
Photo-triggered self-catalyzed fluoroalkylation/cyclization of unactivated alkenes: synthesis of quinazolinones containing the CF2R group†
Jin
Yang
a,
Bin
Sun
*b,
Hao
Ding
a,
Pan-Yi
Huang
a,
Xiao-Li
Tang
a,
Rong-Cheng
Shi
a,
Zhi-Yang
Yan
b,
Chuan-Ming
Yu
 a and
Can
Jin
*ab
a and
Can
Jin
*ab
aCollege of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou, PR China. E-mail: jincan@zjut.edu.cn
bCollaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, PR China. E-mail: sunbin@zjut.edu.cn
First published on 10th December 2020
Abstract
A novel photo-triggered self-catalyzed fluoroalkylation/cyclization of quinazolinones containing unactivated alkenes with various fluoroalkyl bromides has been developed. This transformation exhibits excellent substrate generality with respect to both the coupling partners. Of note is that this is the first example describing the Csp3–Br bond homolysis of alkyl bromides via a substrate (quinazolinones) induced energy transfer process. Additionally, the mild conditions, tolerance to a wide range of functional groups and operational simplicity make this protocol practical for the synthesis of fluorine-containing ring-fused quinazolinones.
Introduction
Ring-fused quinazolinones are usually key core motifs in many pharmaceuticals and biologically active natural products.1 Particularly, both naturally occurring and synthetic ring-fused quinazolinone derivatives exhibit a broad range of pharmacological and biological activities such as antidepressant, antithrombotic, and anticancer activities, and these compounds are even considered as potential inhibitors of COVID-19 (Scheme 1).1c,2 Although various approaches to these polycyclic scaffolds have been established,1c,3 most of these approaches still suffer from the need for complex starting materials, harsh reaction conditions and multiple synthetic steps. To the best of our knowledge, the synthesis of ring-fused quinazolinones through the direct C-2 alkylation of quinazolin-4-ones has rarely been reported.4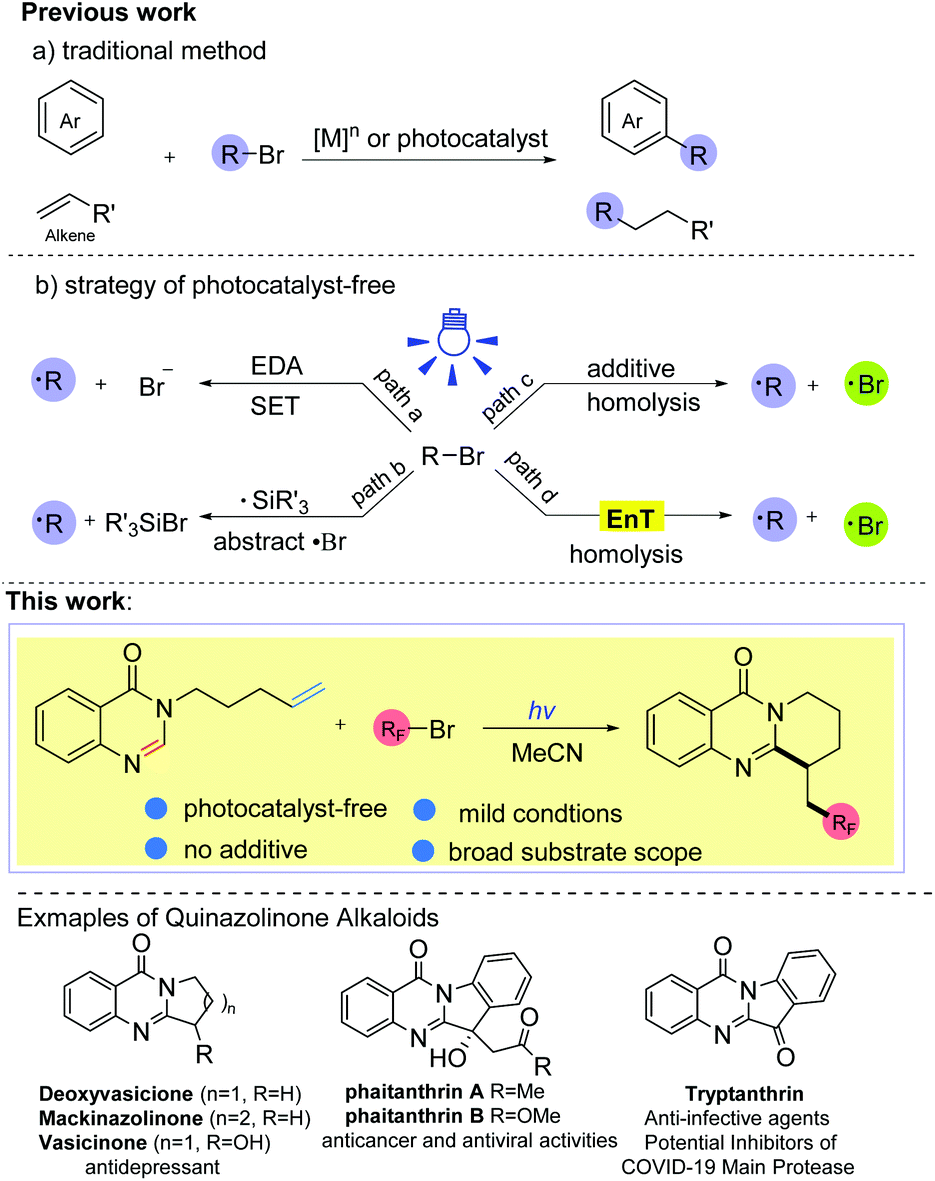 | ||
| Scheme 1 Strategies for the radical generation from alkyl bromides. | ||
1,2-Difunctionalization of alkenes has matured into a powerful tool to connect electron deficient heterocycles and electrophilic radicals in organic synthesis, and this kind of radical tandem reaction is called the polarity reversal strategy.5 Alkyl bromides, which are regarded as alkyl radical precursors, are very appealing due to their easy availability, inexpensiveness and high atom economy. In the past few years, many elegant approaches to the direct alkylation of heteroarenes (or olefins) with alkyl bromides have been established (Scheme 1, a).6 Nevertheless, these methods usually require the assistance of transition metal species ([M]n) or a photocatalyst (Ir, Ru, Au. etc.) to complete the electron transfer (ET) process. Recently, photocatalytic protocols without an exogenous photoredox catalyst have drawn much attention because they could offer a more sustainable and environmentally friendly way to prepare complex molecules as well as achieve various challenging organic transformations. By applying this strategy, some progress has been made in the activation of Csp3–Br bonds to furnish the alkyl radical.7,8,10 For example, synthetic strategies involving electron donor–acceptor complexes have been developed to activate the Csp3–Br bond via a single electron transfer (SET) process (Scheme 1, path a).7 In addition, applying high energy radicals (R3Si˙) to abstract a bromine atom from alkyl bromides is another commonly used protocol to obtain the alkyl radical from alkyl bromides (Scheme 1, path b).8 To date, tremendous efforts have led to direct homolysis of Csp3–X bonds, especially Csp3–I bonds, under irradiation.5f,9,10 However, Csp3–Br bonds are more difficult to cleave under illumination due to their stronger bond energy compared with Csp3–I bonds (for example, 70.8 kcal mol−1 for F3C–Br, 54.3 kcal mol−1 for F3C–I).11 The rarely reported methods of Csp3–Br bond homolysis are limited to a few alkyl bromides with a weak Csp3–Br bond, such as BrCCl3 (BDE = 55.3 kcal mol−1),10b or require strong bases as additives, which may limit the diversity of alkyl sources (Scheme 1, path c). To seek a more efficient and feasible method, we became interested in whether other alkyl bromides with a stronger Csp3–Br bond, such as BrCnF2n+1 (BDE ≈ 66–70 kcal mol−1),11 could undergo the C–Br bond homolysis via an energy transfer (EnT) process (path d). So far, most of the energy transfer processes have needed the catalysis of photocatalysts (Ir, Ru, MesAcr+, etc.),12 while there is no example describing the homolysis of C–Br bonds via a substrate-induced EnT process. With our ongoing studies on the development of green methods for the synthesis of fluorine-containing compounds,13 herein, we disclose an efficient and facile method for the intramolecular fluoroalkyl radical tandem cyclization reaction (RTCR) triggered by the substrate self-induced energy transfer process. The reaction exhibited good tolerance for various difluoro bromides and quinazolinones, providing a facile access to valuable CF2R-containing ring-fused quinazolinones under photocatalyst- and additive-free conditions.
Results and discussion
Optimization of the reaction conditions
To begin our study, 3-(pent-4-en-1-yl)quinazolin-4(3H)-one (1a) and BrCF2CO2Et were chosen as model substrates for the optimization of the reaction conditions. Initially, the reaction was carried out in CH3CN with TMEDA as an additive under the irradiation of a 10 W LED (430–435 nm). Fortunately, the anticipated product was obtained in 32% yield (Table 1, entry 1). Subsequently, the effect of the wavelength of the light source was investigated, and a significant improvement could be obtained when the reaction was conducted under irradiation at 380–385 nm and it gave the desired product in 73% yield (Table 1, entry 4). Other additives, such as Et3N, PMDETA, Cs2CO3, Na2CO3, and K2HPO4, all could not give a better result in this transformation (Table 1, entries 5–9). Surprisingly, the further experiment suggested that the reaction could proceed well under external-additive-free conditions and gave the corresponding product in 82% yield (Table 1, entry 10). Various commonly used solvents, including MeOH, DMSO, EtOAc, DCE and THF, were examined (Table 1, entries 11–15), and acetonitrile proved to be a better solvent. Finally, no reaction occurred when the transformation was conducted under the irradiation of a 450–455 nm LED mainly because it was beyond the absorption range of substrate 1a (Table 1, entry 16). The contrast experiments showed that this transformation also could not proceed well when carried out under air or in darkness (Table 1, entries 17 and 18).|
|
|||
|---|---|---|---|
| Entry | Light source | Additive | Yield 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a Conditions: 3-(Pent-4-en-1-yl)quinazolin-4(3H)-one (1a) (0.2 mmol), BrCF2COOEt (2 equiv.), additive (2.0 equiv.), CH3CN (2 mL), room temperature, N2, 12 h. b Determined by 1HNMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. c MeOH. d DMSO. e EA. f DCE. g THF. h Under air. | |||
| 1 | 430–435 nm | TMEDA | 32 |
| 2 | 400–405 nm | TMEDA | 66 |
| 3 | 380–385 nm | TMEDA | 73 |
| 4 | 365–368 nm | TMEDA | 72 |
| 5 | 380–385 nm | Et3N | 0 |
| 6 | 380–385 nm | PMDETA | 34 |
| 7 | 380–385 nm | Cs2CO3 | 22 |
| 8 | 380–385 nm | Na2CO3 | 18 |
| 9 | 380–385 nm | K2HPO4 | 72 |
| 10 | 380–385 nm | — | 82 |
| 11c | 380–385 nm | — | 0 |
| 12d | 380–385 nm | — | Trace |
| 13e | 380–385 nm | — | 80 |
| 14f | 380–385 nm | — | 56 |
| 15g | 380–385 nm | — | 68 |
| 16 | 450–455 nm | — | 0 |
| 17h | 380–385 nm | — | 0 |
| 18 | — | — | 0 |
With the optimized conditions in hand, we set out to investigate the scope and limitations of quinazolinones and alkyl halides (Table 2). The survey was initiated by investigating the generality of six-membered cyclization products. Substrates containing either electron-donating (methyl, methoxy) or electron-withdrawing (fluoro, chloro, bromo, trifluoromethyl) groups at the 6- or 7-position of the benzene ring were well-tolerated and provided the corresponding ring-fused quinazolinones in 52% to 92% yields (3b–3l). In addition, the optimized conditions were also suitable for 5- and 8-substituted quinazolinones and delivered the desired products 3m and 3n in a yield of 46% and 48% respectively, which indicated that the position of substituents on the phenyl ring has no obvious effect on this reaction. After the scope of quinazolinones was explored, a variety of fluoroalkyl reagents were then examined for this photocatalyst-free system via the radical tandem cyclization reaction with 1a. A variety of α-bromo-α,α-difluorinated esters, such as methyl ester, isopropyl ester and n-butyl ester, were evaluated under standard conditions. Expectedly, the screened substrates were all compatible and gave the corresponding products 4a–4c in moderate to excellent yields. In addition, a sterically encumbered ester derived from 2-adamantanol also gave the desired product 4d in 61% yield. To further extend the substrate scope of this transformation, various α-bromo-α,α-difluorinated amide coupling partners were then investigated under the optimal reaction conditions. The results indicated that both secondary and tertiary amides were tolerated for this transformation, providing the corresponding products 4e–4l in 60% to 75% yields. Further screening revealed that α-bromodifluoroacetylbenzene also exhibited good tolerance, delivering the corresponding product 4m in a yield of 61%. Remarkably, bromodifluoro phosphonate or perfluoroalkyl bromides were also suitable reagents for this reaction and gave the tricyclic products 4n–4p in moderate yields, which demonstrated its wide substrate scope.
| a Conditions: 1 (0.4 mmol), 2 (2.0 equiv.), CH3CN (4 mL), a 10 W LED lamp (380–385 nm), room temperature, N2, 12 h. |
|---|

|
The five-membered ring product was also another important heterocyclic motif existing in numerous bioactive molecules such as mackinazolinone, phaitanthrin B and tryptanthrin. Therefore, the reactions of 3-(but-3-en-1-yl)quinazolin-4(3H)-ones with fluoroalkyl bromides were also examined. A range of quinazolinones bearing electron-withdrawing or electron-donating groups on the aryl ring were well tolerated, resulting in the products 3p–3s in moderate yield. Moreover, the synthetic utility of this approach was further investigated by employing various esters, secondary amides, tertiary amides and phosphates as the coupling partners; to our delight, the corresponding products 4q–4w were obtained in satisfactory yields. In addition, 3-(2-(prop-1-en-2-yl)phenyl)quinazolin-4(3H)-one was also amenable to the transformation, giving the corresponding product 3t in 78% yield. Besides, substrates with electron-donating or electron-withdrawing substituents on the aryl ring were all tolerated well and gave the products 3u–3x in good yields. To showcase the robustness and utility of the developed transformation, several α-bromo-α,α-difluorinated esters from natural molecules were also converted into the corresponding fluorinated cyclization products (4x–4z) under the optimal reaction conditions. Finally, a gram-scale reaction was carried out between 1a (5 mmol) and BrCF2COOEt, and the desired product 3a was finally isolated in 67% yield (1.13 g, see the ESI†).
In order to gain the mechanistic insights of the present reaction, a few control experiments were carried out. Initially, the UV-visible absorption spectra of substrate 1a and BrCF2COOEt showed that only quinazolinones can absorb light of 320–410 nm (Fig. 1.). Furthermore, a mixed solution of 1a and BrCF2COOEt (2.0 equiv.) did not affect the waveform nor the strength of the spectra, suggesting that 1a did not form a complex with BrCF2COOEt. We then performed a series of Stern–Volmer experiments, which revealed that the photoexcited quinazolinone 1a was effectively quenched (Fig. 2.) and linearly correlated with the concentration of BrCF2COOEt, supporting the notion that excited species 1a* contributed to generating a difluoroalkyl radical via energy transfer and acted as a photosensitizer.
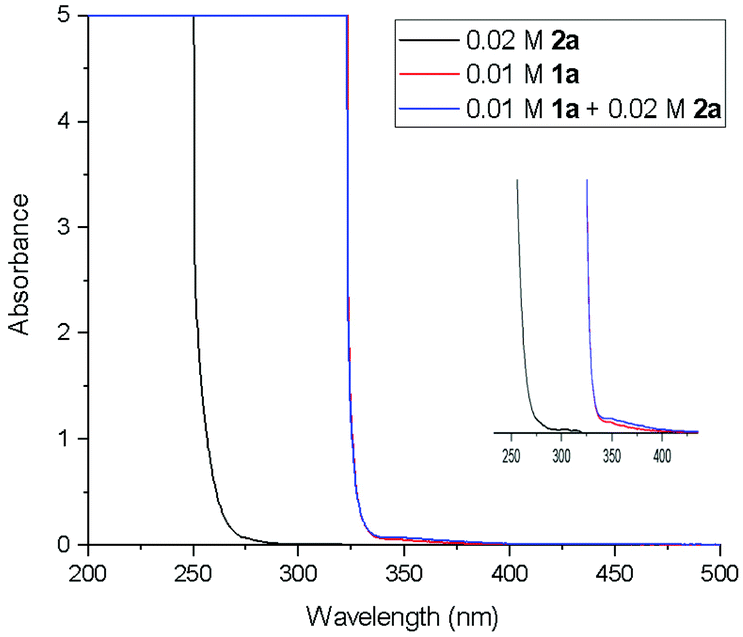 | ||
| Fig. 1 Absorption spectra of 1a and 2a. | ||
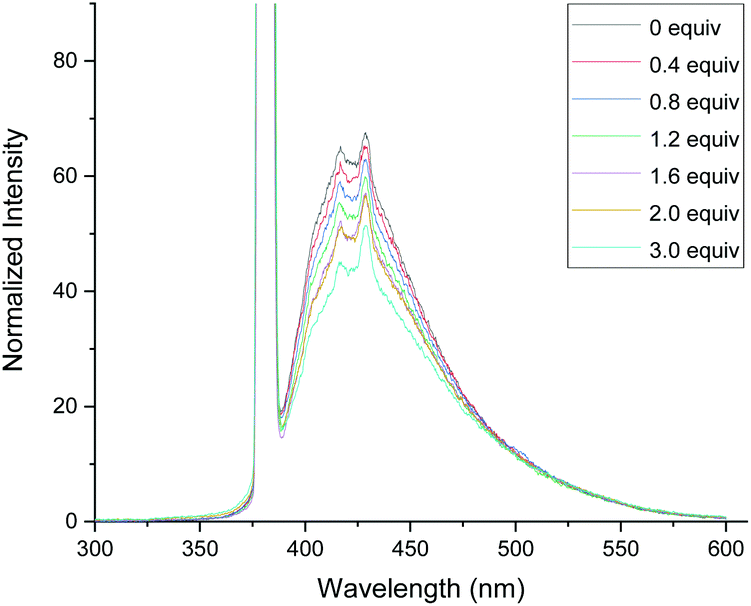 | ||
| Fig. 2 Fluorescence quenching of fluorescein with BrCF2COOEt. | ||
In line with the radical nature of the transformation, the radical scavenger (3.0 equiv.) BHT was firstly added into this reaction system, and the formation of 3a was completely inhibited. To further confirm the information of the fluoroalkyl radical, another radical scavenger was employed in the same reaction system, and the radical adduct 5 was fortunately detected by HRMS. In addition, a photoinduced Heck-type product was detected when 1,1-diphenylethylene (3.0 equiv.) was added into the standard reaction (Scheme 3, a). Subsequently, two atom-transfer radical addition (ATRA) reactions of BrCF2COOEt with nonactivated alkenes (6 and 8) were performed. The results of the formation of the 1,2-difunctionalization product further proved that BrCF2COOEt underwent a rapid C–Br bond homolysis to generate the difluoroalkyl radical (Scheme 2, b). Finally, the quantum yield of this reaction was determined by chemical actinometry, and a high value was obtained (Φ = 14.69, see the ESI†), suggesting that a radical chain mechanism was involved in the reaction pathway. Nevertheless, the “light/dark” experiments showed that the model reaction was interrupted when the irradiation was changed from on to off, and the former reactivity was recovered upon further illumination, suggesting that the reaction needed continuous irradiation.10b,14 According to the above two experimental results and previous reports,10b we reasonably envisioned that the corresponding radical chain might be very short, leading to the situation where the reaction progress could only be observed during the irradiation period.
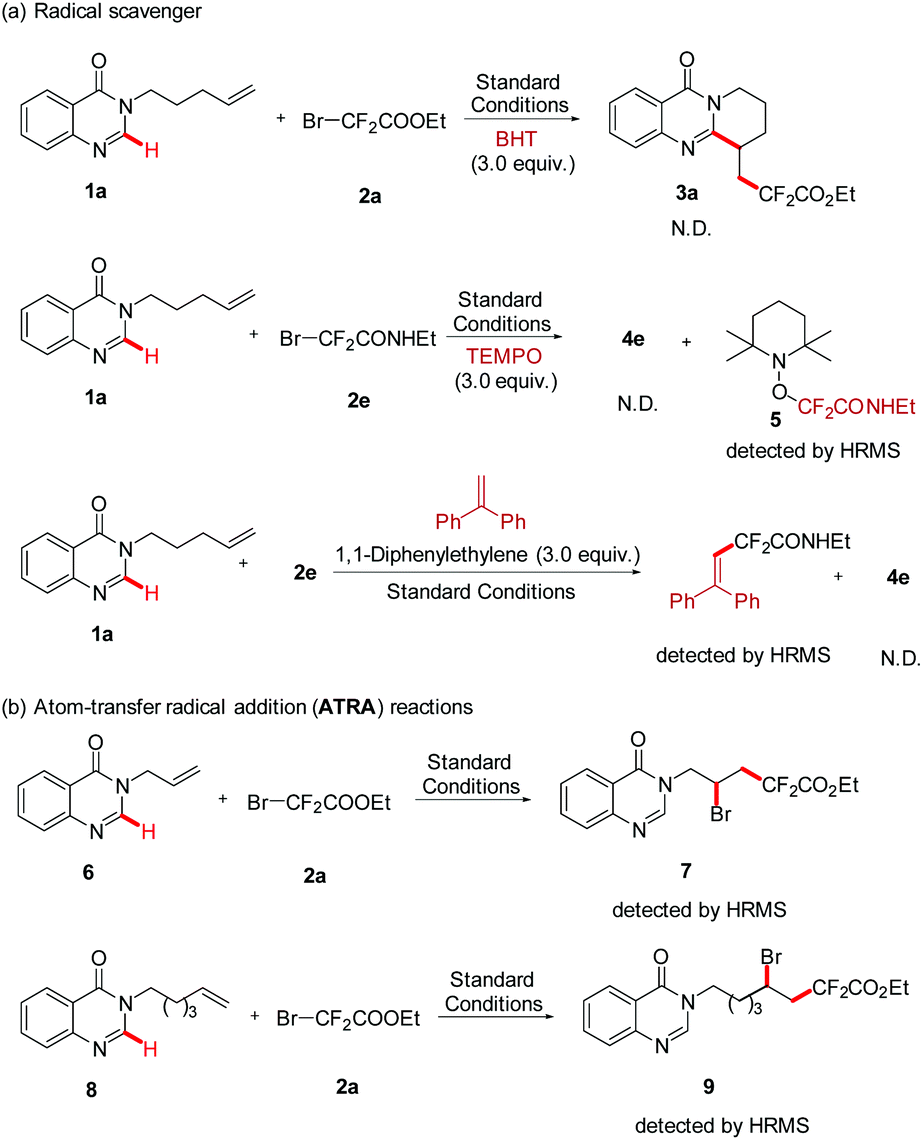 | ||
| Scheme 2 Control experiments. | ||
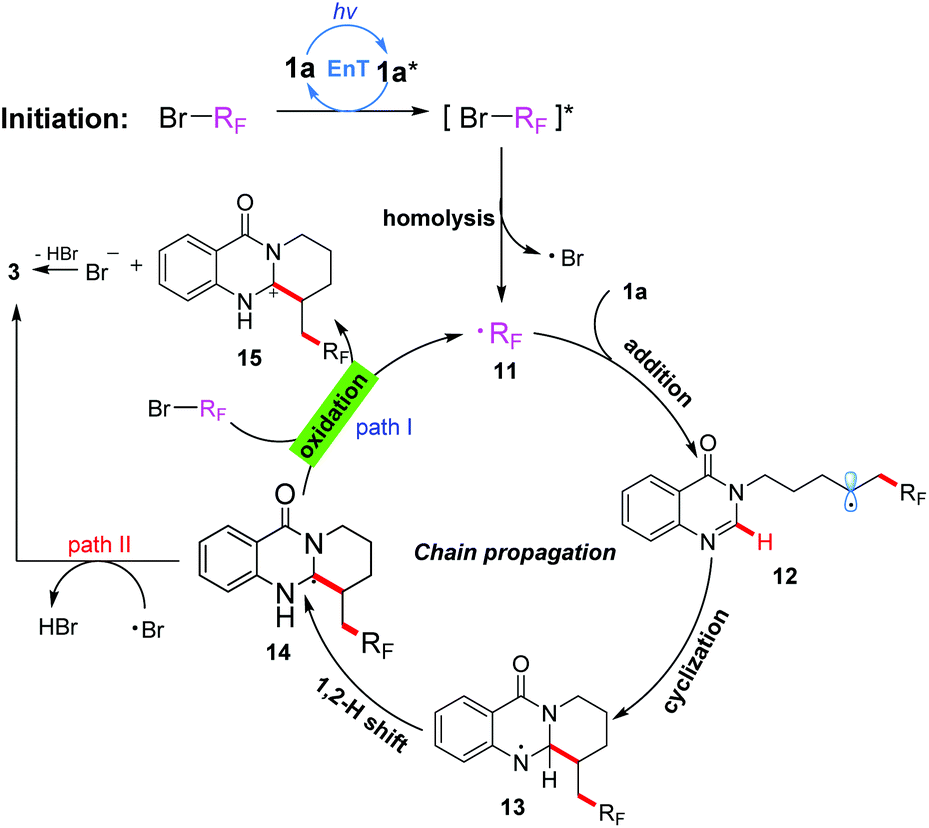 | ||
| Scheme 3 Proposed reaction mechanism. | ||
On the basis of these experimental results, a proposed mechanism is shown in Scheme 3. Initially, under irradiation with 380 nm light, the substrate 1a was transformed into its excited-state 1a*, which then activated the substrate BrRf to generate an excited species [BrRf]*via an energy transfer process. Subsequently, the C–Br bond homolytic cleavage of [BrRf]* immediately took place to afford an alkyl radical 11 (˙Rf) and a bromine radical. The radical 11 preferentially was trapped by an electron-rich alkene to generate another radical intermediate 12, which on subsequent addition to the electron-deficient heterocycle gave the cyclization intermediate 13. The formed intermediate 13 underwent a 1,2-hydrogen shift to produce carbon radical 14, which would be oxidized by BrRf15 to give the carbon cation intermediate 15 and alkyl radical 11 through a SET process. Finally, the product 3 could be obtained after deprotonation by the bromine anion, and alkyl radical 11 initiated a new chain propagation (Path I). Alternatively, the product 3 could also be obtained by a hydrogen atom transfer (HAT) process of intermediate 14 with the bromine radical10b (Path II). Though path II could not be excluded based on the results of the “light/dark” experiments, we are more inclined to believe that the chain propagation process is the main pathway (Path I) of this transformation according to the calculated result of the quantum yield.
Conclusions
In summary, we have developed a green and mild substrate-promoted radical tandem cyclization of quinazolinones containing an unactivated alkene moiety with various difluoro bromides under illumination without any photocatalyst and ligand, which delivered a variety of fluoroalkyl substituted ring fused quinazolinones. The mechanistic studies demonstrated that quinazolinones possessed the photochemical activity, which could enable the homolytic cleavage of the C–Br bond of haloalkanes through energy transfer under illumination. The mild transformation is photocatalyst-free and has an excellent substrate scope, good functional group tolerance and operational simplicity, providing a novel environmentally friendly access to fluorinated molecules.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078299) and the Natural Science Foundation of Zhejiang Province (LQ20B060007, LY21B060005). We are also grateful to the College of Pharmaceutical Sciences, Zhejiang University of Technology and Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals for the financial help.Notes and references
- (a) G. Honda and M. Tabata, Planta Med., 1979, 36, 85–86 CrossRef CAS; (b) H. Danz, D. Baumann and M. Hamburger, Planta Med., 2002, 68, 152–157 CrossRef CAS; (c) N. Mahindroo, Z. Ahmed, A. Bhagat, K. L. Bedi, R. K. Khajuria, V. K. Kapoor and K. L. Dhar, Med. Chem. Res., 2005, 14, 347–368 CrossRef CAS; (d) W. R. Bowman, M. R. J. Elsegood, T. Stein and G. W. Weaver, Org. Biomol. Chem., 2007, 5, 103–113 RSC; (e) K. M. Shakhidoyatov and B. Z. Elmuradov, Chem. Nat. Compd., 2014, 50, 781–800 CrossRef CAS; (f) F. Zheng, M. Zhan, X. Huang, M. D. M. A. Hameed and C.-G. Zhan, Bioorg. Med. Chem., 2014, 22, 538–549 CrossRef CAS.
- (a) Y. Lu, T. Nagashima, B. Miriyala, J. Conde and W. Zhang, J. Comb. Chem., 2010, 12, 125–128 CrossRef CAS; (b) U. V. S. Reddy, M. Chennapuram, K. Seki, C. Seki, B. Anusha, E. Kwon, Y. Okuyama, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Eur. J. Org. Chem., 2017, 3874–3885 CrossRef; (c) R. R. Narkhede, A. V. Pise, R. S. Cheke and S. D. Shinde, Nat. Prod. Bioprospect., 2020, 10, 297–306 CrossRef CAS.
- (a) J. P. Michael, Nat. Prod. Rep., 2004, 21, 650–668 RSC; (b) J.-F. Liu, P. Ye, K. Sprague, K. Sargent, D. Yohannes, C. M. Baldino, C. J. Wilson and S.-C. Ng, Org. Lett., 2005, 7, 3363–3366 CrossRef CAS; (c) W. R. Bowman, M. R. Elsegood, T. Stein and G. W. Weaver, Org. Biomol. Chem., 2007, 5, 103–113 RSC; (d) J. Chen, W. Su, H. Wu, M. Liu and C. Jin, Green Chem., 2007, 9, 972–975 RSC; (e) M.-C. Tseng, H.-Y. Yang and Y.-H. Chu, Org. Biomol. Chem., 2010, 8, 419–427 RSC; (f) L. He, H. Li, J. Chen and X.-F. Wu, RSC Adv., 2014, 4, 12065–12077 RSC; (g) W. Yang, R. Qiao, J. Chen, X. Huang, M. Liu, W. Gao, J. Ding and H. Wu, J. Org. Chem., 2015, 80, 482–489 CrossRef CAS; (h) I. Khan, S. Zaib, S. Batool, N. Abbas, Z. Ashraf, J. Iqbal and A. Saeed, Bioorg. Med. Chem., 2016, 24, 2361–2381 CrossRef CAS; (i) S. H. Kwon, H. A. Seo and C. H. Cheon, Org. Lett., 2016, 18, 5280–5283 CrossRef CAS; (j) A. O. Nasrullaev, Z. I. Islamova, B. Z. Élmuradov, A. M. Bektemirov, S. O. Osipova, Z. A. Khushbaktova, V. N. Syrov and K. M. Shakhidoyatov, Pharm. Chem. J., 2017, 51, 355–360 CrossRef CAS; (k) P. Qian, Y. Deng, H. Mei, J. Han, J. Zhou and Y. Pan, Org. Lett., 2017, 19, 4798–4801 CrossRef CAS; (l) R. Qiao, L. Ye, K. Hu, S. Yu, W. Yang, M. Liu, J. Chen, J. Ding and H. Wu, Org. Biomol. Chem., 2017, 15, 2168–2173 RSC; (m) Y. Zhang, Y. Shao, J. Gong, J. Zhu, T. Cheng and J. Chen, J. Org. Chem., 2019, 84, 2798–2807 CrossRef CAS; (n) J. Guo, Y. Hao, G. Li, Z. Wang, Y. Liu, Y. Li and Q. Wang, Org. Biomol. Chem., 2020, 18, 1994–2001 RSC.
- H. M. Huang, R. W. Adams and D. J. Procter, Chem. Commun., 2018, 54, 10160–10163 RSC.
- (a) Z. Liu and Z. Q. Liu, Org. Lett., 2017, 19, 5649–5652 CrossRef CAS; (b) D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Chem. Sci., 2018, 9, 9012–9017 RSC; (c) J. Q. Buquoi, J. M. Lear, X. Gu and D. A. Nagib, ACS Catal., 2019, 9, 5330–5335 CrossRef CAS; (d) Y. Kumagai, N. Murakami, F. Kamiyama, R. Tanaka, T. Yoshino, M. Kojima and S. Matsunaga, Org. Lett., 2019, 21, 3600–3605 CrossRef CAS; (e) D. Zheng and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 15803–15807 CrossRef CAS; (f) D. Zheng and A. Studer, Org. Lett., 2019, 21, 325–329 CrossRef CAS; (g) T. Li, K. Liang, Y. Zhang, D. Hu, Z. Ma and C. Xia, Org. Lett., 2020, 22, 2386–2390 CrossRef CAS.
- (a) R. Lin, H. Sun, C. Yang, W. Shen and W. Xia, Chem. Commun., 2015, 51, 399–401 RSC; (b) T. L. Andersen, M. W. Frederiksen, K. Domino and T. Skrydstrup, Angew. Chem., Int. Ed., 2016, 55, 10396–10400 CrossRef CAS; (c) T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754–4758 RSC; (d) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS; (e) A. Kaga and S. Chiba, ACS Catal., 2017, 7, 4697–4706 CrossRef CAS; (f) P. Nuhant, M. S. Oderinde, J. Genovino, A. Juneau, Y. Gagné, C. Allais, G. M. Chinigo, C. Choi, N. W. Sach, L. Bernier, Y. M. Fobian, M. W. Bundesmann, B. Khunte, M. Frenette and O. O. Fadeyi, Angew. Chem., Int. Ed., 2017, 56, 15309–15313 CrossRef CAS; (g) V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 CrossRef CAS; (h) A. ElMarrouni, C. B. Ritts and J. Balsells, Chem. Sci., 2018, 9, 6639–6646 RSC; (i) X.-M. Zhang, J. Yang, Q.-B. Zhuang, Y.-Q. Tu, Z. Chen, H. Shao, S.-H. Wang and F.-M. Zhang, ACS Catal., 2018, 8, 6094–6099 CrossRef CAS; (j) G. Barzanò, A. Cheseaux and X. Hu, Org. Lett., 2019, 21, 490–493 CrossRef; (k) L. Dai, Z.-H. Xia, Y.-Y. Gao, Z.-H. Gao and S. Ye, Angew. Chem., Int. Ed., 2019, 58, 18124–18130 CrossRef CAS; (l) J. Dong, X. Lyu, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2019, 10, 976–982 RSC; (m) P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS; (n) X.-L. Lv, C. Wang, Q.-L. Wang and W. Shu, Org. Lett., 2019, 21, 56–59 CrossRef CAS; (o) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS; (p) X.-G. Wang, Y. Li, H.-C. Liu, B.-S. Zhang, X.-Y. Gou, Q. Wang, J.-W. Ma and Y.-M. Liang, J. Am. Chem. Soc., 2019, 141, 13914–13922 CrossRef CAS.
- (a) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485–1489 CrossRef CAS; (b) Y.-q. Yuan, S. Majumder, M.-h. Yang and S.-r. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (c) Z. Li, P. Ma, Y. Tan, Y. Liu, M. Gao, Y. Zhang, B. Yang, X. Huang, Y. Gao and J. Zhang, Green Chem., 2020, 22, 646–650 RSC; (d) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS.
- S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370–11375 CrossRef CAS.
- examples for Csp3–I bond homolysis: (a) M. Silvi, C. Sandford and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 5736–5739 CrossRef CAS; (b) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS; (c) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS; (d) C. Gerleve, M. Kischkewitz and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 2441–2444 CrossRef CAS; (e) M. Kischkewitz, C. Gerleve and A. Studer, Org. Lett., 2018, 20, 3666–3669 CrossRef CAS; (f) X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS.
- examples for Csp3–Br bond homolysis: (a) S. Sumino, A. Fusano and I. Ryu, Org. Lett., 2013, 15, 2826–2829 CrossRef CAS; (b) J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2015, 51, 8280–8283 RSC; (c) Q. Liu, J. Hong, B. Sun, G. Bai, F. Li, G. Liu, Y. Yang and F. Mo, Org. Lett., 2019, 21, 6597–6602 CrossRef CAS; (d) C.-H. Qu, G.-T. Song, J. Xu, W. Yan, C.-H. Zhou, H.-Y. Li, Z.-Z. Chen and Z.-G. Xu, Org. Lett., 2019, 21, 8169–8173 CrossRef CAS; (e) G. Gu, M. Huang, J. K. Kim, J. Zhang, Y. Li and Y. Wu, Green Chem., 2020, 22, 2543–2548 RSC; (f) K. Li, J. Chen, C. Yang, K. Zhang, C. Pan and B. Fan, Org. Lett., 2020, 22, 4261–4265 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- (a) C. W. Kee, K. F. Chin, M. W. Wong and C.-H. Tan, Chem. Commun., 2014, 50, 8211–8214 RSC; (b) J. Xuan, X.-D. Xia, T.-T. Zeng, Z.-J. Feng, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 5653–5656 CrossRef CAS; (c) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796–6802 RSC; (d) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS; (e) Z. Sun, N. Kumagai and M. Shibasaki, Org. Lett., 2017, 19, 3727–3730 CrossRef CAS; (f) S. Yang, P. Li, Z. Wang and L. Wang, Org. Lett., 2017, 19, 3386–3389 CrossRef CAS; (g) Z.-H. Qi and J. Ma, ACS Catal., 2018, 8, 1456–1463 CrossRef CAS; (h) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (i) M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS; (j) W. Ding, C. C. Ho and N. Yoshikai, Org. Lett., 2019, 21, 1202–1206 CrossRef CAS; (k) M. Kudisch, C.-H. Lim, P. Thordarson and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 19479–19486 CrossRef CAS; (l) L. Ma, W.-H. Fang, L. Shen and X. Chen, ACS Catal., 2019, 9, 3672–3684 CrossRef CAS; (m) T. Neveselý, C. G. Daniliuc and R. Gilmour, Org. Lett., 2019, 21, 9724–9728 CrossRef; (n) J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625–13634 CrossRef CAS; (o) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS; (p) D.-F. Chen, C. H. Chrisman and G. M. Miyake, ACS Catal., 2020, 10, 2609–2614 CrossRef CAS; (q) K. A. Rykaczewski and C. S. Schindler, Org. Lett., 2020, 22, 6516–6519 CrossRef CAS; (r) H. Tian, H. Yang, C. Tian, G. An and G. Li, Org. Lett., 2020, 22, 7709–7715 CrossRef CAS; (s) L. Tian, N. A. Till, B. Kudisch, D. W. C. MacMillan and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 4555–4559 CrossRef CAS; (t) J. Zheng, X. Dong and T. P. Yoon, Org. Lett., 2020, 22, 6520–6525 CrossRef CAS.
- J. Wang, B. Sun, L. Zhang, T. Xu, Y. Xie and C. Jin, Chem. – Asian J., 2019, 8, 1942–1946 CAS.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 6019–6019 RSC.
- R. Talukdar, Org. Biomol. Chem., 2020, 18, 8294–8345 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03761c |
| This journal is © The Royal Society of Chemistry 2021 |