated true adsorbed polyAU concentrations in the setups containing iron oxides. | ||
In the first experimental set, we found that at initial NA concentrations ≤10 μg mL−1 all NA molecules added to the batch reactors adsorbed to goethite at both pH 5 and 9 (Fig. 3a–c), similar to the findings at pH 7 (Fig. 2a). With increasing initial NA solution concentration at pH 5 and 9, the adsorbed NA concentration increased. However, above a specific final solution concentration, the adsorbed NA concentration leveled off at well-defined maximum adsorbed concentrations, as previously described for pH 7. As argued above, these plateaus correspond to adsorption capacities of NA on the surfaces of the iron oxides. Consistent with our expectation, the adsorption capacities of gDNA, sDNA, and polyAU on goethite decreased in the order pH 5 > pH 7 > pH 9, presumably due to weakening of NA–goethite electrostatic attraction and inner–sphere complex formation becoming less favorable with increasing pH. We note that the three NAs still had a high affinity to goethite up to pH 9 and thus at pH at which both NAs and goethite surface were net negatively charged. This finding possibly indicates that adsorption at this pH was driven primarily by inner–sphere complex formation reactions rather than NA–goethite electrostatic interactions.
In the first set of experiments, adsorption at an initial NA concentration of 50 μg NA mL−1 resulted in saturation of the surfaces with NAs over the entire tested pH range from 5 to 9 (Fig. 3a–c). We therefore chose this initial NA concentration for the second set of experiments to assess changes in adsorption capacities with solution pH. We found decreasing adsorption capacities of the three NAs with increasing solution pH for all three iron oxides (Fig. 3d–f). There were no buffer-specific offsets in the extents to which NA adsorbed to the iron oxides when we changed the pH buffering species (i.e., between pH 5.5 and 6, pH 7 and 7.5, pH 8 and 8.6), indicating that none of the pH buffers interacted with the iron oxide surfaces in a manner that affected NA adsorption. More importantly, the pH trend further supports NA-oxide electrostatic attraction and inner–sphere complex formation as major drivers for NA adsorption. Electrostatic attraction is weakened as (protonated) hydroxyl groups on the iron oxide surface deprotonate with increasing pH. As argued above, it is likely that with increasing solution pH the relative proportion of inner–sphere complex formation to overall NA adsorption increased.
At low pH and thus strong NA-oxide electrostatic attraction, the NA adsorption capacities to all three oxides decreased in the order: polyAU > sDNA > gDNA, consistent with the size-dependence of NA adsorption discussed at pH 7 in section 3.1. The size-dependence of NA adsorption decreased with increasing pH. At a solution pH > PZC of the iron oxides, the adsorption capacities of the three tested NAs for a given iron oxide were comparable (Fig. 3d and e), presumably due to the larger contribution of NA inner-sphere complexation to total NA adsorption at these solution pH. It is conceivable that inner–sphere complex formation with a limited number of specific iron oxide surface sites at this high pH may have resulted in a smaller size dependence of NA adsorption than under lower pH conditions with NA–iron oxide electrostatic attraction. In the case of hematite, none of the tested NAs adsorbed at pH > 8 (Fig. 3f). We ascribe the absence of adsorption to hematite under these conditions to the net negative surface charge of hematite at these pH values (Section S5, ESI†), in combination with a low density of surface hydroxyl groups that may participate in inner–sphere complex formation.
3.4. Dependence of NA adsorption on solution ionic strength and composition
All experiments above were performed in 10 mM NaCl background electrolyte solutions. Here, we assessed potential effects of solution ionic strength (Fig. 4a–c) and ionic composition (i.e., in the presence of the divalent cation Mg2+ at a total ionic strength of 10 mM; Fig. 4d–f) on NA adsorption to goethite, lepidocrocite, and hematite. We anticipated two potentially opposing effects on NA adsorption from increasing solution ionic strength. On the one hand, from acidic to circumneutral pH, we expected that increases in the ionic strength weakens NA-oxide electrostatic attraction by solution charge screening, possibly leading to decreased adsorption. On the other hand, with increasing ionic strength, we expected NAs to adopt more compact conformations in the adsorbed states due to increasing charge screening of the intramolecular electrostatic repulsion between negatively charged phosphodiester groups in the NA backbone.77–81 More compact conformations and the resulting smaller footprint of individual NAs in adsorbed states at high ionic strength are expected to allow for a denser NA packing on the sorbent surface and thus increased adsorption capacities. We expected a similar increase in adsorption capacity in the presence of Mg2+ due to the formation of cation bridges between negatively charged phosphodiester groups in the NA backbone, resulting in a more compact NA conformation.64,82–84,105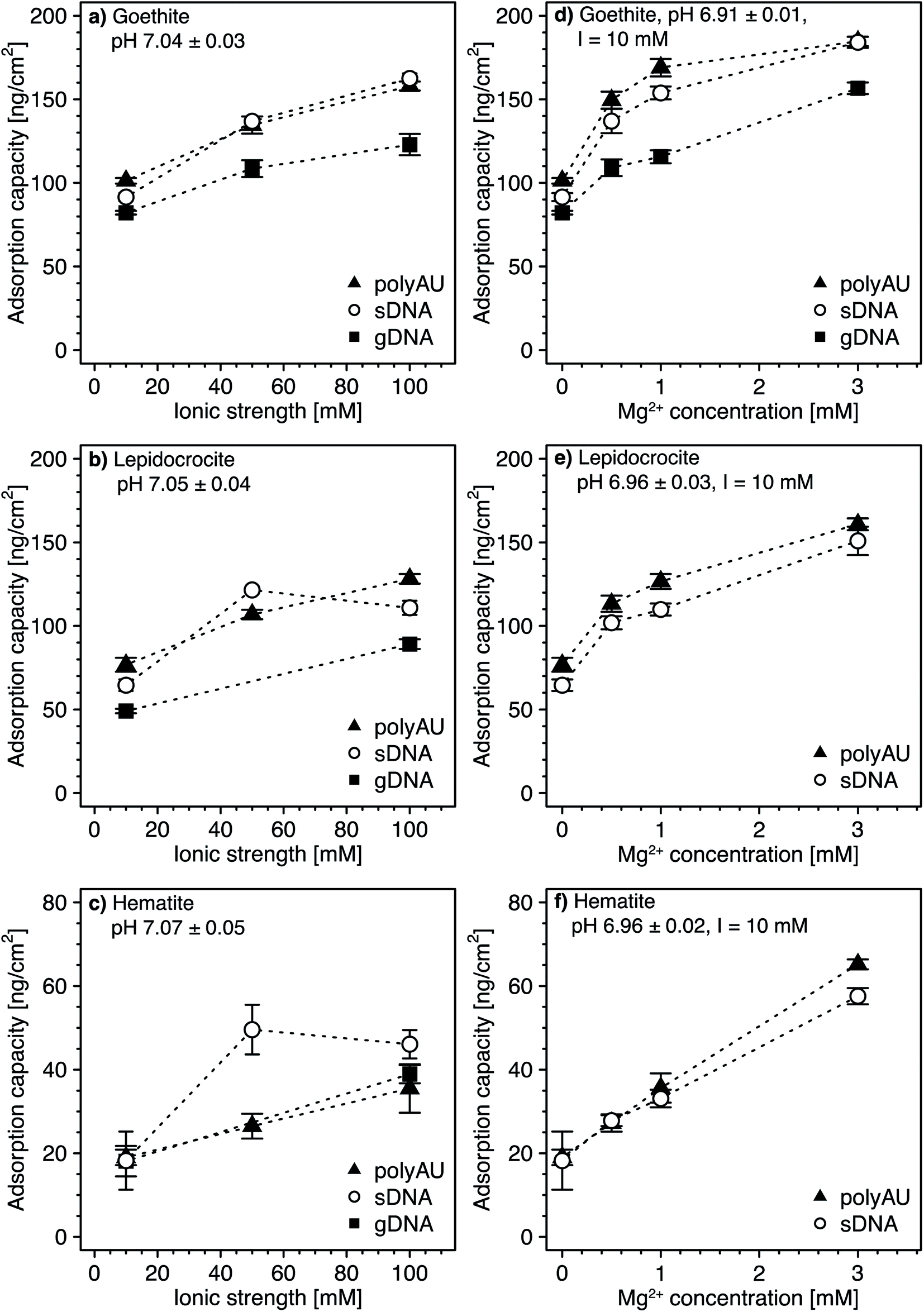 | ||
| Fig. 4 (a–c) Effect of solution ionic strength on adsorption of the model dsRNA (polyAU), sheared genomic DNA (sDNA), and genomic DNA (gDNA) to three iron oxides (goethite (a), lepidocrocite (b), and hematite (c)) at pH 7 (3 mM BisTris) determined by solution depletion in batch equilibration reactors. The adsorption capacity of gDNA to lepidocrocite (b) and hematite (c) was determined only at 10 and 100 mM NaCl. (d–f) Effect of increasing Mg2+ concentration (at a constant total ionic strength of 10 mM, adjusted with NaCl) on the adsorption of gDNA to only goethite (d), and of sDNA and polyAU to all three iron oxides goethite (d), lepidocrocite (e), and hematite (f) in pH 7 buffer (3 mM BisTris), determined by solution depletion in batch equilibration reactors. All experiments were conducted at an initial NA concentration of 50 μg NA mL−1. Data points and error bars represent the mean and the standard deviation of the results of triplicate adsorption experiments run in parallel. | ||
Increasing the ionic strength from 10 to 100 mM NaCl resulted in an increase in the adsorption capacities for all three NAs (i.e., approximately 1.6-fold for goethite (Fig. 4a), 1.7-fold for lepidocrocite (Fig. 4b), and two-fold for hematite (Fig. 4c)). This finding strongly suggests that more compact NA conformations at high ionic strength outweighed the weakening effect of higher ionic strength on NA–sorbent electrostatic attraction. Greaves and coworkers41 obtained similar results when adsorbing DNA to clay minerals and suggested that monovalent ions decrease the effective size of NAs due to screening of intramolecular electrostatic repulsion.
At a Mg2+ concentration of 3 mM, the NA adsorption capacities were approximately 1.9-fold higher for goethite (Fig. 4d), more than two-fold higher for lepidocrocite (Fig. 4e), and about three-fold higher for hematite (Fig. 4f) as compared to adsorption in the absence of dissolved Mg2+. Therefore, only very small concentrations of divalent Mg2+ were needed to increase NA adsorption as compared to the increase in adsorbed NA concentrations by increasing the ionic strength with monovalent Na+ from 10 to 100 mM. We ascribe the larger effect of Mg2+ than Na+ on NA adsorption to Mg2+ being more efficient than Na+ in leading to compact NA conformations because it allows for cation bridging between negatively charged phosphodiester groups in the backbone of the NAs. Furthermore, it is conceivable that Mg2+ additionally increased adsorption by forming cation bridges between negatively charged phosphodiester groups in the NA backbone and negatively charged surface groups of iron oxides (note that the surface of the iron oxides carried negative charges despite its positive net charge at pH 7, as shown by calculated speciation diagrams of hydroxyl groups on the surfaces of the three iron oxides in Section S5, ESI†). Our results are consistent with previous studies reporting increasing DNA adsorption with increasing divalent cation concentration to clay minerals42,45,47 and goethite.34
3.5. Competitive adsorption of NAs and phosphate to goethite
We assessed potential competitive suppression of NA adsorption to goethite by phosphate as a co-solute. In a first set of experiments, we studied the effect of increasing initial phosphate concentrations on NA adsorption at a constant initial NA concentration, with NA and phosphate being added simultaneously to the goethite suspensions (Fig. 5a). In a second set of experiments, we assessed whether the addition order of NA and phosphate affected NA adsorption to goethite (Fig. 5b). We expected competitive adsorption of phosphate and NAs and hence decreasing NA adsorption with increasing phosphate concentration, as previously reported for DNA–phosphate co-adsorption to goethite,35 clay minerals,43 and soils.47 Competition is possible by two pathways. First, at circumneutral pH, phosphate is expected to form inner-sphere complexes with goethite surface hydroxyl groups,106,107 thereby inverting the net surface charge of goethite from positive to negative.107–110 As a result, NAs experience electrostatic repulsion from the negatively charged phosphate-coated goethite surface. Furthermore, the inner–sphere complex formation of phosphate is expected to decrease NA adsorption because it lowers the number of hydroxyl moieties that may also form inner–sphere complexes with the phosphodiester groups in the NA backbone. Second, in addition to phosphate suppressing NAs adsorption through inner–sphere complex formation, phosphate may also electrostatically interact with positively charged hydroxyl groups on the goethite surface, thereby competing with the NAs for remaining positively charged adsorption sites.111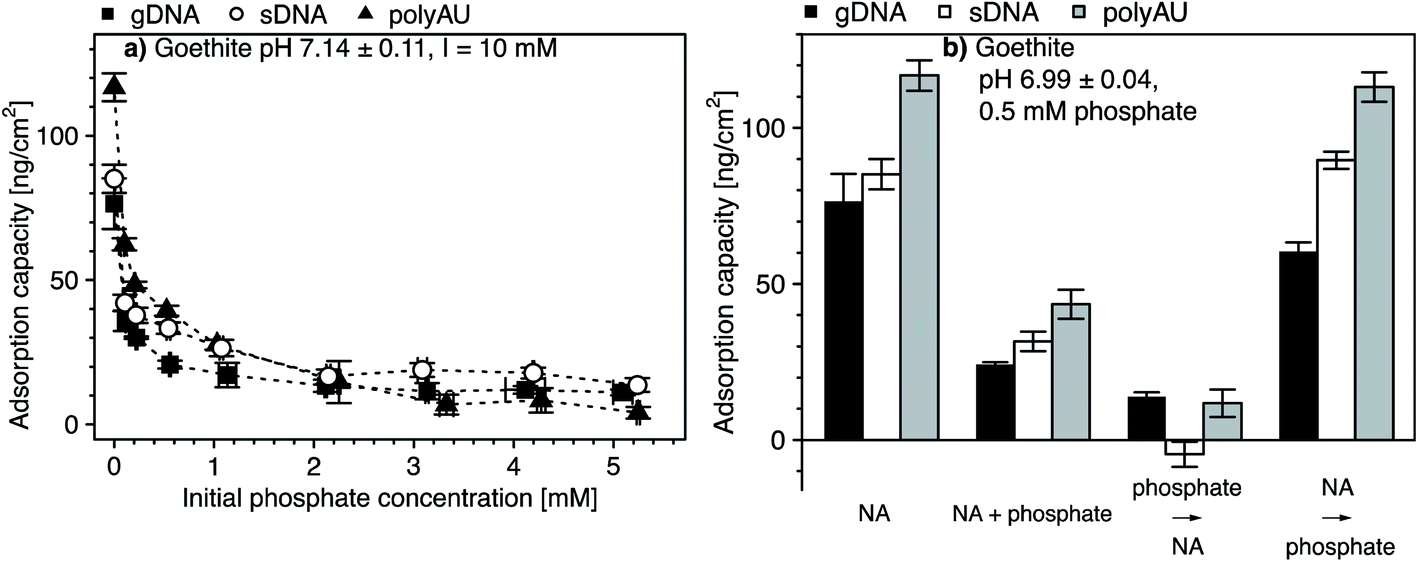 | ||
| Fig. 5 (a) Changes in the adsorption capacities of nucleic acids (NAs) to goethite in the presence of increasing initial phosphate concentrations determined by solution depletion of NAs in batch equilibration reactors. The NAs are the model dsRNA (polyAU), sheared genomic DNA (sDNA), and genomic DNA (gDNA). (b) The effect of addition order of NAs (i.e., polyAU, sDNA and gDNA) and phosphate (at an initial concentration of 0.5 mM) on NA adsorption to goethite determined by solution depletion of NAs in batch equilibration reactors. Where appropriate, the first sorbate was added and equilibrated with the goethite suspension for 2 hours, followed by addition of the second sorbate and equilibration for another 2 hours. Otherwise, both sorbates were added simultaneously and equilibrated for 4 hours. All experiments were conducted at an initial NA concentration of 50 μg NA mL−1, at pH 7 with 3 mM BisTris and 10 mM NaCl as background electrolyte. Data points and error bars represent the mean and the standard deviation of the results of triplicate adsorption experiments run in parallel. In panel (b), the plus (+) indicates simultaneous addition of NA and phosphate whereas the arrow (→) indicates the sequential addition order of NA and phosphate. | ||
In phosphate single-solute adsorption experiments to goethite, phosphate was completely depleted from solution at initial phosphate concentrations below 0.05 mM, demonstrating high affinity of phosphate to goethite surfaces (Section S10, ESI†). Increasing the initial phosphate concentration to 0.1 mM resulted in increased phosphate adsorption which, at higher solution concentrations, leveled off at an adsorbed concentration of approximately 2.7 μmol phosphate m−2. This value is in good agreement with reported phosphate adsorption capacities on goethite.106,111–115 At this concentration, we expect that phosphate occupied all adsorption sites on the goethite surface.
In NA–phosphate co-solute adsorption experiments, the adsorbed concentration of the three NAs decreased with increasing initial phosphate concentration (Fig. 5a). For instance, an initial phosphate concentration of 0.1 mM resulted in a two-fold decrease in the final adsorbed NA concentration as compared to NA adsorbed concentrations in the absence of phosphate. At initial phosphate concentrations above 2 mM, the adsorbed NA concentrations leveled off at low but non-zero values, consistent with the results of a recent study.29 The non-zero adsorbed NA concentrations in the presence of high phosphate concentrations suggests that phosphate competitively displaced NAs only from a fraction of the surface adsorption sites and, thus, that a subset of the total adsorption sites were available only for NA adsorption even at high phosphate concentrations or that NAs effectively outcompeted phosphate from adsorption to these sites. A similar observation was previously reported for DNA adsorption to both goethite35 and to clay minerals.43
The addition order of phosphate and NA largely affected the extent of NA adsorption to goethite surfaces (Fig. 5b). We repeated co-solute adsorption experiments in which we simultaneously added NA (50 μg NA mL−1) and phosphate (0.5 mM). In this case, the adsorption capacities of the three NAs were approximately three-fold smaller than their capacities determined in the respective single-solute NA system (Fig. 5a). When we first added phosphate and subsequently the NAs, the competitive suppression in the adsorption of all three NAs to goethite was even more pronounced: the NA adsorption capacity was more than five-fold smaller than the capacities in the single-solute NA system. This finding is consistent with phosphate adsorbing through inner–sphere complex formation to goethite surfaces,106,107 resulting in its surface charge reversal as well as in blocking potential hydroxyl sites for inner–sphere complex formation with NAs. For agricultural soils, this finding implies that dsRNA may be significantly more mobile and bioavailable if phosphate is adsorbed to iron oxide surfaces.
By comparison, when we first added NAs (followed by NA adsorptive equilibration) and only subsequently phosphate, the adsorbed NA concentration was little affected as compared to the single-solute NA adsorption case (i.e., the decrease in NA adsorption capacity was less than 20% of the capacity in the single-solute NA systems), consistent with the results of a recent study.29 We ascribe the lack of apparent phosphate competition on NA adsorption by kinetically slow displacement of adsorbed NA from the goethite surface by phosphate, likely because phosphate would need to simultaneously break all NA–goethite interactions and inner-sphere complexes for NA desorption to occur.116 It is conceivable that the phosphate equilibration time of 2 hours was too short to result in kinetically slow displacement of pre-adsorbed NAs by phosphate.
