
Biorefinery roadmap based on catalytic production and upgrading 5-hydroxymethylfurfural
Qidong
Hou
 ,
Xinhua
Qi
,
Meinan
Zhen
,
Hengli
Qian
,
Yifan
Nie
,
Chuanyunlong
Bai
,
Shiqiu
Zhang
,
Xinyu
Bai
and
Meiting
Ju
*
,
Xinhua
Qi
,
Meinan
Zhen
,
Hengli
Qian
,
Yifan
Nie
,
Chuanyunlong
Bai
,
Shiqiu
Zhang
,
Xinyu
Bai
and
Meiting
Ju
*
National & Local Joint Engineering Research Center of Biomass Resource Utilization, College of Environmental Science and Engineering, Nankai University, Tianjin 300350, China. E-mail: jumeit@nankai.edu.cn
First published on 12th November 2020
Abstract
Biorefineries, which utilize lignocellulosic biomass as renewable energy source and sustainable carbon feedstock, are a promising solution to alleviate the excessive dependence on the depleting fossil resources and address climate change and other environmental problems. Owing to the recalcitrance and over-functionalized nature of biomass, the conversion of biomass into desirable products requires a series of complex deconstruction, catalytic conversion, separation and purification processes. In the biorefinery roadmap, 5-hydroxymethylfurfural (HMF) stands out as a bridge connecting biomass raw materials to alternative fuels, chemicals and materials, which can displace petroleum-derived products. This review describes the recent advances in the design and development of catalytic systems for the conversion of biomass and their constituent carbohydrates to HMF via hydrolysis, isomerization and dehydration reactions, and the upgrading of HMF towards polymer monomers, fine chemicals, fuel precursors, fuel additives, liquid fuels, and other platform chemicals via hydrogenation, oxidation, esterification, etherification, amination and aldol condensation reactions, with emphasis on how the catalysts, solvents and reaction conditions determine the reaction pathway and product selectivity. We also attempt to provide a conceptual framework on how to evaluate the actual reaction efficiency, reusability, and economic and technical feasibility of different catalytic systems and highlight the key research challenges to be addressed.
 Qidong Hou | Qidong Hou obtained his Bachelor's Degree and Master's Degree in 2013 and 2018, respectively, from the College of Environmental Science and Engineering, Nankai University, China. After he graduated, he worked as the Director of the Biorefinery Center in National & Local Joint Engineering Research Center of Biomass Resource Utilization, China. Dr Hou has wide experience in garbage classification and recycling, pollution control of solid waste, biomass pretreatment, catalytic conversion of biomass to important platform chemicals and their upgradation toward fuels, fine chemicals and degradable plastics through heterogeneous catalysis, advanced oxidation technology, selective oxidation and hydrogenation technology. |
 Xinhua Qi | Dr Xinhua Qi is a Professor at Nankai University, Tianjin, China. His research interests mainly focus on green processes for biomass conversion into value-added materials and chemicals. He has published more than 100 peer reviewed scientific papers, and these papers have been cited over 3200 times with an H index 30. He has also co-authored over 20 patents, 5 books and 3 book chapters on environmental engineering and biomass resources utilization. Prof. Qi has been supported by the National Key Research and Development Program of China, National Natural Science Foundation of China, the Natural Science Fund for Distinguished Young Scholars of Tianjin. |
 Meiting Ju | Meiting Ju is a Professor at the College of Environmental Science and Engineering, Nankai University, China, and Director of the National & Local Joint Engineering Research Center of Biomass Resource Utilization. He is a specialist in pollution control of organic solid waste, valorization of biomass toward fuels, chemicals and materials, industrial ecology, environmental impact assessment and environmental management. He has coauthored more than 100 scientific articles, 10 books and 30 authorized patents. His research focuses on efficient, economic and environmentally friendly biorefinery technologies to promote the establishment of a more sustainable industry. |
1. Introduction
The modern society consumes a significant amount of fossil resources, including petroleum, coal and natural gas, to meet the major demand for energy, chemicals and materials.1 Using the well-established petroleum refinery process, fossil carbon resources can be converted into transportation fuels, fertilizers, fine-chemicals, polymers, plastics, pharmaceuticals, detergents, food additives, electronics, sports equipment, clothes, dyes and agrochemicals.2,3 However, the excessive dependence on unrenewable fossil fuel resources has resulted in a series of economic, social and environmental problems, including shortage of resources and energy crisis, massive emission of environmental pollutants, global climate change, and unsustainable development. Therefore, to reduce the dependence on fossil fuel resources, the exploitation of renewable resources, such as solar, wind, wave power, and biomass is of great importance.4,5Among these renewable resources, biomass is the only renewable carbon-based resource that can serve as both energy and feedstock, with a global production larger than 120 billion tons per annum (the dry basis weight is about 10 billion tons), containing 2.2 × 1021 J of energy.6–10 Moreover, the utilization of biomass is considered to be approximately carbon neutral since the CO2 emission associated with biomass-derived products can be offset by the CO2 capture in photosynthesis.11,12 In fact, biomass was continuously utilized as the predominant energy source and feedstock before the industrial age, and nowadays biomass still ranks the fourth used energy, which is about 10% of the global primary energy source.9 The renaissance of biomass utilization is based on the concept of “biorefinery”, which has been developed to displace the current energy and chemical industry based on petroleum refining, showing a great promise for the realization of sustainable development.
Biomass (Fig. 1) represents the biogenic organic matter originating from carbon dioxide (CO2) and water (H2O) via photosynthesis using energy from sunlight.4 Generally, biomass contains carbohydrates, lignin, fatty acids, lipids, proteins, and other components containing carbon (C), hydrogen (H), oxygen (O), nitrogen (N), phosphorus (P), chlorine (Cl), sulphur (S), and metal elements (K, Na, Ca, and Mg).9,13,14 Lignocellulosic biomass, which is prevalent in the cell walls of plants, accounts for more than 90% of all plant biomass.15 Lignocellulosic biomass is the most abundant and available biomass source for biorefineries, without competing with food reserves.4 Lignocellulosic biomass is mainly composed of cellulose (35–50 wt%), hemicellulose (20–30 wt%) and lignin (20–30 wt%), in which aligned bundles of crystalline and amorphous cellulose microfibrils are embedded in a disordered matrix of hemicellulose and lignin.15 Cellulose is a linear polysaccharide composed of glucose units linked via β-1,4-glycosidic bonds, hemicelluloses are branched polysaccharides consisting primarily of xylose with a small quantity of mannose, galactose, rhamnose, and arabinose via multiple types of glycosidic bonds, and lignin is a three-dimensional amorphous polymer composed of three types of phenylpropanolic monomers (coniferyl alcohol, p-coumaryl alcohol and sinapyl alcohol) linked by carbon–carbon and ether bonds.4,16,17 The natural recalcitrance of lignocellulosic biomass, which largely originates from the tight compaction and aggregation of its partially crystallized cellulose chains and the inherent inaccessibility of cellulose to enzymes and catalysts owing to the surrounding heterogeneous matrix composed of lignin and hemicellulose, is the major obstacle in the cost-effective deconstruction and valorization of biomass.18
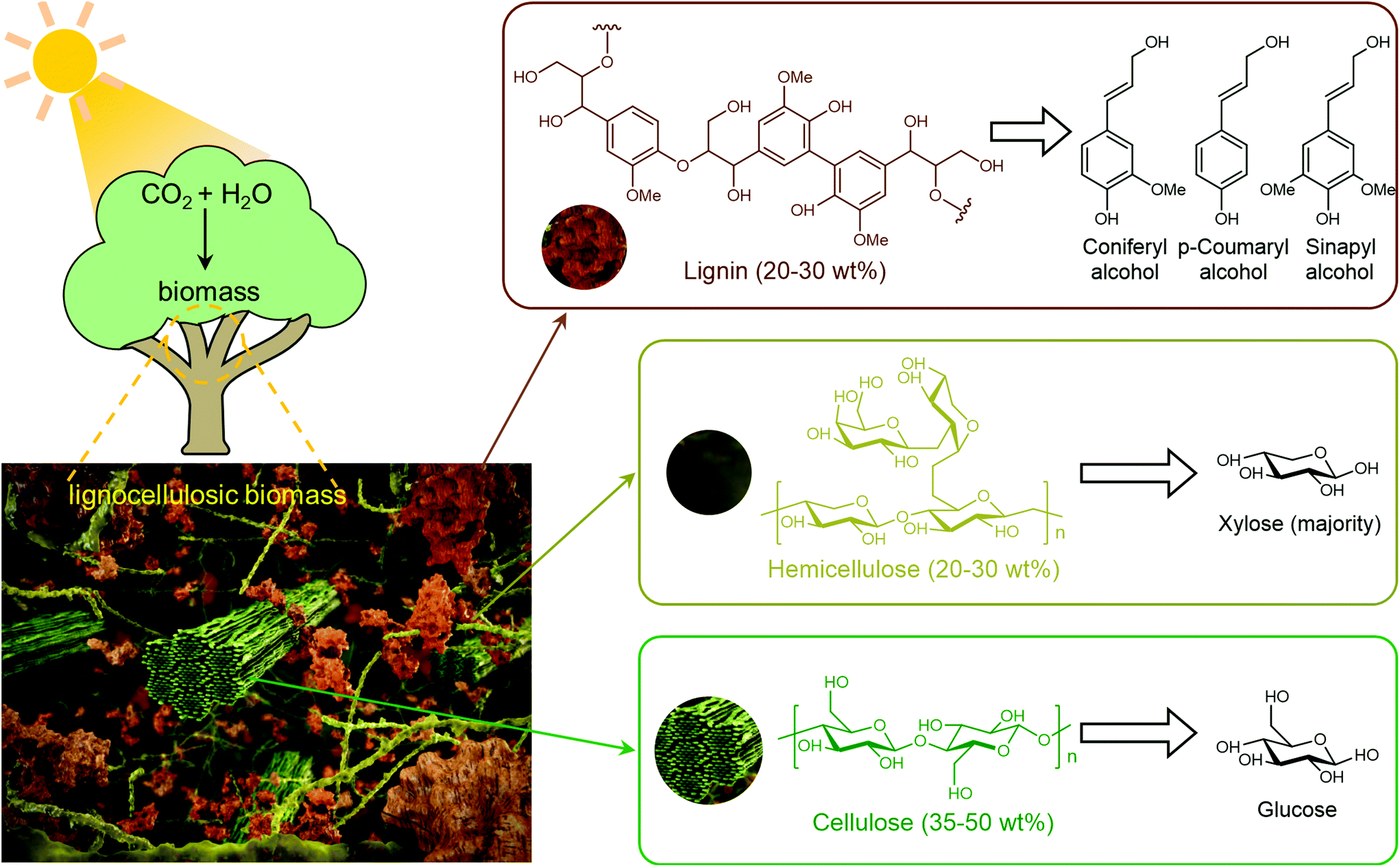 | ||
| Fig. 1 Structure of lignocellulosic biomass and its components. Compiled from ref. 2, 16 and 19. | ||
For a long time, physical, biological, chemical and combined approaches have been investigated to transform lignocellulosic biomass into useful products, forming the roadmap of biorefinery technologies, which have potential to displace petroleum refining.20–22 The direct combustion of lignocellulosic biomass can provide heat, which can be used for the generation of electricity. Alternatively, lignocellulosic biomass can also be converted into biomass briquetting fuel via physical compression. Traditional biological technologies, including aerobic and anaerobic fermentation, mainly produce organic fertilizers, feeds and biogas. In addition, the development of high-performance microbial cell factories can enable the production of more diverse chemicals of interest from biomass-derived carbon sources.22 The efficient biological conversion of lignocellulosic biomass to target products usually requires pretreatment to enhance the accessibility of biomass by altering its three-dimensional (3D) structure, interaction and composition.2,6,16 Ethanol (EtOH), one of the most representative biomass biofuels, is produced industrially from starchy biomass, including corn starch and sugar cane, while its production from lignocellulosic biomass via pretreatment, enzymatic saccharification and fermentation is a more sustainable but challenging approach.2,11 Pyrolysis (or gasification) of lignocellulosic biomass simultaneously produces biochar, bio-oil and gas products, which only consumes approximately 10% of the energy in the original biomass.9,23 However, most biorefinery technologies either provide low-value products or generate a series of complex mixtures, which require tedious separation, purification and upgrading process to obtain the final products.9 Accordingly, the biorefinery strategy aims to convert lignocellulosic biomass to high-quality fine-chemicals, liquid transportation fuels and high-performance structural materials, which can partly or even completely displace the currently indispensable products derived from fossil resources and other unrenewable resources, and thus has attracted great attention.24
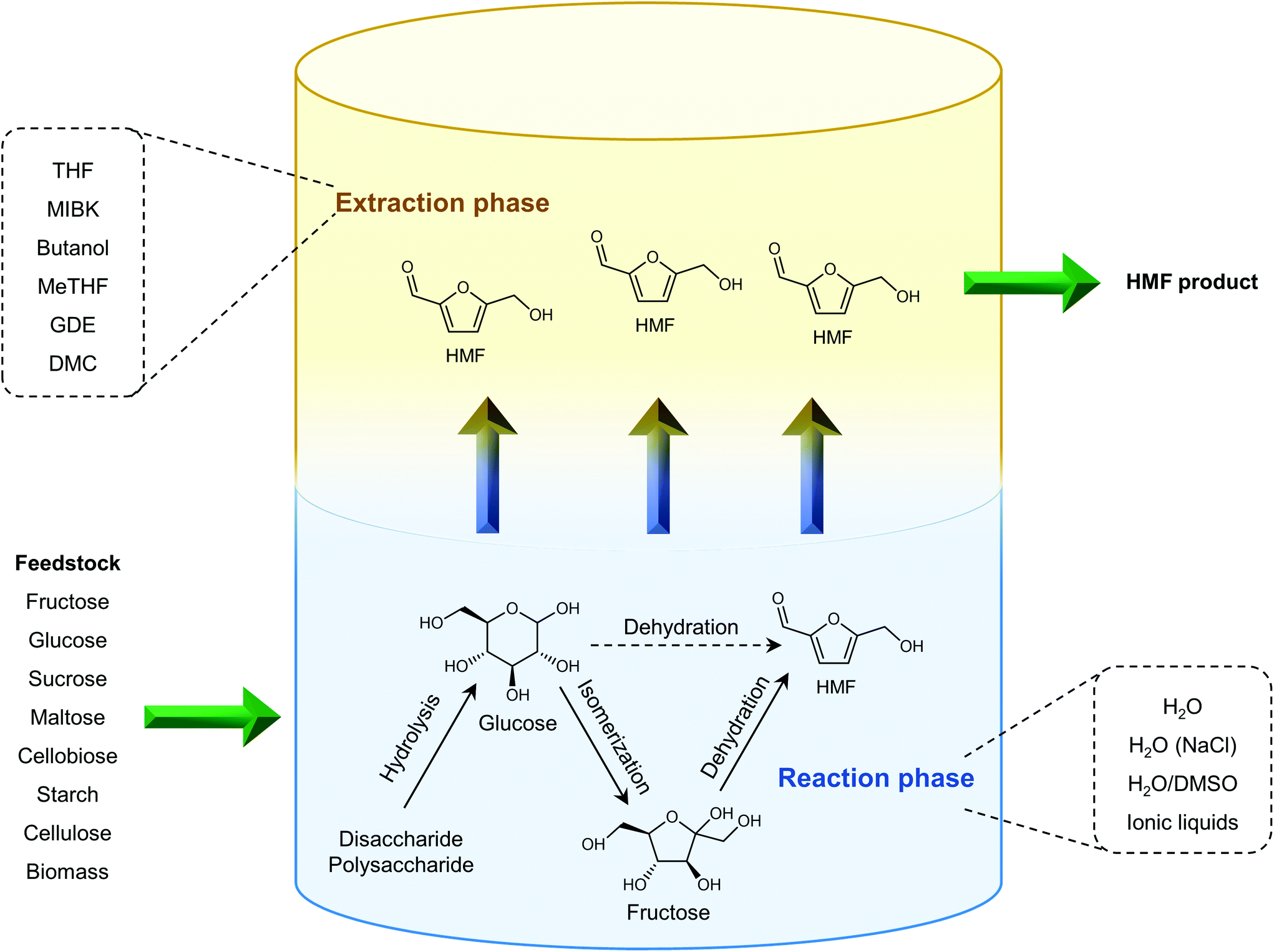
Shifting from the ready-to-use petroleum refinery economy to biorefinery comes with considerable technical and financial challenges since petroleum is under-functionalized relative to the target products, while biomass is over-functionalized.25 The conversion of biomass to final products requires a series of deconstruction, catalytic conversion, separation and purification processes. In these conversion processes, 5-hydroxymethylfurfural (HMF) stands out as a bridge (Fig. 2) connecting biomass raw materials to the biorefinery industry since it not only retains a reasonable proportion of the initial chemical complexity of biomass, but also can be converted into multiple target products, which are promising to displace the corresponding petrochemicals.1,26 In traditional biorefinery processes aiming to obtain valuable products from the carbohydrate fractions, lignin is usually considered as a major obstacle in the efficient utilization of lignocellulosic biomass.27 After pretreatment or fractionation, cellulose-rich material is collected as the predominant feedstock for the biorefinery, some of the hemicellulose is lost and lignin is converted into condensed residue due to its uncontrollable degradation under harsh reaction conditions.28,29 In fact, lignin not only can be used for the generation of heat via combustion, but also to produce high value products as the most abundant resource of bio-aromatics.28,30,31 Glucose can be obtained from cellulose via either enzymatic saccharification or acid-catalyzed hydrolysis. After removing three molecules of water from glucose, glucose can be converted to HMF, a pivotal platform chemical, which can be further converted to versatile products ranging from polymer monomers, fine chemicals, fuel precursors, fuel additives, and liquid fuels to other platform chemicals, covering a broad range of structural complexities and application demands.1,2,26,32 Using a similar but more facile reaction route, hemicellulose can be transformed into furfural, a current commodity chemical with an annual production of about 300 kTon.13,33 Furfural has already been extensively used in the chemical industry.1,33,34 In comparison with furfural, HMF is more attractive owing to the abundance of cellulose feedstock for its synthesis and its versatility of HMF. Therefore, the development of environmentally friendly and cost-effective catalytic technology that can convert cellulose into HMF and its derivates offers great opportunity for the establishment of sustainable biorefineries.
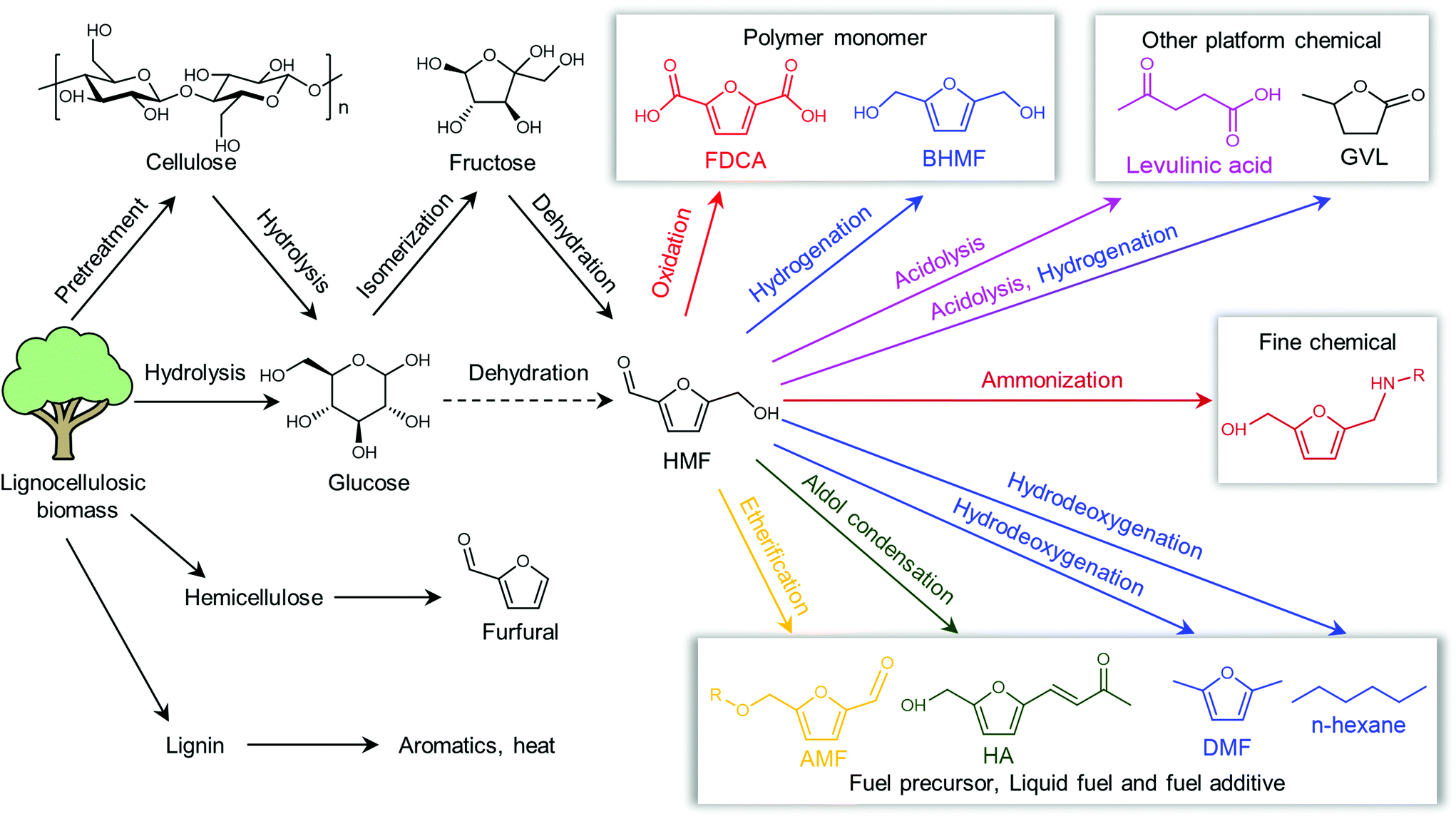 | ||
| Fig. 2 Basic framework of the biorefinery process based on HMF. | ||
The last few decades have witnessed continuously growing interest in the synthesis of HMF and its catalytic upgrading toward biofuels, bio-based chemicals and materials, as evidenced by the number of publications on this topic (Fig. 3). The abundant original research on the transformation of biomass to HMF and its subsequent conversion to value-added chemicals has greatly promoted the development of this field. Although several recent reviews articles, chapters, and books on novel materials and biorefineries presented the synthesis and upgrading of HMF and furfural, in a wider context of materials or biomass valorization,33,35–43 the chemistries, processes and sustainability issues for the synthesis of HMF and upgrading, and upstream and downstream processes have not been systematically summarized and critically evaluated in detail in these works. Also, many interesting results have been recently published on the development and applications of novel materials and solvents for the synthesis and upgrading of HMF.
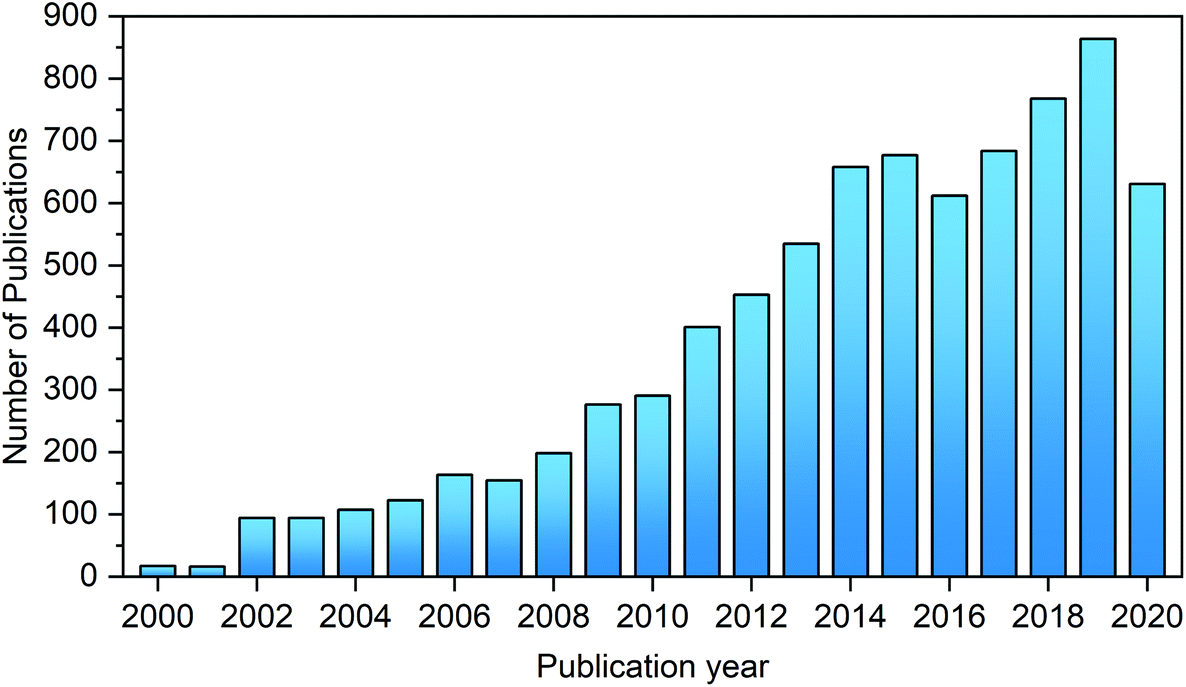 | ||
| Fig. 3 Number of publications on HMF per year (January 2000–July 2020). Source: Web of science (topic: 5-hydroxymethylfurfural, 5-hydroxymethyl-2-furfuraldehyde, 5-hydroxymethyl furfural, 5-HMF or HMF). | ||
Herein, we aim to provide a roadmap for biorefineries based on HMF, covering the important literature since 2007 with focus on the recent development of the state-of-the-art catalytic systems and advanced reaction processes. This review includes two major sections. Section 2 summarizes the advances in the synthesis of HMF using various feedstock, covering the development of homogeneous catalysts, heterogenous catalysts, solvents, reaction pathways and process technology. Section 3 summarizes the upgrading of HMF toward value-added fuels and chemical commodities, including furan derivates, fuel additives, diols, dicarboxylic acids and hydrocarbons via selective oxidation, hydrogenation, etherification, coupling and condensation reaction. Considering the similar roles of furfural and HMF in the biorefinery, the synthesis and upgrading of furfural are also presented in this review, only emphasizing the cutting-edge catalytic technologies. For a more elaborate overview on the synthesis and upgrading of furfural, the reader is referred to the dedicated reviews on this topic.33,34 Throughout, we attempt to reveal the reaction mechanism, highlight the key factors that influence the performance, and systematically evaluate the merits and drawbacks of disparate catalytic systems by comparing their intrinsic reaction efficiency, reusability, economic and technical feasibility and sustainability. The gaps in current research, the drawbacks in analytical and characterization methods, the key research problems and the future trends are also discussed. We hope that this review will serve as a useful reference for the academic research and industrial applications of biorefinery-based HMF, and consequently attract the attention of academic and industrial researchers toward crucial scientific and technical problems, the solution of which can help overcome the main bottlenecks in this field.
Assessing all the catalysts and all the possible HMF derivatives in the literature is beyond the scope of this manuscript since the number of catalysts and potential HMF derivatives are colossal. Therefore, we try to unravel all the types of catalysts, solvents and catalytic systems, and then examine the catalytic systems that fulfil one of the following criteria: (1) the typical catalytic systems that have been confirmed or improved by the following studies; (2) the novel materials and solvents that have been tested preliminarily in the synthesis and upgrading of HMF; and (3) new phenomena and new or general mechanisms observed or confirmed for the catalytic systems. Similarly, we examine the HMF derivatives according to the following criteria: (1) biofuels that can be derived either directly from HMF or from high volume HMF derivatives; (2) commercial chemicals that are produced directly from HMF; (3) non-commercial chemicals that are directly derived from HMF through well-established routes and that present high potential for commercialization as commodities; and (4) commercial products currently derived from petroleum that can be produced from biomass via HMF as the main intermediate.
2. Synthesis of HMF from biomass
Considering the chemical composition of HMF (C6H6O3), it can be considered the dehydration product of a C6 sugar after removing three molecules of water. In thermally processed foods containing a high concentration of sugars and amino acids, HMF is usually generated via the Maillard reaction as an impurity, which is a potential health hazard.44 Structurally, HMF contains a furan ring bearing hydroxymethyl (CH2–OH) and aldehyde (CHO) functional groups. Therefore, the dehydration of carbohydrates to HMF not only can decrease the oxygen content of carbohydrates, but also retain reasonable chemical complexity for subsequent conversion.26 As a pivotal platform in biorefineries, the efficient production of HMF from biomass is prerequisite for the establishment of an HMF-based biorefinery industry. For the historical developments in the synthesis of HMF, the reader is referred to the earlier literature.13,42,45,46 In this section, we aim to present an overview of the reactions, feedstocks, catalysts, solvents and process technologies for the synthesis of HMF, focusing on the development of efficient and selective catalytic systems to improve the intrinsic HMF productivity. For a simple extension of previous work, we only provide a simple description, but the catalytic performance and detailed reaction conditions are summarized in the tables for readers to find useful information readily.2.1. Main feedstocks and reaction processes
Any C6 sugar containing materials either from natural sources or synthetic approaches, including monosaccharides (fructose, glucose and mannose), disaccharides (sucrose, cellobiose and maltose), polysaccharides (inulin, starch and cellulose), food waste, industrial molasses, and lignocellulosic biomass and its hydrolysate can be used as a feedstock for the production of HMF.47,48 The synthesis of HMF from biomass and its components involves a series of complex processes, including pretreatment (or fractionation) of lignocellulosic biomass, hydrolysis of cellulose to glucose, isomerization of glucose to fructose and dehydration of fructose. Meanwhile, these processes are accompanied by a series of side-reactions, including the decomposition of HMF to formic acid and levulinic acid and the condensation of sugars, reaction intermediates and HMF toward humins.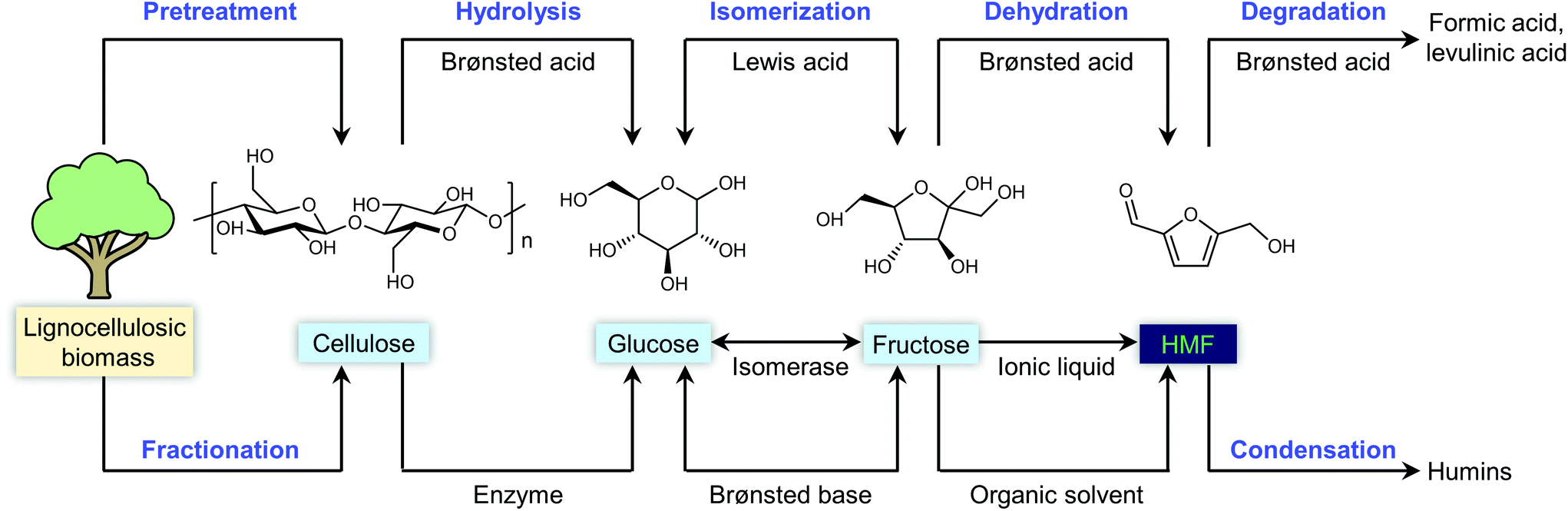 | ||
| Fig. 4 Main reaction for the synthesis of HMF from biomass and its components. Adapted from ref. 49. | ||
| Reaction process | Catalyst | Solvent | Test condition | Activation energy | Ref. |
|---|---|---|---|---|---|
| a Simulated activation energy by DFT. | |||||
| Isomerization of xylose to xylulose | CrCl3 | Water | 105–145 °C, xylose loadings 1 wt% | 64.9 kJ mol−1 | 55 |
| Dehydration of xylulose to furfural | HCl | Water | 105–145 °C, xylulose loadings 1 wt% | 96.7 kJ mol−1 | 55 |
| Dehydration of xylose to furfural | HCl | Water | 105–145 °C, xylose loadings 1 wt% | 133.9 kJ mol−1 | 55 |
| Isomerization of glucose to fructose | CrCl3 | Water | 130–150 °C, glucose loadings 10 wt% | 64.4 kJ mol−1 | 56 |
| Isomerization of glucose to fructose | AlCl3 | Water | 100–120 °C, glucose loadings 0.25 M | 110 kJ mol−1 | 57 |
| Isomerization of glucose to fructose | AlCl3 | Water/THF | 130–160 °C, glucose loadings 0.25 M | 95 kJ mol−1 | 58 |
| Isomerization of fructose to glucose | AlCl3 | Water/THF | 130–160 °C, glucose loadings 0.25 M | 66 kJ mol−1 | 58 |
| Isomerization of glucose to fructose | Sn-Beta | Water | 80–120 °C, glucose loadings 0.52 M | 93 kJ mol−1 | 59 |
| Isomerization of glucose to fructose | Triethylamine | Water | 80–120 °C, glucose loadings 0.52 M | 61 kJ mol−1 | 59 |
| Isomerization of glucose to fructose | Meglumine | Water | 80–110 °C, glucose loadings 10 wt% | 74 kJ mol−1 | 52 |
| Dehydration of fructose to HMF | 5 mM–1 M H2SO4 | Water | 140–180 °C, fructose loadings 0.1–1 M | 123 kJ mol−1 | 13 |
| Dehydration of fructose to HMF | 50 mM H2SO4 | Water/GVL (w/w = 1/3) | 140–180 °C, fructose loadings 0.1–1 M | 84 kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
60 |
| Dehydration of fructose to HMF | 50 mM H2SO4, KCl | Water/GVL (w/w = 1/3) | 140–180 °C, fructose loadings 0.1–1 M | 74 kJ mol−1a |
60 |
| Dehydration of fructose to HMF | 50 mM H2SO4, KCl | Water/GVL (w/w = 1/9) | 140–180 °C, fructose loadings 0.1–1 M | 67 kJ mol−1a |
60 |
| Dehydration of fructose to HMF | EMIMBr | EMIMBr | 57.3 kJ mol−1a |
61 | |
| HMF degradation | 5 mM–1 M H2SO4 | Water | 140–180 °C, HMF loadings 0.1–1 M | 147 kJ mol−1 | 13 |
| Direct dehydration of glucose to HMF | 5 mM–1 M H2SO4 | Water | 140–200 °C, glucose loadings 0.1–1 M | 152 kJ mol−1 | 62 |
| Direct dehydration of glucose to HMF | HAP | EMIMBr | 110–130 °C, glucose loadings 10 wt% | 100.5 kJ mol−1 | 49 |
| Direct dehydration of glucose to HMF | Al-HAP | EMIMBr | 110–130 °C, glucose loadings 10 wt% | 80.7 kJ mol−1 | 49 |
| Direct dehydration of glucose to HMF | Sn-HAP | EMIMBr | 110–130 °C, glucose loadings 10 wt% | 79.2 kJ mol−1 | 49 |
| Direct dehydration of glucose to HMF | Sn/Al-HAP | EMIMBr | 110–130 °C, glucose loadings 10 wt% | 68.4 kJ mol−1 | 49 |
| Hydrolysis of cellulose to glucose | 0.1 M–1 M H2SO4 | Water | 100–200 °C, cellulose loading 1.7 wt% | 170 kJ mol−1 | 63 |
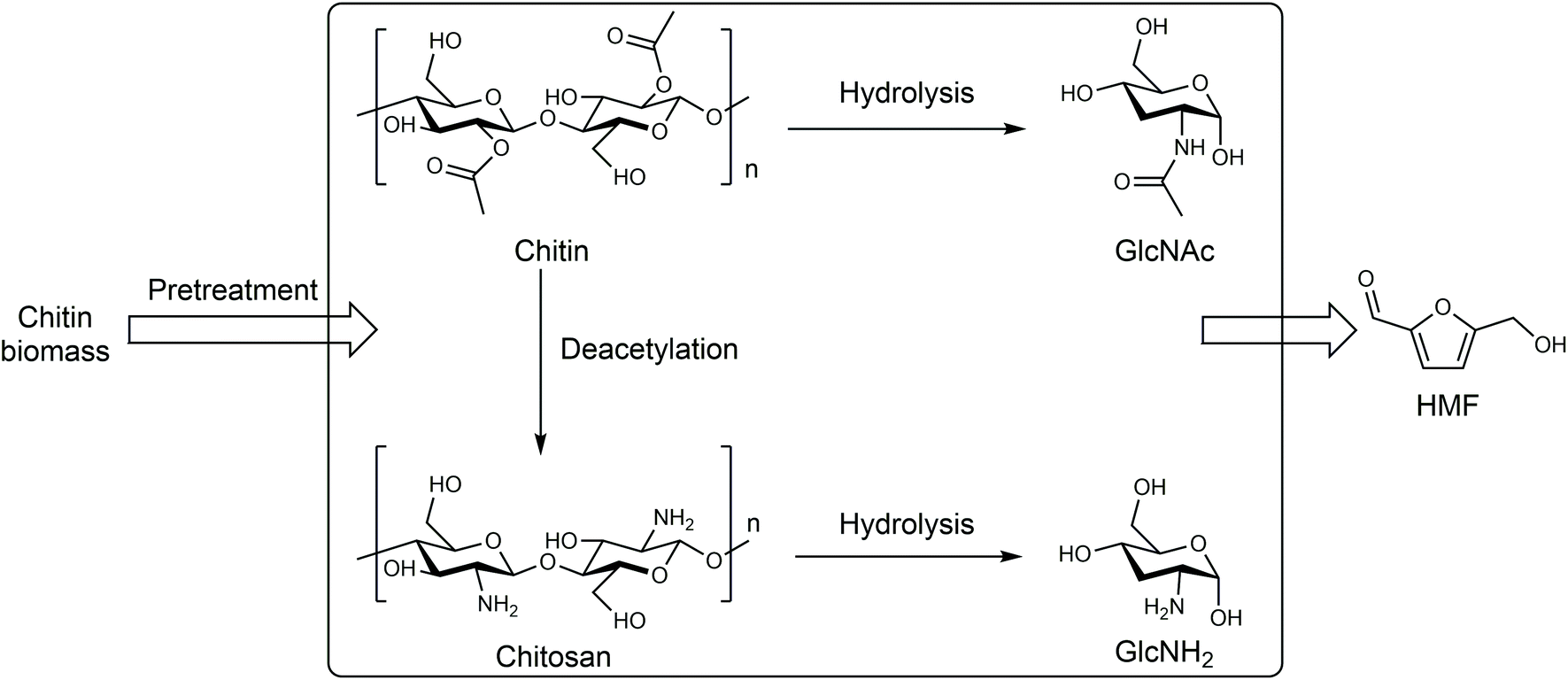 | ||
| Fig. 5 Production of HMF from chitin biomass. Adapted from ref. 66 and 67. | ||
2.2. Reaction media for the production of HMF from biomass
Generally, the dissolution of a reactant and product in a solvent and the dissolution or dispersion of a catalyst in a solvent have predominant influence on reaction processes. In addition, the competing adsorption of solvents with reactants, intermediates, transition states and products on the catalyst surface can affect the reaction pathway.68 The mass transfer between the catalyst surface and liquid phase has a predominant effect on the reaction rates. Besides, solvents can also function as homogeneous catalysts in some cases to alter the reaction pathway. Thus, it is necessary to search for cost-effective and environmentally friendly solvents to avoid the use of hazardous and harmful solvents. The use of ionic liquids, supercritical fluids, fluorous solvents, deep eutectic solvents, and biomass-derived solvents as green solvents in the conversion of lignocellulose has been reviewed in previous reports.17,69,70 In this section, we analyze the influence of different solvents on the production of HMF and discuss their merits and disadvantages from the perspective of green chemistry, with emphasis on green solvents, biomass-derived solvents, and low boiling points solvents.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.79 Several studies attributed the superior catalytic performance of DMSO for the dehydration of fructose to HMF in the presence of O2 to the acidic species, such as CH3SO3H and H2SO4, generated from the decomposition of DMSO at an evaluated temperature,80–82 but the barium chloride precipitation test conducted by Whitaker showed that no sulfate ions were generated at 120 °C and 150 °C under an oxygen atmosphere.78 Moreover, they demonstrated that the formed organic acid could not catalyze the dehydration reaction effectively. Therefore, they concluded that DMSO does not decompose to into a strong acid at a temperature lower than 150 °C and the dehydration of fructose in DMSO is dominated by the solvent effect. Considering these contradictory results, precise control experiments and detailed analysis of the composition of the reaction mixture under various conditions are necessary to eliminate the possible influence of O2 and impurity in the reagents and illuminate the realistic role of DMSO in the conversion of fructose to HMF.
O.79 Several studies attributed the superior catalytic performance of DMSO for the dehydration of fructose to HMF in the presence of O2 to the acidic species, such as CH3SO3H and H2SO4, generated from the decomposition of DMSO at an evaluated temperature,80–82 but the barium chloride precipitation test conducted by Whitaker showed that no sulfate ions were generated at 120 °C and 150 °C under an oxygen atmosphere.78 Moreover, they demonstrated that the formed organic acid could not catalyze the dehydration reaction effectively. Therefore, they concluded that DMSO does not decompose to into a strong acid at a temperature lower than 150 °C and the dehydration of fructose in DMSO is dominated by the solvent effect. Considering these contradictory results, precise control experiments and detailed analysis of the composition of the reaction mixture under various conditions are necessary to eliminate the possible influence of O2 and impurity in the reagents and illuminate the realistic role of DMSO in the conversion of fructose to HMF.
The mechanism of the dehydration of fructose and glucose to HMF in DMSO over different catalysts has also been widely investigated. For example, Zhang et al. studied the mechanism of fructose dehydration to HMF in DMSO over Amberlyst 70, PO43−/niobic acid, and sulfuric acid.8013C, 1H, and 17O NMR, and high-resolution electrospray ionization mass spectrometry (HR ESI-MS) confirmed the formation of intermediates 1 and 3 (Fig. 6), which is consistent with the low energy species calculated from the high level G4MP2 theoretical analysis. Intermediate 2 was also identified by HR ESI-MS and theoretical analysis, but the keto and the enol forms could not be distinguished experimentally. The same intermediates were observed over the three different catalysts, suggesting that there is a common mechanism for the dehydration of fructose to HMF in DMSO, which is independent of the source of protons. Though theoretical analysis, Ren et al. indicated that the H+ from the Brønsted acid prefers to interact with DMSO, leading to the formation [DMSOH]+.79 [DMSOH]+, instead of the original Brønsted acid is the main catalytically active species for the dehydration of fructose to HMF, which can explain why the intermediates are independent of the source of protons. Jia et al. reported that the use of anhydrous DMSO as the reaction medium for the conversion of glucose over chromium trichloride hexahydrate (CrCl3·6H2O) catalyst not only promoted the formation of HMF, but also led to undesirable side-reactions, in particular the dehydration of glucose into cellobiose.83 Yang et al. reported that the formation of a six-coordinated structure (CrCl3–3DMSO) resulted in a low glucose conversion and HMF yield, while N,N-dimethylacetamide (DMA) resulted in a higher HMF yield and selectivity owing to the four-coordinated structures formed between CrCl3 with DMA.84
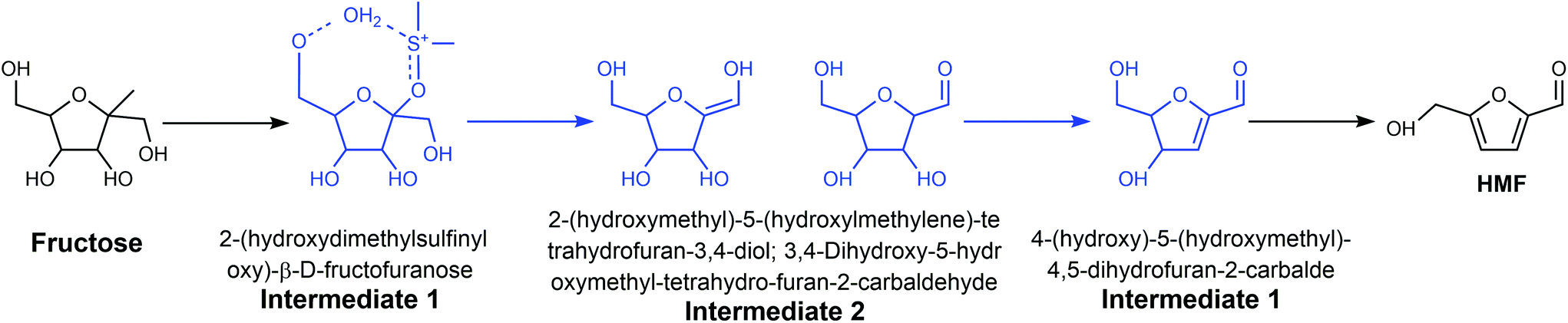 | ||
| Fig. 6 Three intermediates identified in the acid-catalyzed dehydration of fructose to HMF in the medium of DMSO. Adapted from ref. 80. | ||
The products from the conversion of carbohydrates in alcohols depend on the catalyst and structure of the alcohol since alcohols can react with sugars, HMF via etherification or acetylation, and levulinic acid via esterification (Fig. 7). When HCl was used as the catalyst, the use of isopropanol and tert-butanol as the solvent enabled the selective conversion of fructose to HMF.85 In contrast, the use of an ethanol, n-propanol and n-butanol solvent gave a mixture of HMF and 5-alkoxymethylfurfural (AMF), while the use of methanol as the solvent led to a series of ethers with a small amount of HMF. An HMF yield of 68% was obtained from fructose in the medium of isopropanol using NH4Cl as the catalyst.86 Kuo et al investigated the conversion of fructose using TiO2 nanoparticles as the catalyst in various solvents.87 When the reaction was conducted in tetrahydrofuran (THF), CH3CN, N,N-dimethylformamide (N,N-DMF), DMSO, or DMA, the main product was HMF with a yield ranging between 19–54%. The reaction conducted in ethanol, n-propanol and n-butanol primarily gave levulinic ester via either the rehydration of HMF-ether to form levulinic ester, or rehydration of HMF to levulinic acid followed by esterification to form levulinic ester, while the predominant products in the medium of 2-propanol, 2-butanol, and tert-butanol were HMF ether probably owing to the high barrier of the rehydration reaction owing to steric hindrance. The intrinsic reactivity of sugars also influences the reaction pathways and product distributions. For example, psicose and tagatose exhibited higher selectivity toward HMF and 5-(methoxymethyl)furfural (MMF) (combined yield of 55%) than other 2-ketohexoses, including fructose and sorbose, in an H2SO4-catalyzed reaction in methanol.88
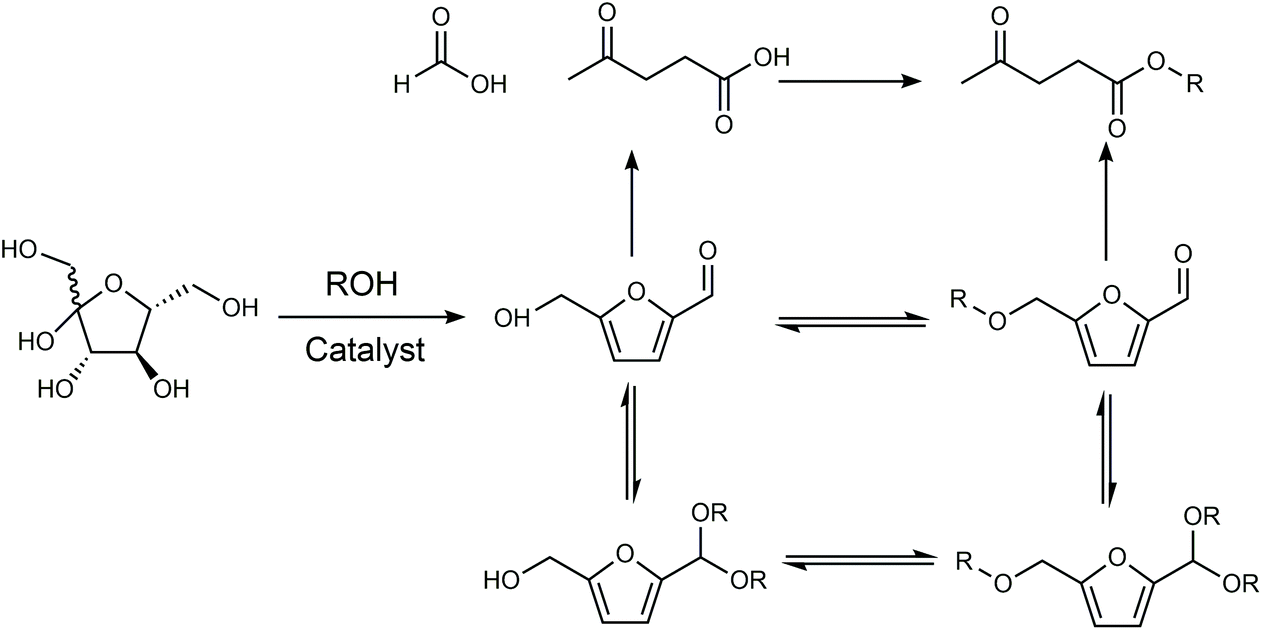 | ||
| Fig. 7 Reaction pathway of carbohydrates in alcohols. Adapted from ref. 87. | ||
The application of biomass-derived γ-valerolactone (GVL) in the Brønsted acid-catalyzed dehydration of sugars and other biomass-derived oxygenates was systematically investigated by the research group of Professor James A. Dumesic, revealing the universal solvent effect in biomass conversion.60,91–103 Mellmer et al. demonstrated that the use of a solvent system consisting of GVL and water in fructose dehydration could lead to a higher reaction rate and HMF selectivity.91 The combination of reaction kinetic studies and ab initio molecular dynamics simulations (AIMD) indicated that the solvation of biomass-derived oxygenates in the solvent system correlates well with the number of vicinal hydroxyl or oxygen-containing groups from these chemicals. Compared with the reaction performed in pure water, the use of the GVL/water solvent system led to a 450-fold increase in the fructose dehydration rate and only 10-fold increase in the HMF degradation rate owing to the better solvation of fructose than HMF. The following research by Mellmer et al. indicated that the addition of catalytic concentrations of inorganic salts, in particular chlorides, to the water/GVL solvent system can enable a further enhancement in the fructose to HMF conversion efficiency with a lower activation energy (Table 1).60 The use of 5 mM chloride salts led to a 10-fold increase in reactivity with an HMF yield of more than 80%. The reaction kinetic results and AIMD analysis indicated that Cl− plays an important role in the stabilization of the deprotonation transition state, thus leading to an enhancement in the fructose dehydration efficiency.
Mellmer et al. reported that the use of a solvent system consisting of water with polar aprotic cosolvents, including GVL, 1,4-dioxane and THF could increase the reaction rates and product selectivity for a series of Brønsted acid-catalyzed reactions, including xylose dehydration to furfural, 1,2-propanediol dehydration to propanal and cellobiose hydrolysis to glucose.92–94 The use of GVL as the solvent promoted the solvation of protons from the catalyst and then reduced the activation energy, thus leading to an increase in the reaction rate. This universal solvent effect was observed for both homogeneous and heterogeneous catalysts, such as H2SO4 and H-BEA zeolite. The reaction kinetics were studied to quantify the effect of polar aprotic organic solvents on the acid-catalyzed conversion of xylose into furfural.92 Among the tested solvents, GVL led to the most significant increase in the reaction rate compared to water, with an improvement in the furfural selectivity.95 The conversion of xylose in the water/GVL solvent system at high temperature (498 K) within around 2 min gave a high furfural yield (>90%), even at industrially relevant xylose concentrations (10 wt%).96 The kinetic model indicated that the improved furfural yield and selectivity at the evaluated temperature are mainly attributed to the reduced bimolecular condensation between xylose and furfural.
Besides the promotion effect for the dehydration of fructose, the GVL/water solvent system also plays an important role in the direct conversion of glucose to HMF. Song et al. observed the formation of moderate yields of HMF, fructose and levoglucosan (LGA) for glucose conversion in a hot-compressed GVL/water (HCGW) solvent system, where the reaction pathway and product selectivity could be tuned by adjusting the GVL concentration.97 Li et al. reported that the combined use of HCl as a catalyst and NaCl as a promoter in GVL/water (v/v = 4) system enabled a high HMF yield (62.45%) from glucose in the absence of additional catalysts responsible for glucose isomerization.104 The water/GVL solvent system could also convert biomass into soluble carbohydrates directly in a packed-bed flow-through reactor owing to the enhanced solubilization of biomass in the mixed solvent.98,99
Similar to the GVL/water system, Cao et al. found that the ratio of water and THF has an important influence on the reaction pathway and the product distribution for cellulose conversion.100,101 When the reaction was performed in pure THF using H2SO4 as the catalyst in the absence of water under relatively harsh reaction conditions (170–230 °C; 5–20 mM H2SO4), cellulose predominantly decomposed to LGA and then LGA was dehydrated to levoglucosenone (LGO), obtaining a maximum LGO yield of 51% (Fig. 8). In contrast, an HMF yield of up to 44% was obtained when the reaction was carried out under mild reaction conditions (140–190 °C; 5 mM H2SO4) in THF in the presence of a small amount of water (<2–3 vol%).102 The dehydration of LGA over a solid Brønsted acid in pure THF gave LGO,105 while the acid-catalyzed isomerization of LGO in the medium of water/THF solvent system gave HMF as the major product,106 which is an alternative reaction pathway for the production of HMF.
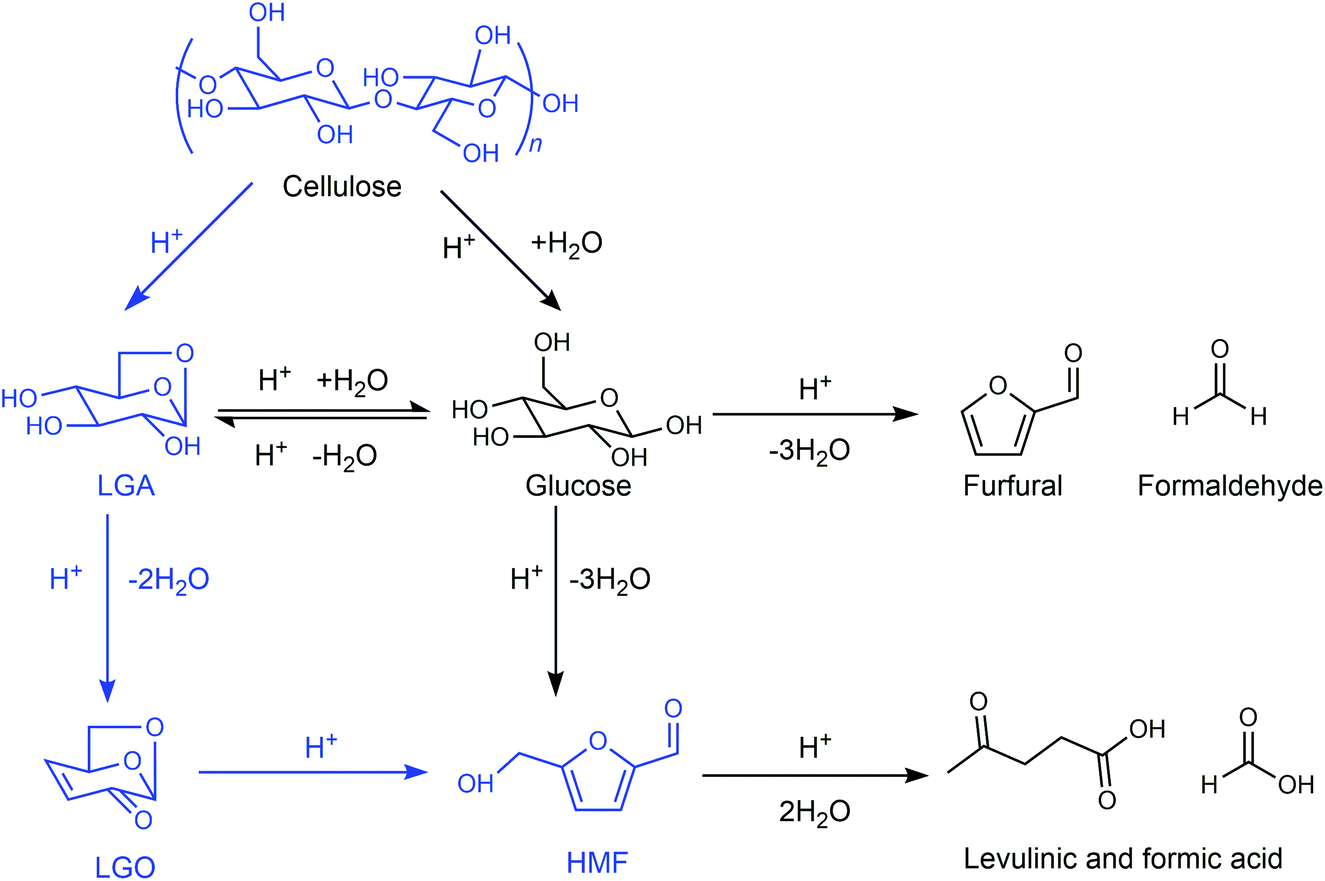 | ||
| Fig. 8 Reaction pathway for cellulose in the medium of THF. Adapted from ref. 100 and 101. | ||
Although GVL can lead to a high reaction rate and improved selectivity, the recycling of GVL is challenging owing to its high boiling point (207–208 °C). As an excellent alternative to water/GVL, the inexpensive solvent system composed of acetone and water also enabled the selective dehydration of fructose to HMF using a common Brønsted acid catalyst, affording an HMF yield of up to 85% even at a fructose loading of 5 wt%.103 Similarly, a mixture of hexafluoroisopropanol (HFIP), which has a low boiling point of 58 °C, and water could also serve as a monophasic reaction medium for the dehydration of fructose.107
Understanding solvent effects in multicomponent systems at the molecular level is of great importance for the screening and establishment of highly effective solvent systems. Combining the advantage of experiments, classical molecular dynamics simulations and machine learning tools, Walker et al. suggested that the screening of mixed solvent systems for biomass conversion over acid catalysts can employ the following workflow: (1) elucidate the reaction network and select candidate solvent systems; (2) compute the reaction rate of different steps; (3) compute the thermodynamic selectivity for the desired product; (4) verify model-predicted solvent systems by experiment; and (5) evaluate the solvent recyclability, cost and avaibility.89
 | ||
| Fig. 9 Conversion of carbohydrates to HMF in biphasic reaction systems. | ||
Owing to the low toxicity, biodegradability, and high partition coefficient of 2-methyltetrahydrofuran (MeTHF), which can be synthesized from biomass-derived furfural or levulinic acid, it is considered as a promising alternative to the petroleum-derived THF.17,45,69 Based on a comprehensive survey on the solvents used in biphasic systems for the synthesis of HMF, Esteban et al. screened 15 promising candidates using a conductor-like screening model for real solvents (COSMO-RS).112 Among them, ethyl acetate (EtOAc), n-propyl acetate and isopropyl acetate were the preferred solvents after considering performance, environmental, health and safety (EHS) impacts integrally. As an organic solvent with a low boiling point, dimethyl carbonate (DMC) is also effective to improve the HMF production and separation efficiency.113,114
| Solvent | Reaction conditions | Substrate | Substrate loadinga | HMF yield | Ref. |
|---|---|---|---|---|---|
| a Relative to the solvent. | |||||
| EMIMCl | 120 °C, 3 h | Fructose | 10 wt% | 74% | 116 |
| BMIMBr | 100 °C, 3 h | Fructose | 40 wt% | 95% | 119 |
| EMIMBr | 100 °C, 3 h | Fructose | 40 wt% | 93% | 119 |
| [GLY(mim)3][OMs]3 | 100 °C, 3 h | Fructose | 40 wt% | 72% | 120 |
| [GLY(mim)3][Cl]3 | 100 °C, 3 h | Fructose | 40 wt% | 37% | 120 |
| [GLY(mim)3][Br]3 | 100 °C, 3 h | Fructose | 40 wt% | 26% | 120 |
| [GLY(mim)3][PF6]3 | 100 °C, 3 h | Fructose | 40 wt% | 19% | 120 |
| [GLY(mim)3][OMs]3 | 100 °C, 3 h | Glucose | 40 wt% | 16% | 120 |
| [GLY(mim)3][OMs]3 | 100 °C, 3 h | Sucrose | 40 wt% | 57% | 120 |
| ChCl/Citric acid | 90 °C, 2 h | Sucrose | 40 wt% | 58.8% | 121 |
| ChCl/Malic acid | 90 °C, 2 h | Sucrose | 40 wt% | 60% | 121 |
| [HMIM]HSO4 | 180 °C, 1 min | Fructose | 3 wt% | 37.4% | 122 |
| [HMIM]HSO4 | 180 °C, 5 min | Glucose | 3 wt% | 17.3% | 122 |
| [HMIM]HSO4 | 180 °C, 10 min | Cellulose | 3 wt% | 12.1–19.8% | 122 |
| [HMIM]HSO4 | 180 °C, 5 min | Glucose | 3 wt% | 17.3% | 122 |
| [HMIM]HSO4 | 180 °C, 30 min, in situ removal of HMF | Fructose | 3 wt% | 77.3% | 122 |
| [HMIM]HSO4 | 180 °C, 5 min, in situ removal of HMF | Glucose | 3 wt% | 76.1% | 122 |
| [HMIM]HSO4 | 180 °C, 10 min, in situ removal of HMF | Cellulose | 3 wt% | 55.9–68.3% | 122 |
The use of a Brønsted acid catalyst in the BMIMCl ionic liquid can improve the fructose conversion efficiency considerably. For example, the use of the sulfonic ion-exchange resin Amberlyst 15 as the catalyst in BMIMCl gave an HMF yield of 82.2% with a fructose conversion of 100% at 120 °C within 1 min.123 The addition of cosolvents including acetone, DMSO, ethanol, methanol, ethyl acetate, and scCO2 to the BMIMCl/Amberlyst 15 catalytic system even enabled the efficient conversion of fructose to HMF at room temperature.124
Br-Containing ionic liquids, including N-methyl-2-pyrrolidone bromide ([NMP]Br), 1-butyl-3-methylimidazolium bromide (BMIMBr) and 1-ethyl-3-methylimidazolium bromide (EMIMBr) are more effective in catalyzing the dehydration of fructose to HMF than the corresponding Cl-based ionic liquids, with a lower activation energy (Table 1).13 Under the optimized conditions, the HMF yield up to 95% and 93% at fructose conversion approaching 100% could be obtained with BMIMBr and EMIMBr, respectively, in the absence of an additional acidic catalyst.119,125,126 Moreover, the reusability of BMIMBr was demonstrated for 6 runs of a recycling experiment. Employing DFT calculations, Li et al. suggested that the superior catalytic performance of BMIMBr for the dehydration of fructose to HMF is mainly attributed to the synergy of the cations and anions in BMIMBr, where the cations function as acid/base catalytic sites, and the anions can stabilize the intermediates and transition states by nucleophile covalent binding or H-bonding with the substrate, and the in situ-formed water serves as an H-shuttle.127
SO3H-based ionic liquids can be used to displace mineral acids to catalyze the dehydration of fructose to HMF.128 The direct conversion of glucose and cellulose in the [HMIM]HSO4 ionic liquid only gave a low HMF yield and selectivity.122 When the formed HMF was in situ removed from the catalytic system by on-line vacuum steam distillation, the conversion of glucose and cellulose gave high HMF yields of 77.3% and 55.9–68.3%, respectively, which is comparable to that obtained with fructose.
Alkaline ionic liquids have also been investigated as catalysts for the dehydration of fructose. For example, a catalytic amount of the alkaline ionic liquid 1-butyl-3-methylimidazolium hydroxide (BMIMOH) was active to catalyze the conversion of fructose to HMF using DMSO as the solvent under a nitrogen atmosphere, obtaining an HMF yield of 91.6% at 160 °C in 8 h, while the use of BMIMBr as the catalyst in DMSO gave a low HMF yield under the same conditions.129 After 3 h reaction at 160 °C, BMIMOH afforded an HMF yield of 54.4% from sucrose in the medium of DMSO.130
The introduction of functional groups in ionic liquids can also influence the conversion of sugars. For example, Siankevich et al. demonstrated that the use of appropriately functionalized IL mixtures could lead to an improved HMF production efficiency from glucose and cellulose over a CrCl2 catalyst under mild conditions with limited side-reactions compared with the single IL.131 The IL mixture consisted of [C2OHmim]Cl and modified IL, which has an additional CH2CH2Ph group on its imidazolium cation, affording an HMF yield of up to 92%. The strong hydrogen bond formed between the hydroxyl group in the [C2OHmim]Cl ionic liquid with glucose weakens and activates the C–O bond, leading to a reduction in the reaction activation energy.132 The interaction between the sugar and CH2CH2Ph group-modified IL led to an increase in the reaction rate for fructose isomerization from the linear to furanosyl form. Consequently, the formation of humins was remarkably suppressed owing to the absence of a carbonyl group, which is required for aldol condensation in the fructouranosyl form. The combination of a sulfonic acid group-modified IL, [C2OHmim]Cl and CH2CH2Ph-group modified IL was effective for the efficient transformation of cellulose into HMF, obtaining an HMF yield of up to 46%.
Electron microscopy revealed at the microscopic level that the nanostructured meshwork formed from the BMIMBF4 ionic liquid and water is beneficial for the high conversion of fructose and HMF selectivity in the H2SO4-catalyzed fructose dehydration reaction.133 In contrast, the use of BMIMBF4 and EMIMBF4 as the reaction medium exhibited an adverse influence on the conversion of carbohydrates to HMF over the SnCl4 catalyst.125 Qu et al. reported that a hydroxy-functionalized ionic liquid, 1-hydroxyethyl-3-methylimidazolium tetrafluoroborate ([AEMIM]BF4), in the medium of DMSO showed higher catalytic activity than [EMIM]OH, [BMIM]OH and [MIMPS]3PW12O40 for the conversion of sucrose to HMF, affording an optimized HMF yield of 68.71% at 160 °C for 6 h.130 Under the same conditions, the conversion of cellobiose only yielded 24.73% of HMF. Under microwave irradiation, 1,1,3,3-tetramethylguanidine tetrafluoroborate ([TMG][BF4]) in DMAc/LiCl exhibited a better catalytic performance than oil heating, obtaining an HMF yield of 28.63% under the optimal conditions.134
The noncoordinating ionic liquid [BMIM][OTf] is not a suitable solvent for the conversion of fructose to HMF owing to the instability of HMF in [Bmim][OTf] under acidic conditions. Ghatta et al. found that adding an optimized amount of water to [Bmim][OTf] could inhibit the side reaction remarkably either using p-toluene sulfonic acid (PTSA) or HCl as the catalyst, thus leading to HMF yields higher than 80% even at a fructose loading of up to 14 wt%.135 Alam et al. reported that the catalytic activity of ILs for the conversion of wood ear mushroom to HMF increases in the following order: [BBIM-SO3][NTf2] > [BBIM-SO3][OTf] ≈ [DMA][CH3SO3] > [NMP][CH3SO3], corelating well with their proton donating ability.136
The separation of HMF from ionic liquids is rather challenging owing to their hydrogen bond interaction, which can be formed either between anions and HMF or between cations and HMF.137 Similar to water–organic biphasic systems, biphasic systems composed of an ionic liquid reactive phase and organic extraction phase (Fig. 9) have been developed to inhibit the degradation of HMF, and then improve the HMF production efficiency, but the extraction of HMF from ionic liquids is difficult due to the strong hydrogen bonds between HMF and ionic liquids. Zhou et al. reported that the addition of functional promoters such as methanol, ethanol and acetonitrile (CH3CN) to the BMIMCl/MIBK biphasic system could disrupt the hydrogen bonding between HMF and BMIMCl, leading to an enhanced HMF extraction efficiency.138,139 In addition, Chen et al. demonstrated that the bubbling effect is necessary for the rapid transfer of HMF from the reaction phase to the extraction phase.139
2.3. Homogenous catalysts for the production of HMF from biomass
A variety of homogeneous catalysts including metal chlorides, mineral acids, organic acids and bases (Table 3) have been investigated for the dehydration of carbohydrates in various reaction media. For the historical development of homogeneous catalytic systems for the synthesis of HMF, the reader is referred to the earlier reviews.17,42,144 In this section, we review the recent advances in homogeneous catalytic systems with emphasis on the underlying mechanism and actual role of various homogeneous species.| Catalyst | Solvent | Reaction conditions | Substrate loadingb | Conversion | HMF yieldg | Ref. |
|---|---|---|---|---|---|---|
| a Relative to monosaccharide. b Relative to total solvent. c Relative to water. d Relative to glycolic acid. e pH = 7.5. f 1 mmol of natural deep eutectic solvents composed of equimolar betaine/HCl, carboxylic acid and H2O combined with 3 mL of ethyl acetate. g HMF yield if unspecified. — Not provided. | ||||||
| AlCl3 10 mol%a | [EMIM]Cl | 120 °C, 6 h | Glucose, 20 wt% | — | 1.6% | 145 |
| MeAlCl2 10 mol%a | [EMIM]Cl | 120 °C, 6 h | Glucose, 20 wt% | — | 7.6% | 145 |
| Et2AlCl 10 mol%a | [EMIM]Cl | 120 °C, 6 h | Glucose, 20 wt% | — | 17% | 145 |
| AlEt3 10 mol%a | [EMIM]Cl | 120 °C, 6 h | Glucose, 20 wt% | — | 51% | 145 |
| LRAlMe2 5 mol%a | [BMIM]Br | 120 °C, 2 h | Glucose, 9.1 wt% | 97% | 58–60% | 146 |
| AlCl3 50 mol%a | LiBr aqueous solution (0.97 g mL−1) /ethyl acetate (v/v = 1/3) | 120 °C, 2 h | Glucose, 0.54 wt% | ∼82% | ∼78% | 147 |
| AlCl3 50 mol%a (3 runs) | LiBr aqueous solution (0.97 g mL−1) /ethyl acetate (v/v = 1/3) | 120 °C, 2 h | Glucose, 0.54 wt% | ∼40% | ∼20% | 147 |
| Al2(SO4)3 0.34 wt%b | GVL/H2O (4/1), 40 g L−1 NaCl | 165 °C, 50 min | Cellulose, 0.97 wt% | — | 43.5% | 148 |
| Al2(SO4)3 0.34 wt%b | GVL/H2O (4/1), 40 g L−1 NaCl | 165 °C, 40 min | Cellulose, 4.32 wt% | — | 36.1% | 148 |
| Al2(SO4)3 0.34 wt%b | GVL/H2O (4/1), 40 g L−1 NaCl | 165 °C, 50 min | Cellulose, 8.64 wt% | — | 19.6% | 148 |
| H2SO4 0.5% w/vb | DMSO/H2O | 150 °C, 2 h | Chlorella sorokiniana, 1.8 wt% | — | 24% | 149 |
| H2SO4 0.5% w/vb | DMSO/LiCl/H2O | 150 °C, 2 h | Chlorella sorokiniana, 1.8 wt% | — | 52% | 149 |
| H2SO4 0.5% w/vb | DMSO/NaCl/H2O | 150 °C, 2 h | Chlorella sorokiniana, 1.8 wt% | — | 44.6% | 149 |
| H2SO4 0.5% w/vb | DMSO/LiCl/H2O | 150 °C, 2 h | Sugar cane bagasse, 1.8 wt% | — | 1% | 149 |
| CrCl3 25 mol%a | [BMIM]Cl | 120 °C, 5 h | Microcrystalline cellulose, 5 wt% | — | 58.4% | 61 |
| CrCl3 25 mol%, AlCl3 2.5 mol%a | [BMIM]Cl | 120 °C, 3 h | Microcrystalline cellulose, 5 wt% | — | 58.3% | 61 |
| CrCl3 25 mol%, AlCl3 2.5 mol%a | [BMIM]Cl | 120 °C, 3 h | Cotton, 5 wt% | — | 59.5% | 61 |
| CrCl3 25 mol%, AlCl3 2.5 mol%a | [BMIM]Cl | 120 °C, 3 h | Filter paper, 5 wt% | — | 54.7% | 61 |
| CrCl3 6.8 mol%a | [BMIM]Cl/toluene (5/22) | 130 °C, 3 h | Microcrystalline cellulose, 1.9 wt% | — | 55% (27% glucose) | 61 |
| ZrOCl2 5 wt%b; [HO2CMMIm]Cl 1.7 wt%b | Isopropanol | 150 °C, 3 h | Glucose, 1.7 wt% | 90% | 43% | 150 |
| [DMA][CH3SO3] 0.5 wt%b | DMA-LiCl | 140 °C, 10 min, microwave | Wood ear mushroom, 5 wt% | — | 44% | 136 |
| [BBIM-SO3][NTf2] 0.5 wt%b | DMA-LiCl | 140 °C, 10 min, microwave | Wood ear mushroom, 5 wt% | — | 44% | 136 |
| SnCl4·5H2O 10 mol%a | EMIMBr | 100 °C, 3 h | Glucose, 10 wt% | 100% | 64.5% | 125 |
| SnCl4·5H2O 40 mol%a | EMIMBr | 100 °C, 1 h | Glucose, 40 wt% | 100% | 56% | 125 |
| SnCl4·5H2O 10 mol%a | EMIMBr/GDE (w/w = 1/4) | 100 °C, 3 h | Glucose, 10 wt% | 100% | 58.7% | 125 |
| SnCl4·5H2O 0.056 Ma | DMSO/H2O (v/v = 1) | Microwave, 140 °C, 40 min | Cooked rice, 5 wt% | — | 22.7 wt% | 151 |
| Cu(NO3)2 0.3 wt%b | H2O, pH = 5.5, 30 bar N2 | 110 °C, 1.5 h | Glucose, 1 wt% | 18% | Fructose, 16% | 152 |
| HCl 0.075 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 36% | 153 |
| H2SO4 0.0375 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 15% | 153 |
| HNO3 0.075 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 16% | 153 |
| Li2SO4 0.113 Mb, H2SO4 0.0375 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 70% | 153 |
| Na2SO4 0.113 Mb, H2SO4 0.0375 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 76% | 153 |
| K2SO4 0.113 Mb, H2SO4 0.0375 Mb | H2O/THF(v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 66% | 153 |
| KCl 0.225 Mb, HCl 0.075 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 44% | 153 |
| KNO3 0.225 Mb, HNO3 0.075 Mb | H2O/THF (v/v = 1/4) | 190 °C, 1 h | Corn stover | — | 0 | 153 |
| LiCl 33.3 mol%c | H2O | 120 °C, 15 min | Glucose, 1 wt% | 9.5% | Fructose, 7.7% | 154 |
| LiBr 33.3 mol%c | H2O | 120 °C, 15 min | Glucose, 1 wt% | 51.8% | Fructose, 30.3% (mannose 1.4%) | 154 |
| LiI 33.3 mol%c | H2O | 120 °C, 15 min | Glucose, 1 wt% | 90.2% | Fructose, 21.4% (mannose 3.8%) | 154 |
| CaBr2 16.7 mol%c | H2O | 120 °C, 15 min | Glucose, 1 wt% | 94.4% | Fructose 22.4%, mannose 2.2% | 154 |
| ZnCl2 33.3 mol%c | H2O | 120 °C, 15 min | Glucose, 1 wt% | 33.9% | Fructose, 0 | 154 |
| [Hnmp]Zn2Cl5 500 mol%a | BMIMCl/DMSO (w/w = 1/7) | 130 °C, 8 h | Cellulose, 1.4 wt% | — | 39.29% | 155 |
| NaCl 4.3 wt%b | H2O/THF (v/v = 1/6) | 200 °C, 8 h | Cellulose, 0.71 wt% | 99.9% | 58.9% | 156 |
| H3BO3 2.75 wt%b | [MIM]Cl | 120 °C, 3 h | Glucose, 10 wt% | 95% | 19% | 157 |
| H3BO3 2.75 wt%b | [EMIM]Cl | 120 °C, 3 h | Glucose, 10 wt% | 95% | 41% | 157 |
| H3BO3 2.75 wt%b | [BMIM]Cl | 120 °C, 3 h | Glucose, 10 wt% | 47% | 14% | 157 |
| H3BO3 2.75 wt%b | [HMIM]Cl | 120 °C, 3 h | Glucose, 10 wt% | 68% | 32% | 157 |
| H3BO3 2.75 wt%b | [OMIM]Cl | 120 °C, 3 h | Glucose, 10 wt% | 63% | 26% | 157 |
| H3BO3 100 g L−1b, NaCl 50 g L−1b |
Water/MIBK (v/v = 0.25) | 150 °C, 1.5 h | Fructose, 6 wt% | 93% | 60% | 158 |
| H3BO3 100 g L−1b, NaCl 50 g L−1b |
Water/MIBK (v/v = 0.25) | 150 °C, 6 h | Glucose, 6 wt% | 41% | 14% | 158 |
| H3BO3 80 mol%d | Choline chloride/glycolic acid (molar ratio = 1) | 140 °C, 2 h | Glucose 8 mol%d | — | 0 | 159 |
| H3BO3 80 mol%d | Dihydrogen citrate/glycolic acid (molar ratio = 1) | 140 °C, 2 h | Glucose 8 mol%d | — | 42% | 159 |
| H3BO3 1 wt%b | [BMIM]Cl | 120 °C, 30 min | Glucose, 10 wt% | — | 1.2% | 160 |
| H3BO3 1 wt%b | [BMIM]Cl | 140 °C, 40 min | Glucose, 10 wt% | — | 0.76% | 161 |
| 12-TPA 1 wt%b | [BMIM]Cl | 140 °C, 40 min | Glucose, 10 wt% | — | 23.5% | 161 |
| 12-TPA 1 wt%b, H3BO3 0.5 wt%b | [BMIM]Cl | 140 °C, 40 min | Glucose, 10 wt% | — | 51.9% | 161 |
| 12-TPA 1 wt%b, H3BO3 0.5 wt%b | TEAC | 140 °C, 40 min | Glucose, 10 wt% | — | 47.6% | 161 |
| 2-Methoxycarbonylphenylboronic acid | DMA | 100 °C, 6 h | Glucose, 10 wt% | — | 22% | 162 |
| MgCl2 200 mol%a, 2-methoxycarbonylphenylboronic acid 100 mol%a | DMA | 120 °C, 4 h | Glucose, 10 wt% | — | 57% | 162 |
| MgCl2 300 mol%a, HCl 0.61%b, 2-methoxycarbonylphenylboronic acid 120 mol%a | [EMIM]Cl | 105 °C, 2 h | Cellulose, 5 wt% | — | 39% | 162 |
| ChCl 2 mMb, H3BO3 4 mMb | H2O (2.6 M NaCl)/MIBK (v/v = 1/5) | 195 °C, 1 h | Starch, 2 wt% | — | 35.9% | 163 |
| ChCl 2 mMb, H3BO3 4 mMb | H2O (2.6 M NaCl)/THF (v/v = 1/5) | 195 °C, 1 h | Starch, 2 wt% | — | 60.3% | 163 |
| ChCl 2 mMb, H3BO3 4 mMb (6 runs) | H2O (2.6 M NaCl)/MIBK (v/v = 1/5) | 195 °C, 1 h | Starch, 2 wt% | — | 19% | 163 |
| ChCl 2 mMb, H3BO3 4 mMb (10 runs) | H2O (2.6 M NaCl) /THF (v/v = 1/5) | 195 °C, 1 h | Starch, 2 wt% | — | 21.7% | 163 |
| Sulfamic acid 0.7 Mb | H2O | 200 °C, 2 min | Chitosan, 3 wt% | — | 21.48% | 164 |
| [PSMIM]HSO4 0.9 wt%b, ZnSO4 0.06 Mb | THF/water (v/v = 20/2) | 160 °C, 1 h | Cellulose, 2.3 wt% | — | 58.8% | 165 |
| NaHSO4 4.9 g L−1b, ZnSO4 18.3 g L−1b |
THF/water (v/v = 20/2) | 160 °C, 1.5 h | Radiata pine hydrolysate | — | 40% (furfural 58%) | 166 |
| H3PO4 1 wt%c | Acetone/H2O (v/v = 2/1), NaCl 35 wt%c | 180 °C, 80 min | Radiata pine hydrolysate | — | 36% | 166 |
| H3PO4 1 wt%c | Acetone/H2O (v/v = 2/1), NaCl, NaCl 35 wt%c | 170 °C, 30 min | Radiata pine hydrolysate | — | Furfural 62% | 166 |
| Na2HPO4/NaH2PO4e | H2O | 110 °C | Glucose, 10 wt% | 52% | Fructose 30% | 167 |
| KH2PO4, 2 wt%b | H2O/MIBK (v/v = 1/4) | 160 °C, 2 h | Sucrose, 5.4 wt% | 100% | 75.5% | 168 |
| KH2PO4, 2 wt%b | H2O | 160 °C, 1 h | Glucose, 1.8 wt% | 49% | Fructose 13.5% | 168 |
| K2HPO4, 2 wt%b | H2O | 120 °C, 1 h | Glucose, 1.8 wt% | 81% | Fructose 34.4% | 168 |
| Betaine/HCl, 1.8 wt%b | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Fructose, 2.7 wt% | — | 66% | 169 |
| Betaine/H2SO4, 2.7 wt%b | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Fructose, 2.7 wt% | — | 68% | 169 |
| Betaine/(C6H5)SO3H, 2.7 wt%b | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Fructose, 2.7 wt% | — | 69% | 169 |
| Betaine/p-CH3(C6H4)SO3H, 3.6 wt%b | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Fructose, 2.7 wt% | — | 66% | 169 |
| Betaine/HCl, 1.8 wt%b | ChCl/H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Fructose, 2.7 wt% | — | 87% | 169 |
| Betaine/H2SO4, 2.7 wt%b | ChCl/H2O/MIBK | 130 °C, 1 h | Fructose, 2.7 wt% | — | 85% | 169 |
| Betaine/(C6H5)SO3H, 2.7 wt%b | ChCl/H2O/MIBK | 130 °C, 1 h | Fructose, 2.7 wt% | — | 85% | 169 |
| Betaine/p-CH3(C6H4)SO3H, 3.6 wt%b | ChCl/H2O/MIBK | 130 °C, 1 h | Fructose, 2.7 wt% | — | 88% | 169 |
| Choline-O-sulfate, 3.6 wt%b | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Fructose, 2.7 wt% | — | 61% | 169 |
| Betaine/HCl, 1.8 wt%b | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Glucose, 2.7 wt% | — | 3% | 169 |
| Betaine/HCl 2.7 wt%b, CuCl2·2H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Glucose, 2.7 wt% | — | 22% | 169 |
| Betaine/HCl 2.7 wt%b, FeCl3·6H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Glucose, 2.7 wt% | — | 29% | 169 |
| Betaine/HCl 2.7 wt%b, MgCl2·6H2O 16.7 wt%a | H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Glucose, 2.7 wt% | — | 4% | 169 |
| Betaine/HCl 2.7 wt%b, SnCl4·5H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK (v/v = 1/10) | 130 °C, 1 h | Glucose, 2.7 wt% | — | 26% | 169 |
| Betaine/HCl 2.7 wt%b, AlCl3·6H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 58% | 169 |
| Betaine/HCl 2.7 wt%b, AlCl3·6H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 64% | 169 |
| Betaine/HCl 2.7 wt%b, NiCl2·6H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 37% | 169 |
| Betaine/HCl 2.7 wt%b, LaCl3·nH2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 39% | 169 |
| Betaine/HCl 2.7 wt%b, CaCl2·2H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 14% | 169 |
| Betaine/HCl 2.7 wt%b, MnCl2·4H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 12% | 169 |
| Betaine/HCl 2.7 wt%b, CoCl2·6H2O 16.7 wt%a | ChCl 2.7 wt%b, H2O/MIBK | 130 °C, 1 h | Glucose, 2.7 wt% | — | 18% | 169 |
| Betaine/HCl 100 mol%c, malic acid 100 mol%c | H2O/ethyl acetatef | Microwave, 140 °C, 11 min | Fructose | — | 94% | 170 |
| Betaine/HCl 100 mol%c, malic acid (5 runs) 100 mol%c | H2O/ethyl acetatef | Microwave, 140 °C, 11 min | Fructose | — | 35% | 170 |
| Betaine/HCl 100 mol%c, malic acid 100 mol%c | H2O/ethyl acetatef | Microwave, 160 °C, 11 min | Sucrose | — | 72% | 170 |
| Betaine/HCl 100 mol%c, L-tartaric acid 100 mol%c | H2O/ethyl acetatef | Microwave, 160 °C, 11 min | Glucose | — | 20% | 170 |
| Betaine/HCl 100 mol%c, L-tartaric acid 100 mol%c | H2O/ethyl acetatef | Microwave, 160 °C, 11 min | Sucrose | — | 20% | 170 |
| EDTA 50 mol%a | H2O/n-butanol (v/v = 1/4) | 160 °C, 2 h | Fructose, 1.8 wt% | — | 61% | 171 |
| EDTA 50 mol%a | H2O/MIBK (v/v = 1/4) | 160 °C, 2 h | Fructose, 1.8 wt% | — | 67% | 171 |
| EDTA 50 mol%a | H2O/MeTHF (v/v = 1/4) | 160 °C, 2 h | Fructose, 1.8 wt% | — | 66% | 171 |
As one of the most outstanding milestones in the production of HMF, Zhang and co-workers first reported that CrCl2 and CrCl3 can convert glucose to HMF directly in the medium of EMIMCl ionic liquid, affording HMF yields of approximately 73% and 45%, respectively, while FeCl3, CuCl, CuCl2, VCl3, MoCl3, PdCl2, PtCl2, PtCl4, RuCl3, RhCl3 and H2SO4 are not effective in catalyzing this conversion.116 They also observed that EMIMCl itself was capable of converting fructose to HMF, but HMF was unstable in pure EMIMCl under the reaction conditions. Fructose was identified as an the main intermediate for the dehydration of glucose to HMF over CrCl2 and CrCl3 in the [EMIM]Cl ionic liquid.172 Therefore, CrCl2 and CrCl3 are not only responsible for the glucose-to-fructose isomerization step, but also play an important role for in the stabilization of HMF in EMIMCl. Compared with EMIMCl and BMIMCl, the use of DBU-based (DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene) ionic liquids as a solvent could further improve the HMF yield and selectivity for the conversion of glucose over CrCl3.173,174 In the medium of water, CrCl3 and AlCl3 are active for the isomerization of glucose to fructose, FeCl3 and CuCl2 are mediocre catalysts, while MgCl2 is inactive.175
The mechanism of CrCl3-catalyzed glucose isomerization to fructose in the medium of the BMMCl ionic liquid was investigated by Jia and co-authors using in situ far-infrared analysis (FIR).117 It was found that the catalytic performances of metal chlorides correlate well with their different coordination structures formed with the oxygen atoms of the substrate. The superior activity of CrCl3 for the conversion of glucose to HMF in comparison with VCl3, FeCl3, and PtCl2 is mainly attributed to its preferential coordination with the glycolaldehyde group of glucose. Moreover, the relatively weak interactions of Cr(III) with the isolated hydroxy and carbonyl groups enable the good stability of HMF in the ionic liquid/CrCl3 system. VCl3 forms strong V–O coordination bonds with oxygens from both glucose and HMF, thus dominantly promoting the formation of humins. PtCl2 cannot convert glucose efficiently owing to its weak coordination ability, while the poor catalytic performance of FeCl3 is mainly ascribed to the strong and irreversible coordination bonds formed indiscriminately between FeCl3 and any oxygen-containing compounds.
13C NMR and 1H NMR were used to elucidate the mechanisms of glucose isomerization, epimerization, and other interconversions over CrCl3 in aqueous solution.176 It was found that the isomerization of glucose to fructose proceeds via a C2–C1 intramolecular hydride transfer process, despite the wide range of catalytic activity. For the epimerization of glucose to mannose, reverse C2–C1 hydride transfer and C1–C2 intramolecular carbon shift mechanisms (the Bilik reaction) occur simultaneously, where the former mechanism dominates the conversion process. Besides, it was observed that the metal chlorides, in particular chromium(III), are also active for the isomerization of glucose to sorbose though the C5–C1 hydride transfer process, which is similar to that reported for Ti-BEA. Jia et al. reported that in aqueous solution, CrCl3·6H2O mainly catalyzed the isomerization of glucose to fructose via the coordination of the chromium species with the end aldehyde group in the chain form of glucose.177 Choudhary et al. identified that the [Cr(H2O)5OH]2+ species formed from CrCl3 hydrolysis is active to catalyze the isomerization of glucose to fructose with an activation energy (64.4 kJ mol−1) lower than many catalysts (Table 1).56
In addition to CrCl2 and CrCl3, other Cr species have also been used for the conversion of glucose to HMF in ionic liquids. For example, a series of Cr(III)-NHC (NHC = N-heterocyclic carbene) complexes were demonstrated to be active for the direct dehydration of glucose to HMF in the BMIMCl ionic liquid and DMSO.178,179 Zhang et al. reported that the small, uniform Cr(0)-nanoparticles formed from Cr(CO)6 either under microwave irradiation (3.6 ± 0.7 nm) or generated in situ via thermolysis (2.3 ± 0.4) during the reaction are strong Lewis acids, which are responsible for the conversion of glucose to HMF.180
The combination of CrCl3 and HCl in water/THF/NaCl afforded a maximum HMF yield of 59% from glucose.56 In the medium of EMIMCl, BMIMCl DMA-LiCl, or EMIMCl-DMA-LiCl ionic liquid, the combination of CrCl2 (or CrCl3) with HCl enabled the direct conversion of cellulose to HMF.181,182 CrCl2 and CrCl3 are also active for the conversion of hemicellulose-derived mannose to HMF in [EMIM]Cl or N,N-dimethylacetamide (DMA), but the conversion efficiency of galactose, galactose, lactose and tagatose are relatively low.183 Under microwave heating, the [BMIM]Cl/CrCl3 catalytic system enabled the fast conversion of glucose, fructose, sucrose, cellobiose, and cellulose, affording HMF yields of 71% and 54% from glucose and cellulose in 30 s and 10 min, respectively.184
Thus, it can be concluded that CrCl3 is capable of converting various carbohydrates to HMF efficiently in aqueous solution, organic solvents, ionic liquids, and biphasic systems, and is compatible with other Lewis acid and Brønsted acid co-catalysts. Compared with CrCl3, other metal chlorides, such as SnCl4 and AlCl3 only show good catalytic performances in certain reaction media. Thus, the selection of suitable solvents and co-catalysts is critical for the improvement in catalytic performance.
SnCl4 can convert glucose, cellobiose, maltose and starch into HMF in the [EMIM]Br and [BMIM]Br ionic liquids, but is ineffective in [EMIM]Cl, [BMIM]Cl, [EMIM]BF4, [BMIM]BF4 and DMSO.125 Moreover, a biphasic system consisting of glycol dimethyl ether or dimethyl carbonate as the extraction phase and EMIMBr/SnCl4 as the reaction phase also enabled the efficient conversion of carbohydrate to HMF, obtaining HMF yields between 49.0% and 65.7%. However, the direct conversion of cellulose in the EMIMBr/SnCl4 catalytic system gives low HMF yield, possible owing to the low solubility of cellulose in EMIMBr. The dissolution of SnCl4 in water produces an acid solution with a pH of about 1.8 owing to the hydrolysis of SnCl4 to the mononuclear species Sn(OH)y(4 − y)+ and H+.185 In aqueous solution, SnCl4 is effective in converting sucrose, maltose, cellobiose, waste cooked rice and bread crusts to monosaccharide and HMF,186 and converting glucose to HMF.185 At evaluated temperature (150 °C), SnCl4 could catalyze the one-pot conversion of inulin and cellulose to levulinic acid, mainly owing to synergistic effect of stannic oxide, H+ and Cl− formed from the hydrolysis of SnCl4.187 In the medium of ZnCl2 molten salt hydrate, the use of SnCl4 also promoted the one-pot conversion of inulin to levulinic acid.188 Besides, the conversion of cellulose in methanol over a tin(II) triflate catalyst yielded a mixture of α-hydroxy esters, including methyl lactate, methyl vinyl glycolate and methyl-4-methoxy-2-hydroxybutanoate, and methyl levulinate.189
When AlCl3 (Table 3) was used to catalyze the conversion of glucose in the medium of EMIMCl, only an HMF yield of 1.6% was obtained.145 Compared with AlCl3, aluminum alkyl or alkoxy compounds, AlEt3 gives a much higher HMF yield. Density functional theory (DFT) calculations indicated that the metal–ligand functionality of the Al(OMe)3 catalyst exhibit both Lewis acid and Lewis base properties, which are responsible for its good catalytic performance.190 Saang'onyo et al. demonstrated that the air-stable dimethyl aluminum complexes LRAlMe2, which contain (aminomethyl)phenolate (LR) ligands could give an HMF yield of 58% from glucose using BMIMBr as the solvent.146
In the AlCl3–H2O(NaCl)/THF biphasic system, the Brønsted acid (H+) resulting from the hydrolysis of AlCl3 is not only responsible for the dehydration of fructose to HMF, but also active for a series of undesirable side-reactions, including the direct degradation of glucose and fructose to formic acid, the polymerization of HMF to humins, and the rehydration of HMF to formic acid and levulinic acid.57 The Lewis acidic [Al(OH)2(aq)]+ species is the predominant catalytic center for the reversible isomerization of glucose to fructose, but its presence also induces the direct degradation of HMF to formic acid.58 Moreover, both Brønsted and Lewis acids can catalyze the polymerization of glucose/fructose to humins and the dehydration of fructose to HMF. A low pH value in aqueous solution improved the rate of AlCl3-catalyzed glucose dehydration in biphasic reaction systems composed of NaCl saturated aqueous solution and 2-sec-butyl phenol (SBP).191 The combination of maleic acid and AlCl3 is more effective than that of HCl and AlCl3 for the conversion of glucose toward HMF and levulinic acid in water.192 The degradation of HMF to humins using maleic acid and AlCl3 as the catalyst was reduced by 50% compared with the catalytic system using HCl and AlCl3 as the catalyst.193 The catalytic complex Al-(MA)2-(OH)2 (aq) formed from maleic acid and AlCl3 has a lower activation energy (95 kJ mol−1) than that (149 kJ mol−1) of HCl and AlCl3 for the glucose-to-fructose isomerization. Simultaneously, the fructose conversion rate is 1.7 times faster than HCl and AlCl3 with 3 times higher selectivity toward HMF.
Under microwave irradiation, AlCl3 in water, water–MIBK biphasic solvent and DMSO gave HMF yields of 37.3%, 43.0%, and 52.4%, respectively, within just 5 min.194 Under the same conditions, the AlCl3/DMSO system could also convert starch and inulin, attaining HMF yields of 30.6% and 39.6%, respectively. In the medium of DMA, the addition of NaF inhibits the AlCl3-catalyzed glucose conversion to HMF, while NaI and NaBr promote the reaction significantly.195 When NaI was used as an additive, the HMF yield improved from 36% to 62% with the glucose conversion increasing from 71% to 86%. In the medium of H2O/DMSO, AlCl3 is more effective than other metal chlorides, such as CrCl3 and SnCl4 for the conversion of underused hexoses to HMF, obtaining HMF yields of 60% and 35% from hemicellulose-derived mannose and galactose, respectively.196,197 In the medium of a DMSO/BMIMCl mixture, the direct conversion of cellulose to HMF could be achieved over AlCl3, giving an HMF yield of 54.9% at 150 °C after 9 h.198 The ball milling of cellulose and Al2(SO4)3 combined with subsequent catalytic reaction in water/GVL/NaCl gave an HMF yield of 43.5%.148 In this process, Al2(SO4)3 is not only helpful for the disruption of the hydrogen bonds in the cellulose molecules during mechanochemical pretreatment, but also serves as a Lewis–Brønsted acid bifunctional catalyst in the conversion of cellulose to HMF.
Cai et al. found that although FeCl3 is not as effective as CrCl3 and AlCl3 for the dehydration of glucose to HMF and the dehydration of xylose to furfural, the use of FeCl3 as a catalyst in water/THF (4:1) mixture achieved a furfural yield of 95% with HMF yield of 51% directly from biomass, which is remarkably higher than that obtained with CrCl3 and AlCl3.199 Yan et al. reported that in the medium of water, transition metal chlorides, including FeCl3, RuCl3, VCl3, TiCl3, MoCl3, and CrCl3 are active to convert cellulose to levulinic acid at the evaluated temperature (220 °C), while in the medium of the water/butanol/NaCl biphasic system, these catalysts, in particular RuCl3, are active for the conversion of cellulose to HMF with a yield as high as 83.3%.200 They proposed that RuCl3 could promote the disruption of hydrogen bonds and C–O–C bonds in cellulose and the subsequent glucose dehydration to HMF, and the water/butanol/NaCl biphasic system could reduce the undesirable degradation of HMF.
Germanium(IV) chloride (GeCl4) is effective to catalyze the conversion of fructose, glucose, sucrose, maltose and cellulose to HMF in the [BMIM]Cl ionic liquid.201 An HMF yield of 92% was obtained from fructose within 5 min at 100 °C. [BMIM]Cl/GeCl4 gave HMF yields of 38.4% and 35% from glucose and cellulose, respectively. In addition, the removal of water from the reaction system with 5 Å molecular sieves further improved the HMF yield from 38.4% to 48.4%. The conversion of glucose, cellobiose and cellulose to HMF was also conducted under mild conditions in the BMIMCl/HfCl4 system (100 °C, 2 h), obtaining HMF yields of 34.5%, 31.7% and 33.4%, respectively.202 It was deduced that the Lewis acidic sites from HfCl4 could disrupt the glycosidic bonds via binding with a glycosidic oxygen atom in a similar manner as protonic acid, resulting in the hydrolysis of polysaccharides to glucose.
Lanthanide-based catalysts, including CeCl3, PrCl3, NdCl3, DyCl3, YbCl3, and Yb(OTf)3 have been investigated for the dehydration of glucose to HMF in the medium of [EMIM]Cl, [BMIM]Cl, [HMIm]Cl and [OMIm]Cl.203 Among the tested catalytic systems, [BMIM]Cl/YbCl3, [HMIm]Cl/YbCl3, [OMIm]Cl/YbCl3 and [BMIM]Cl/Yb(OTf)3 gave HMF yields of around 20%.
Chan et al. reported that WCl6 is more active than CrCl2, CrCl3 and AlCl3 for the dehydration of fructose to HMF in the [BMIM]Cl ionic liquid at a low reaction temperature (22 °C), affording an HMF yield of more than 60%.204
Jerome reported that the use of DMSO and LiCl saturated aqueous solution as a solvent enabled the direct conversion of Chlorella sorokiniana biomass to HMF over H2SO4 catalyst, obtaining an HMF yield (52%) almost 40-fold higher than that obtained with sugar cane bagasse biomass.149 They also found that the use of LiCl led to an HMF selectivity of up to 98.7% with an HMF yield 2-fold higher than that obtained without the use of metal salts.
Mensah et al. identified Cu(OH)+ as an active Lewis-acidic species for glucose isomerization over the Cu(NO3)2 catalyst, which follows the intramolecular 1,2-hydride shift mechanism, attaining a maximum fructose yield of 16% in aqueous solution at a pH of 5.3.152 Zhang et al. found that trace CrCl2 and CuCl2 in the EMIMCl ionic liquid play an important role in the conversion of carbohydrates.205 CrCl2 is responsible for the selective formation and stabilization of HMF, while CuCl2 is active for depolymerization of cellulose and the degradation of formed HMF.205 Similarly, the combined use of CrCl3 with slight AlCl3, which plays an important role in the decomposition of cellulose, achieved an HMF yield (58.3%) comparable to single CrCl3 (58.4%) with a decrease in the reaction time by nearly 1/3.61
Under relatively harsh reaction conditions (200 °C), the combination of concentrated NaCl aqueous solution with THF enabled the direct conversion of cellulose to HMF in high yield (58.9%), and LiCl, KCl, NH4Cl, LiBr, NaBr and KBr also gave HMF yields ranging from 40.7% to 58.9%, while MgCl2 and CaCl2 gave similar HMF yields with a faster rate.156 These results indicate that Cl− itself can promote the hydrolysis of cellulose, isomerization of glucose via the 1,2-hydride shift path, and accelerate the dehydration of fructose. Seawater, which contains Na+, K+, Mg2+, Cg2+, Cl− and SO42−, was more effective for the direct conversion of glucose to HMF than NaCl solution under hydrothermal conditions, giving an HMF yield of 30% with a fructose yield of 4% at 211 °C after 15 min.206 The improved catalytic performance was attributed to the enhanced isomerization of glucose to fructose promoted by seawater. A biphasic reaction system consisting of concentrated seawater (ca. 30 wt% salts) with THF was directly used to produce HMF and furfural from cellulose and hemicellulose fractions, which were obtained by depolymerizing lignin in birch wood over Ru/C, affording an HMF yield of 43.3% with a furfural yield of 40.8%.207 The acidic seawater-catalyzed hydrothermal liquefaction (MHTL) of sugarcane bagasse under microwave irradiation gave the highest HMF yield of 8.1%.208
Cl- and Br-containing salts and ionic liquids play important roles in biorefineries based on HMF. As discussed above, NaCl not only can promote the transfer of HMF from the water phase to organic phase via the salting-out effect,60 but also serve as a co-catalyst to improve the fructose dehydration rate (Fig. 10a).60 HCl is a widely used acid catalyst for the dehydration of fructose to HMF,103 hydrolysis of cellulose to glucose,72 and the direct conversion of cellulose to 5-(chloromethyl)furfural (CMF).209–211 The catalytic effect of Cl−, regardless of the cations, is demonstrated by the direct conversion of cellulose to HMF in NaCl, MgCl2 aqueous solution and seawater.156,206 Similarly, Br− also plays multiple roles in the dissolution and hydrolysis of polysaccharides, isomerization of glucose to fructose, dehydration of fructose and even oxidation of HMF (Fig. 10b). Liu et al. reported that a concentrated LiBr (65 wt%) solution enabled the efficient depolymerization of cellulose to short-chain oligomers and glucose in the absence of any additional acid catalyst, attaining a short-chain oligomer yield of 90.4% under mild conditions (130 °C) even at an initial cellulose concentration of more than 10 wt%.212 Yang et al. reported that [NMP]Br was unable to catalyze the isomerization of glucose to fructose.84 In contrast, Yoo et al. found that a concentrated aqueous solution of LiBr (∼62 wt%) afforded a fructose yield of 30% from glucose under mild conditions (120 °C), which is much higher than that with other salts, demonstrating that Br− is active for the isomerization of glucose to fructose in water.154 The addition of [NMP]Br and [NMP]Cl to DMSO could improve the HMF formation rate significantly from glucose over CrCl3 owing to the improved fructose dehydration to HMF promoted by [NMP]Br and [NMP]Cl.84 For H2SO4-catalyzed fructose dehydration in aqueous solution, the combination of sodium with most anions, including chloride, bromide, iodide, mesylate, tosylate, perchlorate monohydrate, and sulfate led to a higher HMF formation rate compared with the salt-free system, while sodium nitrate led to a decrease in the HMF formation rate.213 As co-catalysts, chlorine and bromide salts work differently in different catalytic systems. For example, the use of bromide salts improves the CrCl3-catalyzed glucose dehydration toward HMF more remarkably in aqueous media than chlorine salt,214 which is mainly attributed to the improved fructose dehydration catalyzed by bromide anions. In contrast, chlorine salt can accelerate the acid-catalyzed fructose dehydration in the GVL/water solvent system, while bromide salt does not lead to an increase in the reaction rate in this catalytic system.60
 | ||
| Fig. 10 Roles of (a) Cl- and (b) Br-containing salts and ionic liquids in biorefineries based on HMF. | ||
The combination of CrCl3 and H3BO3 in [BMIM]Cl gave an HMF yield (69.1%) higher than the sum of the HMF yields (1.4% and 60.3%) obtained with CrCl3 and H3BO3, separately, demonstrating the synergistic effect of CrCl3 and H3BO3.160 Similarly, the combination of tungstophosphoric acid and H3BO3 gave an HMF yield (51.9%) remarkably higher than the separate catalyst.161 The combined catalyst was also effective in tetraethyl ammonium chloride (TEAC), affording an HMF yield (47.6%) higher than that obtained using caprolactam (CPL), DMA, N,N-DMF and DMSO as the solvent. The combination of ortho-carboxyl-substituted phenylboronic acids with hydrated MgCl2 or ionic liquid [BMIM]Cl is also effective to catalyze the direct conversion of glucose to HMF.162,215 Moreover, the combination of MgCl2, HCl, 2-methoxycarbonylphenylboronic acid enabled the one-pot conversion of cellulose to HMF under relatively mild conditions.162 However, further work is needed to confirm the catalytic role of different species in this catalytic system for the conversion of glucose and cellulose to HMF.
Körner et al. investigated the effect of different Brønsted acids, including acetic acid, HCl, H2SO4 and H3PO4, on the hydrothermal conversion of fructose in water.216 They found that the maximum HMF yield is almost independent of the pH or acid type, while a high concentration of H3PO4 has a remarkable accelerating effect beyond donating protons. Similarly, Widsten et al. reported that the use of H3PO4 as a catalyst in an acetone/water solvent system also enabled moderate HMF and furfural yields from radiata pine hydrolysate.166 Fang et al. investigated the influence of Brønsted acids (H2SO4, HCl and HNO3) and alkali metal cations (Li+, Na+, and K+) on the conversion of corn stover in the medium of an H2O/THF system.153 Among the tested catalysts, H2SO4/Na2SO4 gave an HMF yield of 76% at 190 °C for 1 h. They proposed that the presence of alkali metal cations may help inhibit the degradation of HMF not just via the salting-out effect. Similarly, the use of NaHSO4/ZnSO4 as a catalyst in the THF/water biphasic system also gave moderate HMF and furfural yields from the hydrolysate obtained via the prehydrolysis of radiata pine wood chips.166 The biphasic system composed of concentrated NaHSO4–ZnSO4 aqueous solution and THF could convert cellulose to HMF directly, affording a high HMF yield of 53% with a cellulose conversion of 96% at 160 °C in 60 min.217 However, more work is needed to reveal the role of NaHSO4 and ZnSO4 in the conversion of carbohydrate to HMF and the HMF stabilization mechanism.
The HCl-catalyzed degradation of HMF to humins was studied by Tsilomelekis et al. using ATR-FTIR spectroscopy, scanning electron microscopy (SEM) and dynamic light scattering (DLS).218 They proposed that in the medium of water humins is formed either though a ring opening mechanism and/or through nucleophilic attack of the carbonyl group at the α- or β-position of the furan ring. Initially, HMF reacts with HMF molecules and other intermediates via aldol condensation and/or etherification reactions, leading to the formation of soluble oligomers. Subsequently, the humin particles grow via nucleophilic attack. Meanwhile, the direct addition of HMF to the primary particles and the aggregation of the primary particles to large humin particles, which are insoluble in water, also occur in water. They also demonstrated that humins are spatially and chemically heterogeneous materials composed of both insoluble macromolecules and small soluble species, and the soluble fraction increases positively with an increase in the donor number of the solvent.219 Therefore, the use of DMSO as the co-solvent could suppress the nucleophilic attack, thus leading to humins with a smaller size (∼100 nm) than that (3–4 μm) in neat water. Besides HMF, Maruani et al. also identified the water-soluble oligomers of D-glucose (WSO) as the main intermediates for the formation of humins during the hydrothermal conversion of glucose in H2SO4 solution.220 They suggested that WSO can be inserted into the structure of humins via aldol condensation, leading to the formation of humins.
Supercritical CO2 was tested as an acid catalyst by Labauze et al. for the dehydration of fructose.221 The two-phase supercritical CO2–water system gave an HMF yield of 48 mol% after 4 h of reaction at 160 °C under 25 MPa of CO2, which was slightly higher than that (40%) without the use of a catalyst. When ethylenediaminetetraacetic acid (EDTA) was used as the catalyst, an HMF yield of 89% was obtained from fructose in a water–MIBK biphasic system containing PVP and 2-butanol as phase modifiers.171 After five times reuse, EDTA still gave an HMF yield of 88% with 4.3% loss of catalyst. Levulinic acid was used to catalyze the conversion of lignocellulosic biomass such as pinewood and eucalyptus sawdust to HMF and furfural by Seemala et al. in both mono and biphasic solvent systems.222 The maximum HMF yield of 7.4% was obtained in the toluene/water (1:1) mixture at 180 °C for 15 min.
Feng et al. reported that betaine-based catalysts (BX) prepared from the anhydrous betaine derived from the betaine sugar industry with Brønsted acids, such as HCl, H2SO4, (C6H5)SO3H and p-CH3(C6H4)SO3H, as well as choline-O-sulfate (ChOS) derived from ChCl and concentrated sulfuric acid could be used as renewable and sustainable catalysts for the dehydration of fructose in the medium of H2O/MIBK and ChCl/H2O/MIBK biphasic systems, obtaining HMF yields between 61–68%.169 However, these catalysts are not effective for the conversion of glucose to HMF. The combination of BX with AlCl3 in the ChCl/H2O/MIBK biphasic system gave an HMF yield of 64%, which is slightly higher than that obtained (58%) with the AlCl3/ChCl/H2O/MIBK biphasic system.
Homogeneous organic Brønsted bases (Table 4), including primary, secondary, and tertiary amines are more effective than inorganic Brønsted bases for the isomerization of glucose to fructose in water via the proton transfer mechanism (Fig. 11a).223 In particular, triethylamine gave a high fructose yield (32%) and selectivity (63%), which are comparable to Sn-Beta zeolite, with an activation energy (61 kJ mol−1) lower than that with Sn-Beta zeolite (93 kJ mol−1).59 The formation of an enediol intermediate is the rate-limiting step for the isomerization of glucose to fructose. A short reaction time at high temperature and an appropriate pH environment are required to limit the side-reaction and then to afford a high fructose yield. The conversion did not occur at a pH lower than 9, while the fructose yield decreased at a pH higher than 11.5 owing to the degradation of ring fructose and acyclic forms of both sugars (Fig. 11b). Chen et al. conducted a systematic comparison of homogeneous inorganic bases, organic amines, and heterogeneous anion exchange resins for the isomerization of glucose under identical reaction conditions and identified meglumine as the superior homogeneous base, giving a fructose yield of 35% with approximately 80% selectivity, with a relatively low activation energy (74 kJ mol−1).52
 | ||
| Fig. 11 Glucose isomerization to fructose via (a) proton transfer mechanism. Adapted from ref. 59 and 224. (b) Side reactions during the isomerization of glucose over base catalysts. Adapted from ref. 225. (c) Glucose isomerization to fructose via intramolecular hydride shift mechanisms. Adapted from ref. 224. | ||
| Catalysta | Solvent | Reaction conditions | Glucose loadingb | Conversion | Fructose yield | Ref. |
|---|---|---|---|---|---|---|
| a Relative to monosaccharide. b Relative to solvent. | ||||||
| Morpholine 10 mol% | Water | 100 °C, 30 min | 10 wt% | 43% | 17% | 223 |
| Piperazine 10 mol% | Water | 100 °C, 30 min | 10 wt% | 43% | 28% | 223 |
| Ethylenediamine 10 mol% | Water | 100 °C, 30 min | 10 wt% | 43% | 25% | 223 |
| Piperidine 10 mol% | Water | 100 °C, 30 min | 10 wt% | 43% | 29% | 223 |
| Pyrrolidine 10 mol% | Water | 100 °C, 30 min | 10 wt% | 43% | 29% | 223 |
| Triethylamine 12 mol% | Water | 100 °C, 7 min | 9.4 wt% | 50% | 31% | 59 |
2.4. Heterogeneous catalysts for the production of HMF from biomass
Recently, much attention has been paid to replacing traditional liquid acids with solid acid catalysts with the aim of meeting the requirement of green and sustainable chemistry.226,227 Solid acid materials including acid-functionalized carbon-based materials, acid-functionalized polymers, metal oxides, metal phosphates, zeolites, metal–organic frameworks and heteropoly acids (HPAs) have widely investigated as heterogeneous catalysts for acid-catalyzed biomass conversion reactions. The catalytic activity of solid acid catalysts can be manipulated by tuning their structural and acidic properties. The acidic characteristics such as acid type (Brønsted vs. Lewis acidity), concentration, acid intensity and local environment of acid sites have a critical influence on the activity and selectivity for many acid-catalyzed reactions.228 The structural features including morphology, size, specific surface area, pore distribution, location (intra- vs. extra-crystalline) and interfacial effect not only affect the acidic characteristics, but also influence the host–guest interaction, shape selectivity and reaction pathway. Moreover, the spatial proximity of acid sites and pore confinement (environmental effect) also influence the actual catalytic process. In this section, we not only summarize heterogeneous catalysts from the perspective of reaction efficiency, but also emphasize their stability and reusability.| Catalyst | Catalyst loadinga | Solvent | Reaction conditions | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not provided. | |||||||
| MgO | 0.5 wt% | H2O | 90 °C, 0.75 h | Glucose 4 wt% | 44.1% | Fructose 33.4% | 229,230 |
| MgO-biochar | 1 wt% | H2O | 100 °C, 0.5 h | Glucose 10 wt% | 37.5% | Fructose 30% | 231 |
| MgO-biochar (3 runs) | 1 wt% | H2O | 100 °C, 0.5 h | Glucose 10 wt% | 31.9% | Fructose 15% | 231 |
| Bulk-WO3-uc | 1 wt% | H2O/THF | 120 °C, 4 h | Glucose 1 wt% | 100% | 15% | 232 |
| Meso-WO3-uc | 1 wt% | H2O/THF | 120 °C, 2 h | Glucose 1 wt% | 100% | 57% | 232 |
| Meso-WO3-300 | 1 wt% | H2O/THF | 120 °C, 3 h | Glucose 1 wt% | 100% | 56.2% | 232 |
| Meso-WO3-600, HCl | 1 wt% | H2O/THF | 120 °C, 3 h | Glucose 1 wt% | 100% | 40.8% | 232 |
| Meso-WO3-600 | 1 wt% | H2O/THF | 120 °C, 3 h | Glucose 1 wt% | 100% | 30% | 232 |
| Meso-NbW-uc | 1 wt% | H2O/THF | 120 °C, 4 h | Glucose 1 wt% | 100% | 55% | 232 |
| Meso-TiW-uc | 1 wt% | H2O/THF | 120 °C, 4 h | Glucose 1 wt% | 100% | 50% | 232 |
| Meso-ZrW-uc | 1 wt% | H2O/THF | 120 °C, 4 h | Glucose 1 wt% | 100% | 50% | 232 |
| Meso-VW-uc | 1 wt% | H2O/THF | 120 °C, 2 h | Glucose 1 wt% | 100% | 23% | 232 |
| Nb4W4 | 2 wt% | H2O | 120 °C, 2 h | Glucose 4.5 wt% | 36.1% | 18.8% | 233 |
| Nb7W3 | 2.9 wt% | 2-Butanol/H2O (v/v = 2.5) | 140 °C, 2 h | Glucose 0.28 wt% | 100% | 52% | 234 |
| 10CuZr | — | 1-Butanol/H2O (w/w = 2.75) | 200 °C, 5.5 h | Glucose 2.5 wt% | 94.5% | 33.2% | 235 |
| WO3 | 1 wt% | H2O/THF (v/v = 1/9) | 120 °C, 4 h | Glucose 1 wt% | 100% | 15% | 236 |
| NbW5 | 1 wt% | H2O/THF (v/v = 1/9) | 120 °C, 4 h | Glucose 1 wt% | 100% | 60% | 236 |
| TiW5 | 1 wt% | H2O/THF (v/v = 1/9) | 120 °C, 4 h | Glucose 1 wt% | 100% | 55% | 236 |
| HTaWO6 | 4 wt% | DMSO | 140 °C, 0.5 h | Fructose 5 wt% | 99% | 67% | 237 |
| HTaWO6 | 4 wt% | DMSO | 140 °C, 0.5 h | Glucose 5 wt% | 7% | 2% | 237 |
| Nb2O5 | 1 wt% | H2O/THF (v/v = 1/9) | 120 °C, 0.5 h | Glucose, 1 wt% | 100% | 22% | 236 |
| Nb–P–Si oxides | 0.5 g in total recirculation reaction line | H2O/isopropanol (v/v = 5), 3 mL min−1 | 130 °C, 5 h | Fructose 1.8 wt% | 50% | 25% | 238 |
| TiO2 | 1 wt% | H2O/THF (v/v = 1/9) | 120 °C, 0.5 h | Glucose 1 wt% | 100% | 0.6% | 236 |
| Hybrid-TiO2 | 2 wt% | H2O | 130 °C, 7 h | Glucose 2 wt% | 75% | 45% | 239 |
| Inorganic TiO2 | 2 wt% | H2O | 130 °C, 7 h | Glucose 2 wt% | 50% | 8% | 239 |
| NH4AlOHCO3 | 15 wt% | DMSO-BMIMCl | 120 °C, 4 h | Glucose 2 wt% | — | 52.2% | 240 |
| γ-Fe2O3 | 17.2 wt% | DMSO-BMIMCl | 120 °C, 4 h | Glucose 2 wt% | — | 2.5% | 240 |
| γ-Al2O3 | 11.0 wt% | DMSO-BMIMCl | 120 °C, 4 h | Glucose 2 wt% | — | 5.1% | 240 |
| Natural Al2O3 | 10 wt% | EMIMBr | 140 °C, 3 h | Glucose 10 wt% | 100% | 24.0% | 241 |
| Al2O3-b-0.05 | 10 wt% | EMIMBr | 140 °C, 3 h | Glucose 10 wt% | 100% | 49.7% | 241 |
| Al2O3, DIAION® RCP160M | 2 wt%, 1 wt% | H2O–NMP–NaCl/MIBK | 120 °C, 8 h | Glucose 1 wt% | 90.48% | 84.92% | 242 |
| CO2, TiO2, ZrO2 | 5 MPa, 0.7 wt%, 2 wt% | 0.34 M NaCl/THF (v/v = 0.25) | 200 °C, 3 h | Cellulose 2 wt% | — | 48.4% | 243 |
| TiO2, ZrO2 | 0.7 wt%, 2 wt% | 0.34 M NaCl/THF (v/v = 0.25) | 200 °C, 3 h | Cellulose 2 wt% | — | 41.4% | 243 |
| Al2O3–TiO2–W | — | NaCl, H2O/THF (v/v = 0.33) | 170 °C, 2 h | Glucose 2 wt% | — | 70% | 244 |
| Ta7W3 | 2 wt% | H2O/butanol (v/v = 1) | 160 °C, 5 h | Glucose 2 wt% | 100% | 54% | 245 |
| ZrO2 | 0.67 wt% | DMSO | 150 °C, 4 h | Glucose 1.67 wt% | 76.6% | 12.3% | 246 |
| B2O3(20 wt%)/ZrO2 | 0.67 wt% | DMSO | 150 °C, 4 h | Glucose 1.67 wt% | 83.6% | 21.1% | 246 |
| B2O3(20 wt%)/Al2O3 | 0.67 wt% | DMSO | 150 °C, 4 h | Glucose 1.67 wt% | 89.1% | 31.8% | 246 |
| B2O3(20 wt%)/ZrO2–Al2O3 | 0.67 wt% | DMSO | 150 °C, 4 h | Glucose 1.67 wt% | 90.8% | 41.2% | 246 |
| Sn20/γ-Al2O3 | 4 wt% | DMSO/H2O (v/v = 4) | 150 °C, 1 h | Glucose 4 wt% | ∼99% | 27.5% (lactic acid, 16.5%) | 150 |
| 1.5 wt% SnO2/Al2O3-HWT | 2 wt% | DMSO/H2O (v/v = 4) | 110 °C, 4 h | Glucose 4.5 wt% | 59.7% | Fructose + methylfructoside 30.4%, Mannose 11.0% | 247 |
| SA/50 (Si/Al = 50/50) | 1 wt% | DMSO/H2O (v/v = 7/3) | 160 °C, 2 h | Glucose 3 wt% | 70% | 38% | 248 |
| SA/50 (Si/Al = 50/50) | 1 wt% | DMSO/H2O (v/v = 7/3) | 160 °C, 2 h | Glucose 3 wt% | 68% | 37% | 248 |
| ASA (Si/Al = 90/10) | 1 wt% | DMSO/H2O (v/v = 7/3) | 160 °C, 2 h | Glucose 3 wt% | 45% | 18.8% | 248 |
| Cr(III)/CP, H2SO4 | 6.3 wt%, 1 wt% | DMSO/H2O (v/v = 4) | 180 °C | Glucose 3.1 wt% | 92.5% | 64.7% | 249 |
| Cr(III)/CP (5 runs), H2SO4 | 6.3 wt%, 1 wt% | DMSO/H2O (v/v = 4) | 180 °C, 3 h | Glucose 3.1 wt% | 82.3% | 58.2% | 249 |
| Cr(VI)/CP, H2SO4 | 6.3 wt%, 1 wt% | DMSO/H2O (v/v = 4) | 180 °C, 3 h | Glucose 3.1 wt% | 88.5% | 60.7% | 249 |
| TiOSO4 | 1 wt% | BMIMCl | 130 °C, 3 h | Fructose 10 wt% | — | 92% | 250 |
| TiOSO4 | 1 wt% | BMIMCl | 130 °C, 3 h | Glucose 10 wt% | — | 45% | 250 |
| TiOSO4 | 1 wt% | BMIMCl | 130 °C, 3 h | Cellobiose 10 wt% | — | 52% | 250 |
| TiOSO4 | 1 wt% | BMIMCl | 130 °C, 3 h | Cellulose 10 wt% | — | 38% | 250 |
| TiOSO4 (8 runs) | 1 wt% | BMIMCl | 130 °C, 3 h | Cellulose 10 wt% | — | 20% | 250 |
| PAL-SO3H | 1 wt% | GVL/H2O (v/v = 95/5) | 180 °C, 1 h | Xylose 2 wt% | 90.6% | Furfural 87% | 251 |
In the medium of pure water, commercially available γ-Al2O3 was unable to catalyze the conversion of glucose to fructose, while NaAlO2 afforded a fructose yield as high as 52.1% with a glucose conversion of 61.4% under mild conditions (60 °C, 45 min).229 The combination of Al2O3 with metal salts is an effective strategy to improve the HMF production efficiency from glucose. García-Sancho et al. compared the catalytic performance of commercial acidic, neutral and basic Al2O3 for the dehydration of glucose to HMF in the medium of H2O/MIBK.252 Among the tested Al2O3 samples, the acidic Al2O3 exhibited the highest total acidity and the highest catalytic activity. However, the HMF yield under the optimized condition (175 °C, 120 min) was still lower than 20% owing to side reactions. Although CaCl2 could not catalyze the isomerization of glucose to fructose alone, the introduction of CaCl2 to the reaction system promoted the formation of α-D-glucopyranose and then shifted the equilibrium toward HMF with an increase in the reaction rate. Owing to the synergistic effect between CaCl2 and γ-Al2O3, an HMF yield of up to 52% with a glucose conversion of 96% was obtained at 175 °C within 15 min. Similarly, Sampath et al. showed the synergy of Al2O3 with metal salts, including CuCl2, CrCl3 and FeCl3, for the conversion of glucose to HMF in DMSO.253 Even at a glucose loading of up to 30 wt%, the HMF yield of 56% could be obtained by combining CuCl2 and Al2O3. Furthermore, although the activity of Al2O3 decreased rapidly in the recycling experiment, it could be restored after calcination. Pumrod et al. reported that the combination of Al2O3, an ion exchange resin (DIAION® RCP160M) catalyst, and biphasic solvent consisting of water, NMP and MIBK could greatly improve the HMF yield and selectivity compared with the water/MIBK biphasic system.242
The reaction efficiency of glucose conversion to HMF was considerably improved via the combination of the EMIMBr ionic liquid and an Al2O3-based material with low Brønsted acidity and high Lewis acidity (Fig. 12).236 Different from the aqueous solution-based reaction system, the presence of Brønsted acid sites in EMIMBr accelerated the undesirable side-reaction remarkably, leading to a low HMF yield and selectivity. The simple alkaline treatment could block most of the Brønsted acid sites and weak Lewis acid sites, obtaining Al2O3-b-0.05 with a relatively high concentration of Lewis acid sites and low concentration of Brønsted acid sites. The EMIMBr/Al2O3-b-0.05 catalytic system could convert glucose to HMF efficiently, attaining an HMF yield of up to 49.7% even at high glucose concentration (10 wt%), which is close to the most efficient homogeneous catalytic systems, such as EMIMCl/CrCl2 and BMIMCl/CrCl3. The commercially available TiO2 products, P25 and A-TiO2 (anatase), gave HMF yields of 13.8% and 10.9%, respectively, while R-TiO2 (rutile) gave a lower HMF yield.254 In the medium of a BMIMCl/DMSO mixture, γ-Al2O3 gave an HMF yield of only 5.1% from glucose, while ammonium aluminum carbonate hydroxide (NH4AlOHCO3) afforded a much higher HMF yield (52.2%).240
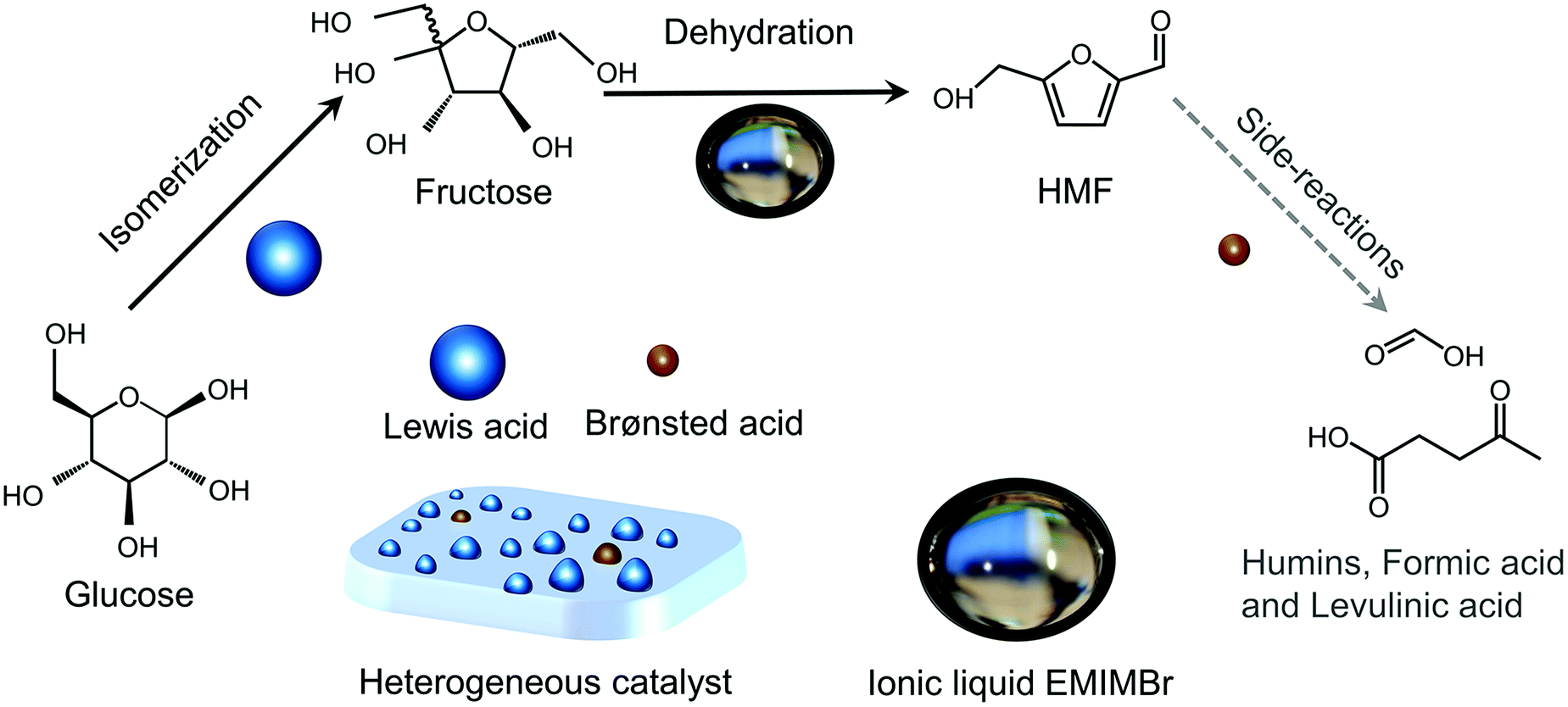 | ||
| Fig. 12 Cooperation effect of a heterogeneous catalyst with ionic liquid EMIMBr for the conversion of glucose to HMF. Reproduced from ref. 236 with permission from Elsevier, Copyright 2018. | ||
Mesoporous Al2O3–B2O3 with an Al/B molar ratio of 1:1, which was prepared from aluminum isopropoxide and boric acid via an evaporation-induced self-assembly method, exhibited abundant Lewis acid sites and undetectable Brønsted acid sites.255 In the medium of DMSO, the mesoporous Al2O3–B2O3 with an Al/B molar ratio of 1:1 gave an optimized HMF yield of 41.4% after 120 min reaction at 140 °C. Han et al. reported that B2O3 supported on ZrO2–Al2O3 (B2O3(20 wt%)/ZrO2–Al2O3) showed an HMF yield of 41.2% from glucose, which is obviously higher than that obtained with B2O3(20 wt%)/ZrO2 and B2O3(20 wt%)/Al2O3.246 Marianou et al. reported that an SnO2 supported on γ-Al2O3 (Sn2O/γ-Al2O3) catalyst gave an HMF of 27.5% from glucose in the medium of DMSO/water.150 They concluded that the introduction of Sn species, either in the oxide or ionic form, on the Al2O3 support, could not only catalyze the conversion of glucose to HMF, but also promote the co-synthesis of lactic acid. Yatoo et al. reported that SnO2 supported on hot water-treated alumina (0.5 wt% Sn/Al2O3-HWT) was effective in catalyzing the isomerization of glucose to fructose and methylfructoside (total yield of 30.4%) in methanol.247 Amorphous silica–alumina materials are typical solid acids widely used for the conversion of biomass, but their catalytic performance is limited due to their lower Brønsted acid strength than corresponding crystalline zeolites. Wang et al. showed that flame-made amorphous silica–alumina (labeled as SA/50, Si/Al = 50/50) with a high Al content exhibited a much higher Brønsted acid strength than the conventional amorphous silica–alumina (ASA, Si/Al = 90/10) prepared via the co-precipitation method due to the synergy of penta- and tetra-coordinated aluminum species, leading to enhanced activity for the conversion of glucose to HMF.248
Niobium oxide (Nb2O5) and niobium phosphate (NbPO) have been extensively investigated for the conversion of various sugars to HMF. Nakajima et al. reported that the commercial niobic acid (Nb2O5·nH2O) with water-tolerant Lewis acid sites can function as a heterogeneous catalyst for the conversion of glucose, attaining an HMF yield of 12.1% at 120 °C in pure water.256 Na+/Nb2O5·nH2O obtained by blocking the Brønsted acid sites on Nb2O5·nH2O with Na+ gave a comparable HMF yield with that of Nb2O5·nH2O, suggesting the formation of HMF from glucose predominantly proceeds on the water-tolerant Lewis acid sites. After treating Nb2O5·nH2O with H3PO4, ca. 70% of the Brønsted acid sites on H3PO4/Nb2O5·nH2O was blocked with PO43−, thus remarkably suppressing undesirable side reactions and then improving the HMF yield to 52.1%. do Prado et al. reported that the dehydration of fructose to HMF over Nb2O5·nH2O in water and DMSO gave optimized HMF yields of 22% and 47%, respectively.257
Niobium oxides with different structures and morphologies have been investigated to establish the relation among structure, acidity and catalytic performance.258,259 It was found that the structure of niobium oxides could control their acidity, and both acid type and strength are closely related to the catalytic activity. Few-layer to monolayer or mesoporous Nb2O5·nH2O exhibited abundant Lewis acid sites, which correspond to the low coordinated NbO5 and in some cases NbO4 sites derived from the oxygen vacancies in thin and flexible NbO6 systems.259 Owing to its low surface area and high structural rigidity, the bulk Nb2O5·nH2O only exhibited a few acid sites. HNb3O8 possessed a small amount of Brønsted acid sites due to its protonic structure, whereas the anhydrous crystalline H-Nb2O5 did not exhibited measurable acid sites. The conversion of fructose to HMF is mainly catalyzed by Brønsted acid, with weaker Brønsted acid sites favoring the selective formation of HMF. Among the Nb-based materials, including bulk HNb3O8, few-layer niobium oxide (hy-Nb), monolayer niobium oxide (hy-Nb-TEOA) and mesoporous niobium oxide, mesoporous niobium oxide gave the highest HMF yield from sucrose owing to the balanced Lewis and Brønsted acid concentration with appropriate acid strength.
Nb2O5 nanosheets synthesized via the bottom-up hydrothermal method in the presence of NH4+ possess predominantly water-tolerant Brønsted acid sites and exhibit high catalytic activity for the hydrolysis of inulin to fructose in water.260 Similarly, the HNb3O8 nanosheets obtained by exfoliating layered HNb3O8 also function as a strong Brønsted acid.261 In situ exfoliation of layered HNb3O8 under microwave irradiation enabled the efficient catalytic conversion of high concentration fructose (10 wt%) to HMF in water, attaining an HMF yield of 55.9% within 18 min.262
Nb2O5·nH2O and NbPO have been used as catalysts to produce HMF from fructose in a continuous reactor at a reaction temperature in the range of 90 °C to 110 °C by Carniti et al., but the HMF selectivity was lower than 40%.263 Interestingly, the HMF selectivity increased with an increase in fructose conversion under the continuous flow condition, which is opposite to the tread usually observed in batch mode. They inferred that the easy deposition of secondary products on the catalyst surface led to the relatively low product selectivity at a low fructose conversion. The possibility for the conversion of glucose to HMF over Nb2O5 in a flow-through reactor under mild conditions (120 °C) was also attempted by Kawamura et al., but the HMF yield was only 0.47%.264
Nb2O5 also plays an important role in the acid-catalyzed depolymerization of disaccharides and the dehydration of other biomass-derived chemicals, including xylose, triose and glycerol. For example, amorphous Nb2O5 and mesoporous Nb2O5 are active for the dehydration of xylose to furfural.265,266 Na+ exchange treatment did not reduce the original catalytic activity, indicating that the conversion proceeds predominantly on the Lewis acid sites. Mesoporous Nb2O5·nH2O prepared using P123 as the structure-directing agent exhibited similar Lewis and Brønsted acid sites with bulk Nb2O5·nH2O, but the former showed much higher catalytic activity for the hydrolysis of cellobiose than the latter, suggesting that the hydrophilic mesopores are crucial for the hydrophilic reaction.267
Similar to Nb2O5, TiO2 is a typical water-tolerant solid acid catalyst, which has been widely investigated for the conversion of biomass.268 The pristine TiO2 mainly contains Lewis acid sites, while partially phosphated TiO2 exhibits higher acidity consisting of both Lewis and Brønsted acid sites. After 2 h reaction over bare TiO2 at 120 °C, 91.5% of glucose was converted to complex by-products, including polymerized species, with the HMF selectivity of just 8.5%.269 The uncalcined hybrid organic–inorganic anatase (hybrid-TiO2), which was synthesized via the hydrothermal method in the presence of citric acid, exhibited an HMF yield as high as 45% with the glucose conversion of 75% in monophasic water, which is much higher than that (8%) obtained with inorganic TiO2 after calcination. This is because the large amount of five-fold coordinatively unsaturated Ti4+ sites in hybrid-TiO2 serve as a Lewis acid to catalyze the isomerization of glucose to fructose.239 Protonated titanate nanotubes, which were synthesized via the hydrothermal treatment of TiO2 in NaOH aqueous solution followed by H+ exchange, serve as a highly active heterogeneous catalyst owing to the presence of both Brønsted and Lewis acid sites.270 The protonated titanate nanotubes gave a much higher HMF yield (14%) from glucose than TiO2 (2%) in water under mild reaction conditions (120 °C, 0.5 h).
DFT study indicated that the conversion of glucose to HMF over TiO2 proceeds via a direct stepwise dehydration mechanism instead of the sequential isomerization–dehydration mechanism owing to the cooperative effect between the Lewis acid derived from tetrahedral Ti4+ and Lewis base of the OH group on the surface of anatase TiO2.271 Isotopic labelling and 13C NMR analysis confirmed this stepwise dehydration mechanism (Fig. 13) for glucose conversion over TiO2 and phosphated TiO2, and identified 3-deoxyglucosone as the main intermediate.272 Firstly, the dehydration of ring-opening glucose at the C3 carbon leads to the formation of a highly reactive enol intermediate and the enol intermediate is rapidly converted to 3-deoxyglucosone via keto-enol tautomerization. Secondly, the intramolecular acetalization of 3-deoxyglucosone and subsequent dehydration result in the formation of HMF.
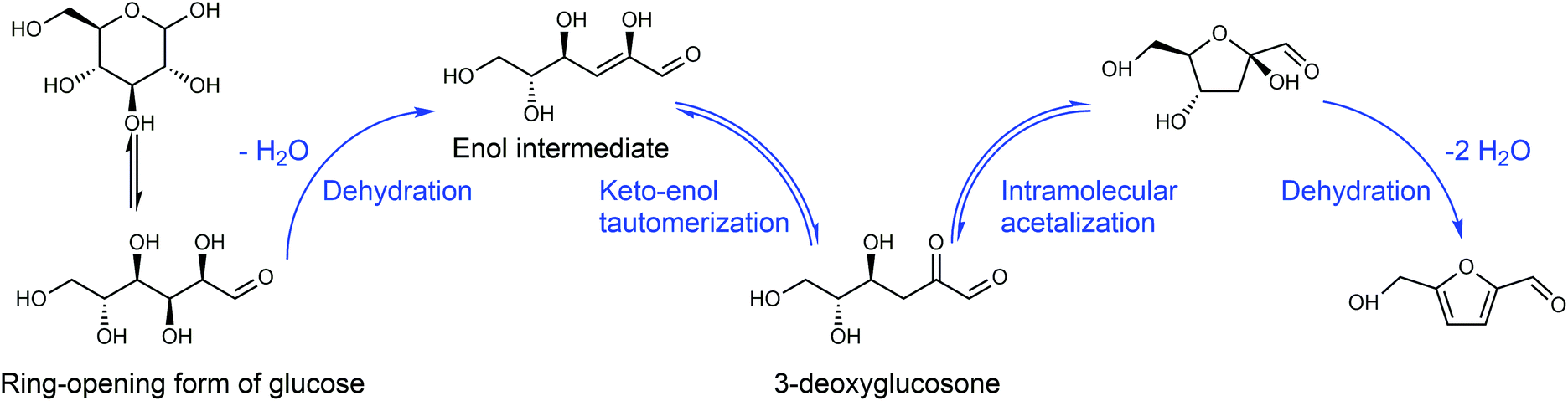 | ||
| Fig. 13 Stepwise dehydration of glucose to HMF over TiO2 and phosphated TiO2. Adapted from ref. 272. | ||
Although mesoporous tungsten oxide without calcination (meso-WO3-uc) gave a higher HMF yield than bulk tungsten oxide, its catalytic performance decreased remarkably after calcination at 600 °C owing to the loss of Brønsted acid sites.232 The effectiveness of WO3 for the conversion of glucose to HMF in a flow-through reactor under mild conditions (120 °C) was investigated by Kawamura et al., but the HMF yield was only 0.32%.264 Periodic DFT calculations suggested that the isomerization of glucose to fructose over tungstite (WO3·H2O) is attributed to the synergistic effect of the Lewis acid sites and neighboring proton donors.273 The Lewis acid site is responsible for the C2–C1 H-shift rate-determining step, while the hydrogen-bond network formed on the catalyst surface promotes the concomitant proton-transfer process.232,236
Doping and recombination of metal oxides is an effective approach to regulate their acidity and alkalinity and then improve their catalytic performance. Periodic DFT calculations suggested that doping of tungstite (WO3·H2O) with Nb5+ and Ti4+ ions can reduce the overall barrier for the isomerization of glucose to fructose.273 Mesoporous Nb–W oxides have both Lewis acid sites and Brønsted acid sites, of which the strong Brønsted acid sites is formed via the isomorphous replacement of Nb5+ ions with higher-valence W6+ ions.274 Layered-type W–Ti–O possesses Brønsted acid sites and strong Lewis acid sites, which transform to Brønsted acid sites in the presence of water, and weak Lewis acid sites, which are water-tolerable.275 Guo et al. investigated the use of ordered mesoporous NbxW(8−x) oxides, which were prepared via an evaporation-induced self-assembly method followed by calcination at 600 °C, for the conversion of glucose to fructose, mannose and HMF.233 The concentration of the Brønsted and Lewis acid distributions in the NbxW(8−x) oxides could be tuned by regulating the Nb/W ratio. Owing to the high concentration of Brønsted and Lewis acid sites, the mesoporous Nb4W4 exhibited a relatively low activation energy (90.2 kJ mol−1) for the conversion of glucose, affording an HMF yield of 18.8% with a mannose yield of 5.3% at just 120 °C. The Lewis acid sites not only catalyze the isomerization of glucose to fructose, but also promote the epimerization of glucose to mannose. Wiesfeld et al. reported that mesoporous tungsten oxide without calcination (meso-WO3-uc) was more active than Nb, Ti, V and Zr-doped WO3 without calcination for the dehydration of glucose.232 Córdova-Pérez et al. found that the Al2O3–TiO2–W material with a W concentration of 5 wt% exhibited an acid–basic site molar ratio of 2.35, which is lower than other Al2O3–TiO2–W materials, and the highest HMF yield from glucose, suggesting that the basic sites also play an important role in the conversion of glucose to HMF.244 Similarly, one-dimensional γ-Al2O3 modified with 0.5–1 wt% of Nb2O5 exhibited strong Lewis acid sites and concentrated Brønsted acid sites, leading to HMF yields between 55.9–59.0% from glucose using DMSO as the solvent.276
In addition to doping and recombination, physical mixing is also an effective approach to regulate the catalytic performance. For example, Jing et al. reported that the combined use of commercial ZrO2 and TiO2 catalyst resulted in an HMF yield of 41.4% from cellulose at 200 °C for 3 h, and the introduction of high-pressure CO2 further improved the HMF yield to 48.4%.243 Cui et al. designed a chromium-based ceramic material via the adsorption of Cr(III) and Cr(VI) by chitosan nanoparticles followed by moulding with SiO2, Al2O3, Na2SiO3, MgO, and CaO (mass ratio of 10:5:5:5:2).249 They found that the combined use of chromium-based ceramic powder as the catalyst and H2SO4 as a co-catalyst enabled the efficient dehydration of glucose to HMF in a water/DMSO system, attaining HMF yields of 64.7% and 60.7% with Cr(III)/CP and Cr(VI)/CP, respectively.
The sulfonation of metal oxides is an effective approach to improve their catalytic performance for the dehydration of fructose to HMF. SnO2–ZrO2 prepared via the sol–gel method gave an HMF yield of around 54% from fructose, while the sulfated SnO2–ZrO2 obtained by impregnating SnO2–ZrO2 with H2SO4 solution gave an HMF yield of up to 75%.277 The catalytic activity of sulfated SnO2–ZrO2 remained almost unchanged in the recycling experiment for five times. The anchoring of MoO3 and SO42− on TiO2 improved their acidity remarkably, especially the quantity of weak to moderate acid sites on TiO2/MoO3-30, thus leading to the improved conversion of cellulose to a series of products, including cellobiose, glucose, fructose, HMF, formic acid and levulinic acid.278 Compared with commercial Nb2O5, the sulfated mesoporous Nb2O5 exhibited much higher Brønsted acidity without an appreciable amount of Lewis acid sites, leading to an HMF yield of up to 88% from fructose in DMSO.279
| Catalyst | Catalyst loadinga | Solvent | Reaction condition | Substrate loadinga | Glucose conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b Fructose yield if unspecified. — Not provided. | |||||||
| Commercial hydrotalcite | 0.71 wt% | Water | 90 °C, 24 h | Glucose 10 wt% | 29% | 27% | 283 |
| Hydrophilic hydrotalcites in carbonate form | 0.71 wt% | Water | 90 °C, 24 h | Glucose 10 wt% | 41% | 31% | 283 |
| Hydrophilic hydrotalcites in hydroxy form | 0.71 wt% | Water | 90 °C, 24 h | Glucose 10 wt% | 71% | 29% | 283 |
| Hydrotalcites (Mg/Al ratio = 3) | 0.71 wt% | Ethanol | 120 °C, 6 h | Glucose 0.71 wt% | 70% | 56% | 225 |
| Hydrotalcites (Mg/Al ratio = 3) | 0.71 wt% | H2O | 120 °C, 2 h | Glucose 0.71 wt% | 28% | 16% | 225 |
| Hydrotalcites (Mg/Al ratio = 3) | 0.71 wt% | Methanol | 120 °C, 2 h | Glucose 0.71 wt% | 55% | 43% | 225 |
| Hydrotalcites (Mg/Al ratio = 3) | 0.71 wt% | 1-Propanol | 120 °C, 2 h | Glucose 0.71 wt% | 56% | 48% | 225 |
| Hydrotalcites (Mg/Al ratio = 3) | 0.71 wt% | DMSO | 120 °C, 2 h | Glucose 0.71 wt% | 24% | 16% | 225 |
| Hydrotalcites (Mg/Al ratio = 3) | 0.71 wt% | N,N-DMF | 120 °C, 2 h | Glucose 0.71 wt% | 48% | 35% | 225 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | 1-Butanol | 120 °C, 5 h | Glucose 10 wt% | 62% | 51% | 284 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | 1-Butanol | 100 °C, 5 h | Glucose 10 wt% | 35% | 32% | 284 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | H2O | 100 °C, 5 h | Glucose 10 wt% | 54% | 30% | 284 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | Ethanol | 90 °C, 5 h | Glucose 10 wt% | 47% | 32% | 284 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | DMF | 100 °C, 5 h | Glucose 10 wt% | 43% | 28% | 284 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | GVL | 100 °C, 5 h | Glucose 10 wt% | 40% | 26% | 284 |
| Hydrotalcites (Mg/Al ratio = 2) | 10 wt% | Toluene | 100 °C, 5 h | Glucose 10 wt% | 50% | 15% | 284 |
| Hydrotalcite, Amberlyst-15 | 3.3 wt%, 3.3 wt% | N,N-DMF | 100 °C, 3 h | Glucose 3.3 wt% | 73% | HMF 42% | 285 |
| Hydrotalcite, Amberlyst-15 | 3.3 wt%, 3.3 wt% | N,N-DMF | 100 °C, 3 h | Cellobiose 3.3 wt% | 52% | HMF 30% | 285 |
The combination of the solid base Mg–Al hydrotalcite and solid acid Amberlyst-15 in N,N-DMF enabled the direct conversion of glucose and cellobiose to HMF via orthogonal tandem catalysis, affording HMF yields of 42% and 30% with glucose conversions of 73% and 53%, respectively, under the optimized conditions.285 Although humins were formed on the surface of the hydrotalcites, these catalysts could be reused three times without an obvious decrease in their catalytic performance after washing with N,N-DMF followed by drying in vacuo.
 | ||
| Fig. 14 Reaction pathway of biomass to HMF over FePO4, MnPO4 and CrPO4. Adapted from ref. 286–288. | ||
| Catalyst | Catalyst loadinga | Solvent | Condition | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not provided. | |||||||
| NbPO | 10 wt% | H2O | 130 °C, 6 h | Fructose, 1 wt% | 87% | 70% | 289 |
| NbPO | 10 wt% | H2O/DMSO (w/w = 2/3) | 130 °C, 6 h | Fructose, 1 wt% | 95% | 90% | 289 |
| NbPO | 10 wt% | H2O | 130 °C, 6 h | Fructose, 10 wt% | 86% | 54% | 289 |
| Mesoporous ZrPO | 0.3 wt% | H2O | 155 °C, 6 h | Glucose, 0.5 wt% | 83.8% | 46.6% | 290 |
| α-ZrPO | 1.7 wt% | DMSO | 120 °C, 3 h | Fructose, 10 wt% | 97.2% | 54.3% | 291 |
| Mesoporous ZrPO | 1.7 wt% | DMSO | 120 °C, 3 h | Fructose, 10 wt% | 95.6% | 52.8% | 291 |
| Amorphous ZrPO | 1.7 wt% | DMSO | 120 °C, 3 h | Fructose, 10 wt% | 95.3% | 54.7% | 291 |
| Amorphous ZrPO-SO42− | 1.7 wt% | DMSO | 120 °C, 3 h | Fructose, 10 wt% | 100% | 73.4% | 291 |
| ZrPO | — | DMSO/H2O (v/v = 9) | 120 °C, 3 h, LHSV: 1 h−1 | Fructose, 5 wt% | 94.5% | 56.3% | 292 |
| ZrPO | — | DMSO/H2O (v/v = 9) | 160 °C, 3 h, LHSV: 1 h−1 | Glucose, 5 wt% | 72% | 32% | 292 |
| HfO(PO4)x | 0.6 wt% | NaCl–H2O/THF | 175 °C, 2.5 h | Glucose, 2 wt% | 99.7% | 90.5% | 293 |
| HfO(PO4)x | 0.6 wt% | NaCl–H2O/THF | 190 °C, 4 h | Cellulose, 2 wt% | 100% | 69.8% | 293 |
| MnPO | 0.5 wt% | H2O (5.4 M NaCl)/THF (v/v = 1:3) |
160 °C, 1.5 h | Glucose, 1.3 wt% | 96% | 59% | 288 |
| MnPO | 0.5 wt% | H2O (5.4 M NaCl)/THF (v/v = 1:3) |
170 °C, 2 h | Cellulose, 10 wt% | — | 44% | 288 |
| MnPO (3 runs) | 0.5 wt% | H2O (5.4 M NaCl)/THF (v/v = 1:3) |
160 °C, 1.5 h | Glucose, 1.3 wt% | 82% | 30% | 288 |
| FePO4 | 1.3 wt% | H2O (6 M NaCl)/THF (v/v = 1:3) |
140 °C, 0.25 h | Glucose, 2.5 wt% | 97.8% | 23.1% | 294 |
| FePO4 | 0.05 wt% | H2O (6 M NaCl)/THF (v/v = 1:3) |
140 °C, 0.25 h | Fructose, 2.5 wt% | 99.9% | 71.5% | 294 |
| FePO4 | 1.3 wt% | H2O (6 M NaCl)/THF (v/v = 1:3) |
160 °C, 1 h | Cellulose, 2.5 wt% | 87.4% | 48.0% | 294 |
| FePO4 | 1.3 wt% | H2O (6 M NaCl)/THF (v/v = 1:3) |
140 °C, 0.25 h | Camellia oleifera shell, 2.5 wt% | — | 7.8% | 294 |
| Branch-like FePO4 | 0.1 wt% | H2O (1.1 M NaCl)/THF (v/v = 1:3) |
160 °C, 1 h | Methyl cellulose, 0.25 wt% | — | ∼48% | 295 |
| Flower-like FePO4 | 0.1 wt% | H2O (1.1 M NaCl)/THF (v/v = 1:3) |
160 °C, 1 h | Methyl cellulose, 0.25 wt% | — | ∼44% | 295 |
| Sphere FePO4 | 0.1 wt% | H2O (1.1 M NaCl)/THF (v/v = 1:3) |
160 °C, 1 h | Methyl cellulose, 0.25 wt% | — | ∼37% | 295 |
| Amorphous FePO4 | 0.1 wt% | H2O (1.1 M NaCl)/THF (v/v = 1:3) |
160 °C, 1 h | Methyl cellulose, 0.25 wt% | — | ∼33% | 295 |
| SnPO | 0.33 wt% | H2O (NaCl) /MIBK | Microwave, 150 °C, 0.33 h | Glucose, 0.33 wt% | 80% | 50% | 296 |
| SnPO | 0.5 wt% | H2O(NaCl)/THF | 175 °C, 1 h | Glucose, 1.25 wt% | 98% | 61% | 297 |
| SnPO | 5 wt% | EMIMBr | 120 °C, 3 h | Glucose, 20 wt% | 94.6 | 52.3% | 254 |
| HAP | 10 wt% | EMIMBr | 130 °C, 4 h | Glucose, 10 wt% | 86.5% | 31.4% | 49 |
| Sn-HAP | 10 wt% | EMIMBr | 130 °C, 6 h | Glucose, 10 wt% | 97.5% | 52.6% | 49 |
| Al-HAP | 10 wt% | EMIMBr | 130 °C, 2 h | Glucose, 10 wt% | 81.6% | 32.5% | 49 |
| Sn/Al-HAP | 10 wt% | EMIMBr | 130 °C, 3 h | Glucose, 10 wt% | 96.0% | 65.6% | 49 |
| Sn/Al-HAP | 20 wt% | EMIMBr | 130 °C, 3 h | Glucose, 10 wt% | — | 46.0% | 49 |
| Sn/Al-HAP | 10 wt% | EMIMBr | 130 °C, 3 h | Starch, 10 wt% | — | 46.5% | 49 |
| 5% P–SnO2 | 0.95 wt% | GVL/H2O (v/v = 20) | 180 °C, 1.5 h | Glucose, 0.95 wt% | — | 46.4% (furfural 18.9%) | 298 |
| SnO2 | 0.95 wt% | GVL/H2O (v/v = 20) | 180 °C, 1.5 h | Glucose, 0.95 wt% | 85.1% | 19.3% (furfural 7.2%) | 298 |
| SnPCP@MnO2-PDA | 1 wt% | DMSO | 150 °C, 5 h | Glucose, 4 wt% | 92.2% | 55.8% | 299 |
| SnPCP@MnO2-PDA | 1 wt% | H2O/THF | 150 °C, 5 h | Glucose, 4 wt% | 73.2% | 41.2% | 299 |
| SiO2 | 4.2 wt% | H2O/acetone (v/v = 5) | 160 °C, 1.67 h | Glucose, 3.3 wt% | 27.6% | 6.0% | 300 |
| H3PO4–SiO2 | 4.2 wt% | H2O/acetone (v/v = 5) | 160 °C, 1.67 h | Glucose, 3.3 wt% | 63.1% | 39.5% | 300 |
| Unsupported FePO4 | 4.2 wt% | H2O/acetone (v/v = 5) | 160 °C, 1.67 h | Glucose, 3.3 wt% | 65.4% | 35.3% | 300 |
| H3PO4–SiO2–FePO4 (0.15) | 4.2 wt% | H2O/acetone (v/v = 5) | 160 °C, 1.67 h | Glucose, 3.3 wt% | 99.9% | 76.3% | 300 |
| H3PO4–SiO2–FePO4 | 4.2 wt% | H2O/acetone (v/v = 5) | 160 °C, 1.67 h | Glucose, 3.3 wt% | 65.8% | 38.8% | 300 |
Similarly, the inexpensive manganese phosphate catalyst (MnPO4) afforded HMF yields of 59% and 44% from glucose and cellulose, respectively.288 The use of FePO4 as a catalyst for the production of HMF was investigated the Zuo's group.301,302 They found that the FePO4 catalyst was active for the conversion of fructose, glucose, cellulose, and Camellia oleifera shell to HMF with moderate to high yields. They proposed that is FePO4 hydrolyzed at the evaluated temperature to H3PO4, which is responsible for the hydrolysis of cellulose and dehydration of fructose and Fe(OH)3, which are responsible for the isomerization of glucose to fructose, and the catalyst could be recovered owing to the reaction of H3PO4 with Fe(OH)3 upon cooling. They observed that although the amorphous FePO4 transformed to the crystalline form after the reaction, the catalyst could be reused in five reaction cycles without loss in its catalytic performance.302 Huang et al. reported that the silica-supported phosphate and iron phosphate heterogeneous catalyst (H3PO4–SiO2–FePO4(0.15)) exhibited a higher HMF yield (76.3%) from glucose than the unsupported FePO4.300
Aluminium phosphate-based materials are effective heterogenous catalysts to catalyze the dehydration reaction. For example, Zhang et al. reported that silicoalumino phosphate (SAPO-34) afforded an HMF yield as high as 93.6% from glucose in the medium of water/GVL at 170 °C for 40 min.303 In addition, they found that the presence of an appropriate amount of water (5–15 wt%) is beneficial for the selective formation of HMF. Romo et al. reported that a SAPO-34/5A zeolite bead catalyst, which was synthesized by growing SAPO-34 zeolite crystals on zeolite 5A beads, exhibited furfural and HMF yields of 45% and 20% from xylose and glucose in the medium of water, respectively.304
Similar to Nb2O5·nH2O, porous niobium phosphate (NbPO), which was prepared via the hydrothermal method using cetyltrimethyl ammonium bromide (CTAB) as the template, could also be used as a water-tolerant solid acid.305 The concentration and ratio of Brønsted and Lewis acid sites could be tuned by regulating the pH value in the synthetic process. Among the NbPO synthesized at different pH, the porous NbPO synthesized at pH = 2 with the highest amount of Brønsted acid sites was the most active for the dehydration of fructose to HMF, obtaining the highest HMF yield of 45% in water.306 In contrast, the porous NbPO synthesized at pH = 7 exhibited the highest amount of Lewis acid sites with a certain amount of Brønsted acid sites, leading to the highest HMF yield of 33.6% from glucose in pure water. The NbPO modified by 0.1 and 10 M HCl solutions gave HMF yields of around 8% after 24 h reaction at 120 °C.307
Hierarchically porous titanium phosphate nanoparticles, which were synthesized from titanium isopropoxide and orthophosphoric acid using P123 as a structure directing agent, exhibited HMF yields of 17%, 22%, and 44% from cellulose, glucose and fructose, respectively, using DMA/LiCl as the solvent under microwave irradiation.308 The use of partially phosphated TiO2 for the selective production of HMF from glucose was investigated by several groups. Atanda et al. reported that nanosized phosphated TiO2 with a bulk Ti/P molar ratio of 10.1 in the medium of water/n-butanol mixture gave the highest HMF yield of 81% with the glucose conversion of 97% at 175 °C in 3 h.309 The same catalyst in a water/THF biphasic system gave an HMF yield of 83%, which is comparable to that in the water/n-butanol mixture.310 The addition of N-methyl-2-pyrrolidone (NMP) to the water/THF biphasic system could further improve the conversion efficiency, affording high HMF yields of 90%, 94%, and 98% from glucose, cellobiose and sucrose, respectively. When mechanocatalytic depolymerized cellulose was used as the feedstock, an HMF yield of up to 74.7% was obtained in this catalytic system.311 Rao et al. reported that the phosphated TiO2 catalyst, which was synthesized via a one-pot sol–gel method only gave an HMF yield of 53% with a glucose conversion of 98% in the water/THF system. The above studies all speculated that the conversion of glucose to HMF proceeds via the widely considered reaction pathway involving the isomerization of glucose to fructose and fructose dehydration steps, but the reaction pathway has not been confirmed by direct evidence to date. In contrast, Noma et al. reported that both TiO2 and phosphoric acid-treated TiO2 gave a low HMF yield (≤34%) with a selectivity lower than 70% under mild conditions (120 °C, 4 h) in the medium of water, water/butanol, water/MIBK or water/2-sec-butylphenol.272 Also, these biphasic systems have been widely demonstrated to be effective to improve the HMF yield over other catalysts by inhibiting side reactions. Moreover, their following work demonstrated that the phosphorylation of TiO2via a simple impregnation method could only anchor two phosphates per nm2, which limits the selective formation of HMF, while photo-assisted phosphorylation could introduce about three phosphates per nm2, thus affording an HMF selectivity of 81% with the glucose conversion of 84% at 135 °C for 4 h in the medium of water/2-sec-butylphenol.312 Isotopic labelling and 13C NMR analysis demonstrated that the conversion of glucose over phosphated TiO2 proceeds via the stepwise dehydration mechanism with 3-deoxyglucosone as the main intermediate (Fig. 13), which is similar to that over TiO2.272,312 They proposed that the low HMF selectivity over the bare TiO2 is attributed to the intense intermolecular reactions between the adsorbed glucose and/or 3-deoxyglucosone molecules due to the strong adsorption of glucose onto the surface of TiO2 (Fig. 15), while the phosphorylation of TiO2 can inhibit these intermolecular side reactions by reducing the adsorption of glucose, and then improve the HMF selectivity.312 In summary, the catalytic performance of TiO2 (section 2.4.1) and titanium phosphate are relatively poor, while partially phosphated TiO2312 and citric acid-modified TiO2239 show much better catalytic performances. Therefore, the precise control of the surface composition of TiO2 and the use of appropriate solvents are crucial for the improvement of the HMF production efficiency.
 | ||
| Fig. 15 Reaction pathway of glucose over TiO2 with and without surface phosphates. Adapted from ref. 312. | ||
Tin phosphate exhibited excellent catalytic activity for the conversion of glucose to HMF in water-containing solvents, including a water/DMSO mixture, water(NaCl)/MIBK, and water(NaCl)/THF biphasic system.296,297,313 Zhang et al. reported that the tin phosphate generated in situ from SnCl4 and (NH4)2HPO4 afforded HMF yields of 71% and 33% from fructose and glucose, respectively.313 Under microwave-assisted heating, large-pore mesoporous tin phosphate (SnPO) exhibited excellent catalytic activity for the direct conversion of fructose, glucose, sucrose, cellobiose, and cellulose in water(NaCl)/MIBK, affording HMF yields of 32–77%.296 The conversion of glucose over SnO2 in a GVL/water mixture gave a low yield of HMF and furfural owing to the concomitant retro-aldol process, while phosphated SnO2 gave an HMF yield of 46.4% with a furfural yield of 18.9%.298
A high concentration glucose (10 wt%) could be converted into HMF effectively in the medium of EMIMBr ionic liquid using SnPO as a heterogeneous catalyst, obtaining an HMF yield if up to 58.3%.254 Compared with bulk SnO2 and small pore tin phosphate (sSnPO), the SnPO catalyst exhibited a lower Brønsted acid concentration and higher Lewis acid concentration, resulting from the tetra-coordinated Sn4+ sites, which are the main active species for the isomerization of glucose to fructose. The excellent conversion efficiency was mainly attributed to the synergistic effect of the SnPO catalyst and EMIMBr ionic liquid. Under the same reaction conditions, CrPO only gave an HMF yield of 14.1%. Similarly, tin(IV) phosphonate (SnBPMA) and zirconium phosphonate (ZrBPMA), which were prepared from SnCl4·5H2O or ZrOCl2·8H2O with N,N-bis(phosphonomethyl)aminoacetic acid, were also investigated for the conversion of fructose, sucrose and inulin to HMF in the EMIMBr ionic liquid by Ning et al.314 An HMF yield of up to 86.5% was obtained from fructose in the EMIMBr/SnBPMA system within 1.5 h at 100 °C, with a reaction rate higher than that obtained with EMIMBr alone. SnBPMA and ZrBPMA gave HMF yields of 40.1% and 48.7% from sucrose and inulin, respectively. However, their catalytic performance for glucose conversion was not investigated. Similarly, the highly efficient transformation of glucose was achieved over an Al and Sn co-modified hydroxyapatite catalyst (Al/Sn-HAP) in the medium of EMIMBr, affording HMF yields of 70.5% and 46.6% at a glucose loading of 10 and 40 wt%, respectively.49 The Al/Sn-HAP catalyst exhibited a remarkably lower activation energy (68.4 kJ mol−1) than HAP (110.5 kJ mol−1), Al-HAP (80.7 kJ mol−1) and Sn-HAP (79.2 kJ mol−1) for the conversion of glucose (Table 1). Moreover, the combination of the Al/Sn-HAP catalyst with EMIMBr enabled the efficient transformation of starch to HMF via tandem catalysis (Fig. 16), obtaining an HMF yield of 46.5% at a relatively high substrate loading.
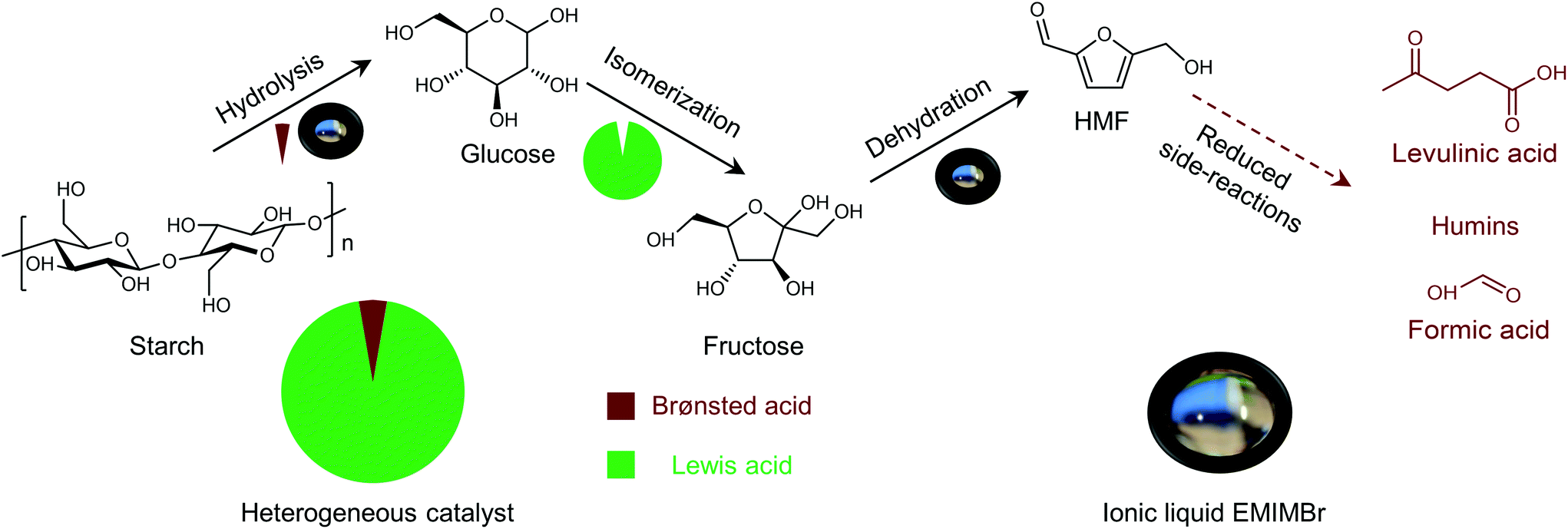 | ||
| Fig. 16 Conversion of starch to HMF via tandem catalysis. Adapted from ref. 49. | ||
Alkaline-earth metal phosphates, such as calcined calcium and strontium phosphate, only afforded HMF yields of around 20% from glucose in hot compressed water at 220 °C for 5 min.315 Zirconium phosphates (ZrPO) showed moderate catalytic activity for the conversion of fructose and glucose to HMF in the medium of DMSO or water/DMSO.291,292
Mesoporous tantalum oxide and tantalum phosphate were tested as heterogeneous catalysts for the conversion of glucose to HMF using a water/MIBK biphasic system as the reaction medium by Jiménez-Morales et al.316,317 Mesoporous tantalum oxide was prepared via the sol–gel method from the Ta(OC2H5)5 precursor using the triblock co-polymer Pluronic L-121 as the structure-directing agent, while the mesoporous tantalum phosphate was prepared by the reaction of tantalum penta-ethoxide with phosphoric acid using hexadecyltrimethylammonium bromide as a surfactant in absolute ethanol. Pyridine-FTIR analysis showed that the concentration of Lewis acid sites in the mesoporous tantalum oxide was 98.1 μmol g−1, while the Brønsted acid sites disappeared after heating at 125 °C under vacuum owing to the removal of coordinated water molecules. Compared with the mesoporous tantalum oxide, the mesoporous tantalum phosphate exhibited much higher acidity (1.48 mmol NH3 g−1) with a Brønsted acid concentration of 309 μmol g−1 and Lewis acid concentration of 97 μmol g−1. The mesoporous tantalum oxide gave an HMF yield of 23% at a glucose conversion of 69% under the optimized conditions (175 °C, 1.5 h). In contrast, the mesoporous tantalum phosphate gave an HMF yield of up to 32.8% at the glucose conversion of 56.3% under the optimized conditions (170 °C, 1 h), and fructose was not observed over this catalyst owing to the rapid dehydration of fructose to HMF over the high concentration of Brønsted acid sites. The leaching of phosphorus or tantalum species was not detected during the reaction process and the catalytic activity could be maintained for three catalytic runs, demonstrating the high stability of mesoporous tantalum oxide and tantalum phosphate. Yang et al. reported that commercial tantalum hydroxide (Ta2O5·nH2O, TA sample) and TA-p (TA treated with 1 M phosphoric acid solution at room temperature for 52 h and then calcined at 300 °C) gave HMF yields of 62% and 90% from fructose in the medium of water/2-butanol mixture, respectively.318 Moreover, an HMF yield as high as 58% with the glucose conversion of 70% was obtained over TA-p at 160 °C in 140 min.
Cao et al. prepared a series of hafnyl phosphates, HfO(PO4)x (x = 1.0, 1.5 and 2.0), as heterogeneous catalysts for the conversion of cellulose and other sugars in the medium of the NaCl–H2O/THF biphasic system.293 An HMF yield as high as 69.8% was achieved from cellulose over HfO(PO4)2 after 240 min reaction at 190 °C. The conversion of fructose, glucose, cellobiose, sucrose, starch and inulin in this reaction system gave high HMF yields of 94.8%, 90.5%, 79.3%, 86.6%, 75.3% and 80.4%, respectively, whereas the direct conversion of wheat straw only gave an HMF yield of 18.6%. They concluded that the introduction of the phosphate group could deactivate the unselective Lewis acid sites in the catalysts and then suppress the HMF decomposition to levulinic acid and formic acid. Moreover, the deposition of humins on the catalyst surface was also remarkably inhibited owing to the poorer HMF adsorption capability (9.3 mg g−1) of HfO(PO4)2. Therefore, the catalyst could maintain its catalytic activity after five catalytic cycles.
In the batch reactor, cerium phosphate afforded a best HMF yield of 52% with the selectivity of 93% from fructose in the medium of water, while in the flow reactor the catalyst only gave an HMF yield of 24% with a selectivity higher than 95%.319 The use of biphasic systems consisting of dimethyl carbonate (DMC) and water as the reaction medium enabled an HMF yield of up to 70% with the selectivity of 93.3% from fructose over cerium phosphate.114 Moreover, the reusability of the cerium phosphate catalyst in water/DMC was better than that in pure water.
Jia et al. reported that the combined use of phosphorus pentoxide (P2O5) and metal chlorides, such as NiCl2 and NaCl, could improve the HMF yield and selectivity remarkably, attaining an HMF yield of 75% with 85% selectivity from fructose after 30 min reaction in DMSO at 80 °C. Under the same conditions, P2O5, H3PO4 and NiCl2 gave low HMF yields and fructose conversion, demonstrating the synergy effect between P2O5 and metal chlorides for the dehydration of fructose to HMF.320
A series of studies on homogeneous metal phosphates indicated that HPO42− plays an important role in the isomerization of glucose. In the aqueous solution of NaH2PO4 and Na2HPO4 at pH = 7.5, glucose can be isomerized to fructose with a yield of 30%.167 The isomerization proceeds via an enediol anion intermediate, similar to the base-catalyzed process. In addition, the aqueous solution of NaH2PO4 and Na2HPO4 is also effective for the isomerization of ribose and lyxose to rare keto-pentoses, ribulose and xylulose, which are important feedstock for the synthesis of commodities and fine chemicals.321 Ma et al. reported that KH2PO4 is active to convert sucrose to HMF in the MIBK/H2O biphasic system, attaining an HMF yield of up to 70% under the optimum conditions.168 Even at a sucrose loading of up to 17.2 wt%, this catalytic system still afforded an HMF yield of 62.5% (higher than 50%), also indicating that HPO42− could catalyze the isomerization of glucose into fructose. The incorporation of a suitable organic moiety in metal phosphates not only can make the catalyst more flexible and robust, but also impart more hydrophobicity on the catalyst surface.322 Therefore, the development of organic–inorganic hybrid metal phosphonates may further broaden the portfolio of metal phosphate catalysts.
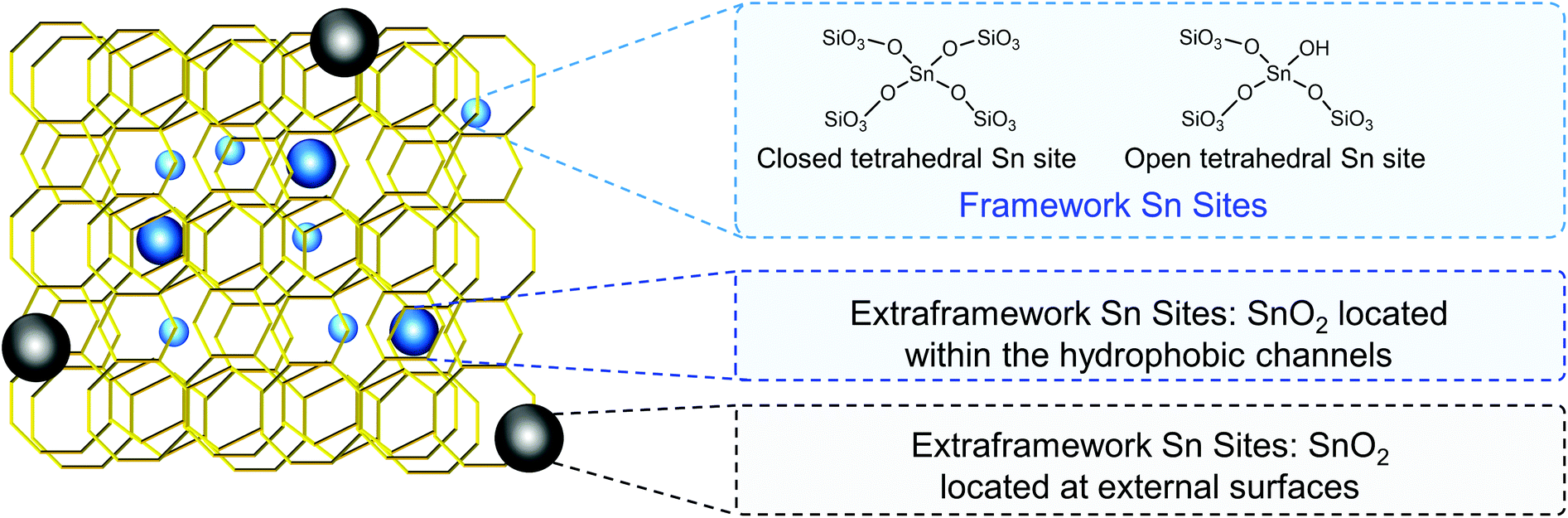 | ||
| Fig. 17 Schematic representation of the structure of Sn-Beta zeolite. Adapted from ref. 325 and 326. | ||
The framework-isolated Sn sites have been proven to be strong Lewis acid sites responsible for most Lewis acid-catalyzed reactions. The open and closed tetrahedral Sn sites in the framework of zeolite can be quantified by FTIR spectroscopy using deuterated acetonitrile as the probe.326 The open Sn sites with proximal silanol groups are responsible for the isomerization of glucose to fructose via an intramolecular hydride shift process.327,328 Tin silsesquioxane with a neighboring SiOH group favors the selective isomerization of glucose to fructose via the intramolecular H-shift mechanism. In contrast, the methyl-ligated tin silsesquioxane without a neighboring SiOH group, a homogeneous model of Sn-Beta, which does not contain “open” sites, prefers the conversion of glucose to mannose via the intramolecular C-shift mechanism, with a much lower reaction rate.328,329 These results also confirm the critical role of the open Sn sites in the formation of fructose. In the medium of methanol, the framework Sn sites mainly dominant the epimerization of glucose to mannose by a Lewis acid-mediated intramolecular carbon shift process.330
SnO2 can be located either at the external surface of the zeolite crystal or within the hydrophobic channel of the zeolite, with the catalytic performance depending on the reaction type and chemical environment.330 The isomerization of glucose to fructose can proceed over the SnO2 cluster located within the hydrophobic channel of the zeolite via a base-catalyzed proton-transfer process, in either water or methanol. In water, the SnO2 particles supported on the external surface of the zeolite crystal or amorphous silica cannot catalyze the isomerization, but in methanol the isomerization can proceed via the proton-transfer process. These results indicate that the contact of Sn sites with bulk water may suppress the isomerization process.
The synthetic process and conditions have an important influence on the Sn sites in Sn Beta zeolite. Compared with hydrophilic zeolite (Sn-Beta–OH), hydrophobic the Sn-Beta zeolite synthesized in fluoride medium (Sn-Beta–F) exhibited a 50 times higher first-order rate constant for glucose isomerization to fructose in water, mainly owing to the reduced kinetic inhibition of the open tetrahedral Sn sites with a few coordinated water molecules.326 To avoid the use of HF, a two-step synthesis process involving the dealumination of commercial Beta zeolite in acid followed by grafting of the Sn precursor in isopropanol under reflux conditions was developed to synthesize an Sn Beta zeolite.325,331 Compared with the conventional synthetic method, this method enabled the fast preparation of highly active Sn Beta zeolite with an Sn loading of up to 2 wt% using a cheap Sn-precursor and industrially available Beta zeolite as the feedstock.
Since Sn-Beta zeolite lacks Brønsted acid sites for dehydration, the combination of Sn-Beta zeolite with a Brønsted acid is usually required for the direct conversion of glucose to HMF (Table 8) in the medium of water or organic solvents.332 The combination of Sn-Beta zeolite (or Ti-Beta) with HCl in a water/THF/NaCl biphasic system gave an HMF selectivity of over 70%, suggesting that Sn-Beta can catalyze the isomerization of glucose in an acidic environment.333 When GVL, GHL, THF:MeTHF (1:1) and THF were used as the solvent, the combined use of Amberlyst-70 and Sn-Beta as the solid acid catalyst in a salt-free reaction system resulted in HMF yields of 59%, 55%, 60% and 63%, respectively, which are comparable to or higher than that obtained with a homogeneous Lewis acid (AlCl3) and Brønsted acid (HCl).334 In addition to obtaining a high HMF yield, this catalytic system could also reduce the use of the homogeneous catalyst and salt. Moreover, the separation and purification of HMF can be readily achieved by distillation owing to the low boiling point of THF.
| Catalyst | Catalyst loadinga | Solvent | Reaction condition | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not provided. | |||||||
| HY zeolite | 5 wt% | [bmpyrr][Cl]0.5[NTf2]0.5 | 80 °C, 5 h | Fructose, 5 wt% | — | 73% | 335 |
| HY zeolite | 5 wt% | [bmpyrr][Cl]0.5[NTf2]0.5 | 120 °C, 4 h | Glucose, 5 wt% | — | 38% | 335 |
| HY zeolite | 5 wt% | [bmpyrr][Cl]0.5[NTf2]0.5 | 120 °C, 3 h | Sucrose, 5 wt% | — | 62% | 335 |
| SBA-15-SO3H | 6.6 wt% | DMSO | 120 °C, 1 h | Fructose, 3.3 wt% | 100% | 96% | 336 |
| SBA-15/5-aminoisophthalic acid | 0.83 wt% | DMSO | Microwave, 135 °C, 20 min | Fructose, 1.1 wt% | — | 74% | 337 |
| SBA-15/5-aminoisophthalic acid | 0.83 wt% | DMSO | Microwave, 135 °C, 20 min | Glucose, 1.1 wt% | — | 64% | 337 |
| SBA-15/5-aminoisophthalic acid | 0.83 wt% | DMSO | Microwave, 135 °C, 20 min | Cellulose, 1.1 wt% | — | 42% | 337 |
| 2SZ@SBA-15-SO3H-NH2 | 2 wt% | BMIMCl | Microwave, 120 °C, 3 h | Cellulose, 5 wt% | 78.1% | 42% | 338 |
| Mesoporous organosilica-SO3H | 1 wt% | BMIMCl | 100 °C, 5 h | Avicel, 5 wt% | — | 10.5% (glucose 40.1%, cellobiose 22.4%) | 339 |
| ZSM-5 | 0.5 wt% | H2O | 150 °C, 1 h | LGO, 1 wt% | 65.3% | 33.7% | 340 |
| ZSM-5 | 0.67 wt% | H2O/THF/NaCl | 170 °C, 1 h | Remnant algal biomass, 4 wt% | 65.3% | 34.4% | 341 |
| Mordenite | 0.5 wt% | H2O | 150 °C, 1 h | LGO, 1 wt% | 16.1% | 4.2% | 340 |
| Nb(0.05)-Beta 18 | 0.6 wt% | H2O | 180 °C, 24 h | Glucose, 3.6 wt% | 41.7% | 17.2% | 342 |
| Nb(0.05)-Beta 18 | 0.6 wt% | NaCl/H2O/MIBK | 180 °C, 12 h | Glucose, 3.6 wt% | 97.3% | 82.1% | 342 |
| Al/MCM-41 | 0.8 wt% | ChCl/H2O/MIBK | 195 °C, 1.5 h | Glucose, 2.5 wt% | — | 57% | 343 |
| Al/MCM-41 | 0.8 wt% | ChCl/MIBK | 195 °C, 1.5 h | Glucose, 2.5 wt% | — | 40% | 343 |
| Al/MCM-41 | 0.8 wt% | H2O/MIBK | 195 °C, 1.5 h | Glucose, 2.5 wt% | — | 30% | 343 |
| Cr/MCM-41 | 0.8 wt% | ChCl/H2O/MIBK | 195 °C, 1 h | Glucose, 2.5 wt% | — | 44% | 343 |
| Zr/MCM-41 | 0.8 wt% | ChCl/H2O/MIBK | 195 °C, 2 h | Glucose, 2.5 wt% | — | 42% | 343 |
| Zr-salen-MCM-41 | 2.5 wt% | DMSO | 140 °C, 4 h | Fructose, 5 wt% | — | 92.0% | 344 |
| Zr-salen-MCM-41 | 2.5 wt% | DMSO | 140 °C, 4 h | Glucose, 5 wt% | — | 36.6% | 344 |
| H-β-zeolite | 5–10 wt% | H2O (HOAc) | 180 °C, 1 h | Chitosan | — | 15.3–28.2% | 345 |
| 2SZ@SBA-15 | 1.5 wt% | iPrOH/DMSO (v/v = 9) | 120 °C, 10 h | Glucose, 3.6 wt% | 89.3% | 57.3% | 346 |
| 5 wt% CeO2-2SZ@SBA-15 | 1.5 wt% | iPrOH/DMSO (v/v = 9) | 120 °C, 10 h | Glucose, 3.6 wt% | 93.9% | 66.5% | 346 |
| 5 wt% CeO2@SBA-15 | 1.5 wt% | iPrOH/DMSO (v/v = 9) | 120 °C, 10 h | Glucose, 3.6 wt% | 65.6% | 14.1% | 346 |
| 5 wt% CeO2-2SZ@SBA-15 | 1.5 wt% | iPrOH/DMSO (v/v = 9) | 120 °C, 10 h | Cellobiose, 3.6 wt% | 85.4% | 53.8% | 346 |
| 5 wt% CeO2-2SZ@SBA-15 | 1.5 wt% | iPrOH/DMSO (v/v = 9) | 120 °C, 10 h | Starch, 3.6 wt% | 75.5% | 45.2% | 346 |
| 5 wt% CeO2-2SZ@SBA-15 | 1.5 wt% | iPrOH/DMSO (v/v = 9) | 120 °C, 10 h | Cellulose, 3.6 wt% | — | ND | 346 |
| Al0.33Nb0.67/SBA-15 | 0.33 wt% | H2O/MIBK (v/v = 1/2) | 170 °C, 6 h | Cellulose, 1.6 wt% | 93.0% | 10.4% | 347 |
| Al0.33Nb0.67/SBA-15 | 0.33 wt% | H2O/MIBK (v/v = 1/2) | 170 °C, 6 h | Cellulose, 1.6 wt% | 93.4% | 55.7% | 347 |
| SO42−/ZrO2@HZSM-5 | 1 wt% | DMSO | 195 °C, 1.5 h | Glucose, 3 wt% | — | 23.6% | 348 |
| SO42−/ZrO2@HZSM-5 | 1 wt% | ChCl/DMSO | 195 °C, 1.5 h | Glucose, 3 wt% | — | 41.3% | 348 |
| SO42−/ZrO2@HZSM-5 | 1 wt% | NaCl/H2O/DMSO | 195 °C, 1.5 h | Glucose, 3 wt% | — | 61.1% | 348 |
| SO42−/ZrO2@HZSM-5 | 1 wt% | NaCl/H2O/ethyl acetate | 195 °C, 1.5 h | Glucose, 3 wt% | — | 40.7% | 348 |
| SO42−/ZrO2@HZSM-5 | 1 wt% | NaCl/H2O/ethyl acetate | 195 °C, 1.5 h | Glucose, 3 wt% | — | 54.7% | 348 |
| Al-Mont | 1.29 wt% | NaCl/H2O/THF | 180 °C, 2.5 h | Glucose, 1.3 wt% | ∼97% | 80.4% | 349 |
| Al-Mont | 1.29 wt% | NaCl/H2O/THF | 180 °C, 2.5 h | Fructose, 1.3 wt% | — | 98.5% | 349 |
| Al-Mont | 1.29 wt% | NaCl/H2O/THF | 180 °C, 2.5 h | Sucrose, 1.3 wt% | — | 86.1% | 349 |
| Al-Mont | 1.29 wt% | NaCl/H2O/THF | 180 °C, 2.5 h | Inulin, 1.3 wt% | — | 81.6% | 349 |
| Al-Mont | 1.29 wt% | NaCl/H2O/THF | 180 °C, 2.5 h | Starch, 1.3 wt% | — | 60.1% | 349 |
| M-Mont (M = Fe, Zn, Zr, Ca) | 1.29 wt% | NaCl/H2O/THF | 180 °C, 2.5 h | Glucose, 1.3 wt% | — | 18–24% | 349 |
| Nb-MMT-900 | 0.8 wt% | NaCl/H2O/MIBK | 170 °C, 3 h | Glucose, 1.2 wt% | 99% | 70.52% | 350 |
| Nb-MMT-900 | 0.8 wt% | H2O | 170 °C, 3 h | Glucose, 1.2 wt% | 63.0% | 18.4% | 350 |
| Nb-MMT-900 | 0.8 wt% | NaCl/H2O/MIBK | 170 °C, 3 h | Cellulose, 1.2 wt% | 54.68% | 10.51% | 350 |
| HY-1 zeolite | 0.7 wt% | H2O/GBL (v/v = 9/105) | 150 °C, 50 min | Fructose, 5 wt% | 99.8% | 69.2% (furfural 7.7%) | 351 |
| HY-2 zeolite | 0.7 wt% | H2O/GBL (v/v = 9/105) | 150 °C, 50 min | Fructose, 5 wt% | 98.8% | 39.1% (furfural 28.6%) | 351 |
| HY-3 zeolite | 0.7 wt% | H2O/GBL (v/v = 9/105) | 150 °C, 50 min | Fructose, 5 wt% | 98.5% | 21.4% (furfural 37.8%) | 351 |
| Hβ zeolite | 1 wt% | H2O/GBL (v/v = 5/95) | 150 °C, 1 h | Fructose, 5 wt% | 99.9% | 14.0% (furfural 50.25%) | 352 |
| Hβ zeolite | 1 wt% | H2O/NMP (v/v = 5/95) | 150 °C, 1 h | Fructose, 5 wt% | 97.4% | 83.3% (furfural 6.7%) | 352 |
| β zeolite, HCl | 0.27 wt%, 200 ppm | H2O | 150 °C, 1 h | Glucose, 3.3 wt% | — | 32.4% | 332 |
| SAPO-34/5A zeolite bead | 1.2 wt% | H2O | 190 °C, 3 h | Glucose, 2 wt% | — | 20% | 304 |
| [BMIM]Br encapsulated H-MOR | 1.1 wt% | H2O (10 wt% NaCl) | 170 °C, 3 h | Glucose, 3.3 wt% | 75% | 39% | 353 |
| [BMIM]Br encapsulated H-MOR (2–5 runs) | 1.1 wt% | H2O (10 wt% NaCl) | 170 °C, 3 h | Glucose, 3.3 wt% | 42–45% | Around 25–30% | 353 |
Owing to the excellent activity of EMIMBr for the dehydration of fructose, an Sn-containing zeolite could also act as a heterogeneous catalyst for the efficient conversion of glucose into HMF in the medium of EMIMBr ionic liquid without the use of an additional Brønsted acid. Sn-MCM-41 gave an HMF yield as high as 70% at 110 °C within 4 h, while MCM-41, Ti-MCM-41, Cr-MCM-41, Zr-MCM-41 and Sn-Beta only gave HMF yields of 4%, 5%, 11%, 6% and 15%, respectively.354 Sn-MCM-41 could be readily recovered and reused for the dehydration of glucose with a slight decrease in its catalytic activity. However, the reason why Sn-MCM-41 exhibited an excellent catalytic performance but Sn-Beta showed a poor catalytic performance was not investigated in this study. Jia and co-authors compared the catalytic performance of Sn/MCM-41, Al/MCM-41 and Cr/MCM-41 for the conversion of mannose into HMF in DMSO.355 They found that Sn/MCM-41 exhibited better catalytic activity than the other two catalysts, resulting in an HMF yield of 45% with glucose conversion of 88% at 150 °C within 60 min. The as-synthesized Sn/MCM-41 catalyst gave HMF yields of 42% and 28% from glucose and galactose, respectively. The control experiment using nano-SnO2 as the catalyst attained a low mannose conversion, suggesting that the Sn species in the framework of Sn/MCM-41, instead of the surface SnO2 are the main catalytic center for sugar conversion.
The Sn montmorillonite (Sn-Mont) catalyst prepared from Ca-Mont and SnCl4·5H2O aqueous solution by ion-exchange contains both Lewis acid and Brønsted acid sites, resulting from Sn4+ and partially hydrolyzed Sn–OH groups, respectively.356 The Sn-Mont catalyst gave an HMF yield of up to 53.5% from glucose at 160 °C for 3 h using a monophasic THF/DMSO mixture as the solvent. Moreover, the one-pot conversion of cellulose to HMF in high yield (39.1%) was also achieved using the THF/H2O-NaCl biphasic system as the reaction medium. Al-doped montmorillonite (Al-Mont) exhibited higher catalytic activity than Fe-, Zn-, Zr- or Ca-doped montmorillonite, obtaining HMF yields of 80.4%, 60.1% and 81.6% from glucose, starch and inulin, respectively.349 Similarly, a niobium-loaded montmorillonite (Nb-MMT) catalyst bearing both Lewis and Brønsted acid sites, which was readily prepared via a cation-exchange method, gave HMF yields of 70.52% and 10.51% from glucose and cellulose, respectively.350
Catalyst deactivation is a serious and universal issue limiting the application of zeolites in biomass conversion. The stability of zeolites depends on many factors, including their framework type (MOR, BEA, MFI or FAU), preparation process (hydrothermal synthesis or post-modification), hydrophobicity, hydrophilicity, solvent (water or alcohol) and reactant. Generally, the deactivation of tin-containing zeolites mainly results from the amorphization of the zeolite framework and leaching, restructuring or fouling of the active tin sites.357 A series of studies showed that the Sn-Beta zeolite deactivates rapidly both in pure water 358,359 and alcohols.360,361 In contrast, Guo et al. reported that the use of a dioxane/water mixture (containing 5 wt% water) as the reaction medium not only greatly improved the stability and reusability of the Sn-Beta zeolite, but also increased the fructose yield to 41.5%.358 Thermogravimetric analysis (TGA) and FTIR analysis showed that the hydrophilization of Sn-Beta is inhibited in the dioxane/water mixture, which can explain the good stability of Sn-beta in this medium.
Similar to Sn-Beta, Ti-substituted zeolite is also a typical Lewis acid, which is active for the isomerization of glucose in water, and the catalytic activity of Ti beta zeolite is lower than that of Sn-Beta owing to its higher activation energy.324,362 Ti-Beta catalyzes the isomerization of glucose to sorbose via the intramolecular C5–C1 hydride shift mechanism and the isomerization of glucose to fructose via the intramolecular C2–C1 hydride shift mechanism simultaneously in both water and methanol, with water favoring the generation of fructose and methanol favoring the generation of sorbose.363 Cordon et al. reported that confining Ti sites within the twelve-membered ring (12-MR) micropores of Ti-Beta could greatly improve the reaction rate of D-glucose isomerization to D-fructose and L-sorbose simultaneously, and a further decrease of micropore size (10-MR pores in Ti-MFI and Ti-CON) resulted in a decrease in the glucose isomerization rate, possible owing to the intrapore reactant diffusion restrictions.364 Moreover, tighter confinement in Ti zeolites led to a remarkable increase in the selectivity toward L-sorbose over D-fructose, attaining an L-sorbose/D-fructose ratio of more than 10 on Ti-MFI.
Xu et al. reported that Cr/β-zeolite is highly effective for the conversion of a series carbohydrates, including fructose, glucose, sucrose, cellobiose and starch to HMF in the H2O/THF/NaCl biphasic system.365 The Lewis and Brønsted acid sites on the Cr/β-zeolite surface work synergistically to catalyze the isomerization of glucose to fructose and dehydration of fructose, obtaining a maximum HMF yield of 72% after 1.5 h reaction at 150 °C. Nb-Modified Beta-zeolite (Nb(0.05)-Beta 18) in the NaCl/H2O/MIBK biphasic system gave an HMF yield of 79.8% at the glucose conversion of 97.4% at 180 °C for 12 h.342
Different from Sn-containing zeolite materials, zeolites with a high Al content usually catalyze the direct conversion of fructose and glucose to HMF owing to the coexistence of sufficient Brønsted acid sites and Lewis acid sites.366 Jia et al. investigated the catalytic performance of MFI, BEA, and Y zeolite for the dehydration of fructose to HMF in different solvents including DMSO, acetone, GVL, and propylene carbonate (PC)/water (1:1 v/v) biphasic system under microwave irradiation.367 They found that in the medium of water, the HMF yield was independent of the particle size of MFI zeolite, and the highest HMF yield of 49.2% with the fructose conversion of 72.4% was obtained over the hydrophobic Y zeolite in DMSO. Jiménez-Morales et al. reported that the mesoporous 10 wt% aluminium-doped MCM-41 silica (10Al-MCM-41) in a water(NaCl)/MIBK biphasic system showed a glucose conversion of 98% and HMF yield of 63% at 195 °C in just 30 min.368 Using the same reaction conditions, the protonated zeolites H-Beta, H-ZSM-5 and H-Y attained HMF yields of 56%, 42% and 42%, respectively.369,370 Jiménez-Morales et al. also investigated the use of Zr-doped MCM-41 silica (Zr-MCM-41) for the conversion of glucose to HMF using similar reaction conditions, obtaining an HMF yield of 23%.371 Feng et al. investigated a series of MCM-41-supported metal catalysts, including Al, Cr, Fe, Cu, Zn and Zr, for the catalytic conversion of glucose to HMF, with the Al/MCM-41 catalyst in the medium of ChCl/H2O/MIBK attaining the highest HMF yield of 57% after 1.5 h reaction at 195 °C.343
An Al-containing zeolite was also effective in catalyzing the conversion of glucose to HMF in the medium of [BMIM]Cl ionic liquid. Hβ-zeolite with a BEA structure and an appropriate Si/Al ratio of 25 showed higher catalytic activity than HY-zeolite, H-mordenite, Hβ-zeolite and HZSM-5, probably owing to the balanced density and strength of Lewis acid sites and Brønsted acid sites.372 An HMF yield of up to 50.3% with 80.6% glucose conversion was obtained over the Hβ-zeolite at 150 °C for 50 min. The catalytic system was also effective for the conversion of other carbohydrates, including sucrose, maltose, cellobiose, starch and cellulose, obtaining HMF yields of 67.6%, 47.8%, 49.3%, 45.4% and 46.5%, respectively.
Velaga et al. reported that H-mordenite zeolite with mesoporosity, which was synthesized using a seed-assisted and template-free method, exhibited a maximum HMF yield of 66% with the glucose conversion of 76% at 180 °C for 1 h in a water(NaCl)/MIBK biphasic system.373 Mordenites modified with NH4Ac, NH4Cl or NH4F were investigated as heterogeneous catalysts for the direct conversion of glucose into HMF using the BMIMBr ionic liquid or water–acetone/ethyl acetate biphasic system as the reaction medium.374 The NH4Cl-modified mordenite afforded 64% HMF yield with 97% glucose conversion in BMIMBr ionic liquid. In contrast, the highest HMF yield (50%) from the biphasic system was obtained with the mordenite treated with 1 M NH4Cl and 2.4 M NH4F, mainly owing to the presence of strong Lewis acid sites.
Alkali metal chlorides, including LiCl, NaCl and KCl, have been demonstrated to be effective to improve the catalytic performance of ionic liquid/zeolite catalytic systems for the conversion of cellulose to HMF.375 An H-form zeolite in [EMIM]Cl ionic liquid only gave a maximum HMF yield of 18.5% after 150 min reaction at 160 °C. The combination of zeolite with LiCl gave the highest HMF yield of 70.3% at 160 °C for 30 min, while zeolite/KCl and zeolite/LiCl afforded the highest total furans yields of 82.0% and 81.2%, respectively, at 140 °C for 75 min. At a relatively mild reaction temperature, zeolite/NaCl-KCl was more effective than zeolite/NaCl and zeolite/KCl, attaining the highest HMF yield of 58.2% with total furan yield reaching 66.2% at 120 °C.
Xia et al. prepared a series of Fe/β-zeolite catalysts with different Fe/Al ratios via a liquid ion-exchange method to catalyze the conversion of glucose to HMF.376,377 Although the introduction of Fe species to the zeolite did not increase the concentration of Lewis and Brønsted acid sites obviously, the Fe/β-zeolite catalyst attained a higher HMF yield than the β-zeolite. Both the HMF yield and Lewis/Brønsted acid ratio vs. Fe/Al ratio exhibited a “volcano relationship”. The control experiment suggested that the extra-framework isolated Fe species bound to the framework Al sites are the main active sites to catalyze the isomerization of glucose to fructose.
In addition to their direct use as catalysts, zeolites are also excellent supports for active components to improve their catalytic performance and stability. Osatiashtiani et al. prepared a ZrO2/SBA-15 catalyst by grafting a sulfated zirconia monolayer on SBA-15.378 The Brønsted acid, Lewis acid and base sites mainly depended on the film thickness and sulfate loading. The presence of base and Lewis acid sites in bilayer ZrO2/SBA-15 contributed to the isomerization of glucose to fructose. Compared with nonporous sulfated zirconia, the bilayer ZrO2/SBA-15 afforded a 3-fold enhancement in the HMF production rate and better hydrothermal stability. Similarly, Zhang et al. synthesized a 5 wt% CeO2-2SZ@SBA-15 catalyst, bearing basic sites, Lewis and Brønsted acid sites simultaneously, by incorporating ceria and sulfated zirconia (SZ) into SBA-15 via the layer-by-layer grafting method followed by the wet impregnation method.346 Consequently, 5 wt% CeO2-2SZ@SBA-15 gave a much higher HMF yield (66.5%) from glucose than 2SZ@SBA-15 and 5 wt% CeO2@SBA-15. Besides, the grafting of basic sites, such as NH2, and Brønsted sites, such as SO3H, was also widely attempted to improve the conversion of carbohydrates to HMF.338 Feng et al. reported that SO42−/ZrO2 supported on HZSM-5 zeolite (SO42−/ZrO2@HZSM-5) was active for the catalytic dehydration of glucose into HMF, obtaining an HMF yield of 61.1%.348
The octahedrally coordinated extra-framework Al sites in H-BEA-25 zeolite could catalyze the isomerization of glucose to fructose effectively with an activation energy ranging between that reported for Ti-BEA and Sn-Beta.379 Although Al-based zeolites are not as effective as Sn-Beta zeolites to isomerize glucose to fructose in water, Al-based zeolites can catalyze the isomerization of glucose to fructose via a two-step process (Fig. 18).380 In the first step, glucose is isomerized to fructose in the medium of alkyl alcohol followed by instantaneous reaction with alkyl alcohol via etherification, leading to the formation of alkyl fructoside. In the second step, fructose is obtained by the hydrolysis of alkyl fructoside in water. Zeolite H-USY (Si/Al = 6) showed higher catalytic activity than the beta, ZSM-5, mordenite and H-USY zeolites with a higher Si/Al ratio, attaining the highest fructose yield of 55% at 120 °C for 1 h. A single-unit-cell Sn-MFI nanosheet with exclusively framework Sn sites was also used for the two-step glucose isomerization process, resulting in a fructose yield (65%) remarkably higher than that obtained with Sn-MCM-41 and Sn-Beta.381
 | ||
| Fig. 18 Isomerization of glucose to fructose over zeolite via a two-step process. Adapted from ref. 380. | ||
The conversion pathway of fructose over zeolite catalysts can be controlled by manipulating the solvent effect or tuning the channel of zeolite. For example, the use of Hβ zeolite in the medium of γ-butyrolactone (GBL)/water mixture led to the reversible tetrahedral-octahedral framework aluminum transformation and promoted the selective formation of furfural (50.25% yield) from fructose (Fig. 19).352 The synergistic effect of GBL with HY-3 zeolite with apertures of 7.4 Å favored the generation of acyclic fructose from cyclic fructose (8.6 Å), while the Brønsted acid sites in the channels of HY-zeolite catalyzed the decomposition of acyclic fructose to xylose and formaldehyde via the selective cleavage of the C–C bond, and the following xylose dehydration, resulting in the formation of furfural.351
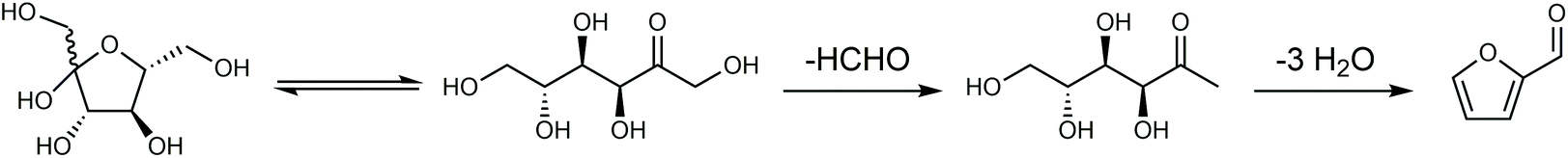 | ||
| Fig. 19 Conversion of fructose to furfural over zeolite. Adapted from ref. 351 and 352. | ||
| Catalyst | Catalyst loadinga | Solvent | Reaction condition | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not available. | |||||||
| Fibrous HPW/TiO2–Al2O3 | 7 wt% | DMSO | 120 °C, 3 h | Fructose, 12 wt% | 89.9% | 80.3% | 383 |
| Powdery HPW/TiO2–Al2O3 | 7 wt% | DMSO | 120 °C, 3 h | Fructose, 12 wt% | 86.7% | 61.3% | 383 |
| ZMPA (30) | 1 wt% | DMSO | 120 °C, 0.5 h | Fructose, 7.2 wt% | 100% | 80.3% | 384 |
| mesoporous-ZrO2 | 1 wt% | DMSO | 120 °C, 0.5 h | Fructose, 7.2 wt% | 90% | 29% | 384 |
| SnCl2-PTA/H β zeolite | 1.25 wt% | NaCl/H2O/THF | 180 °C, 2 h | Wheat straw, 2.5 wt% | — | 30% (furfural 71%) | 386 |
| SnCl2-PTA/H β zeolite (4 runs) | 1.25 wt% | NaCl/H2O/THF | 180 °C, 2 h | Wheat straw, 2.5 wt% | — | 19% (furfural 43%) | 386 |
| [PzS]H2PW | 5.4 mM | 3.1 wt% NaCl, H2O/THF (v/v = 0.2) | 180 °C, 2 h | Glucose, 0.83 wt% | — | 58.2% (LGA, 12.8%) | 387 |
| SiO2-ATS-PTA | 3.3 wt% | Acetone/H2O (v/v = 5) | 160 °C, 140 min | Glucose, 4 wt% | 99.87% | 78.31% | 385 |
| SiO2-ATS-PTA | 3.3 wt% | Acetone/H2O (v/v = 5) | 160 °C, 140 min | Glucose, 4 wt% | 93.36% | 68.35% | 385 |
| SiO2-ATS-PTA | 3.3 wt% | H2O (v/v = 5) | 160 °C, 140 min | Glucose, 4 wt% | 99.77% | 52.21% | 385 |
| PTA | 1.4 wt% | Acetone/H2O (v/v = 5) | 160 °C, 140 min | Glucose, 4 wt% | 75.38% | 52.27% | 385 |
| SiO2-ATS | 1.9 wt% | Acetone/H2O (v/v = 5) | 160 °C, 140 min | Glucose, 4 wt% | 75.38% | 52.27% | 385 |
| HReO4 | 0.5 wt% | DMSO | 140 °C, 1 h | Fructose, 3.6 wt% | 100% | 100% | 388 |
| HReO4 | 0.5 wt% | N,N-DMF | 140 °C, 1 h | Fructose, 3.6 wt% | 64% | 50% | 388 |
| HReO4 | 0.5 wt% | MeCN | 140 °C, 1 h | Fructose, 3.6 wt% | 35% | 25% | 388 |
| HReO4 | 0.5 wt% | THF | 140 °C, 1 h | Fructose, 3.6 wt% | 66% | 30% (levulinic acid 28%) | 388 |
| HReO4 | 0.5 wt% | 1,4-Dioxane | 140 °C, 1 h | Fructose, 3.6 wt% | 100% | 0% (levulinic acid 100%) | 388 |
| HReO4 | 5 mol% | 1,4-Dioxane | 140 °C, 2 h | Xylose, 1.5 wt% | — | Furfural 80% | 389 |
| [MimAM]H2PW12O40 | 2.5 mM | NaCl (0.5 M)/H2O/THF | 160 °C, 7.5 h | Glucose, 2.5 wt% | 99.8% | 53.9% | 390 |
| [MimAM]H2PW12O40 | 2.5 mM | DMSO | 160 °C, 7.5 h | Glucose, 2.5 wt% | 99.8% | 83.2% | 390 |
| ChH2PW12O40 | 0.02 M | H2O/MIBK | 140 °C, 8 h | Cellulose, 1.8 wt% | 87.0% | 75.0% | 391 |
| ChH4CeW12O40 | 5 mM | H2O/DMSO/MIBK (v/v = 1/2/7) | 140 °C, 6 h | Glucose, 1.4 wt% | — | 67.5% | 392 |
The introduction of Lewis acid sites in heteropolyacids is necessary to improve their catalytic performance for the conversion of glucose to HMF. Wang et al. reported that the introduction of aluminum to phosphotungstic acid could form Lewis acid sites owing to the electron-withdrawing effect of terminal WO on hydrated aluminum.393 Al-Doped phosphotungstic acid in H2O/DMSO gave a much higher HMF yield (61.7%) from glucose than that of phosphotungstic acid (around 25%) after 4 h reaction at 170 °C owing to the suitable ratio of Brønsted and Lewis acid sites. The Brønsted–Lewis-surfactant-combined heteropolyacid Cr[(DS)H2PW12O40]3 (DS represents OSO3C12H25 dodecyl sulfate) was used as a heterogeneous catalyst for the one-pot conversion of cellulose to HMF in water, obtaining an HMF yield of 52.7% with the glucose conversion of 77.1% at 150 °C within 2 h, which is much higher than that obtained with CrCl3, Cr(DS)3, H3PW12O40 and Cr[H2PW12O40]3.394 Xu et al. reported that the SnCl2-PTA/H β zeolite, which was prepared by introducing SnCl2 and phosphotungstic acid (PTA) onto H β zeolite, gave an HMF yield of 30% and furfural yield of 71% from wheat straw.386 However, the catalytic activity of the SnCl2-PTA/H β zeolite catalyst decreased remarkably after four cycles owing to the leaching of Sn species.
The design of hybrid catalysts composed of heteropolyacids with ionic liquids or deep-eutectic solvents can regulate the active sites and then improve their catalytic performance. For example, the acid–base bifunctional ionic hybrid catalyst [MimAM]H2PW12O40, which was prepared using an amino-functionalized imidazolium ionic liquid and H3PW12O40via ion exchange, afforded HMF yields of 53.9% and 83.2% from glucose using THF/H2O–NaCl and DMSO as the reaction medium, respectively.390 Similarly, (HOCH2CH2N(CH3)3)H2PW12O40 (abbreviated as ChH2PW12O40) synthesized from choline chloride and H3PW12O40 was more active than H3PW12O40 for the one-pot conversion of cellulose to HMF in water/MIBK, affording an HMF yield up to 75% within 8 h at 140 °C.391 Moreover, the heteropolyacid catalyst could be readily recycled after cooling to room temperature owing to its thermoregulate property.
Numerous carbon-based materials including activated carbon, biochar, carbon nanotubes (CNTs), hydrothermal carbon, graphene, graphene oxide, carbon quantum dots, graphene quantum dots, ordered mesoporous carbon, nanostructured carbon, N-doped carbons, carbon nitride and carbon-based composites have been used as carbon supports or precursors for the preparation of SO3H-containing functional carbon materials via sulfonation. Sulfonated carbon can be prepared either though simultaneous sulfonation and carbonization or post-grafting functionalization approach. For example, the direct hydrothermal treatment of glucose and p-toluenesulfonic acid (TsOH) gave a carbonaceous material with a high density of acid sites (2.0 mmol g−1).396 Both biomass and its derivates, including fructose, glucose,397,398 sucrose, sucralose,399 cellulose, lignin,400,401 yeast cells,402 and cow dung,355 have been used as feedstock for the preparation of SO3H-functionalized carbonaceous solid acids via carbonization followed by sulfonation.
Sulfonated carbon has been widely investigated for the hydrolysis of cellulose to glucose.397,400,403 To improve the reaction efficiency of cellulose hydrolysis, cellulase-mimetic solid acids (Fig. 20) bearing both cellulose-binding sites (such as –OH and –Cl) and catalytic sites (SO3H and COOH) were designed by imitating the catalytic mechanism of cellulase.400,404–406 The binding and catalytic sites work energetically to promote the depolymerization of cellulose, where cellulose firstly attaches onto the catalyst surface via hydrogen bonds and then the β-1,4-glycosidic bond is disrupted by the acidic sites. Besides, the design of a mesoporous structure in carbon materials could promote the adsorption of 1,4-β-D-glucans, thus enabling hydrolysis over the weak acid sites (–COOH).407–409 However, to date, cellulose hydrolysis over these solid acids still require a much higher reaction temperature than the optimum temperature for cellulase, suggesting that the activation energy of cellulose hydrolysis over cellulase-mimetic solid acid is considerably higher than that over cellulase. Thus, it is necessary to deepen our understanding on the catalytic mechanism of cellulase to design real cellulase-mimetic solid acids.
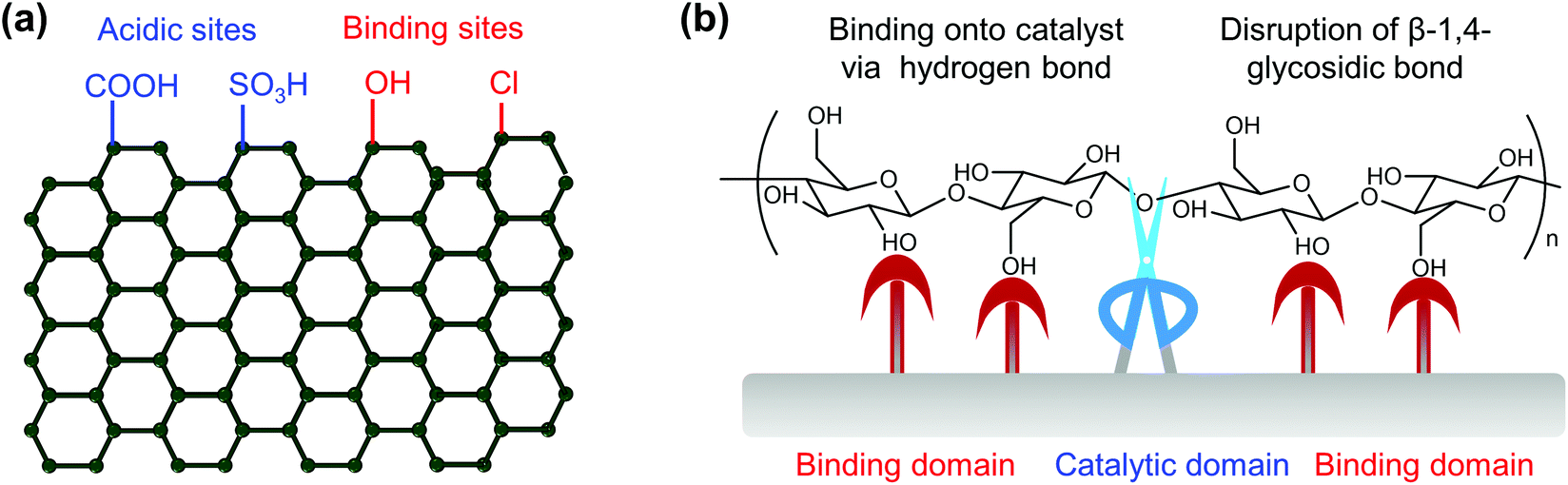 | ||
| Fig. 20 (a) Cellulase-mimetic solid acid catalyst and (b) cellulose hydrolysis process. Adapted from ref. 404. | ||
Numerous SO3H-functionalized carbon materials have been demonstrated to be effective for the dehydration of fructose to HMF. For example, N-rich porous carbonaceous solid acids can be prepared by grafting strong acidic groups on porous carbon obtained by carbonization of polypyrrole in the presence of KOH, leading to higher catalytic activity than Amberlyst 15, H-ZSM-5, H-USY and sulfonic group-functionalized ordered mesoporous silicas for the dehydration of fructose to HMF (Table 10).410 Sulfonic acid-functionalized lignin-derived mesoporous carbon (LDMC-SO3H) with well-ordered two-dimensional hexagonal mesoporous characteristics was synthesized via phenolation and evaporation-induced self-assembly reaction.411 The LDMC-SO3H catalyst gave an HMF yield as high as 98.0% at 140 °C for 2 h in the medium of DMSO. Adsorption kinetic studies suggested that the low adsorption capacity of HMF and high adsorption capacity of fructose on LDMC-SO3H contribute greatly to the high selectivity toward HMF. Li et al. reported that sulfonated graphene quantum dots (SGQDs) could combine the advantages of homogenous and heterogeneous catalysis, affording an HMF yield (51.7%) comparable to homogenous H2SO4 (44.2%) and HCl (60.1%) at a relatively high fructose concentration (6.7 wt% with respect to the reaction medium).412 Besides, sulfonated graphene (SG) also exhibited high catalytic activity for the conversion of corn stalk and xylose into furfural.413
| Catalyst | Catalyst loadinga | Solvent | Condition | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not provided. | |||||||
| 10%TiO2C_S | 1.7 wt% | DMSO | 120 °C, 1 h | Fructose, 8.3 wt% | — | 95% | 414 |
| PC-2-300 | 4 wt% | DMSO | 160 °C, 3 h | Fructose, 5 wt% | — | 93.7% | 415 |
| PC-2-300, 4 wt% | 4 wt% | H2O/2-butanol (v/v = 2/3) | 160 °C, 3 h | Fructose, 5 wt% | — | 80.9% | 415 |
| BC-SO3H-PA | 6.7 wt% | H2O | Microwave (350 W), 90 °C, 1 h | Cellulose, 13.3 wt% | — | 23.1% | 416 |
| BC-SO3H | 6.7 wt% | H2O | Microwave (350 W), 90 °C, 1 h | Cellulose, 13.3 wt% | — | <0.1% | 416 |
| PS-SO3H | 6.7 wt% | H2O | Microwave (350 W), 90 °C, 1 h | Cellulose, 13.3 wt% | — | 0 | 416 |
| PS-NH2-PA | 6.7 wt% | H2O | Microwave (350 W), 90 °C, 1 h | Cellulose, 13.3 wt% | — | 2.5% | 416 |
| Ctobacco stem-SO3H | 1 wt% | GVL/H2O (v/v = 4.7/0.3) | 130 °C, 30 min | Fructose, 2 wt% | ∼100% | 93.7% | 417 |
| Ctobacco stem-SO3H | 1 wt% | GVL/H2O (v/v = 4.7/0.3) | 180 °C, 30 min | Glucose, 2 wt% | ∼96% | 43.8% | 417 |
| Cpolyurethane-SO3H | 0.3 wt% | 1,4-Dioxane | 140 °C, 2 h | Fructose, 0.9 wt% | 100% | 70.1% | 418 |
| SC-CCA | 1.2 wt% | GVL/H2O (v/v = 10) | 130 °C, 20 min | Fructose, 2.4 wt% | 100% | 78.1% | 419 |
| SC-CCA | 1.2 wt% | GVL/H2O (v/v = 10) | 180 °C, 10 min | Glucose, 2.4 wt% | ∼94% | 33.2% | 419 |
| SC-CCA | 1.2 wt% | GVL/H2O (v/v = 10) | 180 °C, 50 min | Cellulose, 2.4 wt% | — | 22.5% | 419 |
| BCSA-IL-X (X = CF3SO3H, HBF4, HPF6 | 0.67 wt% | H2O | 350 W microwave, 80 °C, 3 h | Cellulose, 1.33 wt% | — | 12.70–27.94% | 420 |
| BCSA-IL-Cl | 0.67 wt% | H2O | 350 W microwave, 80 °C, 3 h | Cellulose, 1.33 wt% | — | <0.1% | 420 |
| Bone char | 0.5 wt% | H2O | 90 °C, 3 h | Glucose, 1 wt% | 27.2% | 15% | 421 |
| Bone char; BAIL | 0.25 wt%; 0.25 wt% | H2O | 170 °C, 12 h | Glucose, 2 wt% | 72% | 39% | 421 |
| CS-CaO-800 | 0.6 wt% | H2O | 80 °C, 40 min | Glucose, 5 wt% | 40.9% | 29.2% | 422 |
| CS-CaO-800 | 0.6 wt% | H2O | 80 °C, 40 min | Glucose, 20 wt% | 34.7% | 25.0% | 422 |
| Sibunit-4 | 1 wt% | H2O | 180 °C, 5 h, 1 MPa Ar | Cellulose, 1 wt% | — | 6.6–9.4% (glucose 21.1–25.1%) | 423 |
| Al-Biochar | 2.5 wt% | Acetone/H2O (v/v = 1) | Microwave, 140 °C, 5 min | Glucose, 5 wt% | 29.1% | Fructose 21.5% | 424 |
| Al-Biochar (3 runs) | 2.5 wt% | Acetone/H2O (v/v = 1) | Microwave, 140 °C, 5 min | Glucose, 5 wt% | 19.5% | Fructose 7.8% | 424 |
| Sn-Biochar | 2.5 wt% | H2O | Microwave, 150 °C, 20 min | Glucose, 5 wt% | ∼74% | Fructose 12.1% | 425 |
| SO3H-OAC | 1.25 wt% | THF/H2O (v/v = 3)-saturated NaCl | 160 °C, 3 h | Glucose, 1.25 wt% | 100% | 93% | 426 |
| SO3H-OAC | 1.25 wt% | THF/H2O (v/v = 3) | 160 °C, 3 h | Glucose, 1.25 wt% | 59.5% | 30.6% | 426 |
| SO3H-OAC (5 runs) | 1.25 wt% | THF/H2O (v/v = 3)-saturated NaCl | 160 °C, 3 h | Glucose, 1.25 wt% | — | 74% | 426 |
| PAB2-600 | 2 wt% | DMSO/H2O (v/v = 3) | 180 °C, 20 min | Food waste, 5 wt% | 86.5% | 30.2% | 427 |
| SBC | 5 wt% | DMSO/H2O (v/v = 3) | 180 °C, 30 min | Bread waste, 5 wt% | 86.5% | 30.2% | 428 |
| CNTs-SO3H-NH2-Cr(III) | 2.5 wt% | BMIMCl | 130 °C, 3 h | Glucose, 5 wt% | 86.5% | 58.6% | 429 |
| CNTs-SO3H-NH2-Cr(III) | 2.5 wt% | BMIMCl | 130 °C, 3 h | Cellulose, 5 wt% | — | 40.2% | 429 |
| CNTs-SO3H-NH2-Cr(III) (5 runs) | 2.5 wt% | BMIMCl | 130 °C, 3 h | Cellulose, 5 wt% | — | 36% | 429 |
| CNTs-SO3H-NH2 | 2.5 wt% | BMIMCl | 130 °C, 3 h | Cellulose, 5 wt% | — | 31.1% | 429 |
| N-Doped biochar | 1 wt% | H2O | 120 °C, 20 min, microwave | Glucose, 5 wt% | 12% | Fructose, 10.1% | 430 |
| N-Doped biochar | 1 wt% | H2O | 160 °C, 5 min, microwave | Glucose, 5 wt% | 14.7% | Fructose, 14% | 430 |
| N-Doped biochar | 1 wt% | H2O/acetone (v/v = 1) | 160 °C, 5 min, microwave | Glucose, 5 wt% | 22% | Fructose, 18% | 430 |
| SGQDs | 0.8 wt% | DMSO/water/MIBK/butanol (v/v = 7/3/14/6) | 170 °C, 2 h | Fructose, 6.7 wt% | 91.8% | 51.7% | 412 |
| GQDs | 0.8 wt% | DMSO/water/MIBK/butanol (v/v = 7/3/14/6) | 170 °C, 2 h | Fructose, 6.7 wt% | 91.8% | 31.2% | 412 |
| SGQDs | 0.8 wt% | DMSO/water/MIBK/butanol (v/v = 7/3/14/6) | 170 °C, 2 h | Glucose, 3.3 wt% | 45.1% | 19.5% | 412 |
| SGQDs | 0.8 wt% | DMSO/water/MIBK/butanol (v/v = 7/3/14/6) | 170 °C, 2 h | Cellobiose, 3.3 wt% | 91.2% | 13.2% | 412 |
| SGQDs | 2 wt% | DMSO/water/MIBK/butanol (v/v = 7/3/14/6) | 170 °C, 2 h | Ball-milled cellulose, 0.5 wt% | 78.9% | 22.2% | 412 |
Besides SO3H-functionalized carbon materials, carbon materials only containing weak acidic sites, such as COOH groups398,431 and P–O groups432,433 are also effective in catalyzing the hydrolysis of polysaccharides and the dehydration of carbohydrates to HMF. For example, the carbonaceous microsphere material obtained via the hydrothermal carbonization of glucose was directly used for the dehydration of fructose in BMIMCl, attaining an HMF yield of 88.1% at 100 °C in 90 min.398 A phosphorylated carbonaceous material was also used for the dehydration of fructose to HMF.432,433 It was found that tuning the concentration of P–O groups on the catalyst is an effective approach to improve the catalytic activity and selectivity. Besides, phosphoric acid-activated wood biochar gave an HMF yield of 30.2% from starch-rich food waste.427
Due to the lack of glucose-to-fructose isomerization ability, SO3H-functionalized carbon materials are unable to catalyze the efficient conversion of glucose to HMF. The introduction of basic and Lewis acidic species in carbon materials (Fig. 21) is an effective approach to improve their catalytic activity for glucose isomerization. Bone char, which was prepared via the calcination of bone cattle powder, exhibited obvious catalytic activity for the isomerization of glucose to fructose (19%) owing to its basic sites resulting from Ca (23 wt%), Mg (1.4 wt%) and Na (1.4 wt%) elements.421 The combination of basic bone char and acidic IL 1-methyl-3-(3-sulfopropyl)-imidazolium hydrogen sulfate as the catalyst gave an HMF yield of 39% with the glucose conversion of 72% in the medium of water. Similarly, the carbon/CaO composite (CS-CaO-800) synthesized via the pyrolysis of crab shells was effective in catalyzing the isomerization of glucose to fructose, affording a fructose yield of 25.0% at an ultrahigh glucose loading (20 wt%).422 Yu et al. reported that an aluminium-biochar composite (Al-biochar), which was synthesized via the pyrolysis of AlCl3-treated saw dust, gave a fructose yield of 21.5% in the medium of acetone/H2O at 160 °C for 5 min under microwave heating.424 They considered that the active Al sites contribute to approximately 70% of the isomerization activity and the possible active species are the Al2O3, Al(OH)3, AlO(OH), and Al–O–C moieties in the amorphous phase. However, the fructose yield decreased to 7.8 mol% in the third recycling run. Zhang et al. introduced Al2O3 and TiO2 in sulfonated carbon with hierarchically ordered pores (SCHOP) to strengthen the Lewis acid sites.434 The obtained Al–Ti@SCHOP composite catalyst exhibited higher catalytic activity in DMSO than the mixture of SCHOP, Al2O3 and TiO2, giving a maximum HMF yield of 57.36% from glucose at 130 °C within 5.0 h. After five recycles, the HMF yield only decreased by 6.2%.
 | ||
| Fig. 21 Conversion of glucose to HMF over acid-base bifunctional catalyst. Adapted from ref. 416. | ||
The introduction of N-containing groups is also effective to impart glucose isomerization ability and then promote the conversion of glucose to fructose and HMF (Fig. 21). N-Doped biochar bearing strong basic pyridinic nitrogen, which was obtained via the co-pyrolysis of spent coffee grounds and melamine, was used as a solid base catalyst for the isomerization of glucose, affording the maximum fructose yield of 18% in the medium of water/acetone mixture.430 Chen et al. prepared bamboo-derived biochar bearing both SO3H and polyamide (BC-PA-SO3H) for the direct conversion of cellulose, attaining an HMF yield of 23.10% in water under microwave-assisted conditions.416 In contrast, the PA-free BCSA, PS-SO3H, and microporous amino sulfonic resin (PS-NH2-SO3H) were unable to convert cellulose to HMF. Besides, BCSA-PA and PS-NH2-SO3H gave remarkably higher HMF yields from glucose than that from fructose, which is different from traditional solid acids. They proposed that the superior catalytic performance of BC-PA-SO3H was attributed to the synergistic effect of the acidic and basic sites and the efficient mass transfer between the substrate and catalyst. An acid–base and Cr(III) species multi-functionalized multi-walled carbon nanotube (CNT) catalyst exhibited HMF yields of 58.6% and 40.2% from glucose and cellulose, respectively.429 Nahavandi et al. claimed that the oxidation of activated carbon (OAC) followed by sulfonation is an effective strategy to impart –SO3H, –OH, and –COOH functional groups as well as strong Brønsted base sites, thus leading to excellent catalytic performance for the dehydration of glucose to HMF (yield: 93%).426 However, the presence of Brønsted base sites was only implied by carbon dioxide temperature-programmed desorption (CO2-TPD), and the origin of the Brønsted base sites and their catalytic role were not provided. Since both the catalytic performance and stability of acid–base bifunctional carbon materials have remarkable differences in current reports, more strict control experiments are required to obtain credible experimental results for these bifunctional catalysts.
The use of a GVL/water mixture seems to be effective to promote the direct conversion of glucose and cellulose to HMF over sulfate and sulfonated carbon, without the use of additional active sites responsible for glucose isomerization.417,419 For example, Huang et al. showed that in the medium of water/GVL, biomass-derived mesoporous carbon functionalized with benzenesulfonic acid gave moderate HMF yields of 33.2%, 22.5% and 32.3% from glucose, cellulose and biomass, respectively.419 A similar synergistic effect was also observed for the conversion of glucose to HMF over sulfonated carbon and NaHSO4. For example, Thanh et al. reported that a carbonaceous solid acid catalyst prepared via the incomplete carbonization of Acacia mangium wood sawdust followed by H3PO4 treatment and sulfonation only gave an HMF yield of 26% from the hydrolysate solution of Acacia mangium wood sawdust, which contained 0.05 M citric acid and 0.5 M glucose, while the combined use of the carbonaceous solid acid and NaHSO4 gave a high HMF yield (64%), which is consistent with the phenomenon observed in the NaHSO4/ZnSO4 system.435 However, more work is needed to reveal the synergistic mechanism of GVL with sulfate or sulfonated carbon catalysts.
Since carbon-based materials are generally prepared from organic waste with complex compositions, they may contain various impurities, in particular metal species, and complex functional groups. As discussed in section 2.3.1, trace metal species may play an important role in the conversion of carbohydrates. Therefore, the comprehensive characterization of carbon-based materials, especially the type and content of metal species and functional groups, is of great importance for understanding their actual catalytic species and reaction mechanism.
| Catalyst | Catalyst loading | Solvent | Reaction condition | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not provided. | |||||||
| NUS-6(Hf) | 5 wt% | DMSO | 100 °C, 1 h | Fructose, 5 wt% | 99% | 98% | 437 |
| TFP-DABA | 0.8 wt% | DMSO | 100 °C, 1 h | Fructose, 5 wt% | 99% | 97% | 438 |
| UiO-66(Zr)-MSBDC(20) | 0.33 wt% | H2O | 140 °C, 3 h | Glucose, 10 wt% | 31% | 6% (fructose 22%) | 439 |
| UiO-66(Zr)-MSBDC(20) (3 runs) | 0.33 wt% | H2O | 140 °C, 3 h | Glucose, 10 wt% | 20.6% | 3.4% (fructose 6.9%) | 439 |
| UiO-66-Zr MOF | 2.6 wt% | DMSO | 140 °C, 12 h | Glucose, 2 wt% | — | 49.7% | 440 |
| UiO-66(Zr)-NH2-SO3H | 2.2 wt% | DMSO | 130 °C, 8 h | Glucose, 2 wt% | — | 48.2% | 441 |
| UiO-66(Zr)-NH2-SO3H (5 runs), 2.2 wt% | 2.2 wt% | DMSO | 130 °C, 8 h | Glucose, 2 wt% | — | 38.7% | 441 |
| UiO-66-SO3H/PDA@PU | 1 wt% | DMSO | 120 °C, 2 h | Glucose, 2 wt% | — | 56.6% | 442 |
| UiO-66-SO3H-NH2/PDA@PU | 1 wt% | DMSO | 120 °C, 2 h | Glucose, 2 wt% | — | 70.3% | 442 |
| UiO-66-SO3H-NH2/PDA@PU (5 runs) | 1 wt% | DMSO | 120 °C, 2 h | Glucose, 2 wt% | — | ∼62% | 442 |
| PHs-SO3H@UiO-66(Hf)-NH2 | 2 wt% | BMIMCl | 120 °C, 1 h | Cellulose, 5 wt% | — | 49.6% | 443 |
| PHs-SO3H@UiO-66(Hf)-NH2 (4 run) | 2 wt% | BMIMCl | 120 °C, 1 h | Cellulose, 5 wt% | — | 42.7% | 443 |
Recently, numerous MOFs with Lewis acidity have been synthesized and used as promising adsorbents and catalytic materials owing to their high porosity, tunable functionality and abundant metal sites.444 More importantly, the abundant unsaturated metal centers and electron-deficient groups impart unique Lewis acid sites to MOFs. The development of MOFs with tuned Lewis acidity and Brønsted acidity may improve their catalytic performance for sugar isomerization and dehydration reaction. Su et al. reported the use of a sulfonic acid-functionalized MOF, MIL-101(Cr)-SO3H, as a bifunctional heterogenous catalyst for the conversion of glucose to HMF.50 In this reaction system, the Cr3+ centers in MIL-101(Cr)-SO3H function as Lewis acid sites, which are active for the isomerization of glucose to fructose, while –SO3H serves as Brønsted acid sites responsible for the dehydration of fructose to HMF. The batch reaction in GVL/water (10 wt% water) gave a maximum HMF yield of 44.9% with the selectivity of 45.8%. In addition, the continuous reaction conducted in a fixed-bed reactor produced HMF with a steady yield, demonstrating that the catalyst was highly stable under the tested conditions. Zi et al. showed that the unfunctionalized MOF MIL-53(Al) was effective for the conversion of carboxymethyl cellulose (CMC) to HMF using water as the solvent, attaining an HMF yield of 40.3% with a total reducing sugar (TRS) yield of 54.2% at 200 °C for 4 h.445 They proposed that the Al species in MIL-53(Al) could promote the isomerization of glucose to fructose via the enediol structure formed from the coordination of Al species with the hydroxyl groups of glucose, while the hydrolysis of CMC to glucose and the dehydration of fructose are predominantly catalyzed by the Brønsted acid sites.
The introduction of chromium hydroxide particles on and within MIL(Cr)-101 enabled the efficient two-step isomerization of glucose to fructose (Fig. 18) in consecutive reactions in ethanol and aqueous media, attaining a fructose yield of up to 59.3%.446 The isomerization of glucose to fructose proceeded predominantly over chromium hydroxide via the proton-transfer mechanism, which is same with the base-catalyzed isomerization process, while the ketalization process is mainly catalyzed by Lewis acidic MIL(Cr)-101.
The acidic and basic sites in acid–base bifunctional MOFs can work synergistically to achieve the conversion of glucose to HMF. UiO-66-Zr, UiO-66(Zr)-NH2-SO3H and UiO-66-Zr MOF supported on polyurethane foam (UiO-66-SO3H/PDA@PU) with appropriate acid–base bi-functional sites, which could be readily synthesized by tuning the ratio of organic ligands, afforded HMF yields of 49.7–70.3% from glucose in DMSO.440–442 Zhao et al. reported that the PHs-SO3H@UiO-66(Hf)-NH2 catalyst bearing both acidic and basic sites afforded an HMF yield of 49.6% from cellulose under relatively mild conditions (120 °C, 1 h).443 Burnett et al. reported that the hydrothermally stable ytterbium MOF (Yb6(BDC)7(OH)4(H2O)4) bearing bridging hydroxyl and metal-coordinated water exhibited both Brønsted and Lewis acidity and notable catalytic performance for the direct conversion of glucose to HMF (selectivity of 65% with glucose conversion of 38%) in pure water.447
Besides, MOFs can be used as precursors for the design and synthesis of novel solid acid catalysts. For example, silica-supported Lewis-acidic oxozirconium clusters (Zr6@SiO2) were synthesized via the nanocasting of NU-1000, an MOF with 3 nm channels and oxozirconium clusters, with silica followed by removing the organic linkers at high temperature, to improve the thermal stability of the MOF-derived single-site catalytic clusters.448 NU-1000 after dehydration at 300 °C under vacuum exhibited higher Lewis acidity and catalytic activity than Zr6@SiO2, but its activity vanished after thermal treatment at 500 °C. In contrast, the oxozirconium clusters in Zr6@SiO2 did not aggregate, even upon exposure to air at 600 °C, and the catalytic activity of Zr6@SiO2 for glucose isomerization to fructose (fructose yield: ∼23%) was maintained even after thermal treatment at 500 °C.
| Catalyst | Catalyst loadinga | Solvent | Reaction condition | Substrate loadinga | Conversion | Yieldb | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b HMF yield if unspecified. — Not provided. | |||||||
| HO3S-POPs | ∼0.18 wt% | H2O/dioxane (v/v = 1/4) | Microwave, 140 °C, 15 min | Fructose, 2.5 wt% | 100% | ∼70% | 449 |
| SO3H-PANI-FeVO4 | 2.2 wt% | DMSO | 120 °C, 6 h | Sucrose, 2.5 wt% | — | 87% | 452 |
| MSPFR | 1.2 wt% | GVL/H2O (v/v = 10) | 140 °C, 60 min | Fructose, 2.4 wt% | 99.3% | 82.6% | 453 |
| MSPFR | 1.2 wt% | GVL/H2O (v/v = 10) | 190 °C, 60 min | Glucose, 2.4 wt% | 97.6% | 33.0% | 453 |
| MSPFR | 1.2 wt% | GVL | 190 °C, 100 min | Corn stover, 2.4 wt% | — | 30.7% (furfural 43.4%) | 454 |
| MSPFR | 1.2 wt% | GVL | 190 °C, 100 min | Corn stover, 6.1 wt% | — | 16.7% (furfural 1.5%) | 454 |
| MSPFR | 1.2 wt% | H2O | 190 °C, 100 min | Corn stover, 6.1 wt% | — | <5% (furfural 19.9%) | 454 |
| Polytriphenylamine–SO3H | 0.3 wt% | GVL | 175 °C, 0.5 h | Corncob, 1.25 wt% | — | 32.3% (furfural 73.9%) | 207 |
| H-control-SO3H | 1 wt% | DMSO | 100 °C, 5 h | Fructose, 6.1 wt% | — | 84% | 451 |
| H-control-SO3H (5 runs) | 1 wt% | DMSO | 100 °C, 5 h | Fructose, 6.1 wt% | — | 40% | 451 |
| H-TA-CMP-ASO3H | 1 wt% | DMSO | 100 °C, 5 h | Fructose, 6.1 wt% | — | 85% | 451 |
| H-TA-CMP-ASO3H (5 runs) | 1 wt% | DMSO | 100 °C, 5 h | Fructose, 6.1 wt% | — | 82% | 451 |
| Lignosulfonate-based acidic resin | 1 wt% | DMSO | 120 °C, 1.5 h | Fructose, 1.4 wt% | 100% | 90% | 455 |
| Lignosulfonate-based acidic resin | 1 wt% | DMSO | 120 °C, 2.5 h | Inulin, 1.4 wt% | ∼93% | 73% | 455 |
| Phosphate-functionalized porous organic polymers (B-POP) | 0.75 wt% | DMSO/1,4-dioxane | 130 °C, 0.5 h | Fructose, 5 wt% | 100% | 85% | 456 |
| SPPS | 1.8 wt% | EMIMBr | 140 °C, 4 h | Glucose, 18 wt% | — | 87.2% | 457 |
| SPPS | 1.8 wt% | EMIMBr | 140 °C, 4 h | Glucose, 18 wt% | — | 87.2% | 457 |
| SPPS | 1.8 wt% | EMIMBr | 180 °C, 4 h | Cellulose, 18 wt% | — | 87.2% | 457 |
| SPPS | 1.8 wt% | EMIMBr | 100 °C, 4 h | Glucose, 18 wt% | — | 45.2% | 457 |
| SPPS (5 runs) | 1.8 wt% | EMIMBr | 140 °C, 4 h | Glucose, 18 wt% | — | ∼87% | 457 |
| SPPS | — | EMIMBr/H2O (v/w = 5/0.27) | 80 °C, 72 h | HMF, — | 32% | 68% | 457 |
| Sg-CN | 1.25 wt% | H2O | 100 °C, 0.5 h | Fructose, 12 wt% | — | 96% | 458 |
| Sg-CN | 1.25 wt% | H2O | 150 °C, 8 h | Glucose, 12 wt% | — | Levulinic acid, 41% | 458 |
| SGCN | 0.1 wt% | H2O | 200 °C, 5 h | Glucose, 1 wt% | 100% | 94% | 459 |
| SGCN | 0.1 wt% | H2O | 200 °C, 5 h | Glucose, 1 wt% | 80% | 30% | 459 |
| P-BnNH3Cl | 7 wt% | H2O/DMSO (v/v = 3/22) | 120 °C, 10 h | Glucose, 9 wt% | 89% | 66.4% | 460 |
| P-BnNH3Cl | 7 wt% | H2O/DMSO (v/v = 3/7) | 140 °C, 12 h | Starch, 8.1 wt% | 96% | 41% | 460 |
| P-BnNH3Cl; HCl | 7 wt%; 0.096 M | H2O/DMSO (v/v = 3/7) | 120 °C, 10 h | Cellulose, 8.1 wt% | 100% | 40% | 460 |
| Amberlyst 70 | 1 wt% | H2O | 150 °C, 1 h | LGO, 1 wt% | 46.2% | 29.6% | 340 |
| N-MON-AS (2 wt% SO3H) | 0.92 wt% | DMSO | 140 °C, 20 h | Fructose, 5 wt% | — | 98% | 450 |
| N-MON-AS (2 wt% SO3H) | 0.92 wt% | DMSO | 100 °C, 20 h | Fructose, 5 wt% | — | 91% | 450 |
| N-MON-AS (2 wt% SO3H) | 0.09 wt% | DMSO | 100 °C, 20 h | Fructose, 5 wt% | — | 73% | 450 |
| Micron-sized MON-AS (2 wt% SO3H) | 2.4 wt% | DMSO | 140 °C, 20 h | Fructose, 5 wt% | — | 76% | 450 |
| Micron-sized MON-AS (2 wt% SO3H) | 0.24 wt% | DMSO | 140 °C, 20 h | Fructose, 5 wt% | — | 64% | 450 |
| DICAT-2 | 11.3 wt% | DMSO | MW, 130 °C, 2 min | Fructose, 6.3 wt% | 98% | 90% | 461 |
| DICAT-2 | 11.3 wt% | Isopropanol | MW, 130 °C, 2 min | Fructose, 6.3 wt% | 94% | 85% | 461 |
| DICAT-1 | 6.7 wt% | Isopropanol/DMSO (v/v = 7/3) | 960 °C, 2 h | Sucrose, 9 wt% | — | 77% | 462 |
| Cr-Tanned leather | 10.25 wt% | DMSO | 165 °C, 24 h | Glucose, 5 wt% | — | 28% | 463 |
| COP-SO3H/SB-15 | 2.75 wt% | DMSO | 120 °C, 1 h | Fructose, 5 wt% | 99.5% | 78% | 464 |
| COP-SO3H/SB-15 | 2.75 wt% | DMSO | 120 °C, 1 h | Glucose, 5 wt% | 47.8% | 5% | 464 |
| COP-SO3H/SB-15 | 2.75 wt% | DMSO | 120 °C, 1 h | Glucose, 5 wt% | 70% | 40% | 464 |
| Amberlyst-15 | 0.5 wt% | NaCl/H2O/THF | 160 °C, 1 h | Glucose, 1.25 wt% | 96% | 48% | 465 |
| Fe(III)/Amberlyst-15 | 0.5 wt% | NaCl/H2O/THF | 160 °C, 1 h | Glucose, 1.25 wt% | 97% | 68% | 465 |
| Al2O3/Amberlyst-15 | 1.5 wt% | DMSO | 300 W ultrasound, 25 °C, 1 h | Fructose, 1.5 wt% | ∼63% | 47% | 466 |
| Yb(OTf)2/PhSO3H-MPR | 3.5 wt% | H2O/SBP | 170 °C, 4 h | Glucose, 0.9 wt% | 75% | 52% | 467 |
| Yb(OTf)2/PhSO3H-MPR | 3.5 wt% | H2O/THF | 170 °C, 4 h | Glucose, 0.9 wt% | 35% | 11% | 467 |
| Yb(OTf)2/PhSO3H-MPR | 3.5 wt% | H2O/GVL | 170 °C, 4 h | Glucose, 0.9 wt% | 25% | 13% | 467 |
| SPA-Imd-TinPCP | 1 wt% | DMSO | 160 °C, 5 h | Glucose, 4 wt% | 89.5% | 59.5% | 468 |
| SPA-Imd-TinPCP | 1 wt% | H2O/THF | 160 °C, 5 h | Glucose, 4 wt% | 86.2% | 49.8% | 468 |
| Sn-PIL | 0.5 wt% | H2O/THF | 130 °C, 1 h | Glucose, 1 wt% | 99% | 51.1% | 469 |
| PEG-CF3COO−/SO3H based IL | 0.15 M | CH3CN | 75 °C, 3.5 h | Fructose, 2.5 wt% | 100% | 74.2% | 470 |
| PEG-CCl3COO−/SO3H based IL | 0.15 M | CH3CN | 75 °C, 3.5 h | Fructose, 2.5 wt% | 100% | 73.5% | 470 |
| PEG-CH3COO−/SO3H based IL | 0.15 M | CH3CN | 75 °C, 3.5 h | Fructose, 2.5 wt% | 96.5% | 51.6% | 470 |
| PEG-HSO4−/SO3H based IL | 0.15 M | CH3CN | 75 °C, 3.5 h | Fructose, 2.5 wt% | 100% | 72.6% | 470 |
| PEG-CF3COO−/SO3H based IL (3 run) | 0.15 M | CH3CN | 75 °C, 3.5 h | Fructose, 2.5 wt% | 80% | 51.6% | 470 |
Besides artificial polymers, cellulose and lignin can also be used as precursors for the synthesis of sulfonated polymers. Pawar et al. reported that sulfonic acid-anchored cellulose (DICAT-2), which was prepared via the controlled sulfonation of cellulose by chlorosulfonic acid, functioned as an effective solid acid catalyst for the dehydration of fructose to HMF under microwave irradiation in the medium of isopropanol.461 Lignosulfonate-based acid resin, which was obtained from lignosulfonate via ion-exchange with H2SO4 solution, could also be used as a renewable solid acid for the efficient dehydration of fructose and inulin to HMF in the medium of DMSO.455
The introduction of certain functional groups in SO3H-functionalized polymers can regulate the intensity of acid sites or influence the interaction between the catalyst and substrate and then improve their catalytic activity and selectivity. For example, sulfonated polyaniline (SPAN) gave an HMF yield of up to 71% from fructose, with complete restriction of HMF rehydration (Fig. 22) to levulinic acid and formic acid.471 The SO3H in SPAN preferred to form a stable six-member ring structure with the benzenoid amine (–NH–) and quinoid imine (–N) nitrogen via a hydrogen bond, which limits the Brønsted acidity of the catalyst. SO3H is active for the dehydration of fructose to HMF, while the rehydration of HMF is completely suppressed owing to the decreased Brønsted acidity. Moreover, the stability and recyclability of the SPAN catalyst were improved owing to the formation of hydrogen bonds. Tang et al. reported that acidic resin bearing adjacent SO3H and OH, which was synthesized via the one-pot condensation/sulfation of sodium sulfite, cyclopentanone and formaldehyde followed by ion-exchange with H+ (Fig. 23), gave an optimized HMF yield of 97.1% in the medium of DMSO under mild conditions (120 °C, 1.5 h).472 The improved catalytic performance was attributed to the enhanced adsorption of fructose on the catalyst owing to the hydrogen bonding between OH and fructose. The introduction of SO3H-functionalized ionic liquids with CF3COO−, CCl3COO−, AcO− and HSO−4 anions onto PEG-600 formed acidic catalysts, which were active to convert fructose to HMF in the medium of CH3CN.470
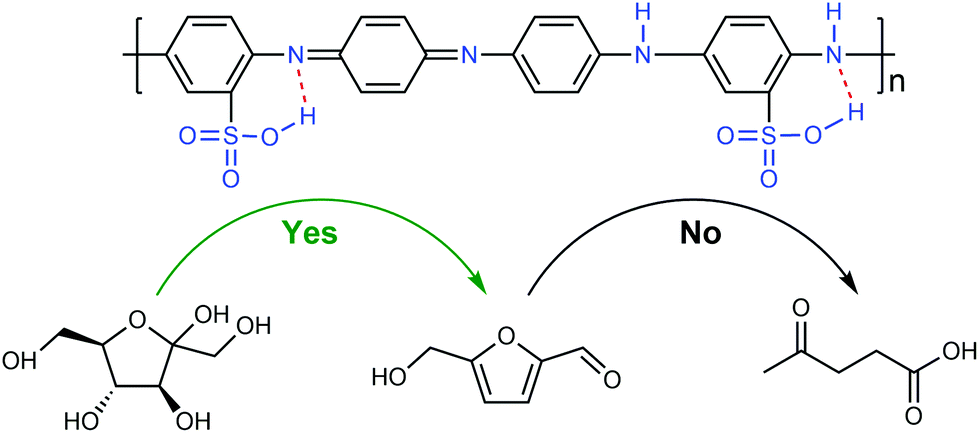 | ||
| Fig. 22 Dehydration of fructose to HMF over SPAN with inhibited HMF rehydration. Adapted from ref. 471. | ||
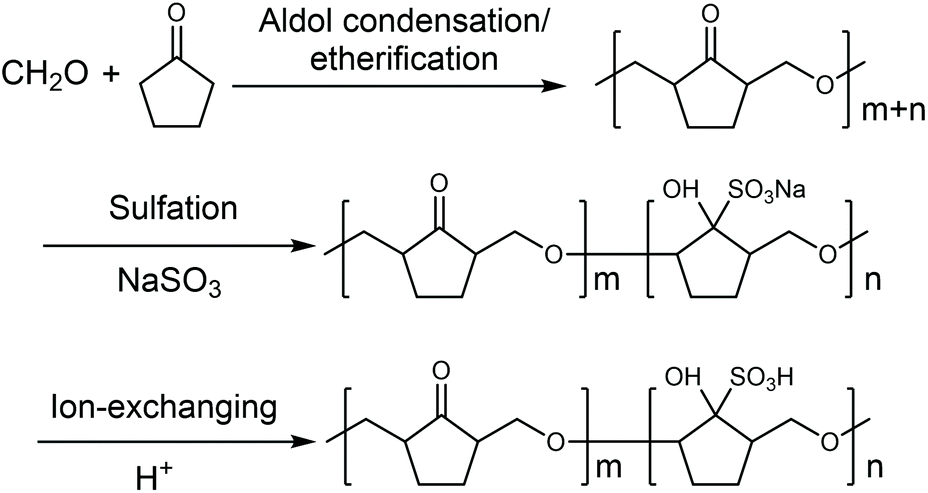 | ||
| Fig. 23 Preparation of acidic resin bearing adjacent SO3H and OH. Adapted from ref. 472. | ||
Johnson et al. investigated the stability of several commercially available sulfonic acid-containing solid acids, including Amberlyst 15, Amberlyst 45 and Nafion for the dehydration of fructose to HMF in a condensed phase flow reactor for a long reaction time of 48 h, revealing that in the medium of water, the catalyst deactivation rate resulting from carbon deposition (fouling) was dramatically faster than that resulting from sulfonic acid leaching.473 After comparing the influence of reactant, solvent, residence time, and feed concentration on the fouling rates, they found that the use of the polar aprotic solvent DMSO with a dilute (50 mM) reactant stream was the only successful approach to inhibit fouling, while operating at relatively mild conditions in aqueous systems did not improve the longevity of the catalyst obviously. The higher HMF yield from DMSO is mainly ascribed to the increased acidity, while the improvement of catalyst lifetime is attributed to the efficient desorption of products owing to the tuned surface polarity.
Currently, most studies on the dehydration of carbohydrate to HMF were carried out at a relatively low substrate loading. The use of a high substrate loading is of great importance for the reduction of solvent usage, energy consumption, reaction volume and operating cost. However, an increase in the substrate loading results in a remarkable decrease in the HMF yield and selectivity owing to the severe side reactions.474 Pyo et al. reported that the use of acidic ion exchange in DR-2030 as a catalyst in the medium of DMSO enabled the highly efficient production of HMF from fructose solution at a fructose concentration as high as 30 wt%, obtaining HMF yields of 85% and 82% at 110 °C in batch and continuous-flow reactions, respectively.475
Besides acid-functionalized polymers, base-functionalized polymers have also been developed for the dehydration of fructose to HMF. For example, Zhu et al. synthesized formyl-modified polyaniline (FS-PAN) as a solid organic-base catalyst (Fig. 24) for the dehydration of fructose to HMF.476 In the medium of DMSO, FS-PAN gave an HMF yield of up to 90.4 mol% at 140 °C for 4 h, which is much higher than that (44.9%) obtained with H2SO4-doped polyaniline (S-PAN). They proposed that the basicity of the polymer is mainly due to the nitrogen atoms incorporated between the phenyl rings in the backbone of the polyaniline chain, and the grafting of electron-withdrawing formyl groups further increased the basicity due to the greater localization of electrons at the amide nitrogen atom. The HMF yield increased linearly from 70% to 90% with an increase in the loading of formyl groups in FS-PAN, suggesting that the amide plays an important role in the synthesis of HMF. Different from S-PAN, the side reactions over FS-PAN, including the rehydration of HMF to levulinic acid and formic acid and the condensation of reaction intermediates to humins are completely inhibited. Therefore, FS-PAN can be reused for at least four runs without obvious loss of catalytic activity. However, since DMSO itself can convert fructose to HMF (yield: 70%) efficiently under the same conditions, it cannot be concluded that amide is the main active site responsible for the conversion of fructose to HMF. One possible explanation for the improvement of HMF yield is that the neutralization of the acidic species with FS-PAN can reduce the decomposition of HMF.
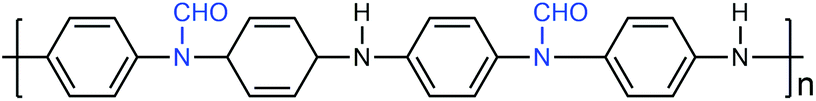 | ||
| Fig. 24 Structure of formyl-modified polyaniline. Adapted from ref. 476. | ||
As discussed in section 2.2.5, almost quantitative dehydration of fructose to HMF can be achieved in the medium of Br-based ionic liquids without the use of an additional catalyst, indicating the important role of Br− in fructose dehydration. Ruby et al. demonstrated that poly(N-alkylvinylpyridinium bromides) could function as acid-free and metal-free heterogeneous catalysts for the dehydration of fructose into HMF, attaining a maximum HMF yield of 77% with the acetals of HMF as the main by-products in the medium of ethanol after 0.5 h at 180 °C.477 Under the same conditions, an I-based polymer gave a comparable HMF yield, while OH-, Cl− and NTf2-based polymers gave much lower fructose conversion and HMF yields than the Br-based polymer. Owing to the absence of Lewis and Brønsted acid sites, both the glucose conversion and HMF yield were low over these catalysts.
Verma et al. reported that the sulfonated carbon nitride (Sg-CN), which was synthesized via the calcination of urea at 500 °C followed by sulfonation using chlorosulfonic acid, exhibited a high HMF yield (96%) in the medium of water under mild conditions (100 °C, 0.5 h) even at a fructose loading as high as 12.4 wt%.458 Glucose could be converted under relatively harsh conditions (150 °C, 8 h) with levulinic acid as the main product. In contrast, another study on sulfonated carbon nitride (S-GCN), which was conducted by Chhabra et al., indicated that glucose, fructose, cellobiose, sucrose and starch can be converted to HMF selectively owing to the presence of Brønsted base and Brønsted acid sites in S-GCN, but the reaction was performed under very harsh conditions (200 °C, 5 h) and low substrate loading (1 wt%).459 Thus, more work is needed to reveal the reason for these conflicting results.
Similar to carbon-based solid acids, cellulase-mimetic sulfonated polymers have also been designed for the depolymerization of cellulose.404 For instance, Yu et al. designed a soluble sulfonated hyperbranched poly(arylene oxindole) with adjacent hydroxyl and sulfonic acid groups to mimic the separate acidic and alkaline active sites in cellulase.478 SHPAO bearing hydroxyl and sulfonic acid groups was more active than H2SO4 and other hyperbranched polymers for the depolymerization of cellulose to glucose, obtaining a glucose yield of 56% with 93% total selectivity toward useful hydrolytic and dehydration products. In contrast, when non-functionalized SHPAO with only sulfonic acid groups was used as a solid acid catalyst, cellulose was mainly converted to levulinic acid.479 The excellent catalytic activity of SHPAO bearing hydroxyl and sulfonic acid groups is ascribed to the presence of neighboring hydroxyl groups and sulfonic acid group. However, similar to the carbon-based solid acids, the stability of cellulase-mimetic sulfonated polymers is a major concern regarding their actual application. For example, Tyufekchiev et al. demonstrated that the good catalytic performance of the sulfonated chloromethyl polystyrene is attributed to the HCl generated from hydrolysis of C–Cl under cellobiose hydrolysis conditions, instead of the previously believed cooperation action of C–Cl moieties to bind cellulose via hydrogen bond and SO3H to catalyze the disruption of glycosidic bond.480
As is consistent with the phenomenon observed with carbon-based solid acids, the use of the GVL/water mixture as the reaction medium can also promote the direct conversion of glucose and cellulose to HMF over sulfonated polymers without specific active sites responsible for glucose isomerization. For example, Li et al. reported that p-hydroxybenzenesulfonic acid-formaldehyde resin (MSPFR), which was prepared from formaldehyde and p-hydroxybenzenesulfonic acid via the hydrothermal method, exhibited HMF yields of 82.6% and 33.0% from fructose and glucose, respectively, in the medium of GVL/H2O mixture.453 Zhang et al. reported that the MSPFR catalyst afforded a furfural yield of 43.4% with an HMF yield of 30.7% at 190 °C within 100 min from raw corn stover.454
The introduction of Lewis acidic metal species and N-containing basic groups in sulfonated polymers is an effective approach to facilitate the conversion of glucose to HMF. Bobbink et al. reported that chromium-tanned leather, a robust Cr-containing material produced via chemical treatment of animal hides by chromium salts in the leather industry, was effective for the dehydration of glucose to HMF, obtaining an HMF yield of 28% with levoglucosenone (LGO) as the main by-product.463 However, the HMF yield dropped to 16% during the second recycling of the catalyst. The Fe3+-modified Amberlyst-15 resin (Fe(III)/Amberlyst-15) exhibited much higher catalytic activity than Amberlyst-15 for the direct conversion of glucose to HMF owing to the synergistic effect of Fe3+ and SO3H, which are active for the isomerization of glucose to fructose and the dehydration of fructose to HMF, respectively.465 Wang et al. prepared an ordered mesoporous phenolic resin (MPR) bearing both Brønsted acid and Lewis acid sites (Yb(OTf)2/PhSO3H-MPR) via sulfonation followed by post-grafting method.467 Lewis acid sites were formed in the pore channels of the ordered phenolic resin owing to the coordination of benzenesulfonic acid with Yb(OTf)3. As expected, this catalyst enabled the one-pot cascade conversion of glucose to HMF with a yield of 52% in the medium of water/SBP biphasic system. The good catalytic performance was attributed to the synergistic effect of uniformly distributed Brønsted and Lewis acid sites. Besides, the catalytic activity could be maintained for five runs. Babaei et al. reported that the sulfonic acid-functionalized triazine covalent organic polymer supported on mesoporous SBA-15 (COP-SO3H/SB-15) afforded HMF yields of 78% and 45% from fructose and glucose in DMSO, respectively, under the optimum reaction conditions.464 Cao et al. reported that a poly-benzyl ammonium chloride resin (PBnNH3Cl) functioned as a bifunctional organocatalyst for the isomerization of glucose and dehydration of fructose (Fig. 25), thus affording good HMF yields from glucose and starch.460 However, additional HCl is required to achieve the effective conversion of cellulose to HMF.
 | ||
| Fig. 25 Direct conversion of glucose to HMF over PBnNH3Cl. Adapted from ref. 460. | ||
Li et al. claimed that the use of sulfonated poly(phenylene sulfide) (SPPS-SO3H) containing just strong Brønsted acid sites as a heterogeneous catalyst in ionic liquid EMIMBr gave HMF yields as high as 87.2% and 68.2% from glucose and cellulose, respectively, in the medium of EMIMBr ionic liquid.457 Several new phenomena, which are contradictory to previously established knowledge, were observed in the SPPS-SO3H/EMIMBr catalytic system. Firstly, SPPS-SO3H containing just strong Brønsted acid sites could directly convert glucose and cellulose into HMF in high yields, but HMF is unstable in this catalytic system even at 80 °C. Secondly, both the HMF yield and selectivity from glucose and cellulose were much higher than that obtained from fructose. With the assistance of DFT calculation, they proposed that the SO3H group in SPPS functions as both a proton donor (Brønsted acid) and proton acceptor (conjugate base) in the conversion of glucose, and the anions and cations of EMIMBr combined with SO3H-SPPS help to stabilize the reaction intermediates and transition states, thus resulting in the facile conversion of glucose to HMF. Thirdly, high temperatures (140 °C and 180 °C) were required to achieve the maximum HMF yields from glucose and cellulose, respectively, while increasing the temperature from 100 °C to 140 °C led to a decrease in the HMF yield.
:1 v/v) gave a fructose yield of 61% with an HMF yield of 30% at 363 K after 24 h reaction.
Various approaches have been attempted to facilitate the recovery and reuse of heterogenous catalysts. For example, the design of heterogenous catalysts with a large bulk size is an effective strategy to facilitate their recovery and reuse. For example, millimeter-size γ-Al2O3 beads bearing propyl sulfonic acid and alkyl groups were used for the dehydration of fructose to HMF.484 It was found that the C16–SO3H–γ-Al2O3 catalyst, which was obtained by calcination at 650 °C, exhibited the highest hydrophobicity and catalytic activity, obtaining an HMF yield of 84% in DMSO/H2O at 110 °C for 4 h. Owing to their hydrophobicity and millimeter size, the reuse of the γ-Al2O3 beads could be readily achieved. The combination of magnetic Fe3O4 with SO3H-functionalized solid acid allowed the magnetic separation of the heterogeneous catalyst from reaction system.485 For instance, the dehydration of fructose to HMF under solvent-free conditions (Fig. 26) was investigated by Shaikh et al. using dicarboxylic acid-modified ferrite as the catalyst.486 The successful surface modification prevented the agglomeration of the ferrite nanoparticles, including Fe3O4 and CoFe2O4. With the assistance of sonication, tartaric acid-modified Fe3O4 gave an HMF selectivity of 87% with the fructose conversion of 98% at 80 °C for 1 h under solvent-free conditions. After the reaction, the catalyst could be recovered via dilution with water followed by separation with an external magnet. Although the recovered catalyst had a larger average size (18 nm) than that (11 nm) of the fresh catalyst, its catalytic performance was maintained for at least five times. They also claimed that GO enabled the efficient dehydration of fructose to HMF under solvent-free conditions.487 Besides the development of heterogeneous catalysts, the design of adsorbents that can adsorb the homogeneous catalyst from the catalytic system is also an effective strategy to achieve the recycling of the catalyst.488
 | ||
| Fig. 26 Dehydration of fructose to HMF over tartaric acid-modified Fe3O4 under solvent-free conditions. Adapted from ref. 486. | ||
The dehydration of fructose in low boiling point organic solvents is highly desirable considering the recycling of the solvent and separation and purification of the HMF product. Since carbohydrates are generally insoluble in low boiling point organic solvents, the direct conversion of carbohydrates in these solvents is difficult. As discussed in section 2.2, fructose can be readily transformed into HMF in the medium of DMSO and several ionic liquids, but the separation of HMF from these solvents is cumbersome and energy-consuming. Sun et al. attempted to create solvation environments similar to solvents (Fig. 27), such as DMSO-, NMP- and Br-containing imidazolium-type ionic liquids in heterogeneous catalysts by incorporating these solvent moieties into porous materials.489 The catalyst bearing both SO3H and ionic liquid moieties enabled the highly efficient conversion of fructose using the readily separable THF as the reaction medium without additional water, obtaining an HMF yield as high as 98.8% at 120 °C within just 10 min. Guo et al. prepared a DMSO-like polymeric solid catalyst, Au@(polythiophene-polythiophene oxides), via the chemical over-oxidative polymerization of thiophene with the assistance of Au nanoparticles. The DMSO-like catalyst exhibited much better catalytic performance than its liquid analog DMSO for the dehydration of fructose to HMF in the medium of low-boiling solvents, such as 1,4-dioxane and water.77 Yang et al. reported that the coating of AlCl3/SiO2 with choline chloride could promote the dissolution of carbohydrates, thus enabling the dehydration of carbohydrates to HMF in MeTHF and MIBK.490 However, the catalytic activity dropped remarkably during the second and third recycling. Similarly, the ionic liquid BMIMBr encapsulated in H-MOR zeolite, which was synthesized via a ship-in-a-bottle strategy, enabled the efficient conversion of glucose to HMF in the water/MIBK/NaCl biphasic system, giving an HMF yield of 42% at 170 °C for 3 h.353
 | ||
| Fig. 27 Dehydration of fructose to HMF in low-boiling points organic solvent based on the collaboration of the solvation environment and SO3H incorporated in a heterogeneous catalyst. Adapted from ref. 489. | ||
2.5. Process technology
To achieve large-scale industrial production and application, process technology should also be developed together with fundamental studies.491 In addition to the development of catalysts and solvents, the scaling up of catalytic technologies from the laboratory to a pilot plant also involves the design and optimization of the reactor system, development of separation and purification process, selection of an energy source and feedstock and evaluation of economic and environmental benefits of an integrated HMF production process. In this section, we give an overview of how to improve the energy utilization efficiency by employing other heating methods, how to achieve the stabilization of HMF and how to achieve the separation and purification of HMF.497,498 and selective oxidation of platform chemicals,499 but also as an energy resource to assist the thermal catalytic dehydration process. Tsutsumi et al. reported the use of a semiconductor material with visible light irradiation to assist the Brønsted acid-catalyzed conversion of fructose HMF.500 Among the investigated semiconductor materials, the silanol group-coated silicon semiconductor (Si–OH) enabled almost quantitative conversion of fructose to HMF in the medium of DMSO/H2O using 4.6 M H3PO4 as an acid catalyst, obtaining an HMF yield of up to 97% at 80 °C. Owing to the selective adsorption of fructose and desorption of HMF on Si–OH (Fig. 28), Si–OH under visible light irradiation could selectively transfer the heat from optical energy to fructose, but not to HMF. Therefore, the presence of Si–OH increased the fructose dehydration rate remarkably but did not increase the HMF degradation rate, thus leading to the selective production HMF from fructose. Similarly, the cellobiose hydrolysis efficiency over the Ir/HY catalyst could also be improved remarkably under visible light illumination owing to the photothermal effect.501
 | ||
| Fig. 28 Visible light irradiation-assisted fructose dehydration to HMF over Brønsted acid. Adapted from ref. 500. | ||
Microwave-assisted processes can improve the reaction efficiency by accelerating the mass and heat transfer. The conversion of sugars and biomass to HMF using homogeneous catalysts, solid acid catalysts and non-catalytic routes under microwave irradiation has been widely studied, demonstrating that microwave heating can improve the reaction efficiency and product selectivity in comparison with conventional heating.502,503 For example, the direct conversion of lignocellulosic biomass (Phyllostachys aureosulcata) in a water/MIBK biphasic reaction system using HCl as the catalyst under microwave irradiation could afford relatively high HMF (42.44%) and furfural (48.90%) yields.504
The artificial introduction of microwave adsorption groups in solid catalysts not only can improve the energy utilization efficiency, but also help to enhance the product selectivity via the oriented transfer of energy to the desired reaction process. For example, Ji et al. reported the use of a CNT–TiO2/SO42− composite catalyst consisting of a carbon nanotube core and acidified TiO2 shell as a microwave-responsive catalyst to promote the catalytic conversion of sugars, including fructose, glucose and sucrose to HMF.505 The inner CNT could generate heat under microwave radiation, and then the generated heat could transfer to the acidified TiO2 shell, thus enabling a six-times higher energy utilization efficiency (4.2 mol kJ−1 L−1) compared with TiO2. The maximal HMF yields of 67%, 52% and 65% were attained from fructose, glucose, and sucrose within 30 min, respectively. Ji et al. also reported that the core/shell structured CNT–polyaniline/SO42− hybrid material could attain an HMF yield of 63% from fructose within 10 min with high energy efficiency (7.6 mol kJ−1 L−1) owing to the localized heating over the catalyst surface under microwave radiation.506 Compared with CNT–TiO2/SO42−, the thinner PANI shell in CNT–polyaniline/SO42− could further improve the overall microwave absorption, thus leading to a higher heat transfer efficiency and improved reaction efficiency. Similarly, the use of Nb-doped TiO2/SO42− (Nb–TiO2/SO42−) as both a catalyst and microwave-responsive material (Fig. 29) could further improve the efficiency of fructose conversion to HMF under microwave heating. Nb doping not only increased the concentration of surface sulfonic groups, but also improved the acid strength of the sulfonic groups owing to the electron transfer from the bulk catalyst to surface sulfonic groups.507 Xiouras et al. demonstrated that although the reaction kinetics of xylose under microwave radiation is similar to that under the conventional heating method, the microwave-assisted method could reduce the energy consumption by 30% on the laboratory scale compared with the conventional heating method.508
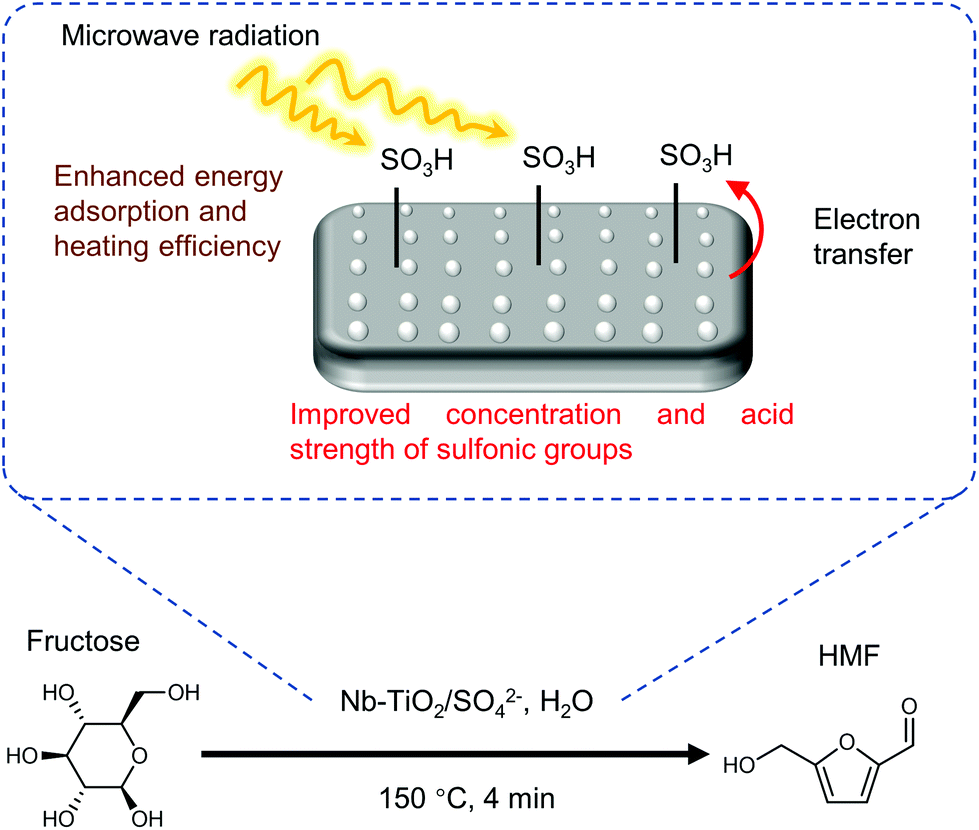 | ||
| Fig. 29 Use of Nb-doped TiO2/SO42− (Nb-TiO2/SO42−) as both a catalyst and microwave-responsive material for the rapid dehydration of fructose to HMF. Adapted from ref. 507. | ||
Under the power of ultrasonic irradiation (300 W), SBA-15 grafted with alumina (Al2O3/SBA-15) enabled the conversion of fructose to HMF at room temperature, affording an HMF yield of 47% in the medium of DMSO within 2.5 h.466 Similarly, sonochemical activation could also accelerate the enzymatic isomerization of glucose to fructose with a considerable reduction in the reaction time and temperature.509
Generally, the introduction of optical energy, microwave radiation and ultrasonic irradiation in a suitable manner can improve the reaction efficiency and product selectivity. However, since optical energy, microwave irradiation and ultrasonic irradiation are usually generated from electricity ultimately resulting from fossil fuel, whether these technologies can improve the actual energy utilization efficiency is still uncertain. Thus, the accurate evaluation of the overall energy utilization efficiency and thorough cost accounting are very important for practical application.
:1 w/w) during the continuous reaction at 90 °C. They inferred that the thiol groups could catalyze the tautomerization of fructose to its furanose form, thus enhancing the HMF production efficiency. Sonsiam et al. reported that the addition of NMP to H2O/MIBK could improve the HMF yield under continuous conditions.513 Weingart et al. reported that an HFIP/water mixture with a low boiling point, which enabled the ready separation of HMF, could also serve as a solvent for the dehydration of fructose in a continuous packed-bed reactor, affording an HMF yield of 76%.514
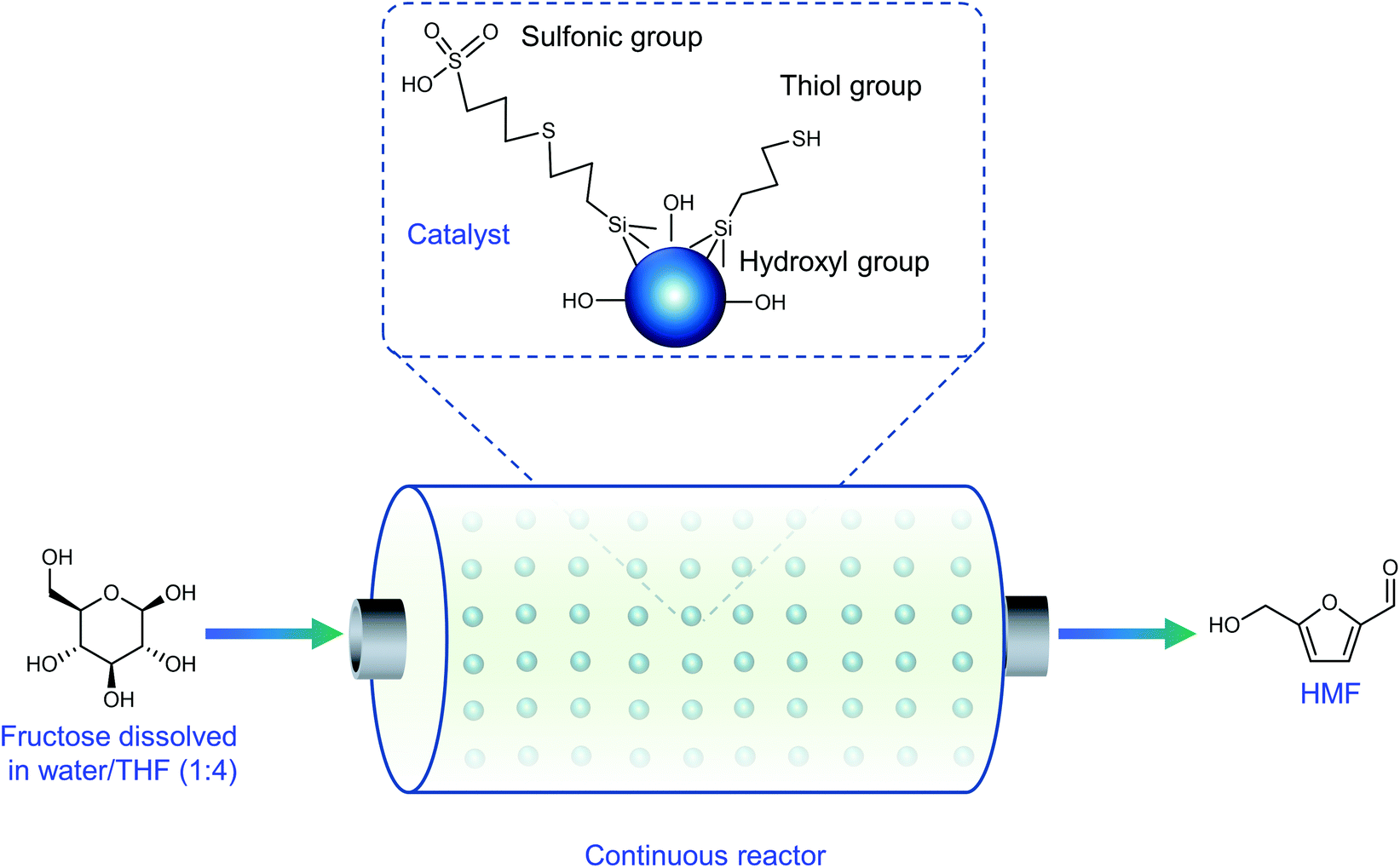 | ||
| Fig. 30 Dehydration of fructose to HMF in a continuous reactor. Adapted from ref. 512. | ||
The development of solid acid catalysts with high stability and reusability is essential for the continuous reaction. Morales-Leal et al. observed that 15% of sulfur from the sulfonic and thiol group-functionalized alumina was loss rapidly at just 70 °C under the continuous reaction.512 Gallo et al. compared the activity and stability of SO3H-functionalized mesoporous carbons (CMK-3 and CMK-5), propylsulfonic acid-functionalized SBA-15 and commercial Nafion SAC-13 for the continuous conversion of fructose to HMF in a fixed bed flow reactor.515 Among the tested catalysts, phenylsulfonic acid-functionalized CMK-5 (CMK-5-PSA) attained the highest HMF production rate. Moreover, the deactivation rate coefficient of CMK-5-PSA (0.002 h−1) was remarkably lower than that of pSO3H-SBA-15 (0.120 h−1) and NafionSAC-13 (0.008 h−1).
The continuous production of HMF from glucose was conducted using Sn-exchanged Amberlyst-15 as the catalyst, which was prepared from Amberlyst-15 and SnCl2 solution via ion-exchange, obtaining an HMF yield of 10.35%.516 The use of a biphasic catalytic system consisting of phosphate buffer saline (PBS) as the reaction phase and 2-sec-butyl phenol as the extraction phase in a continuous microreactor could suppress the side reactions, thus leading to high HMF yields (80.9% and 75.7%) from fructose and glucose, respectively.517 However, the recovery of phosphate buffer saline is difficult and the separation of HMF from 2-sec-butyl phenol is energy-intensive owing to its high boiling point. Thus, the efficient production of HMF from glucose in a continuous reactor will be an important task in the future.
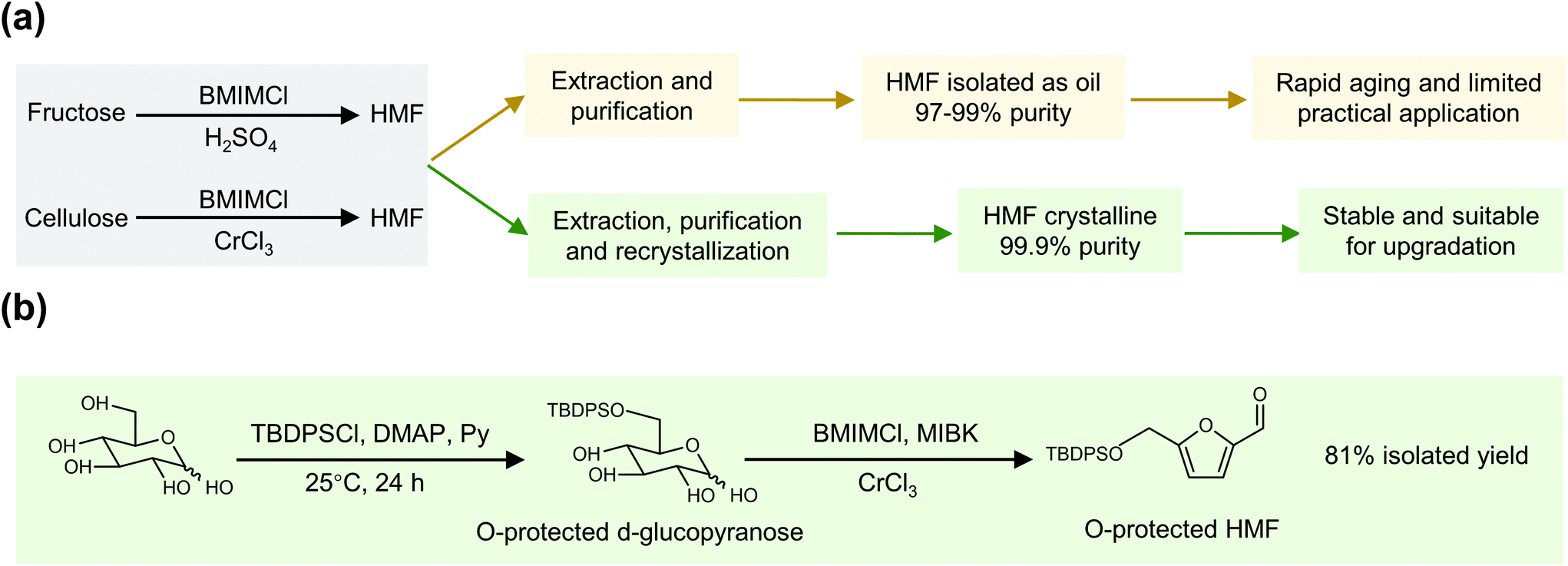 | ||
| Fig. 31 (a) Comparison of oil-like HMF with low purity and crystalline HMF with high purity, and (b) introduction of protecting group at the O(6) position of glucose. Adapted from ref. 518. | ||
The introduction of a protecting group at the O(6) position in glucose enabled the highly selective synthesis of 5-(TBDPS-oxymethyl)furfural (TBDPS = Ph2tBuSi), an HMF derivate, with an isolated yield of up to 81%, which is much higher than the HMF yield (40–50%) obtained with glucose.518 Compared with HMF, 5-(TBDPS-oxymethyl)furfural can be readily separated from the reaction mixture and stored at room temperature for a long time without obvious aging. Gomes et al. demonstrated that the addition of a small amount of sodium dithionite (Na2S2O4) not only improved the stability of HMF, but also inhibited side reactions in a series of processes, thus enabling the high efficiency of HMF distillation, HMF synthesis in a biphasic system, Knoevenagel condensation of HMF with Meldrum's acid, Cannizaro reaction and condensation of HMF with primary diamines toward pyridinium salts.519 Besides the stabilization of HMF during its synthesis and the use of high purity HMF as a feedstock, the stabilization of HMF during its upgrading is also important to avoid its undesirable degradation, and then improve the upgrading efficiency and the selectivity of target product. This aspect will be discussed in detail in section 3.5.
Similarly, the reversible stabilization of xylose, glucose and lignin monomers (Fig. 32) in the depolymerization of biomass by generating acetal with formaldehyde enables the selective depolymerization of cellulose, hemicellulose and lignin to simple sugars and phenolic monomers by inhibiting the degradation of target products, including the dehydration of sugars to furans and their subsequent degradation and lignin condensation.30,520,521 The obtained diformylxylose and diformylglucose products can be utilized as feedstock for the production of fufural and HMF using the catalytic system designed for xylose and glucose dehydration with a similar reaction efficiency, while the hydrogenolysis of formaldehyde-stabilized lignin affords the production of guaiacyl and syringyl monomers at near theoretical yields, which is more efficient than conventional catalytic systems without formaldehyde stabilization.
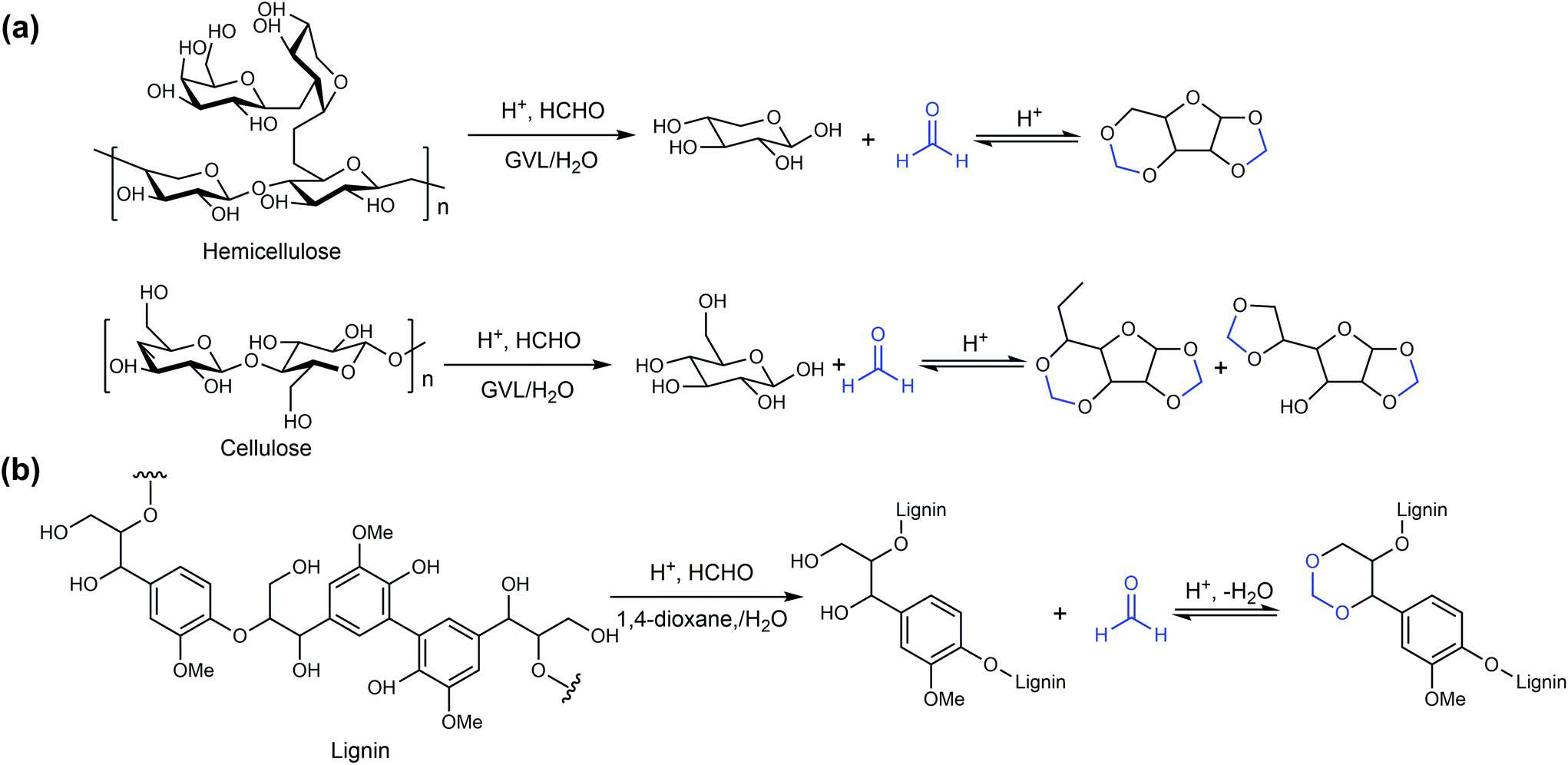 | ||
| Fig. 32 (a) Stabilization of xylose and glucose by generating acetal with formaldehyde during the depolymerization of hemicellulose and cellulose in lignocellulosic biomass. Adapted from ref. 30 and 520. (b) Stabilization of lignin by formaldehyde in extraction of lignin from lignocellulosic biomass. Adapted from ref. 521. | ||
As discussed in sections 2.2.4 and 2.2.5, the construction of water–organic biphasic systems and ionic liquid–organic biphasic systems are not only responsible for the inhibition of HMF degradation, but also promotes the separation of HMF. Introducing a microfiltration membrane in the water/MIBK biphasic system (Fig. 33) could create a microreactor to further improve the liquid–liquid extraction of HMF from the reactive phase (water) to the organic phase (MIBK).522,523 Consequently, the side-reaction was inhibited, resulting in an HMF yield of 93.0% from fructose. Sarwono et al. employed a nanofiltration membrane (NF) to separate IL and HMF from the BMIMCl/CrCl3 catalytic system, as an alternative to the liquid–liquid extraction method (LLE).524 The HMF recovery rate reached 94.87% with an HMF flux rate of 0.2854 L m−2 h−1 in the first run, but the HMF recovery rate and flux rate decreased to 71.65% and 0.2397 L m−2 h−1 in the third run, respectively. Although the complete separation of HMF and IL was not achieved, as indicated by NMR analysis, the NF membrane exhibits great potential for the environmentally friendly separation and purification of HMF. Yan et al. reported that the simultaneous circulation of the reaction phase (water) and extraction phase (MIBK) via pumping, distillation and cooling (Fig. 34) enabled the highly efficient conversion of fructose and glucose to HMF (74% and 90%) in the water/MIBK/HCl and water/MIBK/AlCl3 catalytic systems, respectively.525
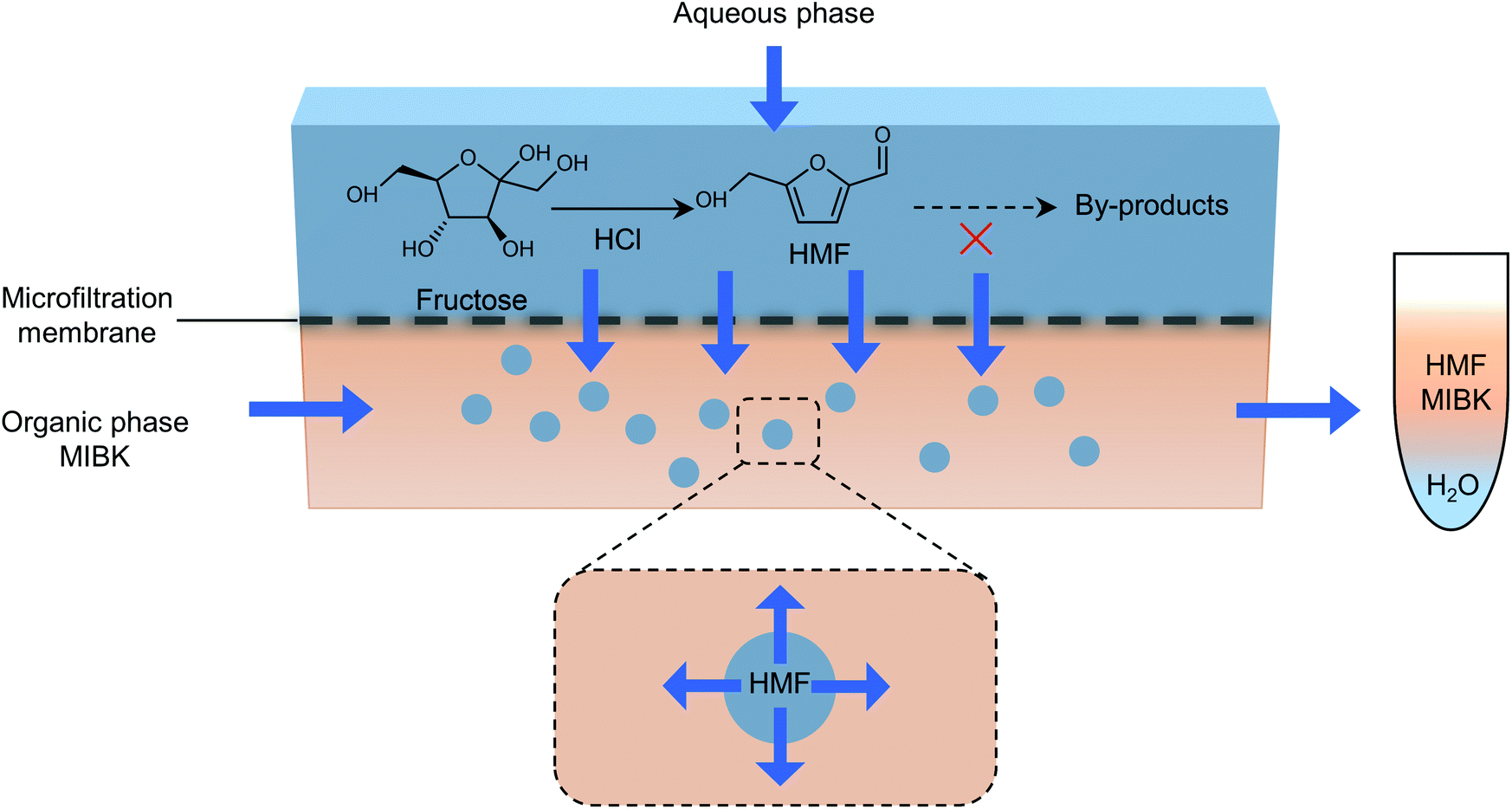 | ||
| Fig. 33 Introduction of microfiltration membrane to improve the liquid-liquid extraction of HMF from water to MIBK. Adapted from ref. 522. | ||
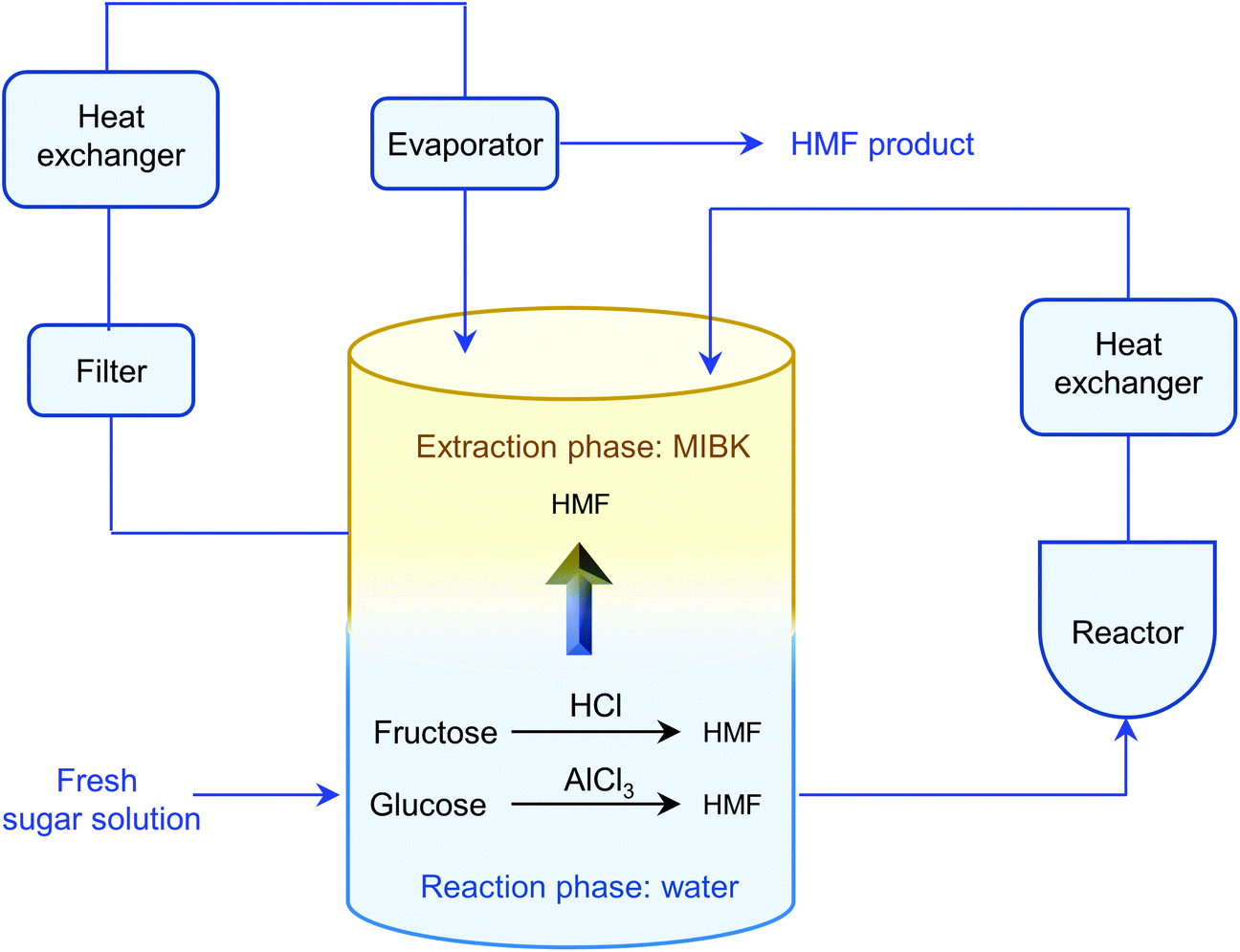 | ||
| Fig. 34 Conversion of fructose and glucose to HMF in water/MIBK biphasic system with the simultaneous circulation of the reaction phase and extraction phase. Adapted from ref. 525. | ||
Based on the systemic investigation of the solubility, saturation and precipitation points, stability, phase separation and phase extraction of mixtures of DMSO with different chemical agents, Gajula et al. designed an integrated strategy to construct solvent systems for the efficient separation of furan derivates from DMSO (Fig. 35), which is challenging due to the high boiling point of DMSO and the instability of the target compounds.526 For instance, the unconverted sugars can be precipitated by adding an organic solvent such as MIBK, and the combination of organic solvents and sugars is beneficial for the extraction of the HMF product from DMSO into the organic solvent. Meanwhile, the combination of water and organic solvent can also promote the phase separation of the organic solvent and DMSO to separate HMF. Water can be used to precipitate 2,5-diformylfuran (DFF), while water or sugar can be used to precipitate 2,5-furandicarboxylic acid (FDCA).
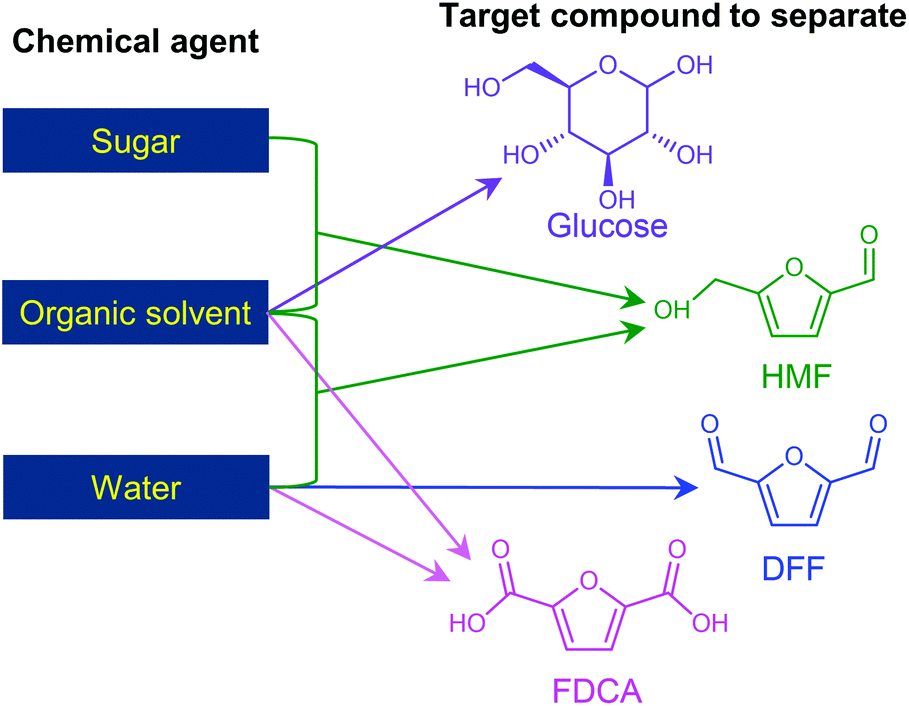 | ||
| Fig. 35 Separation of HMF and its derivates from DMSO by regulating the composition of the catalytic system. Adapted from ref. 526. | ||
To achieve high-yield HMF production from glucose, Alipour et al. devised a novel pathway to decouple the glucose isomerization, fructose dehydration, solvent recycling and HMF separation process (Fig. 36) via a series of liquid–liquid extraction processes.527 In the first step, glucose was converted to fructose by immobilized glucose isomerase enzyme, and then the formed fructose was transferred continuously to the organic phase (octanol) by the lipophilic aryl boronic acid, a ketose-selective binding agent. The simultaneous isomerization and reactive-extraction in turn promoted the isomerization of glucose to fructose by shifting the reaction equilibrium. In the second step, the fructose in the organic phase was extracted to an acidic ionic liquid 1-ethyl-3-methylimidazolium hydrogen sulfate ([EMIM]HSO4). After the second step, the fructose yield reached 89% and nearly a pure solution of fructose in [EMIM]HSO4 was obtained. In the third step, fructose was dehydrated to HMF in the medium of [EMIM]HSO4 using HCl as the catalyst under mild reaction conditions (50 °C, ambient pressure), and simultaneously the formed HMF was extracted to THF. This integrated method enables an HMF yield as high as 80% with prolonged reuse of the catalysts, solvents and reaction media. Moreover, this integrated method has also been proven to be effective for the conversion of corn stover hydrolysate produced via dilute-acid pretreatment. Similarly, Gimbernat et al. developed an integrated cascade process involving the continuous isomerization of glucose to fructose in the aqueous phase, complexing, extracting and transporting fructose to the organic phase (MIBK), and releasing fructose at the interface with the second aqueous phase, and then dehydrating fructose to HMF over acidic resin to avoid the use of costly ionic liquids.528 Nevertheless, the global HMF yield was just 4% with a glucose conversion of 70%.
 | ||
| Fig. 36 Three-step process for the production of HMF from glucose. Adapted from ref. 527. | ||
Various porous materials, including MOFs,529,530 polymers,531,532 activated carbons,533 and zeolites 534 have been used for the selective adsorption of HMF to facilitate the separation of HMF from the catalytic system. For example, Zhang et al. designed a hollow-structured porous aromatic polymer (H-PAP), which had a cavity diameter between 228 ± 11 and 464 ± 15 nm, for the selective adsorption of HMF from aqueous solution, without the adsorption of fructose, levulinic acid and formic acid.535 The apparent adsorption amount of HMF mainly depended on the micropore surface area, micropore volume and cavity diameter, while the selectivity of HMF adsorption was attributed to the surface hydrophobicity. The HMF product with a purity of up to 94.4% could be recovered from the reaction mixture of acid-catalyzed fructose conversion. Yabushita showed that the hydrophobic cavities of p-tert-butylcalix[4]arene macrocycles grafted on amorphous silica (calix/SiO2) were crucial for the selective adsorption of HMF from aqueous solution containing both HMF and glucose.536 They proposed that the selective adsorption of HMF is mainly attributed to the weak dispersive (van der Waals) interactions between the aromatic guests and tert-butyl upper-rim substituents in calixarene hosts.
 | ||
| Fig. 37 Process model for the production of HMF using fructose (a) and glucose (b) as feedstock. Adapted from ref. 103. | ||
Kougioumtzis et al. attempted to scale up the production of HMF from hemicellulose-free biomass via the hydrolysis of biomass to sugars by H2SO4 followed by the dehydration of sugars to HMF by Sn/Al2O3 on an industrial scale (biomass through-put of 1500 kg h−1) using the chemical process optimization software ASPEN PLUS™.539 They obtained an optimized HMF yield of 8% (with respect to cellulose) and by-product yield of 13% (with respect to biomass), with an HMF production rate of 54 kg h−1. They estimated that the total electricity demand is 22.2 MW per tHMF with the heat demand of 218 MW per tHMF.
3. Upgrading of HMF
The upgrading of HMF to high-value fuels, fine chemicals and materials involves a series of complex reaction processes, including selective oxidation, hydrogenation, etherification, coupling, functionalization and condensation reaction and the combination of different reaction networks.4,540,541 The selective tailoring of CO, C–O and furan ring functional groups in HMF toward the desired products requires the development of active and selective catalysts with specific compositions, structures and active sites, the development of suitable reaction media and the regulation of reaction conditions.542 The one-pot synthesis of high-value products from carbohydrates via HMF as an intermediate calls for the elegant manipulation of carbohydrate dehydration and the subsequent HMF upgrading step with good control of the accompanying side reactions.543 In this section, the upgrading of HMF is discussed mainly according to the reaction processes, but also considering the classification of products and the associated catalysts. We not only summarize the recent advances of HMF upgrading technologies, but also highlight the main bottlenecks of current catalytic systems and possible solutions.
3.1. Synthesis of 5-(chloromethyl)furfural and acetoxymethylfurfural
Due to the similarity of 5-(chloromethyl)furfural (CMF) with HMF, CMF is also considered a promising intermediate for the production of biofuels, polymers, fine chemicals, agrochemicals and pharmaceuticals.209,544,545 However, different from HMF, a high yield of CMF can be readily achieved from glucose, sucrose, cellulose and raw biomass in the HCl/dichloroethane/LiCl or HCl/dichloroethane system under mild conditions (Fig. 38), but the synthesis of CMF requires the quantitative consumption of HCl.546 It was proposed that cellulose hydrolysis to glucose, glucose isomerization to fructose and fructose dehydration to HMF all proceed in the aqueous phase containing HCl, while the conversion of HMF to CMF promotes the transfer and sequestration of hydrophobic CMF, thus reducing the further degradation of CMF. Moreover, the separation and purification of CMF are much easier than HMF owing to the lipophilicity and stability of CMF under acidic conditions. Owing to its rich derivative chemistry, CMF is an important starting material for the production of other high-value HMF derivates.546 For example, CMF can be readily converted to levulinic acid in hot water with high yield (Fig. 39).547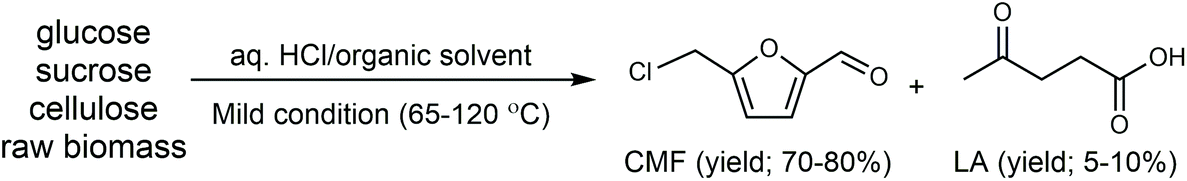 | ||
| Fig. 38 Direct conversion of carbohydrates and biomass to CMF under mild conditions. Adapted from ref. 209 and 544–546. | ||
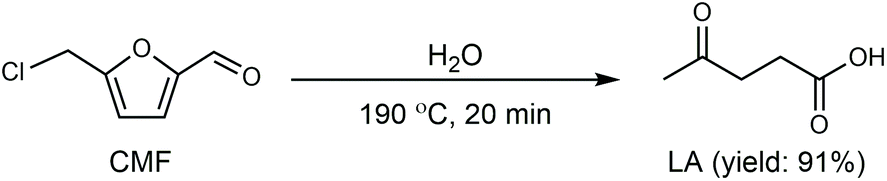 | ||
| Fig. 39 Conversion of CMF to levulinic acid. Adapted from ref. 547. | ||
Acetoxymethylfurfural, the acetylation product of HMF, is also considered a potential intermediate since the acetyl moiety imparts more hydrophobic property and less reactivity to acetoxymethylfurfural.548 Bicker et al. reported that fructose can be converted to acetoxymethylfurfural (Fig. 40) in subcritical acetic acid (scAcOH) with an acetoxymethylfurfural yield of 37% with the fructose conversion of 98%.549 Gavilà et al. reported that the acetolysis of cellulose acetate in the presence of 2 equivalents of acetic anhydride using sulfuric acid as a catalyst gave a maximum acetoxymethylfurfural yield of ca. 40%.550
 | ||
| Fig. 40 Conversion of fructose to acetoxymethylfurfural. Adapted from ref. 549. | ||
3.2. Oxidation of HMF
The oxidation of HMF can give a series of high-value products, including 2,5-diformylfuran (DFF), 2,5-furandicarboxylic acid (FDCA), 5-formyl-2-furan carboxylic acid (FFCA), and 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) (Table 13) via thermochemical, biocatalysis, photocatalysis and electrocatalysis technologies. The reaction pathway (Fig. 41) and product selectivity depend on many factors, including the catalyst, solvent, additive, pH, oxidant and reaction conditions. Generally, thermal aerobic oxidation over metal oxide catalysts in neutral aqueous solution and photocatalytic oxidation in trifluorotoluene (PhCF3) favor the selective oxidation of the alcohol side chain, resulting in the conversion of HMF to DFF.551,552 In contrast, the combination of noble metal catalysts and basic solvents (or basic supports) is prone to oxidize the aldehyde side chain first.553,554 Ag-based catalysts can selectively oxidize HMF to HMFCA, while Au, Pt and Pd-based catalysts not only can oxidize HMF to HMFCA under relatively mild conditions, but also further oxidize HMFCA to FDCA under relatively harsh conditions (high temperature and long reaction time).553,555–559 Since a plethora of original research and review articles have been published, in this section we try to provide a review on the oxidation of HMF with a focus on summarizing the promising reaction pathways, products and catalytic technologies, not aiming to give an exhaustive overview on the results contributed by the scientific community. The oxidation of HMF has already been extensively summarized in previous reviews from different perspectives, and readers can also consult these sources.34,560–566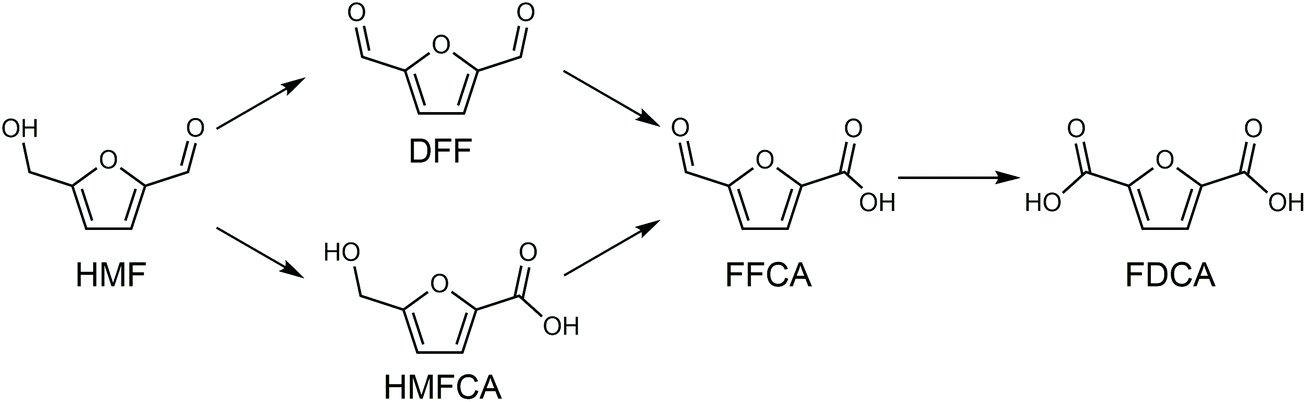 | ||
| Fig. 41 Basic pathways for the oxidation of HMF to FDCA. Adapted from ref. 567. | ||
| Catalysta | Solvent | Conditions | Feedstocka | Conversion | Yield | Ref. |
|---|---|---|---|---|---|---|
| a Relative to solvent. b Reaction conducted in a trickle-bed reactor with weight hourly space velocity (WHSV) of 900 h−1 and gas hourly space velocity (GHSV) of 1 h−1. c Electrochemical oxidation of HMF. d The conversion of carbohydrate to DFF was conducted via one-pot, two-step tandem process and the first step was conducted in the absence of catalyst. — Not provided. | ||||||
| M(NO3)x, 5–7.5 mol% (M = Fe, Cu, Al, Zn or H); TEMPO, 5 mol% | AcOH | 50 °C, 5 h, 0.1 MPa O2 | HMF 6.3 wt% | ∼100% | DFF, 94–99% | 568 |
| M(NO3)x, 5–7.5 mol% (M = Fe, Cu); TEMPO, 5 mol%; NaNO2, 5 mol% | CH3CN | 50 °C, 10–24 h, 0.1 MPa O2 | HMF 6.3 wt% | ∼100% | DFF, 94–99% | 568 |
| Cu/GO, 0.33 wt%; TEMPO, 0.4 wt% | CH3CN | 70 °C, 8 h, 0.4 MPa O2 | HMF, 0.84 wt% | 25.5% | DFF, 18.4% | 569 |
| Cu/G-700H, 0.33 wt%; TEMPO, 0.4 wt% | CH3CN | 70 °C, 8 h, 0.4 MPa O2 | HMF, 0.84 wt% | 12.5% | DFF, 8.6% | 569 |
| Cu/NG4, 0.33 wt%; TEMPO, 0.4 wt% | CH3CN | 50 °C, 8 h, 0.4 MPa O2 | HMF, 0.84 wt% | 99.5% | DFF, 99.2% (FFCA 0.5%) | 569 |
| Cu/NG4, 0.33 wt%; TEMPO, 0.4 wt% | DMSO | 70 °C, 8 h, 0.4 MPa O2 | HMF, 0.84 wt% | 100% | DFF, 92.2% (FFCA 3.2%) | 569 |
| Cu/NG4, 0.33 wt%; TEMPO, 0.4 wt% | CH3CN | 70 °C, 8 h, 0.4 MPa O2 | HMF, 0.84 wt% | 99.5% | DFF, 82.5% (FFCA 1.2%) | 569 |
| NC-950, 0.2 wt% | CH3CN | 100 °C, 14 h, 0.1 MPa air, 14.5 M HNO3 (0.02 mL) | HMF, 0.63 wt% | 100% | DFF, 95.1% | 570 |
| NC-950, 0.2 wt% | CH3CN | 100 °C, 14 h, 0.1 MPa air, 14.5 M HNO3 (0.02 mL) | HMF, 0.63 wt% | 100% | DFF, 93.5% | 570 |
| I2, 10 mol%; NaOH, 20 mol% | TBPH (6 M)/H2O | 70 °C, 36 h, 0.1 MPa air, 14.5 M HNO3 (0.02 mL) | HMF, 10 wt% | — | FDCA, 53% | 571 |
| CuO, 0.5 wt% | 0.04 M NaOH | 40 °C, 2 h, NaClO | HMF, 0.25 wt% | 100% | FDCA, 99.80% | 572 |
| Co3O4, 0.5 wt% | 0.04 M NaOH | 40 °C, 2 h, NaClO | HMF, 0.25 wt% | 100% | FDCA, 95.7% | 572 |
| MnO2, 0.5 wt% | 0.04 M NaOH | 40 °C, 2 h, NaClO | HMF, 0.25 wt% | 100% | FDCA, 34.3% | 572 |
| NiO, 0.5 wt% | 0.04 M NaOH | 40 °C, 2 h, NaClO | HMF, 0.25 wt% | 77.7% | FDCA, 18.1% | 572 |
| FeOx/C, 20 mol% metal | Toluene | 150 °C, 12 h, 1 MPa O2 | HMF, 3.1 wt% | 100% | DFF, 99% | 573 |
| FeNx-900, 1 wt% | N,N-DMF | 100 °C, 10 h, 0.5 MPa O2 | HMF, 0.32 wt% | 99.5% | DFF, 97.3% | 574 |
| Co–Al hydrotalcites, 0.5 wt% | DMSO | 120 °C, 8 h, 0.3 MPa O2 | HMF, 1.26 wt% | 100% | DFF, 95% | 575 |
| Co–Al hydrotalcites, 0.5 wt% | DMSO | 120 °C, 1.5 h, 0.3 MPa O2 | HMF, 1.8 wt% | 100% | DFF, around 10% (HMF 83%) | 575 |
| Co–Al hydrotalcites, 0.5 wt% | DMSO | 120 °C, 8 h, 0.3 MPa O2 | Fructose, 1.8 wt% | 100% | DFF, 77% | 575 |
| Co–Al hydrotalcites (0.5 wt%) | DMSO | 120 °C, 8 h, 0.3 MPa air | Fructose, 1.8 wt% | 100% | DFF, 59% | 575 |
| 10V2O5@MOR(60) 4 wt%, HCl 0.14 M | DMSO | 130 °C, 4 h, 120 °C, 8 h, O2 balloon | Fructose, 2 wt% | 100% | DFF, 94.8% | 576 |
| DICAT-V, 0.34 wt% | CH3CN | 110 °C, 8 h, air 2 MPa | HMF, 1.3 wt% | 92% | DFF, 88% | 577 |
| Without catalyst | 0.1 M NaOH | 130 °C, 2.5 h, 0.5 MPa O2 | HMF, 0.45 wt% | 100% | HMFCA, 4.3%; FDCA 0 | 578 |
| CeO2-cube, 0.39 wt% | 0.1 M NaOH | 130 °C, 2.5 h, 0.5 MPa O2 | HMF, 0.45 wt% | 100% | HMFCA, 4.2%; FDCA 0 | 578 |
| CeO2-oct, 0.41 wt% | 0.1 M NaOH | 130 °C, 2.5 h, 0.5 MPa O2 | HMF, 0.45 wt% | 100% | HMFCA, 4.0%; FDCA 0 | 578 |
| CeO2-rod, 0.22 wt% | 0.1 M NaOH | 130 °C, 2.5 h, 0.5 MPa O2 | HMF, 0.45 wt% | 100% | HMFCA, 6.3%; FDCA 0 | 578 |
| Au/CeO2-rod, 0.22 wt% | NaOH/H2O | 130 °C, 2.5 h, 0.5 MPa O2 | HMF, 0.45 wt% | — | FDCA, 87.4% | 578 |
| AuNPs-sPSB, 2.5 wt% | Cs2CO3/N,N-DMF | 80 °C, 16 h, 1.5 MPa O2 | HMF, 1.1 wt% | 78% | DFF 62.4%, FFCA 15.6% | 579 |
| AuNPs-sPSB, 2.5 wt% | Cs2CO3/N,N-DMF | 80 °C, 16 h, 1.5 MPa O2 | HMF, 1.1 wt% | 78% | DFF 13.9%, FFCA 74.1% | 579 |
| AuNPs-sPSB, 2 wt% | MeOH | 25 °C, 16 h, 1.5 MPa O2 | HMF, 0.88 wt% | 78% | MHFCA 77.2%, FDMC 21.8% | 579 |
| AuNPs-sPSB, 2 wt% | DMA/MeOH(v/v = 4) | 110 °C, 5 h, 3.5 MPa O2 | HMF, 0.88 wt% | 99% | FDCA 99% | 579 |
| Pd–Au/HT, 0.2 wt% | 0.064 M NaOH | 60 °C, 6 h, 60 mL min−1 O2 flow | HMF, 0.4 wt% | 100% | FDCA, 87.4% | 558 |
| Pd/HT, 0.2 wt% | 0.064 M NaOH | 60 °C, 6 h, 60 mL min−1 O2 flow | HMF, 0.4 wt% | 100% | FDCA <3%; HMFCA, 45% | 558 |
| Au/HT, 0.2 wt% | 0.064 M NaOH | 60 °C, 6 h, 60 mL min−1 O2 flow | HMF, 0.4 wt% | 100% | FDCA 0%; HMFCA 5% | 558 |
| AuPd NNWs, 0.05 wt% | 0.033 M Na2CO3 | 60 °C, 6 h, 0.1 MPa O2 | HMF, 0.42 wt% | >99% | FDCA 98% | 580 |
| AuPd NNWs, 0.05 wt% | 0.033 M Na2CO3 | 60 °C, 6 h, 0.1 MPa O2 | HMF, 0.42 wt% | >99% | FDCA 98% | 581 |
| AuPd porous, 0.05 wt% | 0.033 M Na2CO3 | 60 °C, 6 h, 0.1 MPa O2 | HMF, 0.42 wt% | >99% | FDCA 79% (HMFCA, 14%) | 581 |
| AuPd NPs, 0.05 wt% | 0.033 M Na2CO3 | 60 °C, 6 h, 0.1 MPa O2 | HMF, 0.42 wt% | >99% | FDCA 44% (HMFCA, 48%) | 581 |
| AuPd/TiO2, 0.05 wt% | 0.033 M Na2CO3 | 60 °C, 6 h, 0.1 MPa O2 | HMF, 0.42 wt% | >99% | FDCA 61% (HMFCA, 37%) | 581 |
| AuPdt NNWs, 0.05 wt% | 0.033 M Na2CO3 | 60 °C, 6 h, 0.1 MPa O2 | HMF, 0.42 wt% | >99% | FDCA 98% | 581 |
| AuPd-nNiO, metal/HMF molar ratio = 0.01 | H2O | 90 °C, 6 h, 1 MPa O2 | HMF, 0.25 wt% | 95% | FDCA 70% | 582 |
| Ag-PVP/ZrO2, 0.1 wt% | 2.5 g L−1 NaOH | 20 °C, 2 h, 60 mL min−1 O2 | HMF, 0.4 wt% | 100% | HMFCA, 98.2% | 555 |
| Ag2O, 2 wt% | 0.12 mM Na2CO3 | 90 °C, 1 h, 0.2 mL min−1 H2O2 (8.1 wt%) | HMF, 0.378 wt% | 100% | HMFCA, 98% | 556 |
| Ag2O, 2 wt% | 0.12 mM Na2CO3 | 30 W microwave 90 °C, 24 min, 0.2 mL min−1 H2O2 (8.1 wt%) | HMF, 0.378 wt% | 99% | HMFCA, 97% | 556 |
| Ag2O (5 runs), 2 wt% | 0.12 mM Na2CO3 | 90 °C, 1 h, 0.2 mL min−1 H2O2 (8.1 wt%) | HMF, 0.378 wt% | 57% | HMFCA, 42% | 556 |
| Activated MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | >99% | FDCA 74%, FFCA 15% | 583 |
| β-MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | >99% | FDCA 28%, FFCA 66% | 583 |
| β-MnO2-HS, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | — | FDCA 86%, FFCA 1% | 583 |
| λ-MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | >99% | FDCA 12%, FFCA 66% | 583 |
| γ-MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | 93% | FDCA 5%, FFCA 60% | 583 |
| α-MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | >99% | FDCA 59%, FFCA 36% | 583 |
| δ-MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | 96% | FDCA 10%, FFCA 69% | 583 |
| ε-MnO2, 1 wt% | 0.12 M NaHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | >99% | FDCA 63%, FFCA 27% | 583 |
| N-MnO2, 7.5 wt% | Toluene | 25 °C, 6 h, 16 mL min−1 0.1 MPa O2 | HMF, 3.2 wt% | 100% | DFF 99.9% | 552 |
| Co3O4-MnO2 (Co/Mn = 0.25), 1 wt% | 0.1 M NaHCO3 | 120 °C, 5 h, 1 MPa O2 | HMF, 0.63 wt% | 99% | FDCA 95% | 584 |
| MnCo2O4, 2 wt% | 0.012 M KHCO3 | 100 °C, 24 h, 1 MPa O2 | HMF, 0.5 wt% | 99.5% | FDCA, 70.9% | 585 |
| Pd-MnO2, 0.1 wt% | H2O | 100 °C, 6 h, 25 mL min−1 0.1 MPa O2 | HMF, 0.08 wt% | 100% | FDCA 88.1% | 586 |
| PdNP/MnO2, 0.1 wt% | H2O | 100 °C, 6 h, 25 mL min−1 0.1 MPa O2 | HMF, 0.08 wt% | 100% | FDCA 67.8% | 586 |
| MnO2, 0.1 wt% | H2O | 100 °C, 6 h, 25 mL min−1 0.1 MPa O2 | HMF, 0.08 wt% | 42.9% | FDCA 30.1% | 586 |
| Commercial Pd/C, 0.1 wt% | H2O | 100 °C, 6 h, 25 mL min−1 0.1 MPa O2 | HMF, 0.08 wt% | 100% | FDCA 76.6% | 586 |
| Cs/MnOx, 0.2 wt% | N,N-DMF | 100 °C, 12 h, 1 MPa O2 | HMF, 0.6 wt% | 98.4% | DFF, 94.7% | 587 |
| HPMV6, 5 wt% | [BMIM]Cl | 140 °C, 6 h, 1 MPa O2 | HMF, 1.3 wt% | 100% | FDCA, 88.8%; FFCA 1.1% | 588 |
| HPMV6, 5 wt% | [BMIM]Cl | 140 °C, 9 h, 1 MPa O2 | Fructose, 1.8 wt% | — | FDCA, 30.7% | 588 |
| HPMV6, 5 wt% | [BMIM]Cl | 140 °C, 9 h, 1 MPa O2 | Glucose, 1.8 wt% | — | FDCA, 48.6% | 588 |
| Pt-PVB-ACS 800, 0.4 wt% | H2O | 110 °C, 5 h, 1 MPa O2 | HMF, 0.63 wt% | 100% | FDCA, 99% | 589 |
| Pt-ACS 800, 0.4 wt% | H2O | 110 °C, 5 h, 1 MPa O2 | HMF, 0.63 wt% | 100% | FDCA, 75%; FFCA 25% | 589 |
| Pt-PVB-ACS 800 (10 runs), 0.4 wt% | H2O | 110 °C, 5 h, 1 MPa O2 | HMF, 0.63 wt% | 100% | FDCA, 79%; FFCA 21% | 589 |
| Au/HSAG-ox, 0.35 wt% | 0.057 M NaHCO3 | 90 °C, 12 h, 1 MPa O2 | HMF, 0.3 wt% | 45% | HMFCA 43%, FDCA 0 | 590 |
| Au/HSAG, 0.35 wt% | 0.057 M NaHCO3 | 90 °C, 12 h, 1 MPa O2 | HMF, 0.3 wt% | 45% | HMFCA 93%, FDCA 6% | 590 |
| Au/HSAG-H, 0.35 wt% | 0.057 M NaHCO3 | 90 °C, 12 h, 1 MPa O2 | HMF, 0.3 wt% | 45% | HMFCA 56%, FDCA 44% | 590 |
| Au/HSAG-H, 0.35 wt% | 0.057 M NaHCO3 | 90 °C, 12 h, 1 MPa O2 | HMF, 0.3 wt% | 45% | HMFCA 22%, FDCA 75% | 590 |
| Mn6Fe1Ox, 1.2 wt% | N,N-DMF | 110 °C, 5 h, 1.5 MPa O2 | HMF, 2.5 wt% | 97% | DFF, 95% | 591 |
| CeO2, 0.7 wt% | H2O | 110 °C, 15 h, 0.9 MPa O2 | HMF, 0.2 wt% | 21.3% | DFF, 0.1% (HMFCA 0, FFCA 18.9%, FDCA 0) | 592 |
| MnO2, 0.7 wt% | H2O | 130 °C, 12 h, 0.9 MPa O2 | HMF, 0.2 wt% | 99.0% | DFF 0 (HMFCA 0, FFCA 23.3%, FDCA 74.7%) | 592 |
| MgO, 0.7 wt% | H2O | 110 °C, 15 h, 0.9 MPa O2 | HMF, 0.2 wt% | 21.3% | DFF, 0.4% (HMFCA 3.4%, FFCA 0, FDCA 0, formic acid 19.4%) | 592 |
| MgO·MnO2·CeO2, 0.7 wt% | H2O | 110 °C, 10 h, 2 MPa O2 | HMF, 0.2 wt% | 98.8% | DFF 94.1% (HMFCA 0.8%, FFCA 0, FDCA 0) | 592 |
| CuO·MnO2·CeO2, 0.7 wt% | H2O | 130 °C, 4 h, 2 MPa O2 | HMF, 0.2 wt% | 79.0% | DFF 0 (HMFCA 0, FFCA 0, FDCA 77.9%) | 592 |
| Ru/Mn6Ce1Oy, 0.7 wt% | H2O | 150 °C, 15 h, 10 bar O2 | HMF, 1.3 wt% | ≥99.0% | DFF 0 (HMFCA 0, FFCA 0, FDCA 77.9%) | 593 |
| Pt/NC-CeO2, 0.25 wt% | H2O | 110 °C, 8 h, 0.4 MPa O2 | HMF, 0.16 wt% | 100% | FDCA 100% | 594 |
| Ru/ZrO2 H-aero, 0.33 wt% | H2O | 120 °C, 16 h, 10 bar O2 | HMF, 0.63 wt% | 100% | FDCA, 97% | 595 |
| Ru(III)-Fe3O4@SiO2, 0.5 wt% | 0.25 M n-butylamine | 110 °C, 48 h, 10 bar O2 | HMF, 1 wt% | 92% | FDCA, 74.2% | 596 |
| Fe3O4@SiO2-CoOx, 0.5 wt% | 0.78 M NaOH | 110 °C, 8 h, 10 bar O2 | HMF, 1 wt% | 78.6% | Succinic acid, 72.9% | 596 |
| Fe3O4@SiO2-MnOx, 0.5 wt% | 0.25 M n-butylamine | 110 °C, 48 h, 10 bar O2 | HMF, 1 wt% | 5% | FDCA, 3.6% | 596 |
| Fe3O4@SiO2-NH2, 0.5 wt% | 0.25 M n-butylamine | 110 °C, 48 h, 10 bar O2 | HMF, 1 wt% | 35.6% | FDCA, 30.3% | 596 |
| PCNx, 0.2 wt% | 0.1 M K2CO3 | 70 °C, 36 h, O2 flow | HMF, 1.3 wt% | — | DFF, 5% | 597 |
| N-Doped graphene, 0.2 wt% | 0.1 M K2CO3 | 70 °C, 36 h, O2 flow | HMF, 1.3 wt% | — | DFF, 83% | 597 |
| g-C3N4, 0.2 wt% | 0.1 M K2CO3 | 70 °C, 36 h, O2 flow | HMF, 1.3 wt% | — | DFF, 15% | 597 |
| SO3H-PANI-FeVO4, 2.4 wt% | DMSO | 140 °C, 24 h, O2 flow | HMF, 2.5 wt% | 100% | DFF, 99% | 452 |
| RuO2/IL-graphene, 0.33 wt% | Toluene | 100 °C, 12 h, O2 flow | HMF, 2.1 wt% | 60% | DFF, 60% | 598 |
| Rh/C, 0.2 wt% | scCO2 | 150 °C, 2 h, 8 MPa CO2 | HMF, 2.1 wt% | 100% | DFF,100% | 599 |
| VPO (V:P = 0.25), 0.2 wt% |
H2O | 140 °C, 24 h, air | HMF, 0.2 wt% | 22.0% | DFF, 0 | 600 |
| VPO (V:P = 0.25), 0.2 wt% |
Toluene | 140 °C, 24 h, air | HMF, 0.2 wt% | 15.0% | DFF, 15% | 600 |
| VPO (V:P = 0.25), 0.2 wt% |
MIBK | 140 °C, 24 h, air | HMF, 0.2 wt% | 43.1% | DFF, 19.5% | 600 |
| VPO (V:P = 0.25), 0.2 wt% |
DMF | 140 °C, 24 h, air | HMF, 0.2 wt% | 27.4% | DFF, 11.3% | 600 |
| MoV2@CP-5.5–400, 2.3 wt% | DMSO | 120 °C, 4 h, 1 atm O2 | Fructose, 4.5 wt% | — | DFF, 87.3% (HMF, 0.6%) | 601 |
| MoV2@CP-5.5, 1 wt% | DMSO | 130 °C, 3 h, 1 atm O2 | Fructose, 4.5 wt% | — | DFF, 20.0% (HMF, 50.5%) | 601 |
| CeCu(OH)6Mo6O18, 1.7 wt% | p-Chlorotoluene | 130 °C, 8 h, 1 atm O2 | HMF, 0.7 wt% | 99% | DFF, 99% | 602 |
| P(DVPI-Br), 1 wt% | DMSO | 130 °C, 3 h, 1 atm O2 | Fructose, 4.5 wt% | — | DFF, 9.6% (HMF, 60.6%) | 601 |
| MoV2@CP-5.5-400, 2.3 wt% | DMSO | 135 °C, 5 h, 1 atm O2 | glucose, 4.5 wt% | — | DFF, 51.2% (HMF, 0.4%) | 601 |
| MoV2@CP-5.5-400, 2.3 wt% | DMSO | 135 °C, 5 h, 1 atm O2 | sucrose, 4.5 wt% | — | DFF, 20.4% (HMF, 0.2%) | 601 |
| MoV2@CP-5.5-400, 2.3 wt% | DMSO | 135 °C, 5 h, 1 atm O2 | inulin, 4.5 wt% | — | DFF, 61.6% (HMF, 0.6%) | 601 |
| Octahedral MnO2 molecular sieve, 1.2 wt% | DMSO | 120 °C, 10 h, 1 atm O2 | HMF, 2.5 wt% | 99% | DFF, 99% | 551 |
| Without catalyst | DMSO | 120 °C, 10 h, 1 atm O2 | HMF, 2.5 wt% | 99% | DFF, 1.9% | 551 |
| H-Beta zeolite, 1.2 wt% | DMSO | 120 °C, 10 h, 1 atm O2 | HMF, 2.5 wt% | 99% | DFF, 1.9% | 551 |
| V-CS-800, 1.3 wt% | DMSO | 120 °C, 9 h, 20 mL min−1 O2 | HMF, 0.13 wt% | 100% | DFF, 99% | 603 |
| Mo-HNC, 0.4 wt% | DMSO | 150 °C, 9 h, 20 mL min−1 O2 | HMF, 4 wt% | 100% | DFF, 77% | 604 |
| Ru/MgAlO, 1 wt% | H2O | 140 °C, 4 h, 0.62 MPa O2 | HMF, 1.3 wt% | 100% | FDCA 99% (DFF 0.5%, HMFCA 0, FFCA 0.2%) | 605 |
| Ru/MgO, 1 wt% | H2O | 160 °C, 4 h, 0.62 MPa O2 | HMF, 1.3 wt% | 58% | FDCA 90% | 605 |
| Ru/La2O3, 1 wt% | H2O | 140 °C, 4 h, 0.62 MPa O2 | HMF, 1.3 wt% | 4.6% | FDCA 0.8% (DFF 0, HMFCA 0, FFCA 1.4%) | 605 |
| Ru/CeO2, 1 wt% | H2O | 140 °C, 4 h, 0.62 MPa O2 | HMF, 1.3 wt% | 89% | FDCA 7% (DFF 45%, HMFCA 0, FFCA 35%) | 605 |
| Ru/ZrO2, 1 wt% | H2O | 140 °C, 4 h, 0.62 MPa O2 | HMF, 1.3 wt% | 100% | FDCA 0 (DFF 86%, HMFCA 0, FFCA 14%) | 605 |
| Ru/Al2O3b |
0.1 M Na2CO3 | 110 °C, 5 h, 3 MPa O2 | HMF, 1.3 wt% | 64% | FDCA, 63% | 606 |
| Au/CeO2, 0.45 wt% | 0.16 M NaOH | 70 °C, 4 h, 10 bar O2 | HMF, 1 wt% | — | FDCA, 63% | 607 |
| Au/Ce0.50Zr0.50O2, 0.66 wt% | 0.16 M NaOH | 70 °C, 4 h, 10 bar O2 | HMF, 1 wt% | — | FDCA, 70% | 607 |
| Au/Ce0.25Zr0.75O2, 0.66 wt% | 0.16 M NaOH | 70 °C, 4 h, 10 bar O2 | HMF, 1 wt% | — | FDCA, 80% | 607 |
| Au/ZrO2, 0.69 wt% | 0.16 M NaOH | 70 °C, 4 h, 10 bar O2 | HMF, 1 wt% | — | FDCA, 58% | 607 |
| Au/ZrO2, 0.33 wt% | 0.4 M NaOH | 125 °C, 5 h, 10 bar O2 | HMF, 1.3 wt% | 100% | FDCA, 89% | 559 |
| Ag/ZrO2, 0.29 wt% | 0.4 M NaOH | 50 °C, 1 h, 10 bar O2 | HMF, 1.3 wt% | 100% | HMFCA, 98% | 559 |
| PVA-Au/MgF2, 0.5 wt% | H2O | 110 °C, 2 h, 20 bar air | HMF, 0.3 wt% | 7% | FDCA, 0 (DFF 6.7%, HMFCA 0.2%, FFCA 0.07%) | 608 |
| PVA-Au/0.6MgF2-0.4MgO2, 0.5 wt% | H2O | 110 °C, 2 h, 20 bar air | HMF, 0.3 wt% | 77% | FDCA, 14% (DFF 0, HMFCA 42%, FFCA 22%) | 608 |
| PVA-Au/0.4MgF2-0.6MgO2, 0.5 wt% | H2O | 110 °C, 2 h, 20 bar air | HMF, 0.3 wt% | 98% | FDCA, 63% (DFF 0, HMFCA 53%, FFCA 12%) | 608 |
| PVA-Au/MgO2, 0.5 wt% | H2O | 110 °C, 2 h, 20 bar air | HMF, 0.3 wt% | 99% | FDCA, 91% (DFF 0, HMFCA 7%, FFCA 2%) | 608 |
| Pd/C 2.5 mol%; Co(NO3)2 2.5 mol%; Bi(NO3)3, 2.5 mol% | MeOH, K2CO3 (20 mol%) | 60 °C, 14 h, 0.1 MPa O2 | HMF, 3.1 wt% | ∼98% | HMFE, 93% | 609 |
| Heterogeneous PdCoBi/C, 2.5 mol% | MeOH, K2CO3 (20 mol%) | 60 °C, 14 h, 0.1 MPa O2 | HMF, 3.1 wt% | ∼98% | HMFE, 96% | 609 |
| Au–Fe3O4 (2 mol%) | MeOH, 0.1 M K2CO3 | 25 °C, 24 h, 0.1 MPa O2 | HMF, 1.3 wt% | 100% | HMFE, 92% | 610 |
| AuPd–Fe3O4 (2 mol%) | MeOH, 0.1 M K2CO3 | 25 °C, 24 h, 0.1 MPa O2 | HMF, 1.3 wt% | 92% | FDMC, 92% | 610 |
| Co@CN | MeOH | 80 °C, 12 h, 0.1 MPa O2 | HMF, 1 wt% | 100% | FDMC, 95% | 611 |
| Co7Cu3-NC | MeOH | 80 °C, 4 h, 0.2 MPa O2 | HMF, 1 wt% | 100% | FDMC, 95% | 612 |
| Au (3 wt%)–TiO2, 2.5 wt% | 0.13 mL min−1 1.2 M NaOH | 85 °C, 0.6 h, 0.13 mL min−1 10 wt% H2O2, 50 W microwave | HMF, 2.5 wt% | 100% | FDCA, 68% | 613 |
| 10Co@22Nb@MNP, 0.55 wt% | MeCN | 100 °C, 12 h, 70% TBHP | HMF, 0.55 wt% | 96.9% | FDCA 93.5% | 614 |
| Cu/NG, 0.36 wt% | MeCN | 70 °C, 24 h, 70% TBHP (0.1 mL per 2 h) | HMF, 0.6 wt% | 100% | FDCA 95.2% | 615 |
| CdS nanorod, 2.2 wt% | DMSO | 80 °C, 48 h, 70% TBHP | HMF, 1.5 wt% | 91.1% | FFCA 91.1% | 616 |
| AuPd@Co3O4, 0.07 wt% | 0.2 M K2CO3+ 0.1 M NaOH | 90 °C, 1 h, 10% H2O2 (adding with a rate of 1.6 mL h−1) | HMF, 0.26 wt% | 100% | FDCA, 95% | 617 |
| 6-CuO/m-Al2O3, 2.5 wt% | DMSO/H2O (v/v = 3) | 100 °C, 20 h, TBHP | HMF, 0.32 wt% | — | FDMC, 96% | 618 |
| Pd@Beta, 0.3 wt% | 0.15 M Na2CO3 | 90 °C, 24 h, O2 balloon | HMF, 2.5 wt% | 99% | FDMC, 95% | 619 |
| Au (3 wt%)-TiO2, 2.5 wt% | 0.13 mL min−1 1.2 M NaOH | 97 °C, 0.5 h, 0.13 mL min−1 10 wt% H2O2, 50 W microwave | HMF, 2.5 wt% | 100% | FDCA, 99% | 613 |
| Ni3N@C | 1 M KOH (pH 13.6) | 1.45 V vs. RHEc | HMF, 10 mM | ∼100% | FDCA, 98% (FF 99%) | 620 |
| NiCo2O4 | 1 M KOH (pH 13.6) | 1.45 V vs. RHEc | HMF, 10 mM | — | FDCA, 90% (FF 100%) | 621 |
| NiCo2O4 | 1 M KOH (pH 13.6) | 1.5 V vs. RHE; 34.75 Cc | HMF, 5 mM | — | FDCA, 90.4% (FF 87.5%) | 622 |
| NiOOH | 1 M KOH (pH 13.6) | 1.47 V vs. RHE; 4.7 hc | HMF, 5 mM | 99.8% | FDCA, 96.0% (FF of FDCA 96.0%, DFF 0.03%, HMFCA 1.31%, 1.79% FFCA) | 567 |
| CoOOH | 1 M KOH (pH 13.6) | 1.56 V vs. RHE; 22 hc | HMF, 5 mM | 95.5% | FDCA, 35.1% (FF of FDCA 35.1%, DFF 0.56%, HMFCA 13.6%, 30.8% FFCA) | 567 |
| FeOOH | 1 M KOH (pH 13.6) | 1.71 V vs. RHE; 2.3 hc | HMF, 5 mM | 16.0% | FDCA, 1.59% (FF of FDCA 1.59%, DFF 3.64%, HMFCA 2.74%, 4.30% FFCA) | 567 |
| NiCoFe-LDHs | 1 M KOH | 55 °C, 1.71 V vs. RHE; 2.3 hc | HMF, 10 mM | 95.5% | FDCA, 84.9% | 623 |
| NiCoFe-LDHs | 1 M KOH | 55 °C, 1.71 V vs. RHE; 2.3 hc | HMF, 10 mM | 95.5% | FDCA, 84.9% | 623 |
| MoO2–FeP@C | 10 M KOH | 1.486 V vs. RHE | HMF, 10 mM | 100% | FDCA, 98.6% | 624 |
| Sulfonated MoO3–ZrO2, 0.2 wt% | DMSO | 165 °C, 2 h, 20 mL min−1 O2 | Fructose, 4 wt% | 100% | DFF, 74% | 625 |
| MoOx/CS, 0.6 wt% | DMSO | 165 °C, 2 h, 20 mL min−1 O2 | Fructose, 4 wt% | 100% | DFF, 78% | 626 |
| Ru(3%)/H-Beta, 2.4 wt% | DMSO | 120 °C, 1 h, N2d; 140 °C, 24 h, 20 mL min−1 O2 |
Fructose, 2.47 wt% | 100% | DFF, 80% | 627 |
| Ru(3%)/H-Beta, 2.4 wt% | DMSO | 120 °C, 4 h, N2d; 140 °C, 24 h, 20 mL min−1 O2 |
Sucrose, 2.47 wt% | 97% | DFF, 70% | 627 |
| Ru(3%)/H-Beta, 2.4 wt% | DMSO | 120 °C, 6 h, N2d; 140 °C, 24 h, 20 mL min−1 O2 |
Glucose, 2.47 wt% | 97% | DFF, 48% | 627 |
| Ce0.5Fe0.15Zr0.35O2, 1 wt% | [BMIM]Cl | 160 °C, 24 h, 2 MPa O2 | Glucose, 1.26 wt% | 100% | FDCA, 44.2% | 628 |
As a highly effective catalyst for the selective oxidation of alcohols to aldehydes or ketones, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) has been widely investigated for the oxidation of HMF to DFF. For example, Hong et al. reported that the combination of M(NO3)x (M = Fe, Cu, Al, Zn or H) and TEMPO is highly active for the oxidation of HMF to DFF in acetic acid (AcOH), while the combination of M(NO3)x (M = Fe or Cu), TEMPO and NaNO2 is effective for this conversion in CH3CN, giving a DFF yield higher than 95% at 50 °C using pure O2 as the oxidant.568 Even using air as the oxidant, the combination of Fe(NO3)3, TEMPO and NaNO2 still afforded a DFF yield of 88%.
Nitrogen-doped carbon and its composite materials have also been extensively investigated for the selective oxidation of HMF to DFF. Ren et al. developed a metal-free oxidation strategy using a nitrogen-doped carbon material (NC), HNO3 and O2 as the catalyst, co-catalyst and terminal oxidant, respectively, to achieve the conversion of HMF to DFF (Fig. 42) under mild reaction conditions.570 In this catalytic system, the oxidation of HMF was initiated by HNO3 and then HNO3 could be recovered instantaneously by O2. A DFF yield of up to 95.1% was obtained over the nitrogen-doped carbon material with a catalytic amount of HNO3. Similarly, Xu et al. reported that the Anderson-type catalyst CeCu(OH)6Mo6O18 enabled the selective oxidation of HMF to DFF (Fig. 43) via a similar mechanism with oxygenase enzymes.602 In this procedure, Mo5+ species play an important role in the oxidation of HMF to DFF, while Ce4+ species function as electron transfer mediators and [Cu(OH)6Mo6O18]4− anions serve as electron-donors. Lv et al. reported that the use of Cu nanoparticles supported on pyridinic-nitrogen-dominated nitrogen-doped graphene (Cu/NG4) as the catalyst with TEMPO as the co-catalyst enabled the efficient oxidation of HMF to DFF under relatively mild conditions (70 °C, 8 h, 0.4 MPa O2), affording a high DFF yield (99.2%) with a high HMF conversion (99.8%).569 The superior catalytic activity and stability of Cu/NG4 compared with Cu(NO3)2, Cu/GO and Cu/G-900H catalysts was mainly attributed to the strong interaction between the Cu nanoparticles and pyridinic N. Zhang et al. reported that the FeNx/C catalyst afforded a DFF yield of 97.3% from HMF.574 They identified the Fe–N4 species as the main active sites via combined catalyst characterizations and control experiments. However, the FeNx/C catalyst was deactivated rapidly in the recycling experiment owing to the aggregation of the Fe species. Heat treatment under an NH3/N2 flow was required to reactivate the catalyst. Recently, single-atom supported on nitrogen-doped carbon (M–N–C) materials have been intensively developed and exhibit great potential in several fields, in particular CO2 hydrogeneration, CO oxidation and electrocatalysis.207,629 The development of these types of single-atom materials is promising for the selective oxidation of HMF.
 | ||
| Fig. 42 Oxidation of HMF to DFF using NC, HNO3 and O2 as the catalyst, co-catalyst and terminal oxidant, respectively. Adapted from ref. 570. | ||
 | ||
| Fig. 43 Oxidation of HMF to DFF over biomimetic CeCu(OH)6Mo6O18 catalyst. Adapted from ref. 602. | ||
The conversion of carbohydrates to DFF can be either achieved via a one-pot, two-step process or one-pot, one-step process. For example, FeVO4-supported–SO3H-functionalized polyaniline (SO3H–PANI–FeVO4) enabled the effective conversion of carbohydrates to DFF via a one-pot, two-step process, obtaining DFF yields of 80% and 91% from sucrose and fructose, respectively.452 Mo-Containing materials are promising catalysts for the selective oxidation of biomass-derived chemicals.630 Molybdenum oxides supported on carbon spheres (MoOx/CS), which were prepared using glucose and phosphomolybdic via a hydrothermal carbonization method followed by calcination in air, enabled the one-pot conversion of fructose to DFF in DMSO under atmospheric pressure oxygen, obtaining a DFF yield of 78% with the fructose conversion of 100% at 160 °C within 2 h.626 The acid and oxide sites in the MoOx/CS catalyst were responsible for the dehydration of fructose to HMF and aerobic oxidation of HMF to DFF, respectively. Wang et al. designed an MoV2@CP-5.5-400 catalyst via the copolymerization of an ionic liquid bearing carboxylic acid and divinyl benzene and introduction of the heteropolyacid H5PMo10V2O40 through ion-exchange followed by partial carbonization, to achieve the one-pot, one-step conversion of carbohydrates to DFF.601 The catalytic activity for the degradation of fructose to HMF and oxidation of HMF to DFF were both enhanced owing to the increased acid and oxidation property resulting from the partial carbonization. Consequently, the catalyst gave DFF yields of 87.3% and 51.2% from fructose and glucose, respectively. Raut et al. reported that Co–Al hydrotalcite is active for both the dehydration of fructose to HMF and the oxidation of DFF in DMSO, thus enabling the one-pot conversion of fructose with DFF yields of 77% and 59% using 0.3 MPa O2 and air as oxidant, respectively.575 The solvent has an important influence on the conversion of carbohydrates to DFF. For example, Lai et al. demonstrated that DMSO is more suitable than water, toluene, MIBK and N,N-DMF as the reaction medium for the one-pot conversion of fructose to DFF over a vanadium phosphate oxide (VPO) heterogeneous catalyst.600
Several studies have shown that bromide salts play an important role in the oxidation of HMF to DFF and the one-pot conversion of fructose to DFF, but the mechanism is still unclear. Laugel et al. reported that the use of bromides, including HBr and NaBr, in DMSO at the evaluated temperature and prolonged time (150 °C, 23 h) could promote the one-pot conversion of fructose to DFF, obtaining DFF yields of 13% and 67%, respectively.631 They believed that this conversion involves the generation of 5-(bromomethyl)furan-2-car-baldehyde intermediate followed by Kornblum-type reaction, and the main catalytic species are the strong acid species generated from the in situ thermolysis of DMSO. The combination of KBr and TFP-DABA under mild conditions (100 °C, 12 h) gave DFF yields of 65% and 97% from HMF and fructose, respectively, in the medium of DMSO.438 However, these processes led to the stoichiometric consumption of DMSO, which is impractical for industrial production. In contrast, Verma et al. claimed that sulfonated graphitic carbon nitride (Sg-CN) combined with KBr enabled the one-pot conversion of fructose to DFF via the dehydration of fructose to HMF over Sg-CN followed by the oxidation of HMF to DFF by KBr in the medium of water under mild reaction conditions (100 °C, 0.5 h), while the combination of Sg-CN, DMSO and KBr gave 5-methylsulfanylmethyl-furan-2-carbaldehyde (97%) predominantly with a small amount of DFF (3%).458 Nevertheless, it is confusing that KBr is considered an oxidant in this study.
In addition to the oxidation of HMF to DFF with an external oxidant, HMF can also be converted to DFF via the Meerwein–Ponndorf–Verley–Oppenauer (MPVO) reaction or dehydrogenation reaction. For example, DFF and BHMF can be simultaneously produced from HMF (Fig. 44) via the MPVO reaction over the Lewis acid AlMe3, without the use of an external oxidant or reductant.632 In this process, HMF functions as both an oxidant and reductant, obtaining an DFF/BHMF molar ratio approaching 1:1. Under the optimized conditions (microwave irradiation, 80 °C, 1 h), an HMF conversion of up to 44.7% can be obtained within 1 h. Chatterjee et al. reported that the synergistic effect of the Rh/C catalyst with compressed carbon dioxide (scCO2) enabled the efficient dehydrogenation of HMF to DFF under mild conditions without using an additional hydrogen acceptor or oxidant.599 In this catalytic system, the scCO2 not only accelerated the reaction by removing hydrogen, but also shifted the equilibrium toward DFF as a hydrogen acceptor (Fig. 45).
 | ||
| Fig. 44 Simultaneously production of DFF and BHMF from HMF via the MPVO reaction. Adapted from ref. 632. | ||
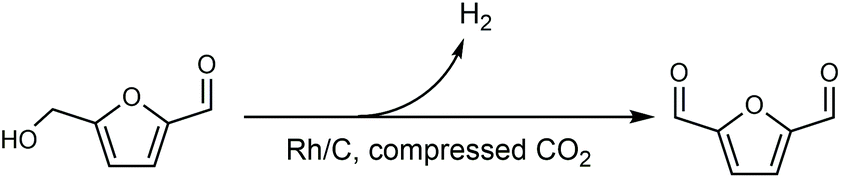 | ||
| Fig. 45 Conversion of HMF to DFF via a dehydrogenation reaction. Adapted from ref. 599. | ||
Photocatalytic oxidation is an effective approach to achieve the conversion of HMF to DFF (Table 14).633,634 Polymeric carbon nitride (C3N4) and C3N4-based materials have been widely investigated as photocatalysts for the oxidation of HMF to DFF. However, bulk C3N4 usually exhibits a low DFF yield and selectivity. H2O2-treated C3N4 exhibited higher selectivity toward DFF than bare C3N4.635–637 Similarly, WO3/g-C3N4, which was synthesized from ammonium tungstate hydrate and melamine via a calcination method, exhibited much better photocatalytic performance than g-C3N4 for the oxidation of HMF to DFF, attaining the maximum HMF conversion of 27.4% with DFF selectivity of 87.2% under visible-light (>400 nm).638 However, the use of the toxic PhCF3 and CH3CN/PhCF3 as the reaction medium is the main disadvantage of these photocatalytic systems. Yang et al. reported that the combination of a tetraalkylammonium decatungstate (TMADT) catalyst with HBr additive enabled the selective photocatalytic oxidation of HMF in the medium of CH3CN, attaining a DFF yield of 67.1% with FDCA yield of 5.8% and HMF conversion of 83.1%.639 They suggested that the photo-excited decatungstate can preferentially oxidize HBr to the Br atom free radical, which is crucial for the improvement the DFF yield.
| Catalyst | Catalyst loadinga | Solvent | Conditions | Feedstocka | Conversion | H2 evolution rate | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Relative to solvent. — Not detected. | ||||||||
| TiO2-m | — | 0.16 M NaOH | 30 °C, 8 h, 1 MPa O2, 300 W Xe-lamp, 250–2500 nm | HMF, 0.3 wt% | 22% | — | DFF 5% (CO2 8%) | 634 |
| Au–TiO2-m | — | 0.16 M Na2CO3 | 30 °C, 8 h, 20 bar air, 300 W Xe-lamp, 250–2500 nm | HMF, 0.3 wt% | 15% | — | DFF 3% (CO2 8%) | 634 |
| AuNPs/TiO2 | 2.5 wt% | 0.1 M Na2CO3 | 30 °C, 8 h, atmospheric air, light (λ = 350–400 nm, 0.3 W cm−2) | HMF, 1.3 wt% | 99% | — | HMFCA 90% | 557 |
| AuNPs/TiO2 | 2.5 wt% | 0.1 M Na2CO3 | 30 °C, 8 h, atmospheric air, light (λ = 420–78 nm, 0.3 W cm−2) | HMF, 1.3 wt% | 99% | — | HMFCA 95% | 557 |
| Nb2O5 | 1 wt% | PhCF3 | >400 nm, 6 h, 10 mL min−1 O2 | HMF, 1.3 wt%, | 19.2% | — | DFF, 17.4% | 640 |
| g-C3N4 | 1 wt% | CH3CN/PhCF3 | >400 nm, 6 h, 10 mL min−1 O2 | HMF, 2.5 wt% | 12% | — | DFF, 7.5% | 638 |
| WO3/g-C3N4 | 1 wt% | CH3CN/PhCF3 | >400 nm, 6 h, 10 mL min−1 O2 | HMF, 2.5 wt%, | 27.4% | — | DFF, 23.9% | 638 |
| H2O | 0.07 wt% | H2O (pH = 7) | Simulated solar light, 6 h, 10 mL min−1 O2 | HMF, 0.006 wt% | 73% | — | DFF, 36% | 641 |
| TMADT | 1 wt% | 6 M HBr, MeCN | Visible light from 35 W of tungsten-bromine lamp, 6 h, 1 atom O2 | HMF, 0.25 wt% | 83.1% | — | DFF, 67.1% (FDCA 5.8%) | 639 |
| CoPz/g-C3N4 | 0.01 wt% | H2O | 300–1000 nm light from Xe light, 20 mL min−1, air, potassium biphthalate/Na2B4O7 buffer solution (pH = 4.01), 14 h | HMF, 0.2 wt% | 100% | — | FDCA, 80% | 642 |
| CoPz/g-C3N4 | 0.01 wt% | H2O | 300–1000 nm light from Xe light, 20 mL min−1, air, potassium biphthalate/Na2B4O7 buffer solution (pH = 9.18), 14 h | HMF, 0.2 wt% | 99.1% | — | FDCA, 96% | 642 |
| ZnxCd1−xS-P | 0.05 wt% | H2O | Visible light from LED (30 × 3 W), Ar, 8 h | HMF, 0.2 wt% | — | 419 μmol h−1 g−1 | — | 643 |
| ZnxCd1−xS-P | 0.05 wt% | H2O | Visible light from LED (30 × 3 W), Ar, 8 h | HMF, 0.2 wt% | 40% | 623 μmol h−1 g−1 | DFF, 26% | 643 |
| Zn0.5Cd0.5S/1%MnO2 | 0.1 wt% | H2O | >400 nm, LED (30 W), N2, 6 h | HMF, 0.2 wt% | — | 25.4 μmol h−1 g−1 | DFF, 14% | 644 |
| Ni/CdS | 0.1 wt% | H2O | Visible light from LED (8 W), N2, 22 h | HMF, 0.2 wt% | 20% | 334 μmol h−1 g−1 | DFF, 20% | 645 |
| NiS/Zn3In2S6 | 0.2 wt% | H2O | >400 nm, 300 W xenon lamp, N2 | HMF, 1.3 wt% | 20% | 120 μmol h−1 g−1 | DFF (selectivity of 94.1%), — | 646 |
| Ni/CdS | 0.1 wt% | 10 M NaOH | Visible light from LED (8 W), N2, 2 h | HMF, 0.2 wt% | 100% | — | FDCA, 100% | 645 |
The photocatalytic oxidation of HMF to valuable chemicals coupled with H2 production via water splitting over semiconductors (Fig. 46) is an effective strategy to improve the utilization efficiency of solar energy. Han et al. investigated the photocatalytic decomposition of HMF and furfural alcohol over an ultrathin CdS nanosheet decorated with nickel (Ni/CdS) catalyst.645 In neutral aqueous solution, HMF was converted to DFF and H2, with a DFF yield (20%) and H2 production rate (334 μmol h−1 g−1) much lower than that (furfural yield 90%, H2 production rate 4970 μmol h−1 g−1) obtained from furfural alcohol. Ye et al. reported that P-doped ZnxCd1−xS with rich S vacancies (ZnxCd1−xS–P) exhibited a remarkably higher hydrogen production rate from HMF aqueous solution in comparison with that from pure water.643 After 8 h of irradiation under visible light, a DFF selectivity of 65% was obtained with the HMF conversion of 40%. However, to date, both the reaction efficiency and DFF selectivity of photocatalytic systems are obviously lower than that of traditional thermal catalytic systems. Therefore, further insight into the mechanism of light adsorption, generation and recombination of electrons and holes (h+), and regulation of reactive oxygen radicals is required to establish more effective photocatalytic systems.
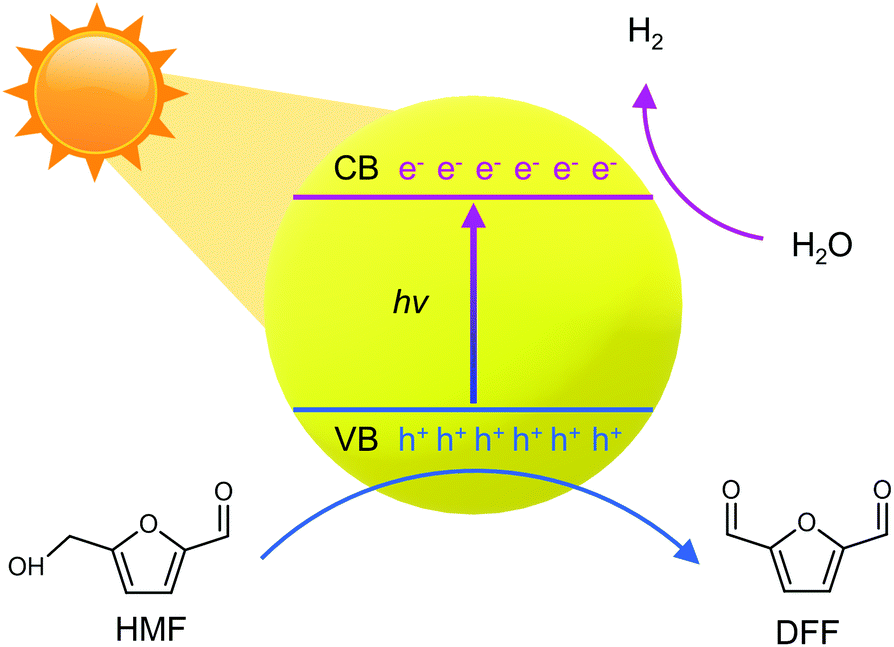 | ||
| Fig. 46 Photocatalytic oxidation of HMF coupled with hydrogen evolution from water splitting. Adapted from ref. 643 and 645. | ||
Brandolese et al. reported that the one-pot, two-step process (Fig. 47) involving the sequential oxidation and self-esterification of HMF into polyester oligomer over a polystyrene-supported triazolium pre-catalyst in the presence of iron(II) phthalocyanine and air, followed by the hydrolysis of the polyester oligomer to HMFCA over a supported base (Ambersep 900 OH) afforded an overall HMFCA yield of 87%.647 This strategy also enabled the efficient production of HMFCA-derived ester and amide via nucleophilic depolymerization of the oligomer intermediate in the presence of methanol and butylamine, respectively.
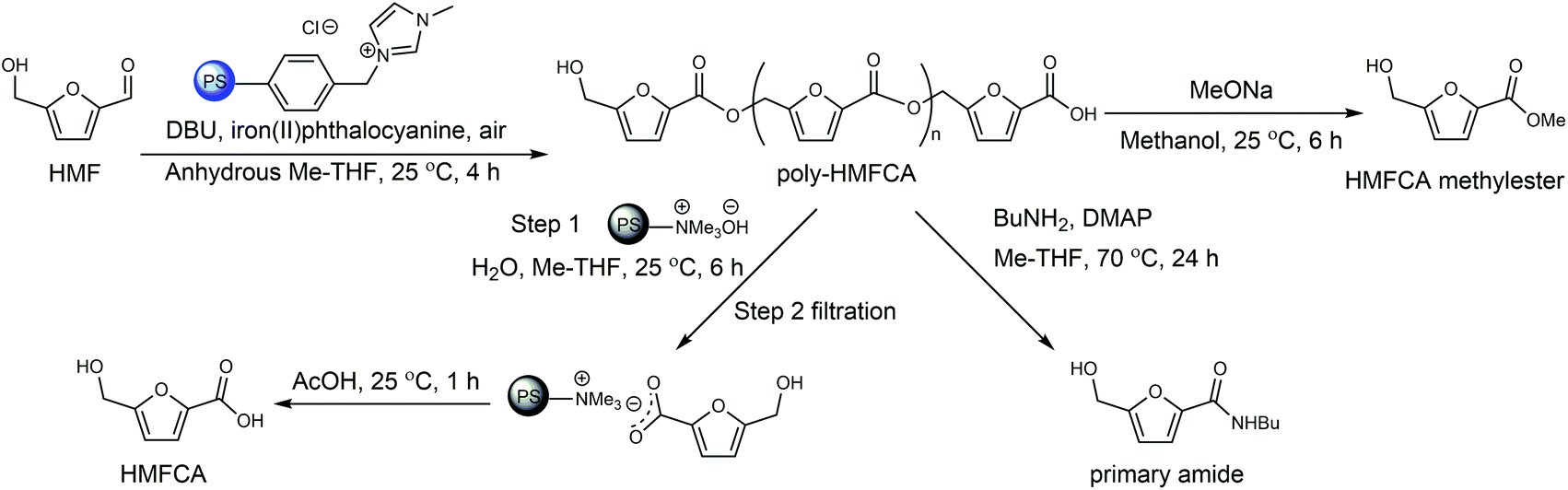 | ||
| Fig. 47 One-pot, two-step conversion of HMF toward HMFCA. Adapted from ref. 647. | ||
Owing to the specificity of enzymes, the highly selective oxidation of HMF to HMFCA can be achieved via biological methods. For example, recombining 3-succinoylsemialdehyde-pyridine dehydrogenase (SAPDH) from Comamonas testosteroni SC1588 into Escherichia coli cells enabled the efficient conversion of HMF to HMFCA (yield of 95%) at 50 °C for 5 h.648 Similarly, resting cells of Gluconobacter oxydans DSM 50049 were active to selectively oxidize HMF into HMFCA, obtaining an HMFCA selectivity of 100% with the HMF conversion of 94%.649
The oxidation of HMF to FDCA can be conducted via thermocatalysis, biocatalysis, electrocatalysis and photocatalysis, and readers can also consult the recent reviews.562,563,657–660 Metal oxides have been intensively investigated as catalysts for the selective oxidation of HMF to FDCA. For example, Hayashi et al. demonstrated that MnO2 can be used as a non-precious-metal catalyst for the selective oxidation of HMF to FDCA in NaHCO3 aqueous solution using oxygen as an oxidant.661 Accordingly, they further showed that the aerobic oxidation of HMF to FDCA over MnO2 depends strongly on its crystal structure and the local environment around the oxygen atoms using combined computational and experimental studies.583 It was observed that the catalytic activity correlates well with the calculated oxygen vacancy formation energy. The reaction rates per surface area for the rate-determining step (FFCA oxidation to FDCA) increased in the order of ε-MnO2 < δ-MnO2 < α-MnO2 ≈ γ-MnO2 < λ-MnO2 < β-MnO2. Moreover, high-surface-area β-MnO2 (β-MnO2-HS) nanoparticles583 and high-surface-area mesoporous β-MnO2 nanoparticles662 were successfully prepared via the simple calcination of the precursor prepared from NaMnO4 and MnSO4 at 400 °C and low-temperature crystallization of solution-derived Mn4+ precursor under template-free conditions, respectively, both affording an FDCA yield (>83%) higher than activated MnO2. Compared with individual MnO2 and Co3O4, the Co–Mn mixed oxide catalyst was more effective for the direct oxidation of HMF to FDCA owing to the enhanced lattice oxygen mobility and presence of different valence states of manganese.584,585 CeO2 and MgO mainly convert HMF to FFCA and formic acid, respectively, MgO·MnO2·CeO2 catalyzes the selective oxidation of HMF to DFF, while CuO·MnO2·CeO2 mainly promotes the oxidation of HMF to FDCA.592
Supported precious metals are also widely used for the selective oxidation of HMF to FDCA. Liao et al. reported that a manganese oxide-supported single-atom Pd (Pd–MnO2) catalyst exhibit an FDCA yield of 88% for the aerobic oxidation of HMF in aqueous solution, with an FDCA productivity (100.91 mmol h−1 gPd−1) much higher than that (45.57 mmol h−1 gPd−1) of Pd nanoparticles supported on MnO2 (PdNP/MnO2).586 Moreover, Pd–MnO2 showed good recyclability after five times reuse, while PdNP/MnO2 showed an obvious decrease in catalytic activity. The influence of the morphology of CeO2, including nanorod, cube and octahedra on the oxidation of HMF over the Au/CeO2 catalyst was investigated by Li et al.578 Au/CeO2-rod showed abundant oxygen vacancies, cationic gold, interfacial Lewis acidic sites and the highest catalytic activity for the selective conversion of HMF to FDCA. The improved reaction efficiency was attributed to the efficient activation of hydroxyl and aldehyde groups and molecular O2 owing to the synergistic effect of the interfacial Lewis acid, homogeneous base and neighboring Au particles. Yu et al. reported that Pt supported on activated porous carbon (Pt-PVB-ACS 800) was more active and durable than Pt supported on inactivated carbon (Pt-CS-800) for the basic additive-free aerobic oxidation of HMF to FDCA, mainly owing to its large surface area, high porosity and low density of oxygen groups.589 Nevertheless, the FDCA yield decreased from 99% to 79% after ten catalytic runs, owing to the aggregation of Pt nanoparticles. Schade et al. reported that Au/ZrO2 exhibited a high yield of FDCA (89%) for the aerobic oxidation of HMF at the evaluated temperature (125 °C) with a maximum productivity of 67 molFDCA h−1 molAu−1, while Ag/ZrO2 exhibited an HMFCA yield higher than 98% under mild conditions (25 °C) with a maximum productivity of 400 molHMFCA h−1 molAg−1.559 Their following study showed that the selective conversion of HMF to HMFCA proceeds through the dehydrogenation of the geminal diol by reduced Ag particles and the reduced Ag particles are released by removing hydrogen with oxygen in the catalytic cycle.663
In addition to precious metals, transition metals have also been investigated for the oxidation of HMF to FDCA. For example, cobalt nanoparticles (NPs) encapsulated in Mn/N-doped graphitic carbon (Co–Mn/N@C) were synthesized by co-pyrolyzing of Co/Mn-lignin complex and dicyandiamide.664 The Co–Mn/N@C catalyst afforded an FDCA yield of 98.8% in Na2CO3 aqueous solution under mild conditions (85 °C, 1 bar O2, 10 h).
The design of bimetallic catalysts is effective to improve the catalytic performance for the selective oxidation of HMF to FDCA.665 Xia et al. reported that bimetallic Pd–Au nanoparticles supported on Mg–Al hydrotalcite are highly effective to catalyze the aerobic oxidation of HMF to FDCA in an NaOH aqueous solution under mild conditions, attaining an FDCA yield of 90% at 60 °C within 6 h.558 In contrast, the monometallic Pd or Au catalyst exhibited a low FDCA yield with HMFCA as the main product since they were unable to further oxidize the intermediate HMFCA under this condition. The superior catalytic activity of the bimetallic catalyst mainly resulted from the electron transfer from Pd to Au, which could promote the adsorption and activation of HMF and then accelerate its oxidation. Zhang et al. reported that bimetallic AuPd and PtPd nanowire networks (NNWs) with abundant structural defects, which were prepared via a salt-mediated emulsion method, exhibited much a higher catalytic performance for the selective oxidation of HMF to FDCA in comparison with AuPd NPs, and core–shell and TiO2-supported AuPd NPs (AuPd/TiO2).581 The enhanced catalytic activity of the NNWs was ascribed to their good ability to generate radicals owing to the presence of abundant structural defects. Bonincontro et al. reported that nanosized NiO-supported Au–Pd nanoparticles (AuPd–nNiO) enabled the oxidation of HMF to FDCA under base-free conditions, attaining a relatively high yield of FDCA (70%) at 90 °C.582
The properties of the support have an important influence on the catalytic activity of metals for the oxidation of HMF. Megías-Sayago et al. investigated the role of acid sites in the conversion of HMF over gold catalysts.607 They observed that the FDCA yield from the oxidation of HMF over Au/CexZr1−xO2 correlates well with the shift in the CO–OH–M4+ bands in its FTIR spectrum using CO as a probe molecule, indicating that the improved surface hydroxyl acidity is beneficial for a higher FDCA yield. They proposed that the addition of an appropriate amount of Zr4+ improves the Brønsted acidity of the catalyst, and then the deprotonated Brønsted sites promote the formation of an alkoxy intermediate, thus improving the FDCA yield. Consequently, the Au/Ce25%Zr75%O2 catalyst with a relatively high hydrophilicity and Brønsted acidity gave the highest activity toward FDCA (80%). Ferraz et al. investigated the influence of basic sites on the support on the oxidation of HMF over PVA-stabilized Au catalysts (PVA-Au/xMgF2–(1 − x)MgO2) in aqueous solution under base-free conditions.608 The Au/MgO catalyst showed a superior performance (FDCA yield of 90%) compared to the other tested catalysts, including PVA-Au/MgF2–MgO2, PVA-Au/MgF2, Au/NiO, Au/TiO2 and Au/CeO2, indicating that the basicity of the support is beneficial for the conversion of HMF to FDCA. However, partial leaching of Mg (89 ppm) occurred under the reaction conditions. Donoeva et al. also demonstrated that the Au nanoparticles on a basic carbon support bearing basic functional groups exhibited better catalytic activity for the oxidation of HMF to FDCA and higher stability than Au nanoparticles supported on acidic carbon support since the positively charged surface groups promoted the adsorption of hydroxyl ions, which function as a cocatalyst to promote the gold-catalyzed dehydrogenation process.590 The influence of MgAlO, MgO, ZrO2, and CeO2 supports on the oxidation of HMF over a supported Ru catalyst in aqueous solution under base-free conditions was investigated by Antonyraj et al.605 Although MgAlO-supported Ru (Ru/MgAlO) exhibited the highest catalytic activity, it was deactivated rapidly in the recycling experiment. In contrast, the magnesium oxide (MgO)-supported Ru catalyst (Ru/MgO) showed good catalytic activity and reusability, affording an FDCA yield of 90% with the HMF conversion of 100%.
The construction of Fe3O4-based composited can impart magnetic property to the catalyst, which is beneficial for its recovery and reuse. Tirsoaga et al. reported that the magnetic-separable cationic Ru(III)–Fe3O4@SiO2 catalyst showed an FDCA yield of 74.2% with the HMF conversion of 92% in the medium of NaOH solution.596 Zhou et al. indicated that the Fe3O4-decorated reduced graphene oxide-supported platinum nanocatalyst (Pt/Fe3O4/rGO) was more active than Pt/rGO for the conversion of HMF to FDCA.666 At a low HMF loading (0.3 wt%), Pt/Fe3O4/rGO enabled the selective conversion of HMF to FDCA in pure water, obtaining an FDCA yield of 98% under mild conditions (95 °C, 8 h, 0.5 MPa O2). They demonstrated that the introduction of Fe3O4 not only promoted the dispersion of Pt, but also promoted the easy magnetic recycling of the catalyst.
Owing to its relatively low reaction rate, a long reaction time (4–24 h) is usually required for the conversion of HMF to FDCA. Ji et al. reported that the continuous control of pH coupled with microwave heating could remarkably accelerate the HMF oxidation rate, obtaining an FDCA yield of 99% within 30 min using 3 wt%Au/TiO2 and H2O2 as the catalyst and oxidant, respectively.613
The avoidance of using a homogeneous acid and base is very important for the actual production of FDCA. Guo et al. reported that Mn–Ce mixed oxide-supported Ru nanoparticles (Ru/Mn6Ce1OY) are highly effective and stable for the aerobic oxidation of HMF to FDCA, affording an FDCA yield higher than 99% in the medium of water with productivity as high as 5.3 molFDCA molRu−1 h−1 in the absence of any base additive.593 The superior catalytic performance was mainly attributed to the well-dispersed metallic Ru nanoparticles and the improved availability and mobility of active oxygen species owing to the high surface concentration of Mn4+ and Ce3+, and the good stability of catalyst was owing to the strong metal–support interaction. Ke et al. reported that the nitrogen-doped carbon-decorated CeO2 supported Pt catalyst (Pt/NC-CeO2) enabled an FDCA yield of 100% in water without the use of a base additive.594
The selective oxidation of HMF toward FDCA can also be performed in a packed bed reactor. Danielli et al. demonstrated the feasibility of using a trickle-bed reactor to convert HMF to FDCA, obtaining an FDCA yield of 98% over Ru/Al2O3 in Na2CO3 solution at 140 °C (GHSV = 900 h−1, WHSV = 1 h−1, 30 bar O2).606 After reactivation via simple hydrothermal treatment, the activity of the Ru/Al2O3 catalyst could be retained for 12 cycles. The oxidation of HMF to FDCA was performed over a heterogenous resin-supported Pt catalyst, affording an FDCA yield of 99% continuously in neat water under continuous flow and base-free conditions (120 °C, residence time of 303 s, O2 flow rate of 1.2 mL min−1 and 7.7 bar O2 pressure).667 In this catalytic system, the FDCA product was obtained with a high space-time-yield of 46.0 g L−1 h−1 without acid/base treatment or purification.
Besides O2 and air, other oxidants have also been tested for the selective oxidation of HMF to FDCA. For example, Ren et al. reported that CuO and Co3O4 exhibit high activity for the oxidation of HMF to FDCA when using NaClO as an oxidant.572 Hazra et al. reported that the use of a metal-free I2/NaOH catalyst enabled the 10 gram-scale synthesis of FDCA from HMF using tert-butyl hydroperoxide (TBHP) as the oxidant.571 Tirsoaga et al. reported that the selective oxidation of HMF over a multi-functional magnetic nano-composite (10Co@22Nb@MNP) catalyst using TBHP as the oxidant resulted in an FDCA selectivity of 96.5% with the HMF conversion of 96.9%.614 However, although these oxidants are more powerful than O2 and air, their large-scale application is limited by their high cost and secondary pollution. Besides, the selective oxidative esterification of HMF in methanol could give furan-2,5-dimethylcarboxylate (FDMC), which is a promising alternative method for the conversion of HMF to FDCA. This reaction will be discussed in detail in section 3.9.
The direct use of the HMF product solution as feedstock reduces the separation and purification steps and then intensifies the process chain of FDCA production. Naim et al. investigated the influence of the HMF synthesis by-products, including unconverted sugars, levulinic acid, formic acid and remaining inorganics on the oxidation of HMF to FDCA over Au/ZrO2 in aqueous medium.668 They found that levulinic acid led to the severe degradation of the formed FDCA due to the poisoning of the catalyst, while other substances decreased the FDCA yield to a lower extent. Therefore, avoiding rehydration of HMF to levulinic acid during the production of HMF is crucial for the subsequent conversion of HMF to FDCA. In a realistic technical scenario, an FDCA yield of up to 74% has been achieved when using impure HMF obtained from unconcentrated sugar syrup as the feedstock.
As discussed in section 3.2.1, the photocatalytic oxidation of HMF not only gives DFF as the main product, but also enables the simultaneous production of H2via water splitting. Also, similar to the thermocatalytic process, the use of a basic reaction medium is an effective strategy to promote the oxidation of HMF toward FDCA. For example, the Ni/CdS catalyst could convert HMF and furfural to their corresponding carboxylates completely in NaOH solution, but in neutral aqueous solution the Ni/CdS catalyst only converted HMF to DFF.645 Xu et al. designed a highly effective photocatalyst by dispersing cobalt thioporphyrazine (CoPz) onto g-C3N4 (abbreviated as CoPz/g-C3N4), which exhibited superior photocatalytic activity for the selective oxidation of HMF toward DFF at low pH (4.01) or FDCA (96.1% yield) at high pH (9.18) under simulated sunlight using air as a benign oxidant. They demonstrated that the poor catalytic performance of single g-C3N4 is owing to the generation of hydroxyl radicals, which could mineralize organics to CO2 and H2O, while the strong interaction between CoPz and g-C3N4 not only inhibited the generation of hydroxyl radicals over g-C3N4, but also accelerated the formation of singlet oxygen (1O2) over CoPz sites, thus improving the catalytic performance remarkably.642 Similarly, a cross-linked conjugated polymer photosensitizer exhibited strong 1O2 generation ability under sunlight and good catalytic performance for the selective oxidation of HMF to FDCA.669,670
Electrochemical oxidation enables the selective oxidation of HMF to FDCA with high yields. Taitt et al. investigated the electrochemical oxidation of HMF to FDCA in 0.1 M KOH solution using NiOOH, CoOOH, and FeOOH as electrodes.567 They found that NiOOH gave an FDCA yield of 96.0% at 1.47 V vs. RHE, while CoOOH and FeOOH were not active for this reaction. The integration of H2 evolution with the oxidation of HMF (Fig. 48) not only avoided the production of the explosive H2/O2 mixture by decoupling the H2 evolution reaction (HER) and O2 evolution reaction (OER), but also gave value-added FDCA and H2 products with a high energy conversion efficiency.671–674 For example, You et al. reported that the use of hierarchically porous Ni3S2/Ni foam as a bifunctional electrocatalyst could reduce the cell voltage by ∼200 mV for integrated H2 production and HMF oxidation to achieve 100 mA cm−2, compared with the pure water splitting, with nearly unity Faradaic efficiency.672 Cha et al. reported that performing the oxidation of HMF at the cathode of a typical hydrogen-producing photoelectrochemical cell (PEC) under AM 1.5 G illumination with TEMPO as the mediator enabled the simultaneous production of H2 and FDCA, with near-quantitative FDCA yield and 100% Faradaic efficiency.671 Li et al. reported that the use of Na4[Fe(CN)6] as a proton-independent electron reservoir is an effective approach to decouple water splitting and organic upgrading, thus enabling efficient HER with the oxidation of HMF to FDCA driven either by electricity or a solar cell under sunlight irradiation with the advantages of great flexibility and safety.673
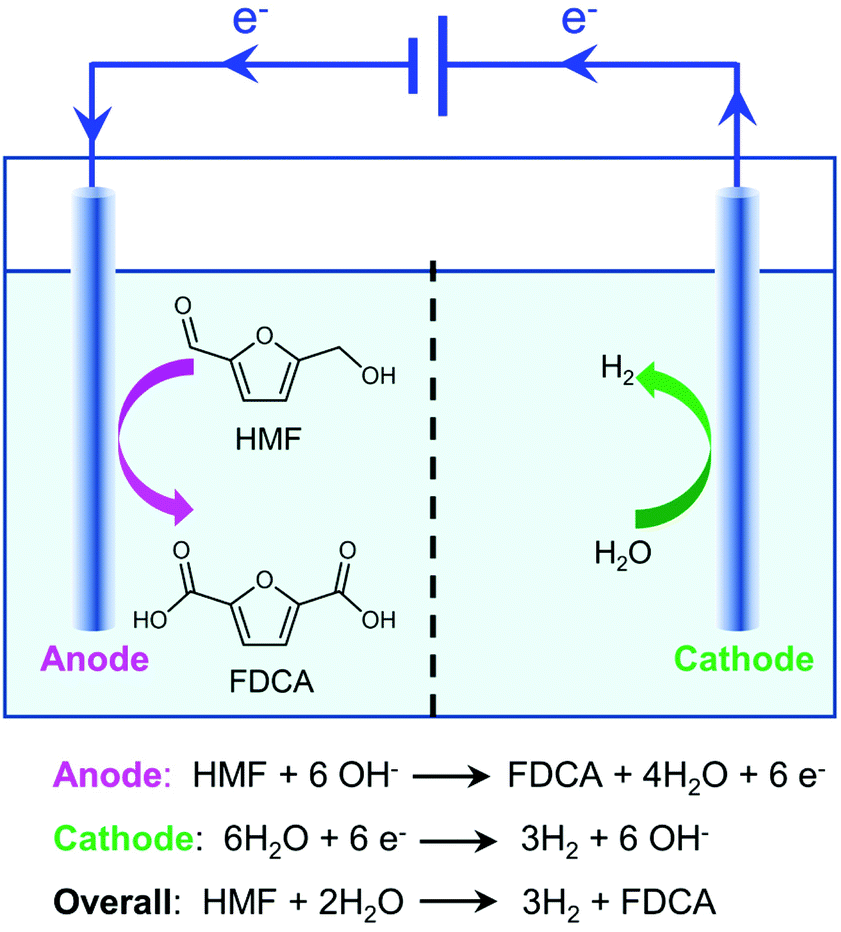 | ||
| Fig. 48 Electrochemical cell for the integration of H2 evolution with the oxidation of HMF to FDCA. Adapted from ref. 672 and 674. | ||
The direct conversion of carbohydrates to FDCA via HMF as an intermediate has also been achieved in recent studies. Motagamwala et al. reported the efficient conversion of high concentration fructose to FDCA in the medium of the GVL/H2O solvent system via a one-pot, two-step process.675 Fructose was converted to HMF at high yield (70%) in the solvent system using FDCA as an acid catalyst. The use of FDCA to replace the corrosive HCl as the catalyst improves the process economics and environmental benefits by eliminating the ion exchange operation. The high solubility of FDCA in the solvent system enabled the effective oxidation of HMF over the Pt/C catalyst at a high HMF concentration (7.5 wt%), in the absence of a homogeneous base. Moreover, the FDCA product with a purity of greater than 99% could be obtained from the solvent system via crystallization. Chen et al. reported that the one-pot, two-step process involving the dehydration of fructose to HMF over Amberlyst-15 in DMSO and oxidation of HMF to FDCA over Pt/C in the H2O(K2CO3)/DMSO medium gave an overall FDCA yield of up to 88.4% from fructose under relatively mild conditions.429 The combination of ionic liquids and heteropoly acids is not only effective for the base-free oxidation of HMF to FDCA, but also enables the one-pot conversion of carbohydrates to HMF.588 Among the tested heteropoly acids and ionic liquids, the combination of HPMV6 (HPM = H3PMo12O40) and BMIMCl gave the highest FDCA yield of 88.8% from HMF. The high solubility of FDCA in BMIMCl is beneficial for the inhibition of the oxidative cleavage of the furan ring. The use of fructose, glucose and cellulose as starting materials gave FDCA yields of 30.7%, 48.6% and 6%, respectively.
Biocatalysis, including enzymatic catalysis (in vitro) and whole-cell catalysis (in vivo), is a promising approach to convert HMF to FDCA.562,660 Biocatalysis can transform HMF to DFF, HMFCA or FFCA efficiently, but the selective enzymatic oxidation of HMF to FDCA is relatively challenging.676–679 Whole cells can transform HMF to FDCA with endogenously produced cofactors without using expensive stoichiometric reagents, but they also catabolize FDCA with the generation of many byproducts.660 In contrast, isolated enzymes allow better control of the reaction pathway and facile product recovery, but complex cofactors, mediators, immobilization, mutation, and cooperation with other enzymes are required to achieve the efficient conversion of HMF to FDCA. Daou et al. reported that the combination of glyoxal oxidase (PciGLOX) isoenzymes obtained from Pycnoporus cinnabarinus and aryl alcohol oxidase (UmaAAO) could enable the cascade oxidation of HMF toward FDCA.680 To avoid the accumulation of HMFCA, HMF was firstly oxidized to DFF and further to FFCA by UmaAAO for 2 h, followed by oxidation with PciGLOX3 in the presence of catalase for 24 h, attaining the maximum FDCA yield of 16%. Yuan et al. demonstrated that selective biocatalytic conversion of HMF to FDCA could be achieved via manipulating the key genes responsible for FDCA synthesis in Raoultella ornithinolytica BF60.681,682 The overexpression of the AldH gene, which is responsible for the oxidation of FFCA to FDCA, combined with depleting adhP3 and alkR genes responsible for the reduction of HMF to BHMF led to an FDCA yield of up to 96.2%. Wang et al. reported that the co-expression of vanillin dehydrogenase (VDH1) and HMF/furfural oxidoreductase (HmfH) in Escherichia coli enabled the cascade catalytic oxidation of HMF to FDCA without the use of an additional sacrificial substrate, attaining an FDCA yield of 96% at an HMF concentration of 150 mM with a productivity of around 0.4 g L−1 h−1.683
The life cycle assessment (LCA) of PEF plastic production from starchy biomass, which involved the conversion of maize grain to fructose via a well-established industrial process, conversion of fructose to HMF in DMA/LiBr using H2SO4 as the catalyst, conversion of HMF to FDCA by KMnO4 in NaOH aqueous solution and polymerization of FDCA with ethylene glycol to PEF, indicated that the predominant environmental impacts (38–49%) are generated at the stage of the conversion of FDCA to polymer owing to the electricity consumption and use of non-renewable chemicals such as dichloromethane.684 Based on LCA, Bello et al. suggested that the search for environmentally friendly substances and the reduction of energy consumption for the production of FDCA from lignocellulosic biomass are necessary to further improve the environmental sustainability.685 Byun et al. suggested that the coproduction of bioethanol with FDCA is a more promising pathway to improve the economic feasibility of the biorefinery process compared with the co-production of ethanol with other products, including adipic acid, caprolactam, pentanediol and phthalic anhydride due to the high output and high value of FDCA.686 The replacement of 25% of ethanol with FDCA produced 15.3–16.7 MJ of FDCA per gasoline gallon equivalent (GGE) of ethanol, resulting in an economic mitigation potential of up to US$2.40–2.48 per GGE.
The carboxylation of furfural is an alternative approach to obtain FDCA from biomass. Banerjee et al. reported that Cs2CO3 molten salts promoted the carboxylation of C–H in 2-furoic acid (Fig. 49a) with CO2, leading to the formation of carboxylates, which could be readily converted to FDCA via protonation.651 In this system, Cs2CO3 molten salts promoted the deprotonation of C–H bonds by CO32− to form carbon-centered nucleophiles followed by reaction with CO2 to generate carboxylates. Similarly, the carbonylation of furfural-derived 5-bromo-furoic acid, which was obtained via the bromination of furoic acid (Fig. 49b), with CO in aqueous solution over a palladium catalyst gave an FDCA yield of up to 98%.687
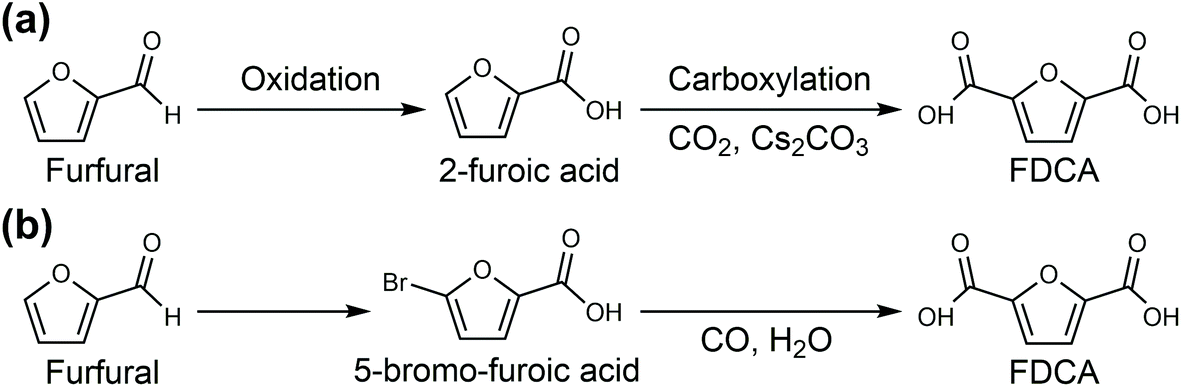 | ||
| Fig. 49 Conversion of furfural to FDCA via 2-furoic acid (a) and 5-bromo-furoic acid (b) as the intermediate. Adapted from ref. 651 and 687. | ||
The feasibility of converting HMF to PTA (Fig. 50) was proven by Pacheco et al.688 The oxidized derivatives of HMF were reacted with ethylene over solid Lewis acid catalysts that do not contain strong Brønsted acids to synthesize precursors of PTA and its equally important diester, dimethyl terephthalate (DMT). The Diels–Alder reaction of HMFCA with high pressure ethylene over Sn-Beta zeolite generated 4-(hydroxymethyl)benzoic acid (HMBA), attaining an HMBA selectivity of 31% with the HMFA conversion of 61% at 190 °C for 6 h. When HMFCA was protected with methanol to form methyl 5-(methoxymethyl) furan-2-carboxylate (MMFC), MMFC could be converted to methyl 4-(methoxymethyl) benzenecarboxylate (MMBC). HMBA and MMBC can be used for the production of PTA and DMT via oxidation, respectively.
 | ||
| Fig. 50 Conversion of HMF to PTA via oxidation and Diels-Alder/dehydration reaction. Adapted from ref. 688. | ||
 | ||
| Fig. 51 Oxidation of HMF toward succinic acid and maleic acid. Adapted from ref. 596. | ||
In addition to the oxidation of HMF, selective oxidation technologies have also been widely used for the conversion of other biomass-derived platform molecules to value-added chemicals. For instance, the oxidation of glucose can produce gluconic acid, gluconates, glucaric acid and formic acid, and the selective oxidation of levulinic acid and furfural can generate maleic acid and anhydride, succinic acid and furanones. These processes have been extensively reviewed, and the reader is referred to these sources.4,34,560,566,691
3.3. Hydrodeoxygenation of HMF
The hydrodeoxygenation of HMF can give a wide variety of products, including 2,5-bishydroxymethylfuran (BHMF), 2,5-bishydroxymethyltetrahydrofuran (BHMTHF), 1,6-hexanediol (1,6-HD), 2,5-dimethylfuran (DMF) and alkanes. Since the hydrodeoxygenation of HMF involves the tailoring of the aldehyde group, hydroxy group and furan ring, complicated and multiple reaction networks may occur during the conversion of HMF. The reaction pathway and product selectivity of HMF hydrodeoxygenation are influenced by the support, metals, additives, solvents, hydrogen donors and reaction conditions (Fig. 52 and Table 15). Here, we summarize the recent advances in HMF hydrodeoxygenation toward valuable products with a focus on identifying the major factors determining the reaction pathway and product selectivity. For a comprehensive overview in this field, readers can also refer to the previous dedicated review articles.1,692–694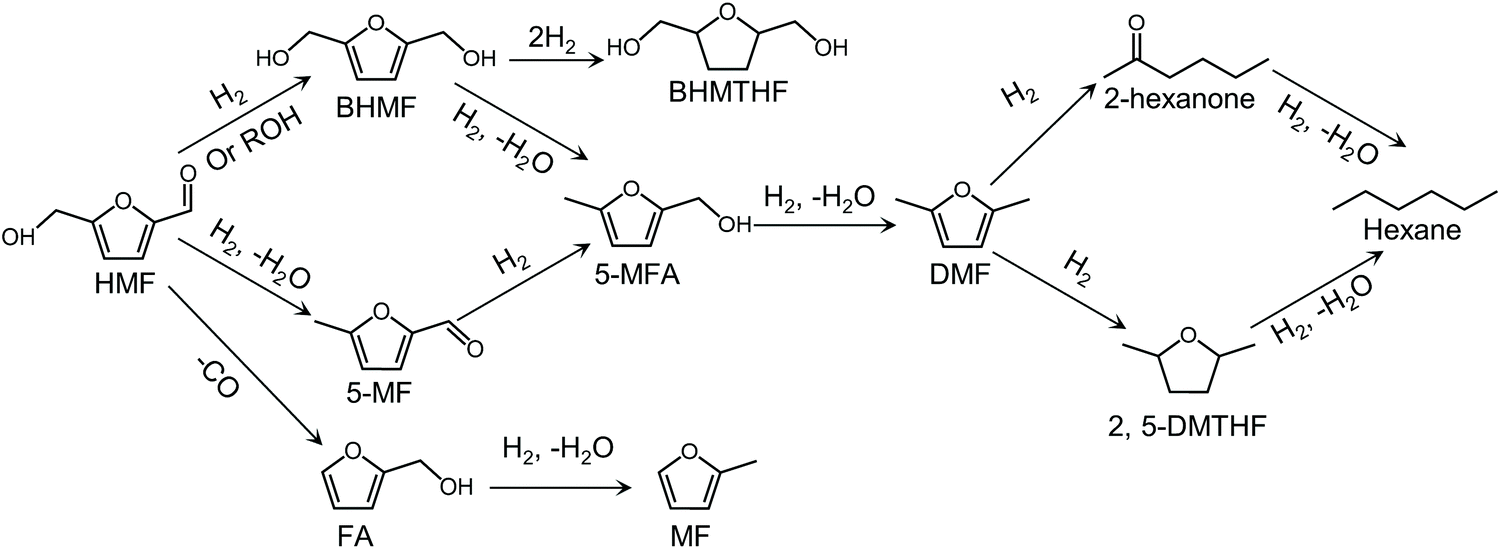 | ||
| Fig. 52 Main reaction pathway for the hydrodeoxygenation of HMF to value-added chemicals. Adapted from ref. 695. | ||
| Catalyst | Catalyst loading | Solvent | Reaction conditions | Feedstock loadinga | Conversion | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. b Relative to HMF. c The reaction was conducted in a fixed reactor. — Not provided. | |||||||
| ZrBa-SBA | 1 wt%a | Isopropanol | 150 °C, 2.5 h | HMF, 1 wt% | 98.3% | BHMF, 96.8% | 696 |
| Zr-Benzylphosphonate | 1.4 wt%a | Isopropanol | 120 °C, 2 h | HMF, 2.5 wt% | >99% | BHMF, 93% | 697 |
| Hf-DTMP | 1 wt%a | 2-Butanol | 130 °C, 4 h | HMF, 2 wt% | 99.1% | BHMF, 96.8% | 698 |
| Hf-lignosulfonate | 1 wt%a | Isopropanol | 100 °C, 2 h | HMF, 1.26 wt% | ∼98% | BHMF, 90% | 699 |
| Hf-lignosulfonate | 1 wt%a | Isopropanol | 120 °C, 10 h | HMF, 1.26 wt% | ∼98% | BHMF, 90% | 699 |
| Ir/SiO2 | 1 mol%b | THF | 60 °C, 5 h, 10 Bar H2 | HMF, — | 70% | BHMF, 58.1% | 700 |
| Ir/SiO2(Cl) | 1 mol%b | THF | 60 °C, 5 h, 10 Bar H2 | HMF, — | 97% | BHMF, 97% | 700 |
| Ir/SiO2, H2SO4 | 1 mol%b | THF | 60 °C, 2 h, 10 Bar H2 | HMF, — | 78% | BHMF 58.1%; DMF 13.3% | 700 |
| Ir/SiO2(Cl), H2SO4 | 1 mol%b | THF | 60 °C, 2 h, 10 Bar H2 | HMF, — | 74% | BHMF 38.5%; DMF 17.8% | 700 |
| NiBi IMCs | 3.3 mM Nia | Isopropanol | 100 °C, 8 h, 2 MPa H2 | HMF, v/v = 1/30a | 100% | BHMF 97.9% | 701 |
| MZCCP | 0.8 wt% | 2-Butanol | 140 °C, 5 h | HMF, 2 wt% | 98.7% | BHMF, 93.4% | 702 |
| MZCCP, 0.8 wt% | 0.8 wt%a | 2-Butanol | 140 °C, 5 h | Fructose, 2 wt% | 98.7% | BHMF, 93.4% | 702 |
| Ni-12Cu/SBA-15 | 1 wt%a | THF | 220 °C, 5 h, formic acid | FMF, 1.8% | 100% | DMF, 71% | 703 |
| Ni-12Cu/SBA-15 | 1 wt%a | THF | 220 °C, 5 h, formic acid | HMF, 1.5% | 100% | DMF, 60.5% | 703 |
| Co/ZrLa0.2Ox | 0.5 wt%a | Water | 40 °C, 10 h, 2 MPa H2 | HMF, 1 wt% | 100% | BHMF, 100% | 704 |
| RuCu@NFC | 0.45 wt%a | Isopropanol | 210 °C, 12 h, isopropanol | HMF, 0.75 wt% | 97.0% | BHMF, 88.8% | 705 |
| Mn-NCA | 1.1 wt%a | Isopropanol | 160 °C, 1.5 h, isopropanol | HMF, 1.3 wt% | 91% | BHMF, 76% | 706 |
| MnO@C–N | 0.5 wt%a | Isopropanol | 170 °C, 1.5 h, isopropanol | HMF, 1.3 wt% | ∼100% | BHMF, 76% | 707 |
| Cu(50)-SiO2 | 0.5 wt%a | 1-Butanol | 90 °C, 1.5 MPa H2, 5 h | HMF, 12 wt% | 100% | BHMF, 97% | 708 |
| NC-Cu/MgAlO | 1 wt%a | Cyclohexanol | 220 °C, 1.5 MPa H2, 0.5 h | HMF, 5 wt% | 100% | DMF, 96.1% | 709 |
| NC-Cu/MgAlO | 1 wt%a | Cyclohexanol | 220 °C, 1.5 MPa H2, 3.5 h | HMF, 5 wt% | 100% | DMTHF, 94.6% | 709 |
| 1.5K-Cu/Al2O3 | 5 gc | Ethanol | 120 °C, 2 MPa H2, WHSV of 1.0 h−1 | HMF, 3 wt% | 99.2% | BHMF, 98.9% | 710 |
| 15CuZr | 5 gc | 1-Butanol | 200 °C, 1.5 MPa H2, WHSV of 0.15 h−1 for 4 h | HMF | 100% | DMF, 30% | 235 |
| 1Ru15CuZr | 5 gc | 1-Butanol | 200 °C, 1.5 MPa H2, WHSV of 0.15 h−1 for 4 h | HMF | 100% | DMF, 45.6% | 235 |
| 1Ru15CuZr | 5 gc | 1-Butanol | 200 °C, 1.5 MPa H2, WHSV of 0.15 h−1 for 4 h | Glucose-derived crude HMF | 77.6% | DMF, 16.4% | 235 |
| K2CO3 | 1.4 wt%a | Me-THF | 25 °C, 2 h, Ph2SiH2 | HMF, 3.2 wt% | 100% | BHMF, 94% | 711 |
| Cu/Al2O3 | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | 100% | DMF, 73.9% | 712 |
| Cu/ZnO | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | 100% | DMF, 59.7% (5-MF 1.5%) | 712 |
| Cu/ZrO2 | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | 100% | DMF, 24.9% | 712 |
| Cu/CeO2 | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | 100% | DMF, 23.6% | 712 |
| Reused Cu/Al2O3 | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | — | DMF, 0% | 712 |
| Reused Cu/ZnO | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | — | DMF, 3.9% | 712 |
| Reused Cu/ZrO2 | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | — | DMF, 3.0% | 712 |
| Reused Cu/CeO2 | 1.4 wt%a | Methanol | 240 °C, 6 h, methanol | HMF, 1.4 wt% | — | DMF, 21% | 712 |
| Cu–Co/Al2O3 | — | H2O | 220 °C, 6 h, 3 MPa H2 | HMF, 0.2 wt% | — | DMF, 87% | 713 |
| Ru/Co3O4 | 1 wt%a | THF | 130 °C, 24 h, 0.7 MPa H2 | HMF, 2.5 wt% | >99% | DMF, 93.4% | 714 |
| Co/rGO | 0.25 wt%a | Ethanol | 200 °C, 1 h, 2 MPa H2 | HMF, 2.5 wt% | 100% | DMF, 94.1% | 715 |
| Co-CoOx | 0.25 wt%a | 1,4-Dioxane | 170 °C, 12 h, 1 MPa H2 | HMF, 1.25 wt% | ∼100% | DMF, 83.3% | 716 |
| 11.8%Co-(ZnO-ZnAl2O4) | 10 wt%a | THF | 130 °C, 24 h, 7 bar H2 | HMF, 2.5 wt% | >99.9% | DMF, 74.2% (polymerization, 22.9%) | 717 |
| CuZnCoOx | 0.43 wt%a | Ethanol | 200 °C, 5 h | HMF, 2.5 wt% | 100% | DMF, 99% | 718 |
| Pd-HCl/C | 0.5 wt%a | THF | 80 °C, 24 h, 2 MPa H2 | HMF, 6.3 wt% | 33.0% | DMF, 33.0% | 719 |
| Pd-AA/C | 0.5 wt%a | THF | 80 °C, 24 h, 2 MPa H2 | HMF, 6.3 wt% | 72.8% | DMF, 72.5% | 719 |
| Pd-EA/C | 0.5 wt%a | THF | 80 °C, 24 h, 2 MPa H2 | HMF, 6.3 wt% | 96.9% | DMF, 93.0% | 719 |
| Pd-GVL/C | 0.5 wt%a | THF | 80 °C, 24 h, 2 MPa H2 | HMF, 6.3 wt% | >99% | DMF, 95.6% | 719 |
| Commercial Pd/C | 0.5 wt%a | THF | 80 °C, 24 h, 2 MPa H2 | HMF, 6.3 wt% | 24.9% | DMF, 13.6% | 719 |
| CuNi | — | 0.2 M sulfate solution | −0.8 V vs. Ag/AgCl, 150 C | HMF, 0.2 g/L | FF 88.0% | DMF 91.1% | 720 |
| Fe–Pd/C | 1 wt%a | THF | 150 °C, 2 h, 2 MPa H2 | HMF, 1.3 wt% | 100% | DMF, 85% | 721 |
| Ru/C-SiO2@m-SiO2 | 0.19 wt%a | THF | 140 °C, 2 h, 1 MPa H2 | HMF, 0.67 wt% | 91% | DMF, 89% | 722 |
| Ru/C-SBA-15 | 0.19 wt%a | THF | 140 °C, 2 h, 1 MPa H2 | HMF, 0.67 wt% | 93% | DMF, 71% | 722 |
| Ru/C-commercial | 0.19 wt%a | THF | 140 °C, 2 h, 1 MPa H2 | HMF, 0.67 wt% | 86% | DMF, 49% | 722 |
| PtCo/GC | 1 wt%a | Ethanol | 180 °C, 2 h, 1 MPa H2 | HMF, 5 wt% | — | DMF, 88% | 723 |
| PtCo/GC | 1 wt%a | Toluene | 180 °C, 2 h, 1 MPa H2 | HMF, 5 wt% | — | DMF, 92% | 723 |
| PtCo/GC | 1 wt%a | RON 95 gasoline | 180 °C, 2 h, 1 MPa H2 | HMF, 5 wt% | — | DMF, 95% | 723 |
| PtCo/GC | 0.8 wt%a | RON 95 gasoline | 180 °C, 2 h, 2.5 MPa H2 | HMF, 10 wt% | — | DMF, 78% | 723 |
| PtCo/GC (4 runs) | 0.4 wt%a | E10 gasoline | 185 °C, 2 h, 2.5 MPa H2 | HMF, 10 wt% | — | DMF, 5% | 723 |
| PtCo/GC | 0.8 wt%a | RON 95 gasoline | 180 °C, 2 h, 2.5 MPa H2 | HMF, 15 wt% | — | DMF, 19% | 723 |
| Pt–Co/MWCNTs | 0.3 wt%a | 1-Butanol | 160 °C, 8 h, 1 MPa H2 | HMF, 1.7 wt% | 100% | DMF 92.3% | 724 |
| RuCo/CoOx | 1 wt%a | 4-Dioxane | 200 °C, 2 h, 0.5 MPa H2 | HMF, 5 wt% | 100% | DMF, 96.5% | 725 |
| 5Ni-7MoS2/mAl2O3 | 0.5 wt%a | Isopropanol | 130 °C, 6 h, 1 MPa H2 | HMF, 1.3 wt% | — | DMF, 95% | 726 |
Illuminating the roles of single atoms, nanoparticles and supports in the hydrodeoxygenation process is of great importance for the construction of highly effective catalysts. Kuai et al. reported that single-atom palladium (Pd1) and Pd nanoparticles (PdNPs) supported on TiO2 worked synergistically for the hydrogenation of ketones and aldehydes to alcohols at room temperature, where PdNPs mainly catalyzed the dissociation of H2 molecules to H atoms, while Pd1 was responsible for the activation of the CO group.728 Owing to the rapid development of single-atom-based materials, this discovery may greatly facilitate the development of superior catalysts for the selective hydrogenation of biomass-derived ketone/aldehydes.
In addition to the hydrogenation of HMF to BHMF using an external hydrogen donor, BHMF can also be obtained via the selective catalytic transfer hydrogenation (CTH) of HMF with alcohol via the Meerwein–Ponndorf–Verley (MPV) reduction reaction (Fig. 53), where alcohols such as 2-butanol or isopropanol function as both the hydrogen donor and solvent.34,698
 | ||
| Fig. 53 Conversion of HMF to BHMF in the medium of alcohol via the MPV reaction. Adapted from ref. 698. | ||
The Lewis acid and base sites are active for the CTH reaction of HMF, while the Brønsted acid sites can catalyze the etherification of HMF or BHMF with alcohol. Zhou et al. designed a monolithic MnOx/N-doped carbon aerogel material (Mn-NCA) as a catalyst for the catalytic transfer hydrogenation of HMF to BHMF, affording a BHMF yield of 76% under mild conditions without agitation.706 They demonstrated that the high catalytic activity of Mn-NCA is attributed to the synergy between the uniformly dispersed MnOx nanoparticles (NPs) and the basic sites resulting from urea and the monolithic three-dimensional hierarchical porous architecture. Owing to its monolithic feature, Mn-NCA could be readily recovered by tweezers and then reused for the catalytic reaction with slight loss in its catalytic performance. Feng et al. also demonstrated that the superior catalytic performance of N-doped carbon-supported MnO catalyst (MnO@C–N) for the CTH reaction is attributed to the MnO and nitrogen-doping since the N species can reduce the activation energy of aldehyde conversion and the energy barrier of acetone desorption.707
A hafnium-based metal–organic coordination polymer catalyst (Hf-DTMP), which was prepared using hafnium tetrachloride (HfCl4) and diethylene triaminepenta(methylene phosphonic acid) (DTMP), exhibited strong acid–base bifunctionality and excellent catalytic activity for the transfer hydrogenation of HMF with 2-butanol, attaining a BHMF yield of up to 96.8%.698 The blocking of both the Lewis acid and base sites led to a remarkable decrease in the BHMF yield, indicating the synergy of the Lewis acid and base sites in the catalytic transfer hydrogenation process. A zirconium-based organic–inorganic coordination polymer (MZCCP) with magnetic property and acid–base bifunctionality, which was synthesized via the reaction of ZrCl4 with cyanuric acid (CA) over the surface of Fe3O4, exhibited a BHMF yield of 93.4% for the CTH reaction of HMF with 2-butanol.702 The reuse of the MZCCP catalyst could be readily achieved by separating it with an external magnet without obvious decrease in its catalytic activity. Wei et al. demonstrated that the introduction of BaO to Zr-SBA could block most of the Brønsted acid sites but retain the Lewis acid sites, which could suppress the further etherification of HMF and BHMF, thus enabling the highly efficient conversion of HMF to BHMF over the ZrBa-SBA catalyst.696 The Hf–lignosulfonate (Hf–LigS) nanohybrids, which were prepared from lignosulfonate with Hf4+via a hydrothermal self-assembly method, exhibited strong Lewis acid–base couple sites and moderate Brønsted acidic sites.699 The Hf–LigS catalyst was not only active for the catalytic transfer hydrogenation of HMF with 2-propanol to BHMF (yield: 90%) under mild conditions (100 °C, 2 h), but also enabled the one-step reductive etherification toward 5-[(1-methylethoxy)methyl]-2-furanmethanol (MEFA) (Fig. 54), affording an MEFA yield of up to 96.4% at 120 °C in 8 h. The CTH reaction led to the stoichiometric consumption of alcohols. Besides, the H2 generated from the in situ decomposition of alcohols can also be used as a hydrogen source for the conversion of HMF to BHMF and the hydrodeoxygenation of HMF to 2,5-dimethylfuran (DMF), as will be discussed in detail in section 3.3.2.
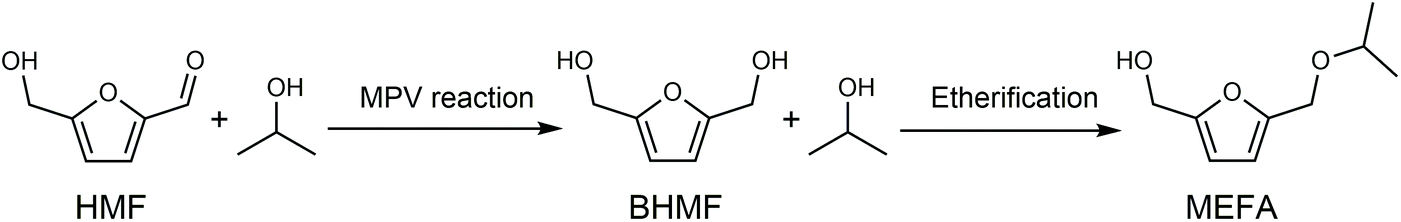 | ||
| Fig. 54 One-pot reductive etherification of HMF with 2-propanol toward MEFA over Hf–LigS. Adapted from ref. 699. | ||
In the presence of an external H2 donor, the selective hydrogenation of HMF toward BHMF can be conducted over supported metal catalysts using only alcohols as the reaction medium. For example, in the presence of external H2 (1.5 MPa), the Cu(50)–SiO2 nanocomposite exhibited both high catalytic activity and stability for the hydrogeneration of HMF to BHMF in the medium of 1-butanol, attaining a BHMF yield of greater than 97% even at an HMF loading as high as 12 wt%.708 Yu showed that the steric hindering effect in NiBi intermetallic compounds (IMCs) due to their well-organized surface atomic arrangement favors the vertical adsorption configuration of unsaturated aldehyde, and then promotes the selective hydrogenation of CO group without converting the CC group in the furan ring, leading to a BHMF yield of 97.9% from HMF.701
Transition metal Co-based catalysts have also been widely studied for the conversion of HMF to DMF. Yang et al. reported that a Co-graphene nanomaterial (Co/rGO), which was prepared via a feasible impregnation–calcination method, was active for the hydrogenation of HMF to DMF, affording a DMF yield of up to 94.1%.715 The excellent catalytic performance was attributed to its superior CO/C–O hydrogenolysis ability and suppressed CC/C–C session ability owing to the synergistic effect of single Co atoms, Co clusters, CoOx nanoparticles and reduced graphene oxide. A bifunctional Co–CoOx catalyst containing both metallic Co and acidic CoOx, which was synthesized via the controlled reduction of Co3O4 under an H2 flow, afforded the selective hydrogenolysis of HMF to DMF with a high yield (83%).716 The excellent catalytic performance was attributed to the synergistic effect of the metallic Co and acidic CoOx sites, where metallic Co functioned as the hydrogenation sites and the Lewis acidic CoOx sites were effective to activate the C–O bonds to accelerate the hydrodeoxygenation process. An et al. reported that the 11.8%Co–(ZnO–ZnAl2O4) catalyst derived from a layered double hydroxide was effective to catalyze the selective hydrogenation of HMF to DMF, affording a DMF yield of 74.2% at 130 °C.717 Gao et al. designed a dandelion-like cobalt oxide microsphere-supported bimetallic catalyst (RuCo/CoOx) for the hydrogenolysis of HMF to DMF, attaining a DMF yield of 96.5% using an HMF/Ru molar ratio of up to 252.7.725 They attributed the superior hydrogenolysis performance to the synergy between the bimetallic RuCo NPs, abundant surface defects, hydrogen spillover effect and dandelion-like superstructure of the catalyst. The strong interaction between the RuCo NPs and the CoOx support imparted excellent stability to the RuCo/CoOx catalyst.
Single-atom catalysts have also been investigated for the selective hydrodeoxygenation of HMF to DMF. For example, Gan et al. reported that the Co-alloyed Pt (Pt1/Co) single-atom alloy catalyst, which was synthesized by a scalable ball milling method on the kilogram level, afforded a DMF yield of 92.9% with the HMF conversion of 100% under 1.0 MPa H2 at 180 °C for 2 h.730 Moreover, the Pt1/Co single-atom alloy catalyst showed excellent stability without aggregation in five recycling runs, indicating the high potential of the single-atom catalyst in hydrodeoxygenation. Using the hydrodeoxygenation of furfuryl alcohol to 2-methylfuran (2-MF) as an example, Fu et al. demonstrated that the introduction of an ultralow loading Pt in a highly dispersed form onto TiO2 could improve the C–O activation rate remarkably, without leading to the bulk reduction of TiO2 or undesirable ring chemistry.731
In addition to the design of the catalyst, the development of suitable reaction media is also very important for the efficient conversion of HMF to DMF. Liu et al. investigated the influence of water content on the hydrogenation and hydrogenolysis of HMF with molecular H2 over the Cu/γ-Al2O3 catalyst in the medium of THF. The reaction kinetic studies indicated that the presence of water in THF remarkably influenced the reaction rate and selectivity. In pure THF, the hydrogenolysis reaction was the dominant process, resulting in the formation of 2-methyl-5-hydroxymethylfuran (MHMF) and DMF.732 In contrast, the addition of 5 wt% H2O to THF inhibited the hydrogenolysis more than the hydrogenation reaction, leading to a relatively high yield of BHMF and low yield of DMF. Nurenberg et al. reported that the hydrodeoxygenation of HMF could be performed over a platinum/cobalt bimetallic alloy supported on graphitic carbon (PtCo/GC) in the medium of commercial gasoline, obtaining a gasoline/DMF mixture, which is promising for direct use in the current internal combustion engine.723 Among the tested solvents, the gasoline/ethanol mixture with a mass ratio of 9:1 (E10) showed the best catalytic performance for the sequential conversion of a high concentration HMF (10 wt%), giving the final product containing 7–20 wt% DMF with improved knock resistance property (higher octane number than E10). However, the DMF yield decreased rapidly in the recycling experiment owing to the deactivation of the PtCo/GC catalyst. Deng et al. found that the product selectivity of furfural hydrogenation over α-MoC depends highly on the alcohol solvents, since the hydrogen donating ability of solvents can control the hydrogenation rate of reactants and the solvent-induced surface modifications of Mo-based catalysts influence the formation of relevant transient states and the final reaction selectivity via enforcing steric hindrance on its surface.156,733
Metal sulfides have also been demonstrated as a promising type of catalyst for the selective hydrogenolysis process. For example, Han et al. reported that the ordered mesoporous alumina-supported nickel–molybdenum sulfide catalyst (5Ni–7MoS2/mAl2O3) enabled the selective hydrogenolysis of HMF to DMF with a yield of 95% at 130 °C under 1 MPa H2 in 2-propanol.726 They attributed the active center to the coordinated unsaturated sites resulting from the substitution of Mo by Ni since the number of coordinated unsaturated sites located at the S-edge correlates well with the proportion of Mo substituted by Ni and a higher proportion of Ni resulted in a higher TOF and lower apparent activation energy (51 kJ mol−1). In this catalytic system, the remarkable synergistic effect of H2 with 2-propanol for the high reaction efficiency was demonstrated. The conversion of HMF to DMF proceeds via (Fig. 55) the HMF → 5-MF → MFA → DMF and the HMF → ethers → DMF reaction pathway simultaneously. The generation of ethers, which are easier to be hydrogenolyzed, could improve the DMF yield obviously. Similar to the 5Ni–7MoS2/mAl2O3 catalyst, Liu et al. reported that the use of MoS2 monolayer sheets decorated with isolated Co atoms as a catalyst could greatly improve the catalytic performance for the hydrodeoxygenation of 4-methylphenol to toluene due to the formation of sulfur vacancies.734
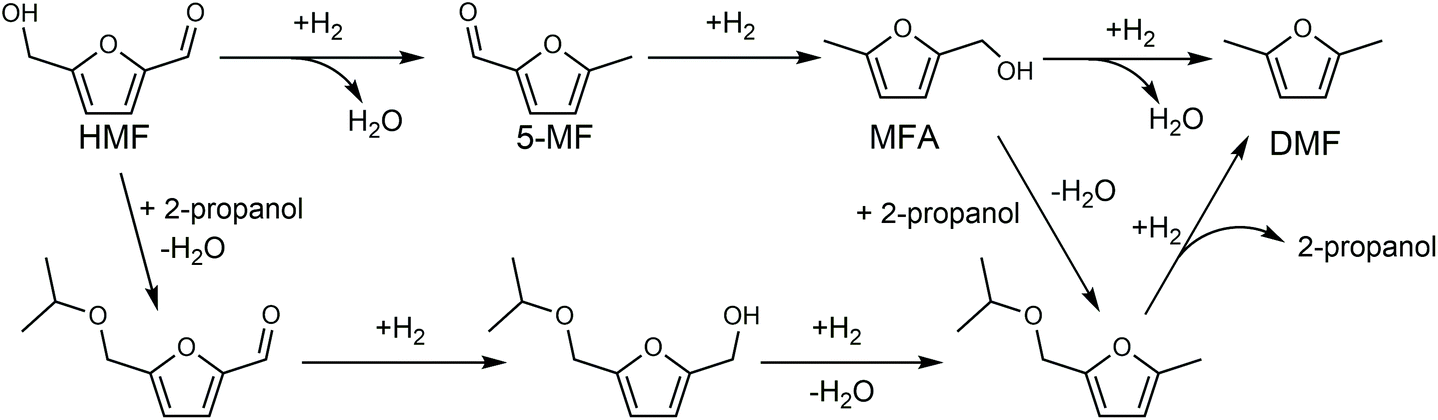 | ||
| Fig. 55 Hydrodeoxygenation of HMF with H2 generated in situ from the decomposition of alcohol. Adapted from ref. 726. | ||
In addition to H2, other hydrogen donors have also been attempted for the conversion of HMF to DMF. For example, Zhang et al. developed a facile process to convert HMF to DMF in ethanol using PdCl2 as the catalyst and non-toxic and low-cost polymethylhydrosiloxane (PMHS) as the hydride source.735 It was demonstrated that the in situ-generated metallic Pd0 species and HCl play important roles in the reaction. This catalytic system enabled a DMF yield as high as 89.7% from HMF at room temperature (25 °C) in 0.5 h. In addition, a DMF yield of 41% was obtained from glucose after 2.5 h reaction at 120 °C.
Besides the catalytic transfer hydrogenation of HMF with alcohol toward BHMF, HMF can also be hydrogenated by the H2 generated in situ from the decomposition of alcohol. For example, the in situ catalytic hydrogenation of HMF to DMF with methanol as the hydrogen donor (Fig. 56) over Cu supported on metal oxide catalysts was investigated by Zhang et al.712 Among the tested catalysts, Cu/Al2O3 gave the maximum DMF yield of 73.9% at 240 °C for 6 h owing to the small Cu crystallite size and strong acidity. However, the catalytic activity of Cu/Al2O3, Cu/ZnO, and Cu/CeO2 was lost almost completely in the recycling experiment, and only Cu/ZrO2 retained a DMF yield of around 21%. Besides, the in situ catalytic hydrogenation does not seem to be cost-effective owing to the consumption of two equivalents of methanol. They also investigated the one-pot hydrogenation of HMF to DMF over the ternary CuZnCoOx catalyst with ethanol as the hydrogen donor, affording a maximum DMF yield of 99%.718 They suggested that the CoOx species catalyzes the in situ decomposition of ethanol to H2 and acetaldehyde effectively (Fig. 57), the zinc oxide promotes the hydrogenation of aldehyde group to hydroxymethyl group, and the Cu–Co alloy promotes the hydrogenation of HMF remarkably. In addition, the catalytic performance of CuZnCoOx could be maintained during six recycling experiment runs.
 | ||
| Fig. 56 Catalytic hydrogenation of HMF to DMF with methanol as the hydrogen donor. Adapted from ref. 712. | ||
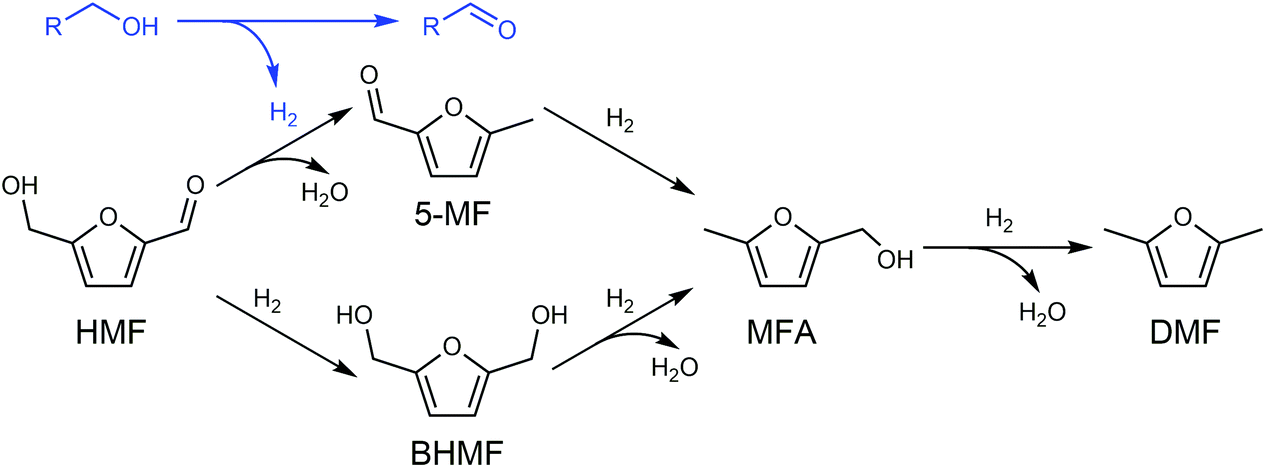 | ||
| Fig. 57 Hydrodeoxygenation of HMF with H2 generated in situ from the decomposition of alcohol. Adapted from ref. 718. | ||
The drop-in coupling of HMF hydrodeoxygenation with the industrially important dehydrogenation reaction may enable the co-production of DMF with commercial products, thus greatly improving the overall benefit and sustainability. Li et al. reported that the simultaneous production of phenol and DMF with unprecedented high yields (>97%) from HMF and cyclohexanol (CHL) or cyclohexanone (CHN) could be achieved via a vapor-phase dehydrogenation–hydrogenation coupling reaction (Fig. 58) at 240 °C in the atmosphere of N2 using a bimetallic Ni–Cu alloy catalyst, without the use of an additional hydrogen and oxygen supply.736 Similarly, Gao et al. reported that the use of a nitrogen-doped carbon (NC)-decorated copper-based catalyst (NC-Cu/MgAlO) enabled the selective synthesis of DMF and DMTHF for the transfer hydrogenolysis of HMF using cyclohexanol as the hydrogen source, obtaining a DMF yield as high as 96.1% at 0.5 h and DMTHF yield of up to 94.6% in 3.5 h.709 They proposed that the surface basic sites activate the alcohol hydroxyl in cyclohexanol and then promote the release of active hydrogen species, while the Cu0 nanoparticles and electrophilic Cu+ species promote the hydrogen transfer and the activation of the carbonyl group and hydroxyl group in HMF, respectively.
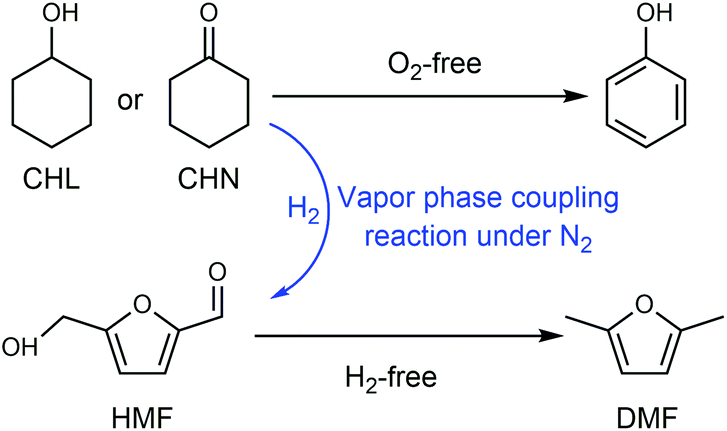 | ||
| Fig. 58 Synchronized production of phenol and DMF via dehydrogenation–hydrogenation coupling process. Adapted from ref. 736. | ||
Electrosynthesis is an efficient and environmentally friendly approach to achieve the oxidation or hydrogenation of HMF to useful chemicals. For example, a non-noble CuNi bimetallic alloy was used as a highly effective and stable electrode for the electro-catalytic hydrogenation of HMF toward DMF, achieving a DMF selectivity of 91.1% with a Faradaic efficiency of 88.0%.720
Owing to the higher stability and hydrophobicity of 5-formyloxymethylfurfural (FMF) than HMF, FMF is considered a promising alternative to HMF for the production of DMF. Sun et al. reported that the bimetallic catalyst 36Ni–12Cu/SBA-15 gave a DMF yield of 71.0% from FMF, which is slightly higher than that (60.5%) obtained from HMF under the same reaction conditions.703
The conversion of sugars to DMF via a two-step process is relatively easy to achieve. For example, Thananatthanachon et al. reported a two-step process involving the dehydration of fructose to the formate ester of HMF with formic acid as both the acid catalyst and solvent at high temperature, and hydrogenation of the formate ester to DMF over the Pd/C catalyst with formic acid as the hydrogen donor.737 The one-pot conversion of fructose to DMF (Table 16) was achieved by Li et al. using a multifunctional heterogeneous catalyst consisting of a carbon-based solid acid shell and CuCo bimetal core.738 The acid sites in the external carbon shell promoted the dehydration of fructose to the intermediate HMF, and then the CuCo bimetal catalyzed the hydrogenation of HMF to DMF synergistically. Among the tested catalysts, CuCo@C-TsOH-IM, which was prepared by impregnating CuCo@C-TsOH-IM with TsOH, exhibited the best catalytic performance, obtaining a DMF yield of 71.1% from fructose in THF at 220 °C under 3 MPa H2 for 10 h. Insyani et al. designed the Pd/UiO-66@SGO catalyst by introducing Pd on a Zr-based metal–organic framework (UiO-66) deposited on sulfonated graphene oxide for the one-pot conversion of sugars, attaining DMF yields of 70.5% and 45.3% from fructose and glucose, respectively, at a relatively low substrate loading (0.5 wt%).739 To produce MF and DMF from hardwood poplar, Seemala et al. constructed an integrated process involving the conversion of poplar wood chips to HMF (66.0%) and furfural (93.5%) using dilute FeCl3 as the catalyst in the medium of THF/water at subpyrolytic temperature, extraction of HMF and furfural from water to an organic phase, which was composed of toluene and 1,4-dioxane, after treatment with Ca(OH)2 and hydrodeoxygenation of HMF and furfural to DMF (yield: 87.8%) and MF (yield: 85.6%) over Cu-the Ni/TiO2 catalyst, attaining an overall fuel and lignin yield of 60%.740
| Catalyst | Catalyst loadinga | Solvent | Conditions | Feedstocka | Conversion | DMF yield | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. — Not provided. | |||||||
| CuCo@C | 0.5 wt% | THF | 220 °C, 10 h, 3 MPa H2 | Fructose, 0.5 wt% | 56.2% | 18.4% | 738 |
| CuCo@C-TsOH | 0.5 wt% | THF | 220 °C, 10 h, 3 MPa H2 | Fructose, 0.5 wt% | >99% | 55.5% | 738 |
| Cu@C-TsOH-IM | 0.5 wt% | THF | 220 °C, 10 h, 3 MPa H2 | Fructose, 0.5 wt% | >99% | 23.1% | 738 |
| Co@C-TsOH-IM | 0.5 wt% | THF | 220 °C, 10 h, 3 MPa H2 | Fructose, 0.5 wt% | >99% | 61.5% | 738 |
| CuCo@C-TsOH-IM | 0.5 wt% | THF | 220 °C, 10 h, 3 MPa H2 | Fructose, 0.5 wt% | >99% | 71.1% | 738 |
| Formic acid; Pd/C | 1.3 wt% | Formic acid; +THF | 150 °C, 2 h, in formic acid; 70 °C, 15 h, in formic acid + THF | Fructose, 12 wt% | — | 51% | 737 |
| 4.8Pd/UiO-66@SGO | 0.5 wt% | THF | 130 °C, 3 h, 1 MPa H2 | HMF, 0.3 wt% | 99.9% | 99.2% | 739 |
| 4.8Pd/UiO-66@SGO | 0.5 wt% | THF | 180 °C, 3 h, 1 MPa H2 | Fructose, 0.50 wt% | 91.8% | 70.5% | 739 |
| 4.8Pd/UiO-66@SGO | 0.5 wt% | THF | 180 °C, 3 h, 1 MPa H2 | Glucose, 0.45 wt% | 87.3% | 45.3% | 739 |
In addition to its application as a drop-in fuel, DMF is also a promising starting material for the production of important commodity chemicals. For example, Tao et al. reported that biorenewable para-xylene (PX), an important petroleum-derived commodity chemical with a global production of nearly 40 million tons per year, which is mainly used for the production of PTA, could be produced from HMF and ethylene (Fig. 59) via an integrated one-pot, two-step process involving the conversion of HMF with formic acid to DMF in n-heptane over the Au^Pd0.2/t-ZrO2 catalyst and Diels–Alder-like coupling reaction of ethylene with DMF to PX, affording an overall PX yield of 85%.741 This process is parallel to the conversion of HMFCA to PTA via the Diels–Alder–dehydration reaction described in section 3.2.3. DMF can be transformed to alkylated tetrahydrofuran via a three-step cascade reaction (Fig. 60), composed of ring opening, aldol condensation and hydrogenation–cyclization process using acid, basic and metal catalyst, respectively.742
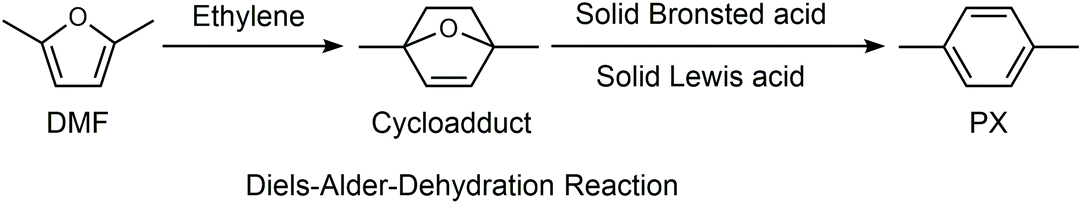 | ||
| Fig. 59 Conversion of DMF to PX. Adapted from ref. 741. | ||
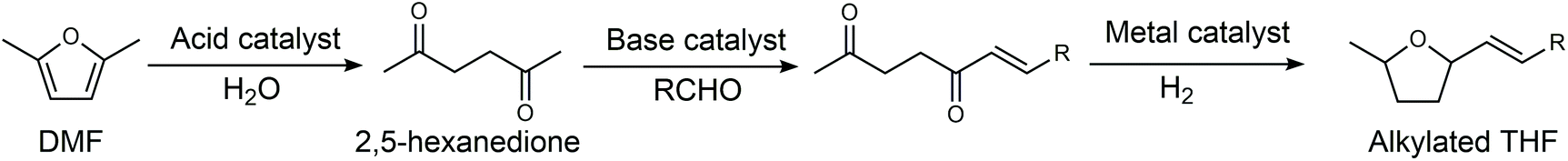 | ||
| Fig. 60 Conversion of DMF to alkylated tetrahydrofuran via a three-step process. Adapted from ref. 742. | ||
:2), HMF was firstly converted to esterified intermediate, (5-formylfuran-2-yl)methyl formate (FFMF) via acid-catalyzed esterification and then converted to 5-MF via hydrogenolysis. In this process, formic acid not only functions as a reactant for the formation of esterified intermediate, but also serves as a hydrogen donor.
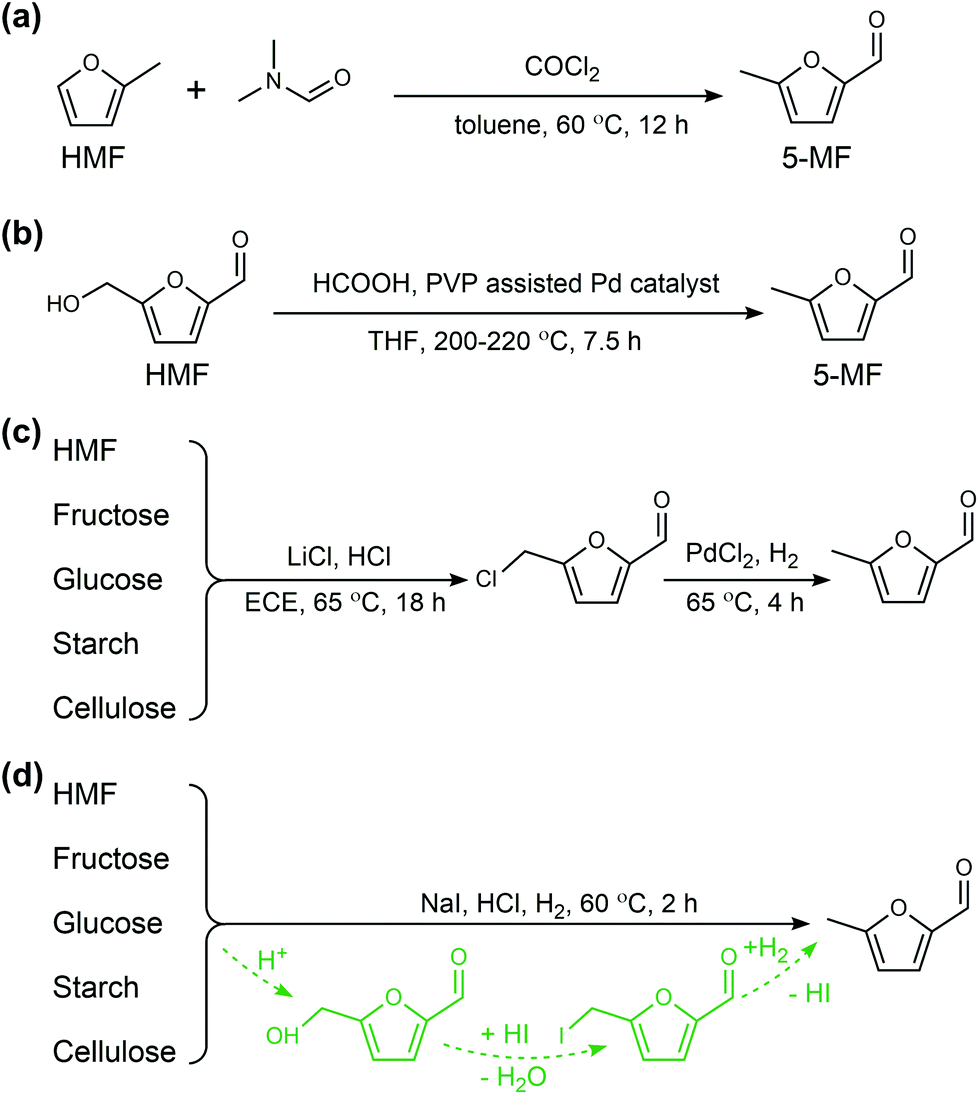 | ||
| Fig. 61 (a) Industrial 5-MF production process. Adapted from ref. 743 and 744. (b) 5-MF production from HMF. Adapted from ref. 744. (c) Conversion of biomass to 5-MF via CMF as the intermediate. Adapted from ref. 210. (d) Direct conversion of biomass to 5-MF. Adapted from ref. 211. | ||
| Catalyst | Catalyst loadinga | Solvent | Conditions | Substrate loadinga | Conversion | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to solvent. — Not provided. | |||||||
| 2.5% Pd-PVP/C (1:2) |
0.5 wt% | THF | 200 °C, 7.5 h, HCOOH, 0.5 MPa N2 | HMF, 2.3 wt% | 87% | 5-MF 80% | 744 |
| 2.5% Pd-PVP/C (1:2) |
0.5 wt% | THF | 200 °C, 7.5 h, HCOOH, 0.5 MPa N2 | ClMF, 2.3 wt% | 87% | 5-MF 80% | 744 |
| NaI; H2SO4 | 0.6 M; 50 mM | H2O/MTHF (v/v = 7/60) | 180 °C, 4 h, 2.1 MPa H2 | HMF, 1.3 wt% | — | 5-MF 80.8% | 211 |
| NaI; HCl | 0.4 M; 36 mM | H2O/MTHF (v/v = 2/3) | 160 °C, 2 h, 2.1 MPa H2 | Fructose, 1.3 wt% | — | 5-MF 30.2% (LA 22.5%, 2-MF 3.1%) | 211 |
| NaI; HCl | 0.4 M; 36 mM | H2O/MTHF (v/v = 2/3) | 160 °C, 2 h, 2.1 MPa H2 | HMF, 1.3 wt% | — | 5-MF 30.1% (LA 23.3%, 2-MF 2.4%) | 211 |
| NaI; HCl | 0.4 M; 36 mM | H2O/MTHF (v/v=2/3) | 160 °C, 2 h, 2.1 MPa H2 | Starch, 1.3 wt% | — | 5-MF 38.0% | 211 |
| NaI; HCl | 0.4 M; 36 mM | H2O/MTHF (v/v = 2/3) | 160 °C, 2 h, 2.1 MPa H2 | Cellulose, 1.3 wt% | — | 5-MF 24.0% (LA 23.7%, 2-MF 2.0%) | 211 |
| Pd/C; HI | 0.5 wt%; 0.79 M | H2O/benzene (v/v = 0.9) | 90 °C, 0.5 h, 2.1 MPa H2 | Fructose, 4.7 wt% | 97% | 5-MF 68% | 210 |
| Pd/C; HI | 0.5 wt%; 0.79 M | H2O/benzene (v/v = 0.9) | 90 °C, 2 h, 2.1 MPa H2 | Glucose, 4.7 wt% | 81% | 5-MF 31% | 210 |
| Pd/C; HI; NaI | 0.5 wt%; 0.03 M, 0.3 M | H2O/benzene (v/v = 0.9) | 115 °C, 2 h, 2.1 MPa H2 | Cellulose, 4.7 wt% | 97% | 5-MF 42% | 210 |
5-MF could also be produced using carbohydrates (including fructose, glucose, cellulose, inulin and starch) and realistic biomass as the starting material via a two-step process, involving the conversion of carbohydrates and biomass to 5-(chloromethyl)furfural (CMF) and hydrodechlorination of CMF to 5-MF (Fig. 61c).209–211 However, this process is not practical owing to the stoichiometric consumption of HCl. Different from HCl, Yang et al. reported that the combined use of HI with Pd/C (or RuCl3) enabled the one-pot conversion of carbohydrates to 5-MF via 5-iodomethylfurfural (IMF) as the main reaction intermediate (Fig. 61d) without consuming HI, attaining 5-MF yields of 68%, 31% and 42% from fructose, glucose, and cellulose, respectively.210 Similarly, Peng et al. reported that the combination of NaI with HCl (or H2SO4) enabled the metal-free hydrogenolysis of biomass to 5-MF, affording 5-MF yields of 80.8% and 38.0% from HMF and starch, respectively.211
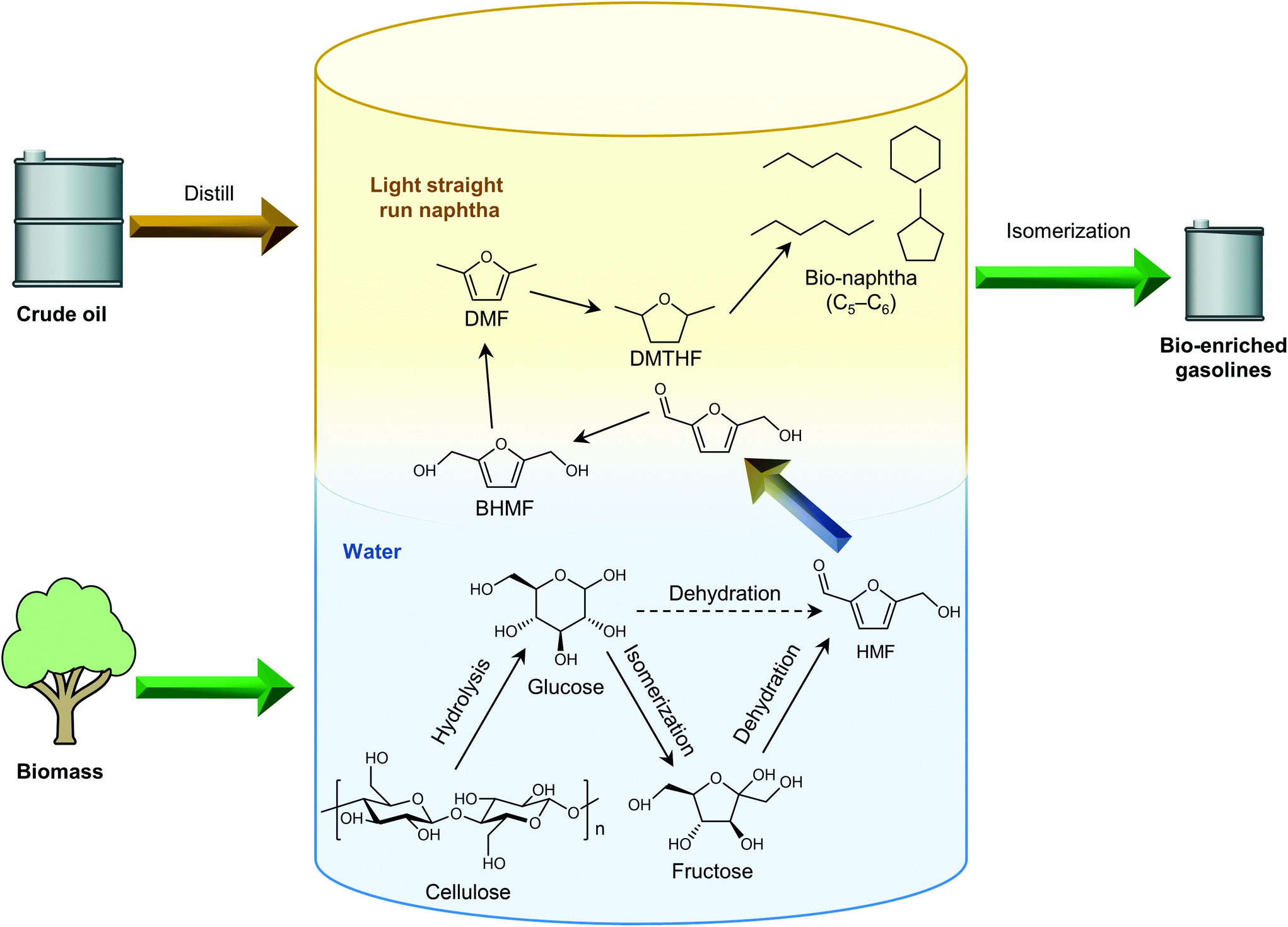 | ||
| Fig. 62 Direct upstream integration of the conversion of cellulose to liquid alkanes with the current light straight run naphtha petrorefinery processes. Adapted from ref. 745. | ||
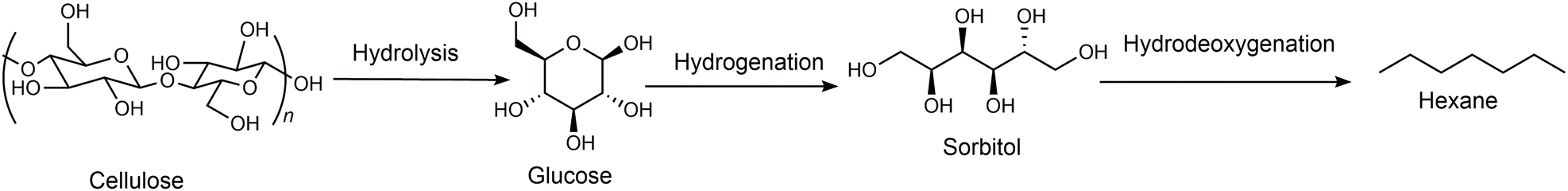 | ||
| Fig. 63 Conversion of cellulose to liquid alkanes via sorbitol as an intermediate. Adapted from ref. 745 and 747. | ||
C5–C6 alkanes require an additional isomerization process to improve their octane number prior to their utilization as gasoline. Alternatively, coupling HMF with carbonyl or hydroxyl group-containing compounds is an effective approach to extend the carbon chain to produce the precursor of liquid alkanes with a high octane number, as will be discussed in detail in section 3.6. Besides, Luo et al. demonstrated that the acceptorless dehydrogenative C–C coupling of biomass-derived DMF (or methylfuran) (Fig. 64) over an Ru-doped ZnIn2S4 catalyst under visible light could produce H2 and C12–16 diesel precursors simultaneously. Subsequently, the desired diesel fuels, straight- and branched-chain alkanes could be synthesized from these precursors via hydrodeoxygenation.8
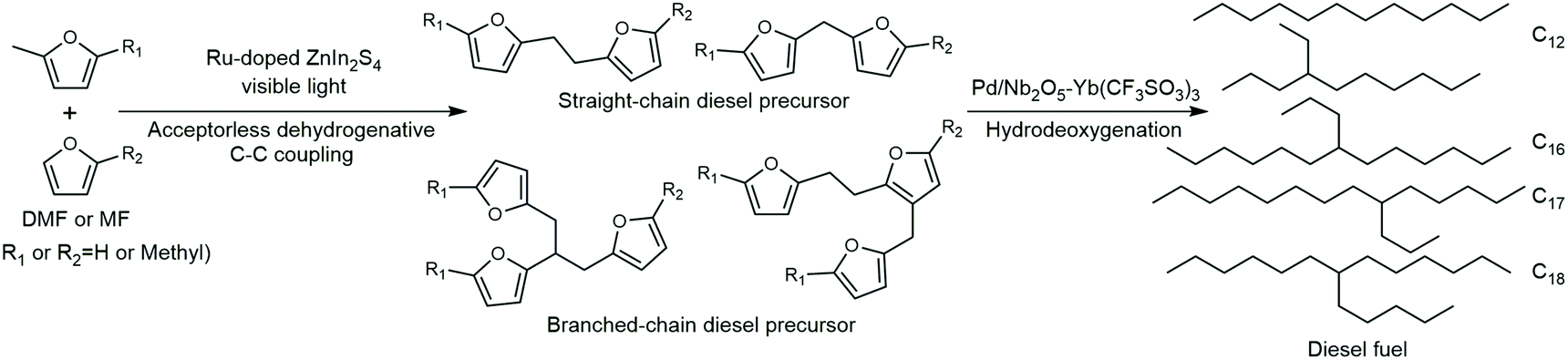 | ||
| Fig. 64 Conversion of DMF (or MF) to diesel fuels. Adapted from ref. 8. | ||
O group on the Pd particles and support, while the special nature of LDH–MgAl–NO3 could prevent the hydrogenation of the CO group on the support by hydrogen spillover, thus resulting in the selective hydrogenation of the furanyl ring without changing the CO group.
 | ||
| Fig. 65 Selective hydrogenation of HMF and furfural to 5-HMTHFF and THFF. Adapted from ref. 749. | ||
Similar to BHMF, bishydroxymethyltetrahydrofuran (BHMTF) is considered a promising building block for bio-based polyesters and promising feedstock for the synthesis of 1,6-hexanediol (1,6-HDO) and 2,5-dimethyltetrahydrofuran (DMTHF).692 Fulignati et al. reported that at a mild temperature (50 °C, 6 h, 30 bar H2), the commercial Ru/C catalyst enabled the selective hydrogenation of HMF to BHMF, while under relatively harsh conditions (100 °C, 6 h, 50 bar H2), HMF was selectively converted to BHMTHF in aqueous solution.727 BHMTHF could be transformed to the high value 1,6-hexanediol (1,6-HDO) over the Pt–WOx/TiO2 catalyst via a ring-opening and hydrogenolysis process (Fig. 66), obtaining a 1,6-HDO yield of up to 70%.750 As an important monomer to produce polyesters and polyurethanes, the market price of 1,6-HDO is $4400 per ton with a market scale of approximately 138000 tons per year.751 Similarly, 1,5-pentanediol (1,5-PDO) can be produced with a high overall yield (80%) from biomass-derived furfural via a consecutive hydrogenation, dehydration, hydration and hydrogenation process.752,753 Based on these studies, the production of 1,5-PDO and 1,6-HDO with 69% and 28% yield from hemicellulose and cellulose via multiple processes was achieved by He et al. using furfural and HMF as the main reaction intermediates, respectively.754 A techno-economic analysis suggested that the MSP of 1,6-HDO and 1,5-PDO is $4090 per ton.754
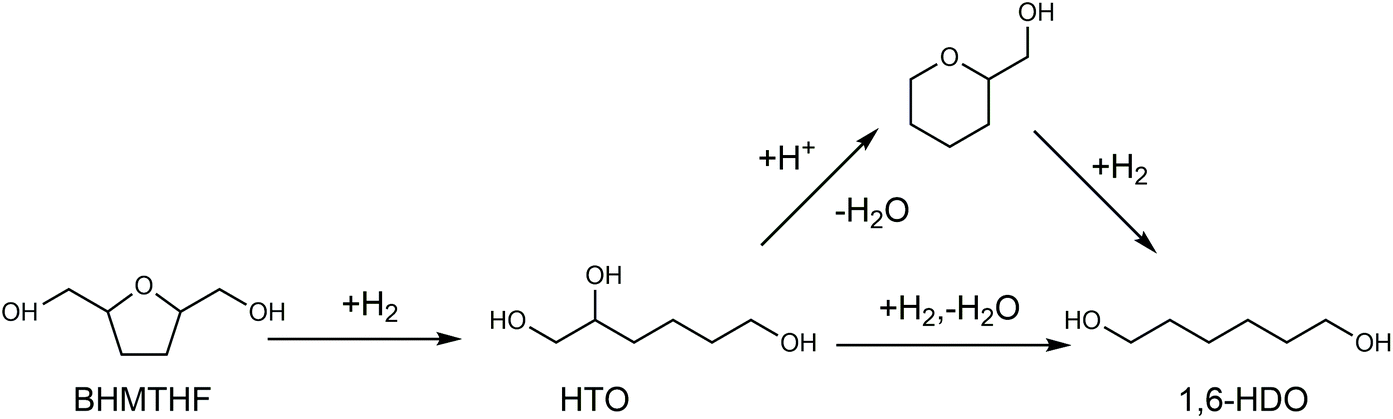 | ||
| Fig. 66 Conversion of BHMTHF to 1,6-HDO. Adapted from ref. 750. | ||
The hydrogenation of HMF in acidic media can yield 1-hydroxyhexane-2,5-dione (HHD) (Fig. 67) via a ring opening and hydrogenation process.755,756 For example, bipyridine-coordinated Cp*–iridium(III) complexes enabled the hydrogenation/hydrolytic ring opening reactions of HMF toward HHD.757,758 Fujita et al. reported that nickel phosphide nanoparticles (Ni2P NPs) were highly effective for the selective hydrogenation of biofuranic aldehydes to diketones in water without using an additive, attaining an HHD yield of up to 84% from HMF, while the conventional Ni(0), NiO, and other metal phosphide NPs are ineffective.759 The spectroscopic analysis showed that the Ni–Ni species function as the H2 activation sites, while P–OH functions as the surface acid sites. Therefore, the Ni2P NPs could combine the hydrogen-activating ability and surface acidity to promote selective conversion of biofuranic aldehydes. HHD can be further converted to methylcyclopent-2-enone (MCP), a valuable edible essence widely applied in foods, beverages and flavors, via an intramolecular aldol condensation over base catalysts.760 Duan et al. achieved the conversion of HMF to MCP via the hydrogenation of HMF to HHD over the Pd/Nb2O5 catalyst and rearrangement of HHD to MCP over the Ca–Al oxide catalyst in the medium of water, attaining an isolated MCP yield of 58%.761 3-Hydroxymethyl-cyclopentone (HCPN) is considered a promising intermediate for the production of high-value fragrances, pesticides and polymers. Ramos et al. reported that the Cu–Al2O3 catalyst is effective to catalyze the conversion of HMF to HCPN under H2 in aqueous solution, while Co–Al2O3 mainly converts HMF to 3-hydroxymethylcyclopentanol (HCPL).762 They observed that the reduced metal phases are responsible for the hydrogenation step. The catalytic activity of Cu–Al2O3 and Co–Al2O3 both decreased remarkably after the first use, but they could be regenerated via calcination at 500 °C in air followed by reduction under H2 at 700 °C. Zhang et al. reported that an MOF-derived bimetallic nickel–copper catalyst gave an HCPN yield of 70.3% with a total rearrangement product yield of 99.8% in water without the use of acidic additives.763 They also demonstrated that water not only functions as the reaction medium and reactant, but also serves as a proton donor to form a slightly acidic condition, which accelerates the ring-rearrangement reaction at high temperature (140 °C). Deng et al. reported that Pd nanoparticles supported on Lewis acidic pyrochlore, including La2Sn2O7, Y2Sn2O7, and Y2(Sn0.7Ce0.3)2O7−δ, are active for the conversion of HMF to HCPN, attaining an HCPN yield of up to 92.5% over the Pd/Y2(Sn0.7Ce0.3)2O7−δ catalyst.764
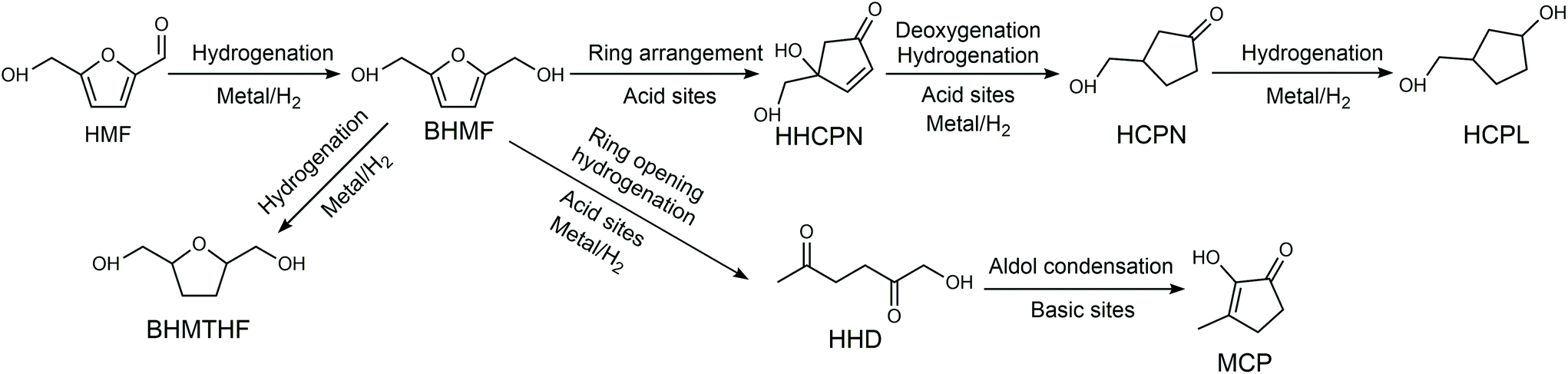 | ||
| Fig. 67 Conversion of HMF to HCPN and MCP. Adapted from ref. 760, 761 and 763. | ||
The use of high-pressure H2 not only consumes a large amount of energy, but also induces potential risk, which is a major limiting factor in the commercialization of the hydrodeoxygenation process. Duan et al. reported that the introduction of high-pressure molecular dinitrogen (N2), which was previously considered as a carrier or inert gas with low cost, is a general strategy to increase the efficiency of catalytic hydrodeoxygenation with a reduced activation energy.765 They suggested that the adsorption and activation of N2 on the metallic ruthenium surface produces N2Hx species, thus providing protic hydrogen to promote the scission of the carbon–oxygen bond and then the hydrogenation of the hydroxy groups. This strategy may be promising to reduce the consumption of H2, leading to a reduction in cost.
3.4. Etherification of HMF with alcohols toward alkoxymethyl furfurals
The etherification reaction of aliphatic alcohols with HMF can generate alkoxymethyl furfurals (AMF), such as 5-(methoxymethyl)furfural (MMF) and 5-(ethoxymethyl)furfural (EMF), which are considered promising fuel additives and precursors of drop-in fuel owing to their high energy density, low toxicity, high stability and suitable flow properties.766 Here, we summarize the state-of-the-art catalytic systems for the conversion of HMF and carbohydrates to AMF with a focus on the heterogenous catalysts.The etherification of aliphatic alcohols and HMF usually proceed over Brønsted acid catalysts with a series of side-reactions (Fig. 7). The product selectivity is influenced by the catalyst, solvent and reaction conditions (Table 18). The acid type and intensity have a critical influence on the reaction pathway of HMF in the medium of ethanol. Generally, a strong Brønsted acid is beneficial for the formation of ethyl levulinate (EL), a strong Lewis acid prefers the generation of ether (EMF), while weak acid favors the generation of the acetal, 5-(alkoxymethyl)furfural dialkylacetal (EMFDEA).767 For example, a high yield of EMF (71%) was attained at mild temperature (100 °C) within 6 h, while a high yield of EL (73%) was attained at an elevated temperature (140 °C, 6 h) for HMF conversion over sulfonic-acid-functionalized carbon nanomaterial (C–SO3H) in the medium of ethanol.768 Similarly, the EL and EMF selectivity from HMF could be tuned by regulating the reaction conditions and pore structure of the ZSM-5 zeolite.769 The use of a high loading of mesoporous ZSM-5 zeolite as the catalyst and n-hexane as the solvent at high temperature is beneficial for the selective formation of EL.
| HMF or carbohydrate loadinga | Catalyst loadinga | Solvent | Reaction conditions | HMF or carbohydrate conversion | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a Relative to solvent. — Not provided. | ||||||
| HMF, 3.2 wt% | Amberlyst-15, 0.9 wt% | Ethanol/ChCl/MeCN | 100 °C, 12 h | 99% | 82% | 770 |
| HMF, 1.6 wt% | [Cu–BTC][HPM], 1 wt% | Ethanol | 140 °C, 12 h | — | 68.4% (EL 20.2%) | 771 |
| HMF, 1.6 wt% | C-SO3H, 1 wt% | Ethanol | 100 °C, 6 h | — | 71% (EL 22%) | 768 |
| HMF, 1.6 wt% | C-SO3H, 1 wt% | Ethanol | 140 °C, 6 h | — | <1% (EL 73%) | 768 |
| HMF | Graphene oxide, 3 wt% | Ethanol | 100 °C, 12 h | — | 92% | 772 |
| HMF, 6.3 wt% | MHGC-SO3H, 2 wt% | GVL/ethanol (v/v = 1.5) | 120 °C, 16 h | 95% | 95% | 773 |
| Fructose, 3.6 wt% | MHGC-SO3H, 2 wt% | GVL/ethanol (v/v = 1.5) | 120 °C, 24 h | 100% | 67.4% | 773 |
| Glucose, 3.6 wt% | MHGC-SO3H, 2 wt% | GVL/ethanol (v/v = 1.5) | 120 °C, 24 h | 98.4% | 3% | 773 |
| Fructose, 6.3 wt% | Graphene oxide, 3 wt% | Ethanol | 100 °C, 24 h | ∼99% | 71% | 772 |
| HMF 6.3 wt% | TaTPA, 3.8 wt% | Ethanol | 120 °C, 45 min | 100% | 88.3% | 774 |
| HMF 6.3 wt% | 30% TaTPA/SnO2 3.8 wt% | Ethanol | 120 °C, 45 min | 99.5% | 90.2% | 774 |
| Fructose 3 wt% | 30% TaTPA/SnO2 3.8 wt% | Ethanol | 120 °C, 8 h | 100% | 68% | 774 |
| HMF, 6.3 wt% | HPW/NbPO, 3.76 wt% | Ethanol | 120 °C, 1 h | 95% | 89% | 775 |
| Fructose, 3.6 wt% | Phosphotungstic acid, 0.72 wt% | Ethanol/DMSO (v/v = 7/3) | 140 °C, 1.5 h | ∼97% | 64% | 776 |
| Inulin, 3.6 wt% | [BMIM][HSO4], 40 wt% | Ethanol/H2O (v/v = 125) | 130 °C, 0.5 h | — | 77% | 777 |
| Glucose, 3.6 wt% | CrCl3, 0.02 M | Ethanol | 140 °C, 10 h | 100% | 15.2% (ethyl glucoside 42.6%) | 778 |
| Glucose, 4 wt% | AlCl3·6H2O 1.5 wt%; PTSA–POM 2 wt% | Ethanol/H2O (v/v = 9) | 150 °C, 0.5 h | 97.9% | 30.6% (HMF 11.5%) | 779 |
| Glucose, 1.8 wt% | AlCl3·6H2O, 0.96 wt%, [BMIM][HSO4], 10 wt% | Ethanol | 130 °C, 0.5 h | — | 36.7% (HMF 2.7%) | 780 |
| Glucose, 3 wt% | H-USY 2.5 wt%; Amberlyst-15 0.5 wt% | Ethanol | 96 °C, 11 h | 91% | 17% (HMF 3%, EL 14 wt%) | 781 |
| Glucose, 3 wt% | H-USY 2.5 wt%; Amberlyst-15 0.5 wt% | Ethanol/DMSO (v/v = 7/3) | 131 °C, 24 h | 98% | 26% (HMF 20%, EL 1%) | 781 |
| HMF, 2.5 wt% | SO3H-PANI-FeVO4, 1 wt% | Ethanol | 120 °C, 6 h | 94.1% | 80% | 452 |
| Fructose, 3.6 wt% | SO3H-PANI-FeVO4, 1 wt% | Ethanol | 120 °C, 2 h | 86.3% | 72.5% | 452 |
| Sucrose, 6.8 wt% | SO3H-PANI-FeVO4, 1 wt% | Ethanol | 120 °C, 24 h | 76.2% | 57.2% | 452 |
| Fructose, 3.6 wt% | HReO4, 0.02 M | Ethanol/THF | 140 °C, 1 h | — | 73% | 388 |
| Fructose, 3.6 wt% | HReO4, 0.02 M | Ethanol | 160 °C, 0.5 h | — | 65% (EL 10%) | 388 |
| Fructose, 3.6 wt% | HReO4, 0.02 M | Ethanol/THF | 160 °C, 16 h | 100% | 0 (EL 80%) | 388 |
| Inulin, 3.6 wt% | HReO4, 0.02 M | Ethanol | 160 °C, 16 h | 77% | — (EL 65%) | 388 |
| Sucrose, 3.6 wt% | HReO4, 0.02 M | Ethanol | 160 °C, 16 h | 62% | — (EL 52%) | 388 |
| Glucose, 3.6 wt% | HReO4, 0.02 M | Ethanol | 160 °C, 72 h | 39% | — (EL 80%) | 388 |
| HMF 5 wt% | Zr-Mont 2 wt% | Isopropanol | Step I: 100 °C, 12 h; step II: 100 °C, 2 h (adding H2O) | 100% | iPMF 84% | 782 |
| HMF 5 wt% | Zr-Mont-400 2 wt% | Isopropanol | Step I: 100 °C, 12 h; step II: 100 °C, 2 h (adding H2O) | 45% | iPMF 34% | 782 |
| HMF 5 wt% | ZrO2 2 wt% | Isopropanol | Step I: 100 °C, 12 h; step II: 100 °C, 2 h (adding H2O) | 8% | iPMF 3% | 782 |
| HMF 5 wt% | Mont 2 wt% | Isopropanol | Step I: 100 °C, 12 h; step II: 100 °C, 2 h (adding H2O) | 17% | iPMF 10% | 782 |
| HMF, 1 wt% | Zr-SBA-UH, 0.51 wt% | Isopropanol | 150 °C, 4 h | — | BPMF 93.6% | 783 |
| HMF, 1 wt% | Zr-SBA-UH, 0.51 wt% | Ethanol | 150 °C, 4 h | — | BEMF 90.8% | 783 |
| HMF, 1 wt% | Zr-SBA-UH, 0.51 wt% | n-Propanol | 150 °C, 4 h | — | BPMF 45.4% | 783 |
| HMF, 1 wt% | Zr-SBA-UH, 0.51 wt% | n-Butanol | 150 °C, 4 h | — | BBMF 27.5% | 783 |
| HMF, 1 wt% | Zr-SBA-UH, 0.51 wt% | Isobutanol | 150 °C, 4 h | — | BPMF 64.8% | 783 |
| HMF, 1 wt% | Zr-SBA-UH, 0.51 wt% | sec-Butanol | 150 °C, 4 h | — | BBMF 5.4% | 783 |
| HMF, 1.3 wt% | Co-400, 0.1 wt% | Methanol | 90 °C, 2 MPa H2, 1 h | ∼100% | BHMF 98.5% | 784 |
| HMF, 2 wt% | MZSM-5, 4.8 wt% | Ethanol/n-hexane (w/w = 1/3) | 150 °C, 12 h | 94.2% | EMF 9.1% (EL 90.8%) | 769 |
| HMF, 2 wt% | MZSM-5, 2.5 wt% | Ethanol/n-hexane (w/w = 3/1) | 150 °C, 12 h | 80.3% | EMF 54.1% (EL45.9%) | 769 |
| HMF, 2 wt% | HZSM-5, 2.5 wt% | Ethanol | 140 °C, 12 h | 61.5% | EMF 57.8% (EL4.7%) | 769 |
| Corn stover, 2 wt% | USY zeolite, 1 wt% | THF/ethanol | 168 °C, 2.9 h | — | EMF, 21.8% | 785 |
| Corn stover, 2 wt% | USY zeolite, 1 wt% | THF/ethanol | 180 °C, 2.5 h | — | EMF 15.1% (HMF 0.3%, EL 0.8%) | 785 |
| Corn stover, 2 wt% | USY zeolite, 1 wt% | DMSO/ethanol | 180 °C, 2.5 h | — | EMF, 12.1% (HMF 6.8%, EL 4.7%) | 785 |
| Corn stover, 2 wt% | USY zeolite, 1 wt% | Cyclohexane/ethanol | 180 °C, 2.5 h | — | EMF, 10.3% (HMF 0.2%, EL 5.1%) | 785 |
| Corn stover, 2 wt% | USY zeolite, 1 wt% | n-Hexane/ethanol | 180 °C, 2.5 h | — | EMF, 4.4% (HMF 0.2%, EL 9.1%) | 785 |
| Corn stover, 2 wt% | USY zeolite, 1 wt% | Ethanol | 180 °C, 2.5 h | — | EMF, 4.4% (HMF 0.3%, EL 0.7%) | 785 |
| HMF, 1 wt% | HCP-1, 0.7 wt% | Ethanol | 100 °C, 10 h | — | EMF, 98.3% (EL 0.5%) | 786 |
| Fructose, 1 wt% | HCP-1, 0.7 wt% | Ethanol/DMSO (v/v = 4) | 105 °C, 8 h | 99.8% | EMF, 78.9% (HMF 15.4%, EL 4.6%) | 786 |
| Fructose, 1 wt% | HCP-1, 0.7 wt%, 5 runs | Ethanol/DMSO (v/v = 4) | 105 °C, 8 h | — | EMF, 75% | 786 |
| Glucose, 1 wt% | HCP-1, 0.7 wt% | Ethanol | 125 °C, 16 h | — | EMF, 18.4% | 786 |
| Sucrose, 1 wt% | HCP-1, 0.7 wt% | Ethanol | 105 °C, 8 h | — | EMF, 28.8% (glucose 7.9%, HMF 14.7%, EL 2.1%) | 786 |
| Inulin, 1 wt% | HCP-1, 0.7 wt% | Ethanol | 105 °C, 8 h | — | EMF, 75.6% (HMF 8.0%, EL 6.5%) | 786 |
| Cellobiose, 1 wt% | HCP-1, 0.7 wt% | Ethanol | 105 °C, 8 h | — | EMF <1% (glucose 1.7%, HMF 3.2%, EL <1%) | 786 |
| Fructose, 0.9 wt% | Cpolyurethane-SO3H, 0.3 wt% | Ethanol/1,4-dioxane (v/v = 7/3) | 140 °C, 2 h | 100% | EMF, 66.0% | 418 |
| Glucose, 0.9 wt% | Cpolyurethane-SO3H, 0.3 wt% | Ethanol/1,4-dioxane (v/v = 7/3) | 140 °C, 4 h | 100% | EMF, 9.2% | 418 |
| HMF 10 wt%, IB 17 wt% | HY zeolite, 3 wt% | GDE | 60 °C, 4 h | 59% | tBMF, 55.5% | 787 |
| HMF 7 wt%, IB 12.4 wt% | Aquivion/m-SiO2 (25 mM H+) | THF | 90 °C, 3 h | 78% | tBMF, 66% (OBMF <4%) | 788 |
| CMF 5 wt% | — | Ethanol | 50 °C, 3 h | 90% | EMF, 90% | 789 |
| BrMF 5 wt% | — | Ethanol | 50 °C, 3 h | 90% | EMF, 90% | 789 |
Since Brønsted acids are also effective to convert fructose and inulin to HMF,777 the one-pot conversion fructose and inulin to AMF are relatively easy to achieve though a one-pot process over Brønsted acid catalysts.775 The conversion of fructose in the medium of alcohols is accompanied by a series of side reactions with EL, resulting in 2-(diethoxymethyl)-5-(ethoxymethyl)furan as the main by-product.790 Using the G4 method, Xiang et al. indicated that ethanol promotes the Brønsted acid-catalyzed dehydration of fructose since the [C2H5OH2]+ species formed by the solvation of H+ on ethanol serves as a more active catalytic species than H+ for the preferential dehydration of fructose to HMF and then the etherification of HMF to EMF.791 The influence of additional organic solvents on the conversion of fructose over Amberlyst-15 catalyst in ethanol was investigated by Guo et al. using combined experimental, spectroscopic and theoretical methods.792 They observed that protonation of the polar additive increases in the following order: THF < acetone < CH3CN < GVL < DMSO, which is proportional to its Kamlet–Taft (KT) π* value, and the preferential protonation of DMSO is beneficial for the selective formation of HMF by preventing the subsequent etherification.
Compared with the one-pot conversion of fructose to EMF, the one-pot conversion of glucose to EMF is more challenging.773 For example, the (HCP-1)-derived hyper-cross-linked polymer exhibited high EMF yields of 98.3%, 78.9% and 75.6% from HMF, fructose and inulin, respectively, whereas the use of glucose resources (glucose and cellulose) as feedstock predominantly gave the ethyl glucoside product with a low EMF yield (18.4%).786 A one-pot, two-step process was developed to improve the EMF production efficiency from glucose. Firstly, glucose is isomerized to fructose by the Lewis acid catalyst. After isomerization, the Lewis acid catalyst is separated from the reaction system and then a Brønsted acid catalyst is added to enable the dehydration of fructose to HMF, and then the etherification of HMF with ethanol to EMF. For example, the combined use of H β zeolite and PSDVB–SO3H in the “two-step, one-pot” process gave an EMF yield of 39.4% with an EL yield of 4.1% from glucose.793 Guo et al. reported that the one-pot, one-step conversion of glucose into EMF could be achieved by combining the acidic ionic liquid 1-butyl-3-methylimidazolium hydrogen sulfate (BMIMHSO4) with the Lewis acid AlCl3, affording a maximum EMF yield of 37%.780 The direct alcoholysis of corn stover to EMF was achieved by Chen et al. using USY zeolite as the catalyst in ethanol/THF, attaining a maximum EMF yield of 21.8% at 168 °C for 2.9 h.785 Compared with other solvents, the use of THF as the solvent could inhibit the formation of EL.
Besides AMF, 2,5-bis(alkoxymethyl)furans (BAMF), a promising biodiesel component or additive, could be synthesized from HMF via reductive etherification (Fig. 68). For example, the use of Zr-SBA prepared via the urea hydrolysis method (Zr-SBA-UH) as a catalyst, with isopropanol as both the solvent and hydrogen source, enabled the highly efficient reductive etherification of HMF to 2,5-bis(isopropoxymethyl)furan (BPMF) with a yield of 93.6%.783 Under 2 MPa H2, the reductive etherification of HMF with methanol over the Co-400 catalyst, which was synthesized from commercially available Co3O4via simple reduction under H2, could give a 2,5-bis(methoxymethyl)furan (BMMF) yield as high as 98.5% at 140 °C within 1 h.784 The catalytic activity of the Co-400 catalyst was retained for use five times without an obvious decrease in the BMMF yield.
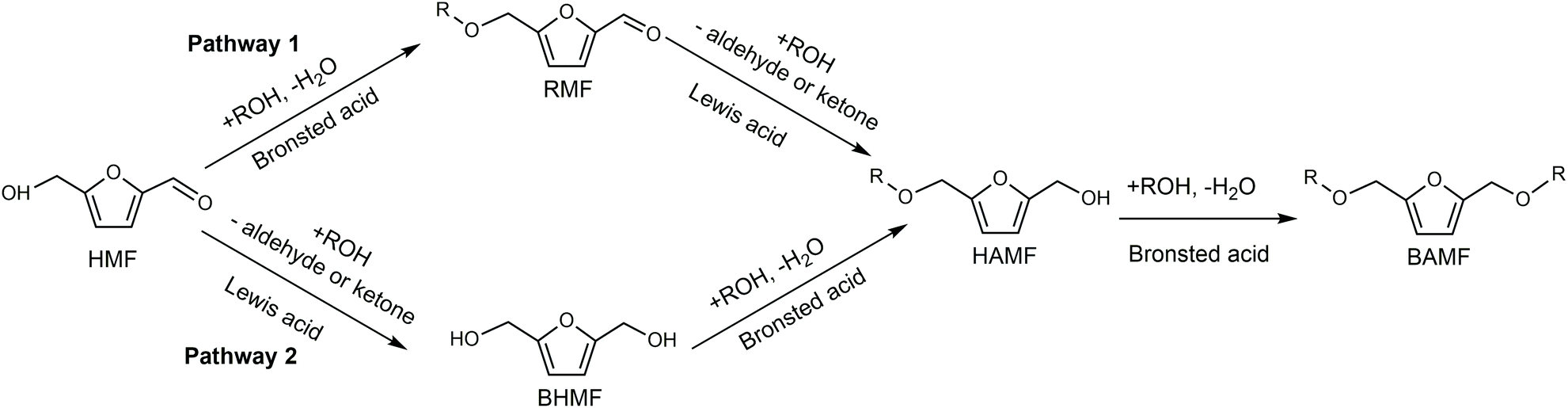 | ||
| Fig. 68 Production of BAMF via the reductive etherification of HMF. Adapted from ref. 783. | ||
The use of 5-(halomethyl)furfural (halo = Cl, Br) to displace HMF (Fig. 69) enables the efficient production of AMFs in excellent isolated yields (>90%) by converting 5-(halomethyl)furfural in monohydric alkyl alcohols, including methanol, ethanol, 1-propanol and 1-butanol under mild conditions without the use of additional catalysts.544,789 The use of higher primary alcohols, such as 1-pentanol and 1-hexanol, and secondary alcohol, such as 2-propanol, requires a base additive, such as N,N-diisopropylethylamine (DIPEA) to afford a high yield of AMFs. In contrast, at a relatively high temperature, the reaction of CMF with ethanol gives levulinic ester as the main product.547
 | ||
| Fig. 69 (a) Production of AMFs from 5-(halomethyl)furfural and alcohols under mild conditions. Adapted from ref. 544 and 789. (b) Production of levulinic ester from 5-(halomethyl)furfural and ethanol at relatively high temperature. Adapted from ref. 547. | ||
Similar to the etherification of HMF with alcohols, Yang et al. reported that tert-butoxymethylfurfural (tBMF), a potential biodiesel additive similar to EMF, could be produced via the etherification of HMF with isobutene (IB) over acid zeolites (Fig. 70). They found that the use of HY zeolite (SiO2/Al2O3 molar ratio = 12) in the medium of glycol dimethyl ether (GDE) could inhibit HMF dimerization and IB oligomerization, thus attaining a tBMF yield of 55.5% with the selectivity of 94% at 60 °C within 3 h.787 Dou et al. reported that the reaction between HMF and IB over an Aquivion perfluorosulfonic acid resin (PFSA) modified mesoporous silica (Aquivion/m-SiO2) solid acid afforded a tBMF yield of 66% with 5,5′-[oxybis(methylene)]bis[2-furaldehyde] (OBMF) yield lower than 4% in the medium of THF.788
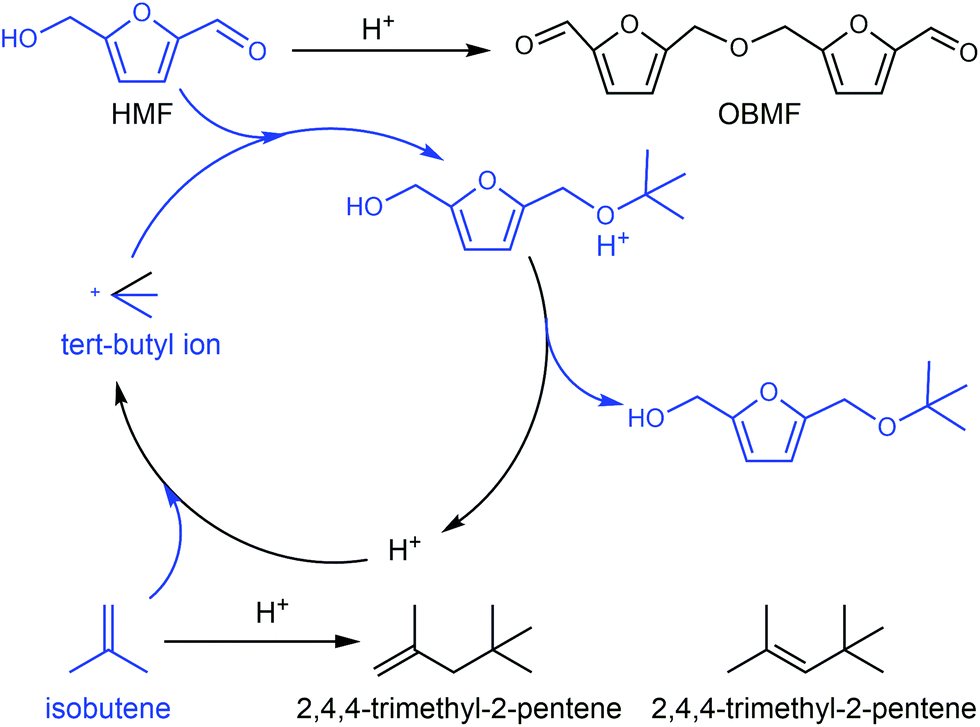 | ||
| Fig. 70 Synthesis of tBMF via the etherification of HMF with isobutene. Adapted from ref. 787. | ||
3.5. Acetalization of HMF with alcohols
The acetalization of HMF with alcohols is a major side reaction accompanying the etherification reaction. Kanai et al. reported that the cerium phosphate (CePO4) catalyst exhibited high catalytic activity for the chemoselective acetalization of HMF with methanol to 5-(dimethoxymethyl)-2-furanmethanol (Fig. 71) in excellent yield (81%) owing to the uniform Lewis acid and weak base sites, which interact with HMF and methanol molecules, respectively.794 The mutual valorization of glycerol with HMF toward diol monomers (Fig. 72), which can be used as a potential solvent, lubricant, antifreeze, personal care product and biodegradable and photodegradable polymer, can be achieved via acetalization reaction using acid catalysts, such as laminar zeolite (ITQ-2) and mesoporous aluminosilicate (MCM-41).795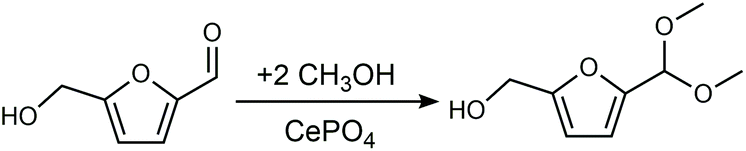 | ||
| Fig. 71 Chemoselective acetalization of HMF with methanol toward 5-(dimethoxymethyl)-2-furanmethanol. Adapted from ref. 794. | ||
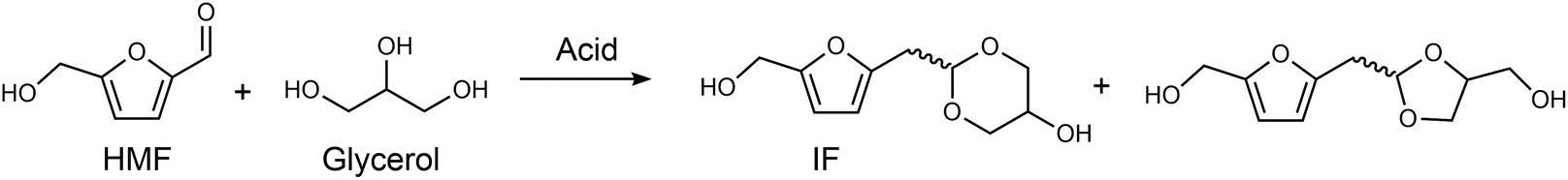 | ||
| Fig. 72 Mutual valorization of HMF with glycerol toward valuable diol monomers. Adapted from ref. 795. | ||
As discussed in section 2.5.4, the stabilization of HMF by protecting group chemistry not only can inhibit the degradation of HMF, but also improve the efficiency and selectivity of upgrading HMF. Recently, a series of studies demonstrated that the protection of the formyl group in HMF via acetylation is an effective strategy to reduce the undesirable oligomerization reactions, thus enabling the upgrading of HMF to high-value products via selective oxidation, oxidative esterification, hydrogenation and Diels–Alder reaction at a high substrate loading with the reduced formation of humins. Kim et al. reported that the acetylation of HMF with 1,3-propanediol (Fig. 73a) gives an HMF acetal derivate (PD-HMF) with obviously higher thermal stability than the corresponding acetal derivates (MeO-HMF, EG-HMF) obtained from HMF with methanol and ethylene glycol, and the aerobic oxidation of PD-HMF (Fig. 73b) over a CeO2-supported Au catalyst in Na2CO3 aqueous solution afforded high FDCA yields (90–95%) even at a high substrate loading (10–20 wt%) without the generation of humins.796 Owing to the six-membered acetal ring structure, the thermal decomposition and self-polymerization of HMF at a high substrate loading were remarkably inhibited. The use of an HMF acetal derivate (PD-HMF) to displace HMF as the starting material was also effective for the selective production of FDCA-derived esters via oxidative esterification at a high substrate loading.797 The aerobic oxidative esterification of a high concentration PD-HMF (10–20 wt%) with methanol and ethylene glycol over an Au/CeO2 catalyst attained 80–95% yields of MFDC and bis(2-hydroxyethyl)furan-2,5-dicarboxylate (HEFDC) (Fig. 73c). Since high yield can be obtained at a high substrate loading, this strategy can greatly improve the reaction efficiency and economics for the subsequent production of PEF.
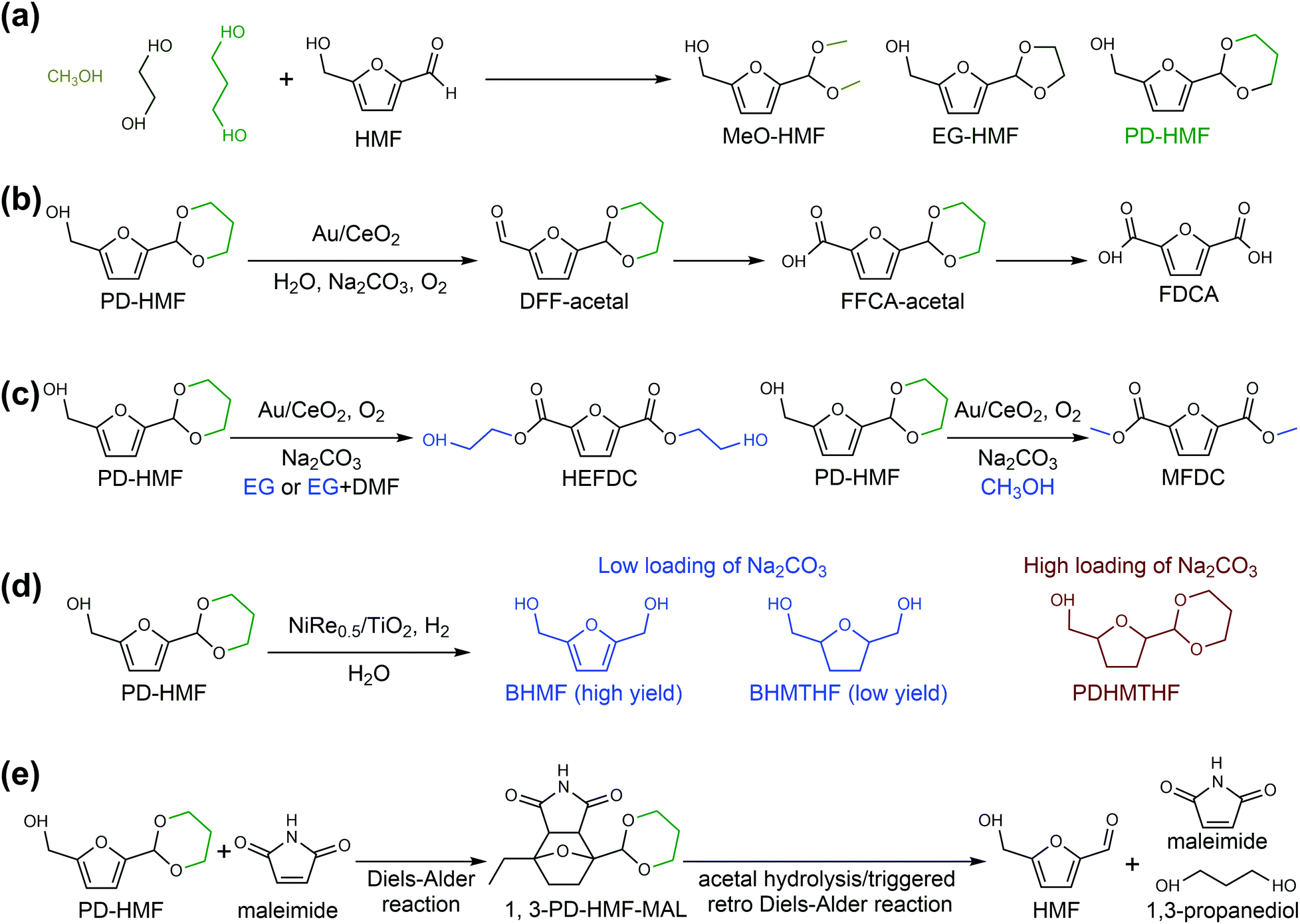 | ||
| Fig. 73 (a) Acetylation of HMF with methanol, ethylene glycol and 1,3-propanediol, and (b) selective oxidation of PD-HMF to FDCA in Na2CO3 aqueous solution. Adapted from ref. 796. (c) Selective oxidation of PD-HMF to bis(2-hydroxyethyl)furan-2,5-dicarboxylate (HEFDC) and methyl furan-2,5-dicarboxylate (MFDC). Adapted from ref. 797. (d) Selective hydrogenation of PD-HMF. Adapted from ref. 798. (e) Selective hydrogenation of PD-HMF to BHMF. Adapted from ref. 799. | ||
Similar to the selective oxidation of PD-HMF, the use of an acetalized HMF derivate to displace HMF for the hydrogenation reaction could also inhibit the undesirable oligomerization. Wiesfeld et al. showed that the hydrogenation of a concentrated PD-HMF aqueous solution over bimetallic Ni–Re supported on TiO2 at a carefully controlled pH (about 10−3 equivalent of Na2CO3 with respect to PD-HMF) gave a BHMF yield of 81–89% (Fig. 73d).798 At neutral pH, a certain amount of unprotected HMF (9%) is formed since the deprotection rate is faster than the hydrogenation rate, while a high pH leads to a low deprotection rate and essentially promotes the undesirable hydrogenation toward 5-hydroxymethyl tetrahydrofuran (PDHMTHF).798
Furthermore, Chang et al. showed that the acetalized HMF derivate could be converted to norcantharimide derivates via Diels–Alder reaction with maleimides, while the hydrolysis of the acetal linkage mismatches the molecular orbital of norcantharimide and then triggers a retro Diels–Alder reaction at ambient temperature, leading to the controlled release of high-value chemicals at 35–60 °C.799 This process is promising for the development of drug delivery systems at human body temperature.
3.6. Aldol condensation of HMF with carbonyl or hydroxyl group-containing compounds
Owing to the low octane number of HMF and furfural-derived liquid alkanes, which are determined by their relatively low carbon chain lengths, they cannot be directly used as transportation fuel. Thus, aldol condensation of furan aldehydes with carbonyl or hydroxyl group-containing compounds offers an effective approach for the desirable chain extension toward desired fuel precursors, which can be further converted into liquid alkanes with sufficient energy density and octane number.Aldol condensation of HMF with acetone using NaOH as a catalyst not only can produce desirable fuel precursors, including (E)-4-[5-(hydroxymethyl)-2-furanyl]-3-butene-2-one (HA) and (E,E)-1,5-bis(5-hydroxymethyl-2-furanyl)-1,4-pentadiene-3-one (HMF-acetone-HMF dimer, HAH) (Fig. 74a), but also lead to undesirable by-products via self- and cross-condensation reactions, resulting in a low selectivity and yield of the desired products.800,801 The undesirable side reactions between HMF and acetone can be inhibited by operating at low temperature (308 K) and low NaOH concentration (∼110 mM), and the selectivity toward HA or HAH can be tuned by regulating the initial concentration of HMF and acetone.802 Chang et al. reported that HAH could be obtained via a two-step process involving the dehydration of fructose to HMF over HCl in water/acetone medium and aldol condensation of HMF with acetone over NaOH, attaining an overall yield of 74.1% at an MSP of $1958 per ton.803 Owing to the low cost, HAH may displace anthraquinone and bisphenol-A as feedstock for the synthesis of organic dye and polyether. The aldol condensation of HMF with acetone toward HA using MgAl and MgZr mixed oxides as catalysts was investigated by Cueto et al.804 Owing to its appropriate acid/base site ratio with medium strength, the MgZr catalyst gave the highest whole yield (37%) of aldol condensation products. After three consecutive runs, the MgAl mixed oxide lost its catalytic activity completely, while the HMF conversion over the MgZr mixed oxide decreased to below 20%. Suttipat et al. reported that the composite of hierarchical faujasite nanosheets and zeolitic imidazolate framework-8 (Hie-FAU-ZIF-8) could function as an effective acid–base bifunctional catalyst for the aldol condensation of HMF with acetone, attaining an HA yield of up to 67% owing to the synergistic effect of the Na+-stabilized zeolite framework and the imidazolate linker bearing basic nitrogen groups.805 Yutthalekha et al. reported that amine-grafted hierarchical basic FAU-type zeolite nanosheets exhibited an excellent catalytic performance for the aldol condensation of HMF and acetone, attaining an HA yield approaching 100%.806 Zhang et al. suggested that the coexistence of acidic silanol and basic alkylamine bifunctional groups on the SBA-15 catalyst may generate a synergistic effect for the aldol condensation of HMF with acetone by combining quantum mechanical and molecular mechanical calculations.807 They proposed that [–SiOH], as a bridge of H shift, promotes the activation of acetone toward enol-acetone, while [–RNH2] mainly catalyzes the aldol condensation of enol-acetone with HMF and the subsequent dehydration. Malkar et al. reported that the aluminum-exchanged dodecatungstophosphoric acid encapsulated inside the cage of ZIF-8 (Al0.66-DTP@ZIF-8) was active for the selective synthesis of HA from HMF and acetone, attaining the maximum HMF conversion of 99% with HA selectively of 84.11%.808 The high HA selectivity is attributed to the kinetically controlled reaction pathway since the activation energy (63.1 kJ mol−1) for the formation of HA is remarkably lower than that (102.1 kJ mol−1) for the formation of HAH.
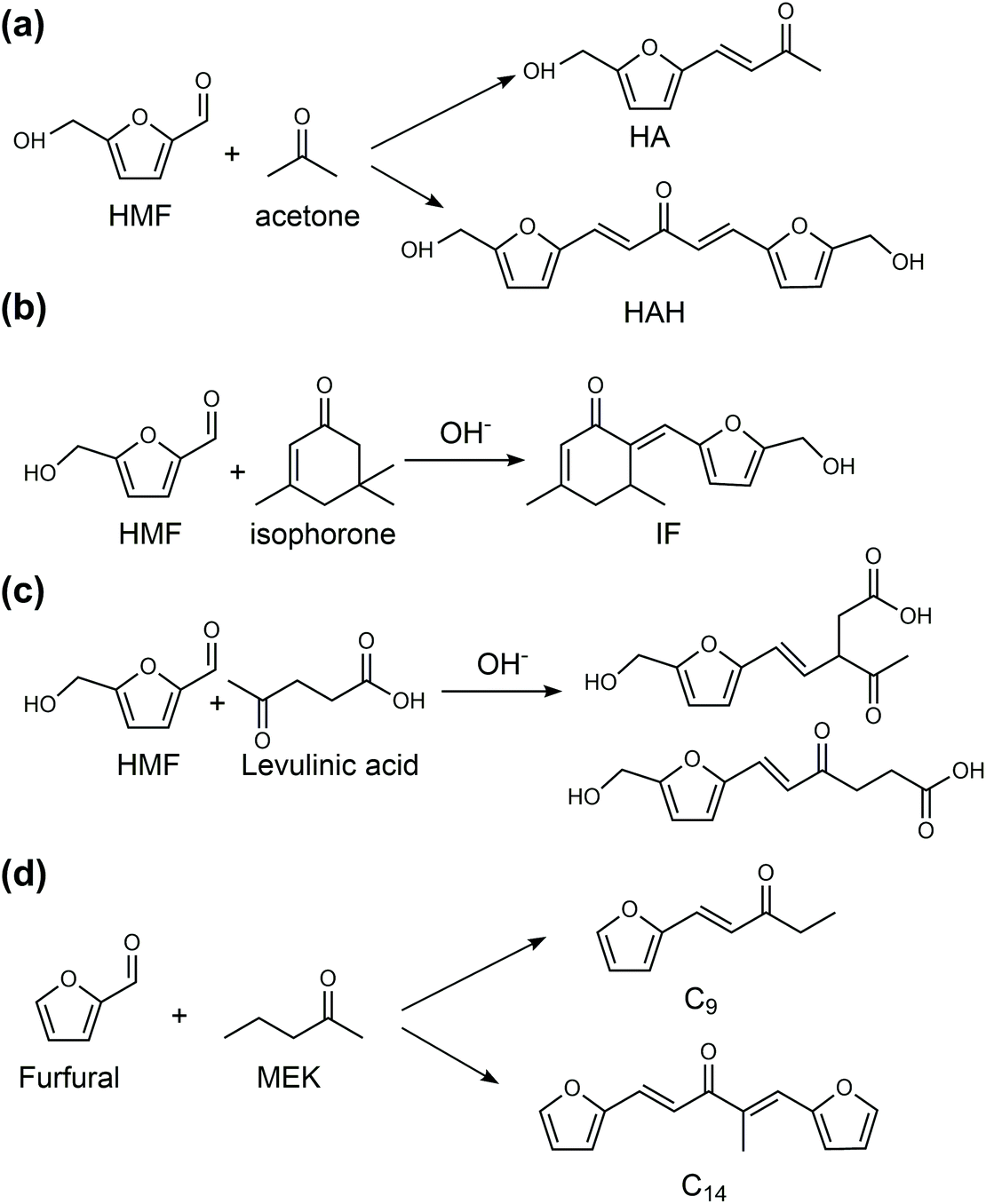 | ||
| Fig. 74 (a) Aldol condensation of HMF with acetone. Adapted from ref. 800. (b) Aldol condensation of HMF with isophorone. Adapted from ref. 809. (c) Aldol condensation of HMF with levulinic acid. Adapted from ref. 810. (d) Aldol condensation of furfural with MEK. Adapted from ref. 811. | ||
In addition to acetone, other carbonyl or hydroxyl group-containing compounds have also been used for the carbon chain extension of HMF. The aldol condensation of isophorone with HMF (Fig. 74b) over NaOH under solvent-free conditions followed by hydrodeoxygenation could give multi-substituted cycloalkanes with high density and low freezing point.809 The aldol condensation between HMF and levulinic acid (Fig. 74c) toward (E)-6-[5-(hydroxymethyl)furan-2-yl]-hex-4-oxo-5-enoic acid and (E)-3-[5-(hydroxymethyl)furan-2-yl]methylene-4-oxo-pentanoic acid can be achieved using NaOH as a catalyst.810,812 Similarly, the selective aldol condensation of levulinic acid and furfural in the aqueous phase was achieved over MgO and ZnO catalysts.813
Similar to HMF, the aldol condensation of furfural with carbonyl or hydroxyl group-containing compounds has also been investigated widely. Jing et al. reported that the Nb2O5 catalyst exhibited good catalytic activity and stability in the aldol condensation of furfural with acetone, 4-heptanone or 2,4-pentanedione, which is superior to other common solid acid and base catalysts, including ZrO2, Al2O3, MgO, CaO and magnesium–aluminum hydrotalcite.814 They proposed that Nb2O5 could activate the CO bond in the carbonyl group-containing molecules, facilitate the formation of a metal enolate intermediate, and then promote the formation of a C–C bond via nucleophilic addition. Moreover, the multifunctional Pd/Nb2O5 catalyst enabled a one-pot, two-step process for the conversion of furfural to liquid alkanes. The carbon chain of furfural can also be extended via aldol condensation with methyl ethyl ketone (MEK) (Fig. 74d), the dehydration product of biomass-derived 2,3-butanediol (BD), under alkaline conditions to obtain C9/C14 fuel precursors.811
The self-coupling reaction is an useful approach to produce value-added chemicals.815 Both HMF and furfural can be converted to dimers though etherification or aldol condensation reaction.816 Amarasekara et al. reported that fructose could be converted to 5,5′-[oxybis(methylene)]bis[2-furaldehyde] (OBMF) in DMSO using Dowex 50 W X8 as a solid acid catalyst (76% yield) via HMF as an intermediate (Fig. 75a).817 Cywar et al. designed polystyrene (PS)-supported benzimidazolium coupled with a dodecyl chain as a catalyst for the self-coupling of furfural and HMF into C10–14 furoins (Fig. 75b), obtaining the highest C10 furoin yield (monool furoin) of 95% from HMF and C14 furoin (triol DHMF) yield of 88% from furfural, respectively.818 The monool furoin obtained with the homo-coupling of furfural is a promising precursor for the production of linear and branched alkanes via hydrodeoxygenation, while HMF-derived triol DHMF can be applied for the synthesis of polyesters and PUs. Wilson et al. reported that the cross-coupling of HMF with furfural over the NHC catalyst, 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene (TPT), could give difuranic C11 diols yield of 43%.819 These difuranic C11 diols are promising feedstocks for the production of linear polyesters and polyurethanes.
 | ||
| Fig. 75 (a) Dimerization of HMF via etherification reaction. Adapted from ref. 817. (b) Homo-coupling and cross-coupling of furfural and HMF via aldol condensation. Adapted from ref. 818 and 819. | ||
The one-pot conversion of cellulose to furanic biocrude involving the conversion of cellulose to HMF and subsequent aldol condensation of HMF toward biocrude were carried out under mild conditions (120 °C, 3.5 h) in the medium of acetone, obtaining furanic biocrude yields of 25.7% and 19.3% with acidic ionic liquids 1-(3-propylsulfonic)-3-methylimidazolium chloride and 1-(4-butylsulfonic)-3-methylimidazolium chloride as catalysts, respectively.820 These furanic compounds are promising feedstock for the production of hydrocarbon fuel using the well-established catalytic hydrodeoxygenation process.
3.7. Reductive amination of HMF
N-Substituted-5-(hydroxymethyl)-2-furfuryl amines, one type of important precursor of pharmaceuticals, can be obtained via the reductive amination of HMF (Fig. 76 and Table 19) with different amines and ammonia under high pressure H2, and the one-pot reaction of HMF with nitrobenzene using Pd/C or NiyAlOx as the catalyst.821,822 Zhu et al. reported that the abundant and cheaply available CO and water could be use as reductants to displace H2 for the reductive amination of HMF toward valuable N-containing compounds using rutile titania-supported gold (Au/TiO2–R) as the catalyst, attaining a broad spectrum of primary and secondary amines and bis(hydroxylmethylfurfuryl)-amines efficiently.823 Gomes et al. reported that δ-lactone-fused cyclopenten-2-ones could be produced from HMF via the activation of HMF with Meldrum's acid followed by reacting with a secondary amine via consecutive furan ring-opening, cyclization and lactonization process.824 The δ-lactone-fused cyclopenten-2-ones with a quaternary carbon have potential in the synthetic and medicinal fields. Besides, 5-(hydroxymethyl)furfurylamine (HMFA) could be synthesized from HMF over immobilized transaminase via amination transfer reaction.825 Similar to HMF, a series of secondary and tertiary amines can be synthesized using furfural, xylose and xylan as the feedstock.389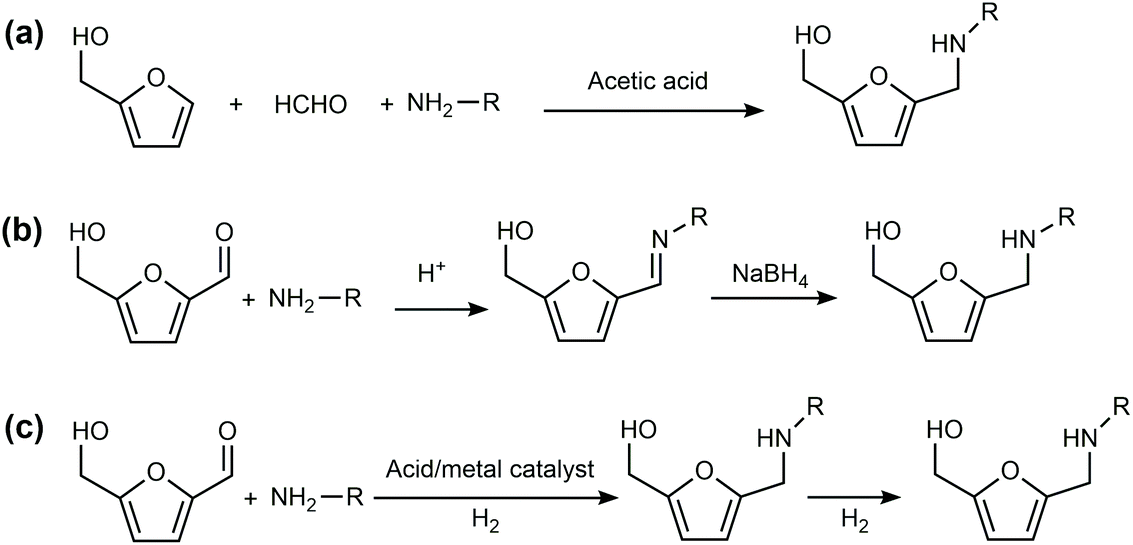 | ||
| Fig. 76 (a) Synthesis of n-substituted-5-(hydroxymethyl)-2-furfuryl amines from furfural via Mannich-type reaction. Indirect (b) and direct method (c) of reductive amination of HMF with primary amines. Adapted from ref. 821. | ||
| Catalyst | Catalyst loading | Solvent | Conditions | Feedstock | Amine Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a Relative to HMF. b Relative to solvent. — Not provided. | ||||||
| Au/TiO2-R | 0.4 mol%a | Methanol/H2O (v/v = 1) | 60 °C, 6 h, 20 bar CO | HMF/R1NH1R2 (molar ratio = 1) | 60–99% | 823 |
| Au/TiO2-R | 0.4 mol%a | Methanol/H2O (v/v = 1) | 60 °C, 6 h, 20 bar CO | HMF, RNH2 (molar ratio = 1), R = n-butyl, n-hexyl or benzyl | bis-(HMF) amines 85–93% | 823 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 1 h, 3 bar H2 | HMF/aniline (molar ratio = 1) | 100% | 821 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 3 h, 3 bar H2 | HMF/2-methylaniline (molar ratio = 1) | 76% | 821 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 1 h, 3 bar H2 | HMF/4-methylaniline (molar ratio = 1) | 98% | 821 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 1 h, 3 bar H2 | HMF/4-methoxyaniline (molar ratio = 1) | 99% | 821 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 3 h, 3 bar H2 | HMF/4-chloroaniline (molar ratio = 1) | 89% | 821 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 3 h, 3 bar H2 | HMF/4-cyanoaniline (molar ratio = 1) | 96% | 821 |
| Pd/C | 0.4 mol%a | Trifluoro toluene | 100 °C, 3 h, 3 bar H2 | HMF/4-acetyl aniline (molar ratio = 1) | 86% | 821 |
| Pd/C | 0.4 mol%a | Methanol | 100 °C, 2 h, 14 bar H2 | HMF/NH3(molar ratio = 1/10) | 94% | 821 |
| Ni1AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 32% | 822 |
| Ni2AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 38% | 822 |
| Ni4AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 85% | 822 |
| Ni6AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 99% | 822 |
| Ni8AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 99% | 822 |
| Ni10AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 96% | 822 |
| NiOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 33% | 822 |
| RANEY® Ni | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 1/49) | 48% | 822 |
| Ni6AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/benzylamine (molar ratio = 10/12) | 76% | 822 |
| Ni6AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/aniline (molar ratio = 10/12) | 85% | 822 |
| Ni6AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/morpholine (molar ratio = 10/12) | 88% | 822 |
| Ni6AlOx | 1.3 wt%b | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 10/12) | 6% | 822 |
| HReO4 | 5 mol% | H2O | 100 °C, 6 h, 1 bar H2 | HMF/NH3 (molar ratio = 10/12) | 6% | 389 |
3.8. Decarbonylation of HMF to furfuryl alcohol
As an important chemical and solvent widely employed in the foundry, refractory, polyester and pharmaceutical industries, the production of furfuryl alcohol utilizes 65% of the global furfural production capacity.33 The production of furfuryl alcohol from HMF via decarbonylation is a potential alternative approach to the industrially applied furfural hydrogenation process.826,827Through theoretical study, Xie et al. suggested that the decarbonylation of HMF to furfuryl alcohol (Fig. 77) over palladium acetate (Pd(OAc)2) involves migratory extrusion, metal-acetate-co-assisted deprotonation, decarbonylation, metal-assisted deprotonation, migratory extrusion and catalyst regeneration.828 Meng et al. reported that adding an appropriate amount of water to an organic solvent could inhibit side reactions, including hydrogenolysis, dehydrogenation and etherification, thus enabling the highly selective decarbonylation of HMF to furfuryl alcohol over the Pd/Al2O3 catalyst.826 Chatterjee et al. reported that the combined use of Pd/Al2O3 as the catalyst and compressed CO2 as the solvent enabled the highly efficient decarbonylation of HMF to furfuryl alcohol, affording a furfuryl alcohol yield as high as 99.4% without the use of any additive, CO surrogate, or organic solvent.829 The excellent catalytic performance is attributed to the synergistic effect between CO2 and Pd/Al2O3, where CO2 shifts the reaction equilibrium toward furfuryl alcohol by accelerating the diffusion of CO and furfuryl alcohol.
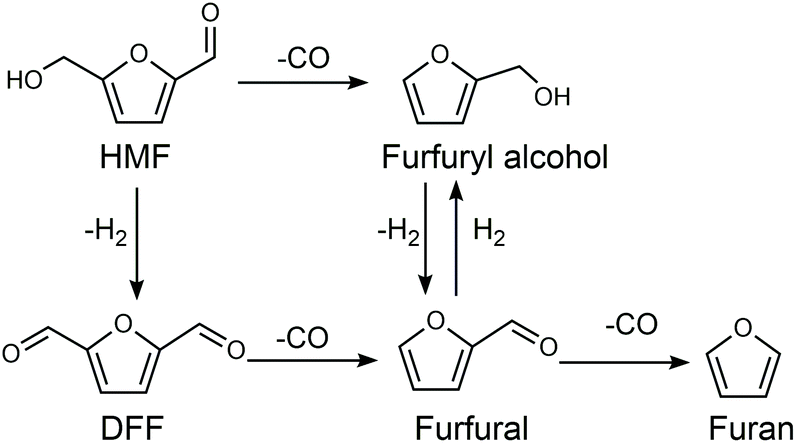 | ||
| Fig. 77 Decarbonylation of HMF to furfuryl alcohol. Adapted from ref. 826 and 829. | ||
3.9. Conversion of HMF to esters
The esterification of HMF and its derivates, including FDCA, BHMF and levulinic acid can provide a series of useful esters products. For example, the combination of catalyst, O2, base and methanol enabled the one-pot oxidative esterification of HMF toward FDMC. Compared with the direct oxidation of HMF to FDCA, the one-pot oxidative esterification of HMF to FDMC could be carried out under milder reaction conditions owing to the change in the reaction pathway (Fig. 78), thus providing an alternative approach for the efficient synthesis of FDCA. In the medium of alcohol, HMF is converted to the hemiacetal intermediate easily, and then the hemiacetal intermediate is oxidized to 5-hydroxymethyl-2-methyl-furoate (HMMF) intermediate rapidly owing to the presence of the active hemiacetal structure. The oxidation of HMMF to 5-formyl-2-methyl-furoate (FMF) is the rate-limiting step because the oxidation of the hydroxyl group to aldehyde group is more difficult than the conversion of the aldehyde group in this system. Once FMF is generated, it can be further converted to FDMC easily. For example, Li et al. reported that the heterogeneous PdCoBi/C catalyst is as effective as Pd/C with Co(NO3)2 and Bi(NO3)3 as homogeneous co-catalysts for the aerobic oxidative esterification of HMF, affording an FDMC yield of 96%.609 Sun et al. reported that the hollow yolk–shell Co@CN catalyst, which was derived from ZIF-67@SiO2@ZIF-8, exhibited an excellent catalytic performance for the oxidative esterification of HMF to FDMC in the medium of MeOH under mild conditions, obtaining an FDMC yield of 95% even at a high HMF loading (2 M).611 Liu et al. reported that an N-doped carbon-supported CoCu bimetallic catalyst (Co7Cu3–NC) was more capable than Co–NC since the doping of Cu imparts stronger O2 activation ability to the Co–Nx species.612 Buonerba et al. showed that the desired oxidation products, including DFF, FFCA, 5-hydroxymethyl furoic acid methylester (HMFE), FDCA and FDMC could be obtained from the selective oxidation of HMF over gold nanoparticles supported on a semi-crystalline nanoporous multiblock copolymer matrix consisting of syndiotactic poly(styrene)-cis-1,4-poly(butadiene) (AuNPs-sPSB) via judicious manipulation of the reaction conditions and solvents.579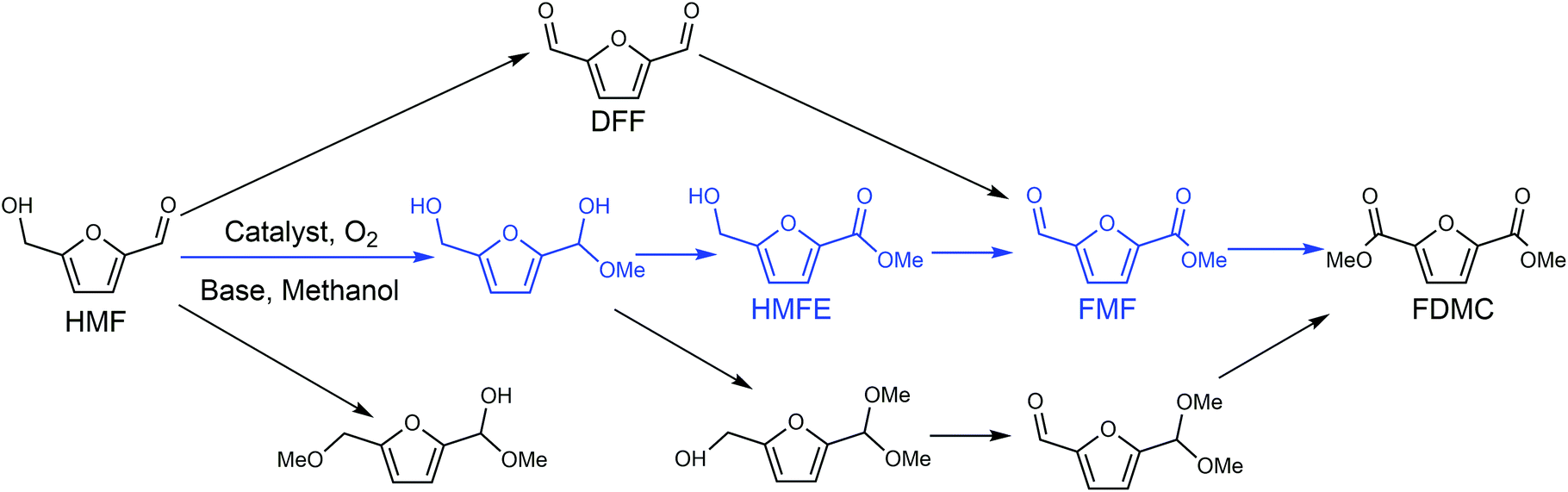 | ||
| Fig. 78 Oxidative esterification of HMF toward FDMC. Adapted from ref. 609–611. | ||
Alloyed AuPd–Fe3O4 nanoparticles attained an FDMC yield of up to 92% under atmospheric O2 at room temperature, while Au–Fe3O4 NPs favored the selective formation of HMFE.610 Kozlov et al. reported that sodium cyanide (NaCN)-promoted oxidative esterification with MnO2 as a recyclable oxygen carrier enabled the efficient conversion of HMF to FDMC in water-free medium and to HMFE in AcOH with 5% water, respectively.830 Gupta et al. reported that the combination of mesoporous alumina nanospheres-embedded with CuO nanoparticles (CuO/m-Al2O3) catalyst with TBHP as both the oxidizing and methylating reagent enabled the efficient oxidative methyl-esterification of HMF toward FDMC, which is an alternative approach to the oxidative esterification of HMF to FDMC in methanol.618
From the viewpoint of reaction, levulinic acid and formic acid can be obtained directly from the Brønsted acid-catalyzed rehydration of HMF (Fig. 12), which is one of the most ubiquitous side reactions during the synthesis of HMF from carbohydrates and biomass.831 Since the Brønsted acid is also involved in the depolymerization of cellulose or lignocellulosic biomass, the direct synthesis of levulinic acid using cellulose or lignocellulosic biomass as a feedstock via a one-pot process generally exhibits higher efficiency than that of converting carbohydrate to HMF followed by decomposition of HMF to levulinic acid and formic acid. Therefore, the use of HMF as the feedstock for the production of levulinic acid and formic acid is not a cost-effective strategy. Accordingly, the transformation of HMF to levulinic acid and formic acid is not specifically discussed in this review. Since levulinic acid is also an important biomass-derived platform chemical for the production of a series of high-value products, including levulinic esters, GVL, valeric acid and ester, butene, 5-nonanone and MeTHF (Fig. 79), the reader is referred to these reviews.832–837 GVL, MeTHF and valeric ester are promising fuel additives, while butene and 5-nonanone can be further upgraded to liquid alkanes. Formic acid is considered a sustainable hydrogen source due to its high volumetric hydrogen density (53 g of H2 per liter).838,839 Readers are referred to the related review on the direct synthesis and application of formic acid from cellulose and biomass.838
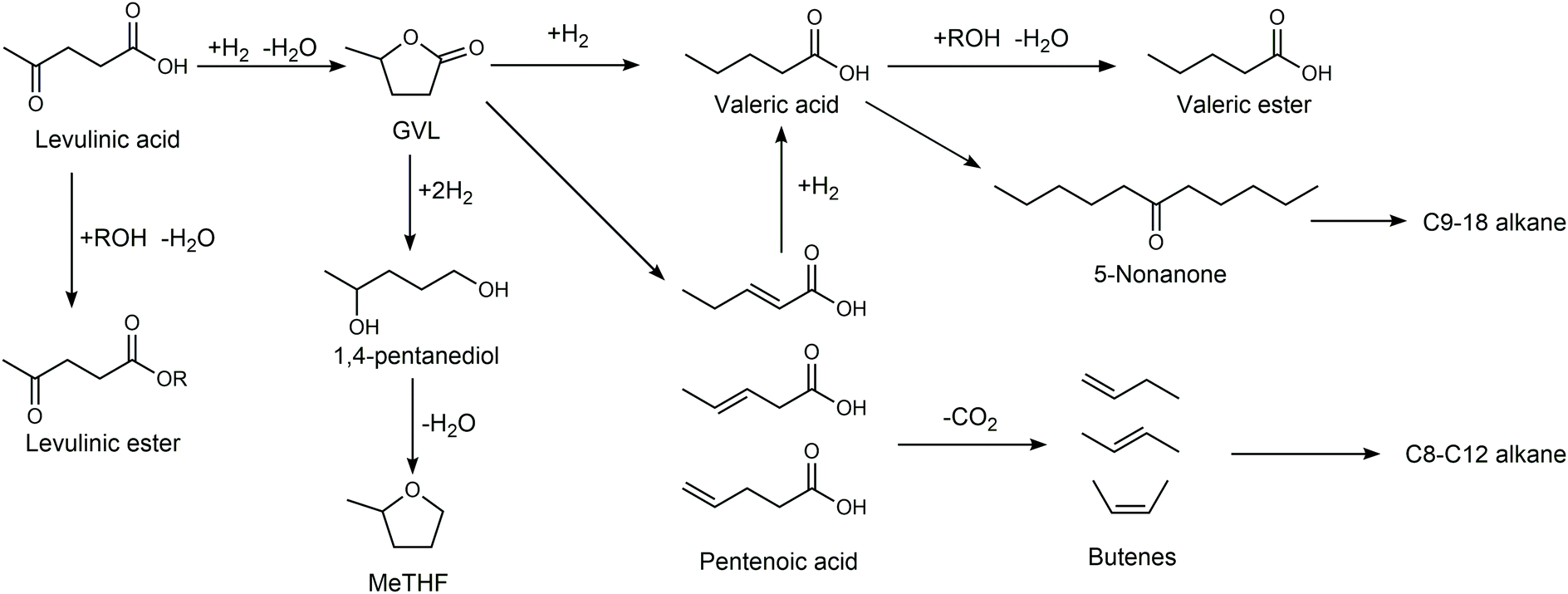 | ||
| Fig. 79 Catalytic upgrading of levulinic acid to chemicals and fuels. Adapted from ref. 836 and 837. | ||
The double esterification of BHMF with fatty acids can give 2,5-bis(hydroxymethyl)furan fatty acid diesters, which are considered promising biodiesel additives. Lăcătuş et al. reported that the lipase Novozym 435 is effective to catalyze the double esterification of BHMF with saturated long-chain fatty acids (Fig. 80a) in the medium of MeTHF with in situ removal of the generated water by molecular sieves, attaining BHMF diester yields between 89% and 99%.840 Based on this, they further indicated that the use of a fatty acid mixture resulting from the hydrolysis of commercial sunflower oil as feedstock enabled the solvent-free biocatalytic synthesis of BHMF diesters, with a conversion higher than 97%.841 In the above catalytic system, the presence of water not only inhibited the activity of the lipases, but also acted as a competitive nucleophilic reagent, thus leading to an obvious decrease in the catalytic performance. Arias et al. reported that the transesterification reaction of BHMF with vinyl esters of acids (Fig. 80b) over lipase not only could avoid the adverse influence of water and fatty acids on the enzymatic activity, but also inhibit the competitive nucleophilic attack, thus leading to a remarkable improvement in the reaction efficiency.842 A BHMF diester yield of up to 97% was obtained within only 0.5 h.
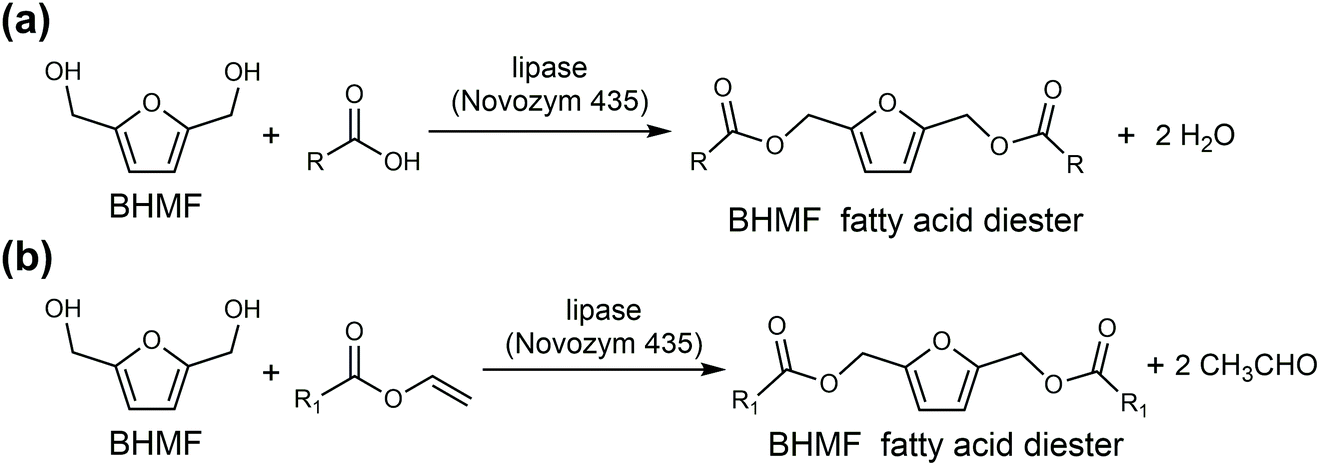 | ||
| Fig. 80 (a) Esterification of BHMF with fatty acids (R2 = H) or fatty acid esters (R2 = vinyl group) toward BHMF fatty acid diesters. Adapted from ref. 840 and 841. (b) Transesterification reaction of BHMF with vinyl esters of acids over lipase. Adapted from ref. 842. | ||
Garcia-Suarez et al. reported that the use of HMF as a CO surrogate for the methoxycarbonylation of 1-hexene to methyl hepatanoate (MH) (Fig. 81) could be conducted in the medium of methanol using Pd-(1,2-bis(di-tert-butylphosphinomethyl)benzene) (DTBPMB) as the catalyst and methanesulfonic acid (MSA) as the co-catalyst.843 In this catalytic system, the conversion of HMF to methyl levulinate (ML) via hydration and following esterification, and the hydrogenation of ML to GVL occurred concomitantly with HMF and methanol as the source of CO and H2, respectively. The MH yield of 50%, ML yield of 12% and GVL yield of 35% were obtained at 120 °C within 20 h. In addition, this catalytic system also enabled the one-pot conversion of glucose, fructose and xylose to ester products, but the yield and selectivity were low.
 | ||
| Fig. 81 Cascade transformation of 1-hexene with HMF to ester products. Adapted from ref. 843. | ||
3.10. Conversion of HMF to other products
The conversion of HMF to vinyl or acetylene-functionalized chemicals can extend the scope of HMF utilization in fine organic synthesis remarkably. For example, Han et al. reported that HMF could be converted to 5-hydroxymethyl-2-vinylfuran (HMVF), a biomass-derived solvent-free adhesive, via the Wittig reaction (Fig. 82).844 After heating or acid treatment at room temperature, the versatile adhesive could bond to a series of substrates, including metal, glass, plastic and rubber via the free radical polymerization of the vinyl group and etherification of the hydroxyl group, with an adhesive performance close to that of Krazy Glue (Loctite 401, cyanoacrylate) and superior to that of white glue (Pattex No. 710, PVA) and Pattex PKME15C epoxy glue. Romashov et al. reported that alkynylation of HMF and DFF (Fig. 83) using Ohira-Bestmann reagent (dimethyl 1-diazo-2-oxopropylphosphonate) gave a high yield of 2-hydroxymethyl-5-ethynylfuran (HMEF) and 2,5-diethynylfuran (DEF), respectively.845 The terminal alkyne units in HMEF and DEF provide facile access to various chemical transformations, including Glaser–Eglinton–Hay reaction, heterocyclization, Sonogashira coupling and polymerization toward high-value furanic pharmaceuticals and conjugated polymers.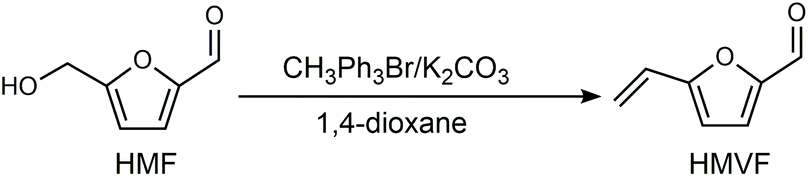 | ||
| Fig. 82 Conversion of HMF to HMVF via Wittig reaction. Adapted from ref. 844. | ||
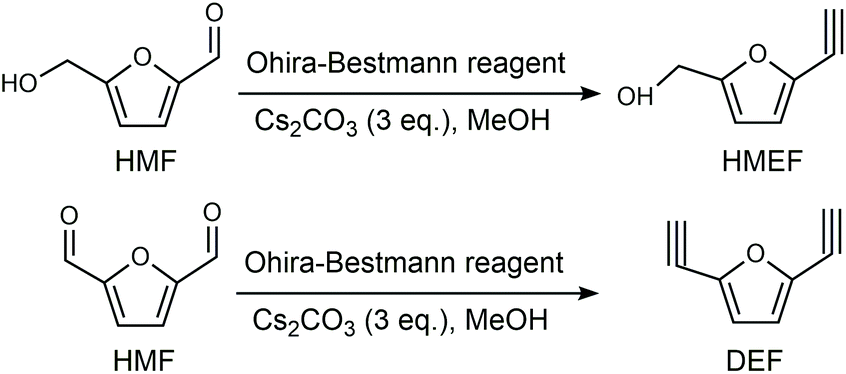 | ||
| Fig. 83 Alkynylation of HMF using Ohira-Bestmann reagent. Adapted from ref. 845. | ||
Besides the conversion of HMF to structurally symmetrical derivates, including FDCA, BHMF, 5,5′-BHMF and HMVF as building blocks of polymers, the direct use of HMF as feedstock for polymer synthesis was achieved via the step-growth copolymerization of HMF (Fig. 84) with dihydrosilane toward linear poly(silyl ether)s, in the presence of B(C6F5)3 and heteroscorpionate zinc hydride complex LZnH [L = (MePz)2CP(Ph)2NPh, MePz = 3,5-dimethylpyrazolyl].846 A series of butenolide derivates of furans with a good color performance in the yellow to red region of the visible spectrum could be fully synthesized using HMF-derived CMF or DFF as feedstock (Fig. 85).847 These butenolide derivates can be used as sustainable synthetic dyes, which are promising to displace polluting petrochemical colorants and expensive plant-based dyes. The Biginelli reaction of HMF, 1,3-dicarbonyl compounds and urea-type building blocks gave moderate to good yields of functionalized dihydropyrimidinones (Fig. 86), indicating the feasibility of using HMF as feedstock in multicomponent reactions (MCRs).848 HMF could also be used as feedstock for the preparation of N-doped graphene-like layered carbon (NG) via a metal-free pyrolysis method.849 More importantly, the HMF-derived NG exhibited superior catalytic activity for the epoxidation reaction owing to its high activation ability for both alkenes and O-2 resulting from its graphitic layered structure and graphitic N species.
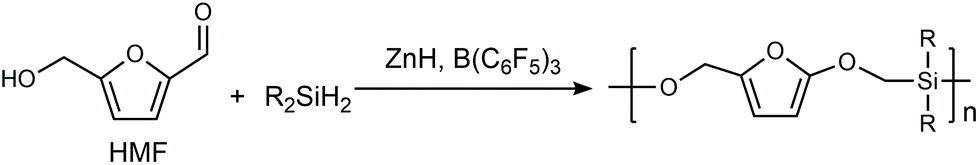 | ||
| Fig. 84 Copolymerization of HMF with dihydrosilanes into linear poly(silyl ether)s. Adapted from ref. 846. | ||
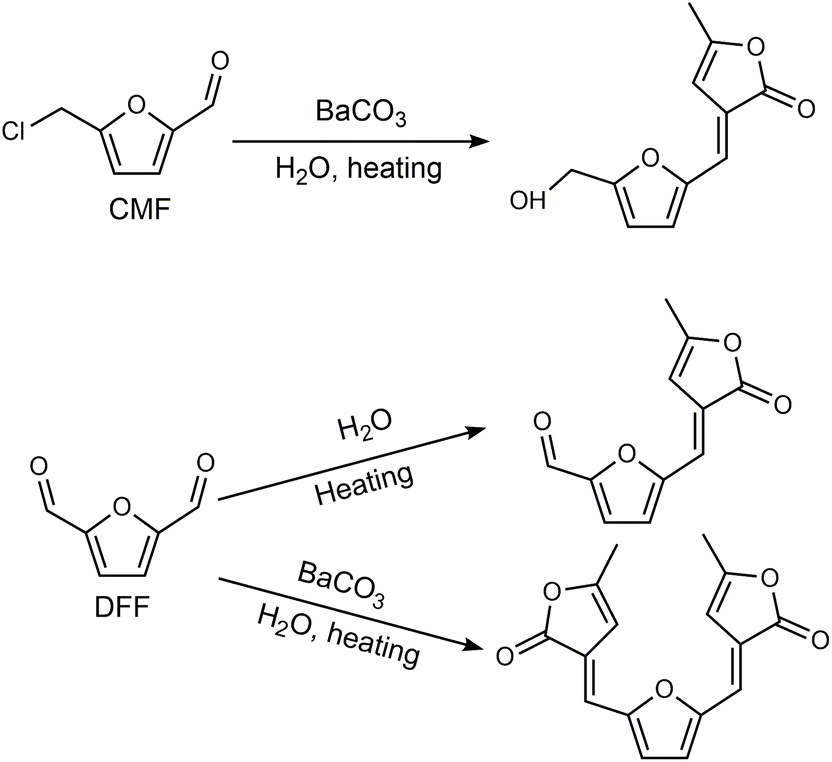 | ||
| Fig. 85 Conversion of CMF and DFF to butenolide derivates. Adapted from ref. 847. | ||
 | ||
| Fig. 86 Conversion of HMF to functionalized dihydropyrimidinones via three-component Biginelli reaction. Adapted from ref. 848. | ||
4. Future perspectives
Although considerable progress has been achieved, the high cost of HMF is still the predominant bottleneck in its commercial production and application. Thus, the economical production of HMF from carbohydrates and biomass should be paid continuous attention. To date, many research still focus on the dehydration of fructose to HMF in the medium of DMSO. In these catalytic systems, a fundamental control experiment is necessary to obtain credible catalytic activity because moderate to high HMF yields from fructose can be readily attained in DMSO under an air atmosphere without the use of additional catalysts. The separation and purification of HMF and recycling of DMSO are difficult owing to the high boiling point of DMSO and the instability of both DMSO and HMF under the reaction conditions. Similarly, the use of certain ionic liquids and biomass-derived GVL as the solvent enables the efficient conversion of fructose to HMF, but the reported high HMF yields from fructose are also likely attributed to the solvents, instead of the solid catalysts.850 The efficient conversion of fructose to HMF in low-boiling point solvents, such as water/acetone, water/THF, water/butanol and water/1,4-dioxane, have been achieved over a few elegant designed solid acid catalysts, showing the feasibility of the facile separation and purification of HMF product and solvent recycling. Therefore, much attention should be paid to the systematic investigation of the catalytic performance, stability and reusability of heterogeneous catalysts in low-boiling point solvents, the separation and purification of HMF, and the recovery and reusability of solvents, in both batch and continuous reaction, to scale up these catalytic systems.Transforming glucose into HMF with high efficiency in green solvents is still challenging.851 The ionic liquid-based homogeneous catalytic systems, such as EMIMCl/CrCl2, BMIMCl/CrCl3 and EMIMBr/SnCl4, are still benchmark catalytic systems for the conversion of glucose to HMF at a high glucose loading (≥10 wt%), with high reaction efficiency, but they suffer from the difficulty of catalyst and ionic liquid recycling and product separation. A few solid catalysts have exhibited both good catalytic performance and reusability for the conversion of glucose to HMF in certain ionic liquids, which can serve as both the reaction medium and highly effective catalyst for the dehydration of fructose, with a reaction efficiency comparable to that of ionic liquid-based homogeneous catalytic systems. Therefore, the search for alternative solvents that can serve as both the solvent and catalyst provides a powerful strategy for the efficient conversion of glucose to HMF. Meanwhile, the development of novel ionic liquid recycling technology is also a promising solution to alleviate the high cost of ionic liquids. In several water–organic biphasic systems, the combination of a Lewis acid (or base and isomerase) with a Brønsted acid has been widely investigated for the conversion of glucose to HMF. The ratio of catalyst, solvent and reaction condition needs to be regulated carefully to inhibit the side reactions, but the degradation of HMF seems to be unavoidable because the activation energy for the degradation of HMF to levulinic and formic acids is usually lower than that of fructose dehydration (Table 1).13,852 A relatively high HMF yield and selectivity can only be achieved at a low substrate loading (≤5 wt%), seriously impeding the improvement of the intrinsic reaction efficiency. Whether or not the combinations of Lewis acid (or base and isomerase) with Brønsted acid can convert glucose to HMF with high intrinsic efficiency in separable low-boiling point solvents, under batch or continuous condition, is still mostly uncharted territory.
Compared with glucose, the use of cellulose as a starting material is attractive but more challenging. The transfer of HMF from the reaction phase to the extraction phase can inhibit the degradation of HMF to some extent, but this degradation under reaction condition cannot be avoided completely since the activation energy for cellulose hydrolysis to glucose63 is remarkably higher than that of glucose isomerization to fructose, fructose dehydration to HMF and HMF degradation (Table 1). Although unrealistically high yields have been claimed in some reports, these results do not seem to be creditable, as is illustrated by the absence of a receivable reaction mechanism and lack of subsequent work to verify or further improve these catalytic systems. Therefore, the systematic evaluation of previously reported catalysts and new catalysts under standardized reaction conditions and their comparison with the widely validated benchmark catalytic systems, such as DMSO/Brønsted acid catalytic system, for the dehydration of fructose to HMF and BMIMCl/CrCl3 catalytic system for the dehydration of glucose to HMF, are necessary. To avoid inconspicuous mistakes, HMF degradation experiments under optimized reaction conditions are highly recommended for any new catalyst and solvent. The stabilization or protection of HMF seems to be a more feasible strategy to inhibit the degradation of HMF. For the direct conversion of lignocellulosic biomass to HMF, the reaction is usually accompanied by the conversion of hemicellulose to furfural, and thus the accurate quantitative analysis of HMF and furfural is required. Furthermore, understanding the influence of inorganic elements, heavy metals, nitrogen, phosphorus, sulfur, and chlorine species9,213,853 on the production and upgrading of HMF is required for the improvement of the reaction efficiency and avoidance of secondary pollution. Besides, food waste is continuously increasing and becoming an important waste biomass resource for the production of HMF.854
The development of multifunctional solid catalytic materials, such as Brønsted–Lewis acid, acid–base, and metal particles–acid or base bi-functional catalysts, is very important to achieve the efficient and environmentally-friendly conversion of glucose and glucose-based polymers to HMF and its derivates.855 Nevertheless, the targeted regulation of Brønsted acid, Lewis acid and base sites and the morphology and pore structure of heterogenous catalysts is difficult. Thus, to guide the rational design of solid acid catalysts, a detailed, quantitative analysis of their functional groups and stability under the reaction conditions is necessary.444 The intensity and nature of acid/base sites, surrounding chemical environment and solvent also have an important influence on the actual catalytic performance.856 Therefore, developing advanced ex situ and/or in situ analysis techniques may be beneficial for the precise quantitative and qualitative characterization of the acid/base sites and analysis of the structural features of solid catalysts.228,857 Based on the detailed characterization of catalysts and carefully designed catalytic experiments, the structure–activity relationship should be established to guide further the design and synthesis of novel catalysts. Besides, the activity, selectivity, durability and reusability of heterogenous catalysts should be tested under actual reaction conditions.
Screening cost-effective, environmentally-friendly and cheap solvents is also important for the efficient production of HMF. Recently, the selective dehydration of fructose to HMF489 and isomerization of glucose to fructose284 in readily separable organic solvents, and the efficient conversion of glucose in pure water239,312,447 have been achieved over a few of well-designed heterogenous catalysts. The effectiveness of the developed heterogenous catalysts and the proposed reaction mechanism should further be tested in low-boiling point solvents. In addition, several solvent systems, especially GVL/water can give moderate HMF yields from glucose with only a Brønsted acid, suggesting that the reaction pathway is different from that in conventional water/organic solvent systems. Therefore, more technologies should be attempted to reveal the actual roles of solvents and their contributions in the production of HMF. Besides, the obvious catalytic effect of trace metal ions in ionic liquids, seawater or biochar for hydrolysis, isomerization and dehydration has been demonstrated extensively. Some anions such as Br−, BF4−, PO43−, HPO42−, H2PO4− and SO42−, which play important roles in the isomerization and dehydration reaction, may result from the catalyst, solvent or biomass.130,134,420 Therefore, the comprehensive quantitative analysis of the composition of the catalyst, solvent and biomass is of great importance for understanding the real catalytic species.
The improvement of these catalytic systems not only need the collaborative innovations of catalyst and solvent, but also involve the establishment of effective methods to evaluate intrinsic catalytic activity and actual reaction efficiency. To accurately analyze the HMF yield and selectivity, the concentration of the products should not be analyzed by UV-vis spectrophotometry. UV-vis spectrophotometry cannot reflect the real HMF concentration since furfural and other chemicals also have a strong adsorption at 284 nm. The comparison of the conversion, yield and selectivity of different catalytic systems should consider the reaction conditions, including temperature, time, substrate concentration, catalyst loading, and solvent composition. For instance, a high HMF yield and selectivity are widely reported in previous reports at a very low substrate loading, but their actual reaction efficiency is low. Although initial reaction rates, turnover frequency (TOF) and activation energy are highly recommended to evaluate the intrinsic catalytic performance for many important reactions, the reliable determination of these parameters in biomass conversion is difficult.858 Consequently, the application of these parameters in the conversion of carbohydrates to HMF is limited. In contrast, the average conversion rate and the HMF formation rate per catalytic system under optimized conditions seem to be more useful indicators to compare the actual efficiency of different catalytic systems. In these complicated catalytic systems, comprehensive control experiments are required to estimate the contribution of different catalyst species, water and organic solvents for polysaccharide depolymerization, glucose isomerization, fructose dehydration and HMF degradation, or at least to identify their actual influence on the above processes.
In addition to the synthesis of HMF from carbohydrates and biomass, the upgrading of HMF to high-value products via hydrodeoxygenation, oxidation, esterification, etherification, amination, and aldol condensation has also achieved huge progress. However, the efficient upgrading of HMF to target products with high yield is usually achieved when using pure HMF as the starting material at a low initial concentration, limiting the actual efficiency. The design and development of metal nanoparticle catalysts with tailored structures, bi-metal or multi-metal catalysts, single atom catalysts with desirable electronic structures and coordination environment and composite catalysts will be beneficial for the improvement in catalytic performance.859 Moreover, HMF stabilization and one-pot catalysis are general strategies to reduce the degradation of HMF during its upgrading, and then to improve the overall yield and efficiency. As an underdeveloped field, understanding the HMF stabilization chemistry and establishing an HMF stabilization toolbox may greatly improve the efficiency and selectivity of HMF upgrading. Currently, the one-pot conversion of carbohydrates and biomass to desired products with HMF as the intermediate only succeeds in limited reaction pathways, such as the conversion of fructose to DFF and AMF. The one-pot conversion of glucose and its polymers suffers from severe side-reactions. Compared with one-pot catalytic systems, the development of tandem catalytic systems 483 consisting of a sequence of precisely tailored catalytic steps over multiple catalysts offers an opportunity to improve the production efficiency and selectivity fundamentally.860 By virtue of the precise control of active sites, the accurate cascade of depolymerization, isomerization, dehydration and upgrading reactions in the appropriate sequence with inhibited side reactions will be achieved in the near future. With the rapid development of metal catalysis, organocatalysis, photocatalysis, electrocatalysis, heterocatalysis and biocatalysis, the combination of different catalytic strategies may provide numerous opportunities to achieve higher yields, higher selectivity, lower costs and more environmental benefits.861,862
The large-scale production and application of HMF and its derivates not only require their efficient production from carbohydrates and biomass, but also upstream and downstream technologies, including biomass pretreatment and fractionation, catalyst synthesis, energy, and by-product utilization.863 For example, digging out low-volume, high-value product streams not only will create new products and market opportunities, but also reduce the overall cost of biorefineries.25,686 In addition, the integration of the biorefinery process with the current industrial catalytic processes provides an effective strategy for the co-production of value-added products with higher efficiency.745 Once one major molecule in the chain can be produced and applied in industry, the economic feasibility of the product tree will improve greatly.1 Although predicting which product or process will be technically, economically, and environmentally viable at an industrial scale is challenging, the principles of green chemistry and green engineering should be considered in the initial design stage of a biorefinery.864 Furthermore, techno-economic analysis, and life cycle assessment combined with uncertainty assessment should be performed to evaluate the feasibility of emerging biorefinery technology.686
5. Conclusions
Although considerable progress has been achieve, the catalytic conversion of biomass and their constituent carbohydrates to HMF and the upgrading of HMF towards value-added products still suffer from low reaction efficiency and high cost in the competition against petroleum-derived chemicals and fuels. The commercial production of HMF from carbohydrates and biomass on a large scale requires the collaborative innovation of catalyst and solvent to improve the intrinsic reaction efficiency, and the establishment of economically feasible HMF separation and purification methods. Meanwhile, the development of highly effective catalytic systems and the exploration of novel reaction pathways and products are still necessary to accelerate the upgrading of HMF towards value-added products. The HMF stabilization and tandem catalysis strategy has shown great promise to reduce side reactions, and consequently improve the reaction efficiency. The integration of upstream and downstream technologies, including biomass pretreatment and fractionation, catalyst synthesis, energy, and by-product utilization are necessary to improve the overall output. Furthermore, more efforts are required to evaluate the actual efficiency, economic and technical feasibility and environmental impact of the integrated biorefinery process from raw materials to final commodities using a standardized evaluation methodology to determine which process can be ultimately applied in practical production. We believe that the development of efficient and economic HMF-based biorefinery technologies will greatly promote the establishment of a more sustainable industry in the near future.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21878163), the China Postdoctoral Science Foundation (2018M640231), the Fundamental Research Funds for the Central Universities, Natural Science Foundation of Tianjin, China (17JCZDJC39500), National Key Research Project (2018YFD080083-03), Science and Technology Demonstration Project of Industrial Integration and Development of Tianjin, China (17ZXYENC00100), National Natural Science Foundation of China (51708301), and Young Elite Scientists Sponsorship Program by Tianjin (TJSQNTJ-2018-06).References
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- X. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS.
- D. E. Resasco, B. Wang and D. Sabatini, Nat. Catal., 2018, 1, 731–735 CrossRef.
- Q. Hou, M. Ju, W. Li, L. Liu, Y. Chen and Q. Yang, Molecules, 2017, 22, 490 CrossRef.
- M. Guo, W. Song and J. Buhain, Renewable Sustainable Energy Rev., 2015, 42, 712–725 CrossRef CAS.
- N. Luo, T. Montini, J. Zhang, P. Fornasiero, E. Fonda, T. Hou, W. Nie, J. Lu, J. Liu, M. Heggen, L. Lin, C. Ma, M. Wang, F. Fan, S. Jin and F. Wang, Nat. Energy, 2019, 4, 575–584 CrossRef CAS.
- W.-J. Liu, W.-W. Li, H. Jiang and H.-Q. Yu, Chem. Rev., 2017, 117, 6367–6398 CrossRef CAS.
- A. R. C. Morais, A. M. da Costa Lopes and R. Bogel-Łukasik, Chem. Rev., 2015, 115, 3–27 CrossRef CAS.
- N. M. Eagan, M. D. Kumbhalkar, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Nat. Rev. Chem., 2019, 3, 223–249 CrossRef CAS.
- X. Zhao, H. Zhou, V. S. Sikarwar, M. Zhao, A.-H. A. Park, P. S. Fennell, L. Shen and L.-S. Fan, Energy Environ. Sci., 2017, 10, 1885–1910 RSC.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- L. Petridis and J. C. Smith, Nat. Rev. Chem., 2018, 2, 382–389 CrossRef CAS.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS.
- B. Mostofian, C. M. Cai, M. D. Smith, L. Petridis, X. Cheng, C. E. Wyman and J. C. Smith, J. Am. Chem. Soc., 2016, 138, 10869–10878 CrossRef CAS.
- P. Langan, L. Petridis, H. M. O'Neill, S. V. Pingali, M. Foston, Y. Nishiyama, R. Schulz, B. Lindner, B. L. Hanson, S. Harton, W. T. Heller, V. Urban, B. R. Evans, S. Gnanakaran, A. J. Ragauskas, J. C. Smith and B. H. Davison, Green Chem., 2014, 16, 63–68 RSC.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS.
- C.-C. Chen, L. Dai, L. Ma and R.-T. Guo, Nat. Rev. Chem., 2020, 4, 114–126 CrossRef.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y.-S. Ko, W. D. Jang and Y.-S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9, 2939–2977 RSC.
- J. Song, C. Chen, S. Zhu, M. Zhu, J. Dai, U. Ray, Y. Li, Y. Kuang, Y. Li, N. Quispe, Y. Yao, A. Gong, U. H. Leiste, H. A. Bruck, J. Y. Zhu, A. Vellore, H. Li, M. L. Minus, Z. Jia, A. Martini, T. Li and L. Hu, Nature, 2018, 554, 224–228 CrossRef CAS.
- T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Rev. Chem., 2018, 2, 35–46 CrossRef CAS.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv., 2018, 4, eaau2968 CrossRef.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- Q. Hou, W. Li, M. Ju, L. Liu, Y. Chen, Q. Yang and J. Wang, Carbohydr. Polym., 2015, 133, 517–523 CrossRef CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- X. Li, R. Xu, J. Yang, S. Nie, D. Liu, Y. Liu and C. Si, Ind. Crops Prod., 2019, 130, 184–197 CrossRef CAS.
- G. P. Perez, A. Mukherjee and M. J. Dumont, J. Ind. Eng. Chem., 2019, 70, 1–34 CrossRef.
- K. Kohli, R. Prajapati and B. K. Sharma, Energies, 2019, 12, 40 CrossRef.
- X. Zou, C. Zhu, Q. Wang and G. Yang, Biofuels, Bioprod. Biorefin., 2019, 13, 153–173 CrossRef CAS.
- B. Agarwal, K. Kailasam, R. S. Sangwan and S. Elumalai, Renewable Sustainable Energy Rev., 2018, 82, 2408–2425 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS.
- J. G. de Vries, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2017, vol. 121, pp. 247–293 Search PubMed.
- L.-L. Zhang, Y. Kong, X. Yang, Y.-Y. Zhang, B.-G. Sun, H.-T. Chen and Y. Sun, J. Sci. Food Agric., 2019, 99, 2340–2347 CrossRef CAS.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- C. Thoma, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544–3564 CrossRef CAS.
- F. Menegazzo, E. Ghedini and M. Signoretto, Molecules, 2018, 23, 2201 CrossRef.
- J. Howard, D. W. Rackemann, J. P. Bartley, C. Samori and W. O. S. Doherty, ACS Sustainable Chem. Eng., 2018, 6, 4531–4538 CrossRef CAS.
- Y. Nie, Q. Hou, C. Bai, H. Qian, X. Bai and M. Ju, J. Cleaner Prod., 2020, 274, 123023 CrossRef CAS.
- Y. Su, G. Chang, Z. Zhang, H. Xing, B. Su, Q. Yang, Q. Ren, Y. Yang and Z. Bao, AIChE J., 2016, 62, 4403–4417 CrossRef CAS.
- J. Wang, J. Xi and Y. Wang, Green Chem., 2015, 17, 737–751 RSC.
- S. S. Chen, D. C. W. Tsang and J.-P. Tessonnier, Appl. Catal., B, 2020, 261, 118126 CrossRef.
- I. Delidovich and R. Palkovits, ChemSusChem, 2016, 9, 547–561 CrossRef CAS.
- H. Li, S. Yang, S. Saravanamurugan and A. Riisager, ACS Catal., 2017, 7, 3010–3029 CrossRef CAS.
- V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS.
- J. Tang, L. Zhu, X. Fu, J. Dai, X. Guo and C. Hu, ACS Catal., 2017, 7, 256–266 CrossRef CAS.
- J. Tang, X. Guo, L. Zhu and C. Hu, ACS Catal., 2015, 5, 5097–5103 CrossRef CAS.
- J. M. Carraher, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2015, 5, 3162–3173 CrossRef CAS.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, K. Ma, W. A. Elliott, P. Bai, R. L. Johnson, T. W. Walker, B. H. Shanks, R. M. Rioux, M. Neurock and J. A. Dumesic, Nat. Commun., 2019, 10, 1132 CrossRef.
- E. W. Leng, M. Mao, Y. Peng, X. M. Li, X. Gong and Y. Zhang, ChemistrySelect, 2019, 4, 181–189 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- F. Shen, S. Sun, J. Yang, M. Qiu and X. Qi, ACS Omega, 2019, 4, 11756–11759 CrossRef CAS.
- A. M. Borrero-López, E. Masson, A. Celzard and V. Fierro, Ind. Crops Prod., 2018, 124, 919–930 CrossRef.
- T.-W. Tzeng, P. Bhaumik and P.-W. Chung, Mol. Catal., 2019, 479, 110627 CrossRef.
- D. Zhou, D. Shen, W. Lu, T. Song, M. Wang, H. Feng, J. Shentu and Y. Long, Molecules, 2020, 25, 541 CrossRef CAS.
- C. Sievers, Y. Noda, L. Qi, E. M. Albuquerque, R. M. Rioux and S. L. Scott, ACS Catal., 2016, 6, 8286–8307 CrossRef CAS.
- A. Farrán, C. Cai, M. Sandoval, Y. Xu, J. Liu, M. J. Hernáiz and R. J. Linhardt, Chem. Rev., 2015, 115, 6811–6853 CrossRef.
- Y. Yang, C. Zhang and Z. C. Zhang, Wiley Interdiscip. Rev.: Energy Environ., 2018, 7, e284 Search PubMed.
- J. Song, H. Fan, J. Ma and B. Han, Green Chem., 2013, 15, 2619–2635 RSC.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516–4521 CrossRef CAS.
- B. R. Caes, T. R. Vanoosbree, F. Lu, J. Ralph, C. T. Maravelias and R. T. Raines, ChemSusChem, 2013, 6, 2083–2089 CrossRef CAS.
- N. Sun, H. Liu, N. Sathitsuksanoh, V. Stavila, M. Sawant, A. Bonito, K. Tran, A. George, K. L. Sale, S. Singh, B. A. Simmons and B. M. Holmes, Biotechnol. Biofuels, 2013, 6, 39 CrossRef CAS.
- A. S. Amarasekara, L. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021–3024 CrossRef CAS.
- H. Kimura, M. Nakahara and N. Matubayasi, J. Phys. Chem. A, 2013, 117, 2102–2113 CrossRef CAS.
- X. Guo, J. Tang, B. Xiang, L. Zhu, H. Yang and C. Hu, ChemCatChem, 2017, 9, 3218–3225 CrossRef CAS.
- M. R. Whitaker, A. Parulkar, P. Ranadive, R. Joshi and N. A. Brunelli, ChemSusChem, 2019, 12, 2211–2219 CrossRef CAS.
- L.-K. Ren, L.-F. Zhu, T. Qi, J.-Q. Tang, H.-Q. Yang and C.-W. Hu, ACS Catal., 2017, 7, 2199–2212 CrossRef CAS.
- J. Zhang, A. Das, R. S. Assary, L. A. Curtiss and E. Weitz, Appl. Catal., B, 2016, 181, 874–887 CrossRef CAS.
- G. S. Svenningsen, R. Kumar, C. E. Wyman and P. Christopher, ACS Catal., 2018, 8, 5591–5600 CrossRef CAS.
- G. R. Akien, L. Qi and I. T. Horváth, Chem. Commun., 2012, 48, 5850–5852 RSC.
- S. Jia, Z. Xu and Z. Zhang, Chem. Eng. J., 2014, 254, 333–339 CrossRef CAS.
- Y. Yang, W. Liu, N. Wang, H. Wang, Z. Song and W. Li, RSC Adv., 2015, 5, 27805–27813 RSC.
- L. Lai and Y. Zhang, ChemSusChem, 2011, 4, 1745–1748 CrossRef CAS.
- J. Liu, Y. Tang, K. Wu, C. Bi and Q. Cui, Carbohydr. Res., 2012, 350, 20–24 CrossRef CAS.
- C.-H. Kuo, A. S. Poyraz, L. Jin, Y. Meng, L. Pahalagedara, S.-Y. Chen, D. A. Kriz, C. Guild, A. Gudz and S. L. Suib, Green Chem., 2014, 16, 785–791 RSC.
- R.-J. van Putten, J. C. van der Waal, M. Harmse, H. H. van de Bovenkamp, E. de Jong and H. J. Heeres, ChemSusChem, 2016, 9, 1827–1834 CrossRef CAS.
- T. W. Walker, A. K. Chew, R. C. Van Lehn, J. A. Dumesic and G. W. Huber, Top. Catal., 2020, 63, 649–663 CrossRef CAS.
- V. Vasudevan and S. H. Mushrif, RSC Adv., 2015, 5, 20756–20763 RSC.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. A. Dumesic, Nat. Catal., 2018, 1, 199–207 CrossRef CAS.
- M. A. Mellmer, C. Sener, J. M. R. Gallo, J. S. Luterbacher, D. M. Alonso and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 11872–11875 CrossRef CAS.
- M. A. Mellmer, D. Martin Alonso, J. S. Luterbacher, J. M. R. Gallo and J. A. Dumesic, Green Chem., 2014, 16, 4659–4662 RSC.
- T. W. Walker, A. K. Chew, H. Li, B. Demir, Z. C. Zhang, G. W. Huber, R. C. Van Lehn and J. A. Dumesic, Energy Environ. Sci., 2018, 11, 617–628 RSC.
- E. I. Gürbüz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270–1274 CrossRef.
- C. Sener, A. H. Motagamwala, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2018, 11, 2321–2331 CrossRef CAS.
- B. Song, Y. Yu and H. Wu, Fuel, 2019, 238, 225–231 CrossRef CAS.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS.
- W. Won, A. H. Motagamwala, J. A. Dumesic and C. T. Maravelias, React. Chem. Eng., 2017, 2, 397–405 RSC.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- J. He, M. Liu, K. Huang, T. W. Walker, C. T. Maravelias, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 3642–3653 RSC.
- R. Weingarten, A. Rodriguez-Beuerman, F. Cao, J. S. Luterbacher, D. M. Alonso, J. A. Dumesic and G. W. Huber, ChemCatChem, 2014, 6, 2229–2234 CrossRef CAS.
- A. H. Motagamwala, K. Huang, C. T. Maravelias and J. A. Dumesic, Energy Environ. Sci., 2019, 12, 2212–2222 RSC.
- M. Li, W. Li, Y. Lu, H. Jameel, H.-M. Chang and L. Ma, RSC Adv., 2017, 7, 14330–14336 RSC.
- O. Oyola-Rivera, J. He, G. W. Huber, J. A. Dumesic and N. Cardona-Martínez, Green Chem., 2019, 21, 4988–4999 RSC.
- S. H. Krishna, T. W. Walker, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 129–138 CrossRef CAS.
- S. Tschirner, E. Weingart, L. Teevs and U. Prüße, Molecules, 2018, 23, 1866 CrossRef.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef.
- J. E. Romo, N. V. Bollar, C. J. Zimmermann and S. G. Wettstein, ChemCatChem, 2018, 10, 4805–4816 CrossRef.
- S. Altway, S. C. Pujar and A. B. de Haan, Fluid Phase Equilib., 2018, 475, 100–110 CrossRef CAS.
- Q. Mei, X. Q. Wei, W. T. Sun, X. H. Zhang, W. Z. Li and L. L. Ma, RSC Adv., 2019, 9, 12846–12853 RSC.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097–2128 RSC.
- P. Tundo, M. Musolino and F. Aricò, Front. Chem., 2019, 7, 300 CrossRef.
- A. Dibenedetto, M. Aresta, L. di Bitonto and C. Pastore, ChemSusChem, 2016, 9, 118–125 CrossRef CAS.
- N. A. S. Ramli and N. A. S. Amin, BioEnergy Res., 2020, 13, 693–736 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597–1600 CrossRef CAS.
- H. Li, W. Xu, T. Huang, S. Jia, Z. Xu, P. Yan, X. Liu and Z. C. Zhang, ACS Catal., 2014, 4, 4446–4454 CrossRef CAS.
- E. A. Khokhlova, V. V. Kachala and V. P. Ananikov, ChemSusChem, 2012, 5, 783–789 CrossRef CAS.
- Y.-N. Li, J.-Q. Wang, L.-N. He, Z.-Z. Yang, A.-H. Liu, B. Yu and C.-R. Luan, Green Chem., 2012, 14, 2752–2758 RSC.
- P. V. Rathod, R. B. Mujmule, W.-J. Chung, A. R. Jadhav and H. Kim, Catal. Lett., 2019, 149, 672–687 CrossRef CAS.
- T. Istasse, L. Bockstal and A. Richel, ChemPlusChem, 2018, 83, 1135–1143 CrossRef CAS.
- K. Enomoto, T. Hosoya and H. Miyafuji, Cellulose, 2018, 25, 2249–2257 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and J. R. L. Smith, Green Chem., 2009, 11, 1327–1331 RSC.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr, ChemSusChem, 2009, 2, 944–946 CrossRef CAS.
- Q. Hou, W. Li, M. Zhen, L. Liu, Y. Chen, Q. Yang, F. Huang, S. Zhang and M. Ju, RSC Adv., 2017, 7, 47288–47296 RSC.
- Q. Wang, K. Su and Z. Li, Mol. Catal., 2017, 438, 197–203 CrossRef CAS.
- J. Li, Y. Yang and D. Zhang, Chem. Phys. Lett., 2019, 723, 175–181 CrossRef CAS.
- S. Marullo, C. Rizzo and F. Danna, Front. Chem., 2019, 7, 15 CrossRef.
- Y.-S. Qu, Y.-L. Song, C.-P. Huang, J. Zhang and B.-H. Chen, Ind. Eng. Chem. Res., 2012, 51, 13008–13013 CrossRef CAS.
- Y. Qu, L. Li, Q. Wei, C. Huang, P. Oleskowicz-Popiel and J. Xu, Sci. Rep., 2016, 6, 26067 CrossRef.
- S. Siankevich, Z. Fei, R. Scopelliti, P. G. Jessop, J. Zhang, N. Yan and P. J. Dyson, ChemSusChem, 2016, 9, 2089–2096 CrossRef CAS.
- S. Siankevich, Z. Fei, R. Scopelliti, G. Laurenczy, S. Katsyuba, N. Yan and P. J. Dyson, ChemSusChem, 2014, 7, 1647–1654 CrossRef CAS.
- A. S. Kashin, K. I. Galkin, E. A. Khokhlova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 2161–2166 CrossRef CAS.
- Y. Qu, Q. Wei, H. Li, P. Oleskowicz-Popiel, C. Huang and J. Xu, Bioresour. Technol., 2014, 162, 358–364 CrossRef CAS.
- A. A. Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallett, ChemSusChem, 2019, 12, 4452–4460 CrossRef CAS.
- M. I. Alam, S. De, T. S. Khan, M. A. Haider and B. Saha, Ind. Crops Prod., 2018, 123, 629–637 CrossRef CAS.
- H. Wang, J. Cui, H. Li, Y. Zhao and J. Wang, J. Mol. Struct., 2019, 1179, 57–64 CrossRef CAS.
- J. Zhou, T. Huang, Y. Zhao, Z. Xia, Z. Xu, S. Jia, J. Wang and Z. C. Zhang, Ind. Eng. Chem. Res., 2015, 54, 7977–7983 CrossRef CAS.
- H. Chen, J. Zhou, J. Mao, J. Yin and S. Li, RSC Adv., 2016, 6, 101485–101491 RSC.
- X. Tang, M. Zuo, Z. Li, H. Liu, C. Xiong, X. Zeng, Y. Sun, L. Hu, S. Liu, T. Lei and L. Lin, ChemSusChem, 2017, 10, 2696–2706 CrossRef CAS.
- P. H. Tran and P. V. Tran, Fuel, 2019, 246, 18–23 CrossRef CAS.
- C. H. J. T. Dietz, F. Gallucci, M. van Sint Annaland, C. Held and M. C. Kroon, Ind. Eng. Chem. Res., 2019, 58, 4240–4247 CrossRef CAS.
- C. H. J. T. Dietz, A. Erve, M. C. Kroon, M. van Sint Annaland, F. Gallucci and C. Held, Fluid Phase Equilib., 2019, 489, 75–82 CrossRef CAS.
- N. Rodriguez Quiroz, A. M. Norton, H. Nguyen, E. Vasileiadou and D. G. Vlachos, ACS Catal., 2019, 9, 9923–9952 CrossRef CAS.
- D. Liu and E. Y. X. Chen, Appl. Catal., A, 2012, 435–436, 78–85 CAS.
- D. Saang'onyo, S. Parkin and F. T. Ladipo, , Polyhedron, 2018, 149, 153–162 CrossRef.
- S. Sadula, O. Oesterling, A. Nardone, B. Dinkelacker and B. Saha, Green Chem., 2017, 19, 3888–3898 RSC.
- F. Shen, S. Sun, X. Zhang, J. Yang, M. Qiu and X. Qi, Cellulose, 2020, 27, 3013–3023 CrossRef CAS.
- J. Amoah, T. Hasunuma, C. Ogino and A. Kondo, Biochem. Eng. J., 2019, 142, 117–123 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, Appl. Catal., A, 2018, 555, 75–87 CrossRef CAS.
- I. K. M. Yu, D. C. W. Tsang, A. C. K. Yip, S. S. Chen, Y. S. Ok and C. S. Poon, Chemosphere, 2017, 184, 1099–1107 CrossRef CAS.
- J. B. Mensah, I. Delidovich, P. J. C. Hausoul, L. Weisgerber, W. Schrader and R. Palkovits, ChemSusChem, 2018, 11, 2579–2586 CrossRef CAS.
- X. Fang, Z. Wang, W. Song, S. Li and W. Lin, J. Taiwan Inst. Chem. Eng., 2019, 97, 105–111 CrossRef CAS.
- C. G. Yoo, N. Li, M. Swannell and X. Pan, Green Chem., 2017, 19, 4402–4411 RSC.
- J. Fang, W. Zheng, K. Liu, H. Li and C. Li, Chem. Eng. J., 2020, 385, 123796 CrossRef.
- Y. Deng, R. Gao, L. Lin, T. Liu, X.-D. Wen, S. Wang and D. Ma, J. Am. Chem. Soc., 2018, 140, 14481–14489 CrossRef CAS.
- T. Stahlberg, S. Rodriguez-Rodriguez, P. Fristrup and A. Riisager, Chem. – Eur. J., 2011, 17, 1456–1464 CrossRef CAS.
- T. S. Hansen, J. Mielby and A. Riisager, Green Chem., 2011, 13, 109–114 RSC.
- H. Matsumiya and T. Hara, Biomass Bioenergy, 2015, 72, 227–232 CrossRef CAS.
- L. Hu, Y. Sun, L. Lin and S. Liu, J. Taiwan Inst. Chem. Eng., 2012, 43, 718–723 CrossRef CAS.
- L. Hu, Y. Sun, L. Lin and S. Liu, Biomass Bioenergy, 2012, 47, 289–294 CrossRef CAS.
- B. R. Caes, M. J. Palte and R. T. Raines, Chem. Sci., 2013, 4, 196–199 RSC.
- A. Mukherjee, M.-J. Dumont and A. Cherestes, Catal. Lett., 2019, 149, 283–291 CrossRef CAS.
- H. S. Kim, M.-R. Park, Y. J. Jeon, S.-K. Kim, Y.-K. Hong and G.-T. Jeong, Energy Technol., 2018, 6, 1747–1754 CrossRef CAS.
- Y. Xuan, R. He, B. Han, T. Wu and Y. Wu, Waste Biomass Valorization, 2018, 9, 401–408 CrossRef CAS.
- P. Widsten, K. Murton and M. West, Ind. Crops Prod., 2018, 119, 237–242 CrossRef CAS.
- I. Delidovich and R. Palkovits, Green Chem., 2016, 18, 5822–5830 RSC.
- H. Ma, C. Liao, P. Yang, Y. Qiao, N. Li and J. Teng, ChemistrySelect, 2018, 3, 12113–12121 CrossRef CAS.
- Y. Feng, M. Li, Z. Gao, X. Zhang, X. Zeng, Y. Sun, X. Tang, T. Lei and L. Lin, ChemSusChem, 2019, 12, 495–502 CrossRef CAS.
- G. R. Gomes and J. C. Pastre, Sustainable Energy Fuels, 2020, 4, 1891–1898 RSC.
- Z. Cao, M. Li, Y. Chen, T. Shen, C. Tang, C. Zhu and H. Ying, Appl. Catal., A, 2019, 569, 93–100 CrossRef CAS.
- Y. Zhang, E. A. Pidko and E. J. M. Hensen, Chem. – Eur. J., 2011, 17, 5210–5210 CrossRef.
- J. Song, B. Zhang, J. Shi, H. Fan, J. Ma, Y. Yang and B. Han, RSC Adv., 2013, 3, 20085–20090 RSC.
- L. Wu, J. Song, B. Zhang, B. Zhou, H. Zhou, H. Fan, Y. Yang and B. Han, Green Chem., 2014, 16, 3935–3941 RSC.
- C. Loerbroks, J. van Rijn, M.-P. Ruby, Q. Tong, F. Schüth and W. Thiel, Chem. – Eur. J., 2014, 20, 12298–12309 CrossRef CAS.
- H. Nguyen, V. Nikolakis and D. G. Vlachos, ACS Catal., 2016, 6, 1497–1504 CrossRef CAS.
- S. Jia, K. Liu, Z. Xu, P. Yan, W. Xu, X. Liu and Z. C. Zhang, Catal. Today, 2014, 234, 83–90 CrossRef CAS.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345–9348 CrossRef CAS.
- F. P. Malan, E. Singleton, P. H. van Rooyen, J. Conradie and M. Landman, New J. Chem., 2018, 42, 19193–19204 RSC.
- J. He, Y. Zhang and E. Y.-X. Chen, ChemSusChem, 2013, 6, 61–64 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS.
- L. Zhou, Y. He, Z. Ma, R. Liang, T. Wu and Y. Wu, Carbohydr. Polym., 2015, 117, 694–700 CrossRef CAS.
- J. B. Binder, A. V. Cefali, J. J. Blank and R. T. Raines, Energy Environ. Sci., 2010, 3, 765–771 RSC.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr, ChemSusChem, 2010, 3, 1071–1077 CrossRef CAS.
- Y. J. Pagán-Torres, T. Wang, J. M. R. Gallo, B. H. Shanks and J. A. Dumesic, ACS Catal., 2012, 2, 930–934 CrossRef.
- I. K. M. Yu, D. C. W. Tsang, A. C. K. Yip, S. S. Chen, Y. S. Ok and C. S. Poon, Bioresour. Technol., 2016, 219, 338–347 CrossRef CAS.
- P. Zhao, C. Zhou, J. Li, S. Xu and C. Hu, ACS Sustainable Chem. Eng., 2019, 7, 5176–5183 CrossRef CAS.
- Y. Wang, C. M. Pedersen, Y. Qiao, T. Deng, J. Shi and X. Hou, Carbohydr. Polym., 2015, 115, 439–443 CrossRef CAS.
- M. Dusselier, R. De Clercq, R. Cornelis and B. F. Sels, Catal. Today, 2017, 279, 339–344 CrossRef CAS.
- Q. Wang, M. Fu, X. Li, R. Huang, R. E. Glaser and L. Zhao, J. Comput. Chem., 2019, 40, 1599–1608 CrossRef CAS.
- T. Wang, J. A. Glasper and B. H. Shanks, Appl. Catal., A, 2015, 498, 214–221 CrossRef CAS.
- X. Zhang, P. Murria, Y. Jiang, W. Xiao, H. I. Kenttämaa, M. M. Abu-Omar and N. S. Mosier, Green Chem., 2016, 18, 5219–5229 RSC.
- X. Zhang, B. B. Hewetson and N. S. Mosier, Energy Fuels, 2015, 29, 2387–2393 CrossRef CAS.
- S. De, S. Dutta and B. Saha, Green Chem., 2011, 13, 2859–2868 RSC.
- Q. Ren, Y. Huang, H. Ma, J. Gao and J. Xu, Chin. J. Catal., 2014, 35, 496–500 CrossRef CAS.
- S. Jia, X. He and Z. Xu, RSC Adv., 2017, 7, 39221–39227 RSC.
- S. Jia, Y. He and G. Wang, ChemistrySelect, 2017, 2, 2356–2362 CrossRef CAS.
- S. Xiao, B. Liu, Y. Wang, Z. Fang and Z. Zhang, Bioresour. Technol., 2014, 151, 361–366 CrossRef CAS.
- C. M. Cai, N. Nagane, R. Kumar and C. E. Wyman, Green Chem., 2014, 16, 3819–3829 RSC.
- L. Yan, R. Ma, H. Wei, L. Li, B. Zou and Y. Xu, Bioresour. Technol., 2019, 279, 84–91 CrossRef CAS.
- Z. Zhang, Q. Wang, H. Xie, W. Liu and Z. Zhao, ChemSusChem, 2011, 4, 131–138 CrossRef CAS.
- Z. Zhang, B. Liu and Z. K. Zhao, Starch – Stärke, 2012, 64, 770–775 CrossRef CAS.
- T. Ståhlberg, M. G. Sørensen and A. Riisager, Green Chem., 2010, 12, 321–325 RSC.
- J. Y. G. Chan and Y. Zhang, ChemSusChem, 2009, 2, 731–734 CrossRef CAS.
- H. Zhao, H. M. Brown, J. E. Holladay and Z. C. Zhang, Top. Catal., 2012, 55, 33–37 CrossRef CAS.
- M. Kammoun, T. Istasse, H. Ayeb, N. Rassaa, T. Bettaieb and A. Richel, Front. Chem., 2019, 7, 12 CrossRef.
- T. Guo, X. Li, X. Liu, Y. Guo and Y. Wang, ChemSusChem, 2018, 11, 2758–2765 CrossRef CAS.
- Y. Shao, D. C. W. Tsang, D. Shen, Y. Zhou, Z. Jin, D. Zhou, W. Lu and Y. Long, Environ. Res., 2020, 184, 109340 CrossRef CAS.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS.
- W. Yang and A. Sen, ChemSusChem, 2011, 4, 349–352 CrossRef CAS.
- Y. Peng, X. Li, T. Gao, T. Li and W. Yang, Green Chem., 2019, 21, 4169–4177 RSC.
- Q. Liu, Q. Ma, S. Sabnis, W. Zheng, D. G. Vlachos, W. Fan, W. Li and L. Ma, Green Chem., 2019, 21, 5030–5038 RSC.
- P. Körner, S. Beil and A. Kruse, React. Chem. Eng., 2019, 4, 747–762 RSC.
- P. Wrigstedt, J. Keskiväli, M. Leskelä and T. Repo, ChemCatChem, 2015, 7, 501–507 CrossRef CAS.
- B. J. Graham and R. T. Raines, Biomass Convers. Biorefin., 2019, 9, 471–477 CrossRef CAS.
- P. Körner, D. Jung and A. Kruse, Green Chem., 2018, 20, 2231–2241 RSC.
- N. Shi, Q. Liu, Q. Zhang, T. Wang and L. Ma, Green Chem., 2013, 15, 1967–1974 RSC.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- Z. Cheng, J. L. Everhart, G. Tsilomelekis, V. Nikolakis, B. Saha and D. G. Vlachos, Green Chem., 2018, 20, 997–1006 RSC.
- V. Maruani, S. Narayanin-Richenapin, E. Framery and B. Andrioletti, ACS Sustainable Chem. Eng., 2018, 6, 13487–13493 CrossRef CAS.
- H. Labauze, S. Camy, P. Floquet, B. Benjelloun-Mlayah and J. S. Condoret, Ind. Eng. Chem. Res., 2019, 58, 92–100 CrossRef CAS.
- B. Seemala, V. Haritos and A. Tanksale, ChemCatChem, 2016, 8, 640–647 CrossRef CAS.
- C. Liu, J. M. Carraher, J. L. Swedberg, C. R. Herndon, C. N. Fleitman and J.-P. Tessonnier, ACS Catal., 2014, 4, 4295–4298 CrossRef CAS.
- Y. Román-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef.
- M. Yabushita, N. Shibayama, K. Nakajima and A. Fukuoka, ACS Catal., 2019, 9, 2101–2109 CrossRef CAS.
- F. Liu, K. Huang, A. Zheng, F.-S. Xiao and S. Dai, ACS Catal., 2018, 8, 372–391 CrossRef CAS.
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- A. Zheng, S. Li, S.-B. Liu and F. Deng, Acc. Chem. Res., 2016, 49, 655–663 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, ChemCatChem, 2016, 8, 1100–1110 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, D. K. Ipsakis, S. A. Karakoulia, K. G. Kalogiannis, H. Yiannoulakis, K. S. Triantafyllidis and A. A. Lappas, ACS Sustainable Chem. Eng., 2018, 6, 16459–16470 CrossRef CAS.
- S. S. Chen, Y. Cao, D. C. W. Tsang, J.-P. Tessonnier, J. Shang, D. Hou, Z. Shen, S. Zhang, Y. S. Ok and K. C. W. Wu, ACS Sustainable Chem. Eng., 2020, 8, 6990–7001 CrossRef CAS.
- J. J. Wiesfeld, R. Gaquere and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2019, 7, 7552–7562 CrossRef CAS.
- J. Guo, S. Zhu, Y. Cen, Z. Qin, J. Wang and W. Fan, Appl. Catal., B, 2017, 200, 611–619 CrossRef CAS.
- B. Guo, L. Ye, G. Tang, L. Zhang, B. Yue, S. C. E. Tsang and H. He, Chin. J. Chem., 2017, 35, 1529–1539 CrossRef CAS.
- J. M. Requies, M. Frias, M. Cuezva, A. Iriondo, I. Agirre and N. Viar, Ind. Eng. Chem. Res., 2018, 57, 11535–11546 CrossRef CAS.
- J. J. Wiesfeld, N. Sommerdijk and E. J. M. Hensen, Catal. Lett., 2018, 148, 3093–3101 CrossRef CAS.
- J. Zhong, Y. Guo and J. Chen, J. Energy Chem., 2017, 26, 147–154 CrossRef.
- A. Gervasini, S. Campisi, P. Carniti, M. Fantauzzi, C. Imparato, N. J. Clayden, A. Aronne and A. Rossi, Appl. Catal., A, 2019, 579, 9–17 CrossRef CAS.
- C. A. S. Lanziano, S. F. Moya, D. H. Barrett, E. Teixeira-Neto, R. Guirardello, F. de Souto da Silva, R. Rinaldi and C. B. Rodella, ChemSusChem, 2018, 11, 872–880 CrossRef CAS.
- Z. Tang and J. H. Su, J. Oleo Sci., 2019, 68, 261–271 CrossRef CAS.
- Q. Hou, M. Zhen, W. Li, L. Liu, J. Liu, S. Zhang, Y. Nie, C. Bai, X. Bai and M. Ju, Appl. Catal., B, 2019, 253, 1–10 CrossRef CAS.
- S. Pumrod, A. Kaewchada, S. Roddecha and A. Jaree, RSC Adv., 2020, 10, 9492–9498 RSC.
- S. S. Jing, X. F. Cao, L. X. Zhong, X. W. Peng, R. C. Sun and J. C. Liu, Ind. Crops Prod., 2018, 126, 151–157 CrossRef CAS.
- G. E. Córdova-Pérez, G. Torres-Torres, F. Ortiz-Chi, S. Godavarthi, A. A. Silahua-Pavon, A. Izquierdo-Colorado, P. Da Costa, N. Hernandez-Como, M. Aleman and C. G. Espinosa-Gonzalez, ChemistrySelect, 2018, 3, 12854–12864 CrossRef.
- B. Guo, L. He, G. Tang, L. Zhang, L. Ye, B. Yue, S. C. E. Tsang and H. He, Chin. J. Catal., 2020, 41, 1248–1260 CrossRef CAS.
- B. Han, P. Zhao, R. He, T. H. Wu and Y. Wu, Waste Biomass Valorization, 2018, 9, 2181–2190 CrossRef CAS.
- M. A. Yatoo and S. Saravanamurugan, Appl. Catal., A, 2019, 582, 117094 CrossRef CAS.
- Z. Wang, T. Li, Y. Jiang, O. Lafon, Z. Liu, J. Trébosc, A. Baiker, J.-P. Amoureux and J. Huang, Nat. Commun., 2020, 11, 225 CrossRef CAS.
- M. Cui, Z. Wu, R. Huang, W. Qi, R. Su and Z. He, Renewable Energy, 2018, 125, 327–333 CrossRef CAS.
- C. Chiappe, M. J. Rodriguez Douton, A. Mezzetta, C. S. Pomelli, G. Assanelli and A. R. de Angelis, ACS Sustainable Chem. Eng., 2017, 5, 5529–5536 CrossRef CAS.
- R. Wang, X. Liang, F. Shen, M. Qiu, J. Yang and X. Qi, ACS Sustainable Chem. Eng., 2020, 8, 1163–1170 CrossRef CAS.
- C. García-Sancho, I. Fúnez-Núñez, R. Moreno-Tost, J. Santamaría-González, E. Pérez-Inestrosa, J. L. G. Fierro and P. Maireles-Torres, Appl. Catal., B, 2017, 206, 617–625 CrossRef.
- G. Sampath and K. Srinivasan, Appl. Catal., A, 2017, 533, 75–80 CrossRef CAS.
- Q. Hou, M. Zhen, L. Liu, Y. Chen, F. Huang, S. Zhang, W. Li and M. Ju, Appl. Catal., B, 2018, 224, 183–193 CrossRef CAS.
- J. Liu, H. Li, Y.-C. Liu, Y.-M. Lu, J. He, X.-F. Liu, Z.-B. Wu and S. Yang, Catal. Commun., 2015, 62, 19–23 CrossRef CAS.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS.
- N. T. do Prado, T. E. Souza, A. R. T. Machado, P. P. Souza, R. S. Monteiro and L. C. A. Oliveira, J. Mol. Catal. A: Chem., 2016, 422, 23–34 CrossRef CAS.
- H. T. Kreissl, K. Nakagawa, Y.-K. Peng, Y. Koito, J. Zheng and S. C. E. Tsang, J. Catal., 2016, 338, 329–339 CrossRef CAS.
- H. T. Kreissl, M. M. J. Li, Y.-K. Peng, K. Nakagawa, T. J. N. Hooper, J. V. Hanna, A. Shepherd, T.-S. Wu, Y.-L. Soo and S. C. E. Tsang, J. Am. Chem. Soc., 2017, 139, 12670–12680 CrossRef CAS.
- W. Fan, Q. Zhang, W. Deng and Y. Wang, Chem. Mater., 2013, 25, 3277–3287 CrossRef CAS.
- A. Takagaki, D. Lu, J. N. Kondo, M. Hara, S. Hayashi and K. Domen, Chem. Mater., 2005, 17, 2487–2489 CrossRef CAS.
- Q. Wu, Y. Yan, Q. Zhang, J. Lu, Z. Yang, Y. Zhang and Y. Tang, ChemSusChem, 2013, 6, 820–825 CrossRef CAS.
- P. Carniti, A. Gervasini, S. Biella and A. Auroux, Catal. Today, 2006, 118, 373–378 CrossRef CAS.
- K. Kawamura, T. Yasuda, T. Hatanaka, K. Hamahiga, N. Matsuda, M. Ueshima and K. Nakai, Chem. Eng. J., 2017, 307, 1066–1075 CrossRef CAS.
- N. K. Gupta, A. Fukuoka and K. Nakajima, ACS Catal., 2017, 7, 2430–2436 CrossRef CAS.
- C. García-Sancho, J. M. Rubio-Caballero, J. M. Mérida-Robles, R. Moreno-Tost, J. Santamaría-González and P. Maireles-Torres, Catal. Today, 2014, 234, 119–124 CrossRef.
- K. Nakajima, T. Fukui, H. Kato, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, Chem. Mater., 2010, 22, 3332–3339 CrossRef CAS.
- P. Sudarsanam, H. Li and T. V. Sagar, ACS Catal., 2020, 10, 9555–9584 CrossRef CAS.
- K. Nakajima, R. Noma, M. Kitano and M. Hara, J. Mol. Catal. A: Chem., 2014, 388–389, 100–105 CrossRef CAS.
- M. Kitano, K. Nakajima, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2010, 132, 6622–6623 CrossRef CAS.
- G. Li, E. A. Pidko, E. J. M. Hensen and K. Nakajima, ChemCatChem, 2018, 10, 4084–4089 CrossRef CAS.
- R. Noma, K. Nakajima, K. Kamata, M. Kitano, S. Hayashi and M. Hara, J. Phys. Chem. C, 2015, 119, 17117–17125 CrossRef CAS.
- G. Li, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2016, 6, 4162–4169 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe, J. N. Kondo, K. Ebitani, S. Hayashi, T. Tatsumi and K. Domen, Angew. Chem., 2010, 122, 1146–1150 CrossRef.
- T. Murayama, K. Nakajima, J. Hirata, K. Omata, E. J. M. Hensen and W. Ueda, Catal. Sci. Technol., 2017, 7, 243–250 RSC.
- H. Jiao, X. Zhao, C. Lv, Y. Wang, D. Yang, Z. Li and X. Yao, Sci. Rep., 2016, 6, 34068 CrossRef CAS.
- Y. Wang, X. Tong, Y. Yan, S. Xue and Y. Zhang, Catal. Commun., 2014, 50, 38–43 CrossRef CAS.
- R. M. de Almeida, N. J. A. de Albuquerque, F. T. C. Souza and S. M. P. Meneghetti, Catal. Sci. Technol., 2016, 6, 3137–3142 RSC.
- E. L. S. Ngee, Y. Gao, X. Chen, T. M. Lee, Z. Hu, D. Zhao and N. Yan, Ind. Eng. Chem. Res., 2014, 53, 14225–14233 CrossRef CAS.
- S. Yu, E. Kim, S. Park, I. K. Song and J. C. Jung, Catal. Commun., 2012, 29, 63–67 CrossRef CAS.
- I. Delidovich and R. Palkovits, J. Catal., 2015, 327, 1–9 CrossRef CAS.
- G. Lee, Y. Jeong, A. Takagaki and J. C. Jung, J. Mol. Catal. A: Chem., 2014, 393, 289–295 CrossRef CAS.
- I. Delidovich and R. Palkovits, Catal. Sci. Technol., 2014, 4, 4322–4329 RSC.
- P. P. Upare, A. Chamas, J. H. Lee, J. C. Kim, S. K. Kwak, Y. K. Hwang and D. W. Hwang, ACS Catal., 2020, 10, 1388–1396 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276–6278 RSC.
- S. Xu, X. Yan, Q. Bu and H. Xia, RSC Adv., 2016, 6, 8048–8052 RSC.
- S. Xu, D. Pan, Y. Wu, X. Song, L. Gao, W. Li, L. Das and G. Xiao, Fuel Process. Technol., 2018, 175, 90–96 CrossRef CAS.
- S. Q. Xu, D. H. Pan, W. Q. Li, P. X. Shen, Y. F. Wu, X. H. Song, Y. L. Zhu, N. N. Xu, L. J. Gao and G. M. Xiao, Fuel Process. Technol., 2018, 181, 199–206 CrossRef CAS.
- D.-M. Gao, B. Zhao, H. Liu, K. Morisato, K. Kanamori, Z. He, M. Zeng, H. Wu, J. Chen and K. Nakanishi, Catal. Sci. Technol., 2018, 8, 3675–3685 RSC.
- K. Saravanan, K. S. Park, S. Jeon and J. W. Bae, ACS Omega, 2018, 3, 808–820 CrossRef CAS.
- W. X. Ni, D. F. Li, X. G. Zhao, W. B. Ma, K. Kong, Q. W. Gu, M. Y. Chen and Z. S. Hou, Catal. Today, 2019, 319, 66–75 CrossRef CAS.
- C. Zhu, C. Cai, Q. Liu, W. Li, J. Tan, C. Wang, L. Chen, Q. Zhang and L. Ma, ChemistrySelect, 2018, 3, 10983–10990 CrossRef CAS.
- Z. Cao, Z. Fan, Y. Chen, M. Li, T. Shen, C. Zhu and H. Ying, Appl. Catal., B, 2019, 244, 170–177 CrossRef CAS.
- L. Yang, X. Yan, S. Xu, H. Chen, H. Xia and S. Zuo, RSC Adv., 2015, 5, 19900–19906 RSC.
- Y. Liu, Z. Li, Y. You, X. Zheng and J. Wen, RSC Adv., 2017, 7, 51281–51289 RSC.
- A. Dutta, D. Gupta, A. K. Patra, B. Saha and A. Bhaumik, ChemSusChem, 2014, 7, 925–933 CrossRef CAS.
- K. T. V. Rao, S. Souzanchi, Z. Yuan, M. B. Ray and C. Xu, RSC Adv., 2017, 7, 48501–48511 RSC.
- W. Li, T. Yang, M. Su and Y. Liu, Catal. Lett., 2020, 150, 3304–3313 CrossRef CAS.
- K. Li, M. Du and P. Ji, ACS Sustainable Chem. Eng., 2018, 6, 5636–5644 CrossRef CAS.
- F. Huang, Y. Su, Z. Long, G. Chen and Y. Yao, Ind. Eng. Chem. Res., 2018, 57, 10198–10205 CrossRef CAS.
- L. Yang, X. Yan, S. Xu, H. Chen, H. Xia and S. Zuo, RSC Adv., 2015, 5, 19900–19906 RSC.
- H. Xia, S. Xu, X. Yan and S. Zuo, Fuel Process. Technol., 2016, 152, 140–146 CrossRef CAS.
- L. Zhang, G. Xi, Z. Chen, Z. Qi and X. Wang, Chem. Eng. J., 2017, 307, 877–883 CrossRef CAS.
- J. E. Romo, T. Wu, X. Huang, J. Lucero, J. L. Irwin, J. Q. Bond, M. A. Carreon and S. G. Wettstein, ACS Omega, 2018, 3, 16253–16259 CrossRef CAS.
- Y. Zhang, J. Wang, X. Li, X. Liu, Y. Xia, B. Hu, G. Lu and Y. Wang, Fuel, 2015, 139, 301–307 CrossRef CAS.
- Y. Zhang, J. Wang, J. Ren, X. Liu, X. Li, Y. Xia, G. Lu and Y. Wang, Catal. Sci. Technol., 2012, 2, 2485–2491 RSC.
- P. Carniti, A. Gervasini, F. Bossola and V. Dal Santo, Appl. Catal., B, 2016, 193, 93–102 CrossRef CAS.
- A. Dutta, A. K. Patra, S. Dutta, B. Saha and A. Bhaumik, J. Mater. Chem., 2012, 22, 14094–14100 RSC.
- L. Atanda, S. Mukundan, A. Shrotri, Q. Ma and J. Beltramini, ChemCatChem, 2015, 7, 781–790 CrossRef CAS.
- L. Atanda, A. Shrotri, S. Mukundan, Q. Ma, M. Konarova and J. Beltramini, ChemSusChem, 2015, 8, 2907–2916 CrossRef CAS.
- L. Atanda, M. Konarova, Q. Ma, S. Mukundan, A. Shrotri and J. Beltramini, Catal. Sci. Technol., 2016, 6, 6257–6266 RSC.
- M. Hattori, K. Kamata and M. Hara, Phys. Chem. Chem. Phys., 2017, 19, 3688–3693 RSC.
- M. Zhang, X. Tong, R. Ma and Y. Li, Catal. Today, 2016, 264, 131–135 CrossRef CAS.
- H. Ning, J. Song, M. Hou, D. Yang, H. Fan and B. Han, Sci. China: Chem., 2013, 56, 1578–1585 CrossRef CAS.
- P. Daorattanachai, P. Khemthong, N. Viriya-empikul, N. Laosiripojana and K. Faungnawakij, Chem. Eng. J., 2015, 278, 92–98 CrossRef CAS.
- I. Jiménez-Morales, A. Teckchandani-Ortiz, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2014, 144, 22–28 CrossRef.
- I. Jiménez-Morales, M. Moreno-Recio, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2014, 154–155, 190–196 CrossRef.
- F. Yang, Q. Liu, M. Yue, X. Bai and Y. Du, Chem. Commun., 2011, 47, 4469–4471 RSC.
- A. Dibenedetto, M. Aresta, C. Pastore, L. Di Bitonto, A. Angelini and E. Quaranta, RSC Adv., 2015, 5, 26941–26948 RSC.
- S. Jia, X. He, J. Ma, Z. Xu, K. Wang and Z. C. Zhang, RSC Adv., 2018, 8, 32533–32537 RSC.
- I. Delidovich, M. S. Gyngazova, N. Sánchez-Bastardo, J. P. Wohland, C. Hoppe and P. Drabo, Green Chem., 2018, 20, 724–734 RSC.
- P. Bhanja and A. Bhaumik, ChemCatChem, 2016, 8, 1607–1616 CrossRef CAS.
- M. Shamzhy, M. Opanasenko, P. Concepcion and A. Martinez, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 6164–6168 CrossRef CAS.
- J. Dijkmans, J. Demol, K. Houthoofd, S. Huang, Y. Pontikes and B. Sels, J. Catal., 2015, 330, 545–557 CrossRef CAS.
- J. W. Harris, M. J. Cordon, J. R. Di Iorio, J. C. Vega-Vila, F. H. Ribeiro and R. Gounder, J. Catal., 2016, 335, 141–154 CrossRef CAS.
- R. Bermejo-Deval, M. Orazov, R. Gounder, S.-J. Hwang and M. E. Davis, ACS Catal., 2014, 4, 2288–2297 CrossRef CAS.
- S. K. Brand, T. R. Josephson, J. A. Labinger, S. Caratzoulas, D. G. Vlachos and M. E. Davis, J. Catal., 2016, 341, 62–71 CrossRef CAS.
- T. R. Josephson, S. K. Brand, S. Caratzoulas and D. G. Vlachos, ACS Catal., 2017, 7, 25–33 CrossRef CAS.
- R. Bermejo-Deval, R. Gounder and M. E. Davis, ACS Catal., 2012, 2, 2705–2713 CrossRef CAS.
- J. Dijkmans, D. Gabriëls, M. Dusselier, F. de Clippel, P. Vanelderen, K. Houthoofd, A. Malfliet, Y. Pontikes and B. F. Sels, Green Chem., 2013, 15, 2777–2785 RSC.
- D. Garcés, L. Faba, E. Díaz and S. Ordóñez, ChemSusChem, 2019, 12, 924–934 Search PubMed.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- S. Marullo, C. Rizzo, A. Meli and F. D'Anna, ACS Sustainable Chem. Eng., 2019, 7, 5818–5826 CrossRef CAS.
- L. Wang, L. B. Zhang, H. Y. Li, Y. B. Ma and R. H. Zhang, Composites, Part B, 2019, 156, 88–94 CrossRef CAS.
- P. Bhanja, A. Modak, S. Chatterjee and A. Bhaumik, ACS Sustainable Chem. Eng., 2017, 5, 2763–2773 CrossRef CAS.
- Y. Zhang, Y. Chen, J. Pan, M. Liu, P. Jin and Y. Yan, Chem. Eng. J., 2017, 313, 1593–1606 CrossRef CAS.
- Q. Wu, F. Liu, X. Yi, Y. Zou and L. Jiang, Green Chem., 2018, 20, 1020–1030 RSC.
- X. Huang, S. Kudo, J. Sperry and J.-I. Hayashi, ACS Sustainable Chem. Eng., 2019, 7, 5892–5899 CrossRef CAS.
- L. K. Rihko-Struckmann, O. Oluyinka, A. Sahni, K. McBride, M. Fachet, K. Ludwig and K. Sundmacher, RSC Adv., 2020, 10, 24753–24763 RSC.
- N. Candu, M. El Fergani, M. Verziu, B. Cojocaru, B. Jurca, N. Apostol, C. Teodorescu, V. I. Parvulescu and S. M. Coman, Catal. Today, 2019, 325, 109–116 CrossRef CAS.
- Y. Feng, G. Yan, T. Wang, W. Jia, X. Zeng, J. Sperry, Y. Sun, X. Tang, T. Lei and L. Lin, ChemSusChem, 2019, 12, 978–982 CrossRef CAS.
- Y. D. Xie, W. W. Yuan, Y. Huang, C. Y. Wu, H. J. Wang, Y. M. Xia and X. Liu, Chin. Chem. Lett., 2019, 30, 359–362 CrossRef CAS.
- N. D. Kalane, R. A. Krishnan, V. D. Yadav, R. Jain and P. Dandekar, Cellulose, 2019, 26, 2805–2819 CrossRef CAS.
- Y. Zhang, Q. Xiong, E. Zhu, M. Liu, J. Pan and Y. Yan, Energy Technol., 2018, 6, 1941–1950 CrossRef CAS.
- Y. Wang, L. W. Zhu, Y. Zhang, H. Y. Cui, W. M. Yi, F. Song, P. P. Zhao, X. Y. Sun, Y. J. Xie, L. H. Wang and Z. H. Li, ChemistrySelect, 2018, 3, 3555–3560 CrossRef CAS.
- Y. C. Feng, M. Zuo, T. Wang, W. L. Jia, X. Y. Zhao, X. H. Zeng, Y. Sun, X. Tang, T. Z. Lei and L. Lin, J. Taiwan Inst. Chem. Eng., 2019, 96, 431–438 CrossRef CAS.
- J. Cao, M. Ma, J. Liu, Y. Yang, H. Liu, X. Xu, J. Huang, H. Yue, G. Tian and S. Feng, Appl. Catal., A, 2019, 571, 96–101 CrossRef CAS.
- G. Qiu, C. Huang, X. Sun and B. Chen, Green Chem., 2019, 21, 3930–3939 RSC.
- L. Wang, H. Guo, Q. Xie, J. Wang, B. Hou, L. Jia, J. Cui and D. Li, Appl. Catal., A, 2019, 51–60 Search PubMed.
- Y. Wang, G. Ding, X. Yang, H. Zheng, Y. Zhu and Y. Li, Appl. Catal., B, 2018, 235, 150–157 CrossRef CAS.
- P. Saxena, B. Velaga and N. R. Peela, ChemistrySelect, 2017, 2, 10379–10386 CrossRef CAS.
- Q. Xu, Z. Zhu, Y. Tian, J. Deng, J. Shi and Y. Fu, BioResources, 2014, 9, 303–315 CAS.
- J. Su, M. Qiu, F. Shen and X. Qi, Cellulose, 2018, 25, 17–22 CrossRef CAS.
- J. Wang, J. Ren, X. Liu, J. Xi, Q. Xia, Y. Zu, G. Lu and Y. Wang, Green Chem., 2012, 14, 2506–2512 RSC.
- G. M. Lari, P. Y. Dapsens, D. Scholz, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2016, 18, 1249–1260 RSC.
- Q. Guo, L. Ren, S. M. Alhassan and M. Tsapatsis, Chem. Commun., 2019, 55, 14942–14945 RSC.
- N. Rajabbeigi, A. I. Torres, C. M. Lew, B. Elyassi, L. Ren, Z. Wang, H. Je Cho, W. Fan, P. Daoutidis and M. Tsapatsis, Chem. Eng. Sci., 2014, 116, 235–242 CrossRef CAS.
- D. Padovan, S. Tolborg, L. Botti, E. Taarning, I. Sádaba and C. Hammond, React. Chem. Eng., 2018, 3, 155–163 RSC.
- D. Padovan, L. Botti and C. Hammond, ACS Catal., 2018, 8, 7131–7140 CrossRef CAS.
- R. Bermejo-Deval, R. S. Assary, E. Nikolla, M. Moliner, Y. Roman-Leshkov, S. J. Hwang, A. Palsdottir, D. Silverman, R. F. Lobo, L. A. Curtiss and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9727–9732 CrossRef CAS.
- R. Gounder and M. E. Davis, ACS Catal., 2013, 3, 1469–1476 CrossRef CAS.
- M. J. Cordon, J. C. Vega-Vila, A. Casper, Z. Huang and R. Gounder, Angew. Chem., Int. Ed., 2020, 132, 19264–19269 CrossRef.
- S. Xu, D. Pan, F. Hu, Y. Wu, H. Wang, Y. Chen, H. Yuan, L. Gao and G. Xiao, Fuel Process. Technol., 2019, 190, 38–46 CrossRef CAS.
- R. Otomo, T. Yokoi and T. Tatsumi, ChemCatChem, 2015, 7, 4180–4187 CrossRef CAS.
- X. Jia, I. K. M. Yu, D. C. W. Tsang and A. C. K. Yip, Microporous Mesoporous Mater., 2019, 284, 43–52 CrossRef CAS.
- I. Jiménez-Morales, M. Moreno-Recio, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2015, 164, 70–76 CrossRef.
- M. Moreno-Recio, I. Jiménez-Morales, P. L. Arias, J. Santamaría-González and P. Maireles-Torres, ChemistrySelect, 2017, 2, 2444–2451 CrossRef CAS.
- M. Moreno-Recio, J. Santamaría-González and P. Maireles-Torres, Chem. Eng. J., 2016, 303, 22–30 CrossRef CAS.
- I. Jiménez-Morales, J. Santamaría-González, A. Jiménez-López and P. Maireles-Torres, Fuel, 2014, 118, 265–271 CrossRef.
- L. Hu, Z. Wu, J. Xu, Y. Sun, L. Lin and S. Liu, Chem. Eng. J., 2014, 244, 137–144 CrossRef CAS.
- B. Velaga and N. R. Peela, Microporous Mesoporous Mater., 2019, 279, 211–219 CrossRef CAS.
- W. Mamo, Y. Chebude, C. Márquez-Álvarez, I. Díaz and E. Sastre, Catal. Sci. Technol., 2016, 6, 2766–2774 RSC.
- H. Abou-Yousef and E. B. Hassan, J. Ind. Eng. Chem., 2014, 20, 1952–1957 CrossRef CAS.
- H. Xia, H. Hu, S. Xu, K. Xiao and S. Zuo, Biomass Bioenergy, 2018, 108, 426–432 CrossRef CAS.
- S. Xu, L. Zhang, K. Xiao and H. Xia, Carbohydr. Res., 2017, 446–447, 48–51 CrossRef CAS.
- A. Osatiashtiani, A. F. Lee, M. Granollers, D. R. Brown, L. Olivi, G. Morales, J. A. Melero and K. Wilson, ACS Catal., 2015, 5, 4345–4352 CrossRef CAS.
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS.
- S. Shunmugavel, M. Paniagua, J. A. Melero and A. Riisager, J. Am. Chem. Soc., 2013, 5246–5249 Search PubMed.
- L. Ren, Q. Guo, P. Kumar, M. Orazov, D. Xu, S. M. Alhassan, K. A. Mkhoyan, M. E. Davis and M. Tsapatsis, Angew. Chem., Int. Ed., 2015, 54, 10848–10851 CrossRef CAS.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Appl. Catal., B, 2015, 174–175, 225–243 CrossRef CAS.
- Y. Tang, Y. Cheng, H. Xu, Y. Wang, L. Ke, X. Huang, X. Liao and B. Shi, Catal. Commun., 2019, 123, 96–99 CrossRef CAS.
- N. Pasha, P. K. Kumari, N. Vamsikrishna, N. Lingaiah, N. J. P. Subhashini and Shivaraj, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2019, 58, 313–320 Search PubMed.
- F. M. Huang, Y. W. Su, Y. Tao, W. Sun and W. T. Wang, Fuel, 2018, 226, 417–422 CrossRef CAS.
- S. Q. Xu, D. H. Pan, Y. F. Wu, N. N. Xu, H. M. Yang, L. J. Gao, W. Q. Li and G. M. Xiao, Ind. Eng. Chem. Res., 2019, 58, 9276–9285 CrossRef CAS.
- P. P. Zhao, H. Y. Cui, Y. Y. Zhang, Y. Zhang, Y. Wang, Y. L. Zhang, Y. J. Xie and W. M. Yi, ChemistryOpen, 2018, 7, 824–832 CrossRef CAS.
- J. R. Bernardo, M. C. Oliveira and A. C. Fernandes, Mol. Catal., 2019, 465, 87–94 CrossRef CAS.
- J. A. T. Caetano and A. C. Fernandes, Green Chem., 2018, 20, 2494–2498 RSC.
- P. Zhao, Y. Zhang, Y. Wang, H. Cui, F. Song, X. Sun and L. Zhang, Green Chem., 2018, 20, 1551–1559 RSC.
- X. Zhang, D. Zhang, Z. Sun, L. Xue, X. Wang and Z. Jiang, Appl. Catal., B, 2016, 196, 50–56 CrossRef CAS.
- F. Lai, F. Yan, P. Wang, S. Wang, S. Li and Z. Zhang, Chem. Eng. J., 2020, 396, 125282 CrossRef CAS.
- X. Wang, T. Lv, M. Wu, J. Sui, Q. Liu, H. Liu, J. Huang and L. Jia, Appl. Catal., A, 2019, 574, 87–96 CrossRef CAS.
- S. Zhao, M. Cheng, J. Li, J. Tian and X. Wang, Chem. Commun., 2011, 47, 2176–2178 RSC.
- L. J. Konwar, P. Mäki-Arvela and J.-P. Mikkola, Chem. Rev., 2019, 119, 11576–11630 CrossRef CAS.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678–2681 RSC.
- H. Guo, X. Qi, L. Li and R. L. Smith, Bioresour. Technol., 2012, 116, 355–359 CrossRef CAS.
- X. Qi, N. Liu and Y. Lian, RSC Adv., 2015, 5, 17526–17531 RSC.
- M. Qiu, C. Bai, L. Yan, F. Shen and X. Qi, ACS Sustainable Chem. Eng., 2018, 6, 13826–13833 CrossRef CAS.
- C. Bai, L. Zhu, F. Shen and X. Qi, Bioresour. Technol., 2016, 220, 656–660 CrossRef CAS.
- X. Qi, L. Yan, F. Shen and M. Qiu, Bioresour. Technol., 2019, 273, 687–691 CrossRef CAS.
- Y. Ni, Z. Bi, H. Su and L. Yan, Green Chem., 2019, 21, 1075–1079 RSC.
- N. V. Gromov, T. B. Medvedeva, O. P. Taran, A. V. Bukhtiyarov, C. Aymonier, I. P. Prosvirin and V. N. Parmon, Top. Catal., 2018, 61, 1912–1927 CrossRef CAS.
- L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889–6894 RSC.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS.
- X. H. Liao, Y. Liu, X. Peng, C. Mi and X. G. Meng, Catal. Lett., 2016, 146, 1249–1255 CrossRef CAS.
- A. Charmot, P. W. Chung and A. Katz, ACS Sustainable Chem. Eng., 2014, 2, 2866–2872 CrossRef CAS.
- P. W. Chung, A. Charmot, O. M. Gazit and A. Katz, Langmuir, 2012, 28, 15222–15232 CrossRef CAS.
- A. T. To, P.-W. Chung and A. Katz, Angew. Chem., Int. Ed., 2015, 54, 11050–11053 CrossRef CAS.
- F. Liu, B. Li, C. Liu, W. Kong, X. Yi, A. Zheng and C. Qi, Catal. Sci. Technol., 2016, 6, 2995–3007 RSC.
- L. Gan, L. Lyu, T. Shen and S. Wang, Appl. Catal., A, 2019, 574, 132–143 CrossRef CAS.
- K. Li, J. Chen, Y. Yan, Y. Min, H. Li, F. Xi, J. Liu and P. Chen, Carbon, 2018, 136, 224–233 CrossRef CAS.
- J. Ma, W. Li, S. Guan, Q. Liu, Q. Li, C. Zhu, T. Yang, A. T. Ogunbiyi and L. Ma, RSC Adv., 2019, 9, 10569–10577 RSC.
- M. M. Songo, R. Moutloali and S. S. Ray, Catalysts, 2019, 9, 15 CrossRef.
- F. Yang, X. Tong, F. Xia, C. Zheng, L. Qin and X. Jiang, Catal. Lett., 2018, 148, 1848–1855 CrossRef CAS.
- Z. Chen, Q. Li, Y. Xiao, C. Zhang, Z. Fu, Y. Liu, X. Yi, A. Zheng, C. Li and D. Yin, Cellulose, 2019, 26, 751–762 CrossRef CAS.
- F. Huang, W. Li, Q. Liu, T. Zhang, S. An, D. Li and X. Zhu, Fuel Process. Technol., 2018, 181, 294–303 CrossRef CAS.
- J. Li, Y. Wang, B. Lu, Y. Wang, T. Deng and X. Hou, Appl. Catal., A, 2018, 566, 140–145 CrossRef CAS.
- F. Huang, W. Z. Li, T. W. Zhang, D. W. Li, Q. Y. Liu, X. F. Zhu and L. L. Ma, Res. Chem. Intermed., 2018, 44, 5439–5453 CrossRef CAS.
- C. Zhang, Z. Cheng, Z. Fu, Y. Liu, X. Yi, A. Zheng, S. R. Kirk and D. Yin, Cellulose, 2017, 24, 95–106 CrossRef CAS.
- B. M. Matsagar, C. Van Nguyen, M. S. A. Hossain, M. T. Islam, Y. Yamauchi, P. L. Dhepe and K. C. W. Wu, Sustainable Energy Fuels, 2018, 2, 2148–2153 RSC.
- F. Shen, J. Fu, X. Zhang and X. Qi, ACS Sustainable Chem. Eng., 2019, 7, 4466–4472 CrossRef CAS.
- K. N. Sorokina, O. P. Taran, T. B. Medvedeva, Y. V. Samoylova, A. V. Piligaev and V. N. Parmon, ChemSusChem, 2017, 10, 562–574 CrossRef CAS.
- I. K. M. Yu, X. Xiong, D. C. W. Tsang, L. Wang, A. J. Hunt, H. Song, J. Shang, Y. S. Ok and C. S. Poon, Green Chem., 2019, 21, 1267–1281 RSC.
- X. Yang, I. K. M. Yu, D.-W. Cho, S. S. Chen, D. C. W. Tsang, J. Shang, A. C. K. Yip, L. Wang and Y. S. Ok, ACS Sustainable Chem. Eng., 2019, 7, 4851–4860 CrossRef CAS.
- M. Nahavandi, T. Kasanneni, Z. S. Yuan, C. C. Xu and S. Rohani, ACS Sustainable Chem. Eng., 2019, 7, 11970–11984 CAS.
- L. C. Cao, I. K. M. Yu, D. C. W. Tsang, S. C. Zhang, Y. S. Ok, E. E. Kwon, H. Song and C. S. Poon, Bioresour. Technol., 2018, 267, 242–248 CrossRef CAS.
- L. Cao, I. K. M. Yu, S. S. Chen, D. C. W. Tsang, L. Wang, X. Xiong, S. Zhang, Y. S. Ok, E. E. Kwon, H. Song and C. S. Poon, Bioresour. Technol., 2018, 252, 76–82 CrossRef CAS.
- G. Y. Chen, L. B. Wu, H. Fan and B. G. Li, Ind. Eng. Chem. Res., 2018, 57, 16172–16181 CrossRef CAS.
- S. S. Chen, I. K. M. Yu, D.-W. Cho, H. Song, D. C. W. Tsang, J.-P. Tessonnier, Y. S. Ok and C. S. Poon, ACS Sustainable Chem. Eng., 2018, 6, 16113–16120 CrossRef CAS.
- X. Qi, Y. Lian, L. Yan and R. L. Smith Jr., Catal. Commun., 2014, 57, 50–54 CrossRef CAS.
- L. Yang, M. Wang, X. Yan, Y. Wang and H. Xia, Asian – J. Chem., 2015, 27, 2979–2982 CrossRef CAS.
- A. Villa, M. Schiavoni, P. F. Fulvio, S. M. Mahurin, S. Dai, R. T. Mayes, G. M. Veith and L. Prati, J. Energy Chem., 2013, 22, 305–311 CrossRef CAS.
- Y. Zhang, J. Wang, J. Wang, Y. Wang, M. Wang, H. Cui, F. Song, X. Sun, Y. Xie and W. Yi, ChemistrySelect, 2019, 4, 5724–5731 CrossRef CAS.
- N. T. Thanh, N. H. Long, L. Q. Dien, G. T. Phuong Ly, P. H. Hoang, N. T. Minh Phuong and N. T. Hue, Energy Sources, Part A, 2019, 1–10 Search PubMed.
- R. Fang, A. Dhakshinamoorthy, Y. Li and H. Garcia, Chem. Soc. Rev., 2020, 49, 3638–3687 RSC.
- Z. Hu, Y. Peng, Y. Gao, Y. Qian, S. Ying, D. Yuan, S. Horike, N. Ogiwara, R. Babarao, Y. Wang, N. Yan and D. Zhao, Chem. Mater., 2016, 28, 2659–2667 CrossRef CAS.
- Y. Peng, Z. Hu, Y. Gao, D. Yuan, Z. Kang, Y. Qian, N. Yan and D. Zhao, ChemSusChem, 2015, 8, 3208–3212 CrossRef CAS.
- R. Oozeerally, D. L. Burnett, T. W. Chamberlain, R. I. Walton and V. Degirmenci, ChemCatChem, 2018, 10, 706–709 CrossRef CAS.
- Y. L. Zhang, B. Li, Y. A. Wei, C. H. Yan, M. J. Meng and Y. S. Yan, J. Taiwan Inst. Chem. Eng., 2019, 96, 93–103 CrossRef CAS.
- Y. Zhang, P. Jin, M. Meng, L. Gao, M. Liu and Y. Yan, Nano, 2018, 13, 1850132 CrossRef.
- Y. Zhang, J. Zhao, K. Wang, L. Gao, M. Meng and Y. Yan, ChemistrySelect, 2018, 3, 9378–9387 CrossRef CAS.
- J. Zhao, Y. Zhang, K. Wang, C. Yan, Z. Da, C. Li and Y. Yan, ChemistrySelect, 2018, 3, 11476–11485 CrossRef CAS.
- Z. Hu and D. Zhao, CrystEngComm, 2017, 19, 4066–4081 RSC.
- G. Zi, Z. Yan, Y. Wang, Y. Chen, Y. Guo, F. Yuan, W. Gao, Y. Wang and J. Wang, Carbohydr. Polym., 2015, 115, 146–151 CrossRef CAS.
- Q. Guo, L. Ren, P. Kumar, V. J. Cybulskis, K. A. Mkhoyan, M. E. Davis and M. Tsapatsis, Angew. Chem., Int. Ed., 2018, 57, 4926–4930 CrossRef CAS.
- D. L. Burnett, R. Oozeerally, R. Pertiwi, T. W. Chamberlain, N. Cherkasov, G. J. Clarkson, Y. K. Krisnandi, V. Degirmenci and R. I. Walton, Chem. Commun., 2019, 55, 11446–11449 RSC.
- C. D. Malonzo, S. M. Shaker, L. Ren, S. D. Prinslow, A. E. Platero-Prats, L. C. Gallington, J. Borycz, A. B. Thompson, T. C. Wang, O. K. Farha, J. T. Hupp, C. C. Lu, K. W. Chapman, J. C. Myers, R. L. Penn, L. Gagliardi, M. Tsapatsis and A. Stein, J. Am. Chem. Soc., 2016, 138, 2739–2748 CrossRef CAS.
- M. Du, A. M. Agrawal, S. Chakraborty, S. J. Garibay, R. Limvorapitux, B. Choi, S. T. Madrahimov and S. T. Nguyen, ACS Sustainable Chem. Eng., 2019, 7, 8126–8135 CrossRef CAS.
- K. Cho, S. M. Lee, H. J. Kim, Y. J. Ko and S. U. Son, Chem. Commun., 2019, 55, 3697–3700 RSC.
- K. Cho, S. M. Lee, H. J. Kim, Y.-J. Ko and S. U. Son, J. Mater. Chem. A, 2018, 6, 15553–15557 RSC.
- A. Kumar and R. Srivastava, Mol. Catal., 2019, 465, 68–79 CrossRef CAS.
- W. Li, T. Zhang, H. Xin, M. Su, L. Ma, H. Jameel, H.-M. Chang and G. Pei, RSC Adv., 2017, 7, 27682–27688 RSC.
- T. Zhang, W. Li, S. An, F. Huang, X. Li, J. Liu, G. Pei and Q. Liu, Bioresour. Technol., 2018, 264, 261–267 CrossRef CAS.
- H. Tang, N. Li, G. Y. Li, W. T. Wang, A. Q. Wang, Y. Cong and X. D. Wang, ACS Sustainable Chem. Eng., 2018, 6, 5645–5652 CrossRef CAS.
- S. Ravi, Y. Choi and J. K. Choe, Appl. Catal., B, 2020, 271, 118942 CrossRef CAS.
- Z. Li, K. Su, J. Ren, D. Yang, B. Cheng, C. K. Kim and X. Yao, Green Chem., 2018, 20, 863–872 RSC.
- S. Verma, R. B. N. Baig, M. N. Nadagouda, C. Len and R. S. Varma, Green Chem., 2017, 19, 164–168 RSC.
- T. Chhabra, A. Bahuguna, S. S. Dhankhar, C. M. Nagaraja and V. Krishnan, Green Chem., 2019, 21, 6012–6026 RSC.
- X. Cao, S. P. Teong, D. Wu, G. Yi, H. Su and Y. Zhang, Green Chem., 2015, 17, 2348–2352 RSC.
- H. Pawar and A. Lali, Ind. Eng. Chem. Res., 2018, 57, 14428–14439 CrossRef CAS.
- A. S. Wagh and H. S. Pawar, Energy Fuels, 2020, 34, 9643–9653 CrossRef CAS.
- F. D. Bobbink, Z. Huang, F. Menoud and P. J. Dyson, ChemSusChem, 2019, 12, 1437–1442 CrossRef CAS.
- Z. Babaei, A. N. Chermahini, M. Dinari, M. Saraji and A. Shahvar, Sustainable Energy Fuels, 2019, 3, 1024–1032 RSC.
- S. Xu, C. Yin, D. Pan, F. Hu, Y. Wu, Y. Miao, L. Gao and G. Xiao, Sustainable Energy Fuels, 2019, 3, 390–395 RSC.
- Z. Babaei, A. Najafi Chermahini, M. Dinari, M. Saraji and A. Shahvar, J. Cleaner Prod., 2018, 198, 381–388 CrossRef CAS.
- K. Wang, C. Liang, Q. Zhang and F. Zhang, ACS Omega, 2019, 4, 1053–1059 CrossRef CAS.
- L. Jiao, S. Sun, X. Meng and P. Ji, Catalysts, 2019, 9, 739 CrossRef CAS.
- G. Qiu, B. Chen, C. Huang, N. Liu and X. Sun, Fuel, 2020, 268, 117136 CrossRef CAS.
- P. Gogoi and R. Borah, J. Chem. Sci., 2018, 130, 170 CrossRef.
- J. Dai, L. Zhu, D. Tang, X. Fu, J. Tang, X. Guo and C. Hu, Green Chem., 2017, 19, 1932–1939 RSC.
- H. Tang, N. Li, F. Chen, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, Green Chem., 2017, 19, 1855–1860 RSC.
- R. L. Johnson, F. A. Perras, M. P. Hanrahan, M. Mellmer, T. F. Garrison, T. Kobayashi, J. A. Dumesic, M. Pruski, A. J. Rossini and B. H. Shanks, ACS Catal., 2019, 9, 11568–11578 CrossRef CAS.
- Y. Nishimura, M. Suda, M. Kuroha, H. Kobayashi, K. Nakajima and A. Fukuoka, Carbohydr. Res., 2019, 486, 107826 CrossRef.
- S.-H. Pyo, M. Sayed and R. Hatti-Kaul, Org. Process Res. Dev., 2019, 23, 952–960 CrossRef CAS.
- L. Zhu, J. Dai, M. Liu, D. Tang, S. Liu and C. Hu, ChemSusChem, 2016, 2174–2181 CrossRef CAS.
- M.-P. Ruby and F. Schüth, Green Chem., 2016, 18, 3422–3429 RSC.
- F. Yu, M. Smet, W. Dehaen and B. F. Sels, Chem. Commun., 2016, 52, 2756–2759 RSC.
- F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Green Chem., 2016, 18, 1694–1705 RSC.
- M. Tyufekchiev, P. Duan, K. Schmidt-Rohr, S. Granados Focil, M. T. Timko and M. H. Emmert, ACS Catal., 2018, 8, 1464–1468 CrossRef CAS.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318–325 CrossRef.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477 CrossRef CAS.
- H. Huang, C. A. Denard, R. Alamillo, A. J. Crisci, Y. Miao, J. A. Dumesic, S. L. Scott and H. Zhao, ACS Catal., 2014, 4, 2165–2168 CrossRef CAS.
- F. Lin, K. Wang, L. Gao and X. Guo, Appl. Organomet. Chem., 2019, 33, e4821 CrossRef.
- B. Liu and Z. Zhang, ACS Catal., 2016, 6, 326–338 CrossRef CAS.
- M. Shaikh, M. Sahu, K. K. Atyam and K. V. S. Ranganath, RSC Adv., 2016, 6, 76795–76801 RSC.
- M. Shaikh, S. K. Singh, S. Khilari, M. Sahu and K. V. S. Ranganath, Catal. Commun., 2018, 106, 64–67 CrossRef CAS.
- R. T. Woodward, M. Kessler, S. Lima and R. Rinaldi, Green Chem., 2018, 20, 2374–2381 RSC.
- Q. Sun, S. Wang, B. Aguila, X. Meng, S. Ma and F.-S. Xiao, Nat. Commun., 2018, 9, 3236 CrossRef.
- J. Yang, K. De Oliveira Vigier, Y. Gu and F. Jérôme, ChemSusChem, 2015, 8, 269–274 CrossRef CAS.
- T. W. Walker, A. H. Motagamwala, J. A. Dumesic and G. W. Huber, J. Catal., 2019, 369, 518–525 CrossRef CAS.
- A. P. S. Brogan, L. Bui-Le and J. P. Hallett, Nat. Chem., 2018, 10, 859–865 CrossRef CAS.
- N. Mimura, O. Sato, M. Shirai and A. Yamaguchi, ChemistrySelect, 2017, 2, 1305–1310 CrossRef CAS.
- N. Sweygers, D. E. C. Depuydt, A. W. Van Vuure, J. Degrève, G. Potters, R. Dewil and L. Appels, Chem. Eng. J., 2020, 386, 123957 CrossRef CAS.
- S. Amirjalayer, H. Fuchs and D. Marx, Angew. Chem., Int. Ed., 2019, 58, 5232–5235 CrossRef CAS.
- F. Shen, X. Xiong, J. Fu, J. Yang, M. Qiu, X. Qi and D. C. W. Tsang, Renewable Sustainable Energy Rev., 2020, 130, 109944 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS.
- X. Liu, X. Duan, W. Wei, S. Wang and B.-J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- K. Tsutsumi, N. Kurata, E. Takata, K. Furuichi, M. Nagano and K. Tabata, Appl. Catal., B, 2014, 147, 1009–1014 CrossRef CAS.
- B. Zhang, J. Li, L. Guo, Z. Chen and C. Li, Appl. Catal., B, 2018, 237, 660–664 CrossRef CAS.
- F. Delbecq and C. Len, Molecules, 2018, 23, 1973 CrossRef.
- N. Sweygers, N. Alewaters, R. Dewil and L. Appels, Sci. Rep., 2018, 8, 7719 CrossRef.
- N. Sweygers, J. Harrer, R. Dewil and L. Appels, J. Cleaner Prod., 2018, 187, 1014–1024 CrossRef CAS.
- T. Ji, R. Tu, L. Mu, X. Lu and J. Zhu, ACS Sustainable Chem. Eng., 2017, 5, 4352–4358 CrossRef CAS.
- T. Ji, R. Tu, L. Mu, X. Lu and J. Zhu, Appl. Catal., B, 2018, 220, 581–588 CrossRef CAS.
- T. Ji, Z. Li, C. Liu, X. Lu, L. Li and J. Zhu, Appl. Catal., B, 2019, 243, 741–749 CrossRef CAS.
- C. Xiouras, N. Radacsi, G. Sturm and G. D. Stefanidis, ChemSusChem, 2016, 9, 2159–2166 CrossRef CAS.
- S. Marullo, A. Sutera, G. Gallo, F. Billeci, C. Rizzo and F. D’Anna, ACS Sustainable Chem. Eng., 2020, 8, 11204–11214 CrossRef CAS.
- R. Galaverna, M. C. Breitkreitz and J. C. Pastre, ACS Sustainable Chem. Eng., 2018, 6, 4220–4230 CrossRef CAS.
- J. Tacacima, S. Derenzo and J. G. R. Poco, Mol. Catal., 2018, 458, 180–188 CrossRef CAS.
- F. J. Morales-Leal, J. Rivera de la Rosa, C. J. Lucio-Ortiz, D. A. De Haro-Del Rio, C. Solis Maldonado, S. Wi, L. B. Casabianca and C. D. Garcia, Appl. Catal., B, 2019, 244, 250–261 CrossRef CAS.
- C. Sonsiam, A. Kaewchada, S. Pumrod and A. Jaree, Chem. Eng. Process., 2019, 138, 65–72 CrossRef CAS.
- E. Weingart, S. Tschirner, L. Teevs and U. Prüße, Molecules, 2018, 23, 1802 CrossRef.
- J. M. R. Gallo, R. Alamillo and J. A. Dumesic, J. Mol. Catal. A: Chem., 2016, 422, 13–17 CrossRef CAS.
- C. Tempelman, U. Jacobs, T. Hut, E. Pereira de Pina, M. van Munster, N. Cherkasov and V. Degirmenci, Appl. Catal., A, 2019, 588, 117267 CrossRef.
- Y. Muranaka, H. Nakagawa, R. Masaki, T. Maki and K. Mae, Ind. Eng. Chem. Res., 2017, 56, 10998–11005 CrossRef CAS.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CrossRef CAS.
- R. F. A. Gomes, Y. N. Mitrev, S. P. Simeonov and C. A. M. Afonso, ChemSusChem, 2018, 11, 1612–1616 CrossRef CAS.
- Y. M. Questell-Santiago, R. Zambrano-Varela, M. T. Amiri and J. S. Luterbacher, Nat. Chem., 2018, 10, 1222–1228 CrossRef CAS.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- C. Zhou, C. Shen, K. Ji, J. Yin and L. Du, ACS Sustainable Chem. Eng., 2018, 6, 3992–3999 CrossRef CAS.
- J. Lueckgen, L. Vanoye, R. Philippe, M. Eternot, P. Fongarland, C. de Bellefon and A. Favre-Réguillon, J. Flow Chem., 2018, 8, 3–9 CrossRef CAS.
- A. Sarwono, Z. Man, A. Idris, A. S. Khan, N. Muhammad and C. D. Wilfred, J. Ind. Eng. Chem., 2019, 69, 171–178 CrossRef CAS.
- P. Yan, M. Xia, S. Chen, W. Han, H. Wang and W. Zhu, Green Chem., 2020, 22, 5274–5284 RSC.
- S. Gajula, K. Inthumathi, S. R. Arumugam and K. Srinivasan, ACS Sustainable Chem. Eng., 2017, 5, 5373–5381 CrossRef CAS.
- S. Alipour, Green Chem., 2016, 18, 4990–4998 RSC.
- A. Gimbernat, M. Guehl, N. Lopes Ferreira, E. Heuson, P. Dhulster, M. Capron, F. Dumeignil, D. Delcroix, J.-S. Girardon and R. Froidevaux, Catalysts, 2018, 8, 335 CrossRef.
- M. Yabushita, P. Li, H. Kobayashi, A. Fukuoka, O. K. Farha and A. Katz, Chem. Commun., 2016, 52, 11791–11794 RSC.
- H. Jin, Y. Li, X. Liu, Y. Ban, Y. Peng, W. Jiao and W. Yang, Chem. Eng. Sci., 2015, 124, 170–178 CrossRef CAS.
- C. Detoni, C. H. Gierlich, M. Rose and R. Palkovits, ACS Sustainable Chem. Eng., 2014, 2, 2407–2415 CrossRef CAS.
- H. S. Pawar, ChemistrySelect, 2020, 5, 6851–6855 CrossRef CAS.
- K. Schute, Y. Louven, C. Detoni and M. Rose, Chem. Ing. Tech., 2016, 88, 355–362 CrossRef CAS.
- M. León, T. D. Swift, V. Nikolakis and D. G. Vlachos, Langmuir, 2013, 29, 6597–6605 CrossRef.
- Y.-B. Zhang, Q.-X. Luo, M.-H. Lu, D. Luo, Z.-W. Liu and Z.-T. Liu, Chem. Eng. J., 2019, 358, 467–479 CrossRef CAS.
- M. Yabushita, N. A. Grosso-Giordano, A. Fukuoka and A. Katz, ACS Appl. Mater. Interfaces, 2018, 10, 39670–39678 CrossRef CAS.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976–2982 CrossRef CAS.
- E. G. L. de Carvalho, F. D. Rodrigues, R. S. Monteiro, R. M. Ribas and M. J. da Silva, Biomass Convers. Biorefin., 2018, 8, 635–646 CrossRef CAS.
- M. A. Kougioumtzis, A. Marianou, K. Atsonios, C. Michailof, N. Nikolopoulos, N. Koukouzas, K. Triantafyllidis, A. Lappas and E. Kakaras, Waste Biomass Valorization, 2018, 9, 2433–2445 CrossRef CAS.
- K. Gupta, R. K. Rai and S. K. Singh, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS.
- F. A. Kucherov, L. V. Romashov, K. I. Galkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2018, 6, 8064–8092 CrossRef CAS.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC.
- B. Liu and Z. Zhang, ChemSusChem, 2016, 9, 2015–2036 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924–7926 CrossRef CAS.
- M. Mascal, ACS Sustainable Chem. Eng., 2019, 7, 5588–5601 CrossRef CAS.
- M. Mascal, ChemSusChem, 2015, 8, 3391–3395 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370–373 RSC.
- E.-S. Kang, Y.-W. Hong, D. W. Chae, B. Kim, B. Kim, Y. J. Kim, J. K. Cho and Y. G. Kim, ChemSusChem, 2015, 8, 1179–1188 CrossRef CAS.
- M. Bicker, D. Kaiser, L. Ott and H. Vogel, J. Supercrit. Fluids, 2005, 36, 118–126 CrossRef CAS.
- L. Gavilà and D. Esposito, Green Chem., 2017, 19, 2496–2500 RSC.
- B. Sarmah and R. Srivastava, Mol. Catal., 2019, 462, 92–103 CrossRef CAS.
- Q. Ke, Y. Jin, F. Ruan, M. N. Ha, D. Li, P. Cui, Y. Cao, H. Wang, T. Wang, V. N. Nguyen, X. Han, X. Wang and P. Cui, Green Chem., 2019, 21, 4313–4318 RSC.
- Y. Liu, H.-Y. Ma, D. Lei, L.-L. Lou, S. Liu, W. Zhou, G.-C. Wang and K. Yu, ACS Catal., 2019, 9, 8306–8315 CrossRef CAS.
- L. Ardemani, G. Cibin, A. J. Dent, M. A. Isaacs, G. Kyriakou, A. F. Lee, C. M. A. Parlett, S. A. Parry and K. Wilson, Chem. Sci., 2015, 6, 4940–4945 RSC.
- J. An, G. Sun and H. Xia, ACS Sustainable Chem. Eng., 2019, 7, 6696–6706 CrossRef CAS.
- D. Zhao, D. Rodriguez-Padron, R. Luque and C. Len, ACS Sustainable Chem. Eng., 2020, 8, 8486–8495 CrossRef CAS.
- B. Zhou, J. Song, Z. Zhang, Z. Jiang, P. Zhang and B. Han, Green Chem., 2017, 19, 1075–1081 RSC.
- H. Xia, J. An, M. Hong, S. Xu, L. Zhang and S. Zuo, Catal. Today, 2019, 319, 113–120 CrossRef CAS.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J.-D. Grunwaldt, Green Chem., 2018, 20, 3530–3541 RSC.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. L. Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- K. R. Hwang, W. Jeon, S. Y. Lee, M. S. Kim and Y. K. Park, Chem. Eng. J., 2020, 390, 13 CrossRef.
- H. B. Yuan, H. L. Liu, J. K. Du, K. Q. Liu, T. F. Wang and L. Liu, Appl. Microbiol. Biotechnol., 2020, 104, 527–543 CrossRef CAS.
- K. Li and Y. J. Sun, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS.
- P. Pal and S. Saravanamurugan, ChemSusChem, 2019, 12, 145–163 CrossRef CAS.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- R. A. Sheldon, Front. Chem., 2020, 8, 132 CrossRef CAS.
- B. J. Taitt, D. H. Nam and K. S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- M. Hong, J. Min, S. Wu, H. Cui, Y. Zhao, J. Li and S. Wang, ACS Omega, 2019, 4, 7054–7060 CrossRef CAS.
- G. Lv, S. Chen, H. Zhu, M. Li and Y. Yang, Appl. Surf. Sci., 2018, 458, 24–31 CrossRef CAS.
- Y. Ren, Z. Yuan, K. Lv, J. Sun, Z. Zhang and Q. Chi, Green Chem., 2018, 20, 4946–4956 RSC.
- S. Hazra, M. Deb and A. J. Elias, Green Chem., 2017, 19, 5548–5552 RSC.
- J. Ren, K. H. Song, Z. Li, Q. Wang, J. Li, Y. X. Wang, D. B. Li and C. K. Kim, Appl. Surf. Sci., 2018, 456, 174–183 CrossRef CAS.
- P. Tan, G. Li, R. Fang, L. Chen, R. Luque and Y. Li, ACS Catal., 2017, 7, 2948–2955 CrossRef CAS.
- J. P. Zhang, S. Nagamatsu, J. M. Du, C. L. Tong, H. H. Fang, D. H. Deng, X. Liu, K. Asakura and Y. Z. Yuan, J. Catal., 2018, 367, 16–26 CrossRef CAS.
- A. B. Raut and B. M. Bhanage, ChemistrySelect, 2018, 3, 11388–11397 CrossRef CAS.
- W. Zhang, T. Meng, J. Tang, W. Zhuang, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 10029–10037 CrossRef CAS.
- H. S. Pawar, ChemistrySelect, 2020, 5, 7417–7426 CrossRef CAS.
- Q. Q. Li, H. Y. Wang, Z. P. Tian, Y. J. Weng, C. G. Wang, J. R. Ma, C. F. Zhu, W. Z. Li, Q. Y. Liu and L. L. Ma, Catal. Sci. Technol., 2019, 9, 1570–1580 RSC.
- A. Buonerba, S. Impemba, A. D. Litta, C. Capacchione, S. Milione and A. Grassi, ChemSusChem, 2018, 11, 3139–3149 CrossRef CAS.
- Q. Wang, W. Hou, S. Li, J. Xie, J. Li, Y. Zhou and J. Wang, Green Chem., 2017, 19, 3820–3830 RSC.
- P. Zhang, X. Zhang, X. Kang, H. Liu, C. Chen, C. Xie and B. Han, Chem. Commun., 2018, 54, 12065–12068 RSC.
- D. Bonincontro, A. Lolli, A. Villa, L. Prati, N. Dimitratos, G. M. Veith, L. E. Chinchilla, G. A. Botton, F. Cavani and S. Albonetti, Green Chem., 2019, 21, 4090–4099 RSC.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 890–900 CrossRef CAS.
- K. T. V. Rao, J. L. Rogers, S. Souzanchi, L. Dessbesell, M. B. Ray and C. Xu, ChemSusChem, 2018, 11, 3323–3334 CrossRef CAS.
- S. Zhang, X. Sun, Z. Zheng and L. Zhang, Catal. Commun., 2018, 113, 19–22 CrossRef CAS.
- X. Liao, J. Hou, Y. Wang, H. Zhang, Y. Sun, X. Li, S. Tang, K. Kato, M. Yamauchi and Z. Jiang, Green Chem., 2019, 21, 4194–4203 RSC.
- Z. Yuan, B. Liu, P. Zhou, Z. Zhang and Q. Chi, Catal. Sci. Technol., 2018, 8, 4430–4439 RSC.
- R. Chen, J. Xin, D. Yan, H. Dong, X. Lu and S. Zhang, ChemSusChem, 2019, 12, 2715–2724 CrossRef CAS.
- H. Yu, K.-A. Kim, M. J. Kang, S. Y. Hwang and H. G. Cha, ACS Sustainable Chem. Eng., 2019, 7, 3742–3748 CrossRef CAS.
- B. Donoeva, N. Masoud and P. E. de Jongh, ACS Catal., 2017, 7, 4581–4591 CrossRef CAS.
- H. Liu, X. J. Cao, J. N. Wei, W. L. Jia, M. Z. Li, X. Tang, X. H. Zeng, Y. Sun, T. Z. Lei, S. J. Liu and L. Lin, ACS Sustainable Chem. Eng., 2019, 7, 7812–7822 CrossRef CAS.
- F. Nocito, M. Ventura, M. Aresta and A. Dibenedetto, ACS Omega, 2018, 3, 18724–18729 CrossRef CAS.
- T. Gao, J. Chen, W. Fang, Q. Cao, W. Su and F. Dumeignil, J. Catal., 2018, 368, 53–68 CrossRef CAS.
- C. Ke, M. Li, G. Fan, L. Yang and F. Li, Chem. - Asian J., 2018, 13, 2714–2722 CrossRef CAS.
- C. M. Pichler, M. G. Al-Shaal, D. Gu, H. Joshi, W. Ciptonugroho and F. Schüth, ChemSusChem, 2018, 11, 2083–2090 CrossRef CAS.
- A. Tirsoaga, M. El Fergani, V. I. Parvulescu and S. M. Coman, ACS Sustainable Chem. Eng., 2018, 6, 14292–14301 CrossRef CAS.
- S. Verma, M. N. Nadagouda and R. S. Varma, Sci. Rep., 2017, 7, 13596 CrossRef.
- J. M. Jeong, S. B. Jin, J. H. Yoon, J. G. Yeo, G. Y. Lee, M. Irshad, S. Lee, D. Seo, B. E. Kwak, B. G. Choi, D. H. Kim and J. W. Kim, Catalysts, 2019, 9, 9 Search PubMed.
- M. Chatterjee, T. Ishizaka, A. Chatterjee and H. Kawanami, Green Chem., 2017, 19, 1315–1326 RSC.
- J. Lai, K. Liu, S. Zhou, D. Zhang, X. Liu, Q. Xu and D. Yin, RSC Adv., 2019, 9, 14242–14246 RSC.
- Q. Wang, W. Hou, T. Meng, Q. Hou, Y. Zhou and J. Wang, Catal. Today, 2019, 319, 57–65 CrossRef CAS.
- J. Xu, T. Su, Z. Zhu, N. Chen, D. Hao, M. Wang, Y. Zhao, W. Ren and H. Lü, Chem. Eng. J., 2020, 396, 125303 CrossRef CAS.
- J. Zhao, X. Chen, Y. Du, Y. Yang and J.-M. Lee, Appl. Catal., A, 2018, 568, 16–22 CrossRef CAS.
- J. Zhao, A. Jayakumar, Z.-T. Hu, Y. Yan, Y. Yang and J.-M. Lee, ACS Sustainable Chem. Eng., 2018, 6, 284–291 CrossRef CAS.
- C. A. Antonyraj, N. T. T. Huynh, K. W. Lee, Y. J. Kim, S. Shin, J. S. Shin and J. K. Cho, J. Chem. Sci., 2018, 130, 9 CrossRef.
- A. Danielli da Fonseca Ferreira, M. Dorneles de Mello and M. A. P. da Silva, Ind. Eng. Chem. Res., 2019, 58, 128–137 CrossRef CAS.
- C. Megías-Sayago, K. Chakarova, A. Penkova, A. Lolli, S. Ivanova, S. Albonetti, F. Cavani and J. A. Odriozola, ACS Catal., 2018, 8, 11154–11164 CrossRef.
- C. P. Ferraz, M. Zielinski, M. Pietrowski, S. Heyte, F. Dumeignil, L. M. Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- F. Li, X.-L. Li, C. Li, J. Shi and Y. Fu, Green Chem., 2018, 20, 3050–3058 RSC.
- A. Cho, S. Byun, J. H. Cho and B. M. Kim, ChemSusChem, 2019, 12, 2310–2317 CrossRef CAS.
- K.-K. Sun, S.-J. Chen, Z.-L. Li, G.-P. Lu and C. Cai, Green Chem., 2019, 21, 1602–1608 RSC.
- H. Liu, N. Ding, J. N. Wei, X. Tang, X. H. Zeng, Y. Sun, T. Z. Lei, H. Y. Fang, T. Y. Li and L. Lin, ChemSusChem, 2020, 13, 4151–4158 CrossRef CAS.
- T. Ji, C. Liu, X. Lu and J. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 11493–11501 CrossRef CAS.
- A. Tirsoaga, M. El Fergani, N. Nuns, P. Simon, P. Granger, V. I. Parvulescu and S. M. Coman, Appl. Catal., B, 2020, 278, 119309 CrossRef CAS.
- C. Yang, X. Li, Z. Zhang, B. Lv, J. Li, Z. Liu, W. Zhu, F. Tao, G. Lv and Y. Yang, J. Energy Chem., 2020, 50, 96–105 CrossRef.
- K. Afroz, M. Ntambwe and N. Nuraje, Inorg. Chem., 2020, 59, 13335–13342 CrossRef CAS.
- Y.-T. Liao, V. C. Nguyen, N. Ishiguro, A. P. Young, C.-K. Tsung and K. C. W. Wu, Appl. Catal., B, 2020, 270, 118805 CrossRef CAS.
- S. S. R. Gupta, A. Vinu and M. L. Kantam, J. Catal., 2020, 389, 259–269 CrossRef CAS.
- W. Zhuang, X. Liu, L. Chen, P. Liu, H. Wen, Y. Zhou and J. Wang, Green Chem., 2020, 22, 4199–4209 RSC.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS.
- L. Gao, Y. Bao, S. Gan, Z. Sun, Z. Song, D. Han, F. Li and L. Niu, ChemSusChem, 2018, 11, 2547–2553 CrossRef CAS.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS.
- J. Zhao, A. Jayakumar and J.-M. Lee, ACS Sustainable Chem. Eng., 2018, 6, 2976–2982 CrossRef CAS.
- C. Zhou, J. Zhao, H. Sun, Y. Song, X. Wan, H. Lin and Y. Yang, ACS Sustainable Chem. Eng., 2019, 7, 315–323 CrossRef CAS.
- B. Sarmah, B. Satpati and R. Srivastava, Catal. Sci. Technol., 2018, 8, 2870–2882 RSC.
- D. Yan, J. Xin, C. Shi, X. Lu, L. Ni, G. Wang and S. Zhang, Chem. Eng. J., 2017, 323, 473–482 CrossRef CAS.
- Y. Cheng, S. Zhao, B. Johannessen, J.-P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm, R. De Marco, H.-M. Cheng, S.-Z. Yang and S. P. Jiang, Adv. Mater., 2018, 30, 1706287 CrossRef.
- Z.-M. Wang, L.-J. Liu, B. Xiang, Y. Wang, Y.-J. Lyu, T. Qi, Z.-B. Si, H.-Q. Yang and C.-W. Hu, Catal. Sci. Technol., 2019, 9, 811–821 RSC.
- C. Laugel, B. Estrine, J. Le Bras, N. Hoffmann, S. Marinkovic and J. Muzart, ChemCatChem, 2014, 6, 1195–1198 CAS.
- G. Li, Z. Sun, Y. Yan, Y. Zhang and Y. Tang, ChemSusChem, 2017, 10, 494–498 CrossRef CAS.
- I. Krivtsov, E. I. García-López, G. Marcì, L. Palmisano, Z. Amghouz, J. R. García, S. Ordóñez and E. Díaz, Appl. Catal., B, 2017, 204, 430–439 CrossRef CAS.
- A. Lolli, V. Maslova, D. Bonincontro, F. Basile, S. Ortelli and S. Albonetti, Molecules, 2018, 23, 2792 CrossRef.
- G. Marcì, E. I. García-López, F. R. Pomilla, L. Palmisano, A. Zaffora, M. Santamaria, I. Krivtsov, M. Ilkaeva, Z. Barbieriková and V. Brezová, Catal. Today, 2019, 328, 21–28 CrossRef.
- M. Ilkaeva, I. Krivtsov, J. R. Garcia, E. Diaz, S. Ordonez, E. I. Garcia-Lopez, G. Marci, L. Palmisano, M. I. Maldonado and S. Malato, Catal. Today, 2018, 315, 138–148 CrossRef CAS.
- M. Ilkaeva, I. Krivtsov, E. I. García-López, G. Marcì, O. Khainakova, J. R. García, L. Palmisano, E. Díaz and S. Ordóñez, J. Catal., 2018, 359, 212–222 CrossRef CAS.
- H. Zhang, Z. Feng, Y. Zhu, Y. Wu and T. Wu, J. Photochem. Photobiol., A, 2019, 371, 1–9 CrossRef CAS.
- B. Yang, W. Hu, F. Wan, C. Zhang, Z. Fu, A. Su, M. Chen and Y. Liu, Chem. Eng. J., 2020, 396, 125345 CrossRef CAS.
- H. Zhang, Q. Wu, C. Guo, Y. Wu and T. Wu, ACS Sustainable Chem. Eng., 2017, 5, 3517–3523 CrossRef CAS.
- E. I. Garcia-Lopez, F. R. Pomilla, E. Bloise, X. F. Lu, G. Mele, L. Palmisano and G. Marci, Top. Catal., 2020, 14 DOI:10.1007/s11244-020-01293-0.
- S. Xu, P. Zhou, Z. Zhang, C. Yang, B. Zhang, K. Deng, S. Bottle and H. Zhu, J. Am. Chem. Soc., 2017, 139, 14775–14782 CrossRef CAS.
- H.-F. Ye, R. Shi, X. Yang, W.-F. Fu and Y. Chen, Appl. Catal., B, 2018, 233, 70–79 CrossRef CAS.
- S. Dhingra, T. Chhabra, V. Krishnan and C. M. Nagaraja, ACS Appl. Energy Mater., 2020, 3, 7138–7148 CrossRef CAS.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS.
- S. Meng, H. Wu, Y. Cui, X. Zheng, H. Wang, S. Chen, Y. Wang and X. Fu, Appl. Catal., B, 2020, 266, 118617 CrossRef.
- A. Brandolese, D. Ragno, G. Di Carmine, T. Bernardi, O. Bortolini, P. P. Giovannini, O. G. Pandoli, A. Altomare and A. Massi, Org. Biomol. Chem., 2018, 16, 8955–8964 RSC.
- S.-S. Shi, X.-Y. Zhang, M.-H. Zong, C.-F. Wang and N. Li, Mol. Catal., 2019, 469, 68–74 CrossRef CAS.
- M. Sayed, S.-H. Pyo, N. Rehnberg and R. Hatti-Kaul, ACS Sustainable Chem. Eng., 2019, 7, 4406–4413 CrossRef CAS.
- J. H. Zhang, Q. D. Liang, W. X. Xie, L. C. Peng, L. He, Z. B. He, S. P. Chowdhury, R. Christensen and Y. H. Ni, Polymers, 2019, 11, 14 Search PubMed.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215 CrossRef CAS.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- S. H. Krishna, K. Huang, K. J. Barnett, J. He, C. T. Maravelias, J. A. Dumesic, G. W. Huber, M. De Bruyn and B. M. Weckhuysen, AIChE J., 2018, 64, 1910–1922 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS.
- F. A. Kucherov, E. G. Gordeev, A. S. Kashin and V. P. Ananikov, Angew. Chem., Int. Ed., 2017, 56, 15931–15935 CrossRef CAS.
- V. A. Klushin, V. P. Kashparova, I. S. Kashparov, Y. A. Chus, A. A. Chizhikova, T. A. Molodtsova and N. V. Smirnova, Russ. Chem. Bull., 2019, 68, 570–577 CrossRef CAS.
- M. Sajid, X. B. Zhao and D. H. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- C. Chen, L. Wang, B. Zhu, Z. Zhou, S. I. El-Hout, J. Yang and J. Zhang, J. Energy Chem., 2021, 54, 528–554 CrossRef.
- A. D. K. Deshan, L. Atanda, L. Moghaddam, D. W. Rackemann, J. Beltramini and W. O. S. Doherty, Front. Chem., 2020, 8, 659 CrossRef CAS.
- D. Troiano, V. Orsat and M. J. Dumont, ACS Catal., 2020, 10, 9145–9169 CrossRef CAS.
- E. Hayashi, T. Komanoya, K. Kamata and M. Hara, ChemSusChem, 2017, 10, 654–658 CrossRef CAS.
- Y. Yamaguchi, R. Aono, E. Hayashi, K. Kamata and M. Hara, ACS Appl. Mater. Interfaces, 2020, 12, 36004–36013 CrossRef CAS.
- O. R. Schade, A. Gaur, A. Zimina, E. Saraçi and J.-D. Grunwaldt, Catal. Sci. Technol., 2020, 10, 5036–5047 RSC.
- H. Zhou, H. Xu and Y. Liu, Appl. Catal., B, 2019, 244, 965–973 CrossRef CAS.
- S. K. Singh, Asian J. Org. Chem., 2018, 7, 1901–1923 CrossRef CAS.
- C. Zhou, W. Shi, X. Wan, Y. Meng, Y. Yao, Z. Guo, Y. Dai, C. Wang and Y. Yang, Catal. Today, 2019, 330, 92–100 CrossRef CAS.
- F. Liguori, P. Barbaro and N. Calisi, ChemSusChem, 2019, 12, 2558–2563 CrossRef CAS.
- W. Naim, O. R. Schade, E. Saraci, D. Wust, A. Kruse and J. D. Grunwaldt, ACS Sustainable Chem. Eng., 2020, 8, 11512–11521 CrossRef CAS.
- W. Wu, S. Xu, G. Qi, H. Zhu, F. Hu, Z. Liu, D. Zhang and B. Liu, Angew. Chem., Int. Ed., 2019, 58, 3062–3066 CrossRef CAS.
- W. B. Wu, D. Mao, S. D. Xu, Kenry, F. Hu, X. Q. Li, D. L. Kong and B. Liu, Chem, 2018, 4, 1937–1951 CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS.
- W. Li, N. Jiang, B. Hu, X. Liu, F. Song, G. Han, T. J. Jordan, T. B. Hanson, T. L. Liu and Y. Sun, Chem, 2018, 4, 637–649 CAS.
- M. Park, M. Gu and B.-S. Kim, ACS Nano, 2020, 14, 6812–6822 CrossRef CAS.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4 CAS.
- Y. Mathieu, W. A. Offen, S. M. Forget, L. Ciano, A. H. Viborg, E. Blagova, B. Henrissat, P. H. Walton, G. J. Davies and H. Brumer, ACS Catal., 2020, 10, 3042–3058 CrossRef CAS.
- L. Hu, A. Y. He, X. Y. Liu, J. Xia, J. X. Xu, S. Y. Zhou and J. M. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS.
- C. Zhang, X. Chang, L. Zhu, Q. Xing, S. You, W. Qi, R. Su and Z. He, Int. J. Biol. Macromol., 2019, 128, 132–139 CrossRef CAS.
- T. K. Godan, R. O. Rajesh, P. C. Loreni, A. Kumar Rai, D. Sahoo, A. Pandey and P. Binod, Bioresour. Technol., 2019, 282, 88–93 CrossRef CAS.
- M. Daou, B. Yassine, S. Wikee, E. Record, F. Duprat, E. Bertrand and C. B. Faulds, Fungal Biol. Rev., 2019, 6, 4 Search PubMed.
- H. B. Yuan, Y. F. Liu, X. Q. Lv, J. H. Li, G. C. Du, Z. P. Shi and L. Liu, J. Microbiol. Biotechnol., 2018, 28, 1999–2008 CrossRef CAS.
- H. Yuan, Y. Liu, J. Li, H.-D. Shin, G. Du, Z. Shi, J. Chen and L. Liu, Biotechnol. Bioeng., 2018, 115, 2148–2155 CrossRef CAS.
- X. Wang, X.-Y. Zhang, M.-H. Zong and N. Li, ACS Sustainable Chem. Eng., 2020, 8, 4341–4345 CrossRef CAS.
- C. Isola, H. L. Sieverding, R. Raghunathan, M. P. Sibi, D. C. Webster, J. Sivaguru and J. J. Stone, J. Cleaner Prod., 2017, 142, 2935–2944 CrossRef CAS.
- S. Bello, I. Salim, P. Mendez-Trelles, E. Rodil, G. Feijoo and M. T. Moreira, Holzforschung, 2019, 73, 105–115 CAS.
- J. Byun and J. Han, Energy Environ. Sci., 2020, 13, 2233–2242 RSC.
- S. C. Zhang, G. F. Shen, Y. Y. Deng, Y. Lei, J. W. Xue, Z. Q. Chen and G. C. Yin, ACS Sustainable Chem. Eng., 2018, 6, 13192–13198 CrossRef CAS.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS.
- G. Lv, S. Chen, H. Zhu, M. Li and Y. Yang, J. Cleaner Prod., 2018, 196, 32–41 CrossRef CAS.
- W. Jia, Z. Si, Y. Feng, X. Zhang, X. Zhao, Y. Sun, X. Tang, X. Zeng and L. Lin, ACS Sustainable Chem. Eng., 2020, 8, 7901–7908 CrossRef CAS.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS.
- X. Tang, J. Wei, N. Ding, Y. Sun, X. Zeng, L. Hu, S. Liu, T. Lei and L. Lin, Renewable Sustainable Energy Rev., 2017, 77, 287–296 CrossRef CAS.
- L. Hu, J. Xu, S. Zhou, A. He, X. Tang, L. Lin, J. Xu and Y. Zhao, ACS Catal., 2018, 8, 2959–2980 CrossRef CAS.
- M. J. Gilkey and B. J. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- Z. Wei, J. Lou, Z. Li and Y. Liu, Catal. Sci. Technol., 2016, 6, 6217–6225 RSC.
- J. Wei, X. Cao, T. Wang, H. Liu, X. Tang, X. Zeng, Y. Sun, T. Lei, S. Liu and L. Lin, Catal. Sci. Technol., 2018, 8, 4474–4484 RSC.
- H. Li, Z. Fang, J. He and S. Yang, ChemSusChem, 2017, 10, 681–686 CrossRef CAS.
- L. Hu, X. Dai, N. Li, X. Tang and Y. Jiang, Sustainable Energy Fuels, 2019, 3, 1033–1041 RSC.
- S. H. Zhou, F. T. Dai, Y. A. Chen, C. Dang, C. Z. Zhang, D. T. Liu and H. S. Qi, Green Chem., 2019, 21, 1421–1431 RSC.
- R. J. Chimentão, H. Oliva, J. Belmar, K. Morales, P. Mäki-Arvela, J. Wärnå, D. Y. Murzin, J. L. G. Fierro, J. Llorca and D. Ruiz, Appl. Catal., B, 2019, 241, 270–283 CrossRef.
- J. Yu, Y. Yang, L. Chen, Z. Li, W. Liu, E. Xu, Y. Zhang, S. Hong, X. Zhang and M. Wei, Appl. Catal., B, 2020, 277, 119273 CrossRef CAS.
- L. Hu, T. Li, J. Xu, A. He, X. Tang, X. Chu and J. Xu, Chem. Eng. J., 2018, 352, 110–119 CrossRef CAS.
- Y. Sun, C. Xiong, Q. Liu, J. Zhang, X. Tang, X. Zeng, S. Liu and L. Lin, Ind. Eng. Chem. Res., 2019, 58, 5414–5422 CrossRef CAS.
- Y. Ma, G. Xu, H. Wang, Y. Wang, Y. Zhang and Y. Fu, ACS Catal., 2018, 8, 1268–1277 CrossRef CAS.
- J. Zhang, W. Xie, Q. Liang, L. Peng and L. He, ChemistrySelect, 2019, 4, 2846–2850 CrossRef CAS.
- S. Zhou, G. Chen, X. Feng, M. Wang, T. Song, D. Liu, F. Lu and H. Qi, Green Chem., 2018, 20, 3593–3603 RSC.
- Y. Feng, S. Long, G. Yan, B. Chen, J. Sperry, W. Xu, Y. Sun, X. Tang, X. Zeng and L. Lin, J. Catal., 2020, 389, 157–165 CrossRef CAS.
- P. P. Upare, Y. K. Hwang and D. W. Hwang, Green Chem., 2018, 20, 879–885 RSC.
- Z. Gao, C. Li, G. Fan, L. Yang and F. Li, Appl. Catal., B, 2018, 226, 523–533 CrossRef CAS.
- D. Hu, H. Hu, H. Zhou, G. Li, C. Chen, J. Zhang, Y. Yang, Y. Hu, Y. Zhang and L. Wang, Catal. Sci. Technol., 2018, 8, 6091–6099 RSC.
- J. X. Long, W. F. Zhao, Y. F. Xu, H. Li and S. Yang, Catalysts, 2018, 8, 12 CrossRef.
- Z. Zhang, C. Wang, X. Gou, H. Chen, K. Chen, X. Lu, P. Ouyang and J. Fu, Appl. Catal., A, 2019, 570, 245–250 CrossRef CAS.
- S. Srivastava, G. C. Jadeja and J. K. Parikh, Int. J. Chem. React. Eng., 2018, 16, 20170197 CAS.
- Y. Zu, P. Yang, J. Wang, X. Liu, J. Ren, G. Lu and Y. Wang, Appl. Catal., B, 2014, 146, 244–248 CrossRef CAS.
- F. Yang, J. B. Mao, S. M. Li, J. M. Yin, J. X. Zhou and W. Liu, Catal. Sci. Technol., 2019, 9, 1329–1333 RSC.
- D. Li, Q. Y. Liu, C. H. Zhu, H. Y. Wang, C. H. Cui, C. G. Wang and L. L. Ma, J. Energy Chem., 2019, 30, 34–41 CrossRef.
- Z. An, W. L. Wang, S. H. Dong and J. He, Catal. Today, 2019, 319, 128–138 CrossRef CAS.
- Z. Zhang, S. Yao, C. Wang, M. Liu, F. Zhang, X. Hu, H. Chen, X. Gou, K. Chen, Y. Zhu, X. Lu, P. Ouyang and J. Fu, J. Catal., 2019, 373, 314–321 CrossRef CAS.
- Y. D. Yang, H. Y. Liu, S. P. Li, C. J. Chen, T. B. Wu, Q. Q. Mei, Y. Y. Wang, B. F. Chen, H. Z. Liu and B. X. Han, ACS Sustainable Chem. Eng., 2019, 7, 5711–5716 CrossRef CAS.
- Y.-R. Zhang, B.-X. Wang, L. Qin, Q. Li and Y.-M. Fan, Green Chem., 2019, 21, 1108–1113 RSC.
- A. D. Talpade, M. S. Tiwari and G. D. Yadav, Mol. Catal., 2019, 465, 1–15 CrossRef CAS.
- R. Xu, L. Kang, J. Knossalla, J. Mielby, Q. Wang, B. Wang, J. Feng, G. He, Y. Qin, J. Xie, A.-C. Swertz, Q. He, S. Kegnæs, D. J. L. Brett, F. Schüth and F. R. Wang, ACS Nano, 2019, 13, 2463–2472 CAS.
- E. Nurenberg, P. Schulze, F. Kohler, M. Zubel, S. Pischinger and F. Schuth, ACS Sustainable Chem. Eng., 2019, 7, 249–257 CrossRef.
- X. Wang, Y. Liu and X. Liang, Green Chem., 2018, 20, 2894–2902 RSC.
- Z. Gao, G. L. Fan, M. R. Liu, L. Yang and F. Li, Appl. Catal., B, 2018, 237, 649–659 CrossRef CAS.
- W. Han, M. Tang, J. Li, X. Li, J. Wang, L. Zhou, Y. Yang, Y. Wang and H. Ge, Appl. Catal., B, 2020, 268, 118748 CrossRef.
- S. Fulignati, C. Antonetti, D. Licursi, M. Pieraccioni, E. Wilbers, H. J. Heeres and A. M. Raspolli Galletti, Appl. Catal., A, 2019, 578, 122–133 CrossRef CAS.
- L. Kuai, Z. Chen, S. Liu, E. Kan, N. Yu, Y. Ren, C. Fang, X. Li, Y. Li and B. Geng, Nat. Commun., 2020, 11, 48 CrossRef.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef.
- T. Gan, Y. Liu, Q. He, H. Zhang, X. He and H. Ji, ACS Sustainable Chem. Eng., 2020, 8, 8692–8699 CrossRef CAS.
- J. Fu, J. Lym, W. Zheng, K. Alexopoulos, A. V. Mironenko, N. Li, J. A. Boscoboinik, D. Su, R. T. Weber and D. G. Vlachos, Nat. Catal., 2020, 3, 446–453 CrossRef CAS.
- Y. Liu, M. A. Mellmer, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2015, 8, 3983–3986 CrossRef CAS.
- Y. Deng, Y. Ge, M. Xu, Q. Yu, D. Xiao, S. Yao and D. Ma, Acc. Chem. Res., 2019, 52, 3372–3383 CrossRef CAS.
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810 CrossRef CAS.
- J. Zhang, K. Dong and W. Luo, Chem. Eng. Sci., 2019, 201, 467–474 CrossRef CAS.
- W. Li, G. Fan, L. Yang and F. Li, Green Chem., 2017, 19, 4353–4363 RSC.
- T. Thananatthanachon and T. B. Rauchfuss, Angew. Chem., 2010, 122, 6766–6768 CrossRef.
- J. F. Li, Z. Song, Y. F. Hou, Z. Y. Li, C. L. Xu, C. L. Liu and W. S. Dong, ACS Appl. Mater. Interfaces, 2019, 11, 12481–12491 CrossRef CAS.
- R. Insyani, D. Verma, S. M. Kim and J. Kim, Green Chem., 2017, 19, 2482–2490 RSC.
- B. Seemala, X. Meng, A. Parikh, N. Nagane, R. Kumar, C. E. Wyman, A. Ragauskas, P. Christopher and C. M. Cai, ACS Sustainable Chem. Eng., 2018, 6, 10587–10594 CrossRef CAS.
- L. Tao, T.-H. Yan, W. Li, Y. Zhao, Q. Zhang, Y.-M. Liu, M. M. Wright, Z.-H. Li, H.-Y. He and Y. Cao, Chem, 2018, 4, 2212–2227 CAS.
- J. Li, E. Muller, M. Pera-Titus, F. Jérôme and K. De Oliveira Vigier, Green Chem., 2019, 21, 2601–2609 RSC.
- M. E. Jung and G. Y. J. Im, J. Org. Chem., 2009, 74, 8739–8753 CrossRef CAS.
- G. Sun, J. An, H. Hu, C. Li, S. Zuo and H. Xia, Catal. Sci. Technol., 2019, 9, 1238–1244 RSC.
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morré, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 RSC.
- A. Deneyer, E. Peeters, T. Renders, S. Van den Bosch, N. Van Oeckel, T. Ennaert, T. Szarvas, T. I. Korányi, M. Dusselier and B. F. Sels, Nat. Energy, 2018, 3, 969–977 CrossRef CAS.
- S. Liu, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2014, 2, 1819–1827 CrossRef CAS.
- Q. Xia, Z. Chen, Y. Shao, X. Gong, H. Wang, X. Liu, S. F. Parker, X. Han, S. Yang and Y. Wang, Nat. Commun., 2016, 7, 11162 CrossRef.
- Y. Yang, Y. Wang, S. Li, X. Shen, B. Chen, H. Liu and B. Han, Green Chem., 2020, 22, 4937–4942 RSC.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef CAS.
- S. P. Burt, K. J. Barnett, D. J. McClelland, P. Wolf, J. A. Dumesic, G. W. Huber and I. Hermans, Green Chem., 2017, 19, 1390–1398 RSC.
- K. Huang, Z. J. Brentzel, K. J. Barnett, J. A. Dumesic, G. W. Huber and C. T. Maravelias, ACS Sustainable Chem. Eng., 2017, 5, 4699–4706 CrossRef CAS.
- Z. J. Brentzel, K. J. Barnett, K. Huang, C. T. Maravelias, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 1351–1355 CrossRef CAS.
- J. He, K. Huang, K. J. Barnett, S. H. Krishna, D. M. Alonso, Z. J. Brentzel, S. P. Burt, T. Walker, W. F. Banholzer, C. T. Maravelias, I. Hermans, J. A. Dumesic and G. W. Huber, Faraday Discuss., 2017, 202, 247–267 RSC.
- B. Wozniak, S. Tin and J. G. de Vries, Chem. Sci., 2019, 10, 6024–6034 RSC.
- Z. Xu, P. Yan and Z. Zongconrad, Chinese J. Org. Chem., 2017, 37, 40–46 CrossRef CAS.
- Z. Xu, P. Yan, W. Xu, X. Liu, Z. Xia, B. Chung, S. Jia and Z. C. Zhang, ACS Catal., 2015, 5, 788–792 CrossRef CAS.
- Z. Xu, P. Yan, H. Li, K. Liu, X. Liu, S. Jia and Z. C. Zhang, ACS Catal., 2016, 6, 3784–3788 CrossRef CAS.
- S. Fujita, K. Nakajima, J. Yamasaki, T. Mizugaki, K. Jitsukawa and T. Mitsudome, ACS Catal., 2020, 10, 4261–4267 CrossRef CAS.
- B. Wozniak, A. Spannenberg, Y. Li, S. Hinze and J. G. de Vries, ChemSusChem, 2018, 11, 356–359 CrossRef CAS.
- Y. Duan, M. Zheng, D. Li, D. Deng, L.-F. Ma and Y. Yang, Green Chem., 2017, 19, 5103–5113 RSC.
- R. Ramos, A. Grigoropoulos, N. Perret, M. Zanella, A. P. Katsoulidis, T. D. Manning, J. B. Claridge and M. J. Rosseinsky, Green Chem., 2017, 19, 1701–1713 RSC.
- S. Zhang, H. Ma, Y. Sun, Y. Luo, X. Liu, M. Zhang, J. Gao and J. Xu, Green Chem., 2019, 21, 1702–1709 RSC.
- Q. Deng, R. Gao, X. Li, J. Wang, Z. L. Zeng, J. J. Zou and S. G. Deng, ACS Catal., 2020, 10, 7355–7366 CrossRef CAS.
- H. Duan, J.-C. Liu, M. Xu, Y. Zhao, X.-L. Ma, J. Dong, X. Zheng, J. Zheng, C. S. Allen, M. Danaie, Y.-K. Peng, T. Issariyakul, D. Chen, A. I. Kirkland, J.-C. Buffet, J. Li, S. C. E. Tsang and D. O'Hare, Nat. Catal., 2019, 2, 1078–1087 CrossRef CAS.
- S. Alipour, H. Omidvarborna and D.-S. Kim, Renewable Sustainable Energy Rev., 2017, 71, 908–926 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- Z. Wang and Q. Chen, Nanomaterials, 2018, 8, 492 CrossRef.
- C. P. A. and S. Darbha, Catal. Commun., 2020, 140, 105998 CrossRef.
- M. Zuo, K. Le, Y. Feng, C. Xiong, Z. Li, X. Zeng, X. Tang, Y. Sun and L. Lin, Ind. Crops Prod., 2018, 112, 18–23 CrossRef CAS.
- Z. Wang and Q. Chen, Green Chem., 2016, 18, 5884–5889 RSC.
- H. Wang, T. Deng, Y. Wang, X. Cui, Y. Qi, X. Mu, X. Hou and Y. Zhu, Green Chem., 2013, 15, 2379–2383 RSC.
- Y.-Y. Bai, S. Su, S. Wang, B. Wang, R.-C. Sun, G. Song and L.-P. Xiao, Energy Technol., 2018, 6, 1951–1958 CrossRef CAS.
- P. K. Kumari, B. S. Rao, D. Padmakar, N. Pasha and N. Lingaiah, Mol. Catal., 2018, 448, 108–115 CrossRef CAS.
- P. K. Kumari, B. S. Rao, D. D. Lakshmi, N. R. S. Paramesh, C. Sumana and N. Lingaiah, Catal. Today, 2019, 325, 53–60 CrossRef CAS.
- H. Wang, T. Deng, Y. Wang, Y. Qi, X. Hou and Y. Zhu, Bioresour. Technol., 2013, 136, 394–400 CrossRef CAS.
- H. X. Guo, X. H. Qi, Y. Hiraga, T. M. Aida and R. L. Smith, Chem. Eng. J., 2017, 314, 508–514 CrossRef CAS.
- X. Yu, X. Gao, R. Tao and L. Peng, Catalysts, 2017, 7, 182 CrossRef.
- H. Xin, T. Zhang, W. Li, M. Su, S. Li, Q. Shao and L. Ma, RSC Adv., 2017, 7, 41546–41551 RSC.
- H. Guo, A. Duereh, Y. Hiraga, X. Qi and R. L. Smith, Energy Fuels, 2018, 32, 8411–8419 CrossRef CAS.
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2016, 18, 726–734 RSC.
- S. Shinde and C. Rode, ChemSusChem, 2017, 10, 4090–4101 CrossRef CAS.
- J. Wei, T. Wang, H. Liu, M. Li, X. Tang, Y. Sun, X. Zeng, L. Hu, T. Lei and L. Lin, Energy Technol., 2019, 7, 1801071 CrossRef.
- X.-L. Li, K. Zhang, S.-Y. Chen, C. Li, F. Li, H.-J. Xu and Y. Fu, Green Chem., 2018, 20, 1095–1105 RSC.
- B. Chen, G. Xu, Z. Zheng, D. Wang, C. Zou and C. Chang, Ind. Crops Prod., 2019, 129, 503–511 CrossRef CAS.
- J. Zhang, K. Dong, W. Luo and H. Guan, Fuel, 2018, 234, 664–673 CrossRef CAS.
- F. Yang, S. Zhang, Z. C. Zhang, J. Mao, S. Li, J. Yin and J. Zhou, Catal. Sci. Technol., 2015, 5, 4602–4612 RSC.
- Y. W. Dou, M. Y. Zhang, S. Zhou, C. Oldani, W. H. Fang and Q. Cao, Eur. J. Inorg. Chem., 2018, 3706–3716 CrossRef CAS.
- S. B. Onkarappa and S. Dutta, ChemistrySelect, 2019, 4, 5540–5543 CrossRef CAS.
- R. Zhong, F. Yu, W. Schutyser, Y. Liao, F. de Clippel, L. Peng and B. F. Sels, Appl. Catal., B, 2017, 206, 74–88 CrossRef CAS.
- B. Xiang, Y. Wang, T. Qi, H.-Q. Yang and C.-W. Hu, J. Catal., 2017, 352, 586–598 CrossRef CAS.
- H. Guo, A. Duereh, Y. Su, E. J. M. Hensen, X. Qi and R. L. Smith, Appl. Catal., B, 2020, 264, 118509 CrossRef CAS.
- L. Zhang, Y. Zhu, L. Tian, Y. He, H. Wang and F. Deng, Fuel Process. Technol., 2019, 193, 39–47 CrossRef CAS.
- S. Kanai, I. Nagahara, Y. Kita, K. Kamata and M. Hara, Chem. Sci., 2017, 8, 3146–3153 RSC.
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS.
- M. Kim, Y. Q. Su, A. Fukuoka, E. J. M. Hensen and K. Nakajima, Angew. Chem., Int. Ed., 2018, 57, 8235–8239 CrossRef CAS.
- M. Kim, Y. Su, T. Aoshima, A. Fukuoka, E. J. M. Hensen and K. Nakajima, ACS Catal., 2019, 9, 4277–4285 CrossRef CAS.
- J. J. Wiesfeld, M. Kim, K. Nakajima and E. J. M. Hensen, Green Chem., 2020, 22, 1229–1238 RSC.
- H. C. Chang, G. W. Huber and J. A. Dumesic, ChemSusChem, 8 DOI:10.1002/cssc.202001471.
- A. D. Sutton, F. D. Waldie, R. Wu, M. Schlaf, L. A. ‘Pete’ Silks III and J. C. Gordon, Nat. Chem., 2013, 5, 428 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
- H. Chang, A. H. Motagamwala, G. W. Huber and J. A. Dumesic, Green Chem., 2019, 21, 5532–5540 RSC.
- H. C. Chang, I. Bajaj, G. W. Huber, C. T. Maravelias and J. A. Dumesic, Green Chem., 2020, 22, 5285–5295 RSC.
- J. Cueto, L. Faba, E. Díaz and S. Ordóñez, Appl. Catal., B, 2017, 201, 221–231 CrossRef CAS.
- D. Suttipat, W. Wannapakdee, T. Yutthalekha, S. Ittisanronnachai, T. Ungpittagul, K. Phomphrai, S. Bureekaew and C. Wattanakit, ACS Appl. Mater. Interfaces, 2018, 10, 16358–16366 CrossRef CAS.
- T. Yutthalekha, D. Suttipat, S. Salakhum, A. Thivasasith, S. Nokbin, J. Limtrakul and C. Wattanakit, Chem. Commun., 2017, 53, 12185–12188 RSC.
- J.-F. Zhang, Z.-M. Wang, Y.-J. Lyu, H. Xie, T. Qi, Z.-B. Si, L.-J. Liu, H.-Q. Yang and C.-W. Hu, J. Phys. Chem. C, 2019, 123, 4903–4913 CrossRef CAS.
- R. S. Malkar, H. Daly, C. Hardacre and G. D. Yadav, ACS Sustainable Chem. Eng., 2019, 7, 16215–16224 CrossRef CAS.
- J. Xie, L. Zhang, X. Zhang, P. Han, J. Xie, L. Pan, D.-R. Zou, S.-H. Liu and J.-J. Zou, Sustainable Energy Fuels, 2018, 2, 1863–1869 RSC.
- A. S. Amarasekara, T. B. Singh, E. Larkin, M. A. Hasan and H.-J. Fan, Ind. Crops Prod., 2015, 65, 546–549 CrossRef CAS.
- X. Cui, X. Zhao and D. Liu, Green Chem., 2018, 20, 2018–2026 RSC.
- L. Zhao, N. Elechi, R. Qian, T. B. Singh, A. S. Amarasekara and H.-J. Fan, J. Phys. Chem. A, 2017, 121, 1985–1992 CrossRef CAS.
- G. Liang, A. Wang, X. Zhao, N. Lei and T. Zhang, Green Chem., 2016, 18, 3430–3438 RSC.
- Y. Jing, Y. Xin, Y. Guo, X. Liu and Y. Wang, Chin. J. Catal., 2019, 40, 1168–1177 CrossRef CAS.
- M. Decostanzi, R. Auvergne, B. Boutevin and S. Caillol, Green Chem., 2019, 21, 724–747 RSC.
- H. Zang, K. Wang, M. Zhang, R. Xie, L. Wang and E. Y. X. Chen, Catal. Sci. Technol., 2018, 8, 1777–1798 RSC.
- A. S. Amarasekara, L. H. Nguyen, N. C. Okorie and S. M. Jamal, Green Chem., 2017, 19, 1570–1575 RSC.
- R. M. Cywar, L. Wang and E. Y. X. Chen, ACS Sustainable Chem. Eng., 2019, 7, 1980–1988 CrossRef CAS.
- J. F. Wilson and E. Y. X. Chen, ACS Sustainable Chem. Eng., 2019, 7, 7035–7046 CrossRef CAS.
- A. S. Amarasekara and C. D. Gutierrez Reyes, Renewable Energy, 2019, 136, 352–357 CrossRef CAS.
- A. Garcia-Ortiz, J. D. Vidal, M. J. Climent, P. Concepcion, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2019, 7, 6243–6250 CrossRef CAS.
- H. K. Yuan, J. P. Li, F. Z. Su, Z. Yan, B. T. Kusema, S. Streiff, Y. J. Huang, M. Pera-Titus and F. Shi, ACS Omega, 2019, 4, 2510–2516 CrossRef CAS.
- M. M. Zhu, L. Tao, Q. Zhang, J. Dong, Y. M. Liu, H. Y. He and Y. Cao, Green Chem., 2017, 19, 3880–3887 RSC.
- R. F. A. Gomes, J. A. S. Coelho and C. A. M. Afonso, ChemSusChem, 2019, 12, 420–425 CrossRef CAS.
- A. Petri, G. Masia and O. Piccolo, Catal. Commun., 2018, 114, 15–18 CrossRef CAS.
- Q. Meng, D. Cao, G. Zhao, C. Qiu, X. Liu, X. Wen, Y. Zhu and Y. Li, Appl. Catal., B, 2017, 212, 15–22 CrossRef CAS.
- S. Zhang, X. Yang, K. Zheng, R. Xiao, Q. Hou, B. Liu, M. Ju and L. Liu, Appl. Catal., A, 2019, 581, 103–110 CrossRef CAS.
- H. Xie, T. Qi, Y. J. Lyu, J. F. Zhang, Z. B. Si, L. J. Liu, L. F. Zhu, H. Q. Yang and C. W. Hu, Phys. Chem. Chem. Phys., 2019, 21, 3795–3804 RSC.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2018, 20, 2345–2355 RSC.
- K. S. Kozlov, L. V. Romashov and V. P. Ananikov, Green Chem., 2019, 21, 3464–3468 RSC.
- J. Li, Y. Jing, C. Liu and D. Zhang, New J. Chem., 2017, 41, 8714–8720 RSC.
- H. C. Genuino, H. H. Van De Bovenkamp, E. Wilbers, J. G. M. Winkelman, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, B. M. Weckhuysen, P. C. A. Bruijnincx and H. J. Heeres, ACS Sustainable Chem. Eng., 2020, 8, 5903–5919 CrossRef CAS.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- D. Ding, J. Wang, J. Xi, X. Liu, G. Lu and Y. Wang, Green Chem., 2014, 16, 3846–3853 RSC.
- D. Ding, J. Xi, J. Wang, X. Liu, G. Lu and Y. Wang, Green Chem., 2015, 17, 4037–4044 RSC.
- A. Seretis, P. Diamantopoulou, I. Thanou, P. Tzevelekidis, C. Fakas, P. Lilas and G. Papadogianakis, Front. Chem., 2020, 8, 221 CrossRef CAS.
- R. G. Weng, Z. H. Yu, J. Xiong and X. B. Lu, Green Chem., 2020, 22, 3013–3027 RSC.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664 RSC.
- P. Zhang, Y.-J. Guo, J. Chen, Y.-R. Zhao, J. Chang, H. Junge, M. Beller and Y. Li, Nat. Catal., 2018, 1, 332–338 CrossRef CAS.
- M. A. Lăcătuş, L. C. Bencze, M. I. Tosa, C. Paizs and F. D. Irimie, ACS Sustainable Chem. Eng., 2018, 6, 11353–11359 CrossRef.
- M. A. Lacatus, A. J. Dudu, L. C. Bencze, G. Katona, F. D. Irimie, C. Paizs and M. I. Tosa, ACS Sustainable Chem. Eng., 2020, 8, 1611–1617 CrossRef CAS.
- K. S. Arias, J. M. Carceller, M. J. Climent, A. Corma and S. Iborra, ChemSusChem, 2020, 13, 1864–1875 CrossRef CAS.
- E. J. Garcia-Suarez, D. Paolicchi, H. Li, J. He, S. Yang, A. Riisager and S. Saravanamurugan, Appl. Catal., A, 2019, 569, 170–174 CrossRef CAS.
- M. Han, X. Liu, X. Zhang, Y. Pang, P. Xu, J. Guo, Y. Liu, S. Zhang and S. Ji, Green Chem., 2017, 19, 722–728 RSC.
- L. V. Romashov and V. P. Ananikov, Chem. – Asian J., 2017, 12, 2652–2655 CrossRef CAS.
- C. Li, L. Wang, M. Wang, B. Liu, X. Liu and D. Cui, Angew. Chem., Int. Ed., 2019, 58, 11434–11438 CrossRef CAS.
- J. Saska, Z. Li, A. L. Otsuki, J. Wei, J. C. Fettinger and M. Mascal, Angew. Chem., Int. Ed., 2019, 58, 17293–17296 CrossRef CAS.
- W. Fan, Y. Queneau and F. Popowycz, Green Chem., 2018, 20, 485–492 RSC.
- G. D. Wen, Q. Q. Gu, Y. F. Liu, R. Schlogl, C. X. Wang, Z. J. Tian and D. S. Su, Angew. Chem., Int. Ed., 2018, 57, 16898–16902 CrossRef CAS.
- Z. Ma, H. Hu, Z. Sun, W. Fang, J. Zhang, L. Yang, Y. Zhang and L. Wang, ChemSusChem, 2017, 10, 1669–1674 CrossRef CAS.
- S. Kunnikuruvan and N. N. Nair, ACS Catal., 2019, 9, 7250–7263 CrossRef CAS.
- K. R. Enslow and A. T. Bell, Catal. Sci. Technol., 2015, 5, 2839–2847 RSC.
- H. Wang, Y. Duan, Q. Zhang and B. Yang, ChemSusChem, 2018, 11, 2562–2568 CrossRef CAS.
- C. Xu, M. Nasrollahzadeh, M. Selva, Z. Issaabadi and R. Luque, Chem. Soc. Rev., 2019, 48, 4791–4822 RSC.
- H. Li, Z. Fang, R. L. Smith and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef.
- K. M. H. Mohammed, A. Chutia, J. Callison, P. P. Wells, E. K. Gibson, A. M. Beale, C. R. A. Catlow and R. Raja, J. Mater. Chem. A, 2016, 4, 5706–5712 RSC.
- Y. G. Kolyagin, A. V. Yakimov, S. Tolborg, P. N. R. Vennestrøm and I. I. Ivanova, J. Phys. Chem. Lett., 2016, 7, 1249–1253 CrossRef CAS.
- M. Zhang, M. Wang, B. Xu and D. Ma, Joule, 2019, 3, 2876–2883 CrossRef.
- C. Mondelli, G. Gozaydin, N. Yan and J. Perez-Ramirez, Chem. Soc. Rev., 2020, 8, 11204–11214 Search PubMed.
- J. Kang, S. He, W. Zhou, Z. Shen, Y. Li, M. Chen, Q. Zhang and Y. Wang, Nat. Commun., 2020, 11, 827 CrossRef CAS.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
- H. Chen, F. Dong and S. D. Minteer, Nat. Catal., 2020, 3, 225–244 CrossRef.
- Z. Cheng, B. Saha and D. G. Vlachos, ChemSusChem, 2018, 11, 3609–3617 CrossRef CAS.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |