
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzyme immobilisation on wood-derived cellulose scaffolds via carbohydrate-binding module fusion constructs†
A. D.
Roberts‡
 ,
K. A. P.
Payne‡
,
S.
Cosgrove‡
,
V.
Tilakaratna‡
,
I.
Penafiel‡
,
W.
Finnigan
,
N. J.
Turner‡
and
N. S.
Scrutton‡
*
,
K. A. P.
Payne‡
,
S.
Cosgrove‡
,
V.
Tilakaratna‡
,
I.
Penafiel‡
,
W.
Finnigan
,
N. J.
Turner‡
and
N. S.
Scrutton‡
*
Manchester Institute of Biotechnology and Department of Chemistry, The University of Manchester, M1 7DN, UK. E-mail: Nigel.Scrutton@manchester.ac.uk
First published on 17th June 2021
Abstract
The development of cost-effective and green enzyme immobilisation techniques will facilitate the adoption of continuous flow biocatalysis (CFB) by industry and academia. In this work, a relatively mild sulfite pulping process was employed to remove lignin and hemicellulose from wood with minimal disruption of its native porous structure, resulting in aligned macroporous cellulosic monoliths termed cellulose scaffolds (CSs). By engineering carbohydrate-binding modules (CBMs) onto the termini of recombinant proteins, the CSs could be employed as low-cost, renewable and biodegradable materials for enzyme immobilisation without any further chemical functionalisation. CBM-tagged fluorescent proteins were initially employed to demonstrate proof-of-principle and to optimise immobilisation conditions; this resulted in initial protein loadings as high as 5.24 wt% and immobilisation efficiencies as high as 97.1%. The process was then translated to a CBM-tagged ω-transaminase (ωTA) from Bacillus megaterium, obtaining enzyme loadings and immobilisation efficiencies as high as 3.99 wt% and 82.4%, respectively. A demonstrative CFB reaction with the immobilised CBM-tagged ωTA displayed ca. 95 ± 5% conversion efficiency relative to the free enzyme in solution under analogous conditions, suggesting that CBM-tagged recombinant enzymes immobilised on wood-derived CSs could potentially compete with other, more complex and costly enzyme immobilisation technologies.
1. Introduction
Global biomass growth generates approximately 1.5 trillion tonnes of cellulose annually, making it a practically inexhaustible renewable resource.1 In addition to hyperabundance, plant-derived cellulose is cheap, biodegradable, non-toxic and inert – and continues to be a considerably active area of research despite having been utilised by humanity for millennia.1–5 Wood-derived cellulose is commonly extracted as a fibrous pulp via the Kraft or sulfite pulping processes, i.e. the base-catalysed hydrolysis of lignin and hemicellulose – the other major components of wood. Relatively mild pulping conditions can, however, remove the majority of lignin and hemicellulose whilst maintaining the native aligned porous structure of the original wood.6–12 This aligned porous structure, which has been optimised by natural selection for the efficient transport of water-based solutions, has recently been exploited for applications such as epoxy resin composites,6 radiative cooling materials7 and water purification10,11 among other applications.12 Aligned porous structures hold certain advantages over non-aligned equivalents, including relatively low back-pressures needed to pass fluids over the active surface – a beneficial property for chromatography and other flow-based applications including continuous flow biocatalysis (CFB).13–16The use of recombinant enzymes as catalysts for chemical reactions is growing in popularity due to their relatively high selectivity and green credentials in comparison to traditional synthesis routes.17,18 Issues such as relatively high cost, poor recyclability and poor stability can, however, make enzyme-based catalysis unviable for some industrial processes.18,19 These issues can be mitigated through enzyme engineering as well as the immobilisation of enzymes onto inert solid supports – which can enhance their physico-chemical stability (allowing them to tolerate harsher reaction conditions) and allow for facile recovery and re-use from batch reactors.20,21 Immobilisation of enzymes onto porous supports or packed particle beds also permits their use as de-facto heterogeneous catalysts for CFB (Fig. 1a).22 In CFB, reactants are passed over an enzyme-immobilised porous support to produce a continuous product stream which can then be further processed or recovered. Several reviews detailing the merits and drawbacks of CFB have been published recently;16,23–25 with drawbacks including relatively high cost and non-reusability of some scaffold materials, and loss of enzyme activity over time.
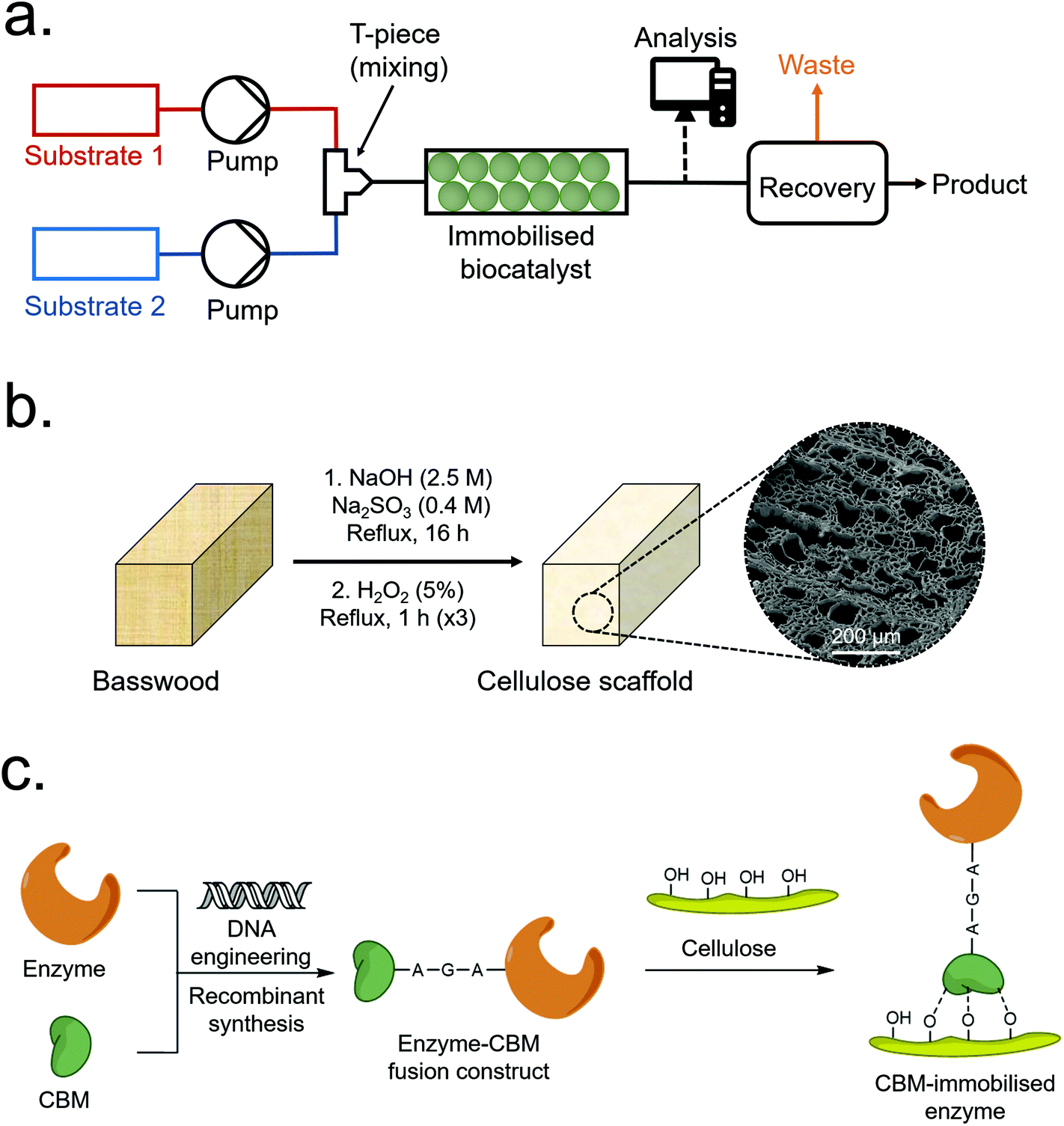 | ||
| Fig. 1 (a) Overview of a typical packed-bed CFB process. (b) Scheme depicting typical CS synthesis. (c) Scheme depicting the immobilisation of a CBM-enzyme construct to a cellulose surface. | ||
Carbohydrate-binding modules (CBMs), previously termed cellulose-binding domains (CBDs), are a class of non-catalytic protein domain which have exceptional affinity binding properties to cellulose and other carbohydrates.26–30 Often found as a sub-domain of carbohydrate-active enzymes such as cellulases or xylenases,26 CBMs have a wide variety of sequences and structures (60+ families of CBMs currently known) which can recognise and bind to different forms of cellulose and other carbohydrates.30 This property has previously been exploited to bind CBM-tagged recombinant proteins to various forms of carbohydrates for applications such as diagnostics, cell immobilisation and bioremediation.27–29 CBMs have also previously been employed for a range of enzyme immobilisation applications, as summarised in Table S1.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30–42 These previous literature reports generally employ commercially obtained microcrystalline cellulose powders as the immobilisation material, with the use of novel cellulose structures remaining largely unexplored. For instance, S. Wang et al. produced CBM-tagged cis-epoxysuccinic acid hydrolase (CESH) chimeras with five different CBMs and evaluated their enzymatic performance immobilised on a commercial crystalline cellulose powder (Avicel® PH-101). Attaining enzyme loadings as high as 2.0%, their best performing CBM-CESH chimera displayed a five-fold improvement in half-life at 30 °C with improved temperature and pH stability.30 Another example is a recent report by B. Estevinho et al., who produced a CBM-enzyme chimera based on CBM2 from Pyrococcus furiosus chitinase and β-galactosidase from Thermotoga maritima (TmLac).38 Employing a bacterial cellulose membrane as their immobilisation support, they achieved enzyme loadings up to 1.85 wt% and immobilisation efficiencies as high as 81 ± 4%, whilst demonstrating enhanced enzyme stability at elevated temperatures.
30–42 These previous literature reports generally employ commercially obtained microcrystalline cellulose powders as the immobilisation material, with the use of novel cellulose structures remaining largely unexplored. For instance, S. Wang et al. produced CBM-tagged cis-epoxysuccinic acid hydrolase (CESH) chimeras with five different CBMs and evaluated their enzymatic performance immobilised on a commercial crystalline cellulose powder (Avicel® PH-101). Attaining enzyme loadings as high as 2.0%, their best performing CBM-CESH chimera displayed a five-fold improvement in half-life at 30 °C with improved temperature and pH stability.30 Another example is a recent report by B. Estevinho et al., who produced a CBM-enzyme chimera based on CBM2 from Pyrococcus furiosus chitinase and β-galactosidase from Thermotoga maritima (TmLac).38 Employing a bacterial cellulose membrane as their immobilisation support, they achieved enzyme loadings up to 1.85 wt% and immobilisation efficiencies as high as 81 ± 4%, whilst demonstrating enhanced enzyme stability at elevated temperatures.
In this work, aligned macroporous cellulose scaffolds (CSs) were produced through the de-lignification of wood via a relatively mild sulfite pulping process (Fig. 1b), before CBM-tagged recombinant proteins were immobilised to the internal surfaces via CBM-cellulose affinity binding (Fig. 1c). The most significant process parameters were identified and optimised via a Design of Experiments (DOE) methodology to maximise total protein loading and immobilisation efficiency, achieving maximum values of 5.24 wt% and 97.1%, respectively. Six different CBMs were initially investigated, and fluorescent proteins were initially employed as reporter tags to facilitate characterisation and optimisation, and to determine if any localised binding was occurring on the scaffolds. Finally, a construct based on an ω-transaminase (ωTA) from Bacillus megaterium and the best-performing CBM (namely, CcCBM2a from C. cellulovorans – EngD) was produced, immobilised onto the CSs, and tested in a CFB setup. The immobilised enzyme displayed ca. 95 ± 5% conversion efficiency relative to the free enzyme in solution under analogous conditions, demonstrating that CS immobilised CBM-tagged enzymes can, in principle, be employed for CFB.
2. Materials and methods
2.1. Characterisation techniques
Wide-angle X-ray diffraction (WAXD) was performed using a PANalytical X'Pert Pro instrument with a Kα radiation source (1.54 Å wavelength), diffraction angle of 5–60° and scanning rate of 1° min−1. Field emission gun scanning electron microscopy (FEG-SEM) was performed using an FEI Quanta 250 instrument with a working distance of 10 mm and accelerating voltage of 10 kV. Acetone-soaked CS samples were air dried overnight before being cut into small portions and adhered to 1 cm2 aluminium stubs with double-sided conductive carbon tape. Samples were sputter coated with 10 nm of an Au/Pd alloy prior to imaging to enhance sample conductivity, and a small amount of Ag paint was also used to improve electrical contact between the sample and carbon tape. UV-visible spectrophotometry was performed using a either a Cary 60 UV-vis spectrophotometer (protein expression and quantification) or a Clariostar Plus plate reader.2.2. Recombinant protein design, expression and purification
Detailed experimental steps for the design, expression and purification of the CBM-fluorescent protein constructs (CBM-FPs) are given in the ESI.† Experimental conditions for the design, expression and purification of the CBM-enzyme construct are given below.An ωTA gene from B. megaterium (SC6394, Bm-ωTA)43 was amplified from a pET21b construct, whilst the pBbA1c-CBM2a backbone was amplified from HisCcCBM2a(EngD)mEGFP using CloneAmp polymerase premix (ClonTech) and the primers detailed in Table S2.† The template was removed by DpnI (New England Biolabs) digest and the DNA purified using a Qiaquick PCR purification kit (Qiagen). The PCR products were cloned together using Infusion HD (Clontech). DNA and transformed into NEB5alpha cells (New England Biolabs). To express the resulting fusion protein LB autoinduction media (formedium) supplemented with 34 μg ml−1 chloramphenicol was inoculated with 5 ml pre-culture and grown at 20 °C for 60 hours. Cells were harvested by centrifugation and stored at −20 °C until required. Protein purification was performed as above except for the addition of 1 mM pyridoxal-5′-phosphate (PLP, Sigma) in the lysis buffer. The protein was quantified using a Cary 60 UV-vis spectrophotometer (Agilent). The A280 extinction coefficient (115740 M−1 cm−1) was calculated from the amino acid sequence using the Protparam tool on the ExPASy proteomics server (https://web.expasy.org/protparam/, accessed 4th March 2021) whilst PLP was quantified by A422 using an extinction coefficient of 6370 M−1 cm−1.44
2.3. Cellulose scaffold synthesis
The protocol for the synthesis of the CSs after DOE-based optimisation is presented below. For context, the initial synthesis protocol (i.e., prior to optimisation) is presented in the ESI.†A block of Tilia Americana basswood (Stockport Hobbycraft Store, UK) was sawn into 10 × 10 × 50 mm portions with the grain of the wood following the long (50 mm) axis (Fig. S1†). Each wood portion weighed approximately 0.9–1 g and was gently sanded by hand with abrasive paper to remove any residual wood flakes. Separately, 150 g NaOH and 25.2 g Na2SO3 were dissolved in DI water to a final volume of 500 ml (7.5 M NaOH, 0.4 M Na2SO3). Note, some of the Na2SO3 remained undissolved at RT as saturation solubility was reached. 25 portions of wood were added to a 1 L round-bottomed flask before addition of the 500 ml NaOH/Na2SO3 solution. The mixture was then refluxed at 140 °C for 16 h with gentle magnetic stirring. The solution was then drained and the wood portions (now CSs) were washed with cold DI water 3× times, before being refluxed with DI water (900 ml) at 125 °C for 1 h 3× times. The CSs were then refluxed in 875 ml H2O2 (20%) for 3 hours at 125 °C a single time. Foaming was no longer an issue and no antifoam 204 was added. The CSs were then washed in cold DI water 3× times before being placed in 200 ml of absolute ethanol overnight. DI water was then added to bring the final volume to 1 L (20% v/v EtOH) where the CSs were then stored at RT until use.
2.4. Protein immobilisation procedures
Transparent PVC tubing (ID 8 mm, OD 12 mm) was cut to a length of approximately 80 mm. A CS would then be trimmed slightly with a razor blade into a cylindrical shape (initially cuboidal) and inserted into the tubing ensuring a tight fit and sitting in the centre of the tubing (Fig. S2a†). A 50 ml plastic Luer-Lock syringe was connected to one end of the PVC tubing using a 3-way Luer Lock valve (for loading/discharging solutions), male/female Luer Lock attachments and poly(vinylidene fluoride) (PVDF) tubing (Fig. S2b†). 30 ml of buffer solution (150 mM TRIS, 50 mM KCl, pH 7.1) was then flushed through the CS at a rate of 250 μL min−1 to flush out residual EtOH. 4 ml of CBM3A-mCherry (10 mg ml−1) was then flushed through the CS at a rate of 5 μL min−1 (Aladin syringe pump, World Precision Instruments) and collected. A further 2 ml of buffer was flushed through and collected to wash out non-adsorbed CBM3-mCherry. The CBM3-mCherry concentration before (10 mg ml−1) and after passing though the CS (including 2 ml additional flush) was measured via UV-visible spectrophotometry (Abs [587], ε = 72000 M−1 cm−1, Mw = 48892.28 g mol−1). The experiments were conducted at room temperature (RT, typically 19 ± 3 °C). Note, to prioritise immobilisation yield over total loading, lower protein concentrations can be employed.
2.5. Activity of CBM2a-ωTA in solution
In order to benchmark the enzymatic activity of the immobilised CBM2a-ωTA construct, its conversion efficiency in solution (i.e., non-immobilised) was first determined. Briefly, to a 1.5 ml Eppendorf tube the following were added: 100 μL potassium phosphate (KPI) buffer solution (1 M, pH 8), 400 μL H2O, 50 μL pyruvate (500 mM in H2O), 250 μL (S)-α-MBA (100 mM in H2O), 100 μL PLP (2 mM in H2O) and 100 μL of CBM2a-ωTA (10 mg ml−1 in 50 mM TRIS pH 7.5, 200 mM KCl in H2O). The final concentrations were therefore: 0.1 M KPI (pH 8), 25 mM pyruvate, 25 mM (S)-α-MBA, 0.2 mM PLP, 20 mM KCl, 5 mM TRIS and 1 mg ml−1 CBM2a-ωTA (14.9 μM). The mixture was briefly agitated and maintained at 30 °C, before 4 μL of solution was removed at defined time points (typically 5 minutes intervals), added to 196 μL of H2O (50× dilution) in a 96 well plate and absorbance at 245 nm measured. The concentration of acetophenone was then determined by referring to a calibration curve of the reactants (pyruvate, α-MBA) and products (acetophenone) at 245 nm. The measurements were taken in triplicate and a parallel control experiment with no enzyme was also conducted as a negative control.2.6. Activity of CS-immobilised CBM2a-ωTA in flow
To determine the activity of CS-immobilised CBM2a-ωTA in a CFB-type set up, the construct was first loaded onto optimised CSs (see section 2.3) under optimised flow conditions (see section 2.4.2). After loading, a running buffer solution consisting of 0.1 M KPI (pH 8), 25 mM pyruvate, 25 mM (S)-α-MBA, 0.2 mM PLP was passed through the CBM2a-ωTA immobilised CS at 30 °C and a rate of 200 μL min−1, with aliquots being sampled every 5 minutes (i.e., every 1 ml of flow through), diluted by a factor of 50 and absorbance measured at 245 nm. Note, the absorbance was measured immediately to prevent activity from any leached enzymes from affecting results significantly. The acetophenone content was then determined by comparison to a calibration curve as in section 2.5.3. Results and discussion
3.1. Design and synthesis of CBM-tagged fusion constructs
For easy visualisation and quantification of CBM binding, the six different CBMs were fused to six different fluorescent proteins. Variants of fluorescent proteins were selected that were reported to be monomeric, these were mECFP, mEGFP, mCitrine, mOrange, mCherry and mNeptune. Three of the CBMs were attached to the N-terminal end of the fluorescent protein whilst the other three were attached to the C-terminus. All CBM-FP fusion constructs were linked either by a glycine/serine rich linker30 or a threonine/proline rich linker derived from T. reesei Cel7A (TrCel6A).45 A summary of the CBM-FP constructs’ design is given in Table S3.† All six constructs were expressed in E. coli and purified by Ni2+ immobilised metal ion affinity (IMAC) as confirmed by UV-vis spectrophotometry (Fig. S3†), with further experimental details provided in the ESI.†
3.2. Initial CS immobilisation scoping trials
After synthesis of the CSs following the initial synthesis protocol, some scoping trials were performed to establish a working set-up to conduct the flow/immobilisation experiments. The details of these scoping trials are presented in the ESI.† These experiments revealed CBM3A-mCherry to have a relatively high expression yield in comparison to the other CBM-FP constructs, along with a relatively high total loading (0.4 wt%) and immobilisation efficiency (59.8%) on the CSs (Table S4†). These features meant it was selected for further optimisation trials over the other CBM-FPs.3.3. Optimisation of CS synthesis
The CSs were initially synthesised following a method adapted from M. Zhu et al., with the protocol given in the ESI.†6 The treatment of cellulosic fibres (e.g., cotton) with concentrated aqueous NaOH is known to increase their capacity to adsorb substances (e.g. dyes) in a process historically known as Mercerisation.1,46 This is attributed to a change from the naturally occurring polymorph Cellulose Iβ to the more thermodynamically favourable Cellulose II polymorph.47 We hypothesised that a similar NaOH treatment could also increase the adsorptive capacity of the CSs and therefore result in a higher loading of the CBM-tagged proteins.
Since NaOH was already employed during the initial CS synthesis (i.e., during the sulfite process), it was hypothesised that a Mercerisation-type effect could be achieved by increasing the NaOH content during this step. Rather than only vary the NaOH concentration however, a number of other variables (deemed to also likely be significant factors in the resulting CS quality) were also varied; namely Na2SO3 concentration, total sulfite processing solution volume, H2O2 concentration and H2O2 solution volume. Due to the large number of variables in the system, and likelihood of complex multi-factor interactions, a Design of Experiments (DOE) Definitive Screening Design (DSD) was employed to obtain a relatively large amount of information from the system with a minimal number of experiments.48,49 A summary of the input variables and fixed variables is presented in Table S5,† with actual experimental conditions employed (calculated using JMP Software from SAS) for each run presented in Table 1. Of the CBM-FPs, CBM3A-mCherry was selected to perform the optimisation due to its relatively good immobilisation performance and expression yield compared to the other CBM-FPs (Table S4†), as uncovered in the initial scoping trials (see ESI† for details). All experiments were conducted with a single CBM3A-mCherry batch to minimise error arising from batch-to-batch variation. Note that the experimental order was randomised to minimise the effect of hidden variables biasing the results, and blocked into two groups – each representing two different hot-plates used. An extra centre run was also included to estimate quadratic effects from the blocked experiments. The measured output factors (or responses) were total protein loading (wt%) and immobilisation efficiency (%).
| Input variables | Output factors | |||||||
|---|---|---|---|---|---|---|---|---|
| Experiment no. | Block | NaOH conc. (M) | Na2SO3 conc. (M) | Sulfite processing vol. (ml) | H2O2 vol. (ml) | H2O2 conc. (%) | Immobilisation efficiency (%) | Loading (wt%) |
| 1 | 1 | 7.5 | 1.2 | 250 | 175 | 4 | 42.3 | 0.708 |
| 2 | 1 | 2.5 | 1.2 | 175 | 100 | 12 | 29.5 | 0.518 |
| 3 | 1 | 5 | 1.2 | 250 | 250 | 12 | 39.7 | 0.640 |
| 4 | 1 | 2.5 | 0.4 | 100 | 175 | 12 | 55.2 | 0.797 |
| 5 | 1 | 5 | 0.8 | 175 | 175 | 8 | 28.6 | 0.354 |
| 6 | 1 | 7.5 | 0.4 | 175 | 250 | 4 | 41.7 | 0.548 |
| 7 | 1 | 5 | 0.4 | 100 | 100 | 4 | 34.5 | 0.598 |
| 8 | 2 | 7.5 | 0.8 | 100 | 250 | 12 | 52.4 | 0.808 |
| 9 | 2 | 7.5 | 0.4 | 250 | 100 | 12 | 42.6 | 0.591 |
| 10 | 2 | 5 | 0.8 | 175 | 175 | 8 | 39.2 | 0.516 |
| 11 | 2 | 2.5 | 0.4 | 250 | 250 | 8 | 15.9 | 0.249 |
| 12 | 2 | 7.5 | 1.2 | 100 | 100 | 8 | 18.4 | 0.258 |
| 13 | 2 | 2.5 | 1.2 | 100 | 250 | 4 | 17.5 | 0.275 |
| 14 | 2 | 2.5 | 0.8 | 250 | 100 | 4 | 11.8 | 0.180 |
| 15 | 1 | 4.5 | 0.2 | 100 | 100 | 14 | 49.4 | 0.826 |
| 16 | 1 | 7.5 | 0.4 | 100 | 175 | 15 | 59.5 | 1.015 |
| 17 | 1 | 7.5 | 0.4 | 100 | 175 | 17.5 | 67.1 | 1.154 |
| 18 | 1 | 7.5 | 0.4 | 100 | 175 | 20 | 70.5 | 1.093 |
After synthesis of the CSs, it could be seen that different CS synthesis conditions resulted in a range of colours from white to light brown – the brown colouration likely being residual lignin (Fig. S5a†). A CS from each experimental condition was soaked in acetone (to induce solvent exchange) and air-dried; some CSs underwent significant shrinkage (i.e., conditions 1, 8, 9 and 12) whilst others remained relatively robust (i.e., conditions 2, 7 and 14) (Fig. S5b†). This appeared to be correlated to the concentration of NaOH during the sulfite processing step, where a high NaOH concentration tended to result in greater shrinkage. FEG-SEM images from cross sections of the CSs revealed the aligned macropores of wood (xylem and phloem) were still intact following treatment (Fig. 2), however at the ends of the CSs (which had greatest contact with the sulfite/H2O2 solutions) this structure broke down into a disordered fibrillar morphology (Fig. S5c†). The porosity of the CSs was determined through wet/dry mass analysis, with porosity varying between ca. 69–78% (Table S6†) and was again correlated with NaOH concentration, with higher NaOH concentrations resulting in lower % porosity (Fig. S5d†).
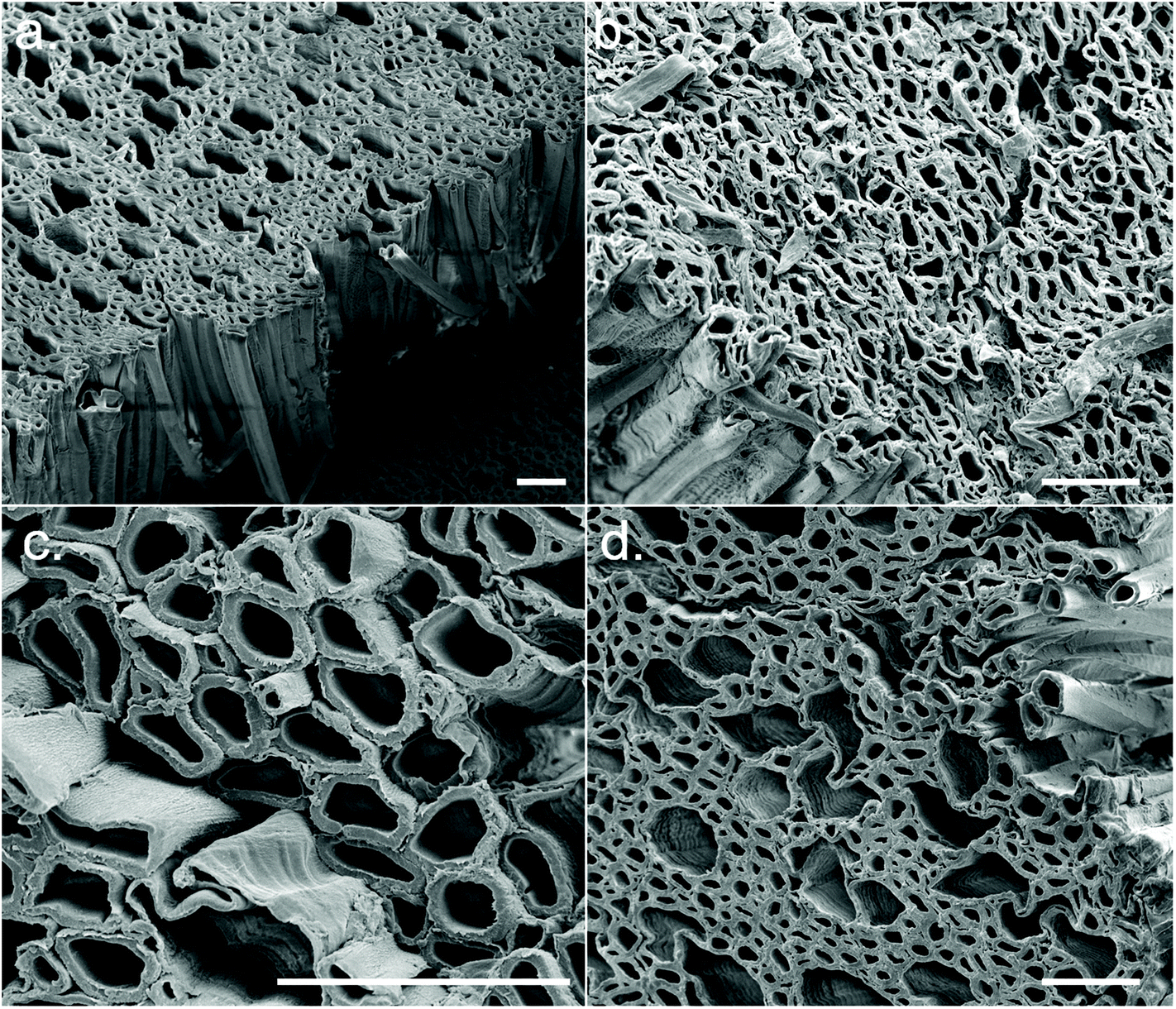 | ||
| Fig. 2 Cross-sectional FEG-SEM images of the CSs displaying aligned microporosity. Representative examples of synthesis conditions (a) 1, (b) 5, (c) 6, and (d) 8. Scale bars = 50 μm. | ||
CBM3A-mCherry solutions were flowed through the synthesised CSs following the non-optimised immobilisation procedure described in the ESI.† The CSs were then cut up and visible light images taken, which showed the distribution of CBM3A-mCherry within the scaffolds (Fig. S6†). There was a noticeable accumulation of the CBM3A-mCherry at either end of the CSs, which had previously been identified as having a fibril-like morphology via FEG-SEM imaging (Fig. S5c†). Measurement of the CBM3A-mCherry concentration before and after flowing through, along with measurement of CS mass, allowed the calculation of the protein loading (wt%) and immobilisation efficiency (%). These data were analysed by JMP software, where the most significant factors from the screening dialogue were selected (using Fit DSD method) to build response surface models (RSMs) for the two output factors (protein loading and immobilisation efficiency). The purpose of the RSMs is to allow predictions (or causal inferences) to be made between the significant input variables and the output factors; i.e., which input variables should be varied in order to maximise the protein loading and immobilisation efficiency. Although there was fairly high noise within the experimental system, including a large variability between blocks and low statistical significance for a number of factors, RSMs could still be constructed (Fig. 3a). The RSM for loading predicted that the highest value (0.896 wt%) would be achieved with the highest concentrations of H2O2, NaOH and Na2SO3, as well as the highest sulfite processing solution volume (Fig. 3a). Interestingly, a similarly high loading (0.882 wt%) was also predicted with the highest H2O2 concentration, but lowest Na2SO3 concentration and sulfite processing volume. A complex interaction such as this would have been missed had a typical one-factor-at-a-time (OFAT) optimisation approach been employed.48 The RSM for immobilisation efficiency predicted that higher efficiencies would be obtained with the highest concentrations of H2O2 and NaOH, a lower Na2SO3 concentration, and the highest H2O2 volume (Fig. 3b).
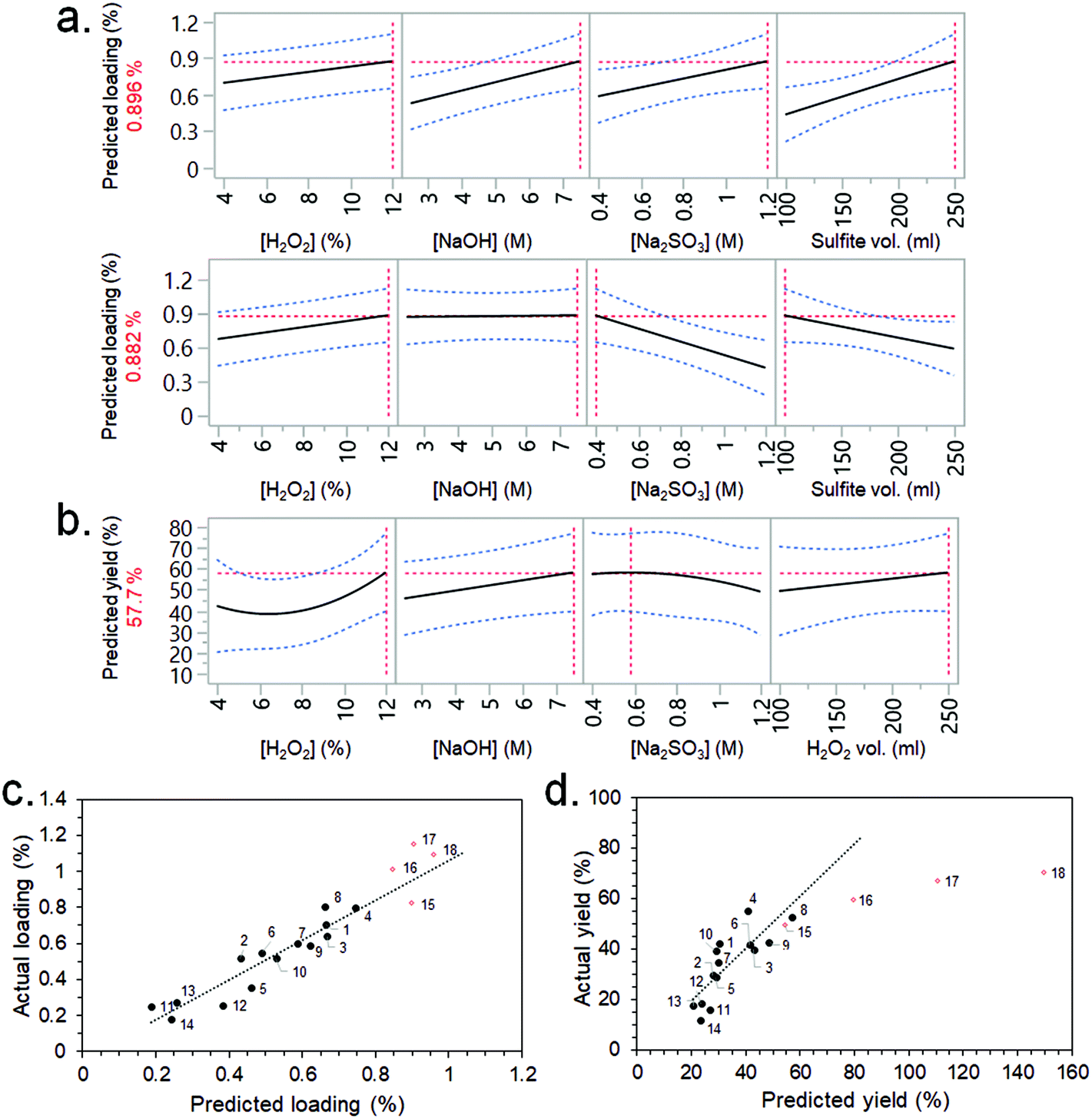 | ||
| Fig. 3 (a) Loading and (b) immobilisation efficiency RSM prediction profilers for the CS synthesis DSD. Blue dashed lines represent confidence intervals and red dashed lines indicate prediction profiler settings. (c) Loading and (d) immobilisation efficiency RSM models calculated from experiments 1–14 (black dots) and validated with experiments 15–18 (red diamonds). | ||
Based on these RSM predictions, four further experiments (experiment no. 15–18) were conducted in order to validate the models and push the output factors (loading and immobilisation efficiency) to higher values. Experiments 16, 17 and 18 employed an increasing concentration of H2O2 (15, 17.5 and 20%, respectively) since there was a strong positive correlation between this factor and both loading and immobilisation efficiency (Fig. 3a and b). The low Na2SO3 concentration (0.4 M) and low sulfite processing volume (100 ml) conditions were selected for these experiments – rather than the high concentration (1.2 M) and volume (250 ml) – since the resulting loading was almost identical (Fig. 3a) but the former conditions required less starting material (i.e., a greener process) and allowed more CSs to be produced per 1 L batch. A lower Na2SO3 concentration also improved immobilisation efficiency slightly (Fig. 3b). The high NaOH concentration of 7.5 M was selected for experiments 16, 17 and 18 since this factor had a positive correlation with immobilisation efficiency (Fig. 3b). A H2O2 volume of 175 ml was selected as this allowed more CSs to be produced per batch than if 250 ml was used (which would have maximised immobilisation efficiency).
The results from these validation experiments are presented in Table 1, Fig. 3c and d. The results agreed fairly well in terms of loading, with experiments 16, 17 and 18 actually having a slightly higher loading than predicted by the RSM (1.01, 1.15 and 1.09%, vs. predicted values of 0.85, 0.90 and 0.96%, respectively) and higher than any of the previous experiments in the DSD (Fig. 3c). The immobilisation efficiency was also improved for experiments 16, 17 and 18 (at 59.5, 67.1 and 70.5%, respectively) – although these did not accurately follow the trend predicted by the model (Fig. 3d).
These results highlight the efficacy and efficiency of employing a DOE-type methodology for the optimisation of an enzyme immobilisation process. Here, the effects and magnitudes of five input variables on a complex system were screened and modelled with only 14 experiments, allowing optimisation of the system to increase the desired output factors (loading and immobilisation efficiency). Increasing the NaOH concentration increased the immobilisation efficiency as was initially hypothesised, but only increased the loading under certain conditions – having no effect when Na2SO3 concentration and sulfite processing volume were low. A traditional OFAT method could therefore have drawn false conclusions depending on the Na2SO3 concentration and sulfite processing volume employed whilst NaOH concentration was the factor being varied.48
WAXD patterns of the CSs produced in the initial DSD (experiments 1–14), as well as untreated Basswood as a control, are presented in Fig. 4. Note that samples were taken from the central regions of the CSs for WAXD analysis where the morphology was relatively aligned (Fig. 2), as opposed to the ends which had a more fibrillar morphology (Fig. S5c†). This was done since samples from the centre were more representative of the bulk CS properties. WAXD patterns from the more fibrillar end regions were not investigated here.
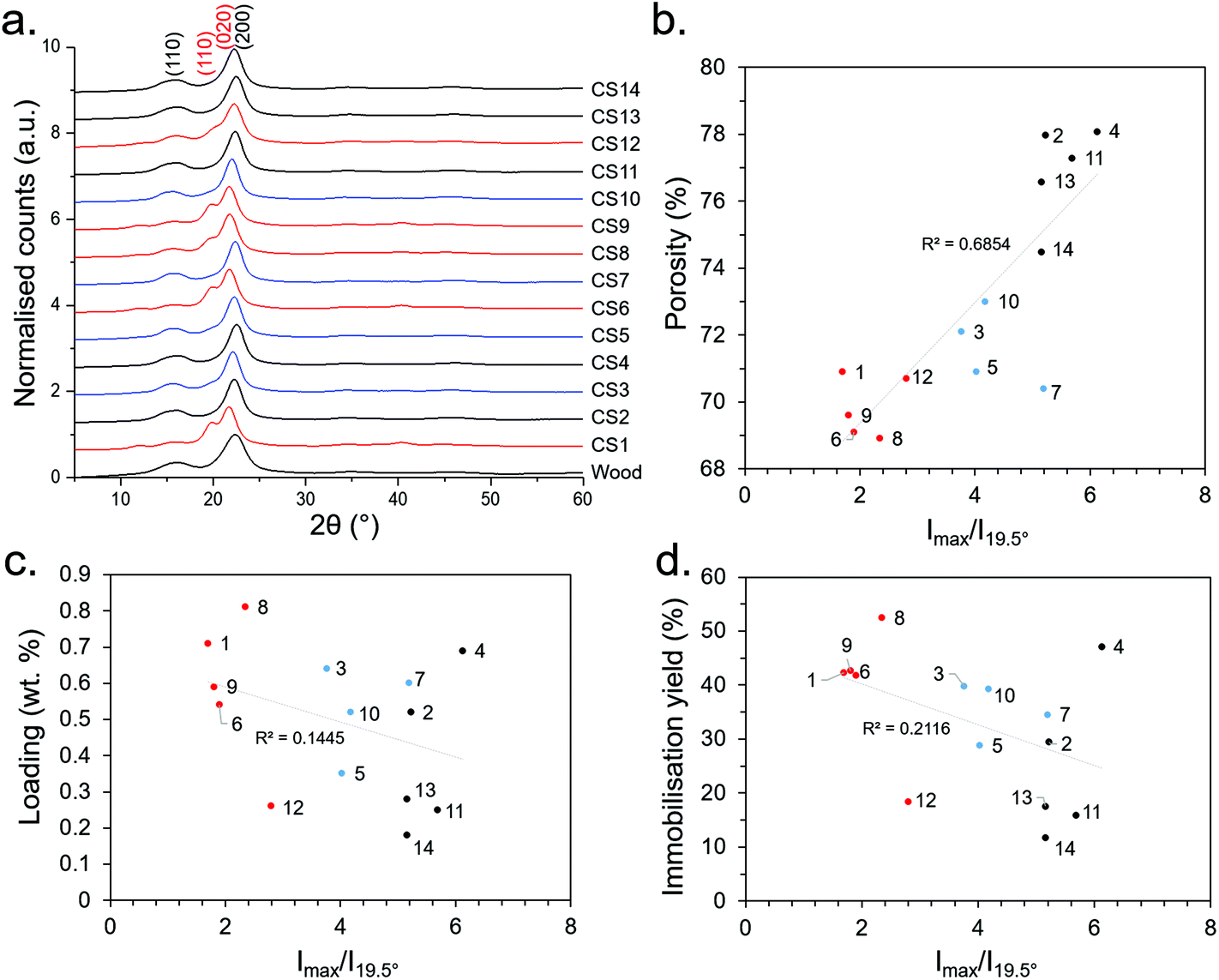 | ||
| Fig. 4 (a) WAXD patterns (b) relationship of Imax/I110 with porosity, (c) Imax/I110 with loading, and (d) Imax/I110 with immobilisation efficiency. Numbers 1–14 represent the synthesis conditions as presented in Table 1. Red, blue and black lines/points represent 7.5, 5 and 2.5 M NaOH concentrations during the sulfite processing step, respectively. | ||
It can be seen from Fig. 4 that most of the CS WAXD patterns have the characteristic 200 and 110 Bragg reflections (at ca. 22° and 16°, respectively) associated with cellulose Iβ,50 as does the untreated Basswood. However, synthesis conditions 1, 6, 8, 9 and 12 have an additional peak at ca. 19.5° and a slightly down-shifted main peak at ca. 21.5°. These were attributed to 110 and 020 Bragg reflections of a cellulose II type structure, suggesting that Mercerisation had indeed occurred under these conditions. Moreover, the conditions which resulted in the cellulose II had the common factor of being treated under the relatively high NaOH concentration of 7.5 M, corroborating our initial hypothesis that Mercerisation could be conducted by increasing the NaOH concentration during the sulfite processing step. Mercerised cellulose is typically a mixture of the cellulose I and II polymorphs (rather than purely cellulose II),50 and it is likely the case here. A useful means to distinguish between the polymorphs quantitatively was to take the ratio of the peak intensity (Imax) and intensity of the 110 peak (I110) at 19.5°; since the ratio becomes more pronounced with increasing cellulose II content. This revealed an unexpected correlation between porosity and cellulose II content (Fig. 4b), however there was no significant correlation between the degree of cellulose II content with loading or immobilisation efficiency (Fig. 4c and d) – suggesting the increasing NaOH content may be increasing loading through another mechanism (e.g., partial hydrolysis of the cellulose polymer backbone). The absence of other pronounced peaks in the WAXD data indicated the removal of salts (e.g., residual NaOH, Na2SO3) and other crystalline impurities from the CS synthesis procedure.
3.4. Optimisation of flow conditions
The flow conditions for the immobilisation of the CBM-FPs had not undergone any parametric optimisation and the employed conditions – which were based on scoping trials with Methyl-red – were likely far from optimal. Therefore, another DOE DSD was conducted, only this time on the factors relating to the flow conditions. Note, the non-optimised CSs were used for this DSD since the results from the CS optimisation were not known at this point (the two DOE optimisations were conducted in parallel for time efficiency).Another characterisation of the significant factors is presented in the ESI (Table S7†), where pH, salt concentration, protein concentration, flow rate and protein volume were chosen as the variables subject to optimisation. Temperature was not controlled as we did not at this point have the equipment for its reliable control, categorical factors (e.g. salt type, buffer type) were set as constants since categorical variables would require additional experimental runs. Before conducting the DSD, an initial scoping experiment where we measured the maximum forcing (Fmax), minimum forcing (Fmin) and centre-point conditions were conducted – this established that the loading (Fmin = 0.024%, Fmax = 1.52%) and immobilisation efficiency (Fmin 33.6%, Fmax = 60.4%) could indeed be significantly improved by varying these factors within the set ranges (Fig. S7†). Note that the buffer concentration was set as a fixed variable (50 mM) for the DSD despite being varied in this scoping experiment (10–300 mM) to simplify the experimental design.
The experimental conditions determined by the DSD are given in Table 2 (experimental order again randomised to reduce bias) along with immobilisation efficiency and protein loading (experiments F1–F13). Using JMP software, the most significant factors were again selected from the screening dialogue to construct RSM models for the output factors (i.e., immobilisation efficiency and protein loading). The RSMs predicted that the optimum conditions for immobilisation efficiency would be the lowest pH (7.1), lowest flow rate (50 μl min−1) and highest protein volume (7.5 ml) (Fig. 5a), and optimum conditions for loading would be the highest protein concentration and volume (the former having a greater effect), lowest flow rate (50 μl min−1) and lowest pH (7.1). Interestingly, salt concentration was not a significant factor – going against our initial understanding that salt concentration would interfere with CBM-based adsorption by affecting non-specific binding.
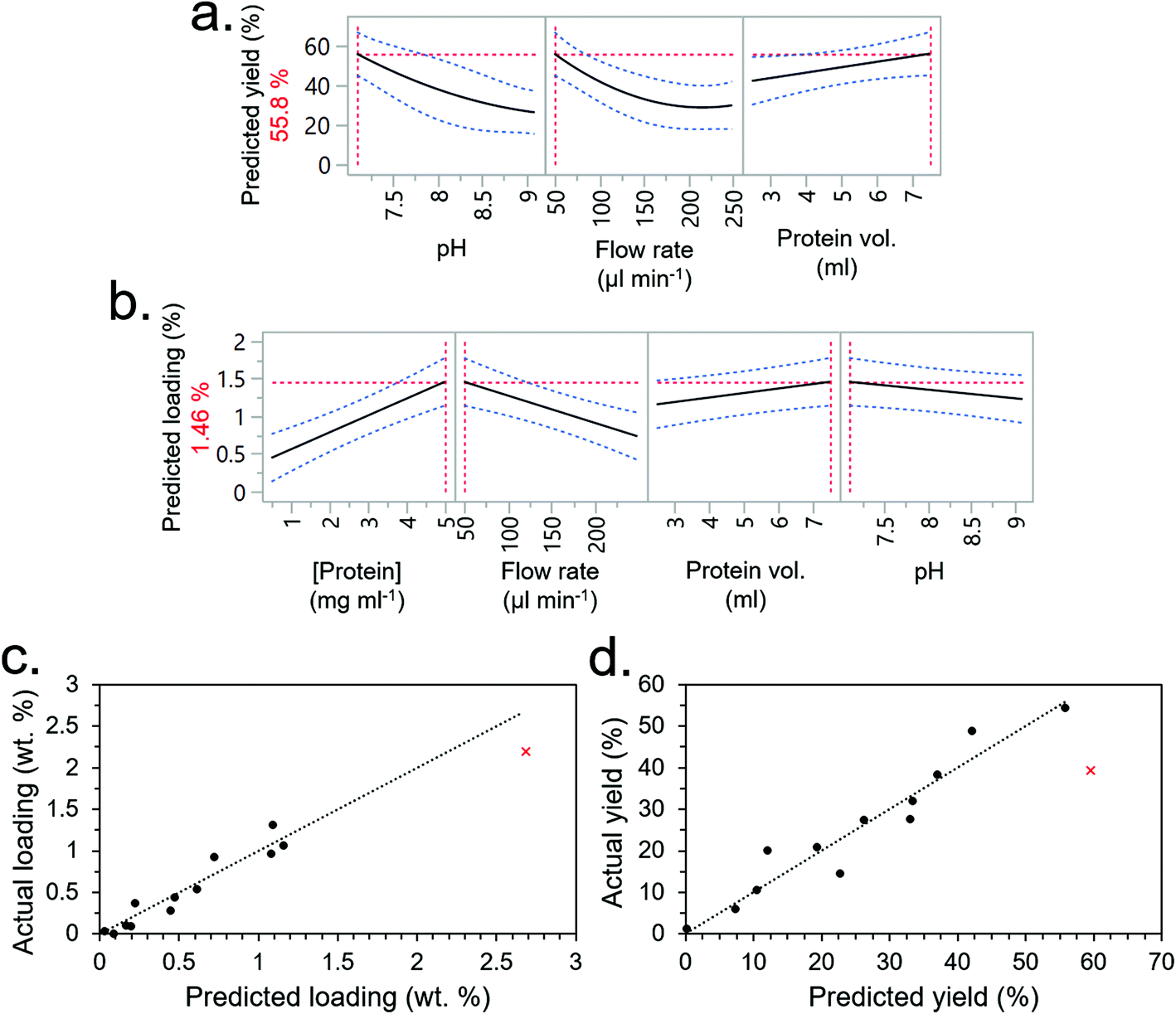 | ||
| Fig. 5 (a) Immobilisation Efficiency and (b) loading RSM prediction profilers for the flow conditions optimisation DSD. Blue dashed lines represent confidence intervals and red dashed lines indicate prediction profiler settings. (c) Immobilisation efficiency and (d) loading RSM models calculated from experiments 1–13 (black dots) and validated with experiment 14 (red cross). | ||
| Input factors | Output factors | |||||||
|---|---|---|---|---|---|---|---|---|
| Experiment no. | pH | Protein conc. (mg ml−1) | Flow rate (μL min−1) | Protein vol. (ml) | Salt conc. (mM) | Buffer conc. (mM) | Immobilisation efficiency (%) | Loading (wt%) |
| F1 | 9.1 | 0.5 | 250 | 7.5 | 10 | 50 | 1.3 | 0.006 |
| F2 | 7.1 | 2.75 | 250 | 2.5 | 10 | 50 | 38.3 | 0.373 |
| F3 | 9.1 | 5 | 250 | 2.5 | 155 | 50 | 6.0 | 0.091 |
| F4 | 9.1 | 2.75 | 50 | 7.5 | 300 | 50 | 27.5 | 0.926 |
| F5 | 8.1 | 5 | 250 | 7.5 | 300 | 50 | 10.6 | 0.541 |
| F6 | 7.1 | 5 | 50 | 2.5 | 300 | 50 | 48.9 | 1.068 |
| F7 | 9.1 | 0.5 | 150 | 2.5 | 300 | 50 | −2.5 | −0.004 |
| F8 | 8.1 | 0.5 | 50 | 2.5 | 10 | 50 | 14.5 | 0.030 |
| F9 | 8.1 | 2.75 | 150 | 5 | 155 | 50 | 20.1 | 0.443 |
| F10 | 9.1 | 5 | 50 | 5 | 10 | 50 | 20.8 | 0.964 |
| F11 | 7.1 | 5 | 150 | 7.5 | 10 | 50 | 27.6 | 1.309 |
| F12 | 7.1 | 0.5 | 250 | 5 | 300 | 50 | 32.0 | 0.103 |
| F13 | 7.1 | 0.5 | 50 | 7.5 | 155 | 50 | 54.5 | 0.278 |
| F14 | 7.1 | 10 | 5 | 4 | 50 | 50 | 39.4 | 2.192 |
| F15 | 7.1 | 10 | 5 | 4 | 50 | 150 | 59.1 | 3.258 |
| F16 | 7.1 | 10 | 5 | 4 | 50 | 250 | 48.5 | 2.596 |
Based on these insights, a 14th experiment (F14) was performed with the intention of both validating the models and extrapolating their predictions to maximise total loading whilst maintaining a high immobilisation efficiency. The conditions for F14, given in Table 2, drastically reduced the flow rate to 5 μL min−1 and increased protein concentration to 10 mg ml−1. Protein volume was reduced to 4 ml to conserve material since it had a lower significance than protein concentration, which was prioritised. The pH was maintained at 7.1 since any lower was out of the buffering range for TRIS. The salt concentration, which was not a significant factor, was set at 50 mM. Under these conditions, a relatively high loading of 2.19 wt% was obtained which roughly followed the prediction of the RSM model (Fig. 5c). Immobilisation efficiency was still high at 39.38% but fell short of the model's prediction of 59.6% (Fig. 5d). Given that the scoping experiment had predicted an even higher loading value, and the only other factor it considered was the buffer concentration, it was inferred that the buffer concentration was indeed a significant factor. To test this hypothesis, experimental condition 14 was repeated but at higher buffer concentrations of 150 and 250 mM (experiments 15 and 16, respectively). This showed a higher loading and immobilisation efficiency with increasing TRIS concentration as hypothesised, peaking at 150 mM (Fig. S8†). More data points at different TRIS concentrations were not conducted due to limited availability of the CBM3A-mCherry protein from that production batch.
3.5. Comparing different CBM-FP constructs under optimised conditions
Since optimisation of the CS synthesis and flow conditions were conducted independently, their combined effect was subsequently investigated. In addition, several of the other produced CBM-FP constructs investigated in the initial scoping experiments (details in the ESI†) were also investigated under the conditions optimised for CBM3A-mCherry to evaluate their relative performance. Note that mECFP-CBM2A and mCitrine-CBM1 were dropped from the study due to their relatively low expression yield and immobilisation efficiency, respectively, in order to reduce workload and simplify the experimental design. Two control experiments were also conduced, namely: (1) testing mCherry with no CBM attached to confirm immobilisation is due to the CBM and not any other effect (e.g., non-specific physical adsorption), and (2) mechanically ablating a CS to destroy the aligned porous structure. The results for these experiments are presented in Fig. 6 and Table S1.† CBM3A-mCherry, CBM30-mOrange and CBM2a-mEGFP and the control experiments were all tested under the conditions optimised primarily to maximise loading (10 mg ml−1 protein concentration), however mNeptune-CBM28 could only be concentrated to ∼5 mg ml−1 so was tested at that concentration instead. CBM2a-mEGFP was also tested at 5 mg ml−1 to have a direct comparison with mNeptune-CBM28, as was mEGFP with no CBM as a further control. After flowing through the protein solutions, 10 ml of buffer (50 mM KCl, 150 mM TRIS, pH 7.1) was flushed through at a rate of 250 μl min−1, measuring the protein concentration in the eluent every 2 ml to assess the extent of protein leaching from the scaffolds.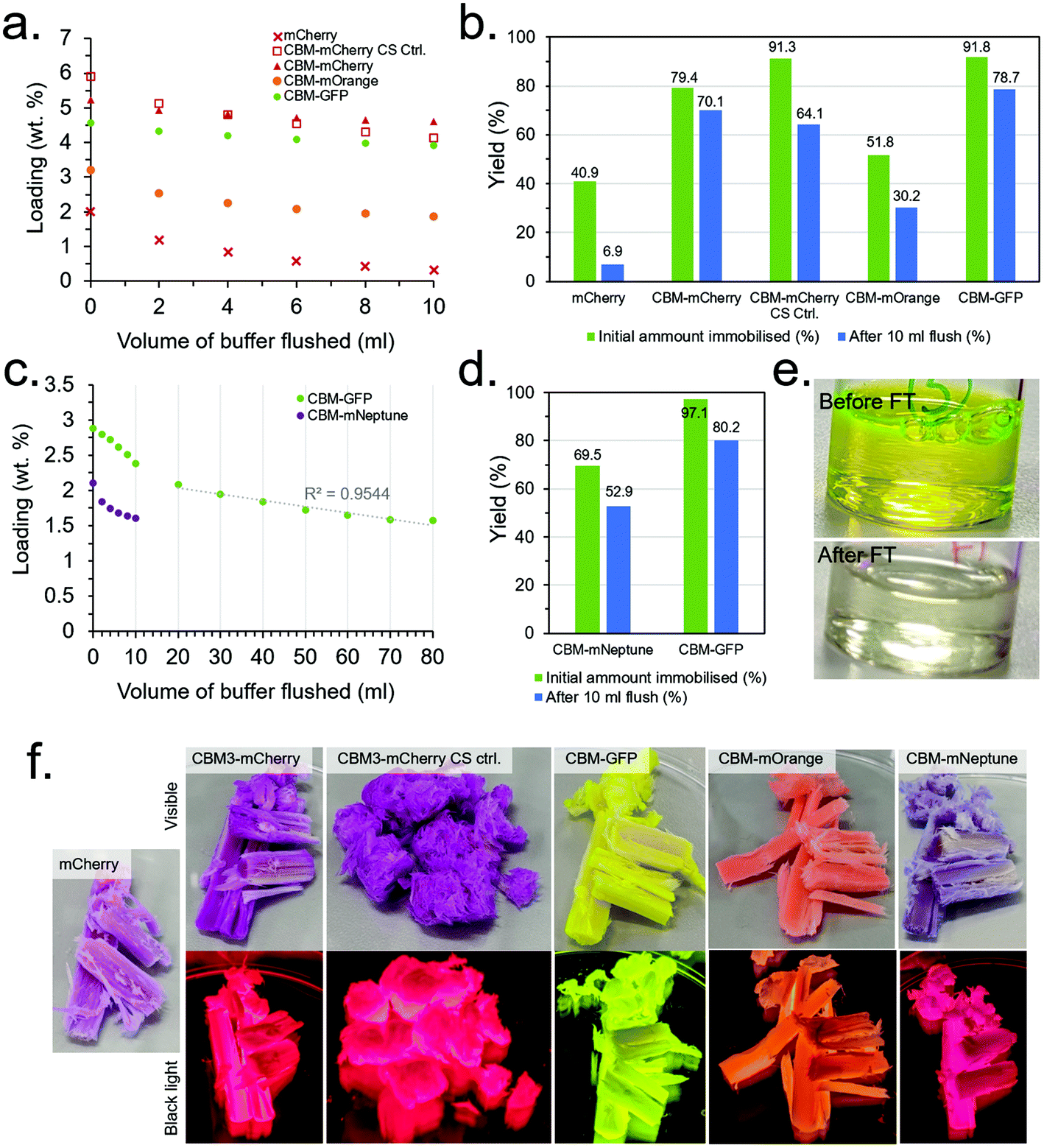 | ||
| Fig. 6 (a) Loading and (b) immobilisation efficiency of the CBM-FPs loaded at ∼10 mg ml−1 before and after a 10 ml buffer flush. (c) Loading and (d) immobilisation efficiency of CBM2a-mEGFP and mNeptune-CBM28 loaded at 5 mg ml−1 before and after a 10 ml flush (extends to 80 ml for CBM2a-mEGFP). (e) CBM2a-mEGFP solution before and after initial loading. (f) Visible light (above) and black light (below) images of CBM-FPs loaded onto CSs (after 10 ml buffer flush). | ||
The results (summarised in Table S1†) found that mCherry with no CBM had poor initial loading and immobilisation efficiency (2.02 wt% and 40.9%, respectively) with very little retained after the 10 ml buffer flush (0.34 wt% and 6.9%), compared with CBM3A-mCherry which had a much higher initial loading and immobilisation efficiency and (5.24 wt% and 79.4%, respectively) with far more retained after the 10 ml buffer flush (4.63 wt% and 70.1%) than without the CBM (Fig. 6a and b). This control was strong evidence that the immobilisation was indeed due to CBM-based binding and not a false positive due to another mechanism. The control experiment with a mechanically ablated CS also had a high initial immobilisation efficiency and loading at 91.3% and 5.94 wt% respectively, however leaching was more pronounced with immobilisation efficiency and loading falling to 64.1% and 4.15 wt% after the 10 ml buffer flush. CBM30-mOrange performed significantly worse than CBM3A-mCherry with an initial immobilisation efficiency and loading of 51.8% and 3.22 wt% respectively, falling to 30.2% and 1.88 wt% after the 10 ml buffer flush. CBM2a-mEGFP performed very well with an initial immobilisation efficiency of 91.8% and initial loading of 4.59 wt%, falling to 78.7% and 3.94 wt% respectively after the 10 ml buffer flush. The mEGFP control (with no CBM) displayed worse performance with an initial loading of 3.43 wt% and immobilisation efficiency of 78.1%, falling to 1.05% and 24.0% after the 10 ml buffer flush.
mNeptune-CBM28, at the lower protein concentration of 5 mg ml−1, performed significantly worse than CBM2a-mEGFP at the same concentration; with CBM2a-mEGFP having a very high initial immobilisation efficiency of 97.1% (Fig. 6c–e). These conditions were therefore taken as optimal for maximisation of immobilisation yield, whereas higher protein concentrations of 10 mg ml−1 were taken as conditions for maximisation of protein loading (Table S1†). Leaching of the CBM-FPs during the 10 ml buffer flush was evidently an issue; therefore a further 70 ml of buffer was flushed through the CBM-GFP loaded CS to assess if the leaching would stabilise over time, however it did not – with the rate of protein leaching remaining roughly constant after the first 10 ml (Fig. 6c). Minimising the extent of leaching through optimisation of the buffer through a DOE-type methodology could be a useful target of a future study.
3.6. Translation to CBM-tagged enzyme constructs for CFB evaluation
Employing the optimised CS synthesis and optimised flow immobilisation conditions (with minor modifications, as detailed in section 2.4.2), the CBM2a-ωTA constructs were immobilised to CSs. Loading and immobilisation efficiency was determined by measuring protein concentration before and after immobilisation via the Bradford method, employing BSA as a standard (Fig. S10†). CBM2a-mEGFP was run in parallel in order to have a direct comparison, with concentration determined by absorbance at 488 nm in addition to the Bradford method. The data, summarised in Table S8,† showed similar initial loadings (0.80 vs. 0.71 wt%) and immobilisation yields (81.4 vs. 95.5%) for the CBM2a-ωTA and CBM2a-mEGFP constructs when concentration was determined via the Bradford assay. However, the concentration of the CBM2a-mEGFP construct was significantly higher when determined via the absorbance at 488 nm (i.e., the mEGFP chromophore) than via the Bradford method (initial concentrations were 4.98 vs. 1.7 mg ml−1, respectively). The concentration determined via absorbance at 488 nm was taken as the more reliable figure, since the Bradford method depends on the amino acid composition of the protein which can differ significantly from the BSA standard.53 Therefore, to correct for this discrepancy and to allow direct comparison of CBM2a-ωTA with the fluorescent protein constructs, the concentration for CBM2a-ωTA was normalised based on the difference in measured concentration for the CBM2a-mEGFP construct via the Bradford method (1.7 mg ml−1) and via its chromophore absorbance at 488 nm (4.98 mg ml−1) (Table S8†). Details of these normalisation factor calculations are given in the ESI.†
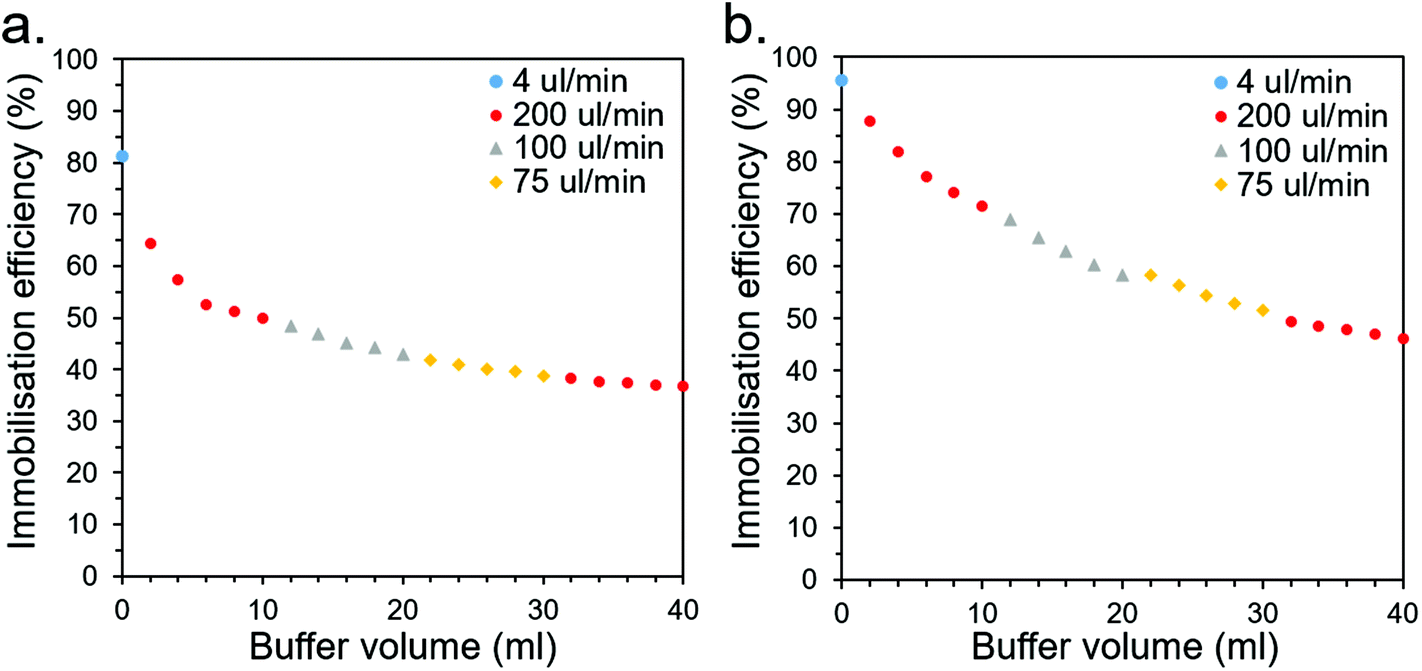 | ||
| Fig. 7 Leaching profiles for CS-immobilised (a) CBM2a-ωTA and (b) CBM2a-mEGFP as buffer is flushed through at various flow rates. | ||
For industrial CFB applications, this rate of leaching would likely be unacceptably high and would therefore need to be improved. This could be addressed through optimisation of the buffer conditions (e.g., salt and buffer type/concentration may be significant factors that affect leaching behaviour), screening for superior CBMs (this study only looked at six), employing multiple CBMs for superior affinity, or employing covalent crosslinking methods in conjunction to robustly anchor the CBMs to the CSs. Other applications, such as bioremediation, may be more suitable than CFB if they have a higher tolerance for leaching and may place a higher priority on cheap, biodegradable materials such as the wood-derived CSs.
Initially, a calibration curve monitoring the change in absorbance of the reactants and products at 245 nm was produced allowing the determination of acetophenone production (Fig. S11†). The activity of the free enzyme in solution (see section 2.5 for experimental details) was determined and found to stabilize at around 50% conversion (50.71% after 300 minutes) (Fig. 8a), which was used as a basis for comparison with the performance of the CS immobilised CBM2a-ωTA in flow.
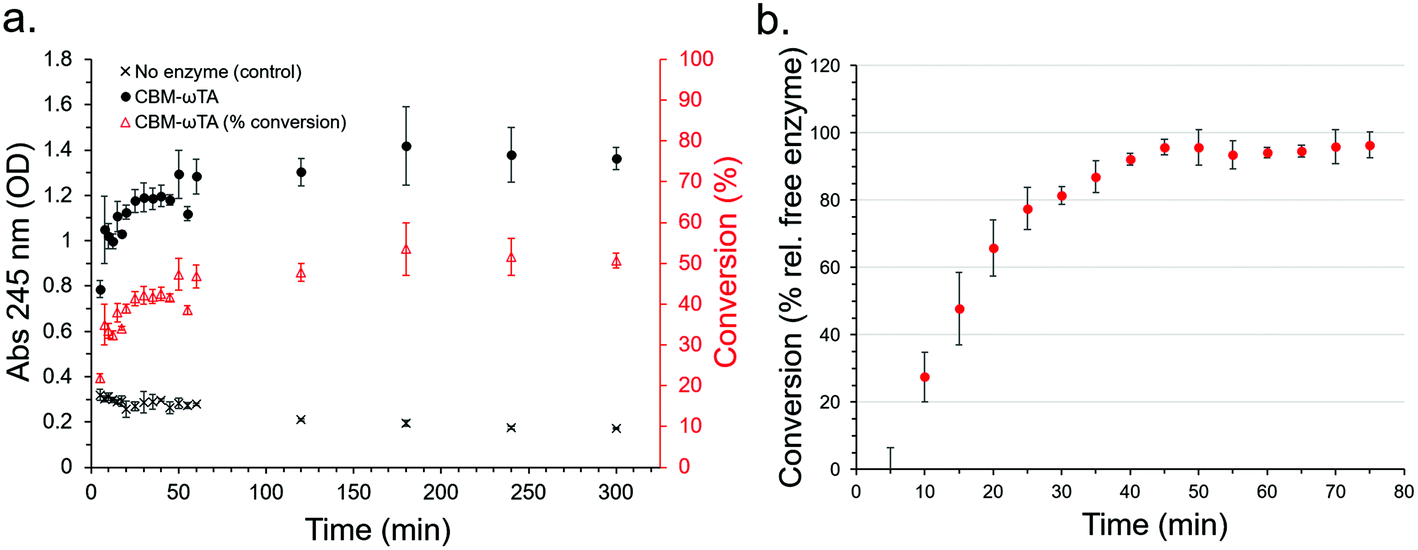 | ||
| Fig. 8 (a) Activity of free CBM2a-ωTA in solution determined through acetophenone assay (absorbance at 245 nm) (b) activity of CS-immobilised CBM2a-ωTA relative to activity of free enzyme in solution. | ||
In three replicate experiments, CBM2a-ωTA was loaded onto the optimised CSs following the optimised procedure as detailed in section 2.6. Note, a higher concentration (ca. 10 mg ml−1 by normalized Bradford assay) was employed than for the leaching study (6.68 mg ml−1) in order to maximise enzyme loading over immobilisation efficiency. Here, the initial enzyme loading on the CSs was 3.99% ± 0.86. After loading, a reactant mixture consisting of 25 mM pyruvate, 25 mM (S)-α-MBA, 0.2 mM PLP and 100 mM KPI buffer at pH 8 was passed through the CSs at a rate of 200 μL min−1, and the production of acetophenone was monitored every 5 minutes (or every 1 mL) at 30 °C (Fig. 8b). Note that acetophenone concentration measurements were conducted immediately to prevent activity from any leached enzymes from significantly affecting the results. The results show that after an initial lag period of about 40 minutes the conversion relative to free enzyme in solution approached 95 ± 5% with good reproducibility between the three replicate experiments. The reason for the significant lag period was unclear, but may have been due to the CSs adjusting to the new buffer environment, or the produced acetophenone being immediately immobilised onto the surface of the CSs until saturation. Extrapolation of this performance corresponds to an acetophenone production rate of 0.09 g h−1, or 2.16 g day−1 – however the long-term stability of the enzyme was not assessed nor have the flow conditions been subject to optimisation in this instance. Higher flow rates than 200 μl min−1 were not explored since the higher backpressures associated with faster rates would often cause leakage with our experimental set-up.
3.7. Testing immobilisation directly from cell lysate
After confirming that a CBM-tagged enzyme could indeed be immobilised to a wood-derived CS and employed in a CFB set-up, we conducted some experiments to further explore the potential of the concept. Firstly, we conducted a scoping experiment to determine if CBM-tagged proteins could be immobilised directly from cell lysate. Enzyme purification can be a relatively expensive and time-consuming step which can limit their appeal as alternatives to traditional chemical catalysts for synthesis. If CBM-tagged enzymes could avoid prior purification and be immobilised directly from lysed cells onto wood-derived CSs it could be a significant cost and time saving, improving their viability for industrial biocatalysis.To test this, a CBM3a-mCherry fusion construct was expressed in E. coli before the cells were collected and subject to lysis via sonication. The lysed cells were briefly centrifuged to remove the majority of the insoluble cell debris – but other soluble impurities remained. This resulted in a cloudy solution with a CBM3a-mCherry concentration of about 3 mg ml−1 – the solution was not subject to further concentration since such a step would also remove the soluble impurities. The solution was flowed through an optimised CS under optimised flow conditions, aside from the concentration being sub-optimal. The protein loading (based on two duplicate experiments) was 1.7 wt%, with an immobilisation yield of 97.6%, falling to 95.8% after a further 5 ml flush of buffer. Higher loadings would likely be possible with higher initial protein concentrations, but would come at the expense of immobilisation yield. As a control, mCherry with no CBM attached was also flowed though in an analogous experiment, although a lower concentration of 0.5 mg ml−1 was employed due to lower expression yield and since it was not possible to concentrate the sample without also removing impurities. This control experiment showed a much lower protein loading of 0.22 wt% and lower immobilisation yield of 77.6%, falling to 61.9% after a 5 ml flush of buffer indicating relatively poor attachment. A visual inspection of the immobilised CS's also suggested that the CBM3a-mCherry CS was unsaturated and could accommodate further protein, whereas the mCherry control appeared saturated (Fig. S12†).
3.8. Testing immobilisation with other wood precursors
In another scoping experiment, we sought to determine if other wood types could also be employed, since American Basswood may not be the most sustainable or economical option – especially in regions where it is not naturally cultivated. Other wood types may also have more optimal pore sizes for certain processes. Here, we tested English Oak due to its relatively small pore size in comparison with Basswood.Briefly, the optimised CS fabrication procedure was employed with Oak wood rather than Basswood to produce Oak-derived CSs. The produced CSs had a similar aspect to Basswood-derived CSs (Fig. S13a†). Then, an aqueous solution of CBM3a-mCherry (3.62 mg ml−1) was flushed through both the Oak-derived and Basswood-derived CSs in parallel, following the optimised procedure. Note that CBM3a-mCherry obtained directly from the cell lysate (i.e., without further purification) was employed to further support the experiment in section 3.7. This meant that the optimal protein concentration of 10 mg ml−1 was not employed as the process of concentration would have removed some impurities. The initial protein loadings for Basswood-derived and Oak-derived CSs were 1.95 and 1.60 wt%, respectively. The initial immobilisation yields were 95.4% and 91.7%, falling to 92.0% and 87.5% after a 5 ml buffer flush, for the Basswood-derived and Oak-derived CSs, respectively. This data show that Oak-derived CSs perform almost as well as Basswood-derived CSs, despite the optimisation being conducted for the latter. Based on this, we believe it is reasonable to infer that many other kinds of wood could also be employed with this process.
3.9. Discussion
Although further investigation is required to explore the full potential of CBM-based enzyme immobilisation on wood-derived CSs for CFB – this proof-of-principle study demonstrates the feasibility of the technique. Parametric optimisation of the CFB conditions (including flow rates, temperatures, pH, ionic conditions and other parameters) to maximise catalytic performance of the system (e.g., turn over number, turn over frequency, space–time-yield etc.) would be a useful objective of a future study. More in-depth WAXD studies would also be insightful, for example a WAXD study to determine if regions which appeared to uptake greater amounts of CBM-FPs (e.g., the relatively fibrillated CS end regions) had a substantially different microstructure would be interesting. Another insightful study would be to investigate the effect of residual lignin on CBM-based immobilisation affinity, since it was apparent from Fig. S5† that different CS synthesis conditions resulted in different levels residual lignin content. It's possible that the residual lignin content could play a significant role in resultant CBM-based immobilisation capacity – if so, the colour of the produced CS's (lighter colour = less residual lignin) could be a good indicator of potential immobilisation capacity. This could allow relatively fast screening of process conditions for future optimisation.The current rate of enzyme leaching is problematic and will need to be addressed if CBM-tagged recombinant enzymes immobilised to CSs are to be employed for industrial applications. Where documented, CBM-based enzyme immobilisation appears to have a persistent problem with leaching – however it is often not clear how much, if any, optimisation was conducted to mitigate the extent of this leaching.30–42 Unfortunately, the field lacks reliable protocols for the measurement of the extent of leaching meaning reliable comparisons between literature reports are unavailable. A detailed study into the factors which effect CBM-based enzymatic leaching would therefore be a helpful future study – especially if it outlines standard conditions for future studies to follow to enable reliable comparisons of leaching behaviour with other reports. It should be noted that other forms of affinity-based immobilisation can achieve remarkably little leaching,51 indicating that CBM-based immobilisation has the potential to overcome this issue. A similar investigation into the stability and longevity of CBM-based immobilised enzymes would also be similarly beneficial.
Compared to other enzyme immobilisation techniques, the use of CSs as an immobilisation material could be considered a relatively low cost, green and scalable alternative. In particular, the aligned macropores of CSs could feasibly be employed as a cheap, scalable parallel microchannel reactor bundles, akin to multichannel microfluidic chip reactors.54 Microfluidic reactors with aligned channels have numerous merits over packed-bed flow reactors – particularly with regard to process intensification – including superior mass and heat transfer profiles, highly symmetric lamellar flow dynamics and relatively high effective contact areas with low back pressures.16,54 High costs associated with the fabrication of such microfluidic reactors have been a barrier to scale up, meaning low cost CSs could be a competitive alternative. Furthermore, the flexible CBM-based affinity immobilisation explored here could, with further development, allow stable and selective tethering of active enzymes to the CS microchannel walls without the need for dirty and costly surface functionalisation chemistries. The in-flow immobilisation technique employed here can also be considered advantageous from a practicality perspective, allowing the CS to be loaded (or re-loaded) with enzyme without dismantling the setup as is often the case for packed-bed flow reactors.51 We also note that although the sulphite pulping process was employed here for the removal of lignin and hemicellulose, other lower-energy and more sustainable processes could be also feasibly be employed. Such processes may also have better recovery of the lignin which could be utilised for other applications, further improving green credentials.
4. Conclusions
In this work, CSs with aligned macroporosity were produced through the de-lignification of wood under relatively mild sulfite pulping conditions. Various CBM-tagged fluorescent proteins were designed, expressed and subsequently immobilised to the CSs, before being employed to efficiently optimise a range of parameters using a DOE-based methodology. Once optimised to maximise protein loading and immobilisation efficiency, a construct based on the best-performing CBM (CBM2a from C. cellulovorans EngD) and an industrially relevant enzyme (ωTA from B. megaterium) was designed, expressed and also immobilised to the CS materials – demonstrating translatability of the process. The initial loadings and immobilisation efficiencies of the CBM2a-ωTA (as high as 3.99 wt% and 82.4%, respectively) were higher than most other values reported in the literature for CBM-based enzyme immobilisation – despite employing a novel, non-commercial, cellulosic scaffold material derived from wood. A proof-of-principle study demonstrated that the CS immobilized CBM2a-ωTA working in a CFB setup maintained its catalytic activity relative to the free enzyme in solution (ca. 95 ± 5% relative activity), meaning such a system, with further development, has the potential to compete with other established enzyme immobilisation technologies – particularly on a cost-performance basis due to the relatively low cost of wood-derived cellulose and the demonstrated ability to immobilise directly from cell lysate. This work establishes a foundation for the further exploration of wood-derived CSs with aligned macroporosity for use as enzymatic microchannel reactors in CFB.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Future Biomanufacturing Research Hub (grant EP/S01778X/1), funded by the Engineering and Physical Sciences Research Council (EPSRC) and Biotechnology and Biological Sciences Research Council (BBSRC) as part of UK Research and Innovation. We acknowledge the University of Manchester Department of Material's X-Ray Diffraction Laboratory and Electron Microscopy Centre for equipment use and support. We would also like to thank the staff in the Faculty of Engineering and Physical Sciences workshops for their assistance. Thanks also go to Dr James Galman for providing the BmTA plasmid.References
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Cellulose: Fascinating Biopolymer and Sustainable Raw Material, Angew. Chem., Int. Ed., 2005, 44(22), 3358 CrossRef CAS PubMed.
- C. Moulherat, M. Tengberg, J.-F. Haquet and B. Mille, First evidence of cotton at Neolithic Mehrgarh, Pakistan: analysis of mineralized fibres from a copper bead, J. Archaeol. Sci., 2002, 29(12), 1393 CrossRef.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Cellulose nanomaterials review: structure, properties and nanocomposites, Chem. Soc. Rev., 2011, 40(7), 3941 RSC.
- T. Huber, J. Müssig, O. Curnow, S. Pang, S. Bickerton and M. P. Staiger, A critical review of all-cellulose composites, J. Mater. Sci., 2012, 47(3), 1171 CrossRef CAS.
- L. Jabbour, R. Bongiovanni, D. Chaussy, C. Gerbaldi and D. Beneventi, Cellulose-based Li-ion batteries: a review, Cellulose, 2013, 20(4), 1523 CrossRef CAS.
- M. Zhu, J. Song, T. Li, A. Gong, Y. Wang, J. Dai, Y. Yao, W. Luo, D. Henderson and L. Hu, Highly Anisotropic, Highly Transparent Wood Composites, Adv. Mater., 2016, 28(26), 5181 CrossRef CAS PubMed.
- T. Li, Y. Zhai, S. He, W. Gan, Z. Wei, M. Heidarinejad, D. Dalgo, R. Mi, X. Zhao, J. Song, J. Dai, C. Chen, A. Aili, A. Vellore, A. Martini, R. Yang, J. Srebric, X. Yin and L. Hu, A radiative cooling structural material, Science, 2019, 364(6442), 760 CrossRef CAS PubMed.
- J. S. Segmehl, V. Studer, T. Keplinger and I. Burgert, Characterization of wood derived hierarchical cellulose scaffolds for multifunctional applications, Materials, 2018, 11(4), 517 CrossRef PubMed.
- S. Vitas, J. S. Segmehl, I. Burgert and E. Cabane, Porosity and pore size distribution of native and delignified beech wood determined by mercury intrusion porosimetry, Materials, 2019, 12(3), 416 CrossRef CAS PubMed.
- H. Guan, Z. Cheng and X. Wang, Highly Compressible Wood Sponges with a Spring-like Lamellar Structure as Effective and Reusable Oil Absorbents, ACS Nano, 2018, 12(10), 10365 CrossRef CAS PubMed.
- S. Vitas, T. Keplinger, N. Reichholf, R. Figi and E. Cabane, Functional lignocellulosic material for the remediation of copper(II) ions from water: Towards the design of a wood filter, J. Hazard. Mater., 2018, 355, 119 CrossRef CAS PubMed.
- T. Keplinger, X. Wang and I. Burgert, Nanofibrillated cellulose composites and wood derived scaffolds for functional materials, J. Mater. Chem. A, 2019, 7(7), 2981 RSC.
- M. Barrow, A. Eltmimi, A. Ahmed, P. Myers and H. Zhang, Frozen polymerization for aligned porous structures with enhanced mechanical stability, conductivity, and as stationary phase for HPLC, J. Mater. Chem., 2012, 22(23), 11615 RSC.
- X. Yan and N. Li, Nanopillar array with multi-scale inter-pillar spacing as chromatography stationary phase support: Theoretical performance evaluation, Chem. Eng. Sci., 2014, 107, 158 CrossRef CAS.
- R. D. Arrua and E. F. Hilder, Highly ordered monolithic structures by directional freezing and UV-initiated cryopolymerisation. Evaluation as stationary phases in high performance liquid chromatography, RSC Adv., 2015, 5(87), 71131 RSC.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification, Trends Biotechnol., 2018, 36(1), 73 CrossRef CAS PubMed.
- A. Illanes, Enzyme biocatalysis, in Princ. Appl, Springer-Verlag New York Inc., United States, 2008 Search PubMed.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Industrial applications of enzyme biocatalysis: Current status and future aspects, Biotechnol. Adv., 2015, 33(7), 1443 CrossRef CAS PubMed.
- S. P. de Souza, I. I. Junior, G. M. A. Silva, L. S. M. Miranda, M. F. Santiago, F. Leung-Yuk Lam, A. Dawood, U. T. Bornscheuer and R. O. M. A. de Souza, Cellulose as an efficient matrix for lipase and transaminase immobilization, RSC Adv., 2016, 6(8), 6665 RSC.
- F. Guo and P. Berglund, Transaminase biocatalysis: optimization and application, Green Chem., 2017, 19(2), 333 RSC.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Improvement of enzyme activity, stability and selectivity via immobilization techniques, Enzyme Microb. Technol., 2007, 40(6), 1451 CrossRef CAS.
- W. Li, J. Shi, Y. Zhao, Y. Tian, Q. Huo, Y. Wu, S. Zhang and Z. Jiang, Bioinspired synthesis of nanofibers on monolithic scaffolds for enzyme immobilization with enhanced loading capacity and activity recovery, J. Chem. Technol. Biotechnol., 2019, 94(12), 3763 CrossRef CAS.
- J. Britton, S. Majumdar and G. A. Weiss, Continuous flow biocatalysis, Chem. Soc. Rev., 2018, 47(15), 5891 RSC.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Biocatalysis Using Immobilized Enzymes in Continuous Flow for the Synthesis of Fine Chemicals, Org. Process Res. Dev., 2019, 23(1), 9 CrossRef CAS.
- M. Romero-Fernández and F. Paradisi, Protein immobilization technology for flow biocatalysis, Curr. Opin. Chem. Biol., 2020, 55, 1 CrossRef PubMed.
- I. Levy and O. Shoseyov, Cellulose-binding domains: Biotechnological applications, Biotechnol. Adv., 2002, 20(3), 191 CrossRef CAS PubMed.
- O. Shoseyov, Z. Shani and I. Levy, Carbohydrate binding modules: biochemical properties and novel applications, Microbiol. Mol. Biol. Rev., 2006, 70(2), 283 CrossRef CAS PubMed.
- P. Tomme, A. Boraston, B. McLean, J. Kormos, A. L. Creagh, K. Sturch, N. R. Gilkes, C. A. Haynes, R. A. Warren and D. G. Kilburn, Characterization and affinity applications of cellulose-binding domains, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 715(1), 283 CrossRef CAS.
- C. Oliveira, V. Carvalho, L. Domingues and F. M. Gama, Recombinant CBM-fusion technology - Applications overview, Biotechnol. Adv., 2015, 33(3), 358 CrossRef CAS PubMed.
- S. Wang, G.-Z. Cui, X.-F. Song, Y. Feng and Q. Cui, Efficiency and Stability Enhancement of Cis-epoxysuccinic Acid Hydrolase by Fusion with a Carbohydrate Binding Module and Immobilization onto Cellulose, Appl. Biochem. Biotechnol., 2012, 168(3), 708 CrossRef CAS PubMed.
- S. Hwang, J. Ahn, S. Lee, T. G. Lee, S. Haam, K. Lee, I.-S. Ahn and J.-K. Jung, Evaluation of cellulose-binding domain fused to a lipase for the lipase immobilization, Biotechnol. Lett., 2004, 26(7), 603 CrossRef CAS PubMed.
- M. Gustavsson, J. Lehtiö, S. Denman, T. T. Teeri, K. Hult and M. Martinelle, Stable linker peptides for a cellulose-binding domain–lipase fusion protein expressed in Pichia pastoris, Protein Eng., 2001, 14(9), 711 CrossRef CAS PubMed.
- M. Jiang and A. Radford, Exploitation of a cellulose-binding domain from Neurospora crassa, Enzyme Microb. Technol., 2000, 27(6), 434 CrossRef CAS PubMed.
- C. Kauffmann, O. Shoseyov, E. Shpigel, E. A. Bayer, R. Lamed, Y. Shoham and R. T. Mandelbaum, Novel Methodology for Enzymatic Removal of Atrazine from Water by CBD-Fusion Protein Immobilized on Cellulose, Environ. Sci. Technol., 2000, 34(7), 1292 CrossRef CAS.
- I. Levy, G. Ward, Y. Hadar, O. Shoseyov and C. G. Dosoretz, Oxidation of 4-bromophenol by the recombinant fused protein cellulose-binding domain-horseradish peroxidase immobilized on cellulose, Biotechnol. Bioeng., 2003, 82(2), 223 CrossRef CAS PubMed.
- R. D. Richins, A. Mulchandani and W. Chen, Expression, immobilization, and enzymatic characterization of cellulose-binding domain-organophosphorus hydrolase fusion enzymes, Biotechnol. Bioeng., 2000, 69(6), 591 CrossRef CAS PubMed.
- J. C. Rotticci-Mulder, M. Gustavsson, M. Holmquist, K. Hult and M. Martinelle, Expression in Pichia pastoris of Candida antarctica Lipase B and Lipase B Fused to a Cellulose-Binding Domain, Protein Expression Purif., 2001, 21(3), 386 CrossRef CAS PubMed.
- B. N. Estevinho, N. Samaniego, D. Talens-Perales, M. J. Fabra, A. López-Rubio, J. Polaina and J. Marín-Navarro, Development of enzymatically-active bacterial cellulose membranes through stable immobilization of an engineered β-galactosidase, Int. J. Biol. Macromol., 2018, 115, 476 CrossRef CAS PubMed.
- S. Lin, Z. Qin, Q. Chen, L. Fan, J. Zhou and L. Zhao, Efficient Immobilization of Bacterial GH Family 46 Chitosanase by Carbohydrate-Binding Module Fusion for the Controllable Preparation of Chitooligosaccharides, J. Agric. Food Chem., 2019, 67(24), 6847 CrossRef CAS PubMed.
- A. Kumar, S. Zhang, G. Wu, C. C. Wu, J. Chen, R. Baskaran and Z. Liu, Cellulose binding domain assisted immobilization of lipase (GSlip–CBD) onto cellulosic nanogel: characterization and application in organic medium, Colloids Surf., B, 2015, 136, 1042 CrossRef CAS PubMed.
- J. Thongekkaew, H. Ikeda and H. Iefuji, Increases thermal stability and cellulose-binding capacity of Cryptococcus sp. S-2 lipase by fusion of cellulose binding domain derived from Trichoderma reesei, Biochem. Biophys. Res. Commun., 2012, 420(1), 183 CrossRef CAS PubMed.
- F. Chang, S. Xue, X. Xie, W. Fang, Z. Fang and Y. Xiao, Carbohydrate-binding module assisted purification and immobilization of β-glucosidase onto cellulose and application in hydrolysis of soybean isoflavone glycosides, J. Biosci. Bioeng., 2018, 125(2), 185 CrossRef CAS PubMed.
- N. van Oosterwijk, S. Willies, J. Hekelaar, A. C. Terwisscha van Scheltinga, N. J. Turner and B. W. Dijkstra, Structural basis of the substrate range and enantioselectivity of two (S)-selective ω-transaminases, Biochemistry, 2016, 55(31), 4422 CrossRef CAS PubMed.
- K. Sopitthummakhun, S. Maenpuen, Y. Yuthavong, U. Leartsakulpanich and P. Chaiyen, Serine hydroxymethyltransferase from Plasmodium vivax is different in substrate specificity from its homologues, FEBS J., 2009, 276(15), 4023 CrossRef CAS PubMed.
- J. M. Fox, P. Jess, R. B. Jambusaria, G. M. Moo, J. Liphardt, D. S. Clark and H. W. Blanch, A single-molecule analysis reveals morphological targets for cellulase synergy, Nat. Chem. Biol., 2013, 9(6), 356 CrossRef CAS PubMed.
- A. C. O'Sullivan, Cellulose: The structure slowly unravels, Cellulose, 1997, 4(3), 173 CrossRef.
- B. J. C. Duchemin, Mercerisation of cellulose in aqueous NaOH at low concentrations, Green Chem., 2015, 17(7), 3941 RSC.
- D. W. Lendrem, B. C. Lendrem, D. Woods, R. Rowland-Jones, M. Burke, M. Chatfield, J. D. Isaacs and M. R. Owen, Lost in space: design of experiments and scientific exploration in a Hogarth Universe, Drug Discovery Today, 2015, 20(11), 1365 CrossRef PubMed.
- B. Jones and C. J. Nachtsheim, A class of three-level designs for definitive screening in the presence of second-order effects, J. Qual. Technol., 2011, 43(1), 1 CrossRef.
- A. D. French, Idealized powder diffraction patterns for cellulose polymorphs, Cellulose, 2014, 21(2), 885 CrossRef CAS.
- W. Böhmer, T. Knaus, A. Volkov, T. K. Slot, N. R. Shiju, K. Engelmark Cassimjee and F. G. Mutti, Highly efficient production of chiral amines in batch and continuous flow by immobilized ω-transaminases on controlled porosity glass metal-ion affinity carrier, J. Biotechnol., 2019, 291, 52 CrossRef PubMed.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Rapid and Sensitive Kinetic Assay for Characterization of ω-Transaminases, Anal. Chem., 2009, 81(19), 8244 CrossRef PubMed.
- N. J. Kruger, Methods Mol. Biol., 1994, 32, 9–15 CAS.
- J. M. Bolivar, J. Wiesbauer and B. Nidetzky, Biotransformations in microstructured reactors: more than flowing with the stream?, Trends Biotechnol., 2011, 29(7), 333 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01008e |
| ‡ EPSRC/BBSRC Future Biomanufacturing Research Hub. |
| This journal is © The Royal Society of Chemistry 2021 |