4. Conclusion
This work systematically assessed similarities in the adsorption characteristics of dsRNA and DNA to iron oxide minerals abundant in soils in the context of environmental risk assessment of dsRNA PIPs. We showed that at slightly acidic to circumneutral pH, typical for most agricultural soils, both dsRNA and DNA adsorb with a high affinity to the iron oxides goethite, lepidocrocite, and hematite, suggesting that adsorption to these minerals will lower transport, decrease bioavailability but possibly increase stability of dsRNA in soils.For both DNA and dsRNA, adsorption capacities to iron oxides decreased with increasing solution pH, consistent with both nucleic acids adsorbing to iron oxide surfaces through electrostatic attraction and inner–sphere complexes formation. Yet, NAs even adsorbed to goethite and lepidocrocite under alkaline solution condition at which electrostatics cease to be attractive, consistent with a greater relative proportion of inner–sphere complex formation to overall NA adsorption at the high pH. While not explicitly discussed, it is conceivable that other attractive forces contributed to NA adsorption particularly at high pH, such as hydrogen bonding between the NAs and the iron oxide surfaces, despite competition from water molecules. Furthermore, we showed increasing dsRNA and DNA adsorption with increasing ionic strength and in the presence of divalent cations in solution, consistent with these changes in solution chemistry leading to more compact conformations of these NAs when adsorbed on the sorbent surface (i.e., have a smaller molecular footprint on the surface and hence a higher number of adsorbed molecules at capacity). The strong enhancement in NA adsorption in the presence of divalent cations strongly advocates that buffers to extract dsRNA from soils contain complexing agents for divalent cations. Both NAs adsorbed with high adsorption capacities to iron oxides over a wide range of pH and ionic strength, suggesting that in agricultural soils planted with RNAi crops with low dsRNA concentrations, the dsRNAs will readily adsorb. Finally, we demonstrated that phosphate largely suppressed the adsorption of dsRNA and DNA to the iron oxides, consistent with competition between these adsorbates.
While shown herein only for phosphate, we expect competitive suppression of dsRNA adsorption also from other negatively charged (bio-macro)molecules, including the so called humic- and fulvic acid components of the soil DOM pool. Any competition leads to decreased dsRNA adsorption in soils, and therefore, is expected to result in enhanced dsRNA mobility in soils, likely higher bioavailability and potentially lower overall stability (by virtue of enhanced enzymatic hydrolysis rates in soil pore waters than in adsorbed states). In this context, future research should investigate the effect of plant-derived biomolecules co-released from RNAi crops on dsRNA adsorption to mineral surfaces.
Our work strongly supports that the adsorption characteristics of dsRNA and DNA are governed by their negatively charged phosphodiester backbone, while base composition plays a minor role. We therefore expect that the results obtained herein with our model dsRNA (i.e., polyAU) can be applied to dsRNA molecules with other base compositions. Direct experimental proof, however, remains to be presented. For fate assessment of dsRNA PIPs, our results strongly suggest that available information on DNA adsorption to mineral surfaces provides valuable guidance for assessing the adsorption behavior of dsRNA to these surfaces and ultimately for predicting the fate of dsRNA molecules in agricultural soils. While similar adsorption characteristics were demonstrated herein in model lab systems with iron oxides, we postulate that the structural similarities of dsRNA and DNA will lead to similar adsorption characteristics also to other sorbents. This study highlights the need to consider dsRNA adsorption to soil particles surfaces in any assessment of the fate of dsRNA molecules in soils. Future studies reporting decreases in dissolved (or extractable) dsRNA molecules in soils (i.e., dsRNA dissipation) should delineate contributions from adsorption and from actual dsRNA breakdown. Future research should investigate dsRNA stability in soils, and more specifically, the effect of dsRNA adsorption to soil particle surfaces on the stability of dsRNA molecules in soils. Based on past work with DNA,18,20,22 it is conceivable that adsorption of dsRNA leads to its stabilization. In this context, our study may aid in the design of buffers to extract dsRNA molecules from soil particle surfaces, a key requirement to monitor dsRNA concentration dynamics in soils planted with RNAi crops or sprayed with dsRNA insecticides.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
K. S and M. S. acknowledge support from an ETH Zürich Research Grant (ETH-14 17-1). K.M.P is supported by the Biotechnology Risk Assessment Grand Program Award 2017-33522-26998 from the U.S. Department of Agriculture. The authors acknowledge assistance from Ralf Kägi in imaging the iron oxides on the electron microscope, Björn Studer in helping with the ion chromatography, Vivian Cheslack in helping with the batch adsorption experiments and Numa Pfenninger for iron oxide surface area analysis. We thank the Genetic Diversity Centre (GDC) at ETH Zurich for access to and help with the QSonica, the QuBit, and the Tapestation.References
- M. Bramlett, G. Plaetinck and P. Maienfisch, RNA-based biocontrols—a new paradigm in crop protection, Engineering, 2020, 6, 522–527 CrossRef CAS.
- J. A. Baum, T. Bogaert, W. Clinton, G. R. Heck, P. Feldmann, O. Ilagan, S. Johnson, G. Plaetinck, T. Munyikwa, M. Pleau, T. Vaughn and J. Roberts, Control of coleopteran insect pests through RNA interference, Nat. Biotechnol., 2007, 25, 1322–1326 CrossRef CAS PubMed.
- Y.-B. Mao, W.-J. Cai, J.-W. Wang, G.-J. Hong, X.-Y. Tao, L.-J. Wang, Y.-P. Huang and X.-Y. Chen, Silencing a cotton bollworm P450 monooxygenase gene by plant-mediated RNAi impairs larval tolerance of gossypol, Nat. Biotechnol., 2007, 25, 1307–1313 CrossRef CAS PubMed.
- L. Shaffer, Inner Workings: RNA-based pesticides aim to get around resistance problems, Proc. Natl. Acad. Sci. U. S. A., 2020, 15, 202024033 Search PubMed.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- R. Bolognesi, P. Ramaseshadri, J. Anderson, P. Bachman, W. Clinton, R. Flannagan, O. Ilagan, C. Lawrence, S. Levine, W. Moar, G. Mueller, J. Tan, J. Uffman, E. Wiggins, G. Heck and G. Segers, Characterizing the mechanism of action of double-stranded RNA activity against western corn rootworm (Diabrotica virgifera virgifera LeConte), PLoS One, 2012, 7, e47534 CrossRef CAS PubMed.
- J. Zhang, S. A. Khan, D. G. Heckel and R. Bock, Next-Generation Insect-Resistant Plants: RNAi-Mediated Crop Protection, Trends Biotechnol., 2017, 35, 871–882 CrossRef CAS PubMed.
- G. P. Head, M. W. Carroll, S. P. Evans, D. M. Rule, A. R. Willse, T. L. Clark, N. P. Storer, R. D. Flannagan, L. W. Samuel and L. J. Meinke, Evaluation of SmartStax and SmartStaxPRO maize against western corn rootworm and northern corn rootworm: efficacy and resistance management, Pest Manage. Sci., 2017, 73, 1883–1899 CrossRef CAS PubMed.
- P. Ramaseshadri, G. Segers, R. Flannagan, E. Wiggins, W. Clinton, O. Ilagan, B. McNulty, T. Clark and R. Bolognesi, Physiological and Cellular Responses Caused by RNAi- Mediated Suppression of Snf7 Orthologue in Western Corn Rootworm (Diabrotica virgifera virgifera) Larvae, PLoS One, 2013, 8, e54270 CrossRef CAS PubMed.
- K. M. Parker and M. Sander, Environmental Fate of Insecticidal Plant-Incorporated Protectants from Genetically Modified Crops: Knowledge Gaps and Research Opportunities, Environ. Sci. Technol., 2017, 51, 12049–12057 CrossRef CAS PubMed.
- M. P. Schmidt and C. E. Martinez, Ironing Out Genes in the Environment: An Experimental Study of the DNA-Goethite Interface, Langmuir, 2017, 33, 8525–8532 CrossRef CAS PubMed.
- S. J. Parikh, F. N. D. Mukome and X. Zhang, ATR–FTIR spectroscopic evidence for biomolecular phosphorus and carboxyl groups facilitating bacterial adhesion to iron oxides, Colloids Surf., B, 2014, 119, 38–46 CrossRef CAS PubMed.
- S. J. Parikh and J. Chorover, ATR-FTIR Spectroscopy Reveals Bond Formation During Bacterial Adhesion to Iron Oxide, Langmuir, 2006, 22, 8492–8500 CrossRef CAS PubMed.
- A. Omoike and J. Chorover, Spectroscopic study of extracellular polymeric substances from Bacillus subtilis: aqueous chemistry and adsorption effects, Environ. Sci. Technol., 2004, 5, 1219–1230 CAS.
- A. Omoike and J. Chorover, Adsorption to goethite of extracellular polymeric substances from Bacillus subtilis, Geochim. Cosmochim. Acta, 2006, 70, 827–838 CrossRef CAS.
- Y. Cao, X. Wei, P. Cai, Q. Huang, X. Rong and W. Liang, Preferential adsorption of extracellular polymeric substances from bacteria on clay minerals and iron oxide, Colloids Surf., B, 2011, 83, 122–127 CrossRef CAS PubMed.
- L. Fang, Y. Cao, Q. Huang, S. L. Walker and P. Cai, Reactions between bacterial exopolymers and goethite: A combined macroscopic and spectroscopic investigation, Water Res., 2012, 46, 5613–5620 CrossRef CAS PubMed.
- G. Stotzky, Persistence and biological activity in soil of insecticidal proteins from Bacillus thuringiensis and of bacterial DNA bound on clays and humic acids, J. Environ. Qual., 2000, 29, 691–705 CrossRef CAS.
- K. M. Parker, V. B. Borrero, D. M. van Leeuwen, M. A. Lever, B. Mateescu and M. Sander, Environmental Fate of RNA Interference Pesticides: Adsorption and Degradation of Double-Stranded RNA Molecules in Agricultural Soils, Environ. Sci. Technol., 2019, 53, 3027–3036 CrossRef CAS PubMed.
- P. Cai, Q. Y. Huang and X. W. Zhang, Interactions of DNA with clay minerals and soil colloidal particles and protection against degradation by DNase, Environ. Sci. Technol., 2006, 40, 2971–2976 CrossRef CAS PubMed.
- F. Scappini, F. Casadei, R. Zamboni, M. Franchi, E. Gallori and S. Monti, Protective effect of clay minerals on adsorbed nucleic acid against UV radiation: possible role in the origin of life, Int. J. Astrobiol., 2004, 3, 17–19 CrossRef CAS.
- N. Mitter, E. A. Worrall, K. E. Robinson, P. Li, R. G. Jain, C. Taochy, S. J. Fletcher, B. J. Carroll, G. Q. M. Lu and Z. P. Xu, Clay nanosheets for topical delivery of RNAi for sustained protection against plant viruses, Nat. Plants, 2017, 3, 16207 CrossRef CAS PubMed.
- U.S. Environmental Protection Agency, A Set of Scientific Issues Being Considered by the Environmental Protection Agency Regarding: RNAi Technology: Program Formulation for Human Health and Ecological Risk Assessment, Arlington, Virginia, 2014, https://www.epa.gov/sites/production/files/2015-06/documents/012814minutes.pdf, accessed 22 December 2020 Search PubMed.
- S. Dubelman, J. Fischer, F. Zapata, K. Huizinga, C. Jiang, J. Uffman, S. Levine and D. Carson, Environmental fate of double-stranded RNA in agricultural soils, PLoS One, 2014, 9, e93155 CrossRef PubMed.
- J. R. Fischer, F. Zapata, S. Dubelman, G. M. Mueller, J. P. Uffman, C. Jiang, P. D. Jensen and S. L. Levine, Aquatic fate of a double-stranded RNA in a sediment-water system following an over-water application, Environ. Toxicol. Chem., 2017, 36, 727–734 CrossRef CAS PubMed.
- V. C. Albright, C. R. Wong, R. L. Hellmich and J. R. Coats, Dissipation of double-stranded RNA in aquatic microcosms, Environ. Toxicol. Chem., 2017, 36, 1249–1253 CrossRef CAS PubMed.
- M. E. S. Joaquim, G. G. Belchior, M. O. de Mello Alves José, F. Zapata, C. Jiang, J. Fischer and G. U. Berger, Dissipation of DvSnf7 double-stranded RNA in brazilian soils, Agric. Environ. Lett., 2019, 4, 1–4 CAS.
- J. R. Fischer, G. R. MacQuarrie, M. Malven, Z. Song and G. Rogan, Dissipation of DvSnf7 RNA from late-season maize tissue in aquatic microcosms, Environ. Toxicol. Chem., 2020, 1–9 CAS.
- K. Zhang, J. Wei, K. E. H. Hartz, M. J. Lydy, T. S. Moon, M. Sander and K. M. Parker, Analysis of RNA Interference (RNAi) Biopesticides: Double-Stranded RNA (dsRNA) Extraction from Agricultural Soils and Quantification by RT-qPCR, Environ. Sci. Technol., 2020, 54, 4893–4902 CrossRef CAS PubMed.
- S. Blum, M. G. Lorenz and W. Wackernagel, Mechanism of retarded DNA degradation and prokaryotic origin of DNases in nonsterile soils, System, Appl. Microbiol., 1997, 20, 513–521 CAS.
- C. M. Gardner and C. K. Gunsch, Adsorption capacity of multiple DNA sources to clay minerals and environmental soil matrices less than previously estimated, Chemosphere, 2017, 175, 45–51 CrossRef CAS PubMed.
- A. Ogram, M. L. Mathot, J. B. Harsh, J. Boyle and C. A. Pettigrew, Effects of DNA Polymer Length on Its Adsorption to Soils, Appl. Environ. Microbiol., 1994, 60, 393–396 CrossRef CAS PubMed.
- A. Ogram, G. S. Sayler, D. Gustin and R. J. Lewis, DNA adsorption to soils and sediments, Environ. Sci. Technol., 1988, 22, 982–984 CrossRef CAS PubMed.
- P. Cai, Q. Huang, X. Zhang and H. Chen, Adsorption of DNA on clay minerals and various colloidal particles from an Alfisol, Soil Biol. Biochem., 2006, 38, 471–476 CrossRef CAS.
- P. Cai, Q. Huang, J. Zhu, D. Jiang, X. Zhou, X. Rong and W. Liang, Effects of low-molecular-weight organic ligands and phosphate on DNA adsorption by soil colloids and minerals, Colloids Surf., B, 2007, 54, 53–59 CrossRef CAS PubMed.
- P. Cai, Q. Y. Huang and X. W. Zhang, Microcalorimetric studies of the effects of MgCl2 concentrations and pH on the adsorption of DNA on montmorillonite, kaolinite and goethite, Appl. Clay Sci., 2006, 32, 147–152 CrossRef CAS.
- M. Khanna and G. Stotzky, Transformation of Bacillus subtilis by DNA bound on montmorillonite and effect of DNase on the transforming ability of bound DNA, Appl. Environ. Microbiol., 1992, 58, 1930–1939 CrossRef CAS PubMed.
- E. Paget, L. J. Monrozier and P. Simonet, Adsorption of DNA on clay minerals: protection against DNaseI and influence on gene transfer, FEMS Microbiol. Lett., 1992, 97, 31–39 CrossRef CAS.
- C. Crecchio, P. Ruggiero, M. Curci, C. Colombo, G. Palumbo and G. Stotzky, Binding of DNA from Bacillus subtilis on montmorillonite–humic acids–aluminum or iron hydroxypolymers, Soil Sci. Soc. Am. J., 2005, 69, 834–841 CrossRef CAS.
- M. Franchi, E. Bramanti, L. M. Bonzi, P. L. Orioli, C. Vettori and E. Gallori, Clay-nucleic acid complexes: characteristics and implications for the preservation of genetic material in primeval habitats, Origins Life Evol. Biospheres, 1999, 29, 297–315 CrossRef CAS PubMed.
- M. P. Greaves and M. J. Wilson, The adsorption of nucleic acids by montmorillonite, Soil Biol. Biochem., 1969, 1, 317–323 CrossRef CAS.
- F. Poly, C. Chenu, P. Simonet, J. Rouiller and L. J. Monrozier, Differences between linear chromosomal and supercoiled plasmid DNA in their mechanisms and extent of adsorption on clay minerals, Langmuir, 2000, 16, 1233–1238 CrossRef CAS.
- G. Pietramellara, M. Franchi, E. Gallori and P. Nannipieri, Effect of molecular characteristics of DNA on its adsorption and binding on homoionic montmorillonite and kaolinite, Biol. Fertil. Soils, 2001, 33, 402–409 CrossRef CAS.
- C. A. Goring and W. V. Bartholomew, Adsorption of mononucleotides, nucleic acids, and nucleoproteins by clays, Soil Sci., 1952, 74, 149–164 CrossRef CAS.
- G. W. Beall, D. S. Sowersby, R. D. Roberts, M. H. Robson and L. K. Lewis, Analysis of oligonucleotide DNA binding and sedimentation properties of montmorillonite clay using ultraviolet light spectroscopy, Biomacromolecules, 2009, 10, 105–112 CrossRef CAS PubMed.
- K. Saeki, M. Sakai and S.-I. Wada, DNA adsorption on synthetic and natural allophanes, Appl. Clay Sci., 2010, 50, 493–497 CrossRef CAS.
- K. Saeki, T. Kunito and M. Sakai, Effects of pH, ionic strength, and solutes on DNA adsorption by andosols, Biol. Fertil. Soils, 2010, 46, 531–535 CrossRef CAS.
- X. Chen, Q. Huang and W. Chen, Adsorption of DNA by bacteria and their composites with minerals, Geomicrobiol. J., 2016, 33, 822–831 CrossRef CAS.
- T. H. Nguyen and M. Elimelech, Adsorption of Plasmid DNA to a Natural Organic Matter-Coated Silica Surface: Kinetics, Conformation, and Reversibility, Langmuir, 2007, 23, 3273–3279 CrossRef CAS PubMed.
- C. Crecchio and G. Stotzky, Binding of DNA on humic acids: Effect on transformation of Bacillus subtilis and resistance to DNase, Soil Biol. Biochem., 1998, 30, 1061–1067 CrossRef CAS.
- M. G. Lorenz and W. Wackernagel, Adsorption of DNA to sand and variable degradation rates of adsorbed DNA, Appl. Environ. Microbiol., 1987, 53, 2948–2952 CrossRef CAS PubMed.
- B. Chamier, M. G. Lorenz and W. Wackernagel, Natural transformation of Acinetobacter calcoaceticus by plasmid DNA adsorbed on sand and groundwater aquifer material, Appl. Environ. Microbiol., 1993, 59, 1662–1667 CrossRef CAS PubMed.
- G. Romanowski, M. G. Lorenz and W. Wackernagel, Adsorption of plasmid DNA to mineral surfaces and protection against DNase I, Appl. Environ. Microbiol., 1991, 57, 1057–1061 CrossRef CAS PubMed.
- T. H. Nguyen, K. L. Chen and M. Elimelech, Adsorption Kinetics and Reversibility of Linear Plasmid DNA on Silica Surfaces: Influence of Alkaline Earth and Transition Metal Ions, Biomacromolecules, 2010, 11, 1225–1230 CrossRef CAS PubMed.
- P. E. Vandeventer, J. S. Lin, T. J. Zwang, A. Nadim, M. S. Johal and A. Niemz, Multiphasic DNA Adsorption to Silica Surfaces under Varying Buffer, pH, and Ionic Strength Conditions, Environ. Sci. Technol., 2012, 116, 5661–5670 CAS.
- P. E. Vandeventer, J. Mejia, A. Nadim, M. S. Johal and A. Niemz, DNA Adsorption to and Elution from Silica Surfaces: Influence of Amino Acid Buffers, Environ. Sci. Technol., 2013, 117, 10742–10749 CAS.
- K. A. Melzak, C. S. Sherwood, R. F. B. Turner and C. A. Haynes, Driving Forces for DNA Adsorption to Silica in Perchlorate Solutions, J. Colloid Interface Sci., 1996, 181, 635–644 CrossRef CAS.
- H. J. I. Cleaves, E. Crapster-Pregont, C. M. Jonsson, C. L. Jonsson, D. A. Sverjensky and R. A. Hazen, The adsorption of short single-stranded DNA oligomers to mineral surfaces, Chemosphere, 2011, 83, 1560–1567 CrossRef PubMed.
- M. Khanna, M. Yoder, L. Calamai and G. Stotzky, X-ray diffractometry and electron microscopy of DNA from Bacillus subtilis bound on clay minerals, Sci. Soils, 1998, 3, 1–10 CrossRef.
- T. H. Nguyen and M. Elimelech, Plasmid DNA Adsorption on Silica: Kinetics and Conformational Changes in Monovalent and Divalent Salts, Biomacromolecules, 2006, 8, 24–32 CrossRef PubMed.
- X. Sheng, C. Qin, B. Yang, X. Hu, C. Liu, M. G. Waigi, X. Li and W. Ling, Metal cation saturation on montmorillonites facilitates the adsorption of DNA via cation bridging, Chemosphere, 2019, 235, 670–678 CrossRef CAS.
- A. Omoike, J. Chorover, A. Kideok D Kwon and J. D. Kubicki, Adhesion of bacterial exopolymers to α-FeOOH: inner-sphere complexation of phosphodiester groups, Environ. Sci. Technol., 2004, 20, 11108–11114 CAS.
- M. D. Frank-Kamenetskiĭ, V. V. Anshelevich and A. V. Lukashin, Polyelectrolyte model of DNA, Phys.-Usp., 1987, 30, 317–330 CrossRef.
- F. T. Hesselink, in Adsorption from Solution at the Solid/Liquid Interface, ed. G. D. Parfitt and C. H. Rochester, London, 1983, pp. 377–412 Search PubMed.
- F. T. Hesselink, On the adsorption of polyelectrolyte macromolecules on a flat interface. An approximate theory for low potentials, J. Electroanal. Chem. Interfacial Electrochem., 1972, 37, 317–325 CrossRef CAS.
- M. Kawaguchi and A. Takahashi, Polymer adsorption at solid-liquid interfaces, Adv. Colloid Interface Sci., 1992, 37, 219–317 CrossRef CAS.
- V. Shubin and P. Linse, Effect of Electrolytes on Adsorption of Cationic Polyacrylamide on Silica: Ellipsometric Study and Theoretical Modeling, Environ. Sci. Technol., 1995, 99, 1285–1291 CAS.
- J. de Reese and J. Plank, Adsorption of polyelectrolytes on calcium carbonate – which thermodynamic parameters are driving this process?, J. Am. Ceram. Soc., 2011, 94, 3515–3522 CrossRef CAS.
- T. G. M. Van De Ven, Kinetic aspects of polymer and polyelectrolyte adsorption on surfaces, Adv. Colloid Interface Sci., 1994, 48, 121–140 CrossRef CAS.
- H. G. M. Van de Steeg, M. A. Cohen Stuart, A. De Keizer and B. H. Bijsterbosch, Polyelectrolyte adsorption: a subtle balance of forces, Langmuir, 1992, 8, 2538–2546 CrossRef CAS.
- M. A. Cohen Stuart, G. J. Fleer, J. Lyklema, W. Norde and J. M. Scheutjens, Adsorption of ions, polyelectrolytes and proteins, Adv. Colloid Interface Sci., 1991, 34, 477–535 CrossRef.
- A. Dobrynin and M. Rubinstein, Theory of polyelectrolytes in solutions and at surfaces, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS.
- A. Takahashi and M. Kawaguchi, in Behavior of Macromolecules, Springer, Berlin, Heidelberg, Berlin/Heidelberg, 1982, vol. 46, pp. 1–65 Search PubMed.
- E. Jenckel and B. Rumbach, Über die Adsorption von hochmolekularen Stoffen aus der Lösung, Z. Elektrochem. Angew. Phys. Chem., 1951, 55, 612–618 CAS.
- F. T. Hesselink, On the theory of polyelectrolyte adsorption: The effect on adsorption behavior of the electrostatic contribution to the adsorption free energy, J. Colloid Interface Sci., 1977, 60, 448–466 CrossRef CAS.
- J. Lipfert, S. Doniach, R. Das and D. Herschlag, Understanding Nucleic Acid–Ion Interactions, Annu. Rev. Biochem., 2014, 83, 813–841 CrossRef CAS PubMed.
- M. I. Zarudnaya, A. L. Potyahaylo and D. M. Hovorun, Dependence of DNA persistence length on ionic conditions, Biopolym. Cell, 2017, 33, 81–91 Search PubMed.
- J. Seils and R. Pecora, Dynamic light scattering study of a monodisperse 2311 base pair circular DNA, Environ. Sci. Technol., 1992, 25, 354–362 CAS.
- M. Salomo, K. Kegler, C. Gutsche, M. Struhalla, J. Reinmuth, W. Skokow, U. Hahn and F. Kremer, The elastic properties of single double-stranded DNA chains of different lengths as measured with optical tweezers, Biophys. Struct. Mech., 2006, 284, 1325–1331 CAS.
- H. Liu, J. Gapinski, L. Skibinska, A. Patkowski and R. Pecora, Effect of electrostatic interactions on the dynamics of semiflexible monodisperse DNA fragments, J. Chem. Phys., 2000, 113, 6001–6010 CrossRef CAS.
- N. Borochov, H. Eisenberg and Z. Kam, Dependence of DNA conformation on the concentration of salt, Biopolymers, 1981, 20, 231–235 CrossRef CAS.
- D. H. Taylor and A. T. Wilson, The adsorption of yeast RNA by allophane, Clays Clay Miner., 1979, 27, 261–268 CrossRef CAS.
- X. Wang, H. Kweon, S. Lee, H. Shin, B. Chua, M. R. Liles, M.-K. Lee and A. Son, Vulnerability of DNA hybridization in soils is due to Mg2+ ion induced DNA aggregation, Soil Biol. Biochem., 2018, 125, 300–308 CrossRef CAS.
- V. K. Misra and D. E. Draper, On the role of magnesium ions in RNA stability, Biopolymers, 1998, 48, 113–135 CrossRef CAS PubMed.
- J. Anastassopoulou, Metal–DNA interactions, J. Mol. Struct., 2003, 651–653, 19–26 CrossRef CAS.
- G. Minasov, V. Tereshko and M. Egli, Atomic-resolution crystal structures of B-DNA reveal specific influences of divalent metal ions on conformation and packing, J. Mol. Biol., 1999, 291, 83–99 CrossRef CAS PubMed.
- B. K. G. Theng, in Developments in Clay Science, Elsevier, 2012, vol. 4, pp. 319–337 Search PubMed.
- T. H. Nguyen and K. L. Chen, Role of Divalent Cations in Plasmid DNA Adsorption to Natural Organic Matter-Coated Silica Surface, Environ. Sci. Technol., 2007, 41, 5370–5375 CrossRef CAS PubMed.
- M. Franchi and E. Gallori, A surface-mediated origin of the RNA world: biogenic activities of clay-adsorbed RNA molecules, Gene, 2005, 346, 205–214 CrossRef CAS PubMed.
- P. Cai, Q. Huang, D. Jiang, X. Rong and W. Liang, Microcalorimetric studies on the adsorption of DNA by soil colloidal particles, Colloids Surf., B, 2006, 49, 49–54 CrossRef CAS.
- NanoDrop 1000 Spectrophotometer V3.8 User's Manual, 3rd edn, http://tools.thermofisher.com/content/sfs/manuals/nd-1000-v3.8-users-manual-8%205x11.pdf, accessed 27 November 2020 Search PubMed.
- W. Saenger, in Principles of Nucleic Acid Structure, Springer, New York, NY, 1984, pp. 253–282 Search PubMed.
- B. Müller and L. Sigg, Adsorption of lead(II) on the goethite surface: Voltammetric evaluation of surface complexation parameters, J. Colloid Interface Sci., 1991, 148, 517–532 CrossRef.
- A. Ruf, PhD thesis, ETH Zürich, 1992.
- K. Pecher, S. B. Haderlein and R. P. Schwarzenbach, Reduction of Polyhalogenated Methanes by Surface-Bound Fe(II) in Aqueous Suspensions of Iron Oxides, Environ. Sci. Technol., 2002, 36, 1734–1741 CrossRef CAS.
- Y. Zhang, L. Charlet and P. W. Schindler, Adsorption of protons, Fe(II) and Al(III) on lepidocrocite (γ-FeOOH), Colloids Surf., 1992, 63, 259–268 CrossRef CAS.
- K. Hellige, K. Pollok, P. Larese-Casanova, T. Behrends and S. Peiffer, Pathways of ferrous iron mineral formation upon sulfidation of lepidocrocite surfaces, Geochim. Cosmochim. Acta, 2012, 81, 69–81 CrossRef CAS.
- R. A. Figueroa and A. A. MacKay, Sorption of oxytetracycline to iron oxides and iron oxide-rich soils, Environ. Sci. Technol., 2005, 39, 6664–6671 CrossRef CAS PubMed.
- S. Bonneville, T. Behrends and P. Van Cappellen, Solubility and dissimilatory reduction kinetics of iron(III) oxyhydroxides: A linear free energy relationship, Geochim. Cosmochim. Acta, 2009, 73, 5273–5282 CrossRef CAS.
- D. O. Jordan, in The Nucleic Acids, ed. E. Chargaff and J. N. Davidson, 1955, vol. 1, pp. 447–492 Search PubMed.
- G. V. Sherbet, M. S. Lakshmi and F. Cajone, Isoelectric characteristics and the secondary structure of some nucleic acids, Biophys. Struct. Mech., 1983, 10, 121–128 CrossRef CAS.
- S. Lagergren, Zur Theorie der sogenannten Absorption gelöster Stoffe, 1898 Search PubMed.
- Y. T. He, J. Wan and T. Tokunaga, Kinetic stability of hematite nanoparticles: the effect of particle sizes, J. Nanopart. Res., 2008, 10, 321–332 CrossRef CAS.
- I. Langmuir, The constitution and fundamental properties of solids and liquids, J. Am. Chem. Soc., 1916, 38, 2221–2295 CrossRef CAS.
- N. V. Hud, Nucleic acid-metal ion interactions, RSC Biomolecular Sciences, 2009 Search PubMed.
- R. L. Parfitt, R. J. Atkinson and R. S. C. Smart, The Mechanism of Phosphate Fixation by Iron Oxides, Soil Sci. Soc. Am. J., 1975, 39, 837–841 CrossRef CAS.
- F. J. Hingston, R. J. Atkinson, A. M. Posner and J. P. Quirk, Specific Adsorption of Anions, Nature, 1967, 215, 1459–1461 CrossRef CAS.
- T. Hiemstra and W. H. Van Riemsdijk, A surface structural approach to ion adsorption: the charge distribution (CD) model, J. Colloid Interface Sci., 1996, 179, 488–508 CrossRef CAS.
- J. Beek and W. H. Van Riemsdijk, in Developments in Soil Science, Elsevier, 1979, vol. 5, pp. 259–284 Search PubMed.
- L. Sigg and W. Stumm, The interaction of anions and weak acids with the hydrous goethite (α-FeOOH) surface, Colloids Surf., 1981, 2, 101–117 CrossRef CAS.
- J. Kim, W. Li, B. L. Philips and C. P. Grey, Phosphate adsorption on the iron oxyhydroxides goethite (α-FeOOH), akaganeite (β-FeOOH), and lepidocrocite (γ-FeOOH): a 31P NMR Study, Energy Environ. Sci., 2011, 4, 4298–4305 RSC.
- J. Antelo, M. Avena, S. Fiol, R. López and F. Arce, Effects of pH and ionic strength on the adsorption of phosphate and arsenate at the goethite–water interface, J. Colloid Interface Sci., 2005, 285, 476–486 CrossRef CAS PubMed.
- O. K. Borggaard, Effect of surface area and mineralogy of iron oxides on their surface charge and anion-adsorption properties, Clays Clay Miner., 1983, 31, 230–232 CrossRef CAS.
- M. I. Tejedor-Tejedor and M. A. Anderson, The protonation of phosphate on the surface of goethite as studied by CIR-FTIR and electrophoretic mobility, Langmuir, 1990, 6, 602–611 CrossRef CAS.
- L. Zeng, X. Li and J. Liu, Adsorptive removal of phosphate from aqueous solutions using iron oxide tailings, Water Res., 2004, 38, 1318–1326 CrossRef CAS PubMed.
- R. T. Podoll, K. C. Irwin and S. Brendlinger, Sorption of water-soluble oligomers on sediments, Environ. Sci. Technol., 1987, 21, 562–568 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1em00010a |
| This journal is © The Royal Society of Chemistry 2021 |