
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering lignin heterogeneity in ball milled softwood: unravelling the synergy between the supramolecular cell wall structure and molecular events†
Ioanna
Sapouna
a and
Martin
Lawoko
 *ab
*ab
aWallenberg Wood Science Center, Department of Fiber and Polymer Technology, School of Chemistry, Royal Institute of Technology, KTH Teknikringen 56–58, 100 44 Stockholm, Sweden
bDivision of Wood Chemistry and Pulp Technology, Department of Fiber and Polymer Technology, School of Chemistry, Royal Institute of Technology, KTH Teknikringen 56–58, 100 44 Stockholm, Sweden. E-mail: lawoko@kth.se
First published on 18th March 2021
Abstract
Mechanical milling of lignocellulose has been used in several studies as a key pretreatment enabling the extraction of lignin from various sources for structural analysis. It is also applied as an alternative to wet chemical methods for lignin valorization. However, the changes caused to the plant cell walls at different hierarchical scales and how they relate to the molecular events are still poorly understood. In this context, we sought to gain deeper insights into molecular heterogeneity in milled cell walls, with a primary focus on lignin. A novel fractionation protocol was developed to enable the advanced analysis (1D and 2D NMR, SEC, XRD) of molecular populations in ball milled fiber walls. The methodology was applied to follow the emergence of such populations through the milling process, and in different milling environments. Lignin heterogeneity in the ball milled fibers was found to consist of distinct populations of small and large fractions of lignin carbohydrate complexes and pure lignin fractions, both with differences in lignin inter-unit abundances. Lignin-carbohydrate bonds of benzyl ester type were unequivocally demonstrated for the first time by combination of HSQC-HMBC NMR analysis. γ-Ester LCC and phenyl glycoside LCCs were also detected. Furthermore, an important branching point in lignin, previously controversial, namely the 4-O-etherified 5-5′ substructure, is unequivocally shown here by HSQC-HMBC analysis of the milled wood isolates, and supported by biomimetic lignin (DHP) to originate from the native structure. Based on the advanced characterization, the origin of lignin heterogeneity in ball milled fibers is proposed to result from the uneven distribution of the applied mechanical energy, where synergistic effects between crystalline and amorphous states play a central role. Accordingly, a plant cell wall model is proposed and a complete mechanism of its disintegration during the milling exercise is presented. The unveiled heterogeneity model of ball milled cell walls can serve as a useful guide for future studies on mechanical fractionation and valorization of lignocellulose based polymers.
Introduction
The nature of wood as a composite material was established by Payen in 1838.1 Thirty years later, Erdman was the first to characterize the only recently then named “lignin” compound as aromatic.2 Ever since, lignin has been the focus of scientists who have tried to extract lignin from wood species in its native state, i.e. without the effect of the extraction process. Braun's “native” lignin was introduced in 1939 and consisted of the extraction of lignin with cold water, ether and ethanol, without any mechanical pre-treatment.3 This admittedly mild approach yielded only a small percentage of lignin in wood, and as a result was not considered representative of the whole lignin population. In his seminal work in 1954, Björkman introduced an analytical extraction process for lignin, thereafter referred to as Björkman's lignin or what is now classically referred to as milled wood lignin (MWL), that was considered the closest representation of lignin in woods amongst isolates.4 The advantage of the MWL protocol has been the use of aqueous dioxane solvent and low temperatures which could not cause condensation- or other reactions on the lignin structure. However, the low yield of MWL (approximately 20–50% of the total lignin) was the driving force for the optimization and further development of the protocol, introducing cellulolytic enzyme digestion as a pre-treatment step.5 This increased the yield of extraction, which depends on the combination of wood species and enzymes. Nevertheless the product still contains an amount of carbohydrates that are not removed with the digestion. Enzymatic mild acidolysis lignin (EMAL) is a carbohydrate-free product, due to the combined effect of enzymes and an acid that hydrolyses lignin-carbohydrate complexes.6 Lignin extraction is limited by solvent accessibility to all levels of the cell structure and diffusion of lignin outside the complex matrix of which it is part of. As a result, the need for circumventing the extraction and studying lignin as a whole is eminent. In this context, whole ball milled cell wall dissolution protocols that enable complete studies on the lignin structure with high resolution, such as solution-state NMR techniques, have been developed.7–9Today, controversy exists regarding the linearity versus branching of MWL. In the work of Adler, biphenyl-(5-5′) and diphenylether- (4-O-5′) couplings formed during lignification could also serve as branching points and as a result Adler's model of MWL is a branched molecule.10 However, the very low amount of these etherified units detected in MWL was key to the work of Crestini and co-workers who recently concluded the linearity of the molecule.7 Additionally, Ralph and co-workers through work with model compounds and characterization of lignin with advanced NMR techniques concluded that 4-O-5′ structures cannot be considered a branching point since they are present in softwoods as free phenolic end groups.8,9 Recent work by Balakshin and co-workers revisited this debate of linearity in MWL and based on combined NMR analyses and size exclusion chromatography studies concluded that MWL is a highly branched and crosslinked polymer.11
All aforementioned protocols of MWL extraction and study have extensively utilized ball milling as a necessary, mechanical pre-treatment. Ball milling reduces the particle size and increases the surface area of the material, which also increases accessibility to the fibers, hence expedite the extraction. Nevertheless, ball milling is considered to cause structural changes. More specifically, β-O-4′ bonds are homolytically cleaved and carbon and oxygen centred radicals are created that further react.12,13 It has also been shown, that the structural changes induced are related to the milling time and intensity.11,14 During ball milling, the wood particles are introduced to a jar and grinding balls are added. Through collisions with the grinding balls and jar, a combination of compression and shear stress forces creates cracks on the substrate. The cracks can propagate either through the cell wall or via the middle lamella. It is suggested that intercellular cracks i.e. through the middle lamella, require less energy, hence they are created first.15 This supports the hypothesis that MWL over-represents lignin located in the middle lamella.14,16 At the same time, the longer the milling, the higher the total energy introduced to the fibers. Consequently, as the wood structure opens up more, lignin located in the cell wall is eventually exposed and extracted. Morphological analysis has shown that MWL exhibits characteristics of secondary cell wall lignin, and hence the origin of MWL is still under debate.17
There is an indisputable need for an in-depth understanding of the ball mill effect not only on the lignin molecule, but also on cellulose and hemicelluloses. Studies that have been performed on individual components, i.e. the ball mill effects on cellulose and cotton are many and important for fundamental understanding.18,19 However, these studies do not reflect the scenario in lignocellulose where synergies between the different polymers in this composite material need to be taken into consideration. The work presented here contributes to this purpose by investigating these synergies. Furthermore, there is limited knowledge on lignin heterogeneity in ball milled wood, as well as how it is related to the aforementioned synergies. The present work investigates these fundamental questions, specifically introducing a green analytical protocol that is combined with state-of-the-art characterization techniques to unravel the relationship between supramolecular and molecular events and their role in lignin heterogeneity. Furthermore, unequivocal evidence is provided for two previously hypothesized structural elements; one with ramifications for molecular branching in lignin and the other in support of lignin carbohydrate bonds. A combination of these two findings supports the existence of lignin carbohydrate networks in plant cell walls.
Materials and methods
Materials and chemicals
All chemicals were purchased from Sigma-Aldrich in analytical grade and used without further purification, unless stated otherwise. Debarked Norway spruce wood chips were used for the experiments.Sample milling
Debarked Norway spruce was milled through a 40-mesh screen using a Wiley Mini Mill 3383-L70 (Thomas Scientific). The product was ball milled with a Retch PM-400 planetary ball mill. For the ball milling, stainless steel jars and grinding balls were used. The ratio of jar volume (L):grinding balls (kg):sample weight (g) was kept constant for all the samples 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.8:40. The ball milling was performed in 1 h intervals with 30 min interval breaks at 300 rpm. The jars of the samples that were ball milled under a nitrogen atmosphere were purged with nitrogen for 1 min before milling. The different samples were ball milled for a total milling time of 1 h, 2 h, 4 h, 8 h, 12 h, 18 h and 24 h.
:0.8:40. The ball milling was performed in 1 h intervals with 30 min interval breaks at 300 rpm. The jars of the samples that were ball milled under a nitrogen atmosphere were purged with nitrogen for 1 min before milling. The different samples were ball milled for a total milling time of 1 h, 2 h, 4 h, 8 h, 12 h, 18 h and 24 h.
Milled wood lignin extraction and purification
Milled wood lignin (MWL) was extracted from 18 h-ball milled samples, milled under both atmospheres. The extraction was according to the protocol by Björkman with some modifications.4 In short, 18 h-ball milled wood (4% w/v) was dispersed in 96% (v/v) aqueous dioxane. The dispersion was mechanically stirred at room temperature for 72 h. After centrifugation, the extract was dried using a rotary evaporator and lyophilized. For the purification of MWL, the protocol by Obst and Kirk was followed.20 The lyophilized sample was dissolved in 90% acetic acid. 20 mL of solvent per g lignin was used. The acetic acid solution was added dropwise in water (220 mL of water per g of lignin). The precipitate was lyophilized, dissolved in 1,2-dichloroethane:ethanol (2:1 v/v, 20 mL solvent per g of lignin) and centrifuged to remove any solids. Afterwards, lignin was precipitated in anhydrous ethyl ether (230 mL per g of lignin). The precipitate was washed three times with fresh ethyl ether and dried.
The yield of purified MWL is almost half of the crude product.
Warm water extraction
10% w/v dispersion of ball milled wood in Milli-Q water was stirred at 80 °C for 4 h. The extract, from now on denoted as WWE, was collected by centrifugation and lyophilized. The wood residue was further extracted as described below.Mild alkaline extraction
10% w/v dispersion of the wood residue of the warm water extraction in 0.1 M sodium hydroxide (NaOH) solution was stirred at room temperature for 3 h. The extract was collected by centrifugation. The wood residue was washed with Milli-Q water until the washing reached pH 6. The washings were combined with the extract. The pH of this solution was set at pH 2 with hydrochloric acid (HCl). The precipitate was collected by centrifugation and washed with Milli-Q water (pH 4 with HCl), twice. The lyophilized precipitate is from now on referred to as the alkaline fraction. The wood residue from this step was used for the next extraction.Ionic liquid/ethanol, HCl extraction
1-Allyl-3-methylimidazolium chloride ([amim]Cl) was added to the wood residue of the alkaline extraction (1:1 weight ratio). The samples were stirred at 80 °C for 2 h. Afterwards, a solution of ethanol:Milli-Q water (8:2) containing 0.1 M HCl was added to produce a 5% w/v dispersion. The sample was stirred at 100 °C for 2 h. After cooling at room temperature, ethanol was evaporated under vacuum in a bath set at 50 °C. The volume was kept constant by the addition of water. After complete ethanol evaporation, the precipitate was collected by centrifugation and washed with Milli-Q water the pH of which was set at 4 with HCl, twice. The lyophilized precipitate is from now on denoted IL/EtOH, H+.
Lignin extraction from WWE and alkaline fractions
5% w/v lyophilized alkaline fraction of the 24 h ball milled sample (milled under normal and nitrogen atmosphere) in a solution of ethanol:Milli-Q water (8:2) containing 0.1 M HCl was mixed at 100 °C for 2 h. The mixture was centrifuged and the ethanol from the lignin-containing supernatant was evaporated under vacuum, at 50 °C at constant volume. Afterwards, the precipitate was collected and washed twice with Milli-Q water (pH 4, HCl). The product is denoted ALK-EtOH, H+. The same process was followed for the WWE. For that fraction (WWE), in addition to the process above, the water phase was extracted with ethyl acetate (EtOAc) twice. Lignin from that fraction was collected after EtOAc evaporated to dryness (WWE-EtOH, H+).
Lignin reduction with sodium borohydride
The reduction of WWE-EtOH, H+, ALK-EtOH, H+ and IL/EtOH, H+ fractions (24 h milled) was performed according to Adler with some modifications.21,22 In short, 1% w/v lignin solution was prepared in 50% aqueous ethanol. 0.1 M NaOH was added to the solution. The amount of the latter was 25% of the lignin solution volume. An amount equal to the lignin in the solution (1:1 w/w) of sodium borohydride (NaBH4) was added to the lignin/NaOH solution in portions. The reaction was left overnight, at room temperature. Thereafter, the pH was adjusted to 4 with HCl and ethanol evaporated with rotary evaporation. The volume of the sample was kept constant by the addition of water. After complete ethanol evaporation, lignin was collected by centrifugation, washed three times with Milli-Q water and lyophilized.
Size-exclusion chromatography in DMSO +0.5 wt% LiBr
The size-exclusion chromatography (SEC) analysis was performed on a SEC-curity 1260 system (Polymer Standards Services, Mainz, Germany). The separation was carried out through two GRAM analytical columns 100 Å and 1000 Å (Polymer Standards Services, Mainz, Germany) in series. The system was coupled to a photodiode array detector and refractive index (RI) detector. The eluent was dimethylsulfoxide (DMSO, HPLC grade) containing 0.5 wt% lithium bromide (LiBr). The flow rate of the eluent was set at 0.5 mL min−1. The columns were thermostated at 60 °C. The standard calibration was performed with pullulan standards with molecular weights ranging from 708 kDa to 342 Da, using the data from an RI detector, at 40 °C. 5 mg of the lyophilized sample was dissolved in 1 mL of DMSO containing 0.5 wt%. LiBr. Before injection, the samples were filtered through a 0.45 μm polytetrafluoroethylene (PTFE) syringe filter.Size-exclusion chromatography in THF
The analysis was performed in a Waters system (Waters Sverige AB, Sollentuna, Sweden). The separation was carried out through Waters Untrastyragel HR4, HR2 and HR0.5 (4.6 × 300 mm) solvent-efficient analytical columns in series with a Styragel guard column (THF, 4.6 × 300 mm). The columns were thermostated at 35°. The system was coupled to a Waters-2998 photodiode array detector operated at 254 nm and 280 nm and an RI detector. Standard calibration was performed with polystyrene standards with molecular weights ranging from 176 kDa to 266 Da, using the data from the 254 nm channel of the UV detector. The eluent was tetrahydrofuran (THF, HPLC grade). Before analysis, the samples were acetylated according to the protocol by Gellerstedt.23 The acetylated sample was dissolved in THF (2 mg mL−1) and filtered through a 0.45 μm PTFE syringe filter before injection.X-ray diffraction
X-ray diffraction (XRD) measurements were performed using an ARL X'TRA powder diffractometer (Thermo Fisher Scientific Inc., USA), using CuKα radiation generated at 45 kV and 44 mA. 2θ was scanned from 5° to 50° with a step size of 0.04°. The raw data were plotted in Origin (version 9.1.0, OriginLab Corporation). The curves were smoothed using the Savitzky–Golay method.1D and 2D nuclear magnetic resonance
Nuclear magnetic resonance (NMR) analysis was performed with a Bruker Avance III HD 400 MHz instrument at 300 K with a BBFO probe equipped with a Z-gradient coil. Sensitivity was improved with the gradient (e/a TAPPI). Additionally, spectra were recorded with a Bruker 400 DMX instrument equipped with a BBO probe. Sample solutions with approximately 12% (w/v) concentration in deuterated DMSO-d6 were analysed. HSQC and HMBC experiments were carried out with the “hsqcetgpsi” and “hmbcgp” pulse program respectively. For the latter, a d6 = 0.06 s delay was selected. A relaxation delay of 1.5 s was selected, with a minimum of 100 scans using 1024 × 256 increments with additionally 16 dummy scans. The F1 direction spectral window was 16 ppm and F1 direction spectral window was 166 ppm. The spectra were processed with MestreNova software (version 9.0.0, Mestrelab Research). The spectra were Fourier transformed and a baseline and manual phase correction was applied to both dimensions. The DMSO-d6 peak at δC/δH = 39.5/2.50 ppm was used as the internal reference. For the quantification, the signals of C2/H2 in lignin were selected as the internal standard.Quantitative 31P NMR analysis was performed following the protocol by Argyropoulos.24 In short, 30 mg of the lyophilized sample was accurately weighed and phosphorylated with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP). The internal standard was endo-N-hydroxy-5-norbornene-2,3-dicarboximide (e-HNDI).
Quantitative 13C experiments were performed with an inverse-gate proton decoupling pulse sequence with a 90° pulse angle and 65000 increments. A relaxation delay d1 = 1.7 s was selected. For the experiments, the relaxation agent, 0.01 M chromium(III) acetylacetonate, was added to the sample. The spectra were processed with MestreNova software (version 9.0.0, Mestrelab Research). The spectra were Fourier transformed and an apodization of 8 Hz was selected. Phase correction was performed manually. Multipoint baseline correction was selected with cubic splines. For the quantification, the integral of the region between 163–100 ppm, was normalized at 6.12. This region contains aromatic and vinylic lignin structures.
For the relative quantification of lignin and hemicelluloses in the fractions, eqn (S1) and (S2)† were used. These equations are explained in detail in the ESI.†
Results and discussion
In order to gain deeper insights into lignin heterogeneity in ball milled wood, we adopted a strategy where lignin populations could be studied in tandem with that of carbohydrate fractions. The reasoning was that since these components are closely associated at the molecular scale, detailed characterization of both components would provide deeper understanding of the mechanisms occurring at different hierarchical scales in the plant cell wall during the milling exercise. Such mechanisms are still poorly understood. In this context, we also studied the role of milling environments, where the milling in air is compared with that under an nitrogen atmosphere, as a function of milling time.For the above purpose, techniques to follow changes in the supramolecular cell wall structure were combined with a newly developed mild, selective and high-yield fractionation protocol to obtain samples that were subjected to advanced molecular level characterization techniques. Analytical methods included XRD, state-of-the art 1D and 2D NMR methods and SEC analyses.
XRD measurement shows progressive decrease in cellulose crystallinity during ball milling
XRD analyses (Fig. 1), showed that wood crystallinity, manifested in cellulose fraction, starts to decrease already in the earlier stages of the ball milling. This is consistent with the literature where ball milling was observed to decrease the crystallinity of cotton cellulose.18,25,26 The X-ray diffractogram pattern of the reference sample i.e. non-ball milled wood, exhibits the characteristic peaks of cellulose I. The broader peaks resulted from the nature of the sample, which is a combination of crystalline and amorphous regions. The crystalline planes (101) and (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) at approximately 2θ = 15° and 2θ = 16.6° cannot be distinguished due to overlap. The plane (200) is visible at 2θ = 22.4° and finally (040) is seen at 2θ = 35°.27,28 The samples milled for longer than 2 h in air and longer than 1 h under an nitrogen atmosphere exhibit a broad, amorphous signal at 2θmax = 20°.
) at approximately 2θ = 15° and 2θ = 16.6° cannot be distinguished due to overlap. The plane (200) is visible at 2θ = 22.4° and finally (040) is seen at 2θ = 35°.27,28 The samples milled for longer than 2 h in air and longer than 1 h under an nitrogen atmosphere exhibit a broad, amorphous signal at 2θmax = 20°.
 | ||
| Fig. 1 X-ray diffractograms of Norway spruce wood ball milled under (a) air and (b) nitrogen atmosphere. The reference sample is non-ball milled spruce. | ||
The observed decrease in crystallinity of cellulose is due to the absorption of mechanical energy which converts it to the amorphous state
This observation is of central importance in the context of understanding lignin heterogeneity and will be re-visited in a later section of this paper. Interestingly, the decrease of crystallinity was faster when the milling was performed under a nitrogen atmosphere than under air atmosphere. This observation suggests that some oxidative mechanisms, more prominent in air milling, retarded the amorphization process, while in the absence of oxygen the process was accelerated. In this context, the oxidative stabilization of radicals resulting from homolytically cleaved glycosidic linkages has been reported.29 Furthermore, the introduction of carbonyl groups on cotton cellulose during ball milling has been demonstrated,18 and is likely due to the presence of oxygen in the system. How these tribochemical events are connected to the amorphization rate differences observed between the two atmospheres here (air and nitrogen milling) remain to be studied. Nevertheless, cellulose amorphization and depolymerization seem to be related.Enabling molecular population studies through greener fractionation protocols
Traditionally, lignins have been isolated following ball milling for analysis. These lignins are extracted by aqueous dioxane to obtain a crude product that is further purified to obtain milled wood lignin (MWL). Unfortunately, the MWL yield is quite low (20–50% on lignin basis) and thus the analysis does not represent the total lignin population. Furthermore, the use of dioxane is no longer appropriate as it is a likely carcinogen and does not biodegrade easily.30,31 Greener fractionation methods that enable lignin population studies are therefore required, as herein, reported (Fig. 2).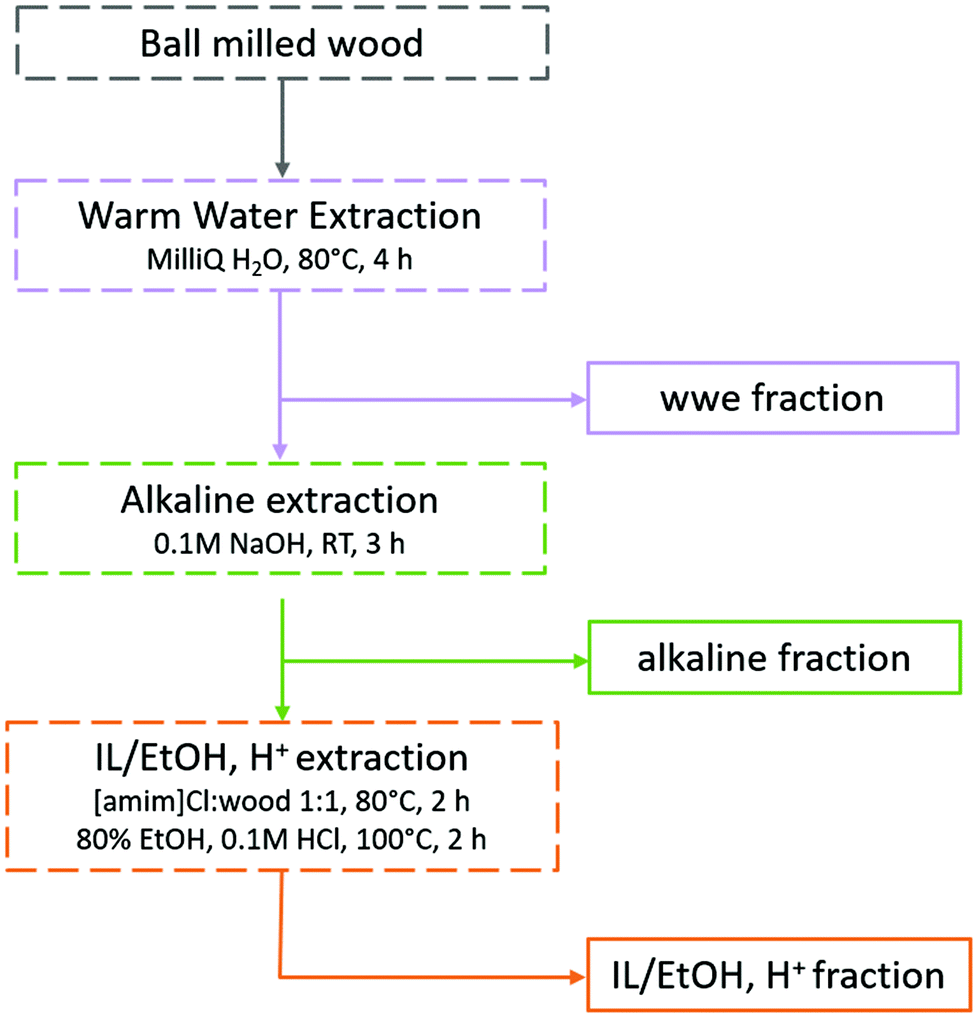 | ||
| Fig. 2 Fractionation protocol for the study of lignin populations. The full analytical protocol is reported in ESI, Fig. S1.† | ||
In the first extraction step, warm water extraction targeting hydrophilic hemicellulose fractions is performed. Next, we applied a mild alkaline extraction of lignin rich fractions. Ionization of phenolic hydroxyls would improve the solubility of phenolic lignins in this solvent system. Based on yields obtained in the first two steps, further extraction steps were required. Reasoning that the recalcitrant polymers were likely locked up in complex networks or entrapped in a complex cellulose matrix, we chose to pre-swell the residue in an ionic liquid ([amim]Cl), then perform a mild and selective acid hydrolysis simultaneously with ethanol extraction of lignin at low temperatures. The conditions for the ethanol acidolysis were chosen carefully not to cause any significant changes to the lignin structure, and was a refinement of our recent work32 where it was shown that etherification of lignin under mild conditions protected its structure. The final residue in the present work was enriched in celluloses, but still contained some lignin and hemicelluloses. Importantly, the order of solvents used in the sequence should not be interchanged. The reason for this is that since this study targets lignin populations, the solvents with a weaker dissolving power for lignin should be applied first in the sequence. In that way, more populations reflective of ball milled wood lignin can be studied.
Material balance demonstrates high recovery yields
A total mass balance of the different fractions shows that the extraction yields increase with milling times irrespective of the milling environment. This is consistent with improved accessibility enabled by cellulose amorphization and depolymerization processes, both of which would enhance the solubility of lignin and hemicelluloses. Tables S1a–c in the ESI† show the relative compositions of the fractions based on NMR studies that are discussed in detail in another section, and Tables S2a and b† present the total yield of extracted lignin and hemicelluloses, respectively. The warm water extract (WWE) consisted, as expected, mainly of hemicelluloses. In Fig. 3(a), the total yield of hemicelluloses is plotted against milling time for both atmospheres. The increase in extraction yield with milling time for both milling atmospheres is clear, with higher yields observed for the nitrogen atmosphere. The increase is not linear due to the presence of recalcitrant hemicelluloses, which is discussed in a later section.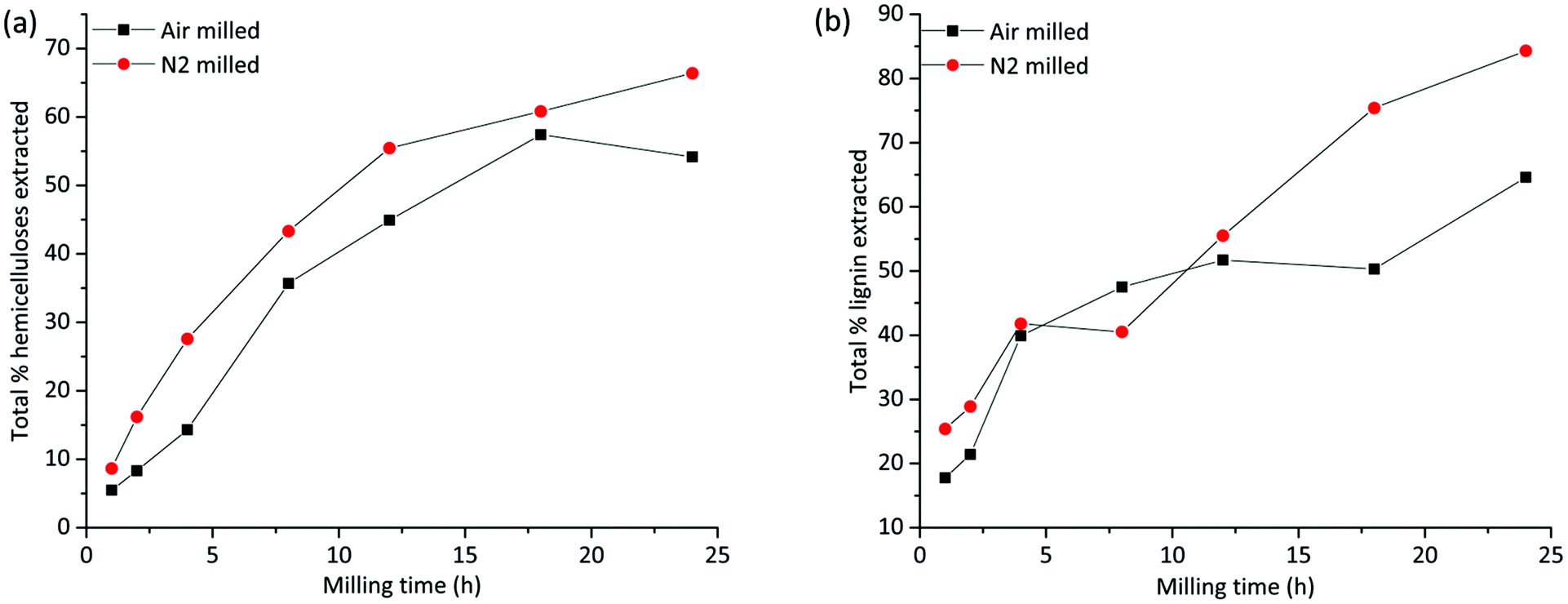 | ||
| Fig. 3 Total yield of extraction of (a) hemicelluloses, from warm water and mild alkaline extraction and (b) lignin from all three extraction steps. | ||
The alkaline dissolved fraction was, as expected, enriched in lignin. Higher extraction yields are observed for the nitrogen atmosphere, which was also the case for the WWE. The lignin yields increased with milling time and varied significantly with the milling environment (Fig. 3(b) and ESI, Fig. S2†) with the higher values reported for the nitrogen atmosphere. Of note, for the alkaline fractionation step, 18–24 h milling resulted in a 32–49% lignin yield in this fraction when the milling was performed in nitrogen and only 17–21% when performed in air atmosphere (ESI, Tables S1a–c†). The increased yield of extraction that is observed in the samples milled under a nitrogen atmosphere is most probably a result of radicals that are formed during milling being responsible for polymer degradation. Contrarily, in air atmosphere, such radicals are likely quenched by oxygen, which is itself a biradical. Hence, less degradation and therefore lower extraction yields are observed in the air atmosphere.
As shown by the analysis, the residue left after alkaline extraction still contained both lignin and hemicelluloses present in an enriched cellulose fraction. Consequently, more lignin was fractionated when this residue was mildly extracted with IL/EtOH, H+. Accordingly, a pure lignin fraction was obtained, and this time the yield was higher for the air atmosphere, which was not surprising since more lignin was extracted for the nitrogen atmosphere in the previous steps. There was simply less extractable lignin left in the residue of the nitrogen milled sample in the final extraction step. The high purity of the lignin in this fraction is due to acid hydrolysis of lignin carbohydrate bonds. The yield of extracted lignin from the IL/EtOH, H+ fractions is presented in ESI, Fig. S2(b).†
Worth noting is that the amount of lignin left in the final residue could not be assessed by the Klason lignin determination method. This was due to incomplete hydrolysis of cellulose, probably caused by hornification during drying.
To put this study in context with conventional studies, we also prepared classical milled wood lignin (MWL) from the 18 h milled wood, for comparison. MWL has been extensively used in the literature as a representative “proximate native lignin” structure. The preparation is described in the material and methods section. We obtained only 17–24% lignin yield for both milling conditions, which is lower than the summative yield of lignin achieved in the present population studies (Fig. 3b). We show therefore that the developed protocol achieves high yields of lignin in different population studies and is therefore exemplary for deeper fundamental studies of the widely applied ball milling pre-treatments.
Lignin molar mass heterogeneity demonstrated through size-exclusion chromatography studies
The size-exclusion chromatography (SEC) of the WWE fractions (ESI, Fig. S3a and b†) shows the successive depolymerization of hemicellulose with increased milling time for both milling environments. Comparing the number average molar mass (Mn) of the WWE between the two milling environments (ESI, Table S3†), it is evident that the depolymerization of hemicelluloses is less in the air environment. This is consistent with the earlier discussed yield of the warm water extracted material being higher for the nitrogen environment. The UV detector signals however, revealed that several lignin populations were co-extracted with the hemicelluloses in the WWE (ESI, Fig. S3a and S3b†). It is observed that the portion of higher molecular weight material containing lignin increased with extended milling time. These lignins were likely dissolved in warm water due to chemical bonds to the relatively more hydrophilic carbohydrate fraction as lignin carbohydrate complexes (LCCs).The depolymerization reactions incurred during milling therefore, affected the porosity of the system enabling improved extraction of higher molar mass fractions constituting LCCs. It is worth noting that native softwood hemicelluloses, specifically galactoglucomannans (GGMs) are suggested to have much higher molar masses. Leppänen et al. reported molar mass in the range of 30 kDa for spruce GGM.33 Thus, the results here evidence that ball milling causes significant depolymerization of hemicelluloses present in LCCs. However, based on the extracted yields of hemicellulose in the WWE, which ranged from low to moderate (ESI, Table S2b†), it is likely that some other hemicelluloses with different chemical properties were left in the residue.
SEC analysis was then performed on the lignin-rich samples. Here, it is worth noting that we studied these on a relative-scale, due to the lack of appropriate standards for lignin SEC. THF-SEC analysis of the acetylated alkaline extracted lignins revealed several interesting features (Fig. 4). Higher molar mass lignin was being extracted with increasing milling time for both environments. This could be explained in two ways; either the higher molar mass lignin became accessible for extraction with extended milling, or lignin condensation reactions occurred during milling and a cumulative effect is observed at extended milling times. Lignin condensation of resonance radicals to homolytically cleaved aryl ether linkages could occur, with the effect that stable C–C bonds are formed. However, this was not supported by structural studies discussed in a later section. Hence, better access to the extraction of larger molecules is the probable explanation for the observed SEC patterns.
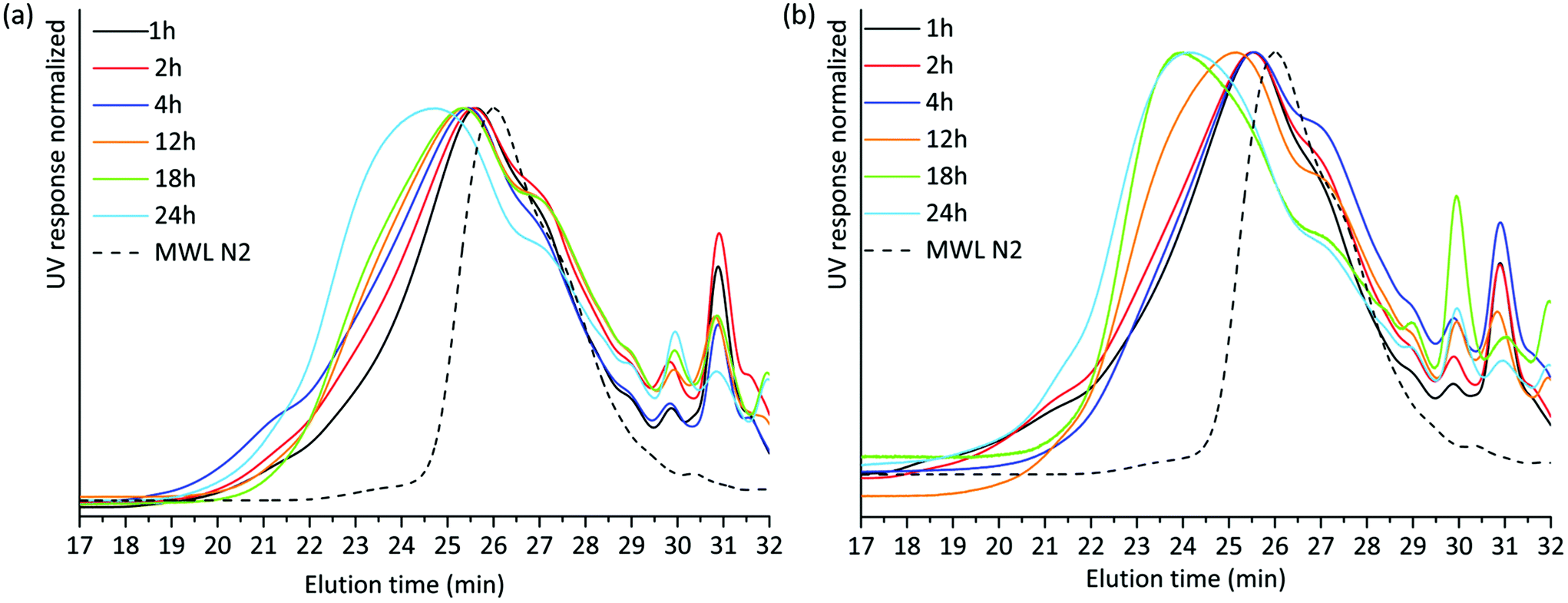 | ||
| Fig. 4 THF SEC overlay of acetylated alkaline extracted lignin milled in (a) air and (b) nitrogen atmosphere. | ||
The THF-SEC of the IL/EtOH, H+ lignins is presented in ESI, Fig. S4a and S4b.† The correlation between milling time and molecular weight of the extract exhibits the same trend in the IL/EtOH, H+ fractions. Irrespective of the milling atmosphere, increasing the milling time yields higher molecular weight fractions.
We then compared the SEC of the lignin fractions obtained through the new protocol with that of MWL obtained from the same sample, all fractions being isolated after 18 h ball milling in nitrogen (Fig. 5). Ocular observations indicate that MWL has a lower molar mass than alkaline and IL/EtOH, H+ fractions. In the case of the alkaline fraction, such a higher molecular weight material could constitute LCC structures, since carbohydrates were present in this fraction. The same cannot be said for the IL/EtOH, H+ fraction which constituted almost pure lignin molecules.
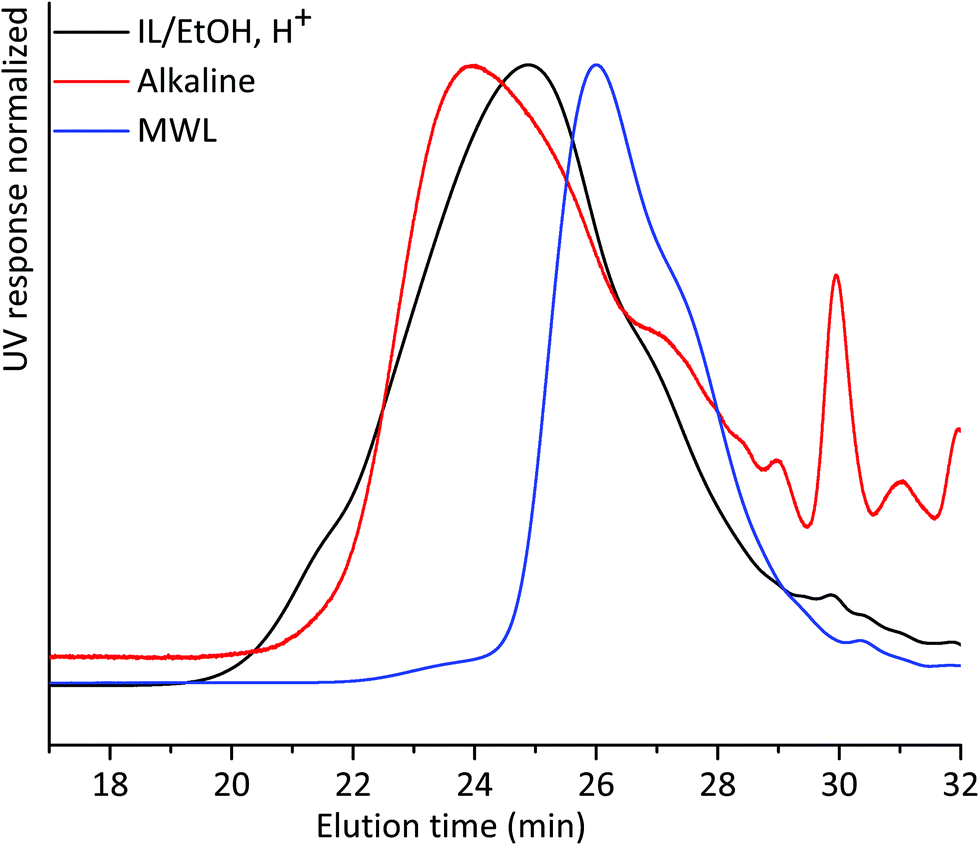 | ||
| Fig. 5 THF-SEC of lignin isolated from alkaline extraction, IL/EtOH, H+ extraction and MWL. All samples were ball milled under a nitrogen atmosphere for 18 h. | ||
The presence of carbohydrates chemically bonded to lignin prevents the assessment of the molecular weight of the lignin part of the copolymer. Therefore, to assess the molecular weight of the lignin moieties in the WWE and alkaline fractions, we performed a mild acid hydrolysis in the presence of ethanol, to hydrolyse the carbohydrates and obtain a purer lignin fraction. In this way, they would be comparable with the IL/EtOH, H+ fraction. The hydrolysed samples are abbreviated WWE-EtOH+, H+ and ALK-EtOH, H+. These samples, as well as the IL/EtOH, H+ fraction, were then reduced with sodium borohydride (NaBH4) for reasons related to NMR analysis discussed later. Consequently, the abbreviated names following the reduction are WWE-EtOH, H+-red, ALK-EtOH, H+-red, and IL/EtOH, H+-red. Prior to SEC analysis, these samples were acetylated. The THF-SEC of these samples and that of acetylated MWL as reference are presented in Fig. 6. It was evident that the molar mass was in the order IL/EtOH, H+-red > ALK-EtOH, H+-red > WWE-EtOH, H+-red, and in support of size heterogeneity of lignin moieties in ball milled wood fibers. Interestingly, MWL shows elution behaviour more similar to WWE-EtOH, H+-red while the other two fractions seem to have a higher molecular weight (lower elution volume) than MWL.
 | ||
| Fig. 6 Overlay of THF SEC of EtOH/H+ extracted and reduced samples milled in (a) air and (b) nitrogen atmosphere. | ||
Molecular structure studies by 1D and 2D NMR methods show distinct structural populations
Two-dimensional NMR (HSQC) was used for detailed structural studies. In WWE fractions of both milling atmospheres (air and nitrogen) a co-extraction of hemicelluloses and lignin is observed (Fig. 8a). Clear signals assigned to anhydro- mannan, glucan, galactan, xylan, arabinan and uronans are observed and substantiate the presence of galactoglucomannan (GGM), and arabinoglucuronoxylan (AGX) as the main hemicelluloses, as well as small amounts of pectins. These compositions are consistent with the carbohydrate analyses performed by HPAEC-PAD on selected samples (ESI Table S2d†). The raw material composition is also reported.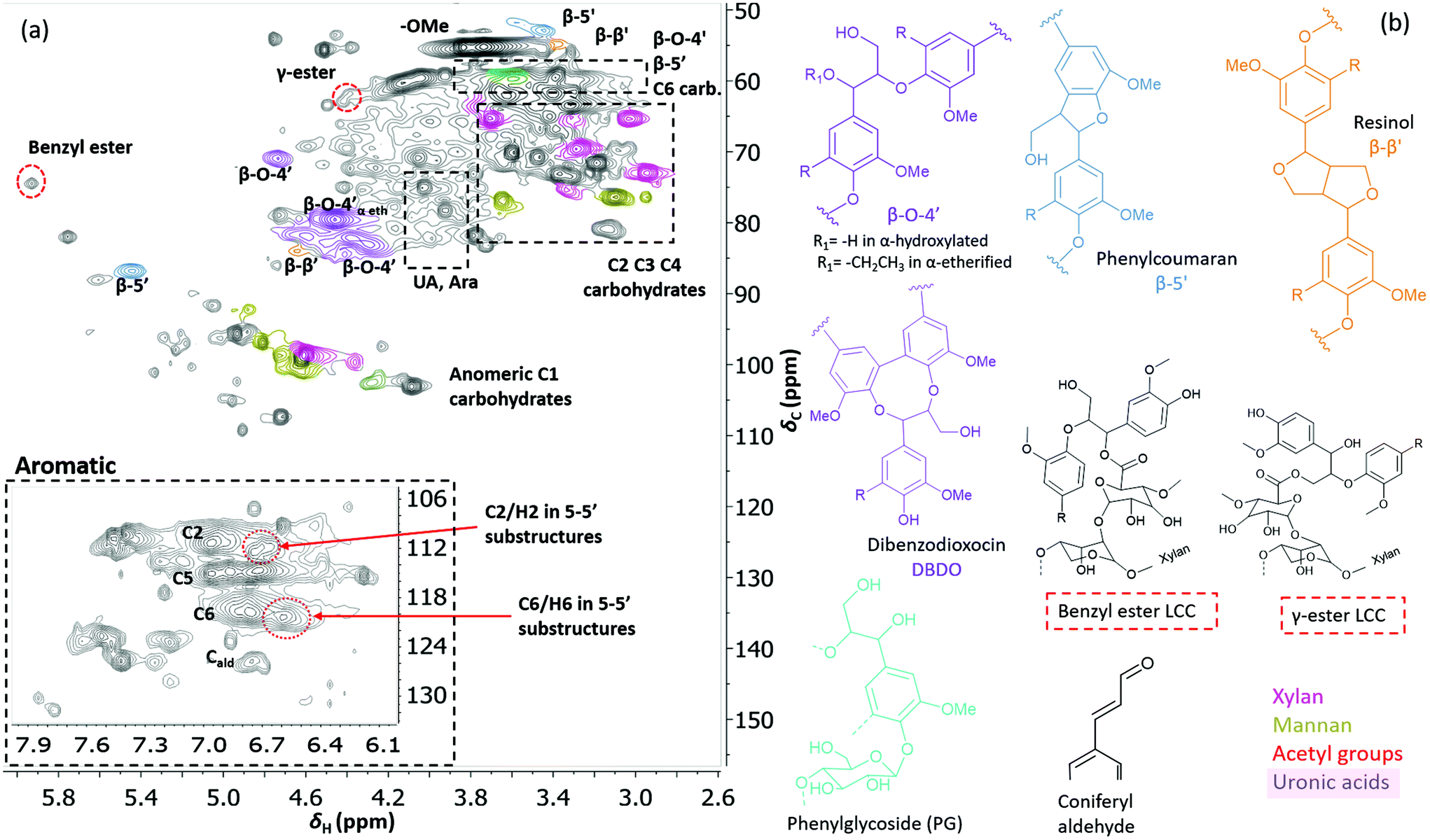 | ||
| Fig. 7 (a) HSQC spectrum in DMSO-d6 of acid hydrolysed WWE fraction (WWE-EtOH, H+). Complete peak assignment can be found in ESI Tables S7 and S8.† The structure of the 4-O-etherified 5-5′ substructure is presented in Fig. 9. (b) Chemical structures and assigned colours for most common lignin inter-unit linkages, LCC structures and hemicelluloses that also appear in Fig. 8 and 12. | ||
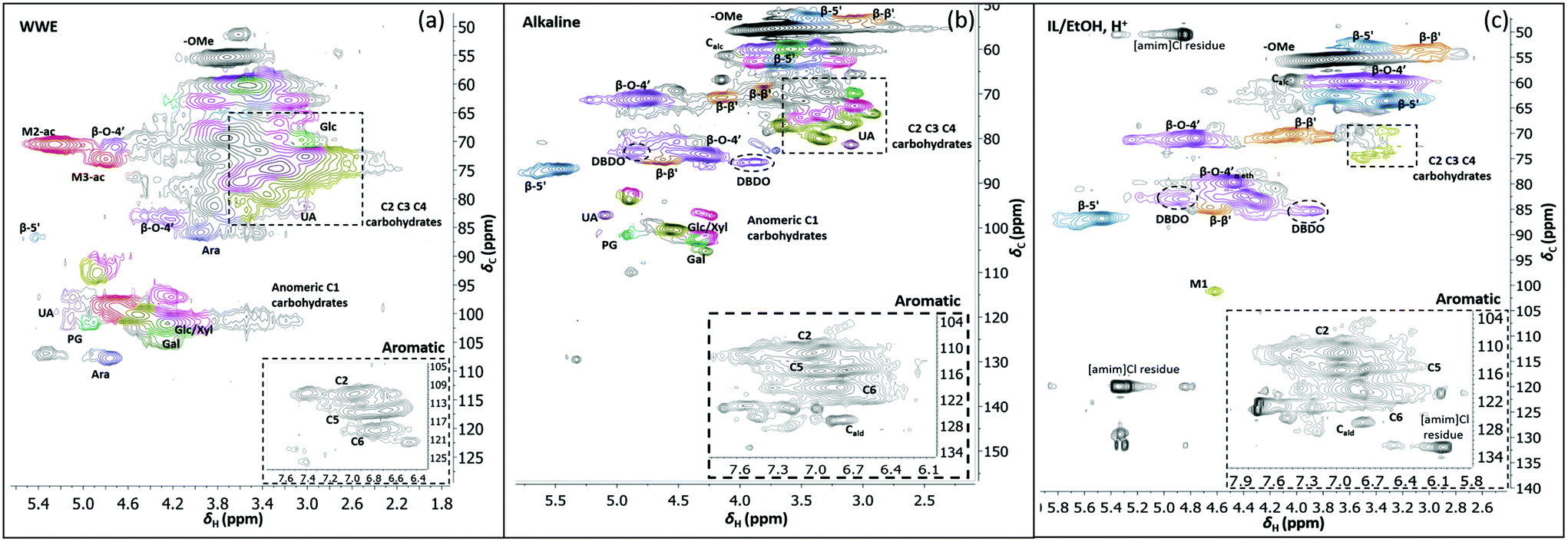 | ||
| Fig. 8 HSQC spectra of WWE (a), alkaline (b) and IL/EtOH, H+ fractions (c), milled in air for 18 h. The spectra were obtained in DMSO-d6. The main structures identified can be found in Fig. 7b and the complete assignment of the signals is presented in ESI, Tables S7 and S8.† | ||
The HSQC also shows the retention of native O-acetyl decorations in mannan units at carbon 2 and carbon 3 hydroxyls resonating at 70.8/5.21 ppm and 73.1/4.80 ppm respectively, in the WWE for both milling environments. Any hemicellulose degradation as suggested by the SEC analysis seems therefore to occur in the main chain. It is thus concluded that the ball milling yielded a fraction containing partially depolymerized hemicelluloses with native branching sugar units and acetyl decorations that were water soluble.
Similarly, typical lignin C–H correlations in the aromatic positions 2, 5 and 6 are observed in the region 105–125 ppm/6–7.5 ppm. The lignin to carbohydrate ratio in the WWE fractions was estimated using the peak integrals and eqn (1) and (2) in the ESI.† The results are reported in ESI, Tables S1a–c.† It is important to note that the carbohydrate and lignin composition analyses by the HSQC method are semi quantitative and need to be validated by classical analysis following acid hydrolysis of the biomass and fractions thereof. This analysis was performed on selected samples only. In this context, HPAEC-PAD was adopted for the monosugar analysis and both Klason and acid soluble lignin were analysed. The results are reported in Tables S2c and d.† In general, the trends are comparable with the NMR results under considerations that the NMR is still semi-quantitative. Hence, for the other samples, NMR was used instead, based on the limitation in sample amounts. It is clear that the ratio of lignin to carbohydrates increases with milling time. This can be explained by the increased solubilization of lignin carbohydrate complexes (LCC) as a result of the continuous depolymerization with milling time. In support of lignin carbohydrate bonds, signals from phenyl glycosides (PG) can be seen in between 100–103/5–5.2 ppm (Fig. 8).34,35 Some of the signals from lignin inter-units in the WWE are unfortunately overlapped with carbohydrate signals in the oxygenated aliphatic regions of the HSQC spectra. However, the β-O-4′ and phenylcoumaran sub-units were estimated using the non-overlapped signals at 83.5/4.22 ppm and 86.9/5.40 ppm respectively, to amount to between 13–23% and 3–5% per 100 aromatic units, for the studied milling times and environments. To better investigate the lignin structure in the WWE, we analysed the WWE-EtOH, H+ fractions, i.e. the same sample but with the bulk of the carbohydrates hydrolysed as discussed in an earlier section. We have previously studied this approach and observed that the lignin structure is protected if the hydrolysis conditions are mild enough and performed with ethanol as the solvent.32 The protection mechanism is based on a capping etherification of the benzylic cation formed under acidic conditions. This prevents the lignin-lignin condensation reactions.
The spectrum of the isolated WWE-EtOH, H+ product is shown in Fig. 7 for the 18-hour milling time in a nitrogen atmosphere. The total β-O-4′ was then estimated using the sum of the integrals at 70.8/4.74 ppm (β-O-4′ α-OH), 79.5/4.46 ppm (α-etherified) and 85.3/3.9 ppm (DBDO) and found to be 35% per 100 aromatic rings.
It is worth noting that lignin-lignin benzyl ethers and lignin carbohydrate benzyl ethers, both present in β-aryl ether structures, exist in milled wood11,36 and it is therefore critical to keep in mind that the etherification with solvent achieved here will result in a signal that overlaps with the original benzyl ethers. The discussed region was also heavily overlapped with carbohydrate signals in the original sample and not included in the β-O-4′ evaluation of the original WWE fraction. This explains why a higher β-O-4′ content is found after the acid hydrolysis in aqueous ethanol.
As expected, the hydrolysis of the bulk of carbohydrates from the WWE yielded upon HSQC analysis, a spectrum indicative of a lignin enriched sample (Fig. 7). The efficiency level of the hydrolysis of glycosidic bonds is manifested in the significant decrease in the intensity of signals assigned to anomeric carbon involved in glycosidic bonds that appear in the region 100–110/4–5.2 ppm. Yet, interestingly some sugar signals are still observed in the recovered lignin enriched fractions. These include signals of mannose, glucose, arabinose, galacturonic acids and glucuronic acid and were probably linked to lignin in LCCs. The phenylglycoside (PG) bonds were also retained. Further investigations of the atom connectivity are to follow but first a few words on why this particular sample is important for deeper analysis, are in order: the HSQC contours were narrow, indicative of smaller oligomers. This is also consistent with SEC analysis discussed earlier (Fig. 6). The oligomeric nature of this fraction has benefits when it comes to HMBC analysis. HMBC analysis requires long mixing times, but if the T2 relaxation times are too short, as in larger molecules which tumble slowly, there will be no signal. Here, we projected that these lignin-carbohydrate oligomers from the acid hydrolysis of WWE would be suitable candidates for HMBC analysis. Indeed, the HSQC spectra showed a number of unusual peaks which could be resolved by HMBC. These are now discussed.
Benzyl ester LCC unequivocally identified for the first time in wood isolates by HSQC-HMBC
Interestingly, we identify, for the first time in wood isolates, a benzyl ester LCC (ESI, Fig. S8† and Fig. 7b, the signal at 74.5/5.92 ppm) only previously reported for biomimetically synthesized lignins.37 The structure was unequivocally assigned by HMBC which clearly showed the connectivity between the two spin systems, i.e. a uronic acid moiety connected to an aryl ether moiety in lignin. The HMBC spectra, together with the complete assignment principle, are shown and described in the ESI (Fig. S11 and S8†). Additional support was derived from the HSQC analysis of dehydrogenation polymer (synthetic lignin) synthesized in the presence of uronic acid (ESI, Fig. S7a†). The mechanism for the formation of benzyl esters is believed to be an addition reaction of the carboxylic functionality in uronic acid moieties to the quinone methide intermediate during β-O-4′ formation. Interestingly, what has previously been detected in wood isolates is the γ-ester LCC instead. The γ-ester LCC is hypothesized to be the result of a uronosyl migration from the benzylic (α) carbon to the γ position, making this LCC type dynamic.37 Indeed, we also further identify this migrant, i.e. γ-ester with the signal at 63/4.4 ppm. The possibility that this signal corresponds to the lignin acetyl ester at the γ position was eliminated by HMBC analysis where the absence of long range HMBC correlation between acetyl protons at 2.0 ppm and the ester carbonyl group was established in the ESI (Fig. S9†). Interestingly, the carbonyl signal in the benzyl ester LCC was more shifted downfield when compared to that of the γ-ester (198 ppm versus 170 ppm, see HMBCs in ESI Fig. S8 and S9†). Clearly, some enhanced deshielding effects of the carbonyl carbon in the benzyl ester were responsible for this observation. We speculate that intramolecular hydrogen bonding could be involved, where the role of the higher mobility of the γ-carbon comes into play. This carbon is flexible and the hydroxyl group bonded to it could easily stretch out as the H-bond donor to the ester carbonyl oxygen in the benzyl ester. In contrast, when the ester bond has migrated to the γ-carbon, the hydroxyl at the α-carbon is limited in mobility due to the α-carbon being directly bonded to the aromatic ring. In effect, no hydrogen bonding occurs.In any case, the dynamic nature of the ester linkage is an interesting phenomenon and the exact role of this dynamism in the cell wall remains to be investigated. For certain, the migration from the α-carbon to the γ-carbon would transfer stress from the rather confined α-carbon region to the more easily accessible γ-carbon. This would enhance the mobility of the xylan or pectin polymers on which the uronic acid moieties are located. One speculation therefore, is that the uronosyl migration is adaptively or responsively triggered in plant cell walls. Dynamic covalent bonds have been applied to polymer chemistry and are shown, for example, to relieve stress in thermosets38 consequently affecting the material properties.
One hypothesis is that the ester migration from the α- to γ-position occurred during the ball milling or even possibly during the extraction process. To address this, we synthesized lignin (dehydrogenation polymer, DHP) in the presence and absence of the uronic acid monomer. The DHP product is simply filtered, washed and dried, hence circumventing the necessity for harsh isolation pre-treatments such as milling and extraction processes. The HSQC spectra are presented in the ESI S7a and b.† The signals from both α- and γ-esters are detected in the sample synthesized in the presence of uronic acid and as expected absent in the reference. We conclude therefore that the uronosyl migration occurs during the lignin polymerization process.
HSQC/HMBC identification of 4-O-etherified 5-5′ biphenyl structures: a branching point in lignin
The debate on whether lignin is linear or branched is still ongoing. The 5-5′ linkages constitute potential branching points in lignin. If they are etherified at the 4-O position to another oligomeric or polymeric lignin moiety, then they constitute a branching point. In contrast, free phenolic 5-5′ structures would simply contribute to the continuity of the lignin backbone making it linear. A branched lignin could confer different properties from a linear lignin with important underpinnings for function in the plant cell wall. In addition, the linearity or branching of the lignin macromolecule is crucial to inform future applications of lignin in polymer systems. Recently, Balakshin and co-workers provided some indirect evidence that the majority of 5-5′ substructures were etherified, in effect making milled wood lignin (MWL) a branched polymer.11 This observation challenges recent literature on the linearity of MWL39 and is contrary to the linear native lignin structure based on lignification theory.9 We thus endeavoured to explore this intricate problem further by performing a deeper analysis of these structures in the lignin fractions obtained in this work. The high yield lignin fractionation combined with population studies could provide additional knowledge on the extent of such branching. As a starting point for this study, we chose the acid hydrolysed WWE fraction (WWE-ETOH, H+) due to its molecular properties being suitable for HMBC analysis, as discussed earlier in detail. Our strategy was to choose the signals of the C2H2 and C6H6 in 5-5′ substructures as starting points. These positions would allow us to study a broader atom connectivity by the HMBC technique (Fig. 9 and ESI Fig. S10†). To the benefit of the analysis, the signals of C2H2 in 5-5′ substructures appearing at 112.1/6.69 ppm, and that of C6H6 in the same structure appearing at 120.6/6.59 ppm were easy to distinguish in the HSQC (Fig. 7a). We have previously assigned these two signals based on model studies.40,41 Balakshin and co-workers suggested that C3 aromatic signals in a 4-O etherified 5-5′ substructure resonated at 151 ppm, in contrast to most C3 resonances between 144 and 149 ppm.11 | ||
| Fig. 9 4-O-Etherified 5-5′ biphenyl structure. R1, R2, R3 represent oligomeric lignin units. | ||
Our strategy going forward was to use HMBC analysis with the H2 and H6 aromatic protons in 5-5′ substructures as starting points. If this structure was indeed etherified, then the aromatic proton H2, should have a three-bond correlation with the C3 aromatic signal in etherified 5-5′ substructures, appearing at 151 ppm (Fig. 9). Indeed, a three-bond correlation was established with a signal at 151 ppm to confirm that the 5-5′ subunits were indeed etherified as recently postulated. The detailed HMBC assignment is described in ESI, Fig. S10.† All atom connectivities shown in Fig. 9 were established and confirmed not only the 4-O etherified 5-5′ connections (connection between A and B rings, Fig. 9) but also the additional connectivity to an α-etherified β-O-4′ substructure (connections between rings A and C, Fig. 9). The α-etherification in this structure could be native lignin-lignin alkyl benzyl ethers,11 or a result of the etherification of Cα hydroxyl in the β-O-4′ substructure with ethanol during the acid hydrolysis step used to obtain the fraction.
Nevertheless, the identification of 4-O-etherified 5-5′ substructures provides support for lignin being branched in milled wood, and that these structures neighboured a β-O-4′ substructure. It is, however, important to note that these structures should be distinguished from dibenzodioxocins if they are to constitute potential branching points. This substantiation is provided in a later section.
Structural studies reveal differences in compositions and lignin inter-unit abundances between the populations
Having evaluated new linkages as discussed above, we set out to study detailed compositions and structures of the lignin populations. First, we compared the compositions of the raw fractions obtained in our protocol, i.e. WWE, alkaline extracts and IL/EtOH, H+ extracts (see protocol, Fig. 2 and ESI, Fig. S1† for fraction identities). It is clear that the WWE and the alkaline fraction contained both lignin and carbohydrates. The carbohydrate structures in these two fractions were quite similar, and as described for the WWE earlier, showing dominance of galactoglucomannan (GGM) and arabinoglucuronoxylan (AGX) and small amounts of pectins manifested in the detection of galacturonic acid. A notable difference, however, is the absence of acetyl decorations in the alkaline fraction (Fig. 7a and b). The reason for this difference is an alkaline hydrolysis of ester linkages.The IL/EtOH, H+ fraction on the other hand had only trace amounts of carbohydrates, as expected, due to the acid hydrolysis. Interestingly, only a single anomeric signal of a mannan unit was observed for this fraction and appeared at 101.9/4.6 ppm. This unit was most probably linked to the lignin in benzyl ether bonds. Benzyl ethers between lignin and C6 in the mannan unit in softwoods was only recently unequivocally established.36 However, the strong signal resulting from etherification with ethanol during the procurement of this fraction would overlap with the signals for lignin carbohydrate benzyl ethers, making the assignment of lignin-carbohydrate benzyl ethers in this work not possible. Furthermore, lignin-lignin benzyl ethers also resonate in this region.11
The total β-O-4′ content in lignin can be approximated by HSQC after setting the signals of C2–Ar to 100 and using the integrals of the HSQC signals in the following equation:

In Fig. 10, the β-O-4′ content for the populations obtained when milling was performed in the two atmospheres is presented as a function of milling time. It is clear that the β-O-4′ is significantly lower in the WWE than in the alkaline and IL/EtOH, H+ samples. To be able to make a fair comparison between the lignin in alkaline and the IL/EtOH, H+ fractions, the alkaline fractions were hydrolysed under the same conditions as the IL/EtOH, H+ for selected fractions, consequently minimizing interfering carbohydrate signals. This treatment does not only remove the bulk of carbohydrates but also etherifies part of the benzylic carbons shifting part of the signals originally assigned to β-O-4′-α-OH, to β-O-4′-α-etherified. In any case, the basis for comparison between the fractions would then be fair following the described hydrolysis approach. Secondly, in the interest of 13C NMR quantitation of the 4-O-etherified 5-5′ inter-unit linkages previously discussed, the purified lignins above were reduced with sodium borohydride. This reduction removes overlapping signals from α- and γ-carbonyls as well as spirodienone that resonate in the same region as the C3 aromatic signal in the 4-O-etherified 5-5′ structures.11 This specific signal is used for the quantitative studies.
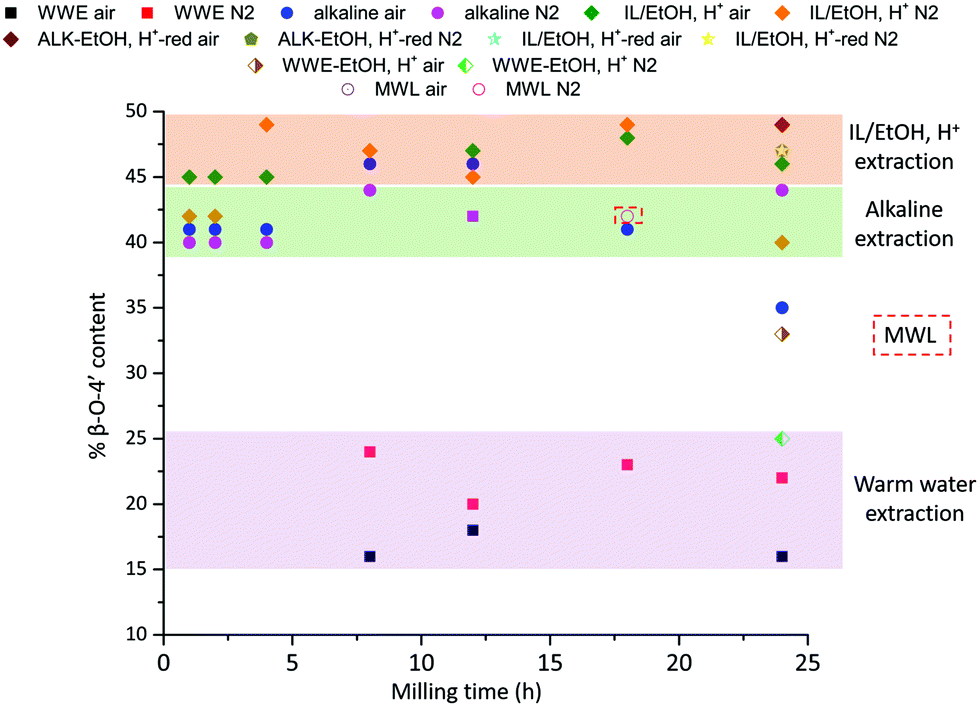 | ||
| Fig. 10 β-O-4′ content of extracted fractions from HSQC and quantitative 13C NMR. The WWE fractions from shorter milling times are not included. The integration was not considered reliable due to the weak signals in the aromatic region. | ||
The purified and reduced lignins were then studied by HSQC (ESI, Fig. S6a and S6b†), which supported the β-O-4′ trends in the raw fractions, i.e. IL/EtOH, H+-red > ALK-EtOH, H+-red (Fig. 10). The alkaline fraction seemed to have the same β-O-4′ content as MWL prepared from the same starting material. Overall, these differences in the β-O-4′ content of lignin populations from the same sample, signal differences in reactivity experienced by different regions of the cell wall during ball milling. The reason for the observed component-specific structural and size heterogeneity is further discussed in a dedicated section at the end of this paper.
Unpredicted base catalysed transesterification supports the presence of acidic and lactone groups in mannan units linked to lignin
As discussed earlier, γ-ester LCC, associated with 4-O-methylglucuronic acid or galacturonic acid, both as integral parts of arabinoglucuronoxylan (AGX) or pectins, were detected in the WWE-EtOH, H+. As evidenced by HMBC, these structures typically appear at 63/4.4 ppm in the HSQC. Surprisingly, γ-esters were also detected in the ALK-EtOH, H+-red and IL/EtOH, H+-red samples (ESI, Fig. S6a and S6b†). The surprise comes from the fact that no uronic acids were detected in these samples and the only detected anomeric sugar signals appeared at 101.9/4.6 ppm and were assigned to the anomeric carbon in mannan units (Fig. 8c and ESI, Table S8†), probably linked to lignin in benzyl ethers through its C6 hydroxyl as previously shown.36 Esters are alkaline labile and our expectation was that they would cleave during the alkaline extraction step or during the alkaline reduction step. Indeed, the absence of γ-ester in the alkaline and IL/EtOH, H+ fractions, i.e. the raw fractions before reduction, was supported by HSQC. Thus, γ-esterification must have occurred during the subsequent reduction step performed under mild alkaline conditions (see materials and methods), or the steps following it.The most likely reaction is a transesterification between the Cγ hydroxyls in lignin and some ester in the carbohydrates, presumably a cyclic ester or lactone. This lactone could have formed during the recovery of the samples performed under slightly acidic conditions.
The lactone, we propose, is formed in two steps consistent with the proposed reaction scheme in Fig. 11.
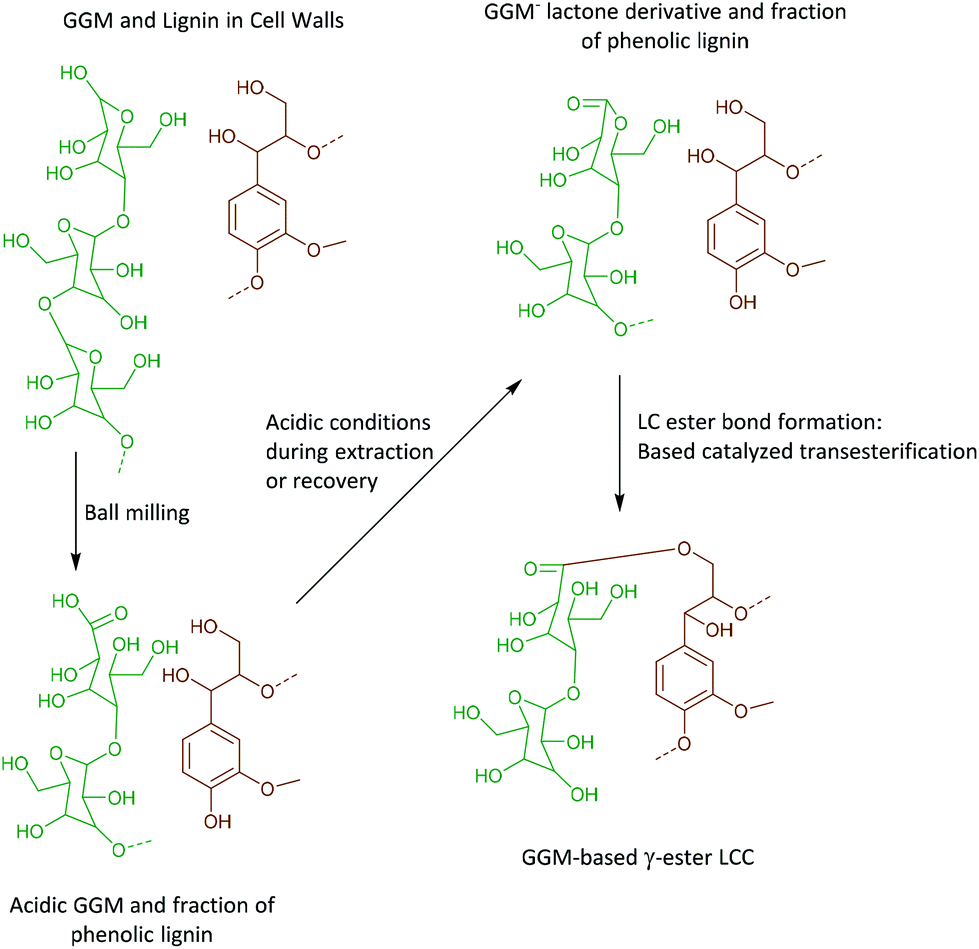 | ||
| Fig. 11 Possible reaction mechanism for the formation of GGM-based γ-ester LCC. | ||
• An oxidation of the aldehydic C1 carbonyl formed through mutarotation during ball milling occurs due to the presence of oxygen in the system. This will also occur in the nitrogen atmosphere used in the present work as it is unlikely nitrogen replaces all the oxygen molecules in the voids present in the fibers.
• Second, the lactonization of the mannonic acid occurs under mildly acidic conditions.
• During the reduction step, which is performed under mild alkaline conditions, the base catalysed transesterification between Cγ hydroxyl and the lactone occurs in a ring opening reaction, subsequently forming a new aliphatic hydroxyl. The esterification is also supported by 13C NMR.
The bigger picture from this reaction is that the oxidative modification of mannose units in close proximity to lignin occurs during milling, and could potentially yield artificial lignin carbohydrate ester bonds under certain precise pre-treatment conditions subsequent to milling.
Quantitative 13C NMR analysis supports the dominance of β-O-4′ and 4-O-etherified 5-5′ subunits in lignin, despite differences between the populations
To quantify the 4-O-etherified 5-5′ sub-structures by 13C NMR, the C3 aromatic signal is used, but the sample has to be reduced first to remove overlapping signals from α-, γ-carbonyls, and spirodienone.11 The results are reported in Table S5.† The 13C spectrum of the IL/EtOH, H+ -red sample is shown in Fig. 12.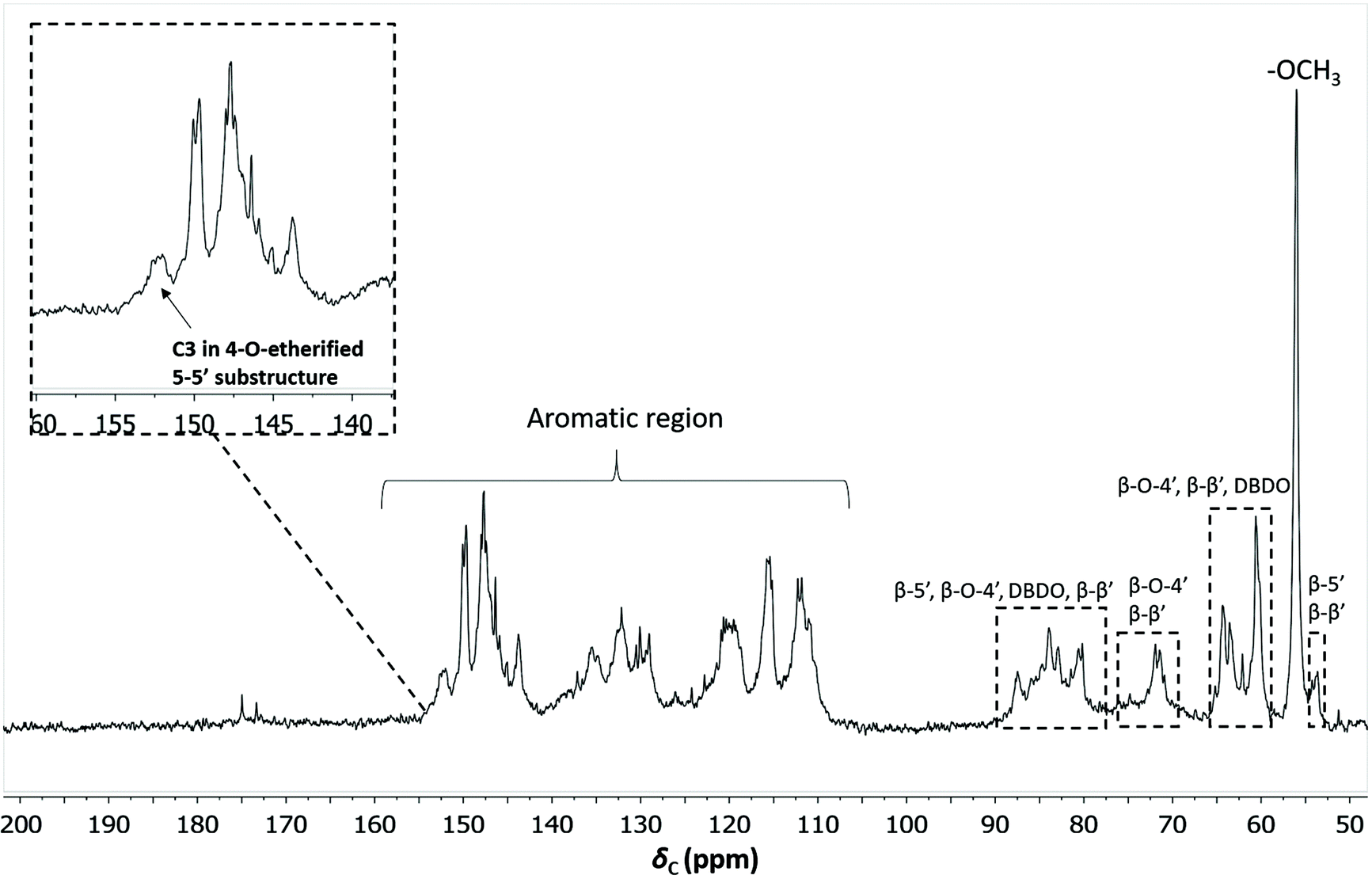 | ||
| Fig. 12 Quantitative 13C NMR of IL/EtOH, H+- red fraction. The chemical structures of the bonds can be found in Fig. 7b. | ||
The β-O-4′ and 4-O-etherified 5-5′ substructures dominate the lignin structures at (42–47%) and (23–26%) respectively. The question is then if these 4-O-etherified 5-5′ substructures constitute branching points in lignin. Dibenzodioxocin (DBDO) substructures fall in the category of these substructures but have been reported to have free phenolic ends 9, meaning that they would not contribute to lignin branching. Our analysis however estimates the amount of DBDO in these fractions at about 2–3%, meaning that the majority of the 4-O-etherified 5-5′ substructures could indeed be branching points in lignin in support of extensive branching in lignin as recently proposed for MWL.11 Indeed, whether the phenyl propane units in these 4-O-etherified 5-5′ substructures further propagate through radical couplings initiated at the free phenolic ends remains to be investigated. Only then can true branching be concluded. From a mechanistic viewpoint, this is a reasonable assumption and would be consistent with endwise polymerization. Yet for unknown reasons, such further coupling has not been observed for dibenzodioxocin structures which exist as free phenolic ends.
4-O-Etherified 5-5′ substructures evidenced in biomimetic synthesized lignin (DHP)
To investigate possibilities of the 4-O-etherified 5-5′ subunits being formed during lignin polymerization and not during the milling, we synthesized lignin (dehydrogenation polymer DHP), which is a bio mimic of lignin polymerization, and studied the DHP by both HSQC and 13C NMR (ESI Fig. S7b and S7c†). From the HSQC, the dibenzodioxocin structure (DBDO), which is a distinguishable and unique eight membered ring in a 4-O-etherified 5-5′ substructure (see Fig. 7b for structure), was quantified at roughly 3%. The 13C NMR of the DHP on the other hand, quantified the cluster consisting of 4-O-etherified 5-5′ substructures, oxidized side chain lignin units and spirodienone at 35%. The oxidized structures such as α-carbonyls were estimated from the HSQC at 3%, coniferyl aldehyde at 2%, the spirodienone structures at 2% and DBDO at 3%. It is therefore evident that the 4-O-etherified 5-5′ substructures excluding DBDO substructures constitute about 25% of the subunits in DHP. Furthermore, signals from lignin-lignin alkyl benzyl ethers were observed in the region 77–80 ppm/4–5 ppm. These were also recently discussed as additional branching points in lignin.11In summary, we confirm that 4-O-etherified 5-5′ substructures different from DBDO are not formed during milling but more likely during lignin polymerization, and substantiate the presence of a potential branching point in native lignins. It is, however, worth noting that DHPs may over represent certain linkages at the expense of others, hence caution should be taken not to put too much into the numerical values.
Deciphering lignin heterogeneity in ball milled wood
Several studies suggest that only lignins of specific morphological origin are accessible for extraction, and this argument has been used to explain the low yields of MWL. In this context, conflicting opinions still exist with some literature studies in support of MWL originating from the secondary cell wall17 and others in support of the middle lamella.16 Furthermore, it has been proposed that lignins from different morphological regions may have different structures. For instance, it is suggested that middle lamella lignin is more condensed than cell wall lignin partly due to the relatively higher content of p-hydroxyl coumaryl units, when compared to cell wall lignins.42 Lignin structure has also been proposed to vary with the environment in which the polymerization occurs. For instance, xylan LCC isolated from wood has been shown to constitute lignin with a lower degree of condensed structures than glucomannan LCC.43 However, synthetic lignins prepared in an environment with xylan show little if any differences from those prepared in the presence of glucomannan, indicating the difficulties in biomimicking exact cell wall environments. Nevertheless, most of these assertions tend to underestimate the effect of ball milling on the molecular structure, which to our knowledge is still poorly studied for lignocellulose.Herein, based on our results, we rationalize lignin heterogeneity in ball milled wood with considerations given to the effect of ball milling. Plant cell wall polymers are assembled to a supramolecular structure consisting of crystalline cellulose aggregated to form microfibrils, and embedded in a matrix of amorphous hemicellulose and lignin, believed to be covalently linked to each other. In this arrangement, it is unlikely that the applied mechanical energy will be distributed equally between crystalline and amorphous regions. Our XRD study shows that cellulose starts to convert to the amorphous state observably after an hour of milling, meaning that a portion of the mechanical energy is absorbed in the process. By then, it is most likely that the amorphous regions further away from cellulose are already more affected due to the higher mobility of the amorphous state, which would more efficiently transfer energy through molecular collisions. Contrarily, lignin and hemicellulose molecules in close proximity to cellulose are less mobile due to close association with the cellulose and thus better protected. This yields a heterogeneous lignin in ball milled lignocellulosic fibers. This is supported by several data which include composition analysis of the fractions, SEC data showing several size populations, and NMR analysis indicating differences in aryl ether linkages between the different lignin populations from the same sample. A schematic presentation of the heterogeneity model is shown in Fig. 13.
 | ||
| Fig. 13 Proposed model for plant cell wall ultrastructure and heterogeneity model of ball milled plant cell wall. Lignin is shown in brown, hemicelluloses in green and cellulose microfibrils in cream. | ||
A retrosynthesis of the data allowed us to propose a plant cell wall model to support the aforementioned heterogeneity model in ball milled fibers. The model (Fig. 13) suggests that some lignin and hemicellulose molecules have close proximity to cellulose microfibrils. These molecules constitute the none-extractable lignins and hemicellulose in the final cellulose rich residue, (Fig. 2 and ESI, Fig. S1†) and some of the extracted ones, but with better preserved structures (in accordance with the structural analyses and molecular weight distributions). The presented plant cell wall model is also consistent with the recent literature on associations of plant cell wall hemicelluloses with cellulose. For instance, two forms of xylan have been shown to exist; a two-fold helix and a three-fold helix. The former has been suggested to dock into cellulose, while the latter is assumed to have a possible role in bonding to lignin.44,45 However, associations between cellulose and lignin are less understood.
The model suggested in the present work would explain lignin heterogeneity in ball milled wood from an energy distribution viewpoint. The WWE consisting of the smallest lignin fragments are representative of molecules farthest from the microfibrils, which due to higher mobility will more efficiently absorb mechanical energy and hence degrade faster. The alkaline and IL/EtOH, H+ fractions represent molecules originally closer to cellulose with more confined mobility, and finally the non-extracted lignin and hemicellulose molecules have the closest proximity to cellulose and are protected by cellulose.
Overall, the results presented herein point toward native lignins (in their none-milled state) being branched molecules crosslinking the hemicelluloses and possibly even hemicelluloses to cellulose fibrils. Furthermore, the results suggest lignin is homogeneously distributed in between cellulose microfibrils in the plant cell wall. If lignin is indeed a molecular glue, this description is consistent with its function. During ball milling, synergies are established between the supramolecular cell wall structure and molecular events that yield preferential protection or degradation of molecules depending on the relative molecular mobilities which dictate the energy transfer efficiency. This yields molecular heterogeneity as seen here.
Outlook
The developed protocol to study lignin populations in ball milled softwood and the heterogeneity model arrived at for ball milled softwood cell walls will need to be investigated for other biomass sources which include hardwoods and gramineous biomass. This will test the universality and inform on any differences between the species.The present work also highlights the analytical challenges of the native lignin structure. Lignin populations in ball milled wood are herein shown to have varying degrees of similarities and differences to the native lignin structure predicted through lignification theory. On the other hand, potential branching in lignin, as demonstrated in this study is not reflective of the lignin structure based on recent lignification theory. To advance native lignin studies therefore, alternative approaches such as biomimetic lignin synthesis need to be more deeply investigated. For the biomimicked lignins to truly reflect native lignin, the natural monolignol biosynthesis pathways should not be decoupled from the system since these pathways regulate monolignol composition, feed rates etc., with underpinnings for the lignin structure. Furthermore, accurate knowledge of the environments in which the lignin polymerization occurs in vivo will be beneficial. In addition, the produced lignin should preferably be readily available for analysis and not undergo any severe extraction processes.
Finally, from a biorefining viewpoint, the heterogeneity model of ball milled cell walls will inform future biomass valorization studies that adopt mechanical milling as a pretreatment strategy. For instance, the differences in accessibility, chemical reactivity, solubility and size heterogeneity of lignin molecules in ball milled substrates are all important considerations for lignin extraction and modification/derivatization.
Conclusions
Ball milling of lignocellulose is widely applied to extract lignin for analysis and is also emerging as an alternative approach to wet chemical methods to valorise lignin. However, the effect of ball milling on lignocellulosic fibers is still poorly understood, and fundamental studies on this will guide future studies on the analytical and applied aspects of lignin. Herein, we investigate the origin of lignin heterogeneity in ball milled spruce. A unique approach was adopted where all the main wood polymers, i.e. cellulose, hemicelluloses and lignin were studied at the same time through the milling process. This approach made it possible to study synergistic effects between the supramolecular cell wall structure and molecular events. Furthermore, the effects of the milling environment (air and nitrogen) were studied. To enable these studies, a novel fractionation protocol was developed, and combined with advanced characterization techniques for supramolecular and molecular structural analyses. The origin of lignin heterogeneity in ball milled wood was found to be a result of synergistic effects between supramolecular and molecular events that occurred during the milling process. More specifically, it is proposed that the proximity of lignin and hemicellulose structures to the cellulose microfibrils played a central role, with molecules closer to cellulose being more structurally preserved than those further away. Based on the results a model of the plant cell wall and its disintegration during ball milling is proposed.Structural studies of the isolated lignins showed, for the first time, unequivocal HMBC evidence for 4-O-etherified 5-5′ diphenyl units in lignin besides the previously known dibenzodioxocin substructures, and in support of previous HSQC assignment of this lignin sub-unit. This is supportive of native lignin having branching points. Furthermore, first-time evidence of lignin carbohydrate bonds of the benzyl ester type in wood isolates is provided. Phenyl glycosides and γ-esters were also detected. In summary, lignin in softwoods is likely a branched polymer, crosslinking hemicelluloses and possibly hemicelluloses to cellulose microfibrils. When subjected to ball milling, this crosslinked structure is degraded and leads to component-specific molecular heterogeneity effected by differences in the absorption of mechanical energy between crystalline and amorphous states which plays a key role.
Abbreviations
| AGX | Arabinoglucuronoxylan |
| Alk | Alkaline extract of milled wood after water extraction |
| Alk ETOH H+ | Purified lignin from the Alk fraction by acid hydrolysis in ethanol/water mixture |
| Alk ETOH H+ red | Sodium borohydride reduced fraction of the Alk ETOH H+ |
| DHP | Dehydrogenation polymer (Synthetic lignin) |
| DBDO | Dibenzodioxocin |
| EMAL | Enzymatic mild acid hydrolysis Lignin |
| GGM | Galactoglucomannan |
| HMBC | Heteronuclear multiple bond correlation NMR spectroscopy |
| HSQC | Heteronuclear single quantum correlation NMR spectroscopy |
| IL ETOH H+ | Lignin fraction procured from the alkaline extracted milled wood residue, by extraction with ionic liquid/ethanol mixture, and in the presence of acid |
| IL ETOH H+ red | The sodium borohydride reduced fraction of the IL ETOH H+ |
| LCC | Lignin carbohydrate complex |
| OMe | Methoxyl group |
| PG | Phenyl glycoside, a type of LCC |
| UA | Uronic acid groups |
| WWE | Warm water extract of milled wood |
| WWE H+ | Enriched fraction of lignin achieved by acid hydrolysis of WWE fraction |
| WWE H+ red | the sodium borohydride reduced fraction of the WWE H+ |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to gratefully acknowledge the Knut and Alice Wallenberg Foundation for financial support. In addition, Dr Nicola Giummarella and Pär Lindén are acknowledged for guidance with experimental work.References
- A. Payen, Comptes Rendus, 1838, 7, 1052–1056 Search PubMed.
- Japan Technical Association of the Pulp and Paper Industry, Sulphite pulp, dissolve pulp [M], Tokyo, Japan Technical Association of the Pulp and Paper Industry, 1966 Search PubMed.
- F. E. Brauns, J. Am. Chem. Soc., 1939, 61, 2120–2127 CrossRef CAS.
- A. Björkman, Nature, 1954, 174, 1057–1058 CrossRef.
- H.-M. Chang, E. B. Cowling and W. Brown, Holzforschung, 1975, 29, 153–159 CrossRef CAS.
- A. Guerra, I. Filpponen, L. A. Lucia and D. S. Argyropoulos, J. Agric. Food Chem., 2006, 54, 9696–9705 CrossRef CAS PubMed.
- C. Crestini, F. Melone, M. Sette and R. Saladino, Biomacromolecules, 2011, 12, 3928–3935 CrossRef CAS PubMed.
- F. Yue, F. Lu, S. Ralph and J. Ralph, Biomacromolecules, 2016, 17, 1909–1920 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- E. Adler, Wood Sci. Technol., 1977, 11, 169–218 CrossRef CAS.
- M. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
- A. Fujimoto, Y. Matsumoto, H.-m. Chang and G. Meshitsuka, J. Wood Sci., 2005, 51, 89–91 CrossRef CAS.
- D. N.-S. Hon, Developments in Polymer Degradation—7, ed. N. Grassie, Springer Netherlands, Dordrecht, 1987, pp. 165–191 Search PubMed.
- G. Zinovyev, I. Sumerskii, T. Rosenau, M. Balakshin and A. Potthast, Molecules, 2018, 23, 2223 CrossRef PubMed.
- P. Karinkanta, A. Ämmälä, M. Illikainen and J. Niinimäki, Biomass Bioenergy, 2018, 113, 31–44 CrossRef CAS.
- K. V. Sarkanen and C. H. Ludwig, Lignins: occurrence, formation, structure and reactions, Wiley, New York, 1971 Search PubMed.
- P. Whiting and D. Goring, J. Wood Chem. Technol., 1981, 1, 111–122 CrossRef CAS.
- Z. Ling, T. Wang, M. Makarem, M. S. Cintrón, H. N. Cheng, X. Kang, M. Bacher, A. Potthast, T. Rosenau, H. King, C. D. Delhom, S. Nam, J. V. Edwards, S. H. Kim, F. Xu and A. D. French, Cellulose, 2019, 26, 305–328 CrossRef CAS.
- C. C. Piras, S. Fernández-Prieto and W. M. De Borggraeve, Nanoscale Adv., 2019, 1, 937–947 RSC.
- J. R. Obst and T. K. Kirk, Methods Enzymol., 1988, 161, 3–12 CrossRef CAS.
- E. Adler, Ind. Eng. Chem., 1957, 49, 1377–1383 CrossRef CAS.
- J. A. M. Erich, Acta Chem. Scand., 1961, 15, 370–383 CrossRef.
- G. Gellerstedt, Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992, pp. 487–497 Search PubMed.
- D. S. Argyropoulos, J. Wood Chem. Technol., 1994, 14, 45–63 CrossRef CAS.
- M. Mattonai, D. Pawcenis, S. del Seppia, J. Łojewska and E. Ribechini, Bioresour. Technol., 2018, 270, 270–277 CrossRef CAS PubMed.
- M. Ago, T. Endo and K. Okajima, Polym. J., 2007, 39, 435–441 CrossRef CAS.
- C. J. Garvey, I. H. Parker and G. P. Simon, Macromol. Chem. Phys., 2005, 206, 1568–1575 CrossRef CAS.
- M. G. Thomas, E. Abraham, P. Jyotishkumar, H. J. Maria, L. A. Pothen and S. Thomas, Int. J. Biol. Macromol., 2015, 81, 768–777 CrossRef CAS PubMed.
- B. Stefanovic, K. Pirker, T. Rosenau and A. Potthast, Carbohydr. Polym., 2014, 111C, 688–699 CrossRef PubMed.
- S. Wilbur, D. Jones and J. F. Risher, et al., Toxicological Profile for 1,4-Dioxane, Agency for Toxic Substances and Disease Registry (US) Appendix D, Health Advisory, Atlanta (GA), 2012, https://www.ncbi.nlm.nih.gov/books/NBK153666/, (accessed November 2020) Search PubMed.
- C. Hogue, c&en-chemical and engineering news, https://cendigitalmagazine.acs.org/2020/11/08/14-dioxane-another-forever-chemical-plagues-drinking-water-utilities-2/content.html, accessed November 2020.
- M. Karlsson, N. Giummarella, P. A. Linden and M. Lawoko, ChemSusChem, 2020, 13, 4666–4677 CrossRef CAS PubMed.
- K. Leppänen, P. Spetz, A. Pranovich, K. Hartonen, V. Kitunen and H. Ilvesniemi, Wood Sci. Technol., 2011, 45, 223–236 CrossRef.
- M. Balakshin, E. Capanema, H. Gracz, H. M. Chang and H. Jameel, Planta, 2011, 233, 1097–1110 CrossRef CAS PubMed.
- Y. Miyagawa, Y. Tobimatsu, P. Y. Lam, T. Mizukami, S. Sakurai, H. Kamitakahara and T. Takano, Plant J., 2020, 104, 156–170 CrossRef CAS PubMed.
- H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 6538 CrossRef PubMed.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, Green Chem., 2019, 21, 1573–1595 RSC.
- L. Chen, L. Zhang, P. J. Griffin and S. J. Rowan, Macromol. Chem. Phys., 2020, 221, 1900440 CrossRef CAS.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- N. Giummarella, P. A. Lindén, D. Areskogh and M. Lawoko, ACS Sustainable Chem. Eng., 2020, 8, 1112–1120 CrossRef CAS.
- L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37–45 CrossRef CAS PubMed.
- N. Terashima, M. Yoshida, J. Hafrén, K. Fukushima and U. Westermark, Holzforschung, 2012, 66, 907–915 CAS.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Biomacromolecules, 2005, 6, 3467–3473 CrossRef CAS PubMed.
- X. Kang, A. Kirui, M. C. D. Widanage, F. Mentink-Vigier, D. J. Cosgrove and T. Wang, Nat. Commun., 2019, 10, 347 CrossRef CAS PubMed.
- O. M. Terrett, J. J. Lyczakowski, L. Yu, D. Iuga, W. T. Franks, S. P. Brown, R. Dupree and P. Dupree, Nat. Commun., 2019, 10, 4978 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc04319b |
| This journal is © The Royal Society of Chemistry 2021 |