
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomass valorisation over polyoxometalate-based catalysts
Jiawei
Zhong
a,
Javier
Pérez-Ramírez
 *ab and
Ning
Yan
*a
*ab and
Ning
Yan
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive, Singapore 117585, Singapore. E-mail: ning.yan@nus.edu.sg
bInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland. E-mail: jpr@chem.ethz.ch
First published on 7th November 2020
Abstract
The efficient utilization of biomass, the only globally available, renewable and abundant carbon-neutral source, is of high significance in green and sustainable chemistry. Polyoxometalates (POMs) and POM-based composites have been widely applied in green catalytic reactions, due to their tunable Brønsted/Lewis-acidity and redox properties enabling high reactivity in a wide range of chemical transformations. This review covers recent advances in the chemocatalytic conversion of biomass into chemicals and fuels over POMs and POM–metal composites. For biomass valorisation over POMs, the advances of acid catalysis including hydrolysis, dehydration, etherification, alcoholysis, transesterification and esterification are summarised. Furthermore, applications in chemical oxidation for the synthesis of organic acids and furan chemicals are discussed. For biomass valorisation over metal–POM composites, an overview of tandem reactions (e.g. hydrolysis–hydrogenation, hydrolysis–oxidation, and hydrogenolysis–hydrodeoxygenation) is highlighted. The future prospects of biomass valorisation over POM-based catalysts are finally presented.
 Jiawei Zhong | Jiawei Zhong completed his Ph.D research in Dalian Institute of Chemical Physics, Chinese Academy of Sciences, and received his PhD degree in industrial catalysis from Dalian University of Technology in 2019, under the supervision of Professor Zhongmin Liu, Chunshan Song and Yingxu Wei. Then he worked as a postdoctoral fellow with Prof. Javier Pérez-Ramírez and Ning Yan at the National University of Singapore. His research interests include heterogeneous catalysis and catalytic materials, catalytic valorization of biomass, as well as C1 chemistry. |
 Javier Pérez-Ramírez | Javier Pérez-Ramírez holds the Chair of Catalysis Engineering at ETH Zurich. His research pursues the design of heterogeneous catalysts and reactor concepts tackling current and future energy, resource, and environmental challenges of society. He directs a National Competence Center of Research in Catalysis in Switzerland and has a visiting appointment at the National University of Singapore within the Flagship Green Energy Program. |
 Ning Yan | Ning Yan received his B.Sc. and PhD degrees in Chemistry from Peking University working with Prof. Yuan Kou. After a Marie Curie Fellowship at the École Polytechnique Fédérale de Lausanne in Switzerland with Prof. Paul Dyson, he joined the Department of Chemical and Biomolecular Engineering at the National University of Singapore as an assistant professor in 2012 and was promoted to a tenured associate professor in 2018. Ning Yan works actively in advanced catalysis, renewable energy, and sustainable chemistry. |
1 Introduction
Since the industrial revolution, the ever-increasing consumption of fossil carbon resources including coal, crude oil and natural gas for energy and chemicals has resulted in enormous emissions of greenhouse gases (GHGs) into the atmosphere. The increasing demand for resources, the diminishing finite fossil reserves as well as the urgent need to reduce GHG emissions have driven the innovative development of alternative energy sources derived from green and sustainable feedstocks in a post-petroleum era.Biomass, including carbohydrates, lignin, and lipids, among others, is an attractive alternative feedstock. It is the only globally available, renewable and abundant carbon-neutral source. Due to the relevant debate regarding “food versus fuel”, lignocellulosic biomass composed of non-edible cellulose (35–50%), hemicelluloses (20–35%) and lignin (10–25%) has attracted significant attention. A biorefinery that provides low-value/high-volume liquid fuels and low-volume/high-value chemicals, in particular from lignocellulosic biomass, has been the subject of intense research efforts.1,2
Polyoxometalates (POMs) are a unique class of anionic polynuclear metal-oxo clusters with structural diversity at the atomic level, with their negative charge being balanced by countercations. In general, POMs are mainly classified into isopolyoxometalates [MxOy]n−, which feature only metal and oxygen atoms [M = typically high-valent (d0 or d1 electronic configuration) group V and VI transition metals (e.g., Mo, W, V)], and heteropolyoxometalates [XxMyOz]n−, containing additionally a heteroelement X (e.g., Si and P).3,4 Due to the tunable Brønsted/Lewis-acidity and redox properties, POMs and POM-based composites have long been applied in heterogeneous catalysis.3–5 In the field of catalytic biomass valorisation, POMs have been employed as environmentally benign acid or oxidation catalysts3,4 in the synthesis of renewable chemicals and fuels including platform chemicals,6–8 organic acids, furans,9–11 and biodiesels.12–14 Similarly, metal–POM composites have also been used for biomass conversion into chemicals and fuels.6,15,16
Many excellent reviews on biosourced chemicals and fuels have been published, devoted to specific feedstock such as carbohydrates,1,9,17 cellulose,8,18,19 lignin,20 C5–C6 sugars,21,22 and glycerol,23,24 focusing on specific reactions such as hydrolysis,7,8,25–27 dehydration,21,23 esterification, transesterification,12–14 oxidation,9,10 hydrogenolysis,24,28 and hydrodeoxygenation,29,30 or dealing with specific catalytic systems such as solid acids,8,14,25,31 metal catalysts,15 functionalised heterogeneous catalysts (e.g., carbon-based catalysts, metal–organic frameworks (MOFs), solid phase ionic liquid based catalysts, and magnetic iron oxide based catalysts).32 An updated and in-depth review on the theme of biomass valorisation over POM-based catalysts, to our knowledge, is not available. The aim of this review is to provide a detailed analysis integrating the state-of-the-art of the chemocatalytic conversion of biomass over POMs and metal–POM composites. While the focus is to show the wide applicability of POM-based catalysts in biorefinery, discussions on the relationship between the catalytic performance and the chemical/electronic/structural properties of the materials, as well as the reaction mechanisms, are also provided. For biomass valorisation over POMs, firstly the advances of acid catalysis including hydrolysis, dehydration, etherification, alcoholysis, transesterification and esterification are examined. Furthermore, applications in chemical oxidation for the synthesis of organic acids and furans are analysed. For biomass valorisation over metal–POM composites, an overview of tandem reactions (e.g. hydrolysis–hydrogenation, hydrolysis–oxidation, and hydrogenolysis–hydrodeoxygenation) is provided.
2 Properties and preparation of POM-based catalysts
Currently, most studies are focused on the Keggin and Wells–Dawson type of POM. The Keggin anion family is based on the [XM12O40]n− anion (X = Si, P, etc.; M = Mo, W, etc.), while the Wells–Dawson anion family bears the chemical formula of [X2M18O62]n−.4 POMs are versatile catalysts due to the multiple active sites, including protons as Brønsted acids, substituted metals with strong Lewis acidity, or metals with redox properties, and thus they are usually applied in acid-catalysed and oxidation reactions.3 POMs with protons as the only countercations are usually called heteropolyacids (HPAs). Phosphotungstic acid (PTA, H3PW12O40, HPW), silicotungstic acid (STA, H4SiW12O40, HSiW), phosphomolybdic acid (PMA, H3PMo12O40, HPMo) and silicomolybdic acid (SMA, H4SiMo12O40, HSiMo) with strong Brønsted acidity are widely applied in acid catalysis. As for oxidation catalysis, the reported POMs [XxMyOz]n− mainly contain P as the heteroelement X, and a combination of Mo and/or V as metal M.4,9,10POMs are normally soluble in both water and polar organic solvents; thus inorganic cations (e.g. Cs+, Ag+, etc.) are usually employed as counter-cations to enhance the insolubility of POMs. In addition to inorganic cations, organic compounds with designable and flexible functional groups (e.g., organic amines, quaternary ammonium ions, organic surfactants, and ionic liquid cations) have also been adopted for the solidification of POMs.3,33 Meanwhile, various heterogeneous porous supports including transition metal oxides, clays, carbons, zeolites, mesoporous silicas, polymers, and MOFs, as well as magnetic nanoparticles (NPs) have been adopted for the immobilization of POMs.3,33 A variety of methods such as impregnation, co-precipitation, deposition precipitation, equilibrium adsorption and ion exchange have been developed.3,5,12,33 Due to the increased thermal/hydrolytic/oxidative stability, these heterogeneous solidified/immobilised POMs have been widely applied as solid catalysts.3–5
Metal–POM composites, containing both the metal sites (Ru, Au, Pd, Pt, etc.) and POM sites, can be prepared by impregnation, encapsulation, physical mixing and other methods.3,15,16 These hybrid materials have also been applied in a variety of one-pot conversions of biomass feedstock in a tandem reaction sequence including the hydrolysis–hydrogenation, hydrolysis–oxidation, and hydrogenolysis–hydrodeoxygenation combinations, owing to the facilitated migration of the substrates/intermediates between the metal sites and POM sites. Since the preparation and characterization of POM-based catalysts have been covered in other excellent reviews,3,5,12,33 in this review, the emphasis is on the functionality of POM-based catalysts in biomass transformations.
3 Biomass valorisation into renewable chemicals and fuels over POMs
3.1 Acid catalysis
3.1.1.1 Monosaccharide and derivative synthesis. As the most abundant and valuable component of lignocellulosic biomass, cellulose is a linear biopolymer consisting of glucose units connected through β-1,4-glycosidic bonds. The hydrolysis of cellulose or raw lignocellulosic biomass by acid catalysts leads to the generation of oligosaccharides or monosaccharides such as glucose.7,8,26 Further dehydration results in the formation of hydroxymethylfurfural (HMF).21,22,34–37 HMF has been widely accepted as an important platform chemical included in the “Top 10 + 4” list revised on the “Top 10” list described by the US Department of Energy (DOE).38 The “Top 10 + 4” list also includes other critical platform chemicals such as furfural (FAL), glycerol, levulinic acid (LA), lactic acid, sorbitol, which will be discussed in this review.
So far, multiple POM-based catalysts have been reported for glucose synthesis.7,8,26 The representative POMs for fructose production are summarised in Table 1. Mizuno et al. found that highly negatively charged HPAs (e.g., H5BW12O40, H5AlW12O40, and H5GaW12O40), in particular H5BW12O40, displayed higher activity than other typical HPAs (e.g., HPW and HSiW) for the saccharification of polysaccharides, due to the high acidity and hydrogen-bonding accepting ability to decrease the crystallinity of polysaccharides (Table 1, entry 1).39 Wang et al. reported that a micellar HPA catalyst [C16H33N(CH3)3]H2PW12O40 displayed a remarkable performance in the hydrolysis of polysaccharides (e.g., cellulose and starch), which is attributed to the facilitated access to catalytic sites by polysaccharide molecules, and the enhanced reaction rate and improved mass transport in the micellar catalyst (Table 1, entries 3 and 4).40 Gedanken et al. prepared HSiW/graphene via a sonochemical method for glucose synthesis from glycogen hydrolysis. The hydrophobic cavities on the graphene surface facilitated the anchoring of glycogen and promoted the attack of glycosidic bonds via protons, which resulted in selective and efficient hydrolysis (Table 1, entry 5).41
| Entry | Substrate | Catalyst | Solvent | Temp. (°C) | Time (h) | Product | Conv.a (%) | Sel.b (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conv. = conversion. b Sel. = selectivity. c Continuous water removal. | ||||||||||
| 1 | Cellulose | H5BW12O40 | H2O | 60 | 48 | Glucose | N/A | N/A | 77 | 39 |
| 2 | Cellulose | Cs1H2PW12O40 | H2O | 160 | 6 | Glucose | N/A | N/A | 27 | 62 |
| 3 | Cellulose | [C16H33(CH3)3]H2PW12O40 | H2O | 170 | 8 | Glucose | 44 | 89 | 39 | 40 |
| 4 | Starch | [C16H33(CH3)3]H2PW12O40 | H2O | 120 | 5 | Glucose | 96 | 86 | 82 | 40 |
| 5 | Glycogen | HSiW/graphene | H2O | 150 | 4 | Glucose | N/A | N/A | 66 | 41 |
| 6 | Fructose | FePW12O40 | DMSO | 120 | 2 | HMF | 97 | 100 | 97c | 63 |
| 7 | Fructose | Cs2.5H0.5PW12O40 | MIBK![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (3:1) :H2O (3:1) |
115 | 1 | HMF | 78 | 95 | 74 | 64 |
| 8 | Fructose | Ag4SiW12O40 | H2O | 120 | 2 | HMF | 98 | 87 | 86 | 65 |
| 9 | Sucrose | Ag4SiW12O40 | H2O | 120 | 2.7 | HMF | 92 | 68 | 63 | 65 |
| 10 | Cellulose | Cr[(OSO3C12H25)H2PW12O40]3 | H2O | 150 | 2 | HMF | 77 | 68 | 53 | 42 |
| 11 | Corn stover | Cr[(OSO3C12H25)H2PW12O40]3 | H2O | 150 | 2 | HMF | 57 | 55 | 31 | 42 |
| 12 | Husk | Cr[(OSO3C12H25)H2PW12O40]3 | H2O | 150 | 2 | HMF | 63 | 57 | 36 | 42 |
| 13 | Fructose | [MIM-PS]3PW12O40 | sec-Butanol | 120 | 2 | HMF | 100 | 99 | 99 | 66 |
| 14 | Fructose | Cs[Cr3O(OOCC2H5)(H2O)3]2[SiW12O40] | H2O | 120 | 1 | HMF | 73 | 46 | 33 | 67 |
| 15 | Cellulose | (HOCH2CH2N(CH3)3)H2PW12O40 | MIBK:H2O (10:1) |
140 | 8 | HMF | 87 | 88 | 75 | 43 |
| 16 | Fructose | Cr[Cr3O(OOCCH2CN)6(H2O)3]3[PW12O40]2·69H2O | CH3OH:toluene (1:2) |
80 | 0.5 | HMF | 35 | 86 | 30 | 68 |
| 17 | Fructose | H3PW12O40/MIL-101 | [EMIM]Cl | 80 | 1 | HMF | 84 | 74 | 63 | 44 |
| 18 | Fructose | H3PW12O40/MCM-41 | DMSO | 120 | 1.3 | HMF | 100 | 80 | 80 | 45 |
| 19 | Cellulose | ChH4AlW12O40 | MIBK:H2O (10:1) |
120 | 10 | LA | 99 | 75 | 75 | 46 |
| 20 | Glucose | (C6H15O2N2)3−xHxPW12O40 | ChCl | 130 | 0.5 | LA | 75 | 71 | 53 | 51 |
| 21 | Cellulose | ChH4PW11TiO40 | MIBK:H2O (10:1) |
130 | 8 | LA | 94 | 81 | 76 | 52 |
A variety of POMs have been evaluated for HMF generation.21,22,34–37 In addition to monophasic systems such as water, organic solvents and ionic liquids, innovative biphasic aqueous/organic systems have also been employed, which efficiently remove the produced HMF from the reactive aqueous phase to the organic phase, thus minimising side reactions.21,34,37 The representative POMs for the hydrolysis of polysaccharides/monosaccharides in monophasic and biphasic systems are summarised in Table 1. Wang and co-workers reported a micellar Brønsted–Lewis-surfactant-combined HPA catalyst Cr[(DS)H2PW12O40]3 (DS represents dodecyl sulfate, OSO3C12H25) showing notable activity and stability for HMF synthesis from unpretreated lignocellulosic biomass (Table 1, entries 10–12). The enhanced performance is mainly attributed to the Brønsted and Lewis acidities, as well as the micellar structure with hydrophobic groups.42 The same authors prepared a series of (HOCH2CH2N(CH3)3)xH3−xPW12O40 (ChxH3−xPW12O40, x = 1, 2 and 3) via choline chloride and HPW, and the highest HMF yield of 75% was achieved over ChH2PW12O40 after 8 h at 140 °C (Table 1, entry 15).43 The hydrophilic head of ChxH3−xPW12O40 concentrates the cellulose substrate, while the hydrophobic tail inhibits further hydration of HMF to by-product LA, thus benefitting HMF production. POMs encapsulated in MOFs (e.g. MIL-10144) or immobilised on porous supports (e.g. MCM-4145) have also been adopted for HMF generation. For instance, Hensen et al. reported PTA/MIL-101–PTA encapsulated in MIL-101 as a composite catalyst to convert sugars. The system achieved the same HMF yield of 63% from fructose in [EMIM]Cl at 80 °C after 1 h (Table 1, entry 17), and in DMSO at 130 °C after 30 min.44
Furthermore, various POMs have been evaluated for the production of LA or levulinate ester from carbohydrates in consecutive reactions.46–61 Novel temperature-responsive HPA nanohybrids [(CH3)3NCH2CH2OH]nH5−nAlW12O40 (ChnH5−nAlW12O40)46 and ChH4PW11TiO40,52 HPA-based ionic liquid catalysts including [PyPS]3PW12O40 [PyPS = 1-(3-sulfopropyl)pyridinium]47 and [C4H6N2(CH2)3SO3H]3−nHnPW12O40,55 HPAs with Brønsted and Lewis acidic sites HnPW11MO39 (M = CuII, ZnII, CrIII, FeIII, SnIV, TiIV, and ZrIV; for Ti and Zr),48,49,52, combination of Zr-MCM-41 and HPW catalysts with tuneable Lewis and Brønsted acidity,50 as well as acid–base bifunctional HPA nanospheres (C6H15O2N2)3−xHxPW12O4051 were developed. Different reaction media including supercritical methanol, water–methanol mixtures,53 and water–methyl isobutyl ketone (MIBK)52,55 were also explored. For example, Wang and co-workers developed novel temperature-responsive ChnH5−nAlW12O40 catalysts, which dissolve at high temperature and precipitate from the reaction medium at room temperature. Among the tested catalysts, ChH4AlW12O40 exhibited the highest LA yield of 74.8% with a cellulose conversion of 98.9% in one pot, due to the synergistic effects of the temperature-stimulus, and dual Lewis and Brønsted acidity (Table 1, entry 19).46 As shown in Fig. 1, the Brønsted acid sites are proposed to facilitate hydrolysis, isomerization and dehydration/rehydration pathways, while Lewis acid sites are beneficial for isomerization of glucose to fructose.
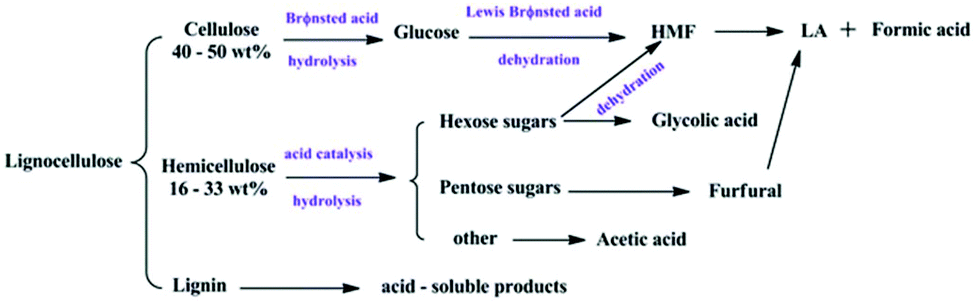 | ||
| Fig. 1 Conversion pathways of cellulose into levulinic acid.46 Reproduced from ref. 46 with permission from The Royal Society of Chemistry, copyright 2016. | ||
3.1.1.2 Acrolein synthesis. The significant surge in biodiesel production via transesterification of triglycerides with methanol is accompanied by a substantial increase of glycerol generation as a by-product (ca. 10 wt%). Acrolein synthesis via the dehydration of glycerol is a promising method for glycerol valorisation, as acrolein is an important intermediate for valued-added chemicals such as acrylic acid and amino-acid DL-methionine.23,69–72 POMs supported on oxides (e.g. titania,73 niobia,74,75 alumina,76,77 silica, alumosilicate,78 and zirconium dioxide77,79) and molecular sieves (MCM-41,80 amino siloxane-functionalised MCM-41,80,81 Zr-MCM-41,82,83 SBA-15,84 and ZSM-585) have been reported for acrolein synthesis from glycerol. For example, Yun et al. prepared the Keggin type H3PW12O40/MCM-41 and Wells–Dawson type H6P2W18O62/MCM-41 for the dehydration of glycerol to acrolein. The selectivity to acrolein increased with the increasing ratio of Brønsted to Lewis acids, while the lower ratio of Brønsted/Lewis acids favoured the production of hydroxyacetone as undesired side products.80
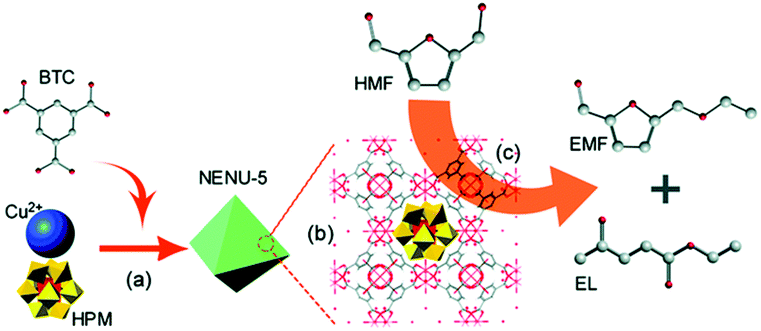 | ||
| Fig. 2 Schematic illustration of the synthesis and catalysis procedure. (a) Synthesis of the catalyst. (b) The crystal structure of the catalyst. (c) The procedure of conversion of HMF into EMF and EL.87 Reproduced from ref. 87 with permission from The Royal Society of Chemistry, copyright 2016. | ||
| Substrate | Catalyst | Solvent | Temp.a (°C) | Time (h) | Conv.b (%) | Sel.c (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Temp. = temperature. b Conv. = conversion. c Sel. = selectivity. | ||||||||
| HMF | H4SiW12O40/MCM-41 | Ethanol | 90 | 2 | 92 | 84 | 81 | 88 |
| HMF | [MIMBS]3PW12O40 | Ethanol | 70 | 24 | 98 | 93 | 91 | 89 |
| HMF | Fe3O4@SiO2-HPW | Ethanol | 100 | 11 | 98 | 86 | 84 | 90 |
| HMF | K-10 clay-HPW | Ethanol | 100 | 10 | 98 | 93 | 92 | 91 |
| HMF | [Cu-BTC][HPM] (NENU-5) | Ethanol | 140 | 12 | N/A | N/A | 68 | 87 |
| Fructose | H3PW12O40 | Ethanol:THF(5:3) |
130 | 0.5 | 98 | 77 | 76 | 92 |
| Fructose | [MIMBS]3PW12O40 | Ethanol | 90 | 24 | N/A | N/A | 91 | 89 |
| Fructose | Fe3O4@SiO2-HPW | Ethanol | 100 | 24 | N/A | N/A | 55 | 90 |
| Fructose | K-10 clay-HPW | Ethanol | 100 | 24 | 100 | 62 | 62 | 91 |
| Fructose | H3PW12O40 | Ethanol:DMSO(7:3) |
140 | 2 | 99 | 64 | 64 | 93 |
3.1.3.1 Biodiesel synthesis. Biodiesel is a non-toxic, biodegradable, environmentally friendly fuel alternative or additive of the petroleum-derived diesel. As a mixture of C12–C22 fatty acid monoalkyl esters (FAMEs), biodiesel is obtained by transesterification of the triacylglyceride (TAG) component and/or esterification of free fatty acids (FFAs) with C1–C2 alcohols in plant oils, animal fat or algae (Fig. 3).12–14,35,94 In contrast to “first-generation” biodiesel derived from edible feedstock (e.g., soybean oil), increasing research interest is focused on the “second-generation” biodiesel from inedible feedstock (e.g. jatropha oil and horn oil) or waste cooking/frying oils.13,35 The fatty acid chain is mainly classified into saturated and unsaturated chains with chain lengths of 16 and 18 carbons, including stearic, oleic, linolenic and palmitic acids.12 For biodiesel production, parameters such as acid strength, structural porosity and surface hydrophobicity exert substantial impact on the catalytic performance.12,14 The representative inorganic cation/organo-solidified POMs and POMs supported on metal oxides, clays, zeolites, mesoporous silica, magnetic materials, and MOFs for biodiesel production are summarised in Table 3.
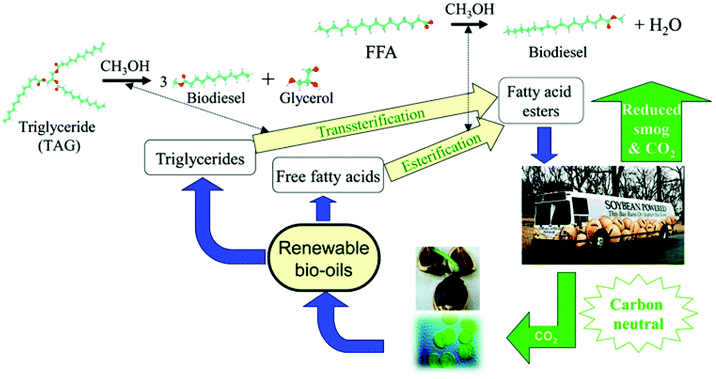 | ||
| Fig. 3 Biodiesel production from renewable bio-oils via transesterification and esterification.13 Reproduced from ref. 13 with permission from The Royal Society of Chemistry, copyright 2014. | ||
| Support | Catalyst | Feedstock | Cat. (wt%) | Alcohol/feedstock mole ratio | Temp.a (°C) | Time (h) | Conv.b/yieldc (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Temp. = temperature. b Conv. = conversion. c Yield. d Assisted by ultrasonic energy. e Microwave. | ||||||||
| Inorganic cation | Zn1.2H0.6PW12O40 | Waste cooking oil | 2.5 | 28 | 65 | 12 | 97b | 98 |
| Cs2.5H0.5PW12O40 | Yellow horn oile | 1 | 12 | 60 | 0.2 | 96c | 99 | |
| Cs1.5H1.5PW12O40 | Jatropha oil | 3 | 25 | 65 | 0.6 | 91c | 100 | |
| Organo-solidified | (C6H15O2N2)2HPW12O40 | Eruca sativa gars (ESG) oil | 9 | 9 | 65 | 12 | 91b | 101 |
| (NH2CH2COOH)H2PW12O40 | Palmitic acid | 6 | 12 | 90 | 3 | 93c | 102 | |
| Ionic liquid | [MIM-PSH]H2PW12O40 | Palmitic acid | 7 | 13 | 80 | 5 | 92c | 103 |
| Oxide | Ta2O5/SiO2-[H3PW12O40/R] (R = Me or Ph) | Soybean oil (20% myristic acid) | 2 | 90 | 65 | 24 | 90c | 104–106 |
| 20%H3PW12O40/ZrO2 | Oleic acid | 10 | 6 | 100 | 4 | 88b | 107 | |
| 25%H3PW12O40/Nb2O5 | Used cooking oil | 3 | 18 | 200 | 20 | 92c | 108 | |
| 15%TPA/SnO2 | Palmitic acid | 1 | 14 | 65 | 4 | 81b | 109 | |
| 25%TPA-Al2O3 | Jatropha oil d | 4 | 19 | 65 | 0.8 | 84c | 110 | |
| H3PW12O40-ZrO2-Si(Et)Si-60 | Eruca sativa gars oil | 5 | 90 | 65 | 24 | 75–99c | 111 | |
| H3PW12O40/ZrO2-Et-HNS-2.0 | Yellow horn oil | 5 | 90 | 65 | 24 | 63–76c | 95 | |
| Clay | 20%HPW/metakaolin flint | Oleic acid | 5 | 30 | 130 | 2 | 97c | 112 |
| 20%H3PW12O40/K10 | Oleic acid | 5 | 8 | 165 | 5 | 100b | 113 | |
| Carbon | H3PW12O40/ACF | Palmitic acid | 1.4 | 97 | 60 | 6 | 88b | 114 |
| 20%TPA-AC | Jatropha oild | 4 | 20 | 65 | 0.66 | 87c | 115 | |
| Zeolite | 30%H3PW12O40/Hβ | Oleic acid | 0.1 | 20 | 60 | 6 | 84b | 116 |
| 30% SiW12/Hβ | Oleic acid | 0.1 | 20 | 60 | 10 | 86b | 117 | |
| Mesoporous silica | 23%H3PW12O40/SBA-15 | Oleic acid | 0.3 | 40 | 40 | 4 | 90b | 118 |
| TSA3/SBA-15 | Waste cooking oil | 0.3 | 8 | 65 | 8 | 86b | 119 | |
| 45%TPA/SBA-15 | Canola oil | 3 | 26 | 200 | 6 | 97c | 120 | |
| TPA3/MCM-41 | Palmitic acid | 0.1 | 40 | 60 | 6 | 100b | 121 | |
| 30%H3PW12O40/MCM-41 | Lauric acid | 2 | 90 | 3 | 95c | 122 | ||
| 30%SiW11/MCM-41 | Oleic acid | 0.1 | 40 | 65 | 16 | 81b | 123 | |
| Magnetic NPs | HPW-PGMA-MNPs | Grease | 4 | 33 | 122 | 24 | 98c | 96 |
| MOF | ZIF-8/HPA | Rapeseed oil | 4 | 10 | 200 | 2 | 98b | 97 |
| AILs/HPW/UiO-66-2COOH | Soybean oil | 10 | 35 | 110 | 6 | 96b | 124 | |
Guo and co-workers prepared novel organic–inorganic hybrid catalysts, HPA and ZrO2 bifunctionalised organosilica hollow nanospheres (H3PW12O40/ZrO2-Et-HNS).95 H3PW12O40/ZrO2-Et-HNS: due to the strong Brønsted and Lewis acidity, unique hollow nanospherical morphology and hydrophobic surface, this material displayed encouraging activity and recyclability for transesterification of yellow horn oil with methanol to biodiesel, as well as esterification of levulinic acid (LA) with methanol to methyl levulinate (discussed in 3.1.3.2). The hollow nanospherical morphology enhanced the accessibility of guest molecules to the acid sites, shortened the diffusion pathway and enhanced the diffusion efficiency. The hydrophobic surface of the hybrid catalysts induced by the incorporation of bridging ethyl groups facilitated the enrichment of the hydrophobic reactant and expulsion of the hydrophilic products, thus accelerating the reaction (Fig. 4).
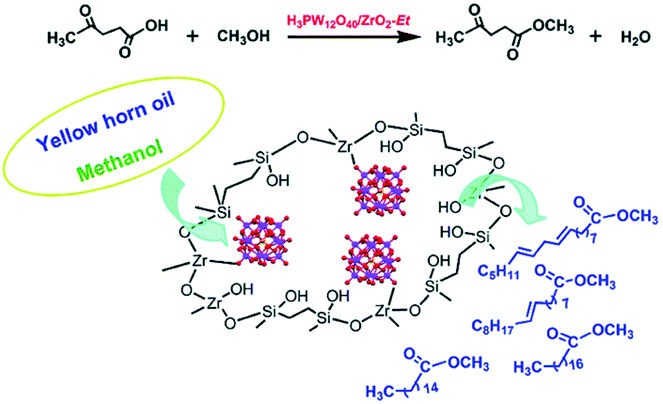 | ||
| Fig. 4 The shell composition of H3PW12O40/ZrO2-Et hollow nanospheres and the processes of levulinic acid esterification and yellow horn oil transesterification catalysed over H3PW12O40/ZrO2-Et hollow nanosphere hybrid catalysts.95 Reproduced from ref. 95 with permission from The Royal Society of Chemistry, copyright 2014. | ||
Li et al. prepared core–shell structured HPW-PGMA-MNPs composed of iron oxide magnetic NPs (MNP) as the core, poly(glycidyl methacrylate) (PGMA) as the shell, and HPW as the surface acid group. HPW-PGMA-MNPs exhibited remarkable activity and good recyclability for the one-pot transformation of waste grease to biodiesel via simultaneous esterification of FFAs and transesterification of TAG with methanol, as a result of the firm attachment of HPW on MNPs via covalent binding, the stable PGMA shell, and the superparamagnetic properties of the MNPs (Fig. 5).96
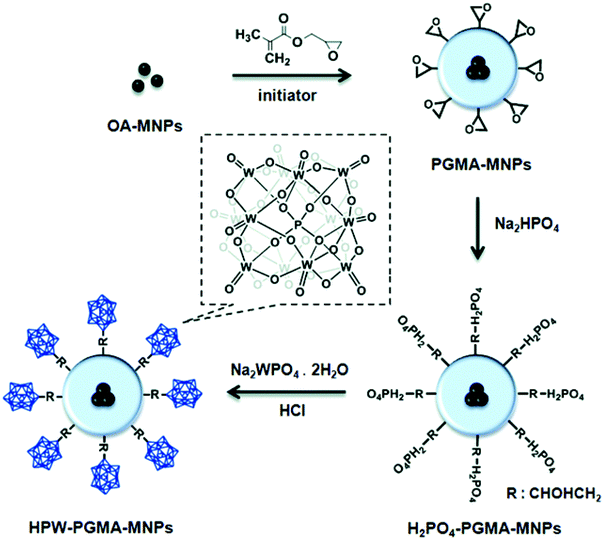 | ||
| Fig. 5 Synthesis of phosphotungstic acid-functionalised iron oxide particles (HPW-PGMA-MNPs) as a magnetic nano-size solid acid catalyst.96 Reproduced from ref. 96 with permission from The Royal Society of Chemistry, copyright 2014. | ||
Shul et al. prepared bifunctional core–shell HPA-functionalised zeolitic imidazolate framework-8 (ZIF-8) NPs via imidazolium medium. The strong O–N bonding indicated the strong interaction between the Keggin structure in HPA and imidazole units in ZIF-8 NPs. The optimised ZIF-8/HPA with both acidic and basic sites, and large surface areas displayed notable activity and recyclability for the transesterification of rapeseed oil with methanol to biodiesel (Fig. 6).97
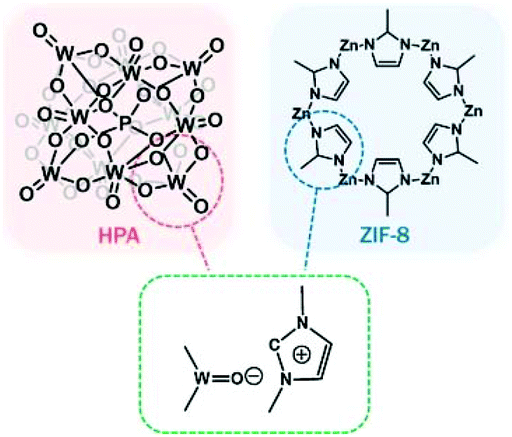 | ||
| Fig. 6 Illustration of the plausible chemical bonding structure for the hybrid ZIF-8/HPA bifunctional catalyst for the transesterification of rapeseed oil.97 Reproduced from ref. 97 with permission from Elsevier, copyright 2019. | ||
3.1.3.2 Synthesis of levulinate esters. Levulinate esters have potential applications in the fragrance and flavouring industry, as blending agents in biodiesel, etc. In particular, ethyl levulinate (EL) is a novel diesel miscible biofuel. Levulinate esters are generally obtained via esterification of levulinic acid (LA), one of the most important platform molecules in the “Top 10 + 4” list mentioned earlier. The typical POM-based catalysts for esterification of levulinic acid to alkyl levulinate are summarised in Table 4.
| Catalysts | Reaction conditionsa | Conv.b/yield (%) | Ref. |
|---|---|---|---|
| a Reaction conditions: Amount of catalyst wt%/molar ratio of alcohol to substrate/reaction temperature in °C/reaction time in h. b Conv. = conversion. | |||
| HPW/desilicated-ZSM-5 | 0.25 wt%/6/78/4 | 94b | 127 |
| H4SiW12O40–SiO2 | 0.1 g/50/65/6 | 73 | 128 |
| H3PW12O40/ZrO2-Si(Ph)Si | 2 wt%/7/65/3 | 99.9 | 129 |
| H3PW12O40/ZrO2-Si(Ph)Si-1.0 | 2 wt%/7/65/3 | 99.9 | 125 |
| H3PW12O40/ZrO2-Si(Et)Si-1.0 | 68 | ||
| H3PW12O40/ZrO2-Si(Ph)-1.0 | 72 | ||
| H3PW12O40/ZrO2-Et-HNS-2.0 | 2 wt%/7/65/1 | 92 | 95 |
| PW12/ZrO2-Si(Et)Si-NTs | 2 wt%/7/65/1.5 | 99.9 | 126 |
| [Cu-BTC][HPM] | N/A/N/A/120/6 | >99 | 130 |
Guo and co-workers prepared ZrO2-based organic–inorganic hybrid catalysts functionalised by Keggin-type HPA and hydrophobic alkyl groups (i.e., benzene-terminally bonded and ethane-/benzene-bridged organosilica moieties), H3PW12O40/ZrO2-Si(Et/Ph)Si and H3PW12O40/ZrO2-Si(Ph).125 H3PW12O40/ZrO2-Si(Et/Ph)Si and H3PW12O40/ZrO2-Si(Ph) exhibited excellent catalytic activity and stability for the esterification of LA. The authors attributed the superior catalytic performance to the strong Brønsted and Lewis acidity, unique textural properties and the hydrophobic surface that inhibit the strong adsorption of the hydrophilic by-products. In particular, the ordered 2D hexagonal mesostructured H3PW12O40/ZrO2-Si(Ph)Si exhibited higher activity than 3D wormhole-like H3PW12O40/ZrO2-Si(Et)Si and H3PW12O40/ZrO2-Si(Ph), due to the higher diffusion efficiency in the ordered mesoporous structure (Fig. 7). The same authors developed HPA and ZrO2 bifunctionalised organosilica nanohybrids PW12/ZrO2-Si(Et)Si with a 1D hollow tubular nanostructure, a 2D hexagonal periodic mesostructure and a 3D interconnected mesostructure.126 Among these, HPW and ZrO2-bifunctionalised organosilica nanotubes PW12/ZrO2-Si(Et)Si-NTs exhibited the highest activity for alkyl levulinates by LA esterification (Fig. 8).
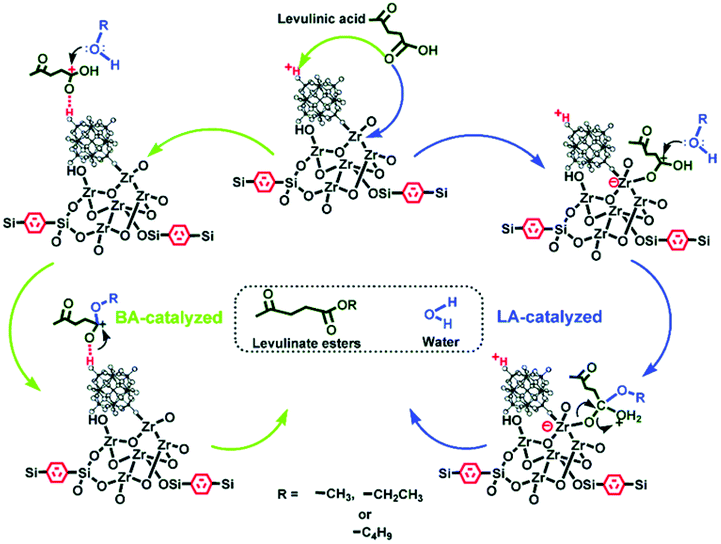 | ||
| Fig. 7 The process of esterification of LA with alcohol to produce levulinate ester over H3PW12O40/ZrO2-Si(Ph)Si hybrid materials. Left part: Brønsted acid site-catalysed reaction; right part: Lewis acid site-catalysed reaction.125 Reproduced from ref. 125 with permission from The Royal Society of Chemistry, copyright 2013. | ||
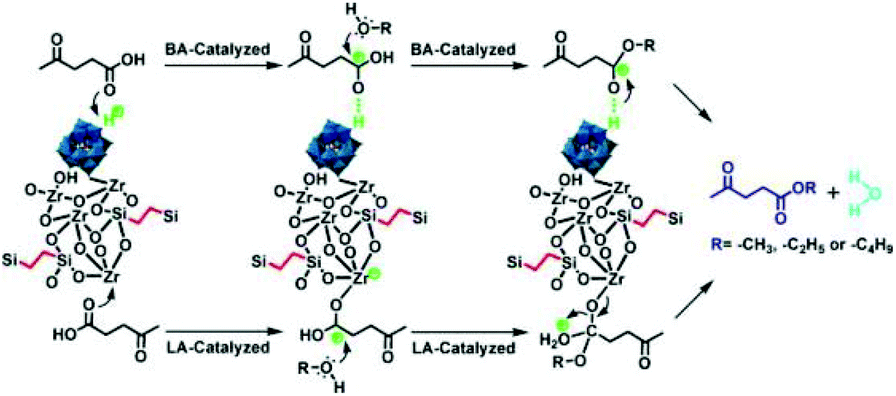
3.2 Oxidation
The carbohydrate feedstock has high oxygen content; thus oxidation into oxygen-containing chemicals or oxygen removal via dehydration, hydrogenolysis, or hydrodeoxygenation is a convenient pathway. The catalytic oxidation of carbohydrates and their derivatives provide a variety of value-added fine chemicals, including organic acids (e.g. formic acid, glycolic acid, gluconic acid, lactic acid, and acrylic acid),4,10,11 as well as furan chemicals (e.g. 2,5-diformylfuran).9,1313.2.1.1 Oxidation of carbohydrates to formic acid. Formic acid (FA) is a versatile chemical finding widespread applications. It is used as a H2 donor in hydrogenation reactions, as well as a hydrogen storage compound in the context of hydrogen economy. Research studies have been focused on sustainable FA generation from oxidation of monosaccharides, oligosaccharides, and bio-polymers such as cellulose, hemicellulose, lignin and even algae, under mild conditions with molecular oxygen or air as an oxidant.9–11,132,133 The oxidative conversion of cellulose includes the hydrolysis of highly polymerised cellulose into glucose over acid sites and the subsequent oxidative C–C bond cleavage of glucose into FA over redox sites, while overoxidation leads to the formation of CO2 which is the thermodynamically favoured product. Keggin-type phosphovanadomolybdates, H3+nPVnMo12−nO40 (HPA-n), with n = 0–6, are typical catalysts for the oxidative conversion of biomass to FA (Fig. 9). The representative H3+nPVnMo12−nO40 (HPA-n) catalysts for the oxidation of carbohydrates to formic acid are summarised in Table 5.
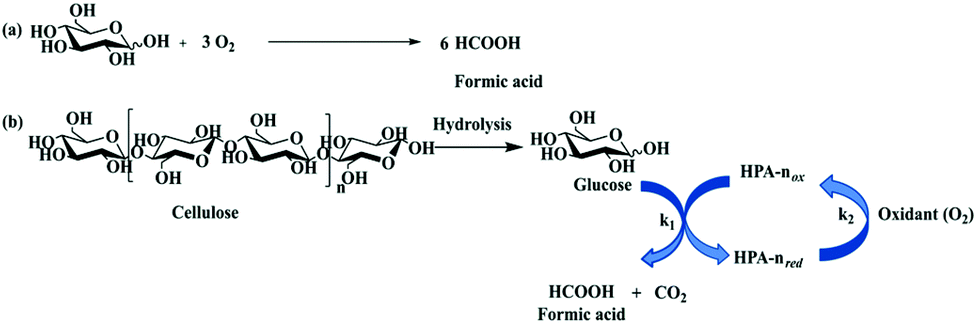 | ||
| Fig. 9 (a) Overall stoichiometric reaction for oxidation of glucose into formic acid; (b) oxidative conversion of carbohydrates (e.g. cellulose) to formic acid over H3+nPVnMo12−nO40 (HPA) catalysts. HPA-nox: The oxidised state of HPA and HPA-nred: the reduced state of HPA.9 Reproduced from ref. 9 with permission from The Royal Society of Chemistry, copyright 2018. | ||
| Entry | Substrate | Feed conc.a (mg mL−1) | Catalyst | Temp.b (°C) | O2 (MPa) | Time (h) | Conv.c (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
|
a Feed conc. = feed concentration.
b Temp. = temperature.
c Conv. = conversation.
d Water:1-hexanol (1:1).
|
|||||||||
| 1 | Glucose | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | >98 | 47 | 144 |
| 2 | Glucose | 25 | H5PV2Mo10O40 | 100 | 5 (air) | 3 | 100 | 52 | 136 |
| 3 | Glucose | 50 | H8PV5Mo7O40 | 90 | 3 | 8 | 100 | 60 | 135 |
| 4 | Glucose | 18 | H8PV5Mo7O40 | 90 | 2 | 48 | 100 | 85a | 141 |
| 5 | Cellobiose | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | >98 | 47 | 134 |
| 6 | Glucose | 72 | [MIMPS]3HPMo11VO40 | 180 | 1 | 1 | 100 | 55 | 140 |
| 7 | Sucrose | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | >98 | 48 | 134 |
| 8 | Sucrose | 34 | H8PV5Mo7O40 | 90 | 2 | 48 | 96 | 76a | 141 |
| 9 | Xylan | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | N/A | 33 | 134 |
| 10 | Xylan | 27 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 24 | 97 | 53 | 138 |
| 11 | Xylan | 50 | H8PV5Mo7O40 + p-TSA | 90 | 3 | 24 | 100 | 58 | 135 |
| 12 | Cellulose | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | N/A | 1 | 134 |
| 13 | Cellulose | 10 | H4PVMo11O40 | 180 | 0.6 | 3 | 100 | 68 | 144 |
| 14 | Cellulose | 10 | H5PV2Mo10O40 + HCl | 170 | 1 | 9 | 100 | 34 | 136 |
| 15 | Cellulose | 27 | H5PV2Mo10O40 + H2SO4 | 180 | 3 | 0.08 | 100 | 61 | 137 |
| 16 | Cellulose | 27 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 66 | N/A | 22 | 139 |
| 17 | Cellulose | 27 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 24 | N/A | 31 | 145 |
| 18 | Cellulose | 50 | H8PV5Mo7O40 + p-TSA | 90 | 3 | 24 | 76 | 28 | 135 |
| 19 | Cellulose | 67 | [MIMPS]3HPMo11VO40 | 180 | 1 | 1 | 93 | 51 | 140 |
| 20 | BM cellulose | 10 | Co0.6H3.8PMo10V2O40 | 160 | 2(O2 + N2) | 5 | N/A | 66 | 146 |
| 21 | Lignin | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | N/A | 14 | 134 |
| 22 | Lignin | 27 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 24 | N/A | 31–32 | 138 and 145 |
| 23 | Lignin | 50 | H8PV5Mo7O40 + p-TSA | 90 | 3 | 24 | 100 | 32 | 135 |
| 24 | Poplar sawdust | 30 | H5PV2Mo10O40 | 80 | 3 | 26 | N/A | 19 | 134 |
| 25 | Pomace | 33 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 24 | N/A | 55 | 139 |
| 26 | Cane trash | 33 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 24 | N/A | 49 | 139 |
| 27 | Beech wood | 27 | H5PV2Mo10O40 + p-TSA | 90 | 3 | 24 | N/A | 35 | 138 |
| 28 | Beech wood | 16 | H8PV5Mo7O40 + p-TSA | 90 | 2 | 48 | N/A | 61d | 141 and 145 |
| 29 | Bagasse | 10 | H4PVMo11O40 | 180 | 0.6 | 3 | 100 | 61 | 144 |
| 30 | Hay | 10 | H4PVMo11O40 | 180 | 2 | 3 | 100 | 55 | 144 |
Wasserscheid et al. firstly reported the oxidation of carbohydrate-based biomass to FA over H5PV2Mo10O40.134 An FA yield of around 50% with nearly full conversion was obtained from monosaccharides (glucose and xylose) and disaccharides (cellobiose and sucrose) at 80 °C and 3 MPa O2 after 26 h (Table 5, entry 1). However, water-insoluble cellulose only offered an FA yield of 1% under the same reaction conditions. Wasserscheid et al. further investigated the activity of H3+nPVnMo12−nO40 (n = 0–6) in selective oxidation of biomass to FA, and found that a higher degree of V-substitution enhanced the catalytic activity. The highest activity was obtained over HPA-5 (Table 5, entry 3),135 probably due to the formation of pervanadyl (VO2+) cations that are known to be a strong oxidative species.
The addition of inorganic or organic acids can further increase the FA yield due to the facilitated hydrolysis of cellulose. Fu et al. reported an FA yield of 34% with full cellulose conversion over HPA-2 combined with HCl at 170 °C and 1 MPa O2 after 9 h (Table 5, entry 14).136 Wu et al. reported an FA yield of 61% with 100% cellulose conversion over HPA-2 and H2SO4 at 180 °C after 5 min (Table 5, entry 15).137 Wasserscheid et al. obtained 22% FA from oxidative conversion of cellulose over HPA-2 and p-toluenesulfonic acid (TSA) at 90 °C and 3 MPa O2 after 66 h.138 The FA generation over a combination of HPA-2 and p-TSA can be expanded to a wide range of feedstocks including lignocellulose (e.g., xylan, lignin, beech wood, pomace, and cane trash), as well as the third-generation biomass (e.g., algae) (Table 5, entries 10, 16, 22, and 25–27).138,139 Similarly, high FA yields from cellulose, xylan, and lignin were obtained over HPA-5 with p-TSA as an additive (Table 5, entries 11, 18 and 23).135
The H+ cation in HPA-n can be exchanged by organic compounds and transition metal cations. Liu et al. prepared a series of heteropolyanion-based ILs with PMo11VO404− anions and –SO3H functionalised cations including –SO3H functionalised methylimidazole (MIMPS), –SO3H functionalised pyridinium (PyPS) and TEAPS.140 The –SO3H functionalised cations are responsible for cellulose hydrolysis to glucose, while the PMo11VO404− anions catalyze the glucose oxidation to FA. [MIMPS]3HPMo11VO40 provided the highest FA yield of 51% from cellulose at 180 °C and 1.0 MPa O2 (Table 5, entry 19).
Novel process technologies such as in situ extraction of FA are adopted to further increase FA yields.141 Albert et al. reported a water–organic biphasic system to effectively boost the FA selectivity compared with monophasic aqueous media.141 FA yields of up to 85% and 61% were obtained from glucose and beech wood over HPA-5 in the biphasic system (Table 5 entries 4, 8 and 28). The in situ extraction of FA with long-chain primary alcohols such as 1-hexanol and 1-heptanol prevented the decrease of pH of the aqueous phase, and resulted in a higher FA selectivity and yield.
In addition to Keggin-type POMs, Lindqvist-type isopolyoxometalates [VnW6−nO19]x− have also been applied in the biomass oxidation to FA. Albert et al. found that K5V3W3O19 selectively oxidised hemicellulose and lignin to FA, while being inactive for cellulose conversion.142,143
3.2.1.2 Oxidation of carbohydrates to glycolic acid. Glycolic acid (2-hydroxy acetic acid) is an important C2 α-hydroxy acid used as a precursor for the biodegradable polymer polyglycolic acid.147 The reaction pathway of carbohydrate conversion to glycolic acid is proposed in Fig. 10. Glucose from the hydrolysis of cellulose undergoes multiple retro-aldol reactions to glycolaldehyde, which is converted to glycolic acid through oxidation. Similarly, fructose from glucose isomerization also undergoes successive retro-aldol reactions and oxidation to provide glycolic acid and FA.148 Han et al. reported heteromolybdic acid H3PMo12O40 exhibiting high activity for one-pot conversion of α-cellulose into glycolic acid. A glycolic acid yield of 49% together with 10% FA were obtained at 180 °C and 0.6 MPa O2 after 1 h.148
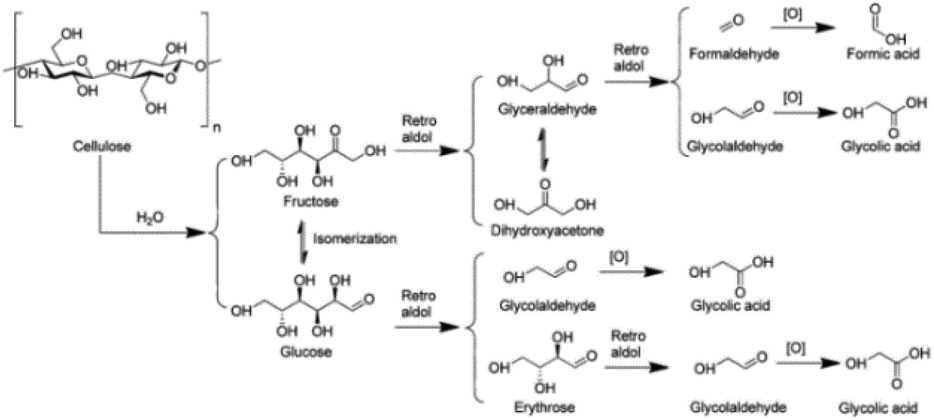 | ||
| Fig. 10 The proposed reaction pathway for the conversion of cellulose to glycolic acid.148 Reproduced from ref. 148 with permission from American Chemical Society, copyright 2012. | ||
3.2.1.3 Oxidation of glycerol to lactic acid. Lactic acid (2-hydroxypropionic acid), as one of the US DOE's top 10 carbohydrate-derived chemicals, has widespread applications in the food, pharmaceutical and chemical industries such as building blocks of biodegradable plastic polylactic acid.18 The reaction pathway of glycerol conversion to lactic acid is shown in Fig. 11. Glycerol undergoes dehydration/oxidation to form dihydroxyacetone (DHA) in equilibrium with glyceraldehyde (GCY). The pyruvaldehyde (PAL) obtained from the dehydration of DHA further undergoes a benzylic acid-type rearrangement to provide lactic acid.149 The oxidation of glycerol into lactic acid in water over representative POM-based catalysts is summarised in Table 6.
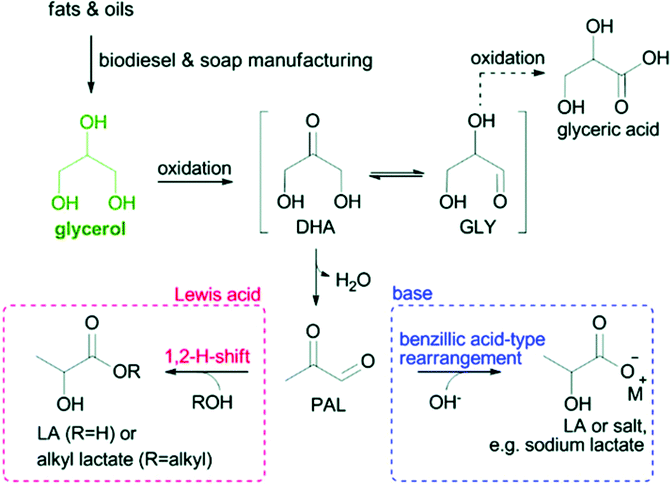 | ||
| Fig. 11 Catalytic conversion of glycerol to lactic acid or alkyl lactates in water or alcohol, respectively.18 Reproduced from ref. 18 with permission from The Royal Society of Chemistry, copyright 2017. | ||
Wang and co-workers reported H3PMo12O40 (HPMo) with optimal Brønsted acidic and redox properties exhibiting a lactic acid selectivity of 90% at 88% glycerol conversion. HPMo loaded on carbonised willow catkins (HPMo/C) achieved an even higher lactic acid selectivity of 94% at 98% glycerol conversion (Table 6).149 The preparation of HPMo/C catalysts is shown in Fig. 12, including (1) surface oxidation of the carbon support; (2) covalent binding of ethanediamine to the surface carboxylic groups; and (3) functionalization with 1-bromodecane and immobilization of HPMo into a lipid-like layer. The inclusion of HPMo in a lipid-like layer prevents HPMo leaching; thus the HPMo/C exhibits high catalyst stability.
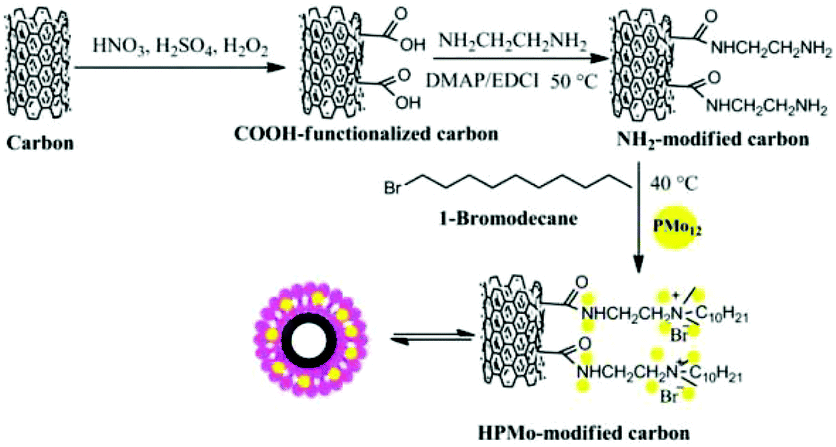 | ||
| Fig. 12 Synthetic progress of HPMo-modified carbon catalysts.149 Reproduced from ref. 149 with permission from WILEY, copyright 2015. | ||
In addition, novel HPMo@lipid(n)/GO catalysts were developed with HPMo embedded in lipid-like bilayers that are covalently bonded to graphene oxide (GO). The coupling of the surface carboxylic groups on functionalised graphene oxide (GO) with diamine provided a covalently linked GO-NH2 monolayer. Then the functionalisation of the GO-NH2 monolayer with 1-bromodecane, followed by embedding of HPMo into the lipid-like bilayer, provided HPMo@lipid(n)/GO hybrid materials (Fig. 13).150 The optimal HPMo@lipid(4)/GO catalyst featured balanced hydrophobic and hydrophilic properties, and the capillary-like microreactor configuration formed by the lipid-like bilayer structure. It achieved a high lactic acid yield of 90% with a glycerol conversion of 97% at 60 °C and 1 MPa O2 after 3.5 h (Table 6). The capillary reactor of HPMo@lipid(n)/GO enhanced the adsorption of glycerol and oxygen molecules, and concentrated them around HPMo, thus improving glycerol conversion. The oxidative ability of HPMo enhanced by GO and optimised by the length of the alkyl chain, combined with the balanced hydrophobicity of the lipid bilayer and the hydrophilicity of HPMo, led to high selectivity to lactic acid.
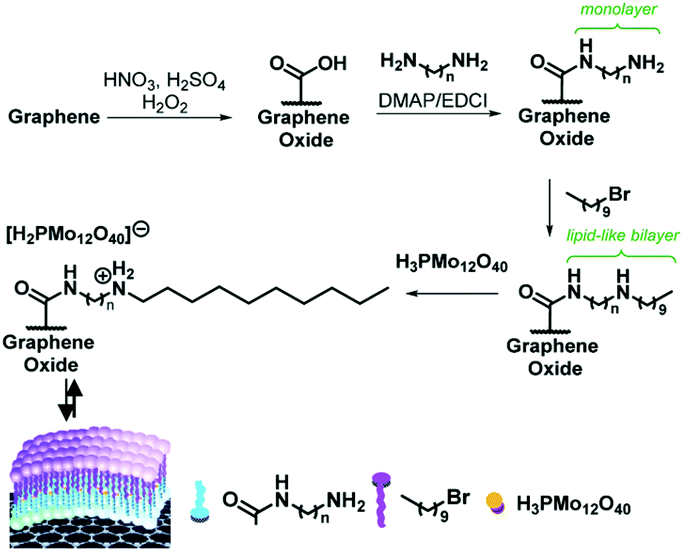 | ||
| Fig. 13 Synthetic strategy for the assembly of HPMo@lipid(n)/GO hybrid catalyst materials, where n represents the length of the diamine carbon chain (n = 2, 4, 6, 8, and 10).150 Reproduced from ref. 150 with permission from The Royal Society of Chemistry, copyright 2017. | ||
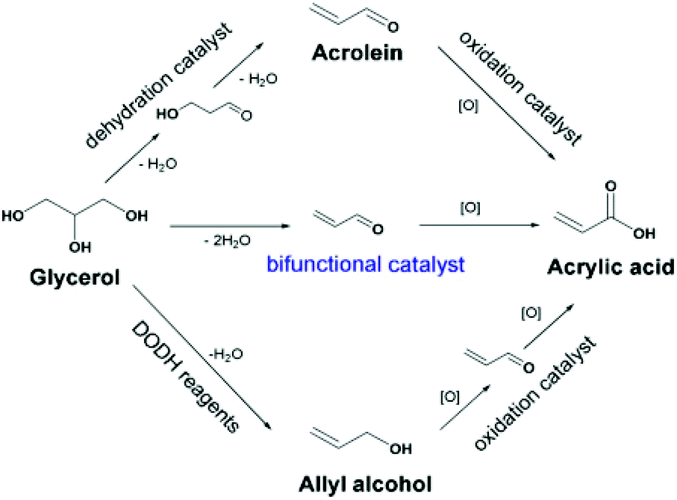
3.2.1.4 Dehydration–oxidation of glycerol to acrylic acid. Acrolein and acrylic acid (AA) are critical chemicals derived from glycerol. AA is generally obtained by a two-step tandem reaction including glycerol dehydration to acrolein over a solid acid and subsequent oxidation of acrolein over Mo- and V-based catalysts (Fig. 14). AA synthesis is also realised via a two-step tandem reaction via allyl alcohol as an intermediate. In this context, the direct oxidative dehydration of glycerol to acrylic acid over bifunctional catalysts is highly desired. Zhang et al. reported that the incorporation of vanadium species into CsPMo and CsPW promoted the formation of acrylic acid from glycerol conversion, due to the synergism between the Keggin anions and the cesium/vanadium species at the secondary structure. The combination of Cs(VO)0.2PMo and Cs(VO)0.2PW as solid solution catalysts changed the oxidation ability and surface acidity, and improved selectivity for acrylic acid. Consequently, H0.1Cs2.5(VO)0.2(PMo12O40)0.25(PW12O40)0.75 displayed the highest acrylic acid yield up to 60% and satisfactory resistance to coke deposition.153
4 Biomass valorisation into renewable chemicals and fuels over metal–POM composites
Metal NP–POM composites are referred to either metal NPs supported on heterogenised POMs or both metal NPs and POMs immobilised on an additional support. Such an intimate contact between the metal and POMs has the advantage of facilitated migration of the substrate/intermediates between the metal and acid sites at a nanometer distance. This section describes how metal–POMs can be used in catalytic biorefinery to achieve a one-pot tandem catalytic process. The use of physical mixtures of metal catalysts and POMs as catalysts is also included to provide readers a more comprehensive overview.4.1 Hydrolysis, hydrogenation and related reactions
Sels and co-workers reported the direct combination of H4SiW12O40/H3PW12O40159 or water-tolerant Cs3.5H0.5SiW12O40 (CsSiW)/Cs2.5H0.5PW12O40 (CsPW)160 with Ru/C to convert cellulose to hexitols. Palkovits et al. adopted physical mixtures of H4SiW12O40/H3PW12O40 and Ru/C for the transformation of cellulose and biomass feedstock spruce to sugar alcohols. High yields of C4–C6 sugar alcohols up to 81% and 65% were obtained at 160 °C starting from cellulose and spruce, respectively.163
Wang and co-workers reported Ru/Cs3PW12O40 catalysts for the transformation of cellobiose and cellulose into sorbitol.162 Ru/Cs3PW12O40 catalysts exhibited full cellobiose conversion, and a high sorbitol yield of 86% at 140 °C. It is proposed that the reversible Brønsted acid sites were generated on the Cs3PW12O40 surface by spillover H species from H2 dissociation on Ru NPs.162 However, the surface area and porosity of CsxH3−xPW12O40 are relatively low, leading to inefficient diffusion of the reactant and product.
Chen et al. encapsulated PTA H3PW12O40 inside the mesoporous cavities of MIL-100(Cr), and developed a Ru-PTA/MIL-100(Cr) catalyst to convert cellulose and cellobiose into sorbitol (Fig. 16).164 The ratio of the acid site density to the number of Ru surface atoms (nA/nRu) in the catalytic performance was analysed in detail. The optimum nA/nRu values between 8.84 and 12.90 led to the maximum conversion of cellulose and cellobiose into sorbitol. The Ru-PTA/MIL-100(Cr) catalysts with optimal acid/metal ratios exhibited high sorbitol yields of 97.1% and 57.9% at full cellobiose and cellulose conversion, respectively. However, the Ru-PTA/MIL-100(Cr) catalysts suffered from poor reusability, which might have resulted from the adsorption of insoluble substances such as oligomeric products on catalysts.
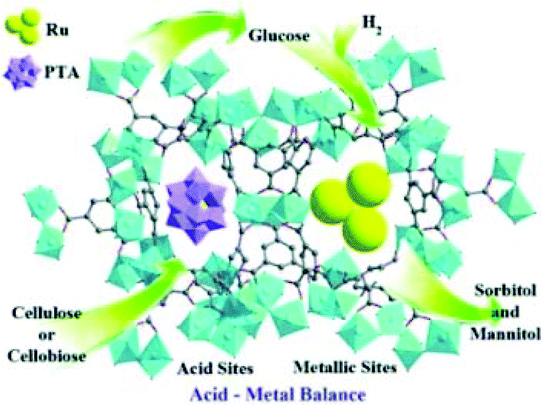 | ||
| Fig. 16 Metal–acid bifunctional catalyst Ru-PTA/MIL-100(Cr) for the conversion of cellulose and cellobiose into sorbitol.164 Reproduced from ref. 164 with permission from WILEY, copyright 2013. | ||
Isosorbide, a precursor of various biopolymers, pharmaceuticals and other chemicals, is obtained from the combined hydrolytic hydrogenation of cellulose to sorbitol and further dehydration of sorbitol.18 Sels and co-workers reported such an approach using a physical mixture of H4SiW12O40 and Ru/C. 52% isosorbide was obtained from purified microcrystalline cellulose, while 63% isosorbide was generated from crude wheat straw pulps obtained by organosolv fractionation.165
The hydrolysis of polysaccharides and subsequent hydrogenolysis with C–C and C–O cleavage can provide high value diols such as ethylene glycol (EG) and propylene glycol (PG).18 For instance, García-Bosch et al. reported a Ru-STA/AC catalyst to achieve a 1,2-PDO yield of 40% and selectivity up to 50% from fructose at 140 °C.166 Zhang and co-workers reported the combination of RANEY® nickel and W species for EG synthesis. The EG yield increased in the following order: H4SiW12O40 < H3PW12O40 < WO3 < H2WO4 (Fig. 17).167 The W species catalysed the cleavage of the C–C bonds in cellulose by a retro-aldol reaction pathway to generate glycolaldehyde, which undergoes further hydrogenation to form EG.167–170 Similarly, Guerrero-Ruiz et al. adopted Ru NPs supported on an HPA-carbon material [activated carbon (AC), high surface area graphite (HSAG)] for the hydrolytic hydrogenolysis of cellulose to alkanediols.171 Among the various POMs tested, Ru/AC-HPA and Ru/HSAG-HPA catalysts based on tungsten-based HPA (PTA and STA) exhibited better performance in terms of EG selectivity.
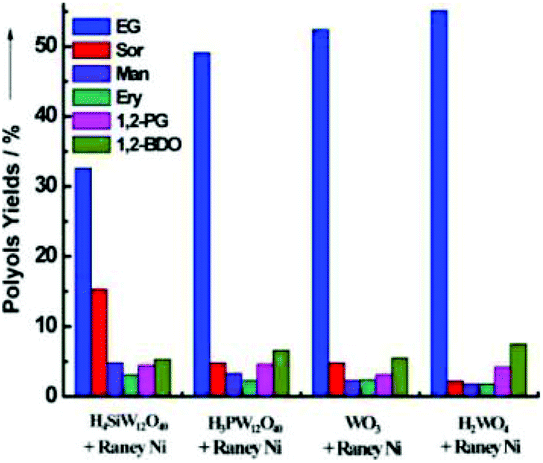 | ||
| Fig. 17 Catalytic performance of different binary catalysts composed of tungsten-based compounds with RANEY® Ni. The reactions were performed with cellulose (0.5 g), water (50 mL), RANEY® Ni (40–50 mg), and a W compound (50 mg) at 245 °C and 6 MPa H2 for 0.5 h.167 Reproduced from ref. 167 with permission from WILEY, copyright 2013. | ||
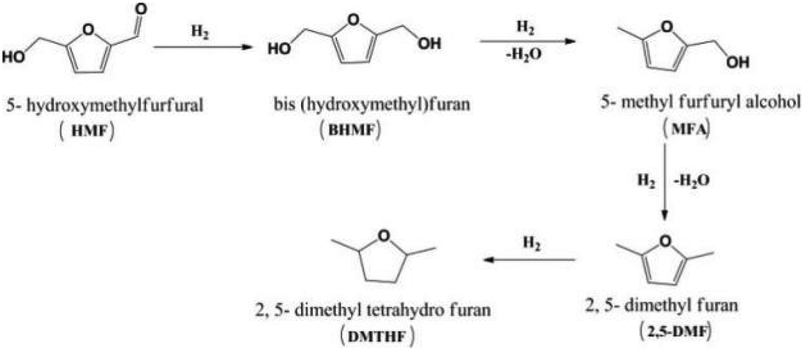 | ||
| Fig. 18 Selective hydrogenation of 5-hydroxymethylfurfural to 2,5-dimethyl furan.172 Reproduced from ref. 172 with permission from American Chemical Society, copyright 2016. | ||
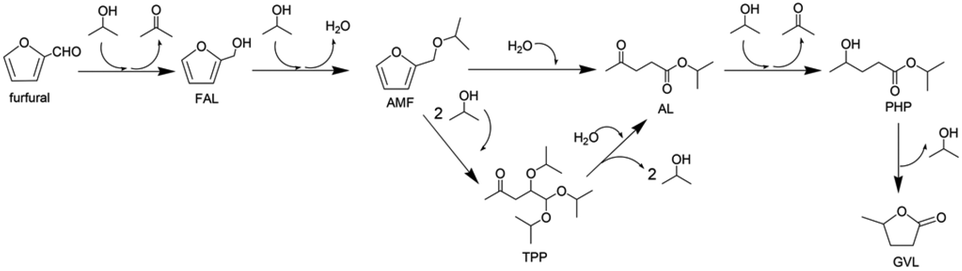
While HMF is a key platform chemical from cellulose, furfural is an important platform molecule derived from hemicellulose for the synthesis of value-added products including furfuryl alcohol (FAL) and alkyl levulinate (AL). Furfural hydrogenation into intermediate FAL and subsequent acid-catalysed alcoholysis of FAL provide AL as an important bio-based fuel additive and a precursor for γ-valerolactone (Fig. 19). The Au-HSiW/ZrO2 catalyst achieved a high AL yield of 80.2% with full furfural conversion at 100 °C.173 The Au surface sites catalysed the transfer hydrogenation reaction of furfural to FAL using 2-propanol as a H-donor, while the acidic HSiW sites catalysed the alcoholysis of FAL to yield AL. Among HSiW/HPW/HPMo impregnated, ZrO2 supported Au catalysts, a linear relationship between the acidity of Au-HPA/ZrO2 catalysts and AL yield was established.
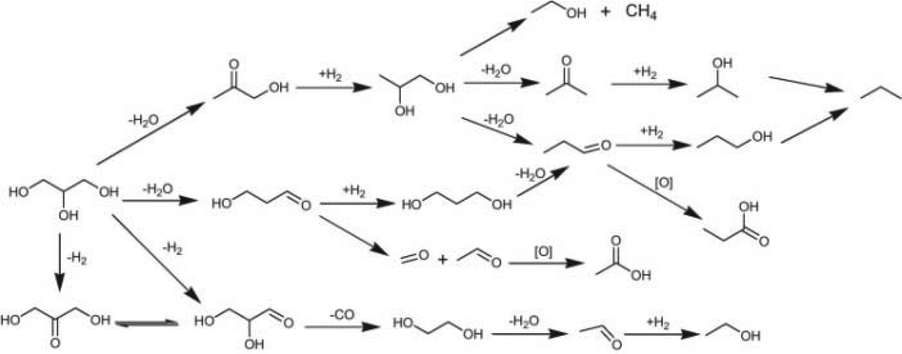 | ||
| Fig. 20 Reaction scheme of glycerol hydrogenolysis and degradation reactions.177 Reproduced from ref. 177 with permission from The Royal Society of Chemistry, copyright 2012. | ||
A series of alkaline metals, including Li, K, Rb and Cs, were used to modify the Pt-H4SiW12O40/ZrO2 catalyst. Li+ exhibited the best promotional effect in glycerol hydrogenolysis, providing 53.6% 1,3-PDO selectivity and 43.5% glycerol conversion at 180 °C, 5 MPa (Table 7).175 A linear relationship between 1,3-PDO yield and concentration of Brønsted acid sites was observed, indicating that the hydrogenolysis of glycerol to 1,3-PDO proceeded via dehydration of glycerol to 3-HPA on Brønsted acid sites followed by hydrogenation of the dehydration product 3-HPA on nearby metal sites. Furthermore, Pt-HSiW/ZrO2 exhibited superior performance in glycerol hydrogenolysis to 1,3-propanediol among HSiW, HPW and HPMo modified Pt/ZrO2 catalysts, due to the higher concentration of Brønsted acid sites.175 In parallel, a close correlation between the 1,2-PDO yield and the concentration of Lewis acid sites was obtained. Thus, the Brønsted acid sites are proposed to facilitate the selective generation of 1,3-PDO, while the Lewis acid sites favor the generation of 1,2-PDO.
| Catalysts | H2 (MPa) | Temp.a (°C) | Time (h) | WHSV (h−1) | Conv.b (%) | Sel.c (1,2-PD) | Sel.c (1,3-PD) | Sel.c (1-PrOH) | Sel.c (acrolein) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Temp. = temperature. b Conv. = conversion. c Sel. = selectivity. | ||||||||||
| 5 wt%Ru/C(3 wt%) + 15 wt%PTA/ZrO2 (6 wt%) | 6 | 180 | 8 | — | 44 | 64.3 | — | — | — | 174 |
| 1 wt%Pt-20 wt%Li2H2SiW/ZrO2 | 5 | 180 | — | 0.09 | 43.5 | 14.2 | 53.6 | 24.1 | — | 175 |
| 2 wt%Pt-15 wt%H4SiW12O40/ZrO2 | 5 | 180 | — | 0.09 | 24.1 | 16.5 | 48.1 | 21.8 | — | 177 |
| 2 wt%Pt-15 wt%H4SiW12O40/ZrO2 | 5 | 200 | — | 0.05 | 99.7 | 5.1 | 0.9 | 80.0 | — | 177 |
| 2 wt%Pt-15 wt%H3PW12O40/ZrO2 | 0.1 | 250 | — | 1.02 | 98.6 | — | — | 97 | — | 178 |
| 0.5 wt%Pd/Cs2.5H0.5PW12O40 | 0.1 | 275 | 5 | 2.80 | 79 | — | — | — | 96 | 182 |
| 1 wt%Pd-30 wt%H3PW12O40/C | 0.1 | 260 | 7 | 0.08 | 90 | — | — | — | 70 | 182 |
| 2 wt%Pd-30 wt%H3PW12O40/Zr-MCM-41 | 0.1 | 320 | 5 | 0.35 | 90 | — | — | — | 83 | 83 |
Zhu and co-workers reported that the Pt-H4SiW12O40/ZrO2 catalyst afforded a high 1-propanol yield of 80% and long-term stability of 160 h at 200 °C and 5 MPa H2 (Table 7).177 The Pt-H4SiW12O40/ZrO2 catalyst exhibited higher activity and propanol selectivity than its counterparts with Pd, Cu, and Ni as metal sites. ZrO2 interacts strongly with Pt sites, and prevents the crystallisation of Pt NPs in calcination at high temperatures. The small Pt particle size with high exposure of the Pt active species on the surface resulted in superior activity of Pt-H4SiW12O40/ZrO2. The Pt-H4SiW12O40/ZrO2 catalysts also displayed high resistance to the impurities in crude glycerol.
Acrolein, an important precursor for the production of adhesives, polymers, and detergents, can be obtained via the dehydration of glycerol over POMs/HPAs. H2 co-feeding and incorporation of platinum-group metals prevent coking and enhance the catalyst stability.73,181 Pd incorporation showed a larger stability enhancement effect than the incorporation of its Pt and Ru counterparts.83,182 For instance, Pd-H3PW12O40/Zr-MCM-41 exhibited high catalytic activity and stability, showing a slight decrease of glycerol conversion from 97% to 87% after 50 h.83
4.2 Oxidation of carbohydrates and derivatives
The hydrolysis of cellulose and cellobiose and further oxidation lead to the generation of gluconic acid, an important food additive and intermediate for the synthesis of fine chemicals and pharmaceuticals.6 For metal NP–POM nanocomposites, the acidic sites facilitate the hydrolysis of cellulose and cellobiose to glucose, while the metal (usually Au) NPs facilitate the oxidation of glucose into gluconic acid (Fig. 21). Han et al. reported a Au/Cs2HPW12O40 acidic/oxidative bifunctional catalyst, achieving a high gluconic acid yield of 96.4% with nearly complete cellobiose conversion.183 Cs2HPW12O40 was inert to the deep oxidation of gluconic acid, and passivated the oxidative activity of Au NPs; thus Au/Cs2HPW12O40 nanocomposites effectively prevented over-oxidation of gluconic acid. XPS analysis and DFT calculations demonstrated that the strong metal–support interfacial interaction effectively modulates the electronic structures of Au NPs, and subsequently the reactivity of the active oxygen species on the Au surface, thus resulting in the selective oxidation to gluconic acid. Several Au/CsxH3−xPW12O40 catalysts were developed for the same reaction. 97% gluconic acid was attained over Au/Cs1.2H1.8PW12O40, which bears stronger acidity and smaller Au NP size compared to other samples, at 145 °C for 3 h.184 Indeed, both the acidity of POMs and mean-size of the Au NPs are crucial parameters in the conversion of cellobiose into gluconic acid. CsxH3−xPW12O40 with stronger acidity not only facilitated the conversion of cellobiose, but also accelerated the desorption of gluconic acid and inhibited the consecutive degradation. The Au NPs with smaller size accelerated the oxidation of glucose, leading to high selectivity for gluconic acid.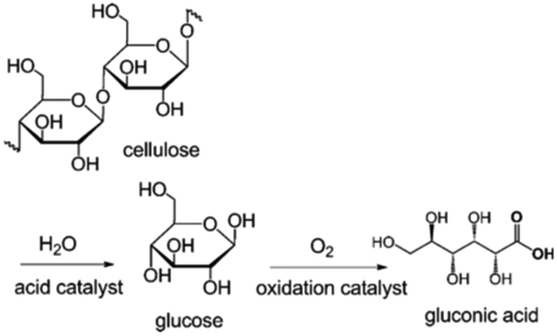 | ||
| Fig. 21 Direct conversion of cellulose into gluconic acid by using hydrolysis and oxidation reactions.184 Reproduced from ref. 184 with permission from WILEY, copyright 2012. | ||
4.3 Hydrodeoxygenation/deoxygenation of biomass-derived oxygenates
Due to the high oxygen content in biomass feedstock, hydrodeoxygenation (HDO) is an effective method to remove (part of) oxygen to convert biomass-derived oxygenates into drop-in bio-fuels and chemicals.20,29,30 The HDO process generally involves a combination of different reactions including hydrolysis, hydrogenation, hydrogenolysis, dehydration, decarboxylation and decarbonylation.29 Based on the structures of substrates, the HDO process can be mainly classified into the HDO of ketones,185,186 ethers,188 carboxylic acids,189 esters,188 angelica lactones,190 furanic compounds,191 carbohydrates,192 and aromatic compounds.186,188For the HDO of aliphatic and aromatic ketones, the reaction pathway involves the hydrogenation of ketones to a secondary alcohol on metal sites (mostly Pt) followed by dehydration of the alcohol to alkene on acid sites, and finally hydrogenation of the alkene to alkane on metal sites.185–187 Kozhevnikov et al. investigated the gas-phase deoxygenation of aromatic ether anisole, aliphatic diisopropyl ether (DPE), and aliphatic ester ethyl propanoate (EP) over bifunctional catalysts comprising Pt, Ru, Ni, and Cu as the metal components and Cs2.5H0.5PW12O40 as the acid component (Fig. 22).188 For the HDO of anisole, the model compound of lignin, consecutive hydrogenation of the aromatic ring, elimination of methanol from methoxylcyclohexane, and further hydrogenation to cyclohexane can be facilitated by bifunctional catalysts. 0.5%Pt/Cs2.5H0.5PW12O40 achieved a high cyclohexane selectivity of 89% with an anisole conversion of 87% under mild conditions (100 °C and 1 bar of H2), and the physical mixture of Pt/C and Cs2.5H0.5PW12O40 (0.35% Pt content) provided 100% yield of cyclohexane. The Pt/C + Cs2.5H0.5PW12O40 physical mixture displayed higher activity and resistance to deactivation than Pt/Cs2.5H0.5PW12O40. For the deoxygenation of DPE, the combined Pt/C + Cs2.5H0.5PW12O40 catalysts achieved an increased DPE conversion of 99% and a propane selectivity of 93%. Similarly, for the deoxygenation of EP, the Pt/Cs2.5H0.5PW12O40 catalyst under H2 showed much better stability compared with Cs2.5H0.5PW12O40, due to the decreased catalyst coking in the presence of Pt and H2.
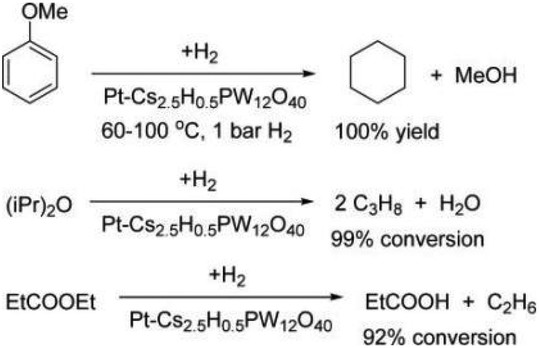 | ||
| Fig. 22 Deoxygenation of ethers and esters over a bifunctional Pt-HPA catalyst in the gas phase.188 Reproduced from ref. 188 with permission from American Chemical Society, copyright 2016. | ||
Recently, interest in the one-pot HDO of furan derivatives and carbohydrates to alkanes has considerably increased. Zhang et al. reported the first one-pot HDO of high carbon furans (HCFs) and δ-furfurylidenelevulinic acid (FDLA) to long-chain alkanes over Pd/C combined with HPW.191 The total yield of alkanes as high as 93.2% including an 89.5% yield of decane was obtained under relatively mild conditions (3 MPa H2, 170 °C, 4 h). The consecutive pathways include (a) the hydrogenation of unsaturated alkenyls, keto-carbonyls and furans of FDLA over metal sites, (b) the formation of a 5-(2-(tetrahydrofuran-2-yl)ethyl)-dihydrofuran-2(3H)-one (DC) intermediate via the intramolecular dehydration reaction, (c) the hydrogenolysis and HDO of furans and lactones over metal–acid sites, and (d) the generation of alkanes via HDO of aldehyde/alcohol via hydrogenation–dehydration–hydrogenation steps (Fig. 23).
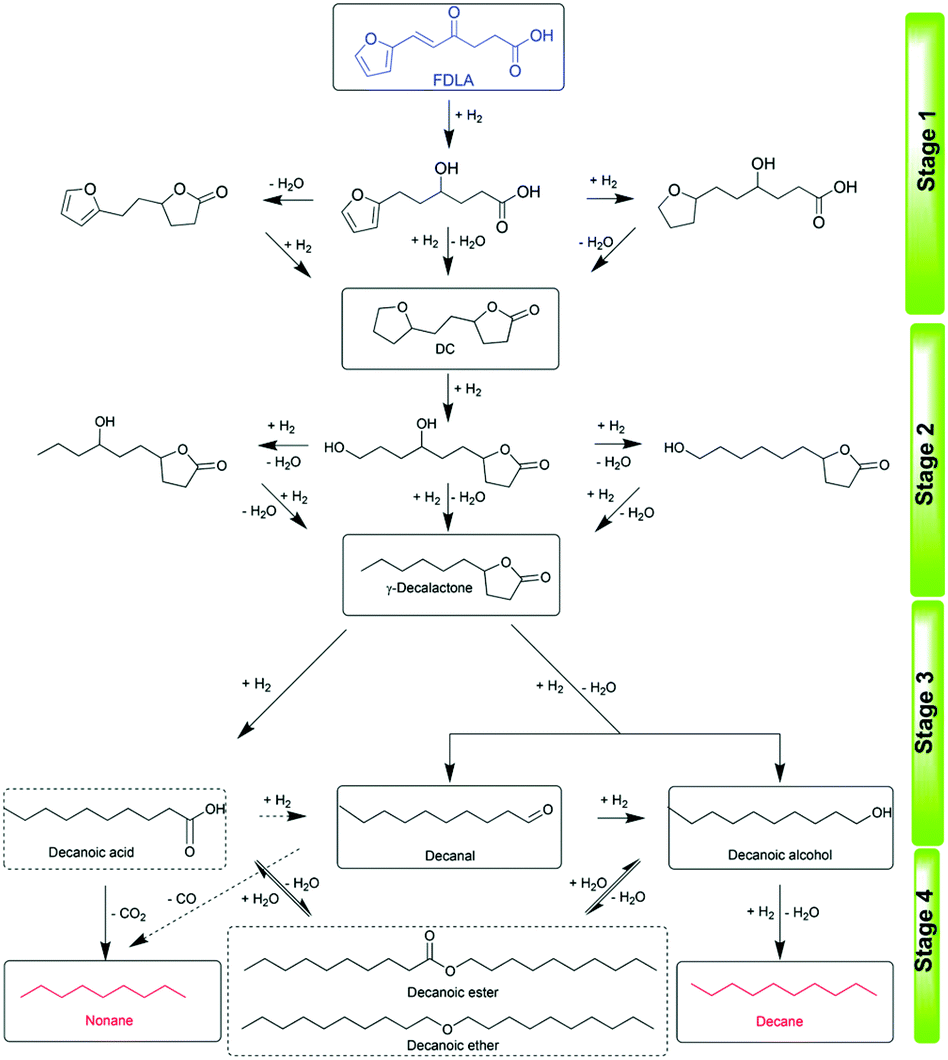 | ||
| Fig. 23 Proposed reaction pathway for the HDO of FDLA.191 Reproduced from ref. 191 with permission from The Royal Society of Chemistry, copyright 2020. | ||
One-pot conversion of cellulose to n-hexane has also been studied. Sels and co-workers reported the combination of tungstosilicic acid (TSA) and hydrothermally TSA-treated Ru/C (ht-STA-Ru/C) catalysts to convert microcrystalline cellulose into 52% n-hexane in a biphasic decane/water system.192 The direct conversion of cellulose to straight-chain alkanes mainly proceeded via HMF rather than sorbitol as an intermediate. This is in contrast to the widely accepted pathway, in which hydrolytic hydrogenation of cellulose to sorbitol and consecutive HDO to n-hexane is proposed. The kinetic study indicated that the hydrothermal treatment of Ru/C with TSA effectively enhanced the selectivity towards HMF hydrogenation and suppressed the glucose hydrogenation ability. The cellulose hydrolysis to glucose, dehydration of glucose to HMF, ring opening hydrolysis of 2,5-dimethylfuran (DMF) to 2,5-hexanedione, as well as ring-opening hydrolysis of 2,5-dimethyltetrahydrofuran (2,5-DMTHF) to 2,5-hexanediol were favoured over TSA in the aqueous phase, while the hydrogenation of the intermediates to the straight-chain alkanes was facilitated over ht-STA-Ru/C (Fig. 24).
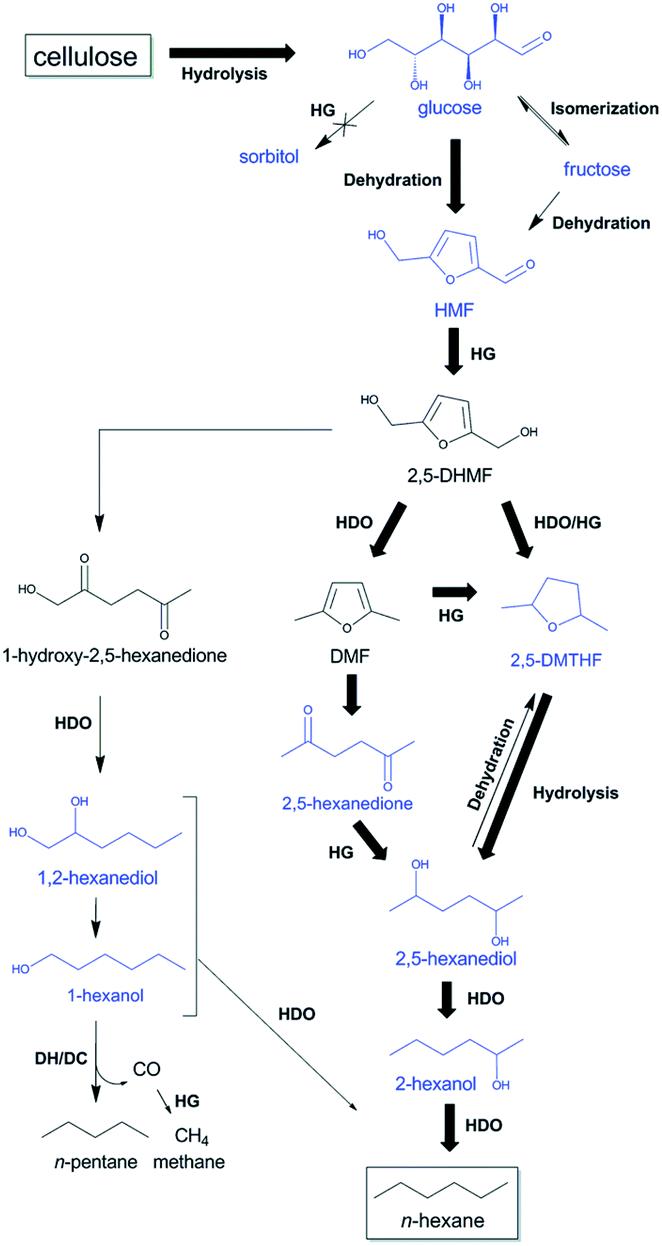 | ||
| Fig. 24 Proposed reaction pathways from cellulose to n-hexane and n-pentane through HMF with TSA and htTSA-Ru/C. The most selective reaction pathway from cellulose to n-hexane is indicated with bold arrows. HDO, hydrodeoxygenation; HG, hydrogenation; and DH/DC, dehydrogenation/decarbonylation. Reproduced from ref. 192 with permission from The Royal Society of Chemistry, copyright 2015. | ||
5 Conclusions and outlook
Recent progress in the chemocatalytic conversion of biomass into chemicals and fuels over POMs and POM–metal composites are summarised. For biomass valorisation over POMs, the advances of acid catalysis and chemical oxidation are discussed. For biomass valorisation over metal–POM composites, an overview of the types of tandem reactions enabled by a metal–POM combination is presented. Despite these encouraging developments, there are still considerable challenges/opportunities ahead for biomass valorisation using POM-based catalysts.As shown in this review, a variety of methods have been adopted to transform POMs into insoluble solids or immobilize POMs on porous supports so that they can be used as recyclable heterogeneous catalysts. However, leaching still seems to be a common issue, thus compromising the catalyst stability. Future efforts should be directed to the detailed study of the POM–support interaction, and the kinetics and thermodynamics of POM adsorption/desorption on various support materials. Furthermore, new combinations of POMs anchored on emerging porous materials, such as nitrides, carbides, covalent organic frameworks (COFs), and porous organic frameworks (POFs), may be studied.
In addition, most of the reported POMs for biomass utilisation are focused on the Keggin and Wells–Dawson-type POMs, and there is still unexplored potential for application of POMs with other structures (e.g., Lindqvist-type). Due to the tunable Brønsted/Lewis-acidity and redox properties, the POMs are generally applied in acid catalysis and chemical oxidation for biomass valorisation, while POM–metal composites are widely applied in the related tandem reactions. Beyond these, attention can be paid to explore other types of reactions in biomass valorisation over POM-based catalysts. For instance, it is envisaged that tungsten-containing POMs are active for retro-aldol condensation to break C–C linkages in carbohydrates. Metal–POM composites should also be effective in making organonitrogen chemicals, which appears to be a new direction in biomass conversion.193–195
POMs have been proven as effective supports to stabilize a range of single atom catalysts (SACs),196,197 exhibiting excellent performance in CO oxidation,198 selective hydrogenation,199,200 and Suzuki–Miyaura coupling reaction.201 Compared to their NP counterparts, SAC-POMs have maximal atom utilization, together with strong and tuneable interactions between single metal atoms and POMs. We put forward SAC-POMs to be potentially efficient catalytic systems to enable both oxidative (e.g., conversion of the –OH group into the –COOH group) and reductive (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation and hydrodeoxygenation) transformations of biomass to value-added fine chemicals under mild conditions.
O hydrogenation and hydrodeoxygenation) transformations of biomass to value-added fine chemicals under mild conditions.
Finally, a deep understanding of the relationship between the catalytic performance and the chemical/electronic/structural properties of the POM-based catalysts will be of significance for biomass utilisation. It is also of high interest to develop a new generation POM-based catalytic system for biomass valorisation in the future. We hope that this review has highlighted the usefulness of POMs in catalytic biorefinery, and will encourage more researchers to delve into this exciting field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National University of Singapore Flagship Green Energy Program (R-279-000-553-646 and R-279-000-553-731) for financial support.Notes and references
- L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2018, 118, 505–613 CrossRef CAS.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS.
- I. A. Weinstock, R. E. Schreiber and R. Neumann, Chem. Rev., 2018, 118, 2680–2717 CrossRef CAS.
- A. Enferadi-Kerenkan, T. O. Do and S. Kaliaguine, Catal. Sci. Technol., 2018, 8, 2257–2284 RSC.
- W. P. Deng, Q. H. Zhang and Y. Wang, Dalton Trans., 2012, 41, 9817–9831 RSC.
- J. J. Wang, J. X. Xi and Y. Q. Wang, Green Chem., 2015, 17, 737–751 RSC.
- L. Hu, L. Lin, Z. Wu, S. Y. Zhou and S. J. Liu, Appl. Catal., B, 2015, 174, 225–243 CrossRef.
- Z. H. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- M. Wang, J. P. Ma, H. F. Liu, N. C. Luo, Z. T. Zhao and F. Wang, ACS Catal., 2018, 8, 2129–2165 CrossRef CAS.
- D. A. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS.
- N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89–107 RSC.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- F. Su and Y. H. Guo, Green Chem., 2014, 16, 2934–2957 RSC.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS.
- H. Li, Z. Fang, R. L. Smith and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- R. De Clercq, M. Dusselier and B. F. Sels, Green Chem., 2017, 19, 5012–5040 RSC.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- T. F. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- X. G. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS.
- B. Katryniok, S. Paul and F. Dumeignil, ACS Catal., 2013, 3, 1819–1834 CrossRef CAS.
- D. Sun, Y. Yamada, S. Sato and W. Ueda, Appl. Catal., B, 2016, 193, 75–92 CrossRef CAS.
- F. Guo, Z. Fang, C. C. Xu and R. L. Smith, Jr., Prog. Energy Combust. Sci., 2012, 38, 672–690 CrossRef CAS.
- Y. B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- L. Vilcocq, P. C. Castilho, F. Carvalheiro and L. C. Duarte, ChemSusChem, 2014, 7, 1010–1019 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS.
- Y. Nakagawa, S. Liu, M. Tamura and K. Tomishige, ChemSusChem, 2015, 8, 1114–1132 CrossRef CAS.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- K.-i. Shimizu and A. Satsuma, Energy Environ. Sci., 2011, 4, 3140–3153 RSC.
- P. Sudarsanam, R. Zhong, S. Van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- Y. Zhou, G. J. Chen, Z. Y. Long and J. Wang, RSC Adv., 2014, 4, 42092–42113 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- S. S. Chen, T. Maneerung, D. C. W. Tsang, Y. S. Ok and C.-H. Wang, Chem. Eng. J., 2017, 328, 246–273 CrossRef CAS.
- B. Agarwal, K. Kailasam, R. S. Sangwan and S. Elumalai, Renewable Sustainable Energy Rev., 2018, 82, 2408–2425 CrossRef CAS.
- C. Xu, E. Paone, D. Rodriguez-Padron, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- Y. Ogasawara, S. Itagaki, K. Yamaguchi and N. Mizuno, ChemSusChem, 2011, 4, 519–525 CrossRef CAS.
- M. X. Cheng, T. Shi, H. Y. Guan, S. T. Wang, X. H. Wang and Z. J. Jiang, Appl. Catal., B, 2011, 107, 104–109 CrossRef CAS.
- M. Klein, A. Varvak, E. Segal, B. Markovsky, I. N. Pulidindi, N. Perkas and A. Gedanken, Green Chem., 2015, 17, 2418–2425 RSC.
- S. Zhao, M. X. Cheng, J. Z. Li, J. Tian and X. H. Wang, Chem. Commun., 2011, 47, 2176–2178 RSC.
- X. Y. Zhang, D. Zhang, Z. Sun, L. F. Xue, X. H. Wang and Z. Jiang, Appl. Catal., B, 2016, 196, 50–56 CrossRef CAS.
- Y. M. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS.
- F. N. D. C. Gomes, F. M. T. Mendes and M. M. V. M. Souza, Catal. Today, 2017, 279, 296–304 CrossRef CAS.
- Z. Sun, L. F. Xue, S. T. Wang, X. H. Wang and J. Y. Shi, Green Chem., 2016, 18, 742–752 RSC.
- C. H. Song, S. J. Liu, X. W. Peng, J. X. Long, W. Y. Lou and X. H. Li, ChemSusChem, 2016, 9, 3307–3316 CrossRef CAS.
- X. Y. Zhang, Y. Li, L. F. Xue, S. T. Wang, X. H. Wang and Z. J. Jiang, ACS Sustainable Chem. Eng., 2018, 6, 165–176 CrossRef CAS.
- B. S. Rao, P. K. Kumari, D. D. Lakshmi and N. Lingaiah, Catal. Today, 2018, 309, 269–275 CrossRef.
- M. M. Wang, L. C. Peng, X. Y. Gao, L. He and J. H. Zhang, Sustainable Energy Fuels, 2020, 4, 1383–1395 RSC.
- Z. Sun, S. T. Wang, X. H. Wang and Z. J. Jiang, Fuel, 2016, 164, 262–266 CrossRef CAS.
- X. Y. Zhang, X. Y. Zhang, N. Y. Sun, S. T. Wang, X. H. Wang and Z. J. Jiang, Renewable Energy, 2019, 141, 802–813 CrossRef CAS.
- F. Rataboul and N. Essayem, Ind. Eng. Chem. Res., 2011, 50, 799–805 CrossRef CAS.
- S. Q. Zhao, G. Z. Xu, C. Chang, S. Q. Fang, Z. Liu and F. G. Du, Catalysts, 2015, 5, 1897–1910 CrossRef CAS.
- Z. Sun, M. X. Cheng, H. C. Li, T. Shi, M. J. Yuan, X. H. Wang and Z. J. Jiang, RSC Adv., 2012, 2, 9058–9065 RSC.
- S. Q. Zhou, X. M. Yang, Y. L. Zhang, L. Y. Jiang, L. P. Zhou, T. L. Lu and Y. L. Su, Cellulose, 2019, 26, 9135–9147 CrossRef CAS.
- Y. X. Zhang, X. L. Chen, X. Lyu, G. L. Zhao, T. T. Zhao, L. J. Han and W. H. Xiao, J. Cleaner Prod., 2019, 215, 712–720 CrossRef CAS.
- P. F. Pinheiro, D. M. Chaves and M. J. da Silva, Cellulose, 2019, 26, 7953–7969 CrossRef CAS.
- X. Y. Li, X. B. Lu, S. X. Nie, M. Liang, Z. H. Yu, B. Y. Duan, J. X. Yang, R. Xu, L. F. Lu and C. L. Si, Ind. Crops Prod., 2020, 145, 112154 CrossRef CAS.
- D. Gupta, C. Mukesh and K. K. Pant, RSC Adv., 2020, 10, 705–718 RSC.
- X. Y. Zhang, H. Z. Zhang, Y. M. Li, M. Bawa, S. T. Wang, X. H. Wang and Z. J. Jiang, Cellulose, 2018, 25, 6405–6419 CrossRef CAS.
- J. Tian, C. Y. Fang, M. X. Cheng and X. H. Wang, Chem. Eng. Technol., 2011, 34, 482–486 CrossRef CAS.
- K.-i. Shimizu, R. Uozumi and A. Satsuma, Catal. Commun., 2009, 10, 1849–1853 CrossRef CAS.
- Q. Zhao, L. Wang, S. Zhao, X. Wang and S. Wang, Fuel, 2011, 90, 2289–2293 CrossRef CAS.
- A. H. Jadhav, H. Kim and I. T. Hwang, Bioresour. Technol., 2013, 132, 342–350 CrossRef CAS.
- Y. Qu, C. Huang, J. Zhang and B. Chen, Bioresour. Technol., 2012, 106, 170–172 CrossRef CAS.
- X. Yi, I. Delidovich, Z. Sun, S. Wang, X. Wang and R. Palkovits, Catal. Sci. Technol., 2015, 5, 2496–2502 RSC.
- T. Yamada, K. Kamata, E. Hayashi, M. Hara and S. Uchida, ChemCatChem, 2019, 11, 3743–3747 Search PubMed.
- B. Katryniok, S. Paul, M. Capron and F. Dumeignil, ChemSusChem, 2009, 2, 719–730 CrossRef CAS.
- B. Katryniok, S. Paul, V. Bellière-Baca, P. Rey and F. Dumeignil, Green Chem., 2010, 12, 2079 RSC.
- L. Liu, X. P. Ye and J. J. Bozell, ChemSusChem, 2012, 5, 1162–1180 CrossRef CAS.
- A. Talebian-Kiakalaieh, N. A. S. Amin and H. Hezaveh, Renewable Sustainable Energy Rev., 2014, 40, 28–59 CrossRef CAS.
- L. Q. Shen, Y. H. Feng, H. B. Yin, A. L. Wang, L. B. Yu, T. S. Jiang, Y. T. Shen and Z. N. Wu, J. Ind. Eng. Chem., 2011, 17, 484–492 CrossRef CAS.
- C. Liu, R. Liu and T. F. Wang, Can. J. Chem. Eng., 2015, 93, 2177–2183 CrossRef CAS.
- R. Liu, T. F. Wang, C. Liu and Y. Jin, Chin. J. Catal., 2013, 34, 2174–2182 CrossRef CAS.
- M. H. Haider, N. F. Dummer, D. Z. Zhang, P. Miedziak, T. E. Davies, S. H. Taylor, D. J. Willock, D. W. Knight, D. Chadwick and G. J. Hutchings, J. Catal., 2012, 286, 206–213 CrossRef CAS.
- A. Talebian-Kiakalaieh, N. A. S. Amin and Z. Y. Zakaria, J. Ind. Eng. Chem., 2016, 34, 300–312 CrossRef CAS.
- H. Atia, U. Armbruster and A. Martin, Appl. Catal., A, 2011, 393, 331–339 CrossRef CAS.
- A. Talebian-Kiakalaieh and N. A. S. Amin, Chin. J. Catal., 2017, 38, 1697–1710 CrossRef CAS.
- T. L. Ma, J. F. Ding, R. Shao, W. Xu and Z. Yun, Chem. Eng. J., 2017, 316, 797–806 CrossRef CAS.
- J. F. Ding, T. L. Ma, R. Shao, W. Xu, P. F. Wang, X. Y. Song, R. F. Guan, K. L. Yeung and W. Han, New J. Chem., 2018, 42, 14271–14280 RSC.
- J. F. Ding, T. L. Ma, M. Y. Cui, R. Shao, R. F. Guan and P. F. Wang, Mol. Catal., 2018, 461, 1–9 CrossRef CAS.
- T. Ma, Z. Yun, W. Xu, L. Chen, L. Li, J. Ding and R. Shao, Chem. Eng. J., 2016, 294, 343–352 CrossRef CAS.
- B. Viswanadham, A. Srikanth, V. P. Kumar and K. V. R. Chary, J. Nanosci. Nanotechnol., 2015, 15, 5391–5402 CrossRef CAS.
- S. Suganuma, T. Hisazumi, K. Taruya, E. Tsuji and N. Katada, Mol. Catal., 2018, 449, 85–92 CrossRef CAS.
- X. F. Liu and R. Wang, Int. J. Chem. Eng., 2018, 316939 Search PubMed.
- Z. H. Wang and Q. W. Chen, Green Chem., 2016, 18, 5884–5889 RSC.
- P. H. Che, F. Lu, J. J. Zhang, Y. Z. Huang, X. Nie, J. Gao and J. Xu, Bioresour. Technol., 2012, 119, 433–436 CrossRef CAS.
- L. Bing, Z. H. Zhang and K. J. Deng, Ind. Eng. Chem. Res., 2012, 51, 15331–15336 CrossRef CAS.
- S. G. Wang, Z. H. Zhang, B. Liu and J. L. Li, Catal. Sci. Technol., 2013, 3, 2104–2112 RSC.
- A. Q. Liu, B. Liu, Y. M. Wang, R. S. Ren and Z. H. Zhang, Fuel, 2014, 117, 68–73 CrossRef CAS.
- Y. Yang, M. M. Abu-Omar and C. W. Hu, Appl. Energy, 2012, 99, 80–84 CrossRef CAS.
- H. L. Wang, T. S. Deng, Y. X. Wang, Y. Q. Qi, X. L. Hou and Y. L. Zhu, Bioresour. Technol., 2013, 136, 394–400 CrossRef CAS.
- K. Wilson and A. F. Lee, Catal. Sci. Technol., 2012, 2, 884–897 RSC.
- F. Su, S. An, D. Y. Song, X. H. Zhang, B. Lu and Y. H. Guo, J. Mater. Chem. A, 2014, 2, 14127–14138 RSC.
- Zillillah, T. A. Ngu and Z. Li, Green Chem., 2014, 16, 1202–1210 RSC.
- Y. Jeon, W. S. Chi, J. Hwang, D. H. Kim, J. H. Kim and Y.-G. Shul, Appl. Catal., B, 2019, 242, 51–59 CrossRef CAS.
- J. Li, X. H. Wang, W. M. Zhu and F. H. Cao, ChemSusChem, 2009, 2, 177–183 CrossRef CAS.
- S. Zhang, Y. G. Zu, Y. J. Fu, M. Luo, D. Y. Zhang and T. Efferth, Bioresour. Technol., 2010, 101, 931–936 CrossRef CAS.
- A. S. Badday, A. Z. Abdullah and K.-T. Lee, Energy, 2013, 60, 283–291 CrossRef CAS.
- Q. Zhao, H. Wang, H. W. Zheng, Z. Sun, W. Shi, S. T. Wang, X. H. Wang and Z. J. Jiang, Catal. Sci. Technol., 2013, 3, 2204–2209 RSC.
- X. X. Han, K. K. Chen, W. Yan, C. T. Hung, L. L. Liu, P. H. Wu, K. C. Lin and S. B. Liu, Fuel, 2016, 165, 115–122 CrossRef CAS.
- X. X. Han, Y. F. He, C. T. Hung, L. L. Liu, S. J. Huang and S. B. Liu, Chem. Eng. Sci., 2013, 104, 64–72 CrossRef CAS.
- L. L. Xu, W. Li, J. L. Hu, K. X. Li, X. Yang, F. Y. Ma, Y. N. Guo, X. D. Yu and Y. H. Guo, J. Mater. Chem., 2009, 19, 8571–8579 RSC.
- L. L. Xu, W. Li, J. L. Hu, X. Yang and Y. H. Guo, Appl. Catal., B, 2009, 90, 587–594 CrossRef CAS.
- L. L. Xu, Y. H. Wang, X. Yang, J. L. Hu, W. Li and Y. H. Guo, Green Chem., 2009, 11, 314–317 RSC.
- C. F. Oliveira, L. M. Dezaneti, F. A. C. Garcia, J. L. de Macedo, J. A. Dias, S. C. L. Dias and K. S. P. Alvim, Appl. Catal., A, 2010, 372, 153–161 CrossRef CAS.
- K. Srilatha, T. Issariyakul, N. Lingaiah, P. S. Sai Prasad, J. Kozinski and A. K. Dalai, Energy Fuels, 2010, 24, 4748–4755 CrossRef CAS.
- K. Srilatha, C. Ramesh Kumar, B. L. A. Prabhavathi Devi, R. B. N. Prasad, P. S. Sai Prasad and N. Lingaiah, Catal. Sci. Technol., 2011, 1, 662–668 RSC.
- A. S. Badday, A. Z. Abdullah and K.-T. Lee, Appl. Energy, 2013, 105, 380–388 CrossRef CAS.
- F. Su, L. Ma, Y. H. Guo and W. Li, Catal. Sci. Technol., 2012, 2, 2367–2374 RSC.
- O. d. S. Lacerda, R. M. Cavalcanti, T. M. d. Matos, R. S. Angélica, G. N. da Rocha Filho and I. d. C. L. Barros, Fuel, 2013, 108, 604–611 CrossRef CAS.
- K. Y. Nandiwale and V. V. Bokade, Ind. Eng. Chem. Res., 2014, 53, 18690–18698 CrossRef CAS.
- J. Alcañiz-Monge, G. Trautwein and J. P. Marco-Lozar, Appl. Catal., A, 2013, 468, 432–441 CrossRef.
- A. S. Badday, A. Z. Abdullah and K.-T. Lee, Renewable Energy, 2014, 62, 10–17 CrossRef CAS.
- A. Patel and N. Narkhede, Energy Fuels, 2012, 26, 6025–6032 CrossRef CAS.
- N. Narkhede and A. Patel, Ind. Eng. Chem. Res., 2013, 52, 13637–13644 CrossRef CAS.
- V. Brahmkhatri and A. Patel, Appl. Catal., A, 2011, 403, 161–172 CrossRef CAS.
- N. Narkhede, V. Brahmkhatri and A. Patel, Fuel, 2014, 135, 253–261 CrossRef CAS.
- C. Baroi and A. K. Dalai, Catal. Today, 2013, 207, 74–85 CrossRef CAS.
- V. Brahmkhatri and A. Patel, Ind. Eng. Chem. Res., 2011, 50, 6620–6628 CrossRef CAS.
- V. Brahmkhatri and A. Patel, Fuel, 2012, 102, 72–77 CrossRef CAS.
- A. Patel and N. Narkhede, Catal. Sci. Technol., 2013, 3, 3317–3325 RSC.
- W. L. Xie and F. Wan, Chem. Eng. J., 2019, 365, 40–50 CrossRef CAS.
- F. Su, Q. Y. Wu, D. Y. Song, X. H. Zhang, M. Wang and Y. H. Guo, J. Mater. Chem. A, 2013, 1, 13209–13221 RSC.
- D. Y. Song, S. An, Y. N. Sun and Y. H. Guo, J. Catal., 2016, 333, 184–199 CrossRef CAS.
- K. Y. Nandiwale, S. K. Sonar, P. S. Niphadkar, P. N. Joshi, S. S. Deshpande, V. S. Patil and V. V. Bokade, Appl. Catal., A, 2013, 460–461, 90–98 CrossRef CAS.
- K. Yan, G. S. Wu, J. L. Wen and A. C. Chen, Catal. Commun., 2013, 34, 58–63 CrossRef CAS.
- F. Su, L. Ma, D. Y. Song, X. H. Zhang and Y. H. Guo, Green Chem., 2013, 15, 885–890 RSC.
- T. M. Guo, M. Qiu and X. H. Qi, Appl. Catal., A, 2019, 572, 168–175 CrossRef CAS.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. L. Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- X. Chen, Y. Liu and J. W. Wu, Mol. Catal., 2020, 483, 110716 CrossRef CAS.
- P. Preuster and J. Albert, Energy Technol., 2018, 6, 501–509 CrossRef CAS.
- R. Wolfel, N. Taccardi, A. Bosmann and P. Wasserscheid, Green Chem., 2011, 13, 2759–2763 RSC.
- J. Albert, D. Luders, A. Bosmann, D. M. Guldi and P. Wasserscheid, Green Chem., 2014, 16, 226–237 RSC.
- J. Li, D. J. Ding, L. Deng, Q. X. Guo and Y. Fu, ChemSusChem, 2012, 5, 1313–1318 CrossRef CAS.
- T. Lu, M. G. Niu, Y. C. Hou, W. Z. Wu, S. H. Ren and F. Yang, Green Chem., 2016, 18, 4725–4732 RSC.
- J. Albert, R. Wolfel, A. Bosmann and P. Wasserscheid, Energy Environ. Sci., 2012, 5, 7956–7962 RSC.
- J. Albert and P. Wasserscheid, Green Chem., 2015, 17, 5164–5171 RSC.
- J. L. Xu, H. Y. Zhang, Y. F. Zhao, Z. Z. Yang, B. Yu, H. J. Xu and Z. M. Liu, Green Chem., 2014, 16, 4931–4935 RSC.
- J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Energy Environ. Sci., 2015, 8, 2985–2990 RSC.
- J. Albert, Faraday Discuss., 2017, 202, 99–109 RSC.
- D. Voss, H. Pickel and J. Albert, ACS Sustainable Chem. Eng., 2019, 7, 9754–9762 CrossRef CAS.
- J. Z. Zhang, M. Sun, X. Liu and Y. Han, Catal. Today, 2014, 233, 77–82 CrossRef CAS.
- J. Albert, A. Jess, C. Kern, F. Pöhlmann, K. Glowienka and P. Wasserscheid, ACS Sustainable Chem. Eng., 2016, 4, 5078–5086 CrossRef CAS.
- N. V. Gromov, O. P. Taran, I. V. Delidovich, A. V. Pestunov, Y. A. Rodikova, D. A. Yatsenko, E. G. Zhizhina and V. N. Parmon, Catal. Today, 2016, 278, 74–81 CrossRef CAS.
- M. Dusselier and B. F. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, 2014, vol. 353, pp. 85–125 Search PubMed.
- J. Z. Zhang, X. Liu, M. Sun, X. H. Ma and Y. Han, ACS Catal., 2012, 2, 1698–1702 CrossRef CAS.
- M. L. Tao, X. Yi, I. Delidovich, R. Palkovits, J. Y. Shi and X. H. Wang, ChemSusChem, 2015, 8, 4195–4201 CrossRef CAS.
- M. L. Tao, N. Y. Sun, Y. M. Li, T. Tong, M. Wielicako, S. T. Wang and X. H. Wang, J. Mater. Chem. A, 2017, 5, 8325–8333 RSC.
- M. L. Tao, D. Zhang, X. Deng, X. Y. Li, J. Y. Shi and X. H. Wang, Chem. Commun., 2016, 52, 3332–3335 RSC.
- M. L. Tao, D. Zhang, H. Y. Guan, G. H. Huang and X. H. Wang, Sci. Rep., 2016, 6, 29840 CrossRef CAS.
- X. K. Li and Y. G. Zhang, ACS Catal., 2016, 6, 2785–2791 CrossRef CAS.
- J. H. Lan, J. C. Lin, Z. Q. Chen and G. H. Yin, ACS Catal., 2015, 5, 2035–2041 CrossRef CAS.
- L. Hu, L. Lin, Z. Wu, S. Y. Zhou and S. J. Liu, Renewable Sustainable Energy. Rev., 2017, 74, 230–257 CrossRef CAS.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. C. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714–8715 CrossRef CAS.
- C. Luo, S. Wang and H. C. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- N. V. Gromov, T. B. Medvedeva, O. P. Taran, M. N. Timofeeva, O. Said-Aizpuru, V. N. Panchenko, E. Y. Gerasimov, I. V. Kozhevnikov and V. N. Parmon, Appl. Catal., A, 2020, 595, 117489 CrossRef CAS.
- M. Liu, W. P. Deng, Q. H. Zhang, Y. L. Wang and Y. Wang, Chem. Commun., 2011, 47, 9717–9719 RSC.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- J. Chen, S. P. Wang, J. Huang, L. M. Chen, L. L. Ma and X. Huang, ChemSusChem, 2013, 6, 1545–1555 CrossRef CAS.
- B. Op de Beeck, J. Geboers, S. Van de Vyver, J. Van Lishout, J. Snelders, W. J. Huijgen, C. M. Courtin, P. A. Jacobs and B. F. Sels, ChemSusChem, 2013, 6, 199–208 CrossRef CAS.
- N. García-Bosch, C. Especel, A. G. Ruiz and I. Rodríguez-Ramos, Catal. Today, 2020, 357, 113–121 CrossRef.
- Z. J. Tai, J. Y. Zhang, A. Q. Wang, J. F. Pang, M. Y. Zheng and T. Zhang, ChemSusChem, 2013, 6, 652–658 CrossRef CAS.
- Z. J. Tai, J. Y. Zhang, A. Q. Wang, M. Y. Zheng and T. Zhang, Chem. Commun., 2012, 48, 7052–7054 RSC.
- A. Q. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS.
- M. Y. Zheng, J. F. Pang, R. Y. Sun, A. Q. Wang and T. Zhang, ACS Catal., 2017, 7, 1939–1954 CrossRef CAS.
- M. Almohalla, I. Rodríguez-Ramos, L. S. Ribeiro, J. J. M. Órfão, M. F. R. Pereira and A. Guerrero-Ruiz, Catal. Today, 2018, 301, 65–71 CrossRef CAS.
- A. B. Gawade, M. S. Tiwari and G. D. Yadav, ACS Sustainable Chem. Eng., 2016, 4, 4113–4123 CrossRef CAS.
- S. H. Zhu, Y. L. Cen, J. Guo, J. C. Chai, J. G. Wang and W. B. Fan, Green Chem., 2016, 18, 5667–5675 RSC.
- M. Balaraju, V. Rekha, P. S. S. Prasad, B. L. A. P. Devi, R. B. N. Prasad and N. Lingaiah, Appl. Catal., A, 2009, 354, 82–87 CrossRef CAS.
- S. H. Zhu, X. Q. Gao, Y. L. Zhu, Y. F. Zhu, X. M. Xiang, C. X. Hu and Y. W. Li, Appl. Catal., B, 2013, 140–141, 60–67 CrossRef CAS.
- S. H. Zhu, Y. N. Qiu, Y. L. Zhu, S. L. Hao, H. Y. Zheng and Y. W. Li, Catal. Today, 2013, 212, 120–126 CrossRef CAS.
- S. H. Zhu, Y. L. Zhu, S. Hao, H. Y. Zheng, T. Mo and Y. W. Li, Green Chem., 2012, 14, 2607–2616 RSC.
- S. S. Priya, V. P. Kumar, M. L. Kantam, S. K. Bhargava, S. Periasamy and K. V. R. Chary, Appl. Catal., A, 2015, 498, 88–98 CrossRef CAS.
- C. T. Q. Mai and F. T. T. Ng, Catal. Today, 2017, 291, 195–203 CrossRef CAS.
- Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2011, 1, 179–190 RSC.
- D. S. Park, B. K. Kwak, N. D. Kim, J. R. Park, J.-H. Cho, S. Oh and J. Yi, ChemCatChem, 2012, 4, 836–843 CrossRef CAS.
- A. Alhanash, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2010, 378, 11–18 CrossRef CAS.
- J. Z. Zhang, X. Liu, M. N. Hedhili, Y. H. Zhu and Y. Han, ChemCatChem, 2011, 3, 1294–1298 CrossRef CAS.
- D. L. An, A. H. Ye, W. P. Deng, Q. H. Zhang and Y. Wang, Chem. – Eur. J., 2012, 18, 2938–2947 CrossRef CAS.
- M. A. Alotaibi, E. F. Kozhevnikova and I. V. Kozhevnikov, Chem. Commun., 2012, 48, 7194–7196 RSC.
- K. Alharbi, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2015, 504, 457–462 CrossRef CAS.
- O. Poole, K. Alharbi, D. Belic, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., B, 2017, 202, 446–453 CrossRef CAS.
- K. Alharbi, W. Alharbi, E. F. Kozhevnikova and I. V. Kozhevnikov, ACS Catal., 2016, 6, 2067–2075 CrossRef CAS.
- M. A. Alotaibi, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2012, 447–448, 32–40 CrossRef CAS.
- O. O. Ayodele, F. A. Dawodu, D. X. Yan, J. Y. Xin and S. J. Zhang, Fuel, 2018, 221, 311–319 CrossRef CAS.
- S. Li, L. Yan, Q. Y. Liu, J. G. Liu, Q. Y. Liu, W. Fan, X. L. Zhao, X. H. Zhang, C. G. Wang, L. L. Ma and Q. Zhang, Green Chem., 2020, 22, 2889–2900 RSC.
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morré, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 RSC.
- X. Q. Ma, G. Gözaydın, H. Y. Yang, W. B. Ning, X. Han, N. Y. Poon, H. Liang, N. Yan and K. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7719–7728 CrossRef CAS.
- Y. Z. Wang, S. Furukawa, S. Song, Q. He, H. Asakura and N. Yan, Angew. Chem., Int. Ed., 2020, 59, 2289–2293 CrossRef CAS.
- S. Song, J. G. Zhang, G. Gözaydin and N. Yan, Angew. Chem., Int. Ed., 2019, 58, 4934–4937 CrossRef CAS.
- S. P. Ding, M. J. Hülsey, J. Pérez-Ramírez and N. Yan, Joule, 2019, 3, 2897–2929 CrossRef CAS.
- M. J. Hulsey, J. G. Zhang and N. Yan, Adv. Mater., 2018, 30, e1802304 CrossRef.
- M. J. Hulsey, B. Zhang, Z. R. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. L. Wu and N. Yan, Nat. Commun., 2019, 10, 1330 CrossRef.
- B. Zhang, G. Sun, S. P. Ding, H. Asakura, J. Zhang, P. Sautet and N. Yan, J. Am. Chem. Soc., 2019, 141, 8185–8197 CAS.
- B. Zhang, H. Asakura, J. Zhang, J. G. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS.
- P. L. He, B. Xu, X. B. Xu, L. Song and X. Wang, Chem. Sci., 2016, 7, 1011–1015 RSC.
| This journal is © The Royal Society of Chemistry 2021 |