
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimethyl sulfide facilitates acid catalysed ring opening of the bicyclic monoterpenes in crude sulfate turpentine to afford p-menthadienes in good yield†
Joshua D.
Tibbetts
 ab and
Steven D.
Bull
*a
ab and
Steven D.
Bull
*a
aDepartment of Chemistry, University of Bath, Bath, BA27AY, UK. E-mail: s.d.bull@bath.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, BA27AY, UK
First published on 16th December 2020
Abstract
The potential of using turpentine for the production of biorenewable chemicals within a biorefinery context is discussed, focussing on the development of practical methodology to convert the bicyclic monoterpene components present in untreated crude sulfate turpentine into mixtures of synthetically useful p-menthadienes (p-MeDs) as feedstocks for chemical production. A simple and scalable biphasic acid catalysed ring-opening (ACRO) protocol employing recyclable 6 M aq. H2SO4 at 90 °C has been developed to transform the major bicyclic monoterpenes (β-pinene, α-pinene, and 3-carene) in untreated commercial crude sulfate turpentine (CST) into mixtures of monocyclic p-MeDs in >70% yield. Investigations using pure bicyclic monoterpenes reveal that the ∼3 mol% Me2S present in CST plays a key role in increasing the rate and yield of these ACRO reactions. Reaction monitoring studies have identified the presence of induction periods and autocatalytic effects, as well as an intriguing time-dependent increase in p-MeD yield after all the bicyclic terpene substrate has been consumed. These effects have been explained using a mechanism that invokes reversible addition of Me2S to the alkene bonds of monoterpenes to produce in situ generated sulfonium salts. These sulfonium salts then function as surfactants to increase the degree of mixing of the aqueous/terpene layers of the biphasic ACRO reaction, resulting in faster protonation of the alkene bonds of the bicyclic terpenes in the rate-determining step. The shorter reaction times of the Me2S facilitated ACRO reactions of CST and gum turpentine enable better yields of p-MeDs to be obtained by minimising p-MeD losses to competing polymerisation pathways that occur at elevated temperatures over time.
Introduction
The non-renewable supply and fluxional price of fossil fuel-derived chemical products means there is a growing demand for biorefineries that can be used to process biomass into biofuels, biopolymers and biorenewable chemicals.1 In this respect, monoterpenes (and monoterpenoids) obtained from plants (and microbes) represent a potentially attractive source of biomass that are available in sufficient quantities to be used for chemical production.2 Monoterpenes are the most energy-dense of the biorenewable feedstocks, whose lightly-oxygenated, unsaturated C10 hydrocarbon structures can be catalytically upgraded using processes that already exist to transform petrochemicals on an industrial scale.3 Annual global biogenic emissions of monoterpenes are estimated to be around 100 million tonnes, although only a relatively small fraction of this total volume is currently available for chemical production.4Turpentine is the largest and cheapest source of monoterpene biomass, with around 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes produced annually as waste by-products of the forestry industry.5 Around 260000 tonnes of crude sulfate turpentine (CST) is generated as a by-product of the Kraft pulping process6 that is used to convert trees (mainly pine and fir) into paper (around 15 kg CST per tonne pulp).7 The remaining 100000 tonnes is produced as gum turpentine (GT), which is obtained through the tapping of trees grown in large plantations. This tapping process involves cutting the trunk of a living tree and harvesting the crude balsam that exudes from the wound, with the resultant oleoresin then distilled to produce GT.5 Turpentine is comprised of a mixture of bicyclic and monocyclic monoterpenes such as α-pinene, β-pinene, 3-carene, camphene, and limonene, as well as small amounts of oxygenated terpenoids and sesquiterpenes. Its composition varies significantly depending on its geographical origin, with North American sources of turpentine rich in α-pinene, β-pinene and limonene, whilst European and Asian sources contain large amounts of pinenes and 3-carene.5a Unfractionated turpentine sources are primarily used as a solvent for paints, resins, coatings and varnishes, whilst 3-carene-rich turpentine has been used as a biorenewable pesticide for protecting forestry plantations against insect infestations.5a,8 Crude turpentine sources can also be used as biofuels, with excess CST often burnt for energy to supply power to Kraft paper pulping mills.5,9
000 tonnes produced annually as waste by-products of the forestry industry.5 Around 260000 tonnes of crude sulfate turpentine (CST) is generated as a by-product of the Kraft pulping process6 that is used to convert trees (mainly pine and fir) into paper (around 15 kg CST per tonne pulp).7 The remaining 100000 tonnes is produced as gum turpentine (GT), which is obtained through the tapping of trees grown in large plantations. This tapping process involves cutting the trunk of a living tree and harvesting the crude balsam that exudes from the wound, with the resultant oleoresin then distilled to produce GT.5 Turpentine is comprised of a mixture of bicyclic and monocyclic monoterpenes such as α-pinene, β-pinene, 3-carene, camphene, and limonene, as well as small amounts of oxygenated terpenoids and sesquiterpenes. Its composition varies significantly depending on its geographical origin, with North American sources of turpentine rich in α-pinene, β-pinene and limonene, whilst European and Asian sources contain large amounts of pinenes and 3-carene.5a Unfractionated turpentine sources are primarily used as a solvent for paints, resins, coatings and varnishes, whilst 3-carene-rich turpentine has been used as a biorenewable pesticide for protecting forestry plantations against insect infestations.5a,8 Crude turpentine sources can also be used as biofuels, with excess CST often burnt for energy to supply power to Kraft paper pulping mills.5,9
Turpentine is also fractionally distilled into its individual components to produce the bicyclic monoterpenes α-pinene, β-pinene, 3-carene, camphene, and monocyclic limonene, as well as small amounts of other monoterpenes of value to the flavour and fragrance industries.8,9 The similar boiling points of these monoterpene components means that multiple distillations are often required to effectively fractionate turpentine into its monoterpene components, which adds significant energy costs to the production process. Untreated CST contains ∼3% volatile sulfur compounds (e.g. MeSH, Me2S, Me2S2) that are present as by-products of the Kraft pulping process. The noxious odor and metal catalyst poisoning properties of these sulfur impurities can be problematic,8 with oxidative treatments and nickel-catalyzed hydrodesulfurization processes often required to reduce the sulfur content of CST to enable it to be transformed using catalytic processes.10 However, these pretreatment processes add production costs, so purified monoterpenes are normally only used to produce high-value products, with up to 50% of the world's CST supply currently consumed for the production of flavours, fragrances and vitamins (Fig. 1).6,9,11
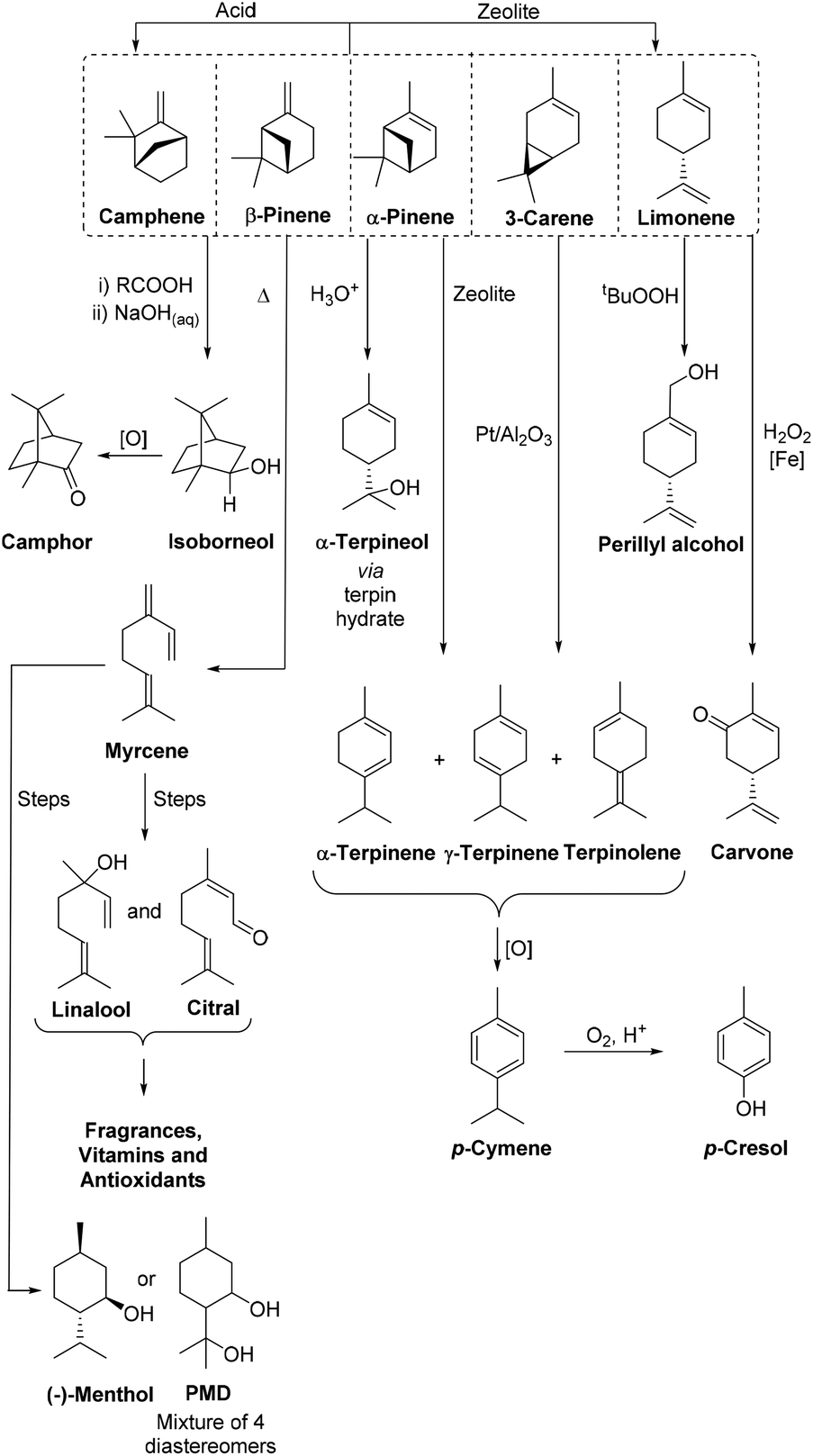 | ||
| Fig. 1 A representative range of scalable catalytic reactions that can be used to transform purified monoterpenes from turpentine into valuable biorenewable chemical products on an industrial scale. | ||
The multiple chemical products produced industrially from turpentine clearly demonstrate that monoterpenes can serve as versatile biorenewable feedstocks for the synthesis of commercially valuable products within a biorefinery context.9 A potential benefit of using monoterpenes as biorenewable feedstocks is the opportunity to transform their cyclic p-menthene skeletons into benzenoid products that are more challenging to access from lignocellulosic feedstocks. The ring structure of p-cymene clearly makes it a highly attractive ‘drop-in’ chemical target for a terpene based biorefinery, particularly as petrochemically sourced p-cymene is currently used as an industrial solvent and as a precursor for the synthesis of a range pesticides, fungicides, flavours and pharmaceuticals.12 Catalytic global oxidation of the methyl and isopropyl sidechains of p-cymene can also be used to transform p-cymene into ‘green’ terephthalic acid (TA),13 which could potentially be used to produce the bulk polymer polyethylene terephthalate (i.e. to prepare bio-PET).14 Importantly, conversion of the chiral bicyclic monoterpenes present in CST into achiral p-menthadienes (p-MeDs) and p-cymene products means that CST feedstocks containing bicyclic monoterpenes with varying enantiopurities and different absolute configurations can be used to prepare p-cymene.15 This would enable batches of CST obtained from different geographical locations to be used as feedstocks, thus providing supply chain flexibility to improve the economics and logistics of the biorefining process.
Numerous one-step (see Table S1†)16,17 and two-step processes (see Table S2†) have been developed to transform the bicyclic monoterpenes present in turpentine into p-cymene. These processes rely on acid catalyzed ring-opening (ACRO) reactions to convert the bicyclic monoterpenes into mixtures of p-MeDs, whose diene-ring systems are then oxidatively isoaromatized to produce the aromatic ring of p-cymene (Fig. 2).18 Many of these one-step processes require the use of high temperatures (>150 °C), access to specialized equipment, extended reaction times, or use catalysts/zeolite additives that are susceptible to poisoning by the sulfur contaminants present in untreated CST. They may also produce mixtures of p-MeDs, p-cymene containing other monoterpenes (e.g. camphene), o/m-cymene and terpene dimers/polymers that require further purification, leading to lower yields and scale-up issues (see Table S1†). This means that two-step processes that employ aqueous acidic conditions in the first-step to convert turpentine into isolable mixtures of p-MeD intermediates, followed by a second isoaromatization step generally proceed in higher yields.19 The ability to produce p-cymene via a two-step process is also potentially attractive from a terpene biorefinery perspective, because it enables access to mixtures of p-MeDs that can be used as versatile synthetic intermediates to produce other value-added biorenewable products, such as fungicides, epoxide monomers, anthelmintic ascaridole and a number of resins and polymers (Fig. 2).20–22
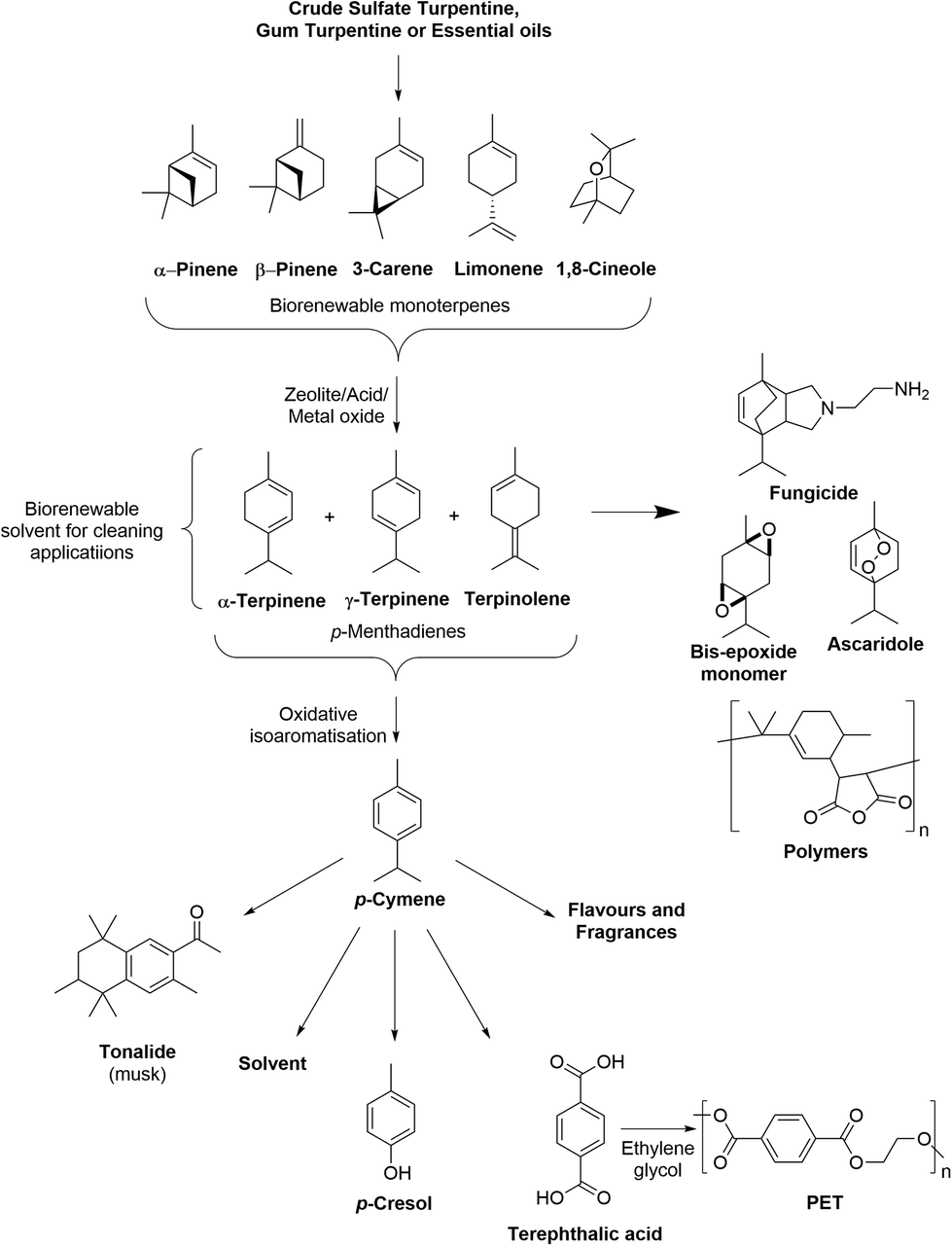 | ||
| Fig. 2 Conversion of biorenewable CST into p-MeD mixtures that can be used as feedstocks to produce p-cymene and other commercially valuable chemical products. | ||
As part of studies directed towards broadening the range of biorenewable products that can be produced in terpene based biorefineries using untreated CST feedstocks,20 we now report development of a practically simple, biphasic ACRO protocol (6 M aq. H2SO4 at 90 °C, 4 h) that enables the bicyclic monoterpenes present in untreated CST to be converted into p-MeD mixtures in >70% yields. Our studies reveal for the first time that Me2S impurities present in CST, that are generally considered to be troublesome noxious contaminants in catalytic upgrading processes,23 play a crucial role in improving the efficiency of industrial CST ACRO processes. This means that the presence of ∼3–5 mol% Me2S as a contaminant in these CST ACRO reactions is essential to produce p-MeDs in good 70–80% yields in short reaction times.
Reaction monitoring studies have revealed the presence of significant induction periods and autocatalytic effects in these ACRO reactions, which have enabled us to propose a working hypothesis to explain how Me2S accelerates these ACRO reactions. It is proposed that reversible nucleophilic addition of Me2S to the alkene bonds of the monoterpene substrates in CST generates terpene sulfonium salts in situ that are responsible for the time-dependent increase in the rate of the ACRO reactions of its bicyclic monoterpene components. The presence of terpene sulfonium salts in these ACRO reactions has been confirmed through HRMS analysis, with these sulfonium salts proposed to function as in situ generated surfactants that improve mixing of the biphasic ACRO reactions, which results in increased mass transfer between the aqueous/organic layers to generate a more acidic interface. This localised increase in interfacial acidity then results in faster protonation of the alkene bonds of the bicyclic monoterpene substrates, which is known to be the rate determining step in these types of ACRO reactions. The higher p-MeD yields produced in the presence of Me2S is due to faster ACRO reactions minimising losses from competing p-MeD polymerisation processes that are problematic in the absence of Me2S. The ability of using ∼3–5 mol% Me2S as an exogenous additive to improve the efficiency of ACRO reactions of other bicyclic monoterpenes has also been demonstrated, with GT and eucalyptus oil (cineole)24 also affording improved p-MeD yields in even shorter reaction times.
Results and discussion
An eco-friendly ACRO process for transforming untreated CST into mixtures of p-MeDs
Initial investigations focused on identifying an economically viable and eco-friendly method for converting commercial untreated CST into mixtures of p-MeDs in good yield. We sought to develop a simple, organic solvent free batch protocol using a cheap, recyclable aqueous Brønsted acid catalyst at ambient pressure and moderate temperature (<100 °C), without the need for any specialised equipment. This ACRO process would ideally afford good yields of p-MeD products that were uncontaminated by any rearranged monoterpene products (e.g. camphene). The biphasic nature of this type of ACRO process would enable clean mixtures of p-MeDs to be isolated using a simple decantation work-up procedure, thus enabling crude p-MeD mixtures to be used in subsequent transformations without the need for purification.A survey of previously reported ACRO reactions of bicyclic monoterpenes25 (ESI, Table S2†) revealed a highly promising Holmen patent which reported that stirring CST (rich in 3-carene) with 6.75 M H2SO4 at 110 °C for 5 h gave a mixture of p-MeDs in 76% yield.25e We opted to trial these biphasic conditions on a commercial sample of untreated CST that was obtained from a paper mill operated by the Södra Forestry Cooperative in Sweden. 1H NMR spectroscopic analysis of this authentic CST revealed that it contained α-pinene (39%), 3-carene (34%), β-pinene (10%), limonene (4%), Me2S (3%) and p-cymene (1%), with the remaining mass balance comprised of a complex mixture of other minor terpenes (∼10%). A brief optimisation study of the original patent conditions25e (time, temperature, volume of acid, stirring rate and reaction work-up) revealed that rapid stirring (500 rpm) of a biphasic mixture of CST and 6 M aq. H2SO4 (20 vol% wrt CST) at 90 °C resulted in >95% of its bicyclic monoterpene fraction being consumed after 4 h (Fig. 3). Reaction monitoring of aliquots revealed that the β-pinene fraction was consumed after 60 min, with its α-pinene fraction taking 2.5 h to fully react. The least reactive 3-carene component took 5 h to be fully consumed (ESI, Fig. S1 and S2†). The initial phase of this ACRO reaction produced a p-MeD fraction that was rich in limonene and terpinolene, which then isomerised to afford a thermodynamic p-MeD mixture of α-terpinene, γ-terpinene and isoterpinolene over time (ESI, Fig. S3†).26 A small amount of p-cymene was also formed, presumably from aerobic oxidation of the cyclohexadiene ring systems of the terpinene fractions. Attempts to employ longer reaction times to drive consumption of the less reactive 3-carene fraction to completion were counterproductive, with competing cationic polymerisation reactions resulting in a decrease in p-MeD yield over time (cf. 70% p-MeD yield after 3.5 h with 50% p-MeD yield after 6 h) (ESI, Fig. S3†).
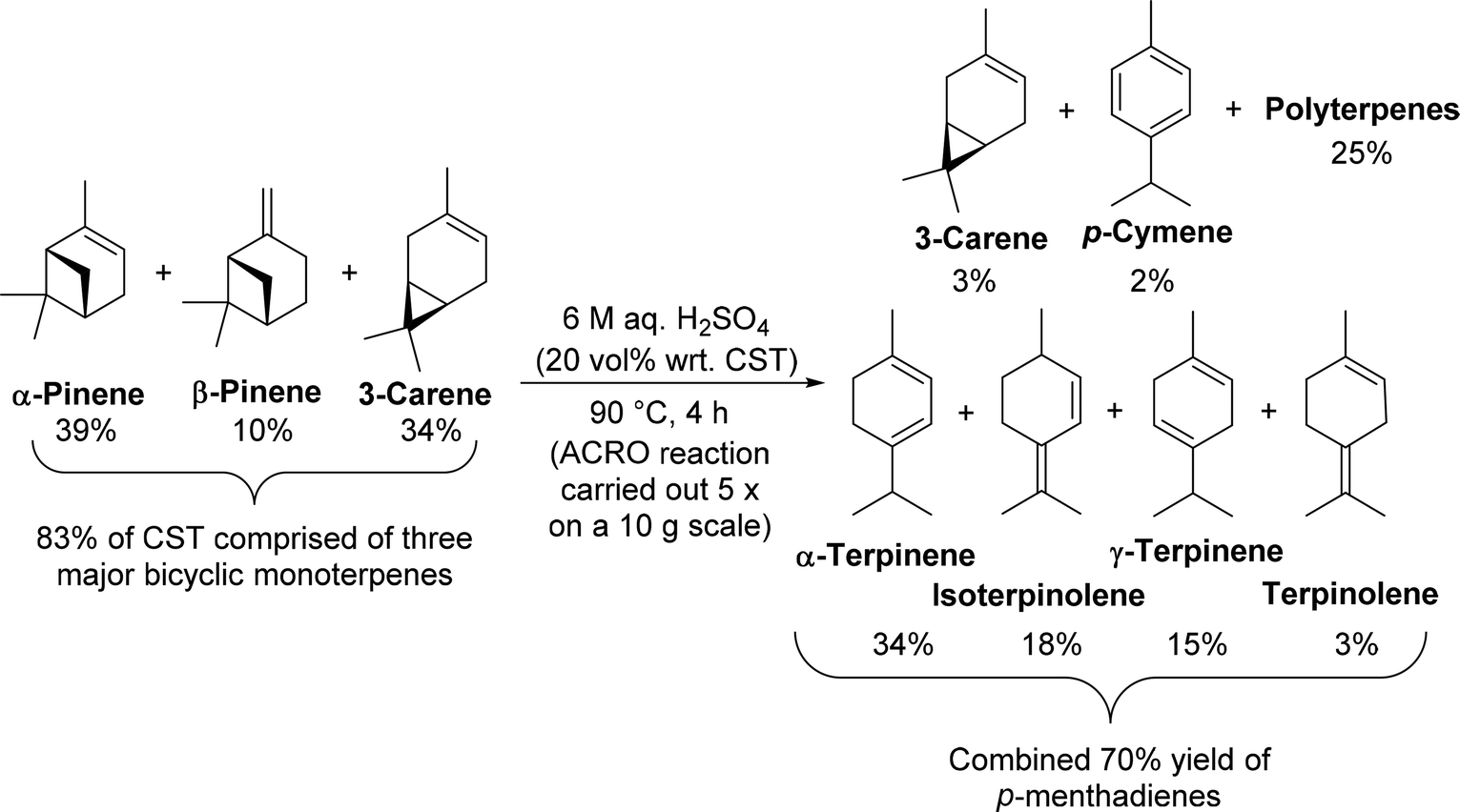 | ||
| Fig. 3 ACRO reactions of the bicyclic monoterpenes present in CST to afford thermodynamic mixtures of p-MeDs and polyterpenes. | ||
The biphasic nature of these ACRO reactions allowed for simple work-up, with the crude p-MeD products isolated by simply decanting off the top organic layer from the bottom aqueous acidic layer. 1H NMR spectroscopic analysis of the monoterpene layer after 4 h at 90 °C revealed that it was comprised of a 34:18:15:3:3:2 mixture of α-terpinene, isoterpinolene, γ-terpinene, terpinolene, unreacted 3-carene, and p-cymene, respectively, with the remaining 25% mass balance comprised of terpene oligomers/polymers (Fig. 3). Distillation of this crude product (b.p. 170–185 °C, 760 mmHg) gave a mixture containing pure p-MeDs (same ratio) and ∼10% p-cymene in 72% isolated yield, with the p-cymene component formed from aromatisation occurring in the distillation step. A viscous red-brown polymeric terpene residue was also recovered from the distillation pot in 22% yield. This polyterpene gum proved to be highly flammable, burning vigorously in air when lit with an open flame, thus demonstrating that it is potentially useful as a liquid biofuel.
Untreated aqueous sulfuric acid layers recovered from the ACRO reaction of CST could be reused as a catalyst in four consecutive ACRO reactions of CST, affording ∼70% yields of p-MeDs, with only a small drop in rate between each successive 4 hours run.27 This ability to reuse the untreated sulfuric acid layer significantly reduces the cost and environmental impact of using 6 M aq. H2SO4 as a cheap Brønsted acid catalyst.
Optimal conditions for the ACRO reaction of CST on a 10 g scale were then established, which involves rapid stirring (500 rpm) of a biphasic mixture of CST with 20 mol% 6 M aq. H2SO4 for 4 h. These conditions reproducibly afforded ∼70% isolated yields of p-MeD products (identical ratio) over five separate runs. Since the four major monoterpenes (α-pinene, 3-carene, β-pinene, limonene) only constitute 87% of the total CST mass, this meant that these ACRO conditions are producing p-MeD products in >80% average yields.
Me2S accelerates the ACRO reactions of the bicyclic monoterpenes present in CST
Aliquot-based reaction monitoring of the ACRO reaction of CST revealed that its pinene fractions were much more reactive than its 3-carene component (ESI, Fig. S2†). This encouraged us to apply our aqueous acidic conditions (6 M aq. H2SO4, 90 °C, no Me2S) to pinene-rich commercial GT (Barrettine Genuine Turpentine; 54:46 mixture of α-pinene/β-pinene). However, we found that the ACRO reactions of the pinene fractions of GT were much slower than the corresponding ACRO reactions of the pinene fractions of CST. For example, it took >5 h for all the β-pinene fraction in GT to be consumed (cf. 1 h for β-pinene present in CST), with >75% of its α-pinene fraction still remaining after 6 h (cf. 2.5 h for total consumption of α-pinene fraction of CST).28 These results implied that something present in untreated CST was functioning to increase the rate of the ACRO reactions of its bicyclic monoterpene components.29
We reasoned that Me2S (typically ∼3 mol%) was potentially responsible for the increased rate of the ACRO reaction, and so decided to compare the reactivity profiles of ACRO reactions of ‘mock’ samples of CST (4:3:1 mixture of α-pinene, 3-carene and β-pinene) in the absence/presence of 5 mol% Me2S. ACRO reaction of ‘mock’ CST using 6 M aq. H2SO4 at 90 °C (no Me2S) resulted in only 33% of its total bicyclic monoterpene content being consumed after 3 h. Conversely, the ‘mock’ sample of CST containing 5 mol% Me2S as an additive reached >95% total consumption after 3 h, under otherwise identical conditions (ESI, Fig. S4†). Furthermore, treatment of pinene-rich GT with 6 M aq. H2SO4 and 5.0 mol% Me2S at 90 °C resulted in faster consumption of its pinene components, with both ACRO reactions complete after only 45 min (ESI, Fig. S5†). Although Me2S has a b.p. of 37 °C, it forms a negative azeotrope30 with the bicyclic monoterpenes present in turpentine (b.p. 156–171 °C), enabling it to persist in the monoterpene layer over the course of these ACRO reactions at 90 °C. This was evidenced by the strong sulfurous smell of Me2S of the terpene layers that remained when their ACRO reactions were complete.
The p-MeD mixtures produced in the Me2S-containing ACRO reactions of ‘mock’ CST and GT were initially rich in limonene and terpinolene, with thermal equilibration then affording a thermodynamic mixture containing α-terpinene, γ-terpinene and isoterpinolene as major components.31 The shorter 2 h reaction time required for full consumption of GT (no 3-carene) in its Me2S-facilitated ACRO reaction enabled ∼90% isolated yields of p-MeDs to be obtained. However, addition of >5 mol% Me2S to either the ‘mock’ CST or GT samples did not produce any further increase in the overall rates of their ACRO reactions.
The optimal ACRO conditions (6 M aq. H2SO4, 5.0 mol% Me2S, 90 °C) developed for GT were then applied to 1,8-cineole (major component of eucalyptus oil) as an alternative biorenewable terpene feedstock. This resulted in 1,8-cineole being rapidly converted into an identical thermodynamic mixture of p-MeDs in 80% yield after 1 h.17i Therefore, these solvent-free ACRO conditions provide a simple, inexpensive method to rapidly transform three readily available biorenewable terpene feedstocks (CST, GT, and eucalyptus oil) into mixtures of p-MeDs in 70–90% yields in an eco-friendly and cost-effective manner.
Unusual reaction profiles in the Me2S facilitated ACRO reactions of bicyclic monoterpenes
The role of Me2S in facilitating the ACRO reactions of the bicyclic monoterpenes in CST was then investigated by carrying out comparative ACRO reactions on pure samples of β-pinene, α-pinene and 3-carene. Reaction of β-pinene with 6 M aq. H2SO4 at 90 °C (no Me2S) took 5 h (ESI, Fig. S6†) to reach completion (cf. 1 h for β-pinene in CST, Fig. S2†). α-Pinene and 3-carene were even less reactive under these ACRO reactions (no Me2S), with only 32% of α-pinene (ESI, Fig. S7†) (cf. 45 min for α-pinene in CST, Fig. S2†) and 34% of 3-carene (ESI, Fig. S8†) (cf. 100 min for 3-carene in CST, Fig. S2†) being consumed after 6 h. Significant amounts of α-pinene were produced in the Me2S-free ACRO reaction of β-pinene (peaking at 9% after 2.5 h), thus indicating that β-pinene could equilibrate into its more thermodynamically-stable α-pinene isomer under these acidic conditions (ESI, Fig. S6†).32As expected, the ACRO reaction of β-pinene in the presence of 5 mol% Me2S was much faster, with a short induction period (∼2 min) followed by its rapid consumption over the next 20 min. This rate of reaction was 3 times faster than when β-pinene was present as a fraction in CST and 15 times faster than when Me2S was absent (Fig. 4a). The ACRO reaction of β-pinene initially gave a complex mixture of p-MeDs (20 min), which then equilibrated to a thermodynamic mixture of p-MeDs after 1 h. Much less α-pinene was formed as an intermediate in the 5 mol% Me2S containing ACRO reaction, with a maximum amount of 3% α-pinene produced after 15 min (Fig. 4a).
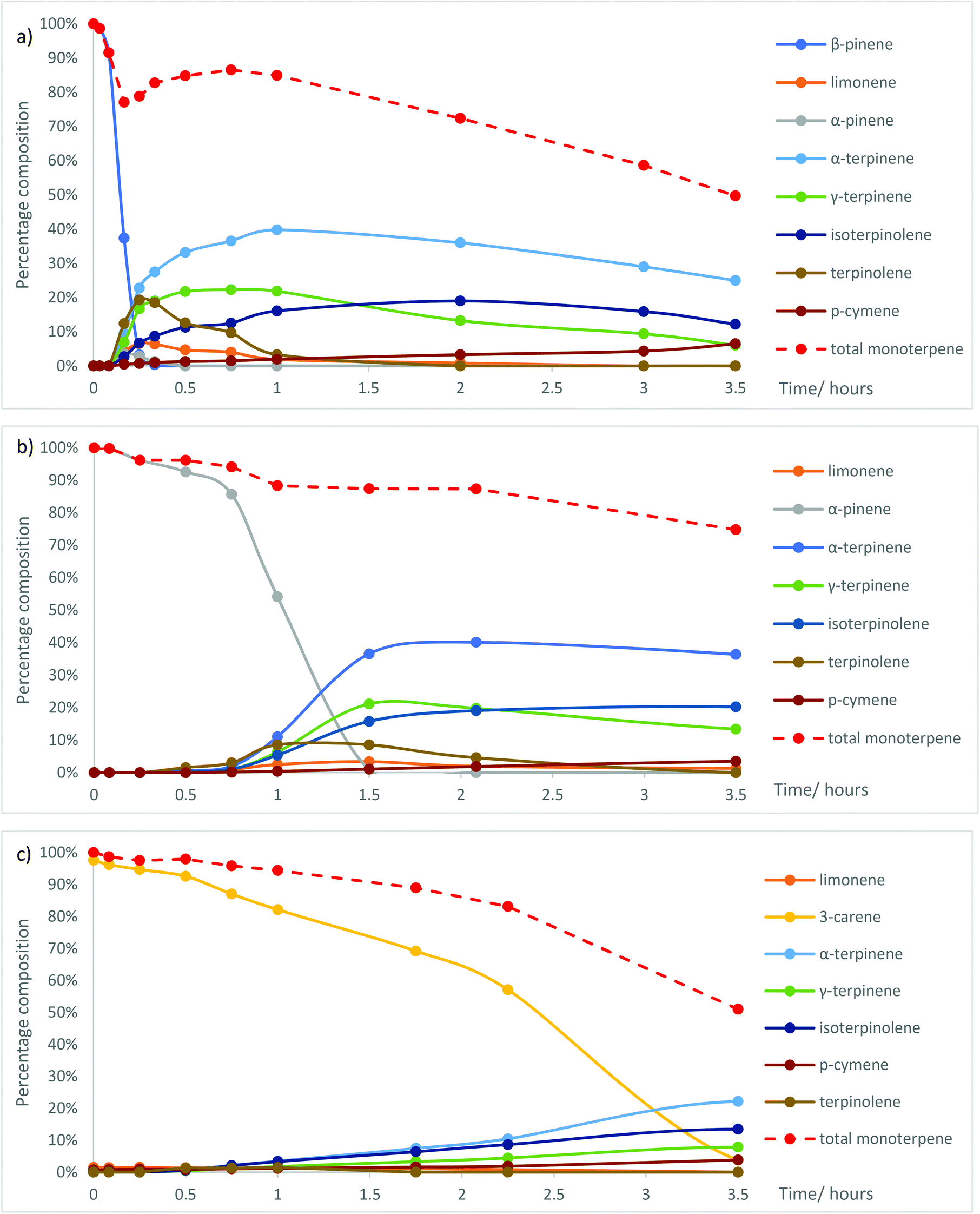 | ||
| Fig. 4 Changes in the ratio of the mixtures of p-MeDs when (a) β-pinene; (b) α-pinene; and (c) 3-carene were reacted with 6 M aq. H2SO4 and 5 mol% Me2S at 90 °C over time. | ||
Examination of reactant to product ratios in the ACRO reaction of β-pinene over the first 45 min (at 90 °C) revealed a curious time-dependent, non-proportional relationship between the amount of β-pinene consumed and the total yield of p-MeD present (Fig. 4a). For example, >95% of the β-pinene was consumed after 15 min to produce a combined 79% yield of p-MeDs, whose total yield then increased by 10% over the next 30 min to give a total p-MeD yield of 87% after 45 min (Fig. 4a). This observation implied that the Me2S-facilitated ACRO reaction of β-pinene was reversibly generating some form of water-soluble terpene-derived species that were not detectable by the aliquot sampling protocol. We reasoned that these water-soluble terpene species were initially being produced under kinetic control (0–15 min) with equilibration over time (from 15–45 min) then generating greater amounts of p-MeDs under thermodynamic control (vide infra). The rates of the competing oligomerisation/polymerisation reactions of the p-MeDs in these ACRO reactions were also found to be greater in the presence of 5 mol% Me2S. For example, the Me2S-facilitated ACRO reaction of β-pinene initially gave an 85% combined yield of p-MeDs after 1 h, with their amount then falling by 35% over the next 2.5 h to give a 50% combined p-MeD yield.
The Me2S-mediated ACRO reaction of α-pinene displayed a significantly longer induction period (only 14% α-pinene consumed after 45 min, Fig. 4b), with more rapid ring-opening then occurring over the next 45 min. This ACRO reaction produced a fully equilibrated mixture of p-MeDs in a combined 85% yield after 90 min (cf. 150 min for α-pinene fraction in CST) which fell to a combined 75% yield after 3.5 h. The 10% loss in p-MeD yield observed for α-pinene from 1.5 to 3.5 h was significantly less than the 25% loss in p-MeD yield for β-pinene over a corresponding time period. This difference indicates that the water-soluble terpene species produced in the ACRO reaction of β-pinene were more efficient at initiating p-MeD polymerisation than the corresponding water-soluble terpene species produced from α-pinene.
The Me2S-mediated ACRO reaction of α-pinene also displayed a significant autocatalytic effect, with the rate of consumption of α-pinene increasing significantly over the first 90 min. For example, only 7% of the α-pinene was consumed in the first 30 min, with 46% consumed after 60 min and >99% consumed after 90 min. This suggested that the terpene-derived water-soluble species generated in these ACRO reactions over time were also likely to be responsible for the corresponding time-dependent increase in the rate of the ACRO reaction of α-pinene. Importantly, the 1.5 h taken to consume pure α-pinene in its Me2S-mediated ACRO reaction was significantly slower than the 45 min required for its total consumption when present as a fraction in GT. Conversely, the 15 min required to consume pure β-pinene was significantly faster than the 30 min required for its consumption when present as a fraction in GT. These results indicated that the water-soluble terpene intermediates that are produced more rapidly from the more reactive β-pinene fraction of GT were capable of accelerating the ACRO reactions of both its pinene fractions.
As expected, 3-carene was the least reactive bicyclic monoterpene investigated, with its Me2S-facilitated ACRO reaction taking between 3.5–4.0 h to proceed to completion to afford a significantly lower 51% yield of p-MeDs (Fig. 4c). Once again, a significant autocatalytic effect was apparent in this Me2S-mediated ACRO reaction, with a relatively slow reaction over the first 30 min, followed by an increased rate of reaction between 30–135 min and an even faster reaction rate after 135 min. The lengthier reaction times and lower p-MeD yields observed in the ACRO reaction of 3-carene confirmed that its presence in CST was responsible for the lower yield of p-MeDs from CST (70% yield, 3.5 h) when compared to GT (95% yield, 2 h). The decreased p-MeD yields from CST are not only caused by the relatively inefficient ACRO reaction of its 3-carene fraction, but are also due to longer reaction times at 90 °C resulting in polymerisation of p-MeDs that originate from its pinene fractions. The relatively large amounts of unreactive 3-carene present in the CST sample used in this study meant that its ACRO reaction needed to be carried out for ∼4.0 h to ensure that >95% of its less reactive 3-carene component was consumed. This time period represents the best compromise for maximising 3-carene consumption, whilst minimising p-MeD losses to polymerisation pathways. However, it should be noted that other sources of pinene-rich CST (e.g. from North American pine) do not contain significant amounts of 3-carene, with these CST sources predicted to produce greater amounts of p-MeD product, similar to the 90% yields obtained in the Me2S-facilitated ACRO reactions of GT. Conversely, prior distillation of 3-carene-rich CST to separate its more volatile α-pinene (b.p. 156 °C)/β-pinene (b.p. 165 °C) components from its higher-boiling 3-carene fraction (b.p. 171 °C) may be beneficial within a biorefinery context. This would enable the more reactive α-pinene/β-pinene fractions to be transformed into mixtures of p-MeDs, with the less reactive 3-carene fraction used to produce other types of useful chemical products.
Mechanism of Me2S free ACRO reactions of β-pinene, α-pinene and 3-carene
The mechanisms of the ACRO reactions (no Me2S) of both pinenes to give p-MeD mixtures under strong aqueous acidic conditions are well established (Fig. 5).32 Rate-limiting protonation of their alkene bonds at the organic–water interface generates a non-classical carbenium ion 1 that fragments with loss of a proton to afford limonene and terpinolene. Virtually no camphene or α-fenchene products are formed, which indicates that carbenium ion 1 is likely to be formed as a tight ion pair. This ion pair does not appear to dissociate appreciatively into more naked carbenium species, since this would favour skeletal rearrangement to produce bicyclic monoterpenes (e.g. camphene) that are not generally observed under these aqueous conditions.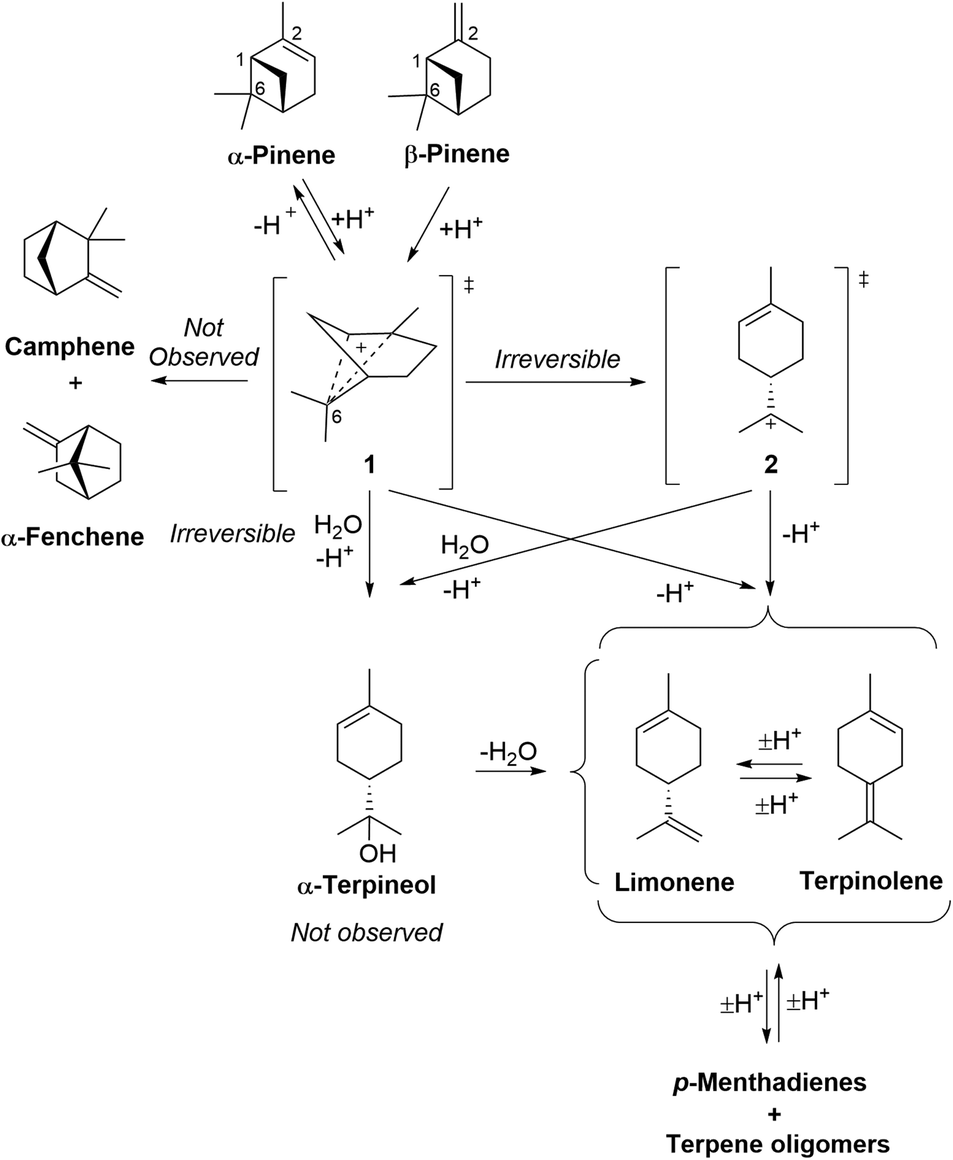 | ||
| Fig. 5 Mechanism of the ACRO reactions of α-pinene and β-pinene to afford limonene and terpinolene. HSO4− counterions of carbenium species omitted for clarity. | ||
The acid-catalysed fragmentation reaction of β-pinene (no Me2S) produces up to 9% of its thermodynamically more stable α-pinene isomer as an intermediate, indicating that the initial alkene protonation steps of both pinenes are fully reversible and rate-determining.32 Irreversible ring-fragmentation of pinane carbenium ion 1 then affords a tertiary p-menthene carbenium ion 2 that can lose a proton to afford limonene or terpinolene. This carbenium rearrangement pathway has been proposed to proceed via a tight ion-pair that is protected from nucleophilic attack by water, with the retained HSO4− counterion potentially acting as a base to facilitate deprotonation of the rearranged p-menthene carbenium ion 2.32
An alternative reaction pathway may also be considered, involving water acting as a nucleophile to carry out SN2 attack at C6 of the non-classical carbenium ion 1 to trigger ring opening to produce α-terpineol (not observed),33 that would then be rapidly dehydrated to give limonene/terpinolene under the strongly acidic conditions. We found that treatment of α-terpineol with 6 M aq. H2SO4 at 90 °C resulted in its rapid conversion into mixtures of limonene and terpinolene within 5 min. This confirms that any α-terpineol formed as an intermediate in the ACRO reactions of CST will be rapidly dehydrated under the strongly acidic conditions and so would be unlikely to be observed. Acid-catalysed fragmentation of α-pinene occurs in a similar manner, with slower protonation of its more stable trisubstituted alkene bond affording the same pinane carbenium ion 1 that then fragments to afford p-menthene carbenium ion 2 (as described for β-pinene).32 Acid-catalysed protonation of the alkene bond of β-pinene has been reported to be ∼10 times faster than the corresponding protonation reaction of α-pinene,32 so this explains why β-pinene is totally consumed after 5 h, with only 35% of the α-pinene transformed after 6 h.
The fragmentation reaction of 3-carene under these ACRO conditions is more complex, because its alkene bond must first isomerise into 2-carene that must then fragment via a relatively unfavoured ACRO pathway (Fig. 6).34 Therefore, acid-catalysed protonation of 3-carene produces a tertiary carbenium ion 3 that eliminates a proton to afford a mixture of 3-carene and 2-carene isomers. Favoured Markovnikoff protonation of the alkene bond of 2-carene will regenerate tertiary carbenium ion 3, however anti-Markovnikoff protonation will produce non-classical secondary carbenium ions 4/5. These disfavoured carbenium ions 4/5 can then fragment to afford p-menthene carbenium ions 6/7 that then eliminate a proton to give isoterpinolene (or isolimonene as a mixture of diastereomers – not observed).
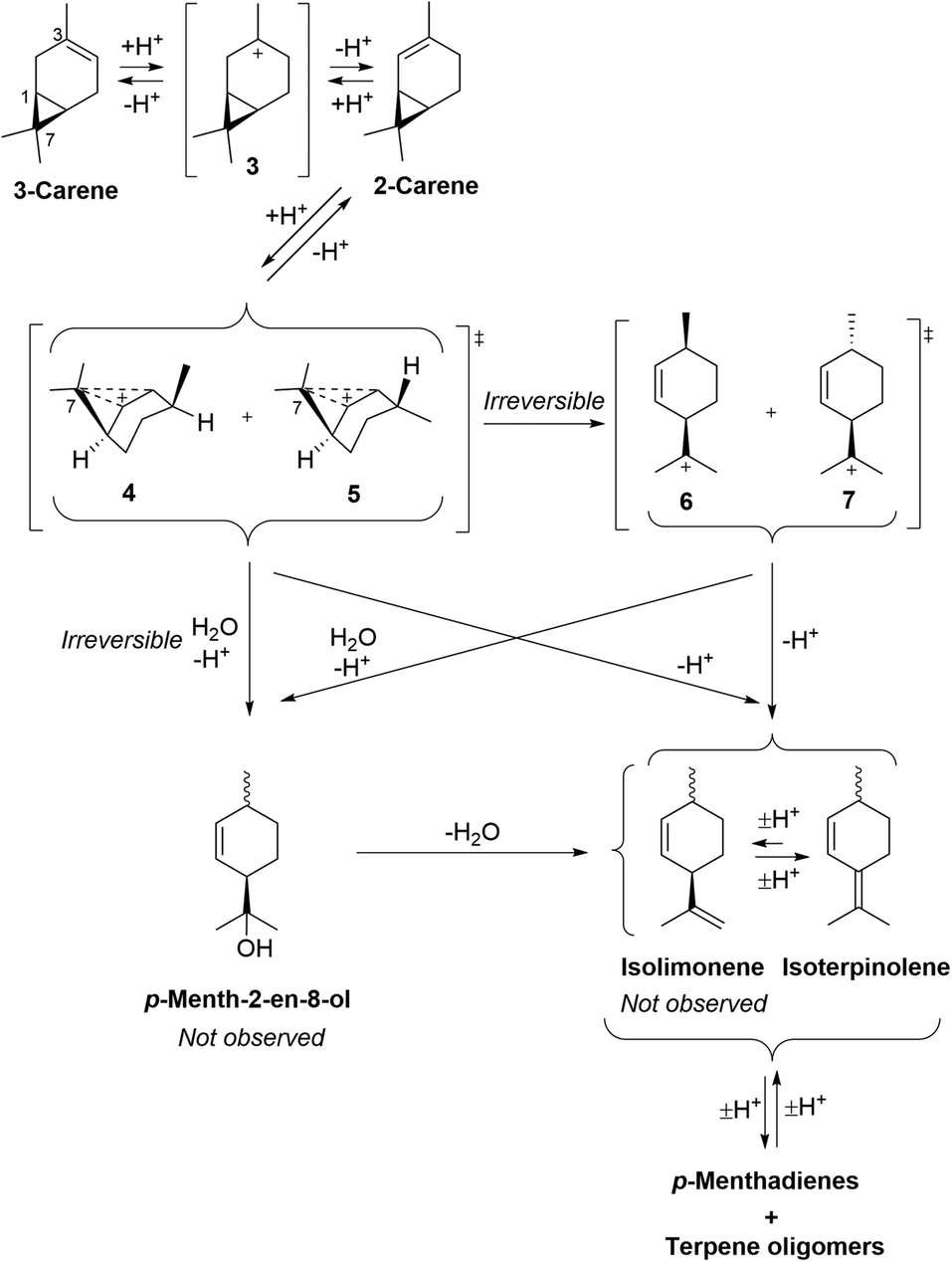 | ||
| Fig. 6 Mechanism of the ACRO reaction of 3-carene to afford isolimonene and isoterpinolene. HSO4− counterions of carbenium species omitted for clarity. | ||
Support for this two-step mechanistic process was found from 1H NMR spectroscopic analysis of the Me2S-mediated ACRO reaction of 3-carene, which showed the appearance of a small amount of 2-carene over time (peaking at 2% after 90 min). Furthermore, treatment of a 55:45 mixture of 3-carene and 2-carene (see ESI† for details) with 6 M aq. H2SO4 at 90 °C resulted in its 2-carene fraction being consumed 1.5 times faster than its 3-carene fraction. These observations indicate that protonation of the alkene bond of 3-carene must be rate-determining, otherwise more significant amounts of 2-carene would be expected to accumulate over time.
Once again, an alternative reaction mechanism may be envisaged whereby water acts as a nucleophile to trigger ring-opening at C7 of the carbenium ions 4/5 to deliver p-menth-2-en-8-ol (mixture of diastereomers). These alcohols would then dehydrate to afford isoterpinolene and isolimonene (not observed) under the strongly acidic conditions. Both proposed mechanisms are consistent with the higher amount of isoterpinolene observed in the Me2S-facilitated ACRO reactions of 3-carene (Fig. 4). No isolimonene was observed in any of the ACRO reactions of 3-carene, which is unsurprising as it would be predicted to rapidly isomerise to its more stable limonene and/or isoterpinolene isomers under strong acidic conditions.
Mechanisms of the acid-catalysed alkene isomerisation and polymerisation reactions of the p-MeD mixtures
The alkene bonds of the limonene/terpinolene products produced from the ACRO reactions of the pinenes can reversibly protonate under the strongly acidic conditions to afford reactive p-menthene carbenium ions 2, 8, 9 (and others not shown) that can then lose protons to generate other p-MeD isomers (Fig. 7). This results in a series of reversible, acid-catalysed alkene isomerisation reactions (via carbenium ion intermediates 2, 8–10) that combine to produce a thermodynamic mixture of p-MeDs over time that contain α-terpinene, γ-terpinene and isoterpinolene as major components.35 A similar reversible protonation mechanism can also operate to catalyse isomerisation of the isolimonene and isoterpinolene products produced from the ACRO reaction of 3-carene to generate identical thermodynamic p-MeD mixtures.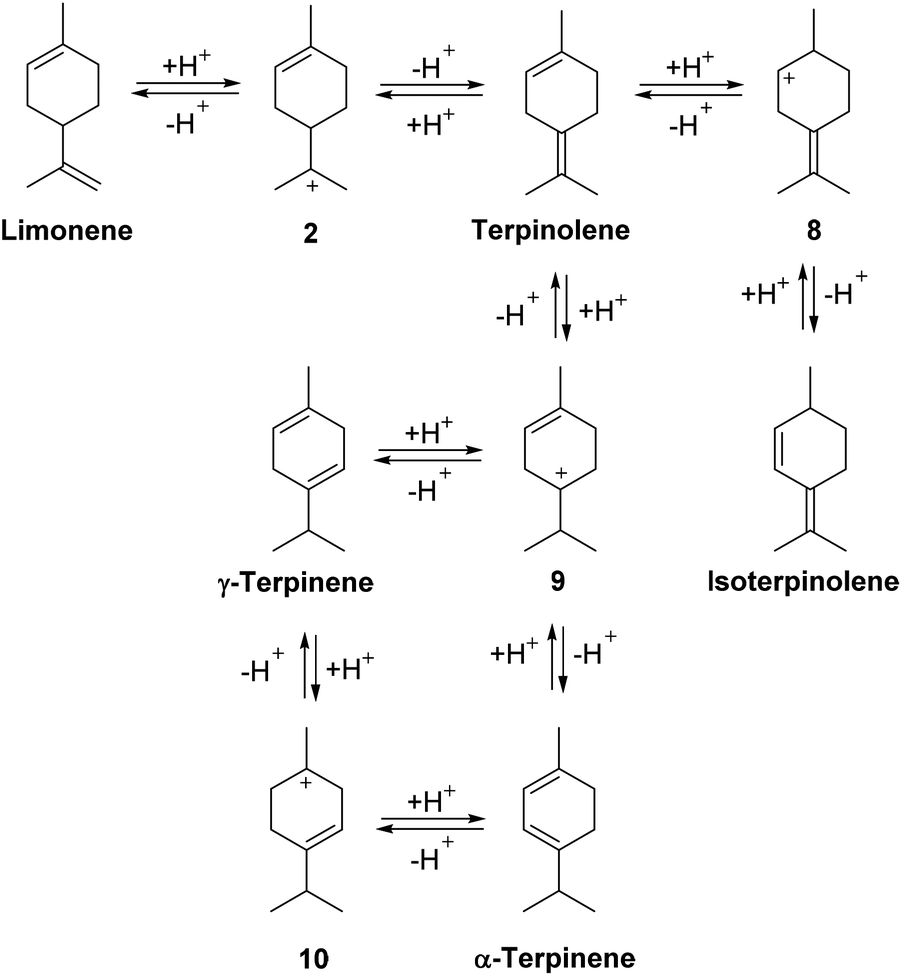 | ||
| Fig. 7 Reversible protonation of the alkene bonds of p-MeDs generates p-menthene carbenium ions 2, 8–10 that equilibrate under thermodynamic control to afford a mixture of p-MeDs that is rich in α-terpinene, γ-terpinene and isoterpinolene. | ||
The carbenium ions 2, 8–10 can also react reversibly with the alkene bonds of other p-MeD donors to afford dimeric carbenium species such as 11 that can eliminate protons to afford dimeric species containing two p-menthene sub-units (Fig. 8).36 Subsequent protonation of an alkene bond of these dimers can reversibly generate carbenium species 11, which can then undergo C–C σ-bond cleavage to produce two monomeric p-MeD species (i.e. depolymerisation), or react further with the alkene bond of a p-MeD-donor to afford trimeric carbenium species 12 (Fig. 8). Multiple repetitions of this type of cationic oligomerisation process, using different p-MeDs and oligomeric terpene carbenium ions as donor and propagating species then results in reversible formation of complex mixtures of oligomeric terpene species over time (Fig. 8).37 Irreversible chain transfer events, cross-polymerisation reactions and carbenium ion rearrangements then combine to produce irreversible oligomerisation steps that lead to increasing amounts of terpene polymer and decreasing amounts of p-MeD over time.
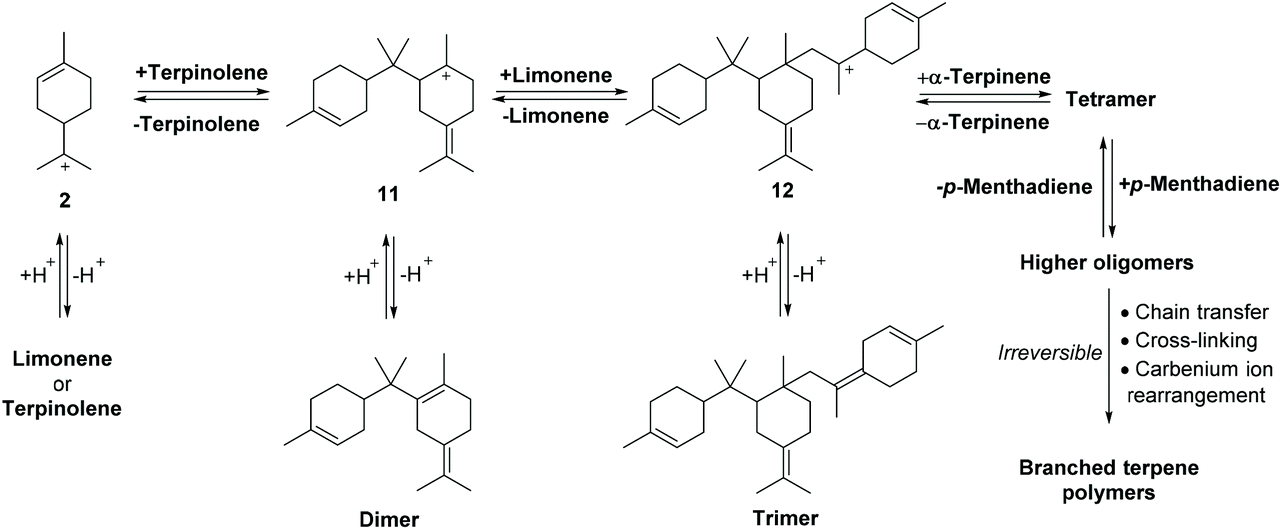 | ||
| Fig. 8 Reversible cationic polymerisation of monomeric/oligomeric carbenium species using different p-MeD donors affords a complex mixture of oligomeric and polymeric terpenoid species over time. Representative oligomerisation process shown for limonene, terpinolene and α-terpinene participating as p-MeD monomers in the oligomerisation process. HSO4− counterions of carbenium species omitted for clarity. | ||
Role of ∼3–5 mol% Me2S in accelerating the ACRO reactions of the bicyclic monoterpenes in CST
We then considered the role that Me2S might be playing in increasing the rate of the ACRO reactions of the pinenes and 3-carene, with the aim of identifying a mechanism that would also explain the anomalous reactivity profiles and unusual time-dependent variation in p-MeD yields.38 The rate-determining step in these ACRO reactions is known to involve initial protonation of the alkene bond of these bicyclic terpene substrates.32Similar p-MeD product ratio profiles were observed in the absence/presence of Me2S in the initial phases of both types of ACRO reaction, which indicated that similar reaction pathways were operating in both cases. A review of the literature revealed two patents describing that use of small amounts of emulsifiers had been used to improve the efficiency of ACRO reactions of pinenes/turpentine for the synthesis of α-terpineol.19b,39 Another key patent described that use of 0.1–1.0% of Polyrad 1100™ (a tertiary amine polymer produced from reaction of dehydroabietyl amine with ethylene oxide) as a cationic surfactant in the biphasic ACRO reaction of 3-carene [35% aq. H2SO4 (∼6 M aq.), 108 °C] was necessary to produce good yields (87%) of p-MeD mixtures after 3 h.40 The reaction conditions and yield/composition of the p-MeDs reported in this surfactant-mediated ACRO reaction of 3-carene were remarkably similar to the results obtained in our Me2S-facilitated ACRO reactions of CST. Consequently, this led us to consider whether a similar surfactant effect was operating to account for the rate acceleration effects in our 5 mol% Me2S-containing ACRO reactions. This would also provide an explanation as to why the rate of these ACRO reactions was not increased when >5 mol% Me2S was used, since surfactant concentrations of 0.1–1.0 mol% Polyrad 1100™ were shown to be optimal in the surfactant-mediated ACRO reactions of 3-carene.40 This surfactant hypothesis was also consistent with a previous report that addition of weak emulsifiers to ACRO reactions of highly purified turpentine reactions is beneficial for producing improved p-MeD yields.28
The pKaH of Me2S is −7.0 which means that it is essentially unprotonated under the strong acidic conditions used in these ACRO reactions. This means that Me2S can potentially act as a nucleophile to intercept terpene carbenium species in situ to reversibly generate water-soluble bicyclic 13, monocyclic 14 and oligomeric 15–18 terpene sulfonium salts (Fig. 9).41 Cationic sulfonium salts are known to be good surfactants that could potentially increase the degree of mixing of the organic and aqueous layers of the biphasic ACRO systems.42 More efficient mixing would lead to better mass transfer between the aqueous and organic layers of the ACRO reactions, resulting in acidity levels at the aqueous–organic interface being relatively increased. Formation of a more acidic interfacial environment would result in faster protonation of the alkene bonds of the bicyclic terpenes, thus accelerating the rate-determining step of these ACRO reactions (Fig. 10). Consequently, high resolution mass spectrometry (HRMS) was used to analyse for the presence of any monomeric and/or oligomeric terpene sulfonium salts in the CST ACRO reactions. Significant accurate mass peaks corresponding to monomeric C12H23S+ (199.1520), dimeric C22H39S+ (335.2775) and even trimeric C32H55S+ (471.4008) sulfonium ion species were detected in both the organic and aqueous layers of the ACRO reaction (ESI, Fig. S18–20†). Crucially, this demonstrates that the terpene sulfonium species can partition between both the aqueous and organic phases, thus enabling them to function as cationic surfactants that promote better mixing, improved mass transfer and faster ACRO reactions. As expected, [M + H]+ ions for the parent bicyclic monoterpenes, p-MeDs and their corresponding oligomers were also observed in the HRMS spectra of both aqueous and organic phases of the ACRO reaction (ESI, Fig. S17†).
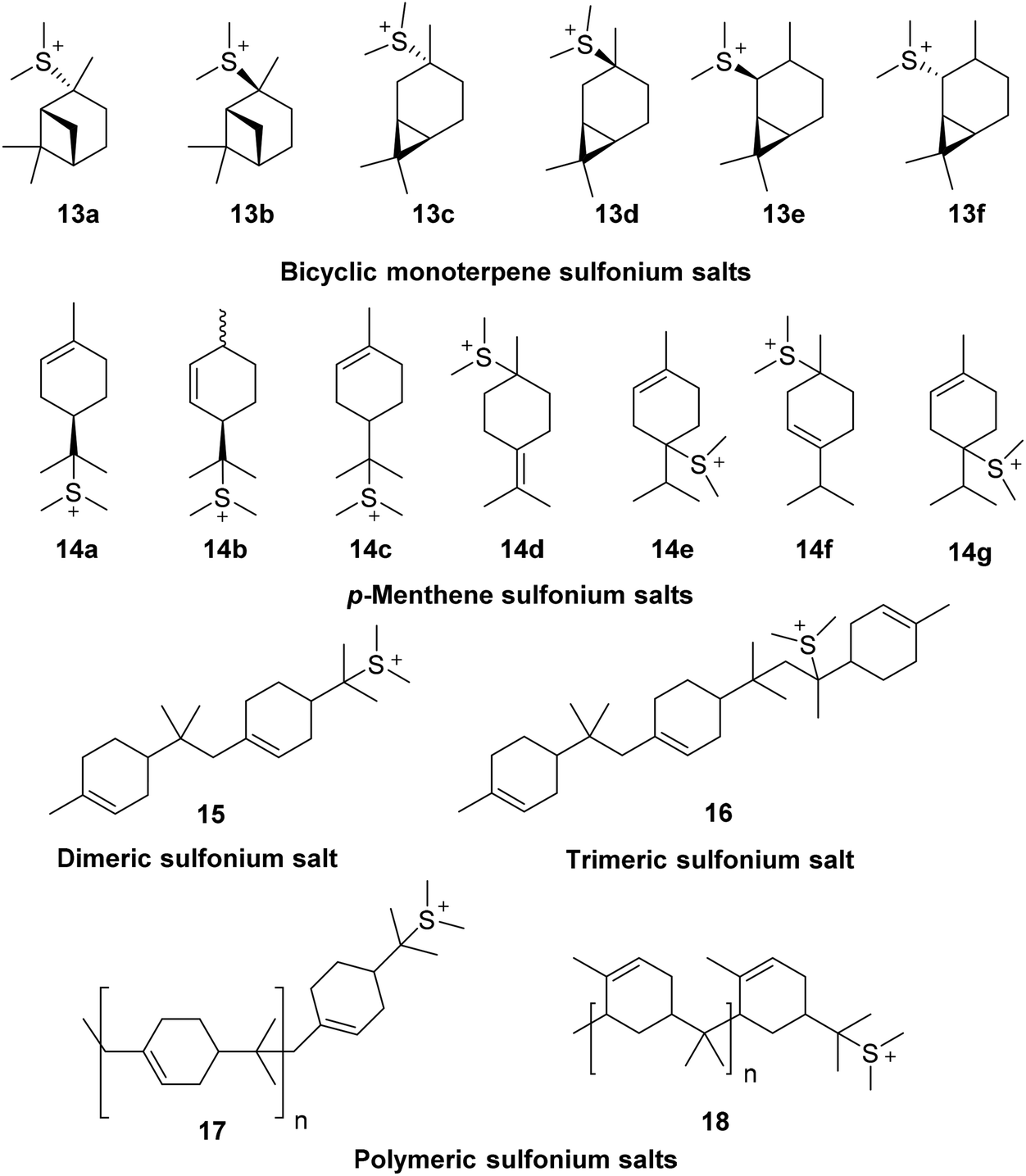 | ||
| Fig. 9 Monomeric, dimeric, trimeric and polymeric terpene sulfonium salts formed from interception of terpene carbenium ions by Me2S in quasi-living cationic polymerisation reactions. Polymeric sulfonium salts drawn as representative homo-polymers of β-pinene and α-pinene for simplicity. HSO4− counterions of carbenium species omitted for clarity. | ||
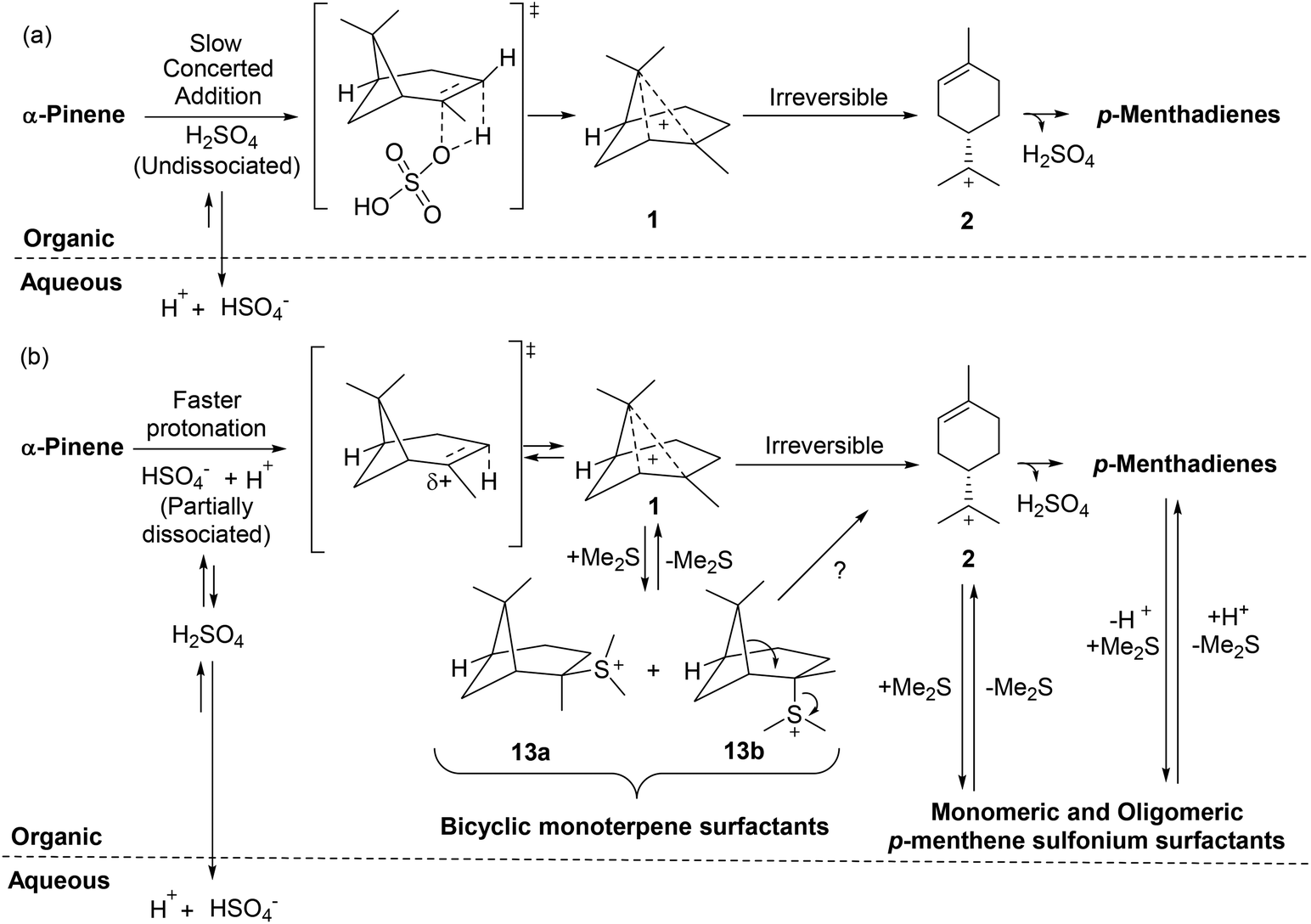 | ||
| Fig. 10 (a) Slow hydrolysis of α-pinene in the absence of Me2S proceeds via an undissociated acid mechanism.45 (b) Faster hydrolysis of α-pinene occurs through a more dissociated acid mechanism caused by formation of terpene sulfonium salts that act as surfactants to increase mixing at the interfacial layer to create a more acidic microenvironment that increases the overall rate of the ACRO reaction. HSO4− counterions of carbenium species omitted for clarity. | ||
In situ formation of terpene sulfonium surfactant species can also be used to explain the latency period observed in the initial phase of these Me2S-mediated ACRO reactions. These delays are likely to be caused by the time taken for a sufficient concentration of terpene sulfonium salts to build up to enable it to facilitate efficient mixing of the water–organic interface.43 Similarly, the time-dependent rate acceleration effects in these ACRO can also be explained by the generation of increasing levels of in situ generated sulfonium surfactants producing a corresponding increase in interfacial acidity levels over time. The increased rate of the ACRO reaction of the α-pinene fraction in GT would be caused by faster ring-opening of its β-pinene fraction resulting in more rapid production of sulfonium surfactants over time. This would lead to a faster net increase in the acidity of the interfacial layer than occurs in the corresponding ACRO reaction of pure α-pinene, thus explaining the observed difference in reactivities.
Reversible formation of oligomeric sulfonium species 15–18 in these ACRO reactions can also explain the ‘time-lag’ effects observed between the degree of consumption of β-pinene and the amount of p-MeD product produced over time. The water soluble oligomeric sulfonium species (e.g.15–18) contain multiple p-menthene monomer units whose structures are stabilised by the presence of a single charged sulfonium head-group. Their reversible formation could explain how the 79% p-MeD yield in the ACRO reaction of β-pinene at full conversion (after 15 min) was found to increase by 8% to afford an 87% yield after 45 min. This would be caused by initially formed kinetic mixtures of oligomeric sulfonium species 15–18 (at 15 min) equilibrating into monomeric p-MeD products under thermodynamic control (from 15–45 min). Perturbation of the equilibria from oligomeric terpene species towards monomeric p-MeD species over this period could potentially be driven by isomerisation of initially-formed limonene/terpinolene ring-opened products into their thermodynamically more stable α-terpinene, γ-terpinene and isoterpinolene isomers.44
The more acidic water/organic interface produced by these types of p-menthene sulfonium surfactants also provide a simple explanation for the rate increases in the p-MeD isomerisation/oligomerisation reactions in the presence of Me2S. This is because the rate-determining steps of both these type of reactions involve protonation of the alkene bonds of monomeric and oligomeric p-menthene species, so a more acidic interfacial region will result in faster reactions in both cases.
The greater rate of polymerisation of the p-MeDs produced in the ACRO reaction of β-pinene over time (when compared to α-pinene) indicate that the structures of the oligomeric species formed can significantly affect the efficiency of subsequent p-MeD oligomerisation/polymerisation processes. Reversible formation of sulfonium species in polyisobutylene reactions has previously been used to control the efficiency of living chain polymerisation reactions of isobutylene to produce uniform polymers with narrow molecular weight distributions.46 Therefore, similar quasi-living effects may also be operating in the oligomerisation reactions of the p-MeDs, with reversible formation of stable terpene sulfonium intermediates resulting in more controlled oligomerisation pathways. These terpene sulfonium intermediates may also increase the chances of non-reversible reactions (e.g. chain transfer, cross-linking and carbenium ion rearrangement) occurring to favour polymer formation in the presence of Me2S over extended periods of time.
Conclusions
This paper describes the development of a highly practical acid-catalysed method (6 M aq. H2SO4, 90 °C, 4 h) for converting the bicyclic monoterpenes present in untreated CST into mixtures of p-MeDs in good 70–75% yields. Mechanistic investigations have revealed that the ∼3% Me2S present as a contaminant in CST plays a key role in accelerating the acid-catalysed ring-opening reactions of its bicyclic terpene components. Reaction monitoring and HRMS analysis of these ACRO reactions suggests that Me2S reacts with the alkene bonds of monoterpenes to generate in situ terpene sulfonium salts that can serve as surfactants to improve mixing of the aqueous and organic layers of these biphasic ACRO reactions. Increased mass transfer effects then produce a more acidic aqueous–organic interface, facilitating faster protonation of the alkene bonds of the bicyclic terpenes in the rate-determining step of their ACRO reactions. Addition of 5 mol% Me2S to the ACRO reactions of GT and cineole has been used to facilitate their efficient conversion into mixtures of p-MeDs in similarly good yields and shorter reaction times. Ongoing studies directed towards using unpurified p-MeD mixtures generated in these sulfurous ACRO reactions as biorenewable feedstocks for the production of bio-p-cymene and a range of useful chemical products within a terpene biorefinery context will be reported in due course.Experimental
General experimental details
All reagents were purchased from commercial suppliers apart from CST which was obtained from a Swedish paper mill owned by Södra Forestry and Barrettine Genuine Turpentine that was purchased from a local hardware store. All reactions were carried out under air. Nuclear magnetic resonance spectra were recorded using either a Bruker Avance 300, 400 or 500 MHz spectrometer, or an Agilent Technologies 500 MHz spectrometer. All 13C spectra were proton decoupled (13C{1H}) and all spectra run in CDCl3. Chemical shifts (δ) are reported in parts per million (ppm) and are referenced to residual solvent peaks. HRMS analysis was carried out via direct injection using a Bruker MaXis HD ESI-QTOF mass spectrometer. Representative NMR spectra used to analyse and characterise the mixtures of p-MeDs produced in this study, as well as HRMS spectra used to identify the presence of terpene sulfonium species, can be found in the ESI.†Reaction monitoring experiments
A mixture of CST (5.0 mL, 4.35 g, 32.0 mmol) and 1,2,4,5-tetramethylbenzene (0.43 g, 3.2 mmol, 10 mol%, inert) was heated to 90 °C and stirred at 500 rpm. Aq. H2SO4 (1.0 mL, 6 M aq.) was added in one portion and the reaction stirred at 90 °C for 7 h. Stirring was periodically stopped and the two layers allowed to separate and settle before an aliquot of the organic layer was taken and diluted with CDCl3 for 1H NMR analysis. This reaction monitoring protocol was repeated using pure α-pinene, β-pinene, 3-carene and GT, both with and without addition of Me2S (0.12 mL, 0.10 g, 1.6 mmol, 5 mol%) to the organic layer at the start of the reaction.Preparative ACRO reactions of CST
CST (12.0 mL, 10.44 g, 76.8 mmol) was heated to 90 °C and stirred at 500 rpm. Aq. H2SO4 (2.4 mL, 6 M aq.) was added in one portion and the reaction stirred at 90 °C for 4 h. Stirring was then stopped and the organic and aqueous layers allowed to separate and cool to rt. The organic layer was then decanted off and distilled under reduced pressure to afford a distillate (b.p. = 170–185 °C, 760 mmHg) containing a mixture of monomeric p-MeDs [7.52 g, 55.3 mmol, major components = α-terpinene, γ-terpinene, isoterpinolene and p-cymene] in 72% yield and a brown terpene oligomer/polymer residue in 22% yield. The acidic aqueous layer could be recovered and reused in subsequent ACRO reactions as required.Preparative ACRO reactions of GT
GT (12.0 mL, 10.44 g, 76.8 mmol) and Me2S (0.28 mL, 0.24 g, 3.8 mmol, 5 mol%) were heated to 90 °C and stirred at 500 rpm. Aq. H2SO4 (2.4 mL, 6 M aq.) was added in one portion and the reaction stirred at 90 °C for 4 h. Stirring was then stopped, and the organic and aqueous layers allowed to separate and cool to rt. The organic layer was then decanted off and distilled under reduced pressure to afford a distillate (b.p. = 170–185 °C, 760 mmHg) containing a mixture of monomeric p-MeDs [9.03 g, 66.4 mmol, major components: α-terpinene, γ-terpinene, isoterpinolene and p-cymene] in 87% yield and a brown terpene oligomer/polymer residue in 11% yield. The acidic aqueous layer could be recovered and reused in subsequent ACRO reactions as required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the EPSRC for funding through the Centre for Doctoral Training in Sustainable Chemical Technologies (EP/L016354/1). Södra Forestry Cooperative are thanked for supplying an authentic industrial sample of CST.Notes and references
- (a) B. Holmbom and T. Salmi, Catal. Rev., 2007, 49, 197–340 CrossRef; (b) A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC; (c) C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC; (d) R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS.
- N. Tsolakis, W. Barn, J. S. Srai and M. Kumar, J. Cleaner Prod., 2019, 222, 802–822 CrossRef CAS.
- W. Schwab, C. Fuchs and F. C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3–8 CrossRef CAS.
- A. Arneth, R. K. Monson, G. Schurgers, Ü. Niinemets and P. I. Palmer, Atmos. Chem. Phys. Discuss., 2008, 8, 7017–7050 CrossRef.
- (a) M. GScheidmeier and H. Fleig, Turpentines, in Ullman's encyclopedia of industrial chemistry, Wiley, 2012, pp. 537–550 Search PubMed; (b) R. Patt, O. Kordsachia and R. Suttinger, Pulp, in Ullman's encyclopedia of industrial chemistry, Wiley, 2012, pp. 477–540 Search PubMed; (c) E. Sjöström, Wood chemistry, fundamentals and applications, Academic Press Inc., London, 2nd edn, 1993 Search PubMed.
- The Kraft paper pulping process involves high pressure (7–13 bar) heating of wood chips at 150–180 °C in the presence of a caustic solution of ‘white liquor’ comprised of aqueous sodium hydroxide, sodium sulfide, sodium carbonate and small quantities of other sodium salts (e.g. sodium sulfate and sodium bisulfite). This process affords a black liquor filtrate that may be recycled for further pulp processing, or separated into its individual alkali lignin (500 kg per tonne pulp), oligosaccharides/sugars (up to 400 kg per tonne pulp), tall oil (fatty acids, rosin, and sterols) (up to 100 kg per tonne pulp) and CST (15 kg per tonne pulp) components. All of these fractions can potentially be used for the production of biorenewable chemical products. See: G. J. H. Buisman and J. H. M. Lange, in Industrial Biorenewables: A Practical Viewpoint, John Wiley & Sons, 2016, pp. 21–62 Search PubMed.
- For an excellent review on the production/uses of CST see: E. S. Izmest'ev, S. A. Rubtsova and A. V. Kutchin, Theor. Appl. Ecol., 2019, 12–22 Search PubMed.
- A. J. D. Silvestre and A. Gandini, in Terpenes: Major Sources, Properties and Applications in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, ch. 2, pp. 17–38 Search PubMed.
- R. Höfer, in The Pine Biorefinery Platform Chemicals Value Chain in Industrial Biorefineries & White Biotechnology, ed. A. Pandey, R. Höfer, M. Taherzaden and K. M. Nampoothiri, Elsevier, 2006, ch. 3b Search PubMed.
- P. Knuuttila, Fuel, 2013, 104, 101–108 CrossRef CAS.
- C. Sell, in The Chemistry of Fragrances: From Perfumer to Consumer, RSC, 2006, pp. 52–131 Search PubMed.
- (a) M. Eggersdorfer, Terpenes, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2000, pp. 29–43 Search PubMed; (b) J. H. Clark, D. J. MacQuarrie and J. Sherwood, Green Chem., 2012, 14, 90–93 RSC; (c) P. Osswald, R. Whitsand, J. Schaffer and M. Kohler, Fuel, 2017, 187, 43–50 CrossRef CAS.
- F. Neatu, G. Culica, M. Florea, V. I. Parvulescu and F. Cavani, ChemSusChem, 2016, 9, 3102–3112 CrossRef CAS.
- R. A. Sheldon, I. Arends and U. Hanefeld, Introduction: Green Chemistry and Catalysis, Wiley, 2007, pp. 165–166 Search PubMed.
- (a) F. H. Thurber and L. J. Roll, Ind. Eng. Chem., 1927, 19, 739–742 CrossRef; (b) E. Kugler and E. sz. Kováts, Helv. Chim. Acta, 1963, 46, 1480–1513 CrossRef.
- M. Golets, S. Ajaikumar and J. P. Mikkola, Chem. Rev., 2015, 115, 3141–3169 CrossRef CAS.
- (a) J. A. Linnekoski, M. Asikainen, H. Heikkinen, R. K. Kaila, J. Räsänen, A. Laitinen and A. Harlin, Org. Process Res. Dev., 2014, 18, 1468–1475 CrossRef CAS; (b) F. Al-Wadaani, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2009, 363, 153–156 CrossRef CAS; (c) M. Golets, S. Ajaikumar, M. Mohln, J. Wärnå, S. Rakesh and J. Mikkola, J. Catal., 2013, 307, 305–315 CrossRef CAS; (d) H. Jaramillo, L. A. Palacio and L. Sierra, Stud. Surf. Sci. Catal., 2002, 1, 1291–1298 CrossRef; (e) T. Kawahara, Y. Henmi, N. Onda, T. Sato, A. Kawai-Nakamura, K. Sue, H. Iwamura and T. Hiaki, Org. Process Res. Dev., 2013, 17, 1485–1491 CrossRef CAS; (f) L. Wang, X. Chen, X. Xu, X. Wei, L. Wang and Z. Tong, Shipin Gongye Keji, 2010, 31, 197–199 CAS; (g) A. G. Heochst, Ger. Pat, 19521225A1, 1996 Search PubMed; (h) D. M. Roberge, D. Buhl, J. P. M. Niederer and W. F. Hölderich, Appl. Catal., A, 2001, 215, 111–124 CrossRef CAS; (i) B. A. Leita, A. C. Warden, N. Burke, S. O. Shea and D. Trimm, Green Chem., 2010, 12, 70–76 RSC.
- Significant volumes of biorenewable p-cymene were historically available as a major by-product of the sulfite process used to transform wood chips into pulp. This process involves digestion of wood chips with calcium bisulfite and SO2 (acid), magnesium bisulfite (pH 4.0), or sodium sulfite/ammonia (neutral). However, the sulfite process has largely been superseded by the Kraft process and so p-cymene is not now generally available from this source. See: (a) Ref. 5; (b) J. Poerschmann, A. Goerig, H. J. Schoenert and K. Guenther, Chem. Tech., 1990, 42, 73–74 CAS.
- A number of industrial processes currently employ alternative acidic conditions to convert turpentine/α-pinene feedstocks into commercially valuable α-terpineol and/or camphene that invariably produce significant amounts of up to 30% of p-MeD mixtures (e.g. limonene and terpinolene) as waste by-products that are sold as technical grade dipentene for solvent/biofuel applications. (a) R. W. Charlton and A. R. Day, Ind. Eng. Chem., 1936, 29, 92–95 CrossRef; (b) Haarmann & Reimer GMBH, Ger. Pat, DE4111900A1, 1992 Search PubMed; (c) V. T. Murakami, I. O. Marques and R. Cella, ChemistrySelect, 2019, 4, 8800–8806 CrossRef CAS; (d) N. Wijayati, H. D. Pranowo, H. D. Pranowo and T. Jumina, Indones. J. Chem., 2013, 13, 59–65 CrossRef CAS; (e) N. Wijayati, H. D. Pranowo, H. D. Pranowo and T. Jumina, Int. J. Chem. Eng. Appl., 2013, 4, 178–182 CAS; (f) Heyden Newport Chemical Corp., US Pat, US2866826, 1958 Search PubMed.
- W. B. Cunningham, J. D. Tibbetts, M. Hutchby, K. A. Maltby, M. G. Davidson, U. Hintermair, P. Plucinski and S. D. Bull, Green Chem., 2020, 22, 513–524 RSC.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS.
- (a) L. Liu, J. Liao, W. Duan, G. Lin and F. Lei, Lett. Org. Chem., 2015, 17, 283–289 CrossRef; (b) Y. P. Yen, M. J. Yeh and W. F. Hsiao, J. Pestic. Sci., 2007, 32, 49–52 CrossRef CAS.
- D. M. Roberge, D. Buhl, J. P. M. Niederer and W. F. Hölderich, Appl. Catal., A, 2001, 215, 111–124 CrossRef CAS.
- Around 7000 tonnes of eucalyptus oil (primarily cineole) is harvested annually from Eucalyptus and Camphor Laurel trees. J. Panten and H. Surburg, Flavors and Fragrances, 4. Natural Raw Materials, in Ullman's encyclopedia of industrial chemistry, Wiley, London, 2012, pp. 1–58 Search PubMed.
- (a) R. Rachwalik, M. Hunger and B. Sulikowski, Appl. Catal., A, 2012, 427–428, 98–105 CrossRef CAS; (b) A. I. Bokin, B. I. Kutepov, A. N. Khazipova, E. A. Travkin, N. A. Shchadneva, R. I. Khusnutdinov and U. M. Dzhemilev, Russ. J. Appl. Chem., 2003, 76, 234–237 CrossRef; (c) M. C. C. Costa, R. A. W. Johnstone and D. Whittaker, J. Mol. Catal. A: Chem., 1996, 104, 251–259 CrossRef; (d) N. Nuttens, D. Verboekend, A. Deneyer, J. Van Aelst and B. F. Sels, ChemSusChem, 2015, 8, 1197–1205 CrossRef CAS; (e) A. B. Holmen, Wo. Pat, WO2015023225, 2015 Search PubMed; (f) M. Akizuki and Y. Oshima, Ind. Eng. Chem. Res., 2017, 56, 6204–6212 CrossRef CAS; (g) T. Szuppa, A. Stolle and B. Ondruschka, Org. Biomol. Chem., 2010, 8, 1560–1567 RSC.
- No evidence of any m-menthadienes (>3%) formed from competing fragmentation of the C6 cyclopropyl of 3-carene and/or the C6 cyclobutyl bonds of α-pinene/β-pinene were observed in the ACRO reactions carried out in this study.
- The color of the aqueous H2SO4 layer darkened between each successive ACRO run, however this colour change did not affect the 3.5 h reaction time taken to consume CST between each run.
- The results of these Me2S-free ACRO reactions are consistent with results of previous ACRO reactions of α-pinene using different amounts of 50–60 mol% H2SO4 at 50–60 °C over 7–9 h periods, which produced p-MeD mixtures containing 5–30% unreacted α-pinene. See: Heyden Newport Chemical Corp., US Pat, 2799717, 1957 Search PubMed.
- The increased reactivity of untreated CST over pure turpentine fractions in ACRO reactions has been noted previously (no explanation given), see: A. G. Hoechst, UK Pat, GB789809, 1958 Search PubMed.
- For example, batch distillation of CST (10000 kg) containing 0.26% MeSH (b.p. 6 °C, 0.13% Me2S (b.p. 37 °C) and 0.10% Me2S2 (110 °C) [∼0.35% Combined Sulfur Compounds (CSC)] at 760 mmHg gave: a head fraction (100 kg) distilling between 34–90 °C containing 18.5% CSC; an intermediate fraction (200 kg) distilling between 90–150 °C containing 0.5% CSC; a product fraction (6700 kg) distilling between 150–170 °C containing 0.02% CSC, and a still residue (3000 kg) containing 0.5% CSC. See: I. I. Efishev, A. V. Prokhorov and A. P. Matyushkina, Bum. Prom-st., 1954, 29, 23–25 CAS.
- R. B. Bates, E. S. Caldwell and H. P. Klein, J. Org. Chem., 1969, 34, 2615–2617 CrossRef CAS.
- C. M. Williams and D. Whittaker, J. Chem. Soc. B, 1971, 668–672 RSC.
- Precedent that water can undergo nucleophilic SN2 type attack at gem-dimethyl centres to trigger ring-fragmentation reactions of bicyclic terpene carbenium ions is available from studies on acid-catalysed hydration reactions of optically active thujene and sabinene that are reported to afford optically active terpinen-4-ol. See: (a) T. Norin and L. A. Smedman, Acta Chem. Scand., 1971, 25, 2010 CrossRef CAS; (b) I. Cristea, E. Kozma and A. Ritiu, Stud. Univ. Babes-Bolyai, Chem., 2000, 45, 257–261 CAS.
- A. Y. Sidorenko, A. Aho, J. Ganbaatar, D. Batsuren, D. B. Utenkova, G. M. Sen'kov, J. Warna, D. Y. Murzin and V. K. Agabekov, Mol. Catal., 2017, 443, 193–202 CrossRef CAS.
- Gas-phase isomerisation of α-terpinene at 250 °C over an Al2O3 catalyst afforded an equilibrium mixture containing α-terpinene (44%), isoterpinolene (22%) and γ-terpinene (16%) as major components. See: Z. A. Filippenko, O. M. Baranov, G. N. Roganov and G. Y. Kabo, Khim. Prir. Soedin., 1985, 51–54 CAS.
- H. A. Meylemans, R. L. Quintana and B. G. Harvey, Fuel, 2012, 97, 560–568 CrossRef CAS.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- An alternative mechanism involving aerobic oxidation of Me2S to DMSO that might then act as a polar solvent to increase mixing of the organic/aqueous layers of these ACRO reactions was discounted because this process does not explain the time-dependent variations in p-MeD yield and the increase in rate of the ACRO reaction of α-pinene in the presence of β-pinene in GT.
- Krems Chemie GmbH, Eur. Pat, EP0035703A1, 1982 Search PubMed.
- L. L. C. Hercules, US Pat, US3422029A, 1969 Search PubMed.
- For a report of acid-catalysed addition of Me2S to the alkene bonds of terpenes to afford stable sulfonium salts, see: B. Badet, M. Julia, J. M. Mallet and C. Schmitz, Tetrahedron, 1988, 44, 2913–2924 CrossRef CAS.
- B. Z. Putlitz, H. P. Hentze, K. Landfester and M. Antonietti, Langmuir, 2000, 16, 3214–3220 CrossRef.
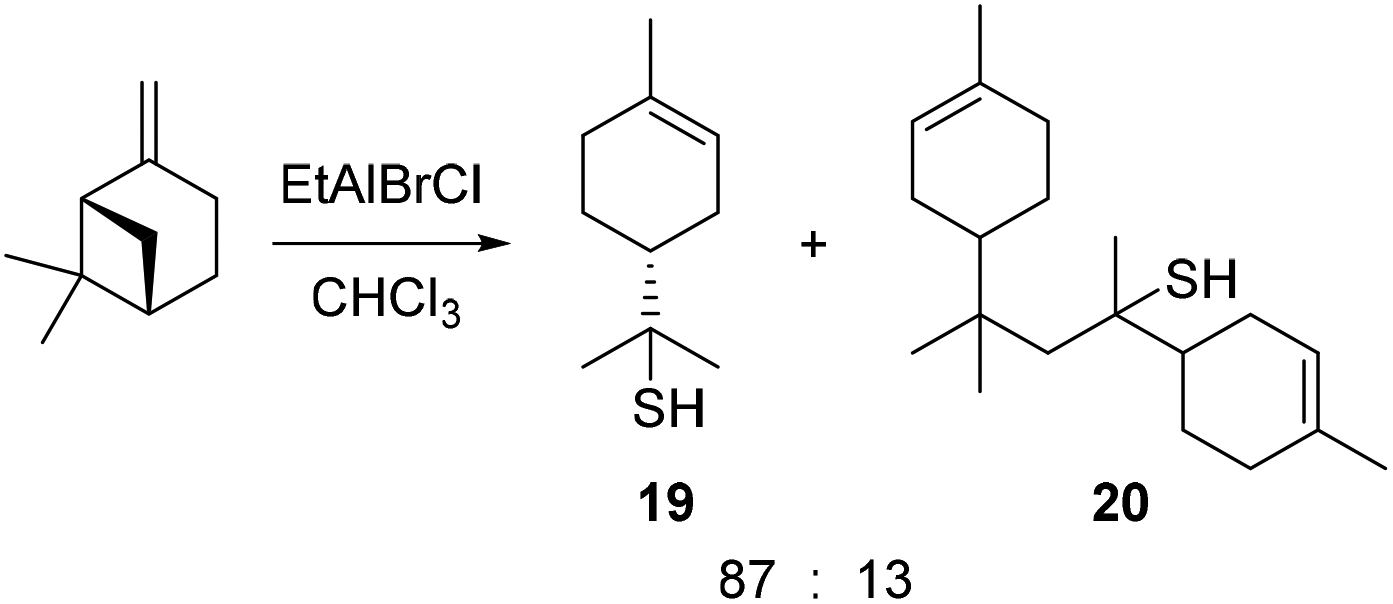
- Reaction of β-pinene with H2S and EtAlBrCl gave pinane thiol 19 and a dimer 20, thus demonstrating that p-menthene carbenium ions can be intercepted by thiol nucleophiles, see: G. A. Tostikov, F. Y. Kanzafarov, U. M. Dzhemilev, U. M. Kantyukova and L. M. Zelenova, Zh. Org. Khim., 1983, 19, 2075–2081 Search PubMed .
. - The reversibility of these type of cationic polymerisation pathways has been demonstrated for the acid-catalysed depolymerisation of isobutylene in the presence of a suitable arene acceptor, see: C. B. Watson, D. Tan and D. E. Bergbreiter, Macromolecules, 2019, 52, 3042–3048 CrossRef CAS.
- C. M. Williams and D. Whittaker, J. Chem. Soc. D, 1970, 960–961 RSC.
- D. L. Morgan, C. D. Stokes, M. A. Meierhoefer and R. F. Storey, Macromolecules, 2009, 42, 2344–2352 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03452e |
| This journal is © The Royal Society of Chemistry 2021 |