
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous hydrogenation of saturated bicarbonate slurry to formates using multiphase catalysis†
Christophe
Rebreyend
,
Evgeny A.
Pidko
 * and
Georgy A.
Filonenko
*
* and
Georgy A.
Filonenko
*
Inorganic Systems Engineering, Department of Chemical Engineering, Delft University of Technology, van der Maasweg 9, Delft, 2629 HZ, The Netherlands. E-mail: e.a.pidko@tudelft.nl; G.A.Filonenko@tudelft.nl
First published on 3rd September 2021
Abstract
Formic acid and formate salts are key intermediates along the pathways for CO2 utilization and hydrogen storage. Herein we report a highly efficient multiphase catalytic system utilizing a ruthenium PNP pincer catalyst for converting supersaturated bicarbonate solutions and slurries to aqueous formate solutions up to 12 M in molarity. The biphasic catalytic system delivers turnover frequencies up to 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and remains stable for up to 474000 turnovers once reaction conditions are optimized.
000 h−1 and remains stable for up to 474000 turnovers once reaction conditions are optimized.
Current CO2 emissions are a concern regarding climate change.1,2 Consequently, efforts have been made to capture CO2 efficiently from the atmosphere and to utilize it for various goals.3 Among the various CO2 utilization strategies, its chemical conversion to C1 building blocks has been the focus of intense research effort over the last decades.4–6 The common utilization routes rely on CO2 reduction that can be performed electrochemically7,8 or utilize molecular hydrogen as the reducing agent.9,10 The latter can be further split into heterogeneous processes that mainly target methanol as a main product and homogeneous reduction protocols that typically produce formic acid and its salts.11–27 Although in recent years there have been significant progress in base metal catalysts for CO2 hydrogenation, namely iron,28–32 cobalt33–37 and manganese,38–40 the state-of-the-art in homogeneous CO2 hydrogenation is dominated by noble metal catalysts. Fig. 1 depicts several representative examples of such complexes. One of the first highly active CO2 hydrogenation catalysts reported by Nozaki and co-workers27 (2, Fig. 1) shows exceptional turnover frequencies (TOFs) up to 150
000 h−1 in hydrogenating CO2 in the presence of aqueous KOH/THF mixtures at 120–200 °C. A family of iridium catalysts reported by Himeda and co-workers11,12,16,17,41,42 with complex 3 (Fig. 1) as a representative example were also noted for their CO2 hydrogenation activity pronounced in aqueous solutions at room temperature and 1 bar of hydrogen pressure. Importantly, operation of these complexes could be switched between CO2 hydrogenation and formic acid dehydrogenation by controlling the pH of the reaction solution. Finally, our group reported a highly active ruthenium catalyst (1, Fig. 1), which was active for both CO2 hydrogenation and formate dehydrogenation albeit in organic solvents in the presence of organic bases.
 | ||
| Fig. 1 Representative homogeneous catalysts for the hydrogenation of CO2 to formates (1–3) and examples of catalysts operating in biphasic systems (4, 5), allowing for easy product separation. | ||
From the process standpoint, homogeneous CO2 hydrogenation should fulfil several requirements to be practical. Firstly, the catalyst should be sufficiently stable to provide high, economically relevant productivity. Secondly, the catalyst should be separable form the formate product that can easily be dehydrogenated once the hydrogenation reactor is decompressed. The latter has recently been addressed by immobilizing homogeneous catalysts on solid supports43 and encapsulating them into porous carriers.44,45
While this approach greatly simplifies catalyst separation, it has a drawback of consistently lower activity of the immobilized catalyst compared to its free molecular predecessor.
An approach alternative to immobilization makes use of biphasic solvent systems,46,47 in which the catalyst and reactants reside in different phases. Formate salts typically reside in the aqueous phase while the organometallic catalyst species remain in the organic phase and the catalyst and product(s) separation in the biphasic system can be done by simple decantation. Successful utilization of biphasic catalyst systems for CO2 hydrogenation has been demonstrated with catalysts 446 and 547 shown above and the development of highly active biphasic CO2 hydrogenation is our primary goal in this work.
Herein we report the development of a highly active multiphase catalyst system based on our Ru-PNP catalyst 1 enabling the hydrogenation of supersaturated aqueous bicarbonate to formate with outstanding efficiency. While complex 1 is a highly competent catalyst in fully organic media, e.g. DMF/DBU system utilized in earlier studies13 it performed poorly in aqueous environments. Our initial trials using 1 at 40 bar of equimolar H2/CO2 in water in the presence of KOH base provided very low formate yields (Table 1, entries 3 and 6) with a maximal TON of 1573 at 90 °C. Assuming the low solubility of 1 to be the major factor limiting catalytic performance in the aqueous media, we performed CO2 hydrogenation tests in the presence of an additional organic solvent. Both water-miscible DMF and immiscible toluene addition could improve the formate yields (Table 1); however, the KOH conversion remained limited to a maximum of 26%. We envisioned that such limitation might stem from the poor transport of ionic species across the phase boundary and employed a phase-transfer catalyst (PTC, methyltrioctylammonium chloride) to enhance it. Our choice of PTC was methyltrioctylammonium chloride which combines several features critical for practical applications – its wide commercial availability, low price and high affinity for organic solvents that prevents the loss of PTC to the aqueous phase. The latter is a main drawback of some alternative PTCs, e.g. tetrabutylammonium salts, that are water soluble and can additionally contaminate the resulting formate product. Addition of PTC resulted in a significant increase of formate yields in biphasic catalytic systems (Table 2).
| Entry | Solvent | KOH (mmol) | TON | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: 2 mL of total solvent (1/1 in the case of mixed solvent systems), 0.107 μmol catalyst 1, T = 90 °C, p = 40 bar (pH2 = pCO2 = 20 bar), t = 3 h. | ||||
| 1 | H2O/DMF | 2.5 | 5627 | 26 |
| 2 | H2O/toluene | 2.5 | 3125 | 13 |
| 3 | H2O | 2.5 | 745 | 3.3 |
| 4 | H2O/DMF | 5.0 | 7610 | 18 |
| 5 | H2O/toluene | 5.0 | 2992 | 7.5 |
| 6 | H2O | 5.0 | 1573 | 3.7 |
| Entry | PTC (mmol) | KOH (mmol) | TON | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: 2 mL of total solvent (H2O/toluene = 1/1), 0.107 μmol catalyst 1, T = 90 °C, p = 40 bar (pH2 = pCO2 = 20 bar), PTC is methyltrioctylammonium chloride, t = 3 h. | ||||
| 1 | No | 2.5 | 3125 | 13 |
| 2 | No | 5.0 | 2992 | 7.5 |
| 3 | 0.025 | 5.0 | 20060 |
52 |
| 4 | 0.29 | 5.0 | 17732 |
46 |
| 5 | 0.052 | 14 | 16509 |
14 |
In the same screening experiment we noted that elevated base concentrations consistently resulted in lower formate yields, implying the limited stability of 1 in a highly alkaline medium (Table 2). Circumventing this, we opted to perform the direct hydrogenation of bicarbonates – a process with additional advantage of using hydrogen as the only gaseous feedstock. Similar to the H2O/KOH system, the hydrogenation of H2O/KHCO3 shows a limited efficiency in pure water or in the absence of a PTC (Table 3). However, a biphasic medium with PTC provides excellent TON values >100000 with yields significantly higher than those obtained with potassium hydroxide base (Table 3). In addition, this catalytic systems retains its composition after catalysis with negligible extent of Ru leaching into the aqueous phase as evidenced by ICP-MS analysis of the spent product mixtures (see the ESI†). While the water-toluene biphasic medium provided good catalytic performance, catalysis can readily be performed using other organic solvents that have higher affinity to water compared to toluene. For example, ethereal solvents, 1,4-dioxane and THF were found to form a biphasic mixture in the presence of a high formate salt content with THF-water medium providing a performance similar to that of toluene (see Table 3 and S16, 17 of the ESI† for details).
| Entry | Solvent | PTC (mmol) | KHCO3 (mmol) | TON | Yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 2 mL of total solvent (1/1 in the case of mixed solvent systems), 0.107 μmol catalyst 1, T = 90 °C, p = 40 bar (pH2 = 40 bar), t = 3 h, PTC = methyltrioctylammonium chloride. | |||||
| 1 | H2O | No | 10 | 3963 | 4.5 |
| 2 | H2O/toluene | No | 10 | 4263 | 4.8 |
| 3 | H2O/toluene | 0.055 | 10 | 66495 |
78 |
| 4 | Toluene | 0.055 | 5.0 | 289 | 0.6 |
| 5 | H2O/toluene | 0.055 | 5.0 | 26897 |
65 |
| 6 | H2O/toluene | 0.055 | 14 | 102593 |
86 |
| 7 | H2O/THF | 0.055 | 5.0 | 29923 |
71 |
| 8 | H2O/dioxane | 0.055 | 5.0 | 6096 | 14 |
In sharp contrast with hydroxide bases, a gradual increase of KHCO3 loading leads to the marked increase in both observed TON and final formate yield. For example, we could utilize KHCO3 loadings of up to 14 mmol mLH2O−1 and obtain final yields in the extent of 86% corresponding to a TON of 102593 (Table 3, entry 6). Remarkably, even at the temperature of 90 °C, the reaction mixture is saturated in potassium bicarbonate with the main fraction of KHCO3 remaining solid. In line with the solubility of potassium formate being significantly higher than that of bicarbonate our approach effectively transforms bicarbonate slurries to >50%wt formate solutions with no need for product concentration.
The trend of increasing formate yield as a function of initial bicarbonate loading was confirmed in a separate study depicted in Fig. 2. Upon the increase of KHCO3 loading from 2 to 14 mol per litre water, the formate yield increased from 63 to 86%. Interestingly, the catalytic reaction apparently accelerated upon the addition of bicarbonate as the registered TON values for hydrogenation also increased from 13373 to 102593. Contrary to the case of potassium bicarbonate, NaHCO3 does not impact catalysis favourably upon the loading increase. Sodium formate yields decrease and TON values calculated as the number of turnovers per that of catalyst molecules remain similar. These results might stem from the significantly higher solubility of potassium bicarbonate compared to NaHCO3 that would result in a higher ionic strength of the aqueous phase throughout catalysis. Considering that identical catalyst loadings were used for all experiments depicted in Fig. 2, our data implies that the selection of the alkali cation is crucial for producing efficient catalytic systems.
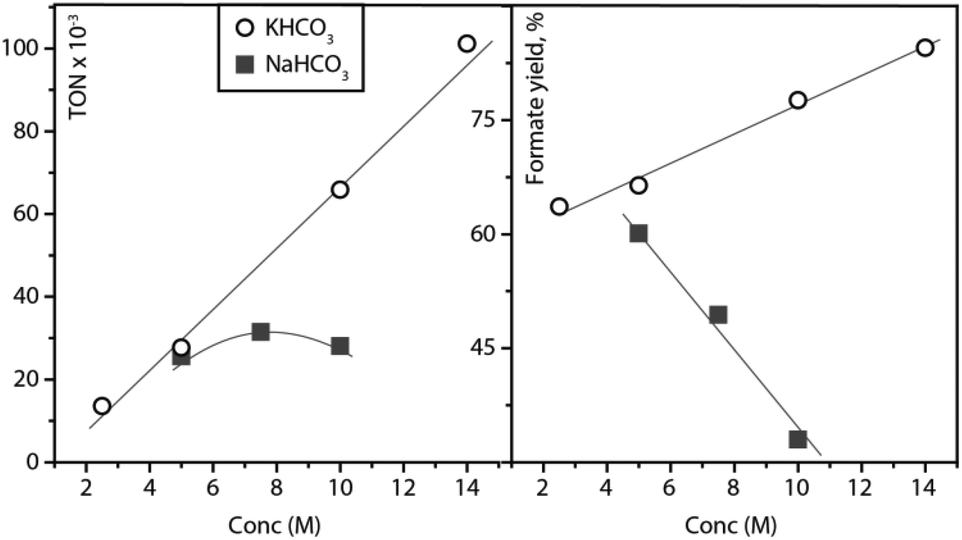 | ||
| Fig. 2 Concentration effects of KHCO3 and NaHCO3 on the formate yield and TONs. Reaction conditions: 2 mL of solvent total (H2O/toluene = 1/1), 0.107 μmol catalyst 1, 0.055 mmol methyltrioctylammonium chloride, T = 90 °C, p = 40 bar (pH2 = 40 bar), t = 4.5 h, concentrations are considered as M/H2O. | ||
Finally, we examined the impact of reaction conditions on the performance of the biphasic catalytic system. Varying the reaction temperature and pressure over a series of experiments (Fig. 3 and ESI†) we confirmed that biphasic hydrogenation exhibits behavior similar to that of single solvent systems. Namely, the increase of the reaction temperature from 65 to 120 °C leads to a marked drop in formate yield, while the increase of H2 pressure from 5 to 60 bar H2 results in increased formate yield. This is consistent with the expected thermodynamic behavior of the system as well as recent reports detailing this reaction.48
 | ||
| Fig. 3 Effect of temperature and pressure on the formate yield and TONs in the hydrogenation of KHCO3. Reaction conditions: 2 mL of solvent, 0.107 μmol catalyst 1, 55 mM methyltrioctylammonium chloride, t = 16 h, KHCO3 = 5 mmol, T and p are varied. | ||
Examining the kinetics of bicarbonate hydrogenation we found that 1 is capable of developing high turnover frequencies (TOFs) with no apparent inhibition often observed in organic media.49 Tracking hydrogen consumption in the course of hydrogenation we estimated the initial TOF to be in the extent of 73000 h−1 (Fig. 4) with kinetic traces following a regular monoexponential trendline. These experiments could also confirm the ease of scaling this reaction that was performed at the 50 mmol scale as compared to a five-fold lower scale used in screening.
 | ||
| Fig. 4 Conversion versus time in the hydrogenation of KHCO3. Reaction conditions: 10 mL of solvent (H2O/toluene = 1/1), 0.535 μmol catalyst 1, methyltrioctylammonium chloride, T = 90 °C, p = 50 bar (pH2 = 50 bar), KHCO3 = 50 mmol (10 M in H2O). Inside the graph are the stirred reaction mixtures before (left) and after (right) reaction, showing the net conversion of the bicarbonate suspension into a clear solution at RT. | ||
Finally, we probed the performance limits of the biphasic catalytic system based on 1. In order to maximize TON per batch we varied the catalyst concentration and found no significant productivity drop with the catalyst concentration as low as 2 ppm with respect to the bicarbonate substrate (Table 4). Under these conditions, TON values up to 300000 can be achieved with similar formate yields in the extent of 70%, producing clear formate solutions at the end of the catalytic reaction.
| Entry | Cat. (μmol) | TON | Yield (%) |
|---|---|---|---|
| a Reaction conditions: 2 mL of solvent, 5 mmol KHCO3, T = 90 °C, pH2 = 40 bar, 0.055 mmol methyltrioctylammonium chloride, t = 18 h. | |||
| 1 | 0.006 | 474026 |
55 |
| 2 | 0.012 | 296756 |
70 |
| 3 | 0.029 | 109224 |
64 |
| 4 | 0.059 | 56996 |
67 |
| 5 | 0.12 | 27728 |
67 |
Conclusions
To conclude, in this work we developed a potent catalytic system based on ruthenium complex 1. Making use of its biphasic composition, our system is capable of converting supersaturated bicarbonate slurries to formate solutions close to their saturation point. We demonstrate the generation of ca. 50%wt aqueous formate solutions from commercial potassium bicarbonate with no substrate purification required prior to catalysis. With a maximum TON of 474 × 103 catalyst 1 appears very promising for developing intense hydrogenation processes in aqueous medium free of the limitations associated with conventional organic solvents.Author contributions
CR performed investigation and data analysis and developed the methodology; GA and EAP contributed to project management, supervision and data analysis; EAP provided resources; all authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Nitto Denko Corporation for financial support of the project.Notes and references
- M. Fasihi, O. Efimova and C. Breyer, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schütz, M. Hill and L. F. Dumée, Mater. Horiz., 2017, 4, 345–361 RSC.
- J. W. Maina, J. M. Pringle, J. M. Razal, S. Nunes, L. Vega, F. Gallucci and L. F. Dumée, ChemSusChem, 2021, 14, 1805–1820 CrossRef CAS PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- S. Chatterjee, I. Dutta, Y. Lum, Z. Lai and K.-W. Huang, Energy Environ. Sci., 2021, 14, 1194–1246 RSC.
- M.-Y. Lee, K. T. Park, W. Lee, H. Lim, Y. Kwon and S. Kang, Crit. Rev. Environ. Sci. Technol., 2020, 50, 769–815 CrossRef CAS.
- D. H. Apaydin, S. Schlager, E. Portenkirchner and N. S. Sariciftci, ChemPhysChem, 2017, 18, 3094–3116 CrossRef CAS PubMed.
- CO2 Hydrogenation Catalysis, ed. Y. Himeda, Wiley-VCH, Weinheim, 2021 Search PubMed.
- W.-H. Wang, X. Feng and M. BaoHomogeneously Catalyzed CO2 Hydrogenation to Formic Acid/Formate by Using Precious Metal Catalysts, in, CO2 Hydrogenation Catalysis, ed. Y. Himeda, Wiley-VCH, Weinheim, 2021, ch. 2 Search PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- N. Onishi, S. Xu, Y. Manaka, Y. Suna, W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, Inorg. Chem., 2015, 54, 5114–5123 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- E. Graf and W. Leitner, J. Chem. Soc., Chem. Commun., 1992, 623–624 RSC.
- H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714–2715 RSC.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara, H. Arakawa and K. Kasuga, Organometallics, 2004, 23, 1480–1483 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef CAS.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- D. Jantke, L. Pardatscher, M. Drees, M. Cokoja, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2016, 9, 2849–2854 CrossRef CAS PubMed.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS.
- F. Joó, F. Joó, L. Nádasdi, J. Elek, G. Laurenczy and L. Nádasdi, Chem. Commun., 1999, 971–972 RSC.
- J. Kothandaraman, M. Czaun, A. Goeppert, R. Haiges, J.-P. Jones, R. B. May, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2015, 8, 1442–1451 CrossRef CAS PubMed.
- S.-M. Lu, Green Chem., 2016, 18, 4553–4558 RSC.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 RSC.
- S. Oldenhof, J. I. van der Vlugt and J. N. H. Reek, Catal. Sci. Technol., 2016, 6, 404–408 RSC.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed.
- F. Bertini, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2016, 6, 2889–2893 CrossRef CAS.
- J. Choi and Y. Lee, Inorg. Chem. Front., 2020, 7, 1845–1850 RSC.
- C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem. – Eur. J., 2012, 18, 72–75 CrossRef CAS PubMed.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef CAS PubMed.
- J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS PubMed.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef CAS PubMed.
- F. Bertini, M. Glatz, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Chem. Sci., 2017, 8, 5024–5029 RSC.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- R. Kanega, M. Z. Ertem, N. Onishi, D. J. Szalda, E. Fujita and Y. Himeda, Organometallics, 2020, 39, 1519–1531 CrossRef CAS.
- S. Xu, N. Onishi, A. Tsurusaki, Y. Manaka, W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, Eur. J. Inorg. Chem., 2015, 5591–5594 CrossRef CAS.
- B. Chen, M. Dong, S. Liu, Z. Xie, J. Yang, S. Li, Y. Wang, J. Du, H. Liu and B. Han, ACS Catal., 2020, 10, 8557–8566 CrossRef CAS.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed.
- C. Wu, F. Irshad, M. Luo, Y. Zhao, X. Ma and S. Wang, ChemCatChem, 2019, 11, 1256–1263 CrossRef CAS.
- M. Scott, B. Blas Molinos, C. Westhues, G. Franciò and W. Leitner, ChemSusChem, 2017, 10, 1085–1093 CrossRef CAS PubMed.
- C. Guan, Y. Pan, E. P. L. Ang, J. Hu, C. Yao, M.-H. Huang, H. Li, Z. Lai and K.-W. Huang, Green Chem., 2018, 20, 4201–4205 RSC.
- A. Weilhard, S. P. Argent and V. Sans, Nat. Commun., 2021, 12, 231 CrossRef CAS PubMed.
- G. A. Filonenko, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02246f |
| This journal is © The Royal Society of Chemistry 2021 |