
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-dehydration of a reactive epichlorohydrin-containing organic mixture using a defect-free thin carbon molecular sieve composite membrane†
Seong-Joong
Kim‡
 a,
YongSung
Kwon‡
ab,
DaeHun
Kim
a,
Hosik
Park
a,
Young Hoon
Cho
a,
Seung-Eun
Nam
*a and
You-In
Park
*a
a,
YongSung
Kwon‡
ab,
DaeHun
Kim
a,
Hosik
Park
a,
Young Hoon
Cho
a,
Seung-Eun
Nam
*a and
You-In
Park
*a
aGreen Carbon Research Center, Korea Research Institute of Chemical Technology (KRICT), Gajeong-ro 141, Daejeon 34114, Korea. E-mail: senam@krict.re.kr; yipark@krict.re.kr; Fax: +82-42-860-7283; Tel: +82-42-860-7241 Tel: +82-42-860-7243
bDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, South Korea
First published on 16th March 2021
Abstract
A high-performance thin carbon molecular sieve (CMS) composite membrane was prepared using a drop-coating process for dehydration of a ternary mixture (water/IPA/ECH) by a pervaporation process. The amount of polymer solution dripped onto an alumina disc was varied during the drop-coating process to optimize the formation of the desired active-carbon layer. A defect-free CMS membrane was obtained with a dripped amount of 110 μL, resulting in high gas permeance and optimized selectivity. Furthermore, the CMS membrane was carbonized at 650–800 °C, leading to higher selectivity at elevated temperature with the decrease of gas permeance due to the shrinkage of its graphene-like carbon structure. For separation of the water/IPA/ECH ternary mixture, the highest separation performance (total flux 1.05 kg m−2 h−1) and separation factor (158![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 692) were obtained from the CMS membrane carbonized at 700 °C. On the other hand, the CMS membrane showed a unique separation property: the water flux increased with increasing water content in the feed, without remarkable reduction of the separation factor (still >150000). This is attributed to the rigid pore structure of the CMS membrane, which provides a molecular sieving separation mechanism. Furthermore, increasing the feed-solution temperature led to an increase of both the total flux and separation factor. The CMS membrane performance exhibited the highest separation factor (when compared with other membranes) for dehydration of the water/IPA binary mixture via pervaporation. Moreover, it was much higher than that of a polymeric membrane for dehydration of the water/IPA/ECH ternary mixture.
692) were obtained from the CMS membrane carbonized at 700 °C. On the other hand, the CMS membrane showed a unique separation property: the water flux increased with increasing water content in the feed, without remarkable reduction of the separation factor (still >150000). This is attributed to the rigid pore structure of the CMS membrane, which provides a molecular sieving separation mechanism. Furthermore, increasing the feed-solution temperature led to an increase of both the total flux and separation factor. The CMS membrane performance exhibited the highest separation factor (when compared with other membranes) for dehydration of the water/IPA binary mixture via pervaporation. Moreover, it was much higher than that of a polymeric membrane for dehydration of the water/IPA/ECH ternary mixture.
1. Introduction
Epoxy resins are a group of reactive compounds with epoxy rings and have the characteristics of excellent mechanical strength, high adhesion, high heat and electrical resistance, and durability.1 Therefore, these resins have been commercially available for use in adhesives, paints and coatings, electronic materials, and composite materials.2 Furthermore, in 2019, the epoxy-resin world market was valued at more than $5.9 billion and this is expected to increase to $10.3 billion by 2027 (CAGR of 7.0%).3Epoxy resins are generally synthesized by polymerization of epichlorohydrin (ECH) and phenol, with isopropyl alcohol (IPA) as a solvent.2 To control the molecular weight of the epoxy resin, an excess of ECH is included in the raw material. After the reaction, water remains as a by-product, along with the epoxy resin (product), a chlorinated impurity, and the unreacted initial raw materials (IPA and ECH).4 For the reuse of ECH from the unreacted and residual mixture, the chlorinated impurity can be removed via a simple distillation process. However, the IPA/ECH mixture forms an azeotrope with water during the distillation process, which hinders purification of the mixture into a high-purity product.5,6 Accordingly, the water/IPA/ECH mixture remaining after the distillation process has typically just been reused. This causes a gradual reduction in the reaction efficiency and eventually demands disposal because the composition becomes inadequate for reuse.7 Therefore, if water (high heat capacity) is preferentially removed through a membrane process, and the IPA/ECH components are separated and purified through a distillation process, the energy efficiency of the epoxy-resin manufacturing process can be improved by reusing unreacted materials. In addition, it is possible to implement an environment-friendly epoxy-resin production process by preventing the emission of ECH, which is an environmentally harmful material.8
Pervaporation is a unique separation process in which the feed mixture is supplied as a liquid; then, the liquid feed is converted to the vapor phase during the separation process through decompression of the membrane permeate side. This process is commonly used for dehydration and removal of substances from an organic aqueous solution and for separation of organic–organic liquid mixtures.9–12 In particular, many studies have been done to overcome the limitations of distillation processes such as the separation of components in an azeotropic mixture (water–alcohol).10,13 Furthermore, the pervaporation process has the advantages of low operating cost, as well as being environment-friendly and safe.14,15 Indeed, it is a relatively lower-energy consumption process than distillation because it operates under mild heating conditions.16,17 Most of the energy used for pervaporation is required to maintain the vacuum state on the permeate side of the membrane.
The separation mechanism of a pervaporation membrane follows a solution–diffusion model based on the chemical potential gradient between the membrane material and penetrants.12 In particular, hydrophilic polymers such as polyvinyl alcohol (PVA),18,19 chitosan,20 Nafion,21 and polyacrylonitrile (PAN)22 have been widely used for dehydration involving an organic aqueous solution. However, these polymeric membranes come with difficulties related to relatively low chemical and thermal stability, swelling at high temperature, and water in the feed, as well as a performance trade-off between permeation flux and selectivity.23–26 Recently, for the dehydration of water/IPA/ECH mixtures, Chaudhari et al. studied the stability and separation efficiency of PVA/PVAm (polyvinyl amine) blended membranes.7 This membrane was stable for up to 15 days in the reactive ECH-containing mixture solution, but it was relatively less stable than a crosslinked PVA membrane. Furthermore, they modified the surface of PVA/TEOS (tetraethyl orthosilicate) membranes with PVAm/STA (silicotungstic acid). With these, an initial water content drop to 96.23% was observed in the permeate. The membrane was then operated for 168 h without major deviation.27
Alternatively, microporous ceramic membranes including silica, zeolites, and carbon molecular sieves (CMS) have been investigated as promising candidates for dehydration of organic mixtures due to their superior chemical and thermal stability. Nevertheless, there are drawbacks: silica has a limited chemical and hydrothermal stability;23,28 with zeolites it is difficult to synthesize a defect-free membrane and it also exhibits acid sensitivity.28,29 In addition, most inorganic membranes have problems related to their high material cost and to the complexity of their synthesis or manufacturing.28
The CMS membrane is attractive for the pervaporation process due to its excellent separation properties and chemical and thermal stability with a simple manufacturing process,30 but it has seldom been used for pervaporation processes. The CMS membrane with an ultra-micro pore structure (3–6 Å) can offer high water permeation flux and a high separation factor through its molecular sieving separation mechanism. Moreover, a CMS membrane possesses an anti-swelling property in water-containing mixtures even at elevated temperatures.31 Indeed, the excellent pervaporation performance and stability of CMS membranes were observed for water/alcohol mixtures (ethanol, methanol, and IPA),28,32–36 water/acetic acid,32 and benzene/cyclohexane.37 Furthermore, the brittleness of the initial CMS membranes was overcome by depositing a carbon layer on a porous substrate such as alumina and stainless steel.35 However, depositing a polymer solution on a substrate is quite complex, costly, and a time-consuming process due to the need for the coating process, which, in some case, needs an intermediate layer and repetitive coating process. The intermediate layer has frequently been employed to deposit a thin, uniform active layer without defects by decreasing the pore size and narrowing the pore-size distribution of the support surface.28 However, a negative influence of the support and intermediate layer on the membrane performance has been often reported in terms of its diffusion resistance.38,39 Hence, to achieve the maximized performance of a CMS membrane supported by a porous substrate, the number of intermediate layers should be minimized, and the manufacturing process simplified as well.
In this study, to investigate defect-free high-performance CMS composite membranes, a simple one-step drop-coating process was employed on an alumina disc without an additional intermediate layer. These CMS membranes were used for the dehydration of ECH-containing aqueous mixtures. To the best of our knowledge, the dehydration of an ECH-containing aqueous mixture using a pervaporation process with a CMS membrane has been never reported. Moreover, CMS-membranes have rarely been prepared using a drop-coating method.
Commercial Matrimid polyimide was employed as a precursor, and has the properties of high glass-transition temperature, high carbon content, and thereby, attractive permeance and selectivity. The CMS membranes were deposited on alumina discs by varying the amount of polymer solution dripped during drop-coating, and then carbonized in the temperature range of 650–800 °C to tailor the separation performance. Finally, the CMS membranes were evaluated for the separation of a water/IPA/ECH mixture as well as for the separation of gases such as He, H2, CO2, N2, and C3H8. Furthermore, the temperature and concentration of the feed mixtures were varied during the pervaporation tests.
2. Experimental
2.1 Materials
Alumina powder (CR15) and Matrimid 5218 were purchased from Baikowski and Huntsman Chemical Company, respectively. They were dried at 60 °C for more than 1 week before use. All the solvents of n-methyl-2-pyrrolidone (NMP, ≥99.5%), isopropyl alcohol (IPA, 99.9%), and epichlorohydrin (ECH, >99.0%) were supplied by Samchun Chemicals. For the gas permeation test, He (99.999%), H2 (99.99%), CO2 (99.999%), N2 (99.999%), and C3H8 (99.99%) were used.2.2 Preparation of a supported CMS membrane on an alumina disc
An alumina disc was fabricated by the powder-pressing method to use as a substrate. A portion of alumina powder (1.5 g) was used to fill a die of 22 mm diameter, and then compacted using upper and lower punches at 100 bar. For calcination of an ejected alumina disc, the sample was put in a muffle furnace, and the temperature was increased to 600 °C at a heating rate of 2 °C min−1 and a soaking time of 1 h. Then the temperature was raised to 1180 °C at a ramping rate of 1 °C min−1. After remaining for 1 h at 1180 °C, the furnace was cooled by natural convection, and an alumina disc with a diameter of 21 mm, a thickness of 2.22 mm, and a porosity of 13.3 ± 0.76% was finally obtained. The alumina disc was polished using sand paper (1500 grit) and kept in an oven at 60 °C for at least 6 h prior to the polymer coating.The drop-coating procedure with polymer solution is illustrated in Fig. 1. A polymer solution was prepared by dissolving 5 wt% Matrimid in NMP at 60 °C, and then stirred vigorously for 24 h. Various amounts of Matrimid solution (in the range of 80–130 μL) were dripped onto the alumina disc kept in the oven at 60 °C. Then, the Matrimid solution was spread uniformly using tweezers. The alumina disc with a uniform layer of polymer solution was dried in the oven at 60 °C for 6 h. The polymer-coated alumina disc was annealed again at 150 °C under vacuum for more than 24 h to remove any residual solvent.
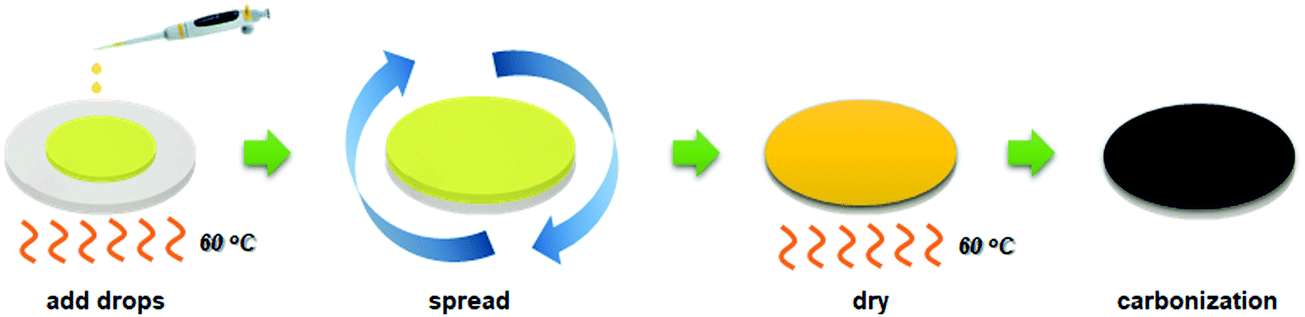 | ||
| Fig. 1 Deposition of a defect-free thin CMS composite membrane on the alumina substrate via the drop-coating method. | ||
For the carbonization of the polymer-coated alumina disc, the sample was placed on an alumina ceramic plate, and pyrolyzed in a tubular furnace under ultra-high purity helium gas (99.9999%) with a flow rate of 50 cc min−1 after purging the tube inside at a flow rate of 1000 cc min−1 for 30 min. The carbonization temperature (650–800 °C) was reached by heating at a rate of 2.8 °C min−1, given 1 h of soaking time, and then cooled to <50 °C by natural convection.
2.3 Characterization
The functionalities of the CMS membranes after pyrolysis were characterized using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR, Bruker ALPHA-P FTIR spectrometer). An X-ray photoelectron spectrometer (XPS, AXIS NOVA, KRATOS Analytical) was utilized to investigate the chemical change of the CMS membranes. The XPS analysis was carried out using a monochromatic Al-Kα X-ray source (15 keV) at a pass energy of 160 and 40 eV for survey analysis and high-resolution analysis, respectively. The pressure in the measurement chamber was maintained at <1.0 × 10−8 torr. The structural change of the CMS membranes was studied using X-ray diffraction (XRD) patterns from a Rigaku D/Max-2200V diffractometer attached to a Cu tube and a graphite monochromator at 40 kV. Raman spectra were recorded with a SENTERRA spectrometer (Bruker) using an excitation source with 532 nm wavelength. To monitor the surface and cross-section of the CMS membrane and the carbon layer deposited on the alumina disc, a scanning electron microscope (SEM, SEC SNE-4500M) and an energy dispersive X-ray spectroscope (EDS, XFlash6160, Bruker) equipped with a SEM (MAGNA FEG, Tescan) were employed. The thermal decomposition behavior of the Matrimid polyimide was characterized using thermo-gravimetric analysis (TGA, TGA Q5000, TA Instruments, Inc.) at a heating rate of 10 °C min−1 under nitrogen gas. The surface properties of CMS membranes were measured using contact angle (CA, DSA30S, KRÜSS GmbH). Deionized water was dripped onto the surface of the CMS membrane, and this was repeated five times to ensure reliability.2.4 Gas permeation test
To identify the CMS membrane with optimized performance during drop-coating and to assess the effective pore size of the CMS membranes carbonized at various temperatures, pure-gas permeation tests were performed using an aluminum module with dead-end flow. High purity gases (i.e., He, H2, CO2, N2, and C3H8) were fed into the side of the carbon layer. The gas was thoroughly purged prior to measurement to replace completely the previous gas with the one being measured. The operating pressure was adjusted using a gas regulator and monitored using a pressure gage. The volumetric flow rate on the permeate side was measured using a bubble flow meter. All gas permeation tests were implemented at 2 bar and 25 °C. The gas permeance was indicated in gas permeation units (GPU) as in the following equation:40| 1 GPU = 10−6 cm3 [STP]/(cm2 s cmHg) |
2.5 Pervaporation test
The feed solutions, which included pure water and water/IPA and water/IPA/ECH mixtures, were prepared by mixing each component by weight ratio. The water content in the feed solutions was kept in the range of 5–20 wt%. For the water/IPA/ECH mixture, the weight ratio of ECH to IPA was always fixed at 5 to 3. A portion of each feed solution (2 L) was kept in a SUS double jacket at specific temperatures (30–60 °C) to maintain the feed solution at a consistent temperature. The pervaporation test was carried out in a customized cross-flow system. The mixture solution was fed onto the carbon layer at a flow rate of 0.5 L min−1 using a Teflon-coated gear pump. No extra pressure was applied on the feed side. The opposite (permeate) side of the CMS membrane was kept under vacuum (1.5 torr). The penetrants were condensed in a cold trap using liquid nitrogen and then weighed. The weight fraction in the solution was measured using an FID detector-equipped gas chromatograph (GC, DS 6200, Donam Co.). The detection limit of the GC was 5 ppm for each gradient. The pervaporation experiment was repeated three times, and the membrane performance was determined by averaging the data. The permeation flux (J) and separation factor (α) were calculated as shown in the following equations:41where W is the permeated weight, A is the effective membrane surface area, and t is the permeation time.
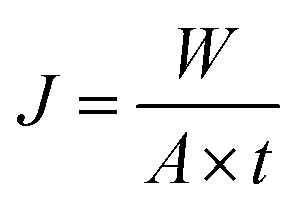
where p is the weight fraction of the permeate side and f is the weight fraction of the feed.
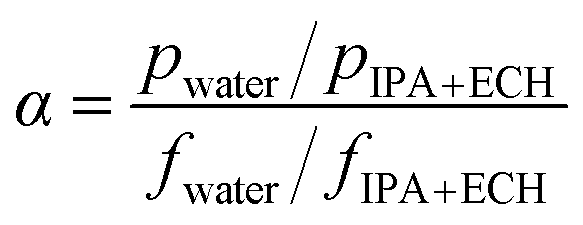
3. Results and discussion
Drop-coating is a simple process used to form a uniform layer on a porous support and is cost-effective due to its ease of operation.42 B. Zhang et al. utilized drop-coating for the deposition of a polymer layer with polyetherimide and resorcinol-formaldehyde resin for a carbon membrane.43 However, the derived carbon layer was non-uniform, rough, and thick (>50 μm), leading to undesirable gas-separation performance. Furthermore, to achieve acceptable separation performance, the drop and drying processes were repeatedly carried out (several times).44In this study, drop-coating with polyimide solution provided a defect-free, uniform carbon layer in a single step. The polyimide was suitable for forming a desirable coating layer even with sub-micro thickness due to its high thermal stability, which minimizes the deformation (e.g., cracks, delamination, and absorption into the substrate) of the coating layer during pyrolysis without any further post-treatment process (e.g., oxidation and crosslinking). The coating process was carried out in an oven at 60 °C because low temperature (<40 °C) caused constant absorption of polymer solution into the surface pores of the alumina substrate over a long retention time. Higher temperature (>80 °C) induced fast evaporation of the solution, resulting in an absorbed and defective coating layer. The drop-coating process applied in this study has shown high reproducibility of the coating thickness (see S2, ESI†).
3.1 Optimization of the drop-coating process
The drop-coating process was optimized by varying the amount of polymer solution dripped onto the substrate. The SEM/EDS images of the CMS membrane are shown in Fig. 2. The aluminum component is indicated in the carbon layer, in particular for the membranes coated with 80 and 90 μL. This indicates that a small drop-amount induces the absorption of polymer solution into the substrate. The CMS membranes with drop-amounts >100 μL seem to be uniform and defect free. This phenomenon is also observed visually in photographs of the polymer and CMS membranes (see S1, ESI†). An area of the partially absorbed polymer on the porous alumina support is observable in the membranes coated with small drop-amounts (80–90 μL), and this leads to a lack of uniformity and an unacceptable carbon layer after pyrolysis. S3 and S4 (ESI†) show the SEM images of polymeric and CMS membranes prepared with different drop-amounts of polymer solution, respectively. The polymeric layer on the alumina disc was deposited to a thickness of 6.91–12.1 μm. After pyrolysis, the thickness of the CMS layer is then decreased to the range of 2.38–6.19 μm, which is a relatively thin layer compared with CMS membranes prepared in other studies using the drop-coating method.42–44 The polymeric layer is shrunk about 50% due to the decomposition and release of volatile compounds. However, the reduction ratio of the CMS layer coated with the polymer solution of 80 μm was about 75%, attributed to the severe absorption of polymer solution into the porous alumina disc. In particular, a lot of voids are observed at the bottom of the polymeric layer, which may give rise to severe reduction during the carbonization process. | ||
| Fig. 2 SEM/EDS analysis of CMS composite membranes deposited via the drop-coating method. | ||
To optimize the drop-amount of the polymer solution and to verify the integrity of the CMS membranes, gas permeation tests were performed with He, H2, CO2, and N2 gases, the results of which are listed in Table 1. The gas permeance of all the CMS membranes are as follows: H2 (2.89 Å) > CO2 (3.30 Å) > He (2.60 Å) > N2 (3.64 Å), which corresponds to the kinetic diameter of the gas molecules, except for helium due to its inert property.45 The CMS membranes consist of micropores (5–10 Å) and ultra-micropores (<5 Å), which serve in selective surface flow and molecular sieving separation, respectively.46 Thus, condensable CO2 can strongly interact with the CMS membrane, resulting in higher permeance than for helium.
| Drop amount (μL) | Gas permeance (GPU) | Ideal selectivity | |||||
|---|---|---|---|---|---|---|---|
| He | H2 | CO2 | N2 | H2/N2 | He/N2 | CO2/N2 | |
| 80 | 488.9 | 1847 | 1347 | 185.3 | 9.967 | 2.638 | 7.269 |
| 90 | 329.9 | 1051 | 901.3 | 56.35 | 18.65 | 5.854 | 15.99 |
| 100 | 256.6 | 895.0 | 747.6 | 40.74 | 21.96 | 6.298 | 18.35 |
| 110 | 199.0 | 550.2 | 327.5 | 17.25 | 31.89 | 11.53 | 18.98 |
| 120 | 176.2 | 477.7 | 294.3 | 14.73 | 32.43 | 11.96 | 19.97 |
| 130 | 161.4 | 431.8 | 265.4 | 13.14 | 32.86 | 12.28 | 20.19 |
The gas permeance for all gases decreased with increasing drop-amount of polymer solution, but the selectivity increased. This is attributed to the thicker and defect-free carbon layer of the CMS membranes coated with the larger drop-amounts of polymer solution, as mentioned above. A noticeable increase of selectivity was observed up to the drop-amount of 110 μL while the gas permeance of the CMS membranes steadily decreased. It is believed that defect-free CMS membranes prepared using the drop-coating process can be achieved if the drop-amount is 110 μL. For the polymer composite membrane, the optimal amount of polymer solution was somewhat different compared to the CMS composite membrane (see S5, ESI†). The optimized selectivity could be obtained with the drop amount of polymer solution more than 90 μL while the gas permeance of all the membranes constantly decreased. This different optimal amount between the polymer and CMS layers is attributed to the thicker polymer layer (see S3 and S4, ESI†). The thicker layer can provide a more reliable and defect-less pore structure. However, the absorbed and deposited polymer layer onto the porous alumina disc is constantly decomposed and shrunk during pyrolysis, which may cause a defective mesopore or micropore (∼1 nm). Therefore, to extend the measurement of gas permeation and pervaporation, a CMS membrane coated with a drop-amount of 110 μL was adopted.
3.2 Membrane characterization
The CMS membranes were carbonized at various pyrolysis temperatures (650–800 °C), and then characterized using TGA, ATR-FTIR, Raman spectra, and XRD, as shown in Fig. 3.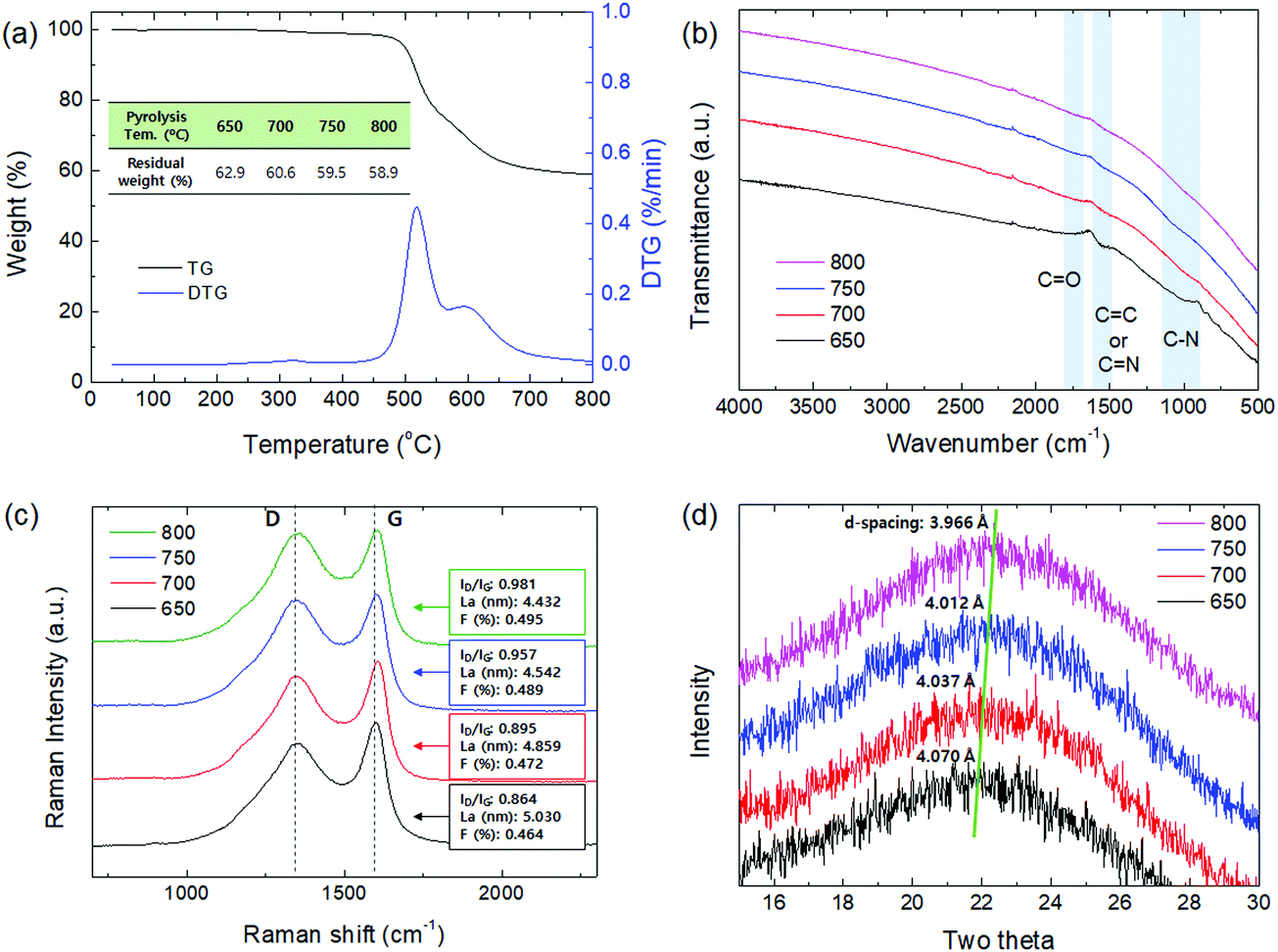 | ||
| Fig. 3 (a) TGA, (b) ATR-FTIR, (c) Raman spectra, and (d) XRD analysis of CMS composite membranes carbonized at a temperature of 650–800 °C. | ||
Fig. 3(a) indicates the result from TGA analysis of polyimide, which was implemented in the range of 30–800 °C. The decomposition of polyimide was initiated near 420 °C and continued until 800 °C. The decomposition area indicated in derivative thermogravimetry (DTG) was divided into two areas at 520 and 600 °C. The former is assigned to pore creation while the latter is assigned to pore compaction and reduction.47 It is believed that significant development and shrinkage of the pores occurred when the applied pyrolysis temperature was more than 600 °C. The residual weight then steadily decreased with an increase of the pyrolysis temperature, implying that the CMS pore structure became more compact as the pyrolysis temperature increased.
The chemical structure of the CMS membrane is indicated in Fig. 3(b). The overall peaks in all the CMS membranes are indistinguishable because during pyrolysis, the polymer precursor undergoes decomposition of its volatile compounds and rearrangement of its pore structure. Nevertheless, three broad, weak peaks were observed at 1750, 1556, and 1020 cm−1 (corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC or CN, and C–N, respectively).34,48 With increasing pyrolysis temperature, the intensity of the peaks was clearly weaker, and then after membrane carbonization at 800 °C, no CMS membrane peaks were detected. After the evolution of polyimide during pyrolysis, the CMS membrane was mainly composed of C–C (sp3) and CC (sp2) bonds with minor chains such as CO, CN, and C
O, CC or CN, and C–N, respectively).34,48 With increasing pyrolysis temperature, the intensity of the peaks was clearly weaker, and then after membrane carbonization at 800 °C, no CMS membrane peaks were detected. After the evolution of polyimide during pyrolysis, the CMS membrane was mainly composed of C–C (sp3) and CC (sp2) bonds with minor chains such as CO, CN, and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, as shown in S6 (ESI†). The higher carbonization temperature caused the transformation of more C–C (sp3) into CC (sp2) bonds. Moreover, reduction of the peak intensity in the N1s spectra of the CMS membranes carbonized at higher temperature was observed in S7 (ESI†), indicating more decomposition of the nitrogen-containing compounds.
N, as shown in S6 (ESI†). The higher carbonization temperature caused the transformation of more C–C (sp3) into CC (sp2) bonds. Moreover, reduction of the peak intensity in the N1s spectra of the CMS membranes carbonized at higher temperature was observed in S7 (ESI†), indicating more decomposition of the nitrogen-containing compounds.
Raman spectroscopy was carried out to investigate the structure of CMS membranes prepared at different pyrolysis temperatures, as indicated in Fig. 3(c). We confirmed two peaks corresponding to the disordered D-band (∼1300 cm−1) and graphitic G-band (1600 cm−1), which represent amorphous carbon and ideal graphene sheets, respectively.32 The CMS membrane is a turbostratic structure in which two structures coexist. With an increase in the pyrolysis temperature, the ID/IG ratio (intensity of D-/G-bands) increases, which indicates a more amorphous structure. Moreover, we can calculate the average in-plane crystallite size (La) and fractional value (F) for the amount of disordered carbon using the following equations:49
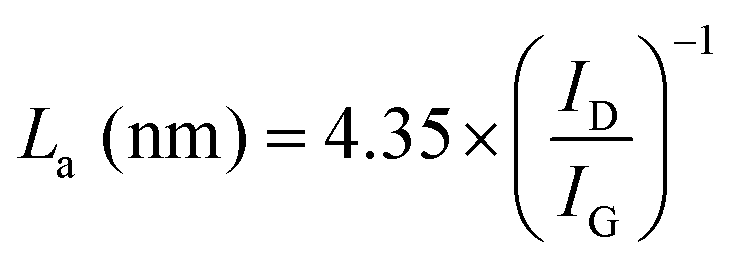
Increasing the pyrolysis temperature offers smaller crystallite size and a higher fractional value of disordered carbon due to the more severe decomposition of volatile compounds at higher temperature. This causes constant rearrangement of the carbon structure, resulting in the presence of a more disordered carbon structure.
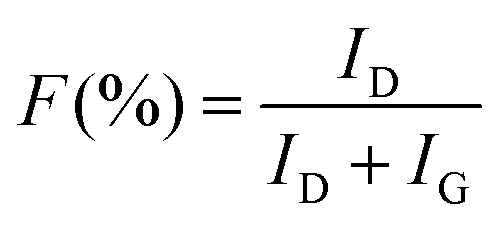
On the other hand, the constant evolution during pyrolysis can lead to greater compaction and shrinkage of the carbon structure,50 and this was reflected in the XRD patterns in Fig. 3(d). The amorphous peaks in the range of 22–23° appeared and shifted slightly to higher two-theta values at higher pyrolysis temperatures, indicating reduction of the d-spacing. This is attributed to the constant decomposition of polymer chains and rearrangement of graphitic carbon layers, leading to a smaller in-plane size and more severe densification of the carbon layer with increasing pyrolysis temperature.
As shown in Fig. 4, the contact angle of the CMS membranes was measured to determine their hydrophilicity, which is a property important for water permeation in the pervaporation process. In general, a hydrophilic surface has good affinity with water, thus enabling water to spread and penetrate better. The contact angle of the CMS membrane carbonized at 650 °C was 62.7°; however, the contact angle increased to 80.6° with an increase in the pyrolysis temperature to 800 °C. This is attributed to the higher carbon content and carbon–carbon bonds of CMS membranes carbonized at higher temperature due to the constant decomposition of volatile compounds (see S6 and S7 (ESI†), and Fig. 3b).
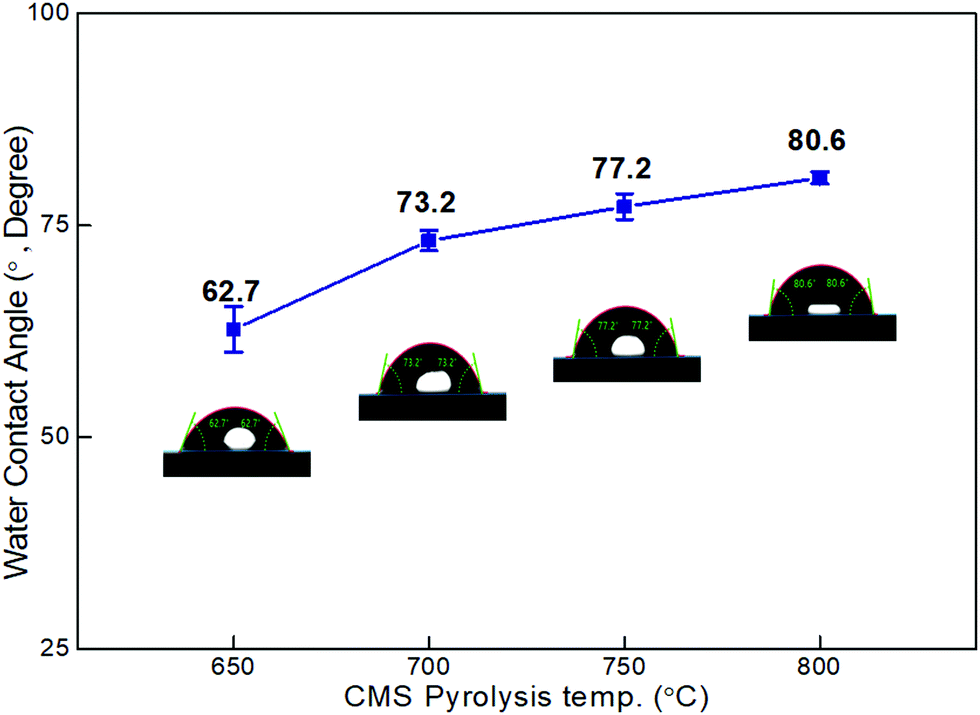 | ||
| Fig. 4 Contact angle of CMS composite membranes carbonized at temperatures within the range of 650–800 °C. | ||
3.3 Gas permeation properties
Gas permeation tests of the CMS membranes were carried out using He, H2, CO2, and N2 gases (which presented different kinetic diameters), as shown in Fig. 5. As mentioned earlier, the gas permeance of the CMS membrane carbonized at 650 °C in decreasing order was: H2 > CO2 > He > N2 > C3H8. The permeation of C3H8 was only observed in the CMS membrane carbonized at 650 °C, and even then, its permeance was very low (0.98 GPU). The CMS membranes carbonized at >700 °C completely obstruct the diffusion of the C3H8 molecule into the pores by sieving separation. The N2 permeance of the CMS membrane carbonized at 700 °C was remarkably lower (0.61 GPU) than those for the other gases. This implies the formation of a pore structure selective for molecules larger than 3.64 Å. This led to high performance in the selection of H2 and CO2 against N2, as shown in Fig. 5(b). The selectivity for H2 from H2/N2 and CO2 from CO2/N2 is 468 and 259, respectively. However, after carbonization at 750 °C, the CMS membrane undergoes further compaction and pore reduction, resulting in more extreme reduction of CO2 permeance than that of H2 and He. Eventually, He and CO2 permeance was reversed at a pyrolysis temperature of 800 °C. The H2/N2 selectivity of the CMS membrane was maximized at 750 °C, but was reversed at 800 °C due to the severe reduction of pore size, which even hinders the diffusion of H2. However, H2/CO2 selectivity was constantly increased and reached 18.1 (with 109 GPU of H2 permeance) in the CMS membrane carbonized at 800 °C because the reduction ratio of CO2 permeance with increasing pyrolysis temperature was always more severe than that of H2. Consequently, the CMS membrane carbonized at higher temperature could provide more selectivity according to the sieving separation mechanism. Apart from this, the gas separation performance of CMS membranes was outstanding (see S8, ESI†).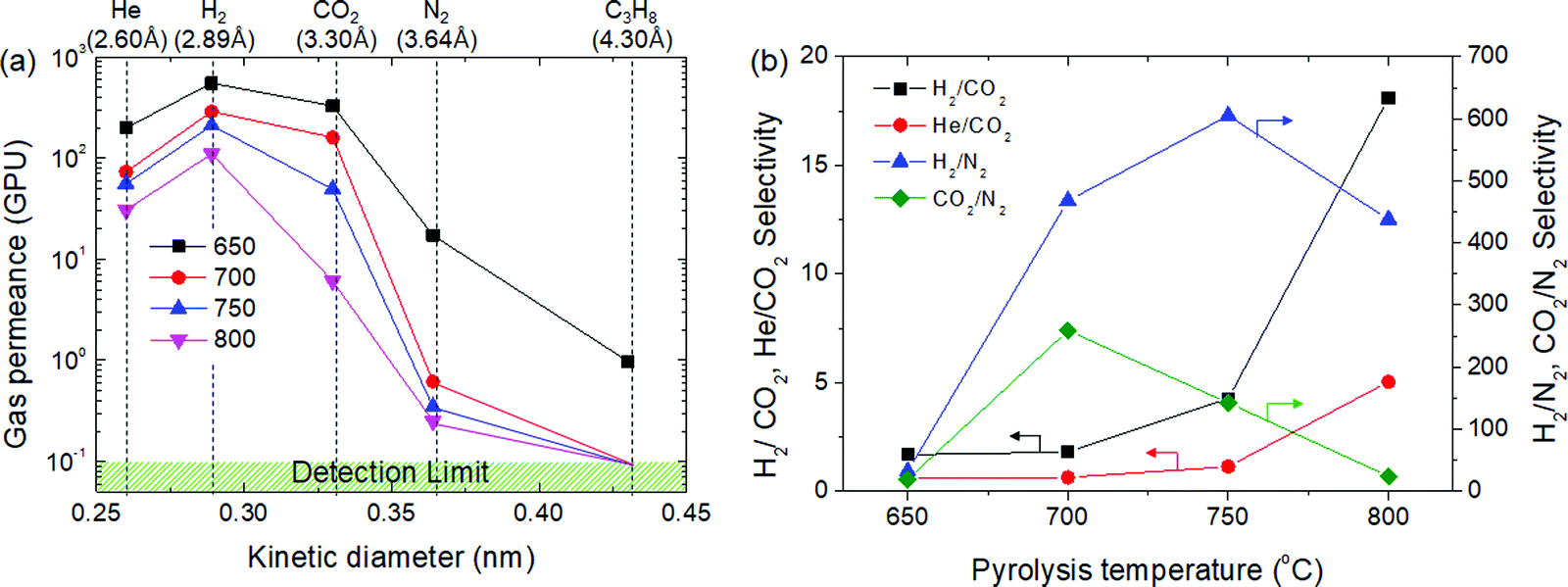 | ||
| Fig. 5 (a) Gas permeance and (b) selectivity of CMS composite membranes carbonized at temperatures in the range of 650–800 °C. | ||
3.4 Pervaporation performances
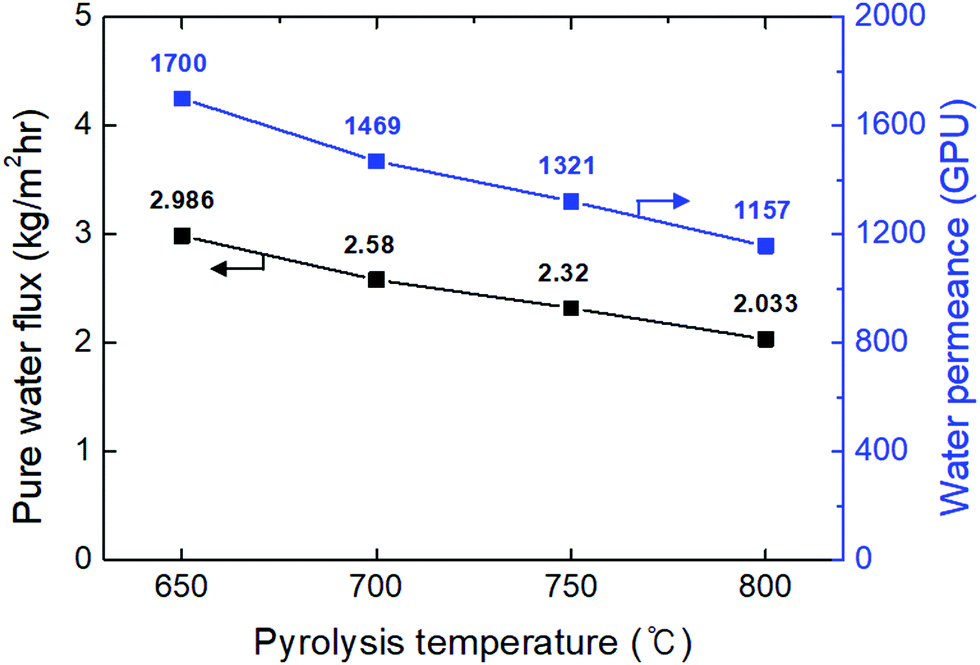 | ||
| Fig. 6 Pure water flux and permeance of the CMS composite membranes carbonized at temperatures of 650–800 °C, with a feed solution temperature of 60 °C. | ||
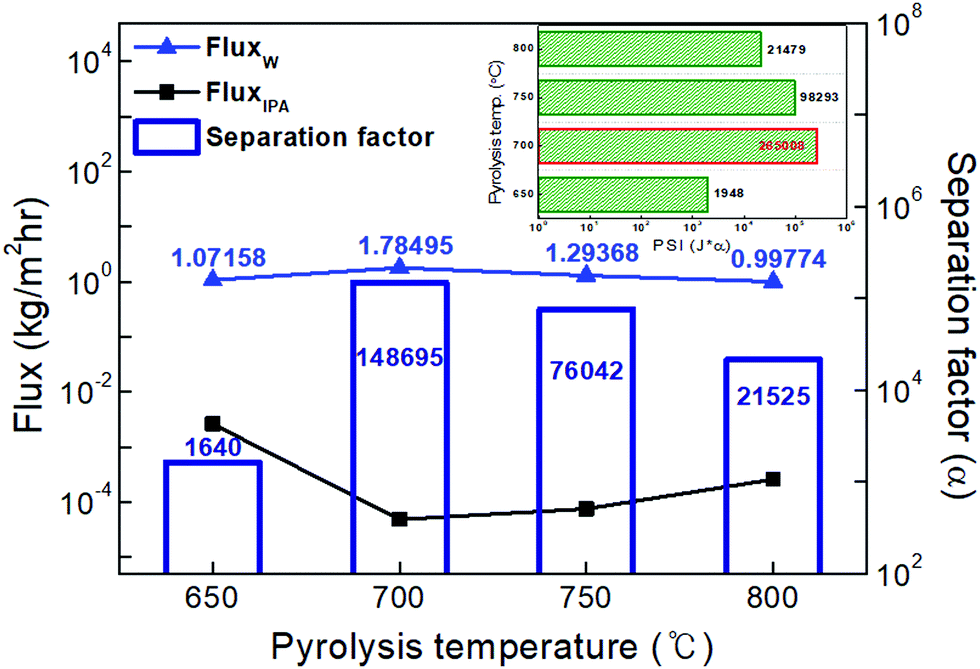 | ||
| Fig. 7 Dehydration performance of CMS composite membranes carbonized at various temperatures (650–800 °C) for a binary mixture of water/IPA (20/80 wt%) and a feed temperature of 60 °C. The small box in the figure presents a Pervaporation Separation Index (PSI, water flux (J) × separation factor α). | ||
The highest water flux and separation factor were observed with the CMS membrane carbonized at 700 °C. This is attributed to the blockage of the CMS membrane to penetration by IPA because the membrane pore size was smaller than that of an IPA molecule. Indeed, the permeance of N2 (kinetic diameter of 3.64 Å) was extremely low (0.62–0.25 GPU). Therefore, the competition between water and IPA molecules penetrating the pores is negligible, leading to water-dominant permeation without hindrance by IPA, resulting in both higher water flux and a higher separation factor. However, the competition between water and IPA can still occur at the membrane surface, resulting in lower water flux in the feed mixture than for pure water flux.
On the other hand, the water flux steadily decreased with increasing pyrolysis temperature. This can be attributed to the severe compaction of the carbon structure, as mentioned in Fig. 3(c and d). Moreover, a CMS membrane prepared at higher pyrolysis temperature was more hydrophobic, which weakens the interaction between water and the membrane surface. Therefore, the retention of water on the membrane surface is less, leading to the reduction of both water flux and the separation factor. To compare the pervaporation performance of the CMS membranes with the gas permeance, the water and IPA flux was transformed to the permeance unit (GPU) as shown in S12 (ESI†). The water permeance of 568–1016 GPU was always higher than for gases such as He, H2, and CO2 while the IPA permeance of 0.027–0.31 GPU was remarkably lower than even nitrogen (0.25–17.25 GPU). This reflects that the sieving separation is predominant during the pervaporation process with CMS membranes.
The pervaporation performance of the CMS membranes when varying the feed composition is indicated in S13 (ESI†). As the water ratio in the feed was increased, the water flux increased while the separation factor decreased. This is attributed to the more water-dominant sorption to the membrane surface with higher water content. Furthermore, it has been often reported that water permeation can be hindered by IPA more than vice versa.32 Nevertheless, it should be noted that the IPA flux of each CMS membrane (regardless of the carbonization temperature) was similar across all the feed compositions due to the size-exclusion property of the CMS membrane.
726, 91112, and 86576) at pyrolysis temperatures of 700, 750, and 800 °C. These are performances that exceeded the high purity of 99.99% for water (see S15, ESI†).
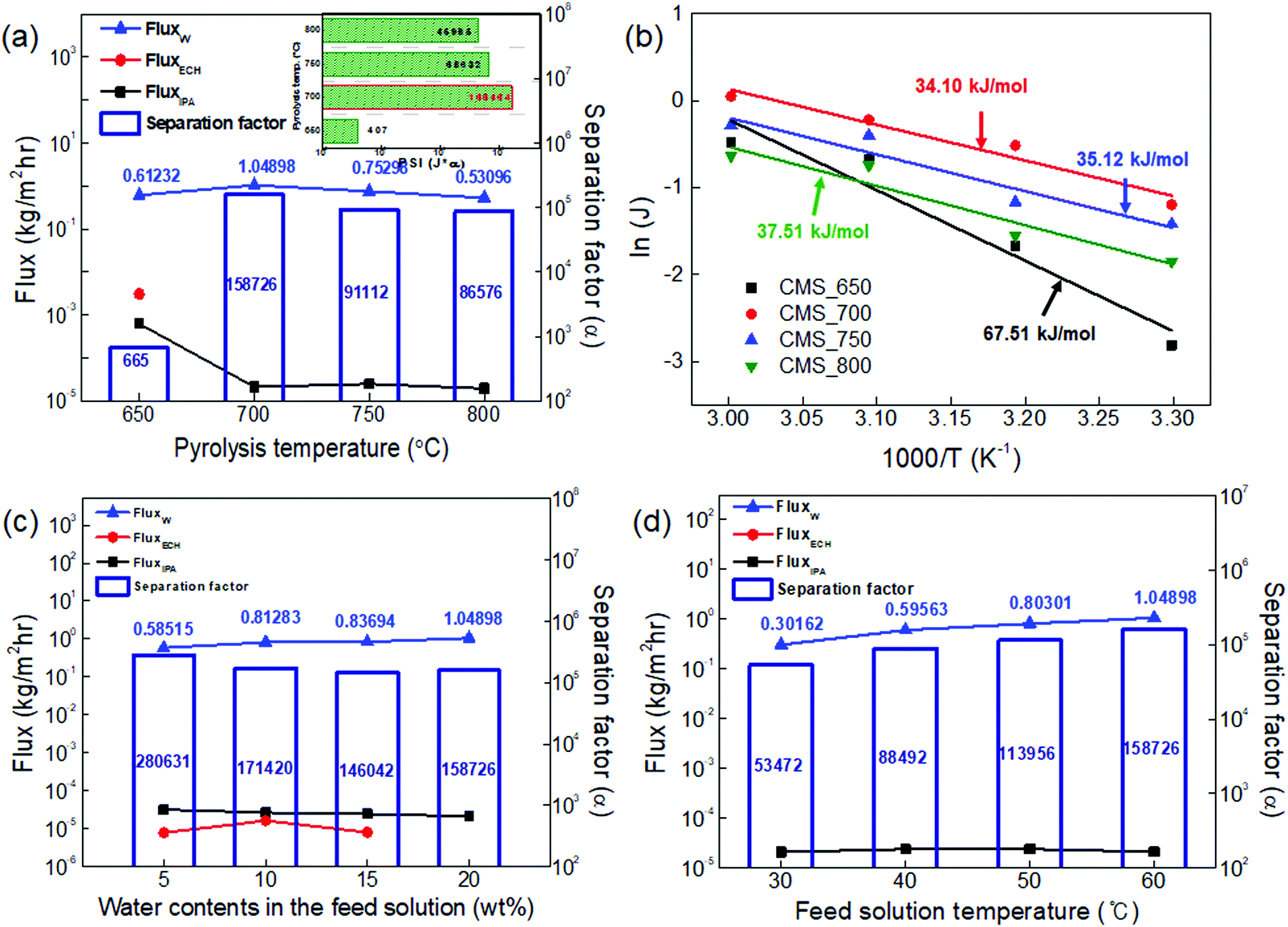 | ||
| Fig. 8 (a) Dehydration performance and (b) activation energy of CMS composite membranes carbonized at a temperature of 650–800 °C for the water/IPA/ECH (20/30/50 wt%) ternary mixture (at a feed temperature of 60 °C). The small box included in the figure represents a PSI, and the effect of (c) water content and (d) temperature in the feed solution on the dehydration performance of the CMS composite membrane carbonized at 700 °C for the water/IPA/ECH mixture. | ||
The activation energy (E) for permeation of the CMS membranes carbonized at temperatures in the range of 650–800 °C was calculated using the Arrhenius equation and is illustrated in Fig. 8(b).55
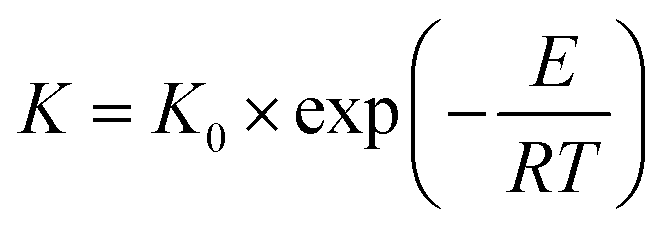
As shown in Fig. 8(c), the influence of the water content in the feed on the pervaporation performance was examined using the CMS membrane carbonized at 700 °C, which showed the most ideal membrane performance. The water flux increased and the separation factor decreased with an increase of the water content in the feed mixture, as mentioned above (S9, ESI†). The separation factor of more than 150000 at a water content of 20 wt% was still high, even though its trend relative to the increase of water content was slightly decreased. The diffusion of IPA and ECH, which have relatively larger molecular size, is hindered due to the rigid pores of the CMS membrane. Therefore, the flux of IPA and ECH slightly decreased with an increase in the water content of the feed mixture, while the water flux steadily increased. This property is advantageous for the pervaporation process. The free volume identified as the diffusive path in polymeric membranes can easily be expanded by swelling at high operating temperature and high water content in the feed solution. This combination gives rise to a significant increase of the total flux and a decrease of the separation factor.56 Similar results for dehydration of a water/alcohol mixture using various polymeric membranes have frequently been reported.9,57,58
Fig. 8(d) shows the performance of the CMS membrane with a feed-solution temperature of 30–60 °C. The water flux and separation factor of the CMS membranes simultaneously increased with an increase in the feed-solution temperature. The relatively low water flux and separation factor at lower temperatures can be attributed to the low vapor-pressure driving force on the feed side. This is a unique property of the CMS membrane associated with the sieving separation mechanism. In contrast, the polymeric membrane typically has a trade-off between the total flux and separation factor because the free volume of the polymer is expanded by higher segmental polymer chain motion at elevated temperature, leading to the effect of increased swelling, as mentioned above.53,59
On the other hand, it should be noted that for the CMS membrane, no stability issue (e.g., swelling, dissolution, cracking, or delamination) was caused by highly reactive ECH during the pervaporation test with the water/IPA/ECH mixture. For the separation of ECH-containing mixtures using a polymeric membrane, the stability of the membrane is one of the main problems to be solved.7,27 Moreover, polymeric membranes undergo swelling, which can cause reduction of the separation factor. In contrast, such problems did not occur with the CMS membrane due to its high chemical and thermal stability and rigid pores.
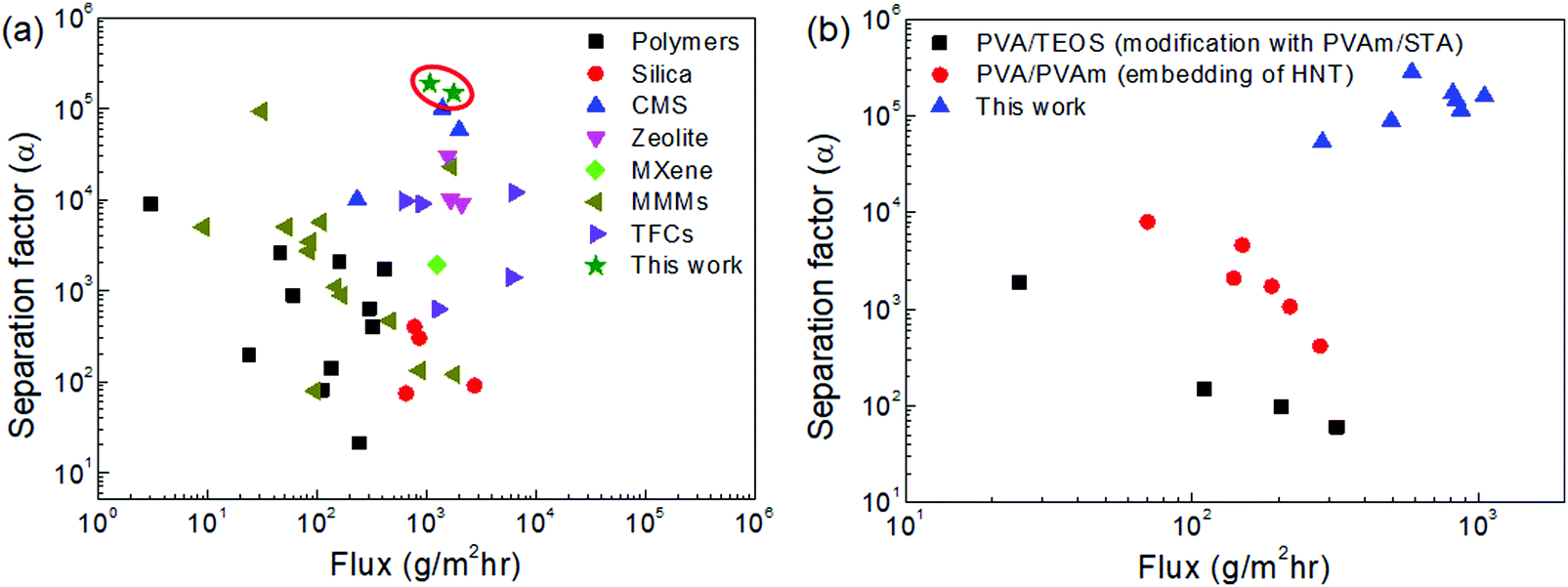 | ||
| Fig. 9 Comparison of the dehydration performance of the CMS membranes studied in this work and other membranes reported in the literature for (a) binary (water/IPA) and (b) ternary (water/IPA/ECH) mixtures (all references are indicated in S16 and 17, ESI†). | ||
Fig. 9(b) and Fig. S17 (ESI†) show the trade-off between the water flux and separation factor of the CMS membranes studied in this work and of the polymer membranes reported in the literature for the pervaporation of a water/IPA/ECH mixture. The CMS membranes remarkably surpassed the performance of the polymer membranes (3–10 times for water flux, and 10 times for the separation factor). This outstanding performance is attributed to the sieving separation property of the CMS membrane due to its rigid pores that prevent deformation of the pore structure such as that occurring in polymer membranes.
4. Conclusions
A thin, defect-free carbon active layer was successfully prepared on each alumina disc using a single-step drop-coating process without an intermediate layer. For the pervaporation process, the highest pure water flux was achieved in the CMS membrane carbonized at 650 °C because its pores were the largest. However, for the dehydration of the feed mixture, this performance could not be properly implemented due to the competition among the penetrants (i.e., water, IPA, and ECH). Otherwise, the CMS membranes carbonized at temperatures higher than 700 °C possessed smaller pores (>3.64 Å) that correspond to the kinetic diameter of N2. Pores of this size block the penetration of relatively larger molecules such as IPA and ECH, resulting in water of high purity (>99.99%) from the dehydration of binary (water/IPA) and ternary (water/IPA/ECH) mixtures. Furthermore, the rigid pore structure of the CMS membranes gave rise to an increase of both water flux and separation factor at elevated feed temperature, following molecular sieving separation. This has advantages over the use of polymeric membranes because in these, the solution-diffusion mechanism is predominant. This means that polymeric membranes commonly undergo significant swelling and expansion of their free volume, resulting in the severe reduction of the separation factor. Finally, the CMS membranes studied in this work achieved the highest separation factor, along with high flux, during dehydration of the water/IPA mixture. Moreover, their separation performance for the water/IPA/ECH mixture surpassed that of the polymeric membranes, more than 3–10 and 10 times for the water flux and separation factor, respectively.On the other hand, the elevated pyrolysis temperature reduces the hydrophilicity of the CMS membrane, and leads to reduction of the water flux. Therefore, it is expected that increasing the hydrophilicity of a CMS membrane, in particular the membrane surface, will give rise to improvement of the water flux and the separation factor. For improved technology using CMS membranes in a pervaporation process, the modification of CMS membranes in terms of their hydrophilic properties has to be studied more in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Council of Science & Technology (NST) grant by the Korea government (MSIP) (No. CRC-14-01-KRICT) and the New & Renewable Energy Core Technology Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) granted financial resource from the Ministry of Trade, Industry and Energy, Republic of Korea (No. 20172010106170).References
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- B. Ellis, Chemistry and Technology of Epoxy Resins, Springer, 1993 Search PubMed.
- A. Choudhary and E. Prasad, Allied Market Research, 2020 Search PubMed.
- S. Chaudhari, D. Chang, K. Cho, M. Shon, Y. Kwon, S. Nam and Y. Park, J. Taiwan Inst. Chem. Eng., 2020, 114, 103–114 CrossRef CAS.
- H.-G. Kim, H.-R. Na, H. R. Lee, M. I. Kim, C.-S. Lim and B. Seo, Sep. Purif. Technol., 2021, 254, 117678 CrossRef CAS.
- S.-J. Wang, D. S.-H. Wong, I. J. Q. Lim, Y.-T. Chen and C.-C. Huang, Ind. Eng. Chem. Res., 2018, 57, 6926–6936 CrossRef CAS.
- S. Chaudhari, Y. Kwon, M. Shon, S. Nam and Y. Park, RSC Adv., 2019, 9, 5908–5917 RSC.
- G. W. Olsen, S. E. Lacy, S. R. Chamberlin, D. L. Albert, T. G. Arceneaux, L. F. Bullard, B. A. Stafford and J. M. Boswell, Am. J. Ind. Med., 1994, 25, 205–218 CrossRef CAS PubMed.
- A. Svang-Ariyaskul, R. Y. M. Huang, P. L. Douglas, R. Pal, X. Feng, P. Chen and L. Liu, J. Membr. Sci., 2006, 280, 815–823 CrossRef CAS.
- R. Castro-Muñoz, J. Buera-González, O. de la Iglesia, F. Galiano, V. Fíla, M. Malankowska, C. Rubio, A. Figoli, C. Téllez and J. Coronas, J. Membr. Sci., 2019, 582, 423–434 CrossRef.
- Y. Li, L. H. Wee, J. A. Martens and I. F. J. Vankelecom, J. Mater. Chem. A, 2014, 2, 10034–10040 RSC.
- B. Smitha, D. Suhanya, S. Sridhar and M. Ramakrishna, J. Membr. Sci., 2004, 241, 1–21 CrossRef CAS.
- X. H. Zhang, Q. L. Liu, Y. Xiong, A. M. Zhu, Y. Chen and Q. G. Zhang, J. Membr. Sci., 2009, 327, 274–280 CrossRef CAS.
- W. Z. A. W. Jusoh, S. A. Rahman, A. L. Ahmad and N. M. Mokhtar, Applications of Nanotechnology for Green Synthesis, 2020, pp. 97–124 Search PubMed.
- C. Arregoitia-Sarabia, D. González-Revuelta, M. Fallanza, D. Gorri and I. Ortiz, Sep. Purif. Technol., 2020, 117101 CrossRef CAS.
- A. M. Eliceche, M. C. Daviou, P. M. Hoch and I. O. Uribe, Comput. Chem. Eng., 2002, 26, 563–573 CrossRef CAS.
- S. S. Hosseini, H. Pahlavanzadeh and M. Tamadondar, Iran. Chem. Eng. J., 2014, 13, 76–84 Search PubMed.
- M. E. Dmitrenko, A. V. Penkova, A. I. Kuzminova, M. Morshed, M. I. Larionov, H. Alem, A. A. Zolotarev, S. S. Ermakov and D. Roizard, Appl. Surf. Sci., 2018, 450, 527–537 CrossRef CAS.
- A. V. Penkova, M. E. Dmitrenko, N. A. Savon, A. B. Missyul, A. S. Mazur, A. I. Kuzminova, A. A. Zolotarev, V. Mikhailovskii, E. Lahderanta and D. A. Markelov, Sep. Purif. Technol., 2018, 204, 1–12 CrossRef CAS.
- M. Ghazali, M. Nawawi and R. Y. M. Huang, J. Membr. Sci., 1997, 124, 53–62 CrossRef.
- K. S. Sportsman, J. D. Way, W.-J. Chen, G. P. Pez and D. V. Laciak, J. Membr. Sci., 2002, 203, 155–166 CrossRef CAS.
- H.-A. Tsai, Y.-L. Ye, K.-R. Lee, S.-H. Huang, M.-C. Suen and J.-Y. Lai, J. Membr. Sci., 2011, 368, 254–263 CrossRef CAS.
- J. Sekulić, M. W. J. Luiten, J. E. Ten Elshof, N. E. Benes and K. Keizer, Desalination, 2002, 148, 19–23 CrossRef.
- C. Yu, C. Zhong, Y. Liu, X. Gu, G. Yang, W. Xing and N. Xu, Chem. Eng. Res. Des., 2012, 90, 1372–1380 CrossRef CAS.
- V. S. Praptowidodo, J. Mol. Struct., 2005, 739, 207–212 CrossRef CAS.
- T. A. Peters, C. H. S. Poeth, N. E. Benes, H. Buijs, F. F. Vercauteren and J. T. F. Keurentjes, J. Membr. Sci., 2006, 276, 42–50 CrossRef CAS.
- S. Chaudhari, Y. Kwon, M. Shon, S. Nam and Y. Park, J. Ind. Eng. Chem., 2020, 81, 185–195 CrossRef CAS.
- P. H. T. Ngamou, M. E. Ivanova, O. Guillon and W. A. Meulenberg, J. Mater. Chem. A, 2019, 7, 7082–7091 RSC.
- S. Sommer and T. Melin, Chem. Eng. Process., 2005, 44, 1138–1156 CrossRef CAS.
- Z. Pan, F. Yu, L. Li, M. Liu, C. Song, J. Yang, H. Li, C. Wang, Y. Pan and T. Wang, Sep. Purif. Technol., 2020, 116948 CrossRef CAS.
- N. Tahri, I. Jedidi, S. Ayadi, S. Cerneaux, M. Cretin and R. Ben Amar, Desalin. Water Treat., 2016, 57, 23473–23488 CrossRef CAS.
- S. Tanaka, T. Yasuda, Y. Katayama and Y. Miyake, J. Membr. Sci., 2011, 379, 52–59 CrossRef CAS.
- Y.-R. Dong, M. Nakao, N. Nishiyama, Y. Egashira and K. Ueyama, Sep. Purif. Technol., 2010, 73, 2–7 CrossRef CAS.
- K.-S. Liao, Y.-J. Fu, C.-C. Hu, J.-T. Chen, D.-W. Lin, K.-R. Lee, K.-L. Tung, Y. C. Jean and J.-Y. Lai, Carbon, 2012, 50, 4220–4227 CrossRef CAS.
- P. S. Tin, H. Y. Lin, R. C. Ong and T.-S. Chung, Carbon, 2011, 49, 369–375 CrossRef CAS.
- M. Yoshimune, K. Mizoguchi and K. Haraya, J. Membr. Sci., 2013, 425, 149–155 CrossRef.
- Y. Sakata, A. Muto, M. A. Uddin and H. Suga, Sep. Purif. Technol., 1999, 17, 97–100 CrossRef CAS.
- H. Nagasawa, N. Matsuda, M. Kanezashi, T. Yoshioka and T. Tsuru, J. Membr. Sci., 2016, 498, 336–344 CrossRef CAS.
- J. Shao, Z. Zhan, J. Li, Z. Wang, K. Li and Y. Yan, J. Membr. Sci., 2014, 451, 10–17 CrossRef CAS.
- S.-J. Kim, P. S. Lee, J.-S. Chang, S.-E. Nam and Y.-I. Park, Sep. Purif. Technol., 2018, 194, 443–450 CrossRef CAS.
- S. Chaudhari, M. Baek, Y. Kwon, M. Shon, S. Nam and Y. Park, Appl. Surf. Sci., 2019, 493, 193–201 CrossRef CAS.
- Y. Wu, F. Wang, B. Zhang, D. Zhao, T. Wang and J. Qiu, Asia-Pac. J. Chem. Eng., 2018, 13, e2251 CrossRef.
- B. Zhang, Y. Shi, Y. Wu, T. Wang and J. Qiu, J. Appl. Polym. Sci., 2014, 131, 39925 Search PubMed.
- X. Zhang, B. Zhang, Y. Wu, D. Wang and T. Wang, J. Appl. Polym. Sci., 2017, 134, 44889 Search PubMed.
- C. A. Scholes and U. K. Ghosh, Membranes, 2017, 7, 9 CrossRef PubMed.
- Y. Xiao, M. L. Chng, T.-S. Chung, M. Toriida, S. Tamai, H. Chen and Y. C. J. Jean, Carbon, 2010, 48, 408–416 CrossRef CAS.
- B. T. Low and T. S. Chung, Carbon, 2011, 49, 2104–2112 CrossRef CAS.
- A. K. Itta and H.-H. Tseng, Int. J. Hydrogen Energy, 2011, 36, 8645–8657 CrossRef CAS.
- C.-L. Liu, W.-S. Dong, J.-R. Song and L. Liu, Mater. Sci. Eng., A, 2007, 459, 347–354 CrossRef.
- S. Tanaka, N. Nakatani, A. Doi and Y. Miyake, Carbon, 2011, 49, 3184–3189 CrossRef CAS.
- F. Y. Li and T.-S. Chung, Int. J. Hydrogen Energy, 2013, 38, 9786–9793 CrossRef CAS.
- D. Van Baelen, B. Van der Bruggen, K. Van den Dungen, J. Degrève and C. Vandecasteele, Chem. Eng. Sci., 2005, 60, 1583–1590 CrossRef CAS.
- Y. Kwon, S. Chaudhari, C. Kim, D. Son, J. Park, M. Moon, M. Shon, Y. Park and S. Nam, RSC Adv., 2018, 8, 20669–20678 RSC.
- P. D. Chapman, T. Oliveira, A. G. Livingston and K. Li, J. Membr. Sci., 2008, 318, 5–37 CrossRef CAS.
- S. G. Adoor, B. Prathab, L. S. Manjeshwar and T. M. Aminabhavi, Polymer, 2007, 48, 5417–5430 CrossRef CAS.
- X. Feng and R. Y. M. Huang, Ind. Eng. Chem. Res., 1997, 36, 1048–1066 CrossRef CAS.
- C.-Y. Tu, Y.-L. Liu, K.-R. Lee and J.-Y. Lai, J. Membr. Sci., 2006, 274, 47–55 CrossRef CAS.
- H. G. Premakshi, A. M. Sajjan and M. Y. Kariduraganavar, J. Mater. Chem. A, 2015, 3, 3952–3961 RSC.
- M. Amirilargani, M. A. Tofighy, T. Mohammadi and B. Sadatnia, Ind. Eng. Chem. Res., 2014, 53, 12819–12829 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00899k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |