
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-throughput optofluidic screening for improved microbial cell factories via real-time micron-scale productivity monitoring†
Matthew
Rienzo‡
 a,
Ke-Chih
Lin‡
b,
Kellen C.
Mobilia
b,
Eric K.
Sackmann
b,
Volker
Kurz
b,
Adam H.
Navidi
a,
Jarett
King
a,
Robert M.
Onorato
b,
Lawrence K.
Chao
a,
Tony
Wu
a,
Hanxiao
Jiang
a,
Justin K.
Valley
a,
Troy A.
Lionberger‡
*b and
Michael D.
Leavell‡
*a
a,
Ke-Chih
Lin‡
b,
Kellen C.
Mobilia
b,
Eric K.
Sackmann
b,
Volker
Kurz
b,
Adam H.
Navidi
a,
Jarett
King
a,
Robert M.
Onorato
b,
Lawrence K.
Chao
a,
Tony
Wu
a,
Hanxiao
Jiang
a,
Justin K.
Valley
a,
Troy A.
Lionberger‡
*b and
Michael D.
Leavell‡
*a
aResearch and Development, Amyris, Inc., 5885 Hollis St., Suite 100, Emeryville, CA 94608, USA. E-mail: leavell@amyris.com
bTechnology and Business Development, Berkeley Lights, Inc., 5858 Horton St., Unit 320, Emeryville, CA 94608, USA. E-mail: troy.lionberger@berkeleylights.com
First published on 23rd June 2021
Abstract
The industrial synthetic biology sector has made huge investments to achieve relevant miniaturized screening systems for scalable fermentation. Here we present the first example of a high-throughput (>103 genotypes per week) perfusion-based screening system to improve small-molecule secretion from microbial strains. Using the Berkeley Lights Beacon® system, the productivity of each strain could be directly monitored in real time during continuous culture, yielding phenotypes that correlated strongly (r2 > 0.8, p < 0.0005) with behavior in industrially relevant bioreactor processes. This method allows a much closer approximation of a typical fed-batch fermentation than conventional batch-like droplet or microplate culture models, in addition to rich time-dependent data on growth and productivity. We demonstrate these advantages by application to the improvement of high-productivity strains using whole-genome random mutagenesis, yielding mutants with substantially improved (by up to 85%) peak specific productivities in bioreactors. Each screen of ∼5 × 103 mutants could be completed in under 8 days (including 5 days involving user intervention), saving ∼50–75% of the time required for conventional microplate-based screening methods.
Introduction
Over the past several decades, metabolic engineering has developed microbial cell factories as sustainable alternatives to chemical synthesis from petroleum feedstocks or harvesting of animals and plants.1–9 These fermentation processes employ natural and engineered enzymes to give elegant one-pot alternatives to conventional manufacturing. The gold-standard method for selection of a microbial strain for manufacturing is the lab-scale bioreactor, which represents a costly and labor-intensive commitment, as thousands or more candidate genotypes are often considered. Most high-throughput strain improvement campaigns have heavily relied on hit selection from microplate-based culture models,10,11 growth-coupled selection schemes,12 and more recently droplet microfluidics,13 with varying success in the prediction of scalable bioreactor phenotypes.A shortcoming of microplate- and droplet-based culture models is that they often do not replicate the conditions observed in feedback-regulated bioreactor systems.10 Indeed, it is infeasible to individually and dynamically control the chemical composition of thousands of wells or droplets in a typical screening scheme, as is done in low-throughput bioreactors. The Beacon® optofluidic system developed by Berkeley Lights is unique in that it addresses this challenge by spatially isolating strains in incubation chambers (“NanoPen™ chambers” or “pens”) within a microfluidic device, the OptoSelect™ chip, while continuously controlling the extracellular environment by perfusion of fresh media.14 This approach has the potential to more closely mimic a bioreactor by tightly controlling nutrient levels, oxygenation, and pH balance during growth. Previous reports of similar “microchemostat” systems have established a proof of concept for this style of microbial culture and in situ assay,15–19 but such approaches have not demonstrated the throughput, automation, robustness, and operational feasibility that are possible with the Beacon system and/or are not intended to screen for improved metabolic flux.
The operation of the Beacon system is illustrated in Fig. 1. Briefly, the system can hold up to four chips, each of which includes a set of channels that traverse the full area of the chip in a serpentine fashion. Cells are loaded into the chip by syringe pump from a sample vessel and allowed to collect throughout the chip within the channels. 10× bright-field imaging is used to visualize the cells, and a proprietary algorithm is used to select cells of appropriate roundness, size, and distance from other cells for seeding into pens. The device uses light-actuated cell positioning, termed OptoElectroPositioning (OEP™), which entails irradiating the silicon base of the chip with visible light while applying an electrical potential across the device. This induces a negative dielectrophoretic force that can be used to individually manipulate single cells by moving a square “light cage” surrounding each targeted single cell,20 a process that has been automated at Berkeley Lights using image recognition software. The cells are moved into separate pens to be cultured as individual colonies. The chip is perfused with culture media as cells multiply. Cells within pens are not disturbed by media flow, but passive diffusion causes rapid exchange with fresh media such that nutrient levels are kept constant and waste is carried away rapidly. The phenotype of each colony can then be assessed by monitoring the growth and secretions using bright-field imaging and fluorescence-based assays. In this manner, the Beacon system can allow simultaneous culture of ∼104–5 segregated colonies of differing genotypes. Once colonies bearing desirable phenotypes are identified, OEP can be used to reposition clumps of cells from a colony of interest into the channel using sequences of moving light bars, as shown in Fig. 1e. Subsequently, cells in the channels can be exported into microplates for further study through media perfusion. Charts illustrating some typical screening workflows are shown in Fig. S1 and S2.†
 | ||
| Fig. 1 The Beacon® optofluidic system. (a) Bright-field image of an entire OptoSelect™ chip with channels indicated in blue. (b) Magnified image of a portion of a channel following injection of suspended cells by syringe pump. Single cells (green) and multiples (red) are highlighted. (c) Each channel branches off into 102–103 “NanoPen™ chambers” of volume ∼0.25–1.7 nL, depending on chip design. (d) Fluorescence images can be collected periodically in addition to bright-field images, allowing identification of clones with desirable phenotypes. (e) Desired clones are exported into microplates for propagation and further study using Opto-ElectroPositioning (OEP) force, followed by media perfusion. | ||
Here, we describe two novel methods for identifying improved small molecule-producing strains using the Beacon system. The methods rely on fluorescence-based detection of secreted product, from which per cell productivities can be inferred. Although similar assays have previously been used to identify mammalian cell lines for antibody production, this is the first demonstration that the Beacon system can be applied to screening microbial strains for efficient small-molecule production. To our knowledge, it is also the first example of high-throughput (>103 strains per week) strain screening based on small-molecule secretion rate under steady-state culture conditions. Using strains that produce a fluorescent product as a test case, we show that Beacon system's assay scores correlate well with peak strain performance in 0.5- and 2.0 L pulse-fed bioreactors. We also demonstrate the ability of the platform to rapidly isolate strains from random whole-genome mutagenesis libraries that exhibit improved specific productivities in ambr250 bioreactors. The optimized two-tiered screening workflow reduced the timeline of library screening by 2-fold, obviated solid culture and colony-picking, and eliminated >95% of material waste produced in conventional microplate screening.
Results
Bioreactor performance metrics and screening strategy
In metabolic engineering, specific productivity (qp; mol product per mol biomass per hour) and growth rate (μ; per hour) are two key variables intrinsic to the genotype of a strain under a set of fixed culture conditions. If a scaled-down screening system effectively models strain physiology in a lab-scale bioreactor, estimates of qp and μ can be used to predict aspects of bioreactor performance. In this study, microfluidic culture conditions were empirically selected by optimizing the correlation between strain qp on the Beacon system and in a gold-standard lab-scale fermentation platform. The assays used allow measurement of biomass and product secretion, which together enable inference of both qp and μ. Note that in this work, conditions were not selected to optimize correlation of μ between the Beacon system's culture model and the lab-scale bioreactor.The values of qp and μ for a strain are industrially relevant because they are determinants of yield (Y; mol product per mol sugar fed) and volumetric productivity (P; mol product per L per hour), which are two key factors in the cost of production during manufacturing. In this work, Beacon assays were evaluated by comparing assay values for strains to qp, Y, and P values observed from lab-scale bioreactor fermentations. These fermentations typically begin with a period of rapid growth, followed by a later stage of pseudo-steady-state production when nutrients are more restricted. The Beacon system's assays attempt to model the latter stage. Thus, the relevant performance metrics were taken from the interval 24–48 hours following bioreactor inoculation, after the end of the rapid growth phase.
Bright-field intensity integration for biomass measurement
Most previously reported applications of the Beacon system have used mammalian cells, which are large enough to count explicitly via digital image processing. Because microbial cells are generally smaller and can grow in multilayers on the OptoSelect chip, counting is not feasible for hosts like S. cerevisiae. Instead, bright-field images were automatically processed to determine colony area and cell density. For the latter method, pixel intensity was integrated over the area of each NanoPen chamber, using reference measurements from nearby empty pens to calibrate the local background signal. The resulting background-corrected assay value is hereafter termed “OD score”.To validate OD score as a biomass measurement, NanoPen chambers in an OptoSelect chip were seeded with varying numbers of S. cerevisiae cells from a mixture of strains, which were then cultured on the Beacon system until a range of colony sizes was visible. After imaging the chip, cells were packed tightly into the NanoPen chambers via centrifugation to achieve uniform density before reimaging (Fig. 2). The area of the resulting packed colony was assumed to be a linear reflection of biomass. The OD score of the colonies measured before centrifugation was proportional to this packed colony area, indicating that it was linear over a wide range of colony sizes (Fig. 2), with typical CV < 15% for colonies > 3500 μm2 in area. In contrast, unpacked colony area did not linearly predict packed colony area, likely because larger colonies accumulated biomass in multilayers rather than strictly by increasing in area (Fig. S3†).
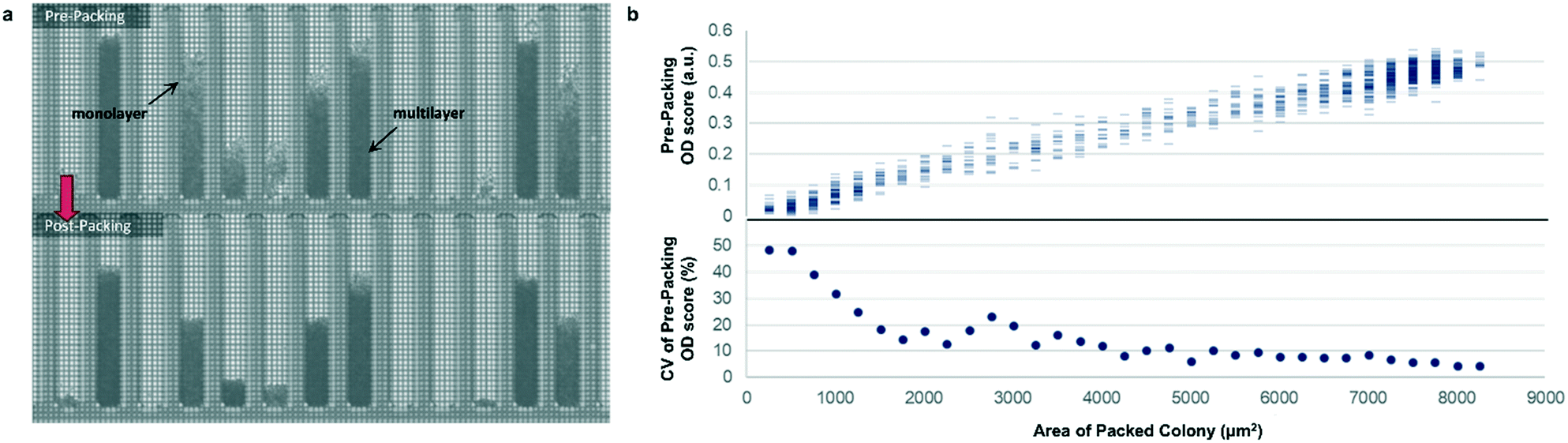 | ||
| Fig. 2 Bright-field density biomass measurement. (a) Representative image of a chip before and after centrifugation packing, illustrating the effect of packing on colony area and bright-field image density. (b) Top, relationship between unpacked colony OD score and packed colony area, illustrating assay linearity for the corrected biomass measurement. Bottom, relative variability (coefficient of variation, CV) of biomass sample measurement as a function of colony size, assuming perfect linearity of post-packing colony area with biomass. | ||
Sealed-pen productivity and yield inference
The Beacon system is configured to acquire fluorescence images of OptoSelect chips during strain cultivation. Although there are many ways to transduce a fluorescent signal in response to the local concentration of an analyte, detection is easiest when the product is intrinsically fluorescent.21,22 A set of S. cerevisiae strains engineered to produce a fluorescent small molecule was thus chosen to demonstrate the feasibility of screening for a secretion phenotype.Previous work on the Beacon system focused on secretion of macromolecules, which diffuse slowly enough to allow accumulation and visualization using fluorescent readouts.23 For low-molecular-weight compounds (<1 kD) like that tested here, rapid diffusion from the NanoPen chamber was initially a concern. A method was therefore devised to seal the NanoPen chambers using a hydrophobic oil with high respiratory gas solubility, but low product and carbohydrate solubility. After sealing, each pen effectively becomes a miniature batch reactor with fixed carbon source, accumulating product, and continuous gas exchange. When the assay is complete, the oil can be replaced with aqueous media to allow export of desired strains.
To determine whether strain performance in the sealed-pen assay aligns with lab-scale bioreactor performance, six strains previously tested in 0.5 L bioreactors were selected. The strains were sequentially seeded into NanoPen chambers on OptoSelect chips on the Beacon system in high replication (n = 40–50). Strains were cultured for ∼18 hours before sealing and then incubated for an additional 20 minutes with periodic fluorescence imaging (Fig. 3a), allowing inference of the relative P for each pen from the slope of the intensity curve for each colony during the 20 minute interval. To compare the per cell metabolic fluxes generated by each strain, relative specific productivities for the cells in each pen were calculated by normalizing the slope of the fluorescence intensity curve to the measured OD score, giving what is hereafter referred to as the “qp score”. Note that the precise definition of qp includes generation of both secreted and intracellular product; however, the strains tested in this work secrete the vast majority of the fluorescent product (Jiang et al., unpublished results), and so rough relative inferences of qp can be made from the biomass-normalized measurement of secreted product in this context.
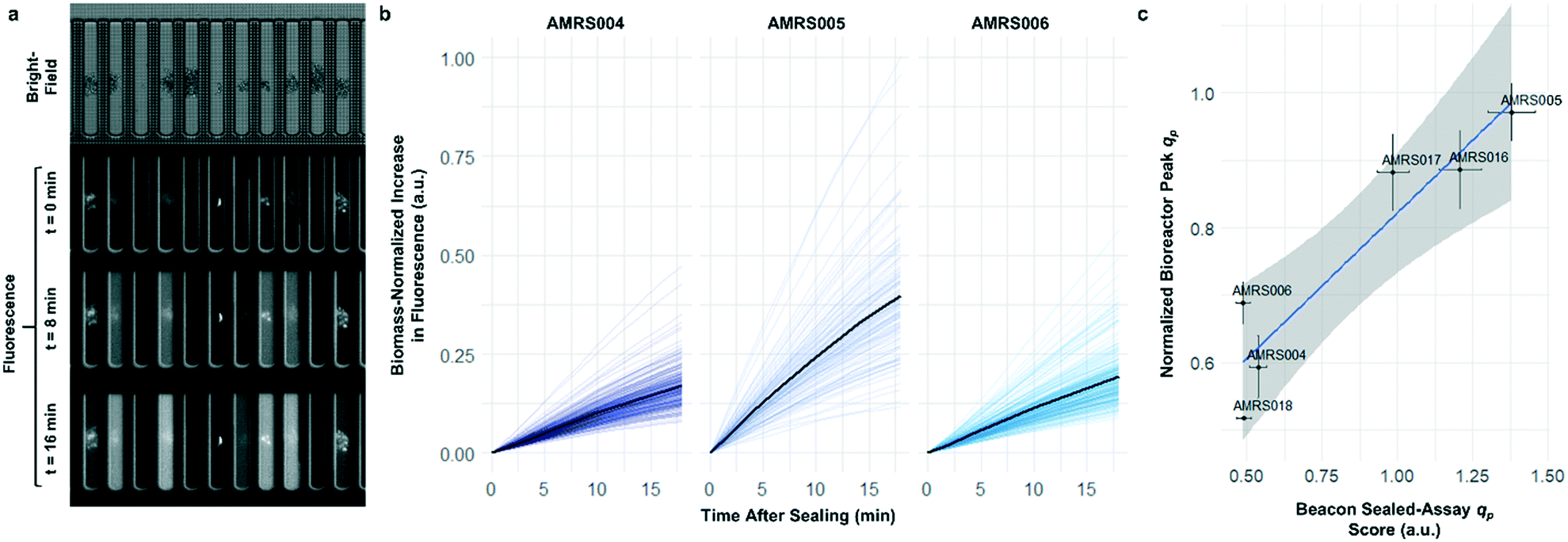 | ||
| Fig. 3 Sealed-pen productivity assay. (a) Illustration of the sealed-pen assay, showing bright-field and time-lapse fluorescence images. (b) Examples of biomass-normalized time-course plots of NanoPen chamber fluorescence during the assay period for three strains (AMRS004, AMRS005, and AMRS006). Each faint line represents an individual pen, with the average value for each strain indicated with a dark line. (c) Correlation between average sealed-pen assay qp scores and average lab-scale bioreactor qp measured during the 24–48 hour interval. The Beacon system qp score for each pen was calculated by normalizing the average rate of increase in fluorescence to the OD score (corresponding to the slopes of the faint lines in b). Bioreactor data plotted are the mean of several biological replicates per strain (n = 2–5). Error bars in both directions show the 95% confidence interval of the mean. The least-squares linear regression line (blue) and its 95% confidence band (gray) are also shown (r2 = 0.87, p = 0.006). | ||
The Beacon qp scores clearly distinguished the three top performing strains from the three weaker producers (Fig. 3c). Mean qp scores from the Beacon assay showed positive correlation (r2 > 0.85) with the qp measured in bioreactor experiments in the 24–48 hour interval. This encouraging result indicated that the Beacon system could provide a relevant culture model for the selection of the most promising strains for bioreactor testing.
In addition to the 20 minute productivity measurement, the sealed-pen method also has the potential to assess the average yield of product with respect to feedstock. In early experiments, we demonstrated that 24 hour sealed-pen cultures of four strains gave endpoint fluorescence values that ranked them consistently with the relative titers attained in microplate models after carbon sources had been exhausted (Fig. S6†). Although the sealed-pen assays were not pursued as a primary strategy for rapid screening, this preliminary result suggests a complementary approach for assessing strain resilience to product and byproduct accumulation.
Real-time productivity monitoring via steady-state product gradient assay
The rapid development of fluorescence signal in the sealed-pen assay prompted investigation of an alternative method, in which pens are left unsealed throughout the experiment. In this assay, local product concentration within each pen is mainly governed by the rate of production by the colony and diffusion away from the colony. As a result of the effective boundary condition just outside of each pen (product concentration ≈ 0), the concentration gradient in the cell-free gradient measurement area at steady state will be proportional to the rate of secretion of the analyte (Fig. S8†). This allows for inference of the productivity in each pen in the few seconds preceding a single fluorescence image of the chip by simply extracting the slope of the linear fit of signal intensity with respect to position in the pen (Fig. 4). If images are acquired periodically, one can easily collect time-dependent productivity data for every colony throughout the experiment.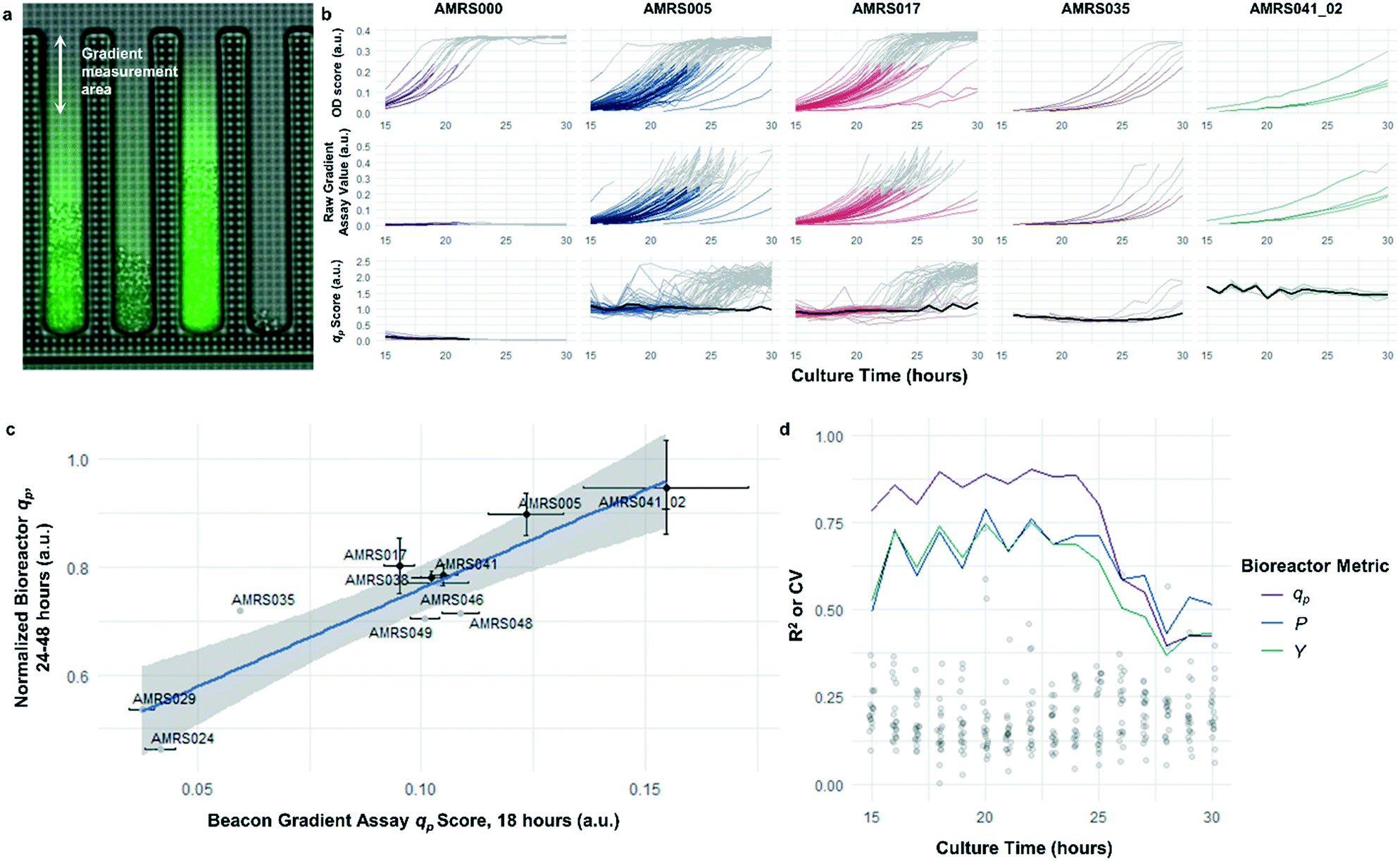 | ||
| Fig. 4 Open-pen gradient productivity assay. (a) Illustration of the diffusion gradient productivity assay, with the fluorescence image superimposed over the bright-field image. Images are periodically acquired in the bright field and fluorescence channels while the chips are continuously perfused with fresh culture media; the open pens allow product to diffuse out of the pens and into the channel, where it is rapidly carried away by the flowing media. Raw assay score is calculated by taking the average slope of the fluorescence intensity in the pen with respect to distance from the channel, after background correction. Refer to Materials and methods section for details on image processing. (b) Example data illustrating average time-dependent biomass measurements, raw assay scores (i.e. fluorescence slope), and qp scores within each genotype. Data shown in color were included in calculation of average qp. Data shown in gray were collected from colonies exceeding OD score of 0.25 and were excluded from average qp calculations. NanoPen chambers above this threshold OD score frequently contained biomass within the measurement area of the pen, resulting in spurious spikes in the fluorescence gradient measured. In the qp plots, average qp across all pens within the accepted OD score range is plotted in black. (c) Correlation between open-pen Beacon assay qp score at 18 hours of incubation and peak bioreactor qp. Error bars in both directions show the 95% confidence interval of the mean. The least-squares linear regression line (blue) and its 95% confidence band (gray) are also shown (r2 = 0.82, p = 0.00012). (d) Assay performance as a function of culture duration. Each line plot shows the time dependence of the R2 values for linear correlations like those shown in c, between mean bioreactor performance metrics (qp, volumetric productivity, yield) from the 24–48 hour interval for each strain and mean Beacon qp score. Points show the distribution of coefficients of variation for each of the strains tested over the course of the experiment. | ||
Strain productivity was high enough to enable quantitation of productivity within hours of initiating incubation. To check the comparability of the sealed- and open-pen assays, the same set of six strains tested in the sealed-pen assay were subjected to the new format using identical culture media. The mean qp scores for each strain from the two assays correlated extremely well (r2 > 0.98, Fig. S9†) and the variability was comparable, with most strains showing CVs around 15–20%.
Next, an expanded set of 11 strains that had exhibited varying performance in 0.5 L bioreactors was selected to help identify optimal chip culture conditions for screening (Fig. S10, Table S1†). Using the real-time data made available by the gradient productivity assay, several media compositions and assay durations were evaluated to maximize correlation of qp score to values of Y, P, and qp measured from bioreactor fermentations. In each experiment, an r2 value was computed for the linear regression of the bioreactor metric with respect to the mean chip score at a given time. A plot was then constructed to show the evolution of the correlation strength over time to aid in the selection of assay duration (Fig. 4). Ultimately, the conditions that optimized prediction of bioreactor performance resulted in a strong positive correlation (r2 = 0.82) between mean qp score and 24–48 hour interval bioreactor qp for a set of 11 diverse engineered strains. As a comparison, endpoint titers in microplate culture model tests of the same strains yielded at best a correlation of r2 = 0.73, also with respect to bioreactor qp (Fig. S11†).
There are several advantages offered by the open-pen assay when compared with the sealed-pen assay: (1) the perfusion-based culture system likely provides a closer approximation of common fed-batch commercial processes, wherein nutrients are constantly provided and waste is removed; (2) low concentrations of feed source and other nutrients, which might be quickly depleted in a sealed assay, can be used in order to avoid the problem of overflow metabolism often observed in batch processes; (3) the export of desired clones is simplified and accelerated; (4) the open-pen assay gives a more direct readout for rate of conversion of sugar into product, since secreted intermediates like ethanol may diffuse out of the pen before being reconsumed; (5) productivity data can be aggregated based on colony size at different time points, rather than requiring all data to be collected at a single time point with possibly wide-ranging OD scores.
Productivity-based screening identified hits exhibiting increased peak qp in lab-scale bioreactors
Following the optimization described above, four strain libraries were generated by random whole-genome chemical mutagenesis of strains containing proprietary engineering for overproduction of the fluorescent target. As a point of reference, these parent strains had previously been shown to produce the target at titers reaching 15–22 g L−1 in lab-scale bioreactors. Libraries were screened using a two-tiered scheme. In tier 1, four chips were dedicated to screening each clone at n = 1, with a total throughput of ∼4000 clones per library (Fig. 5 and S12 and S13†). After selection and export of 44 high-scoring clones (hits), strains were cultured to accumulate biomass in a 96-well plate. Hits were then submitted to tier 2 screening on two additional chips, in which each strain was cultured in higher replication (typically n ∼ 50–100) to identify strains for promotion to bioreactors with higher confidence. Details on the throughput and data variability for each screen are available in Table S2.† These workflows can be operationalized highly efficiently, allowing at least two tier-1 or tier-2 screens to be scheduled on each Beacon system within each 5 day work week (Fig. S1 and S2†).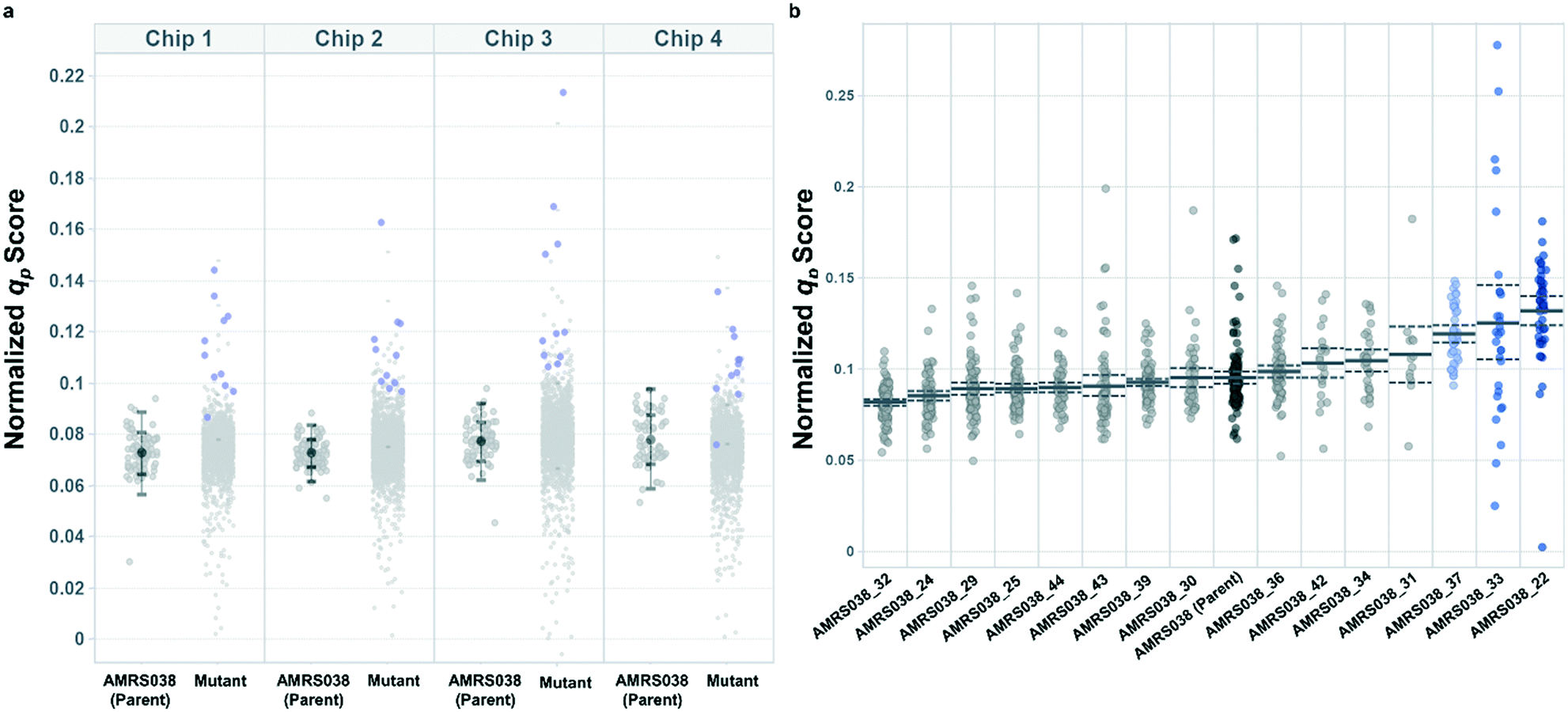 | ||
| Fig. 5 Hit selection in tiered Beacon system screening. (a) Representative tier 1 assay data, showing normalized qp scores from AMRS038 and its mutants on four OptoSelect chips. Strains promoted to tier 2 are highlighted in blue and were selected based on score magnitude and consistency over several time points. Error bars show one and two standard deviations above and below the mean for the parent strain on each chip. (b) Representative tier 2 assay data, showing qp scores from biological replicate colonies (n = 20–100) of strains promoted from tier 1. Error bars (dashed lines) represent the 95% confidence interval of the population mean. Strains highlighted in blue were statistically improved over AMRS038 by a two-tailed Dunnett's test of multiple comparisons (p < 0.05). | ||
Each tier-2 screen identified at least one mutant with mean qp score improved over the parent strain by >20%. Seven mutants improved by varying extents were then tested alongside their parent strains in ambr250 bioreactors to assess performance at lab scale. Excitingly, four of the seven strains exhibited peak bioreactor qp values improved over parent by 10–85% (Fig. 6 and S14†). These gains were aligned with or even larger than the magnitude of increases observed in the nanoliter culture model. The strain with the largest bioreactor qp improvement (AMRS046_12) additionally achieved a 20% increase in average P over a 6 day fermentation, making it a lead candidate for further engineering.
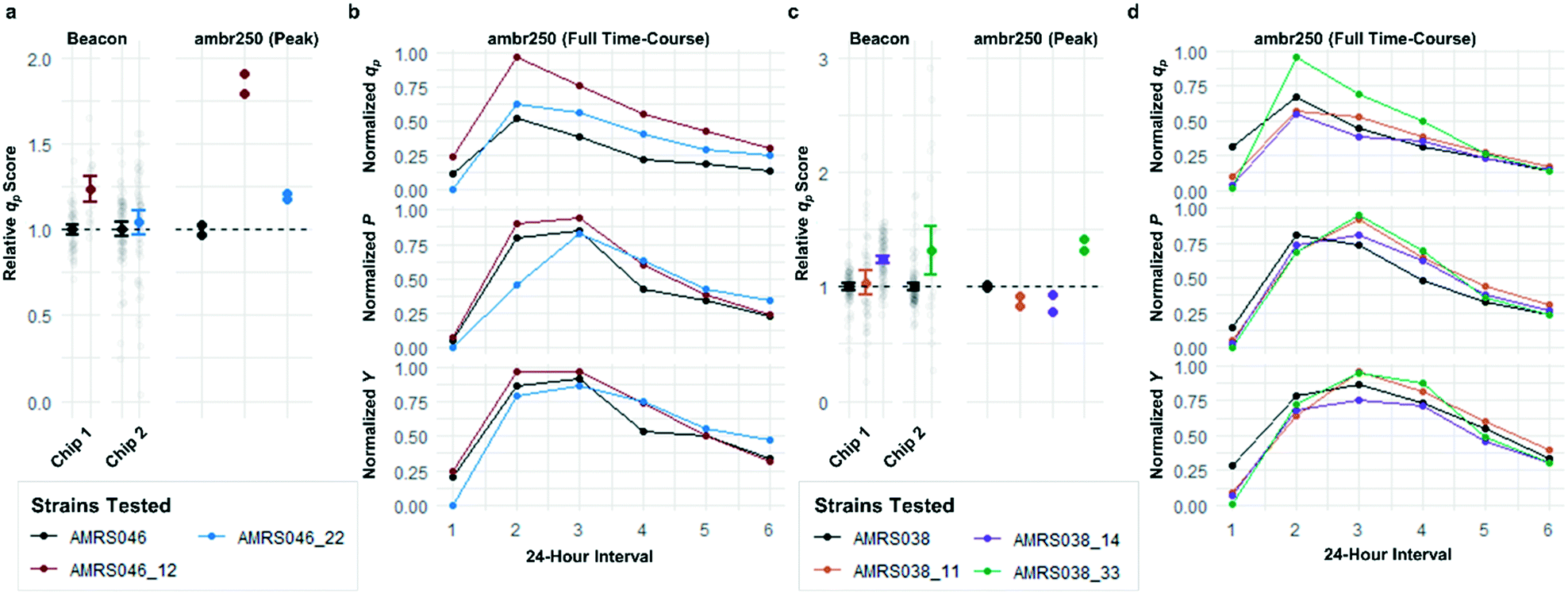 | ||
| Fig. 6 Translation of improved flux phenotype into lab-scale bioreactors. Performance of mutants selected for analysis in lab-scale bioreactors from 2 of the 4 library screens. (a and c) Comparison of the qp scores measured in the Beacon instrument's open-pen assay and the peak (interval 2) qp measured from each ambr250 bioreactor run. Strains are color-coded, with black representing the parent strain in each screen. Hits in the screens shown were promoted based on performance on two separate OptoSelect chips within the screen. Beacon scores for mutants were normalized to the parent strain analyzed on the same chip, shown to their left in black. Bioreactor experiments were performed in duplicate, with each biological replicate shown explicitly and each point normalized to the mean of the peak qp values measured for the parent strain. (b and d) Time-course data for ambr250 fermentations run with each parent strain and mutant. Each point indicates the performance metric (specific productivity qp, volumetric productivity P, or yield with respect to sugar Y) calculated over a 24 hour interval. The bioreactor measurements are normalized to the highest value shown in each graph. | ||
Conclusions
The Beacon system represents an extremely promising opportunity for the high-throughput screening of microbial cell factories. Remarkably, the platform can identify strains with improved qp at a volume 109-fold smaller than the bioreactor it models. Strain productivity on the Beacon system correlated with peak bioreactor performance parameters as well as or better than data from conventional microplate culture models. The improvement in data quality may be partly attributed to the direct readout for qp, as opposed to endpoint yield measurements from carbon-exhausted cultures or error-prone plate-based productivity measurements. It is likely also a consequence of maintaining a roughly constant chemical environment, in which strains are held in a stable metabolic state throughout the experiment.The two-tiered libraries above were processed without microplates, agar trays, liquid handling robotics, or extraction solvents. Complete results for both tiers were generated in under ten days – approximately half of the time typically used for a microplate library screen of comparable size with sophisticated automation. The importance of reducing this cycle time cannot be overstated, as each round of strain design in an engineering campaign depends on results from the previous round. Together, the rapid data turnaround and high data quality showcased here could reduce time-to-market for a metabolic engineering product by months or more. We note that some alternative microfluidic approaches, such as culture within droplets in an emulsion, can encapsulate and sort based on fluorescence readouts at much higher throughputs (>104 droplets per second). However, the flexible feed style and productivity monitoring described here are important advancements that are not yet attainable through droplet-based screening.
As stated above, strain improvement efforts are motivated by the need to reduce manufacturing costs, which are influenced by several factors. Here we have focused on screening for strains with elevated peak qp, but the same assays could be used to improve other phenotypes as well. For example, screens could be designed to find strains more likely to sustain peak qp throughout a longer fermentation by (1) choosing strains with less selective pressure for mutation, (2) identifying strains with resistance to feedback inhibition by products or intermediates, or (3) uncoupling growth rate from metabolic pathway flux.24
As can be seen in Fig. 6, some strains with highly improved bioreactor qp values exhibit relatively smaller improvements in P and Y, which must be increased to reduce manufacturing cost. The Beacon system's assays offer the exciting opportunity to monitor qp and μ simultaneously, which can in theory allow one to screen for strains with improved P and Y as well. These variables can be written as:

This study has established the power of the Beacon system for predictive modeling of microbial molecule fermentations and suggests that its utility will extend to other small-molecule targets. However, we acknowledge the need for generalizable approaches to detection, as the reliance on fluorescence detection currently restricts the analytes that may be measured. With the introduction of abilities such as quantitation of feedstock or respiratory gases, the platform could simplify the process of screening based on multiple metabolic indicators. The low-volume chip offers flexibility by enabling assays that require precious reagents or dynamic control of the cellular environment. Potential applications may include cultivation under selective pressure to circumvent endogenous regulatory mechanisms and overcoming cellular toxicity associated with product accumulation. The rich data afforded by this platform may also be advantageous for the application of artificial intelligence to metabolic engineering, with the potential to dramatically accelerate progress in the field.
Materials and methods
Media and reagents
Chemicals were purchased from Thermo Fisher Scientific unless indicated otherwise. All media components were filter sterilized, with the exception of yeast extract, Bacto Peptone, and agar, which were autoclaved before addition of other components.Solid media contained yeast extract (10 g L−1), Bacto Peptone (20 g L−1), agar (20 g L−1), maltose (30 g L−1), and lysine (2 g L−1). A modified solid medium containing glucose (20 g L−1) and maltose (10 g L−1) instead of 30 g L−1 maltose was used to prepare biomass for ambr250 bioreactor experiments.
Rich growth media was used for biomass accumulation prior to some experiments and for recovery from whole-genome random mutagenesis. The rich growth media contained yeast extract (10 g L−1), Bacto Peptone (20 g L−1), maltose (30 g L−1), and lysine (2 g L−1).
A chemically defined basal media called bird seed medium (BSM, a modification of bird medium25) was used in the Beacon system and microplate culture models and contained KH2PO4 (8 g L−1), (NH3)2SO4 (15 g L−1), MgSO4 (25 mM), succinic acid (50 mM), EDTA (400 μM), ZnSO4 (200 μM), CuSO4 (20 μM), MnCl2 (16 μM), CoCl2 (20 μM), Na2MoO4 (20 μM), FeSO4 (100 μM), CaCl2 (200 μM), biotin (0.6 mg L−1), p-aminobenzoic acid (2.4 mg L−1), calcium pantothenate (12 mg L−1), nicotinic acid (12 mg L−1), myoinositol (300 mg L−1), thiamine·HCl (12 mg L−1), and pyridoxine·HCl (12 mg L−1), adjusted to pH 5.0. sucrose, glucose, fructose, maltose, and/or lysine were included in various concentrations in optimization experiments.
Vitamin stock solution for lab-scale bioreactor experiments contained biotin (0.8 g L−1), p-aminobenzoic acid (0.8 g L−1), nicotinic acid (4 g L−1), myoinositol (10 g L−1), thiamine·HCl (4 g L−1), pyridoxine·HCl (4 g L−1), and calcium pantothenate (4 g L−1).
Trace metal stock solution for lab-scale bioreactor experiments contained 80 mM EDTA, ZnSO4·7H2O (11.5 g L−1), CuSO4 (0.64 g L−1), MnCl2·4H2O (0.64 g L−1), CoCl2·6H2O (0.94 g L−1), FeSO4·7H2O (5.6 g L−1), CaCl2·2H2O (5.8 g L−1), and Na2MoO4·2H2O (0.96 g L−1).
Seed media used to build biomass for lab-scale bioreactor inoculation contained sucrose (20 g L−1), maltose (40 g L−1), KH2PO4 (8 g L−1), (NH3)2SO4 (15 g L−1), MgSO4 (25 mM), succinic acid (50 mM), vitamin stock solution (3 mL L−1), and trace metal stock solution (5 mL L−1) adjusted to pH 5.0.
Lab-scale bioreactor starting media contained maltose (7 g L−1), NH4H2PO4 (9 g L−1), MgSO4·7H2O (4 g L−1), KH2PO4 (3 g L−1), vitamin stock solution (3.75 mL L−1), and trace metal stock solution (6.25 mL L−1), adjusted to pH 5.0.
A concentrated nutrient supplement called post-sterile addition (PSA), added once during each fill/draw cycle (described below) of a lab-scale bioreactor, contained NH4H2PO4 (64.8 g L−1), MgSO4·7H2O (27.8 g L−1), KH2PO4 (23.2 g L−1), vitamin stock solution (28 mL L−1), and trace metal stock solution (36 mL L−1).
Feedstock added to lab-scale bioreactors was a Brazilian cane syrup (Amyris Brasil) with a total glucose equivalent concentration of 740 g L−1, with 57.68% total reducing sugars by mass.
Yeast transformation and DNA assembly
All strains used in this study were derived from CEN.PK113-7D, which was a gift from the Delft University of Technology.26 DNA constructs for yeast transformation were generated using assembly methods as previously described.27,28 Standard methods were implemented for transformation.29,30 Strains AMRS002-AMRS041 were rationally designed and constructed using these methods.Strain storage and preparation of agar cultures
Strains were stored long-term at −70 °C after diluting stationary phase precultures 1.67-fold with 50% glycerol. To recover strains, glycerol stocks were held on dry ice and streaked using sterile wooden rods onto solid media in Petri dishes (Corning) and incubated at 28 °C for 72 h or until colonies were 2–3 mm in diameter.Microplate culture model and assay
Single colonies were transferred with sterile pipet tips from agar plates into chemically defined preculture medium containing 2% maltose in BSM (360 μL well−1) in 1.1 mL round-well 96 well plates (Axygen). Plates were sealed with breathable seals and incubated at 28 °C with shaking at 1000 rpm and 80% humidity for 72 h. Strains were then subcultured by diluting 6.6 μL of whole broth into production medium containing either 4% sucrose in BSM (150 μL well−1) in 1.6 mL round-well 96 well plates (Axygen). Plates were again sealed with breathable seals and incubated at 33.5 °C with shaking at 1000 rpm and 80% humidity for 72 h in darkness.For spectroscopic analysis of titers of the fluorescent product made by the engineered strains, production plates were shaken at 1000 rpm for 30 s before addition of DMSO (900 μL well−1) and then again shaken at 1000 rpm for 30 s. Plates were centrifuged at 500 × g for 30 seconds at ambient temperature, and then 10 μL well−1 of the supernatant was transferred for dilution into 90 μL well−1 of DMSO in a 300 μL flat-bottom black fluorescence assay plate (Corning). Assay plates were shaken at 1500 rpm for 30 s and then analyzed for full-well fluorescence using an M5 plate reader (Molecular Devices).
Random whole-genome mutagenesis
Single colonies were transferred using a sterile wooden rod into rich liquid growth media (3 mL) in a 14 mL polypropylene culture tube with a loose-fitting cap (Corning). Cultures were shaken at an angle at 28 °C for 18 h until reaching stationary phase. A 1 mL aliquot of the starter culture was then diluted into a 250 mL baffled PETG flask (ThermoFisher) containing rich growth media (40 mL) followed by an additional 24 h incubation at 28 °C with agitation.After outgrowth, cells were pelleted by centrifugation (3000 × g, 3.5 min, room temperature) and washed once in 45 mL phosphate-buffered saline (PBS). Cell density was then determined by counting and a 1 mL aliquot was prepared at 6 × 107 cells mL−1 in PBS. To this suspension was added the chemical mutagen N-ethyl-N-nitrosourea to a final concentration of 20 mM. The reaction was incubated for an exposure time of 30 min with agitation before neutralization of the mutagen by addition of 15% sodium thiosulfate (500 μL). Cells were then pelleted again by centrifugation and washed twice with 1 mL PBS before resuspending in 2 mL rich growth medium. The cells were recovered overnight (∼18 h) at 28 °C with agitation in a 14 mL polypropylene culture tube with a loose-fitting cap, then stored at 4 °C and submitted for Beacon system screening within 48 h. Strains with names notated in the format “AMRS###_##” were generated using this procedure and identified through Beacon system screening, where the first 3 digits of the number indicate the mutagenized parent strain and the last two digits are used to index the hits. Strains AMRS042-AMRS050 were also generated through random mutagenesis but were selected through conventional microplate screening methods.
Beacon system experiments
Following sterilization, 4 OptoSelect chips were loaded onto the Beacon system for a sequence of pre-workflow operations. The first step was chip wetting and priming. The Wetting Solution was introduced to the chip to eliminate air pockets and was then incubated for surface functionalization. DI water was then flushed through each chip to remove the wetting solution.
To enable the sealed-pen productivity assay (see section below), a specialized differential wetting procedure was implemented in place of the typical wetting procedure. In this case, a functionalization that renders surfaces hydrophobic was implemented to wet the surface along the channels exclusively, while the standard wetting solution was used to wet the surface in the NanoPen chambers. Differential wetting allowed for different surface properties between the channels and the NanoPen chambers, enhancing the sealing performance in the sealed-pen productivity assay.
After wetting and priming, the Beacon system automatically located the fiducial markers on the chips for x–y stage and focus calibration. Lastly, two types of reference imaging were performed sequentially. The bright-field high-magnification (10× objective) images were taken across the chips as a reference for the quantification of colony size during cell culture. Subsequently, the fluorescence images (4× objective, FITC channel) were captured before and after equilibrating 50 mg L−1 of the final product throughout all OptoSelect chips. The fluorescence images taken before and after equilibration were used as the background reference and the normalization reference, respectively, for the quantification of product concentration during the productivity assay.
For each strain to be loaded into the NanoPen chambers, 25 μL of cell suspension was imported from the well-plate incubator to the channels of the OptoSelect chips. Single cells were identified automatically by the Beacon instrument control software using a convolutional neural network algorithm optimized for yeast cells. A positioning strategy was then automatically implemented to maximize loading throughput using the OEP technology. Residual cells in the channels were flushed to waste after loading. In general, it took roughly 4–5 hours to load the cells across all 4 chips in a single library screen workflow.
The OD score of each colony i at a given time t (ODi,t) was quantified by comparing the intensity of the bright-field images within each pen (Ii,t) to that of the reference images taken during the system preparation process (Ii,Ref), with the same illumination conditions. The percentage difference in brightness was proportional to the amount of biomass in the pen. Brightness values were normalized to the average pixel brightness of all empty pens in same image to correct bight field intensity fluctuation from image to image. The OD score was calculated from the formula:
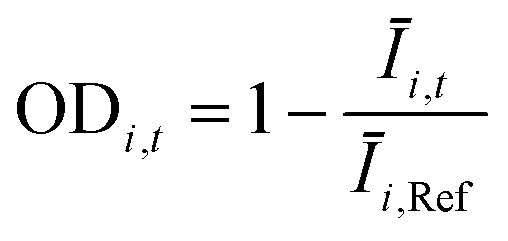
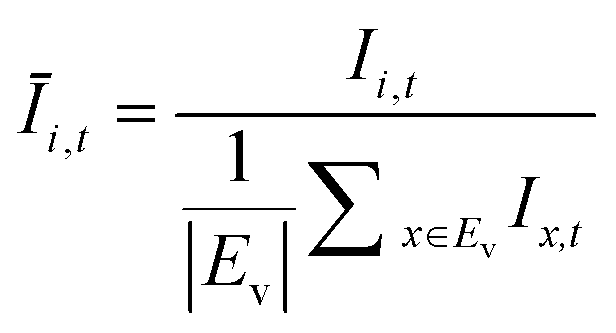
where Ev is the set of empty pens in image v, and the denominators represent the average raw brightness of empty pens.

The product concentration at pixel i at time t (Ci,t) was calculated from the following formula:

In a library screen workflow, the top producers were selected based on assay scores with customizable criteria. For instance, in the library screens for the mutagenesis of AMRS038 and AMRS043, the exported colonies maintained the highest qp scores over 3 consecutive assay time points, equivalent to a 3 hour duration.
After the series of clonality de-risking operations, the OEP unload sequence was performed to export the chosen best clones identified in the assay. Each colony of interest was unpenned by OEP into the channels and delivered into the 96 well export plate (Corning), where 150 μL of rich growth media was loaded in each well. Blank exports and colony exports were performed alternately to assess clonality control. The 96 well export plate was kept at 4 °C in the well-plate incubator during the export process and switched to room temperature at the end of the workflow. The entire colony export process took about 15 hours for 4 chips with 48 colonies retrieved in total, but can be easily optimized by a wide margin for higher efficiency. The export colonies were then transferred to a 1.6 mL round-well 96 well plate (Axygen) for outgrowth.
Lab-scale bioreactor experiments
For the 0.5- and 2.0 L fermentations, the glass bioreactors were configured with dual Rushton impellers and autoclaved before use. Air flow was kept constant at 0.5 L min−1 during each run. Temperature began at 28 °C for the first 12 hours and increased at 0.5 °C hour−1 to a setpoint of 30 °C. Dissolved oxygen was measured by submerged probes and maintained at 30% through agitation control for the first 8 feed pulses. The target pH was set at 5.0 with one-sided control using ammonium hydroxide (20% v/v). Load cells were in place to record weights of vessels, feed, and base in real time. L81 PEG-PPG-PEG block copolymer was used to control foaming.
The ambr250 system uses filterless microbial disposable bioreactors with dual impellers. Temperature was kept at 28 °C for first 12 hours and increased at 0.5 °C hour−1 to a setpoint of 30 °C. Dissolved oxygen is measured with an optode and drives the same control scheme as described for the 0.5 L systems. Off-gas was measured using a gas analyzer custom-built for the ambr250 system (BlueSens 001-0GSR52). The target pH was set at 5.0 with one-sided control using ammonium hydroxide (20% v/v). The ambr250 system was also configured with an integrated ViCell XR cell counter (Beckmann) for biomass quantification. L81 PEG-PPG-PEG block copolymer was used to control foaming. Feed consumption was recorded by tracking added volume.
The 0.5 L, 2.0 L, and 0.25 L fermentations all used an identical fed-batch process with daily drawdowns (removal of culture broth to avoid overflows). To begin the initial growth phase, the fed-batch process delivered a fixed bolus of 10 g L−1 of syrup at a starting rate of 6 g L−1 hour−1. This early phase was marked by rapid biomass production and ethanol accumulation. Following sugar depletion and re-consumption of ethanol produced, a rapid rise in dissolved oxygen triggered the system to deliver another 10 g L−1 bolus of syrup. The feed rate automatically increased by 2.5% if the total time taken to consume the previous bolus was less than 30 minutes. This feed strategy was repeated for 8 boluses in the growth phase. During the growth phase, an agitation ramp was enabled to keep the dissolved oxygen above 30% saturation. Once 8 boluses were fed, the bolus size was increased to 15 g L−1 and two-sided feed control was enabled. Consumption times less than 10 minutes triggered a 2.5% increase; greater than 70 minutes, a decrease. Samples were collected every 24 hours for measurement of titer and biomass. Broth was removed at collection times until 50% of the vessel volume remained, and excess broth was discarded. Following broth removal, a volume of PSA was added equal to the volume of broth removed × 0.054.
Yield (Y), volumetric productivity (P), and specific productivity (qp) in any interval i were calculated respectively from the equations:
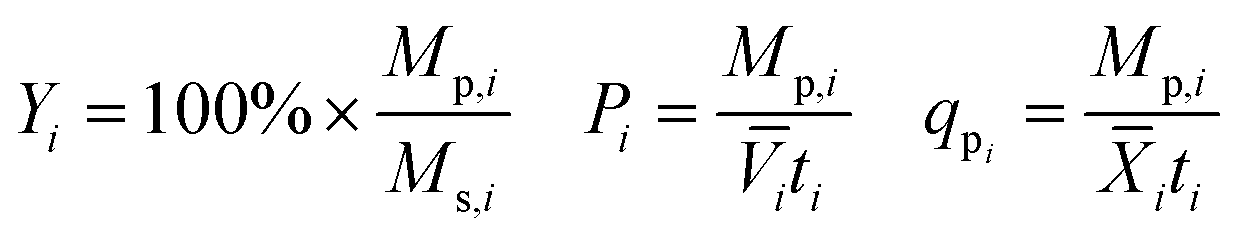
![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) i and
i and ![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) i are the time averages of, respectively, the liquid volume and the dry biomass content of the bioreactor over interval i, and ti is the duration of interval i. For each bioreactor run, the values of Y, P, and qp calculated from the 24–48 hour interval were used for correlation analyses.
i are the time averages of, respectively, the liquid volume and the dry biomass content of the bioreactor over interval i, and ti is the duration of interval i. For each bioreactor run, the values of Y, P, and qp calculated from the 24–48 hour interval were used for correlation analyses.
Conclusions
The conclusions section should come in this section at the end of the article, before the acknowledgements.Author contributions
MDL, TAL, AHN, and JKV conceived the study. TW and HJ designed and constructed the parent strains. RMO and VK wrote the image-processing software. EKS, VK, MR, KCL, KCM, and LKC designed the experiments. KCL, KCM, VK, MR, JK, and LKC performed the experiments. MR, KCL, KCM, VK, JK, and LKC performed the data analysis. MR, KCL, LKC, and JK wrote the manuscript.Conflicts of interest
Authors are employees of Amyris, Inc., or Berkeley Lights, Inc.Acknowledgements
We thank Sunil Chandran, Kirsten Benjamin, Dana Nadler, and Stefan Moser for helpful discussion.References
- J. Nielsen and J. D. Keasling, Cell, 2016, 164, 1185–1197 CrossRef CAS PubMed.
- B. M. Woolston, S. Edgar and G. Stephanopoulos, Annu. Rev. Chem. Biomol. Eng., 2013, 4, 259–288 CrossRef CAS PubMed.
- P. Hill, et al. , J. Ind. Microbiol. Biotechnol., 2020, 47, 965–975 CrossRef CAS PubMed.
- A. Burgard, M. J. Burk, R. Osterhout, S. Van Dien and H. Yim, Curr. Opin. Biotechnol., 2016, 42, 118–125 CrossRef CAS PubMed.
- D. Jullesson, F. David, B. Pfleger and J. Nielsen, Biotechnol. Adv., 2015, 33, 1395–1402 CrossRef CAS PubMed.
- V. Chubukov, A. Mukhopadhyay, C. J. Petzold, J. D. Keasling and H. G. Martín, npj Syst. Biol. Appl., 2016, 2, 1–11 Search PubMed.
- Y.-J. Wee, J.-N. Kim and H.-W. Ryu, Food Technol. Biotechnol., 2006, 44, 163–172 CAS.
- National Science Foundation, Catalyzing Commercialization: Fermentation Process Makes Malonates from Sugar, Not Cyanide, Chem. Eng. Prog., 2016, 16 Search PubMed.
- B. Dumas, C. Brocard-Masson, K. Assemat-Lebrun and T. Achstetter, Biotechnol. J., 2006, 1, 299–307 CrossRef CAS PubMed.
- M. Rienzo, et al. , Metab. Eng., 2020, 63, 102–125 CrossRef PubMed.
- M. Wehrs, et al. , J. Ind. Microbiol. Biotechnol., 2020, 47, 913–927 CrossRef PubMed.
- J. A. Dietrich, A. E. McKee and J. D. Keasling, Annu. Rev. Biochem., 2010, 79, 563–590 CrossRef CAS PubMed.
- E. K. Bowman and H. S. Alper, Trends Biotechnol., 2020, 38, 701–714 CrossRef CAS PubMed.
- A. Mocciaro, et al. , Commun. Biol., 2018, 1, 41 CrossRef PubMed.
- N. Dénervaud, et al. , Proc. Natl. Acad. Sci., 2013, 110, 15842–15847 CrossRef PubMed.
- J. Park, J. Wu, M. Polymenis and A. Han, Lab Chip, 2013, 13, 4217–4224 RSC.
- M. Kim, J. Bae and T. Kim, Anal. Chem., 2017, 89, 9676–9684 CrossRef CAS PubMed.
- F. K. Balagaddé, L. You, C. L. Hansen, F. H. Arnold and S. R. Quake, Science, 2005, 309, 137–140 CrossRef PubMed.
- A. E. Vasdekis and G. Stephanopoulos, Metab. Eng., 2015, 27, 115–135 CrossRef CAS PubMed.
- P. Y. Chiou, A. T. Ohta and M. C. Wu, Nature, 2005, 436, 370–372 CrossRef CAS PubMed.
- G. H. An, J. Bielich, R. Auerbach and E. A. Johnson, Bio/Technology, 1991, 9, 70–73 CAS.
- J. M. Wagner, et al. , Metab. Eng., 2018, 47, 346–356 CrossRef CAS PubMed.
- K. Le, et al. , Biotechnol. Prog., 2018, 34, 1438–1446 CrossRef CAS PubMed.
- P. Rugbjerg and L. Olsson, J. Ind. Microbiol. Biotechnol., 2020, 47, 993–1004 CrossRef CAS.
- R. Metzenberg, Fungal Genet. Rep., 2004, 51, 19–20 CrossRef.
- J. F. Nijkamp, et al. , Microb. Cell Fact., 2012, 11, 36 CrossRef CAS.
- S. de Kok, et al. , ACS Synth. Biol., 2014, 3, 97–106 CrossRef CAS PubMed.
- Z. Serber, R. Lowe, J. A. Ubersax and S. S. Chandran, Compositions and methods for the assembly of polynucleotides, US Pat. No. 8221982 B2, US Pat. and Trademark Office, 2012 Search PubMed.
- C. Guthrie and G. R. Fink, Methods in enzymology: guide to yeast genetics and molecular biology, Academic Press, 1991 Search PubMed.
- M. D. Rose, F. Winston and P. Hieter, Open Biol., 1990, 6, 14 Search PubMed.
- J. Ansel, et al. , PLoS Genet., 2008, 4, e1000049 CrossRef PubMed.
- M. Ackermann, Nat. Rev. Microbiol., 2015, 13, 497–508 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00389e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |