
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct and oxidative dehydrogenation of propane: from catalyst design to industrial application
James H.
Carter
*a,
Takudzwa
Bere
a,
Jack R.
Pitchers
a,
Daniel G.
Hewes
a,
Bart D.
Vandegehuchte
b,
Christopher J.
Kiely
c,
Stuart H.
Taylor
 a and
Graham J.
Hutchings
*a
a and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: carterj5@cf.ac.uk; hutch@cf.ac.uk
bTotalEnergies One Tech Belgium, Seneffe B-7181, Belgium
cDepartment of Materials Science and Engineering, Lehigh University, Bethlehem, PA 18015, USA
First published on 15th November 2021
Abstract
The direct formation of propene from propane is a well-established commercial process, which on the basis of energy consumption, is environmentally preferred to the current large-scale sources of propene from steam cracking and fluid catalytic cracking. Propene is a major platform chemical with applications in the polymer and chemical industry and its place in modern society is guaranteed over the coming decades. At present, sources of propane are mostly non-renewable, but the development of technologies to produce renewable “green” propane are gaining traction, which coupled with new catalytic processes will provide the platform to produce green propene. In this critical review, we examine the significant advances made in the development of dehydrogenation catalysts, evaluating the technological and environmental merits. Currently, non-oxidative direct dehydrogenation (DDH) is the only commercialised process, and this is reflected in the high space–time yield commonly reported over the most active catalysts which are typically Pt or Cr based. However, the formation of coke over even the most selective catalysts necessitates multi-reactor cycling to facilitate regeneration. Oxidative dehydrogenation using O2 (ODH-O2) does not suffer from coke formation, but can lead to overoxidation, limiting the yield of propene. While no commercial processes have yet been developed, a promising new class of ODH-O2 catalysts has emerged in recent years which use boron as the active component. These catalysts are amongst the most active and selective reported to date for this reaction. The use of CO2 as a soft oxidant (ODH-CO2) has also gained interest due to the environmental advantages of utilising CO2. Although this is an attractive prospect with considerable potential, the propene yields reported over these catalysts are an order of magnitude lower than the most active DDH and ODH-O2 catalysts. Despite significant advances in the past decade, current ODH-CO2 catalysts remain far from displaying the activity levels necessary to be considered for commercial application. The specific requirements of catalyst design for each sub-reaction are discussed and we identify that, regardless of the method of dehydrogenation, the balance of acid and base sites on the catalyst surface is of paramount importance. Future catalyst design in DDH and ODH-O2 should focus on improving selectivity to propene, while ODH-CO2 catalysts are limited by their low intrinsic activity with respect to CO2. The scarcity of some common catalytic elements is also discussed, with recommendations focusing on more abundant chemical elements. Future research should focus on the low temperature activation of CO2 as a priority. With further research and development of lower energy routes to propene based on the dehydrogenation of green propane, it should be possible to transform the manufacturing landscape of this key chemical intermediate.
 James H. Carter | James H. Carter obtained his PhD under the supervision of Professors Graham Hutchings and Stan Golunski, and currently works as a Post-Doctoral Research Associate at Cardiff Catalysis Institute in the Hutchings group. His research focuses on the synthesis, characterisation and evaluation of novel heterogenous catalysts for wide-ranging applications, including carbon-free energy, environmental catalysis, sustainable manufacturing and selective oxidation. His research is centred on advancing the fundamental understanding of the chemistry and materials, and involves collaboration with universities and industry nationally and internationally. |
 Takudzwa Bere | Takudzwa Bere is a PhD candidate at Cardiff Catalysis Institute under the supervision of Professor Graham Hutchins and Professor Stuart Taylor. His primary research encompasses building fundamental structure–activity relationships through catalyst design and advanced characterisation with a focus on the design, synthesis and functionalization of novel amorphous catalytic materials, synthesised using the supercritical antisolvent (SAS) precipitation method. Target reactions include non-oxidative propane dehydrogenation reaction and alcohol dehydration for alternative fuels. |
 Jack R. Pitchers | Jack Pitchers is a PhD candidate at the Cardiff Catalysis institute, based at Cardiff university, under the supervision of Dr Graham Hutchings and Dr Stuart Taylor. Jack acquired a MSci in natural sciences at Lancaster university in 2018 and subsequently acquired a MRes as part of the catalysis CDT programme at Cardiff University. His primary interests are focused on the design, synthesis, and characterisation of novel metal oxide catalysts for selective oxidation. |
 Daniel G. Hewes | Daniel Hewes is a postgraduate PhD student at Cardiff Catalysis institute under the supervision of Professor Graham Hutchings and Professor Chris Kiely. His primary research focuses on the advanced characterisation techniques relating to Electron Microscopy and X-ray Absorption Spectroscopy, and how these techniques apply to the structure–activity relationships of novel catalytic materials. |
 Bart D. Vandegehuchte | Bart Vandegehuchte (Ghent, Belgium, 1985) obtained his Master degree in Chemical Engineering at Ghent University in 2008. In 2014 at the same university, he obtained his PhD at the Laboratory of Chemical Technology with Prof. Joris W. Thybaut in microkinetic modeling and hydrocracking. In 2015, he joined Total Refining & Chemicals as a research engineer in the Catalysis Refining & Base Chemicals department. His initial focus was on bottom-of-the-barrel conversion and selective hydrogenation of acetylenics. Nowadays, his main research has shifted to oxidative coupling of methane, life cycle analysis, and metal-based nanocatalysis for conventional and emerging technologies. |
 Christopher J. Kiely | Christopher Kiely is the Harold B. Chambers Senior Professor of Materials Science and Engineering at Lehigh University, USA. He received his B.Sc and Ph.D degrees from Bristol University, UK. His research involves the development and application of analytical electron microscopy techniques for the study of nanoscale features in catalyst and other particulate materials. |
 Stuart H. Taylor | Stuart Taylor completed his PhD at the University of Liverpool in 1994, and then took up an academic position at Cardiff University School of Chemistry in 1997. Promoted to Professor in 2013, currently he is the Director of Research for the School and has more than 29 years of experience in heterogeneous catalysis research, publishing over 320 papers and patents. He has wide-ranging expertise in experimental studies of fundamental catalysis, which impacts fuels, sustainability, energy and the environment. His research receives support from a wide range of funding bodies and industry and collaborates extensively with universities and industry alike. |
 Graham J. Hutchings | Graham Hutchings is Regius Professor of Chemistry at Cardiff University. His early career was in industry with ICI and AECI Ltd where he became interested in gold catalysis. In 1984 he moved to academia and has held chairs at the Universities of Witwatersrand, Liverpool and Cardiff. He has published over 900 papers and patents. He was elected a Fellow of the Royal Society in 2009 and was awarded the Davy Medal in 2013, the ENI Award for Advanced Environmental Solutions in 2017, the RSC Faraday Lectureship and Prize in 2018, a CBE in the Queen's Birthday Honours List in 2018 and the Michel Boudart Award in 2021. |
1. Introduction
1.1. The importance of propene in modern society
Propene is a major platform chemical with a myriad of uses in the manufacturing and chemical industry. The majority of propene is used to manufacture polypropene, a recyclable thermoplastic polymer which is second only to polyethene in the scale of production. Polypropene has favourable physical properties such as high chemical resistance and good elasticity, which makes it useful in a broad range of applications, including packaging, manufacturing and in construction materials. Propene is also the precursor to many other polymer-related chemicals, including propene oxide (used to make polyurethane), acrylic acid (used to make acrylic polymers) and acrylonitrile (used to make polypropenonitrile). Additionally, large scale chemicals such as isopropanol and cumene (used to manufacture phenol and acetone) are made from propene. Annual propene production was around 130 megatonnes in 20191 and is predicted to increase to 191 megatonnes by 2030. Significantly, the expected growth in propane demand cannot be met by existing processes, which are based on steam-cracking (SC) and fluid catalytic cracking (FCC). Instead, direct or on-purpose processes are being developed to fill the so-called ‘propene gap’. The focus of this critical review is on the dehydrogenation of propane.Direct dehydrogenation can be performed either with or without an oxidant present, and O2, CO2 and N2O can all participate as oxidants to form propene from propane. The discovery of active and selective catalysts is central to the development of these on-purpose technologies and is the focus of the current review. Research into propane dehydrogenation is an ever-growing field and to date there are several review articles available on direct dehydrogenation (DDH) (Sattler,2 2013), soft oxidation with CO2 (ODH-CO2) (Atanga,3 2018) and oxidative dehydrogenation (ODH-O2) (Carrero,4 2014 and Shi,5 2018). Otroshchenko et al. recently reviewed the literature on metal oxide catalysts for each dehydrogenation reaction, including reaction-engineering and catalyst design, but omitted supported nanoparticle catalysts. To our knowledge there are no comprehensive literature reviews that compare and contrast non-oxidative and oxidative dehydrogenation reactions.
The scope of this article is to critically review the literature on DDH, ODH-CO2 and ODH-O2 to identify over-arching themes and common rules for catalyst design. We do not try to capture aspects of reaction design and engineering, which itself is a vibrant field of research. We attempt to compare the performance of catalysts in the different sub-reactions by calculating a space time yield (STY), (measured in molC3H6 kgcat−1 h−1), which is based on the initial activity of the catalysts. This enables a meaningful comparison between different research groups and reaction conditions. Additionally, we emphasise the environmental and industrial perspectives of each reaction and the prospects for industrial application.
1.2. Propene production processes – an environmental perspective
Traditionally, the industrial production of light alkenes, such as propene, has been carried out via either SC, or FCC of naphtha and diesel feedstocks. These methods typically provide a range of products including ethene, propene and gasoline, amongst others.6 As of 2016, these routes accounted for 81% of propene production in industry, with 50% coming from SC and 31% from FCC. The remaining 19% was from newer technologies, such as direct dehydrogenation.6SC, which involves the heating of fossil-fuels in the absence of oxygen to produce alkenes, requires temperatures up to 750–875 °C.7 As an established process, naphtha SC has a high carbon efficiency, producing almost no CO2 from the cracking process itself.7 Although naphtha SC has an effective carbon efficiency, the process still relies on high temperatures for alkene production, which itself produces a significant amount of CO2 from energy consumption.7,8 These factors taken together result in steam cracking being responsible for the global emission of more than 300 million tonnes of CO2 per year, as well as naphtha SC producing ozone-depleting CFCs.7 FCC provides a way of producing light alkenes at lower temperatures compared to SC, by employing a catalyst into the process. However, despite its modestly lower operating temperatures, the FCC process produces many by-products. Many reduced S and N species are formed, as well as significant CO and HCN, which start forming at as low as 450 °C. Partial burn FCC reactors, (i.e., reactors which form CO, rather than CO2) will incorporate a CO burner downstream.9 Feedstocks involved in FCC processes typically include high molecular weight hydrocarbons, breaking them down into more valuable light alkenes; however, it often struggles to process lighter feedstocks such as naphtha.9 Both processes are designed to produce a wide array of products, as well as often generating by-products that require additional energy to process. In addition, while propene demand is increasing, processing plants are tending towards higher ethene production due to shale gas production, leading towards a global shortage of propene.6,10 Overall, cracking of naptha and other petrochemical feedstocks to produce propene is unsustainable and leads to significant CO2 emissions. Alternative indirect routes to propene, from syn gas and via methanol, has also gained interest in recent years.7 The feedstock for this process can be biomass, shale gas or coal. Using coal as a feedstock has gained widespread use in China but poses a huge environmental burden.
The environmental and economic aspects of different existing propene production technologies are important considerations when reviewing the feasibility of newer technologies based on dehydrogenation. Green chemistry principles, such as atom economy, the use of toxic chemicals and the renewability of the feedstock, are especially relevant. This is summarised in Table 1, which compares steam cracking, MTO (from coal and natural gas) and dehydrogenation. Atom economy is a measurement of the percentage of atomic mass in the starting reagents that ends up in the final product. It serves as a useful indicator of how chemically efficient a process could be. It is important to remember that in practice, the real atom economy is determined by the selectivity of the catalyst, and the prevalence of side-reactions. Although it is not possible to precisely calculate the atom economy of SC or MTO due to the complexity of the process and uncertainty in the composition of the feedstock, it is apparent that both of these technologies are inferior to dehydrogenation. In the case of SC a wide range of chemicals are made, and while these are ultimately utilised in the production of other chemicals, the % of propene formed from such a process is low. Equally, in the MTO process from coal or natural gas, the atom economy is limited by the generation of H2O as a by-product. In contrast, in the case of dehydrogenation only H2 is generated as a by-product, meaning an atom economy of 95% is possible. With respect to the toxicity of the chemicals, SC forms various carcinogenic compounds, and the toxicity of methanol is well-established. In contrast, propane is non-hazardous. At present each of the feedstocks used in the main methods of propene production tend to be non-renewable. In the case of SC and coal-based MTO, this is unavoidable due to the nature of the feedstock. However, natural gas and propane can be produced from biomass. In these cases, the feedstock is renewable and so these processes are potentially much more sustainable than when using fossil fuels. Rodríguez-Vallejo et al. carried out life cycle analyses of naphtha cracking, propane dehydrogenation and MTO (using coal or natural gas as feedstocks).11 In this analysis, the global warming potential (GWP), resource scarcity, impact on human health and the impact on the ecosystem was considered and quantified as a cost per kg of C3H6 produced. These data are shown in Table 1. In terms of the global warming potential of each process, the MTO based reactions are clearly worse than the naphtha cracking or direct dehydrogenation reactions, but the CO2 emissions of the coal-based feedstock is almost an order of magnitude higher than naphtha cracking. A similar trend was observed in the ecosystem category. This was measured in terms of local species lost integrated over time and underlines the gross negative impact of utilising coal as a feedstock in this chemistry. Resource scarcity was calculated based on the monetary burden of future mineral and fossil fuel extraction and shows that natural gas has around double the cost of that associated with propane, coal and naphtha. Finally, the cost to human health was estimated in terms of disability-adjusted life years, and naphtha cracking and propane dehydrogenation came out favourably compared to the MTO processes. Based on these metrics, it is clear that MTO from coal or natural gas has a larger environmental burden than SC or dehydrogenation. Opportunities for improving the sustainability of the dehydrogenation process were recently highlighted by Agarwal et al. who investigated various intensification and integration strategies. It was shown that such strategies, which involved additional waste heat recovery and off-gas recycling, up to 70% reductions in CO2 emissions were possible.12
| Feedstock | Processes | Atom economy | Toxic reactants | Renewability of feedstock | GWP (kg CO2 per kg C3H6)a | Resource scarcity (USD per kg C3H6)a | Human health (DALY per kg C3H6)a | Ecosystem (species year per kg C3H6)a |
|---|---|---|---|---|---|---|---|---|
| SC = steam cracking, GWP = global warming potential, DALY = disability-adjusted life years, MTO = methanol to olefins.a Values from ref. 11. | ||||||||
| Naphtha | SC | Low | Crude oil/naptha streams | Non-renewable | 1.8 | 0.61 | 0.25 | 0.05 |
| Coal | Syn gas, methanol synthesis, MTO | Low | Methanol | Non-renewable | 16.5 | 0.60 | 4.0 | 1.1 |
| Natural gas | Syn gas, methanol synthesis, MTO | Low | Methanol | Mostly non-renewable | 4.5 | 1.10 | 0.8 | 0.2 |
| Propane | DH | High | None | Mostly non-renewable | 2.5 | 0.55 | 0.4 | 0.1 |
The increased interest in direct propane dehydrogenation has been enhanced by the fall in propane price, which is the direct result of the discovery of vast shale gas reserves in North America. Although propane is a minor component in shale gas, it is facile to separate and therefore represents a valuable, but non-renewable source of propane. Increasing public pressure to move away from such non-renewable feedstocks continues to gain momentum and the propane production industry has responded accordingly. The production of renewable ‘green’ propane (also called bio-propane and bioLPG) is an emerging sector which is already being operated on a large scale. TotalEnergies operates a bio-refinery in La Mede, which has a design capacity for bio-propane of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes per year.13 Additionally, Neste recently opened a renewable propane production plant, which will produce 40000 tonnes per year.14 The propane generated is then sold on to be used in direct propane dehydrogenation.15 While this is an important start, it represents less than 1% of annual propane capacity. Bio-propane can be produced from the processing of biomass, notably as a side-product in the production of bio-diesel16 as well as glycerol17 and other substrates.18 With increasing pressure to source chemical feedstocks sustainably, we can expect that new interest will focus on renewable and green routes to the key platform chemicals currently required by modern society, and that green propane will play an important role in this.
000 tonnes per year.13 Additionally, Neste recently opened a renewable propane production plant, which will produce 40000 tonnes per year.14 The propane generated is then sold on to be used in direct propane dehydrogenation.15 While this is an important start, it represents less than 1% of annual propane capacity. Bio-propane can be produced from the processing of biomass, notably as a side-product in the production of bio-diesel16 as well as glycerol17 and other substrates.18 With increasing pressure to source chemical feedstocks sustainably, we can expect that new interest will focus on renewable and green routes to the key platform chemicals currently required by modern society, and that green propane will play an important role in this.
Overall, direct propane dehydrogenation satisfies many principles of green chemistry in comparison to SC and MTO benchmarks: it is highly atom-economic, does not use toxic reactants (or generate toxic by-products) and can in principle, utilise renewable feedstocks. However, in practice dehydrogenation will only be desirable as a means of producing propene if the reaction can operate efficiently, with minimal side-products and maximum yield. These factors are governed by the choice of catalyst, the main focus of the current review. The development of an active, selective and stable catalyst is essential for dehydrogenation to be considered as a viable process in the near- and long-term future.
2. Thermodynamics and environmental aspects of different dehydrogenation processes
Each dehydrogenation reaction has different thermodynamic, environmental and economic aspects, which are important to recognise when designing catalysts and sustainable processes. In section 1.2, the environmental implications of cracking and MTO were discussed and compared with DDH. The majority of analyses focus on well-established processes, where more information and data are available, but no such analyses were found for oxidative dehydrogenation. As a result, the environmental impact of these novel reactions can only be speculated upon. The following section focuses on the thermodynamic and environmental aspects of each sub-reaction, drawing on available literature whenever possible, and when not possible suggesting the most important aspects that could affect the sustainability of each process.2.1 DDH
DDH is an endothermic reaction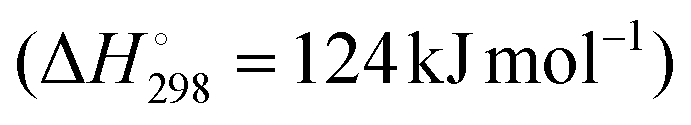 , which is equilibrium-limited at higher partial pressures of propane and lower reaction temperatures. As a result, typical reaction temperatures for industrial DDH processes are 550–750 °C. Another key challenge in catalyst design for DDH is the fact that coke is also a favoured product under the conditions required for high propene yield and its formation is assumed to be initiated by deep dehydrogenation. Fig. 1 illustrates the typical reaction pathways observed in DDH. Catalytic and thermal (gas-phase radical) cracking reactions lead to the formation of methane and ethene. Ethene can then hydrogenate to ethane. The hydrogenolysis of propane is also possible, which forms methane and ethane.
, which is equilibrium-limited at higher partial pressures of propane and lower reaction temperatures. As a result, typical reaction temperatures for industrial DDH processes are 550–750 °C. Another key challenge in catalyst design for DDH is the fact that coke is also a favoured product under the conditions required for high propene yield and its formation is assumed to be initiated by deep dehydrogenation. Fig. 1 illustrates the typical reaction pathways observed in DDH. Catalytic and thermal (gas-phase radical) cracking reactions lead to the formation of methane and ethene. Ethene can then hydrogenate to ethane. The hydrogenolysis of propane is also possible, which forms methane and ethane.
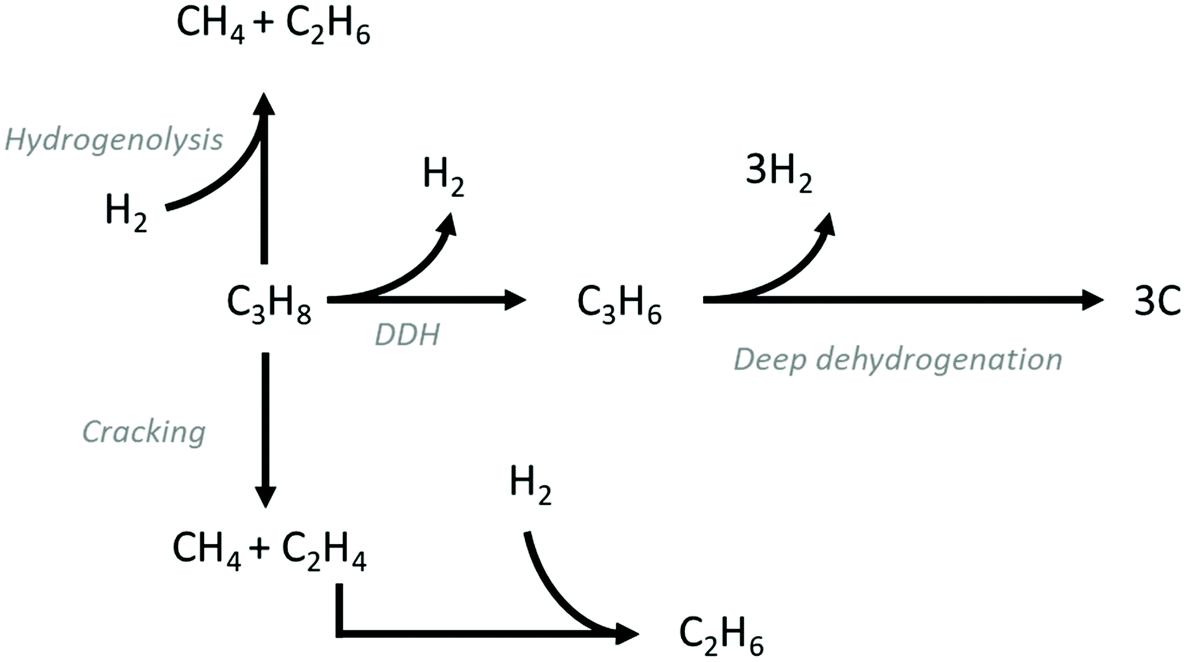 | ||
| Fig. 1 Reaction pathways commonly observed during the DDH of propane. In addition to DDH, unfavourable deep dehydrogenation to coke, hydrogenolysis and cracking are also observed. | ||
As described above, the atom economy for DDH is high (95%) and the only by-product (in theory) is molecular hydrogen, which is a valuable resource with many uses and applications, not least as a carbon-free fuel. In practice however, frequent regeneration steps are required to remove coke, which involves combustion to form carbon dioxide. This means that the environmental impact of this process will depend to some extent on the prevalence of coke formation on the catalyst surface. As a feedstock, C3H8 is non-toxic, unlike methanol in the MTO process. The high flammability of C3H8 may present a safety risk in a chemical plant, but such a risk is present in all forms of C3H6 production.
2.2 ODH-O2
ODH-O2 is exothermic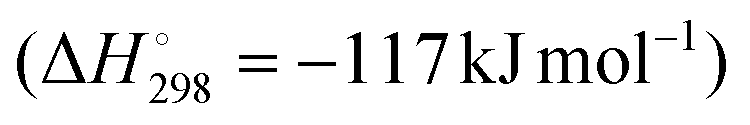 and conversion is not limited by thermodynamic equilibrium. Additionally, the formation of coke is suppressed substantially, with overoxidation constituting the main competing pathway. Fig. 2 shows the typical reaction pathways that occur over ODH-O2 catalysts. Overoxidation is the main drawback, with CO and particularly CO2 reported as by-products. Cracking products are also reported, but generally as minor products. In common with ODH-CO2, water is formed via oxidative dehydrogenation. CO2 can also be formed via direct combustion of C3H8.19
and conversion is not limited by thermodynamic equilibrium. Additionally, the formation of coke is suppressed substantially, with overoxidation constituting the main competing pathway. Fig. 2 shows the typical reaction pathways that occur over ODH-O2 catalysts. Overoxidation is the main drawback, with CO and particularly CO2 reported as by-products. Cracking products are also reported, but generally as minor products. In common with ODH-CO2, water is formed via oxidative dehydrogenation. CO2 can also be formed via direct combustion of C3H8.19
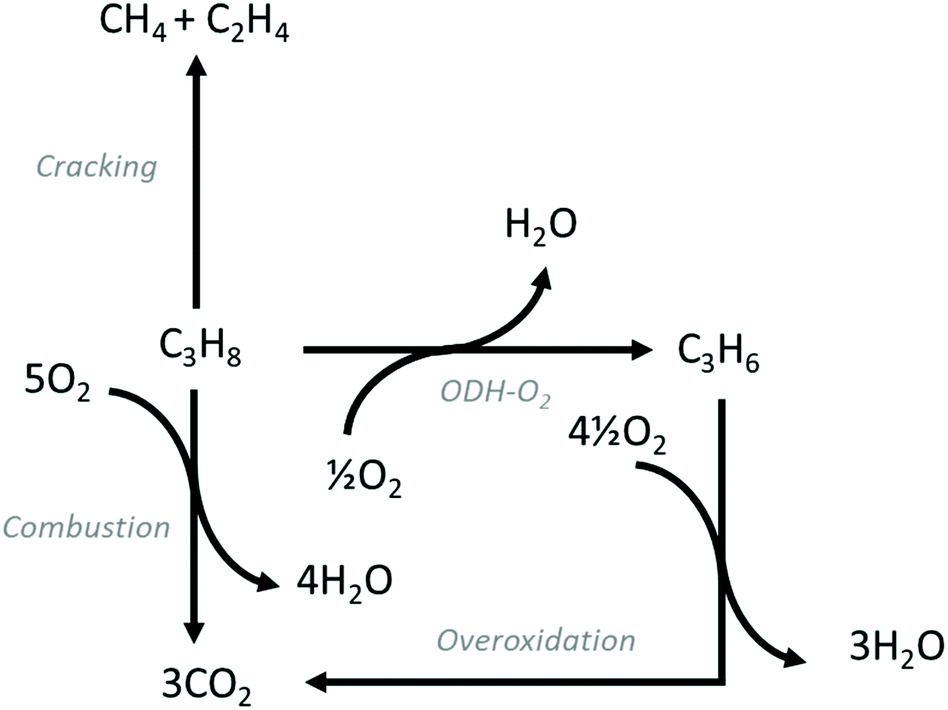 | ||
| Fig. 2 Reaction pathways commonly observed in ODH-O2 reactions. In addition to ODH-O2, overoxidation, combustion and cracking are often observed. | ||
The addition of O2 enables lower operating temperatures to be utilised (typically 450–550 °C) and is exothermic, meaning the energy costs of sustaining the reaction temperature are less than those in DDH and ODH-CO2. However, the atom economy of ODH-O2 is 70% and H2O is generated as a by-product rather than H2. While CO2 may be generated as a by-product, this depends on the catalyst, and the overall CO2 generated may be comparable to a DDH process. There will be increased energy consumption associated with supplying the oxidant in sufficient purity and volume. If air is used, downstream separation of N2 will be necessary, which would increase process energy consumption. The reactants and products are non-toxic, but the use of O2 and C3H8 introduce an elevated safety risk. Although an in-depth environmental analysis of an ODH-O2 process for C3H8 has not yet been carried out, it may be concluded that ODH-O2 is comparable, if not more environmentally-friendly than DDH due to the lower expected costs associated with maintaining the reaction temperature. However, the performance and composition of the catalyst will be a crucial factor in this comparison. The economic aspects of ODH-O2 have not been analysed for C3H6 production. However, the oxidative dehydrogenation of C2H6 has been investigated extensively, and such an analysis is available. Gaffney and Mason compared ODH-O2 of C2H6 with thermal cracking and showed that ODH-O2 has the potential to significantly reduce the plant capital costs and the overall cost of production.20 Given the similarities in the cracking and dehydrogenation of ethane and propane, it is reasonable to conclude that the economic factors associated with the ODH-O2 of C3H8 are also favourable compared with steam cracking.
2.3 ODH-CO2
ODH-CO2 is even more endothermic than DDH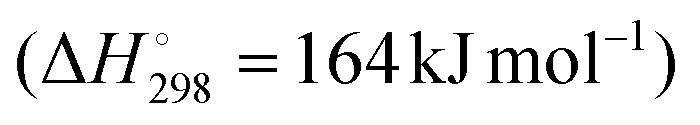 , due to the contribution of the mildly endothermic reverse water–gas shift (RWGS) reaction (Fig. 3). While it seems as though the addition of CO2 would be detrimental on this basis, the removal of hydrogen via the RWGS reaction favourably shifts the thermodynamic equilibrium to enable higher propene yields. Hence, ODH-CO2 can be carried out at lower reaction temperatures than DDH (450–600 °C). Although the reaction can proceed via a one-step oxidative dehydrogenation pathway or a two-step DDH + RWGS reaction, the net reaction is the same. A key competing reaction pathway in ODH-CO2 is dry reforming of propane (DRP) (Fig. 3), which can occur under the same conditions as ODH-CO2.21 In addition to DRP, CO2 can react with coke to generate CO via the reverse Boudouard reaction, which is reported to proceed under typical reaction conditions for ODH-CO2 (500–600 °C). Zangeneh et al.22 carried out a full thermodynamic analysis of the ODH-CO2 reaction, considering coke-forming and dry reforming side-reactions. The inclusion of these reactions in the thermodynamic model strongly impacted the equilibrium product of propene, increasing the equilibrium conversion of propane at the expense of propene selectivity. Dry reforming of propane was favoured at higher temperatures, while coke-formation was favoured at lower temperatures and at lower CO2:C3H8 ratios. It was also pointed out that the reported experimental data showed conversions well below equilibrium values, suggesting rapid catalyst deactivation. This analysis highlighted the need for kinetic control of dry reforming and coke forming reactions in order to maximise propene yield. Instead of hydrogen, water is formed either as the primary oxidative dehydrogenation product or as a result of DDH followed by the RWGS reaction.
, due to the contribution of the mildly endothermic reverse water–gas shift (RWGS) reaction (Fig. 3). While it seems as though the addition of CO2 would be detrimental on this basis, the removal of hydrogen via the RWGS reaction favourably shifts the thermodynamic equilibrium to enable higher propene yields. Hence, ODH-CO2 can be carried out at lower reaction temperatures than DDH (450–600 °C). Although the reaction can proceed via a one-step oxidative dehydrogenation pathway or a two-step DDH + RWGS reaction, the net reaction is the same. A key competing reaction pathway in ODH-CO2 is dry reforming of propane (DRP) (Fig. 3), which can occur under the same conditions as ODH-CO2.21 In addition to DRP, CO2 can react with coke to generate CO via the reverse Boudouard reaction, which is reported to proceed under typical reaction conditions for ODH-CO2 (500–600 °C). Zangeneh et al.22 carried out a full thermodynamic analysis of the ODH-CO2 reaction, considering coke-forming and dry reforming side-reactions. The inclusion of these reactions in the thermodynamic model strongly impacted the equilibrium product of propene, increasing the equilibrium conversion of propane at the expense of propene selectivity. Dry reforming of propane was favoured at higher temperatures, while coke-formation was favoured at lower temperatures and at lower CO2:C3H8 ratios. It was also pointed out that the reported experimental data showed conversions well below equilibrium values, suggesting rapid catalyst deactivation. This analysis highlighted the need for kinetic control of dry reforming and coke forming reactions in order to maximise propene yield. Instead of hydrogen, water is formed either as the primary oxidative dehydrogenation product or as a result of DDH followed by the RWGS reaction.
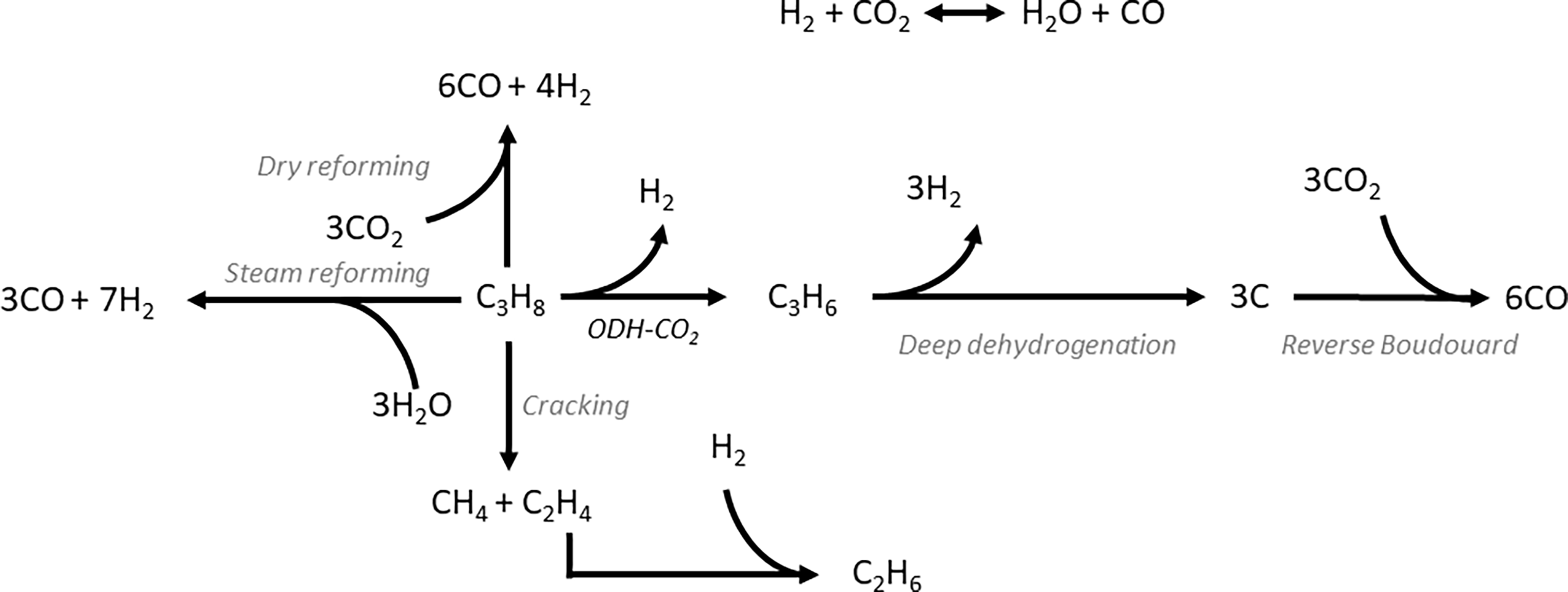 | ||
| Fig. 3 Reaction pathways commonly observed in ODH-CO2 reaction. In addition to ODH-CO2, deep dehydrogenation, cracking and in some catalysts dry reforming are observed. The reverse Boudouard reaction is also frequently reported to occur under typical reaction conditions. | ||
From an environmental perspective, ODH-CO2 has the greatest potential in the context of a circular economy. Although the formation of water is less desirable than hydrogen, ODH-CO2 has the benefit of consuming CO2 in the process, and this is often cited as the motivation for investigating this route. Chemical CO2 utilisation is an attractive prospect in the context of combatting climate change: if CO2 can be consumed in large scale processes, the overall carbon footprint of certain products/technologies could be significantly reduced. However, it is important to consider how CO, which is formed from ODH-CO2, could be used in a sustainable way to justify the apparent greenness of ODH-CO2. If the CO is used in the water–gas shift (WGS) reaction for hydrogen production, then CO2 is immediately formed as a result, and the process becomes carbon neutral. Equally, if CO is combined with H2 to be used in various syn-gas processes, e.g. methanol or Fischer–Tropsch synthesis, then it is likely that the carbon will be burned as a fuel and ultimately returned to CO2. While this may not be considered to be as desirable as sequestering CO2 from the atmosphere, the utilisation of CO2 in this technology would form part of a ‘renewable carbon’ cycle, and avoids additional CO2 emissions. This concept of carbon avoidance forms an important part of the short and medium term strategies to curb CO2 emissions, and carbon capture and storage technologies are a fundamental aspect of this.23 The source of CO2 is also an important consideration. CO2 may be sequestered from the environment at additional process costs, or may be recovered from other CO2 forming processes. In comparison to other dehydrogenation reactions, ODH-CO2 has the lowest atom economy (48%), but compared to cracking processes, where C3H6 is a minor product, this is respectable. The overall efficiency of the reaction will also depend on the catalyst, and if the above undesirable pathways can be sufficiently minimised.
Overall, each dehydrogenation reaction has unique operating conditions and challenges, which are dictated by their thermodynamics. In the case of DDH, the formation of coke remains the biggest challenge, as it is thermodynamically favoured under conditions that enable high propane conversion. In ODH-CO2, dry reforming is also a competing pathway under typical reaction conditions and in the case of ODH-O2, overoxidation is prevalent due to CO2 being the thermodynamically favoured product. The thermodynamic tendency to form undesirable by-products means that such reactions must be carefully kinetically controlled. Careful catalyst design can inhibit and even switch-off some of these side-reactions, as will be discussed in the subsequent sections.
3. Commercial processes
The commercial dehydrogenation of propane is exclusively carried out in the absence of an oxidant. There are several technologies that currently operate, using various catalysts based on Pt or Cr, as shown in Table 2.| Technology Name | Oleflex | Dow FCdh | Uhde STAR Process | CATOFIN | K-PRO |
|---|---|---|---|---|---|
| Licensor/developer | UOP LLC Honeywell | Dow | Krupp-Uhde | ABB Lummus | KBR, inc. |
| Reactor type | 4 moving-bed reactors + 1 regeneration unit | Fluidised circulating reactor and regenerator | Adiabatic | Fixed bed parallel reactors | Orthoflow FCC continuous + continuous catalyst regeneration |
| Catalyst | Alkaline promoted Pt–Sn/Al2O3 | Pt–Ga/Al2O3 | Pt–Sn/ZnAl2O3/CaAl2O3 | (Na/K) promoted CrOx/Al2O3 | Proprietary – precious metal and Cr free |
| Catalyst Life | 1–3 years | Unknown | Unknown | 2–3 years | Unknown |
| Temperature/°C | 650 | Unknown | 550–590 | 560–650 | Unknown |
| Pressure/bar | 2–3 | 1.3–1.7 | 5–6 | 0.5 | 1.5 |
| Cycle Time | 5–10 days | <2 min | 7 h (+1 h) regeneration | 15 min | <1 min |
| Conversion/% | 30–40 | 43–53 | ∼40 | 48–53 | 45 |
| Selectivity/% | 84 (at 40% conversion) | 92–96 | ∼89 | 88 | 87–90 |
The Oleflex process from UOP employs alkali metal-promoted Pt–Sn/Al2O3 catalysts in a reaction system composed of four moving bed reactors. In this process, the catalyst is continuously regenerated in a separate regeneration circuit. The reactors are connected in series with gas flow pre-heaters operating at pressures between 1 and 3 bar in the region of 650 °C. The typical catalyst in the Oleflex process consists of spherical pellets of γ-Al2O3 (surface area ca. 100 m2 g−1) containing Pt (<1 wt%) promoted with Sn and alkali metals.
The Dow FCDh technology utilises a fluidised reactor system coupled with a fluidised regeneration reactor, which enables continuous operation/regeneration.24 The catalyst is comprised of a commercially available Al2O3 support that is impregnated with Ga and Pt. The catalyst can be fully recoverable after the regeneration step, which involves high temperature oxidative treatment to remove retained carbon species i.e. coke. Compared with the Oleflex process, which requires a complicated regeneration protocol involving Cl2, O2 and then H2, the Dow FCDh technology enables a much simpler regeneration cycle.
The Steam Active Reforming (STAR) process from Phillips Petroleum, developed by Uhde employs a Sn promoted Pt catalyst, (0.2 to 0.6 wt%), dispersed on a zinc–aluminate support, with a calcium/magnesium aluminate binder. The operating pressures and temperatures are between 6 and 9 bar and 550 to 590 °C, respectively.2 Also, due to its non-acidic nature, the support does not promote undesired side reactions, such as cracking, isomerization or coke formation and contains Sn as a promoter to reduce coke formation and increase selectivity. Catalyst development for the STAR process continues: BASF and Thyssenkrupp announced a joint development agreement in 2020, which aims to reduce CO2 emissions, reduce feedstock consumption and the operating costs of the process. This underlines the opportunities in catalyst design in improving even already-commercialised processes.
Chromium oxide supported on aluminium oxide (CrOx/Al2O3) is commonly employed industrially in the CATOFIN process and is composed of 18–20 wt% chromium oxide supported on 1–2 wt% alkaline metal (Na or K) promoted aluminium oxide.2,26 The alkali promoter influences both the activity and selectivity of the catalyst by reducing the surface acidity of alumina, thus suppressing undesired side reactions. In the context of the commercialised CATOFIN process, improvements in the catalyst design and process conditions have been made, however challenges still exist relating to their activity, selectivity and stability. To boost the yield of propene and reduce emissions, the addition of a heat-generating material (HGM) has been employed, a notable example of which was developed by Clariant.27 The exact composition of the HGM is likely to include copper oxide and additional promotors such as MnO2, supported on α-Al2O3.28 This is loaded with the catalyst in the reactor, and is reduced during the reaction, which generates heat and therefore offsets the heat consumption that occurs during the DDH reaction. Since its rollout in 2015 in Ningbo Haiyue New Material Co., who are based in Ningbo City, China, the same HGM technology has been implemented in additional CATOFIN processes and Ineos recently awarded Clariant a long-term contract to supply the HGM.29
K-PRO was developed by KBR and is the latest process to be commercialised; the first contract was awarded in January 2020, around 1 year after the company introduced the technology and since then a licence has been agreed with JS Energy Ltd.30 The significance of this process is that the catalyst does not contain precious metals or chromium, although the active component(s) remain proprietary information. The reactor is based on KBR's FCC orthoflow reactor, which is an up-flowing vertical reactor riser, conceptually similar to the Dow FCDh process in that the reactor is a fluidised bed, but the regeneration section is different.25
The commercial interest in further developing dehydrogenation technologies is evident from a simple examination of the patent literature. Fig. 4a shows the number of patents filed annually since 1980 that disclose inventions of novel catalysts for propane dehydrogenation. There is a marked increase over time, which has accelerated in the last decade. This growing interest and development are driven by the demand for propene itself, but also by the opportunity to further improve dehydrogenation catalysts. It is also evident from the sub-reaction breakdown that the majority (86%) of filed patents are for DDH catalysts, followed by ODH-O2 (12%) and finally ODH-CO2 (2.4%). Fig. 4b shows the breakdown of these patents by the 14 most prolific companies. China Petroleum & Chemical Corporation (SINOPEC) have published more patents than the next 13 companies combined, underlining the interest in direct propane dehydrogenation in the East Asia. It is also clear from this data that the interest in propane dehydrogenation is global, with companies from the USA, Europe and Asia all filing multiple patents.
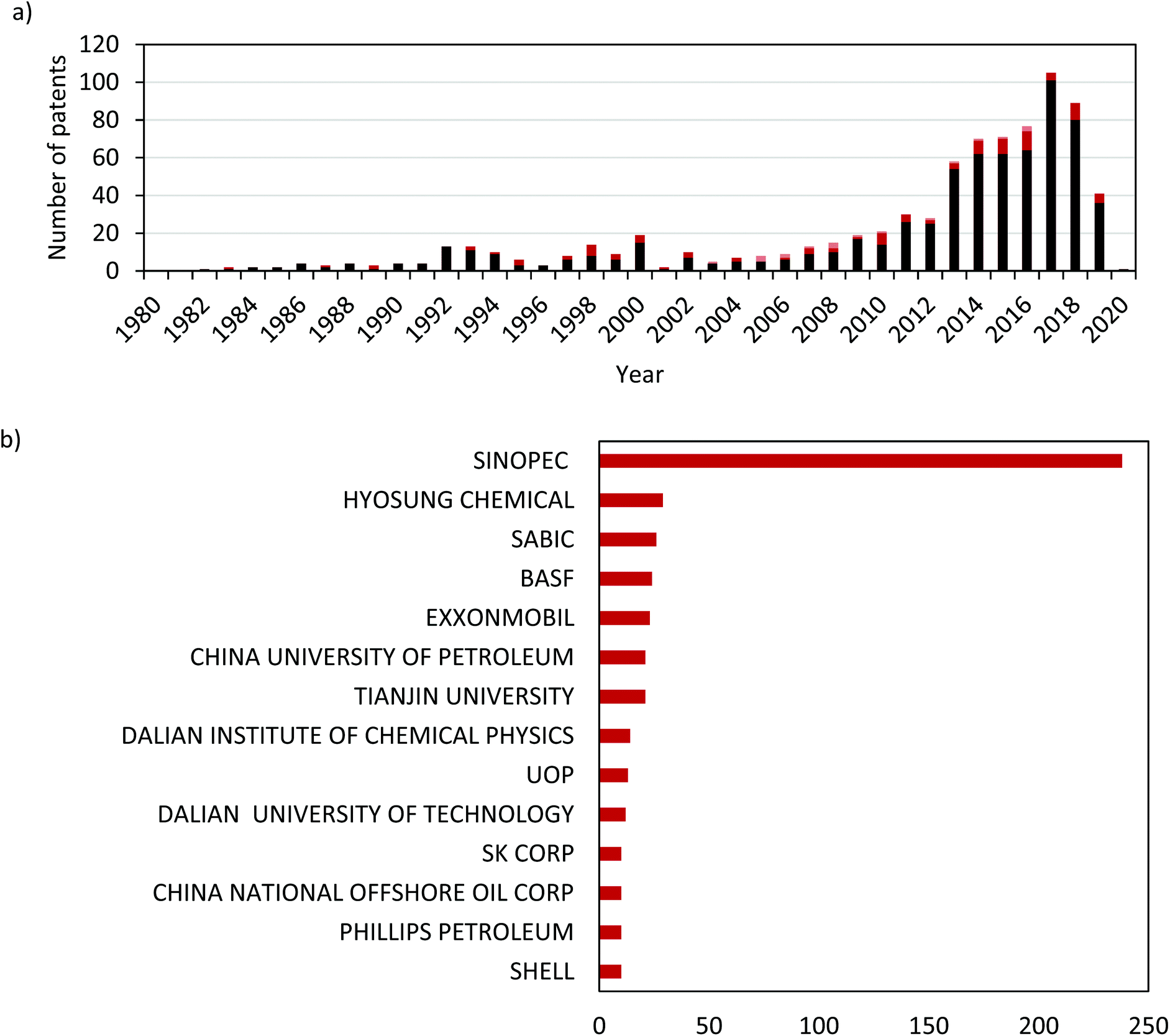 | ||
| Fig. 4 (a) The number of patents published per year from 1980–2020 including the breakdown based on sub-reaction (DDH, dark red; ODH-O2, medium red and ODH-CO2, light red) and (b) the breakdown of patents filed per company for the top 14 companies active in this area. | ||
4. Direct dehydrogenation
4.1. Pt-Based DDH catalysts
Platinum is the primary component in many dehydrogenation catalysts due to its ability to activate C–H bonds, coupled with its low activity for the breaking of C–C bonds. As shown above in Table 1, Pt is present in three commercial processes. In this section, the most significant publications in the field of Pt-catalysed DDH are discussed.The superiority of Pt as a DDH catalyst over other transition metals was recently illustrated by Araujo-Lopez et al. who used DFT calculations to measure the transition state energy for non-oxidative (and oxidative) C–H bond activation.31 It was shown that Pt has the lowest value out of a range of transition metals, consistent with Pt typically outperforming other such metals experimentally. Interestingly, while Ag and Au did not facilitate non-oxidative C–H bond activation, in the presence of hydroxyl groups the energy barrier was much lower. These calculations help to explain the success of Pt catalysts for DDH and also highlight that other elements should be considered in ODH reactions.
The reaction mechanism for DDH has been the subject of much research. In describing catalytic dehydrogenation reactions, the most commonly quoted mechanism is the Horiuti–Polanyi mechanism, proposed in 1934.32 The mechanism proceeds as follows:
i. Dissociative adsorption of the alkane (cleaving the first C–H bond).
ii. C–H cleavage of a second hydrogen from C3H7.
iii. Formation of a hydrogen molecule.
iv. Desorption of both hydrogen and the corresponding alkene.
The dissociative adsorption of propane and the C–H cleavage steps have been suggested as being rate-limiting,33 although this depends on the catalyst. Due to the reaction conditions, side reactions frequently compete with the dehydrogenation reaction, reducing catalyst activity and selectivity. At high reaction temperatures, side reactions such as hydrogenolysis, cracking, aromatization and isomerization are thermodynamically and kinetically favoured.34–36
DDH is possible over almost all Pt sites, but the intrinsic selectivity and activity depends on Pt particle size: atomic steps and kink sites i.e. coordinatively-unsaturated species are responsible for C–H and C–C bond cleavage, which can take place here with near zero activation energy, leading to coke formation and thus catalyst deactivation.37–39 While turnover frequency (TOF) increases as the Pt particle size decreases, selectivity is high in very small Pt species (<1 nm) and larger Pt nanoparticles (>3 nm).40 A similar trend was also observed for stability, which is likely related to less selective Pt NPs forming coke more readily and blocking active sites.
One of the most fruitful areas of development in Pt-catalysed DDH is in the use of promotors. Specifically, Sn and Ga have been very successful, and this is reflected in their application in commercial processes, as discussed in section 3. The origin of the promotional effect of Sn has been discussed in detail in previous review articles.2,41 Practically, the addition of Sn results in higher propene selectivity, lower support acidity, and increased diffusion of coke away from the active Pt site. Both geometric and electronic effects have been cited to explain these observations. Regarding geometric effects, Sn can modify the Pt particle size, which as previously discussed, strongly influences the reactivity of propane.42 Specifically, Sn (oxide) can partially obscure the surface Pt species, which in turns creates smaller Pt ensembles. Regarding electronic effects, evidence for electron transfer from Sn to Pt has also been reported by DFT43 and XPS analyses.44 In an early report of Sn promoting Pt catalysts, Bariås et al. demonstrated that the effect is support sensitive, in comparing Pt/Al2O3 and Pt/SiO2.45 The addition of Sn to Pt/Al2O3 was wholly beneficial, improving Pt dispersion and selectivity, while maintaining specific activity. Over Pt/SiO2, however, the Sn was more readily reduced and although an improvement in Pt dispersion and stability was noted, it came at the expense of specific activity. In fact, the oxidation state of Sn in the active catalyst is still debated and a range of different species may co-exist from Sn4+ through to Sn0 before and during the DDH reaction.41
Pt promotion using Ga has also yielded highly active catalysts, and the role of Ga has been compared to that of Sn, i.e. due to both geometric and electronic effects. Wang et al. prepared a highly active Pt3–Ga/Al2O3 catalyst and showed it could be improved by the addition of CeO2: Pt3–Ga/CeO2–Al2O3 yielded an initial STY of 87 molC3H6 kgcat−1 h−1 (compared to 83 molC3H6 kgcat−1 h−1 without CeO2) (Table 3, entries 12 and 15).
| Entry | Catalyst | T/°C | Feed composition | WHSV /h−1 | X C3H8/% | S C3H6/% | STY /molC3H6 kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| T = reaction temperature; feed composition = molar ratio of C3H8/H2/carrier in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. Hd = Hydrothermally synthesized. | ||||||||
| 1 | Pt/Mg(ln)(Al)Ox | 550 | C3H8 = 100 | 1.6 | 24.2 | 98.2 | 7.8 | 54 |
| 2 | Pt–Sn/B–ZrO2 | 550 | C3H8/H2/N2 = 1:1:8 |
1.8 | 36 | 97 | 13 | 69 |
| 3 | Pt/Silicalite-1 | 550 | C3H8/N2 = 1:1 |
2.8 | 35 | 95 | 20 | 59 |
| 4 | Pt3–Mn/SiO2 | 550 | C3H8/N2 = 1.25:98.75 |
29.5 | 6.8 | 95 | 41 | 65 |
| 5 | Pt–Sn/Mg(3Zn)AlO | 550 | C3H8/N2 = 1:4 |
3.8 | 55.2 | 99.4 | 47 | 56 |
| 6 | Pt–Ga/Hd-Al2O3 | 580 | C3H8/N2 = 1:9 |
2.4 | 65.5 | 97 | 32 | 51 |
| 7 | Pt/Al2O3–600 | 590 | C3H8/H2/He = 1:1.25:4 |
5.2 | 48.5 | 96.9 | 51 | 50 |
| 8 | Pt–Sn/Al2O3sheet | 590 | C3H8/H2/N2 = 1:1.25:4 |
9.5 | 48.7 | 99.1 | 95 | 49 |
| 9 | Pt–Cu/h-BN | 600 | C3H8/H2/N2 = 1:1:3.9 |
4.0 | 24 | 97.3 | 20 | 70 |
| 10 | Pt–ln/LaAlO3/γ-Al2O3 | 600 | C3H8/H2/N2 = 8:7:35 |
2.6 | 47 | 96 | 26 | 53 |
| 11 | Pt/TiO2–Al2O3-10 | 600 | C3H8/H2/N2 = 1:1:1.85 |
10.2 | 47.3 | 78 | 80 | 52 |
| 12 | Pt3–Ga/Al2O3 | 600 | C3H8/H2/N2 = 1:1:1.85 |
10.2 | 39.4 | 98 | 83 | 79 |
| 13 | Pt–Zn@Silicalite-1 | 600 | C3H8/N2 = 1:1.73 |
6.5 | 45 | 99.8 | 66 | 63 |
| 14 | Pt/TiO2–Al2O3-20 | 600 | C3H8/H2/N2 = 1:1:1.85 |
10.2 | 45.5 | 88 | 86 | 52 |
| 15 | Pt3–Ga/CeO2–Al2O3 | 600 | C3H8/H2/N2 = 1:1:1.85 |
10 | 41.1 | 98.5 | 87 | 79 |
| 16 | Pt/ln/Mg(Al)Ox | 620 | C3H8/H2/Ar = 8:7:35 |
3.3 | 69 | 98 | 45 | 55 |
Ge was recently shown to also boost the performance of Pt catalysts: Rimaz et al. prepared various Ge loadings, followed by reduction at different temperatures.46 An initial propene yield of 53.8% was achieved on a Pt–Ge/Al2O3 catalyst, comprising 0.5 wt% Pt and 1.5 wt% Ge reduced at 600 °C. The improvement of the bimetallic catalyst was ascribed to the electron withdrawing Ge promotor, which aided C–H activation. Isolated single Pt atoms in a Cu matrix (10 wt% Cu and 0.1 wt% Pt) supported on Al2O3 simultaneously displayed enhanced catalyst activity, selectivity and stability over a 120 h period.47 In producing supported single atom materials, undesired side-reactions were suppressed. Thus, isolated Pt atoms dispersed on Cu nanoparticles were identified as active centres with high propene selectivity; dramatically enhancing the desorption of surface bound propene, prohibiting its further dehydrogenation.
Numerous Pt-based catalysts supported on alumina (Al2O3) have been reported. The development of new catalysts with simultaneously enhanced activity, selectivity and stability is the ultimate goal. Gamma-alumina (γ-Al2O3) is characterized by a high number and density of medium-strong acid sites, which can lead to undesired side reactions. Brønsted and Lewis acid sites on the support surface promote cracking and surface acid sites result in faster dehydrogenation for propene than propane, leading to deep dehydrogenation. Limited support acidity is needed to avoid undesirable side reactions, such as catalytic cracking and coke formation. However, Lewis acid sites and amphoteric –OH groups can function as nucleation sites for Pt, enhancing dispersion. This, coupled with the high specific surface areas frequently reported for γ-Al2O3 explain its popularity as a catalyst support.48 Recent advances in the use of numerous supports and catalysis synthesis methods for Pt-based catalysts and the underlying structure–activity correlations are considered below.
The importance of Al speciation in improving catalyst performance and stability was investigated by Shi et al. The effect of surface penta-coordinated (AlV), Al3+ sites on a Pt–Sn/γ-Al2O3 nanosheet catalyst was investigated and after a 24 h reaction, a 4.1% drop in propane conversion with 45% propene yield was observed, corresponding to a STY of 95 molC3H6 kgcat−1 h−1 (Table 3, entry 8).49 By comparison, the conventional Pt–Sn/γ-Al2O3 catalyst rapidly deactivated, retaining only 65% of its initial activity. The Pt–Sn/γ-Al2O3 nanosheet catalyst also exhibited enhanced activity due to the high proportion of AlV sites and their ability to uniformly disperse and stabilise Pt–Sn clusters.
Jang et al. explored calcination process parameters, namely temperatures of 600 °C and 750 °C, to modulate the surface properties of Pt–Sn/γ-Al2O3 catalysts.50 It was shown that calcination at 600 °C resulted in a 41.2% propene yield over a 20 h DDH reaction, equivalent to a STY of 51 molC3H6 kgcat−1 h−1 (Table 3, entry 7). The increased sintering resistance of the metal was ascribed to stabilization from the larger fraction of AlV, strong Lewis acid sites, which enhanced the metal–support interactions and suppressed coke deposition.
Yu et al. investigated the influence of sol–gel (Sg), hydrothermally synthesized (Hd) and commercially (C) obtained Al2O3 in tuning the coordination structure of Al3+ species in Pt–Ga/Al2O3 catalysts.51 At 580 °C, initial propene yields of 63.5% and 57.5% were observed for Pt–Ga/Hd-Al2O3 and Pt–Ga/Sg-Al2O3, respectively. Pt–Ga/Hd-Al2O3 achieved a STY of 32 molC3H6 kgcat−1 h−1 (Table 2, entry 6). It was shown that AlV sites facilitate high dispersion of the active Pt and Ga ions, and result in strong metal–support interactions. Additionally, hydrogen spillover was more effective due to the increased dispersion, which aided the desorption of H2 from the catalyst. This study demonstrated the importance of careful support preparation and the importance of the support surface in determining support metal nanoparticle dispersion and catalytic activity.
The modification of Al2O3 with metal oxides, such as TiO252 has been undertaken with some promising results. In the case of TiO2, it was shown that the addition of 10 and 20 wt% TiO2 to Al2O3 greatly enhanced the activity of the catalyst. The most active catalyst, a Pt/TiO2–Al2O3 catalyst (with a TiO2 loading of 20 wt%) exhibited an initial STY of 86 molC3H6 kgcat−1 h−1 (Table 3, entry 14). The enhanced performance was ascribed to electron transfer and acid site modulation effects resulting in changes to the adsorption properties of Pt. Similarly, Chen et al. reported a Pt–ln/LaAlO3/γ-Al2O3 catalyst with an initial STY of 26 molC3H6 kgcat−1 h−1 (Table 3, entry 10).53
Alternative supports, such as calcined hydrotalcite, Mg(Al)Ox, have also been extensively investigated. Pt/Mg(Al)Ox modified with ln was prepared using the co-precipitation method by Tolek et al. At 550 °C, the catalyst achieved an initial propane conversion of 24.2% and 98.2% selectivity to propene.54 This corresponded to a STY of 7.8 molC3H6 kgcat−1 h−1 (Table 3, entry 1). The Pt/Mg(ln)(Al)O catalyst performance was attributed to a high fraction of metallic state ln (In0) coupled with a high Pt dispersion with an average Pt particle size of 0.91 nm. Shen et al. prepared Pt/Mg(ln)(Al)O catalysts, where promising results were obtained by altering the calcination temperature and Mg/Al ratio.55 An optimised Mg/Al ratio of 4 led to an initial STY of 45 molC3H6 kgcat−1 h−1 at 620 °C (Table 3, entry 16). These optimised Pt/Mg(Al)O catalyst materials were characterised by having the lowest fraction of strong acid sites and highest specific surface areas. Wu et al. studied zinc-modified Pt–Sn/Mg(Al)Ox, which exhibited an initial STY of 47 molC3H6 kgcat−1 h−1 (Table 3, entry 5).56 Doping the support with an optimal amount of Zn resulted in smaller, and more uniformly dispersed metal particles distributed on the Mg(Al)Ox support. Overall, there have been several attempts to enhance catalyst activity through introducing a second metal to Al2O3 to form a mixed metal oxide support, or by preparing Al containing layered double hydroxides. Generally these have been shown to modestly boost catalyst performance, and the role of the second metal in most cases is to either enhance the dispersion of Pt, or adjust the acid–base properties of the support to inhibit side reactions.
Zeolites have been studied as potential supports for Pt-based catalysts due to their high surface areas, well-defined pore architectures and tuneable acid properties.57,58 Wannapakdee et al. explored silicate-1 and Al2O3 based monometallic Pt catalysts.59 The performance over 10 h on-stream differed between the Pt/silicalite-1 and Pt/Al2O3 materials, shown by >99% and 60% propene selectivity, respectively. The modified hierarchical pore structure, high specific surface area and presence of weak acid sites on Pt/silicalite-1 was concluded to be beneficial. Other zeolites which have been listed in patent literature for DDH of propane include Ta incorporated MCM-68, and H-ZSM-5.60,61 In addition, non-zeolite porous silicates have been successfully applied to Pt nanoparticles.62
Pt–Zn nanoclusters encapsulated in silicalite-1 was investigated by Wang et al., An initial STY of 66 molC3H6 kgcat−1 h−1 was measured, along with excellent cyclic stability during four DDH cycles at 600 °C (Table 3, entry 13).63 Xu et al. evaluated a series of Pt–Sn/Si-beta catalysts with different Pt/Sn ratios at 570 °C and reported high initial propane conversions, ca. 50%, irrespective of Pt/Sn ratio.64 The Pt–Sn2/Si-beta demonstrated an initial propene yield of 47.5%, which decreased by 3.4% over a 48 h reaction period, in contrast to the unmodified Pt/Si-beta catalyst which displayed a 47% reduction in the propene yield under identical reaction conditions. Through extensive characterisation the enhanced performance and stability was attributed to ultra-small, uniformly dispersed particles, ranging in size from 1.0 to 1.4 nm, consistent with previous studies that observed structure-sensitivity. It was found that embedding Sn in the zeolite framework was an effective geometric diluter for Pt ensembles resulting in smaller, isolated Pt clusters which increase propene selectivity.
Wu et al. investigated the importance of sub-surface layers in supported nanoparticles. They reported that a Pt3–Mn/SiO2 catalyst was enhanced by the presence of a Pt3–In subsurface layer (compared with a Pt subsurface layer). The optimised catalyst exhibited an initial STY of 41 molC3H6 kgcat−1 h−1 (Table 3, entry 4).65 Computational and experimental results suggested the role of the subsurface was to facilitate the activation of propane on the surface. Continuous testing for a week revealed long-term stability, and particle size distributions in the spent catalysts that were similar to those of the fresh catalysts. While the results in this work reveal a crucial role in the sub-surface composition of the catalyst, this aspect of catalyst design is often overlooked. Another sometimes neglected area of catalyst design is the role of heat treatments in modifying the nanostructure of the supported phase. Deng et al. reported highly selective Pt/SiO2 and Pt–Sn/SiO2 catalysts prepared under oxidative, reductive and inert atmospheres.66 The Pt–Sn/SiO2 materials prepared in inert and reductive atmospheres displayed initial propene yields of 17.8% and 25.7%, respectively. This was attributed to the reductive treatment and the presence of Sn which induced electronic and surface modifications of Pt nanoparticles following direct reduction in an H2 atmosphere at temperatures in excess of 500 °C.67,68
Pt-Based catalysts supported on a boron-modified ZrO2 have been reported by Miao et al.69 The most active and stable catalyst, Pt–Sn/B-ZrO2, achieved a STY of 13 molC3H6 kgcat−1 h−1, that was attributed to the moderate surface acidity of B-ZrO2 (Table 3, entry 2). Wang et al. explored the use of hexagonal boron nitride (h-BN) nanosheets as a support for Pt–Cu clusters (with 0.01 wt% Pt and 1.0 wt% Cu loadings).70 This afforded a STY of 20 molC3H6 kgcat−1 h−1 at 600 °C, respectively (Table 3, entry 10). The role of Cu in the catalyst was to dilute the Pt species and promote desorption of propene, thereby enhancing selectivity. When tested under identical reaction conditions, this catalyst showed enhanced STY and stability compared to analogous catalysts supported on MgAl2O4, H-ZSM-5, SBA-15, SiC and Al2O3. The h-BN nanosheets provided abundant stacking fault edges terminated with B–O defects that are associated with unpaired electrons, which strongly interact with active metal species, helping to strengthen the interaction between the metal and support.71,72
Finally, other support materials which have been included in patent literature over the years have tried to improve upon the catalytic activity of those currently used in industrial processes. Support materials such as carbon nanotubes and nanodiamonds tend to impart good dispersion and high stability of Pt nanoparticles,73–75 whereas a support made entirely of TiO2 was shown to exhibit high activity while reducing carbon deposition.76 Au and Sn supported Pt has also been investigated, as well as various metallic sulfide materials, all designed to increase the activity of the catalyst.77,78
In summary, supported Pt catalysts constitute a major category of DDH catalysts, and have been studied extensively. Improvements in performance have been realised by tuning the particle size, promotion with other elements and selecting appropriate supports. Structure–activity relationships have been observed with regards to particle size, composition (presence of promotor/metal), as well as metal–support interactions. Control of these variables has been shown to influence the catalytic properties, and the reaction mechanism. Table 3 compares the activity of Pt catalysts over a wide range of conditions and with different supports and promotors. Pt–Sn and Pt–Ga supported on Al2O3 (often but not always γ-Al2O3) are the most active in this class of DDH catalyst, which is unsurprising given their application in commercial catalysts. Additionally, the incorporation of Ti into an Al2O3 support appears to boost activity significantly. Finally, Pt–Zn/silicalite-1 also exhibited a high STY, but Zn has not received the same attention as other promotors. Further studies may reveal that this bimetallic catalyst can compete with the current commercial catalysts.
4.2. Cr-Oxide based DDH catalysts
Chromium oxide-based catalysts have been extensively studied for DDH since the pioneering work by Frey and Huppke in 1933.80 This proved the importance of Cr2O3 as a key catalyst component for the dehydrogenation of light alkanes to their corresponding alkenes at relatively high temperature and low pressure. Since the discovery of Cr-based dehydrogenation catalysts, several industrial companies have developed commercial dehydrogenation processes and catalysts.Understanding the nature of the active chromium sites is fundamental to optimizing catalyst properties and limiting catalyst deactivation. Considerable research efforts have been undertaken to understand the nature of active sites and factors influencing catalytic performance in CrOx/Al2O3 catalysts.81,82 Spectroscopic analysis reveals a number of surface species with different oxidation states (Cr6+, Cr5+, Cr3+, Cr2+) and speciation (chromates, polychromates, crystalline and amorphous phases).83–86 Weckhuysen et al. demonstrated that two types of surface chromium oxide species exist with differences in their reducibility: monomeric and polymeric species.87 The relative concentration of different Cr surface species is strongly influenced by chromium loading and specific surface area of the selected support. Various studies have shown that low Cr loadings contribute to the dispersion of monomeric Cr6+ species, with some Cr3+ species and coordinative vacancies also being observed.88–91 Meanwhile, at high Cr loadings, polymeric Cr3+ species exist as clusters and/or crystalline Cr2O3. This limits the accessibility of the reactants to the Cr sites, thus limiting catalytic activity.
Therefore, different surface Cr species are known to coexist in the Cr-oxide based supported catalysts, causing differences in the catalytic performance for DDH. In particular, it has been reported that the redox cycle between active Cr6+ and Cr3+ species are responsible for the activity in the dehydrogenation reaction, but the reduction of Cr6+ to Cr3+ is rapid under reaction conditions.91–93 Thus, dehydrogenation activity has been mostly attributed to monomeric Cr3+ and less so to polymeric Cr3+ and Cr2+ sites produced in the initial steps of the DDH reaction. This is in agreement with De Rossi et al. who reported a correlation between DDH activity and the concentration of the coordinatively unsaturated Cr3+ species over CrOx/Al2O3 and CrOx/SiO2 catalysts.94 Additionally, through IR spectroscopy studies of CO and NO adsorption, the authors concluded that the most active Cr3+ species had two coordinative vacancies.
As described above, the Horiuti–Polanyi mechanism is widely accepted as the mechanism for the DDH reaction. However, an alternative dehydrogenation mechanism for Cr-based catalysts has been suggested,95,96 which proceeds as follows:
i. The formation of Cr3+–C3H8 species.
ii. Heterolytic activation of the C–H bond of Cr3+–C3H8 results in a Cr3+–C3H7 intermediate.
iii. The Cr3+–C3H7 intermediate undergoes β-H elimination to form Cr3+–C3H6 + H− species.
iv. Propene desorption and formation of Cr–H species.
v. Desorption of H2 due to proton transfer, completing the reaction cycle.
During the DDH process, the rate determining steps are the activation of the propane molecule to form the adsorbed Cr3+–C3H7 intermediate, and the β-H transfer to form the Cr3+–C3H6 hydride surface species which can significantly affect the DDH performance. In this instance, it is proposed that Cr3+–O surface species can activate propane via sigma-bond metathesis by generating a Cr–alkyl intermediate that subsequently undergoes β-hydride elimination to form propene.92,95
As a catalyst support component, Al2O3 has been widely used in alkane dehydrogenation processes due to its appropriate chemical properties, high structural stability and low cost. However, an inherent and major problem of the CrOx/Al2O3 catalyst system is rapid deactivation, which occurs as a consequence of coke formation. Cr nanoparticles dispersed on a series of rod-shaped porous alumina supports, followed by calcination at various temperatures (T), denoted Cr–Al-T, were prepared by Gao et al.97 Catalytic tests, at 600 °C, revealed that Cr–Al-800 afforded an initial propene yield of 30%, from which it was inferred that the reducibility and lower surface acidity of the Cr–Al-800 catalyst were advantageous, leading to a higher proportion of DDH active Cr3+ species and better anti-coking ability. Over the course of five dehydrogenation–regeneration cycles, the Cr–Al-800 catalyst demonstrated superior regenerative ability, and the initial STY was 60 molC3H6 kgcat−1 h−1 (Table 4, entry 9), which is comparable to many of the Pt catalysts reported in the literature.
| Entry | Catalyst | T/°C | Feed composition | WHSV /h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| T = reaction temperature; feed composition = molar ratio of C3H8/H2/carrier in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. | ||||||||
| 1 | Zn0.3Cr | 480 | C3H8/Ar = 1:19 |
0.5 | 21 | 94 | 2.1 | 108 |
| 2 | Cr2.5Ni5.0/Al2O3 | 550 | C3H8/Ar = 1:9 |
1.2 | 47 | 95 | 11 | 101 |
| 3 | Cr10Zr90/SiO2 | 550 | C3H8/N2 = 2:3 |
4.3 | 15 | 95 | 13 | 103 |
| 4 | Cr2Zr30/SiO2-600 | 550 | C3H8/N2 = 2:3 |
7.4 | 20 | 95 | 29 | 104 |
| 5 | K–CrZr5Ox | 550 | C3H8/He = 1:14 |
1.2 | 54 | 95 | 13 | 107 |
| 6 | Cs–P/CrZrOx | 550 | C3H8/N2 = 2:3 |
5.9 | 30 | 97.5 | 38 | 106 |
| 7 | CrOx/H-ZSM-5 (260) | 580 | C3H8/N2 = 1:19 |
0.6 | 60.8 | 78.2 | 5.9 | 102 |
| 8 | CrOx/Al2O3 | 600 | C3H8/He = 1:14 |
1.2 | 41.8 | 89.8 | 9.3 | 99 |
| 9 | CrOx/Al2O3 | 600 | C3H8 | 9.5 | 33.2 | 90.4 | 60 | 97 |
| 10 | Cr7.5/Al2O3 | 600 | C3H8/Ar = 1:4 |
2.4 | 62.5 | 88.5 | 27 | 98 |
| 11 | Ce–CrOx/Al2O3 | 630 | C3H8/N2 = 1:6.1 |
1.2 | 86 | 78 | 17 | 100 |
Lang et al. compared the Cr loading in a number of CrOx/Al2O3 catalysts.98 The 7.5 wt% Cr/Al2O3 catalyst exhibited superior catalytic performance, exhibiting an initial propene selectivity and yield of 88.5% and 55.3%, respectively. This corresponded to a STY of 27 molC3H6 kgcat−1 h−1 (Table 4, entry 11). The surface Cr6+/Cr3+ ratios produce differences in the reducibility of the catalysts and a direct correlation between increasing Cr metal loading and Cr6+/Cr3+ ratios exists, leading to increased activity, decreased propene selectivity and excellent regenerative ability.
Węgrzyniak et al. investigated the influence of both mesoporous alumina and chromium metal loading in the range of 1–30 wt%.99 The yield of propene achieved at 550 °C increased from 10.4 to 31.7% with 1 to 20 wt% Cr loading, however, further increase in Cr loading caused a decrease in the propene yield. The highest STY was achieved over the 20wt% CrOx/Al2O3 catalyst, which achieved 9.3 molC3H6 kgcat−1 h−1 (Table 4, entry 8), with a Cr concentration of 6.6 Cr atoms nm−2 which corresponded to 18% of Cr redox species. In agreement with catalytic activity, beyond this concentration of Cr the formation rate declines rapidly with the subsequent drop-in activity correlating well with the appearance of α-Cr2O3 crystallites located outside of the mesopore system. The catalytic performance after four dehydrogenation–regeneration cycles of the 20 wt% CrOx/Al2O3 catalyst did not show any significant differences, demonstrating good long-term stability. The irreversible initial deactivation in the consecutive DDH cycles was attributed to either: (i) structural changes of active species (such as agglomeration), or (ii) collapse of pore structure.
Numerous efforts have been made to improve the performance of the CrOx/Al2O3 catalyst using different promoting elements. Notably, Zhang et al. reported the modification of a commercial CrOx/Al2O3 catalyst with Ce, which decreased the amount of inactive isolated Cr6+ sites.100 This increased the amount of oxygen vacancies on the catalyst, thus promoting the interaction between Ce and Cr species. Consequently the Ce–CrOx/Al2O3 catalyst, when tested at 630 °C, displayed an STY of 17 molC3H6 kgcat−1 h−1 (Table 4, entry 12) and better regeneration ability over four cycles than that of the corresponding CrOx/Al2O3 catalyst over a single regeneration cycle.
Ni-Modified CrOx/γ-Al2O3 catalysts (5 wt% Ni and Cr loading) exhibited enhanced activity and selectivity to monometallic Ni/γ-Al2O3 and CrOx/γ-Al2O3, displaying an initial conversion of 47%, propene selectivity of 95% and yield of 44.7% at 550 °C. This equates to a STY of 11 molC3H6 kgcat−1 h−1 (Table 4, entry 2).101 The presence of Ni promoted the formation of oligomeric Cr species and specifically Cr3+ active sites.
Other metal oxides have been utilised as supports for Cr-based catalysts in an attempt to improve the catalyst stability, including materials such as ZrO2, mesoporous SiO2 and nanocarbons. He et al. investigated the effect of Cr loading on mesoporous MCM-41 with the CrOx/MCM-41 (10 wt% loading) catalyst displaying a propene yield of 47.5% at 630 °C.93 FT-IR analysis of this material suggested that high Cr loadings depleted surface hydroxyls, which were postulated to be essential for suppressing coke deposition.
The influence of surface acidity on activity has been investigated more in-depth by Hu et al. who prepared SiO2, Al2O3 and H-ZSM-5 (SiO2/Al2O3 = 28, 43, 80 and 260) supported Cr (5 wt%) catalysts.102 Under identical reaction conditions, the CrOx/H-ZSM-5 (nominal SiO2/Al2O3 ratio = 260) materials displayed better catalytic performance and stability than a commercial CrOx/Al2O3 catalyst, maintaining ca. 32.6% conversion and 94.2% propene selectivity after 50 h on stream. The authors attributed this enhanced performance to a high dispersion of Cr species, moderate acidity and strong metal–support interactions in the CrOx/H-ZSM-5(260) catalyst. Fig. 5 illustrates the effect of surface acidity and Cr loading on the propene formation rate. Additionally, it was found that the CrOx/H-ZSM-5(260) catalyst possessed strong anti-coking capability over long-term testing.
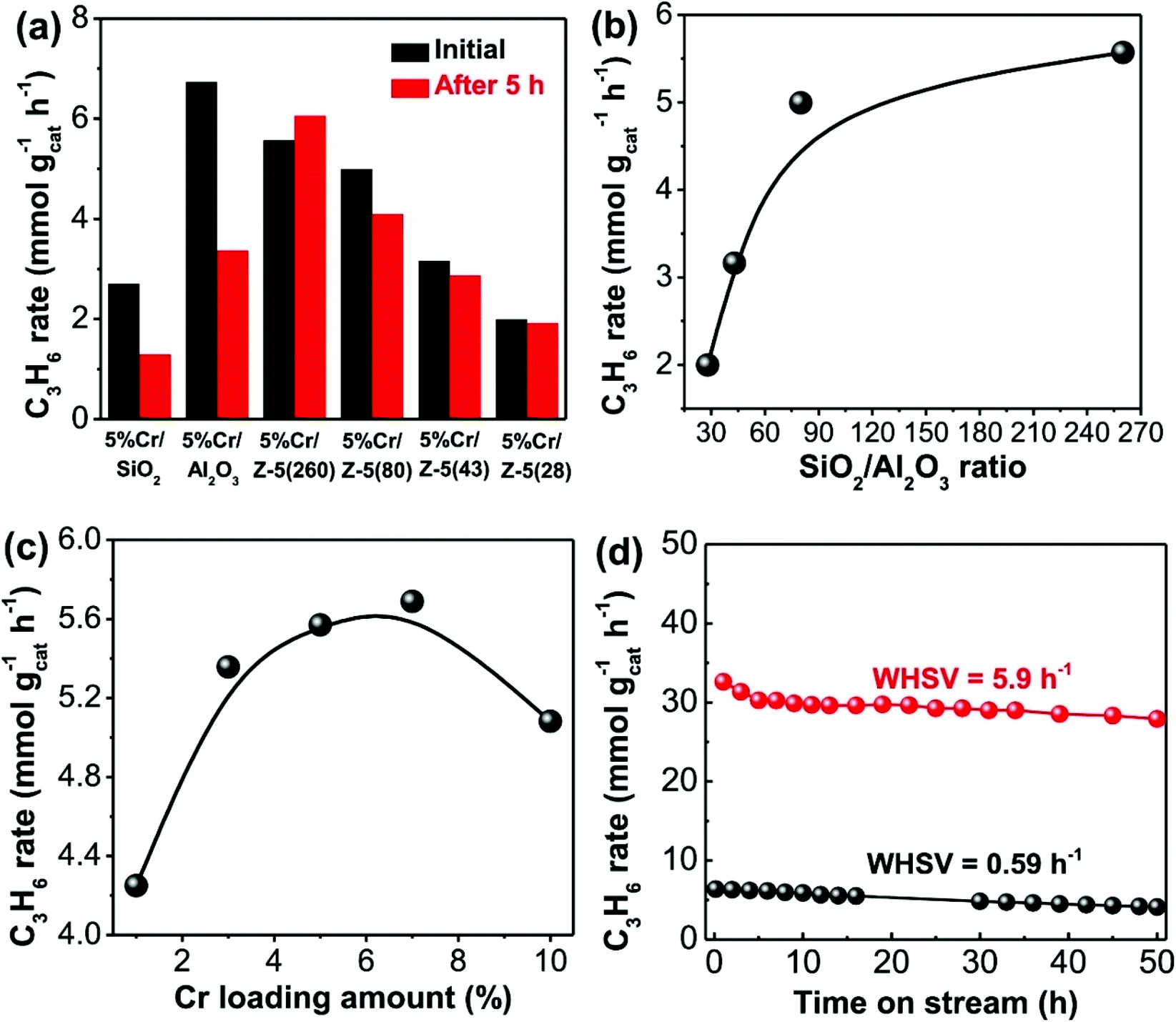 | ||
| Fig. 5 Catalytic performance of the CrOx catalysts on different supports. (a) Propene formation rates of the supported CrOx catalysts before and after 5 h on-stream. (b) Initial propene formation rates of 5% CrOx/H-ZSM-5 catalysts as a function of SiO2/Al2O3 ratio. (c) Initial propene formation rates of CrOx/H-ZSM-5(260) catalysts as a function of Cr loading amount. (d) Stability testing (propene formation rates) of 5% CrOx/H-ZSM-5(260) catalyst at a WHSV of 0.59 and 5.9 h−1 for 50 h time-on-stream. The tests in (a–c) were performed at a WHSV of 0.59 h−1. Reproduced from ref. 102 with permission from Elsevier. | ||
Moreover, Han et al. introduced Zr as a promoting element for Cr-catalysts on a number of supports (Al2O3, AlSiOx, SiTiO2 and SiO2).103 The Zr addition improved the turnover frequency for all Cr loadings when compared to analogous Cr-only catalysts and it was found that the greatest promotional effect was observed over the smallest ZrO2 crystallites. The order of reactivity was: CrZr/SiO2 > CrZr/AlSiOx > CrZr/SiTiOx. Likewise, the degree of crystallinity in CrZr/SiO2 catalysts proved to be an important structural property affecting both the rates of propene and coke formation.104 With increasing calcination temperature (450–600 °C) the catalysts displayed enhanced activity, selectivity and long-term stability (150 h) over consecutive dehydrogenation/regeneration cycles. However, the catalyst calcined at 600 °C exhibited a STY of 13 molC3H6 kgcat−1 h−1 (Table 4, entry 3).
Fujdala and Tilley reported CrOx/Si/Al/Ox and CrOx/Si/Zr/Ox catalysts exhibiting propene yields in excess of 33% at 450 °C.105 Further improvements were observed when basic promoters (Cs and Ca) were added to CrZr/Al2O3 catalysts which displayed a superior propene formation rate of 38 molC3H6 kgcat−1 h−1 after modification of the support with an acidic P-based modifier (Table 4, entry 6).106 The positive effect was attributed to the promoters’ ability to block the sites responsible for coke formation and negligible contribution to undesirable secondary reactions by the additional acid sites generated by the P-based modifier.
Mixed metal oxide catalysts have also been widely studied in the context of Cr-based catalysts. WÈ©grzyniak et al. demonstrated that ordered mesoporous Cr–Zr–O and Cr–Zr–K–O catalysts had improved catalytic performance compared to CrOx/Al2O3 materials and other promising catalysts in the DDH process.107 It was found that all the catalysts modified with K displayed respectable regeneration ability as well as an improvement in the catalyst's STY, which was measured to be 13 molC3H6 kgcat−1 h−1 (Table 4, entry 5).
Another series of binary Zn–Cr mixed oxides with different Zn/Cr molar ratios (0–0.5) proved advantageous for the DDH reaction.108 An optimal Zn/Cr ratio of 0.3 produced a highly active catalyst which displayed conversion, propene selectivity and propene yield values of 31.3%, 94% and 29.4%, respectively. The activity of the binary-component oxide catalyst was attributed to the defect-rich spinel structure, which generated a higher concentration of active Cr3+ species.
In summary, the dehydrogenation performance of CrOx-based catalysts is largely influenced by the catalyst preparation method, identity of the support, as well as the promoting elements and active sites present in the resultant catalyst. The aforementioned factors influence activity, selectivity and stability and can be enhanced by optimizing elements of the catalyst design process. The most common deactivation pathway is coking, which can cause sintering in both the support and Cr. Efforts to improve catalyst design include using supports with ordered mesoporosity and moderate surface acidity, both of which aid mass transfer and avoid undesirable secondary reactions. Numerous Cr deposition methods have been explored, with novel surface organometallic chemistry methods emerging as viable preparation methods for ensuring isolated CrOx species, although these may not be appropriate for large-scale application. Promoting elements have been found to cause beneficial geometric and electronic effects on both the supports and CrOx species. However, recent advances in unsupported, binary metal oxides have been successful in yielding catalysts with respectable activity, selectivity and resistance to coking. However, significant environmental and safety concerns still exist in the preparation, usage and disposal of CrOx-based catalysts due to the associated toxicity. In comparison to Pt catalysts, CrOx catalysts generally exhibit lower STYs, although the most active CrOx catalysts (typically supported on Al2O3) are as active as moderate supported Pt catalysts.
4.3. Ga-Based DDH catalysts
Historically, a bifunctional Ga/H-ZSM-5 material has been applied as a catalyst for the commercial process for propane aromatization, developed jointly by UOP and BP as the Cyclar Process.109,110 More recently, the Dow FCDh process was developed which uses a Pt–Ga based catalyst. Gallium oxide (GaOx)-based catalysts have been the subject of intensive research as candidate catalysts and/or supports for the DDH reaction. This is because Ga is another non-noble metallic element known to catalyse the dehydrogenation of propane, usually through oxidic, tetrahedrally coordinated Ga3+ species, which are considered to be active sites for C–H activation.111,112 However, several surface gallium species (e.g., Ga2O3, GaO+ and Gan+) can exist and are considered active constituents exhibiting different performance for the DDH reaction. Furthermore, the types of surface Ga species present are dependent on the identity of the support, Ga loading, as well as the preparation method.Numerous studies have shown that Ga2O3 dispersed on TiO2, MgO, Al2O3, ZrO2, H-ZSM-5, ZSM-8 and MWW zeolites display distinctly different catalytic properties for alkane dehydrogenation.113–116 Sattler et al. reported a Pt-promoted Ga/γ-Al2O3 species (Pt3Ga/γ-Al2O3) as a highly promising catalyst for propane dehydrogenation, which showed remarkably high activity, selectivity, and long-term stability.117 For catalysts containing both Ga and a low amount of Pt (∼0.1 wt%), the observed conversion was close to the equilibrium value, 55% at 620 °C with the addition of Pt as a promoter facilitating recombination of hydrogen atoms into H2. By modulating the support acidity further with a K-dopant, the resultant Pt3GaK/γ-Al2O3 catalyst demonstrated enhanced stability, displaying a propane conversion of 31.1% and a selectivity for propene of 92.6% after approximately 150 cycles or 14 days of operation. The authors postulated the main active site for C–H activation was Ga3+ embedded within the γ-Al2O3 framework.
Further investigations by Searles et al. showed that a Gaδ+Pt0/SiO2 catalyst, featuring a highly dispersed GaxPt alloy structure, had a long-term stability evident in unchanged propene selectivity (>99%) over the course of a 20 h DDH reaction.118 Both the moderate surface acidity of the support and good dispersion of the generated gallium single-sites proved beneficial. Additionally, the monometallic Ga3+/SiO2 catalyst also displayed minimal deactivation over a 20 h DDH reaction, retaining a high propene selectivity (94%) throughout.
A comparative approach to study the effect of the support acidity was taken by Szeto et al. through which marked differences were observed in the reactivity of silica and alumina supported Ga catalysts.119 An initial conversion and selectivity of 24% and 79%, respectively, were reported for Ga/Al2O3. The STY was 5.9 molC3H6 kg−1cat h−1 (Table 5, entry 4). On the other hand, the Ga/SiO2 catalyst was reported to have low activity (8% conversion initially) and moderately high selectivity to propene (90%) under identical testing conditions. This highlights the importance of selecting an appropriate support for different active centres.
| Entry | Catalyst | T/°C | Feed composition | WHSV /gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| T = reaction temperature; feed composition = molar ratio of C3H8/H2/carrier in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. | ||||||||
| 2 | Ga125-Rh | 450 | C3H8/He = 1:9 |
0.88 | 29 | 95 | 5.0 | 126 |
| 3 | Ga/SiO2 | 550 | C3H8/Ar = 1:4 |
2.4 | 10 | 90 | 4.5 | 49 |
| 4 | Ga/Al2O3 | 550 | C3H8/Ar = 1:4 |
1.5 | 24 | 79 | 5.9 | 119 |
| 5 | Ga-MFI | 600 | C3H8/Ar = 1:19 |
0.79 | 41 | 75 | 3.7 | 123 |
| 6 | Ga-MFI | 600 | C3H8/Ar = 1:19 |
2.4 | 12.2 | 82 | 4.9 | 122 |
| 7 | Ga/SBA-15 | 620 | C3H8/Ar = 1:19 |
0.6 | 29.7 | 92 | 3.3 | 121 |
| 8 | Ga/Al2O3 | 620 | C3H8/Ar = 1:19 |
0.6 | 46 | 95.2 | 5.5 | 121 |
| 9 | Ga/H-ZSM-5 | 620 | C3H8/Ar = 1:19 |
0.6 | 78.1 | 60.2 | 5.9 | 121 |
Cybulskis et al. reported that the preparation strategy could greatly influence the structure of Ga/SiO2 catalysts.120 Utilizing a pH-controlled incipient-wetness impregnation method the resultant catalyst contained a high proportion of isolated Lewis acidic Ga3+ sites dispersed on SiO2, with the ability to activate hydrocarbon C–H bonds with up to 99% selectivity through a non-redox pathway at 550 °C. A variety of spectroscopic characterisation methods further verified isolated four-coordinate Ga3+–O centres as catalytically relevant sites in these Ga/SiO2 catalysts. Meanwhile the Gaδ+–H and Gaδ+–O species, which were identified as spectators, remained stable in inert and reductive atmospheres from room temperature to 550 °C, before reversibly decomposing in the presence of C3H6 under reaction conditions to regenerate the Lewis acidic Ga3+–O centres.
Other supports (H-ZSM-5, SBA-15, γ-Al2O3 and SiO2) and the effect of Ga loading have also been investigated. Shao et al. established an optimal Ga loading of 5 wt%, reporting an initial propene yield on different supports that followed the trend: Ga/H-ZSM-5 (48%) > Ga/γ-Al2O3 (43%) > Ga/SBA-15 (28%) > Ga/SiO2 (9%) (Table 5, entries 7–9).121 This was attributed to differences in surface acidity between supports, with strong acid sites largely determining the dispersion of Ga species, as well as inducing adverse side reactions. Zeolite supports can in principle hinder the formation of large hydrocarbon molecules due to their unique framework and channel structure, which are beneficial to the catalytic stability. Consequently, it was found that the Ga/H-ZSM-5 catalyst displayed the highest initial activity but lowest stability. Factors such as the highest specific surface area and lowest acidity were associated with the superior catalytic stability of the Ga/SBA-15 catalyst over the 30 h DDH reaction at 620 °C.
Further improvements reported by Kim et al. explored the catalytic function of Ga-based catalysts supported on bulk and hierarchical MFI-type zeolites, where Ga was used in place of Al.122 Catalytic testing at 600 °C revealed substantial activity, selectivity and stability improvements in hierarchical Ga/MFI relative to the corresponding bulk Ga/MFI catalyst. The authors hypothesized that the enhanced activity and selectivity of the hierarchical catalysts arose from their quasi-2D structure and lower surface acidity. Generally, it is accepted that lowering the surface acidity can further enhance the catalytic performance of a DDH catalyst. The addition of 3-mercaptopropyl-trimethoxysilane (MPS) reduces Brønsted and increases Lewis acid sites in the preparation Ga/MFI catalysts, which resulted in improved performance.123 When compared to a CrOx/Al2O3 catalyst, the Ga/MFI catalyst, although less selective, was more stable and retained the moderate propene selectivity over the 12 h DDH reaction. Acidity characterization techniques confirmed a high concentration of strong Lewis acid sites and a reduction in the concentration of Brønsted acid sites in the Ga-MFI catalyst (Table 5, entries 5 and 6).
Schreiber et al. further verified the active sites for MFI supported Ga catalysts with moderate acidity, reporting an optimal Ga/Al ratio of 0.5, which enhanced the dehydrogenation rate.124 The authors attributed high Ga/Al ratios (>0.5) with the formation of additional GaxOy clusters which negatively affected the dehydrogenation rate and thus catalytic performance. Therefore, for Ga/MFI zeolites a synergy between Ga+ species and a Brønsted acid site (BAS) (optimised to one Ga+ per BAS) exists forming a Lewis–Brønsted acid pair during the dehydrogenation of propane. DFT calculations indicated that the generated Lewis–Brønsted acid pair is much more active than an isolated Ga+, due to the large increase in Lewis acidity upon protonation of Ga+ by the BAS, which facilitates heterolytic C–H bond activation. The BAS also significantly reduces the activation barrier for H2 elimination by recombination with a hydride. This was in agreement with findings by Phadke et al.125
Using a different approach, Raman et al. reported the synthesis of Ga–Rh supported catalytically active liquid metal solutions.126 A correlation between alloy composition and specific activity was observed with Ga125–Rh exhibiting STYs of 5.0 molC3H6 kgcat−1 h−1 (Table 5, entry 1). Bauer et al. reported that an alternative liquid metal solution, Ga37–Pt/Al2O3, exhibited propane conversion as low as 350 °C.127 The Ga–Pt alloy melts at temperatures between 270 °C and 330 °C.20 At 400 °C and 450 °C, selectivity to propene was ca. 95% and 90%, respectively and activity was attributed to isolated Pt atoms in the supported liquid Ga–Pt alloy.
In summary, GaOx based catalysts are promising propane dehydrogenation candidates. However, these catalysts are limited by deactivation through coke formation and the sintering of GaOx species which are dependent on the support, Ga loading and preparation method. Studies have also shown the advantages of incorporating promoter elements and/or the formation of intermetallic alloys in enhancing the dehydrogenation performance. Ga catalysts generally exhibit STYs that are one order of magnitude lower than Pt catalysts, and the most active CrOx catalysts, but have the advantage of being benign and inexpensive.
4.4. V-Based DDH catalysts
Vanadium-based catalysts have garnered increased interest as alternative materials to Pt- and Cr-based catalysts for the DDH reaction, to their comparative low cost and toxicity. Numerous studies have elucidated the effect of the preparation method, vanadium loading, promoter and support characteristics on the surface chemistry and catalyst performance of vanadium species.128–131 Generally, the oxidation state of vanadium (i.e., V3+, V4+, V5+) determines the catalytic activity.132 In addition, the distribution of V oxidation states is controlled by the V loading, which in turn influences the degree of polymerization. It has been reported that V5+ is the least active, whereas V4+ and V3+ species (generated from the reduction of V5+) are more active in the DDH reaction.133–137 Additionally, these studies suggest that a number of VOx species exist including monovanadate (isolated V), polyvanadate (oligomeric Vn+) and V2O5 crystallites.The nature of the support is an essential parameter to consider as several possible VOx can species exist. It is generally accepted that the metal–support (V–O–support) interactions affect the resultant catalyst performance.138 It is proposed that V–O–support bonds are active sites, as lattice oxygen in V–O bonds directly catalyses C–H activation forming H2. The activity is dependent on the bond strength of the V–O–support bond. Therefore, it is expected that catalyst performance is affected when different supports, such as SiO2, Al2O3 and ZrO2 are utilised.
From a comparison of VOx/MCM-41, CrOx/MCM-41 and Pt–Sn/Al2O3 catalysts, it was shown that VOx/MCM-41 exhibited superior propene selectivity (above 90%) and stability during several successive dehydrogenation–regeneration cycles.90 Subsequent studies expounded on the importance of the support surface acidity on a range of aluminosilicates with varying SiO2 contents.139 Sokolov et al. investigated the effect of the support on the stability of Al2O3, SiO2 and AlSiOx supported VOx catalyst. It was reported that at 1 wt% V/AlSiOx (with 10 wt% SiO2 in the support) was only around 25% less active than a Pt–Sn/Al2O3 comparison sample. However, the STY of this most active catalyst at 550 °C was 5.1 molC3H6 kg−1cat h−1 (Table 6, entry 1), which is an order of magnitude below the values commonly reported for supported Pt catalysts tested at 600 °C, suggesting that more active Pt–Sn catalysts can be prepared than the one reported in this work. The catalyst deactivated quickly due to carbon deposition, but retained its initial activity over the course of ten successive dehydrogenation–regeneration cycles. The authors observed a positive correlation between the rate of coke formation and the acidity of the support, as well as reporting that coke can actually catalyse the DDH reaction. Additional studies attributed the dependence of coke formation on the degree of polymerization of VOx species over catalysts supported on a range of aluminosilicates, with differing SiO2 contents varying from 1 to 100 wt%.140 It was concluded that coke preferentially formed on larger V2O5 crystallites rather than small or isolated VOx species.
| Entry | Catalyst | T/°C | Feed composition | WHSV/gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| T = reaction temperature; feed composition = molar ratio of C3H8/H2/carrier in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. DMSN = dendritic mesoporous silica nanoparticles. | ||||||||
| 1 | V/SiO2 | 550 | C3H8/N2 = 2:3 |
0.95 | 30 | 85 | 5.1 | 139 |
| 2 | VZrO2 | 550 | C3H8 /H2/N2 = 1:1:5.1 |
2.06 | 25 | 85 | 9.2 | 149 |
| 3 | VZrO2 | 550 | C3H8/Ar = 1:6.5 |
4.7 | 25 | 98 | 24 | 150 |
| 4 | V-SiO2 | 580 | C3H8/Ar = 1:10 |
0.59 | 64 | 90 | 7.9 | 145 |
| 5 | VOx/SiO2 | 580 | C3H8/Ar = 1:10 |
0.6 | 55 | 91 | 6.2 | 147 |
| 6 | 7VOx/Si-Beta | 600 | C3H8/N2 = 1:19 |
0.6 | 40 | 90 | 4.4 | 148 |
| 7 | VOx–Mg/Al2O3 | 600 | C3H8 /H2/N2 = 1:1:1.6 |
1.3 | 31 | 85 | 7.8 | 143 |
| 8 | V-DMSN | 600 | C3H8/N2 = 1:7.5 |
6.7 | 18.1 | 91 | 22 | 146 |
| 9 | VOx/γ-Al2O3 | 600 | C3H8/H2/N2 = 1:1:1.6 |
3.3 | 15 | 94 | 15 | 132 |
| 10 | VOx/Al2O3 | 610 | C3H8/N2 = 4:1 |
2.8 | 30 | 85 | 15 | 141 |
The cation–anion double hydrolysis approach (CADH), used for the synthesis of mesoporous Al2O3 with high surface area and a pure γ-Al2O3 phase, was undertaken with the crystallisation temperature varied between 25–120 °C.141 It was found that the crystallisation temperature of the support directly influenced the catalytic performance of the VOx/Al2O3 catalysts, which was a consequence of the increase in Lewis acid sites with increasing crystallization temperatures up to 100 °C. This in turn affected the distribution of different surface V species. After an induction time of 1 h, the VOx/Al2O3 catalyst (crystallized at 100 °C) achieved the best performance giving a propene yield of 60%, equivalent to a STY of 15 molC3H6 kg−1cat h−1 (Table 6, entry 10).
Similar studies investigating V loading (1–20 wt%) and its effect on the surface V species present in VOx/Al2O3 catalysts reported a volcano plot-type trend between V loading and catalytic performance, with maximum activity being observed at 12 wt% V loading.132 The authors concluded that the relative proportions of V3+, V4+ and V5+ species present on the catalyst surface were affected by V loading and at the optimal loading of 12 wt%, a high concentration of isolated V3+ species was detected. In situ DRIFTS analysis elucidated the mechanistic pathway, whereby a vanadium propyl intermediate is formed after propane activation and rapidly transforms to propenyl-vanadium. Propene is formed from propenyl-vanadium or propyl-vanadium and desorbs from the catalyst surface.
Gu et al. reported that P modification could greatly influence the structure and activity of VOx/Al2O3 catalysts.142 More specifically, P modification decreased the degree of VOx polymerization, which had positive implications on the catalyst stability, as evidenced by the retention of high activity (ca. 45% propane conversion) after an 8 h DDH reaction in all P-modified catalysts. This was almost twice the conversion reached with the unmodified catalyst. The combined and synergistic effect of the surface acidity modifications, their influence on the active V3+ species, and weakened metal–support interactions facilitated desorption of propene and inhibited the deep dehydrogenation that leads to coke precursors.
Subsequent studies on a VOx/Al2O3 catalysts (with 12 wt% V loading) modified with Mg revealed the anti-coking capability and improved catalytic performance evident through propene yields of 19–27% over a 6 h DDH reaction.143 It was found that the dispersing action of Mg modification to V2O5 crystallites and an increase in active V3+ species reduced the amount and polymerization of coke deposits. However, the acidity decreases sharply with excessive Mg addition (above 1 wt%) as the excess MgO covers the surface VOx species, which leads to the loss of DDH activity. The highest STY achieved was 7.8 molC3H6 kg−1cat h−1 (Table 6, entry 7).
In addition to alumina, other supports have been investigated. Earlier reports by Ovsitser et al. described a silica supported VOx catalyst which exhibited high propene yield (86%), high stability and minimal deactivation over a 20 h DDH reaction.144 Hu et al. explored vanadium doped porous silica (V–SiO2) with varying V:Si mass ratios of vanadium to silicon of 1:24, 1:12, 1:6, and 1:3, respectively.145 The V/SiO2 catalyst (with mass ratio of 1:12) displayed the optimal catalytic performance, achieving a superior initial propene yield of 59.5% and a stable propene selectivity of ∼90% over 6 h at 580 °C. This was equivalent to a STY of 7.9 molC3H6 kg−1cat h−1 (Table 6, entry 4). Results from successive dehydrogenation–regeneration cycles indicated that the catalyst reached a stable state displaying excellent regeneration stability after two dehydrogenation cycles. In common with VOx/Al2O3 catalysts, moderate V loadings were concomitant with increased activity, which was attributed to more vanadium active sites in the channels of the porous silica with increased doping. The moderate vanadium doping level was conducive to highly dispersed vanadium species in the form of isolated and low-polymerized VOx species.
As previously discussed for other DDH catalysts, innovative techniques have also been applied in the preparation of V/SiO2 catalysts. Vanadium-containing dendritic mesoporous silica nanoparticles (V-DMSNs) exhibited an initial propane conversion and propene selectivity of 18.1% and 91% respectively. The STY was calculated to be 22 molC3H6 kg−1cat h−1 (Table 6, entry 8), which is one of the highest VOx containing catalysts reported to date.146 Compared to analogous catalysts synthesized via wet impregnation, the direct co-assembly process for the V-DMSNs catalysts contributed to enhancing the interaction between the V5+ species and the silica support. Highly dispersed and stable VOx species, and an absence of V2O5 crystallites, were conducive to high catalytic activity and stability. When tested for both ODH-O2 and DDH reactions, the V-DMSNs catalysts displayed superior performance, signifying the importance of the preparation conditions and synthesis strategy to form well-defined structure of the support and sufficient concentration of highly dispersed and isolated VO4 active sites.
Hu et al. investigated the influence of gelation and calcination temperatures on the properties and catalytic performance.147 Interestingly, enhancement to catalytic performance in terms of propane conversion, propene selectivity and yield of propene were observed for VOx/SiO2 catalysts (gelation temperature: 60 °C, calcination temperature: 580 °C). The VOx/SiO2 catalyst exhibited an initial propane conversion of 55% and stable propene selectivity of ca. 91% over 4 h, with excellent reusability displayed over eight successive reaction-regeneration cycles. The STY was 6.2 molC3H6 kg−1cat h−1 (Table 6, entry 5), due to the low GHSV so while the catalyst achieved high selectivity at reasonable propane conversion, it was not as intrinsically active as other supported VOx catalysts.
Dealuminated beta zeolites (Si-beta) are promising catalyst supports due to their high surface area and thermal stability as mentioned before. Chen et al. synthesized numerous VOx/Si-beta zeolites with various V loadings (in the 0.5–10 wt% range) with the initial propane conversion shown to linearly increase with the amount of acid sites, Fig. 6.148 This demonstrated the dependency of catalytic activity on total Lewis and Brønsted acid sites generated by VOx on silica, with Brønsted acid sites disappearing after high temperature treatment due to decomposition of hydrated VOx. The most active catalyst, V/Si-beta (having 3 wt% V loading) displayed an initial propene yield of 33.4% and retained activity over multiple dehydrogenation–regeneration cycles. Thus, only weak and medium strength sites were generated and ascribed to mono- and polymeric VOx species, respectively, both of which aided the adsorption and activation of propane. A correlation between increasing V loading, increased degree of polymerization and a subsequent increase in strong surface acid sites was reported. At 7 wt% VOx/Si-beta, the STY was 4.4 molC3H6 kg−1cat h−1 (Table 6, entry 6), which is 5–6 times less active than the most active VOx catalysts reported to date, suggesting that Si-beta does not facilitate a high concentration of active V sites.
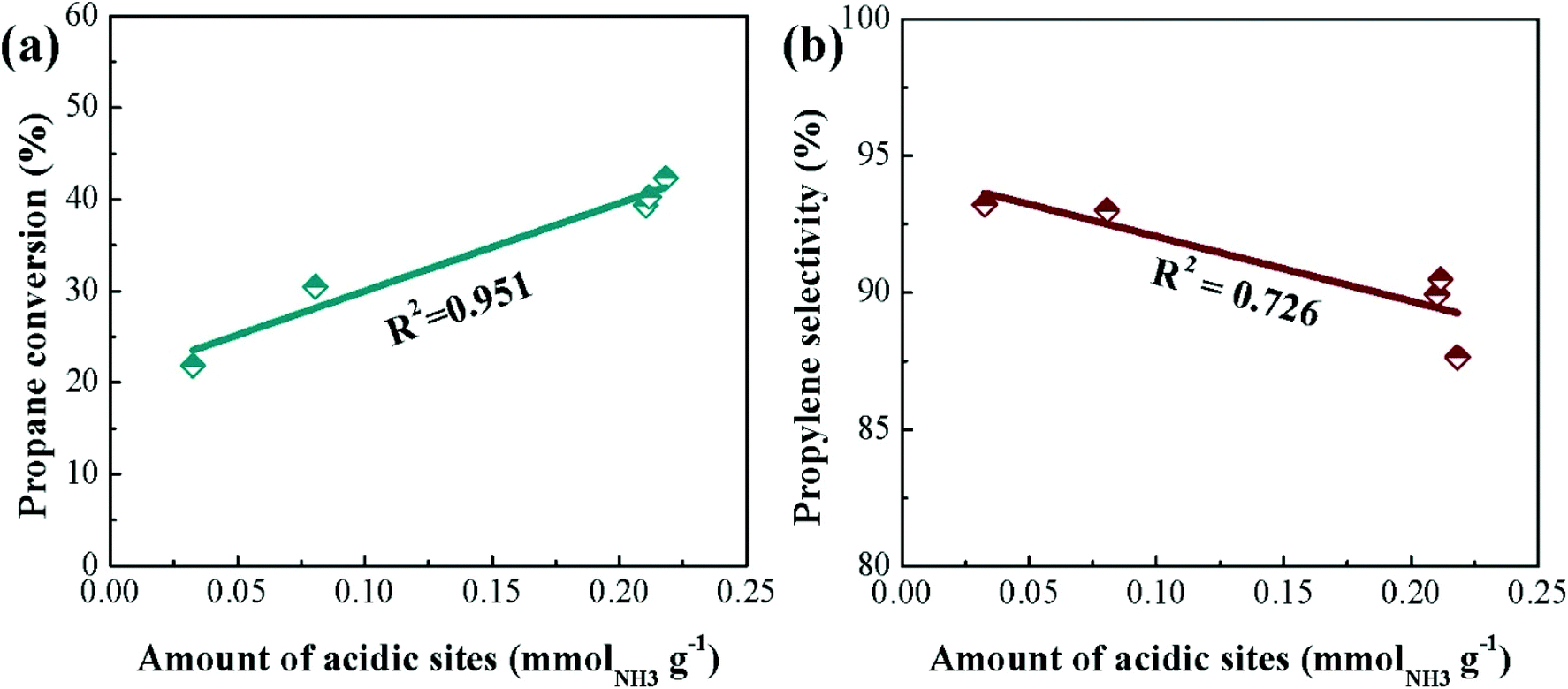 | ||
| Fig. 6 Correlations between the (A) initial propane conversion and (B) propene selectivity as a function of the total amount of acidic sites, created by the interaction sites between V species and Si-Beta support in V/SiBeta catalysts. Reproduced from ref. 148 with permission from Elsevier. | ||
Alternatively, ZrO2 can be used as a support for VOx. It was found that VOx/ZrO2 is approximately five times more active than VOx/Al2O3 and pure ZrO2, exhibiting a STY of 9.2 molC3H6 kg−1cat h−1 (Table 6, entry 2).149 The authors proposed that the enhanced performance of VOx/ZrO2 results from the facile reduction of V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, V–O–V and V–O–Zr bonds, thus producing higher proportions of the active V3+ species. In view of this, Jeon et al. investigated various loadings of V on VOx/ZrO2 catalysts.150 For the most active catalyst, with 8 wt% V loading, the initial yield of propene increased from 24% at 550 °C to 28% at 575 °C with a negligible (<1%) increase at 600 °C. This corresponded to a STY of 24 molC3H6 kg−1cat h−1 (Table 6, entry 3), which is one of the most active VOx-based catalysts reported. As discussed later in section 4.5, ZrO2 is active by itself and therefore this result likely reflects the contribution from an active ZrO2 support rather than an optimised or improved VOx catalyst. Indeed, the authors alluded to a correlation between activity and the formation of a higher concentration of coordinatively unsaturated (Zrcus) sites, created by the addition of the V dopant into the Zr lattice, increasing the density of Lewis acid sites. As discussed below, the STY of ZrO2 without VOx is actually much higher than in the current work.
O, V–O–V and V–O–Zr bonds, thus producing higher proportions of the active V3+ species. In view of this, Jeon et al. investigated various loadings of V on VOx/ZrO2 catalysts.150 For the most active catalyst, with 8 wt% V loading, the initial yield of propene increased from 24% at 550 °C to 28% at 575 °C with a negligible (<1%) increase at 600 °C. This corresponded to a STY of 24 molC3H6 kg−1cat h−1 (Table 6, entry 3), which is one of the most active VOx-based catalysts reported. As discussed later in section 4.5, ZrO2 is active by itself and therefore this result likely reflects the contribution from an active ZrO2 support rather than an optimised or improved VOx catalyst. Indeed, the authors alluded to a correlation between activity and the formation of a higher concentration of coordinatively unsaturated (Zrcus) sites, created by the addition of the V dopant into the Zr lattice, increasing the density of Lewis acid sites. As discussed below, the STY of ZrO2 without VOx is actually much higher than in the current work.
To date, many studies have investigated VOx-based catalysts, which in some cases have been said to exhibit a catalyst performance similar to that of Pt- and CrOx-based catalysts. However, comparing the typical STY values for VOx with those of CrOx or Pt, it is clear that the VOx catalysts reported in the literature are not as active as supported CrOx or Pt catalysts. In view of catalyst design, the nature of the support is an essential consideration, due to the importance of metal–support interactions and the influence of acid sites on the formation of different surface VOx species. Of particular importance is the role of the support in altering stabilization and electronic effects of the active sites. Much like the industrial (Pt- and Cr-based) catalysts, the surface acidity and selectivity of the catalyst is affected by the nature of the support. Although the most active V species has been identified, the relative proportion and stability of this surface V species can be tuned by varying the preparation method. On the other hand, the role of the other V species on the DDH reaction remains unclear so further work to elucidate this could yield more active and stable DDH catalysts. The range in performance of V-based catalysts varies quite significantly, with the most active catalysts exhibiting STYs that are around 3–4 time less active than the most active Pt catalysts.
4.5. Other DDH catalysts
As previously discussed, the industrial application of Pt- and CrOx-catalysts is well established because they offer superior activity, selectivity and stability for the direct dehydrogenation of propane. However, these catalysts are limited in two main areas: economically, by the high cost of Pt and environmentally, by the toxicity of Cr6+ species. Thus, a definite need to develop alternative catalyst formulation exists.Commonly, coordinatively unsaturated metal cations with neighbouring oxygen vacancies serve as adsorption sites for light alkanes, whereby surface oxygen sites abstract hydrogen from the adsorbed alkane. Armed with this knowledge, researchers continue to focus their efforts on catalyst design with the aim of enhancing dehydrogenation performance, creating both economic and environmental benefits.
Bulk ZrO2, characterized with coordinatively unsaturated Zr cations (Zrcus4+) as active sites, has emerged as a highly active and selective unsupported catalyst for the direct dehydrogenation of propane. Kondratenko and co-workers first reported that ZrO2 is active in its own right for DDH and subsequently demonstrated the importance of crystallite size and phase composition in determining activity.151,152 The rate of propene formation was inversely proportional to the crystallite size, but crucially, amorphous ZrO2 was very poorly active. Time on-stream conversions dropped significantly over time due to carbon deposition, but regenerative cycles were completed to examine if the activity could be regained. At 550 °C, ZrO2 was durable after 20 cycles but at 600 °C the ZrO2 crystallites sintered, causing a loss in activity. The initial performance corresponded to an STY of 64 molC3H6 kg−1cat h−1 (Table 7, entry 13), which is one of the highest non-Pt values reported.
| Entry | Catalyst | T/°C | Feed composition | WHSV/gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY /molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
| T = reaction temperature; feed composition = molar ratio of C3H8/H2/carrier in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. | ||||||||
| 1 | Pd3–Fe | 510 | C3H8/H2/Ar = 1.5:1.5:97 |
8.0 | 22 | 87 | 32 | 165 |
| 2 | Zn/ZrTiOx | 550 | C3H8/H2/N2 = 8:1:11 |
4.7 | 30 | 95 | 55 | 157 |
| 3 | OMC-2 | 600 | C3H8/N2 = 1:19 |
0.6 | 66 | 71 | 5.7 | 169 |
| 4 | Co–Al2O3 | 590 | C3H8/H2/N2 = 1:0.8:3.2 |
3.2 | 25 | 97 | 16 | 160 |
| 5 | Sn-HMS | 600 | C3H8/C3H6 = 99.87:0.13 |
0.4 | 40 | 90 | 3.0 | 163 |
| 6 | PT-MCN | 600 | C3H8/Ar = 1:19 |
0.6 | 33 | 85 | 3.4 | 171 |
| 7 | CMSC700 | 600 | C3H8/Ar = 1:19 |
0.6 | 41 | 89 | 4.5 | 168 |
| 8 | Ni3–Ga/Al2O3 | 600 | C3H8/Ar = 1:9 |
2.4 | 13 | 94 | 6.1 | 166 |
| 9 | ZrO2 | 600 | C3H8/Ar = 1:7.5 |
2.8 | 22 | 99 | 22 | 153 |
| 10 | CuZrOx | 600 | C3H8/Ar = 1:7.5 |
2.8 | 26 | 99 | 26 | 153 |
| 11 | ZnO/CN-silicalite-1 | 600 | C3H8:H2:N2 = 1:1:5 |
0.54 | 52 | 89 | 6.3 | 156 |
| 12 | ZnO/H-ZSM-5 | 600 | C3H8/Ar = 19:1 |
0.54 | 70 | 80 | 6.3 | 155 |
| 13 | ZrO2 | 600 | C3H8/Ar = 1:1.5 |
11.7 | 28 | 86 | 64 | 151 |
Jeon et al. reported the modification of ZrO2 with a vanadium dopant that exhibited an initial propene yield of 28%, excellent recyclability over ten cycles with no change in the active sites, and twice the activity observed in bulk un-doped ZrO2.150 This was attributed to an increase in Lewis acidic (Zrcus4+) active sites, that are a consequence of structural transformation. Additionally, the authors discovered that regardless of the vanadium content, all the catalysts exhibited a propene selectivity in excess of 98%.
Similarly, a positive effect from the modification of ZrO2 with a Cu dopant was reported by Jeon et al.153 The optimal CuZrOx catalyst displayed improved activity and selectivity at 600 °C compared to conventional (1 wt%) Pt/Al2O3, (0.5 wt% Pt-1.5 wt% Sn) Pt–Sn/Al2O3 and (5 wt% CrOx/Al2O3). The authors established a correlation between the specific activity of CuZrOx catalysts and the number of weak acid sites, resulting from the substitution of Cu in the Zr sub-lattice generating oxygen vacancies and Zrcus4+ ions. The STY of the CuZrOx catalyst was 26 molC3H6 kg−1cat h−1, compared to 22 molC3H6 kg−1cat h−1 in the unmodified catalyst (Table 7, entries 9 and 10). While this improvement is promising, the STY was lower than half of an optimised ZrO2 catalyst previously reported in ref. 151, suggesting that there is still room for improvement in the CuZrOx preparation, or that it is possible to promote activity through careful catalyst preparation rather than adding metals.
The concentration of dopant atoms, and the temperature of reduction were other contributing factors that affected the catalytic behaviour for ZrO2-based catalysts.3 Numerous binary YZrOx and LaZrOx-based catalysts displayed high activity, selectivity and stability as well as comparable performance to K–CrOx/Al2O3 catalysts over the course of 60 DDH cycles at 550 °C, 600 °C and 625 °C under industrially relevant conditions. The experimental parameters had a beneficial effect on the concentration of Zrcus4+ ions, and the concentration of surface lattice oxygen, which in tandem play an active role in propane activation.
As discussed in the above sections, Zn has been used as a promotor in Cr and Pt catalysts. Sun et al. also investigated it as a promotor for Nb catalysts.154 However, its use as a catalyst in its own right has recently been reported, notably by Yuan and co-workers. It was shown that ZnO nanoclusters supported in dealuminated zeolite-beta exhibited an initial propane conversion and propene selectivity of 53% and 97%, respectively at 600 °C. The initial STY was 4.2 molC3H6 kg−1cat h−1, and the conversion dropped to 40% after 6 h on-stream due to coking. ZnO/H-ZSM-5 was also investigated by the same group and it was shown that high SiO2:Al2O3 ratio supports yielded more selective DDH catalysts.155 The presence of strong acid sites is known to promote coke formation via deep dehydrogenation and therefore this trend is expected. The highest STY yield was observed over 10 wt% ZnO/ZSM-5 where the SiO2:Al2O3 molar ratio was 280, which produced 6.3 molC3H6 kg−1cat h−1 (Table 7, entry 12).
Kondratenko and co-workers reported the activity of ZnO supported on composites of N-doped carbon in silicalite-1.156 Initial propane conversion and propene selectivity of the catalyst was ca. 52% and 89% respectively at. This corresponds to a STY of 6.3 molC3H6 kg−1cat h−1 (Table 7, entry 11). Although the catalyst was active in the absence of the N-doped carbon composite, i.e. ZnO/silicalite-1, the activity was over 3 times lower. The role of the N-doped carbon was to encapsulate the ZnO nanoparticles and maintain high dispersion of the active component.
While the above studies indicated that supported Zn catalysts can be selective DDH catalysts, the STY values were an order of magnitude below typical Pt and CrOx based catalysts. However, this gap in performance was recently closed by Han et al., who investigated a range of Zr-containing metal oxides as supports for Zn, including ZrO2, TiZrOx and CeZrOx (as well as control catalysts without ZrO2 such as Al2O3 and SiO2).157 High catalyst activity was observed when Zr was present in the support, which is expected given that it is active by iteslf.151 However, the origin of the high activity could also be linked to the speciation of Zn. In the most active catalyst, 4 wt% Zn/TiZrOx, the Zn was present exclusively as isolated tri-coordinated Zn2+Ox. This catalyst produced a STY of 55 molC3H6 kg−1cat h−1 at 550 °C (Table 7, entry 2). Long term stability studies over 250 h and 10 regeneration cycles showed that a small drop in propane conversion and no loss in propene selectivity takes place. The drop in propane conversion was confirmed by ICP analysis to be due to loss of Zn from the catalyst. Temporal analysis of products studies were carried out to probe the rate-determining step of the reaction over supported Zn catalysts. Interestingly, it was shown that the formation of H2 was rate-limiting in the catalyst, not C–H bond activation. Additionally, the presence of Ti in the catalyst support was shown to promote H2 formation, which suggests that Ti in general is a good choice for a propane dehydrogenation catalyst. The observation that Zn–Zr catalysed DDH is not limited by C–H bond activation is a significant finding. As observed over similar catalysts, the rate of coke formation was dependent on acid and base sites, in this case present on the support. However, the best predictor of coke formation rate was the number of basic sites on the catalyst, which showed a strong positive correlation.
In section 3, the current commercial processes for propane dehydrogenation were presented and discussed. With the exception of the K-PRO process by KBR, all of the commercial catalysts have been disclosed. It is only known that the catalyst does not consist of Pt or Cr. We note that a patent was filed in the US in 2020 by Mukherjee et al. at Exelus, inc. (who developed the catalyst that is licensed to the K-PRO process)158 for alkane dehydrogenation using a catalyst consisting of Zn, Zr and Ti oxides.159 The optimal loadings and formulations disclosed in the patent are very similar to those reported by Han et al.157 and therefore we tentatively suggest that the catalyst in the K-PRO process is likely to be based on Zn–Zr–Ti–Ox.
Co-Based catalysts have also been demonstrated as potential alternatives to Pt- and Cr-based catalysts. Dai et al. reported a Co/Al2O3 catalyst with a high stability and selectivity (>97% propene selectivity), and a corresponding STY of 16 molC3H6 kg−1cat h−1 (Table 7, entry 4).160 Through extensive characterization it was observed that the Al2O3 sufficiently stabilized isolated Co2+, preventing surface reconstruction of Co species under reaction conditions. As a result, the catalyst efficiently facilitated propene desorption, inhibiting the formation of coke and other by-products. The application of a reductive-reoxidation treatment positively affected dehydrogenation performance of Co/Al2O3 catalysts.161 An initial propene yield of 28% and high selectivity to propene (>93%) was obtained over the course of the reaction. The authors attributed this to increased dispersion of Co metal species, a consequence of the mild reoxidation conditions resulting in markedly smaller (6 nm) Co metal nanoparticles, contrary to those observed in the fresh (34 nm) and reduced counterparts (25 nm).
As an efficient promoter of Pt-based catalysts, Sn was previously thought to be an inactive metal for dehydrogenation reactions. Surprisingly, metallic Sn supported on SiO2 has proven to be an efficient, dehydrogenation catalyst, exhibiting a sustained propene yield of 30% over the course of a 70 h DDH reaction at 600 °C.162 The incorporation of a hydrogenation component (Pd) can improve the stability of this catalyst, however, loss of Sn metal under reaction conditions, as a consequence of its low melting point, requires further investigation into more suitable supports than SiO2. Hexagonal mesoporous silicas have emerged as possible candidates due to Sn-HMS exhibiting high propane conversion (ca. 40%) and enhanced stability after multiple oxidative regenerations and 170 h on-stream.163 The improved stabilization is a result of two factors: firstly, Sn incorporation into the lattice of HMS forms strong Sn–O–Si bonds, which inhibit Sn reduction to low melting point Sn0, and secondly, additional stabilization of the Sn species is induced by the mesoporous structure of the HMS support.
Wang et al. revealed the dependency of the nature and distribution of SnOx species on the Sn loading, and through advanced characterization revealed uniform distribution and stabilization of isolated Sn4+ species in fresh samples, whilst being stabilized in the Sn2+ state after reduction on SiO2 due to strong metal–support interactions.164 Superior long-term stability, as well as recovery of the initial activity upon oxidative regeneration, suggests that these materials could be very promising DDH catalysts.
Pd and Pd3–Fe supported on SiO2 exhibited propene yields of 4.5% and 9.4% respectively at 510 °C. The bimetallic catalyst exhibited a much higher propene yield than Pd-only, despite both catalysts having a similar average nanoparticle size (ca. 2 nm).165 The STY of the Pd3–Fe catalysts was 32 molC3H6 kg−1cat h−1 (Table 7, entry 1). This was partly due to a geometric effect in the formation of the Pd3–Fe alloy nanoparticles, breaking the ensemble of Pd, thus enhancing the selectivity and suppressing structure sensitive C–C hydrogenolysis and coking reactions. Electronic effects, often thought to promote high selectivity in Pt dehydrogenation catalysts, were not concluded to strongly influence the selectivity of Pd3–Fe/SiO2.
Recent advances in catalyst design have yielded non-noble-based intermetallic compound (IMC) catalysts, which present unique surface and catalytic chemistry that may be promising for propene production. He et al. explored highly selective and stable phase-pure Ni3–Ga IMC nanoparticles with Ga-rich surfaces supported on Al2O3. These exhibited high selectivity (∼94%), good stability over long term (82 h) testing and regeneration ability at 600 °C, resulting in a STY of 6.1 molC3H6 kg−1cat h−1 (Table 7, entry 8).166 In order to compare with industrial-type catalysts, Pt–Sn/Al2O3 (Oleflex) and CrOx/Al2O3 (CATOFIN) catalysts were prepared and tested under the same reaction conditions. It was shown that the Ni3–Ga/Al2O3 catalyst exhibited comparable conversion to Pt–Sn/Al2O3 and inferior conversion to CrOx/Al2O3, although after 14 h on-stream the CrOx/Al2O3 had deactivated to a similar conversion as Ni3–Ga/Al2O3. Although the authors claim that Ni3–Ga/Al2O3 exhibited comparable performance to the commercial catalysts, there is no indication that the Pt–Sn/Al2O3 and CrOx/Al2O3 catalysts prepared in this work would be as active as the actual commercial catalysts. Furthermore, the STY of all of the catalysts was an order of magnitude below the literature values associated with Pt–Sn and CrOx catalysts (Tables 3 and 4, respectively).
Typically, oxygenic functional groups on the surface of carbonaceous materials, high surface area, stable structure, tuneable nanoscale dimensions and porosity are all important parameters which determine the efficiency of metal free carbon-based dehydrogenation catalysts. The induced activity of the oxygenic functional groups is a consequence of localization of π-electrons from defects, edges and vacancies.167 Meanwhile, their porous structures are key determinants in achieving efficient mass and diffusion transfer.
Hu et al. recently reported the exploitation of biomass-derived porous carbon materials as dehydrogenation catalysts prepared by a simple alkali (KOH) activation method.168 The carbon catalyst activated with KOH at 700 °C exhibited the best catalytic activity, displaying an initial propene yield of 36.6% and a STY of 4.5 molC3H6 kg−1cat h−1 (Table 7, entry 7). The activity was attributed to the carbon catalysts possessing high surface area, hierarchical porous structure, and abundant oxygenated functional groups. Interestingly, no carbon deposition was observed with a carbon balance of 100% being maintained over 10 h of reaction time, rendering high stability signified by negligible loss in surface area and pore volume.
Recent interest into mesoporous carbons has yielded numerous dehydrogenation catalysts, with the large and well-ordered porosity proving beneficial for mass transport while long term stability (of up to 100 h) has also been shown, which exhibited an STY of 5.7 molC3H6 kg−1cat h−1 (Table 7, entry 3).169 As discussed above, it has been previously mentioned that coke formed in the DDH reaction can catalyse the reaction itself, so it is expected that carbonaceous catalysts would exhibit some activity. Of course the obvious drawback of such catalysts is the inability to burn off the coke, as would be typically done over mixed metal oxide or supported Pt catalysts in order to regenerate them. Mesoporous carbons prepared via a soft templating method and carbonized at 600 °C, 700 °C and 800 °C were investigated by Hu et al.170 The increase in carbonization temperature positively influenced the propene selectivity of the aforementioned catalysts to 85.6%, 88.7% and 93.5%, respectively. The authors observed tuneable concentrations of the CO, OC–O and –OH functional groups by adjusting the carbonization temperature. As such, low carbonization temperatures (600 °C) enhanced deep cracking due to surface acidic sites (e.g. OC–O), hindering propene desorption. The subsequent increase in carbonization temperature (up to 700–800 °C) caused the decomposition of the acidic sites, thus increasing propene selectivity. Consequently, a linear relationship between the concentration of CO groups and specific activity was established. The application of resorcinol-formaldehyde and Pluronic F127 also induced different pore ordering, with highly ordered mesoporous carbons displaying improved catalytic performance as a result of more accessible active sites and favourable mass transport properties.
Moreover, Pan et al. reported the modification of carbon with phosphorus from various sources, which introduced defects, forming numerous surface functional groups.171 It was found that P modification of mesoporous carbon with triethyl phosphate had a promoting effect relative to (NH4)2HPO4- and H3PO4-modified mesoporous carbon displaying initial propene yields of 27.2%, 18.7% and 17.2%, respectively. The most active catalyst exhibited a STY of 3.4 molC3H6 kg−1cat h−1 (Table 7, entry 6). The improved catalyst performance was attributed to larger specific surface area, pore volumes, and pore sizes, as well as an increased number of carbonyl/quinone groups and a higher degree of graphitization.
There are many non Pt, Cr, Ga and V containing catalysts in the literature that have not been given the same attention as the aforementioned elements. Notable examples include Zn/ZrTiOx and ZrO2, which in terms of STY, outperform many Pt and Cr-based catalysts. These catalysts consist of non-toxic, abundant elements and are good candidates for commercialisation. Based on an analysis of the patent literature and commercial press releases, we suggest that the catalyst in the K-PRO process consists of a mixed metal oxide of Zr, Zn and Ti. Despite very impressive catalyst performance, investigations into Zn and Zr catalysts are very few. In contrast to Pt, Cr, Ga and V catalysts, which have been extensively studied and are fairly well-understood, Zn and Zr catalysts should be studied in greater detail to better understand the active species involved and how the performance can be further improved.
In conclusion, the direct dehydrogenation of propane can be catalysed by a broad range of metal oxides. Pt and Cr-based catalysts constitute the most commonly reported catalysts and are amongst the most active, which is not surprising given that both of these have been implemented in commercial processes. However, improvements in this sub-field of catalyst research has been incremental for some time and it is unlikely that a step-change in activity will be realised by further investigation. In contrast, Zr and Zn based catalysts are relatively immature and there are many opportunities to prepare highly active, stable DDH catalysts using these elements. Despite numerous investigations into coke formation over various catalysts, all DDH catalysts are prone to coking to some extent, and this remains the frontier in DDH catalyst design. More often than not, the origin of coke formation is due to strong acid/base sites on the support, or specific structures of the active component. Careful preparations and appropriate promotors can help to neutralise coke formation, but this can come at the expense of propene selectivity or propane conversion.
5. Oxidative dehydrogenation with oxygen
The oxidative dehydrogenation of propane using oxygen (ODH-O2) is an attractive route to propene due to favourable thermodynamics, theoretically being unlimited and exothermic is highly beneficial for industrial scale-up. Oxygen is cheap and non-toxic, but conditions must be controlled to ensure that the oxygen/propane ratio is not flammable. Regarding safety and industrially relevant processes, if selectivity is improved further for ODH-O2, then the reaction could be an exciting and useful process. In general, productivities are not currently high enough to be of interest for industrial application due to the low selectivity of many metal oxide catalysts, but more recent developments in the field may change this, with boron-based catalysts forming ethene as a secondary product in contrast to COx products over metal oxide catalysts.ODH-O2 reaction pathways are much simpler than DDH and ODH-CO2, with fewer side reactions and products forming over typical catalysts. Overoxidation is the main limitation in ODH-O2. For metal oxide-based catalysts, gaseous oxygen is utilised via a Mars-Van Krevelen mechanism, with lattice oxygen abstracting hydrogen from propane. The following section describes the most significant literature reports on catalysts for ODH-O2.
5.1. Vanadia based ODH-O2 catalysts
The first vanadia-based catalysts for oxidative dehydrogenation were V–Mg–O catalysts applied for the ODH-O2 of butane. In 1988, V–Mg–O catalysts were found to be active and selective to propene when compared to V2O5 and MgO.172 Since this early discovery, there have been many investigations into V-based catalysts for ODH-O2, which have focussed on catalyst optimisation as well as understanding the nature of the active site.Sam et al.173 analysed Mg2V2O7, which in contrast to other V–Mg–O phases tested, contained vanadium species with a VO bond. Mg2V2O7 exhibited a conversion of 6.9% and propene selectivity of 53.5% at 550 °C, which was much more productive than Mg2V2O8 and Mg2V2O6. This early work highlighted the importance of the VO bond in producing effective ODH-O2 catalysts. Corma et al. later examined the influence of supports for supported vanadia catalysts.174 It was shown that VO2+ pairs present in V2O5 are active for ODH-O2 but were also the source of overoxidation to COx products. More acidic supports, such as silica, decreased dispersion and therefore more V2O5 was detected, compared to more basic supports such as MgO and TiO2. It was also found that if the VO bonds were adjacent to V–O sites, then overoxidation occurred. Limiting the formation of V2O5 and forming V–O sites with tetrahedral coordination was found to be crucial to forming a selective catalyst.
Subsequent reviews of the literature by Kung and Kung,175 and Blasco and López Nieto176 further highlighted that VO bonds were the active sites for ODH-O2 in these materials. These reviews have guided modern investigations to focus on preparing isolated V species on metal oxide supports, rather than Mg-containing mixed oxide materials. It was shown that ODH-O2 rates initially increase with VOx surface density but decrease when V2O5 crystallites form at high V loadings, due to agglomeration of particles. It was also found that the formation of V2O5 crystallites significantly reduced the selectivity to propene, as the surface propyl species will react over two V atoms.177 The formation of V2O5 was shown to occur at lower V loadings for more acidic supports, such as SiO2, whilst using a high surface area silicate, MCM-41, was found to maintain a high STY due to a larger area for dispersion.178 A general trend can be shown across the literature in which isolated VO4 species (sometimes denoted as monomolecular or monomeric) are predominant at low vanadium loadings, and as the VOx surface density increases, the predominant V species become V2O7 dimers, forming polymeric vanadia across the surface (also commonly denoted in the literature as polyvanadate, V2O5-like, 2-dimensional or more generally, VOx). With higher V loadings, V2O5 crystallites become present (also denoted as 3-dimensional species), as shown in Fig. 7. This has been shown to be the case for many supports including SBA-15,179 TiO2180 and γ-Al2O3181
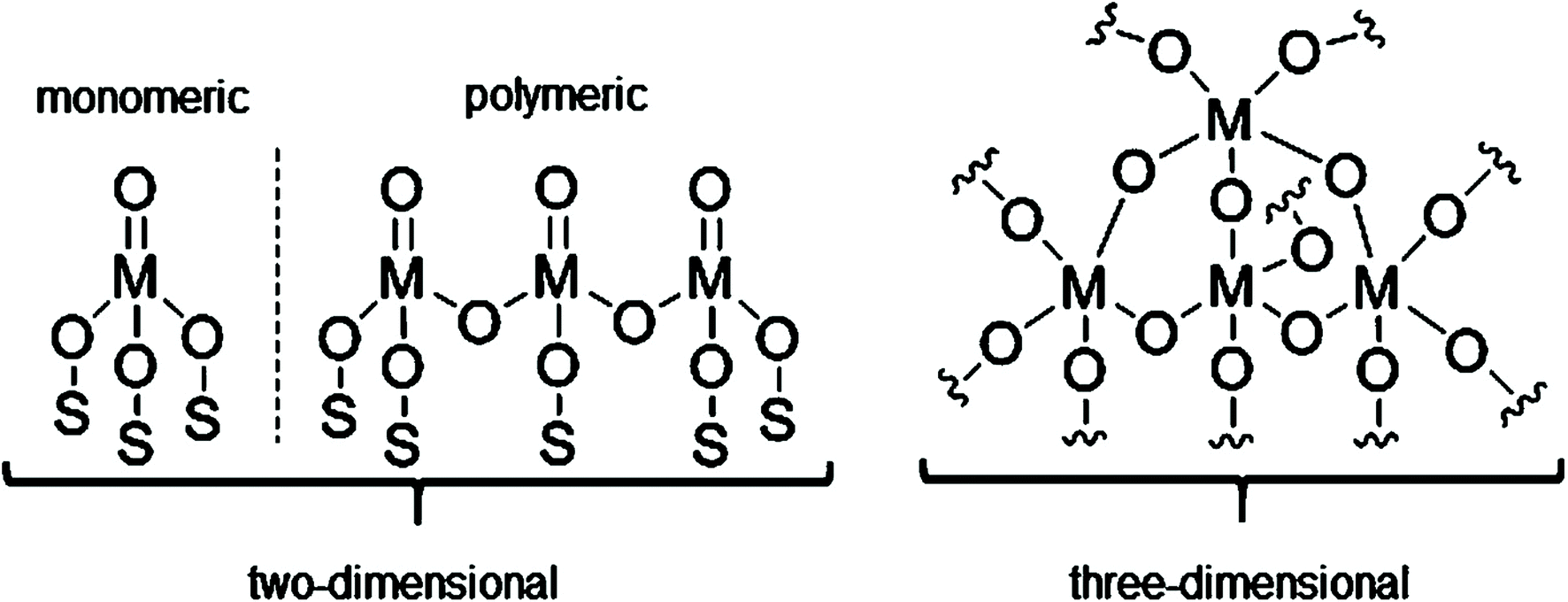 | ||
| Fig. 7 Two dimensional (monomeric and polymeric) and three-dimensional structures for supported metal catalysts. S = support atom, M = support metal.182 | ||
The density of isolated V sites can also be influenced by the nature of the support, and some supports can sustain a higher density of isolated V sites compared to others. For example, a higher density of isolated V sites was achieved on SBA-15, when compared to conventional SiO2 and MCM-41, due to a lower surface acidity.183 The loading of vanadia required to sustain only monomeric sites is 8–9 V nm−2 on most supports, with SiO2 showing a maximum loading of 3.3 V nm−2 for monomer only coverage.184 SiO2 tends to have a poor interaction with the vanadia precursor species in general due to similarities in the acidity of both, so dispersion of V atoms is lower. Depending on the preparation method, monomolecular or polymeric species can exist with high dispersion, or 3-dimensional V2O5 crystallites can form even below monolayer coverage.
An in-depth review of vanadia based catalysts for ODH-O2 was undertaken by Carrero et al.4 in 2014. The review highlighted that the activity of supported vanadia catalysts can be explained by one scaling parameter: the energy of oxygen defect formation. This in turns defines the reducibility of the site and therefore the redox potential. In practice, the formulation of the oxide support, the V loading (and subsequently, the dispersion and speciation) all affect the final catalyst structure and give rise to the wide-range of performances reported. The review also highlighted the complexity in the relationship between V species and catalytic activity: not all VO4 units are equally active; the local geometry and support affect the reducibility and therefore activity. In fact, the authors concluded that despite many reports in advancing the synthesis of supported VOX catalysts, only a minority of surface species are highly active. Since 2014, several studies have further improved vanadia dispersion and subsequently, the catalyst performance. The most significant advances in this endeavour are discussed below.
The use of acidic supports has been probed, as acidity has been shown to increase the activity of ODH-O2 catalysts. Supporting VOx supported on H-ZSM-5 was shown not to enhance performance since, despite the acidity of the support increasing propane conversion for low V loadings. The support influence was detrimental to selectivity and overall lowered the propene yield. The isolated VOx species present at low V loadings became reduced at a lower temperature compared to polyvanadate species and V2O5, suggesting that isolated species can facilitate oxidation of propane more effectively.185
In contrast, vanadia has been supported on Al2O3 that was doped with varying concentrations of MgO to induce basic site functionality.186 Increasing the Mg content led to a decrease in strong acid sites, decreasing activity, consistent with the fact that strong acid sites can activate propene. An Mg/V ratio of 0.8 showed the highest yield of propene, as the ratio of Mg/V balanced the effects of strong acid sites for propane conversion, whilst limiting overoxidation through the addition of Mg resulting in a change of surface species and acidity. The strength of the V–O bond was also influenced by Mg doping, with the binding energy of V–O bonds increasing with increasing Mg content. This effect increased selectivity by decreasing overoxidation due to the inhibited lability of lattice oxygen.
Zhang and Liu187 also investigated V–Mg–O catalysts with the aim of increasing the loading whilst maintaining a high level of dispersion on SBA-15, which has previously been show V2O5 crystallite formation even at low loading levels. To limit agglomeration of vanadia, co-impregnation of V and Mg was undertaken which resulted in the suppression of 3D V2O5 crystallite formation. In this investigation, it was shown that with increasing V content, the V environments went from isolated VO4 species to Mg3V2O8, and no crystalline V2O5 was detected, even with V loading levels of 25 wt% when using co-impregnation of V and Mg. The Mg3V2O8 phase was shown to be intrinsically more selective than dispersed VO4, and at 20% propane conversion, propene selectivity was found to be higher for Mg–V–O catalysts, than V–O catalysts, with some dependence on crystallite size (a 20–30 nm crystallite size gave the highest propene selectivity of ∼59%). Small Mg3V2O8 crystallites showed preferential kinetics (i.e., a higher rate of propene formation compared to rate of combustion and rate of propene formation) when compared with dispersed VO4 species, since the latter have an increased preference for propene oxidation compared to Mg3V2O8. This is interesting as H2-TPR showed a lower level of reducibility for Mg3V2O8 when compared to VO4 species, suggesting that former catalyst was sufficiently reducible to achieve oxidation of propane, but not to overoxidise to COx products.
Supporting vanadia on SBA-15 via wet impregnation has been explored and a loading of 6.6 wt% V achieved a surface density of 1.6 V nm−2, the highest surface density achieved on SBA-15 without V2O5 crystallite formation; coverage was increased by rehydrating the support before impregnation, aiding dispersion.188 The increased dispersion led to an increased selectivity when compared to similar catalysts. Zhu et al. investigated pre-treatment of the support, with the simple idea that modifying the support before loading V could control the dispersion.189 A SiO2 support was treated at different temperatures before grafting VOCl3 onto the surface. Higher temperature treatments (700 °C to 1000 °C) resulted in the removal of neighbouring silanols and only isolated silanols were detected using IR spectroscopy. Isolated silanols can anchor VOx species, and isolated silanols would be expected to produce monomeric VO4. Due to the silanol site concentration decreasing, the loading of vanadia decreased with increasing support pre-treatment temperature. However, the increase in pre-treatment temperature also led to an increase in monomeric vanadia sites compared to dimeric sites. The catalysts with the highest abundance of isolated monomers showed the highest propene selectivity of 90% at 500 °C and 68% at 600 °C (Table 8, entry 4), leading to the conclusion that monomeric sites are more selective than the polymeric structure. This study highlighted the difference in selectivity between different VOx species. It must be noted that no 3D V2O5 crystallites were detected when using VOCl3 grafting on heat-treated SiO2 supports, and therefore selectivity to propene was maintained.
| Entry | Catalyst | T/°C | O2/C3H8 | WHSV/h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature O2/C3H8 = molar ratio of CO2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. Value next to element/phase represent wt%. |
||||||||
| 1 | 5V-Zr–O fibres | 400 | 0.5 | 70.8 | 4.0 | 64.6 | 38 | 192 |
| 2 | 0.4Na+–V–SiO2 | 490 | 0.5 | 19.3 | 6.5 | 66 | 17 | 182 |
| 3 | 3V/γ-Al2O3 | 500 | 0.5 | 4.7 | 12.5 | 75.9 | 9.4 | 194 |
| 4 | VOx/SiO2−(1000) | 500 | 1 | 4.7 | 30 | 60 | 18 | 189 |
| 5 | 3V-DMSN | 500 | 0.5 | 6.3 | 10.4 | 66.5 | 9.1 | 146 |
| 6 | V/SiO2 (SOMC) | 525 | 0.5 | 118 | 5 | 66 | 82 | 190 |
| 7 | 1.8V-KIT-6 | 550 | 1 | 4.7 | 46.2 | 64.5 | 30 | 193 |
To probe the influence of monomer sites, Barman et al.190 grafted monomeric V species onto SiO2 using surface organometallic chemistry (SOMC). This approach represents another example of changing the preparation technique to increase dispersion. It was found in this study that monomeric vanadia species are intrinsically more selective. The incipient-wetness impregnation technique formed catalysts which were more selective than SOMC catalysts below 500 °C, but above this temperature, the SOMC catalysts exhibited an equivalent or even higher selectivity. A STY of 82 molC3H6 kgcat−1 h−1 (Table 8, entry 6) was measured over V/SiO2 prepared via SOMC.
In addition to high dispersion and control of oxygen environments of the vanadia, tailored support properties are crucial in optimising ODH-CO2 performance. A widely debated topic in catalysis is the effect of support porosity on catalytic properties. The preparation route of Al2O3 was varied to form flower-like, sphere-like and bulk-like morphologies to investigate the influence of varying porosities. Bulk-like morphology only exhibited intraparticle pores, and the monolithic architecture of γ-Al2O3 (formed by a roasting method) achieved the highest productivity for ODH-O2, despite the surface vanadia species being consistent across all architectures, with the degree of polymerisation being fairly similar for all Al2O3 structures as (shown by adsorption edge energy analysis).191 The bulk-like morphology showed a lower resistance to external mass transfer, and reduced the residence time of propene through the catalyst bed, lowering the tendency for further reactions.
Mesoporosity was also explored when forming fibrous vanadia catalysts via electrospinning. A comparison between fibrous catalysts which presented mesoporosity, and particle-like catalysts without apparent mesoporosity, indicated no influence on catalytic performance due to porosity,192 consistent with the aforementioned morphology analysis. However, it is interesting to note that a change in preparation technique changed performance. A one-step synthesis to form VZrOx catalysts was found to form the most active catalyst with 5 wt% V loading, when compared to incipient wetness impregnation catalysts. Incipient wetness impregnation with 5 wt% V loading showed the formation of 3D-V2O5 crystals, known to be detrimental to selectivity, whilst the one-step process did not show evidence of V2O5 formation and improved the dispersion of isolated VOx sites. It is also noteworthy that the VZrOx catalysts from the research by Ternero-Hidalgo et al. operate at a lower temperature, namely 400 °C, when compared to other catalysts in the field (Table 8, entry 1) and achieved a STY of 38 molC3H6 kgcat−1 h−1.
Vanadia supported on dendritic mesoporous silica nanoparticles (V-DMSN) was formed via a one-step synthesis as DMSN materials were found to have large pore volumes and a larger surface area than conventional mesoporous silica.146 The one step synthesis, similar to the VZrOx catalysts discussed previously, led to higher dispersion and a higher stability of vanadia on the DMSN support when compared to incipient wetness impregnation (denoted V/DMSN). A surface V density of 0.55 V nm−2 (2 wt% V), gave the highest STY of 9.1 molC3H6 kgcat−1 h−1 (Table 8, entry 5). H2-TPR analysis revealed that the isolated V species were more reducible, which is a strong predicter of activity in ODH-O2.
Despite SiO2 having a large surface area, and that many different types of silica supports having been tested, the low reactivity of silanol groups towards V precursors means that a relatively low loading of isolated V sites are reported. Doping the SiO2 surface with 0.4 wt% Na+ enhanced the V dispersion to a level corresponding to monolayer coverage (i.e., 8.6 V nm−2 for 6.2 wt% V loading) (Table 8, entry 2), which is equivalent to many other metal oxide supports.182 It was hypothesised that Na+ ions provide a source of Si–O–Na+ surface species, which facilitate increased monomeric vanadia species, boosting the propane conversion. However, it should be noted that the intrinsic activity of the V/SiO2 catalysts did not increase; the calculated TOFs were similar for V supported on promoted and unpromoted SiO2. Instead, the presence of Na+ simply facilitated a greater density of isolated V sites. The activity of the Na+ promoted V/SiO2 was sustained for 4.5 days and post-reaction Raman spectroscopy showed no formation of 3D V2O5 crystallites. This is an excellent example of doping having a beneficial effect. However, it is sometimes found that doping has deleterious effects on redox properties. For instance, doping with K+ was found to decrease the selectivity and activity when compared to V-only supported catalysts.193 When loaded onto KIT-6 supports, the V began to segregate with the addition of K+ ions because VOx and K+ have a strong interaction, which led to the formation of unselective K+ and V mixed phases such as potassium vanadate. The interaction between K+ and VOx species decreased the reducibility of the catalyst. Interestingly, K+ incorporation led to an increase in the ethene to propane ratio, but the alkene yield was decreased overall. The incorporation of basic metal oxides should be done cautiously, as the benefit of enhanced propene desorption can be outweighed by the detrimental effects on the redox properties of the catalysts.
In conclusion, vanadia catalysts have been extensively investigated for ODH-O2, with the main focus being on maximising the number of isolated vanadia species, which is fundamental to increasing propene selectivity. A more precise view of vanadia species in recent studies has shown isolated vanadia sites to be more selective than polymeric species which contain V–O–V bridging bonds. Therefore, high activity is associated with high V dispersion. Acidic supports inherently decrease V dispersion, thus decreasing selectivity and overall productivity despite increasing activity. More frequently, there are reports of dispersion being influenced by the preparation technique, with grafting vanadia onto the support surface showing promise. However, a one-step synthesis of support and vanadia, in contrast to analogous surface impregnation catalysts, has been shown to improve catalyst productivity. The one-step synthesis method was shown to improve productivity for different support materials, morphologies and loadings when compared to analogous catalysts formed with more traditional surface loading techniques. Research into one-step synthesis methods is far from exhausted and shows considerable potential for further development. In contrast, incipient-wetness techniques can still be improved, with hydration of the surface or doping being effective strategies. Doping has been shown to influence the intrinsic reactivity of the active sites, but an increase in activity is usually accompanied by a decrease in selectivity. Increasing selectivity via dispersion techniques may be deemed a more promising route to improving catalytic performance. The choice of support material composition and chemical properties is more influential than morphology on overall catalytic performance; many studies comparing mesoporous catalysts with bulk-like morphologies found mesoporosity to have either a detrimental or negligible effect on productivity. Porosity can, however, increase specific surface area and therefore support a higher loading of isolated vanadia sites.
5.2. Boron-based ODH-O2 catalysts
Boron based catalysts are relatively new to the field of dehydrogenation catalysis. Hexagonal boron nitride (h-BN) has historically been used a catalyst diluent due to its high thermal stability under oxidative conditions. The 2-D structure of h-BN also allows for high thermal conductivity via efficient phonon transmission across the sheets of BN.195 High thermal conductivity is appealing with regards to industrial application, as this limits the formation of hot spots, and selectivity becomes more controllable. The discovery that h-BN catalyses propane dehydrogenation was first reported in 2016196 and since then there has been significant developments in the understanding of the active centres and reaction mechanisms. In the initial investigation, h-BN (Table 9 entry 8) was observed to catalyse ODH-O2, with similar activity to V/SiO2 catalysts. However, boron nitride nanotubes (BNNTs) displayed higher productivity, selectivity and yield than conventional V-SiO2 catalysts with the BNNT having a STY of 110 molC3H6 kgcat−1 h−1 (Table 9, entry 1), which is ∼7 times greater than the analogous V/SiO2 materials tested.196Fig. 8 illustrates this difference in performance. In contrast to supported VOx catalysts that produce COx, the major by-product is typically ethene, which is of course more valuable than carbon oxides. | ||
| Fig. 8 Comparisons of propene productivity plotted as a function of C3H8 conversion between V/SiO2, h-BN, and BNNT. V/SiO2: 5 to 15 kg-cat s mol C3H8–1; h-BN: 15 to 40; BNNT: 2 to 5; T = 490 °C, PO2 = 0.15 atm, PC3H8 = 0.3 atm. Reproduced from ref. 196 with permission from The American Association for the Advancement of Science. | ||
| Entry | Catalyst | T/°C | O2/C3H8 | WHSV/h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; O2/C3H8 = molar ratio of O2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. Values next to element/phase represent wt%. |
||||||||
| 1 | BNNT | 490 | 0.5 | 31.7 | 6.5 | 76 | 110 | 196 |
| 2 | BN/DFNS | 490 | 1 | 1.2 | 18 | 59.4 | 2.4 | 198 |
| 3 | Elemental B | 490 | 2 | 152.7 | 9.8 | 83 | 280 | 199 |
| 4 | 10B2O3/SBA-15 | 500 | 1.5 | 9.5 | 31.5 | 64 | 43 | 207 |
| 5 | BN-OH | 530 | 1.5 | 37.6 | 20.6 | 80.2 | 130 | 197 |
| 6 | Macroporous BPO4 | 535 | 1.5 | 93.9 | 20.8 | 78.1 | 320 | 211 |
| 7 | SiB6 | 545 | 1.5 | 9.5 | 31.1 | 72.1 | 48 | 201 |
| 8 | h-BN | 560 | 0.5 | 4.0 | 14 | 79 | 11 | 196 |
Such a different product distribution hints at a distinct reaction mechanism from other metal oxide ODH-O2 catalysts, and investigations into the reaction mechanism have suggested as much. First order dependence on O2 partial pressure and second order dependence on propane partial pressure was noted by Grant et al.196 It was also suggested that molecular oxygen forms an “armchair” from B to N at the platelet edges to form a B–O–O–N active site, but subsequent studies found catalysts without any N present were also highly active.
Edge-hydroxylated boron nitride (BN-OH) was subsequently reported to have the highest productivity of any previously reported catalyst for the dehydrogenation of propane.197 BN-OH catalysts were also shown to be stable over 300 h on-stream (with no significant reduction or change in selectivity at 530 °C). The BN-OH materials were catalytically active after O2 activation, with DFT calculations suggesting that neighbouring B-OH sites formed B–O–B linkages once both B-OH groups are dehydrated. The sites could be reduced back to BN-OH using steam. Clear discrepancies were seen between previously proposed reaction mechanisms and explanations on how structure influences catalytic activity, showing that in situ edge functionalisation is key to catalytic activity. When comparing supported BN and supported B2O3 on dendritic fibrous nanosilica (termed BN/DFNS and B2O3/DFNS respectively), BN/DFNS showed improved productivity relative to B2O3/DFNS, although both were active.198 This finding is noteworthy, as it shows that BN on the surface can be oxidised to B2O3 during the reaction. The as-prepared BN catalysts however already contained a large proportion of hydroxyl groups, underlining the importance of hydroxylated B in catalysing ODH-O2. This observation is consistent with the activity trends noted for edge-hydroxylated BN (Table 9, entry 2).
Subsequently, the Hermans group probed the influence of heteroatoms on activity and selectivity of boron-based catalysts using a range of metal borides, including B4C, TiB2, elemental B (Table 9, entry 3) and WB amongst others. Elemental boron showed a remarkably high STY of 280 molC3H6 kgcat−1 h−1,199 which represented an improved productivity over BN-OH catalysts that had been previously described in the literature. In a subsequent review, it was noted that the propene selectivity was the same at isoconversion for every boron material that was tested,200 suggesting that the boron heteroatom is the source of the active site. Metal borides with a high oxygen and boron surface content showed the highest productivities and the presence of BOx species have been identified as essential for catalytic activity. The catalytic activity of various B compounds depended on the concentration of B at the surface. Under ODH-O2 conditions, the B is oxidised to BOx, which is the active site. It was also shown that the activation of O2 is the rate-limiting step for boron-based catalysts in ODH-O2. These new insights disproved the previously suggested mechanism that required N.
Yan et al. reported that silicon boride was active for the oxidative dehydrogenation of propane, ethane and isobutane.201 At 545 °C, SiB6 exhibited a STY of 48 molC3H6 kgcat−1 h−1 (Table 9, entry 7) and subsequent stability tests at 545 °C showed that a propane conversion of 20% and propene selectivity of 80% could be maintained over the 100 h experiment.
An extended literature review by Shi et al.5 in 2018 outlined recent advances in testing and further understanding of structure–performance relationships. Key comparisons were drawn between traditional V catalysts and the highly promising B catalysts. In particular, COx selectivity for edge hydroxylated boron catalysts was shown to be 0.5%, in comparison to 10–50% for V-based catalysts, whilst productivities for boron catalysts can be maintained for 300 h. Mechanistic studies showed oxygen to have a reaction order of 0.5, suggesting that dissociative adsorption of oxygen is of importance to facilitate the reaction, whilst in the absence of oxygen catalysts had no activity. In situ infrared analysis has demonstrated that the presence of OH groups led to the interaction of molecular oxygen with the surface. Interestingly, alkane partial pressure was shown to have a reaction order of 2,197 with two propane molecules needed during the reduction phase of the redox cycle, to form the two adjacent BN-OH sites in the original catalyst (Fig. 9).
 | ||
| Fig. 9 Proposed reaction mechanism for ODH-O2 over boron nitride catalysts for the oxidative dehydrogenation of propane. Reproduced from ref. 202 with permission from Elsevier. | ||
As described above, there are differences in the reaction mechanism between boron-based catalysts when compared to supported metal oxide catalysts. For example, h-BN and other boron-based catalysts do not function for non-oxidative propane dehydrogenation,199 whilst metal oxides do function initially and do not require an induction period. This shows that boron-based catalysts form the necessary active sites under reaction conditions, and that the initial catalysts can be perceived as precursors to the active phase. Propane partial pressures exhibit a second order rate dependence for h-BN, whereas in metal oxides it is first order. Metal oxide catalysts such as VOx and MoOx used for ODH-O2 operate using via a Mars-Van Krevelen mechanism, whilst h-BN cannot do this due to the absence of lattice oxygen.200 Therefore, in order to optimise boron-based catalysts, a more complete understanding of the reaction mechanism must be developed to tailor the nanostructure to the process.
DFT studies of optimised structures of h-BN have been undertaken to probe the ODH-O2 reaction mechanism. The dissociative O2 adsorption was shown to be favoured on B terminated edges, rather than N terminated edges, leading to B atom terminated platelet edges being more active. DFT calculations also showed that the dehydrogenation reaction at B atom terminated edges is favoured over C–C bond breaking due to kinetic and thermodynamicfactors.203 Other DFT studies have built upon the understanding that boron is the active centre for ODH-O2 and suggest that C–H abstraction is undertaken by heterolytic cleavage over a B–O pair in a BO3 active structure. It has been suggested that O2 abstracts the hydrogen from the newly formed O–H group, reducing O2 to HO2, and subsequently forms B-OOH and B-C3H7 on neighbouring BO3 sites. The HO2 group then abstracts hydrogen from the propyl surface intermediate to form propene, whilst H2O2 breaks down to form H2O and the BO3 active site is regenerated.204
Experimental studies comparing h-BN and VOx/Al2O3 once more highlighted the different reaction mechanisms at work. It has been shown h-BN exhibited a C2/C1 product ratio of 2, whilst the VOx catalyst shows almost no C2 products, and complete oxidation to COx dominated.205 This implies that different reaction pathways dominate, with only the first C–C bond cleavage being favourable for h-BN catalysts, leaving the methyl group to have many different reaction pathways. Interestingly, an increase in C2H4 selectivity was recorded with increasing temperature. Methyl radicals generated from C–C bond cleavage over h-BN catalysts can undergo four secondary reactions which include oxidative coupling of the methyl groups to produce C2 products. The oxidative coupling leads to a better yield of C2 products, whilst other processes such as hydrogenation of methyl to form methane and selective oxidation to CHxOy serve to lower the COx selectivity. Although the study did not undertake extensive surface characterisation techniques, the origin of selectivity is explained and may lead to improving conditions and structural properties by mapping the various reaction pathways. For example, isoconversion studies would extend understanding of the reaction mechanism and form a better comparison between h-BN and VOx/Al2O3.
Advanced characterisation of gas-phase radicals was undertaken by Zhang et al.204 to develop an understanding of reaction pathways of methyl radicals which are formed during C–C bond cleavage. Using synchrotron radiation vacuum ultraviolet photoionization mass spectroscopy (SVUV-PIMS), methyl radicals were detected under reaction conditions (600 °C, ODH-O2 conditions with BN treated via gas exfoliation to increase surface area to 72.7 m2 g−1). The detection of gas phase methyl radicals linked with the kinetic data showed that methyl radicals contribute to C1 and C2 products. The selective route to propene was expected to be a surface-controlled pathway as no propyl radicals were detected by SVUV-PIMS, despite the group being able to demonstrate a high sensitivity for propyl radicals. Therefore, control of conditions to maximise surface-controlled processes may increase the selectivity to propene and limit C1 and C2 products. Both high surface BN and supported oxidised boron catalysts were tested, with both showing formation of gas-phase radicals.
Venegas et al.206 also highlighted the importance of gas-phase reactions in the reaction mechanism over B-based catalysts and suggested that the propyl radicals are not bound to the surface and react with HOO˙ radicals in the gas-phase. The hypothesis is that if propane reaction pathways were surface-mediated, then similar selectivity-conversion trends to metal oxides would be seen. This is a direct contradiction of the analysis of radicals undertaken using SVUV-PIMS shown previously, which did not detect propyl radicals despite previously showing the capability, and so further elucidation may be needed.
Lu et al. investigated various B2O3/SBA-15 catalysts. 10 wt% of B2O3 yielded the highest conversions at 500 °C, while conversion drops above this loading.207 The supported boron oxide catalysts showed activity at 405 °C, much lower than previous examples of boron-based catalysts. Analysis by 11B NMR detected four different B environments in fresh and spent catalysts: namely B3O9/2 (boroxol ring), BO3/2 (non-ring), tri-coordinated planar B species and tetra-coordinated B species. The latter species were fused into the silica matrix. The presence of boroxol ring and non-ring sites was proposed as the reason for activity at lower temperature, but the data presented does not differentiate between the two. Comparison of fresh and spent catalysts showed an increase in the boroxol ring environment and a decrease in BO3/2. This suggests a structural change due to oxidative conditions, rather than temperature, as the materials were prepared at 700 °C.199
Amorphous B(OH)xO3−x was identified using 11B solid state NMR, SEM and XAS after h-BN and BNNTs were exposed to ODH-O2 conditions. Boron species with different levels of hydroxylation were detected using 1H–11B double resonance experiments208 to show a wide range of oxidation states associated with boron. A range of oxide bridges, hydroxyl and oxide species were present, which result from oxidation and hydrolysis of BN catalysts. The study clearly demonstrates the complexity of these catalysts under ODH-O2 conditions, and that in situ characterisation of the working catalyst is crucial to understanding the reactivity.
Boron oxide was supported on SiO2 to probe how structural control of the surface influences the catalytic performance of boron-containing catalysts. The preparation method employed (calcination of tri-isopropyl borate on an amorphous silica support at 550 °C) formed BO3 chains, which cannot be replenished, unlike h-BN and BNNT. The BOx/SiO2 material showed a ∼10% lower selectivity for a similar conversion when compared to h-BN, but the loading of B was comparatively low, and the bare SiO2 may lower the selectivity.209 Dual or single boron sites were present with varying levels of oxidation and hydroxylation after exposure to ODH-O2 conditions, and the active centre of the catalyst may be one or more of these species. The difficulty in fully understanding the active site in this system is due to the co-existing boron species detected after exposure; BO3 and B-OH have both been implicated but conclusive evidence has not been found. The appearance of metaborate and BO3OH species in the spent catalysts was detected, but there is no data suggesting that these are desirable. It is clear that restructuring of the surface occurs, with a dynamic active site that undertakes ODH-O2. It is also reasonable to expect that a hierarchy of activity exists, where some B species are more active than others, but a multitude of oxidised or hydroxylated boron surface sites can catalyse the reaction. The preparation of single-site boron catalysts would be instructive to understand the active phase.
A h-BN/cordierite monolithic catalyst exhibited a productivity increase of 55% when compared to elemental B. This is among the most active B catalysts reported to date. It was shown that optimising conditions for a high WHSV can lower CO2 selectivity to 0%, which impressive even by the standards of h-BN catalysts.210 It was hypothesised that the high WHSV did not allow for deep oxidation and therefore limited the formation of COx products. The monolith surface was characterised using FT-IR to confirm the presence of BN, but the B speciation was not determined. Wang et al. speculated that the linear channel structure of cordierite allowed for gas-phase radical chemistry to be enhanced and therefore showed improvement over h-BN powders. The h-BN/cordierite material was prepared by the chemical vapour deposition process. The geometry of the support was essential in this case, as well as the support forming strong oxygen bridge bonds with the catalytic phase.
When comparing macroporous BPO4 to bulk BPO4, it was found that the macroporous structure increased the performance significantly. The STY of macroporous BPO4 was 320 molC3H6 kgcat−1 h−1 (Table 9, entry 6).211 When using the same WHSV, macroporous BPO4 exhibited three-times the conversion of bulk BPO4 despite being tested at a 30 °C lower temperature, whilst achieving roughly the same product distribution. The surface area normalised results revealed that both catalysts were equally active, suggesting that the same tri-coordinated B–O species is the active, but an increased number of active sites were formed in the macroporous structure. The interconnected macroporous structure was found to achieve outstanding productivity when exposed to a large WHSV by facilitating efficient mass transfer through the catalyst. As macroporous BPO4 and h-BN/cordierite both exhibited a very high STY, it is implied that optimisation of mass transfer via engineering porosity has significant impact on the resulting catalyst. These data suggest that the reaction proceeds in the gas-phase, and as long as it can be rapidly initiated by a large surface area, then a large WHSV will limit surface intermediate contact time, and increase selectivity and turnover frequency.
Overall, it is clear that an important and rapidly developing area of ODH-O2 catalysis is the application of boron-based materials. Many reports corroborate that B–O and B-OH species, or a mix between the two (i.e., B(OHx)O3−x) are the active sites for h-BN based catalysts. While many reports examine catalyst design, the specific active site and phase is not well understood. This is to be expected with such a new generation of catalysts, but many papers suggesting different active sites and phases of boron exist, resulting in confusion of how to progress with catalyst improvement. Attempts to understand the mechanism have not yet been validated, and a different range of structures have been concluded to be the active sites. Supported catalysts have been formed and show high productivities and potential advancement in the field, but the fundamental understanding of in situ phases and mechanistic processes is still somewhat lacking. It may be beneficial to explore applications of boron-based catalysts in other oxidation reactions and start to build up structure–activity relationships. For example, focussing on bulk and layered materials as well as high surface area materials, may be advantageous in developing an understanding of the intrinsic properties which are desirable when using boron-based catalysts for ODH-O2. Controlling the structure by a consistent synthesis method seems viable, as shown by the formation of nanoporous hexagonal boron nitride nanosheets (h-BNNS) from amorphous h-BN precursors. The h-BNNS catalysts prepared by Chen et al.212 exhibited a surface area of 347 m2 g−1, as well as high purity, high crystallinity and increased thermal stability when compared to the amorphous h-BN materials. The dehydrogenation activity was measured using dodecahydro-N-ethylcarbazole as the substrate. The h-BNNS catalyst achieved a 34% increase in yield when compared to amorphous h-BN. To build on this, nanoporous h-BNNS could be applied to propane oxidative dehydrogenation, and if this is successful, begin to build structure–activity relationships using these highly crystalline materials.
5.3. Other ODH-O2 catalysts
While the majority of ODH-O2 catalysts are based on BOx or VOx, a multitude of other elements have been demonstrated to be active for ODH-O2. In this section the most significant reports are critically reviewed and compared to the state-of-the-art VOx- and BOx-based ODH-O2 catalysts. Cavani et al. compiled an extensive literature review in 2007, where MoOx-based catalysts were shown to have a lower catalyst performance when compared to analogous vanadia based catalysts for the for ODH-O2 reaction.213 A direct comparison of kinetic parameters for V2O5/Al2O3 and MoO3/Al2O3 in the ODH-O2 reaction showed that vanadia-based catalysts had higher selectivity at isoconversion, and were more reducible than the analogous MoOx-based catalysts.214 The V and Mo catalysed ODH-O2 reaction proceed via similar mechanisms. When comparing V-based catalysts to Mo-based catalysts, both supported on ZrO2, it was found that Mo was less active as the catalyst showed a higher activation energy.215MoO bonds were found to be active for C–H abstraction, with the strength of the MoO bond influencing the activity: the stronger the bond, the lower the activity. Furthermore the strength of the MoO bond was shown to be influenced by MoOx loading. The bridging oxygen bond between Mo and the support was found to be active in the complete combustion of propane.216 The reactivity of Mo–O–M anchoring bonds is influenced by the cation electronegativity in the support material; a low support cation electronegativity leads to increased basicity of the bridging oxygen, and therefore has a higher activity for redox processes.217 Analysing speciation of MoOx on ZrO2 supports found that two dimensional polymolybdate species dominated the surface up to loadings of 5 Mo atoms nm−2 and that further increasing Mo concentration led to a lower activity per Mo atom, ascribed to either limiting access to active sites or strengthening of the MoO bond.216
The selectivity of MoOx/ZrO2 can be increased by the addition of alkali metal dopants to reduce Lewis acidity of the Mo6+ centres allowing propene to desorb more effectively, thus decreasing the rate of consecutive oxidation reactions.218 However, the overall performance of the catalyst was lower due to decreased reducibility, which inhibited the Mo6+ centre from undertaking C–H abstraction. A typical conversion selectivity trend was seen, where selectivity decreases with an increase in conversion. When MoOx was supported on Al2O3, MoO3 crystallites formed above 4.5 Mo atoms nm−2. Below a surface density of 4.5 Mo atoms nm−2, the polymolybdate regions were abundant and ODH-O2 rates were high. Above monolayer surface coverage (4.5 Mo atoms nm−2), the formation of MoOx crystallites occurred which limited the number of exposed Mo–O sites. Unlike VOx catalysts where V2O5 crystallites reduce selectivity, the presence of MoO3 crystallites were not detrimental to selectivity, just the intrinsic activity. Bridging oxygen bonds between the support and Mo for both Al2O3 and ZrO2 were found to be active for propane combustion to COx, in a similar manner to VOx/Al2O3 catalysts.
The above studies of Mo-based catalysts have shown that they are limited when compared to analogous V-based catalysts. A selectivity increase is found for increasing molybdenum loadings up to 10 wt%, indeed suggesting that polymeric MoOx up to monolayer coverage gives the best selectivity. However, selectivity was slightly lower at the same conversion when compared to 2 wt% V catalysts.219 Reports involving Mo containing catalysts often concern mixed metal oxide catalysts containing both V and Mo. When comparing Mo-only to mixed Mo and V oxide catalysts formed via flame spray pyrolysis, a Mo-only loading of 10 wt% on Al2O3 (10Mo/Al2O3) gave a yield of 9.4%, compared to a mixed catalyst containing 4 wt% Mo and 2 wt% V (4Mo2V/Al2O3) reaching a yield of 10% (Table 10, entries 8 and 10). The relative rate of oxidation of propene to COx products, when compared to oxidative dehydrogenation of propane, was found to be lower for 4Mo2 V/Al2O3 than 10Mo/Al2O3 between 350–500 °C. A Mo/V ratio of 2 gave the highest yield for the mixed Mo/V catalysts, and the synergistic nature of the two metals leads to the suppression of overoxidation. This research was limited in that it did not contain any in situ characterisation to probe the synergistic nature of the metal species. Interestingly, V catalysts formed via co-impregnation with Mo exhibited a V–O binding energy increase which limited overoxidation as the oxygen became less labile.220 Mo also increased the concentrations of V4+ and V3+species, which are more selective than V5+ entities. The strength of the V–O bond can therefore be carefully controlled by level of Mo doping, which may be the origin of synergistic properties which were observed for 4Mo2 V/Al2O3 catalysts.
| Entry | Catalyst | T/°C | O2/C3H8 | WHSV/h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; O2/C3H8 = molar ratio of O2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. The value next to element/phase represents wt% loading. |
||||||||
| 1 | Co3O4/ZIF-8 | 250 | 1 | 11.8 | 7.1 | 40.8 | 7.2 | 232 |
| 2 | NiCeOx | 375 | 1 | 3.0 | 30.3 | 39.4 | 7.4 | 230 |
| 3 | NiO-polyoxometalate | 450 | 4 | 0.47 | 44 | 45 | 2.0 | 227 |
| 4 | 5CrOx–MgO | 450 | 0.5 | 5.9 | 10.8 | 84.1 | 11 | 234 |
| 5 | NiO-SPP | 450 | 1 | 11.8 | 21 | 50 | 26 | 228 |
| 6 | B-NiMoO4 | 475 | 1.25 | 3.2 | 18.2 | 63.8 | 8.4 | 243 |
| 7 | Mesoporous NiMoO4 | 500 | 2 | 2.4 | 11.8 | 60.1 | 3.5 | 225 |
| 8 | 10Mo/Al2O3 | 500 | 5 | 3.9 | 32.0 | 29.4 | 7.8 | 219 |
| 9 | NiO–CeO2 | 500 | 1 | 10.0 | 52 | 72 | 79 | 229 |
| 10 | 4Mo2V/Al2O3 | 500 | 5 | 15.6 | 34.0 | 29.4 | 33 | 219 |
| 11 | g-C3N4 | 515 | 0.25 | 1.4 | 23.9 | 58.5 | 4.0 | 238 |
| 12 | 0.5Pt–5Sn/Si-beta | 550 | 0.5 | 4.7 | 48 | 79 | 40 | 241 |
| 13 | NiMoO4 with Ni/Mo ratio 0.4 | 600 | 1 | 2.4 | 32.5 | 48 | 7.7 | 223 |
The cycling of Mo-only catalysts has also been explored.221 Interestingly, propene selectivity increased by 15% for the second ODH-O2 phase, at a similar propene conversion, leading to an overall propene yield increase of 5%. In situ XANES showed that the oxidation state of Mo in the second ODH-O2 phase was between +6 and +4 (corresponding tetrahedral and octahedral coordination respectively), with the change from the initial +6 environment happening under the DDH conditions. The distorted structure between +6 and +4 oxidation state of Mo is seemingly more selective than the tetrahedral Mo moieties in the original catalyst. In situ characterisation has thus highlighted a key structure–property relationship which will be useful in guiding future ODH-O2 catalysts based on Mo.
Mo supported on titanate nanotubes (Mo-TNT) is a rare example of a supported Mo catalyst being developed in recent years. Mo-TNT was explored due to MoOx/TiO2 showing high dispersion,217 an effect that was limited by anatase titania having a low surface area. Doping with K, according to a Mo/K ratio of 10, led to increased dispersion of MoOx across the TNT surface, achieving a conversion of 21.2% and selectivity of 53.3%.222 The increased dispersion caused by doping the titanate with K led to an increase in propene yield by 20% when compared to undoped Mo-TNT catalysts, despite a lower conversion.
Transition metal binary molybdates have also been investigated for ODH-O2 activity. For instance, nickel molybdate catalysts have been shown to be active for ODH-O2.223 Increased selectivity was found to be directly correlated with an increase in Mo/Ni ratio. Further investigation of phases present found that NiO, whilst showing complete conversion, favoured overoxidation, whereas α-NiMoO4 was more active for propane conversion, and β-NiMoO4 was more selective. A Mo/Ni ratio of 0.4 gave the highest propane yield of 15.6% (Table 10, entry 13), which was ascribed to the high ratio of β-NiMoO4/α-NiMoO4, the mesoporous structure and redox properties. Although the activity was identified as being sensitive to the phases present, the underlying properties of each phase were not individually explored.
Chen et al. reported that nickel molybdates were active for ODH-O2 and in particular the α-NiMoO4 phase exhibited an enhanced yield compared to NiO and β-NiMoO4.224 Physical grinding of Mo and Ni precursors also led to the absence of NiO in α-NiMoO4 when compared to the analogous co-precipitation technique. It was shown that NiMoO4 had enhanced ODH-O2 performance when comparing mesoporous to bulk catalysts, but only due to the enhanced number of active sites related to the increased surface area, and not the improved mass transfer properties that usually accompany mesoporosity.225 The mesoporous catalysts had a lower specific productivity due to the loss of MoO3 from the surface of the catalysts during synthesis. This confirmed that MoO3 exhibits favourable activity to NiMoO4 for the ODH-O2 reaction (Table 10, entry 7).
NiO has been found to be detrimental to ODH-O2 selectivity, favouring overoxidation products. Nb was found to be a superior dopant for NiO based catalysts, due to a reduction in the surface concentration of electrophilic O species and increased surface acidity. However, propene was still not the major product over NiO at around 10% conversion,226 suggesting that NiO-based catalysts are limited in their application for ODH-O2. In comparison to Nb-NiO catalysts, α-NiMoO4 showed a selectivity of ∼75% at roughly 10% conversion, with propene being the major product at similar conversions to Nb-NiO. The NiO-polyoxometalates have been shown to stabilise the adsorbed oxygen at the surface, which would limit overoxidation,227 leading to the conclusion that NiO oxygen is too labile to achieve a high selectivity. When undertaking surface polymeric phosphate modification of NiO nanoparticles (NiO-SPP), an increase in selectivity of 2–3 times for NiO-SPP's at isoconversion was shown (Table 10, entry 5).228 This is due to the polymeric phosphates weaker interaction with propene when compared to NiO nanoparticles, leading to a higher desorption rate and therefore higher selectivity.
CeO2 was found to be a selective catalyst when HCl was added to the gas feed with O2.229 O2 was hypothesised to have filled the oxygen vacancies at the surface, forming O22− species, which mediated the oxidation of Cl− to Cl radicals and resulted in high conversion and selectivity to propene. Once again, radical driven routes showed a high selectivity of 72% and conversion of 52% when the CeO2 was combined with 8 wt% NiO as NiO was shown to enhance the Cl surface coverage which in turn enhanced the route to selective conversion of propane (Table 10, entry 9). Ni was confirmed to increase oxygen vacancies when incorporated into the CeO2 lattice, leading to an increase in active sites.230 XPS studies showed O vacancies doubled in concentration over 250 min, causing the selectivity to drop to 25.2%. Whilst oxygen vacancies are necessary to activate oxygen, they decreased selectivity in the absence of HCl,231 which is why CeO2 was shown to have a much lower selectivity without the presence of HCl; a change of active species from oxygen vacancies to Cl radical like species, has been shown to increase selectivity without a large detrimental effect on conversion. However, the space time yields observed are significantly lower than the most active ODH-O2 catalysts.
Co-Based catalysts have been investigated for ODH-O2, but their performance is also limited and vastly inferior to V- or B-based catalysts. However, Wang et al. developed a Co3O4/ZIF-8 catalyst that could operate at 250 °C, which is about 200 °C lower than the operating temperature for many ODH-O2 catalysts.232 Such low temperature activity warrants further investigation, but the reported space time yield of 7.2 molC3H6 kgcat−1 h−1 (Table 10, entry 1) is relatively low for an ODH-O2 catalyst. Tanasoi et al. investigated various mixed metal oxides derived from layered double hydroxide precursors. CoMgAlOx exhibited the highest propene yield, but the propene selectivity was limited due to an increase in cracking, which then led to overoxidation.233 There was no correlation between reducibility and catalyst performance, suggesting that redox properties are not rate-determining in these ODH-O2 catalysts. Unfortunately, the balance of acid and base sites was not measured, so a possible correlation in this respect was not identified.
Although CrOx-based catalysts have been widely investigated for DDH and ODH-CO2, there is limited research available for CrOx-catalysts for ODH-O2. Some CrOx-type catalysts have been shown to form propene, with the selective phase being Cr5O12 when supported on nano-crystalline MgO.234 The space time yield was calculated to be 11.3 molC3H6 kgcat−1 h−1 (Table 10, entry 4), which is significantly less than Cr catalysts for DDH and about equivalent to the performance of CrOx catalysts for ODH-CO2. When supported on carbon nanotubes, the presence of Cr2O3 increased the CO active sites on the carbon nanotubes, whilst also increasing propene desorption,235 suggesting that Cr promotes the functionality of the carbon active centre.
Nanocarbons are an emerging field of catalysts for alkane dehydrogenation. Frank et al. showed that propane can be activated by surface oxygen groups on oxidised carbon nanotubes, noting that sp2 hybridised carbon species are selective and stable for ODH-O2, and that carbon nanotubes provide a balance between reactivity (sp3) and stability (sp2).236 Nanocarbons also show high selectivity to alkenes when compared to metal oxides, but the intrinsic activity is lower. And additional feature of nanocarbons is that they are tuneable; N or B doping within the lattice leads to a change in electronic structure which can influence intrinsic catalytic activity, whilst changing surface functionality can influence selectivity. For example, adding B2O3 to the surface inhibits the activity of electrophilic oxygen species which promote overoxidation. Interestingly, this is one of the first examples of using B2O3 for ODH-O2, where it acts to inhibit the activity through functionalisation of the surface, leading to an increased selectivity. More recently, Qi et al. confirmed the ketonic carbonyl groups to be the active site for general ODH reactions, whilst also confirming the effects of B2O3.237 The unique chemical structure of nanocarbons means that the reaction mechanism does not operate under a Mars-van Krevelen or Langmuir–Hinshelwood route. In situ characterisation and measurements proved vital to finding the reaction mechanism for nanocarbons; in situ experiments for the oxidative dehydrogenation of ethylbenzene using nanocarbons showed the redox cycle of carbonyl-hydroxyl groups being the basis for the reaction mechanism.
Recently, Cao et al. discovered that graphitic carbon nitride (g-C3N4) are highly selective to propene (75%) at a moderate conversion of 12.8%. Additionally, selectivity to ethene was 15%, meaning total olefin selectivity was around 90%. The highest STY was measured at 515 °C to be 4.0 molC3H6 kgcat−1 h−1 (Table 10, entry 11). g-C3N4 is analogous to graphite in structure, as is h-BN. The rate dependence of propane was first order, which is different to h-BN, but it was found to be the same as h-BN regarding the rate dependence of oxygen. DFT calculations suggested this first-order rate dependence of propane was due to simultaneous H abstraction by carbonyl species, which are similarly distant from one another when compared with the distance between the C–H bonds in propane. The simultaneous H abstraction would therefore lead to a lower concentration of the C3H7* reaction intermediate, as well as an increased O2/O* ratio and therefore a lower likelihood of overoxidation.238 This new route to propene shows promise and enhances our understanding of selectivity patterns. The materials were shown to be activated in a similar way to h-BN, forming the proposed active site, i.e., carbonyl bonds, on the surface under oxidative conditions. Under these oxidative conditions, the surface area increased from 8.9 m2 g−1 to 78.1 m2 g−1, due to the formation of mesopores at the surface. The large increase in surface area when compared to the starting material, was a large factor in obtaining the high activity.
While supported precious metal clusters are amongst the most researched class of catalyst for DDH (and are used in some industrial processes) their application in ODH-O2 has been relatively limited. Preliminary research into supported Pt metal clusters for ODH-O2 was reported in the early 90s, with studies comparing particle-support interactions and the influence of supports on the reaction. Notably, Hubbard et al. reported Pt/ZrO2 showed at least one order of magnitude higher activity than Pt/γ-Al2O3.239 This is due to γ-alumina seemingly deactivating the Pt, which was confirmed due to the small Pt particles on γ-alumina having a higher reduction temperature when compared to bulk Pt.
To achieve high activity in supported metal catalysts, the dispersion of the supported metal must be optimised in order to maximise the number active centres and allow efficient use of the precious metal itself, which is often the most expensive component of the catalyst. Additionally, particle size and activity may not scale linearly. Vajda et al. reported that supported under-coordinated Pt8–10 clusters were highly active for ODH-O2.240 The activity of the catalyst in this study was shown to be much larger than the more commonly used vanadia-based catalysts, whilst showing a higher selectivity. The key to an increased activity was due to the under-coordinated Pt cluster strongly adsorbing the propane when compared to an extended Pt surface, confirmed by catalytic data which was in agreement with DFT. DFT showed a significant weakening of the C–H bond when propane interacts with under-coordinated Pt clusters in comparison to Pt(111) surface.
Combining Pt clusters with Sn showed increased productivity due to the electron transfer between the Pt and Sn which were in close proximity. 0.5Pt–5Sn/Si-beta exhibited a STY of 40 molC3H6 kgcat−1 h−1 (Table 10, entry 12).241 Pt–Sn catalysts were characterised via C3H8-TPD and C3H6-TPD. In comparison to 0.5Pt/Si-Beta and 5Sn/Si-beta, 0.5Pt–5Sn/Si-beta showed that the combination of the Pt and Sn led to a stronger adsorption of propane to allow for increased activity and propene desorbed more readily, leading to the understanding that propene would not be further oxidised to COx products due to a higher rate of desorption. It was also found that Pt–Sn/SAPO-34 catalysts showed an increased conversion when oxygen was introduced into the gas feed (compared to DDH conditions).242 This was due to the oxygen reacting with hydrogen to form water and driving the equilibrium towards propene formation. Conversion largely depended on the C3H8:O2 ratio: an increase in propane conversion was found with an increase in O2 concentration relative to propane concentration, but too high an oxygen concentration caused overoxidation to COx. The activity and selectivity were much higher for Pt–Sn/SAPO-34 – a yield of 72.4% as compared to a ∼10–15% yield for most V based catalysts. Introducing oxygen into the feed also reduced the formation of coke, which is a feature of instability in DDH. Water formed via ODH-O2 was found to reduce stability, by increasing sintering of Pt due to a weaker metal–support interaction.242 Pt–Sn catalysts are of course widely studied for DDH and are found in industrial catalysts, as described above.
In summary, a variety of different catalysts have been shown to be active and selective for ODH-O2. While vanadia- and boron-based catalysts comprise a large proportion of the reports to date, Ni-based catalysts have shown promise, particularly NiO-CeO2. Mo-Based catalysts also prominently feature but are currently limited in comparison to analogous metal oxide catalysts, with low propene selectivity at moderate propane conversion. Graphitic carbon nitride, g-C3N4, although much less active than NiOx per kg, exhibited high selectivity to olefins at moderate propane conversion and may warrant further investigation to maximise performance. High surface area, porous C3N4 may be a strong candidate to produce an active and selective catalyst.
ODH-O2 offers many inherent advantages as the reaction is exothermic, thermodynamically unlimited and avoids formation of coke. Overall, extensive research into VOx-based catalysts has been undertaken, with the clear limitation being overoxidation to COx products, but recent developments with one-step preparations and grafting techniques have improved catalyst performance to be comparable with the most active DDH Pt- and CrOx catalysts. Deep oxidation remains a challenge for VOx and other supported metal oxides. However, it is clear than B-based catalysts, although recently developed and less understood in both structure and mechanism, have clear potential for commercialisation due to the high selectivity at high propane conversion. Additionally, ethene is formed as a secondary product. Many B-based catalysts have similar, if not higher, STYs compared to DDH catalysts. Furthering the understanding of the reaction mechanism is essential to developing more efficient catalysts, and this appears to be the focus of recent research in the literature.
6. Oxidative dehydrogenation with carbon dioxide
The so-called ‘soft-oxidation’ of propane using CO2 was proposed as an alternative process to using O2 in order to decrease overoxidation to products such as CO and CO2. In the context of the global effort to reduce anthropogenic CO2 emissions, the ODH-CO2 reaction has gained a renewed interest. CO2 utilisation technologies are of general interest to develop a circular economy, but it is important to acknowledge that the current demand for propene (ca. 130 million tonnes per annum1) is a small fraction of the annual CO2 emissions from fossil fuels (35 Gt).244 Therefore the ODH-CO2 process is unlikely to make a significant impact on global emissions. Additionally, there is an increased cost associated with obtaining concentrated CO2 compared to using air, as could be envisaged in an ODH-O2 process. It could be argued that commercial ODH-CO2 processes would significantly reduce the carbon footprint of an individual chemical or industrial plant that would otherwise release CO2 into the atmosphere, but this would depend on how CO is used as a by-product in the reaction. CO2 was initially utilised in the dehydrogenation of ethylbenzene,245 but since then has been reported for other alkane substrates, including ethane and butane.246 The addition of CO2 in the dehydrogenation reaction has multiple effects: As discussed in Section 2, it facilitates the RWGS reaction which removes H2 from the system and shifts the thermodynamic equilibrium limit to favour propene production. Additionally, CO2 can dissociate on the surface of a catalyst and provide O species to dehydrogenate propane directly. Finally, it can react with coke via the reverse-Boudouard reaction to produce CO, which can potentially extend the lifetime of the catalyst.As an acidic molecule, carbon dioxide will preferentially adsorb on basic sites, which is in contrast to propane which will adsorb on acidic sites. Therefore, the correct balance of acidic and basic sites is a key consideration in catalyst design. Equally, the ratio of carbon dioxide to propane is important to take in account, as it has kinetic and thermodynamic implications. Thermodynamically, higher CO2:C3H8 ratios limit coke forming side-reactions.22 Interestingly, the addition of carbon dioxide has been found in some cases to be detrimental to the catalyst performance;247 this underlines the dependency of catalytic reactivity on the structure of the catalyst.
Generally, there are two proposed reaction pathways that describe the soft oxidation of propane to propene. These are described as the direct and indirect pathways:
| Direct pathway: C3H8 + CO2 ⇌ C3H6 + H2O + CO |
| Indirect pathway: C3H8 ⇌ C3H6 + H2 |
| H2 + CO2 ⇌ H2O + CO |
In the direct pathway, oxygen from CO2 abstracts the H from propane, forming H2O as a primary product. Conversely, in the indirect pathway the dehydrogenation occurs in the absence of an oxidant, and the role of CO2 is to remove H2via the RWGS reaction. It should be noted that regardless of the pathway, the RWGS reaction must mediate the balance of H2O, CO, CO2 and H2, although the contribution of RWGS to the final product distribution is often overlooked. In the following sub-sections, the literature reports concerning catalysts for ODH-CO2 are reviewed and discussed.
6.1. Chromium-based ODH-CO2 catalysts
Chromium oxide is one of the most widely investigated catalysts for the DDH reaction and as described above, is a key component of the industrial CATOFIN process. It is also one of the earliest reported transition metal oxide catalysts for the soft oxidation of propane to propene. Generally, the studies of Cr2O3-catalysed ODH-CO2 reactions are carried out under similar conditions to the DDH process, but with the addition of CO2 to the gas-feed, typically supplied as a 1:1 ratio or in excess of the partial pressure of propane. The performance and reaction conditions of some reported catalysts are shown below in Table 11.
| Entry | Catalyst | T/°C | CO2/C3H8 | WHSV/gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; CO2/C3H8 = molar ratio of CO2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. AC = activated carbon. |
||||||||
| 1 | RuCrOx/SiO2 | 496 | 1 | 7.9 | 9 | 85 | 13 | 266 |
| 2 | 5%Cr2O3/ZrO2 | 500 | 2 | — | 15 | 83 | — | 267 |
| 3 | Cr2O3-(15 wt%)Al2O3 | 550 | 1 | — | 4.7 | 91.3 | — | 248 |
| 4 | 10%Cr2O3-ZrO2 | 550 | 2 | 0.30 | 51.1 | 81.7 | 2.6 | 251 |
| 5 | CrOx/Na-ZSM-5 | 550 | 2 | 0.30 | 48 | 86 | 2.5 | 261 |
| 6 | CrOx-mesoporous ZrO2 | 550 | 2 | 0.30 | 68 | 60 | 2.5 | 252 |
| 7 | CrOx/SBA-15 | 550 | 2 | 0.30 | 24.2 | 83.9 | 1.9 | 250 |
| 8 | CrOx/ZrO2 | 550 | 2.6 | 0.3 | 40 | 88 | 2.2 | 253 |
| 9 | 5%CrOx/AC | 550 | 5 | 3.0 | 39.8 | 86.7 | 21 | 260 |
| 10 | CrOx/MCM-41 | 550 | 5 | 1.2 | 40 | 90 | 8.9 | 88 |
| 11 | Cr2O3/beta-zeolite | 550 | 5 | 1.2 | 33 | 81.6 | 6.6 | 263 |
| 12 | 7% CrOx/SBA-1 | 550 | 5 | 1.2 | 37.7 | 85.8 | 7.9 | 85 |
| 13 | 3.4% Cr2O3/SBA-1 | 550 | 5 | 1.2 | 33.2 | 87.9 | 7.2 | 84 |
| 14 | 0.5Ni-CrOx/SBA-15 | 600 | 1 | 1.8 | 25 | 90 | 8.3 | 264 |
| 15 | CrOx/B-MFI | 600 | 1 | 1.1 | 22.1 | 91 | 4.7 | 262 |
| 16 | CrOx/SiO2 | 600 | 4 | 1.2 | 70 | 79 | 14 | 258 |
| 17 | CrOx/SiO2 | 600 | 5 | 1.2 | 41 | 65 | 6.6 | 256 |
| 18 | CrOx/SiO2 | 600 | 7 | 0.89 | 36.3 | 92 | 6.2 | 254 |
| 19 | CrOx/SiO2 | 600 | 1 | 1.8 | 40 | 90 | 13 | 259 |
| 20 | CrOx/SBA-1 | 600 | 5 | 1.2 | 39.4 | 86.6 | 8.4 | 257 |
The first study of Cr2O3-based catalysts for ODH-CO2 was reported by Takahara et al. in 1996 using Cr2O3 supported on SiO2, activated carbon (AC) or Al2O3. The catalyst tests were performed at 600 °C with a feed of propane and argon or carbon dioxide.248 It was found that over 5 wt% Cr2O3/SiO2, the propene yield increased from 6.5% to 9.1% when CO2 was co-fed instead of Ar. In contrast, the alumina supported catalyst was inhibited by the addition of CO2, as shown by the drop in propene yield from 12.7% to 3.3%. The 5 wt% Cr2O3/AC material was unaffected by CO2 addition. The Cr2O3/SiO2 catalyst deactivated rapidly, retaining just 66% of its original activity after 4 h on-stream. The stability was noted to the same with or without CO2 in the reactant feed. Not only did this study establish Cr2O3 as an active ODH-CO2 catalyst, it also captured one of the recurring themes in catalyst design for ODH-CO2: some catalysts are inhibited by the addition of CO2 while others perform much better.
Relative to many other metal oxides, Cr2O3 is considered to be one of the most promising catalysts for ODH-CO2. Takehira et al. compared the catalytic performance of several d block and p block oxides (namely Cr, Ga, Ni, V, Fe, Mn and Co) supported on MCM-41, at 550 °C with a CO2:C3H8 molar ratio of 5.6.91 After 30 min on-stream, the propene yield of the Cr-based catalyst was 15% whereas for the Ga-based counterpart it was 5%. The remaining catalysts all exhibited lower propene yields (in the 1–4% range). However, as the catalysts were not characterised, it is unclear if the dispersion of these catalysts was comparable, and therefore if the results reflect the true intrinsic activity of each metal. It was also noted that the order of reactivity resembled the trends observed by Nakagawa et al. for diamond-supported metal catalysts in the DDH reaction.249 This suggests that there are commonalities in catalyst design between DDH and ODH-CO2 which most likely relate to the interaction of the alkane with the catalyst.
Contrary to this, Zhang et al. compared ZrO2, SBA-15 and γ-Al2O3 as supports for Cr2O3 in the DDH, ODH-CO2 and ODH-O2 reactions.250 They found that the highest propene yield was observed with different catalyst systems under different dehydrogenation reactions, i.e., the activity trends in DDH did not translate to ODH-CO2 catalysts. Specifically, Cr2O3/ZrO2 catalyst was the most active for the DDH reaction, while the highest yield of propene for the ODH-CO2 and ODH-O2 reactions was observed over Cr2O3/SBA-15 (Table 11, entry 7). Across all the catalysts however, the highest propene yields were generally observed in the DDH reaction, ascribed to the production of carbon oxides in the ODH-O2 and ODH-CO2 reactions. However, it was reported that Cr2O3/ZrO2 was more stable on-stream in ODH-CO2 than in the DDH reaction. Wu et al. also observed increased stability of Cr2O3/ZrO2 in the presence of CO2 (Table 11, entry 4).251 They reported that the CO formation rate was higher than the CO2 consumption rate and the excess CO was suggested to originate from the Boudouard reaction (CO2 + C → 2CO); this could explain the increased stability of this catalyst. It is possible that some dry reforming of propane also took place, which would produce H2 and CO. Zhao et al. recently reported the facile preparation of Cr supported in mesoporous ZrO2 using an evaporation-induced self-assembly method (Table 11, entry 6).252 The authors suggested that propene could react with carbon dioxide to form carbon monoxide, methane and water. Other studies have reported a stronger inhibition effect for Cr2O3/ZrO2 catalysts in the presence of CO2. For instance, Ferreira et al. studied the adsorption properties of CO2 and concluded that it binds strongly to active Cr2O3 sites, lowering the activity and stability of the catalyst,253 whereas Xie et al. suggested that coke formation could originate from CO2 hydrogenation over Cr2+ species. These conflicting reports may relate to the structural polymorph of ZrO2 present in the catalyst support; this factor is often overlooked, but recent studies from Kondratenko et al. revealed that nanocrystalline ZrO2 is an active DDH catalyst in its own right151,152 under similar reaction conditions. The ZrO2 crystal phase and crystallite size were strongly correlated to the catalyst activity: smaller crystallites were more active but amorphous ZrO2 had very low intrinsic activity. Moreover, monoclinic ZrO2 was more active than tetragonal ZrO2.
Cr2O3-catalysed ODH-CO2 has been examined over different polymorphs of SiO2,254–256 including various structured porous oxides, including SBA-1,84 SBA-15,250,257 MCM-41,91 amorphous silica84 and mesoporous silica spheres.258 Notably, Michorczyk et al. reported a detailed comparison of different silica supports, coupled with an in-depth characterisation study.84 It was shown that in each case, the same active Cr species was formed, namely Cr6+, but higher surface area silicas (e.g., SBA-1 and SBA-15) were able to facilitate a higher number of such sites. A clear correlation was found between the rate of propene formation and the wt% of Cr6+, as determined using X-ray photoelectron spectroscopy (XPS). In DDH, while Cr6+ is considered active, under reaction conditions Cr3+ is rapidly formed and considered the principle active species.93,94 The selectivity to propene was similar across the different silica supports (88–93%), with the observed differences being attributable to differences in propane conversion rather than catalytic properties. These Cr2O3/SiO2 catalysts were highly active at 550 °C and the most active catalyst (Cr2O3/SBA-1) achieved a STY of 7.9 molC3H6 kgcat−1 h−1, as shown in Table 11, entry 12. It should be noted that the experiments were carried out with a CO2:C3H8 ratio of 5, which as discussed above, has been shown to enable a higher equilibrium yield of propene. Therefore, in studies where high conversions are generally targeted and carbon dioxide is in excess of propane, higher propene yields are reported.
Takehira et al. carried out X-ray absorption spectroscopy (XAS) on CrOx/MCM-41, confirming that the reduction of Cr6+ occurs during the reaction.91 Specifically, they identified Cr6+O4 tetrahedra as the active species. These Cr3+O6 octahedra were formed after reduction, which were considered to be less active. It was shown that CO2 could re-oxidise the Cr, which suggests that the redox cycle between Cr6+ and Cr3+ is an important process during the reaction. Yi et al. discriminated between isolated Cr6+ and polymeric Cr6+, finding that after reduction, isolated Cr3+ was more active than polymeric Cr3+. This underlines the importance of Cr dispersion in these catalysts.259
In a more detailed examination of Cr2O3/SBA-1, the relationship between the physicochemical properties of the catalyst and the observed activity were explored.85 Michorczyk et al. found a strong positive correlation between the initial propene yield and the reducibility of the catalyst, specifically the H2 consumption. In situ UV-vis measurements revealed that Cr6+ is rapidly reduced to Cr3+ or Cr2+ in the presence of C3H8 under reaction conditions. The reaction proceeds through a Mars-Van Krevelen mechanism, via the Cr2+/3+ redox couple. Rapid deactivation, typical of Cr2O3 catalysts, was observed, although the majority of the initial activity could be recovered after regeneration in air. The remainder was ascribed to pore collapse and/or the formation of inactive Cr sites, although the loss of surface Cr sites through agglomeration was not considered.
Despite early reports that activated carbon (AC) was not a suitable support for Cr2O3, subsequent studies have shown that above 550 °C, the activity of Cr2O3/AC in the presence of CO2 was higher than in the DDH reaction.260 The enhancement in propane conversion due to the presence of CO2 increased at higher temperatures (i.e., above 600 °C), although this was chiefly due to propane dry reforming proceeding, generating CO and H2 rather than propene. While this Cr2O3/AC exhibited the highest STY of propene out of all CrOx-based catalysts (21 molC3H6 kgcat−1 h−1, Table 11, entry 9), it rapidly deactivated on-stream and due to the support being carbonaceous, cannot be regenerated using air. This severely limits the utility of this catalyst.
Zeolites have also been considered as catalyst supports for Cr2O3. Zhang et al. prepared CrOx/Na-ZSM-5 via incipient wetness.261 Although the initial conversion and selectivity was high (48% and 86%, respectively) (Table 11, entry 5), the catalysts deactivated rapidly and the reaction conditions were such that the STY was only 2.5 molC3H6 kgcat−1 h−1 at 550 °C with a CO2:C3H8 ratio of 2. Zhu et al. investigated a range of MFI supports, including silicalite-1 and H[B]MFI.262 CrOx/H[B]MFI was found to be the most active catalyst, giving a STY of 4.7 molC3H6 kgcat−1 h−1 at 600 °C with a CO2:C3H8 of 1 (Table 11, entry 15). Although the as-prepared catalyst exhibited typically poor stability for Cr catalysts, steam treating the catalyst resulted in a marked improvement. After 60 h on-stream, the as-prepared catalyst retained only 25% of its initial conversion, while the steam treated catalyst retained ca. 74%. The authors ascribed the effect to stabilised Cr3+ formed in the steam treatment. Presumably the reduction of Cr6+ during the reaction results in an inactive or less active Cr3+ species. It should also be noted that the as-prepared catalyst was initially almost twice as active as the steam treated catalyst, which leaves the possibility that at higher conversions, increased coke formation may have occurred which would exaggerate the deactivation compared to the steam treated catalyst. Ideally such stability comparisons would be made at isoconversion, however this discovery is one of only a few that shows improved stability on-stream. Recently, Michorczyk et al. prepared a Cr-β-zeolite and investigated the effect of dealumination, which clearly elucidated the effect of acidity on the catalytic performance.263 It was reported that the dealuminated catalyst, Cr-SiBeta, exhibited significantly higher propene yields (up to 27%), compared to the Al-containing catalyst, Cr-AlBeta (2%). The presence of acid sites in the Cr-AlBeta catalyst resulted in cracking pathways that led to increased production of methane and ethene. Moreover, the Cr–Albeta catalyst performed less well in the presence of CO2 than in the DDH reaction. This was explained by the strong adsorption of CO2 on the catalyst, similar to the inhibition observed over Cr2O3/Al2O3 catalysts. The stability of the dealuminated catalyst was also improved compared with Cr–Albeta, but significant loss in activity was observed over the 4 h reaction. In considering the redox properties of the catalysts in this work, and their previous investigations, the authors also identified a strong correlation between the H2 consumption from H2-TPR studies and the reported propene yield, as shown in Fig. 10.
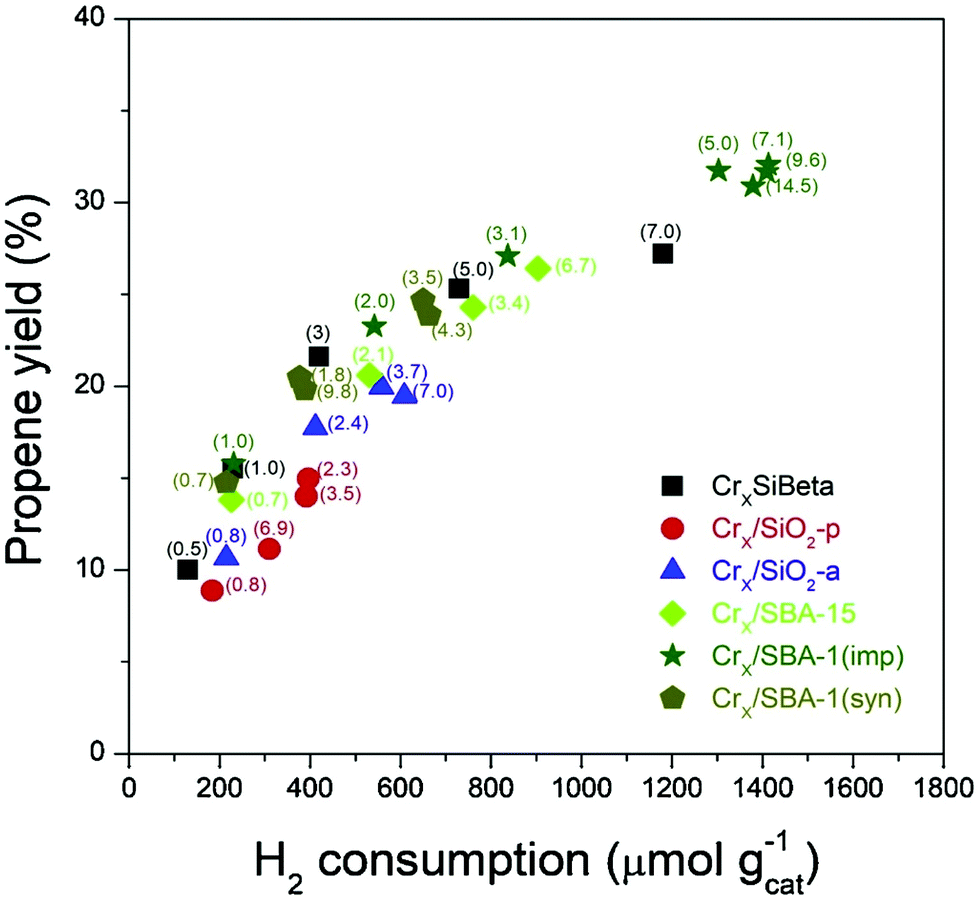 | ||
| Fig. 10 The relationship between the reducibility of various supported Cr catalysts, and the reported propene yield. Reproduced from ref. 263 with permission from Elsevier. | ||
While the majority of investigations on ODH-CO2 Cr2O3 catalysts have focused on support modifications, the addition of a promotor has also been shown to be beneficial. Yun et al. demonstrated that the addition of 0.5 wt% Ni to a 10 wt% CrOx/SBA-15 catalyst increased the propane and CO2 conversion, as well as the on-stream stability.264 After 1 h on-stream, the Ni-promoted catalyst was 50% more active and the conversion and selectivity was almost maintained over 12 h on-stream. In contrast, the less active unpromoted catalyst retained only half of the initial activity over the same time period. Using three-stage H2-TPR experiments (where the catalyst is reduced twice under H2 and then re-oxidised in CO2 before the final H2-TPR program), the authors showed that the addition of Ni facilitated the re-oxidation of Cr during the reaction. This is consistent with previously reported mechanisms of deactivation over Cr-based catalysts, although coking is also often reported to cause deactivation. Interestingly, the promoted catalyst was marginally more prone to coking. This strongly suggests that overreduction of active Cr sites is the key deactivation mechanism over these catalysts. There is a lot of potential to develop promoted Cr catalysts that could enhance the redox activity of the catalyst; precious metals including Pd and Pt are often supported on reducible supports for this purpose.265 Crucially, it was found that above 0.5 wt% Ni, propane reforming was prevalent, which underlines the importance of appropriate loadings of promotors.
O'Brien et al. investigated the effect of a Ru promoter to a 10 wt% CrOx/SiO2 catalyst. Consistent with Yun et al. it was shown that the propene formation rate was increased. A 1 wt% Ru loading was optimal and doubled the activity. Interestingly, this is approximately the same molar ratio reported as the optimum by Yun et al. for Ni-promoted CrOx/SBA-15 (Table 11, entry 14).264 Another similarity was the propensity of reforming to dominate at high loadings (>1 wt%). The enhanced stability seen in Ni-promoted Cr catalysts was not observed in this work and the activity could not be fully recovered after regeneration. Consequently, aggregation of Cr species was suggested as an additional deactivation mechanism in addition to coke formation and overreduction of Cr. Rather than aid Cr re-oxidation in a one-step oxidative dehydrogenation, the authors concluded that the Ru promotes the RWGS reaction, which removes H2 and increases the equilibrium conversion of propane. However, the results of three-stage H2-TPR reactions indicated that Cr can be more easily oxidised in the presence of Ru, which suggests that Ru can participate in a similar role as Ni-promoted Cr catalysts.
Overall supported chromium oxide catalysts represent one of the most active classes of ODH-CO2 catalysts, with STY values in the range of 2–22 molC3H6 kgcat−1 h−1. The role of the support is simply to maximise the active Cr6+ surface area, rather than add any chemical functionality, hence this is the reason why chemically-unreactive supports such as SiO2 and activated carbon yield the most active CrOX catalysts. The most significant barrier to commercial application is the poor on-stream stability exhibited by all chromium-based catalysts. While incremental improvements in the activity of Cr catalysts have been reported since their initial discovery, significant advances in catalyst stability have been relatively few.
6.2. Gallium-based ODH-CO2 catalysts
Gallium oxide is the second most widely studied element for ODH-CO2 catalysis, owing to its successful application for DDH, as discussed above. It was first identified as an efficient DDH catalyst in 1988 in the context of dehydrocyclodimerisation,268 but since the turn of the century it has been the subject of more intensive research.The first application of gallium catalysts in ODH-CO2 was carried out using ethane as a substrate.269 Although ODH-CO2 using ethane is beyond the scope of the current review, it is instructive to consider the catalytic activity of Ga2O3 in the early reports from Nakagawa et al. TiO2, Al2O3 and SiO2 were selected as catalyst supports for Ga2O3 and it was found that Ga2O3/TiO2 was the most active catalyst.270 Additionally, it was found that co-feeding steam was beneficial to maintain high activity, although rapid deactivation was still apparent. Consistent with the literature on supported CrOx catalysts, the addition of CO2 to Ga2O3/Al2O3 was shown to be detrimental to ethene yield. This strongly suggests that Al2O3 is the origin of the inhibition observed in CrOx/Al2O3 catalysts.248 It is known that the balance of acid and base sites is important to mediate the activation of the alkane and the desorption of the alkene. The addition of CO2 is likely to result in competitive adsorption on basic sites, and this may result in longer surface lifetimes of alkyl intermediates, resulting in undesirable side-reactions.
Zheng et al. investigated ODH-CO2 of propane over different polymorphs of gallium oxide and found that β-Ga2O3 (Table 12, entry 2) was the most active, even outperforming Cr2O3 at 500 °C.271 Interestingly, this was not the case for the DDH reaction, where Cr2O3 was the most active catalyst. However, the activity of all the Ga2O3 polymorphs was lower in the presence of CO2. Subsequently, Michorczyk et al. investigated CrOx/Ga2O3 for ODH-CO2 and showed that at elevated temperatures (>570 °C), the addition of CO2 is beneficial (compared to DDH).272 At 550 °C, the STY was 5.3 molC3H6 kgcat−1 h−1. A non-oxidative reaction pathway was suggested to proceed via an initial heterolytic dissociation of propane, form a gallium alkoxide and a gallium hydride species. Then proton abstraction occurs, leaving H+ and H− on the surface of the gallium oxide and propene. The abstraction of the second H atom is slow and considered rate-limiting. The authors suggested that this can be enhanced by a secondary equilibrium involving the surface migration of a surface proton. This equilibrium is as follows:
| H− + C3H7+ + H+-support ⇌ H− + H+ + C3H7+-support |
| Entry | Catalyst | T/°C | CO2/C3H8 | WHSV/gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; CO2/C3H8 = molar ratio of CO2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. |
||||||||
| 1 | Ga2O3–Al2O3 | 500 | 2 | 0.15 | 49.7 | 91.7 | 1.4 | 274 |
| 2 | β-Ga2O3 | 500 | 2 | 0.15 | 23 | 94 | 0.7 | 271 |
| 3 | Ga2O3–Al2O3 | 550 | 5 | 1.2 | 19 | 91.6 | 4.3 | 273 |
| 4 | CrOx/β-Ga2O3 | 550 | 7 | 0.89 | 33.7 | 84.7 | 5.3 | 272 |
| 5 | Ga2O3/Al2O3 | 600 | 2 | 0.30 | 26 | 94 | 1.5 | 114 |
| 6 | GaN/SiO2 | 600 | 2 | 1.8 | 31 | 93 | 11 | 276 |
| 7 | Ga2O3–Al2O3 | 600 | 2 | 0.15 | 49.7 | 91.7 | 1.6 | 275 |
| 8 | Ga2O3/ZSM-48 | 600 | 2 | 0.30 | 52.6 | 42.2 | 1.5 | 113 |
This results in the formation of propene via:
| C3H7+-support ⇌ C3H6 + H+-support |
These reactions are facilitated by Lewis acid sites, specifically tetrahedral Ga ions. The role of CO2 in ODH-CO2 is to assist the above equilibrium by removing H2via the RWGS reaction.
The effect of the support was closely examined for propane ODH-CO2 by Xu et al.114 It was found that Ga2O3 supported on TiO2, Al2O3, and ZrO2 exhibited high activity, while SiO2 and MgO resulted in poorer performing catalysts. However, consistent with previous reports it was found that the addition of CO2 to the reaction feed was detrimental when Al2O3 and ZrO2 were used as supports and higher propane conversion was observed when using lower CO2:C3H8 ratios. Despite this, the propene yield was highest over Ga2O3/Al2O3 in ODH-CO2, the equivalent of 1.5 molC3H6 kgcat−1 h−1 (Table 12, entry 5). The low activity of Ga2O3/MgO and Ga2O3/SiO2 was ascribed to the absence of medium and strong acid sites. In addition to the acid–base properties of the support, the authors considered the interaction between the Ga and the support to be of importance. In the Ga2O3/TiO2 catalyst, a significant proportion of the Ga (45%) was reduced. This catalyst exhibited the highest propane conversion, but with a propene selectivity of 73% it did not result in the highest propene yield.
Although the addition of Al2O3 to ODH-CO2 catalysts is typically not beneficial,114,273 Chen et al. investigated spinel-type Ga2O3–Al2O3 mixed oxides and showed that they can function as stable catalysts.274,275 At 600 °C, although the initial propane conversion was similar for DDH and ODH-CO2, after 8 h on-stream the stability of the catalyst was markedly improved in the presence of CO2, where Ga8Al2O15 retained 66% of its initial activity, compared to just 44% in the absence of CO2. The origin of stability was linked to the low rate of coking associated with the spinel catalyst. Using NH3-TPD analysis, the authors concluded that weak Lewis acid sites were conducive to high activity. These acid–base properties most likely contribute to the enhanced stability as well; it is known that stronger acid sites promote oligomerisation pathways that produce coke. While many previous studies on ODH-CO2 have shown Al2O3 to be detrimental to catalyst performance (compared with DDH activity), this investigation highlights the sensitivity of the catalyst performance to the structure of the surface, and the acid–base properties therein.
Ren et al. studied the effect of acidity and pore geometry in Ga2O3 supported on ZSM-48 and ZSM-5.113 At 600 °C, the most active catalyst, Ga2O3/ZSM-48 with a Si:Al ratio of 130, demonstrated a 22.2% propene yield, although the selectivity to propene was only 42% (Table 12, entry 8). Aromatics (including retained carbonaceous products) accounted for 40% of the converted carbon. An increase in the Si:Al ratio resulted in an increase in propene selectivity on both H-ZSM-5 and ZSM-48 supports. Compared with DDH, in ODH-CO2 the propane conversion was slightly higher, but the propene selectivity was lower, resulting in a marginally decreased propene yield. The increased performance of Ga2O3/ZSM-48 was attributed to the abundance of weak and medium strength acid sites.
Recently, it has been shown that gallium nitride can function as a highly active catalyst for ODH-CO2 when supported on porous SiO2.276 At 600 °C with a CO2:C3H8 ratio of 2, the catalyst exhibited a propane conversion of 31% and a propene selectivity of 93%, equivalent to a STY of 11 molC3H6 kgcat−1 h−1 (Table 12, entry 6), the highest value reported for any Ga-based catalyst. Although some deactivation was observed (ca. 83% of initial steady state activity retained after 8 h on-stream), the catalysts can be considered to be amongst the most stable reported to date. While the authors did not investigate the reaction mechanism of the supported GaN, it is instructive to consider the observed induction period in each catalyst whereby the propane conversion increases over the first 1–2 h; for the most active catalyst it increases from 20% to 30% in this timeframe. A similar induction period has been observed over BN catalysts for the ODH-O2 of propane by Hermans et al., amongst others,196,202,204 which was ascribed to the oxidation of BN to form active B-OH groups. It is reasonable to suggest that a similar oxidation/hydroxylation of the GaN surface occurs here, although it is unclear how, or even if, CO2 could facilitate this in situ. XPS of the as-prepared samples showed a large O 1s signal, but this was ascribed to adsorbed H2O.
Overall, Ga2O3 can be considered to be moderately active for ODH-CO2, with reasonable propene yields, but generally poor stability. In most cases, the inclusion of CO2 is not beneficial for the reaction. Compared with CrOX, Ga typically results in lower propene yield and comparable stability. The recent breakthrough regarding GaN, however, is very noteworthy. With a similar STY to the most active CrOX catalysts and relatively good on-stream stability at high conversion (ca. 30%), this new class of Ga catalysts has real potential. Future research into GaN catalysts should seek to understand the reaction mechanism in order to identify the active sites and improve catalyst performance further.
6.3. Metal oxide based ODH-CO2 catalysts
In addition to chromium and gallium, a selection of other oxide catalysts, mostly of transition metals, have been explored for their ODH-CO2 activity. This section details the most significant advances made to date with respect to these other metal oxides.Bulk and supported iron oxides have been investigated by Michorczyk et al.277 Unsupported Fe2O3 exhibited reasonable activity at 600 °C when using a CO2:C3H8 ratio of 5, with a propene yield of 7.5%. The activity was four times higher than in the absence of CO2, and the propene selectivity increased from 84 to 92.5%. The dispersion of Fe2O3 on activated carbon significantly improved the conversion of propane from 8.2 to 29.7%, which corresponds to a STY of 16.1 molC3H6 kgcat−1 h−1 (Table 13 entry 7). In contrast, using Al2O3 as a support for the Fe2O3 inhibited the performance of the catalyst. The reaction mechanism was concluded to proceed via a redox pathway, whereby the lattice oxygen associated with the iron oxide directly participates in the reaction.
| Entry | Catalyst | T/°C | CO2/C3H8 | WHSV/gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; CO2/C3H8 = molar ratio of CO2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. AC = activated carbon. |
||||||||
| 1 | 10 mol%Fe2O3–CeO2 | 550 | 1 | 0.59 | 18 | 46 | 1.0 | 287 |
| 2 | 8 wt% In2O3/H-ZSM-5 | 580 | 4 | 1.2 | 13 | 81 | 2.5 | 282 |
| 3 | 5 wt% ZnO/H-ZSM-5 | 600 | 2 | 0.30 | 68 | 47 | 2.0 | 278 |
| 4 | V-MCM-41 | 600 | 4 | 1.0 | 58 | 90 | 11 | 286 |
| 5 | In2O3–Al2O3 | 600 | 4 | 0.15 | 35 | 75 | 0.81 | 279 |
| 6 | In2O3/Al2O3 | 600 | 4 | 0.15 | 14 | 80 | 0.36 | 281 |
| 7 | Fe2O3/AC | 600 | 4.9 | 2.77 | 3 | 83 | 16 | 277 |
A 5 wt% ZnO/H-ZSM-5 catalyst was investigated by Ren et al. using a broad range of SiO2/Al2O3 ratios (60, 120, 160, 201 and 242) at 600 °C.278 While all of the prepared catalysts exhibited some activity (with conversion ranging from 40–86%), the highest propene yield was observed over ZnO/H-ZSM-5 (160), which was 46.8% (Table 13, entry 3). In each catalyst, the main competing pathway was to aromatics, which is consistent with the use of ZSM-5 in aromatisation catalysis.247 It was recognised that CO2 both promotes the RWGS reaction over the supported ZnO catalysts, but also blocks active sites. The stability was improved in the presence of CO2 (compared to DDH); ca. 74% of the original activity was retained after 10 h on-stream, which is relatively good compared to Ga- and Cr-based catalysts.
Indium oxide and various mixed-metal oxides containing In have been considered as ODH-CO2 catalysts in recent years. Chen et al. initially demonstrated that In2O3 was active, but also showed that the performance is improved as a binary oxide of In2O3–Al2O3.279 Specifically, In20Al80OX exhibited the highest propene yield under the reaction conditions used (600 °C, CO2:C3H8 = 4:1), which was ca. 27% (Table 13, entry 5). Interestingly, each of the mixed metal catalysts exhibited an induction period over the first 3 h, followed by moderate deactivation. In contrast, Al2O3 and In2O3 alone deactivated much more rapidly on-stream and without an induction period. The origin of this induction period was found to be reduction of In2O3 to In0 nanoclusters, which suggests the active structure is more of a supported nanoparticle type catalyst than a mixed-metal oxide. Chen et al. also considered a wide range of secondary metal oxides for this reaction, using sol–gel or co-precipitation to prepare In2O3–MOx, where x = Al, Zn, Zr, Ti, Fe, Mg, Si and Ce.280 It was shown that the In2O3–Al2O3 combination was the most active, due in part to the high dispersion of In in this particular catalyst. Further investigation into the composition of the support revealed that compared with ZrO2 and SiO2, Al2O3 was the most effective support for In2O3, although ZrO2 was also active.281 The basicity of the In/ZrO2 system was suggested to promote the RWGS reaction, which in turn boosted the ODH-CO2 activity. This conclusion was supported by the substantial increase in the activity observed in ODH-CO2 compared with DDH. Tian et al. recently investigated In/H-ZSM-5.282 At 580 °C and a CO2:C3H8 ratio of 4:1, the highest propene yield was 10%. Although the propane conversion was similar without In present, the selectivity was significantly increased from 28% for H-ZSM-5 to 81% for 4 wt% In/H-ZSM-5 (Table 13, entry 2).
Supported vanadia catalysts have also been considered in ODH-CO2 having been widely applied in various reactions, including propane ODH-O2,4 toluene oxidation283 and formaldehyde oxidation.284 Vanadia catalysts for ODH-CO2 were first reported by Nakagawa et al. and in this early study, supported vanadia was found to outperform Ga2O3.249 Despite this, few studies have been reported since.285 Notably, Ascoop et al. investigated the role of CO2 in the reaction over composite WOX–VOX/SiO2 catalysts. Using D2 as a co-feed, it was shown that 45% of the resultant water contained D2O. This confirmed a Mars-Van Krevelen mechanism as well as a two-step DDH + RWGS mechanism proceeds over this catalyst. It was found that CO2 could partially re-oxidise the catalyst and that coke formation was significantly suppressed in the presence of CO2. Han et al. examined a range of VOX loadings using an MCM-41 as a support.286 The most active catalyst, 6.8 wt% VOx/MCM-41, exhibited an initial propene yield of 51% at 600 °C with a CO2:C3H8 ratio of 4:1. However, the catalyst retained only 65% of its initial activity after 2 h on-stream. The initial STY of this catalyst was 10.7 molC3H6 kgcat−1 h−1 (Table 13, entry 4), which is amongst the most active reported to date, but requires significant improvements to its on-stream stability.
Tsilomelekis and Wang recently investigated mixed metal oxides comprising Fe and Ce for the ODH-CO2 reaction. It was found that 10 mol% Fe in CeO2 exhibited the highest propene yield. The propene selectivity of this catalyst was ca. 45% at 550 °C with a propane conversion of ca. 18%. The major side-reactions observed were dry reforming and cracking. They showed that reforming pathways were more prevalent at higher CO2:C3H8 ratios, such as 2.5:1 and 5:1. However, the catalysts were relatively stable on-stream over 20 h, retaining 80% of their initial activity. Despite the low selectivity, the Fe–CeO2 catalysts exhibited a comparable STY to supported In2O3 (Table 13) and some Ga2O3 catalysts (Table 12).
6.4. Supported nanoparticle based ODH-CO2 catalysts
Despite forming an active component in various industrial DDH processes and being extensively reported in the academic literature for DDH, the number of studies carried out over Pt-based catalysts for ODH-CO2 is comparatively low. This is also true of other noble metals such as Pd, Au and Ru, where only a handful of studies are available. In this section, the available literature on supported nanoparticle (NP) catalysts, including noble metals, is discussed.The earliest report of utilising a supported metal NP in ODH-CO2 was by Guo et al. in 2005 who compared Ga and Re supported on H-ZSM-5 (Table 14, entry 4).247 It was shown that at 550 °C, the addition of CO2 inhibited the propane conversion significantly and did not greatly affect the selectivity to propene (5.7%). As described above, aromatisation tends to be the dominant pathway when H-ZSM-5 is used.
| Entry | Catalyst | T/°C | CO2/C3H8 | WHSV /gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kg−1cat h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; CO2/C3H8 = molar ratio of CO2:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. |
||||||||
| 1 | Pd/CeZrAlOx | 500 | 1 | 3.3 | 9.5 | 93 | 4.9 | 291 |
| 2 | Fe3–Ni1/CeO2 | 550 | 1 | 11.8 | 2.7 | 58.2 | 3.9 | 21 |
| 3 | Co3–Pd1/CeO2 | 550 | 1 | 11.8 | 2.8 | 25.8 | 1.8 | 290 |
| 4 | Re/H-ZSM-5 | 550 | 2.3 | 0.70 | 57.6 | 5.65 | 0.48 | 247 |
| 5 | Mo2C | 550 | 0.2 | 0.96 | 0.5 | 99 | 0.11 | 295 |
Supported gold catalysts, which are widely used as catalysts for oxidation reactions288 including the epoxidation of propene,289 were studied by Tóth et al. When Au NPs supported on MgO, ZnO or Al2O3 were investigated over a broad temperature range (500–750 °C), only Au/ZnO exhibited reasonable catalytic activity. The propene selectivity was 56% at 50% conversion at 600 °C. The Au/ZnO material was shown to be very unreactive towards propene and evidence for propane activation at 25 °C was found. Despite this promising initial data, Au NP catalysts have not been investigated further.
Gomez et al. investigated various mono- and bi-metallic NP catalysts using CeO2 as a support at a reaction temperature of 650 °C and a CO2:C3H8 ratio of 1:1 (Table 14, entries 2 and 3).21,290 Among the wide range of catalysts tested were Ni1, Fe1 and Fe3-Ni1 (where the subscript number indicates the molar ratio). While Ni1 was active mostly for dry reforming to CO + H2, Fe1 was inactive. However, Fe3-Ni1 exhibited much higher selectivity towards propene. In situ XAS measurements revealed that under reaction conditions Fe was oxidised and Ni was metallic. While the overall performance of Fe3-Ni1 was moderate (58% selectivity to propene at 2.7% propane conversion, equivalent to a STY of 3.9 molC3H6 kgcat−1 h−1), the findings demonstrate the sensitivity of reaction pathways to the nanostructure of the catalyst and suggest further optimisation is possible. Subsequent studies on Fe–Co and Fe–Pd catalysts revealed similar trends, whereby the addition of Fe to Co or Pd boosted the selectivity to propene compared with the corresponding monometallic catalyst. However, in all cases the dry reforming reaction competed with the dehydrogenation pathway, significantly limiting the propene selectivity.290
Nowicka et al. carried out a detailed investigation into Pd/CeZrAlOX catalysts.291 It was shown that CO2 dissociation on the catalyst support was facilitated by the presence of Pd. At 500 °C the catalyst achieved 9.5% conversion and 93% selectivity to propene. Although the STY was 4.9 molC3H6 kgcat−1 h−1, (Table 14, entry 1) the reaction temperature was 500 °C, which is low for ODH-CO2 catalysts. Above this temperature, the selectivity to propene rapidly drops and methane and carbon monoxide production increase. It should also be noted that propene selectivity was calculated based on observed products, rather than propane consumption and therefore the contribution of dry reforming was not measured.
Metal carbides have been intensively investigated in recent years for a broad range of reactions and are often described as having properties analogous to precious metals.292–294 To date, there have been two reports of metal carbides catalysing ODH-CO2, both of which focus on Mo2C.295,296 It was found that in addition to dehydrogenation, hydrogenolysis and reforming played important roles in the reaction network. Overall, the performance of the carbide catalysts was poor compared to typical metal oxide or supported nanoparticle catalysts (Table 14, entry 5).
The number of studies on supported nanoparticle catalysts for ODH-CO2 are relatively few and those available indicate that this field of research has great potential. The major challenge is in understanding and inhibiting side-reactions such as reforming, which are known to proceed over this class of catalyst, unlike in Cr- and Ga-based catalysts. Careful consideration of the nanostructure and composition has shown that the selectivity can be readily tuned to favour different pathways, and further investigation in this area may well be able to harness the high activity of supported nanoparticles as well as favour dehydrogenation pathways.
Despite the fact that ODH-CO2 of light alkanes has been known for over twenty years, catalyst design has largely been limited to a handful of metal oxide catalysts, namely chromia and gallia. The most promising of these catalysts are supported metal oxides, which typically result in high initial propene yields. However, all catalysts suffer from rapid deactivation on-stream, which is typically attributed to coke formation blocking active sites. However, few investigations have studied the long-term stability of the catalyst beyond 12 h on-stream. In many cases, the catalyst can be regenerated periodically, by burning off the coke using O2. As discussed above, catalyst regeneration is commonplace in commercial DDH processes and therefore isn't necessarily a barrier to commercialisation. However, the best STYs reported are, with few exceptions, in the range of 5–10 molC3H6 kgcat−1 h−1, whereas for the DDH reaction, values over 100 molC3H6 kgcat−1 h−1 are possible. As a result, further advances in the activation of propane over ODH-CO2 catalysts are required. Despite dozens of reports over several decades on Cr- and Ga- based catalysts, the improvements in catalyst performance have been incremental. The knowledge gained from these studies on the importance of reducibility and the balance of acid and base sites should be applied to supported NP catalysts, which are a promising and somewhat neglected category of ODH-CO2 catalyst.
7. Oxidative dehydrogenation with nitrous oxide
O2 and CO2 are currently the most commonly reported oxidants in the oxidative dehydrogenation of propane. However, other oxidants have also been shown to be feasible. Sulfur dioxide and halogen-based compounds have largely been ignored in recent decades due to their corrosive properties, while H2O2 has had its commercialisation deferred due to difficulties in its synthesis.297 Another relevant oxidant, however, is N2O. It should be noted that N2O is a greenhouse gas which has a global warming potential approximately 310 times higher than that of CO2.298,299 Therefore, processes that remove N2O gas from the atmosphere are highly desirable. The industrial sector produces N2O as a by-product from various processes, including nitric and adipic acid production as well as fossil fuel combustion sources which make up a total of 15% of anthropogenic sources.298 Many production plants will have N2O abatement in place already, so the possibility for flowing propane through this process exists. Despite this, research into the ODH-N2O reaction of propane is rather limited, mostly due to the cost of N2O and product dilution in N2. Furthermore the research that does exist is reserved for elucidating structure–activity relationships, which can be used to better understand the ODH reaction in general.3007.1. Fe-Zeolite based ODH-N2O catalysts
In 1998 Panov et al.301 reported a new type of O species observed in Fe-ZSM-5 when it was exposed to N2O. The species was referred to as α-oxygen and is able to perform highly selective dehydrogenation and oxidation reactions of hydrocarbons such as the oxidation of benzene to phenol, which occurred with nearly 100% selectivity at conversions of 97–98%. In addition, dehydrogenation of ethane and methane oxidation reactions are also possible using N2O. Panov further detailed the characteristics of α-oxygen. It is typically associated with isolated Fe ions or small complexes found within the zeolite's micropores, as opposed to the tetrahedral Fe ions within the crystal lattice, or the finely dispersed metal oxide phase of the zeolite surface. It has been suggested that this oxygen species could be useful in the oxidative dehydrogenation of propane.Other examples of ZSM-5 modified catalysts on top of Fe include doping with metals such as Mn and Co.302 The authors examined the selectivity, conversion and yield of propene of these three materials. While all three showed similar selectivity (between 70–80%), Fe-ZSM-5 was reported to exhibit the highest conversion and therefore best yield towards propene. The α-oxygen species is formed via the interaction between N2O and framework stabilised Fe2+ ions, resulting in the dissociation of N2O and the formation of mono- and bi-nuclear Fe3+–O− species, (α-oxygen). These α-oxygen species react with propane by providing atomic oxygen, producing propanol, which is subsequently dehydrated to propene.303,304 It is from this mechanism that the selectivity originates, because once the hydrocarbon's interaction with α-oxygen is complete, the molecule would need to vacate the zeolite channel in order for a new N2O molecule to enter and generate an new α-oxygen site, therefore not allowing for immediate further oxidation.301 It is also evident from this mechanism that coordination of oxygen to an Fe2+ species is required, and therefore does not occur on tetrahedral framework Fe species due to a lack of vacant sites in the Fe complex.303
By considering simply the activation of N2O, Groen et al.305 enhanced the activation by performing an alkaline treatment on Fe-ZSM-5, which increased the number of Fe2+ sites, decreased the amount of oligonuclear clusters and increased the porosity of the zeolite. The calcined catalysts were treated in 0.2 M NaOH for 2 h, before undergoing an ion-exchange in NH4NO3 solution. Where the calcined Fe-ZSM-5 catalyst reached complete conversion of N2O at 552 °C, the catalysts which had undergone alkaline treatment and subsequent ion-exchange reached total conversion at slightly under 527 °C. This increase in porosity and highly active Fe sites shows potential for the dehydrogenation reaction of propane, however little research effort has extended into this area.305,306
Further to this, Pérez-Ramírez et al.303,307 reported highly active catalysts after the steam treatment of Fe-ZSM-5 zeolites, which produced an initial STY of 180 molC3H6 kgcat−1 h−1 (Table 15, entry 2) when carried out at 525 °C. This high productivity is ascribed to the generation of extra-framework Fe sites during the steam treatment.303,307,308 However, this high productivity is hindered by a rapid deactivation, with the authors observing a minimum decrease in propene yield of ca. 3% over 3 h at 452 °C, and a maximum decrease in propene yield of ca. 12% over the same timescale at 525 °C. This rapid deactivation, which will be discussed further, is the most significant issue facing the use of N2O as an oxidant to date.
| Entry | Catalyst | T/°C | N2O/C3H8 | WHSV /gC3H8 gcat−1 h−1 | X C3H8/% | S C3H6/% | STY/molC3H6 kgcat−1 h−1 | Ref. |
|---|---|---|---|---|---|---|---|---|
|
T = reaction temperature; N2O/C3H8 = molar ratio of N2O:C3H8 in the reaction, XC3H8 = initial propane conversion, SC3H6 = initial propene selectivity, STY = space time yield. Values next to element/phase is the loading in wt%. |
||||||||
| 1 | FeH-MFI-1200-HT | 450 | 1 | 3.0 | 64 | 29 | 12 | 309 |
| 2 | Steam-activated Fe-ZSM-5 | 450 | 1 | 39.4 | 48 | 45 | 180 | 303 |
| 3 | VOx/MCM-41 | 475 | 1 | — | — | — | 29 | 313 |
| 4 | Steam-activated Fe-ZSM-5 | 500 | 1 | 2.4 | 29.2 | 68.8 | 10 | 311 |
| 5 | In2O3–Al2O3 mixed oxide | 600 | 4 | 0.15 | 43.6 | 63.1 | 6.8 | 316 |
The main deactivation pathway of this catalyst is ascribed to the build-up of carbonaceous deposits at the Brønsted acid sites in the zeolite pores, therefore blocking the pores and active Fe sites. It has been reported that blocking or removing the Brønsted acid sites lessens the impact of deactivation.303,309 Despite being the major deactivation pathway, compared to other zeolite examples, ZSM-5 shows significant resistance to deactivation, despite the finding that they produce more coke than other examples. This behaviour is attributed to the location of the coke formation, which occurs at Al-protonic sites, away from the extra-framework Fe-active sites.303 However, the use of N2O as an oxidant invariably leads to rapid deactivation despite the positive effect of the zeolite, when compared to other oxidants, such as O2. Using a mixture of oxidant gases, O2 and N2O, was reported to decrease the deactivation of Fe-ZSM-5 further via oxidising the carbon deposits.309 The limitation of this being the inclusion of too much O2, leads to overoxidation of the desired products and therefore lower selectivity towards propene. Bulánek et al. reported that a mixture of N2O and O2 over a steam-treated Fe-ZSM-5 catalyst and found that with the inclusion of oxygen the conversion consistently increased; however, the selectivity consistently decreased (Table 15, entry 1).309 The problem remains, that when comparing oxidant properties for the ODH reaction of propane, N2O has a significant tendency for rapid deactivation.
In 2010, Kowalska-Kuś et al.310 investigated the role of Brønsted acid sites in Fe-ZSM-5 and their effect on the ODH-N2O activity. It was found that in the presence of a high number of protonic sites, cracking and ODH reactions of propane occurred simultaneously, and would often lead to high selectivity towards total oxidation products. Conversely, removal of protonic sites via doping with sodium reduced the oxidative activity of the zeolite and modified the reaction to favour the ODH pathway. Subsequently, the propene would undergo further cracking reactions. Therefore, shorter contact times result in higher propene selectivity. This method led to propene selectivities higher than previously reported for this reaction, (over 80%), but the limitation of this approach is the significantly lowered conversion due to the lower oxidative activity of the zeolite.
It is clear that adjusting the acidity of the zeolite greatly influences the conversion and selectivity of the ODH-N2O reaction. As mentioned earlier, steam treatment has been found to increase the number of Fe2+ active sites. This, as well as dealuminating the zeolites, leads to a higher presence of weak and medium acid sites, which enhance selectivity towards propene.304,311 Steam treatment was also found to increase stability towards coking in the zeolites.311 With these methodologies implemented, Ates et al.311 were able to produce a catalyst with a STY of 10 molC3H6 kgcat−1 h−1 (Table 15, entry 4).
In summary, while the ODH-N2O reaction is hindered significantly by its tendency to deactivate rapidly without the presence of O2, Fe-ZSM-5 as a catalyst is very resistant to deactivation and its use for the ODH-N2O reaction is contingent on several factors. The oxidation of extra-framework Fe2+ sites to Fe3+–O− sites is a crucial step in the mechanism, and therefore producing a material with a large number of these sites would increase the yield of propene. The Brønsted acidity of the Fe-ZSM-5 zeolite both decreases the selectivity to propene, due to a high number of protonic sites leading to selectivity towards total oxidation, and increases the rate of deactivation, allowing for the generation of carbonaceous deposits which block the zeolite pores. The most promising materials currently are steamed Fe-ZSM-5, as the steam treatment has been found to produce more Fe2+ sites and dealuminate the framework, causing an increase in weak and medium acid sites. With fewer strong protonic sites, the reaction mechanism shifts towards favouring the dehydrogenation of propane, therefore resulting in a consecutive propane dehydrogenation-propene cracking pathway which allows for a higher selectivity to propene with shorter contact times. Despite these advancements, recent research on Fe-ZSM-5 for ODH-N2O has been minimal due to their rapid deactivation, the high cost of N2O and the presence of N2 in the product stream.
7.2. Other ODH-N2O catalysts
Other catalysts reported to be active for the ODH-N2O reaction of propane include supported vanadium oxides. In 2001 Kondratenko et al.312 reported that VOx/γ-Al2O3 was active for ODH of propane with O2 and N2O and investigated the effect of vanadia loading. It was found that, similar to Fe-ZSM-5 catalysts, on replacing O2 with N2O the overall activity of the catalyst decreased, but the selectivity increased. This trend was ascribed to the lower oxidising potential of N2O.Further to this, DFT studies were carried out to elucidate the role of the oxidant in the ODH reaction.300 These calculations first revealed that propene is formed via successive removal of two hydrogen atoms via a redox mechanism over VOx active sites. The active sites are reduced from V5+ to V3+ and V4+ during hydrogen removal from the propane molecule, which then require re-oxidation before removal of a second hydrogen. The N2O was found to only oxidise V3+, whereas O2 was able to oxidise both V3+ and V4+. Upon re-oxidation of the VOx active sites with O2, peroxovanadates were formed; however these species were not formed with N2O as the oxidant. Peroxovanadates are highly active for the total oxidation of propane. The formation of these peroxovanadates is a key explanation for the more selective nature of N2O compared to O2. In the most active catalyst, the STY of VOx/MCM-41 was 29 molC3H6 kgcat−1 h−1 (Table 15, entry 3), which is less than the most active steam-activated Fe-ZSM-5 catalysts, but more active than some reports over Fe-MFI catalysts.
In addition, V/γ-Al2O3, V/MCM-41, and V/amorphous SiO2 catalysts have also been prepared and characterised in order to elucidate the structure activity relationships between the catalyst and the ODH-N2O reaction of propane.313–315 It was found that the selectivity for propene was lower over V/γ-Al2O3; a result which was ascribed to the acidic nature of the alumina support, over which dehydrogenated propene adsorbed to the support surface where it further oxidised to COx species. This effect is less prominent on less acidic support surfaces such as SiO2 and MCM-41.
The vanadia loading on the support materials was also found to influence the selectivity.312–314 On the materials V/MCM-41, and V/SiO2 materials, the nature and distribution of the V remained consistent up to a loading of ca. 5 wt%, above which three-dimensional V2O5 clusters began to form, and these have unreactive bulk V sites. At lower loadings however, the support surface is more exposed, allowing for adsorption of propene and total oxidation. Therefore, a loading of ca. 5 wt% is optimal to get a high surface coverage, without forming three-dimensional clusters. Kondratenko et al. were able to achieve selectivities of over 90% at conversions limited to 2 and 10% with such materials.315
Finally, in 2010, Chen et al. reported the use of In2O3-Al2O3 as a novel catalyst for the ODH-CO2.279 In 2011 the same group reported the same catalyst's potential for the ODH-N2O reaction.316 The activity of the In2O3–Al2O3 catalyst shows a significant dependence on the concentration of In0 in the material. The catalyst exhibited a low STY of 6.8 molC3H6 kgcat−1 h−1 (Table 15, entry 5). Despite this, the In2O3–Al2O3 catalyst shows potential to surpass the more commonly researched Fe-zeolites in terms of stability. Whereas previous examples of Fe-zeolites have displayed significant deactivation over 3 h time-on-stream, this In-based catalyst was observed to lose 6% propene yield over a total of 12 h on stream. Considering that deactivation is the most significant issue plaguing the use of N2O as an oxidant in the ODH-N2O reaction of propane, the In2O3-Al2O3 has good potential to be an active and stable catalyst.
In summary, although Fe-ZSM-5 has the most potential for the ODH-N2O reaction for propane, several other catalysts have been tested. Vanadia based catalysts have been shown to be highly active for the reaction, which were found to be more selective when supported on less acidic, silica-based support materials, and at loading levels of lower than 5 wt%. Indium based catalysts have also been suggested for the ODH-N2O reaction, with the potential for a highly stable catalyst compared to Fe-ZSM-5, which displays significant deactivation and is likely to be the underlying reason behind the relatively low research effort put into the N2O route compared to the other potential oxidants described in this review article.
8. Summary of propane dehydrogenation and over-arching themes in catalyst design
In the above sections, the literature on the catalytic dehydrogenation of propane has been critically reviewed. Each sub-reaction of propane dehydrogenation has specific kinetic and thermodynamic requirements, and this is reflected in the diverse variety of catalysts that have been developed in recent decades. Throughout this review we have attempted to compare the productivity of catalysts for DDH and the various ODH reactions by measuring space time yield of the catalyst (i.e., the moles of propene produced per kg of catalyst per hour). This is an imperfect metric with which to compare catalysts, but the wide range of reaction conditions and catalyst types explored means that calculating turnover frequencies for each catalyst is not possible. One assumption when comparing productivity is that catalysts are tested under conditions to maximise their performance. This is of course not the case for all studies, especially those where kinetic measurements were of interest, or where there were other constraints present. However, by comparing the range of values associated with a single class of catalyst, it is possible to identify the highest performing catalysts in each class and broadly assess the potential of each. Fig. 11 below compares a selection of literature catalysts (presented in Tables 2–14) for each reaction, colour-coded to highlight the performance of different elements in different reactions. The horizontal dashed line is marked at the equivalent of 5 kgC3H6 kgcat h−1, which has been considered to be space time yield that may warrant consideration of commercial application.196,213 Such a value is difficult to pinpoint, but given that this is where the more active Pt and CrOX catalysts for DDH are found, it is reasonable.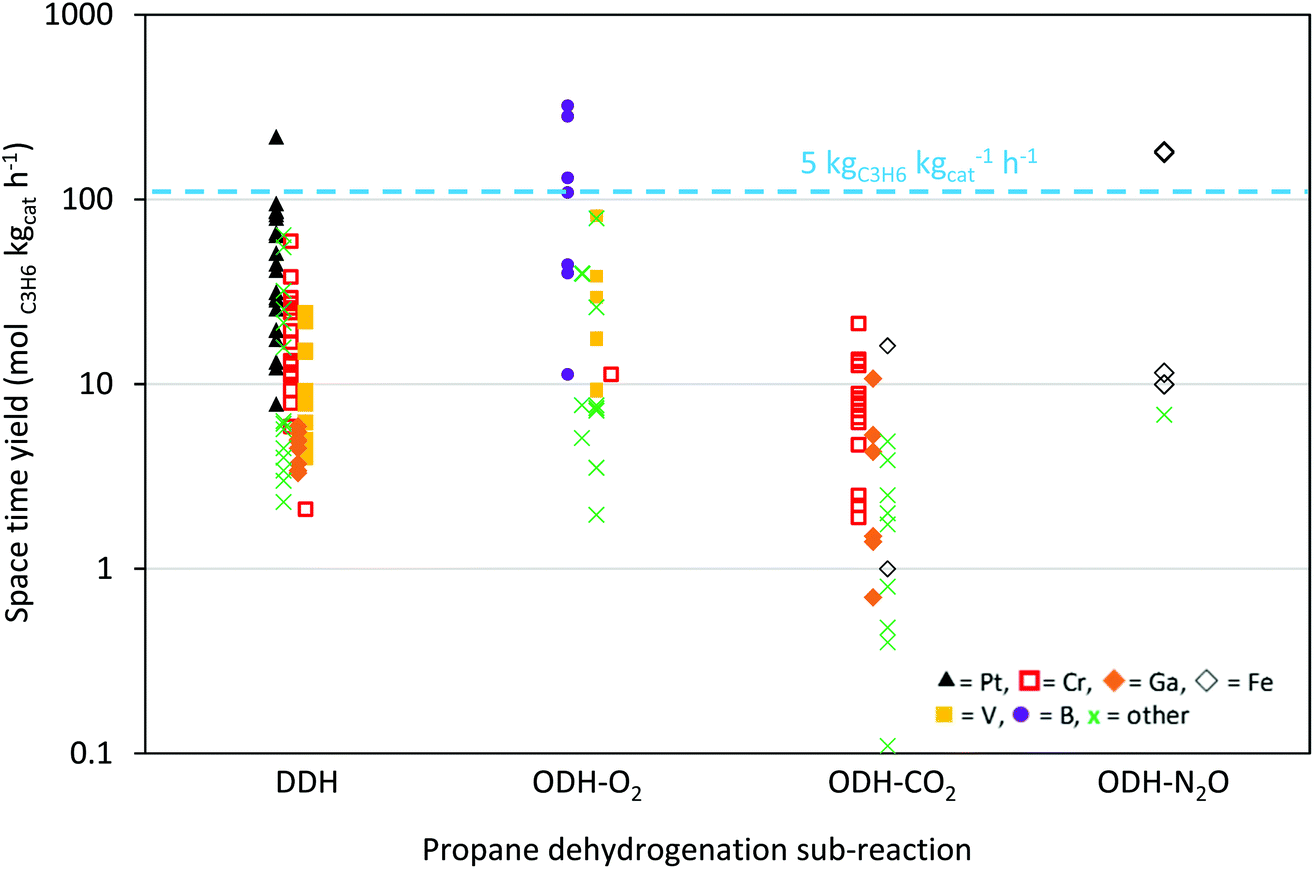 | ||
| Fig. 11 Space time yield plot of a selection of DDH, ODH-CO2 and ODH-O2 catalysts, showing the relative performance of catalysts in each sub-reaction. | ||
It is clear that for the DDH reaction, Pt- and Cr-based catalysts dominate the highest space time yield measurements. This is expected given their widespread use in industrial processes. Beyond this, ZrO2 and ZnO/ZrO2 catalysts perform strongly, and their low cost and low toxicity make them well-suited for commercial application. These materials have only recently been reported in the literature and with further development they may match or outperform Pt- and Cr-based catalysts. It is also possible that they are already present in the K-PRO process.
In ODH-O2, B-based catalysts are the best performing, with STY values in excess of 70 molC3H6 kgcat−1 h−1 reported. Some boron catalysts exceeded 200 molC3H6 kgcat−1 h−1, well above the 5 kgC3H6 kgcat h−1 threshold and underscores their potential for commercial application. Although ODH-O2 produces H2O as a side-product (rather than H2), ethene is typically the major by-product in B-catalysed reactions rather than coke formation. As an exothermic reaction, large-scale implementation may be preferable to the endothermic DDH reaction, which requires specific heat-generating technology to be efficient. Therefore the commercialisation of a ODH-O2 process using B catalysts may be realistic.
ODH-CO2 catalysts do not yield more than ca. 20 molC3H6 kgcat−1 h−1, which suggests that CO2 activation remains a key challenge. While CrOx based catalysts yield the highest STY, only incremental advances have been made in recent years and it will take a step-change in catalyst development before ODH-CO2 can be meaningfully compared to DDH and ODH-O2 reactions, as a mean to produce propene. Interestingly, some of the highest STY values reported are CrOX/activated carbon and FeOX/activated carbon. The obvious drawback of activated carbon as a support is that oxidative regenerations will combust the support, but clearly its chemical properties are desirable as a catalyst support.
In ODH-N2O, Fe-ZSM-5 are the most active catalysts known, and in one example a STY of 178 molC3H6 kgcat−1 h−1 was reported. ODH-N2O is the least studied sub-reaction and it is thought that the relatively high cost and low availability of N2O may be the most limiting aspect. N2O is a by-product in various chemical processes including adipic acid and nitric acid production, but there may be few other opportunities for large-scale sequestration and utilisation of N2O on an industrial scale. It is also apparent that the selectivity to propene is often below 70% and the catalysts rapidly deactivate on-stream.
Table 16 below shows a summary of the typical chemical and catalytic properties associated with each reaction. While the temperature range of each reaction overlaps, generally ODH-O2 and ODH-N2O are carried out at lower temperature than DDH, while ODH-CO2 typically lies in between or nearer to DDH reaction temperatures. In terms of side-reactions, cracking is the only common source of by-products in each sub-reaction. Coking remains a major challenge in DDH, ODH-CO2 and ODH-N2O, while overoxidation is generally observed in the ODH-O2 and ODH-N2O processes. Redox sites play an active role in each of the oxidative dehydrogenation reactions, while in DDH they are not required. The importance of acid and base sites have been frequently examined across each sub-reaction, and interestingly remain the only catalytic properties that are applicable to each sub-reaction. Acid sites are widely known to activate alkanes, but if the acid sites are too strong, the alkane will be retained on the surface and lead to coking, or overoxidation in the case of ODH-N2O and ODH-O2 reactions. Basic sites on the other hand, are known to facilitate alkene desorption and in the case of ODH-CO2, to activate CO2. Therefore, a balance of these sites is necessary to promote sufficient propane activation and propene desorption. Prevalent deactivation mechanisms also vary between sub-reactions, with coking frequently cited as a cause for loss of on-stream activity in DDH, ODH-CO2 and ODH-N2O. The ODH-O2 catalysts on the other hand are generally much more stable on-stream. It is noteworthy however, that despite relatively fast deactivation of DDH catalysts, the activity can be regenerated after an oxidative treatment, significantly extending the catalyst lifetime.
| Reaction | Reaction temperature range /°C | Significant side reactions | Role of redox sites | Role of acid sites | Role of basic sites | Typical deactivation mechanisms |
|---|---|---|---|---|---|---|
| DDH | 550–630 | Coking, cracking | Not directly involved | Alkane activation, strong acid sites promote coking | Alkene desorption | Coking |
| ODH-CO2 | 400–600 | Dry reforming, coking, cracking | CO2 activation | Alkane activation, strong acid sites promote coking | Alkene desorption, CO2 activation | Coking, overreduction of catalyst |
| ODH-O2 | 400–600 | Overoxidation, cracking | O2 activation | Alkane activation | Alkene desorption | Sintering |
| ODH-N2O | 400–600 | Overoxidation, coking, cracking, | N2O activation | Alkane activation, strong acid sites promote coking | Alkene desorption | Coking |
Regarding catalyst design for each reaction, there are catalysts that can efficiently operate for DDH, ODH-CO2 and ODH-O2, while there are some that are reaction-specific. This is exemplified by supported chromium oxides, which have been reported for DDH, ODH-CO2 and ODH-O2. On the other hand, Fe-ZSM-5, which is highly active for ODH-N2O has a unique dimeric Fe2+ site that can activate N2O but not O2. Therefore, it would not be applicable to ODH-O2 or DDH. The ability of Cr to catalyse multiple sub-reactions is likely to be related to its stability (and activity) in multiple valence states. In DDH, Cr3+ sites are considered the active species, while in ODH-CO2 and ODH-O2, the redox cycle of Cr to Cr4+ and Cr3+ all facilitate the activation of the oxidant. Supported vanadia catalysts can also exhibit multiple valence states, and are also active in DDH, ODH-CO2 and ODH-O2.
Aside from the active element, catalyst supports also comprise a vital component of any propane dehydrogenation catalyst. The primary role of the support is to enhance and maintain high dispersion of the active phase. This has been demonstrated multiple times over metal oxides such as those of Ga, Cr, V and Fe, where SiO2, Al2O3, and ZrO2 have been widely implemented as supports. The acid and base sites of the support also affect the performance of the catalyst and can be adjusted with promotors. This strategy has been widely applied in DDH catalysts. The support may also play a more active role in catalysis, by activating propane, CO2 or O2.
A multitude of chemical elements including precious metals, rare earth metals, base metals and non-metals have all been discovered to play a role in catalysing propane dehydrogenation in various contexts. Any sustainable process should aim to use abundant and renewable raw materials, and this applies to the catalyst as well as the starting reactants. It is clear that across the more active propane dehydrogenation catalysts known, there is a range in the abundance and production of the chemical elements required. Table 17 summarises the availability of a range of chemical elements commonly applied in propane dehydrogenation catalysts.
| Chemical element | Common mineral form | Annual production in 2019/103 tonnes | Estimated world reserves/103 tonnes |
|---|---|---|---|
| a From ref. 317 which uses data for 2014. Unless otherwise stated, the values were reproduced from ref. 318. | |||
| Boron | B2O3, other borates | >3500 | >1200000 |
| Chromium | chromite | 44000 |
570000 |
| Gallium | Trace element in other minerals (<50 ppm) | 0.44a | 560a |
| Palladium | Metallic | 0.21 | Unknown |
| Platinum | Metallic | 0.18 | Unknown |
| Vanadium | Oxidic form | 73 | >63000 |
| Zinc | Spharelite | 13000 |
1900000 |
| Zirconium | ZrSiO4 | 1400 | 64000 |
In the DDH reaction, the most active elements are typically Pt and Cr, which are already implemented in commercial processes, and more recently Zr and Zn-based alternatives have been used. While chromium is relatively abundant as a chemical element, Pt is very scarce and production limited to a few countries, namely South Africa. The global reserves of individual platinum group metals (PGMs) is not possible to accurately quantify, but it is estimated that the total global reserve of PGMs is over 100000 tonnes meaning that Pt and Pd are easily the most scarce chemical elements commonly used for propane dehydrogenation. While supported nanoparticle catalysts use a fraction of the quantity of metal that would be required in a metal oxide catalyst, it would be desirable for any DDH process to use an abundant, non-toxic element as the main component of the catalyst. This makes the recent discovery of Zr/TiZnOx all the more promising.157 The ODH-O2 reaction has recently been dominated by the discovery of boron-based catalysts, which are highly abundant compared to most other ODH-O2 catalysts such as V. Accordingly it should be considered a good candidate for commercialisation from the point of view of sustainability. ODH-CO2 catalysts are typically Cr or Ga based. As described above, Cr is highly abundant but also toxic. Gallium is present in the Earth's crust at around 17 ppm (ref. 319) and is typically found as an impurity in minerals such as bauxite, sphalerite and germanite. Although present as a trace element in these minerals, the abundance of bauxite is vast and it is estimated that the amount of gallium contained in bauxite alone is 560000 tonnes, which places it well above platinum in abundance. The field of research into DDH and ODH-O2 is very diverse, and in recent years a number of novel discoveries has expanded the range of catalytically active elements known for this reaction. This means that the reliance on scarce elements like Pt, or toxic ones like Cr is limited. Future sustainable DDH and ODH-O2 processes should capitalise on this knowledge and implement abundant elements such as B, Zn, V or Zr. With regards to ODH-CO2, the performance of the most active catalysts is still well below what would be considered for commercialisation, but the research community should look to the successes of the abovementioned abundant elements for inspiration in designing novel ODH-CO2 catalysts.
9. Outlook
While the current focus of government policy is based on net zero carbon, it is clear that society will still need a wide range of products based on carbon-containing molecules. It is now essential that the chemical industry focusses on accessing such carbon from sustainable sources. It is likely that for the near- and mid-range future the current key platform molecules such as propene will still be required. Against this background, the dehydrogenation of propane will remain a major industrial process and its future importance is guaranteed due to the scale and breadth of propene applications in modern society. Currently, the majority of propane is sourced from non-renewable feedstocks, but the industry has recognised the need to change and steps have been taken to develop processes for renewable propane to be generated. It may be many years before so-called bio-propane can compete with non-renewable propane sources in terms of supply, but the catalytic processes described in this review will be required to enable this technology to be used. Therefore, the development of catalysts for the direct dehydrogenation of propane in the presence or absence of an oxidant will continue to attract attention from academia and industry alike, as significant advances have been made in recent years. An analysis of the sustainability of current C3H6 production methods, namely steam cracking and methanol-to-olefins, reveals that direct dehydrogenation has several advantages in efficiency when a selective catalyst is applied. Life cycle analyses of oxidative dehydrogenation reactions using O2 and CO2 have not been carried out, due to the fact that these reactions have not been commercialised yet. However, in principle the processes have the potential to offer lower temperature routes to forming C3H6 without lengthy catalyst regeneration steps.DDH is to date the only reaction that has been operated commercially. The most active catalysts reported in the literature are based on chromium oxide or platinum promoted with tin oxide, but even these require frequent regeneration due to coking. Therefore, the future of DDH catalyst design should focus on reducing deep dehydrogenation reactions that lead to coking. Irrespective of catalyst, common themes have been established in efforts to improve C–H bond activation, reducing propane and hydrogen adsorption on surface sites. Of these, supports with weak and moderate surface acidity coupled promoting elements (B, Ga, Cu and Ni to name a few) result in lower Pt utilisation, which is desirable for potentially industrially applicable catalysts. Additionally, a number of noble, non-noble based intermetallic alloys and numerous mixed metal oxides are emerging as viable alternatives to chromium oxide or platinum-based catalysts; and this presents an economic and environmental benefit. In doing so, the frequency of regeneration will be reduced, meaning that fewer CO2 forming regeneration cycles will be required, thus reducing the carbon footprint of propane production.
ODH-O2 offers a pathway that avoids the formation of coke entirely, but until recently, the STY typically reported in ODH-O2 catalysts such as supported vanadia were, with few exceptions, an order of magnitude below that of DDH catalysts. The advent of boron-based catalysts for this reaction has led to a significant increase in the prospects for commercial application. As a relatively stable, non-hazardous and abundant element, boron catalysts may well play a major role in future propene production processes. Furthermore, they appear to be stable on-stream in laboratory tests, although industrially relevant stability tests have not yet been reported.
ODH-CO2 has attracted interest, mostly in academia,320 due to the prospect of utilising CO2 and developing carbon neutral or carbon-negative processes. This is determined by the downstream use of CO, the primary product of CO2 dissociation. Although ODH-CO2 catalysts are not known to overoxidise propane, the drawbacks associated with DDH, i.e., coking, often remain and the inertness of CO2 means that low STYs are frequently reported. The majority of investigations thus far have focussed on Ga- or Cr-based catalysts and improvement in these catalysts has been incremental. Supported nanoparticles on the other hand, have received little attention despite showing some promising results. In this class of catalysts, minimising competing reactions such as reforming, is essential. Future catalyst design should aim to move away from Cr- and Ga-based catalysts to focus on low-temperature CO2 activation and have controlled redox and acid–base properties. For this reaction to become significant beyond academic interest, a step-change in catalyst performance is needed.
ODH-N2O is the least studied reaction and progress has not been substantial in recent years. This may be due to the unfavourable economic costs associated with using N2O as an oxidant, although in an integrated facility where N2O is produced as a by-product this could be a useful approach. In terms of catalyst performance, the most active catalyst, Fe-ZSM-5, can achieve a similar STY to the more active DDH and ODH-O2 catalysts. However, the intrinsic selectivity to propene is often below 70% and is undoubtedly the main disadvantage. If propene selectivity can be improved to 90%+, the catalyst would out-perform some of the most active DDH and ODH-O2 catalysts reported in the literature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out as part of the Consortium on Metal Nanocatalysis funded by TotalEnergies. The authors would like to acknowledge the contributions of Moritz Schreiber, Luis Robles and Didier Borremans from Total who were involved in the early conception of the work, and in collating relevant press releases and patent literature.References
- Global Data, Global Propylene Capacity and Capital Expenditure Outlook to 2030 – Asia and Middle East to Lead Globally in Terms of Propylene Capacity Additions, 2021.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
- M. A. Atanga, F. Rezaei, A. Jawad, M. Fitch and A. A. Rownaghi, Appl. Catal., B, 2018, 220, 429–445 CrossRef CAS.
- C. A. Carrero, R. Schloegl, I. E. Wachs and R. Schomaecker, ACS Catal., 2014, 4, 3357–3380 CrossRef CAS.
- L. Shi, Y. Wang, B. Yan, W. Song, D. Shao and A. H. Lu, Chem. Commun., 2018, 54, 10936–10946 RSC.
- A. Akah, J. Williams and M. Ghrami, Catal. Surv. Asia, 2019, 23, 265–276 CrossRef CAS.
- I. Amghizar, L. A. Vandewalle, K. M. Van Geem and G. B. Marin, Engineering, 2017, 3, 171–178 CrossRef CAS.
- M. Ghanta, D. Fahey and B. Subramaniam, Appl. Petrochem. Res., 2014, 4, 167–179 CrossRef CAS.
- M. O. Xunhua, B. De Graaf and P. Diddams, HCN emissions in fluid catalytic cracking, 2013, vol. 18 Search PubMed.
- Global petrochemical trends: H2 2020 | S&P Global Platts.
- D. F. Rodríguez-Vallejo, G. Guillén-Gosálbez and B. Chachuat, ACS Sustainable Chem. Eng., 2020, 8, 3072–3081 CrossRef.
- A. Agarwal, D. Sengupta and M. El-Halwagi, ACS Sustainable Chem. Eng., 2018, 6, 2407–2421 CrossRef CAS.
- Formule-verte, Biocarburants: la bioraffinerie de Total à La Mède opérationnelle en 2018, https://www.formule-verte.com/biocarburants-la-bioraffinerie-de-total-a-la-mede-operationnelle-en-2018/.
- Neste, Neste delivers first batch of 100% renewable propane to European market - Green Car Congress, https://www.neste.com/releases-and-news/renewable-solutions/neste-delivers-first-batch-100-renewable-propane-european-market%0Ahttps://www.greencarcongress.com/2018/03/20180319neste.html, (accessed 3 March 2021).
- Borealis, Borealis and Neste begin strategic co-operation to accelerate circularity and bioeconomy in plastics, https://www.borealisgroup.com/news/borealis-and-neste-begin-strategic-co-operation-to-accelerate-circularity-and-bioeconomy-in-plastics, (accessed 4 May 2021).
- UK Dept of Energy & Climate Change, Atlantic Consulting and Menecon Consulting, RHI Evidence Report : Biopropane for Grid Injection, 2014.
- D. Taher, M. E. Thibault, D. Di Mondo, M. Jennings and M. Schlaf, Chem. – Eur. J., 2009, 15, 10132–10143 CrossRef CAS PubMed.
- N. Menon, A. Pásztor, B. R. K. Menon, P. Kallio, K. Fisher, M. K. Akhtar, D. Leys, P. R. Jones and N. S. Scrutton, Biotechnol. Biofuels, 2015, 8, 61 CrossRef.
- K. Chen, A. Khodakov, J. Yang, A. T. Bell and E. Iglesia, J. Catal., 1999, 186, 325–333 CrossRef CAS.
- A. M. Gaffney and O. M. Mason, Catal. Today, 2017, 285, 159–165 CrossRef CAS.
- E. Gomez, S. Kattel, B. Yan, S. Yao, P. Liu and J. G. Chen, Nat. Commun., 2018, 9, 1398 CrossRef.
- F. T. Zangeneh, A. Taeb, K. Gholivand and S. Sahebdelfar, Chem. Eng. Commun., 2016, 203, 557–565 CrossRef CAS.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CrossRef CAS.
- M. Pretz, B. Fish, L. Luo and B. Tears, Shaping the future of on-purpose propylene production, https://www.hydrocarbonprocessing.com/magazine/2017/april-2017/special-focus-petrochemical-developments/shaping-the-future-of-on-purpose-propylene-production, (accessed 13 May 2021).
- S. Sarkaer, in Global Refining and Petrochemicals Congress, 2019 Search PubMed.
- Clariant, CATOFIN® Propane/Butane Dehydrogenation, http://www.cbi.com/What-We-Do/Technology/Refining/Propane-Butane-Upgrading/Propane-Butane-Dehydrogenation.
- Clariant, Clariant's breakthrough hgm concept for propane dehydrogenation unit - a proven success in China, https://www.clariant.com/en/Corporate/News/2015/03/Clariants-breakthrough-HGM-concept-for-Propane-dehydrogenation-Unit-a-proven-success-in-China, (accessed 6 May 2021).
- US Pat., USO09725380B2, 2017.
- Inside Industry, Ineos selects clariant's catofin® catalysts for europe's largest propane dehydrogenation plan, https://www.insideindustry.com/index.php/news2/chemicals-pharmaceuticals/897-ineos-selects-clariant-s-catofin-catalysts-for-europe-s-largest-propane-dehydrogenation-plant, (accessed 6 May 2021).
- Hydrocarbon Engineering, KBR signs agreement for propane dehydrogenation project, https://www.hydrocarbonengineering.com/petrochemicals/14042021/kbr-signs-agreement-for-propane-dehydrogenation-project/, (accessed 6 May 2021).
- E. Araujo-Lopez, B. D. Vandegehuchte, D. Curulla-Ferre, D. I. Sharapa and F. Studt, J. Phys. Chem. C, 2020, 124, 27503–27510 CrossRef CAS.
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC.
- A. Iglesias-Juez, A. M. Beale, K. Maaijen, T. C. Weng, P. Glatzel and B. M. Weckhuysen, J. Catal., 2010, 276, 268–279 CrossRef CAS.
- A. Farjoo, F. Khorasheh, S. Niknaddaf and M. Soltani, Sci. Iran., 2011, 18, 458–464 CrossRef CAS.
- Q. Li, Z. Sui, X. Zhou, Y. Zhu, J. Zhou and D. Chen, Top. Catal., 2011, 54, 888–896 CrossRef CAS.
- Z. J. Zhao, C. C. Chiu and J. Gong, Chem. Sci., 2015, 6, 4403–4425 RSC.
- R. M. Rioux, H. Song, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 2192–2202 CrossRef CAS PubMed.
- R. T. Vang, K. Honkala, S. Dahl, E. K. Vestergaard, J. Schnadt, E. Lægsgaard, B. S. Clausen, J. K. Nørskov and F. Besenbacher, Nat. Mater., 2005, 4, 160–162 CrossRef CAS PubMed.
- M. Santhosh Kumar, D. Chen, J. C. Walmsley and A. Holmen, Catal. Commun., 2008, 9, 747–750 CrossRef.
- W. Zhang, H. Wang, J. Jiang, Z. Sui, Y. Zhu, D. Chen and X. Zhou, ACS Catal., 2020, 10, 12932–12942 CrossRef CAS.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- S. Chen, C. Pei, G. Sun, Z.-J. Zhao and J. Gong, Acc. Mater. Res., 2020, 1, 30–40 CrossRef CAS.
- M.-L. Yang, Y.-A. Zhu, X.-G. Zhou, Z.-J. Sui and D. Chen, ACS Catal., 2012, 2, 1247–1258 CrossRef CAS.
- G. J. Siri, J. M. Ramallo-López, M. L. Casella, J. L. G. Fierro, F. G. Requejo and O. A. Ferretti, Appl. Catal., A, 2005, 278, 239–249 CrossRef CAS.
- O. A. Bariås, A. Holmen and E. A. Blekkan, J. Catal., 1996, 158, 1–12 CrossRef.
- S. Rimaz, L. Chen, S. Kawi and A. Borgna, Appl. Catal., A, 2019, 588, 1172663 CrossRef.
- G. Sun, Z. J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef PubMed.
- R. M. Mironenko, O. B. Belskaya, V. P. Talsi, T. I. Gulyaeva, M. O. Kazakov, A. I. Nizovskii, A. V. Kalinkin, V. I. Bukhtiyarov, A. V. Lavrenov and V. A. Likholobov, Appl. Catal., A, 2014, 469, 472–482 CrossRef CAS.
- L. Shi, G. M. Deng, W. C. Li, S. Miao, Q. N. Wang, W. P. Zhang and A. H. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994–13998 CrossRef CAS PubMed.
- E. J. Jang, J. Lee, H. Y. Jeong and J. H. Kwak, Appl. Catal., A, 2019, 572, 1–8 CrossRef CAS.
- Q. Yu, T. Yu, H. Chen, G. Fang, X. Pan and X. Bao, J. Energy Chem., 2020, 41, 93–99 CrossRef.
- F. Jiang, L. Zeng, S. Li, G. Liu, S. Wang and J. Gong, ACS Catal., 2015, 5, 438–447 CrossRef CAS.
- X. Chen, M. Ge, Y. Li, Y. Liu, J. Wang and L. Zhang, Appl. Surf. Sci., 2019, 490, 611–621 CrossRef CAS.
- W. Tolek, K. Suriye, P. Praserthdam and J. Panpranot, Catal. Today, 2020, 358, 100–108 CrossRef CAS.
- L. L. Shen, K. Xia, W. Z. Lang, L. F. Chu, X. Yan and Y. J. Guo, Chem. Eng. J., 2017, 324, 336–346 CrossRef CAS.
- X. Wu, Q. Zhang, L. Chen, Q. Liu, X. Zhang, Q. Zhang, L. Ma and C. Wang, Fuel Process. Technol., 2020, 198, 106222 CrossRef CAS.
- Y. Zhang, Y. Zhou, M. Tang, X. Liu and Y. Duan, Chem. Eng. J., 2012, 181–182, 530–537 CrossRef CAS.
- M. Santhosh Kumar, A. Holmen and D. Chen, Microporous Mesoporous Mater., 2009, 126, 152–158 CrossRef CAS.
- W. Wannapakdee, T. Yutthalekha, P. Dugkhuntod, K. Rodponthukwaji, A. Thivasasith, S. Nokbin, T. Witoon, S. Pengpanich and C. Wattanakit, Catalysts, 2019, 9, 174 CrossRef.
- Z. Guofu and D. Shengxin, CN Pat., CN104607235, 2015 Search PubMed.
- C. Zunzhong and S. Luyang, CN Pat., CN105056990, 2015 Search PubMed.
- S. Barri, EU Pat., EP0212850, 1985 Search PubMed.
- Y. Wang, Z. P. Hu, X. Lv, L. Chen and Z. Y. Yuan, J. Catal., 2020, 385, 61–69 CrossRef CAS.
- Z. Xu, Y. Yue, X. Bao, Z. Xie and H. Zhu, ACS Catal., 2020, 10, 818–828 CrossRef CAS.
- Z. Wu, B. C. Bukowski, Z. Li, C. Milligan, L. Zhou, T. Ma, Y. Wu, Y. Ren, F. H. Ribeiro, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, J. Am. Chem. Soc., 2018, 140, 14870–14877 CrossRef PubMed.
- L. Deng, T. Shishido, K. Teramura and T. Tanaka, in Catalysis Today, 2014, vol. 232, pp. 33–39 Search PubMed.
- L. Deng, H. Miura, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, J. Catal., 2018, 365, 277–291 CrossRef CAS.
- L. Deng, H. Miura, T. Shishido, S. Hosokawa, K. Teramura and T. Tanaka, Chem. Commun., 2017, 53, 6937–6940 RSC.
- Z. Ji, D. Miao, L. Gao, X. Pan and X. Bao, Chin. J. Catal., 2020, 41, 719–729 CrossRef CAS.
- L. Wang, Y. Wang, C. W. Zhang, J. Wen, X. Weng and L. Shi, Catal. Sci. Technol., 2020, 10, 1248–1255 RSC.
- J. Wu, L. Wang, X. Yang, B. Lv and J. Chen, Ind. Eng. Chem. Res., 2018, 57, 2805–2810 CrossRef CAS.
- C. Meng, Y. Li, H. Wu, W. Wei, Y. Ning, Y. Cui, Q. Fu and X. Bao, Phys. Chem. Chem. Phys., 2018, 20, 11013–11020 RSC.
- L. Jie, R. Junfeng, M. Aizeng, D Zhijian, L. Changcheng and X. Jingxin, CN Pat., CN106607017, 2017 Search PubMed.
- T. Fengya, S. Qing, M. Zhangxi, S. Yifan and W. Yangdong, CN Pat., CN107970919, 2018 Search PubMed.
- F. Zhigui, M. Changxi, W. Wenhai, Z. Tieqiang, W. Xing and J. Dongyu, CN Pat., CN110898833, 2020 Search PubMed.
- Q. Botao, G. Yalin, H. Jiahui, Z. Junying and L. Fang, CN Pat., CN110237840, 2019 Search PubMed.
- P. E. Højlund Nielsen, N. Munksgård and L. J. Lemus-Yegres, Int. Pat., WO2017162427A.1, 2017 Search PubMed.
- L. A. Petrov, Y. A. Alhamed, A. A. Al-Zahrani, M. A. Daous, A. A. Khamis and M. H. Al-Hazmi, Int. Pat., WO/2014/181289, 2014 Search PubMed.
- T. Wang, F. Jiang, G. Liu, L. Zeng, Z. J. Zhao and J. Gong, AIChE J., 2016, 62, 4365–4376 CrossRef CAS.
- F. E. Frey and W. F. Huppke, Ind. Eng. Chem., 1933, 25, 54–59 CrossRef.
- A. Hakuli, A. Kytökivi and A. O. I. Krause, Appl. Catal., A, 2000, 190, 219–232 CrossRef CAS.
- B. M. Weckhuysen, J. Catal., 2002, 210, 418–430 CrossRef.
- A. Hakuli, M. E. Harlin, L. B. Backman and A. O. I. Krause, J. Catal., 1999, 184, 349–356 CrossRef CAS.
- P. Michorczyk, J. Ogonowski and K. Zeńczak, J. Mol. Catal. A: Chem., 2011, 349, 1–12 CrossRef CAS.
- P. Michorczyk, P. Pietrzyk and J. Ogonowski, Microporous Mesoporous Mater., 2012, 161, 56–66 CrossRef CAS.
- B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2003, 5, 4351–4360 RSC.
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Chem. Rev., 1996, 96, 3327–3349 CrossRef CAS PubMed.
- P. Michorczyk, J. Ogonowski, P. Kuśtrowski and L. Chmielarz, Appl. Catal., A, 2008, 349, 62–69 CrossRef CAS.
- M. Santhosh Kumar, N. Hammer, M. Rønning, A. Holmen, D. Chen, J. C. Walmsley and G. Øye, J. Catal., 2009, 261, 116–128 CrossRef CAS.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, J. Catal., 2012, 293, 67–75 CrossRef CAS.
- K. Takehira, Y. Ohishi, T. Shishido, T. Kawabata, K. Takaki, Q. Zhang and Y. Wang, J. Catal., 2004, 224, 404–416 CrossRef CAS.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 223–232 CrossRef CAS.
- D. He, Y. Zhang, S. Yang, Y. Mei and Y. Luo, ChemCatChem, 2018, 10, 5434–5440 CrossRef CAS.
- S. De Rossi, G. Ferraris, S. Fremiotti, E. Garrone, G. Ghiotti, M. C. Campa and V. Indovina, J. Catal., 1994, 148, 36–46 CrossRef CAS.
- M. P. Conley, M. F. Delley, F. Núnez-Zarur, A. Comas-Vives and C. Copéret, Inorg. Chem., 2015, 54, 5065–5078 CrossRef CAS PubMed.
- C. Liu, G. Li, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 344, 570–577 CrossRef CAS.
- X. Q. Gao, W. D. Lu, S. Z. Hu, W. C. Li and A. H. Lu, Chin. J. Catal., 2019, 40, 184–191 CrossRef CAS.
- W. Z. Lang, C. L. Hu, L. F. Chu and Y. J. Guo, RSC Adv., 2014, 4, 37107–37113 RSC.
- A. WÈ©grzyniak, S. Jarczewski, A. WÈ©grzynowicz, B. Michorczyk, P. Kuśtrowski and P. Michorczyk, Nanomaterials, 2017, 7, 249 CrossRef PubMed.
- Y. Zhang, S. Yang, J. Lu, Y. Mei, D. He and Y. Luo, Ind. Eng. Chem. Res., 2019, 58, 19818–19824 CrossRef CAS.
- P. P. Li, W. Z. Lang, K. Xia, L. Luan, X. Yan and Y. J. Guo, Appl. Catal., A, 2016, 522, 172–179 CrossRef CAS.
- Z. P. Hu, Y. Wang, D. Yang and Z. Y. Yuan, J. Energy Chem., 2020, 47, 225–233 CrossRef.
- S. Han, Y. Zhao, T. Otroshchenko, Y. Zhang, D. Zhao, H. Lund, T. H. Vuong, J. Rabeah, U. Bentrup, V. A. Kondratenko, U. Rodemerck, D. Linke, M. Gao, H. Jiao, G. Jiang and E. V. Kondratenko, ACS Catal., 2020, 10, 1575–1590 CrossRef CAS.
- S. Han, T. Otroshchenko, D. Zhao, H. Lund, N. Rockstroh, T. H. Vuong, J. Rabeah, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, Appl. Catal., A, 2020, 590, 117350 CrossRef CAS.
- K. L. Fujdala and T. D. Tilley, J. Catal., 2003, 218, 123–134 CrossRef CAS.
- S. Han, T. Otroshchenko, D. Zhao, H. Lund, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, Catal. Commun., 2020, 138, 105956 CrossRef CAS.
- A. WÈ©grzyniak, A. Rokicińska, E. HÈ©drzak, B. Michorczyk, K. Zeńczak-Tomera, P. Kuśtrowski and P. Michorczyk, Catal. Sci. Technol., 2017, 7, 6059–6068 RSC.
- J. Liu, Y. Liu, Y. Ni, H. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2020, 10, 1739–1746 RSC.
- P. C. Doolan and P. R. Pujado, Hydrocarbon Process., 1989, 68, 72–74 CAS.
- A. Bhan and W. N. Delgass, Catal. Rev. - Sci. Eng., 2008, 50, 19–151 CrossRef CAS.
- K. Searles, G. Siddiqi, O. V. Safonova and C. Copéret, Chem. Sci., 2017, 8, 2661–2666 RSC.
- J. Im and M. Choi, ACS Catal., 2016, 6, 2819–2826 CrossRef CAS.
- Y. Ren, J. Wang, W. Hua, Y. Yue and Z. Gao, J. Ind. Eng. Chem., 2012, 18, 731–736 CrossRef CAS.
- B. Xu, B. Zheng, W. Hua, Y. Yue and Z. Gao, J. Catal., 2006, 239, 470–477 CrossRef CAS.
- B. Xu, T. Li, B. Zheng, W. Hua, Y. Yue and Z. Gao, Catal. Lett., 2007, 119, 283–288 CrossRef CAS.
- J. Wang, F. Zhang, W. Hua, Y. Yue and Z. Gao, Catal. Commun., 2012, 18, 63–67 CrossRef CAS.
- J. J. H. B. Sattler, I. D. Gonzalez-Jimenez, L. Luo, B. A. Stears, A. Malek, D. G. Barton, B. A. Kilos, M. P. Kaminsky, T. W. G. M. Verhoeven, E. J. Koers, M. Baldus and B. M. Weckhuysen, Angew. Chemie, 2014, 126, 9405–9410 CrossRef.
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS PubMed.
- K. C. Szeto, Z. R. Jones, N. Merle, C. Rios, A. Gallo, F. Le Quemener, L. Delevoye, R. M. Gauvin, S. L. Scott and M. Taoufik, ACS Catal., 2018, 8, 7566–7577 CrossRef CAS.
- V. J. Cybulskis, S. U. Pradhan, J. J. Lovón-Quintana, A. S. Hock, B. Hu, G. Zhang, W. N. Delgass, F. H. Ribeiro and J. T. Miller, Catal. Lett., 2017, 147, 1252–1262 CrossRef CAS.
- C. T. Shao, W. Z. Lang, X. Yan and Y. J. Guo, RSC Adv., 2017, 7, 4710–4723 RSC.
- W. G. Kim, J. So, S. W. Choi, Y. Liu, R. S. Dixit, C. Sievers, D. S. Sholl, S. Nair and C. W. Jones, Chem. Mater., 2017, 29, 7213–7222 CrossRef CAS.
- S. W. Choi, W. G. Kim, J. S. So, J. S. Moore, Y. Liu, R. S. Dixit, J. G. Pendergast, C. Sievers, D. S. Sholl, S. Nair and C. W. Jones, J. Catal., 2017, 345, 113–123 CrossRef CAS.
- M. W. Schreiber, C. P. Plaisance, M. Baumgärtl, K. Reuter, A. Jentys, R. Bermejo-Deval and J. A. Lercher, J. Am. Chem. Soc., 2018, 140, 4849–4859 CrossRef CAS PubMed.
- N. M. Phadke, E. Mansoor, M. Bondil, M. Head-Gordon and A. T. Bell, J. Am. Chem. Soc., 2019, 141, 1614–1627 CrossRef CAS PubMed.
- N. Raman, S. Maisel, M. Grabau, N. Taccardi, J. Debuschewitz, M. Wolf, H. Wittkämper, T. Bauer, M. Wu, M. Haumann, C. Papp, A. Görling, E. Spiecker, J. Libuda, H. P. Steinrück and P. Wasserscheid, ACS Catal., 2019, 9, 9499–9507 CrossRef CAS PubMed.
- T. Bauer, S. Maisel, D. Blaumeiser, J. Vecchietti, N. Taccardi, P. Wasserscheid, A. Bonivardi, A. Görling and J. Libuda, ACS Catal., 2019, 9, 2842–2853 CrossRef CAS PubMed.
- J. Keränen, A. Auroux, S. Ek and L. Niinistö, Appl. Catal., A, 2002, 228, 213–225 CrossRef.
- F. Roozeboom, M. C. Mittelmeijer-Hazeleger, J. A. Moulijn, J. Medema, V. H. J. De Beer and P. J. Gellings, J. Phys. Chem., 1980, 84, 2783–2791 CrossRef CAS.
- V. Murgia, E. M. F. Torres, J. C. Gottifredi and E. L. Sham, Appl. Catal., A, 2006, 312, 134–143 CrossRef CAS.
- A. Klisińska, S. Loridant, B. Grzybowska, J. Stoch and I. Gressel, Appl. Catal., A, 2006, 309, 17–27 CrossRef.
- G. Liu, Z. J. Zhao, T. Wu, L. Zeng and J. Gong, ACS Catal., 2016, 6, 5207–5214 CrossRef CAS.
- Z. J. Zhao, T. Wu, C. Xiong, G. Sun, R. Mu, L. Zeng and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 6791–6795 CrossRef CAS PubMed.
- F. Klose, T. Wolff, H. Lorenz, A. Seidel-Morgenstern, Y. Suchorski, M. Piórkowska and H. Weiss, J. Catal., 2007, 247, 176–193 CrossRef CAS.
- M. E. Harlin, V. M. Niemi and A. O. I. Krause, J. Catal., 2000, 195, 67–78 CrossRef CAS.
- U. Rodemerck, M. Stoyanova, E. V. Kondratenko and D. Linke, J. Catal., 2017, 352, 256–263 CrossRef CAS.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128–2191 CrossRef CAS PubMed.
- D. E. Keller, S. M. K. Airaksinen, A. O. Krause, B. M. Weckhuysen and D. C. Koningsberger, J. Am. Chem. Soc., 2007, 129, 3189–3197 CrossRef CAS PubMed.
- S. Sokolov, M. Stoyanova, U. Rodemerck, D. Linke and E. V. Kondratenko, Catal. Sci. Technol., 2014, 4, 1323–1332 RSC.
- S. Sokolov, V. Y. Bychkov, M. Stoyanova, U. Rodemerck, U. Bentrup, D. Linke, Y. P. Tyulenin, V. N. Korchak and E. V. Kondratenko, ChemCatChem, 2015, 7, 1691–1700 CrossRef CAS.
- P. Bai, Z. Ma, T. Li, Y. Tian, Z. Zhang, Z. Zhong, W. Xing, P. Wu, X. Liu and Z. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 25979–25990 CrossRef CAS PubMed.
- Y. Gu, H. Liu, M. Yang, Z. Ma, L. Zhao, W. Xing, P. Wu, X. Liu, S. Mintova, P. Bai and Z. Yan, Appl. Catal., B, 2020, 274, 119089 CrossRef CAS.
- T. Wu, G. Liu, L. Zeng, G. Sun, S. Chen, R. Mu, S. Agbotse Gbonfoun, Z. J. Zhao and J. Gong, AIChE J., 2017, 63, 4911–4919 CrossRef CAS.
- O. Ovsitser, R. Schomaecker, E. V. Kondratenko, T. Wolfram and A. Trunschke, Catal. Today, 2012, 192, 16–19 CrossRef CAS.
- P. Hu, W. Z. Lang, X. Yan, X. F. Chen and Y. J. Guo, Appl. Catal., A, 2018, 553, 65–73 CrossRef CAS.
- Q. Liu, Z. Yang, M. Luo, Z. Zhao, J. Wang, Z. Xie and L. Guo, Microporous Mesoporous Mater., 2019, 282, 133–145 CrossRef CAS.
- P. Hu, W. Z. Lang, X. Yan, L. F. Chu and Y. J. Guo, J. Catal., 2018, 358, 108–117 CrossRef CAS.
- C. Chen, M. Sun, Z. Hu, Y. Liu, S. Zhang and Z. Y. Yuan, Chin. J. Catal., 2020, 41, 276–285 CrossRef CAS.
- Y. Xie, R. Luo, G. Sun, S. Chen, Z. J. Zhao, R. Mu and J. Gong, Chem. Sci., 2020, 11, 3845–3851 RSC.
- N. Jeon, H. Choe, B. Jeong and Y. Yun, Catal. Today, 2020, 352, 337–344 CrossRef.
- Y. Zhang, Y. Zhao, T. Otroshchenko, H. Lund, M.-M. Pohl, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, Nat. Commun., 2018, 9, 3794 CrossRef PubMed.
- Y. Zhang, Y. Zhao, T. Otroshchenko, S. Han, H. Lund, U. Rodemerck, D. Linke, H. Jiao, G. Jiang and E. V. Kondratenko, J. Catal., 2019, 371, 313–324 CrossRef CAS.
- N. Jeon, H. Choe, B. Jeong and Y. Yun, Appl. Catal., A, 2019, 586, 117211 CrossRef.
- Y. N. Sun, C. Gao, L. Tao, G. Wang, D. Han, C. Li and H. Shan, Catal. Commun., 2014, 50, 73–77 CrossRef CAS.
- C. Chen, Z. P. Hu, J. T. Ren, S. Zhang, Z. Wang and Z. Y. Yuan, Mol. Catal., 2019, 476, 110508 CrossRef CAS.
- D. Zhao, Y. Li, S. Han, Y. Zhang, G. Jiang, Y. Wang, K. Guo, Z. Zhao, C. Xu, R. Li, C. Yu, J. Zhang, B. Ge and E. V. Kondratenko, iScience, 2019, 13, 269–276 CrossRef CAS PubMed.
- S. Han, D. Zhao, T. Otroshchenko, H. Lund, U. Bentrup, V. A. Kondratenko, N. Rockstroh, S. Bartling, D. E. Doronkin, J.-D. Grunwaldt, U. Rodemerck, D. Linke, M. Gao, G. Jiang and E. V. Kondratenko, ACS Catal., 2020, 10, 8933–8949 CrossRef CAS.
- M. Mukherjee, Hydrocarbonprocesssing.com https://www.hydrocarbonprocessing.com/authors/e/exelus-inc/mukherjee-m, (accessed 27 May 2021).
- M. Mukherjee, V. M. Vadhri, N. Joshi and G. Brock, US Pat., 2020/0101445A1, 2020 Search PubMed.
- Y. Dai, J. Gu, S. Tian, Y. Wu, J. Chen, F. Li, Y. Du, L. Peng, W. Ding and Y. Yang, J. Catal., 2020, 381, 482–492 CrossRef CAS.
- X. Li, P. Wang, H. Wang and C. Li, Appl. Surf. Sci., 2018, 441, 688–693 CrossRef CAS.
- G. Wang, H. Zhang, H. Wang, Q. Zhu, C. Li and H. Shan, J. Catal., 2016, 344, 606–608 CrossRef CAS.
- G. Wang, H. Zhang, Q. Zhu, X. Zhu, X. Li, H. Wang, C. Li and H. Shan, J. Catal., 2017, 351, 90–94 CrossRef CAS.
- H. Wang, H. Huang, K. Bashir and C. Li, Appl. Catal., A, 2020, 590, 117291 CrossRef CAS.
- C. Yang, Z. Wu, G. Zhang, H. Sheng, J. Tian, Z. Duan, H. Sohn, A. J. Kropf, T. Wu, T. R. Krause and J. T. Miller, Catal. Today, 2019, 323, 123–128 CrossRef CAS.
- Y. He, Y. Song, D. A. Cullen and S. Laursen, J. Am. Chem. Soc., 2018, 140, 14010–14014 CrossRef CAS PubMed.
- Z. Zhao, G. Ge, W. Li, X. Guo and G. Wang, Chin. J. Catal., 2016, 37, 644–670 CrossRef CAS.
- Z. P. Hu, H. Zhao, C. Chen and Z. Y. Yuan, Catal. Today, 2018, 316, 214–222 CrossRef CAS.
- L. Liu, Q. F. Deng, B. Agula, X. Zhao, T. Z. Ren and Z. Y. Yuan, Chem. Commun., 2011, 47, 8334–8336 RSC.
- Z. P. Hu, J. T. Ren, D. Yang, Z. Wang and Z. Y. Yuan, Chin. J. Catal., 2019, 40, 1385–1394 CrossRef CAS.
- S. F. Pan, J. L. Yin, X. L. Zhu, X. J. Guo, P. Hu, X. Yan, W. Z. Lang and Y. J. Guo, Carbon, 2019, 152, 855–864 CrossRef CAS.
- M. A. Chaar, D. Patel and H. H. Kung, J. Catal., 1988, 109, 463–467 CrossRef CAS.
- D. Siew Hew Sam, V. Soenen and J. C. Volta, J. Catal., 1990, 123, 417–435 CrossRef.
- A. Corma, J. M. López-Nieto, N. Paredes, M. Pérez, Y. Shen, H. Cao and S. L. Suib, Stud. Surf. Sci. Catal., 1992, 72, 213–220 CrossRef CAS.
- H. H. Kung and M. C. Kung, Appl. Catal., A, 1997, 157, 105–116 CrossRef CAS.
- T. Blasco and J. M. López Nieto, Appl. Catal., A, 1997, 157, 117–142 CrossRef CAS.
- A. Khodakov, B. Olthof, A. T. Bell and E. Iglesia, J. Catal., 1999, 181, 205–216 CrossRef CAS.
- B. Solsona, T. Blasco, J. M. López Nieto, M. L. Peña, F. Rey and A. Vidal-Moya, J. Catal., 2001, 203, 443–452 CrossRef CAS.
- G. Du, S. Lim, M. Pinault, C. Wang, F. Fang, L. Pfefferle and G. L. Haller, J. Catal., 2008, 253, 74–90 CrossRef CAS.
- A. Christodoulakis, M. Machli, A. A. Lemonidou and S. Boghosian, J. Catal., 2004, 222, 293–306 CrossRef CAS.
- M. D. Argyle, K. Chen, A. T. Bell and E. Iglesia, J. Catal., 2002, 208, 139–149 CrossRef CAS.
- J. T. Grant, C. A. Carrero, A. M. Love, R. Verel and I. Hermans, ACS Catal., 2015, 5, 5787–5793 CrossRef CAS.
- Y. M. Liu, Y. Cao, N. Yi, W. L. Feng, W. L. Dai, S. R. Yan, H. Y. He and K. N. Fan, J. Catal., 2004, 224, 417–428 CrossRef CAS.
- B. Schimmoeller, Y. Jiang, S. E. Pratsinis and A. Baiker, J. Catal., 2010, 274, 64–75 CrossRef CAS.
- A. Z. Varzaneh, M. S. Moghaddam and J. T. Darian, Pet. Chem., 2018, 58, 13–21 CrossRef CAS.
- T. S. Kharlamova, K. L. Timofeev, M. A. Salaev, V. A. Svetlichnyi and O. V. Vodyankina, Appl. Catal., A, 2020, 598, 117574 CrossRef CAS.
- S. Zhang and H. Liu, Appl. Catal., A, 2019, 573, 41–48 CrossRef CAS.
- G. Mitran, R. Ahmed, E. Iro, S. Hajimirzaee, S. Hodgson, A. Urdă, M. Olea and I. C. Marcu, Catal. Today, 2018, 306, 260–267 CrossRef CAS.
- H. Zhu, S. Ould-Chikh, H. Dong, I. Llorens, Y. Saih, D. H. Anjum, J. L. Hazemann and J. M. Basset, ChemCatChem, 2015, 7, 3332–3339 CrossRef CAS.
- S. Barman, N. Maity, K. Bhatte, S. Ould-Chikh, O. Dachwald, C. Haeßner, Y. Saih, E. Abou-Hamad, I. Llorens, J. L. Hazemann, K. Köhler, V. D'Elia and J. M. Basset, ACS Catal., 2016, 6, 5908–5921 CrossRef CAS.
- Q. X. Luo, X. K. Zhang, B. L. Hou, J. G. Chen, C. Zhu, Z. W. Liu, Z. T. Liu and J. Lu, Catal. Sci. Technol., 2018, 8, 4864–4876 RSC.
- J. J. Ternero-Hidalgo, J. Torres-Liñán, M. O. Guerrero-Pérez, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2019, 325, 131–143 CrossRef CAS.
- Q. Liu, M. Luo, Z. Zhao and L. Guo, Catal. Lett., 2019, 149, 1345–1358 CrossRef CAS.
- J. Guo, X. Li, Y. Tang and J. Zhang, ChemistrySelect, 2019, 4, 13576–13581 CrossRef CAS.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- J. T. Grant, C. A. Carrero, F. Goeltl, J. Venegas, P. Mueller, S. P. Burt, S. E. Specht, W. P. McDermott, A. Chieregato and I. Hermans, Science, 2016, 354, 1570–1573 CrossRef CAS PubMed.
- L. Shi, D. Wang, W. Song, D. Shao, W. P. Zhang and A. H. Lu, ChemCatChem, 2017, 9, 1788–1793 CrossRef CAS.
- R. Belgamwar, A. G. M. Rankin, A. Maity, A. K. Mishra, J. S. Gómez, J. Trébosc, C. P. Vinod and O. Lafon, ACS Sustainable Chem. Eng., 2020, 8, 16124–16135 CrossRef CAS.
- J. T. Grant, W. P. McDermott, J. M. Venegas, S. P. Burt, J. Micka, S. P. Phivilay, C. A. Carrero and I. Hermans, ChemCatChem, 2017, 9, 3623–3626 CrossRef CAS.
- J. M. Venegas, W. P. McDermott and I. Hermans, Acc. Chem. Res., 2018, 51, 2556–2564 CrossRef CAS PubMed.
- B. Yan, W. C. Li and A. H. Lu, J. Catal., 2019, 369, 296–301 CrossRef CAS.
- L. Shi, D. Wang and A. H. Lu, Chin. J. Catal., 2018, 39, 908–913 CrossRef CAS.
- H. Li, J. Zhang, P. Wu, S. Xun, W. Jiang, M. Zhang, W. Zhu and H. Li, J. Phys. Chem. C, 2019, 123, 2256–2266 CrossRef CAS.
- X. Zhang, R. You, Z. Wei, X. Jiang, J. Yang, Y. Pan, P. Wu, Q. Jia, Z. Bao, L. Bai, M. Jin, B. Sumpter, V. Fung, W. Huang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 8042–8046 CrossRef CAS PubMed.
- J. Tian, J. Tan, M. Xu, Z. Zhang, S. Wan, S. Wang, J. Lin and Y. Wang, Sci. Adv., 2019, 5, 1–7 Search PubMed.
- J. M. Venegas, Z. Zhang, T. O. Agbi, W. P. McDermott, A. Alexandrova and I. Hermans, Angew. Chem., Int. Ed., 2020, 59, 16527–16535 CrossRef CAS PubMed.
- W. D. Lu, D. Wang, Z. Zhao, W. Song, W. C. Li and A. H. Lu, ACS Catal., 2019, 9, 8263–8270 CrossRef CAS.
- A. M. Love, B. Thomas, S. E. Specht, M. P. Hanrahan, J. M. Venegas, S. P. Burt, J. T. Grant, M. C. Cendejas, W. P. McDermott, A. J. Rossini and I. Hermans, J. Am. Chem. Soc., 2019, 141, 182–190 CrossRef CAS PubMed.
- A. M. Love, M. C. Cendejas, B. Thomas, W. P. McDermott, P. Uchupalanun, C. Kruszynski, S. P. Burt, T. Agbi, A. J. Rossini and I. Hermans, J. Phys. Chem. C, 2019, 123, 27000–27011 CrossRef CAS.
- Y. Wang, W. C. Li, Y. X. Zhou, R. Lu and A. H. Lu, Catal. Today, 2020, 339, 62–66 CrossRef CAS.
- W. D. Lu, X. Q. Gao, Q. G. Wang, W. C. Li, Z. C. Zhao, D. Q. Wang and A. H. Lu, Chin. J. Catal., 2020, 41, 1837–1845 CrossRef CAS.
- H. Chen, Z. Yang, Z. Zhang, Z. Chen, M. Chi, S. Wang, J. Fu and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 10626–10630 CrossRef CAS PubMed.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113–131 CrossRef CAS.
- Z. Qiao, Z. Wang, C. Zhang, S. Yuan, Y. Zhu and J. Wang, AIChE J., 2012, 59, 215–228 CrossRef.
- K. Chen, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2000, 104, 1292–1299 CrossRef CAS.
- K. Chen, S. Xie, E. Iglesia and A. T. Bell, J. Catal., 2000, 189, 421–430 CrossRef CAS.
- G. Tsilomelekis, A. Christodoulakis and S. Boghosian, Catal. Today, 2007, 127, 139–147 CrossRef CAS.
- K. Chen, S. Xie, A. T. Bell and E. Iglesia, J. Catal., 2000, 195, 244–252 CrossRef CAS.
- M. Høj, T. Kessler, P. Beato, A. D. Jensen and J. D. Grunwaldt, Appl. Catal., A, 2014, 472, 29–38 CrossRef.
- S. Chen, L. Zeng, R. Mu, C. Xiong, Z. J. Zhao, C. Zhao, C. Pei, L. Peng, J. Luo, L. S. Fan and J. Gong, J. Am. Chem. Soc., 2019, 141, 18653–18657 CrossRef CAS PubMed.
- S. K. Matam, C. Moffat, P. Hellier, M. Bowker, I. P. Silverwood, C. R. A. Catlow, S. D. Jackson, J. Craswell, P. P. Wells, S. F. Parker and E. K. Gibson, Catalysts, 2020, 10, 1–13 CrossRef.
- E. Goudarzi, R. Asadi, J. T. Darian and A. Shahbazi Kootenaei, RSC Adv., 2019, 9, 11797–11809 RSC.
- X. Fan, D. Liu, Z. Zhao, J. Li and J. Liu, Catal. Today, 2020, 339, 67–78 CrossRef CAS.
- M. Chen, J. L. Wu, Y. M. Liu, Y. Cao, L. Guo, H. Y. He and K. N. Fan, J. Solid State Chem., 2011, 184, 3357–3363 CrossRef CAS.
- X. Fan, J. Li, Z. Zhao, Y. Wei, J. Liu, A. Duan and G. Jiang, J. Energy Chem., 2014, 23, 171–178 CrossRef CAS.
- D. Delgado, R. Sanchis, B. Solsona, P. Concepción and J. M. López Nieto, Top. Catal., 2020, 63, 1731–1742 CrossRef CAS.
- Q. Zhang, C. Cao, T. Xu, M. Sun, J. Zhang, Y. Wang and H. Wan, Chem. Commun., 2009, 2376–2378 RSC.
- K. Du, M. Hao, Z. Li, W. Hong, J. Liu, L. Xiao, S. Zou, H. Kobayashi and J. Fan, Chin. J. Catal., 2019, 40, 1057–1062 CrossRef CAS.
- Q. Xie, H. Zhang, J. Kang, J. Cheng, Q. Zhang and Y. Wang, ACS Catal., 2018, 8, 4902–4916 CrossRef CAS.
- K. Fang, L. Liu, M. Zhang, L. Zhao, J. Zhou, W. Li, X. Mu and C. Yang, Catalysts, 2018, 8, 19 CrossRef.
- J. Liu, M. Hao, C. Chen, K. Du, Q. Zhou, S. Zou, L. Xiao and J. Fan, Appl. Surf. Sci., 2020, 528, 147025 CrossRef CAS.
- L. Wang, C. Ao, Y. Zhai, B. Feng, J. Duan, S. Qian, W. Zhao, L. Zhang and F. Liu, Inorg. Chem. Commun., 2020, 112, 107725 CrossRef CAS.
- S. Tanasoi, G. Mitran, N. Tanchoux, T. Cacciaguerra, F. Fajula, I. Sndulescu, D. Tichit and I. C. Marcu, Appl. Catal., A, 2011, 395, 78–86 CrossRef CAS.
- F. Ma, S. Chen, Y. Li, H. Zhou, A. Xu and W. Lu, Appl. Surf. Sci., 2014, 313, 654–659 CrossRef CAS.
- F. Ma, S. Chen, H. Zhou, Y. Li and W. Lu, RSC Adv., 2014, 4, 40776–40781 RSC.
- B. Frank, J. Zhang, R. Blume, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 6913–6917 CrossRef CAS PubMed.
- W. Qi, P. Yan and D. S. Su, Acc. Chem. Res., 2018, 51, 640–648 CrossRef CAS PubMed.
- L. Cao, P. Dai, L. Zhu, L. Yan, R. Chen, D. Liu, X. Gu, L. Li, Q. Xue and X. Zhao, Appl. Catal., B, 2020, 262, 118277 CrossRef.
- C. P. Hubbard, K. Otto, H. S. Gandhi and K. Y. S. Ng, J. Catal., 1993, 139, 268–276 CrossRef CAS.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- L. Sun, Y. Chai, W. Dai, G. Wu, N. Guan and L. Li, Catal. Sci. Technol., 2018, 8, 3044–3051 RSC.
- Z. Nawaz and F. Wei, Ind. Eng. Chem. Res., 2013, 52, 346–352 CrossRef CAS.
- B. Farin, M. Devillers and E. M. Gaigneaux, Microporous Mesoporous Mater., 2017, 242, 200–207 CrossRef CAS.
- R. B. Jackson, J. G. Canadell, C. Le Quéré, R. M. Andrew, J. I. Korsbakken, G. P. Peters and N. Nakicenovic, Nat. Clim. Change, 2016, 6, 7–10 CrossRef.
- S. Sato, M. Ohhara, T. Sodesawa and F. Nozaki, Appl. Catal., 1988, 37, 207–215 CrossRef CAS.
- B. M. Reddy, S. C. Lee, D. S. Han and S. E. Park, Appl. Catal., B, 2009, 87, 230–238 CrossRef CAS.
- J. Guo, H. Lou, H. Zhao, L. Zheng and X. Zheng, J. Mol. Catal. A: Chem., 2005, 239, 222–227 CrossRef CAS.
- I. Takahara and M. Saito, Chem. Lett., 1996, 973–974 CrossRef CAS.
- K. Nakagawa, C. Kajita, N. Ikenaga, M. Nishitani-Gamo, T. Ando and T. Suzuki, Catal. Today, 2003, 84, 149–157 CrossRef CAS.
- X. Zhang, Y. Yue and Z. Gao, Catal. Lett., 2002, 83, 19–25 CrossRef CAS.
- R. Wu, P. Xie, Y. Cheng, Y. Yue, S. Gu, W. Yang, C. Miao, W. Hua and Z. Gao, Catal. Commun., 2013, 39, 20–23 CrossRef CAS.
- Z. Xie, Y. Ren, J. Li, Z. Zhao, X. Fan, B. Liu, W. Song, L. Kong, X. Xiao, J. Liu and G. Jiang, J. Catal., 2019, 372, 206–216 CrossRef CAS.
- J. F. S. de Oliveira, D. P. Volanti, J. M. C. Bueno and A. P. Ferreira, Appl. Catal., A, 2018, 558, 55–66 CrossRef CAS.
- P. Michorczyk and J. Ogonowski, React. Kinet. Catal. Lett., 2005, 87, 177–183 CrossRef.
- M. A. Botavina, Y. A. Agafonov, N. A. Gaidai, E. Groppo, V. Cortés Corberán, A. L. Lapidus and G. Martra, Catal. Sci. Technol., 2016, 6, 840–850 RSC.
- L. Li, W. Zhu, L. Shi, Y. Liu, H. Liu, Y. Ni, S. Liu, H. Zhou and Z. Liu, Chin. J. Catal., 2016, 37, 359–366 CrossRef CAS.
- P. Michorczyk, J. Ogonowski and M. Niemczyk, Appl. Catal., A, 2010, 374, 142–149 CrossRef CAS.
- H. M. Wang, Y. Chen, X. Yan, W. Z. Lang and Y. J. Guo, Microporous Mesoporous Mater., 2019, 284, 69–77 CrossRef CAS.
- J. Baek, H. J. Yun, D. Yun, Y. Choi and J. Yi, ACS Catal., 2012, 2, 1893–1903 CrossRef CAS.
- P. Kuśtrowski, P. Michorczyk, L. Chmielarz, Z. Piwowarska, B. Dudek, J. Ogonowski and R. Dziembaj, Thermochim. Acta, 2008, 471, 26–32 CrossRef.
- F. Zhang, R. Wu, Y. Yue, W. Yang, S. Gu, C. Miao, W. Hua and Z. Gao, Microporous Mesoporous Mater., 2011, 145, 194–199 CrossRef CAS.
- Q. Zhu, M. Takiguchi, T. Setoyama, T. Yokoi, J. N. Kondo and T. Tatsumi, Catal. Lett., 2011, 141, 670–677 CrossRef CAS.
- P. Michorczyk, K. Zeńczak-Tomera, B. Michorczyk, A. WÈ©grzyniak, M. Basta, Y. Millot, L. Valentin and S. Dzwigaj, J. CO2 Util., 2020, 36, 54–63 CrossRef CAS.
- D. Yun, J. Baek, Y. Choi, W. Kim, H. J. Lee and J. Yi, ChemCatChem, 2012, 4, 1952–1959 CrossRef CAS.
- M. Veronica Ganduglia-Pirovano, J. De, L. Figuera, V. Matolin, J. I. Flege, P. Luches, G. Gasperi, M. Sauerbrey, S. Valeri and J. Falta, Front. Chem., 2019, 7, 57 CrossRef PubMed http://www.frontiersin.org .
- R. Jin, J. Easa, D. T. Tran and C. P. O'Brien, Catal. Sci. Technol., 2020, 10, 1769–1777 RSC.
- X. Du, B. Yao, S. Gonzalez-Cortes, V. L. Kuznetsov, H. Almegren, T. Xiao and P. P. Edwards, Faraday Discuss., 2015, 183, 161–176 RSC.
- N. S. Gnep, J. Y. Doyement and M. Guisnet, J. Mol. Catal., 1988, 45, 281–284 CrossRef CAS.
- K. Nakagawa, M. Okamura, N. Ikenaga, T. Suzuki and T. Kobayashi, Chem. Commun., 1998, 1025–1026 RSC.
- K. Nakagawa, C. Kajita, K. Okumura, N. Ikenaga, T. Suzuki, K. Nakagawa, T. Ando, T. Suzuki, M. Nishitani-Gamo, T. Ando and T. Kobayashi, J. Catal., 2001, 203, 87–93 CrossRef CAS.
- B. Zheng, W. Hua, Y. Yue and Z. Gao, J. Catal., 2005, 232, 143–151 CrossRef CAS.
- P. Michorczyk, K. Góra-Marek and J. Ogonowski, Catal. Lett., 2006, 109, 195–198 CrossRef CAS.
- P. Michorczyk, P. Kuśtrowski, A. Kolak and M. Zimowska, Catal. Commun., 2013, 35, 95–100 CrossRef CAS.
- M. Chen, J. Xu, Y. M. Liu, Y. Cao, H. Y. He, J. H. Zhuang and K. N. Fan, Catal. Lett., 2008, 124, 369–375 CrossRef CAS.
- M. Chen, J. Xu, F. Z. Su, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Catal., 2008, 256, 293–300 CrossRef CAS.
- L. Zhang, Z. Y. Wang, J. Song, Y. Lang, J. G. Chen, Q. X. Luo, Z. H. He, K. Wang, Z. W. Liu and Z. T. Liu, J. CO2 Util., 2020, 38, 306–313 CrossRef CAS.
- P. Michorczyk, P. Kuśtrowski, L. Chmielarz and J. Ogonowski, React. Kinet. Catal. Lett., 2004, 82, 121–130 CrossRef CAS.
- Y. Ren, F. Zhang, W. Hua, Y. Yue and Z. Gao, Catal. Today, 2009, 148, 316–322 CrossRef CAS.
- M. Chen, J. Xu, Y. Cao, H. Y. He, K. N. Fan and J. H. Zhuang, J. Catal., 2010, 272, 101–108 CrossRef CAS.
- M. Chen, J. Xu, Y. M. Liu, Y. Cao, H. Y. He and J. H. Zhuang, Appl. Catal., A, 2010, 377, 35–41 CrossRef CAS.
- M. Chen, J. L. Wu, Y. M. Liu, Y. Cao, L. Guo, H. Y. He and K. N. Fan, Appl. Catal., A, 2011, 407, 20–28 CrossRef CAS.
- H. Tian, J. Liao, F. Zha, X. Guo, X. Tang, Y. Chang and X. Ma, ChemistrySelect, 2020, 5, 3626–3637 CrossRef CAS.
- K. Srivastava, V. Devra and A. Rani, Fuel Process. Technol., 2014, 121, 1–8 CrossRef CAS.
- E. V. Danilevich, G. Y. Popova, T. V. Andrushkevich, V. V. Kaichev, I. G. Danilova, Y. A. Chesalov, V. A. Rogov, V. I. Bukhtiyarov and V. N. Parmon, Appl. Catal., A, 2014, 475, 98–108 CrossRef CAS.
- J. Kang, A. D. Czaja and V. V. Guliants, Eur. J. Inorg. Chem., 2017, 2017, 4757–4762 CrossRef CAS.
- Z. F. Han, X. L. Xue, J. M. Wu, W. Z. Lang and Y. J. Guo, Chin. J. Catal., 2018, 39, 1099–1109 CrossRef CAS.
- H. Wang and G. Tsilomelekis, Catal. Sci. Technol., 2020, 10, 4362–4372 RSC.
- J. H. Carter, X. Liu, Q. He, S. Althahban, E. Nowicka, S. J. Freakley, L. Niu, D. J. Morgan, Y. Li, J. W. H. Niemantsverdriet, S. Golunski, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2017, 56, 16037–16041 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- E. Gomez, Z. Xie and J. G. Chen, AIChE J., 2019 DOI:10.1002/aic.16670.
- E. Nowicka, C. Reece, S. M. Althahban, K. M. H. Mohammed, S. A. Kondrat, D. J. Morgan, Q. He, D. J. Willock, S. Golunski, C. J. Kiely and G. J. Hutchings, ACS Catal., 2018, 8, 3454–3468 CrossRef CAS.
- R. Hou, K. Chang, J. G. Chen and T. Wang, Top. Catal., 2015, 58, 240–246 CrossRef CAS.
- A. C. Lausche, J. A. Schaidle, N. Schweitzer and L. T. Thompson, in Comprehensive Inorganic Chemistry II (Second Edition): From Elements to Applications, Elsevier Ltd, 2013, vol. 7, pp. 371–404 Search PubMed.
- T. H. Nguyen, T. V. Nguyen, C. G. Cooper, A. A. Adesina, A. C. Lausche, J. A. Schaidle, N. Schweitzer and L. T. Thompson, in New Developments and Application in Chemical Reaction Engineering, ed. H.-K. Rhee, I.-S. Nam and J. M. B. Park, Elsevier, 2006, vol. 159, pp. 781–784 Search PubMed.
- M. M. Sullivan and A. Bhan, J. Catal., 2018, 357, 195–205 CrossRef.
- B. Frank, T. P. Cotter, M. E. Schuster, R. Schlögl and A. Trunschke, Chem. – Eur. J., 2013, 19, 16938–16945 CrossRef CAS PubMed.
- J. C. Védrine, Catalysts, 2016, 6, 22 CrossRef.
- M. Konsolakis, ACS Catal., 2015, 5, 6397–6421 CrossRef CAS.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef CAS.
- X. Rozanska, E. V. Kondratenko and J. Sauer, J. Catal., 2008, 256, 84–94 CrossRef CAS.
- G. I. Panov, A. K. Uriarte, M. A. Rodkin and V. I. Sobolev, Catal. Today, 1998, 41, 365–385 CrossRef CAS.
- K. Nowińska, A. W
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) cław and A. Izbińska, Appl. Catal., A, 2003, 243, 225–236 CrossRef.
cław and A. Izbińska, Appl. Catal., A, 2003, 243, 225–236 CrossRef. - J. Pérez-Ramírez and A. Gallardo-Llamas, J. Catal., 2004, 223, 382–388 CrossRef.
- P. Sazama, N. K. Sathu, E. Tabor, B. Wichterlová, Š. Sklenák and Z. Sobalík, J. Catal., 2013, 299, 188–203 CrossRef CAS.
- J. C. Groen, A. Brückner, E. Berrier, L. Maldonado, J. A. Moulijn and J. Pérez-Ramírez, J. Catal., 2006, 243, 212–216 CrossRef CAS.
- I. Melián-Cabrera, S. Espinosa, J. C. Groen, B. V. D. Linden, F. Kapteijn and J. A. Moulijn, J. Catal., 2006, 238, 250–259 CrossRef.
- J. Pérez-Ramírez and E. V. Kondratenko, Chem. Commun., 2003, 3, 2152–2153 RSC.
- J. Pérez-Ramírez and A. Gallardo-Llamas, Appl. Catal., A, 2005, 279, 117–123 CrossRef.
- R. Bulánek, B. Wichterlová, K. Novoveská and V. Kreibich, Appl. Catal., A, 2004, 264, 13–22 CrossRef.
- J. Kowalska-Kuś, A. Held and K. Nowińska, Catal. Lett., 2010, 136, 199–208 CrossRef.
- A. Ates, C. Hardacre and A. Goguet, Appl. Catal., A, 2012, 441442, 30–41 CrossRef.
- E. V. Kondratenko and M. Baerns, Appl. Catal., A, 2001, 222, 133–143 CrossRef CAS.
- E. V. Kondratenko, M. Cherian, M. Baerns, D. Su, R. Schlögl, X. Wang and I. E. Wachs, J. Catal., 2005, 234, 131–142 CrossRef CAS.
- E. V. Kondratenko, M. Cherian and M. Baerns, in Catalysis Today, Elsevier, 2006, vol. 112, pp. 60–63 Search PubMed.
- O. Ovsitser, M. Cherian and E. V. Kondratenko, J. Phys. Chem. C, 2007, 111, 8594–8602 CrossRef CAS.
- M. Chen, J. L. Wu, Y. M. Liu, Y. Cao and K. N. Fan, Catal. Commun., 2011, 12, 1063–1066 CrossRef CAS.
- C. Rongguo, G. Juan, L. Yin, H. Dieter and L. Maren, Supply and Demand of Lithium and Gallium, 2016 Search PubMed.
- U.S. Geological Survey, Mineral commodity summaries 2020, 2020 Search PubMed.
- J. D. Burton, F. Culkin and J. P. Riley, Geochim. Cosmochim. Acta, 1959, 16, 151–180 CrossRef.
- N. E. T. Laboratory, Doe invests 17 million dollars to advance carbon utilization projects, https://netl.doe.gov/node/9789#:~:text=DOEInvests%2417Millionto,Projects%7Cnetl.doe.gov&text=TheU.S.DepartmentofEnergy's,developmentprojectsforcarbonutilization, (accessed 14 May 2021).
| This journal is © The Royal Society of Chemistry 2021 |