
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting Li–S battery performance using an in-cell electropolymerized conductive polymer†
Xiguang
Gao
a,
Chenyang
Guo
a,
Zhong
Ma
 a,
Guan
Xi
ab,
Yuezhong
Meng
b and
Yuning
Li
*a
a,
Guan
Xi
ab,
Yuezhong
Meng
b and
Yuning
Li
*a
aDepartment of Chemical Engineering and Waterloo Institute for Nanotechnology (WIN), University of Waterloo, 200 University Ave West, Waterloo, Ontario N2L 3G1, Canada. E-mail: yuning.li@uwaterloo.ca; Fax: +1-519-888-4347; Tel: +1-519-888-4567 ext. 31105
bThe Key Laboratory of Low-Carbon Chemistry & Energy Conservation of Guangdong Province, State Key Laboratory of Optoelectronic Materials and Technologies, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: mengyzh@sysu.edu.cn
First published on 21st December 2020
Abstract
Lithium–sulfur (Li–S) batteries represent a promising high-density energy storage technology. The use of conductive polymers to enhance the performance of Li–S batteries has received much attention. In this work, a convenient and low-cost in-cell electropolymerization method is developed for the preparation of a conductive polymer, poly(3,4-ethylenedioxythiophene) (PEDOT), as a cathode binder inside a Li–S battery. The cell with this electropolymerized PEDOT (ePEDOT) shows notably improved specific capacity, cycling stability, and rate performance in comparison with the cell using the chemically synthesized commercial PEDOT:PSS (cPEDOT:PSS) as the binder. The performance enhancement is attributed to the formation of tightly integrated interfaces between ePEDOT and other components in the sulfur cathode. Moreover, this study revealed that the electrochemical dedoping of PEDOT is likely to occur during the Li–S battery cycling and thus the contribution of PEDOT (and some other conductive polymers) to the enhancement of the electrical conductivity of the sulfur cathode may not be as significant as expected.
Introduction
The increasing energy demand and environmental issues caused by the combustion of fossil fuels have stimulated global interest in exploiting clean and sustainable energy sources, such as solar, wind, and geothermal energy. However, due to their intermittent nature, a large portion of energy harvested from these energy sources needs to be stored for later use by utilizing some energy storage systems such as rechargeable batteries, supercapacitors, and compressed air and pumped hydroelectric energy storage (PHES) facilities. Among them, rechargeable batteries are of great importance due to their compact size, high efficiency, long cycle life, and pollution-free operation.1 Lithium-ion batteries (LIBs) have demonstrated great commercial success in portable electronics and electric vehicles since their invention in 1990s. However, the specific capacities of conventional intercalation-type cathode materials including lithium transition metal oxides (LiCoO2 and LiNixCoyMnzO2) and phosphates (LiFePO4) are unsatisfactory even at their theoretical limits.2 Therefore, there is an urgent need to develop batteries with a higher energy density in order to meet the emerging demands of electric vehicles and grid applications. With a high theoretical specific capacity of 1672 mA h g−1, a high specific energy of 2500 W h kg−1, an energy density of 2800 W h L−1, natural abundance (of sulfur), and environmental friendliness, lithium–sulfur (Li–S) batteries represent a promising technology to replace conventional LIBs.3 However, certain issues such as the large volume change during the charge/discharge of the sulfur cathode, the low conductivity of sulfur and sulfides, and the shuttling of lithium polysulfides result in poor cell performance, hindering the practical applications of Li–S batteries.4,5 To solve these issues, sulfur was embedded in a variety of inorganic conductors to form composites, including ordered mesoporous carbon (CMK-3),6 CNTs,7 graphene,8 and MXene.9 These conductive host materials can enhance the conductivity of the cathode to improve the sulfur utilization, while the micro/nanostructures or surface functionalities of the hosts can physically or chemically trap polysulfides to suppress the polysulfide shuttle effect.Conductive polymers (CPs), e.g., poly(3,4-ethylenedioxythiophene) (PEDOT), polypyrrole (PPy), and polyaniline (PANI), have also attracted considerable attention for improving the performance of Li–S batteries. CPs have numerous advantages including mild synthesis and processing conditions, chemical and structural diversity, high conductivity (up to ∼102–103 S cm−1), and excellent mechanical properties.10,11 Moreover, some CPs such as PEDOT and PPy possess abundant polar heteroatoms (oxygen and nitrogen, respectively), which have better polysulfide absorption capabilities compared to non-polar carbon-based conductors.12 CPs have been employed in Li–S batteries as cathode binders13–15 and conductive coatings on sulfur particles,16 sulfur/carbon composites,17,18 the top surface of sulfur cathodes,19,20 current collectors21 and separators.21,22 Improved stability and in some cases enhanced rate performance of the sulfur cathode have been achieved due to the incorporation of CPs.
Electropolymerization (e-polymerization) has been a widely adopted method to produce various CPs due to its simplicity and ease of implementation. A rechargeable battery, which is essentially an electrochemical device, may be used as a reactor for the in situ polymerization of a monomer to form a CP inside the battery. This “in-cell” (inside a battery) polymerization method would greatly simplify the incorporation of a CP into a battery and result in more tightly integrated interfaces with other components of the battery. Recently, in-cell ring-opening polymerization of a liquid electrolyte solvent to form a solid or gel polymer electrolyte was reported, which significantly lowered the interfacial resistances and promoted uniform lithium deposition.23–26 However, to the best of our knowledge, the utilization of in-cell e-polymerization to form a CP inside a battery has not been reported yet.
In this study, we report the in-cell e-polymerization of 3,4-ethylenedioxythiophene (EDOT) to form a CP, ePEDOT, as a binder at the cathode inside a Li–S battery. The battery with ePEDOT:PSS (poly(styrene sulfonate)) showed enhanced specific capacity and cycling stability in comparison to the battery with the commercial PEDOT:PSS (cPEDOT:PSS) binder, which is attributed to the tightly integrated interfaces of ePEDOT:PSS with other components in the sulfur cathode. Our study demonstrated the feasibility of using in-cell e-polymerization to produce CPs in rechargeable batteries to boost the battery performance.
Moreover, our study revealed that the initially doped conductive PEDOT is de-doped to become less conductive during the battery discharging process and could not be fully re-doped during the following charging process within the typical potential window of 1.7–2.8 V for Li–S batteries. Our finding has provided new insights into the role of CPs during battery cycling, where the CPs may not contribute to the improvement of the electrical conductivity of the cathode as significantly as expected previously.
Experimental
Materials and instrumentation
Sublimed sulfur (Fluka), Super P (TIMCAL), carbon paper (Toray, TGP-H-060), transparent and conductive indium-doped tin oxide (ITO) coated glass substrates (Delta Technologies), poly(4-styrenesulfonic acid) (H-PSS, Sigma-Aldrich, Mw ∼75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 18 wt% in H2O), 3,4-ethylenedioxythiophene (EDOT, Sigma-Aldrich), high conductivity commercial PEDOT:PSS (cPEDOT:PSS) (PH 1000, 1.1 wt% dispersion in H2O, Ossila), and the battery electrolyte (1 M LiTFSI in 1:1 (v:v) 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) with 2 wt% LiNO3, Suzhou Fosai) were obtained from commercial sources and used as received. Poly(4-styrenesulfonate) lithium salt (Li-PSS) was prepared by neutralizing H-PSS with lithium hydroxide. EDOT was dissolved in the electrolyte (1 M LiTFSI in 1:1 (v:v) DOL/DME with 2 wt% LiNO3) to prepare a 60 mM EDOT-containing electrolyte solution. Cyclic voltammetry (CV) and chronoamperometry (CA) measurements were performed using a CHI CHI604E electrochemical workstation or a VMP3 Bio-Logic potentiostat. The electrochemical impedance spectroscopy (EIS) measurement was carried out using a VMP3 Bio-Logic potentiostat with a 5 mV amplitude in the frequency range from 100 KHz to 100 mHz. The EIS data were fitted using the ZView software. The ultraviolet–visible–near infrared (UV–Vis–NIR) spectra were recorded using an Agilent Cary 7000 spectrophotometer. Galvanostatic cycling of batteries was performed using a LAND CT2001A battery tester. The peel test was performed using a Universal Macro-Tribometer (UNMT-2MT, Centre for Tribology, Inc.) equipped with a 1 kg load cell.
000, 18 wt% in H2O), 3,4-ethylenedioxythiophene (EDOT, Sigma-Aldrich), high conductivity commercial PEDOT:PSS (cPEDOT:PSS) (PH 1000, 1.1 wt% dispersion in H2O, Ossila), and the battery electrolyte (1 M LiTFSI in 1:1 (v:v) 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) with 2 wt% LiNO3, Suzhou Fosai) were obtained from commercial sources and used as received. Poly(4-styrenesulfonate) lithium salt (Li-PSS) was prepared by neutralizing H-PSS with lithium hydroxide. EDOT was dissolved in the electrolyte (1 M LiTFSI in 1:1 (v:v) DOL/DME with 2 wt% LiNO3) to prepare a 60 mM EDOT-containing electrolyte solution. Cyclic voltammetry (CV) and chronoamperometry (CA) measurements were performed using a CHI CHI604E electrochemical workstation or a VMP3 Bio-Logic potentiostat. The electrochemical impedance spectroscopy (EIS) measurement was carried out using a VMP3 Bio-Logic potentiostat with a 5 mV amplitude in the frequency range from 100 KHz to 100 mHz. The EIS data were fitted using the ZView software. The ultraviolet–visible–near infrared (UV–Vis–NIR) spectra were recorded using an Agilent Cary 7000 spectrophotometer. Galvanostatic cycling of batteries was performed using a LAND CT2001A battery tester. The peel test was performed using a Universal Macro-Tribometer (UNMT-2MT, Centre for Tribology, Inc.) equipped with a 1 kg load cell.
General procedure for fabricating coin cells
CR2032 coin cells composed of the cathode (on ITO, carbon paper, or carbon-coated Al substrate), Celgard 2500 separator, electrolyte, and lithium foil anode were assembled inside an argon (Ar)-filled glovebox with both O2 and H2O levels below 0.1 ppm. Characterization and e-polymerization of the cells were carried out outside the glove box under ambient conditions. Some of the cells were disassembled inside the Ar-filled glovebox for further experiments.Electrolyte stability studies using linear sweep voltammetry (LSV)
A coin cell composed of carbon paper, a lithium foil, and 40 μL of the electrolyte (1 M LiTFSI in 1:1 (v:v) DOL/DME with 2 wt% LiNO3) was assembled inside an Ar-filled glovebox. The cell was scanned in the LSV mode from the open circuit potential (OCP) to 5 V vs. Li/Li+ at a scan rate of 1 mV s−1.
In-cell e-polymerization of EDOT in the absence of sulfur
:1 was ground in a water/ethanol (w:w = 9:1) mixture to form a uniform slurry, which was coated on a carbon-coated Al foil by doctor blading. The substrate was dried at 50 °C overnight and then cut into disks (∼12 mm in diameter), which were used to assemble coin cells with Celgard 2500, a lithium foil, and 20 μL of 60 mM EDOT-containing electrolyte for each cell. The cells were subjected to e-polymerization using the CA method at 4.1 V for 800 s. After e-polymerization, the cell was disassembled, and all the cell parts were soaked in 3 mL of DME. Then, a known amount of the supernatant was evacuated using a rotovap to remove the majority of the DME solvent. The residue was diluted with 3 mL of deionized water, which was subjected to the UV–vis absorption measurement. The amount of unreacted EDOT in the cell after polymerization was then calculated using the pre-determined molar absorptivity of EDOT.
To collect the CV data, the cell containing the e-polymerized ePEDOT:H-PSS was measured directly without being disassembled in the potential range of 1.7–3.9 V vs. Li/Li+ at a scan rate of 1 mV s−1. For comparison, a Super P/cPEDOT:PSS (weight ratio of 4:1) substrate was also fabricated by coating the slurry on a carbon-coated Al foil to assemble a coin cell with the EDOT-free electrolyte. The cell was tested using the same potential range and scan rate.
Preparation and characterization of Li–S batteries
An S/Super P composite was prepared by mixing sublimed sulfur with Super P in a weight ratio of 65:35 in an agate mortar, followed by heating at 155 °C for 17 h in a sealed Teflon-lined stainless steel container. The S/Super P composite, additional Super P, and Li-PSS or H-PSS binder in a weight ratio of 77:13:10 were ground in a water/ethanol (w:w = 9:1) mixture to form a uniform slurry, which was coated on a carbon-coated Al foil by doctor blading. For comparison, a sulfur cathode containing 10 wt% cPEDOT:PSS as the binder was also prepared using the same method. The electrode films were then dried at 50 °C overnight and cut into disks with a diameter of ∼12 mm. The weight ratio of sulfur:Super P:binder in the cathode was 5:4:1. The areal sulfur loading in the electrode was 0.9–1.1 mg cm−2. Coin cells were then assembled using the above cathode, 20 μL of the electrolyte (with or without EDOT) for each cell, and the lithium foil anode. The e-polymerization of EDOT inside the cell was performed using the CA method at 4.10 V vs. Li/Li+ for 800 s. The EIS data of the cell were acquired before and immediately after e-polymerization. The CV data of sulfur cathodes were measured in the potential range of 1.7–2.8 V at a scan rate of 0.1 mV s−1. Galvanostatic cycling of Li–S cells was performed in the potential range of 1.7–2.8 V at room temperature.
180° peel tests on ePEDOT:H-PSS and cPEDOT:PSS binders
Sulfur cathodes using the ePEDOT:H-PSS or cPEDOT:PSS binder were prepared as described above. The cathode film disk (diameter: 12 mm) was attached to a glass slide using a double-sided tape. Then, a one-sided tape (width: 8 mm) was firmly attached to the cathode film. Then, the tape was pulled at an angle of 180° with a constant speed of 100 μm s−1 using a mechanical tester. The middle square part (8 mm × 8 mm) of the cathode film was used to obtain the load force vs. displacement curve. Before and after the peel test, optical images of the cathode film and the tape were taken using a microscope for comparison.Study of the conductivity–potential dependence of ePEDOT using combined CV and EIS
An ITO substrate was paired with a lithium foil to fabricate a coin cell, which was filled with 20 μL of 60 mM EDOT-containing electrolyte. e-Polymerization was performed using the CA method at 4.10 V vs. Li/Li+ for 800 s. Then, the cell was subjected to CV cycling between 1.7 and 3.36 V vs. Li/Li+ for 20 cycles until the CV diagram was stabilized. Afterwards, four more CV cycles were applied to the same cell. During each cycle, the cell was switched from the CV testing mode to the EIS testing mode at potentials of 1.7 and 3.36 V, respectively. Finally, such testing was conducted for an additional CV cycle by switching from the CV to the EIS mode at more potential points of 1.7, 2.34, 2.43, 2.8, and 3.36 V during the oxidation process and 2.8, 2.28, 1.93, and 1.7 V during the reverse reduction process. All the obtained EIS spectra were fitted to obtain the Rct values using ZView. The CV and EIS measurements and automatic switching from CV to EIS were performed using a VMP3 Bio-Logic potentiostat.Results and discussion
e-Polymerization of EDOT via cyclic voltammetry
Cyclic voltammetry (CV) and chronoamperometry (CA)28,29 are two commonly used electrochemical methods for the polymerization of EDOT to prepare PEDOT (and other CPs). First, the CV method was employed in this study. Prior to e-polymerization, the electrochemical stability of the electrolyte (1 M LiTFSI in 1:1 (v:v) DOL/DME with 2 wt% LiNO3) was examined using the linear sweep voltammetry (LSV) method using a coin cell with carbon paper and a lithium foil as electrodes in the potential range from the open circuit potential to 5 V vs. Li+/Li. The current starts to increase abruptly at 4.56 V due to electrolyte decomposition, which signifies the upper potential limit for e-polymerization (Fig. S1 in the ESI†).
Next, a conductive ITO-coated glass substrate was paired with a lithium foil in a coin cell for e-polymerization of EDOT, in which the transparent ITO substrate would allow the UV–vis measurement of the formed polymer, ePEDOT. A 60 mM EDOT solution was prepared by adding EDOT to 1 M LiTFSI in 1:1 (v:v) DOL/DME with 2 wt% LiNO3. The coin cell loaded with the EDOT-containing electrolyte was scanned in a potential window between the open circuit voltage (∼3 V) and 4.2 V vs. Li+/Li in the CV mode at a scan rate of 20 mV s−1 for up to 10 cycles. For comparison, another coin cell loaded with the EDOT-free electrolyte was tested under the same conditions. The cyclic voltammograms for the 1st cycles of the two cells are shown in Fig. 1a. A notable oxidation process is observed for the cell using the EDOT-containing electrolyte, in stark contrast to the irresponsive flat line obtained for the cell containing the EDOT-free electrolyte. For the cell with the EDOT-containing electrolyte, the current starts to increase sharply when the potential is above ∼4.1 V, indicating the start of the oxidation (e-polymerization) of EDOT. It is noteworthy that the current is higher during the early reverse scan than that during the forward scan, leading to a crossover at 3.99 V. This phenomenon indicates that the deposition of the polymer proceeds through a nucleation-and-growth mechanism, which has been observed in e-polymerizations for preparing various CPs.30,31 The electrochemical impedance spectroscopy (EIS) spectra of the electrode after different numbers of CV cycles are shown in Fig. 1b. Before e-polymerization, Warburg diffusion behaviour (an oblique line) dominates in the EIS spectrum. Once the deposition of PEDOT begins, a semicircle together with an oblique tail shows up. By fitting the EIS spectra using the equivalent circuit model shown in Fig. 1c, Rs, Rct, Wo and CPE, which represent the solution resistance, charge transfer resistance, Warburg diffusion impedance and constant phase element, respectively, can be obtained.32,33 The Rct values after different numbers of CV cycles are plotted in Fig. 1c. As Rct is strongly related to the conductivity of the electrode, the change in Rct can be related to the deposition of the PEDOT layer on the ITO substrate. As the number of CV cycles is increased from 1 to 5, the Rct continuously drops from 244 to 61 Ω, i.e., the conductivity increases. This can be explained by the gradual growth of PEDOT from the initially formed isolated PEDOT nuclei to the final continuous PEDOT film on the ITO substrate, leading to a gradual increase in conductivity with increasing the number of CV cycles.34 The Rct of the electrode at the 10th CV cycle is similar to that measured at the 5th CV cycle, which indicates that the polymerization is almost completed after 5 CV cycles to form the polymer ePEDOT. The ePEDOT-bearing ITO substrate after 10 CV cycles was taken out of the coin cell, washed with DME and isopropanol, dried with N2 gas, and subjected to the UV–Vis–NIR spectroscopy measurement. As shown in Fig. 1d, the as-prepared ePEDOT shows a strong, flat absorption profile starting from 540 nm and extending into the near-IR region, indicating the formation of doped PEDOT on the ITO substrate. The broad bands centered at 970 nm and from 1360 to 2000 nm are attributed to the polaron (radical cation) and bipolaron (dication) charge carriers, respectively.35–37 Next, the as-prepared ePEDOT on the ITO substrate was dedoped by treatment with ethylenediamine. The dedoped ePEDOT shows a distinct peak at 572 nm, which can be ascribed to the characteristic π–π* electronic transition of the neutral PEDOT,35 as well as decreased polaron and bipolaron absorption bands. For comparison, the UV–Vis–NIR spectra of the doped (as-coated) and dedoped cPEDOT:PSS were also measured (Fig. 1d). The as-coated cPEDOT:PSS on the ITO substrate exhibits a sloping absorption curve from 350 to 2000 nm, indicating that bipolarons are the main charge carriers. The difference in the distribution of polarons and dipolarons between the as-prepared ePEDOT and the coated cPEDOT:PSS may be due to the difference in counterion (TFSI−vs. PSS) and polymer molecular weight. The dedoped cPEDOT showed a π–π* electronic transition peak at 615 nm, an intensified polaron absorption band centered at 982 nm, and a weakened bipolaron band from 1360 to 2000 nm, which is consistent with the literature.36,37 The aforementioned UV–Vis–NIR data confirm the formation of doped PEDOT, ePEDOT, after e-polymerization.
 | ||
| Fig. 1 (a) CV curves of the two-electrode coin cells with the EDOT-containing or EDOT-free (blank) electrolyte for the 1st cycle scanned between 3 and 4.2 V at a scan rate of 20 mV s−1. The arrows indicate the scan direction. An ITO-coated glass substrate was used. (b) EIS spectra of the same coin cell shown in (a) with the EDOT-containing electrolyte before and after CV scanning for different numbers of cycles. (c) The change of Rct with increasing the number of CV cycles. The inset shows the equivalent circuit for the fitting of EIS data shown in (b). (d) UV–Vis–NIR spectra of the as-prepared and dedoped ePEDOT, and the as-coated and dedoped cPEDOT:PSS on ITO-coated glass substrates, where the ePEDOT was e-polymerized by the CV method (between 3 and 4.2 V for 10 cycles with a scan rate of 20 mV s−1). | ||
Optimization of e-polymerization of EDOT via chronoamperometry
To have better control over the e-polymerization process, the chronoamperometry (CA) technique was adopted since the amount of charge injected into the cell for polymerization can be precisely controlled by the applied potential and time. A carbon paper substrate, which has a porous structure for growing a larger amount of ePEDOT, was used to replace the ITO-coated glass substrate. A constant potential of 3.80, 3.90, 4.00, 4.05, 4.10, 4.15, 4.20, 4.30, or 4.40 V was applied to coin cells containing 60 mM EDOT in the electrolyte for 800 s. The current vs. time responses at different potentials (chronoamperograms) are shown in Fig. 2a. The current remains low at a potential below 3.90 V, indicating that the applied potentials are inadequate to initiate the polymerization. At a potential of 4.00 V, the curve starts to show a very broad peak between 200 s and 400 s. A current peak appears earlier and sharper with further increasing the potential to 4.10 V, which agrees with the CV data (Fig. 1a). As the potential exceeds 4.15 V, two peaks start to appear with both becoming sharper and earlier with increasing potential. The features of these CA curves are similar to those observed in the electrosynthesis of PEDOT35 and poly(anthraquinone) derivatives38 reported previously. The area under the current vs. time curve is used to obtain the amount of charge injected into the cell during e-polymerization. The charge vs. potential plot is shown in Fig. 2b. As 40 μL of the EDOT-containing electrolyte was used for each cell, the maximum charge required for the polymerization (oxidation) of all the monomer molecules and the p-doping of the resultant polymer at a doping level of 0.4 (the typical doping level for electropolymerized CPs is between 0.25 and 0.4)39 is 0.55C (see the ESI† for detailed calculations). From Fig. 2b, the effective charge injected into the cell for e-polymerization at 4.10 V is 0.56C, which agrees well with the calculated value. A larger amount of charge injected at a higher potential (>4.10 V) may over-oxidize or over-dope the polymer, which possibly leads to the appearance of the 2nd peak in the corresponding chronoamperograms (Fig. 2a). The optical images of a series of carbon papers with ePEDOT deposited at different potentials (Fig. S3, ESI†) clearly show that a potential higher than 4.10 V would result in non-uniform deposition of the polymers (large polymer aggregates). It should be mentioned that the ePEDOT prepared by the CV and CA methods are very similar as the UV–Vis–NIR spectra of the ePEDOT prepared by these two methods are almost identical (Fig. S4, ESI†).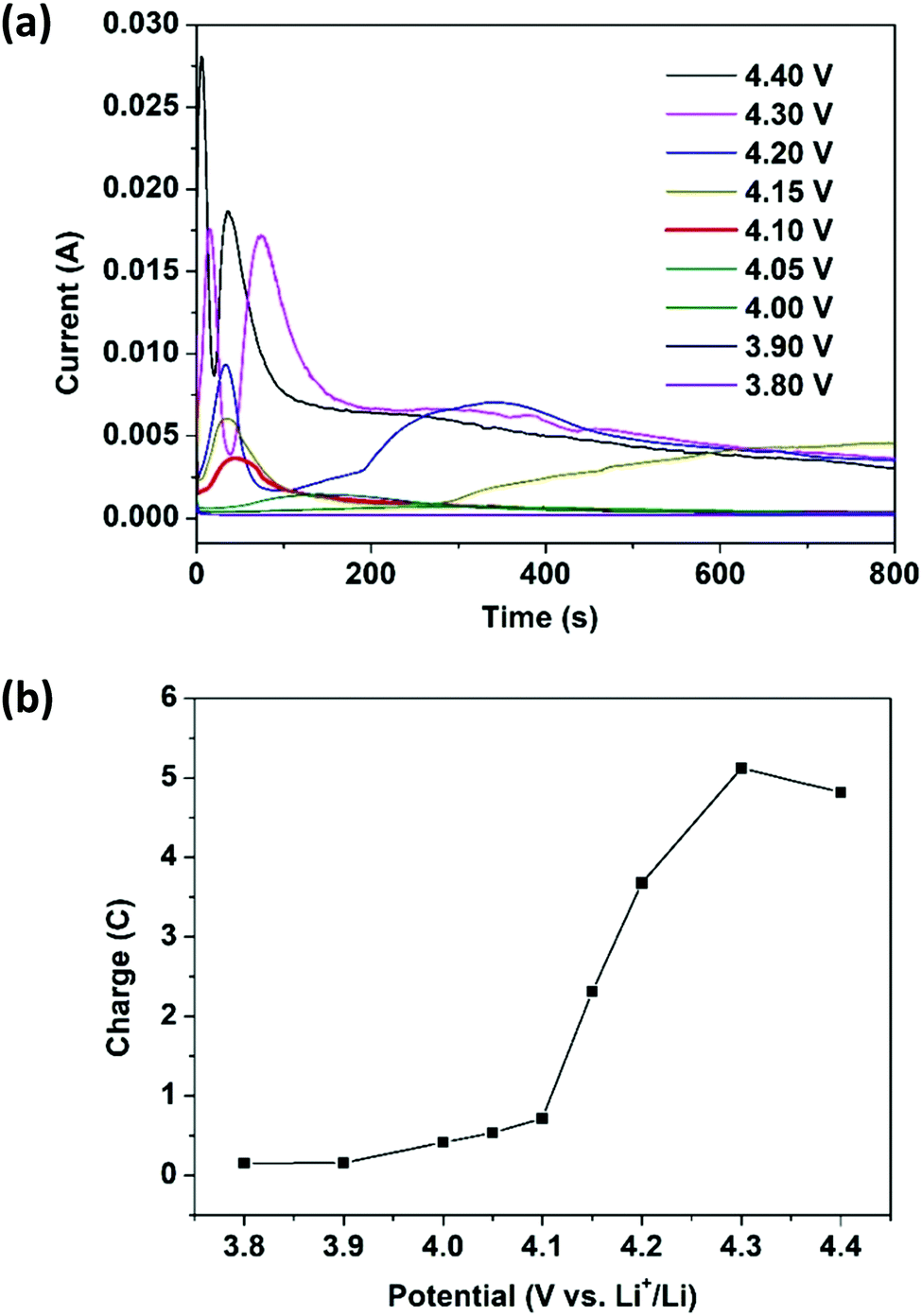 | ||
| Fig. 2 (a) Chronoamperograms for the e-polymerization of EDOT at different potentials from 3.8 to 4.4 V. Carbon paper substrates were paired with lithium foils to construct two-electrode coin cells. (b) Total charges injected into the coin cells at different potentials for 800 s. | ||
To determine the yield of e-polymerization, the following experiment was performed. At first, a sulfur-free Super P/H-PSS (weight ratio of 4:1) substrate was prepared by coating the slurry on a carbon-coated Al foil, which has similar structure and composition to those of the sulfur cathode. The substrate was assembled into a coin cell by pairing with a lithium foil and using 20 μL of 60 mM EDOT-containing electrolyte. e-Polymerization was performed in the CA mode at 4.10 V for 800 s. The cell was disassembled and the parts were washed with DME to dissolve the unreacted EDOT for the UV–vis measurement. It was found that 86% of EDOT was converted to the polymer ePEDOT (see Fig. S5 and S6, ESI,† for details).
Based on the aforementioned results, a potential of 4.10 V vs. Li/Li+ and a reaction time of 800 s are chosen as the optimal conditions for the e-polymerization of EDOT using the CA method.
Fabrication and characterization of Li–S batteries with the ePEDOT binder prepared via in-cell e-polymerization
A schematic illustrating the formation of ePEDOT on the sulfur cathode via the in-cell e-polymerization method is shown in Fig. 3. Specifically, poly(4-styrenesulfonic acid) (H-PSS) or poly(lithium 4-styrenesulfonate) (Li-PSS) is used as a pre-binder in the sulfur cathode, which is expected to form a doped ePEDOT:PSS that is similar to the commercial one, cPEDOT:PSS, in the cathode. The Li–S batteries with the 60 mM EDOT-containing electrolyte were assembled and in-cell e-polymerized using the CA method at 4.10 V for 800 s. The chronoamperograms of the sulfur cathodes are similar to those obtained using carbon paper (without sulfur) as the substrate (Fig. S7, ESI†) since sulfur is inert under the e-polymerization (4.1 V vs. Li/Li+) conditions, suggesting the successful formation of ePEDOT at the sulfur cathode. As a comparison, batteries using the EDOT-free electrolyte were also fabricated. The CV curves of the sulfur cathodes using the H-PSS or Li-PSS binder with and without ePEDOT are shown in Fig. 4a–d. Comparing the two cells with only H-PSS and Li-PSS binders (Fig. 4a and b), the former shows a very weak and delayed 2nd reduction peak, which corresponds to the conversion of lithium polysulfides to Li2S2 and Li2S2 to Li2S, in the 1st and 2nd cycles. In the subsequent cycles, this peak becomes stronger, but the peak position shifts to a much lower potential compared to the Li-PSS based cell. This indicates that the sulfonic acid (–SO3H) groups in H-PSS may strongly interact with lithium polysulfides and impede their further reduction (a delayed second reduction peak). In other words, this suggests that the –SO3H groups may have a better trapping capability than the lithium sulfonate (–SO3Li) groups in Li-PSS. After e-polymerization, the cell with the resultant ePEDOT:H-PSS (Fig. 4c) shows similar CV profiles with delayed second reduction peaks compared to the cell with the H-PSS binder. Interestingly, after e-polymerization, the cell with the ePEDOT:Li-PSS binder (Fig. 4d) also displays delayed second reduction peaks, resembling closely those of the cells with H-PSS and ePEDOT:H-PSS binders (Fig. 4a and c). This may be attributed to the generation of protons (during the e-polymerization of EDOT) that convert the –SO3Li groups into –SO3H groups that have a stronger trapping effect on lithium polysulfides. Delayed second reduction peaks in the 1st and 2nd cycles are also observed for the commercial cPEDOT:PSS binder (Fig. S8a, ESI†) since cPEDOT:PSS is acidic (pH = ∼2 for the 1.1 wt% aqueous dispersion).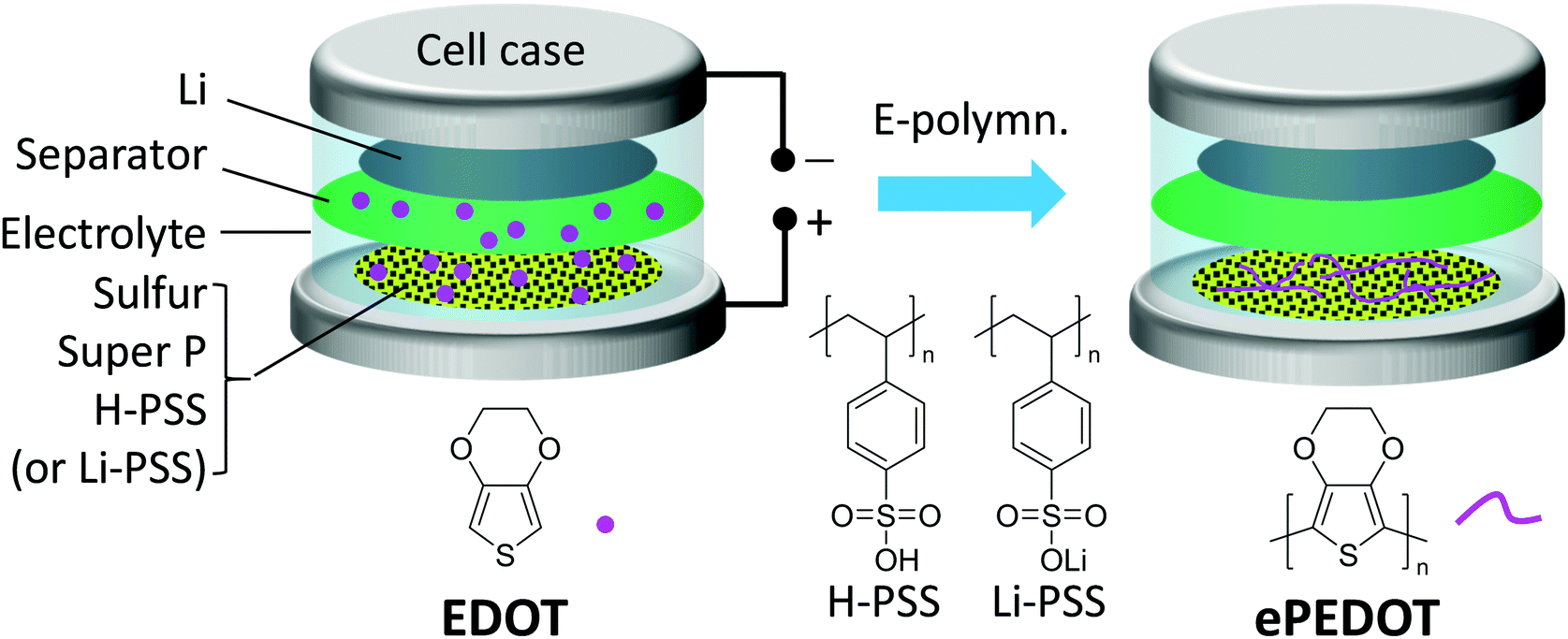 | ||
| Fig. 3 Schematic illustrating the formation of ePEDOT in the sulfur cathode inside a Li–S battery by in-cell e-polymerization. | ||
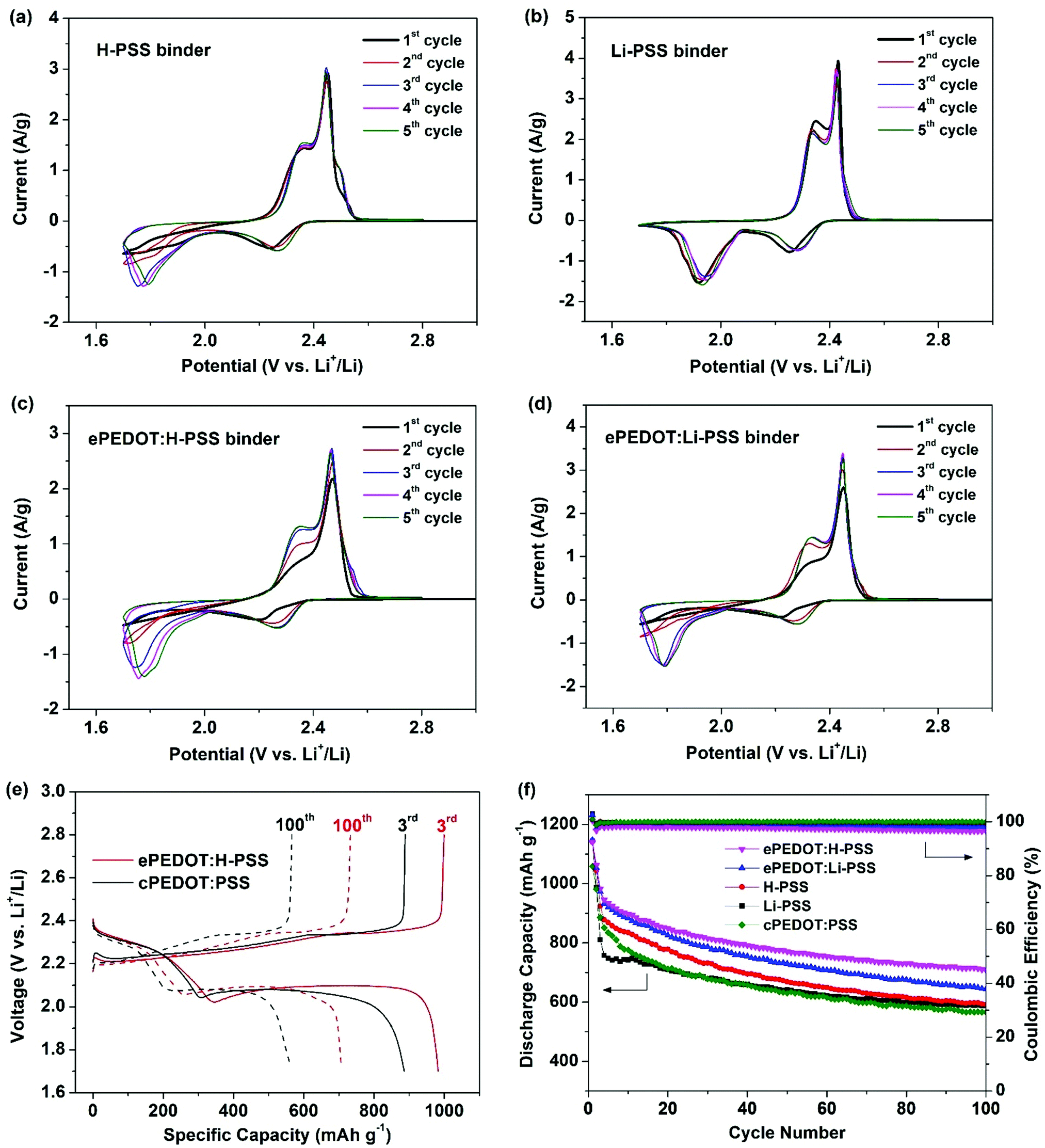 | ||
| Fig. 4 Cyclic voltammograms of sulfur cathodes using (a) H-PSS, (b) Li-PSS, (c) ePEDOT:H-PSS, and (d) ePEDOT:Li-PSS as binders at a scan rate of 0.1 mV s−1. (e) Charge/discharge profiles of sulfur cathodes using ePEDOT:H-PSS and cPEDOT:PSS binders at 0.2C. (f) Cycling performance of sulfur cathodes using ePEDOT:H-PSS, ePEDOT:Li-PSS, H-PSS, Li-PSS, and cPEDOT:PSS binders. The batteries were activated at 0.1C for 2 cycles and cycled at 0.2C afterwards. | ||
The 3rd and 100th charge/discharge profiles of the sulfur cathodes at 0.2C are compared in Fig. 4e and Fig. S8b (ESI†). In the 3rd cycles at 0.2C (Fig. S8b, ESI†), the polarization potentials (ΔE) of the cells with different binders follow the order of H-PSS (204 mV) < ePEDOT:H-PSS (210 mV) < Li-PSS (235 mV) < cPEDOT:PSS (237 mV) < ePEDOT:Li-PSS (243 mV). The galvanostatic cycling performance of sulfur cathodes using different binders is shown in Fig. 4f. The initial capacities of sulfur cathodes with the ePEDOT:H-PSS and ePEDOT:Li-PSS binders are very close, i.e. 1142 and 1147 mA h g−1, respectively. These values are almost identical to those of the cathodes with the H-PSS (1141 mA h g−1) and Li-PSS (1145 mA h g−1) binders, respectively. However, the cell with the Li-PSS binder shows an abrupt decay in capacity in the first few cycles compared to the cell with the H-PSS binder. This might be explained by the weaker trapping capability of the Li-PSS binder to lithium polysulfides than the H-PPS binder as discussed previously. After the 3rd cycle, the cell with H-PSS decays at a faster rate than the cell with Li-PSS, while both cells show exactly the same specific capacity of 596 mA h g−1 at the 100th cycle. The faster decay of the cell with H-PSS between the 3rd cycle and the 100th cycle might be due to the fact that even though H-PSS has a stronger affinity to lithium polysulfides, the latter can still gradually escape from the cathode to the bulk electrolyte, resulting in a reduction in sulfur utilization. Consequently, the capacity retentions of the H-PSS and Li-PSS based cells after 100 cycles with respect to their initial specific capacities are very similar, i.e. 52% and 51%, respectively.
On the other hand, the cells with the ePEDOT:H-PSS and ePEDOT:Li-PSS binders show much improved cycling stability with capacity retentions of 62% and 56%, respectively. The results demonstrate that ePEDOT has an additional benefit to the suppression of the polysulfide shuttle effect due to its strong polysulfide absorption capability.12 The sulfur cathode using the commercial cPEDOT:PSS binder shows an initial capacity of 1059 mA h g−1 and a capacity retention of 53% after 100 cycles, both of which are lower than those of the batteries with the ePEDOT:H-PSS and ePEDOT:Li-PSS binders, indicating that ePEDOT formed via in-cell e-polymerization leads to an improved initial specific capacity and better cycling stability. Moreover, the cell with the ePEDOT:H-PSS binder also results in improved rate performance compared to that with the cPEDOT:PSS binder (Fig. S9, ESI†). The improved specific capacity, cycling stability, and rate performance of the cells with the ePEDOT:H-PSS and ePEDOT:Li-PSS binders might originate from the tightly integrated interfaces between ePEDOT and Super P (where e-polymerization occurs), where the trapped lithium polysulfides are in close proximity to Super P for more efficient charge transfer. To further confirm the trapping capability of PEDOT toward lithium polysulfides, the cells using the sulfur cathodes with different binders were disassembled after 100 cycles, and all the cell components were soaked in 5 mL of DOL/DME. Then, the UV–Vis spectra of the supernatants were recorded (shown in Fig. S10, ESI†). Compared with the sulfur cathode using the H-PSS binder, those using ePEDOT:PSS and cPEDOT:H-PSS binders show a notable reduction in the absorbance of lithium polysulfides (260, 280 and 310 nm for Li2S6,40 420 nm for Li2S441), suggesting that both ePEDOT and cPEDOT have obvious polysulfide trapping abilities. Considering the ease of fabrication and the enhanced cell performance, the in-cell e-polymerized ePEDOT:H-PSS and ePEDOT:Li-PSS binders are advantageous over the cPEDOT:PSS binder.
Adhesion evaluation of ePEDOT:PSS by the 180° peel test
As a glue for bonding conductive carbon particles and sulfur particles and for bonding the entire cathode film to Al current collectors, the binder is an important component of Li–S batteries. The adhesion strengths of cPEDOT:PSS and ePEDOT:H-PSS binders were evaluated by the 180° peel test using a mechanical tester. The load force vs. displacement curves are shown in Fig. S11b and c (ESI†). The adhesion strengths (calculated by dividing the load force with the width of the tape) of ePEDOT:H-PSS and cPEDOT:PSS binders are 0.039 ± 0.008 and 0.028 ± 0.003 N mm−1, respectively, indicating that the ePEDOT:H-PSS binder has better adhesion compared to the cPEDOT:PSS binder. The optical images of the cathode film and the tape before and after the peel test were also compared. As shown in Fig. S12 (ESI†), before the peel test, the sulfur cathode film using the cPEDOT:PSS or ePEDOT:H-PSS adhesive is completely covered on the carbon-coated aluminum foil. After the peel test, large pieces were peeled off from the sulfur cathode with the cPEDOT:PSS binder, while only small pieces were peeled off from the one with the ePEDOT:H-PSS binder, which further confirmed that the adhesion of the ePEDOT:PSS binder is superior to that of the cPEDOT:H-PSS binder. The better adhesion strength of the ePEDOT:H-PSS binder can also explain its better battery performance than the cPEDOT:PSS binder.Investigation of the doping/dedoping of PEDOT via CV and EIS
CPs are conductive in the doped state, while they are semiconducting or insulating in the undoped (or dedoped) state. When CPs are used in rechargeable lithium batteries, the enhanced battery performance has been often linked to the high conductivity of CPs in the literature. However, the initially doped and conductive CPs may be electrochemically dedoped and become less conductive or even insulating during the battery cycling process. This potential issue has not been discussed and investigated in previous studies. On the other hand, the CV data of CPs reported in the literature, which can reveal the doping and dedoping behaviours at different potentials, have been reported with reference electrodes such as the SCE and Ag/AgCl that are different from Li+/Li and in electrolytes that are different from conventional battery electrolytes, which makes it rather difficult to gauge the doping and dedoping behaviours of CPs when CPs are employed in lithium batteries.Here we investigated the aforementioned issue that might be associated with ePEDOT:H-PSS during the discharge/charge processes of Li–S batteries via CV and EIS measurements. A sulfur cathode was prepared using Li-PSS as the binder in order to rule out the influence of the binder (H-PSS) on the CV profiles as discussed earlier, while an electrode with Super P/ePEDOT:H-PSS (sulphur-free) on the carbon-coated Al substrate was prepared in order to obtain the intrinsic redox characteristics of ePEDOT:H-PSS. The CV curves of both electrodes were measured using Li+/Li as the reference electrode in the same LiTFSI-based electrolyte for a direct comparison (Fig. 5a). The electrochemical reaction associated with each peak position is shown in Fig. 5b. For the sulfur cathode, the two reduction (discharge) peaks at 2.28 and 1.93 V are attributed to the conversion of S8 to lithium polysulfides (Li2Sn, n = 4–8) and lithium polysulfides to Li2S2/Li2S, respectively. The two oxidation (charge) peaks at 2.34 and 2.43 V are attributed to the conversion of Li2S2/Li2S to lithium polysulfides and lithium polysulfides to S8, respectively.
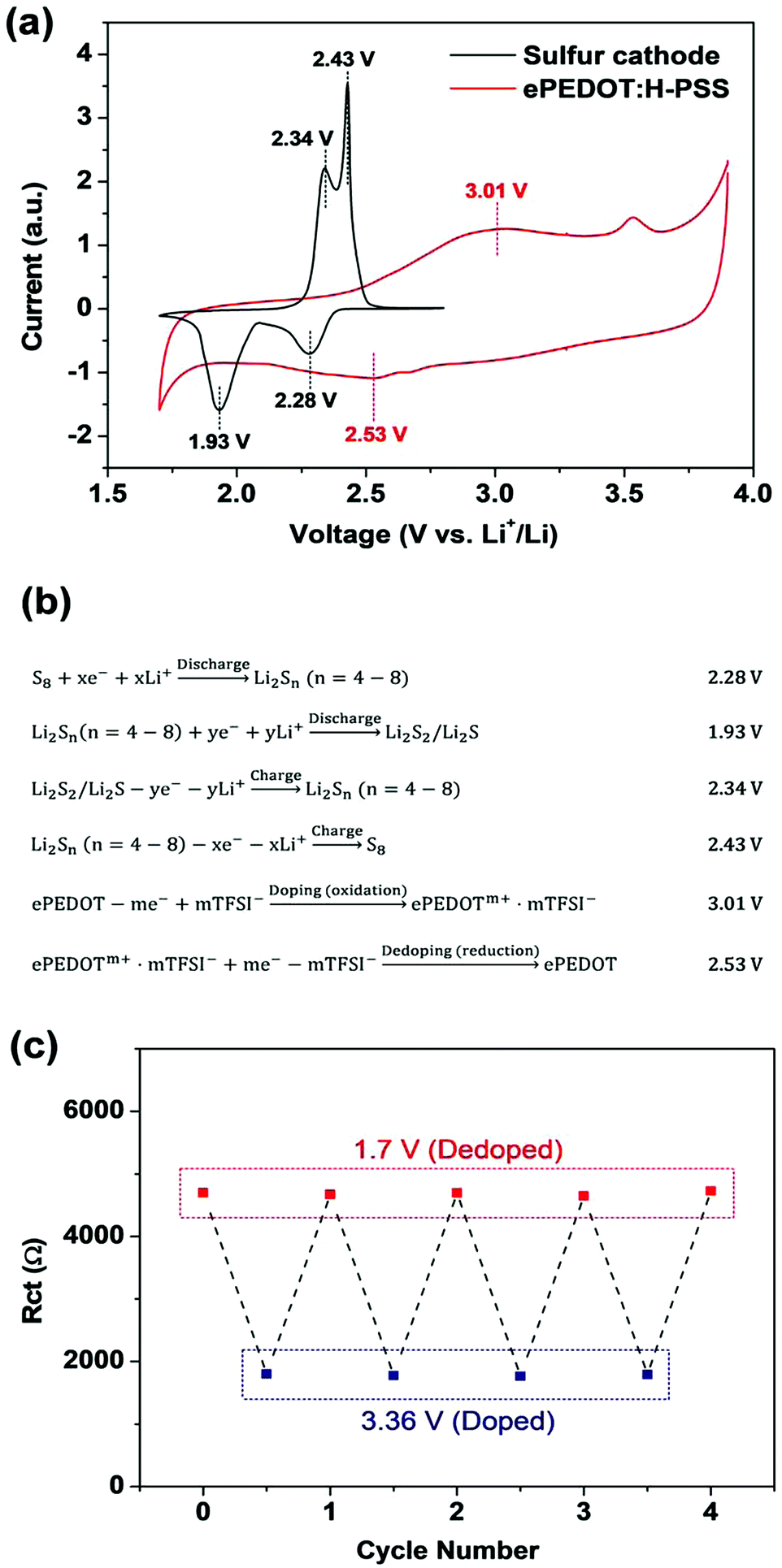 | ||
| Fig. 5 (a) CV curves of the sulfur cathode and ePEDOT:H-PSS. The sulfur cathode was made using Li-PSS as the binder, while ePEDOT:H-PSS was formed on the Super P/H-PSS substrate via in-cell e-polymerization using the CA method at 4.10 V for 800 s. The potential ranges for the sulfur cathode and ePEDOT:H-PSS are 1.7–2.8 and 1.7–3.9 V, respectively. (b) Electrochemical processes involved in the charging/discharging of Li–S batteries and the doping/dedoping of ePEDOT. (c) Rct values for ePEDOT deposited on an ITO-coated glass substrate at high (3.36 V vs. Li+/Li) and low (1.7 V vs. Li+/Li) potentials. | ||
For ePEDOT:H-PSS, the broad reduction peak at 2.53 V and the oxidation peak at 3.01 V correspond to the dedoping and doping processes, respectively (Fig. 5b). The peak at 3.54 V is due to the oxidation of lithium nitrite (LiNO2) that is generated by the reaction of lithium nitrate with lithium metal,42,43 which disappeared when a LiNO3-free electrolyte was used (Fig. S13, ESI†). From the CV curve of ePEDOT:H-PSS (Fig. 5a), it can be seen that doping starts at 2.42 V, reaches a peak at 3.01 V, and finishes at 3.36 V, while dedoping starts at 2.86 V, reaches a peak at 2.53 V, and finishes at 1.95 V. This suggests that the dedoping of ePEDOT:H-PSS occurs simultaneously with the discharge of the sulfur cathode and that ePEDOT:H-PSS becomes mostly dedoped after the sulfur cathode is discharged to 1.7 V. After recharging the sulfur cathode to 2.8 V, the doping level of ePEDOT:H-PSS can only be partially restored with respect to its original state. Therefore, ePEDOT:H-PSS may only be highly conductive and contribute to the improvement of the conductivity of the sulfur cathode during the 1st discharging process of the sulfur cathode. In the subsequent 1st charging process in the typical voltage range of 1.7–2.8 V, ePEDOT:H-PSS cannot be fully doped. Therefore, ePEDOT:H-PSS would make smaller contributions to the improvement of the conductivity of the cathode in the subsequent discharge/charge cycles. Since cPEDOT:PSS shows similar reduction/oxidation profiles compared to ePEDOT:H-PSS (Fig. S13, ESI†), the dedoping of cPEDOT:PSS is also likely to occur when it is used in the sulfur cathode during the discharging/charging processes.
It should be mentioned that other cathode materials having higher cycling potential windows, e.g. 3–4.2 V for lithium cobalt/manganese oxide and 2–4.2 V for lithium iron phosphate, PEDOT would be more heavily doped during the cell operation. In this regard, it is expected that PEDOT would contribute more to improving the conductivity of the cathode when it is used for these high voltage batteries. On the other hand, the doping/dedoping windows of different CPs vary. The cathode conductivity and performance of Li–S batteries may be further improved if an appropriate monomer is selected to make a CP that has lower oxidation and reduction potentials compared to the sulfur cathode.
The dependence of the conductivity of ePEDOT on the cell potential was further studied by combined CV and EIS measurements. To eliminate the interference of carbon materials (Super P and carbon paper) with the EIS results, a bare ITO-coated glass substrate was used to grow ePEDOT by e-polymerization using the CA method at 4.10 V vs. Li+/Li for 800 s. After in-cell e-polymerization, the cell was scanned using CV for 20 cycles to obtain a stabilized CV diagram (Fig. S14a, ESI†) and then subjected immediately to the EIS measurement at 1.7 and 3.36 V for four consecutive CV cycles (Fig. S14b and c, ESI†). The obtained EIS spectra were fitted using the equivalent circuit shown in Fig. 1c to obtain the Rct values, which are plotted in Fig. 5c. The average Rct at 3.36 V vs. Li+/Li (ePEDOT in the doped state) is 2.22 × 103 Ω, which is much smaller than the value of 4.92 × 103 Ω at 1.7 V vs. Li+/Li (ePEDOT in the dedoped state). Similar Rct values are maintained at 1.7 and 3.36 V for different cycles demonstrating reversible switching between the doped and dedoped states of ePEDOT during the cell cycling process. More data points were collected during one CV cycle at additional potential points between 1.7 V and 3.36 V vs. Li+/Li, which are critical for Li–S batteries (Fig. S14d–f, ESI†). The results show a more detailed resistance-potential dependence of ePEDOT, where the Rct decreases with increasing cell potential during the oxidation (doping) process and increases with decreasing cell potential during the reverse reduction (dedoping) process.
Conclusions
In-cell e-polymerization of EDOT to produce a conductive polymer ePEDOT as a binder in the cathode of a Li–S battery is explored as a simple approach to improve the battery performance. It was found that the e-polymerization of an EDOT-containing electrolyte at a potential of 4.10 V vs. Li+/Li for 800 s using chronoamperometry could form an ePEDOT:H-PSS or ePEDOT:Li-PSS binder in the sulfur cathode in the presence of an H-PSS or Li-PSS pre-binder. The Li–S batteries with the ePEDOT:H-PSS or ePEDOT:Li-PSS binder prepared by in-cell e-polymerization showed notably improved specific capacity, cycling stability, and rate performance compared to the cell prepared using the analogous commercial cPEDOT:PSS binder. The enhanced performance is attributed to the tightly integrated interfaces between the ePEDOT and other components including Super P in the cathode, where the trapped lithium polysulfides by ePEDOT might be in close proximity to Super P for efficient charge transfer.Through a comparison of the redox profiles of the sulfur cathode and ePEDOT:H-PSS under identical conditions, it was found that ePEDOT might be dedoped within the typical Li–S battery cycling potential window of 1.7–2.8 V vs. Li+/Li. The EIS measurements of ePEDOT further confirmed that the conductivity decreases with decreasing potential in the range of 1.7–2.8 V vs. Li+/Li. These results indicate that PEDOT (ePEDOT or cPEDOT) and most other CPs may not contribute as much to the enhancement of the electrical conductivity of the cathode as previously expected in Li–S batteries. However, this issue may be alleviated in batteries with higher charge/discharge potentials or a conductive polymer with relatively low oxidation (doping) and reduction (dedoping) potentials. Nonetheless, this study offered a novel, facile, and low-cost in-cell e-polymerization method to produce a conductive polymer binder to boost the performance of Li–S batteries and potentially other types of rechargeable batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Discovery Grants (RGPIN-2016-04366) and Strategic Partnership Grants for Projects (STPGP 506317-17; STPGP 521458) of the Natural Sciences and Engineering Research Council of Canada (NSERC).Notes and references
- B. Dunn, H. Kamath and J. Tarascon, Science, 2011, 334, 928–936 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS.
- R. Fang, S. Zhao, Z. Sun, D.-W. Wang, H.-M. Cheng and F. Li, Adv. Mater., 2017, 1606823 CrossRef.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1–54 Search PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS.
- Y. S. Su, Y. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 14495–14499 RSC.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS.
- T. H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef.
- Z. Yu, Y. Xia, D. Du and J. Ouyang, ACS Appl. Mater. Interfaces, 2016, 8, 11629–11638 CrossRef CAS.
- W. Li, Q. Zhang, G. Zheng, Z. W. Seh, H. Yao and Y. Cui, Nano Lett., 2013, 13, 5534–5540 CrossRef CAS.
- J. Pan, G. Xu, B. Ding, Z. Chang, A. Wang, H. Dou and X. Zhang, RSC Adv., 2016, 6, 40650–40655 RSC.
- L. Yan, X. Gao, J. P. Thomas, J. Ngai, H. Altounian, K. T. Leung, Y. Meng and Y. Li, Sustainable Energy Fuels, 2018, 2, 1574–1581 RSC.
- Z. Wang, Y. Chen, V. Battaglia and G. Liu, J. Mater. Res., 2014, 29, 1027–1033 CrossRef CAS.
- Y. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910–8915 CrossRef CAS.
- Y. Yang, G. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. Bao and Y. Cui, ACS Nano, 2011, 5, 9187–9193 CrossRef CAS.
- G. C. Li, G. R. Li, S. H. Ye and X. P. Gao, Adv. Energy Mater., 2012, 2, 1238–1245 CrossRef CAS.
- N. Nakamura, T. Yokoshima, H. Nara, T. Momma and T. Osaka, J. Power Sources, 2015, 274, 1263–1266 CrossRef CAS.
- N. Nakamura, Y. Wu, T. Yokoshima, H. Nara, T. Momma and T. Osaka, J. Electrochem. Soc., 2016, 163, A683–A689 CrossRef CAS.
- F. Li, M. R. Kaiser, J. Ma, Z. Guo, H. Liu and J. Wang, Energy Storage Mater., 2018, 13, 312–322 CrossRef.
- Y. Li, W. Wang, X. Liu, E. Mao, M. Wang, G. Li, L. Fu, Z. Li, A. Y. S. Eng, Z. W. Seh and Y. Sun, Energy Storage Mater., 2019, 23, 261–268 CrossRef.
- C. Z. Zhao, Q. Zhao, X. Liu, J. Zheng, S. Stalin, Q. Zhang and L. A. Archer, Adv. Mater., 2020, 32, 1–8 Search PubMed.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- H. Zhou, H. Liu, Y. Li, X. Yue, X. Wang, M. Gonzalez, Y. S. Meng and P. Liu, J. Mater. Chem. A, 2019, 7, 16984–16991 RSC.
- Q. Liu, B. Cai, S. Li, Q. Yu, F. Lv, F. Kang, Q. Wang and B. Li, J. Mater. Chem. A, 2020, 8, 7197–7204 RSC.
- T. P. A. Van Der Pol, S. T. Keene, B. W. H. Saes, S. C. J. Meskers, A. Salleo, Y. Van De Burgt and R. A. J. Janssen, J. Phys. Chem. C, 2019, 123, 24328–24337 CrossRef CAS.
- J. G. Ibanez, M. E. Rincón, S. Gutierrez-Granados, M. Chahma, O. A. Jaramillo-Quintero and B. A. Frontana-Uribe, Chem. Rev., 2018, 118, 4731–4816 CrossRef CAS.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- W. J. Albery, F. Li and A. R. Mount, J. Electroanal. Chem., 1991, 310, 239–253 CrossRef CAS.
- A. J. Downard and D. Pletcher, J. Electroanal. Chem., 1986, 206, 147–152 CrossRef CAS.
- J. F. Rubinson and Y. P. Kayinamura, Chem. Soc. Rev., 2009, 38, 3339–3347 RSC.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS.
- D. Wei, P. Espindola, T. Lindfors, C. Kvarnström, J. Heinze and A. Ivaska, J. Electroanal. Chem., 2007, 602, 203–209 CrossRef CAS.
- C. Li and T. Imae, Macromolecules, 2004, 37, 2411–2416 CrossRef CAS.
- M. Wieland, C. Malacrida, Q. Yu, C. Schlewitz, L. Scapinello, A. Penoni and S. Ludwigs, Flex. Print. Electron., 2020, 5, 014016 CrossRef CAS.
- T. A. Yemata, Y. Zheng, A. K. K. Kyaw, X. Wang, J. Song, W. S. Chin and J. Xu, RSC Adv., 2020, 10, 1786–1792 RSC.
- T. Liu, K. C. Kim, B. Lee, S. Jin, M. J. Lee, M. Li, S. Noda, S. S. Jang and S. W. Lee, ACS Appl. Energy Mater., 2020, 3, 3728–3735 CrossRef CAS.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS.
- J. He, Y. Chen and A. Manthiram, iScience, 2018, 4, 36–43 CrossRef CAS.
- L. Hu, C. Dai, J. M. Lim, Y. Chen, X. Lian, M. Wang, Y. Li, P. Xiao, G. Henkelman and M. Xu, Chem. Sci., 2018, 9, 666–675 RSC.
- W. Walker, V. Giordani, J. Uddin, V. S. Bryantsev, G. V. Chase and D. Addison, J. Am. Chem. Soc., 2013, 135, 2076–2079 CrossRef CAS.
- V. S. Bryantsev, J. Uddin, V. Giordani, W. Walker, G. V. Chase and D. Addison, J. Am. Chem. Soc., 2014, 136, 3087–3096 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00797h |
| This journal is © The Royal Society of Chemistry 2021 |