
Organic photoredox catalyzed C–H silylation of quinoxalinones or electron-deficient heteroarenes under ambient air conditions†
Changhui
Dai
,
Yanling
Zhan
,
Ping
Liu
* and
Peipei
Sun
 *
*
School of Chemistry and Materials Science, Jiangsu Provincial Key Laboratory of Material Cycle Processes and Pollution Control, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, Nanjing Normal University, Nanjing 210023, People's Republic of China. E-mail: sunpeipei@njnu.edu.cn; pingliu@njnu.edu.cn
First published on 12th December 2020
Abstract
Direct C–H silylation of quinoxalinones was achieved by the combination of organic photoredox catalysis and hydrogen atom transfer (HAT) under ambient air conditions. Transition metal- and external oxidant-free conditions were the major features of this protocol. A series of silylated quinoxalinones with broad functional groups had been synthesized in moderate to high yields. This methodology was also applicable for the C–H silylation of some electron-deficient heteroarenes.
There has been growing interest in the biological activity evaluation of organosilanes because of their high lipophilicity and other special properties.1 Organosilicon molecules also play important roles in the field of functional materials.2 It is well-known that heteroarene skeletons exist in a large number of marketed drugs;3 therefore, the introduction of a silicon motif to heteroarenes to access heteroarylsilanes has continuously gained much attention in the past decades. The conventional methodologies for the introduction of silyl substituents to heteroarenes involved transition-metal-catalyzed cross-couplings of aryl halides4 or aryl esters5 with hydrosilanes or disilanes (Scheme 1a). The direct C–H silylation has emerged as one of the most attractive alternatives without the need for the prefunctionalization of heteroarenes. A very early example involved the use of a silicon electrophile Me3SiOTf to carry out the Friedel–Crafts C–H silylation of heteroarenes (Scheme 1b).6 These reactions could be promoted effectively by a Lewis acid7 or Brønsted acid.8 In 2015, Grubbs and co-workers discovered that the catalytic C–H bond silylation of aromatic heterocycles could occur in the presence of hydrosilanes and a strong base KOtBu (Scheme 1c).9a–b Moreover, a recent example of the site-selective sp2 C–H silylation of (poly)azines was demonstrated by Martin's group using a silicon reagent R3SiBPin and a strong base KHMDS (Scheme 1c).9c Transition-metal complexes based on Ir,10a–d Rh,10e and Ru10f–h have also been proved to be effective in C–H/Si–H cross-coupling to form C–Si bonds (Scheme 1d). Despite the significant advances such as the above examples having been made, several challenges, for instance, reducing the amount of strong acids or bases and developing more transition-metal-free conditions, still remain for chemists.
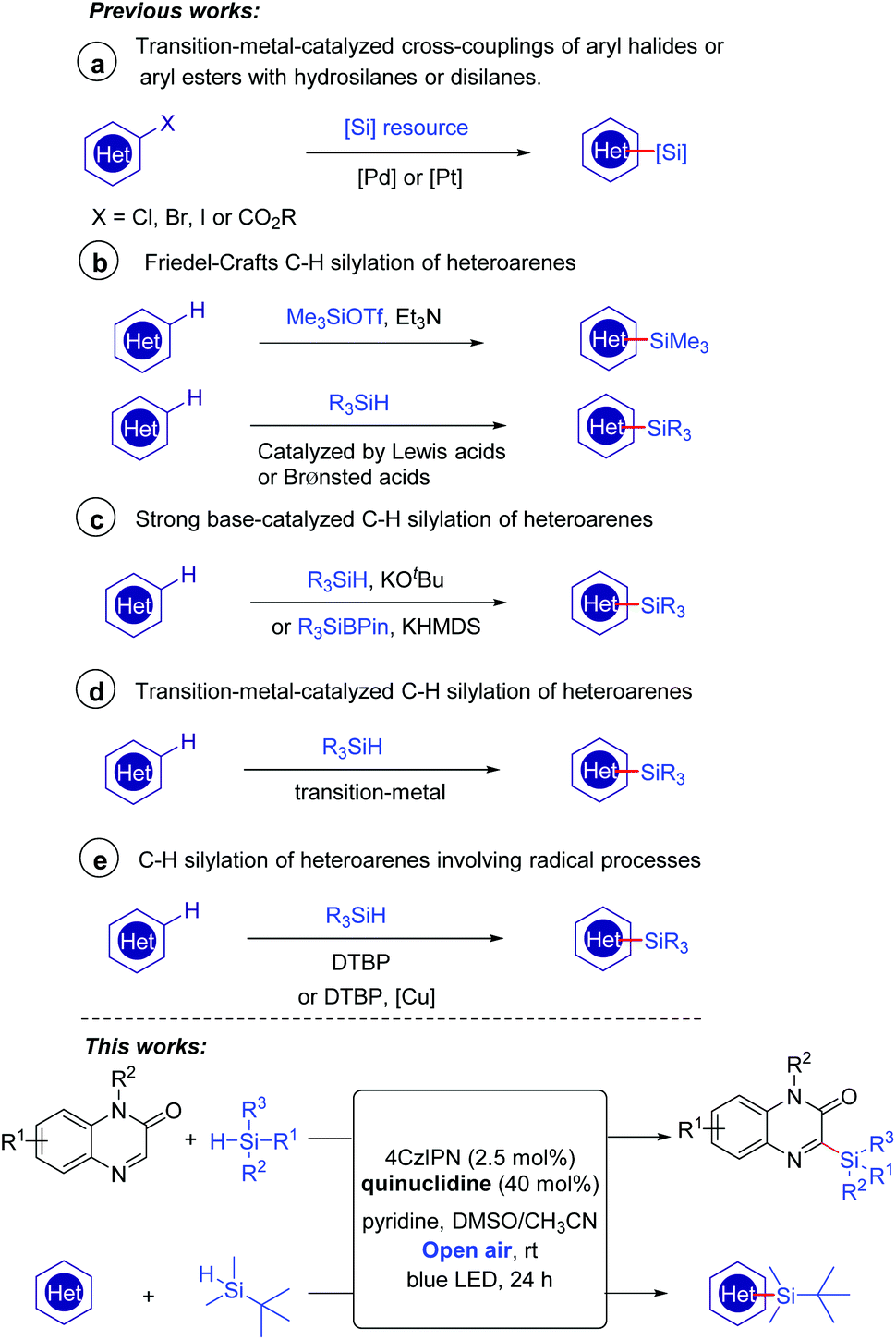 | ||
| Scheme 1 Strategies for the direct silylation of heteroarenes. | ||
Silyl radicals, which could be generated from hydrosilanes in the presence of peroxides,11 were used in the difunctionalization of alkenes12 or radical addition cascade cyclization13 to synthesize silylated products. However, the direct C–H silylation of heteroarenes through a radical pathway has rarely been reported. Recently, DTBP or DTBP/copper salts were utilized to mediate the C–H silylation of heteroaromatic compounds including furans, thiophenes, coumarins, indole, pyridine, and quinoline under heating conditions by the groups of Cai, Liu and Maruoka, respectively (Scheme 1e).14 In our recent works, a selective C-5 oxidative radical silylation of imidazopyridines was developed by using DTBP as the oxidant.15 The visible-light-induced photoredox catalysis of radical reactions has evoked much attention in the past decade since these reactions can occur under very mild reaction conditions.16 In 2019, Wang, Zhang and co-workers developed a photocatalytic C–H silylation of heteroarenes by using trialkylhydrosilanes as the silyl source in the presence of [Ir(ppy)2(dtbbpy)]PF6 and Na2S2O8.17 Both electron-deficient and -rich heteroarenes were tolerated in this transformation. Organic dyes are typically less expensive and less toxic and, therefore, have been used as attractive alternatives to transition-metal complexes in photoredox catalysis.18 Organic photoredox catalysis in combination with aerobic oxidation provides environmentally benign and efficient synthesis procedures using O2 as the sole oxidant.19 As part of our continuing efforts on the direct C–H functionalization of heteroarenes,15,20 herein, we disclose the C–H silylation of quinoxalinones and some other electron-deficient heteroarenes by the combination of organic photoredox catalysis and hydrogen atom transfer under ambient air conditions.
Initially, we selected 1-methylquinoxalin-2(1H)-one (1a) and tert-butyldimethylsilane (2a) as the model substrates to evaluate the photochemical reaction (Table 1; for details, see the ESI†). Using Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst, quinuclidine (A) as the hydrogen atom transfer (HAT) catalyst and K2CO3 as the base, the model reaction was performed in CH3CN under blue light irradiation and open air. We were delighted to obtain the silylated product 3-(tert-butyldimethylsilyl)-1-methylquinoxalin-2(1H)-one (3a) in 31% yield after 24 h (entry 1). The reaction could not be carried out in the absence of a photocatalyst (entry 2). Among the several photocatalysts we examined, the organic photoredox catalyst 4CzIPN was the most appropriate one, providing 3a in 47% yield (entries 1, 3 and 4). The presence of the HAT catalyst is necessary for the formation of silyl radicals in the photochemical reaction.21a When triisopropylsilylthiol (B) was used as the HAT catalyst instead of quinuclidine, the desired product 3a was not observed (entry 5). To improve the reaction yield, another inorganic base Cs2CO3 as well as organic bases 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and pyridine were examined (entries 6–8). To our delight, the reaction efficiency was obviously improved when pyridine was employed, and the yield of 3a was further increased to 73% with 2 equiv. pyridine being added (entry 9). The results revealed that both light irradiation and O2 were necessary to promote the reaction (entries 10 and 11). In addition, the mixed solvent of DMSO and CH3CN with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio could increase the solubility of the substrates and give 3a in 77% yield (entry 12).
:1 volume ratio could increase the solubility of the substrates and give 3a in 77% yield (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Base (equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (1.0 mmol, 5.0 equiv.), photocatalyst (2.5 mol%), HAT catalyst (40 mol%), base and solvent (4 mL) were stirred under irradiation (12 W blue LEDs) at room temperature under open air for 24 h. b Isolated yield based on 1a. c Triisopropylsilylthiol (B) was used as the HAT catalyst. d Without light. e Under Ar (1 atm). | ||||
| 1 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | K2CO3 (1) | CH3CN | 31 |
| 2 | None | K2CO3 (1) | CH3CN | 0 |
| 3 | Ir[ppy]2(dtbbpy)PF6 | K2CO3 (1) | CH3CN | 0 |
| 4 | 4CzIPN | K2CO3 (1) | CH3CN | 47 |
| 5c | 4CzIPN | K2CO3 (1) | CH3CN | 0 |
| 6 | 4CzIPN | Cs2CO3 (1) | CH3CN | 32 |
| 7 | 4CzIPN | DBU (1) | CH3CN | 41 |
| 8 | 4CzIPN | Pyridine (1) | CH3CN | 65 |
| 9 | 4CzIPN | Pyridine (2) | CH3CN | 73 |
| 10d | 4CzIPN | Pyridine (2) | CH3CN | 0 |
| 11e | 4CzIPN | Pyridine (2) | CH3CN | 0 |
| 12 | 4CzIPN | Pyridine (2) |
DMSO/CH
3
CN (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1, v/v) :1, v/v)
|
77 |
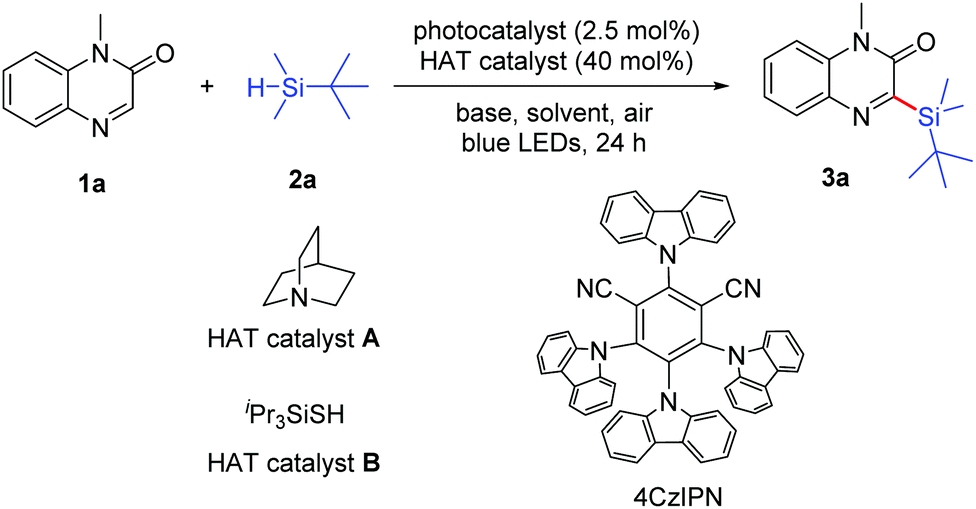
With the optimal reaction conditions in hand, we then investigated the scope of 1-methylquinoxalin-2(1H)-ones 1, and the results are summarized in Scheme 2. This reaction proceeded smoothly with a wide range of substituted quinoxalin-2(1H)-ones by employing tert-butyldimethylsilane as the reaction partner. The 1-methylquinoxalin-2(1H)-ones substituted with electron-donating (Me) or electron-withdrawing groups (F, Cl, Br, and NO2) were tolerated and the corresponding products 3b–3h were obtained. However, the 1-methylquinoxalin-2(1H)-ones bearing Br or NO2 (1c, 1d and 1h) gave the products 3c, 3d and 3h in relatively low yields under the same conditions, which might be attributed to the poor solubility and low conversion of the starting materials. In addition, 3-(tert-butyldimethylsilyl)-1-methylbenzo[g]quinoxalin-2(1H)-one (3i) could also be obtained in 66% yield. A variety of quinoxalin-2(1H)-ones (1j–1u) with N-protected groups such as n-butyl, cyclopropylmethyl, ester, allyl, propargyl, aryl, benzyl, etc. were well compatible in the reaction, and gave the corresponding products 3j–3u in 51–81% yields. It is worth mentioning that unsaturated allylic and propargylic substrates were tolerated under aerobic conditions, affording the desired products 3m and 3n in 51% and 55% yields, respectively.
 | ||
| Scheme 2 Substrate scope of 1-methylquinoxalin-2(1H)-ones 1. Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2a (1.0 mmol, 5.0 equiv.), 4CzIPN (2.5 mol%), quinuclidine (40 mol%), pyridine (0.4 mmol, 2.0 equiv.), and DMSO/CH3CN (3:1 (v/v), 4 mL) were stirred under irradiation (12 W blue LEDs) at room temperature under open air for 24 h. aIsolated yield based on 1. | ||
To further extend the scope of this reaction, we tested the applicability of several other heteroaromatic substrates (Scheme 3). The results showed that a variety of heterocycles, including quinoxalines (4a and 4b), benzothiazole (4c), pyridazine (4d), imidazo[1,2-b]pyridazine (4e), coumarin (4f), benzooxazinone (4g), ethyl isonicotinate (4h), quinoline-4-carbonitrile (4j) and isoquinoline-4-carbonitrile (4k), could provide the corresponding silylated products under the same reaction conditions. Moreover, the electron-deficient 1,4-dicyanobenzene (4i) could also be employed as a substrate and it generated the product 5i in satisfactory yield (65%).
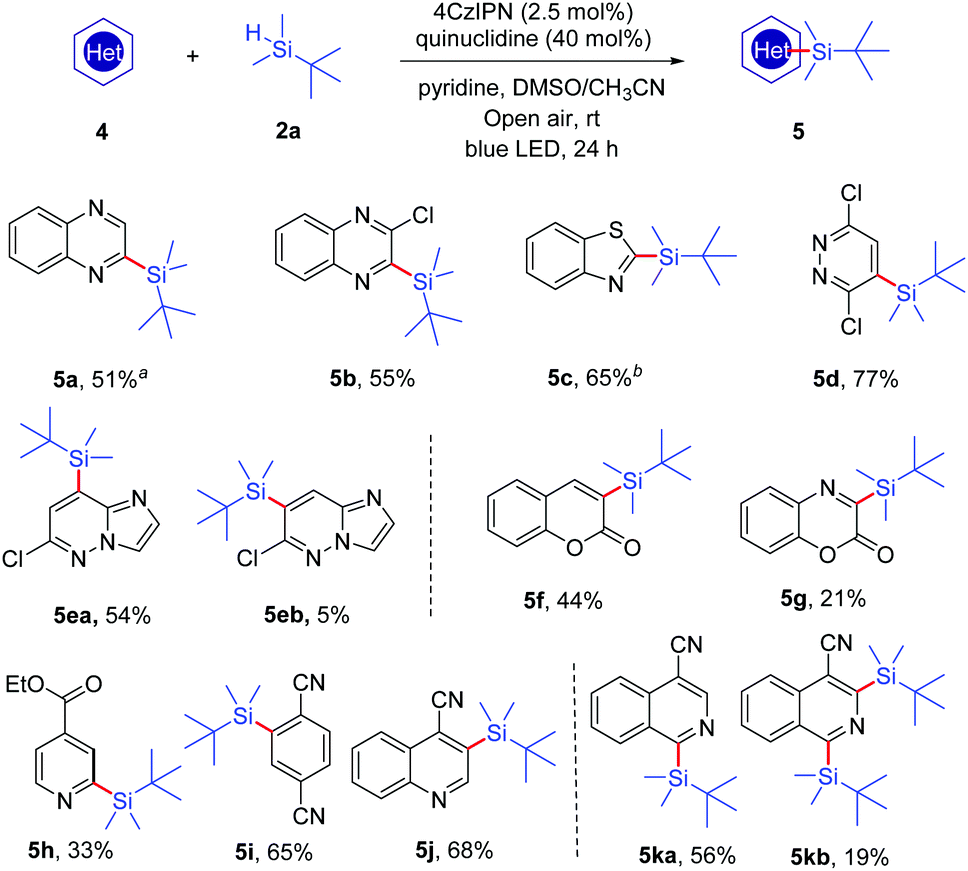 | ||
| Scheme 3 Substrate scope of heteroarenes 4 and electron-deficient arene 4i. Reaction conditions: 4 (0.2 mmol, 1.0 equiv.), 2a (1.0 mmol, 5.0 equiv.), 4CzIPN (2.5 mol%), quinuclidine (40 mol%), pyridine (0.4 mmol, 2.0 equiv.), and DMSO/CH3CN (3:1 (v/v), 4 mL) were stirred under irradiation (12 W blue LEDs) at room temperature under open air for 24 h. aIsolated yield based on 4. b2a (2.0 mmol, 10.0 equiv.) and quinuclidine (80 mol%). | ||
Finally, the procedure was extended successfully to other trialkylsilanes (Scheme 4). The results showed that aliphatic silanes, such as triethylsilane (2b) and triisopropylsilane (2c), were compatible under ambient air conditions to yield the silylated products 6a and 6b in 72% and 66% yields, respectively. However, a limitation was that arylsilanes, such as triphenylsilane, diphenylsilane, phenylsilane and methyldiphenylsilane, were incompatible under our reaction conditions and the substrate 1a could be recovered completely. We also failed to obtain the desired product from di-tert-butylsilane.
 | ||
| Scheme 4 Substrate scope of silanes 2. Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2 (1.0 mmol, 5.0 equiv.), 4CzIPN (2.5 mol%), quinuclidine (40 mol%), pyridine (0.4 mmol, 2.0 equiv.), and DMSO/CH3CN (3:1 (v/v), 4 mL) were stirred under irradiation (12 W blue LEDs) at room temperature under open air for 24 h. aIsolated yield based on 1a. b2 (2.0 mmol, 10.0 equiv.) and quinuclidine (80 mol%). | ||
Next, gram scale synthesis was performed to prove the applicability of the reaction, and the product 3a was obtained in 63% yield (1.04 g, 3.8 mmol) under the irradiation of blue LEDs (Scheme 5a). In addition, the product 3a could be used for further transformation. When 3a was treated with PhI(OAc)2 (1.5 equiv.) and Pd(OAc)2 (5 mol%) in AcOH (1 mL) at 100 °C for 24 h, followed by the addition of water (1 mL) and heating at 100 °C for another 24 h, 1-methyl-1,4-dihydroquinoxaline-2,3-dione (7) was obtained in 53% yield (Scheme 5b).
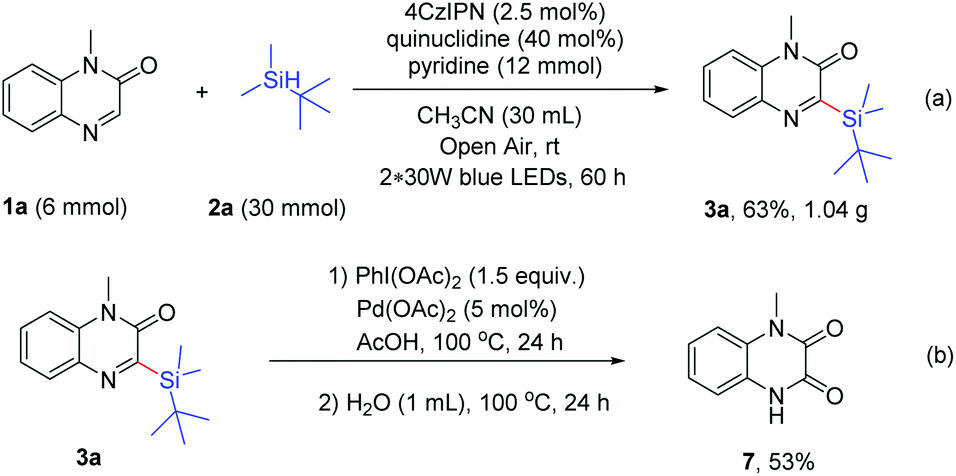 | ||
| Scheme 5 Scaling-up synthesis and further transformation of the product 1a. | ||
To understand the details of this C–H silylation reaction, several control experiments were conducted. The reaction was obviously suppressed in the presence of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy, 3 equiv.) under the standard conditions, and meanwhile, the adduct 8 was detected by LC-MS (Scheme 6-a). When 1,1-diphenylethylene (3 equiv.) was added, the reaction was also inhibited (Scheme 6-b). These results revealed that the silyl radical might be a pivotal intermediate in this transformation. The reaction was completely inhibited by the addition of a singlet oxygen quencher, such as 1,4-diazabicyclo[2.2.2]octane (DABCO), indicating that singlet oxygen probably played a role in this catalytic cycle (Scheme 6-c).21c The value of kH/kD (1.27) from two parallel reactions of 1a with 2c and 2c–d respectively showed that the Si–H bond cleavage might not be the kinetically rate-determining step in this reaction (Scheme 6-d). The results of the Stern–Volmer experiment indicated that the luminescence of 4CzIPN could be gradually quenched with the increase of the concentration of quinuclidine (Scheme 6-e and f, see the ESI† for details).
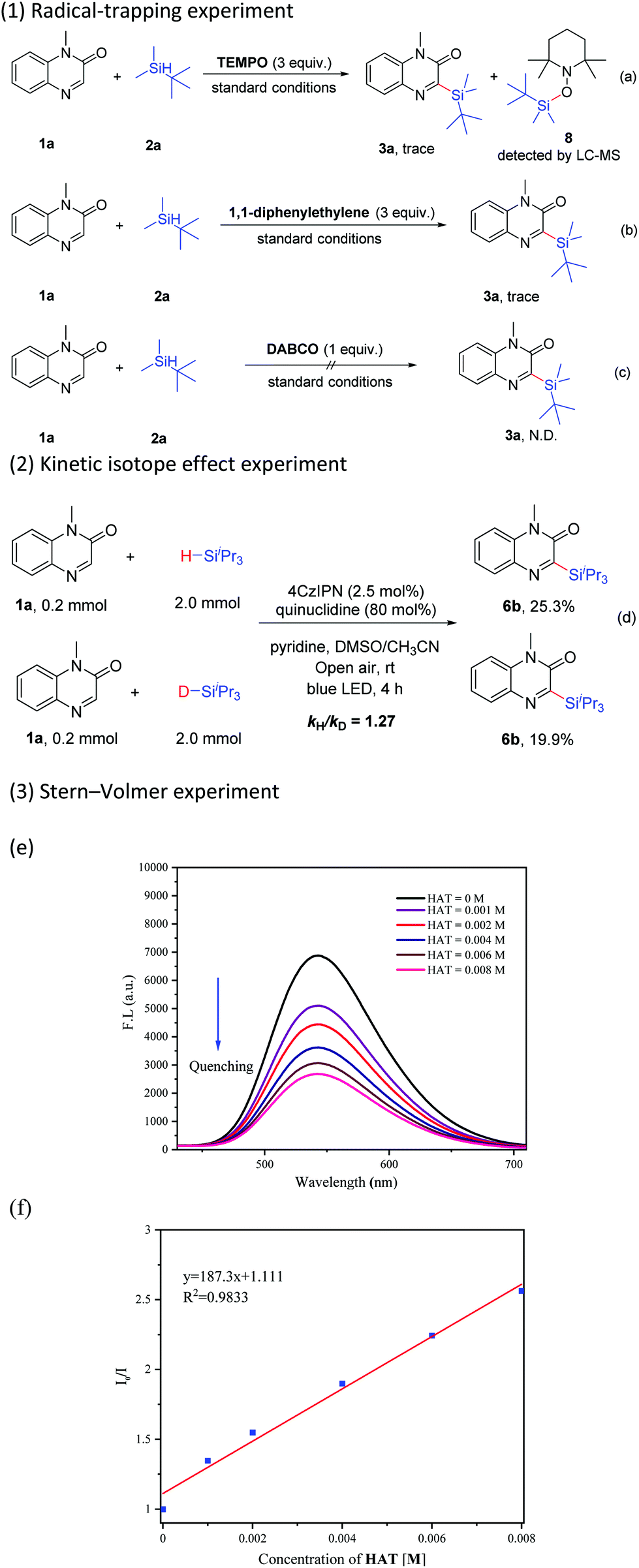 | ||
| Scheme 6 Control experiments. (a) Radical-trapping experiment with TEMPO. (b) Radical-trapping experiment with 1,1-diphenylethylene. (c) The addition of a singlet oxygen quencher DABCO. (d) Control experiments on kinetic isotope effects. (e) Emission spectra of 4CzIPN at different concentrations of quinuclidine. (f) Stern–Volmer plot of 4CzIPN at different concentrations of quinuclidine. | ||
Based on the above observations and some previous literature reports, a possible mechanism of this reaction was proposed (Scheme 7). Initially, the photocatalyst 4CzIPN was excited into 4CzIPN* under the irradiation of blue LEDs. A single electron was transferred from quinuclidine to 4CzIPN*, resulting in the generation of a quinuclidinium radical cation and a 4CzIPN˙− radical anion.21a–b Meanwhile, 1O2 was generated from 3O2 with activated 4CzIPN* under light irradiation.21c Subsequently, another single-electron-transfer (SET) process occurred between 1O2 and 4CzIPN˙−, giving O2˙− and regenerating 4CzIPN for the next photoredox cycle.21d–e Then, the resulting quinuclidinium radical cation served as the HAT catalyst to produce the t-butyldimethylsilyl radical by abstracting the hydrogen atom from t-butyldimethylsilane. After that, the addition of the t-butyldimethylsilyl radical to 1a formed the intermediate I. Finally, the intermediate I underwent direct hydrogen atom transfer to O2˙− to give the desired product 3a.
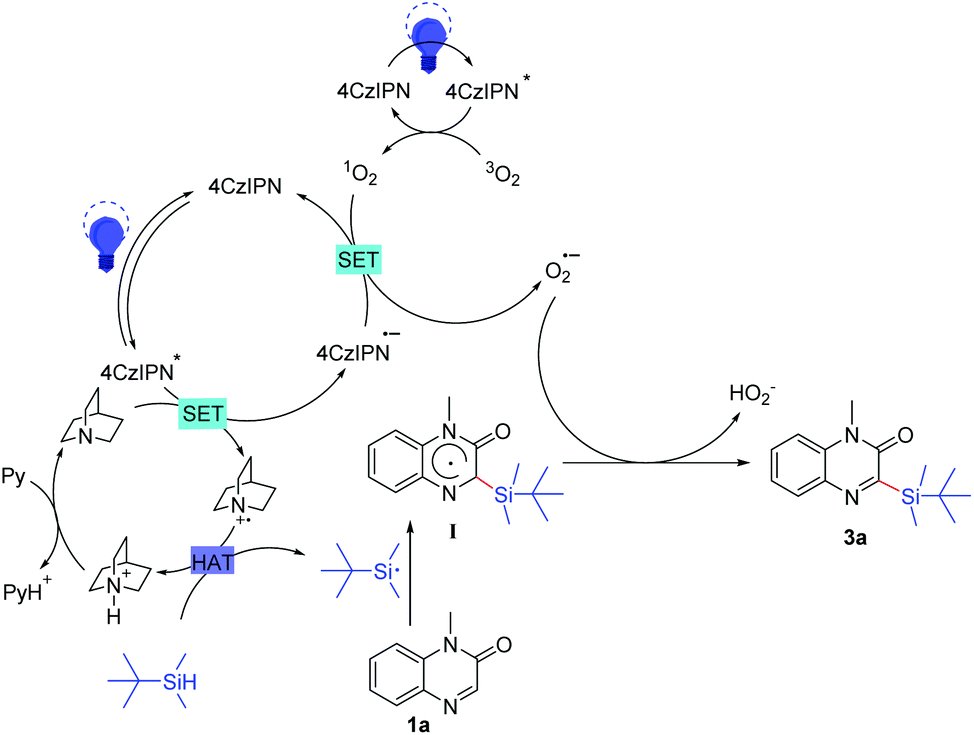 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In conclusion, a direct and efficient strategy has been developed for the C–H silylation of quinoxalinones by using a combination of the organic photocatalyst 4CzIPN and the hydrogen atom transfer reagent quinuclidine under ambient conditions. In this transition-metal-free process, the silyl radicals generated in situ were regioselectively added to quinoxalinones or a series of electron-deficient heteroarenes with high functional group tolerance. This methodology reported herein offered an environmentally friendly way for the synthesis of silylated heteroarenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Projects 21672104 and 21502097) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- For selected reviews, please see: (a) P. K. Pooni and G. A. Showell, Mini-Rev. Med. Chem., 2006, 6, 1169–1177 CrossRef CAS; (b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS; (c) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS.
- (a) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC; (b) T. A. Su, H. Li, R. S. Klausen, N. T. Kim, M. Neupane, J. L. Leighton, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Acc. Chem. Res., 2017, 50, 1088–1095 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS.
- (a) S. E. Denmark and J. M. Kallemeyn, Org. Lett., 2003, 5, 3483–3486 CrossRef CAS; (b) A. Hamze, O. Provot, M. Alami and J.-D. Brion, Org. Lett., 2006, 8, 931–934 CrossRef CAS; (c) E. McNeill, T. E. Barder and S. L. Buchwald, Org. Lett., 2007, 9, 3785–3788 CrossRef CAS.
- L. Guo, A. Chatupheeraphat and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 11810–11813 CrossRef CAS.
- U. Frick and G. Simchen, Synthesis, 1984, 929–930 CrossRef CAS.
- For selected examples and reviews, please see: (a) H. T. Klare, M. Oestreich, J. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312–3315 CrossRef CAS; (b) Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663–3666 CrossRef CAS; (c) Q. Yin, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 3204–3207 CrossRef CAS; (d) Y. Han, S. Zhang, J. He and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 7399–7407 CrossRef CAS; (e) S. Bähr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52–59 CrossRef.
- Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868–7871 CrossRef CAS.
- (a) A. A. Toutov, W.-B. Liu, K. N. Betz, B. M. Stoltz and R. H. Grubbs, Nat. Protoc., 2015, 10, 1897–1903 CrossRef CAS; (b) A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef CAS; (c) Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127–132 CrossRef CAS.
- (a) B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508–7510 CrossRef CAS; (b) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592–595 CrossRef CAS; (c) T. Wakaki, M. Kanai and Y. Kuninobu, Org. Lett., 2015, 17, 1758–1761 CrossRef CAS; (d) M. Murai, N. Nishinaka and K. Takai, Angew. Chem., Int. Ed., 2018, 57, 5843–5847 CrossRef CAS; (e) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS; (f) S. Wübbolt and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 15876–15879 CrossRef; (g) K. Devaraj, C. Sollert, C. Juds, P. J. Gates and L. T. Pilarski, Chem. Commun., 2016, 52, 5868–5871 RSC; (h) H. Fang, L. Guo, Y. Zhang, W. Yao and Z. Huang, Org. Lett., 2016, 18, 5624–5627 CrossRef CAS.
- C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS.
- (a) Y. Yang, R.-J. Song, X.-H. Ouyang, C.-Y. Wang, J.-H. Li and S. Luo, Angew. Chem., Int. Ed., 2017, 56, 7916–7919 CrossRef CAS; (b) Y. Lan, X.-H. Chang, P. Fan, C.-C. Shan, Z.-B. Liu, T.-P. Loh and Y.-H. Xu, ACS Catal., 2017, 7, 7120–7125 CrossRef CAS.
- (a) Z. Yan, J. Xie and C. Zhu, Adv. Synth. Catal., 2017, 359, 4153–4157 CrossRef CAS; (b) Y. Yang, R.-J. Song, Y. Li, X.-H. Ouyang, J.-H. Li and D.-L. He, Chem. Commun., 2018, 54, 1441–1444 RSC.
- (a) J. Gu and C. Cai, Chem. Commun., 2016, 52, 10779–10782 RSC; (b) Z. Xu, L. Chai and Z.-Q. Liu, Org. Lett., 2017, 19, 5573–5576 CrossRef CAS; (c) R. Sakamoto, B.-N. Nguyen and K. Maruoka, Asian J. Org. Chem., 2018, 7, 1085–1088 CrossRef CAS.
- Y. Li, K. Shu, P. Liu and P. Sun, Org. Lett., 2020, 22, 6304–6307 CrossRef CAS.
- For selected reviews on visible light photocatalysis, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS; (c) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS; (d) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS.
- S. Liu, P. Pan, H. Fan, H. Li, W. Wang and Y. Zhang, Chem. Sci., 2019, 10, 3817–3825 RSC.
- For selected reviews on organic photoredox catalysis, see: (a) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (c) M. Uygur and O. G. Mancheño, Org. Biomol. Chem., 2019, 17, 5475–5489 RSC.
- (a) L. Wang, Z.-G. Ma, X.-J. Wei, Q.-Y. Meng, D.-T. Yang, S.-F. Du, Z.-F. Chen, L.-Z. Wu and Q. Liu, Green Chem., 2014, 16, 3752–3757 RSC; (b) X. Liu, X. Ye, F. Bureš, H. Liu and Z. Jiang, Angew. Chem., Int. Ed., 2015, 54, 11443–11447 CrossRef CAS; (c) H. Cui, W. Wei, D. Yang, Y. Zhang, H. Zhao, L. Wang and H. Wang, Green Chem., 2017, 19, 3520–3524 RSC; (d) Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512–5516 RSC.
- (a) P. Liu, Y. Gao, W. Gu, Z. Shen and P. Sun, J. Org. Chem., 2015, 80, 11559–11565 CrossRef CAS; (b) Y. Gao, W. Lu, P. Liu and P. Sun, J. Org. Chem., 2016, 81, 2482–2487 CrossRef CAS; (c) Q. Chang, Z. Liu, P. Liu, L. Yu and P. Sun, J. Org. Chem., 2017, 82, 5391–5397 CrossRef CAS.
- (a) J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS; (b) R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 16621–116625 CrossRef CAS; (c) A. Guerrero-Corella, A. M. Martinez-Gualda, F. Ahmadi, E. Ming, A. Fraile and J. Alemán, Chem. Commun., 2017, 53, 10463–10466 RSC; (d) Q. Zhang, Y. Huang, L.-W. Zhan, W.-Y. Tang, J. Hou and B.-D. Li, Org. Lett., 2020, 22, 7460–7464 CrossRef CAS; (e) M. Shee, S. S. Shah and N. D. P. Singh, Chem. – Eur. J., 2020, 26, 14070–14074 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03697h |
| This journal is © The Royal Society of Chemistry 2021 |