
Facile and sustainable etherification of ethyl cellulose towards excellent UV blocking and fluorescence properties
Bowen
Li
,
Chaoqun
Xu
,
Liang
Liu
,
Juan
Yu
 * and
Yimin
Fan
*
* and
Yimin
Fan
*
Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Jiangsu Key Lab of Biomass-Based Green Fuel and Chemicals, Key Laboratory of Forestry Genetics and Biotechnology of Ministry of Education, College of Chemical Engineering, Nanjing Forestry University, Longpan Road 159, Nanjing 210037, China. E-mail: yujuannjfu@njfu.edu.cn; fanyimin@njfu.edu.cn
First published on 23rd November 2020
Abstract
Chemical modification of cellulose to prepare functional cellulose derivatives could enable the utilization of cellulose in various promising applications. However, the conventional methods for cellulose functionalization suffer from severe drawbacks, especially the environmental aspects, due to the use of toxic reagents and the generation of large amounts of waste. Herein, a more sustainable way, the novel hydroxyl–yne click reaction, is proposed to synthesize cellulose derivatives. The hydroxyl groups of ethyl cellulose (EC) were reacted with the alkyne groups of 1-phenyl-2-propargyl-1-ketone (PPK) at room temperature for preparing new kinds of ethyl cellulose phenyl propylene ketone ether derivatives (ECPPKs). More than 80% substitution was rapidly completed within the first 5 min and 82% of the residual hydroxyl groups (–OHC3) in EC could be substituted, demonstrating the high efficiency of the hydroxyl–yne click reaction for the modification of cellulosic materials. FTIR and NMR results proved the successful grafting of PPK via a vinyl ether linkage (–C–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–). The introduction of PPK moieties widened the temperature window for the melt processing of cellulose, which is beneficial for the thermo processing of cellulosic materials. Furthermore, owing to the existence of the newly formed vinyl ether linkage, ECPPKs showed almost 100% shielding ratio for UV light and visible light (420 nm) excited fluorescence properties. This facile and efficient method provides a more sustainable strategy for the functionalization of cellulosic materials, expanding its application in UV-blocking and fluorescent material fields.
C–). The introduction of PPK moieties widened the temperature window for the melt processing of cellulose, which is beneficial for the thermo processing of cellulosic materials. Furthermore, owing to the existence of the newly formed vinyl ether linkage, ECPPKs showed almost 100% shielding ratio for UV light and visible light (420 nm) excited fluorescence properties. This facile and efficient method provides a more sustainable strategy for the functionalization of cellulosic materials, expanding its application in UV-blocking and fluorescent material fields.
1. Introduction
Cellulose, the most significant structural component in plants, is recognized as an almost inexhaustible renewable polymer in the world.1,2 Due to its fascinating structure and properties such as sustainability, degradability, biocompatibility and good mechanical properties, cellulose has been widely used in various industries including paper, textile and food.3,4 Cellulose is composed of linear β-(1–4) linked D-glucose chains with a large number of hydroxy groups (three vacant hydroxy groups/anhydro glucose unit (AGU)), which can undergo chemical reactions to afford high value-added products.5,6 Indeed, the chemical utilization of cellulose dates back over 170 years with the synthesis of cellulose nitrate.5 Since then, various synthesis methods have been developed for the chemical modification of cellulose in order to extend its utilization to highly sophisticated applications.In general, these methods can be classified into two major approaches: one is polymer grafting which aims at grafting polymer chains onto the backbone of cellulose, conferring a variety of novel properties on cellulose. Free radical polymerizations, including ionic and ring-opening polymerization, nitroxide mediated polymerization (NMP), atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer polymerization (RAFT), have been widely reported in the literature to prepare cellulose graft copolymers with valuable properties.3,7,8 However, the cellulose contents of these as-prepared cellulose graft copolymers are very low which limits the application efficiency of the cellulose resource. Besides, these methods suffer from the disadvantages of the need for large amounts of chemicals, complex post-treatment processes and rigorous reaction conditions.9 The second approach is to introduce functional groups into the structure of cellulose via etherification, esterification, oxidation, silylation, etc.10 The obtained cellulose derivatives such as ethers,11,12 esters,13,14 silylates15,16 and amines17 have been reported to be used in various applications, including coatings, membranes, composites, optical films, and medical applications.18 But toxic and corrosive chemicals are generally involved in these synthetic processes. For example, acid anhydrides, acids or acyl chlorides, which are dangerous and need to be handled with extra caution, are commonly used to prepare cellulose esters. Furthermore, the shortcomings of these methods namely the requirements of high temperature and long reaction time and the production of waste products seriously limit the manufacture of cellulose derivatives and their applications. Therefore, a greener or more sustainable approach to synthesize cellulose derivatives is needed.13,19
Owing to its striking advantages of high efficiency, good selectivity, mild reaction conditions, strong functional group tolerance, simple post-processing steps, and no byproducts or benign byproducts, click chemistry has been developed rapidly since it was first published by Sharpless et al.20–23 CuI-and RuII-catalyzed azide–alkyne click polymerizations (AACPs),24–26 metal-free azide–alkyne click polymerizations (MFCPs),27,28 and thiol–yne click polymerization29,30 have been widely used as efficient tools for the chemical modification of cellulose. However, these click reactions suffer from the drawbacks of residual catalyst, unsafe azide monomers and strong pungent aroma of thiol compounds. Recently, Tang's group established a novel phenol–yne click polymerization, in which aromatic diols and diynes could be rapidly polymerized by simply mixing the diynes and diphenols in THF in the presence of 4-dimethylamino-pyridine (DMAP) under ambient conditions.31 The remarkable advantages of this click polymerization are the avoidance of expensive transition-metal catalysts, mild reaction conditions, high efficiency, and absence of byproducts. Furthermore, the reaction between n-butyl alcohol, hydroxypropyl cellulose (HPC) and N-(4-ethynylcarbonylphenyl) diphenylamine (alkyne-TPA) was successfully examined under similar conditions, confirming that the click reaction between the hydroxyl group of the natural polymer and active acetylene could happen.32
Herein, driven by the critical need to develop an alternative more sustainable method for the modification of cellulose, we applied the novel hydroxyl–yne click reaction to synthesize a new kind of cellulose derivative (cellulose ketone ether) as shown in Scheme 1. In the present work, the click reaction between aromatic acetylene ketones (1-phenyl-2-propargyl-1-ketone, PPK) and ethyl cellulose (EC) with a small amount of 4-dimethylaminopyridine (DMAP) was firstly investigated as a proof-of-concept. FTIR and NMR were used to demonstrate the successful click reaction of EC and acetylene ketones and to verify the high reaction efficiency. Furthermore, ultraviolet (UV) blocking ability, fluorescence performance and thermal stability of the prepared ethyl cellulose phenyl propylene ketone ethers (ECPPKs) were also investigated.
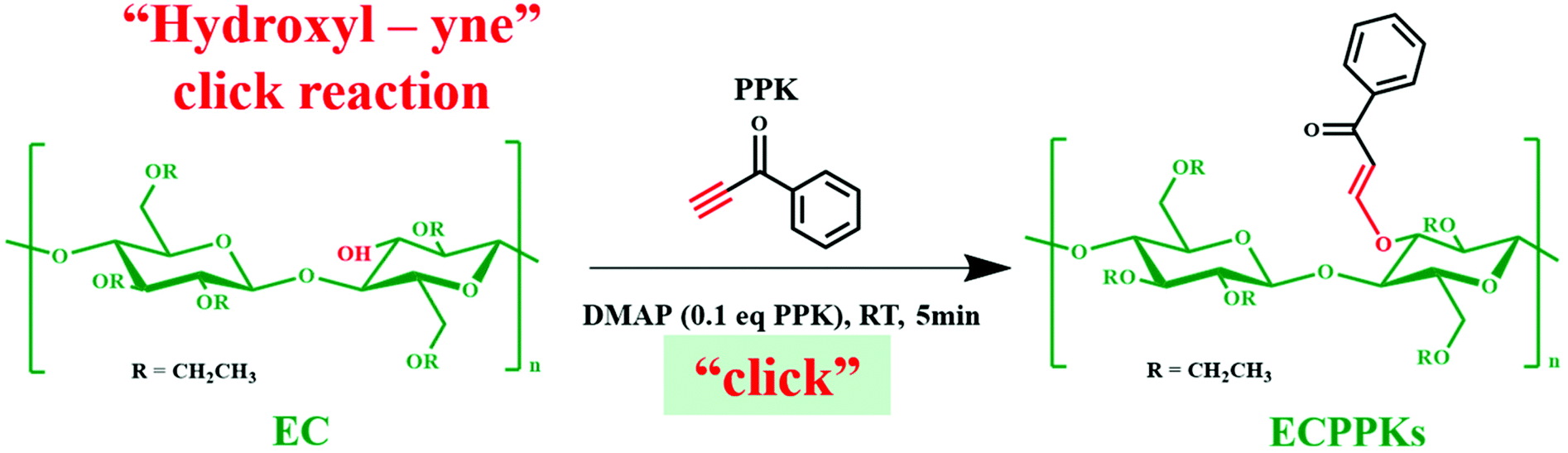 | ||
| Scheme 1 Modification of ethyl cellulose by the hydroxyl–yne click reaction. | ||
2. Experimental section
2.1. Materials
Ethyl cellulose (EC, 9–11 mPa s) purchased from Aladdin and was vacuum dried under 40 °C for 2 h before use. The degree of substitution of ethyl (DSethyl) in EC was determined by 1H NMR spectroscopy using p-nitrobenzaldehyde as the internal standard. According to eqn (1), DSethyl of EC is 2.5. 1-Phenyl-2-propargyl-1-alcohol and 4-dimethylaminopyridine (DMAP) were purchased from Aladdin. Acetone, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), dichloromethane (DCM) and petroleum ether (60–90 °C) were AR grade reagents and other chemicals of analytical grade were obtained from Sinopharm Chemical Reagent Limited Corporation, China, and used without further purification. 1-Phenyl-2-propargyl-1-ketone (PPK) was prepared according to ref. 33, and a yellow solid was obtained with a yield of 86%. 1H NMR for PPK (600 MHz, CDCl3, 25 °C): δ = 3.46 (s, 1H,![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 7.47 (m, 2H, Ar–H), 7.66 (m, 1H, Ar–H), 8.19 (m, 2H, Ar–H). FT-IR for PPK: v(cm−1) = 3300 (C–H stretching), 2092 (CC stretching), 1640 (CO stretching).
CH), 7.47 (m, 2H, Ar–H), 7.66 (m, 1H, Ar–H), 8.19 (m, 2H, Ar–H). FT-IR for PPK: v(cm−1) = 3300 (C–H stretching), 2092 (CC stretching), 1640 (CO stretching).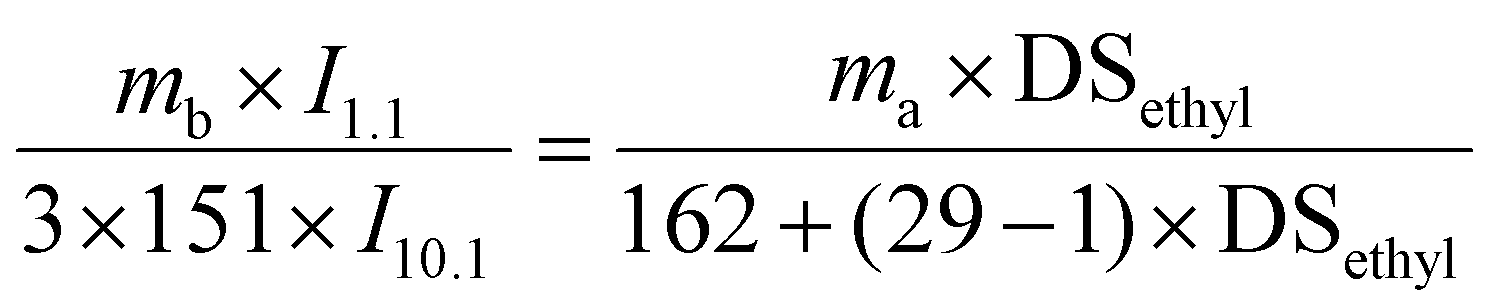 | (1) |
2.2. Synthesis of ethyl cellulose phenyl propylene ketone ethers (ECPPKs)
Ethyl cellulose phenyl propylene ketone ethers (ECPPKs) were prepared according Scheme 1. In a typical procedure, 0.93 g ethyl cellulose (EC, 2 mmol –OH) was dissolved in THF, then 0.26 g 1-phenyl-2-propargyl-1-ketone (PPK, 2 mmol) and 0.024 g 4-dimethylaminopyridine (DMAP, 0.2 mmol) were injected into the EC solution under vigorous stirring. The mixture was allowed to react at room temperature for 2 h. The resulting products were precipitated with an excess volume of petroleum ether (60–90 °C) and then filtered. After being dried under vacuum at 40 °C, 0.96 g of yellow solid ECPPKs were obtained. The reaction temperature, time and the OH/PPK molar ratio were controlled in the range of 25–60 °C, 5 min–24 h and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:5, respectively, with the aim of optimizing the reaction efficiency.
:1–1:5, respectively, with the aim of optimizing the reaction efficiency.
2.3. Characterization
FT-IR spectra were recorded using an attenuated total reflection (ATR) mode with a Thermo Fisher Nicolet iS 5. All spectra were acquired from 4000 to 500 cm−1 using 16 scans.NMR spectra were acquired on a Bruker Biospin 600 MHz. The sample was dissolved in DMSO-d6 at room temperature. The DS values of PPK (DSPPK) in ECPPKs were determined by 1H NMR spectroscopy according to eqn (2):34,35
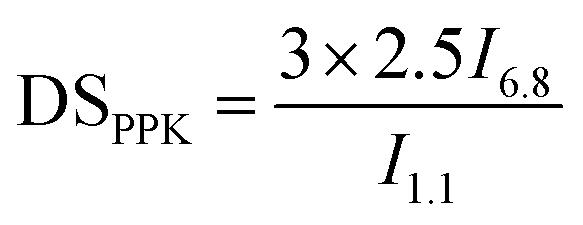 | (2) |
UV-Vis (ultraviolet and visible spectrophotometry): the optical performances of EC and ECPPK films were studied using a Ultrospec 2100 pro UV-Vis spectrophotometer in the wavelength range 200–800 nm. For the UV absorption peak test, ECPPKs were dissolved in N,N-dimethylformamide (DMF) and scanned in the range of 200–600 nm.
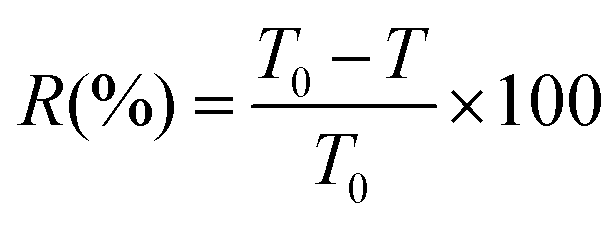 | (3) |
Fluorescence spectra were recorded on a PerkinElmer LS55 spectrofluorometer under the excitation of 420 nm. ECPPKs were dissolved in N,N-dimethylformamide (DMF) at different concentrations.
TGA analysis was conducted with a NETZSCH TGA 209 F1. EC and ECPPKs were dried in a vacuum overnight and heated from 30 to 600 °C at a heating rate of 20 °C min−1 under N2. Then thermal degradation behavior was estimated based on the weight loss against temperature.
Differential scanning calorimetry (DSC) measurements of ECPPKs were recorded using a NETZSCH DSC214. Every sample of about 5 mg was heated to 200 °C at a scanning rate of 20 °C min−1 under N2. All the glass transition temperatures (Tg) were calculated from the second scan.
3. Results and discussion
3.1. Synthesis of ECPPKs
As shown in Scheme 1, the aromatic ring was attached to the backbone of EC by a new vinyl ether bond after the hydroxyl–yne click reaction. Firstly, the FT-IR spectra were used to verify the change in the chemical structure of EC (Fig. 1). The intensive peaks appearing at 3300 cm−1, 2092 cm−1 and 1640 cm−1 in the spectrum of PPK were due to the stretching vibrations ofC–H, CC and CO in PPK. However, the characteristic peaks of 3300 cm−1 and 2092 cm−1 could not be found in the spectra of ECPPKs, indicating that the alkynyl groups in PPK have reacted. Besides, the presence of several important characteristic peaks at 1655 cm−1, 1610 cm−1, 1580 cm−1, 1537 cm−1, 780 cm−1 and 700 cm−1 and the slight decrease of the peak intensity at 3400 cm−1 in the spectra of ECPPKs also confirmed that EC was successfully modified to ECPPKs. In detail, the strong absorption peaks at around 1655 cm−1 are related to the stretching vibrations of CO in ECPPKs, and the relatively sharp peak at 1610 cm−1 corresponds to the stretching vibrations of –CC–.36 This further demonstrated the formation of a new vinyl ether bond during the hydroxyl–yne click reaction. The characteristic peaks at 1580 cm−1 and 1537 cm−1 are related to the skeleton vibration peak of the benzene ring which comes from the PPK moieties, while the absorption bands of the –C–H surface bending vibration peak at around 780 cm−1 and 700 cm−1 are attributed to the singly substituted benzene ring in ECPPKs. It is obvious that the intensity of the peaks ranging from 1700–1530 cm−1 and 650–820 cm−1 both increased with the increase in the OH/PPK molar ratio from 1:1 to 1:5. This indicates that the DSPPK of ECPPKs could be controlled by varying the OH/PPK molar ratio and will be further discussed in the following reaction efficiency section.
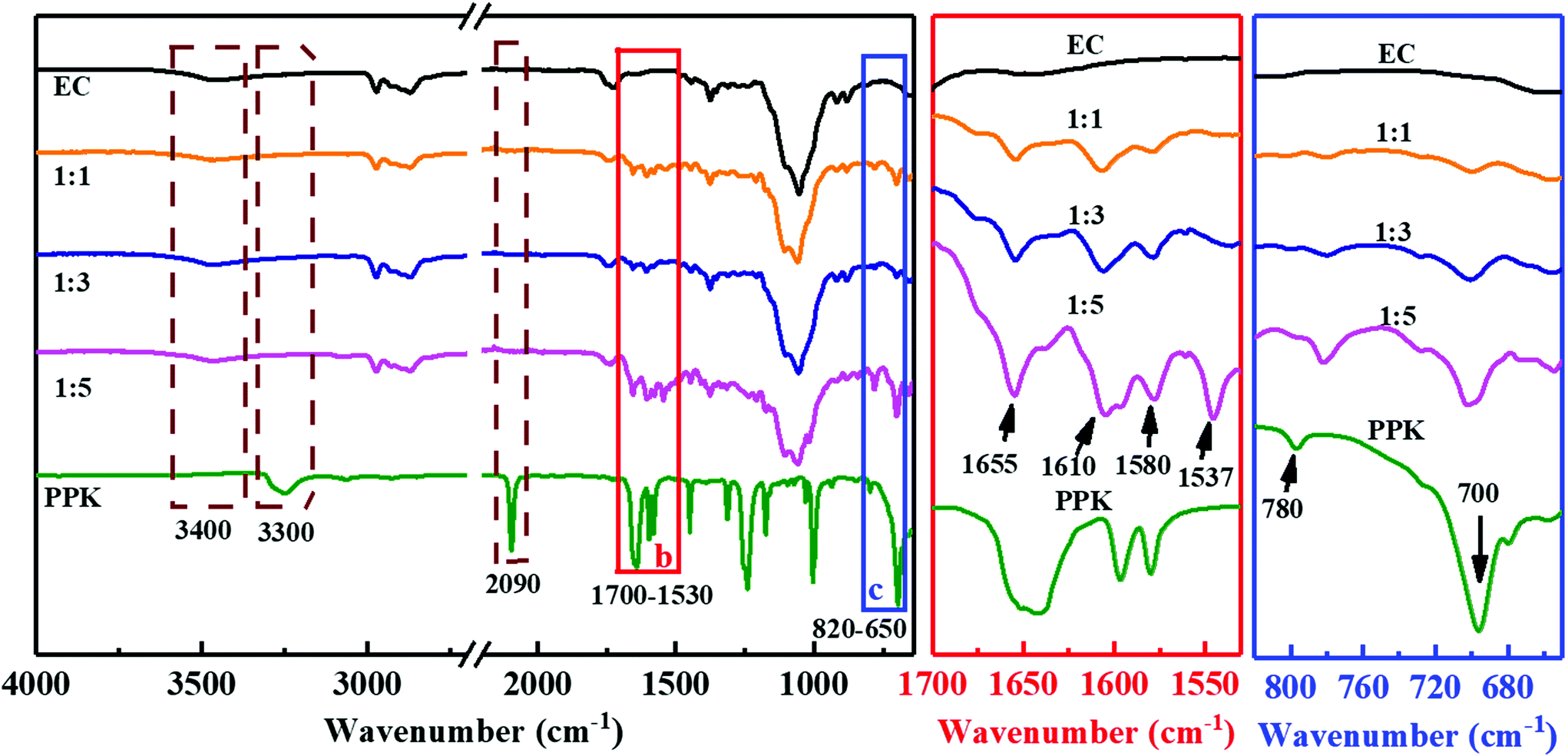 | ||
| Fig. 1 FTIR spectra of PPK, EC and ECPPKs with different OH/PPK molar ratios (reaction time: 2 h). | ||
The chemical structure of ECPPKs was further confirmed by the NMR spectra. As shown in Fig. 2a, the protons of the cellulose backbone of EC showed overlapping signals in the range of 2.8–4.5 ppm. The unimodal signal at 1.1 ppm corresponds to the methyl protons (protons 8′) in the ethyl of EC. In comparison with EC and PPK, the single peak at 3.46 ppm which comes from the protons on the C–H of PPK is absent. However, there are new peaks appearing at around 8.2 ppm and 6.8 ppm which can be ascribed to the protons on the new vinyl groups (protons 1 and 2, Fig. 2b). This confirmed the fact that triple bonds in PPK were transformed into double bonds in ECPPKs. Three peaks at around 8.2 ppm (protons 3′ and 7′), 7.6 ppm (protons 5′) and 7.5 ppm (protons 4′ and 6′) were clearly assigned in the spectrum of PPK (Fig. 2a), while these five aromatic protons in ECPPKs presented five different peaks at around 7.9 ppm (protons 3 and 7), 7.8 ppm (protons 7 and 3), 7.7 ppm (protons 5), 7.6 ppm (protons 4 and 6) and 7.5 ppm (protons 6 and 4) (Fig. 2b). This may be caused by the different chemical surroundings when the phenyl structure is anchored to the backbone of EC. Besides, 13C NMR and HSQC NMR analysis were also conducted to confirm the structure of ECPPKs. As shown in Fig. 2b, 107 ppm and 143 ppm, respectively, correspond to the new vinyl groups, and the peak at around 195 ppm corresponded to the carbonyl group on the ECPPKs.31 The result from the HSQC NMR spectrum of ECPPKs which clearly marked the position of the new vinyl groups (Fig. 2b) also provided solid support for the successful preparation of ECPPKs. These results further demonstrated the conclusion drawn from the FTIR spectra that ECPPKs were successfully prepared by the hydroxyl–yne click reaction.
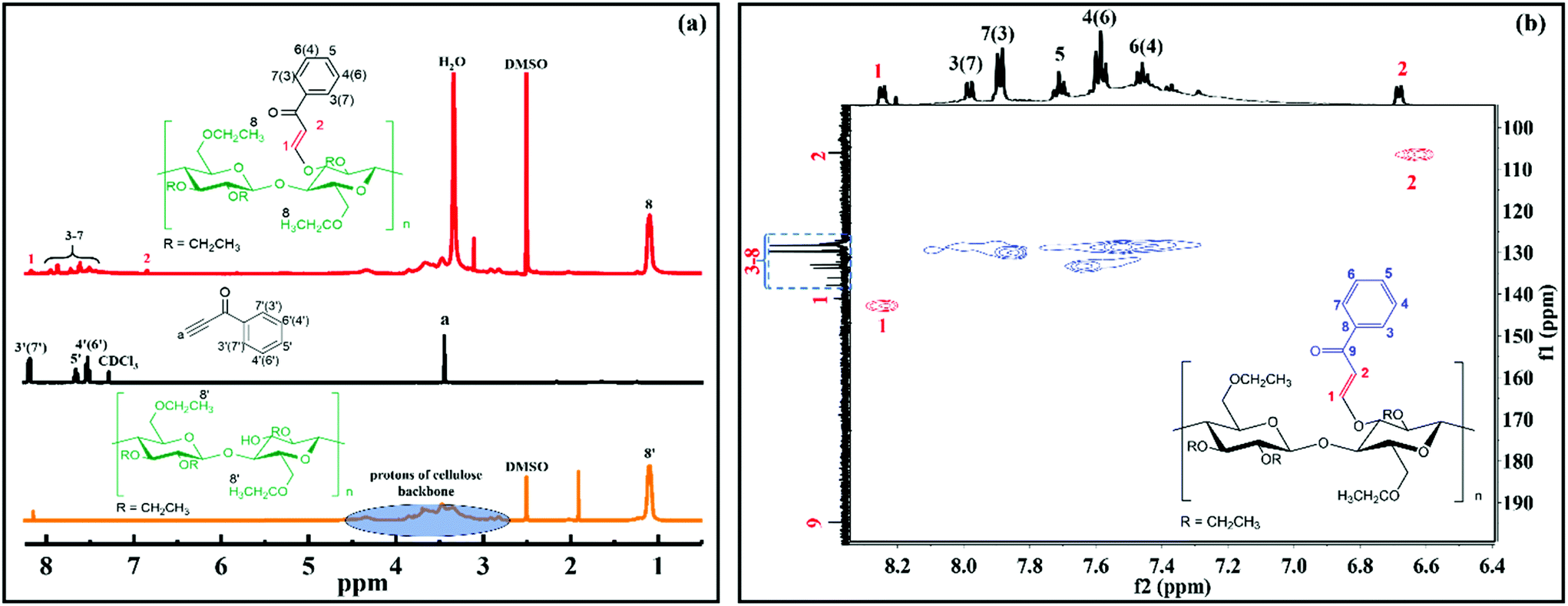 | ||
| Fig. 2 1H NMR spectra of EC, PPK and ECPPK (a), and HSQC NMR spectra of ECPPK in CDCl3 (b). | ||
3.2. Reaction efficiency of hydroxyl–yne click reaction
Usually, the reactivity of the hydroxyl groups in cellulose, two secondary alcohols present at C2 and C3 positions in the AGU and one primary alcohol position at C6 in the AGU, follows the order: OHC6 ≫ OHC2 > –OHC3.37 Therefore, with the aim to examine the high modification efficiency of the hydroxyl–yne click reaction for cellulosic materials, ethyl cellulose (EC) with a DSethyl of 2.5, in which only a few hydroxyl groups (on average one –OHC3 for every two AGUs) with the lowest reactivity remained, was chosen as the raw material. To our delight, the hydroxyl groups (–OHC3) of EC can easily react with PPK at room temperature, as shown by the above results from FTIR and NMR.To quantify the reaction efficiency of this novel cellulosic modification method, the DSPPK of ECPPKs was determined according to eqn (2) (ref. 34 and 35) and the DSPPK of the prepared ECPPKs is displayed in Fig. 3a. The effect of the reaction temperature on the hydroxyl–yne click reaction was firstly investigated. When the OH/PPK molar ratio is 1:1 and reaction time is 5 min, the DSPPK of ECPPKs reached only 0.10 at 60 °C which slightly increased compared with 0.09 at 25 °C and 0.09 at 45 °C. However, as shown in Fig. 3a, the DSPPK of ECPPKs can reach as high as 0.13 by extending reaction time to 2 h under the same OH/PPK molar ratio of 1:1. This means that the reaction time is a more determining factor than reaction temperature during this click reaction. As revealed by the FTIR spectra (Fig. 3b), with the extension of the reaction time from 5 min to 2 h, the new peaks ranging from 1700–1530 cm−1 and 650–820 cm−1 gradually grew in the spectrum of ECPPKs indicating that a higher content of PPK was introduced to EC. However, the intensity of the new peaks did not grow after 2 h. And when compared with the effect of the OH/PPK molar ratio on DSPPK of ECPPKs, the role of reaction time was also found to be limited. It was clearly shown that the DSPPK of ECPPKs gradually shifted to a high value with the increase of the OH/PPK molar ratios (Fig. 3a) which is consistent with the results shown in the FTIR spectra in Fig. 1. An OH/PPK molar ratio of 1:5 resulted in a DSPPK of 0.40 at 2 h at 25 °C, but the value of DSPPK was limited to 0.26 at 24 h at an OH/PPK molar ratio of 1:3. Overall, the influence of the reaction conditions on the reaction efficiency follows the order: OH/PPK molar ratios > reaction time > reaction temperature.
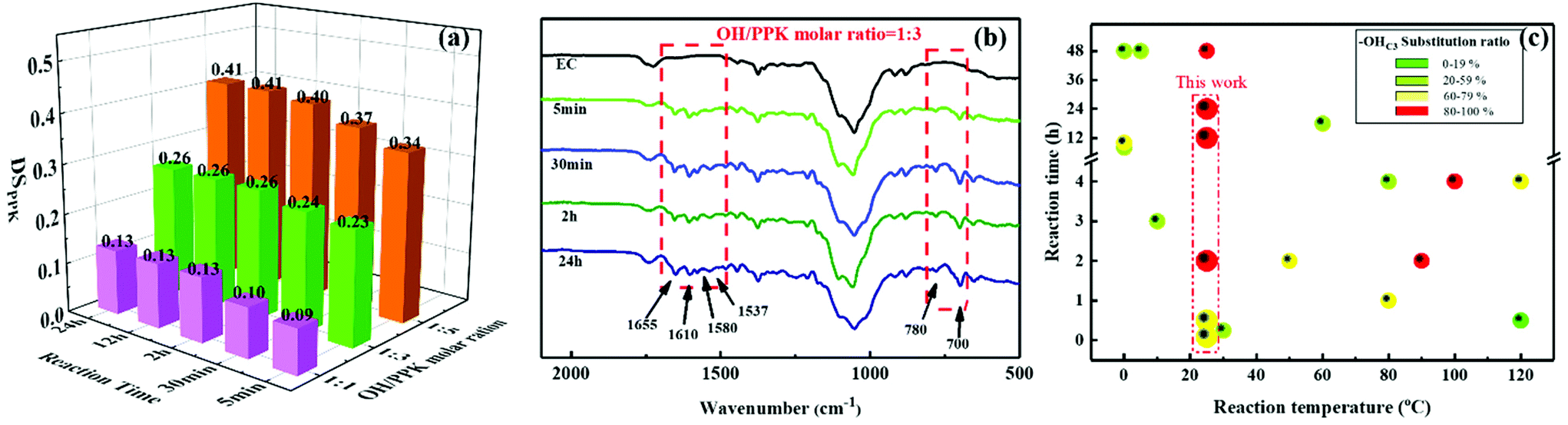 | ||
| Fig. 3 The DSPPK of ECPPKs with different grafting ratios and reaction times (a) and FTIR spectra of ECPPKs with different reaction times at 25 °C (b) and the distribution of –OHC3 substitution ratio in reported (ethyl)cellulose derivatives and this work (c). | ||
Furthermore, it is worth mentioning that PPK can quickly link to EC within just 5 minutes showing the new peaks at around 1655 cm−1 and 700 cm−1 in the FTIR spectrum (Fig. 3b). In detail, when the OH/PPK molar ratio was 1:3, the DSPPK of ECPPKs could respectively reach as high as 0.23 within 5 min and 0.26 within 24 h (Fig. 3a). This indicates that more than 88% substitution was rapidly completed within the first 5 min. And similar results were also obtained with an OH/PPK molar ratio of 1:5, in which the DSPPK increased from 0.34 to 0.41 as the reaction time increased from 5 min to 24 h. This further demonstrated the excellent efficiency of the hydroxyl–yne click reaction on the cellulosic material. Another result that should be noted is that the DSPPK of EC could be as high as 0.41 by adjusting the OH/PPK molar ratios (1:5) and reaction time (24 h). Given that the DSethyl of the raw material EC is 2.5, and the DS of residual –OH groups in EC is only 0.5, the DSPPK of ECPPKs is 0.41, indicating that 82% of the hydroxyl groups (–OHC3) in EC were substituted by PPK during the hydroxyl–yne click reaction process. It is well known that –OHC3 usually displays the lowest reactivity among all hydroxyl groups in all cellulosic materials and is difficult to substitute. As shown in Table 1 (entries 1, 2, 4, 6, 10, 11, 13, 14, 15 and 17), the–OHC3 substitution ratio of these 10 materials could not reach 60% even with excess derivatization reagents or high temperature or long reaction time in the published works. Therefore, the high substitution ratio of –OH groups in EC further confirmed that the hydroxyl–yne click reaction is an efficient modification tool for cellulosic materials. Compared to other reported relevant works (listed in Table 1) with this work, either esterification of (ethyl)cellulose or etherification of (ethyl)cellulose, a higher reaction temperature or longer reaction time was needed to obtain a good conversion (more than 60%) of –OHC3 (Fig. 3c). Besides, waste by-products such as HCl, HBr and CH3CHO also existed in these methods. However, trimethylsilyl cellulose ether40 (Table 1, entry 3) and cellulose succinates44 (Table 1, entry 9) could also be prepared without by-products with the –OHC3 substitution ratio higher than 60% in no more than 1 h. But the molar ratio of AGU/derivatization reagents of these two products is almost twice that in this work. N-α-t-Butoxycarbonyl-L-glycine ester of (ethyl)cellulose50 (Table 1, entry 17) was reported to obtain 100% substitution ratio of –OHC3 under a low molar ratio of AGU/derivatization reagents of 1:1.5 without by-products, but a reaction time of 48 h was needed. Therefore, the high substitution ratio (more than 60%) of –OH groups in EC at room temperature in just 5 min further confirmed that the hydroxyl–yne click reaction is an efficient modification tool for cellulosic materials. Overall, the mild reaction conditions and the efficient reaction process make the hydroxyl–yne click reaction a truly sustainable modification method for cellulosic materials.
| Entry | Starting material | Products | Derivatization reagents | Temperature (°C) | Molar ratio of AGU/derivatization reagents | Time | Byproducts | –OHC3 substitution ratioa (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a HMDS: 1,1,1,3,3,3-hexa-methyldisilazane; 2-BiBr: 2-bromoisobutyryl bromide; M4C: 4-(4-methoxyazobenzene-4′-yloxy) butyric acid; conversion of –OHC3: calculated from the DS of cellulosic derivatives. | |||||||||
| 1 | Cellulose | Glycidyl cellulose ether | Glycidol | 60 | 1:6 |
18 h | None | 20 | 38 |
| 2 | Cellulose | Cyanoethyl cellulose ether | Acrylonitrile | 0 | 1:10 |
8 h | None | 39 | 39 |
| 3 | Cellulose | Trimethylsilyl cellulose ether | HMDSa | 80 | 1:5 |
1 h | None | 73 | 40 |
| 4 | Cellulose | Benzyl cellulose ether | Benzyl bromide | 10 | 1:9 |
3 h | None | 50 | 41 |
| 5 | Cellulose | Cellulose benzoates | Benzoyl Chloride | 100 | 1:10 |
4 h | HCl | 100 | 35 |
| 6 | Cellulose | Cellulose benzoates | Vinyl benzoate | 80 | 1:3 |
4 h | CH3CHO | 40 | 42 |
| 7 | Cellulose | Cellulose acetate | Acetyl chloride | 50 | 1:10 |
2 h | HCl | 79 | 43 |
| 8 | Cellulose | Cellulose acetate | Vinyl acetate | 90 | 1:35 |
2 h | CH3CHO | 86 | 14 |
| 9 | Cellulose | Cellulose succinates | Succinic anhydride | 25 | 1:6 |
30 min | None | 62 | 44 |
| 10 | Cellulose | Cellulose levulinate esters | α-Angelica lactone | 120 | 1:1.67 |
30 min | None | 10 | 45 |
| 11 | Cellulose | cellulose hexanoate ester | Vinyl hexanoate | 30 | 1:6.5 |
15 min | CH3CHO | 34 | 13 |
| 12 | Cellulose | Cellulose laurate ester | Vinyl laurate | 120 | 1:6 |
4 h | CH3CHO | 74 | 46 |
| 13 | Ethyl cellulose | EC-Br ester macroinitiator | 2-BiBra | 0 | 1:3 |
48 h | HBr | 57 | 47 |
| 14 | Ethyl cellulose | Ethyl cellulose azobenzene mesogen ester | M4Ca | 0 | 1:6.8 |
10 h | None | 49 | 48 |
| 15 | Ethyl cellulose | Azido-ethyl cellulose ester | 6-Azidohexanoic acid | 5 | 1:0.4 |
48 h | None | 38 | 25 |
| 16 | Ethyl cellulose | Pentafluorobenzoyl ethyl cellulose ester | Pentafluorobenzoyl chloride | 25 | 1:1 |
24 h | HCl | 74 | 49 |
| 17 | Ethyl cellulose | N-α-t-Butoxycarbonyl-L-glycine ester of EC | N-α-t-Boc-L-glycine | 25 | 1:1.5 |
48 h | None | 100 | 50 |
| 18 | Ethyl cellulose | Ethyl pent-4-enyl cellulose ether | 5-Bromo-pent-1-ene | 50 | 1:4.3 |
96 h | HBr | 90 | 11 |
| 19 | Ethyl cellulose | ECPPKs | PPK | 25 | 1:2.5 |
5 min | None | 68 | This work |
| 30 min | 74 | ||||||||
| 2 h | 80 | ||||||||
| 12 h | 82 | ||||||||
| 24 h | 82 | ||||||||
3.3. Thermal properties of ECPPKs
The thermal stability of EC and ECPPKs was estimated by TGA under a continuous nitrogen atmosphere. TGA and DTG curves of the raw material EC and ECPPKs with different DSPPK were recorded and are illustrated in Fig. 4a. The temperature at the maximum thermal decomposition rate (Td,max) of the raw material EC is ∼355 °C. However, ECPPKs with DSPPK of 0.13 showed an increased Td,max of 361 °C. And the Td,max of ECPPKs increased gradually with DSPPK. This is probably because the better thermal stability of aromatic rings is passed on to ECPPK molecules.51,52 With DSPPK of 0.40, the Td,max of ECPPKs reached 363 °C, which was an 8 °C increment from that of pristine EC. In addition, the ECPPKs with DSPPK above 0.40 showed a high thermal stability above 600 °C with about 20% mass left, which is obviously higher than that of EC with only 7% mass left. Overall, the thermal stability of ECPPKs has been improved due to the presence of PPK moieties.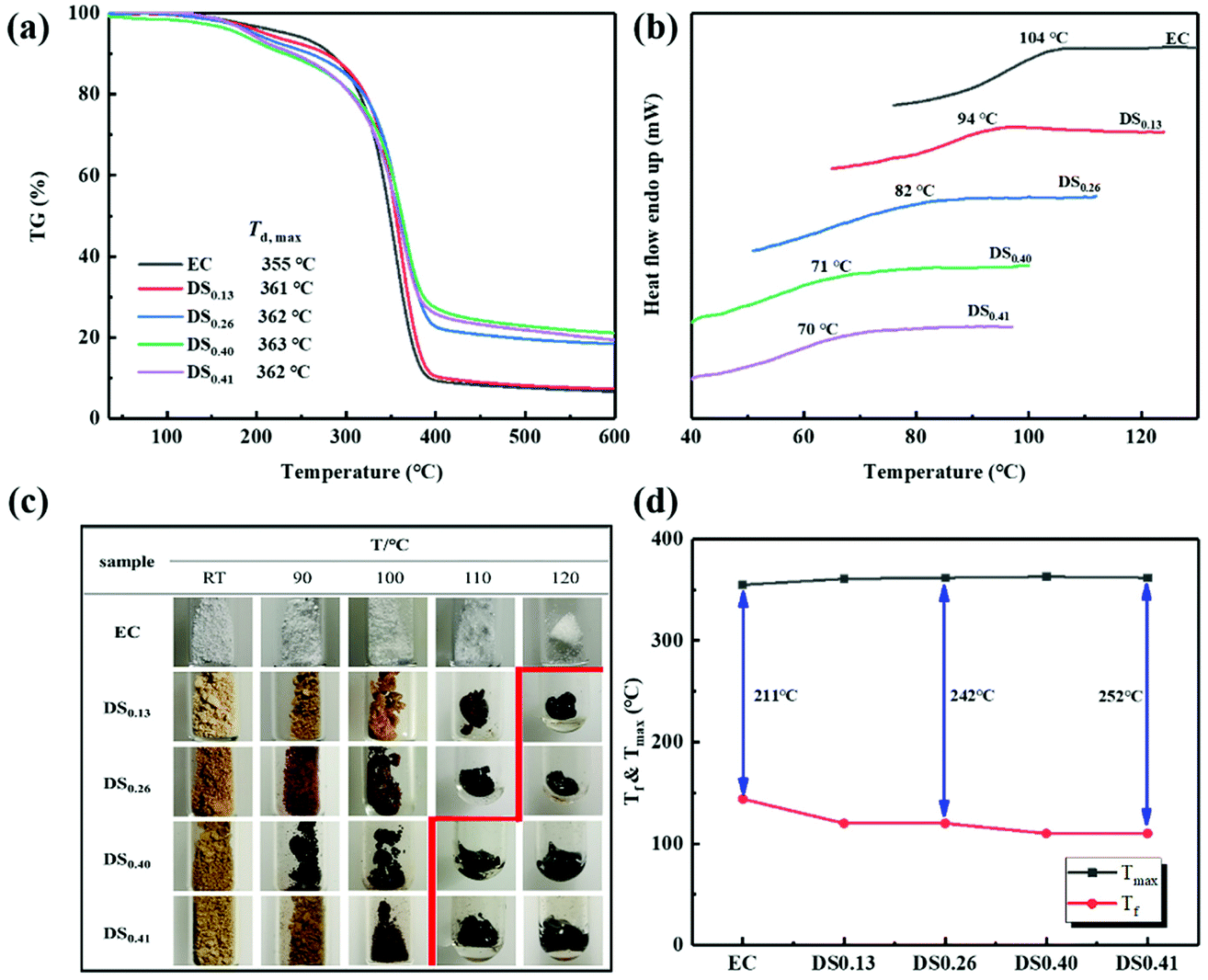 | ||
| Fig. 4 TGA thermograms (a) and DSC curves (b) of unmodified EC and ECPPKs with different DSppk. (c) Picture of unmodified EC and different ECPPKs at different temperatures. (d) Tf and Tmax of unmodified EC and ECPPKs. | ||
The glass transition temperature (Tg) of EC and ECPPKs with various DSPPK was characterized by DSC, as shown in Fig. 4b. It is clearly shown that both EC and ECPPKs have obvious glass transition temperature (Tg). The raw material EC has a Tg of 104 °C; however, the Tg of all ECPPKs shifted toward lower values and decreased dramatically with the increase of DSPPK of ECPPKs. This is ascribed to two reasons: (1) the hydroxyl groups of EC were substituted after the hydroxyl–yne click reaction, thus, the strong intermolecular H-bond interactions was disrupted, and the limitation effect on the movement of EC chain segments were decreased;51 (2) the substituent segments in ECPPKs can act as internal plasticizers which could improve the mobility of the EC chains.53 Furthermore, when DSPPK of ECPPKs reached 0.41, the Tg was as low as 70 °C, which was a 34 °C decrease from that of pure EC. This confirmed that the substituents in ECPPKs with soft middle segments (–O–CC moieties) and bulky terminal moieties (phenyl) played an important role in decreasing the Tg of EC.53 Overall, the results of DSC highlight that the hydroxyl–yne click reaction is an effective approach for adjusting the Tg of cellulosic materials.
Thermal processability is one of the most important factors for the application of cellulose derivatives. As shown in Fig. 4c, the thermal flow behaviours of EC and ECPPKs at different temperatures were recorded. With the increase of DSPPK, flowing temperature (Tf) of ECPPKs decreased from 120 °C to 110 °C, which was 34 °C lower than that of pure EC (144 °C (ref. 54)). This confirmed that PPK moieties were introduced into the EC and increased the distance between the EC chains that improved the mobility of the EC chains. From Fig. 4d, it is clearly seen that the Tf of the ECPPKs decreased upon the introduction of PPK moieties, which is beneficial for the thermo processing of cellulosic materials using this method, while the temperature at the maximum thermal decomposition rate (Tmax) of ECPPKs increased upon the introduction of PPK moieties. The temperature window (Tmax–Tf) was widened with the increase of PPK moieties for thermal processability. This suggested that PPK substituents played a significant role in improving the thermal processability of EC.
3.4. UV-blocking properties of ECPPKs
It is well known that one common route to improve the UV-blocking properties of cellulosic materials is to introduce aromatic ring structures.19,51 As stated above, the aromatic ring could be easily linked to the EC backbone. Therefore, ECPPKs are expected to show excellent UV absorption performance. The UV-vis adsorption spectra of EC, PPK and ECPPKs with different DSPPK are shown in Fig. 5a. No peak appeared in the spectrum of EC in the range from 200–600 nm. The absorption peaks at 266 nm and 338 nm in the spectrum of PPK correspond to π → π* and n → π* electronic transitions of the phenyl ring and the carbonyl group.55 And these peaks remained in all the spectra of ECPPKs, indicating the existence of the phenyl ring and carbonyl group in the ECPPK structure. In addition, a new peak at around 420 nm emerged on the ECPPK spectra, which could be assigned to the new vinyl ether linkage (–C–O–CC–) formed by the click reaction of terminal alkyne and the hydroxyl group. A similar phenomenon was reported when aromatic ester was grafted onto phenol-containing poly(disubstituted acetylenes) via the phenol-yne click reaction.56 Obviously, the intensity of the absorption peak at around 338 nm and 420 nm increased with the DSPPK of ECPPKs, which further confirmed the fact that the UV absorption properties were imparted into ECPPKs by the PPK moieties. Overall, the successful modification of EC to ECPPKs can be clearly concluded from the UV-vis absorption spectra which is consistent with the FTIR and 1H NMR spectra.
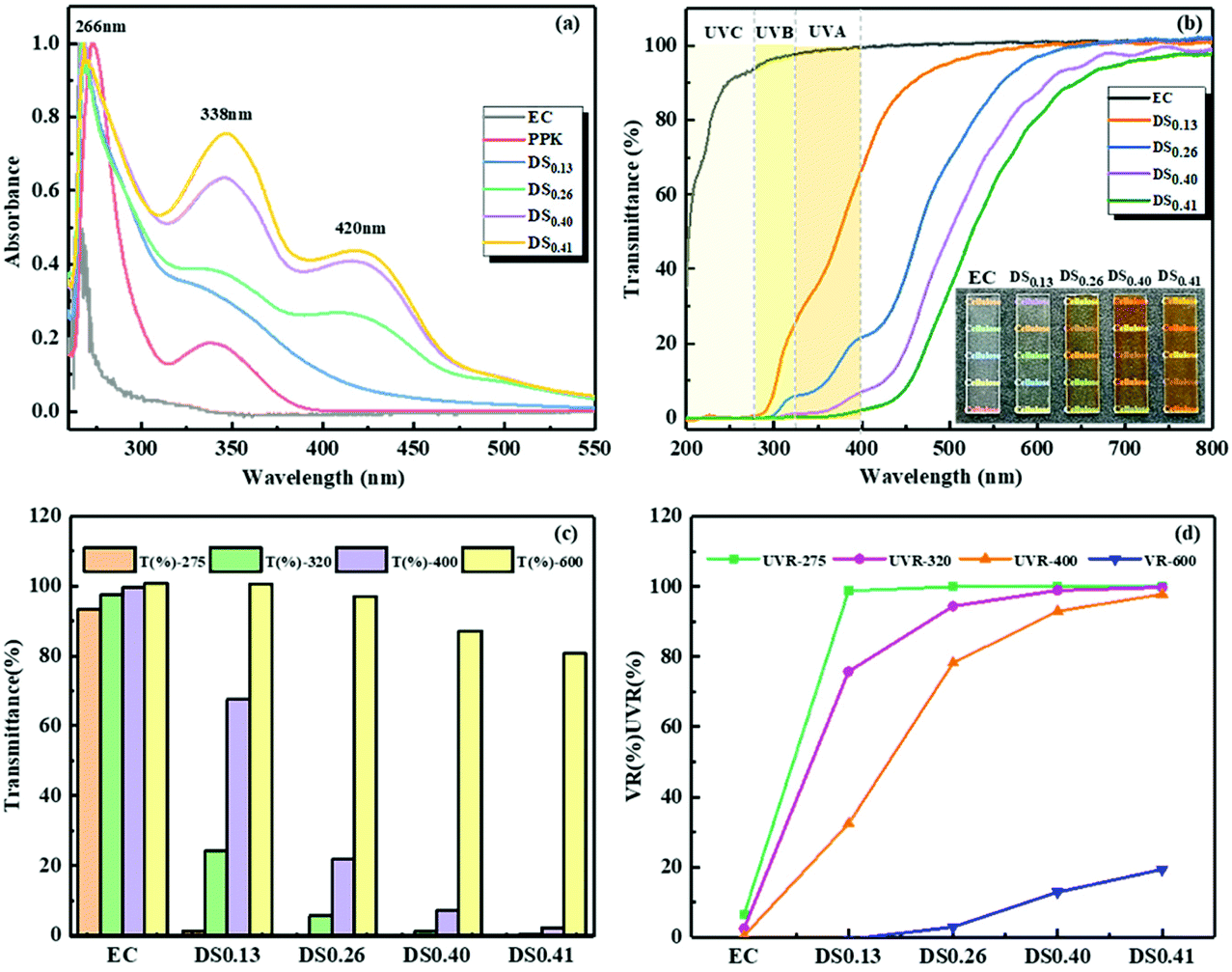 | ||
| Fig. 5 The absorbance (a), transmittance (b) and transmittance values as well as shielding ratios at 275 nm, 320 nm, 400 nm and 600 nm (c and d) of EC, PPK and ECPPKs with different DSPPK. | ||
The optical properties of ECPPKs and EC films were recorded in the wavelength range from 0 to 800 nm. As shown in Fig. 5b, high transmittance was detected for the pure EC film in the wavelength range of both the UV and visible spectrum, which means that EC film has poor UV-shielding performance. However, with a DSPPK of only 0.13, the ECPPK films shielded 100% of the UVC (200–275 nm), most of the UVB (320–275 nm) and over 50% of UVA light. When the DSPPK of ECPPKs was increased to 0.40–0.41, nearly 100% of UVA light was also shielded. However, most cellulosic UV-absorbing films made by the incorporation of phenolic structures could only efficiently block UVB and UVC light below 320 nm, while showing limited shielding ability of UVA light (<50%).55,57–59 This excellent UV-blocking performance of ECPPKs was attributed to the phenolic structures as well as the newly formed –O–CC moieties in ECPPKs, which perform strong UV absorption as revealed in Fig. 5a.
The highly transparent UV-absorption materials are desirable for the applications of transparent packaging and UV-shielding materials.51 Thus, to synthetically analyse both the optical transparency and UV-shielding performance of ECPPK films, the transmittance at 600 nm, 320 nm and 275 nm is displayed in Fig. 5c. The shielding ratio of visible light (VR600 at 600 nm) and UV light (UVR at 320 nm and 275 nm) was evaluated through eqn (3) (ref. 60) and displayed in Fig. 5d. It is shown that the light brown color was introduced into ECPPK films by the introduction of PPK (inset picture of Fig. 5b). Despite this, all ECPPK films maintained good transparency in the visible light region, and presented high transmittance in the visible region (more than 80% at 600 nm) with the VR-600 less than 20% (Fig. 5c). However, when the DSPPK increased from 0.13 to 0.41, the UVR presented a gradually growing trend at 275 nm, 320 nm and 400 nm. And it is easy to find that UVR (at 275 nm, 320 nm and 400 nm) and VR (at 600 nm) increase gradually as DSPPK increased. This suggests that ECPPK films have good UV-shielding properties and highly transparent properties at the same time which could find promising application in UV-blocking materials.
3.5. Fluorescence properties
The fluorescence properties of ECPPKs and EC were studied using N,N-dimethylformamide (DMF) as the solvent (Fig. 6). No characteristic emission peaks were found in the fluorescence spectra of EC and PPK when excited at 420 nm (according to the UV-visible spectra in Fig. 5a). Interestingly, the newly prepared ECPPKs (when DSPPK is above 0.26) exhibited good fluorescence properties, exhibiting characteristic emission at 530 nm upon 420 nm excitation (Fig. 6a). In addition, Tang and co-workers reported a similar phenomenon wherein poly(disubstituted acetylenes) (PDSAs) showed stronger fluorescence after the attachment of aromatic rings by the highly efficient “phenol–yne click reaction”.56 Given that PPK molecules didn't show fluorescence properties, the reason why ECPPKs exhibited good fluorescence properties could be the conjugation effect between carbon–carbon double bond in aromatic rings of PPK and the newly formed vinyl ether linkage by the hydroxyl–yne click reaction.61–65 In addition, it is clearly showed in Fig. 6a that the higher the DSPPK, the stronger the fluorescence intensity of ECPPKs and the intensity increases linearly with DSPPK. This indicated that the concentration of PPK has an influence on the fluorescence properties of ECPPKs.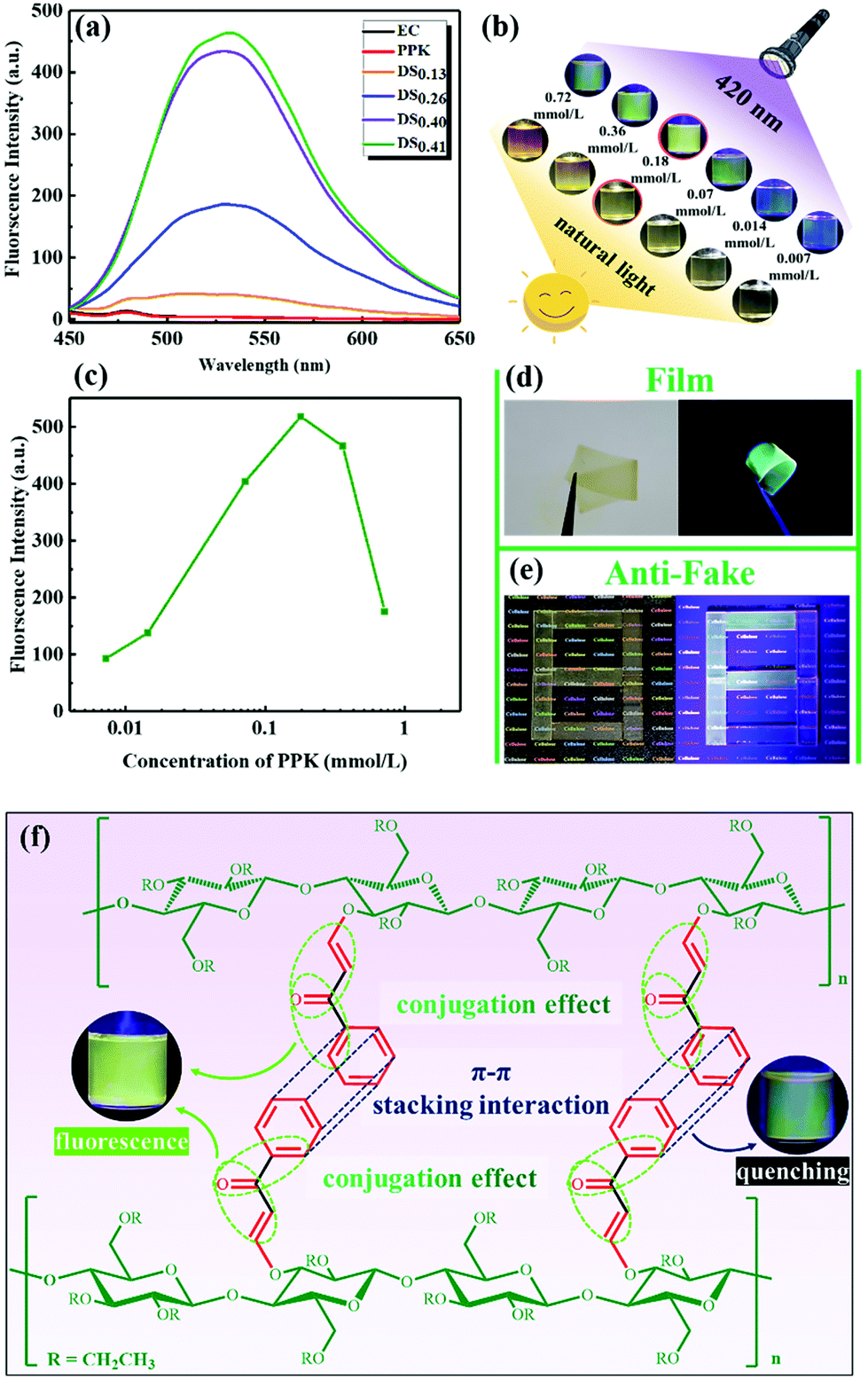 | ||
| Fig. 6 Fluorescence properties of ECPPKs. (a) Fluorescence spectra of EC, PPK and ECPPKs with different degrees of substitution (0.1 mg ml−1); fluorescence photographs (b) and (c) for the intensity of ECPPKs with different PPK concentrations; (d and e) fluorescence photographs of the ECPPK polymer composite membrane and fluorescent anti-counterfeiting under 420 nm excitation. (f) Images of possible fluorescence mechanism. | ||
To further demonstrate the fluorescence properties of ECPPKs, the effect of PPK concentration on the fluorescence intensity of ECPPKs (DSPPK is 0.41) was further investigated as shown in Fig. 6b and c. When the PPK concentration increased from 0.007 mmol L−1 to 0.18 mmol L−1, the fluorescence intensity showed an increasing trend. However, the fluorescence intensity dramatically decreased when the concentration increased from 0.18 mmol L−1 to 0.72 mmol L−1. This phenomenon is consistent with the conventional fluorescence aggregation-caused quenching (ACQ). The π–π stacking interaction of the aromatic ring between different fluorescent molecules is the common reason for ACQ,66,67 therefore the aggregation of PPK could be the reason for the ACQ properties of ECPPKs. Given that the PPK molecule was anchored to the ECPPK chains, the aggregation of PPK could happen in the same or different ECPPK chains. Because ECPPK chains can move freely in the solution state, the increase of the PPK concentration could increase the aggregation possibility of PPK molecules in the same or different ECPPK chains resulting in a decreased fluorescence intensity.68 Therefore, the ACQ effect of ECPPKs may be eliminated by suppressing the movement of ECPPK chains. To verify this assumption, ECPPKs were mixed with the polylactic acid (PLA) matrix to prepare solid composite films. As expected, the ECPPK solid composites showed good fluorescence properties under the excitation of 420 nm which means that the fluorescence quenching of the ACQ effect was really suppressed under these conditions. This is because the movement of ECPPK chains was limited and the distance between different ECPPK chains could be enlarged by the PLA matrix in the solid composite films. Therefore, the aggregation chance of PPK molecules was reduced, weakening the fluorescence quenching of the ACQ effect. In addition, the visible light excited fluorescence properties which avoided the disadvantage of UV excitation made these new kinds of cellulose derivative a promising candidate in the field of fluorescence films and anti-counterfeiting (Fig. 6d and e).69
Based on the above results, the fluorescence properties of ECPPKs could be influenced by two effects: (1) the conjugation effect between the carbon–carbon double bond in aromatic rings of PPK and the newly formed vinyl ether linkage by the hydroxyl–yne click reaction, which is positive for the fluorescence intensity; (2) the π–π stacking interaction of the aromatic ring between ECPPK chains, which leads to the quenching phenomenon (Fig. 6f). Therefore, as the concentration of the solution increases, the aggregation possibility of PPK moieties between different EC chains increases, therefore the π–π stacking interaction enhanced and the fluorescence intensity weakened. In addition, the good fluorescence properties of the ECPPK composite could be ascribed to the anchoring effect of the cellulose backbone which could tackle the fluorescence quenching of ACQ in the solid state.68
4. Conclusions
In summary, a novel hydroxyl–yne click reaction was applied as an efficient and sustainable tool to generate new cellulose derivatives. The reaction offers significant merits of mild reaction temperature (room temperature and heat free), no by-products, fast reaction rate (5 min) and high reaction efficiency (substituting 82% of hydroxyl groups with –OHC3). ECPPKs showed excellent thermoplastic properties with an improved thermal stability and extended thermal processing temperature range when compared with the raw material EC. Besides, owing to the newly formed vinyl ether linkage, the prepared ethyl cellulose phenyl propylene ketone ether (ECPPK) film achieved nearly 100% shielding ratio of UVA light while the high transparency in the visible range was preserved. Moreover, the ECPPKs showed fluorescence properties with the emission peaks at around 530 nm upon visible light excitation (420 nm). In brief, the successful fabrication of ECPPKs based on the alkyne and hydroxyl group of the EC via hydroxyl–yne click chemistry strategy provides a new avenue for (ethyl)cellulose modification and further expands the potential application value for cellulosic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 31901275), the Natural Science Foundation of Jiangsu Province (No. BK20170924), the Youth Science and Technology Innovation Fund of Nanjing Forestry University (No. CX2017006), the Project Funded by the National First-class Disciplines (PNFD), the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Overseas Research and Study Program of Jiangsu Universities.References
- R. F. S. Barbosa, A. G. Souza, F. F. Ferreira and D. S. Rosa, Carbohydr. Polym., 2019, 218, 208–217 CrossRef CAS.
- T. Elschner, M. Kotteritzsch and T. Heinze, Macromol. Biosci., 2014, 14, 161–165 CrossRef CAS.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- A. Carlmark, E. Larsson and E. Malmström, Eur. Polym. J., 2012, 48, 1646–1659 CrossRef CAS.
- C. Schönbein, Ber. Naturforsch. Ges. Basel, 1847, 7, 27 Search PubMed.
- A. Isogai, T. Hänninen, S. Fujisawa and T. Saito, Prog. Polym. Sci., 2018, 86, 122–148 CrossRef CAS.
- M. Tizzotti, A. Charlot, E. Fleury, M. Stenzel and J. Bernard, Macromol. Rapid Commun., 2010, 31, 1751–1772 CrossRef CAS.
- S. Kalia, A. Dufresne, B. M. Cherian, B. S. Kaith, L. Avérous, J. Njuguna and E. Nassiopoulos, Int. J. Polym. Sci., 2011, 2011, 1–35 Search PubMed.
- L. Zhou, H. He, M. C. Li, S. Huang, C. Mei and Q. Wu, Carbohydr. Polym., 2018, 189, 331–341 CrossRef CAS.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS.
- Y. Dong, L. I. Mosquera-Giraldo, L. S. Taylor and K. J. Edgar, Biomacromolecules, 2016, 17, 454–465 CrossRef CAS.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS.
- J. Ding, C. Li, J. Liu, Y. Lu, G. Qin, L. Gan and M. Long, Carbohydr. Polym., 2017, 157, 1785–1793 CrossRef CAS.
- M. Chen, R.-M. Li, T. Runge, J. Feng, J. Feng, S. Hu and Q.-S. Shi, ACS Sustainable Chem. Eng., 2019, 7, 16971–16978 CrossRef CAS.
- F. Z. Khan, T. Sakaguchi, M. Shiotsuki, Y. Nishio and T. Masuda, Macromolecules, 2006, 39, 6025–6030 CrossRef CAS.
- G. Chantereau, N. Brown, M.-A. Dourges, C. S. Freire, A. J. Silvestre, G. Sebe and V. Coma, Carbohydr. Polym., 2019, 220, 71–78 CrossRef CAS.
- N. C. Ellebracht and C. W. Jones, Cellulose, 2018, 25, 6495–6512 CrossRef CAS.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- L. Rong, M. Zeng, H. Liu, B. Wang, Z. Mao, H. Xu, L. Zhang, Y. Zhong, J. Yuan and X. Sui, Carbohydr. Polym., 2019, 209, 223–229 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- W. Wu, R. Tang, Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- P. Fuchs, P. Vana and K. Zhang, J. Polym. Sci., 2020, 58, 1535–1543 CrossRef CAS.
- A. A. Nada, F. H. H. Abdellatif, E. A. Ali, R. A. Abdelazeem, A. A. S. Soliman and N. Y. Abou-Zeid, Carbohydr. Polym., 2018, 199, 610–618 CrossRef CAS.
- M. G. Roberts, Q. Yu, R. Keunen, J. Liu, E. C. N. Wong, C. K. Rastogi, R. M. Reilly, C. Allen and M. A. Winnik, Biomacromolecules, 2020, 21, 2014–2023 CrossRef CAS.
- G. Xiao, C. Ding, F. Song, X. Qian and X. An, Cellulose, 2016, 24, 591–607 CrossRef.
- A. Qin, Y. Liu and B. Z. Tang, Macromol. Chem. Phys., 2015, 216, 818–828 CrossRef CAS.
- G. J. W. Aalbers, C. E. Boott, F. D'Acierno, L. Lewis, J. Ho, C. A. Michal, W. Y. Hamad and M. J. MacLachlan, Biomacromolecules, 2019, 20, 2779–2785 CrossRef CAS.
- P. Tingaut, R. Hauert and T. Zimmermann, J. Mater. Chem., 2011, 21, 16066–16076 RSC.
- Y. Shi, T. Bai, W. Bai, Z. Wang, M. Chen, B. Yao, J. Z. Sun, A. Qin, J. Ling and B. Z. Tang, Chemistry, 2017, 23, 10725–10731 CrossRef CAS.
- X. Hu, X. Zhao, B. He, Z. Zhao, Z. Zheng, P. Zhang, X. Shi, R. T. K. Kwok, J. W. Y. Lam, A. Qin and B. Z. Tang, Research, 2018, 2018, 3152870 CrossRef.
- H. Dong, R. Zheng, J. W. Lam, M. Häussler, A. Qin and B. Z. Tang, Macromolecules, 2005, 38, 6382–6391 CrossRef CAS.
- P. Xiao, J. Zhang, Y. Feng, J. Wu, J. He and J. Zhang, Cellulose, 2014, 21, 2369–2378 CrossRef CAS.
- J. Zhang, J. Wu, Y. Cao, S. Sang, J. Zhang and J. He, Cellulose, 2008, 16, 299–308 CrossRef.
- E. Stern, G. G. Muccioli, L. Hamtiaux, R. Millet, J. H. Poupaert, J.-P. Hénichart, P. Depreux, J.-F. Goossens and D. M. Lambert, J. Med. Chem., 2007, 50, 5471–5484 CrossRef CAS.
- F. Joubert, O. M. Musa, D. R. Hodgson and N. R. Cameron, Chem. Soc. Rev., 2014, 43, 7217–7235 RSC.
- T. Kakibe, S. Nakamura, W. Mizuta and H. Kishi, Chem. Lett., 2017, 46, 737–739 CrossRef CAS.
- W. Li, R. Liu, H. Kang, Y. Sun, F. Dong and Y. Huang, Polym. Chem., 2013, 4, 2556–2563 RSC.
- M. Kostag, S. Köhler, T. Liebert and T. Heinze, Macromol. Symp., 2010, 294, 96–106 CrossRef CAS.
- M. Abe, K. Sugimura, Y. Nishiyama and Y. Nishio, ACS Sustainable Chem. Eng., 2017, 5, 4505–4510 CrossRef CAS.
- L. P. Hinner, J. L. Wissner, A. Beurer, B. A. Nebel and B. Hauer, Green Chem., 2016, 18, 6099–6107 RSC.
- C. Achtel and T. Heinze, Macromol. Chem. Phys., 2016, 217, 2041–2048 CrossRef CAS.
- Z. Söyler, K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, Green Chem., 2018, 20, 214–224 RSC.
- M. Pei, X. Peng, Y. Shen, Y. Yang, Y. Guo, Q. Zheng, H. Xie and H. Sun, Green Chem., 2020, 22, 707–717 RSC.
- X. Wen, H. Wang, Y. Wei, X. Wang and C. Liu, Carbohydr. Polym., 2017, 168, 247–254 CrossRef CAS.
- D. Shen, H. Yu and Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4099–4108 CrossRef CAS.
- T. Hu, H. Xie, J. Xiao, H. Zhang and E. Chen, Cellulose, 2010, 17, 547–558 CrossRef CAS.
- F. Z. Khan, T. Sakaguchi, M. Shiotsuki, Y. Nishio and T. Masuda, Macromolecules, 2006, 39, 9208–9214 CrossRef CAS.
- Y. Ikeuchi, F. Z. Khan, N. Onishi, M. Shiotsuki, T. Masuda, Y. Nishio and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3986–3993 CrossRef CAS.
- X. Zhang, W. Liu, D. Yang and X. Qiu, Adv. Funct. Mater., 2019, 29, 1806912 CrossRef.
- J. Yu, Y. Liu, X. Liu, C. Wang, J. Wang, F. Chu and C. Tang, Green Chem., 2014, 16, 1854–1864 RSC.
- Z. Chen, J. Zhang, P. Xiao, W. Tian and J. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 4931–4939 CrossRef CAS.
- Y. Fukumori, H. Ichikawa, Y. Yamaok, E. Akaho and Y. Takeuchi, Chem. Pharm. Bull., 1991, 39, 164–169 CrossRef CAS.
- Z. Zhang, G. Sèbe, X. Wang and K. C. Tam, ACS Appl. Nano Mater., 2018, 1, 632–641 CrossRef CAS.
- W. Wang, Y. Shi, X. Wang, A. Qin, J. Z. Sun and B. Z. Tang, Polym. Chem., 2017, 8, 2630–2639 RSC.
- X. Niu, Y. Liu, G. Fang, C. Huang, O. J. Rojas and H. Pan, Biomacromolecules, 2018, 19, 4565–4575 CrossRef CAS.
- J. A. Sirviö, M. Visanko, J. P. Heiskanen and H. Liimatainen, J. Mater. Chem. A, 2016, 4, 6368–6375 RSC.
- Y. Tan, H. Wu, T. Xie, L. Chen, S. Hu, H. Tian, Y. Wang and J. Wang, Int. J. Biol. Macromol., 2020, 155, 1325–1332 CrossRef CAS.
- J. Luo, M. Zhang, B. Yang, G. Liu, J. Tan, J. Nie and S. Song, Carbohydr. Polym., 2019, 203, 110–118 CrossRef CAS.
- X. Ji, Y. Yao, J. Li, X. Yan and F. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS.
- A. Kaeser and A. P. Schenning, Adv. Mater., 2010, 22, 2985–2997 CrossRef CAS.
- W. Shi and H. Ma, Chem. Commun., 2012, 48, 8732–8744 RSC.
- Y. Yamaguchi, Y. Matsubara, T. Ochi, T. Wakamiya and Z.-I. Yoshida, J. Am. Chem. Soc., 2008, 42, 13867–13869 CrossRef.
- X. Zhang, J. Y. Liu, W. W. Ma and M. L. Yang, J. Mater. Chem. B, 2016, 4, 6662–6669 RSC.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, J. Chem. Educ., 2015, 93, 345–350 CrossRef.
- Y. Huang, J. Xing, Q. Gong, L. C. Chen, G. Liu, C. Yao, Z. Wang, H. L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef.
- W. Tian, J. Zhang, J. Yu, J. Wu, H. Nawaz, J. Zhang, J. He and F. Wang, Adv. Opt. Mater., 2016, 4, 2044–2050 CrossRef CAS.
- Y.-L. Wang, C. Li, H.-Q. Qu, C. Fan, P.-J. Zhao, R. Tian and M.-Q. Zhu, J. Am. Chem. Soc., 2020, 142, 7497–7505 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |