
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasmonic nanocatalysts for visible-NIR light induced hydrogen generation from storage materials
Priyanka
Verma
 ab,
Kohsuke
Mori
ac,
Yasutaka
Kuwahara
acd,
Robert
Raja
b and
Hiromi
Yamashita
*ac
ab,
Kohsuke
Mori
ac,
Yasutaka
Kuwahara
acd,
Robert
Raja
b and
Hiromi
Yamashita
*ac
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.jp
bSchool of Chemistry, University of Southampton, University Road, Highfield, Southampton, SO17 1 BJ, UK
cUnits of Elements Strategy Initiative for Catalysts and Batteries, Kyoto University, Katsura, Kyoto 615-8520, Japan
dJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 19th December 2020
Abstract
Solar-to-chemical conversion processes, assisted by localised surface plasmon resonance (LSPR), are a fast-growing field of plasmonics that efficiently utilise solar energy due to their unique catalytic and optical responses. Plasmonic nanostructures can harvest abundant sunlight by concentrating the incident light energy in the nanoscale regime to drive chemical reactions on the surface of nanoparticles. The advantages of employing LSPR in catalysis include higher conversion rates and improved selectivity under mild reaction conditions. This review highlights the latest progress on harnessing plasmonic photocatalysts involving noble metal nanoparticles (Au, Ag) and doped semiconductor (WO3−x, MoO3−x) nanostructures for their application in the enhanced hydrogen evolution from ammonia borane (AB) under visible-NIR light irradiation. An overview of multi-metallic heterostructures, in combination with plasmonic nanoparticles, which are available at the current stage in plasmon chemistry has been included. The rational design, tuning of plasmonic absorption and their underlying mechanisms for enhanced catalytic activities have also been reviewed. Finally, the current challenges and future perspectives have been discussed to further improve the efficiency of plasmon-mediated heterogeneous catalysis to achieve more practical applications in the near future.
 Priyanka Verma | Priyanka Verma is currently working as a Royal Society-Newton International fellow at the University of Southampton. She received the Institute Gold medal for academic excellence in MSc degree at the Indian Institute of Technology, Hyderabad, India (2014). Her PhD degree at Osaka University was supported by the FRIENDSHIP scholarship of Japan International Corporation Agency (2014–2017). Later, she worked as an assistant professor (2017–2019) in the research group led by Prof. Hiromi Yamashita at Osaka University. Her research interests include the design and development of visible-light responsive plasmonic nanomaterials for energy applications, mainly solar-powered hydrogen generation. |
 Kohsuke Mori | Kohsuke Mori received his PhD degree from the Graduate School of Engineering Science at Osaka University under the supervision of Prof. K. Kaneda in 2003. In 2004, he joined Prof. T. Don, Tilley's group at University of California, Berkeley, as a postdoctoral fellow. He moved to the current faculty in 2005 and then became an associate professor in 2009. His current research interests focus on hybrid photocatalysts based on visible-light-responsible metal complexes, metal and alloy nanoparticle catalysts to establish clean and environmentally-friendly chemical processes, and diesel automobile catalysts. |
 Yasutaka Kuwahara | Yasutaka Kuwahara received his PhD degree in engineering from Osaka University in 2011. He was a postdoctoral research fellow at Georgia Institute of Technology, USA, in 2011. He was a researcher at National Institute of Advanced Industrial Science and Technology (AIST), Japan, in 2012–2014. He was appointed as an assistant professor in 2014–2019 and has been appointed as a lecturer since 2019 in Osaka University. His current research interests include the design of nanostructured catalysts with multi-functionalities utilizing porous materials and their applications in green chemical reactions and the conversion of energy and resources. |
 Robert Raja | Robert Raja is a professor of materials chemistry and catalysis at the University of Southampton, UK and his research group has a proven international reputation, both in academia and industry, for the discovery and design of novel catalytic materials that address fundamental challenges in the chemical, environmental and energy landscape. His career includes appointments at the University of Cambridge (1999–2006); Bayer Chemicals, Germany (2001–2003); and 1851 Exhibition at the Royal Institution of Great Britain (1997–1999). Notable accolades include the Erskine Fellowship and the Barrer Award from the Royal Society of Chemistry ‘for outstanding contributions to materials chemistry and industrial catalysis’. |
 Hiromi Yamashita | Hiromi Yamashita has been a professor at Osaka University since 2004. He received a PhD degree from Kyoto University in 1987. He was an assistant professor at Tohoku University and an associate professor at Osaka Prefecture University. He was also a visiting research fellow at the California Institute of Technology. He is now the president of Catalysis Society of Japan (2019–2020) and the president of Asia-Pacific Association of Catalysis Societies (2019–2022). His research interests include the design of single-site photocatalysts and nanostructured catalysts. |
1. Introduction
Photocatalysis has been at the forefront of emerging sustainable technology for providing solutions to the present energy and environmental challenges, such as water purification, organic transformation, solar fuel production by water splitting, renewables and other related research.1 The generation of electrons and holes in photocatalysts upon light irradiation leads to a redox reaction, resulting in the conversion of solar-to-chemical energy. In the last decade, a new technique to efficiently exploit solar energy in chemical transformations has emerged due to the localised surface plasmon resonance (LSPR) effect, which is usually exhibited by coinage metals, alkali metals and a few semiconductors.2–4 LSPR plays a remarkable role in various fields of technologies such as surface-enhanced Raman spectroscopy, biosensors, energy transfer, photonics and solar cells.5–7 It can be defined as the collective oscillation of free electrons establishing a resonance when the frequency of incident radiation matches the natural frequency of the oscillating electrons against the positively charged nucleus.8–10 The frequency of oscillations can be tailored by a change in the surrounding medium, the nature of metals, and the size and morphology of nanostructures.11,12 Mathematically, the plasma frequency (ωp) can be expressed as shown in eqn (1), where N is the metal carrier concentration, meff is the effective mass of carriers and e is the electronic charge.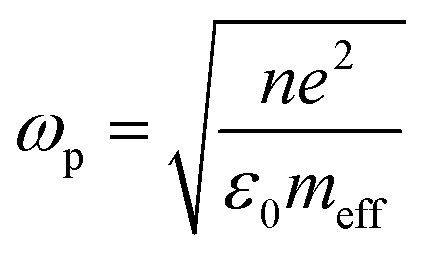 | (1) |
The LSPR of noble metal nanoparticles (NPs), especially Au18 and Ag, has been investigated with significant efforts for various chemical transformation reactions.19–21 The unique catalytic and optical properties of noble metal plasmonic nanostructures play a significant role in the solar-to-chemical energy conversion.22 Pioneering research work on plasmonic catalysis was reported by Tatsuma et al. in the early 2000s, demonstrating the LSPR-induced oxidation of methanol over Au NPs on TiO2 films.23 Azuwa et al. coined the term ‘plasmonic photocatalysis’ for the visible light promoted decomposition of methylene blue dye over Ag NPs.24 In addition to the mono metallic NPs, the bimetallic combination of plasmonic NPs with active metals has also been extensively studied because the coupling with various metals can improve the overall light absorption and charge separation efficiencies.25–27
The fast-growing field of plasmon-mediated heterogeneous catalysis has found its applications in various important catalytic reactions such as hydrogen production, water purification, carbon–carbon coupling, and hydrogenation and oxidation reactions.5,28,29 This phenomenon is not only limited to noble metal NPs but has also been reported by some heavily doped semiconductor oxides such as WO3−x, MoO3−x and Cu2−xS.30 The LSPR absorption occurs due to the presence of free carriers introduced via doping effects which display absorption in a much higher near infrared (NIR) region.31 Hence these plasmonic semiconductors have been found very useful in various photothermal therapy and bioimaging techniques.8
Due to the continuous increase in the global energy consumption and the generation of large amounts of greenhouse gases, the quest for environmentally-friendly energy carriers has greatly increased.32–34 The use of hydrogen as a clean fuel has attracted much attention because it produces water as the oxidation product and it can be synthesised from water as well using photocatalytic processes.35 The current energy crisis in the world can potentially be solved by using hydrogen (H2) as a clean fuel because of its abundance and significantly higher energy density.36–39 It has been anticipated that the use of H2 as a clean fuel will bring the desired shift from non-renewable fossil fuels towards sustainable energy in the future. However, the extremely low boiling point of H2 makes it difficult to store in compressed or liquid form, hence its safe storage and transportation are the major challenges to establish a hydrogen economy for wider applications. The main goal is to develop a system in which H2 can be stored and released in a controlled fashion at temperatures below 350 K in order to use the heat generated in fuel cells. Therefore, efforts are being made by researchers around the world to explore hydrogen storage materials with high gravimetric and volumetric densities which can release hydrogen at reasonable temperatures.40–43 Various hydrogen storage materials, for example, formic acid, metal hydrides, borohydrides, hydrazine, methanol and ammonia borane (NH3BH3; AB) have been investigated in recent years. AB (Mw = 30.8 g mol−1; ρ = 0.78 g cm−3 at 298 K) has gained significant attention because it has the highest hydrogen storage content of 19.6 wt% along with strong interactions between boron and hydrogen atoms.44–46 It is isoelectronic with ethane (CH3CH3) but exhibits exceptionally different properties due to heteropolar dihydrogen bonding. The presence of three protic (Hδ+) and three hydridic (Hδ−) hydrogen introduces a polarity which assists in hydrogen generation by intra- and inter-molecular interactions. It is a white coloured solid powder, non-toxic, and stable enough to store and transport under ambient conditions and its high solubility in polar solvents (e.g. water) makes it a promising candidate for hydrogen storage.47–49 AB is also potentially consistent with the 2025 target decided by the US Department of Energy (DOE), a gravimetric capacity of 5.5 (g H2) kg−1 equivalent to 5.5 wt% H2, for onboard hydrogen storage in light-duty fuel cell vehicles. The release of hydrogen can take place in two possible ways: thermolysis or solvolysis. The thermolysis pathway has drawbacks such as the requirement of high temperatures, long induction times and the formation of a variety of by-products (borazine (B3N3H6) and ammonia (NH3)) during the reaction. Hydrolysis is one of the practical ways used in fuel cells, but it is relatively stable against hydrolysis compared to thermolysis in aqueous solutions at neutral or basic pH values. AB undergoes rapid hydrolysis under acidic conditions promoted by metals or solid acid catalysts.50,51 It can release 3 moles of H2 per mole of AB only in the presence of a suitable catalyst even at near room temperature as shown in eqn (2).52–54
| NH3BH3(aq) + 2H2O(l) → NH4BO2(aq) + 3H2(g) | (2) |
| NH4B(OCH3)4 + NH4Cl + LiAlH4 → NH3BH3 + NH4Al(OCH3)4 + LiCl + H2 | (3) |
| xB(OCH3)3 + [NH4+]x[Xx−] + xLiAlH4 → xNH3BH3 + xAl(OCH3)4 + LixX + xH2 | (4) |
Quite significant conversion efficiency have been witnessed in recent years using noble, non-noble metals and semiconductors under visible light irradiation.55–59
Photocatalytic fuel production enables the conservation of solar energy in chemical bonds which can be released later to avoid storage issues. It is interesting to be able to exploit renewable solar energy to produce fuels for energy without generating any toxic by-products. Fig. 1 displays the increasing number of papers published on hydrogen production from ammonia borane and plasmonic catalysis in the last 15 years. There have been several review publications based on the fundamentals and theory of the LSPR phenomenon which can be referred to for a better understanding.7,19,29,60 Xiong et al. reviewed the tailoring of the physical parameters of plasmonic materials for energy and environmental applications.22 Linic et al. discussed the underlying mechanisms for the enhanced catalytic activity by noble metal NPs.16 Yamashita et al. studied the recent advances in single-active plasmonic nanocatalysts and highlighted their structure–property relationships.8
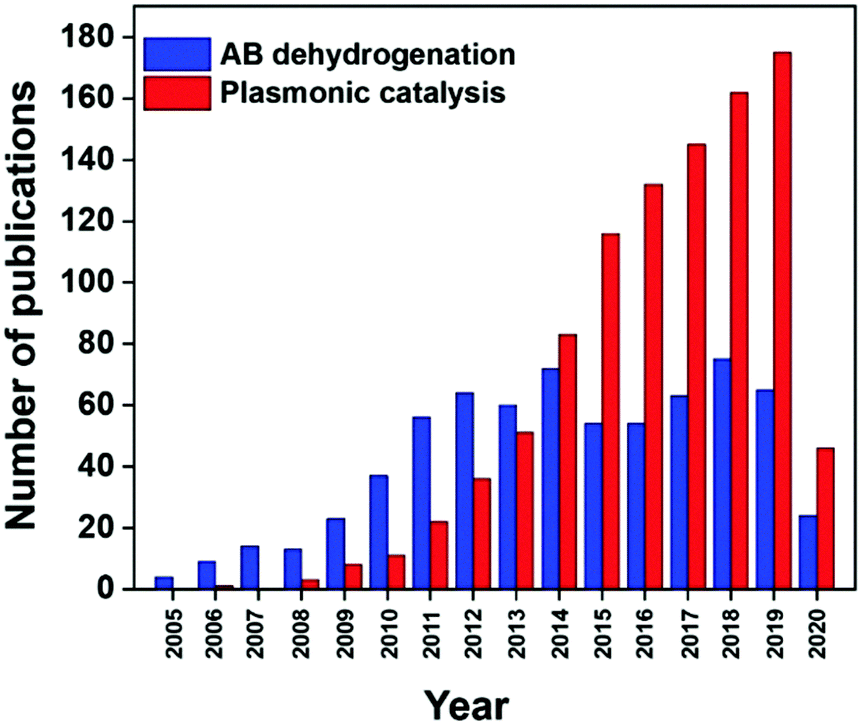 | ||
| Fig. 1 Number of publications in the last 15 years found on Scopus for the entry ‘ammonia borane dehydrogenation’ and ‘plasmonic catalysis’. | ||
In this review article, we highlight the recent progress in the design and characterisation of noble metal (Au, Ag) and doped semiconductor-based (WO3−x, MoO3−x) photocatalysts for their applications in plasmon-enhanced hydrogen production in the last seven years. The first report on the plasmon-mediated hydrogen production from AB was published in 2013 using size and colour-controlled Ag NPs confined within the channels of mesoporous silica.61 Since then, many efforts have been made to enhance the efficiency of this reaction by exploring the different combinations of metal–semiconductor nanocatalysts under visible light irradiation.62 The latest progress in the design of plasmonic catalysts from 2013 to 2020 along with a special emphasis on the comparison of catalytic activities under various reaction conditions has been presented. The advantages of this approach demonstrate higher reaction rates with improved selectivity under mild conditions.
This review article also sheds light on the design and coupling of plasmonic nanostructures with insulator and semiconducting support materials along with their underlying mechanistic pathways under light irradiation conditions. Fig. 2 displays a schematic illustration of plasmonic catalysts which can be classified into four main categories: (i) plasmon-induced electron transfer, (ii) plasmon enhancement of light absorption, (iii) direct plasmonic catalysis, and (iv) activation by hybrid plasmonic catalysis. The research reports corresponding to each classification will be elaborated including plasmonic NPs in combination with semiconductors and active metal species. An in-depth discussion on hot electron driven plasmonic catalysis has been done to bridge the gap in understanding the mechanistic pathway. The mechanism of the plasmonic photocatalytic process is a complex phenomenon in which both noble metal and semiconducting plasmonic catalysts can interact with the incident light for solar hydrogen generation. Amongst several possible energy transfer pathways, charge separation, hot electron injection, photothermal, electric field enhancements, plasmon-induced resonance energy transfer (PIRET), desorption induced by electronic transition (DIET), scattering and plasmonic heating are the most prominent ones, which will be discussed in this report. This review article has been written with an objective to gather the most recent information on unique plasmonic nanomaterials which would assist in the development of new catalytic systems with improved efficiency for solar light utilisation.
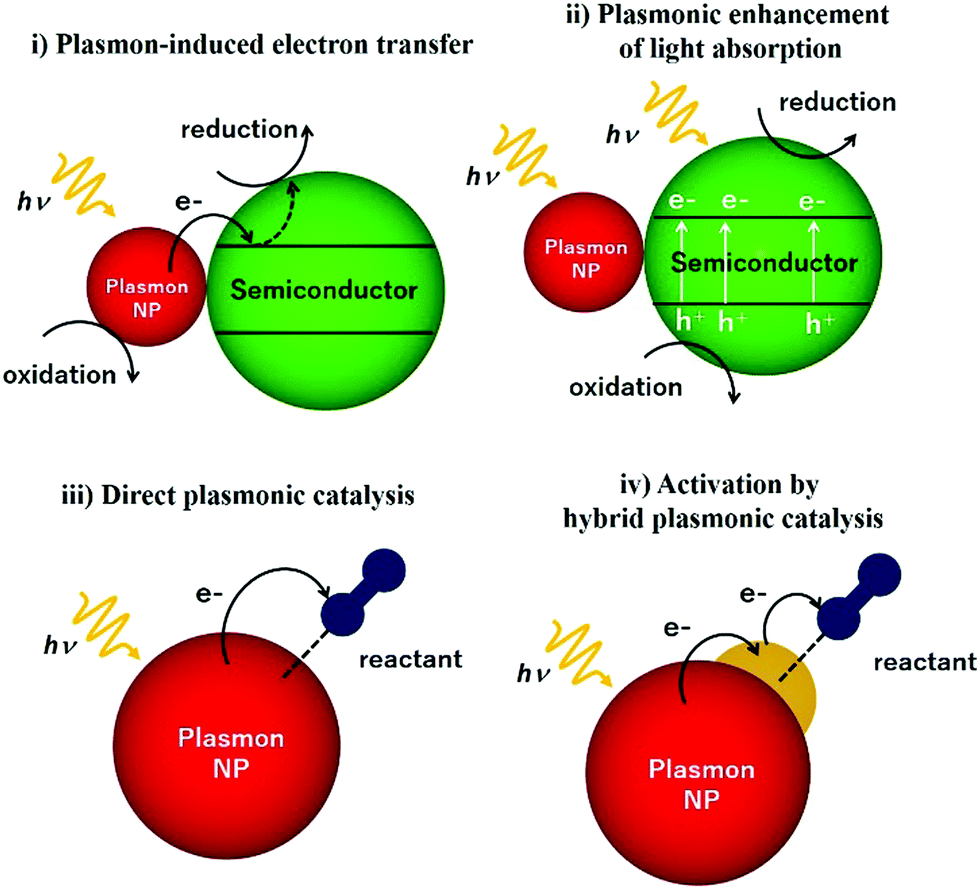 | ||
| Fig. 2 Schematic illustration of different types of plasmonic catalysts. | ||
2. Noble metal-based plasmonic catalysts
Traditional semiconductor materials like TiO2, CdS, and C3N4 are often used as photocatalytic materials to generate photoexcited electrons and holes under UV-vis light irradiation.49,63 Charge carriers can participate in various chemical reactions like water splitting to produce hydrogen, decomposition of organic pollutants or CO2 reduction. These photocatalytic materials often suffer from low stability, faster recombination rate of charge carriers and lower light absorption. The wide bandgap of n-type semiconductors displays strong absorption in the UV region, which limits the efficient utilisation of solar energy accounting for the major visible-near infrared region. One of the strategies is to combine semiconductor materials with plasmonic NPs like Ag and Au to extend their light absorption in the visible region.64,65 The bare plasmonic nanomaterials or those coupled with semiconductors exhibit strong light absorption in the visible-near infrared region and display an intense localized electric field with improved transfer of charge carriers under illumination conditions. In this section, we will discuss two of the most popular noble metal-based catalysts (Au and Ag) for efficient plasmon-mediated catalysis.2.1. Plasmonic gold hybrid nanocatalysts on insulating and semiconducting supports
Au based plasmonic catalysts in combination with catalytically active metals like Pd, Co, Rh, etc. have been extensively studied and reported for the AB dehydrogenation reaction. Table 1 summarises the specific case studies discussed in this report along with a comparison of the catalytic activities in the dark and under visible light irradiation.| Catalysts | Synthesis method | Metal loadings | Light source | Catalytic activity | Ref. | |
|---|---|---|---|---|---|---|
| Dark | Light | |||||
| Au/TiO2 | Photo-assisted deposition | 1.0 wt% Au on TiO2 | 500 W Xe > 420 nm | 0.2 μmol min−1 | 0.36 μmol min−1 | 66 |
| Au–D NPs | Seed-mediated growth | Colloidal | 300 W Xe 420–780 nm | — | TOF = 203 min−1 | 67 |
| Au–Co/CN | Wet impregnation | 30 wt% (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co = 4:96) :Co = 4:96) |
150 W Xe >420 nm | TOF = 1704 mol H2 mol−1 metal h−1 | TOF = 2987 mol H2 mol−1 metal h−1 | 68 |
| Au–Pd THH | Seed-mediated growth | Colloidal | 500 W Xe 200–800 nm | TOF = 142 mol H2 mol catalyst−1 min−1 | TOF = 426 mol H2 mol catalyst−1 min−1 | 69 |
| AuPd/NH2-MIL-101 | Impregnation + H2 reduction 200 °C | Au: 2.5 wt%; Pd: 0.5 wt% | 500 W Xe > 420 nm | 0.89 mL min−1 | 1.6 mL min−1 | 70 |
| Au–Rh-CP1 | Impregnation + NaBH4 reduction | Au: 10%; Rh: 90% on polymer | 400–780 nm | TOF = 780 min−1 | TOF = 980 min−1 | 71 |
To increase the visible-light responsiveness of TiO2, Au has been a popular choice because of its electron storage effect and LSPR characteristics. Au NPs on TiO2 can act as an electron trap site which assists in reducing the electron–hole recombination rate and increases the quantum yield. In a similar research direction, Jo et al. studied the synthesis of varied amounts of plasmonic Au NPs on a TiO2 support (0.5, 1.0, 2.0 and 3.0 wt%) by the photo-assisted deposition (PAD) method.66 The optimum amount of Au was found to be 1.0 wt% for efficient hydrogen generation from AB because the excessive loading of Au NPs facilitated the recombination of charge carriers under visible light irradiation, as evidenced by time-resolved photoluminescence spectroscopy. The size of Au NPs as estimated from the TEM images was found to be 9.5 ± 1.5 nm along with an absorption maximum observed at 539 nm for Au(1.0 wt%)/TiO2 as shown in Fig. 3(a). The catalytic activity was studied as a function of irradiation wavelength using a monochromator and the maximum hydrogen was obtained at 540 nm which coincides well with the LSPR absorption maximum of Au/TiO2 as shown in Fig. 3(b).
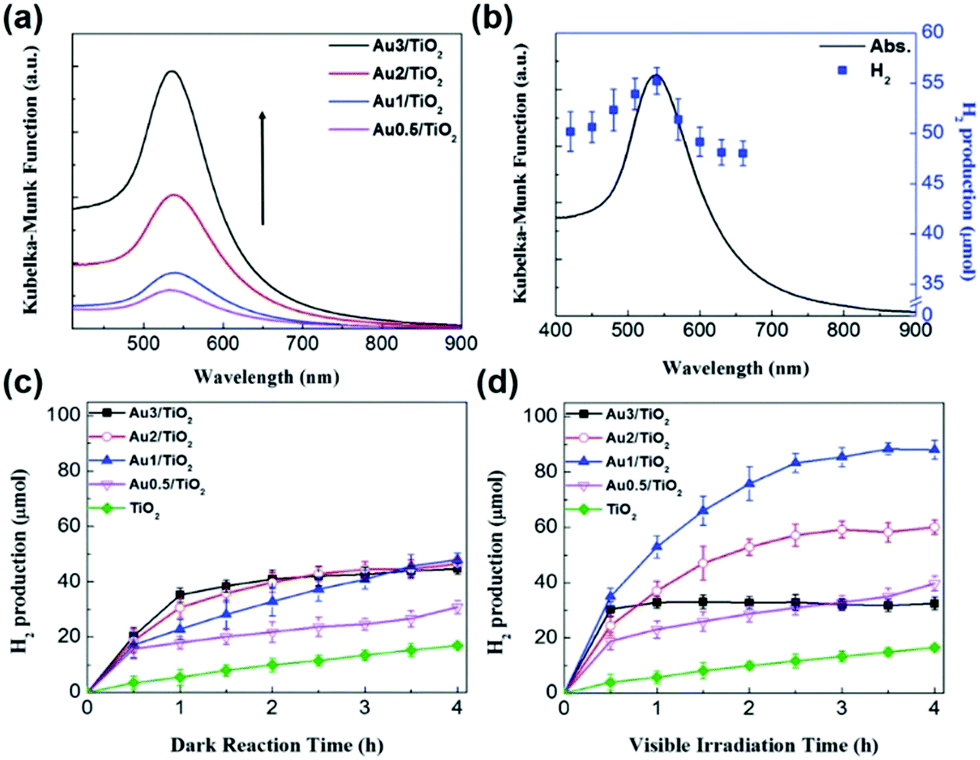 | ||
| Fig. 3 (a) UV-visible absorption spectra of Au/TiO2 with varied Au content; (b) UV-visible absorption spectrum of Au1/TiO2 and H2 generation by photocatalytic AB decomposition in 4 h on Au1/TiO2 as a function of irradiation wavelength (controlled by using a monochromator); (c) time profile of H2 generation from AB on Au/TiO2 in (c) the dark and (d) under visible light irradiation. [Cat.]0 = 3.6 g L−1, [AB]0 = 3.6 mM and λ > 420 nm for (d). Reproduced with permission from ref. 66 Copyright 2017 The Royal Society of Chemistry. | ||
The amount of hydrogen generation over Au/TiO2 in 4 h was 48 and 88 μmol in the dark and under visible light irradiation (λ > 420 nm), respectively, as shown in Fig. 3(c and d). Bare TiO2 did not show an enhancement in the catalytic performance because of its large bandgap values.66 The amount of hydrogen generation over TiO2 was 18 μmol in the dark and under visible light irradiation conditions for a period of 4 h. Similar sized Au NP colloids (∼10 nm) were also synthesised by the citrate stabilization method and the Au content was adjusted similar to that of Au NPs deposited on TiO2. Unlike Au/TiO2, the bare Au colloid NPs did not show any optimum Au concentration for efficient hydrogen generation. The activity increased on increasing the Au content from 0.5 to 3.0 wt% because at higher Au NP concentrations, the LSPR excitation absorbs more photons leading to the generation of a large number of charge pairs. The maximum catalytic performances over 3 wt% Au NPs in the dark and under visible light irradiation generated 10 and 31 μmol of hydrogen in 4 h. A physical mixture of Au colloid NPs and TiO2 displayed a little higher catalytic activity of 47 μmol under visible light irradiation. The different photocatalytic behaviour of Au/TiO2 and Au NPs suggests their different mechanistic pathways and other competitive factors affecting the photocatalytic activity of Au/TiO2. The mechanistic behaviour was investigated by studying the effect of scavenger addition in the reaction mixture under visible light irradiation. For example, when Cr(VI) was added as an electron scavenger, the observed photocatalytic performance was significantly decreased, which confirms the important role of LSPR-excited electrons in hydrogen generation. The existence of charge carriers was supported by photoluminescence spectroscopy in which the PL emission intensity at 400 nm was gradually decreased on increasing the Au content in the catalyst, Au/TiO2. This is due to the efficient charge separation of photoexcited carriers in the Au/TiO2 catalyst and hence displaying enhanced catalytic activity under light irradiation conditions.
Larger sized nanostructures tend to display lower catalytic performance than small sized NPs due to the exposed number of atoms. However, the compensation effect of LSPR can present excellent catalytic performances for larger sized NPs as a remedy for the loss in catalytic activity due to size enlargement. This can be explained by the higher heating power (Ps) and turnover frequency (TOF) of the enlarged nanostructures. Sun's group has recently reported the synthesis of different morphologies of Au nanoparticles including spherical (S-Au NPs), rod (R-Au NPs) and dumbbell-shaped (D-Au NPs) by a seed-mediated growth process and studied the photocatalytic dehydrogenation of AB.67 The catalytic trend in the volume of H2 produced followed the order: S-Au > D-Au > R-Au; the smallest NPs displayed the maximum catalytic activity due to the larger number of exposed surface atoms. The extinction ratio (calculated by the ratio of longitudinal resonance and transverse resonance) can be used as a parameter to estimate the photocatalytic performance of Au NPs. D-Au with an extinction ratio of 2.92 exhibits great potential as a photocatalyst when compared with R-Au (2.74) and S-Au (0.00) NPs, in terms of their ability to absorb photons. The Ps and TOF of D-Au was 52.5 and 3.89 times higher than those of S-Au NPs because of the presence of a larger number of surface atoms on small sized S-Au NPs. Moreover, D-Au NPs displayed the highest temperature variation during the photocatalytic test. Therefore, despite the higher catalytic activity of S-Au NPs, the strengthened LSPR compensation effect leads to superior Ps and TOF values for D-Au NPs. Further, authors have also investigated the role of H+ ions in the hydrolysis of AB in which lower pH solution (<6.0) improved the hydrogen production ability of Au nanostructures.
The combination of plasmonic NPs with catalytically active metals to form bimetallic nanostructures has attracted significant research attention because of their unique optical, electrical and catalytic properties in comparison to their monometallic counterparts. Ren et al. reported the use of graphitic carbon nitride (g-C3N4; CN) to design multifunctional Au–Co NPs for the room temperature photocatalytic dehydrogenation of AB.68 CN was chosen as a support material because the work functions of many transition metals including Au and Co lie in between the valence and conduction band of CN, which assists in the creation of a Mott–Schottky heterojunction. The wt% of the metal was optimised to be 30 wt% with a molar ratio of 4:96 (Au/Co) as shown in Fig. 4(a). A complete dehydrogenation of AB was observed within 5 min in the dark with a remarkable TOF value of 1704 mol H2 mol−1 metal h−1. The catalytic activity was attributed to the Mott–Schottky effect which is the electron enrichment of the metal NPs by an electron flow from the conduction band of C3N4 to the metal NPs leading to the formation of a potential barrier.
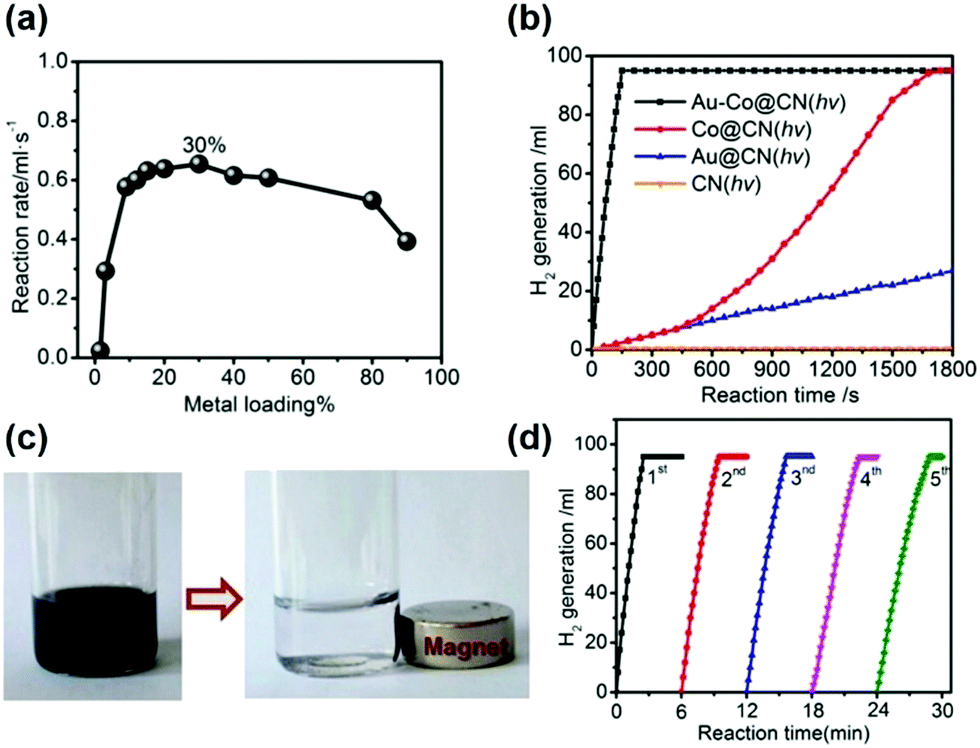 | ||
| Fig. 4 (a) Hydrogen generation from aqueous AB solution catalyzed by Au–Co@CN-x% (x: weight percentages of metal contents, x = 1.5–90) under photoirradiation; (b) hydrolysis of aqueous AB solution catalyzed by Au–Co@CN, Co@CN and Au@CN with photoirradiation (λ ≥ 400 nm); (c) photographs of the Au–Co@CN dispersions before and after magnetic separation; (d) hydrolysis of aqueous AB solution catalyzed by Au–Co@CN from the 1st to 5th cycle under photoirradiation. Typical conditions: 0.26 M aqueous AB solution (5 mL), molar ratio of catalyst/AB = 0.02, ambient atmosphere = 298 K. Reproduced with permission from ref. 68 Copyright 2015, American Chemical Society. | ||
Under visible light irradiation (λ ≥ 400 nm), the reaction rate was significantly enhanced and complete conversion was achieved in 150 s as shown in Fig. 4(b) (TOF = 2987 mol H2 mol−1 metal h−1). This was due to the enhanced charge separation due to the injection of more electrons from carbon nitride to the metal NPs. Fig. 4(b) also displays that the bare CN support was inactive for AB dehydrogenation under visible light irradiation. The plasmonic absorption of Au NPs owing to the LSPR effect, as observed in the UV-vis spectra, was also attributed to the enhanced photocatalytic activity in the hydrogen generation from AB. Additionally, the catalyst also exhibited superparamagnetic characteristics and hence can easily be separated from the reaction media using an external magnet for further recycling/stability tests up to five cycles (Fig. 4(c and d)).
The efficiency of plasmonic photocatalysis in hydrogen generation can be drastically improved by employing core–shell heterostructures as the plasmonic core can modify the surface properties of the active metal and enhance overall the light absorption. Huang et al. reported the synthesis of polyhedral Au–Pd core–shell nanostructures and studied the facet effects on the catalytic efficiency.69 Different morphologies of nanocrystals like tetrahexahedra (THH), octahedra and cube were compared for their efficiency in hydrogen generation in the dark and under visible-light irradiation conditions. Fig. 5 displays the SEM image of Au–Pd core–shell THH, octahedral and cubic nanocrystals. Au–Pd THH nanocrystals displayed superior catalytic performance (TOF = 142 mol H2 mol catalyst−1 min−1) because of the presence of the high-index {730} facet involving a higher fraction of surface atoms. A three-fold increment in the catalytic activity was obtained under visible light irradiation with a TOF value of 426 mol H2 mol catalyst−1 min−1. The broader range of absorption in the UV-vis spectra of Au–Pd THH with an LSPR peak maxima at 450 nm extending from the visible to near-infrared regime accounts for the enhanced catalytic performance under visible light irradiation. The mechanistic pathway involves the transfer of hot electrons from the Au core to the Pd shell under visible light irradiation and this charge heterogeneity weakens the B–N bond in AB.
 | ||
| Fig. 5 (a1–a3) SEM images of THH Au–Pd core–shell nanocrystals at different magnifications. (b1–b3) SEM images of (b1) Au–Pd core–shell nanocubes, (b2) Pd nanocubes, and (b3) Au nanocubes. (c1–c3) SEM images of (c1) Au–Pd core–shell octahedra, (c2) Pd octahedra, and (c3) Au octahedra. All scale bars are 100 nm. Particle models are shown. Reproduced with permission from ref. 69 Copyright 2016, Wiley-VCH. | ||
The generated hot electrons prefer to position themselves near the edges and corners of the nanocrystals and hence THH displays efficient charge heterogeneity and superior catalytic performance due to the highest number of edges and corners in the structure followed by cubic and octahedral arrangement.
Apart from a core–shell type morphology, plasmonic bimetallic alloy NPs have also been extensively reported for improving the rate of hydrogen generation. Wen et al. reported the integration of plasmonic PdAu alloy NPs on a chromium-based amine-functionalized metal–organic framework (MOF), the NH2-MIL-101 material, in order to enhance the overall activity of the Pd-catalyzed reaction by the assistance of visible light.70 Characterization using techniques like X-ray diffraction (XRD), UV-vis and extended X-ray absorption fine structure (EXAFS) analysis confirmed the successful incorporation of bimetallic NPs within the MOF. The Au LIII-edge and Pd K-edge EXAFS analysis confirmed the contiguous metal–metal bonding. The UV-vis spectra exhibit intense absorption at 200–300 nm attributed to the π → π* transition of the linker in the MOF. The heterogeneity created at the Au–Pd interface enhanced the visible light absorption owing to the LSPR of the Au NPs. The following trend in the catalytic activity for AB dehydrogenation was observed in the dark: Au/NH2-MIL-101 < Pd/NH2-MIL-101 < AuPd/NH2-MIL-101. Under visible light irradiation, all the samples exhibited enhanced activities and AuPd/NH2-MIL-101 displayed superior activity with a hydrogen generation rate of 1.6 mL min−1 which was twice higher than its original conversion in the absence of light irradiation (0.89 mL min−1). Many reports have studied the effect of using MOF materials as photo-active components behaving as a semiconductor. Upon visible light irradiation, the organic linker produces photo-excited electrons and transfers them to the nodes of NH2-MIL-101 via the linker-to-cluster charge-transfer mechanism (LCCT). These excited electrons are further transferred to the deposited metal NPs and hence form electron-rich species as the active site for the reaction. The observed enhancement of bimetallic AuPd alloy NPs was attributed not only to the photoactive MOF component but also to the LSPR effect of Au NPs. The energetic hot electrons on the surface of Au NPs upon visible light irradiation gets transferred to the Pd NPs due to their lower work function values and hence create electron-rich Pd species to catalyse the reaction efficiently. Finally, the synergistic effect between AuPd NPs and the MOF support was suggested to be the reason for efficient catalytic activities. To test the wide applicability of the hybrid catalyst, an attempt was made to catalyse the Suzuki–Miyaura coupling reaction. Significant yields of 35% and 68% of biphenyl were obtained in the dark and under visible light irradiation, respectively, for the AuPd/NH2-MIL-101 alloy NPs.
Metal NPs supported on polymers often exhibit lower catalytic activities due to the long diffusion length for the substrate molecules to reach the active site of the catalyst. Hence, it is highly desired to reduce the diffusion length in order to improve the catalytic activity of polymer supported metal NPs. Abundance in the techniques to prepare polymers and their relatively large size can demonstrate the compatibility in continuous flow reactors. Liu et al. studied the use of polymers to encapsulate metal NPs for efficient methanolysis of AB as shown in eqn (5).71 In this study, Au–Rh NPs were encapsulated in cyclic polysiloxane (CP) hosts with donut-like assembly for efficient catalysis under visible light irradiation. The cyclic shape leads to the reduced diffusion length of the assembly and was further modified with 6-mercapto-1-hexanol to stabilise the metal NPs through thiol-metal interaction, which was labelled as CP1. A TEM image of the CP1-Au0.1Rh0.9 catalyst displaying the circular shape of NPs along the polymer backbone is shown in Fig. 6A. The bimetallic Au–Rh NPs were fused together due to the co-reduction of NaAuCl4 and RhCl3 precursors which was confirmed by the zerovalent state of Rh NPs in the XPS spectra as shown in Fig. 5B. The bimetallic NPs displayed a TOF value of 780 min−1 in the dark and a higher TOF of 980 min−1 under visible light irradiation for the methanolysis of AB. Speculation of enhanced activity under visible light irradiation was made in two ways including charge separation and localized heating effects.
| NH3BH3(aq) + 4CH3OH(l) → NH4B(OCH3)4(aq) + 3H2(g) | (5) |
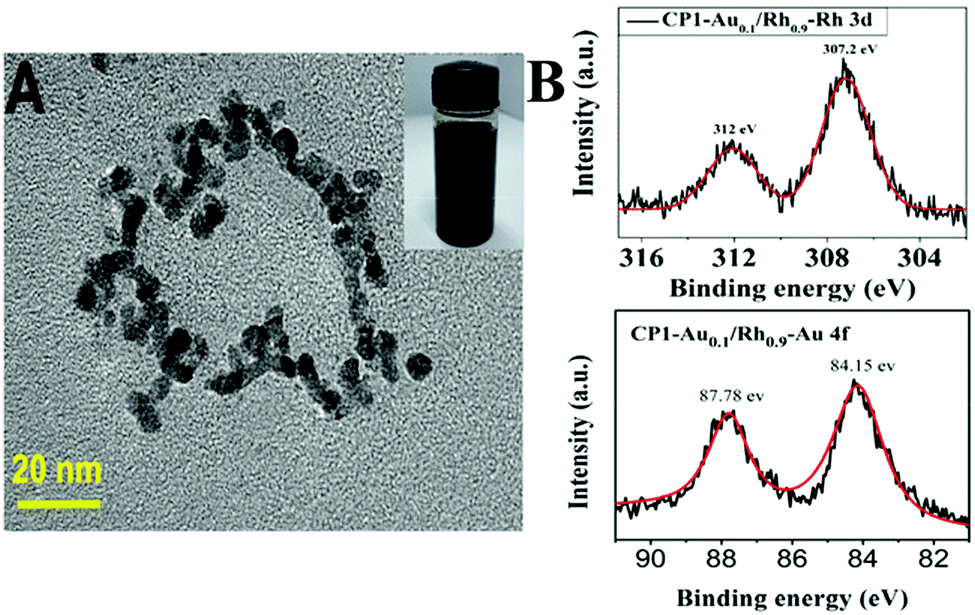 | ||
| Fig. 6 (A) TEM imaging of CP1-Au0.1Rh0.9 on the carbon support with the inset showing its methanol solution (2 mg ml−1). (B) XPS analysis of CP1-Au0.1Rh0.9 showing peaks corresponding to the 3d5/2 (307.2 eV) and 3d3/2 (312 eV) electrons of the Rh element. XPS analysis of CP1-Au0.1Rh0.9 showing the peaks corresponding to the 4f7/2 (84.15 eV) and 4f5/2 (87.78 eV) electrons of the Au element. Reproduced with permission from ref. 71 Copyright 2019 Wiley-VCH. | ||
2.2. Plasmonic silver hybrid nanocatalysts on insulating and semiconducting supports
The creation of surface plasmons by the excellent light absorption ability of Ag nanostructures in comparison to other noble metals makes them extremely useful in photocatalysis. Hence, under visible light irradiation, the plasmonic Ag can drive chemical reactions at a lower temperature than the conventional counterparts. Table 2 lists the plasmonic Ag-based catalytic systems along with their synthetic method and catalytic activity which have been discussed for the hydrogen generation in this section.| Catalysts | Synthesis method | Metal loadings | Light source | Catalytic activity (rate) | Ref. | |
|---|---|---|---|---|---|---|
| Dark | Light | |||||
| Ag/SBA-15 (B) | MW-assisted deposition | 1.0 wt% Ag on SBA-15 | 500 W Xe > 420 nm | 0.13 μmol min−1 | 0.67 μmol min−1 | 61 and 72 |
| Pd/Ag/SBA-15 (B) | LSPR-assisted deposition | 0.1 wt% Pd; 1.0 wt% Ag | 500 W Xe > 420 nm | 0.74 μmol min−1 | 2.28 μmol min−1 | 17 and 73 |
| Ru/Ag/SBA-15 (B) | LSPR-assisted deposition + NaBH4 reduction | 0.5 wt% Ru; 1.0 wt% Ag | 500 W Xe > 420 nm | 2.70 μmol min−1 | 3.60 μmol min−1 | 78 |
| Ag/Ti-SBA-15 | MW-assisted deposition | 0.5 mol % Ti; 1.0 wt% Ag | 500 W Xe 200–780 nm | 2.4 μmol min−1 | 3.3 μmol min−1 | 79 |
| Ag/TiO2 | Colloidal deposition | Ag: 0.06 wt% | 100 W LED 450 nm | — | 2.5 μmol min−1 | 81 |
| Ag–C–Co | Ethylene glycol reduction | Colloidal | — | 6.48 × 103 mL min−1 g−1 | 86 | |
A classic example of tuning the LSPR absorption of morphology controlled plasmonic NPs was reported by Fuku et al. in which size and colour-controlled Ag NPs were synthesised within the mesoporous channels of SBA-15 silica.61 The nanocatalysts were prepared by a microwave (MW)-assisted alcohol reduction method in which 1-hexanol was used as a solvent and a reducing agent in the presence or absence of sodium laurate (Lau) as a surface directing agent followed by microwave heating for 3–5 min.72Fig. 7(a–c) displays the TEM micrographs and the sample photographs of three catalysts. The formation of uniformly dispersed spherical yellow NPs with an average diameter of 4 nm was denoted as Ag/SBA-15 (Y). The red and blue nanorods were formed in the absence of a surface directing agent and were labelled as Ag/SBA-15 (R) and Ag/SBA-15 (B). The morphology of the nanostructures plays an important role in the LSPR absorption of plasmonic materials. A sharp absorption peak at 420 nm was obtained for spherical Ag/SBA-15 (Y) and two peaks were observed in the Vis-NIR region for red and blue nanorods because of the transverse and longitudinal modes of nanorods. SBA-15 displayed no absorption in the visible region and was found to be inactive for the AB dehydrogenation reaction.73 Ag/SBA-15 (Y) was found to display the highest catalytic activity (0.94 mol% min−1) because of the smallest size NPs followed by Ag/SBA-15 (R) and Ag/SBA-15 (B). Under visible light irradiation, the enhancement follows the order of Ag/SBA-15 (B) > Ag/SBA-15 (R) > Ag/SBA-15 (Y). The significant enhancement shown by Ag/SBA-15 (B) is evident because of its light absorption in the visible-NIR region of the spectrum. The wavelength dependence of catalytic activities was also found to be in accordance with the plasmonic absorption of Ag NPs. To eliminate the photothermal effects, a catalytic reaction was also carried out at a higher temperature in the absence of light illumination. It was found that the reaction rate under thermal conditions was higher than that in the dark but significantly lower than that under light irradiation conditions.73 The three catalysts were also tested for the p-nitrophenol (p-NP) reduction to p-aminophenol (p-AP) using AB as the source of hydrogen. Interestingly, the same trend in the catalytic activity and enhancements to that of AB dehydrogenation was observed. Moreover, the catalysts were successfully recovered after the reaction and they retained their colour and morphology after the reaction without any leaching of NPs. The charge polarization on the surface of Ag NPs due to the LSPR effect was ascribed to the enhanced catalytic activities under visible light irradiation.
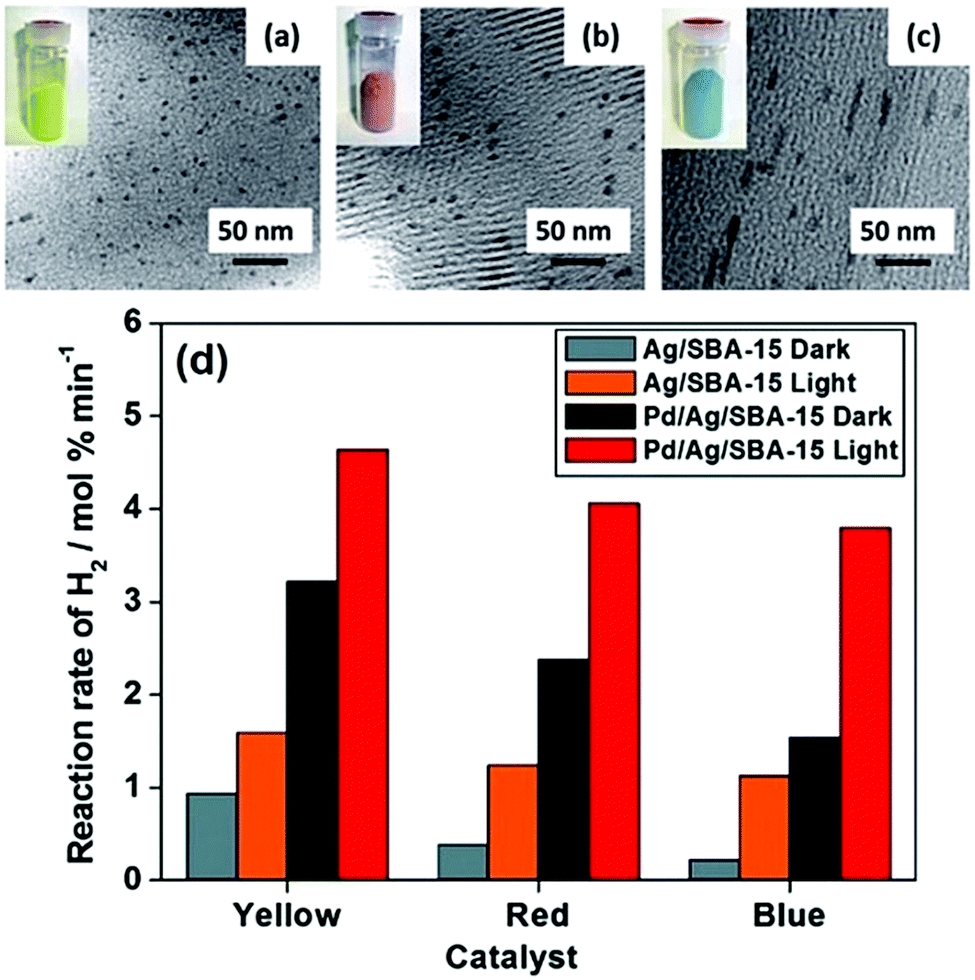 | ||
| Fig. 7 TEM images and sample photographs (inset) of (a) Ag/SBA-15 (Y), (b) Ag/SBA-15 (R), (c) Ag/SBA-15 (B) and (d) comparison of the initial rates of hydrogen production from AB for monometallic Ag/SBA-15 and bimetallic Pd/Ag/SBA-15 in the dark and under light irradiation conditions (λ > 420 nm). Reproduced with permission from ref. 73 Copyright 2015 The Royal Society of Chemistry. | ||
The combination of the plasmonic metal with catalytically active metal NPs is one of the recently explored approaches in order to improve the optical and catalytic properties especially for noble metal-based NPs. Majima et al. reported the specific coverage of the tips of Au NPs with Pt nanoclusters for hydrogen evolution from water using methanol as the charge scavenger.74 Verma et al. studied the PdAg bimetallic nanostructures on size and colour-controlled Ag/SBA-15 nanostructures. An aqueous suspension of Ag NPs along with a Pd precursor, Pd(OAc)2, was photoirradiated for 2 h under inert atmospheric conditions.73,75 Uniform surface deposition was carried out in the absence of a reducing agent and the galvanic displacement of Pd2+ ions with Ag leads to the formation of bimetallic nanostructures labelled as Pd/Ag/SBA-15 (Y), Pd/Ag/SBA-15 (R) and Pd/Ag/SBA-15 (B). A clear absorption peak at λ = 420 nm even after Pd deposition confirmed the plasmonic behaviour of Pd/Ag/SBA-15 catalysts. A comparative decrease in the absorption intensity compared to Ag/SBA-15 samples indicated the absorption interference by Pd NPs. All bimetallic Pd/Ag/SBA-15 catalysts displayed superior catalytic performance in comparison to their monometallic counterparts as summarised in Fig. 7(d). Pd/Ag/SBA-15 (Y) exhibits a 3-fold enhancement in the reaction rate (3.22 mol% min−1) compared to Ag/SBA-15 (Y) (0.94 mol% min−1). The trend in the catalytic performance follows the order of Pd/Ag/SBA-15 (Y) > Pd/Ag/SBA-15 (R) > Pd/Ag/SBA-15 (B) and even a higher rate of reaction under visible light irradiation. Pd/Ag/SBA-15 (B) showed the maximum enhancements due to its wide absorption in the visible-NIR region.
The morphology and color controlled Ag/SBA-15 and Pd/Ag/SBA-15 catalysts displayed different catalytic and enhancement trends. Smaller size yellow NPs displayed a superior reaction rate followed by rods of a shorter aspect ratio and then blue nanorods of a longer aspect ratio because of their size control and change in the number of exposed surface atoms. However, in the presence of visible light irradiation, the trend in the catalytic activity enhancement was the opposite as it did not depend on the surface area but on the intensity of LSPR absorption. The stronger the absorption, the more significant the enhancement in the catalytic response: blue > red > yellow. A similar effect was observed in the case of bimetallic Pd/Ag/SBA-15 morphology-controlled catalysts. This observation can be related to the concept of the LSPR compensation effect which accounts for the enhanced photocatalytic performance of enlarged nanostructures, as a remedy for the loss in the catalytic activity, due to size enlargement. It has been demonstrated that enlarged nanostructures (blue nanorods in this case) have a superior ability to generate higher heating power when compared to smaller nanostructures (yellow spherical NPs) and hence are capable of displaying superior enhancement effects under visible light irradiation conditions.
This study was further extended by investigating a systematic comparison in the catalytic and optical behaviour of PdAg and PdAu nanostructures supported on mesoporous SBA-15 silica.76 The Pd/Ag/SBA-15 and Pd/Au/SBA-15 catalysts displayed characteristic LSPR absorption at 400 and 520 nm, respectively. The uniformly dispersed bimetallic NPs with an average diameter of 4.2 nm and 4.9 nm for Pd/Ag/SBA-15 and Pd/Au/SBA-15, respectively, were also characterised by XPS analysis. The corresponding shifts in the binding energy values provide strong evidence of the creation of a heterojunction by different metals situated adjacent to each other. Further, the appearance of a shoulder peak and a shorter Pd–Pd distance than Pd foil, in the Pd K-edge EXFAS analysis, confirmed the existence of heteroatomic bonding. Pd/Ag/SBA-15 was found to be more active in AB dehydrogenation than Pd/Au/SBA-15 under visible light irradiation with a reaction rate of 4.64 mol% min−1. Furthermore, the bimetallic catalysts were tested in the Suzuki–Miyaura coupling reaction between iodobenzene and phenylboronic acid to form biphenyl as the product, in which Pd/Au/SBA-15 was found to exhibit significantly superior catalytic performances compared to Pd/Ag/SBA-15 in the dark and under visible light irradiation conditions. A plausible mechanism for the enhanced catalytic activity of bimetallic NPs under visible light irradiation was proposed as follows. The charge carriers generated on the surface of plasmonic NPs are transferred to the active metal species and get accumulated in the LUMO of the reactant molecules (AB) adsorbed on the surface. The addition of hot electrons leads to B–N bond elongation and enhances the overall reaction rate as illustrated in Scheme 1.
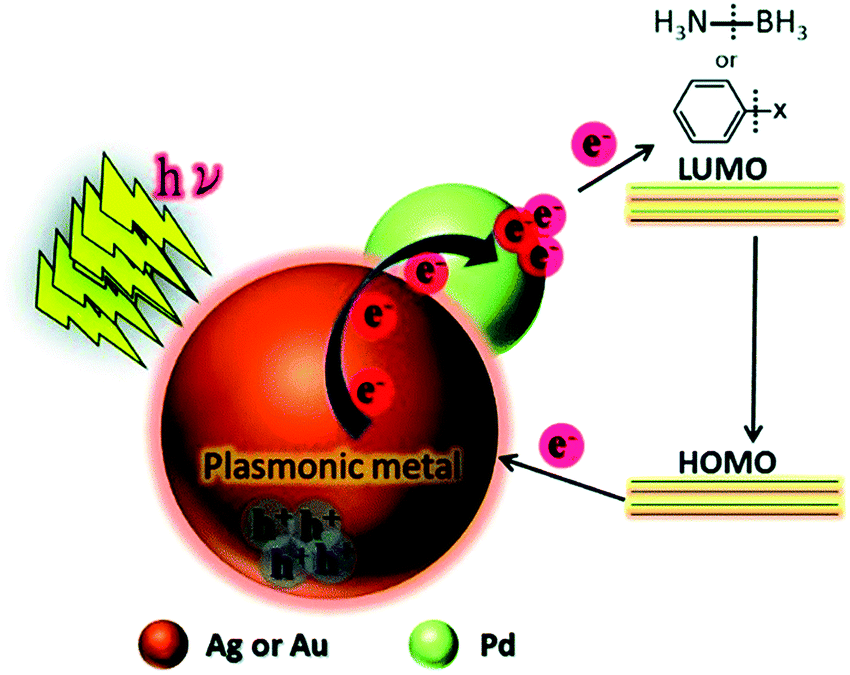 | ||
| Scheme 1 Schematic illustration of the plausible mechanistic pathway for the enhanced catalytic activities under visible light irradiation over plasmonic metal NPs. Reproduced with permission from ref. 76 Copyright 2016 The Royal Society of Chemistry. | ||
To further explore and develop visible light sensitive catalysts, a series of active metals (Ru, Ni, Co) were deposited on a blue Ag nanorod catalyst. This unique combination of Ag nanorods with active metal components was not reported until then.77,78 The bimetallic nanorods displayed 2 absorption peaks at 370 nm and 980 nm, confirming the optical signature for the formation of nanorods. Ru/Ag/SBA-15 was found to be the most active with a reaction rate of 3.60 μmol min−1; however, Pd/Ag/SBA-15 displayed a maximum enhancement (approximately two-fold) under visible light irradiation. The selection of active metal deposition on Ag nanorods was based on their work function values which assist in the suitable positioning of the Fermi energy level for an efficient charge transfer process.
A pioneering approach of merging single-site Ti-oxide moieties and plasmonic NPs for efficient hydrogen generation was reported for the first time in 2017 by Verma et al.79 A new type of photocatalytic system was designed by introducing titanium oxide moieties and Ag NPs within the framework of mesoporous silica, in order to link together Vis-active plasmonic and UV-active single-site photocatalysts. The Ti-oxide species were introduced during the sol–gel synthesis of SBA-15 and MW assisted deposition was used to grow highly dispersed Ag NPs. The isolated and tetrahedrally coordinated heteroatoms (Ti, Cr, V, Mo) doped within the framework of mesoporous silica is often termed as single-site photocatalysts. They are capable of displaying superior adsorption and catalytic properties than the conventional oxide materials. The purpose of combining UV active Ti-oxide moieties and visible light sensitive Ag NPs was to explore the broad spectral range of the solar spectrum. Ti-SBA-15 was synthesised by using P123 as the surface directing agent, and titanium tert-butoxide (TBOT) and tetraethyl orthosilicate (TEOS) were used as the precursor for Ti and Si, respectively.80 The uniform dispersion of small-sized Ag NPs (3.2 nm) (Fig. 8(a)) was achieved by MW heating for 3 min in the presence of sodium laurate. An intense absorption owing to the Ag-plasmon was observed in the visible region of the spectrum at 400 nm. Fig. 8(c) displays the Ti K-edge X-ray absorption near edge structure (XANES) spectra of Ag/Ti-SBA-15, Ti-SBA-15, titanium tetrabutoxide (TBOT) and TiO2. The single pre-edge peak at 4968 eV confirmed the existence of single-site isolated Ti-oxide moieties. Fig. 8(d) shows the time profile of hydrogen production from AB under light irradiation conditions. A trace amount of hydrogen was produced using the Ti-SBA-15 catalyst in the dark (0.1 μmol min−1) and under light irradiation (0.2 μmol min−1) conditions.79 Ag/Ti-SBA-15 displayed an unexpected higher rate of reaction (3.3 μmol min−1) in comparison to its counterparts: Ag/SBA-15, Ag/TiO2/SBA-15 and a physical mixture of Ag/SBA-15 + Ti-SBA-15. The charge transfer excited state is formed by the excitation of Ti-oxide moieties upon UV light absorption (200–270 nm). There occurs a charge relay from Ti-oxide to Ag which helps in the B–N bond elongation of AB adsorbed on its surface, hence leading to the enhanced catalytic activity as illustrated in Fig. 8(b). This can be cited as one of the classic examples of the synthesis of hybrid plasmonic materials with unique UV-vis broad-spectrum sensitivity.
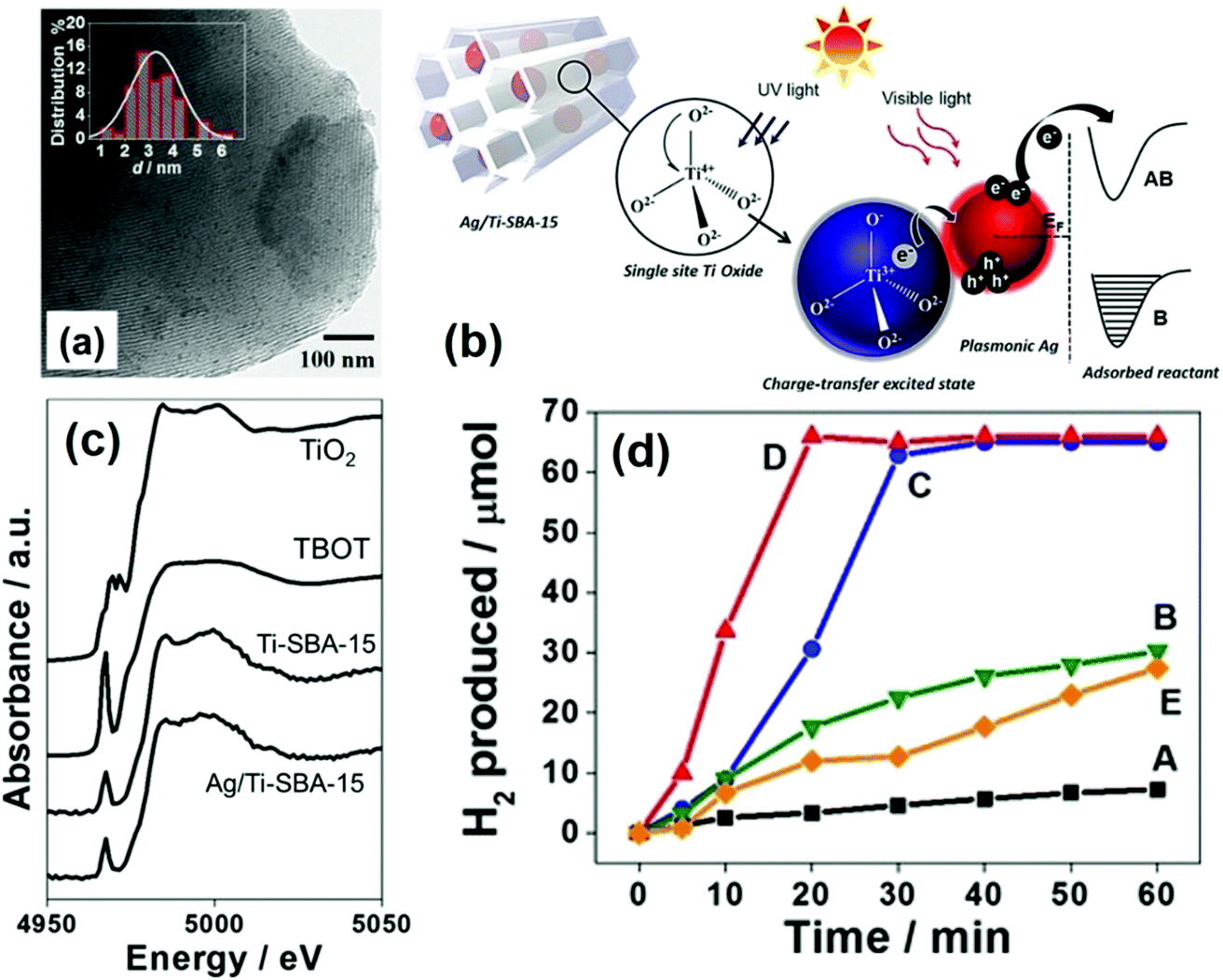 | ||
| Fig. 8 (a) TEM image and size distribution histogram (inset) of Ag/Ti-SBA-15; (b) schematic illustration of the light-induced charge-transfer pathway on the catalyst under UV and visible-light irradiation (B: bonding AB: antibonding); (c) Ti K-edge XANES spectra of Ag/Ti-SBA-15, Ti-SBA-15, TBOT, TiO2; (d) time profile for hydrogen production from AB under light irradiation for (A) Ti-SBA-15, (B) (Ag + Ti)/SBA-15, (C) Ag/SBA-15, (D) Ag/Ti-SBA-15 and (E) Ag/TiO2/SBA-15. Reproduced with permission from ref. 79 Copyright 2017 Wiley-VCH. | ||
As discussed in the previous section about the Au/TiO2 system, Simagina et al. very recently reported Ag deposition on TiO2 as an important strategy to extend the UV light absorption of the TiO2 photocatalyst. Anatase TiO2 with a crystallite size of 10–11 nm and a specific surface area of 347 m2 g−1 was synthesised by hydrothermal treatment with KOH at 120 °C. The extremely low content of 0.06 wt% of colloidal Ag was loaded onto TiO2 by an incipient wetness impregnation method.81 The Ag NPs can create efficient charge transfer in the photocatalyst owing to the LSPR effect. An intense absorption band at 420 nm was observed with an average particle size of about 36 nm. The prepared catalyst was employed for the photocatalytic hydrothermolysis of AB at 90 °C. Approximately 150 μmol h−1 of H2 was evolved using the Ag/TiO2 catalyst under visible light irradiation using an LED of λ = 450 nm and power of 100 W.
The use of AB was not only employed as a direct source of hydrogen generation but also as an in situ source of molecular hydrogen to carry out selective hydrogenation reactions. In our previous report, a series of 1.0 wt% plasmonic Ag NPs were prepared on CeO2 coated SBA-15 by the MW-assisted alcohol reduction method.82 AB was used as the in situ source of hydrogen to selectively reduce the nitro functional group to the amino group. The amounts of CeO2 were varied from 0.0, 0.5, 1.0, 2.0 to 5.0 wt% on SBA-15 by using Ce(NO3)3·6H2O as the cerium precursor.
An intense plasmonic absorption band at 400 nm was observed for Ag/SBA-15. The increase in the coating amount of CeO2 decreased the intensity of absorption, which might be due to the optical hindrance arising because of the possible interaction between Ag and CeO2. The electronic structure of Ag NPs was studied by XAFS analysis which confirmed the existence of Ag in the zero oxidation state. Ag 3d XPS displayed a peak shift in the binding energies towards higher values due to the charge transfer from CeO2 to Ag owing to the difference in their electronegativities.
The catalytic performance was evaluated in the hydrogen production from AB and by utilising the in situ generated H2 for chemoselective –NO2 reduction in nitrostyrene (p-NS) to the –NH2 group in aminostyrene (p-AS) without affecting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The reduction of p-NS is commercially an important process but still very few reports explore the chemoselective reduction process under ambient conditions.83 The possible hydrogenation products by the complete and incomplete reduction of p-NS are p-AS (desired), p-nitroethylbenzene (p-NEB), and p-aminoethylbenzene (p-AEB) as shown in Scheme 2. All catalysts displayed activity in the reduction to form p-AS and by-products in the dark and under visible light irradiation. The obtained yield of p-AS in the dark was 17% by Ag/SBA-15 and 72% by an optimum coating of CeO2 in Ag/CeO2(2.0)/SBA-15. Under visible light irradiation, the enhanced yield of 32% and 90% of p-AS was obtained by Ag/SBA-15 and Ag/CeO2(2.0)/SBA-15, respectively. The increased activity under visible light irradiation was attributed to the efficient transfer of the H+/H− pair towards the polar –NO2 group instead of non-polar CC. The charge polarization of plasmonic Ag NPs leads to the easy adsorption of polar molecules on their surface under light illumination conditions. Further, the effects arising from metal–support interaction and the synergistic catalytic effects may contribute to obtaining the increased yields of the desired products.
C bond. The reduction of p-NS is commercially an important process but still very few reports explore the chemoselective reduction process under ambient conditions.83 The possible hydrogenation products by the complete and incomplete reduction of p-NS are p-AS (desired), p-nitroethylbenzene (p-NEB), and p-aminoethylbenzene (p-AEB) as shown in Scheme 2. All catalysts displayed activity in the reduction to form p-AS and by-products in the dark and under visible light irradiation. The obtained yield of p-AS in the dark was 17% by Ag/SBA-15 and 72% by an optimum coating of CeO2 in Ag/CeO2(2.0)/SBA-15. Under visible light irradiation, the enhanced yield of 32% and 90% of p-AS was obtained by Ag/SBA-15 and Ag/CeO2(2.0)/SBA-15, respectively. The increased activity under visible light irradiation was attributed to the efficient transfer of the H+/H− pair towards the polar –NO2 group instead of non-polar CC. The charge polarization of plasmonic Ag NPs leads to the easy adsorption of polar molecules on their surface under light illumination conditions. Further, the effects arising from metal–support interaction and the synergistic catalytic effects may contribute to obtaining the increased yields of the desired products.
 | ||
| Scheme 2 Selective reduction of p-nitrostyrene (p-NS) to p-aminostyrene (p-AS) and other by-products. | ||
The morphology controlled monometallic Ag/SBA-15 (Y), (R), (B) and bimetallic Pd/Ag/SBA-15 (Y), (R), (B) systems were also tested to study the chemoselective reduction of p-NS to p-AS using AB as an in situ source of hydrogen.84Fig. 9(A–C) summarises the UV-vis spectra and the obtained reaction results in the dark and under visible light irradiation. The UV-vis spectra of the three different Ag catalysts responded differently with visible light irradiation as shown in Fig. 9(A). The Pd deposition decreased the absorption intensity and broadened the spectral peaks for bimetallic catalysts (Fig. 9(B)), which can be attributed to the surface coverage of plasmonic Ag NPs. Fig. 9(C) depicts the results of the chemoselective reduction reaction of p-NS to p-AS in the dark and under visible light irradiation. The reduction reaction proceeds via the formation of nitroso and hydroxylamine intermediates as shown in eqn (6) below:
| R–NO2 + 2e− → R–NO + 2e− → R–NH(OH) + 2e− → R–NH2 | (6) |
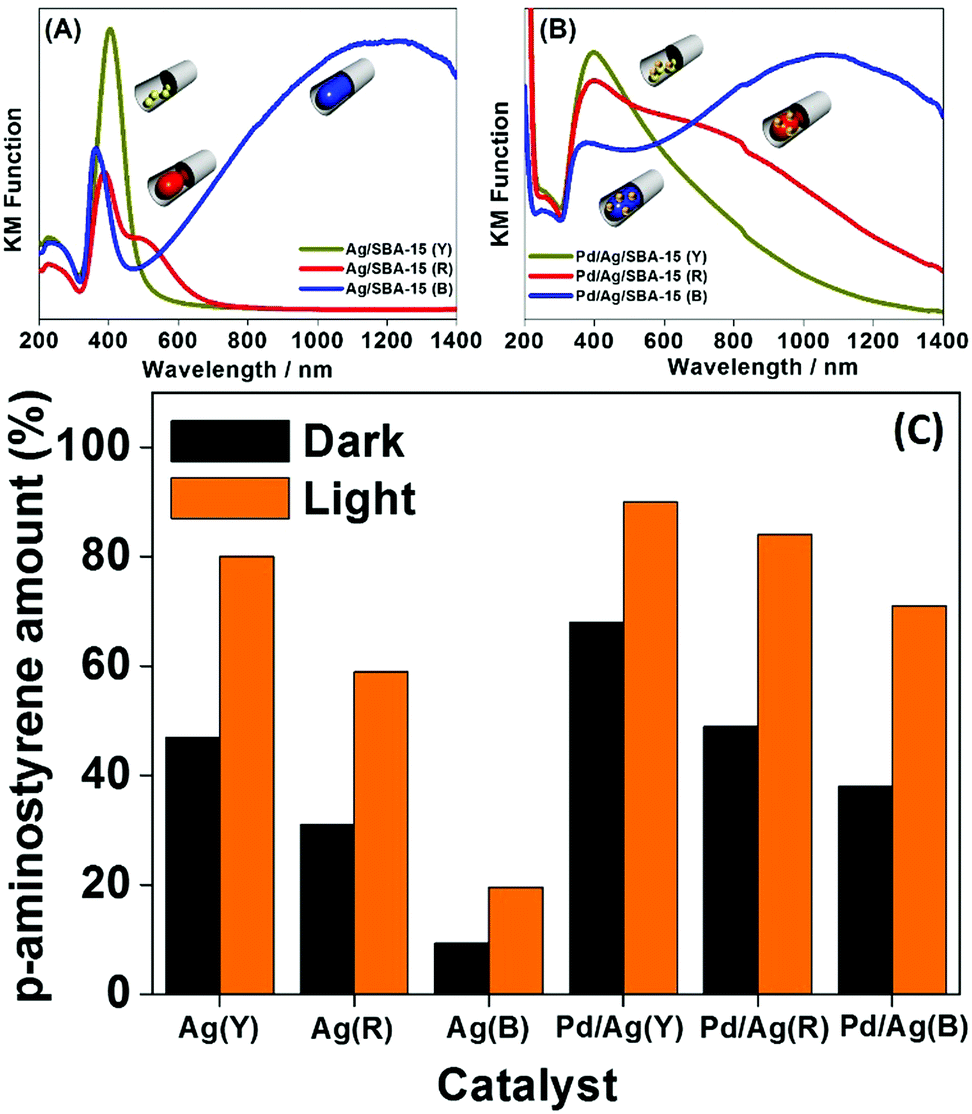 | ||
| Fig. 9 UV-vis spectra of (A) Ag/SBA-15 (Y), Ag/SBA-15 (R), Ag/SBA-15 (B), (B) Pd/Ag/SBA-15 (Y), Pd/Ag/SBA-15 (R), and Pd/Ag/SBA-15 (B) and (C) a comparison of the yields of p-AS in the dark and under visible light irradiation (λ > 420 nm). Reproduced with permission from ref. 84 Copyright 2017 Springer. | ||
No product formation was observed in the absence of catalyst. The monometallic catalysts were found to be less active than the bimetallic ones. The obtained trend in the production of p-AS follows the order: Pd/Ag/SBA-15 (Y) > Pd/Ag/SBA-15 (R) > Pd/Ag/SBA-15 (B) > Ag/SBA-15 (Y) > Ag/SBA-15 (R) > Ag/SBA-15 (B). The higher catalytic performance of smaller yellow NPs indicates that the reaction takes place on the surface of the exposed surface atoms and hence follows the same trend as in the AB dehydrogenation reaction. Under visible light irradiation, Pd/Ag/SBA-15 (Y) exhibits a superior conversion efficiency of 89% compared to 68% in the dark. The synergistic effect of PdAg NPs arising due to the significant enhancement and superior catalytic performance under visible light irradiation, when compared with monometallic Pd and Ag, enhances the hydrogen generation and plasmonic Ag assist in efficient charge separation under visible light irradiation. The higher chemoselective performance was assigned to the efficient charge transfer of the H+/H− pair to the polar –NO2 groups in p-NS.
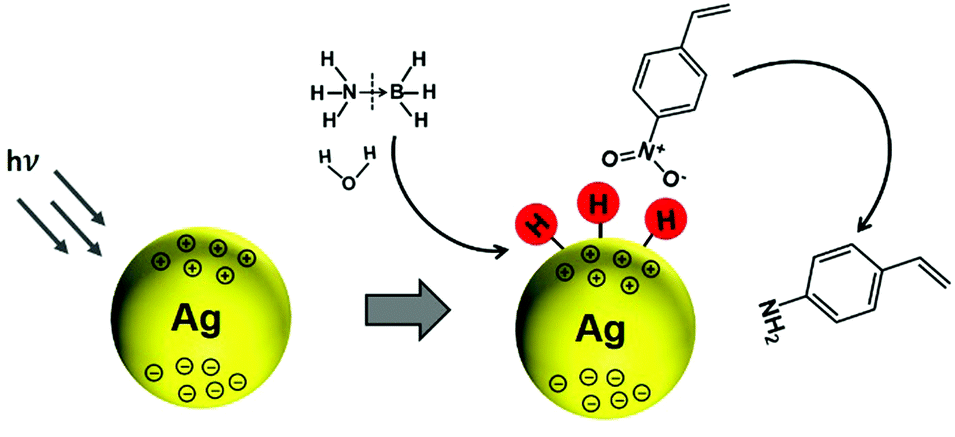
In another interesting research study, Yamashita et al. reported Ag/MxOy (TiO2, ZrO2, Al2O3 and CeO2) plasmonic catalysts for the photocatalytic reduction of p-NS at RT and atmospheric pressure and AB as an in situ source of hydrogen.85 The catalysts were prepared by a simple impregnation method using AgNO3 as the silver precursor. The size of Ag NPs was found to be in the range of 5–20 nm with the smallest (6.8 nm) and the largest (20.7) sized NPs formed by Ag/ZrO2 and Ag/Al2O3, respectively. Fig. 10(a) displays a TEM micrograph along with a size distribution histogram (inset) of Ag/TiO2 with an average diameter of 11.8 ± 3.4 nm. All the catalysts exhibit intense absorption at 400–500 nm due to the LSPR effect of Ag NPs, as shown in Fig. 10(b). The prepared catalysts exhibit complete conversion with varying selectivity for the formation of p-AS in the dark and under visible light irradiation (λ > 420 nm). Ag/TiO2 displayed a superior catalytic performance with 100% conversion and 47% and 81% selectivity in the dark and under visible light irradiation, respectively. Different wavelengths of LED light (blue – 470 nm, green – 530 nm and red – 627 nm) with a power density of 66.7 mW were used in order to understand the mechanism of enhancement by plasmonic Ag NPs. Fig. 10(c and d) shows the effect of using an LED and the wavelength-dependent action spectrum of Ag/TiO2. As expected, the selectivity values change significantly with different wavelengths of light. The catalytic performance of Ag/TiO2 was found to be highly consistent with the LSPR absorption intensity and the obtained trend was green LED > red LED > blue LED > dark. The effect of charge scavenger addition was also studied to investigate the important role of the photogenerated electrons and holes in the hydrogen evolution from AB under visible light irradiation. The proposed mechanism was based on the preferred alignment of the polar –NO2 group towards the dipole generated on plasmonic Ag and the non-polar CC facing away from the dipole, leading to enhanced chemoselectivity (Scheme 3).
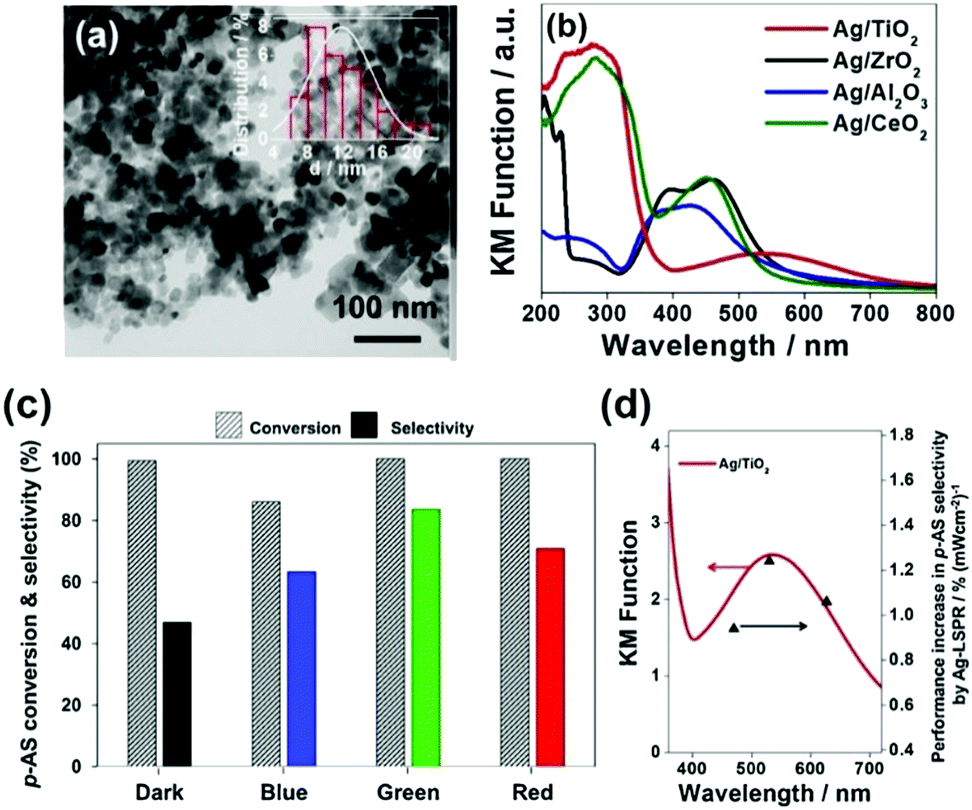 | ||
| Fig. 10 (a) TEM micrograph of Ag/TiO2; (b) UV-vis absorption spectra of Ag NPs deposited on metal oxide support materials; (c) effect of using different wavelengths of LED (power = 66.7 mW) in the reaction conversion and selectivity utilizing Ag/TiO2 (impregnation); (d) Wavelength dependent performance increase in the p-AS selectivity over Ag/TiO2 catalysts upon irradiation with LED light (blue light λ = 470 nm, green light λ = 530 nm and red light λ = 627 nm). Reaction conditions: catalyst (5 mg), p-NS (20 mM), biphenyl (20 mM), AB (60 mM) in ethanol (11 mL) for 3 h under an inert argon atmosphere. Reproduced with permission from ref. 85 Copyright 2019 Elsevier. | ||
 | ||
| Scheme 3 Schematic illustration of the preferred alignment of the polar –NO2 group towards plasmonic Ag NPs for chemoselective p-NS under visible light irradiation (λ > 420 nm). Reproduced with permission from ref. 85 Copyright 2019 Elsevier. | ||
The development of such types of photocatalysts will further assist in opening a new avenue for tuning the reaction selectivity in visible-light-driven catalytic reactions.
Significant efforts have been devoted in recent years to fabricate multi-component nanostructures to improve the electronic and intrinsic properties of noble metals. The application performance of carbon isolated core shell nanostructures has rarely been reported. Zhou et al. synthesised a Ag–C–Co sandwiched structure in which the Co shell assembles around the Ag–C spherical core nanostructure.86 Damping of Ag surface plasmon resonance was observed because of the surface covering with the Co shell. It was observed that the catalyst displayed ferromagnetic properties and hence can be separated from the reaction mixture using an external magnet. The rate of hydrogen generation from AB was 6.48 × 103 mL min−1 g−1 using Ag–C–Co nanospheres, which was remarkably higher than those of Ag–Co, C–Co and Co nanospheres of comparable sizes as shown in Fig. 11(A). The enhanced catalytic activity was attributed to the Ag SPR effect and electronic synergistic effects. The 11B NMR spectra of the freshly prepared AB solution and after completion of the reaction were measured as shown in Fig. 11(B). The presence of a signal at δ = 7.5 ppm and disappearance of the strong signal at δ = −23.9 ppm indicate the formation of a new B–O bond and breaking of B–H and B–N bonds in AB, respectively. A schematic illustration of AB hydrolysis over the sandwiched nanostructure is displayed in Fig. 11(C). The activation energy, Ea of the reaction by using the Arrhenius equation, was calculated to be 34.87 kJ mol−1 as shown in Fig. 11(D). The easy recovery due to the ferromagnetic nature and high catalytic activity induced by synergistic effects explains the potential application of the prepared catalyst in hydrogen fuel and chemical industries.
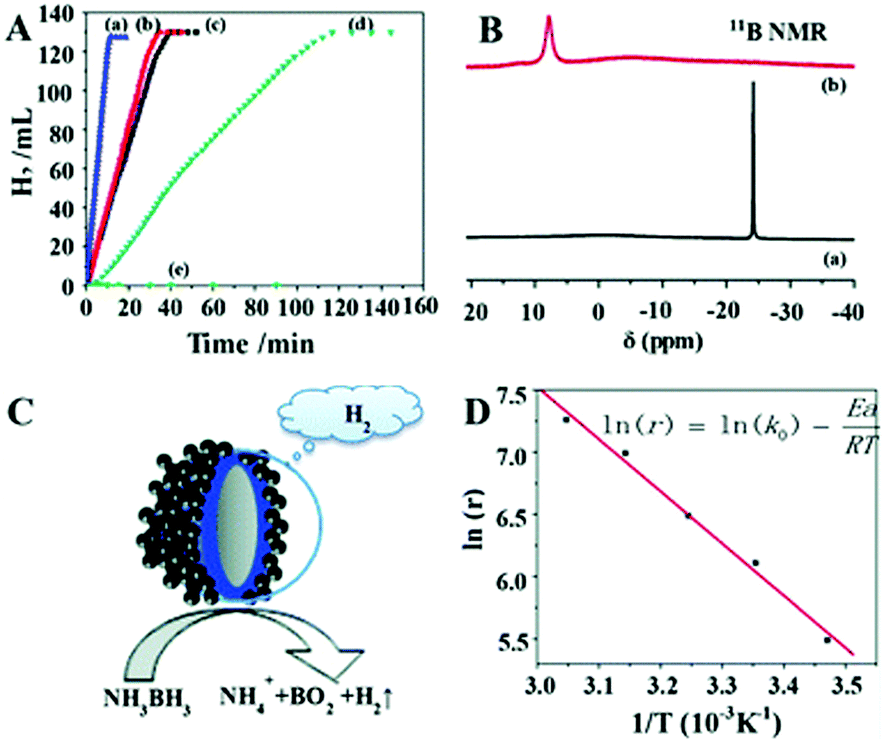 | ||
| Fig. 11 (A) Hydrogen generation from AB aqueous solution (0.26 M, 10 mL) catalyzed by (a) Ag–C–Co nanospheres, (b) Ag–Co nanospheres, (c) C–Co nanospheres, (d) Co nanospheres, (e) Ag–C nanospheres, (f) isolated Ag nanospheres, and (g) isolated C nanospheres at RT; (B) 11B NMR spectra of the aqueous solution measured at different stages of hydrolysis: (a) freshly prepared AB (0.13 M) solution, (b) after the completion of the reaction in the presence of Ag–C–Co sandwiched sphere structures; (C) schematic view of the catalytic dehydrogenation reaction; (D) Arrhenius plot for the generation of hydrolysis of AB solution using 200 nm Ag–C–Co nanospheres as catalysts. (In the inserted equation, r represents the reaction rate (mL min−1 g−1), k0 is the reaction constant (mL min−1 g−1), Ea is the activation energy of the reaction (kJ mol−1), R is the universal gas constant, and T (K) is the reaction temperature; and catalyst/AB = 0.035.) Reproduced with permission from ref. 86 Copyright 2013. Royal Society of Chemistry. | ||
The plasmonic noble metal nanostructures discussed in this section involved Ag and Au based monometallic and bimetallic (core–shell and alloy) catalysts incorporated on mesoporous silica, TiO2, single-site Ti oxide modified SBA-15, MOFs and polymers. The first example of plasmon-mediated catalysis over Ag/SBA-15 catalysts for efficient hydrogen generation from AB has also been illustrated.87 These nanostructures have been prepared mainly by photo-assisted deposition, wet-impregnation, seed-mediated growth and the LSPR-assisted deposition method. The integration of plasmonic nanostructures with catalytically active species (Pd, Ru, Pt and Co) has been an important strategy for the design of such materials. An interesting case study of in situ hydrogen generation over Ag-based catalysts has also been reviewed for chemoselective reactions.
2.3. Mechanism of plasmonic enhancement over noble metal nanostructures
In this section, the mechanism of enhancement over noble metal NPs in combination with insulating or semiconducting supports will be discussed. Fig. 12(a) depicts the possible pathways during the course of reaction on metal NPs under visible light irradiation. An energy diagram displaying the SPR effect on NPs is shown in Fig. 12(b). As discussed in several reports earlier,76,77,79 the plasmonic excitation of Ag, Au NPs under light irradiation at a suitable wavelength can generate charge carriers popularly known as hot electrons and hot holes. The different pathways of plasmonic excitation upon visible light irradiation will be discussed briefly in the following sub sections.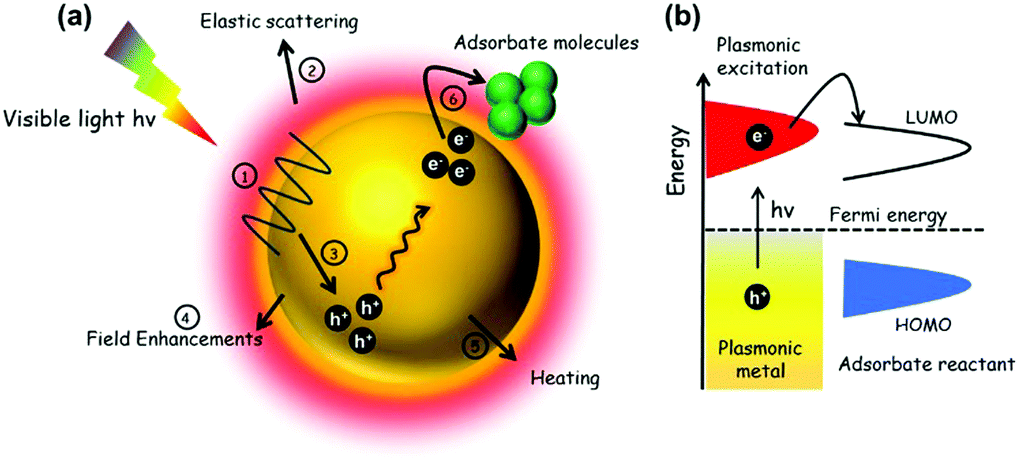 | ||
| Fig. 12 (a) Schematic representation of mechanistic pathways on metal NP upon visible light irradiation (1) surface plasmon, (2) relaxation through scattering, (3) creation of hot electrons and holes, (4) field enhancements, (5) plasmonic heating, and (6) transfer of hot electrons to the reactant molecules; (b) energy diagram showing SPR on pure plasmonic metal NP and electron transfer to the LUMO of adsorbate/reactant molecules. | ||
| P = Iσabs | (7) |
| δT = P/(4πkR) | (8) |
3. Semiconductor-based plasmonic catalysts
The hot electrons generated on noble metal nanostructures can directly be transferred to the hydrogen carrier molecules, which enhances the photocatalytic activity.17,92 However, the electron–phonon relaxation in the noble metal NPs inhibits both the process of generation and transfer of hot electrons. The combination of two or more plasmonic NPs can enhance the LSPR absorption to a significant strength but the high cost of such noble metal NPs restricts their practical applications. Hence the quest for finding non-metal plasmonic nanostructures was initiated to find more economical materials for LSPR coupling. Some recently explored doped plasmonic semiconductors are WO3−x, MoO3−x and Cu2−xS. The SPR excitation in such non-metal-based nanostructures arises due to the charge oscillations from ion-doping or lattice vacancies resulting in strong and broad absorption in the visible and infrared regions. In this section, we will describe two of the most researched semiconductor-based plasmonic materials i.e. WO3−x and MoO3−x for the hydrogen generation reaction.3.1. Plasmonic WO3−x hybrid nanocatalysts
The plasmonic WO3−x nanocatalyst is a blue coloured crystalline solid with an intense LSPR peak in the NIR region of the absorption spectrum. We came across interesting reports on plasmon-mediated catalysis of WO3−x integrated with Au, Ag, Rh, and Ni2P NPs and carbon heterostructures. The synthesis process is usually done by solvothermal treatment at 120–160 °C and summarised in Table 3 along with the details of several catalytic systems described in this section.| Catalysts | Synthesis method | Metal loadings | Light source | Catalytic activity | Ref. | |
|---|---|---|---|---|---|---|
| Dark | Light | |||||
| WO3−x | Solvothermal synthesis | Colloidal | 300 W Xe > 420 nm | 0.26 μmol min−1 | 1.32 μmol min−1 | 93 |
| Au/WO3−x | Solvothermal synthesis | Atom ratio Au:W = 10 |
300 W Xe > 420 nm | 0.016 μmol min−1 | 0.1 μmol min−1 | 38 |
| Ag/WO3−x FTO | Solvent evaporation method | 0.6 mmol Ag | 300 W Xe > 750 nm | — | 0.18 μmol min−1 | 94 |
| Rh/WO3−x | Solvothermal synthesis | 1.2 wt% Rh | 300 W Xe > 400 nm | TOF = 500 molH2 molRh−1 min−1 | TOF = 800 molH2 molRh−1 min−1 | 95 |
| WO3−x/Ni2P | Glovebox and Schlenk technique | Mass ratio WO3−x:Ni2P = 4 |
300 W Xe > 400 nm | — | 1.38 × 105 μmol g−1 h−1 | 96 |
| W18O49/C | Solvothermal synthesis | Mass ratio W(CO)6:C fibre = 2.5 |
300 W Xe > 750 nm | — | 0.04 μmol min−1 at λ = 1500 nm | 97 |
| NaYF4:Yb–Er@W18O49 | Solvothermal synthesis | Colloidal | Laser diode 980 nm | — | 0.035 μmol min−1 at λ = 980 nm | 98 |
| TiO2:Er-Yb@W18O49 | Solvothermal synthesis | Colloidal | Laser diode 980 nm | — | 0.02 μmol min−1 at λ = 980 nm | 99 |
The first report on the surfactant-free synthesis of WO3−x nanowires for the LSPR assisted hydrogen evolution from AB was investigated by Xue et al.93 Blue-coloured WO3−x nanowires with strong absorption in the Vis-NIR region due to the presence of abundant oxygen vacancies were synthesised by solvothermal treatment of the W(CO)6 precursor in ethanol at 160 °C for 12 h as shown in Fig. 13(a). The colour and the absorption peak disappeared upon treatment with H2O2 due to the passivation of oxygen vacancies (Fig. 13(a)). The XRD pattern of the obtained nanowires was indexed as a monoclinic crystal structure as shown in Fig. 13(b). The HR-TEM image displayed nanowires of lengths ranging from several hundred nanometres to a few micrometres and diameters of about 10 nm. The growth of nanowires along the 〈010〉 direction was confirmed by the lattice fringes with an interplanar d-spacing of 0.376 nm. The effect of the reaction temperature in the plasmonic absorption of nanowires was studied by performing synthesis at 120, 140 and 160 °C. A much weaker plasmonic absorption for the sample prepared at 120 and 140 °C in comparison to 160 °C indicates the significance of the reaction temperature for the creation of oxygen vacancies. Quantitative analysis of the W4f XPS spectra shown in Fig. 13(c and d) revealed the presence of two pairs of peaks (W6+ and W5+ with 26% and 74% content) in WO3−x in comparison to one set of peaks in standard WO3. The actual formula was estimated to be WO2.87 with an x value of 0.13 in WO3−x.
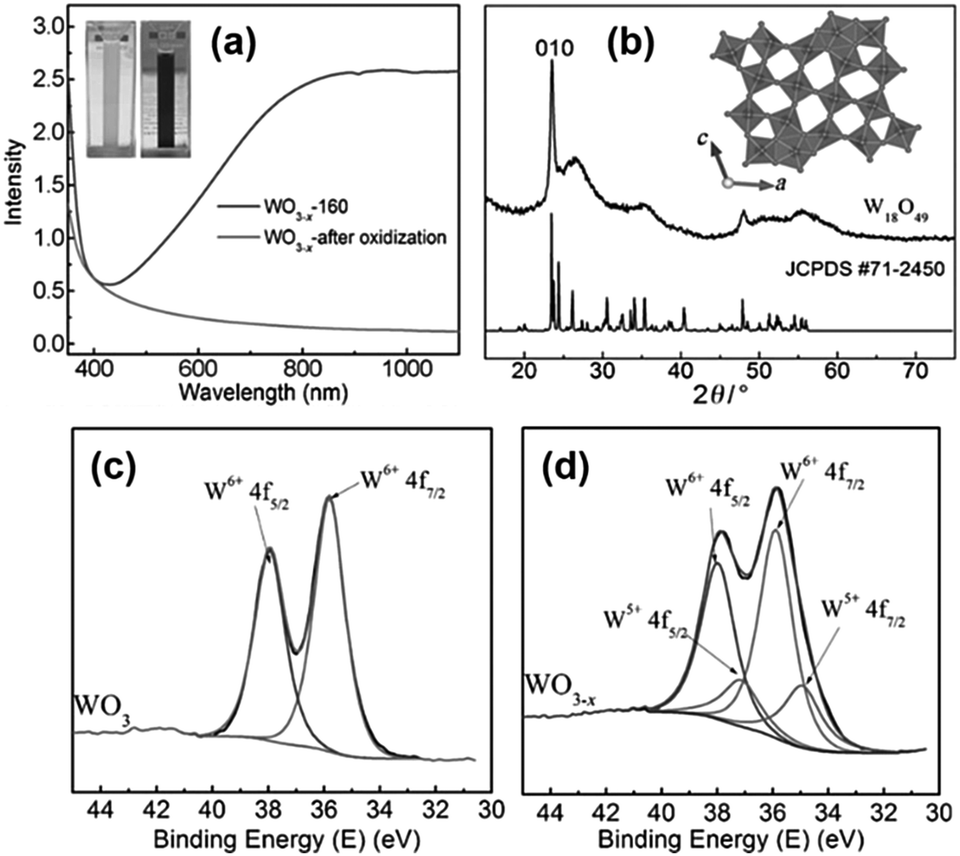 | ||
| Fig. 13 (a) UV-vis absorption spectra, (b) XRD pattern of plasmonic W18O49 and standard oxide and (c and d) W 4f XPS spectra of WO3 and WO3−x. Reproduced with permission from ref. 93 Copyright 2015 Wiley-VCH. | ||
The catalytic performance of WO3−x nanowires was compared with commercial WO3 in the dark and under visible light irradiation conditions (λ > 420 nm). As expected, no hydrolysis took place in the absence of catalysts. The superior catalytic performance of WO3−x (15.8 μmol h−1) compared with WO3 (9.5 μmol h−1) in the dark was attributed to the presence of more active sites in WO3−x nanowires. Under visible light irradiation (λ > 420 nm), a five-fold enhancement over WO3−x nanowires (79.5 μmol h−1) and 1.6 times enhancement over commercial WO3 (14.8 μmol h−1) were achieved. A significant enhancement over WO3−x nanowires was attributed to the wide LSPR absorption in the visible and NIR regions from 500 to 1500 nm. The mechanistic pathways under illumination conditions induce the generation of hot electrons for enhanced charge transfer and create localized heating effects due to the non-radiative decay of electronic oscillations as shown in Scheme 4.
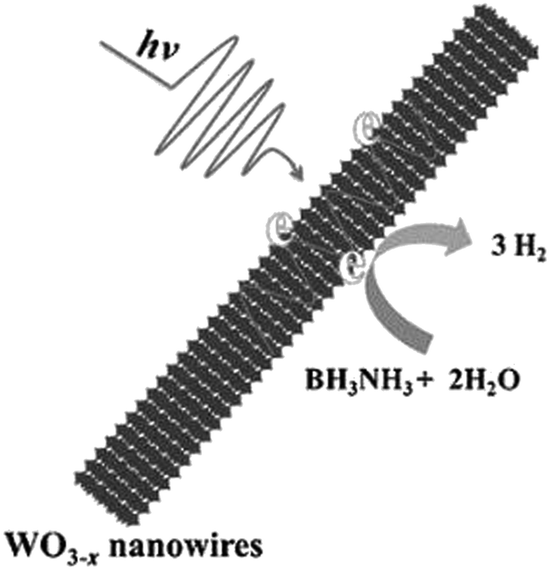 | ||
| Scheme 4 Schematic illustration of LSPR enhanced H2 generation from AB using WO3−x nanowires. Reproduced with permission from ref. 93 Copyright 2015 Wiley-VCH. | ||
The quasi-metallic behaviour of WO3−x displays a similar charge carrier density to the conventional plasmonic (Ag, Au) NPs. The optimization of strong light absorption in the NIR region can further be enhanced by integrating WO3−x with Au for higher photocatalytic efficiency. Wang et al. reported the design of a Au@WO3−x heterostructure with dual plasmon resonance for photocatalytic hydrogen generation from AB.38 The material was synthesised without using any surfactant and WO3−x nanowires were grown on the Au NP core by the solvothermal method. The obtained urchin-like Au@WO3−x core–shell nanostructure (sized 1 μm) in which the Au core NPs were of 40 nm was synthesised by a seed-mediated method, using HAuCl4 and citrate solution. The tungsten precursor (W(CO)6) was mixed with Au NPs in ethanol and was treated hydrothermally at 180 °C for 12 h.
The presence of ethanol partially reduced W6+ to W5+ which led to the creation of oxygen defects in WO3−x. The core–shell structure was distinguished based on the atomic ratio of Au and W in the nanostructure. For example, the formation of a compact structure when the atom ratio of W:Au was 10, denoted as Au@WO3−x (10) in which Au core NPs were completely enclosed by the WO3−x shell. On the other hand, when the atom ratio (W:Au) is equivalent to 2, the Au core structure is visible with a loosened WO3−x shell, denoted as Au@WO3−x (2). The plasmonic absorption of Au@WO3−x arising from Au NPs in the visible region showed a decrease in the intensity along with a blue shift upon increasing the W to Au atomic ratio. However, the absorption component arising from WO3−x in the NIR region displayed an increase in the peak intensity in Au@WO3−x (10) due to the energy transfer from Au NPs to WO3−x. The heterojunction creation was further confirmed by the shift towards the lower binding energy values in XPS analysis for Au@WO3−x compared to WO3−x. The Au@WO3−x (10) catalyst displayed superior catalytic performance in the AB dehydrogenation followed by Au@WO3−x (2), Au NPs and WO3−x. Under visible light irradiation (λ > 420 nm), all catalysts show enhancement following the trend similar to that in the dark. As discussed earlier, the bare WO3−x also displays catalytic activity in the dark (0.3 μmol h−1) and can show enhancement under visible light irradiation (1.3 μmol h−1) due to their strong light absorption in the vis-NIR region. Again, Au@WO3−x (10) displayed superior catalytic performance with an enhancement factor of 2.85 under visible light irradiation. Further, the wavelength dependence of catalytic activity was studied by using monochromatic light (λ = 475, 550, 600, 650, 700 and 780 nm) and the observed increase in hydrogen generation was found to be in accordance with the absorption spectra. A probable mechanism for the enhanced catalytic activity was proposed as follows. The hot electrons generated by the plasmon excitation of Au NPs under visible light irradiation get transferred to the CB of WO3−x and also the electron–hole pair generated in the CB and VB of semiconductor WO3−x simultaneously. The accumulated electrons finally assist in the H2 generation from AB. The presence of the Au core can drastically affect the hetero-morphology and plasmon of WO3−x for enhancing the photocatalytic efficiency. This work can provide a basic understanding of designing metal–semiconductor hybrids with dual plasmon resonances for hydrogen generation.
Synergistic catalysis, in combination with photo and thermochemical conversion, has developed as a promising approach to enhance the catalytic performance by solar technologies and LSPR effects. An IR-driven plasmonic coupling of metal/non-metal heterostructures was reported for the first time by Dong et al. The coupling of non-stoichiometric W18O49 nanostructures with Ag nanorice particles supported on fluorine-doped tin oxide (FTO) was investigated to enable an efficient charge transfer process during plasmonic excitation.94 Ag nanorice was synthesised by using polyvinyl pyrrolidone (PVP) as the surfactant, AgNO3 as the precursor and ethylene glycol as the reductant and solvent. The prepared Ag nanorice particles were assembled onto FTO glass loaded W18O49 nanowires (NWs) by a solvent evaporation method along with the photoreduction process (Fig. 14). Unlike spherical NPs, the strong LSPR absorption of nanorice particles in the infrared region has potential applications in sensor technology and in efficient catalytic conversions. The TEM image displayed the exfoliation of Ag/W18O49 heterostructures from the FTO surface with different contact types and angles. The UV spectra of the heterostructure film displayed two absorption bands at 400 and 1250 nm originating from the transverse and longitudinal modes of Ag, respectively. Moreover, the intensity of absorption was much stronger than that of the individual Ag nanorice or W18O49 NWs indicating the synergistic plasmon coupling between the two components. 3D simulations were performed using a finite element method (FEM) and the predicted maximum enhancement factor (|E|2/|E0|2) of the electric field intensity was 5.15 × 104 for tip-on-side arrangement at an angle of 90° under an excitation wavelength of 1400 nm. The photocatalytic hydrolysis of AB under IR light irradiation exhibited a superior hydrogen evolution rate of 0.18 μmol min−1 for the Ag/W18O49 film in comparison to the W18O49 (0.02 μmol min−1) and Ag nanorice films (0.022 μmol min−1). Much lower catalytic activities were observed under visible light irradiation indicating IR-driven strong coupling for boosting the hydrogen generation from AB. Further, the photothermal effect was studied by using thermal imaging techniques to monitor the temperature of the reaction solution under IR light irradiation. A significant increase in the located temperature of the Ag/W18O49 film from 22 to 98 °C confirmed the existence of a plasmonic-coupling-improved photothermal effect for enhancing the catalytic performance. The contribution of hot electrons was also studied by fixing the temperature of the reaction solution at 15 °C and the obtained catalytic activity under IR light irradiation was 0.048 μmol min−1. An average value of apparent quantum efficiency (AQE) of 4.03% was achieved using the heterostructure film under 1250 nm excitation. Fig. 15(A) shows the transient absorption (TA) spectra for the Ag/W18O49 heterostructure to detect the presence of charge carriers under an excitation wavelength of 400 nm. The blue-shifted TA signal in comparison to the bare Ag nanorice and W18O49 NWs confirms the existence of photoexcited charge transfer. The time constant (τ) obtained from the kinetic curves shown in Fig. 15(B and C) explains the relaxation process of hot electrons from an excited state to the ground state. Interestingly, the separation of hot electrons in the heterostructure film was confirmed by performing an IR-excited photoreduction of Pt4+ ions on the surface of Ag/W18O49. The deposition of Pt0 NPs upon SPR excitation at the interface of Ag and W18O49 is shown in the TEM image in Fig. 15(E). The mechanistic pathway of AB hydrolysis is explained by a schematic illustration shown in Fig. 15(D). Under IR light irradiation, the plasmonic coupling of Ag/W18O49 induces a localized electric field and generates hot electrons at the interface. These hot electrons get accumulated on Ag and facilitate the adsorption of the AB molecule. The adsorbed AB undergoes charge polarization and forms an activated transition state to ultimately release hydrogen by interacting with ·OH radicals from water molecules. The heterostructure film catalyst was successfully reused up to three catalytic runs without any loss of activity. Such types of metal/non-metal heterostructure films offer enormous potential for their applications in photoelectronic devices with ultra-broad photoabsorption.
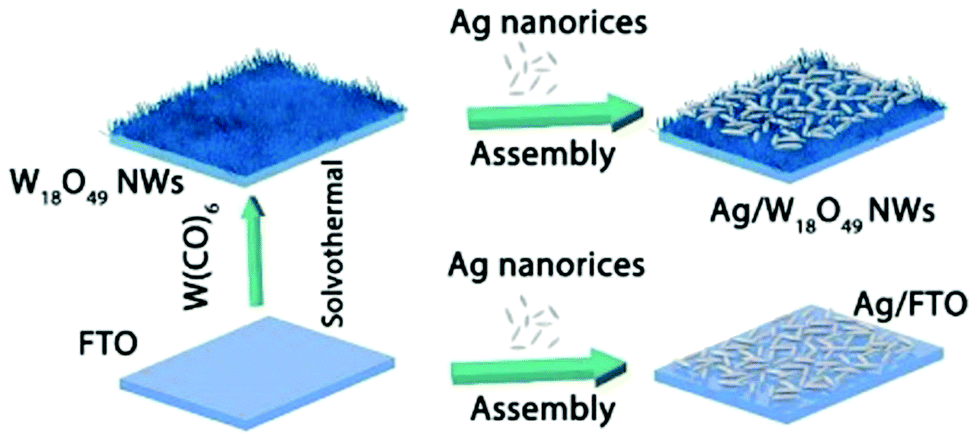 | ||
| Fig. 14 Fabrication process of Ag nanorice, W18O49 NWs and Ag/W18O49 heterostructure films. Reproduced with permission from ref. 94 Copyright 2019 Elsevier. | ||
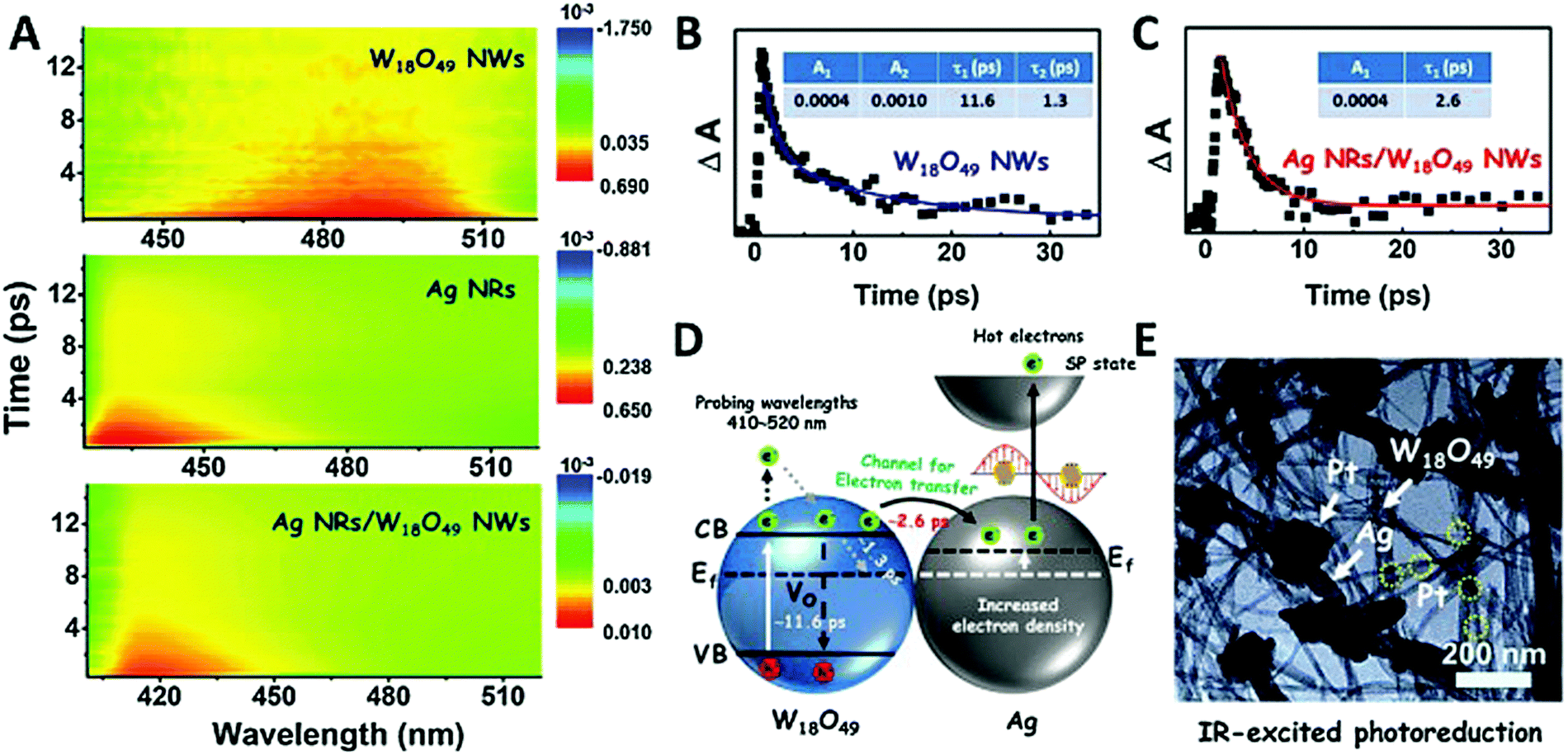 | ||
| Fig. 15 (A) Ultrafast transient absorption (TA) spectra of W18O49 NWs, Ag NRs, and Ag/W18O49 heterostructures; TA kinetics curves of (B) W18O49 NWs and (C) Ag/W18O49 heterostructures with a probing wavelength at 480 nm; (D) schematic of the kinetics process of the interfacial electron transfer in the Ag/W18O49 heterostructures; (E) TEM image of the Pt nanoparticle-loaded Ag/W18O49 heterostructures fabricated via an IR-excited photoreduction reaction. Reproduced with permission from ref. 94 Copyright 2019 Elsevier. | ||
Among all metal-based catalysts, Rh has been reported to be the most active catalyst for AB dehydrogenation. However, owing to its high cost and scarce abundance much more effort is required to enhance its catalytic properties and utilization efficiency. Zhang et al. reported a one-pot synthesis to prepare Rh/WO3−x nanocatalysts using Rh2(OAc)4 and W(CO)6 as precursors in benzyl alcohol suspension followed by solvothermal treatment at 200 °C for 12 h.95 The ultra-small Rh NPs (sized 1.7 nm) were anchored on WO3−x nanowires with an approximate loading of 1.2 wt% as determined by ICP-AES measurements. XPS analysis confirmed the increase in the electron density of Rh by displaying a negative shift in the binding energy values of the Rh 3d spectra. Further, the W 4f spectra indicated the non-stoichiometric nature by showing two different oxidation states of W (W5+ and W6+) in WO3−x (WO2.72). WO3−x and Rh/WO3−x also displayed strong absorption in the UV and Vis-NIR regions due to the presence of oxygen vacancies and LSPR associated with WO3−x nanowires. This distinct absorption possibly assisted in enhancing the catalytic performance under light irradiation conditions. The catalytic activities were evaluated for Rh/WO3−x in comparison with the bare WO3−x, Rh/C and the physical mixture of Rh + WO3−x catalysts for AB dehydrogenation at 298 K. Rh/WO3−x displayed superior catalytic performance with TOF values of 500 and 805 molH2 molRh−1 min−1 in the dark and under visible light irradiation (λ > 400 nm), respectively. However, the authors did not observe any catalytic activity on bare WO3−x in the dark and under visible light irradiation. The lower activation energy, Ea, values of 45.2 kJ mol−1 under visible light irradiation compared to in the dark (50.5 kJ mol−1) suggest a lower energy barrier from their corresponding Arrhenius plots. Fig. 16 illustrates the mechanistic pathway under visible light irradiation in which the photogenerated hot electrons of WO3−x get quickly transferred to the Rh NPs via an interface between them.
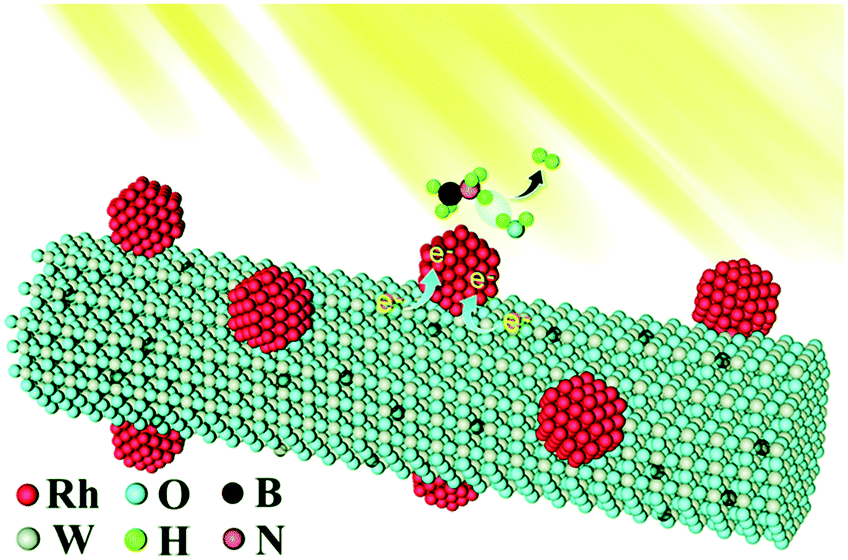 | ||
| Fig. 16 Schematic illustration of the Rh/WO3−x nanowire catalyst for hydrogen generation from AB under visible light irradiation. Reproduced with permission from ref. 95 Copyright 2019 The Royal Society of Chemistry. | ||
The presence of oxygen vacancies created new energy levels in the WO3−x nanowires and Rh NPs act as an electron sink to reduce the recombination rate of photoexcited hot electrons and hence facilitated hydrogen evolution from AB. The reusability tests could be carried out up to five cycles without any significant loss in catalytic activity. This work summarises an interesting strategy to prepare highly efficient reusable heterogeneous photocatalysts for AB dehydrogenation.
To date, various noble metal nanostructures in combination with WO3−x have been discussed. Lou et al. reported a novel combination of non-noble Ni2P NPs in combination with WO3−x nanorods as a novel strategy to develop plasmonic catalysts.96 The controlled nanostructure of WO3−x ultra-thin nanorods exhibits oxygen vacancies and assists in the transfer of charge carriers. Ni2P is a low-cost and more widely available catalyst, which in combination with WO3−x nanorods displayed ten-times enhanced hydrogen generation than with bare Ni2P NPs. The catalytic performance of plasmonic WO3−x (923 μmol g−1 h−1) was observed to be much higher than that of commercial WO3 (23.8 μmol g−1 h−1) but one order lower than that of WO3−x–Ni2P nanohybrids. The TEM image revealed that Ni2P NPs were closely attached and sandwiched between the layers of WO3−x nanorods in the hybrid catalyst. The X-ray diffraction pattern demonstrated the preservation of the crystalline structure of WO3−x even after higher mass loading of Ni2P NPs. The absorption spectrum of the WO3−x/Ni2P nanohybrid catalyst displayed wide absorption in the Vis-NIR region but with lower intensities than the bare WO3−x due to the surface coverage by Ni2P NPs. WO3−x/Ni2P (mass ratio WO3−x:Ni2P = 4) demonstrated hydrogen evolution of 1.38 × 105 μmol g−1 h−1 which is 10–13 times more than the catalytic performance of Ni2P NPs under visible light irradiation. The catalyst also exhibited excellent stability even after 5 catalytic cycles without any loss of catalytic activity. The mechanistic pathway of the nanohybrid catalyst was elucidated by adding charge scavengers for electrons (NaNO3), holes (NaHCO3) and hydroxyl radicals (2-propanol) in the reaction mixture. A significant decrease in the catalytic activity with the electron scavenger in comparison to other charge carriers was observed, indicating the contribution of photogenerated electrons in the AB dehydrogenation. According to the proposed mechanism, the hot electrons are generated by the LSPR excitation of WO3−x nanorods and facilitate the electron transfer to Ni2P NPs as shown in Fig. 17. The Ni2P NPs act as an electron trapping site and the hydrolysis of AB takes place on its surface. The holes and hydroxyl radicals also participate in the reaction with water to enhance the hydrogen evolution. The nanohybrid catalyst developed in this report presents a strategy to develop new plasmonic catalysts for the efficient harvesting of solar energy. The unique noble-metal-free hybrid catalyst can further be tested for diverse and challenging applications such as water splitting or CO2 reduction.
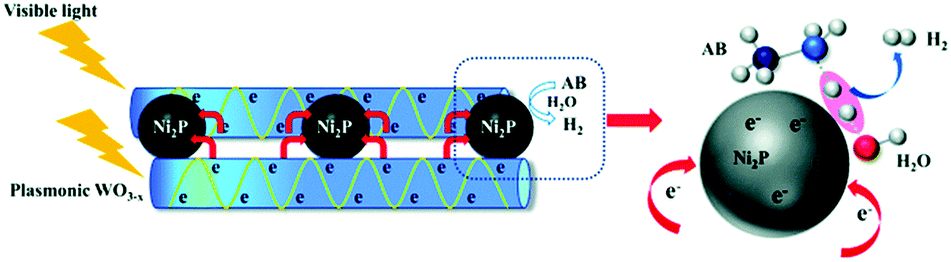 | ||
| Fig. 17 Schematic illustration of the WO3−x/Ni2P nanohybrid catalyst for hydrogen evolution. Reproduced with permission from ref. 96 Copyright 2018 The Royal Society of Chemistry. | ||
Another approach of designing noble-metal-free photocatalysts was attempted by Zhang et al. who constructed a W18O49/C heterostructure by directly growing W18O49 onto carbon fibres in three simple steps.97 The use of 1-D carbon fibres provides abundant active sites due to the presence of surface functional groups like hydroxyl, carboxyl and carbonyls along with a fine conductivity of 0.003–0.007 Ω cm which can boost the charge separation efficiency. At first, polyvinylpyrrolidone (PVP) fibres were synthesised using electrospinning techniques followed by the carbonisation of as-synthesised fibres at 900 °C under a N2 atmosphere. Finally, the growth of plasmonic W18O49 nanowires was carried out on carbon fibres through solvothermal synthesis creating a heterostructure. The Raman and Fourier transform infrared (FT-IR) spectrum exhibited characteristics peaks of carbon fibres and W18O49 nanowires indicating the successful assembly of both the components in the W18O49/C heterostructure. X-ray photoelectron spectroscopy (XPS) revealed the presence of two oxidation states of W including W5+ and W6+, hence confirming the formation of non-stoichiometric plasmonic tungsten oxide nanowires. Fig. 18(A) displays an intense and broad absorption peak in the Vis-NIR region extending up to 2400 nm due to the collective oscillations of excess electrons generated by oxygen vacancies. The hydrogen evolution on W18O49/C heterostructures was studied at a fixed temperature of 15 °C using a reflux condenser to weaken the contribution arising from the photothermal effect. A 1.75-fold rate enhancement in the hydrogen generation was observed for W18O49/C (0.14 μmol min−1) in comparison to W18O49 (0.08 μmol min−1) under IR-light irradiation (λ > 750 nm) as shown in Fig. 18(B). The carbon fibres did not produce any hydrogen even under IR light irradiation but their presence in the heterostructure enhanced the overall catalytic activity. This observation indicates that carbon fibres act as an electron sink to receive electrons generated by plasmonic W18O49 and to enhance the charge separation and the hydrogen production activity.
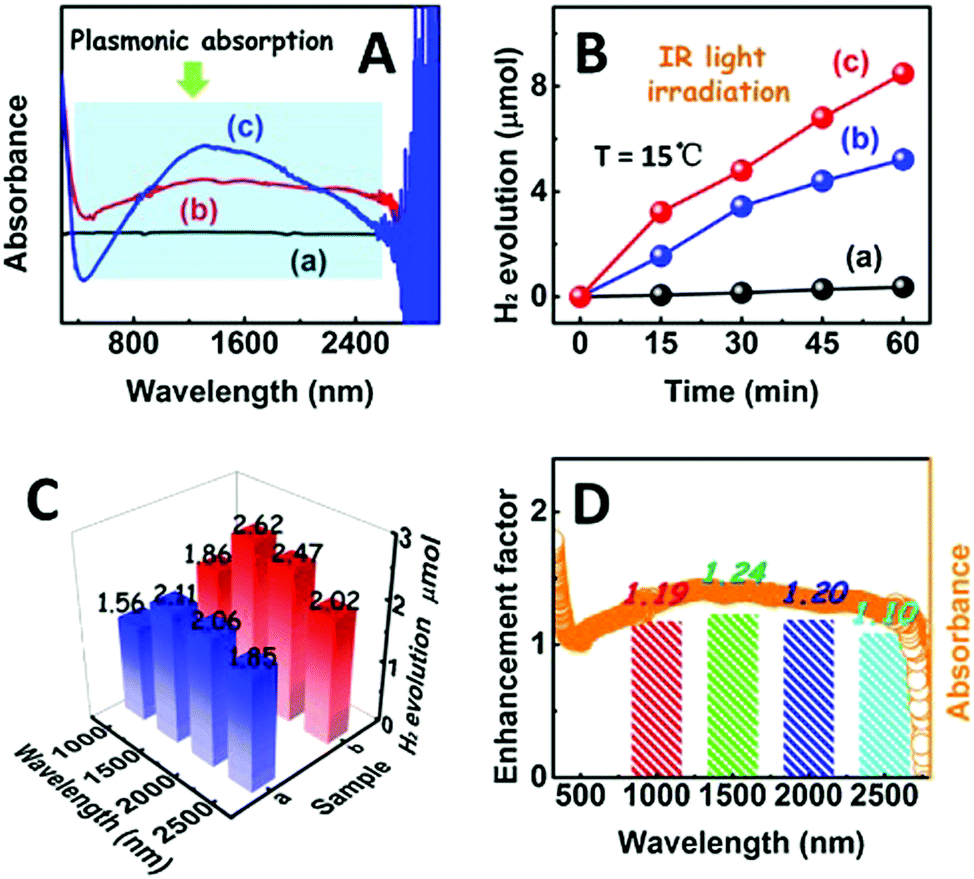 | ||
| Fig. 18 (A) UV-vis-IR absorption spectra of the as-fabricated (a) carbon fibres; (b) W18O49/C heterostructures; (c) W18O49 nanowires; (B) time-dependent H2 production from NH3BH3 solution at 15 °C over different samples upon IR-light irradiation: (a) carbon fibres; (b) W18O49 nanowires; (c) W18O49/C heterostructures; (C) H2 production amount from NH3BH3 solution at 15 °C over (a) W18O49 nanowires and (b) W18O49/C heterostructures under irradiation at the indicated wavelengths for 1 h; (D) wavelength-dependent enhancement factors and plasmonic absorption of W18O49/C heterostructures. Reproduced with permission from ref. 97 Copyright 2018 Elsevier. | ||
Further, the wavelength-dependent catalytic activity under monochromatic light irradiation of λ = 1000, 1500, 2000 and 2500 nm was studied. A maximum catalytic performance of 2.62 μmol h−1 was observed at an incident photon energy of 1500 nm which also corresponds to the absorption maxima in the Vis-NIR spectrum (Fig. 18(C)). The enhancement factor (ratio of H2 evolution over W18O49/C and W18O49 nanowires) was also consistent with the absorption spectrum, as shown in Fig. 18(D). This proves that the photocatalytic enhanced activity was induced by the LSPR effect of plasmonic tungsten oxide. The in-depth understanding of the charge separation was also studied by finite-difference-time-domain (FDTD) simulations and ultrafast transient absorption (TA) spectroscopy which proved the existence of IR generated hot electrons in W18O49 and the electron mediator nature of carbon fibres. It was revealed that the timescale of hot electron transfer from W18O49 nanowires to carbon fibres was within 50 fs which is much shorter than the relaxation time of hot electrons generated by surface plasmon in W18O49 (5.5 ps). Hence, enhanced charge separation was greatly promoted leading to higher hydrogen production from AB. This work can be considered as an outstanding example of an efficient IR-driven plasmonic catalytic system combined with low-cost carbon heterostructures.
A new class of low-cost NIR responsive hierarchical heterostructure was reported by Dong et al. in which a quasi-core shell heterostructure consisting of plasmonic WO3−x and Ln3+-doped NaYF4 NPs was developed for hydrogen evolution from AB.98 The unique combination of the two components is because of their significant overlapping of LSPR absorption arising from W18O49 with the excitation and emission spectrum of NaYF4:Yb–Er NPs to induce a significant enhancement in the NIR-plasmonic energy. The spherical catalyst with a sea-urchin like morphology was synthesised by the solvothermal growth of WO3−x nanowires onto NaYF4:Yb–Er NPs to form a core–shell NaYF4:Yb–Er@W18O49 nanostructure. The XRD patterns displayed the cubic and monoclinic structures of NaYF4 and W18O49, respectively, along with an intense LSPR absorption in the Vis-NIR region. The rate of hydrogen evolution for NaYF4:Yb–Er@W18O49 under irradiation of 980 nm diode was 2.11 μmol h−1 which was 3.5 times higher than that of the bare W18O49 nanowires (0.60 μmol h−1) under similar reaction conditions. Such a unique combination of Ln3+-doped NaYF4 and WO3−x may further lead to developing NIR-responsive catalysts for applications in green and sustainable energy sources.
The same research group has also very recently reported Ln3+-doped TiO2@W18O49 quasi-core/shell nanostructures prepared via a facile solvothermal synthesis procedure.99 The quasi-core/shell TiO2:Er–Yb@W18O49 exhibited a photocatalytic hydrogen evolution rate of 1.1 μmol h−1 which was 2.5 times higher than that of the WO3−x nanowires (0.4 μmol h−1). The creation of a heterojunction and the transfer of hot electrons between TiO2:Er–Yb nanosheets and W18O49 nanowires are responsible for the enhanced hydrogen evolution from AB. Such types of novel photocatalyst materials can show great potential in solving the increasing energy demand issues.
3.2. Plasmonic MoO3−x hybrid nanocatalysts
MoO3 is an n-type semiconductor with photochromic and electrochromic properties. The layered structure allows the intercalation of small ions to form molybdenum bronze structures which display intense colour and semiconducting properties. MoO3−x displays intense blue colour due to the presence of oxygen vacancies. Table 4 summarises the MoO3−x-based catalytic systems discussed in this section along with their reaction rates for AB dehydrogenation.| Catalysts | Synthesis method | Metal loadings | Light source | Catalytic activity | Ref. | |
|---|---|---|---|---|---|---|
| Dark | Light | |||||
| MoO3−x | Solvothermal synthesis | Colloidal | 500 W Xe > 420 nm | 0.85 μmol min−1 | 3.44 μmol min−1 | 101 |
| Nanoflower MoO3−x-F | Solvothermal synthesis | Colloidal | 300 W Xe > 420 nm | 2.26 μmol min−1 | 6.0 μmol min−1 | 102 |
| MoO3−x-50 | Aqueous phase at RT | Colloidal | 500 W Xe > 420 nm | 2.10 μmol min−1 | 2.49 μmol min−1 | 103 |
| MoO3−x-200 | Evaporation induced self-induced process | Colloidal | 500 W Xe > 420 nm | 2.21 μmol min−1 | 3.88 μmol min−1 | 104 |
| Pd/MoO3−x | Impregnation + NaBH4 reduction | 3.0 wt% Pd | 500 W Xe > 420 nm | 0.5 mL min−1 | 2.0 mL min−1 | 105 |
| Er–Yb:TiO2/MoO3−x | Solvothermal synthesis + ultrasonic dispersion | Colloidal | Laser diode 980 nm | — | 0.13 μmol min−1 | 106 |
An initial report to understand the synthetic strategy and tunable plasmonic behaviour of MoO3−x hybrids was reported by Wang et al. in 2012. The MoO3−x–oleylamine hybrids can transform from blue nanosheets to white nanotubes by oxidation.100 This change takes place due to the oxidation of Mo5+ ions to Mo6+ as revealed by spectroscopic measurements. The absorption spectra and TEM image of blue nanosheets and white nanotubes are shown in Fig. 19(a–c). The strong ESR signal at g = 1.92 confirms the presence of a single electron which indicates the existence of Mo5+ species with a d1 configuration, as shown in Fig. 19(d). XPS analysis also displayed the absence of Mo5+ species in white nanosheets as depicted in Fig. 19(e and f). During this oxidation process, a tuneable LSPR can be observed upon a change in the morphology from blue nanosheets to white nanotubes by converting Mo5+ to Mo6+. Further, the intensity of LSPR absorption gradually decreases on air oxidation at room temperature, hence displaying the tuneable LSPR properties of MoO3−x. The MoO3−x nanosheets were used to study the photothermal effects using a 980 nm laser with a power density of 1.4 W cm−2. The temperature increment from 25.2 to 52.2 °C within 15 min displayed the obvious photothermal effects of the nanosheets which can find applications in tumour therapy.
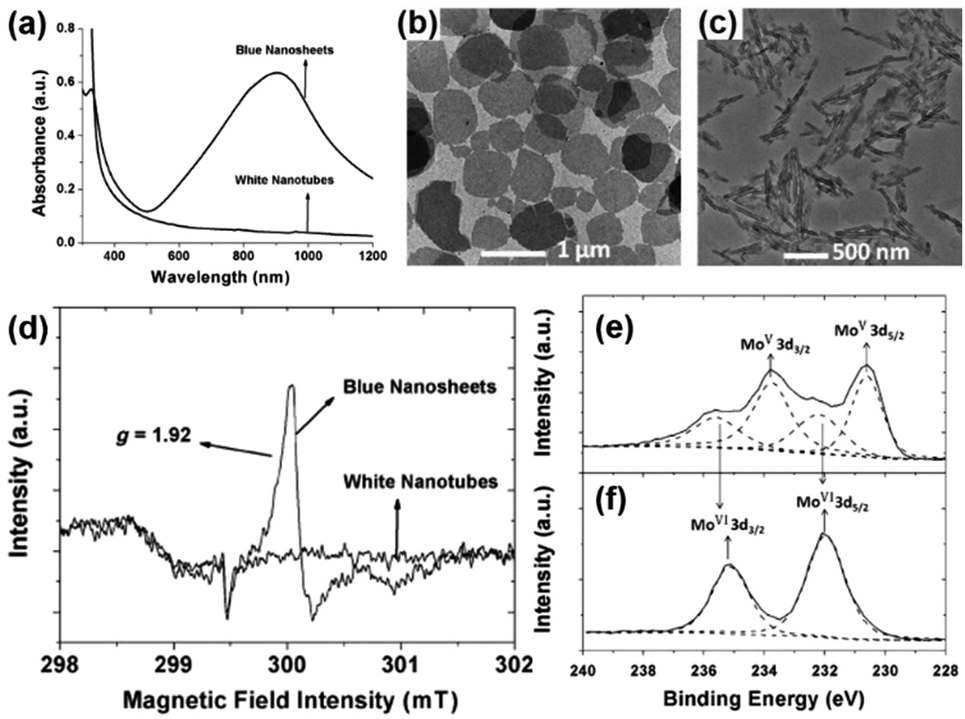 | ||
| Fig. 19 (a) UV-vis-NIR absorption spectra of MoO3−x nanosheets and MoO3−x nanotubes; (b) TEM image of ultrathin MoO3−x nanosheets; (c) TEM image of white multiwalled nanotubes; (d) electron spin resonance (ESR) analysis of the blue nanosheets and white nanotubes dispersed in cyclohexane at liquid nitrogen temperature; (e) Mo 3d X-ray photoelectron spectroscopy (XPS) spectra of blue nanosheets and (f) white nanotubes. Reproduced with permission from ref. 100 Copyright 2012 Wiley-VCH. | ||
The pioneering work on exploring the plasmonic catalysis over MoO3−x nanosheets was demonstrated by Cheng et al. The surfactant-free synthesis of nanosheets for hydrogen generation from AB under visible light irradiation was investigated.101 A variety of different surfactants such as oleylamine and trioctylphosphine oxide had previously been used during synthesis to allow morphological control of such nanostructures; however, their presence adversely affected the catalytic performances. The plasmonic nanosheets were synthesised by reacting molybdenum metal powder with H2O2 followed by solvothermal treatment in ethanol at 160 °C for 12 h. The obtained blue coloured nanosheets display an intense LSPR peak with an absorption maximum at 680 nm. On decreasing the synthesis temperature to 140 °C, a less intense absorption peak red-shifted to 950 nm was obtained with smaller sized MoO3−x nanosheets. The role of ethanol in synthesis is to not only act as a solvent but also as a reducing agent to generate oxygen vacancies in the semiconductor oxide. The Mo 3d XPS spectra exhibited that both Mo6+ and Mo5+ exist in MoO3−x, whereas only Mo6+ was present in MoO3. Quantitative analysis revealed the average oxidation state of 5.23 in MoO3−x which was found to be in accordance with the valence state determined by the energy shift of the XANES spectra.
The catalytic performance in the hydrogen evolution from AB was reported over plasmonic MoO3−x and commercial MoO3 in the dark and under light irradiation (λ > 420 nm) conditions. The initial rate of catalysis for MoO3−x was found to be 1.43 mol% min−1 which was about 3-times faster than the MoO3 sample (0.54 mol% min−1) in the dark. Under visible light irradiation, the rate of hydrogen evolution over MoO3−x was 5.74 mol% min−1 which was 3.7 times higher than that of the MoO3 sample (1.55 mol% min−1). Furthermore, the enhancement comparison in the dark was approximately four times over the MoO3−x nanocatalyst. An intense visible light LSPR absorption and a facile surfactant-free synthesis can receive increasing attention in various applications such as bioimaging, sensors and photovoltaics.
There have been several reports investigating the morphology dependent catalytic behaviour of noble metal NPs; however, tuning the morphology of semiconductor heterostructures for establishing the structure–property relationship is rare. Gong et al. recently reported the morphology dependent catalytic activity of MoO3−x (flower (F), schistose (S) and nanorod (R)) for AB dehydrogenation in the dark and under visible light irradiation.102 The morphology was controlled by varying the reaction temperature and the solvent composition during the solvothermal treatment of Mo(VI) oxide bis(2,4-pentanedionate) in an acetic acid/water mixture at 180–220 °C for 20 h reaction time. The obtained plasmonic MoO3−x-S, MoO3−x-F and MoO3−x-R had surface area values of 20.08, 9.83 and 26.16 m2 g−1, respectively. XPS analysis revealed the presence of two pairs of peaks due to the presence of Mo5+ and Mo6+ cations and the ratio of Mo5+ was determined to be 8.84%, 6.96% and 3.95% for MoO3−x-S, MoO3−x-F and MoO3−x-R, respectively. Interestingly, the trend in the intensity of UV-vis absorption (MoO3−x-S > MoO3−x-F > MoO3−x-R) was in accordance with the mass ratio of Mo5+ in the catalysts as determined from XPS. MoO3−x-F exhibited superior catalytic performance in the AB dehydrogenation followed by MoO3−x-R and MoO3−x-S. This trend was due to the synergistic balance between the surface area and the LSPR absorption intensity in MoO3−x-F. A three-fold enhancement in the hydrogen evolution over MoO3−x-F was observed under visible light irradiation with a reaction rate of ∼6.0 μmol min−1 in comparison to that in the dark (2.26 μmol min−1). The catalyst could be successfully recycled up to 3 catalytic runs and its structural morphology was preserved as confirmed by XRD. This study summarises the effect of using morphologically controlled plasmonic MoO3−x for the efficient photocatalytic hydrogen evolution from AB.
Most reports envisaged to date have employed solvothermal treatment within the temperature range of 100–200 °C to prepare semiconductor-based plasmonic nanostructures. For the first time, Shi et al. reported the room-temperature aqueous phase synthesis of a plasmonic MoO3−x-Y nanocatalyst (Y denotes the molar ratio of Mo6+/Mo5+) by a simple stirring of varying compositions of Mo precursors, (NH4)6Mo7O24·4H2O and MoCl5, for 48 h.103 The low-cost and easy operation synthetic strategy can be useful for practical applications related to solar energy utilization. The deep blue coloured MoO3−x-50 exhibits an intense LSPR absorption maxima at 682 nm in the UV-vis spectra. However, unlike previous reports, the MoO3−x synthesised in this report was amorphous and disordered. XPS confirmed the presence of Mo6+ and Mo5+ cations in MoO3−x-50 accounting for 60.1 and 39.9% of the total Mo atoms with an average oxidation state of 5.6. The initial rate of hydrogen evolution over MoO3 and MoO3−x-50 in the dark was found to be 0.74 and 2.10 μmol min−1, respectively. Under visible light irradiation (λ > 420 nm), the rate of hydrogen evolution can reach 2.49 μmol min−1 for MoO3−x-50 and 0.87 μmol min−1 for MoO3.
The mechanistic pathway of plasmon driven catalysis was further studied by performing experiments on the effect of wavelength dependence, photothermal effects and scavenger addition. The effect of wavelength dependence was studied by probing the catalytic reaction with several monochromatic light irradiation of wavelengths, λ = 440, 480 and 627 nm. The rate of hydrogen generation increased on increasing the wavelength of incident light irradiation. A maximum enhancement was observed with λ = 627 monochromatic light, which was found to be consistent with the optical absorption of the MoO3−x-50 catalyst, affirming the enhancement due to the LSPR absorption of MoO3−x. To study the influence of the photothermal effect on the hydrogen generation ability, catalysis was carried out at 40 °C in the dark. The rate of thermal catalysis was found to be slightly higher than that in the dark but much lower than the results obtained under visible light irradiation, hence confirming the critical role of LSPR excitation of the MoO3−x plasmonic catalyst. Further, the effect of a positive charge scavenger was studied by adding NaHCO3 during the catalytic reaction. It was speculated that HCO3− species would adsorb on the surface of the catalyst and react with the positive charges generated during plasmonic excitation. The hydrogen generation rate was significantly reduced under visible light irradiation than in the dark, suggesting that the charge separation and AB activation were drastically inhibited. This report was also the first example of room temperature synthesised plasmonic semiconductor catalysts tested in photocatalytic AB dehydrogenation.
In general, doped semiconductors display low specific surface area values which can be detrimental for catalytic reactions taking place on the exposed surface atoms. To overcome this shortcoming, Yin et al. reported the synthesis of crystalline high surface area plasmonic MoO3−x by an evaporation induced self-assembly (EISA) process in which F127 was used as an organic template followed by H2 reduction at 200 °C, denoted as MoO3−x-200 °C.104 The obtained nanocatalyst was compared with commercial MoO3 powder and previously reported plasmonic MoO3−x nanosheets synthesised by the solvothermal method. The specific surface area, as determined by N2 physisorption analysis, was found to be 30 m2 g−1 which is approximately 22 and 9 times higher than those of commercial MoO3 and MoO3−x nanosheets, respectively. The catalytic performance of MoO3−x-200 °C in the hydrogen evolution from AB was observed to be 2.21 and 3.88 μmol min−1 in the dark and under visible light irradiation (λ > 420 nm). The obtained catalytic performance was much higher than that of the commercial MoO3 in the dark (0.44 μmol min−1) and under visible light irradiation (0.92 μmol min−1) because of the intense LSPR absorption in the UV-vis spectra. The effective adsorption of AB molecules on MoO3−x-200 °C because of its higher surface area values leads to the enhanced catalytic activity compared to other plasmonic catalysts.
Single component plasmonic nanostructures are often restricted in heterogeneous catalytic applications because of weaker light absorption and poor catalytic efficiency. The multicomponent system can exhibit synergistic coupling effects; hence, it is important to explore the development of MoO3−x-based hybrid materials for improved solar harvesting. The catalytic activity of the bare MoO3−x was enhanced by coupling it with active metal (Pd) NPs.105 The synthesis procedure was modified by thermally decomposing the Mo precursor, (NH4)6Mo7O24·4H2O, to MoO3 orthorhombic plates. Subsequently, H2PdCl4 was impregnated onto MoO3 plates followed by NaBH4 reduction which reduces MoO3 and Pd2+ into MoO3−x and Pd0 to form a hybrid Pd/MoO3−x plasmonic catalyst as illustrated in Fig. 20. NaBH4 was used as the reducing agent because of the more positive reduction potential of PdCl42−/Pd0 (0.591 V vs. SHE) than H+/H2 (0 V vs. SHE) to deposit Pd NPs sized 10.3 nm on MoO3−x. The addition of polyvinylpyrrolidone (PVP) assisted in the synthesis of highly dispersed and much smaller NPs sized 3.3 nm as shown in Fig. 21(a). Fig. 21(b) displays the LSPR absorption of Pd/MoO3−x at 640 nm and a much stronger red-shifted absorption of Pd/MoO3−x (PVP) at 670 nm. MoO3 displays absorption only in the UV region for wavelengths less than 400 nm. The partial reduction of MoO3−x was further confirmed by XPS and XANES analysis using Mo foil and MoO3 as the reference materials. The rate of hydrogen evolution of Pd/MoO3−x was studied and compared with the MoO3−x nanosheets and Pd/SiO2 in the dark and under visible light irradiation (λ > 420 nm) as shown in Fig. 21(c and d). Pd/MoO3−x exhibited higher catalytic performances than MoO3−x nanosheets and Pd/SiO2 and hence revealed the synergistic effect between Pd NPs and the MoO3−x support. The hydrogen generation rate over Pd/MoO3−x was found to be 2.0 mL min−1 under visible light irradiation which is four-fold higher than that in the dark (0.5 mL min−1). The injection of hot electrons from LSPR-excited MoO3−x to the Pd NPs due to the interfacial electron transfer facilitates enhanced hydrogen evolution from AB. The catalysts also exhibited unique reversible tunability upon air oxidation and NaBH4 reduction processes.
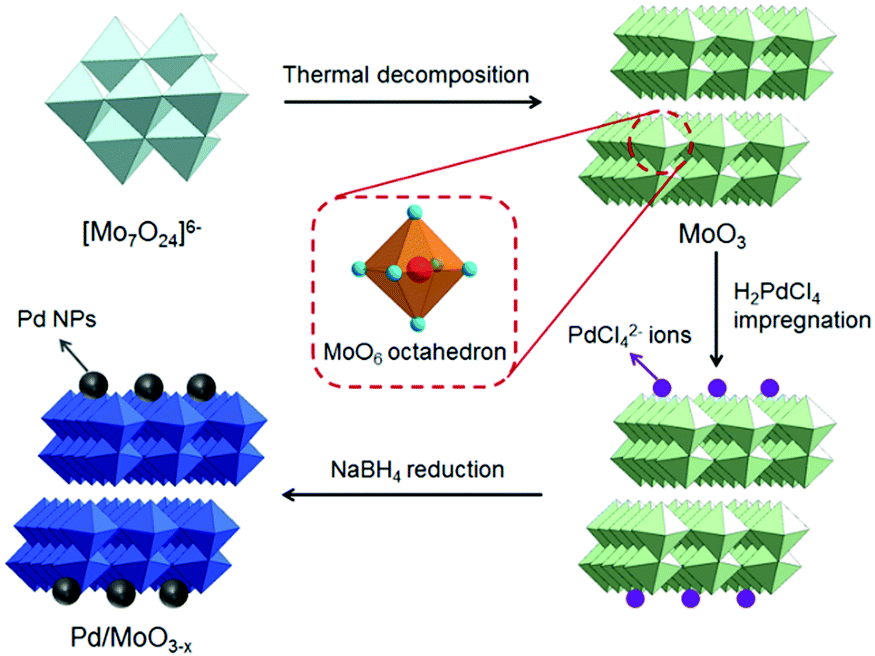 | ||
| Fig. 20 Schematic illustration of the synthesis process of the Pd/MoO3−x hybrid catalyst. Reproduced with permission from ref. 105 Copyright 2015 Wiley-VCH. | ||
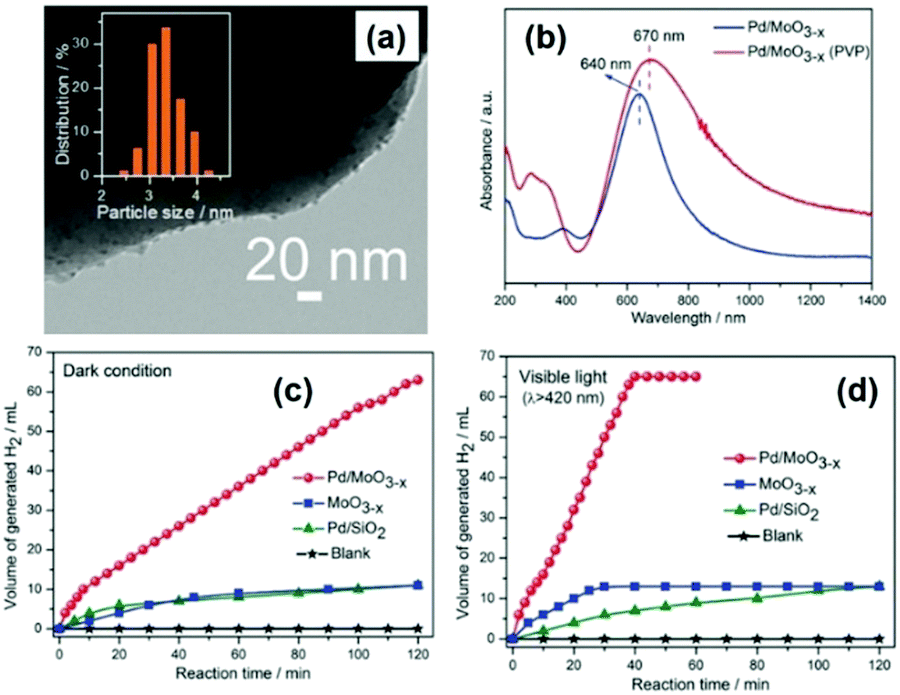 | ||
| Fig. 21 (a) TEM image of the Pd/MoO3−x hybrid and the (inset) particle size distribution of Pd NPs; (b) UV-vis-NIR diffuse reflectance spectra of the Pd/MoO3−x hybrid and pristine MoO3; (c) plots of evolved H2 gas as a function of reaction time from an aqueous AB solution (1 mmol in 10 mL water) hydrolysis over different catalysts in the dark and (d) under visible light irradiation (λ > 420 nm). Reproduced with permission from ref. 105 Copyright 2015 Wiley-VCH. | ||
Another interesting approach of preparing multi-component semiconductor heterostructures was reported by Shang et al. in 2019. The crystalline hybrid consisted of Er–Yb:TiO2 nanosheets on the surface of plasmonic MoO3−x nanosheets with a 2D contact with each other.106 The semiconductor hybrid exhibits an intense LSPR absorption in the visible and NIR regions as shown in Fig. 22(a). Under the excitation of 980 nm laser (power density = 7 W cm−2), the hydrogen evolution over Er–Yb:TiO2/MoO3−x was observed to be 6.7 μmol h−1 which is 1.5 times higher than that using the bare MoO3−x nanosheets (4.4 μmol h−1) as summarised in Fig. 22(b). To further exploit the larger absorption in the solar spectrum, simulated sunlight with a power density of 0.25 W cm−2 was used. The larger absorption of MoO3−x enhanced the hydrogen production activity to approximately 8.0 μmol h−1. Fig. 22(c) depicts a schematic illustration of hot electron transfer for hydrogen evolution from AB under visible light irradiation conditions. The proposed synthetic method assists in strengthening the LSPR absorption for highly efficient solar to chemical energy conversion.
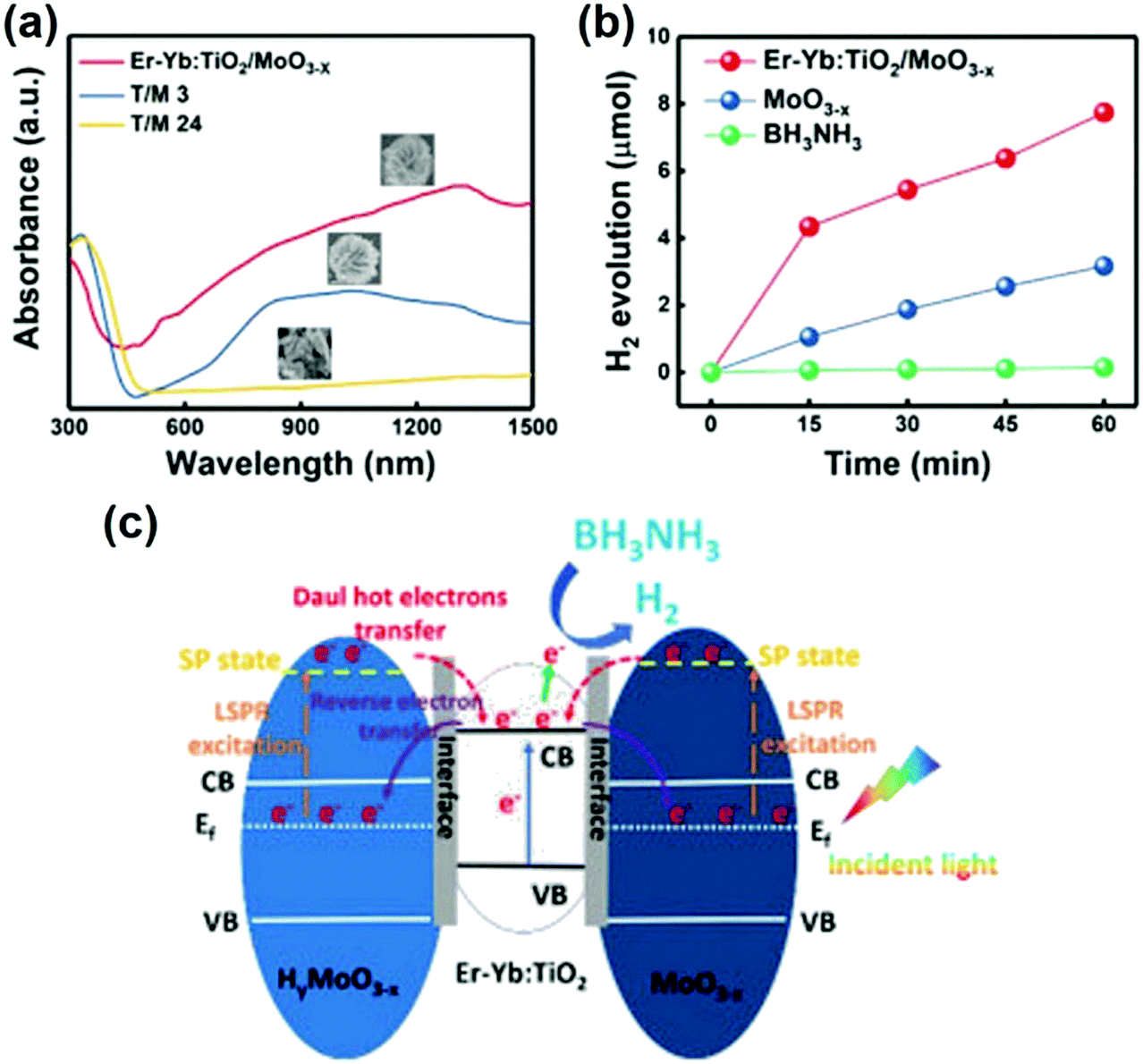 | ||
| Fig. 22 (a) Absorption spectra for Er–Yb:TiO2/MoO3−x NSH, T/M 3 and T/M 24; the insets show the corresponding morphology of each sample; (b) H2 yield of Er–Yb:TiO2/MoO3−x NSH, MoO3−x NTs and BH3NH3 under the excitation of simulated sunlight; (c) the schematic diagram of the hot electron transfer process for H2 generation from BH3NH3. Reproduced with permission from ref. 106 Copyright 2019 Elsevier. | ||
The nanocatalysts discussed in this section involved WO3−x and MoO3−x based plasmonic materials, prepared mainly by the solvothermal method of synthesis. WO3 and MoO3 that merely displayed absorption in the UV region responded strongly to the visible light in MoO3−x and WO3−x. The unique behaviour and strong LSPR absorption were attributed to the presence of oxygen vacancies, as evidenced by XPS spectroscopic analysis. The first report on plasmon-mediated catalysis was published in 2014 and since then many hybrid nanostructures have been discovered. Several interesting and creative approaches have been developed in the last five years to enhance the efficiency of these catalysts. For example, the unique combination with Ni2P, carbon, and Er–Yb led to the synthesis of highly active and visible light responsive catalysts.
3.3. Mechanism of plasmonic enhancement over doped semiconductors
In this section, the relevant mechanistic pathways of doped semiconducting plasmonic catalysts and their integration with plasmonic metals are discussed in order to have a better understanding of the plasmonic effect for efficient solar light harvesting. The main aspects of mechanisms including light absorption and scattering, charge carrier vacancy, photothermal, plasmon-induced resonance energy transfer (PIRET) and field enhancement effects have been elaborated.107–1104. Conclusions and perspective
Plasmon-mediated catalysis is a recently emerging field of heterogeneous catalysis as an alternative to the conventional semiconductor-based photocatalysis. The semiconductor photocatalysts mainly absorb UV light along with the partial absorption of visible light photons, whereas plasmonic photocatalysts absorb low energy photons in the visible and NIR regions to drive chemical reactions. The generation of hot electrons and localised heating effects owing to the LSPR effect of plasmonic nanostructures are responsible for efficient solar-to-chemical conversion reactions. In this review article, we have discussed significant improvements in heterogeneous catalysis by two main types of plasmonic nanostructures namely noble metal-based and semiconductor-based photocatalysts integrated with catalytically active nanostructures such as Pd, Rh, Ru, Co, Ni2P NPs, carbon heterostructures, C3N4 and metal–organic frameworks. The prepared heterostructures have been evaluated for their efficient catalysis in the hydrogen generation from AB in the dark and under visible-NIR irradiation. The first example of plasmon-mediated catalysis over noble metal (in 2013) and semiconductor-based (in 2014) nanostructures has been highlighted in the respective sections. Several research reports have employed the use of high-intensity specific wavelength lasers to excite the plasmons of metal nanostructures. Most research reports 100% conversion of AB with enhanced reaction rates under illumination conditions. However, it is difficult to compare the efficacy of the most active catalytic system because of varying metal content ratios, plasmon absorption intensities and the presence of dopants and defects. Interestingly, AB has not only been used as a solar fuel but also to generate in situ hydrogen for chemoselective hydrogenation reactions. The mechanism of enhancement by the generation of charge carriers and localised electric field effects has also been summarised.Significant research efforts in recent years have established AB as an excellent hydrogen storage material because of its high purity and enhanced rate of hydrogen generation. This stable molecule can undergo hydrolytic dehydrogenation in polar solvents in the presence of a suitable catalyst. However, there are still some challenges related to AB dehydrogenation. The regeneration of AB from its hydrolysis by-product, metaborate, is difficult and cost ineffective. There have been reports exploring the partial regeneration of AB from dehydrocoupling of by-products, the details of which are beyond the scope of this review article. Also, the formation of NH3 from NH4BO2 can poison the functioning of fuel cells. The use of solvent can also affect the gravimetric capacity of hydrogen. Despite the aforementioned challenges, the stability of AB in polar solvents and its fast release of high purity hydrogen makes it a promising and reliable candidate for next generation hydrogen storage materials. The current state-of-the-art indicates the use of AB for future technological advancements in various applications. It has been observed that bare supports such as insulators, SBA-15 and semiconductors like TiO2 and C3N4 are unable to boost the evolution of hydrogen from AB. However, their integration with active metals like Rh, Ru and Pd along with plasmonic metals such as Ag and Au proved to be an efficient strategy for enhancing the catalytic performance under visible light irradiation. The use of doped semiconductors was found to display higher enhancement effects due to their extended visible-NIR light absorption. Overall, it is difficult to compare the level of performances of most active catalytic systems because of the different ratios of active species, but, based on their ability of light-harvesting, we can conclude that the following, bare supports < noble-metal based hybrids < doped hybrid semiconductors, are capable of showing significant enhancements in hydrogen generation. The LSPR enhancement of nanostructures can be varied by controlling factors such as size, morphology, concentration of the free charge carriers and refractive index of the medium in order to efficiently harness the solar energy. Doped semiconductors can exhibit strong LSPR absorption in the Vis-NIR region even for small sized nanostructures, which is not usually shown by noble-metal based nanostructures. It is possible to design Ag, Au NPs which can display strong IR absorption (for example, nanorod, cubic or prism morphology) but at the expense of their high surface area values which can lead to a decrease in the overall catalytic performances. Other than the optical range of tunability offered by doped-semiconductors, the amount of doping levels can create a resonance between plasmonic excitation and band-gap transitions, which can drastically enhance the rate of charge carrier generation and hence the photocatalytic performance in hydrogen generation. Continuous research efforts and investigation are still needed to address the issues of cleaving the strong B–O bond for AB regeneration and preventing the formation of gaseous products to reduce its application cost making it viable for practical applications.
The direct utilisation of solar energy to produce fuels has many advantages but their efficiency is still very low for practical applications. Significant efforts are required to enhance the efficiency of more challenging and practical photochemical applications such as water splitting and CO2 reduction. One of the ways to achieve more practical artificial photosynthesis is to develop co-catalyst materials for plasmonic nanostructures using non-precious metals such as MoS2, graphene, and Zn or Sn-based materials. Integration with co-catalysts or alloying with nanometals by carefully controlling the morphology of nanostructures is expected to be the future direction for the advancement in this field. Another aspect is to develop materials with stronger absorption for visible light. This can be achieved by increasing the size of the nanostructures which shifts the absorption band to higher wavelengths but also strengthens the overall intensity of the LSPR band. The strategies to design and develop visible light-sensitive plasmonic materials should be envisaged. Moreover, the mechanism of plasmonic enhancement is inconsistent within the field. It is a complex phenomenon that accounts for many factors including the size and morphology of plasmonic nanostructures, positioning of the Fermi energy level, absorption intensity, bandgap, the effect of dopant addition and defects in semiconductors. A comparison of catalytic results, while considering all the aspects, is difficult due to entirely different preparative techniques and properties. An in-depth evaluation of catalytic pathways along with theoretical simulation studies is required to understand the effects of enhancements and lower the activation barrier to facilitate the chemical conversion processes. Overall, we anticipate that the recent research progress summarised in this review will assist in providing the latest guide to researchers for designing photoactive plasmonic nanomaterials for hydrogen generation. We envisage that these significant developments will foster future technological advancements in solar powered hydrogen generation from AB for practical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the International Joint Research Promotion Program of Osaka University. PV acknowledges the Royal Society-Newton International Fellowship (NIF\R1\180185) for her postdoctoral research funding at the University of Southampton. The present work was partially supported by Grants-in-Aid for Scientific Research (No. 19K15311, 26220911, 18K14056, and 19H00838) from the Japan Society for the Promotion of Science (JSPS) and MEXT. YK, KM and HY acknowledge the MEXT program “Element Strategy Initiative” (No. JPMXP0112101003).References
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O’Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- F. Wang, C. Li, H. Chen, R. Jiang, L. D. Sun, Q. Li, J. Wang, J. C. Yu and C. H. Yan, J. Am. Chem. Soc., 2013, 135, 5588–5601 CrossRef CAS.
- N. Wu, Nanoscale, 2018, 10, 2679–2696 RSC.
- T. Yoshii, Y. Kuwahara, K. Mori and H. Yamashita, J. Phys. Chem. C, 2019, 123, 24575–24583 CrossRef CAS.
- A. Agarwal, M. S. Vitiello, L. Viti, A. Cupolillo and A. Politano, Nanoscale, 2018, 10, 8938–8946 RSC.
- A. Gellé, T. Jin, L. De La Garza, G. D. Price, L. V. Besteiro and A. Moores, Chem. Rev., 2020, 120, 986–1041 CrossRef.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- H. Cheng, K. Fuku, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2015, 3, 5244–5258 RSC.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- E. Kazuma and Y. Kim, Angew. Chem., Int. Ed., 2019, 58, 4800–4808 CrossRef CAS.
- H. Jing, Q. Zhang, N. Large, C. Yu, D. A. Blom, P. Nordlander and H. Wang, Nano Lett., 2014, 14, 3674–3682 CrossRef CAS.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- M. J. Landry, A. Gellé, B. Y. Meng, C. J. Barrett and A. Moores, ACS Catal., 2017, 7, 6128–6133 CrossRef CAS.
- S. Lee, Y. W. Lee, H. Ahn, J. H. Kim and S. W. Han, Curr. Opin. Electrochem., 2017, 4, 11–17 CrossRef CAS.
- Y. Li, Z. Li, C. Chi, H. Shan, L. Zheng and Z. Fang, Adv. Sci., 2017, 4, 1–25 Search PubMed.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Bull. Chem. Soc. Jpn., 2019, 92, 19–29 CrossRef CAS.
- P. Verma, K. Mori, Y. Kuwahara, S. J. Cho and H. Yamashita, Catal. Today, 2020, 352, 255–261 CrossRef CAS.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS.
- S. Sarina, H. Y. Zhu, Q. Xiao, E. Jaatinen, J. Jia, Y. Huang, Z. Zheng and H. Wu, Angew. Chem., Int. Ed., 2014, 53, 2935–2940 CrossRef CAS.
- D. Wang, S. C. Pillai, S. H. Ho, J. Zeng, Y. Li and D. D. Dionysiou, Appl. Catal., B, 2018, 237, 721–741 CrossRef CAS.
- R. Long, Y. Li, L. Song and Y. Xiong, Small, 2015, 11, 3873–3889 CrossRef CAS.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS.
- M. A. Mahmoud and M. A. El-Sayed, Nano Lett., 2011, 11, 946–953 CrossRef CAS.
- S. Zhang, M. Li, J. Zhao, H. Wang, X. Zhu, J. Han and X. Liu, Appl. Catal., B, 2019, 252, 24–32 CrossRef CAS.
- M. Dhiman, J. Mater. Chem. A, 2020, 8, 10074–10095 RSC.
- T. Bora, D. Zoepfl and J. Dutta, Sci. Rep., 2016, 6, 1–10 CrossRef.
- X. Zhang, X. Ke and J. Yao, J. Mater. Chem. A, 2018, 6, 1941–1966 RSC.
- H. Yin, Y. Kuwahara, K. Mori, C. Louis and H. Yamashita, Catal. Sci. Technol., 2020, 10, 4141–4163 RSC.
- Y. Kuwahara, Y. Yoshimura, K. Haematsu and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 9203–9210 CrossRef CAS.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111–1116 CrossRef CAS.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS.
- P. Verma, D. J. Stewart and R. Raja, Catalysts, 2020, 10, 1176 CrossRef CAS.
- T. Shimbayashi and K. ichi Fujita, Tetrahedron, 2020, 76, 130946 CrossRef CAS.
- D. A. Bulushev, M. Zacharska, E. V. Shlyakhova, A. L. Chuvilin, Y. Guo, S. Beloshapkin, A. V. Okotrub and L. G. Bulusheva, ACS Catal., 2016, 6, 681–691 CrossRef CAS.
- D. B. Burueva, L. M. Kovtunova, V. I. Bukhtiyarov, K. V. Kovtunov and I. V. Koptyug, Chem. – Eur. J., 2019, 25, 1420–1431 CrossRef CAS.
- K. Chen, L. L. Gong, S. J. Ding, J. Liu, S. Ma, J. H. Wang, D. J. Yang, G. M. Pan, Z. H. Hao, L. Zhou and Q. Q. Wang, Plasmonics, 2020, 15, 21–29 CrossRef CAS.
- Y. Chen, Z. Huang, Z. Ma, J. Chen and X. Tang, Catal. Sci. Technol., 2017, 7, 4250–4258 RSC.
- S. Akbayrak and S. Özkar, Int. J. Hydrogen Energy, 2018, 43, 18592–18606 CrossRef CAS.
- P. Bhattacharya, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 11153–11161 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190–194 CrossRef CAS.
- W. Chen, J. Ji, X. Feng, X. Duan, G. Qian, P. Li, X. Zhou, D. Chen and W. Yuan, J. Am. Chem. Soc., 2014, 136, 16736–16739 CrossRef CAS.
- U. B. Demirci, Int. J. Hydrogen Energy, 2017, 42, 9978–10013 CrossRef CAS.
- W. W. Zhan, Q. L. Zhu and Q. Xu, ACS Catal., 2016, 6, 6892–6905 CrossRef CAS.
- N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509–521 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS.
- T. Kamegawa and T. Nakaue, Chem. Commun., 2015, 51, 16802–16805 RSC.
- K. Mori, K. Miyawaki and H. Yamashita, ACS Catal., 2016, 6, 3128–3135 CrossRef CAS.
- A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS.
- U. Sanyal, U. B. Demirci, B. R. Jagirdar and P. Miele, ChemSusChem, 2011, 4, 1731–1739 CrossRef CAS.
- M. A. Esteruelas, A. M. López, M. Mora and E. Oñate, ACS Catal., 2015, 5, 187–191 CrossRef CAS.
- S. Masuda, K. Shun, K. Mori, Y. Kuwahara and H. Yamashita, Chem. Sci., 2020, 11, 4194–4203 RSC.
- M. Navlani-García, P. Verma, K. Mori, Y. Kuwahara and H. Yamashita, J. Chem. Sci., 2017, 129, 1695–1703 CrossRef.
- M. Gao, Y. Yu, W. Yang, J. Li, S. Xu, M. Feng and H. Li, Nanoscale, 2019, 11, 3506–3513 RSC.
- H. Göksu, H. Can, K. Şendil, M. S. Gültekin and Ö. Metin, Appl. Catal., A, 2014, 488, 176–182 CrossRef.
- Q. Wang, C. Xu, M. Ming, Y. Yang, B. Xu, Y. Wang, Y. Zhang, J. Wu and G. Fan, Nanomaterials, 2018, 8, 280–290 CrossRef.
- Q. Yang, Y. Z. Chen, Z. U. Wang, Q. Xu and H. L. Jiang, Chem. Commun., 2015, 51, 10419–10422 RSC.
- H. Zhang, X. Gu, J. Song, N. Fan and H. Su, ACS Appl. Mater. Interfaces, 2017, 9, 32767–32774 CrossRef CAS.
- S. Gao, K. Ueno and H. Misawa, Acc. Chem. Res., 2011, 44, 251–260 CrossRef CAS.
- K. Fuku, R. Hayashi, S. Takakura, T. Kamegawa, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2013, 52, 7446–7450 CrossRef CAS.
- J. Fernández-Catalá, M. Navlani-García, P. Verma, Á. Berenguer-Murcia, K. Mori, Y. Kuwahara, H. Yamashita and D. Cazorla-Amorós, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.05.032.
- M. Navlani-García, P. Verma, Y. Kuwahara, T. Kamegawa, K. Mori and H. Yamashita, J. Photochem. Photobiol., A, 2018, 358, 327–333 CrossRef.
- A. Tanaka, K. Nakanishi, R. Hamada, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 1886–1891 CrossRef CAS.
- A. Tanaka, K. Fuku, T. Nishi, K. Hashimoto and H. Kominami, J. Phys. Chem. C, 2013, 117, 16983–16989 CrossRef CAS.
- S. Jo, P. Verma, Y. Kuwahara, K. Mori, W. Choi and H. Yamashita, J. Mater. Chem. A, 2017, 5, 21883–21892 RSC.
- M. Zhu, Y. Dai, W. Fu, Y. Wu, X. Zou, T. You and Y. Sun, Nanotechnology, 2018, 29, 165707 CrossRef.
- L. T. Guo, Y. Y. Cai, J. M. Ge, Y. N. Zhang, L. H. Gong, X. H. Li, K. X. Wang, Q. Z. Ren, J. Su and J. S. Chen, ACS Catal., 2015, 5, 388–392 CrossRef CAS.
- S. Rej, C. F. Hsia, T. Y. Chen, F. C. Lin, J. S. Huang and M. H. Huang, Angew. Chem., Int. Ed., 2016, 55, 7222–7226 CrossRef CAS.
- M. Wen, Y. Kuwahara, K. Mori and H. Yamashita, Top. Catal., 2016, 59, 1765–1771 CrossRef CAS.
- J. Yu, G. Li, H. Liu, Z. Li and Y. Liu, ChemCatChem, 2019, 11, 5536–5542 CrossRef CAS.
- K. Mori, P. Verma, R. Hayashi, K. Fuku and H. Yamashita, Chem. – Eur. J., 2015, 21, 11885–11893 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2015, 3, 18889–18897 RSC.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, 22-Mesoporous silica supported Ag-based plasmonic photocatalysts, Current Development in Photocatalysis and Photocatalytic Materials, Elsevier, 2020, pp. 353–368. ISBN: 9780128190005 Search PubMed.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2016, 4, 10142–10150 RSC.
- P. Verma, K. Yuan, Y. Kuwahara, K. Mori and H. Yamashita, Appl. Catal., B, 2018, 223, 10–15 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Sci. Technol., 2017, 7, 2551–2558 RSC.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Chem. – Eur. J., 2017, 23, 3616–3622 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori, R. Raja and H. Yamashita, Nanoscale, 2020, 12, 11333–11363 RSC.
- V. I. Simagina, O. V. Komova, A. M. Ozerova, O. V. Netskina, G. V. Odegova, N. L. Kayl and T. N. Filippov, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.04.070.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Today, 2019, 324, 83–89 CrossRef CAS.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2012, 51, 136–139 CrossRef CAS.
- P. Verma, M. Navlani-García, Y. Kuwahara, K. Mori and H. Yamashita, J. Chem. Sci., 2017, 129, 1661–1669 CrossRef CAS.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Catal. Today, 2020, 355, 620–626 CrossRef CAS.
- B. Zhou, M. Wen and Q. Wu, Nanoscale, 2013, 5, 8602–8608 RSC.
- M. Navlani-García, P. Verma, D. Salinas-Torres, R. Raja, K. Mori and H. Yamashita, ACS Symposium Series, 2020, 1360(2), 151–188 CrossRef.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852–14853 CrossRef CAS.
- J. G. Smith, J. A. Faucheaux and P. K. Jain, Nano Today, 2015, 10, 67–80 CrossRef CAS.
- S. Mubeen, G. Hernandez-Sosa, D. Moses, J. Lee and M. Moskovits, Nano Lett., 2011, 11, 5548–5552 CrossRef CAS.
- P. Zhang, T. Wang and J. Gong, Adv. Mater., 2015, 27, 5328–5342 CrossRef CAS.
- H. Yamashita, K. Mori, Y. Kuwahara, T. Kamegawa, M. Wen, P. Verma and M. Che, Chem. Soc. Rev., 2018, 47, 8072–8096 RSC.
- Z. Lou, Q. Gu, L. Xu, Y. Liao and C. Xue, Chem. – Asian J., 2015, 10, 1291–1294 CrossRef CAS.
- Y. Liu, Z. Zhang, Y. Fang, B. Liu, J. Huang, F. Miao, Y. Bao and B. Dong, Appl. Catal., B, 2019, 252, 164–173 CrossRef CAS.
- X. Li, Y. Yan, Y. Jiang, X. Wu, S. Li, J. Huang, J. Li, Y. Lin, D. Yang and H. Zhang, Nanoscale Adv., 2019, 1, 3941–3947 RSC.
- Y. Lou, J. He, G. Liu, S. Qi, L. Cheng, J. Chen, Y. Zhao and J. J. Zhu, Chem. Commun., 2018, 54, 6188–6191 RSC.
- N. Lu, Z. Zhang, Y. Wang, B. Liu, L. Guo, L. Wang, J. Huang, K. Liu and B. Dong, Appl. Catal., B, 2018, 233, 19–25 CrossRef CAS.
- Z. Zhang, Y. Liu, Y. Fang, B. Cao, J. Huang, K. Liu and B. Dong, Adv. Sci., 2018, 5, 1800748 CrossRef.
- X. Xu, G. Fang, J. Shang, K. Liu, Y. Bao, Y. Yang, Y. Liu and B. Dong, J. Mater. Sci., 2020, 55, 2958–2966 CrossRef CAS.
- Q. Huang, S. Hu, J. Zhuang and X. Wang, Chem. – Eur. J., 2012, 18, 15283–15287 CrossRef CAS.
- H. Cheng, T. Kamegawa, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2014, 53, 2910–2914 CrossRef CAS.
- J. Gong, Z. Li, T. Zhang, R. Chen, X. Zheng and G. Zhang, Funct. Mater. Lett., 2017, 10, 1750079 CrossRef CAS.
- J. Shi, Y. Kuwahara, M. Wen, M. Navlani-García, K. Mori, T. An and H. Yamashita, Chem. – Asian J., 2016, 11, 2377–2381 CrossRef CAS.
- H. Yin, Y. Kuwahara, K. Mori, H. Cheng, M. Wen and H. Yamashita, J. Mater. Chem. A, 2017, 5, 8946–8953 RSC.
- H. Cheng, X. Qian, Y. Kuwahara, K. Mori and H. Yamashita, Adv. Mater., 2015, 27, 4616–4621 CrossRef CAS.
- J. Shang, X. Xu, K. Liu, Y. Bao, Yangyang and M. He, Ceram. Int., 2019, 45, 16625–16630 CrossRef CAS.
- J. A. Faucheaux, A. L. D. Stanton and P. K. Jain, J. Phys. Chem. Lett., 2014, 5, 976–985 CrossRef CAS.
- I. Kriegel, F. Scotognella and L. Manna, Phys. Rep., 2017, 674, 1–52 CrossRef CAS.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS.
- Y. Zhao, H. Pan, Y. Lou, X. Qiu, J. Zhu and C. Burda, J. Am. Chem. Soc., 2009, 131, 4253–4261 CrossRef CAS.
- P. K. Jain, W. Huang and M. A. El-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |