
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA binary mixed polymer brush coating with adjusted hydrophobic property to control protein adsorption†
Chaoshi
Chen
a,
Muhammad
Atif
 a,
Kang
He
a,
Miao
Zhang
a,
Lijuan
Chen
b and
Yanmei
Wang
*a
a,
Kang
He
a,
Miao
Zhang
a,
Lijuan
Chen
b and
Yanmei
Wang
*a
aCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: wangyanm@ustc.edu.cn
bCollege of Materials and Chemical Engineering, West Anhui University, Luan 237012, P. R. China
First published on 14th February 2021
Abstract
In this work, binary mixed polymer brushes based on poly[(2-methyl-2-oxazoline)-random-glycidyl methacrylate] (PMOXA-r-GMA) and poly(glycidyl methacrylate)-block-poly(N-isopropyl acrylamide)-block-polystyrene (PGMA-b-PNIPAM-b-PSt) were used as thermoresponsive coating to control protein adsorption. Firstly, PMOXA-r-GMA was synthesized via cationic ring opening polymerization of 2-methyl-2-oxazoline followed by its copolymerization with glycidyl methacrylate (GMA); PGMA-b-PNIPAM-b-PSt was synthesized by reversible addition–fragmentation chain transfer polymerization of GMA, N-isopropyl acrylamide, and styrene sequentially. Then the mixed brushes were fabricated successfully by spin coating the mixture of PMOXA-r-GMA and PGMA-b-PNIPAM-b-PSt solutions onto silicon or glass substrates followed by an annealing protocol using GMA segments as an anchor. Afterwards, the investigation for adjusting the hydrophobic property of mixed brushes was performed. The results showed that when the environmental temperature is lower than the lower critical solution temperature (LCST) of PNIPAM, the tethered PNIPAM blocks are swollen and could bring the hydrophobic PSt block with optimum molar mass to the surface; when the environmental temperature is higher than the LCST of PNIPAM, the PNIPAM blocks collapse and could bring the hydrophobic block inside the hydrophilic PMOXA grafted polymer layer. Finally, the protein adsorption on mixed polymer brushes was investigated using a fluorescein isothiocyanate-labelled protein assay and ellipsometrys. The results showed that under the optimum molar mass of PSt block, mixed brushes could adsorb proteins in large quantities below the LCST of PNIPAM, and display resistance to protein adsorption above the LCST of PNIPAM.
Introduction
Controlling the hydrophobic interaction between surfaces and proteins in an aqueous environment is important for it could control protein adsorption which is the first step of surface fouling.1–9 Stimuli-responsive surfaces are usually used to control the level of hydrophobic interactions between surfaces and proteins due to its switching of hydrophobic/hydrophilic properties upon environment stimulus.10–14 The responsive surfaces could be obtained by grafting one end of polymer chains to the surface forming polymer brushes. Among the polymer brushes, binary mixed polymer brushes composed of a hydrophilic part and an environmental responsive part have drawn more attention as its antifouling ability can be adjusted by switching the surface property between hydrophilic and hydrophobic behavior by changing the environmental stimuli (temperature, pH, ionic strength (I), etc.).15–22 Hoy and coworkers prepared mixed polymer brushes made of a hydrophilic homopolymer, poly(ethylene oxide) (PEO, also known as poly(ethylen glycol) (PEG)), and an amphiphilic block copolymer, polystyrene-b-poly(acrylic acid) (PSt-b-PAA).23 In which, the PSt-b-PAA copolymer was grafted to the surface via the reactive PAA part, and the hydrophobic block, PSt as a hydrophobic probe, is immiscible with PAA and PEG. They demonstrated that the hydrophobic property of the created mixed brushes provided by the PSt probe can be adjusted via PSt domains up or down with respect to the top surface through variation of the PAA swelling depending on external stimuli (pH, I, and the presence of Ca2+), and the actuation of the hydrophobic domains in the brushes can be used to tune bovine fibrinogen adsorption on and off.Although PEO and its corresponding derivatives have been used widely as antifouling materials because of their great hydrophilic properties, PEO is known to undergo degradation especially in a physiological environment.24 Recently, poly(2-methyl-2-oxazoline)(PMOXA), a peptidomimetic polymer, has attracted interest for its similar properties (e.g. hydrophilicity and biocompatibility) to PEG and less degradation than PEG.25–29 Cavallaro and coworkers reported that the films developed via plasma deposition of 2-methyl-2-oxazoline (MOXA) could resist the adsorption of Staphylococcus epidermidis and cells by more than 90% regardless of the type of substrate used.30 Bai and coworkers developed poly(2-methyl-2-oxazoline-random-glycidyl methacrylate) (PMOXA-r-GMA) comb copolymers.31 The PMOXA based coating could then be prepared by a simple annealing protocol of PMOXA-r-GMA on silicon/glass surfaces by using GMA segments as the anchor, because the epoxide groups of glycidyl methacrylate (GMA) segments can not only react with functional groups (i.e., Si–OH) on the surface of silicon/glass, but perform self-crosslinking with each other as well.32–34 The prepared coatings possess strong stability and excellent bovine serum albumin (BSA) resistant ability as well. The stimuli-responsive polymer could undergo a rapid conformational change in response to an external stimulus (e.g., temperature, light, redox signal, magnetic field, pH, ionic strength, or a specific molecule), resulting in the switching of the surface functionality on/off.11,14,35–37 Among them, poly(N-isopropyl acrylamide) (PNIPAM), a thermoresponsive polymer, has received extensive attention due to its lower critical solution temperature (LCST) of 32 °C close to body temperature. When the environmental temperature is lower than 32 °C, PNIPAM chains swell due to the strong hydrogen bond formed between the water molecules and the PNIPAM chains. When the environmental temperature is higher than 32 °C, the thermal dissociation of water molecules from the hydrated polymer chains can lead to the intrinsic affinity of PNIPAM chains, leading to the lessening of the hydrogen bond between water molecules and the PNIPAM chains; as result, PNIPAM chains shrink and display hydrophobic behavior.38–43
Inspired by the stability and low fouling ability of PMOXA based coatings and swelling/deswelling behaviors of PNIPAM chains with temperature, herein our purpose is to create binary mixed polymer brushes, on which the adsorption of protein could be adjusted through the change of the hydrophobic/hydrophilic behavior of mixed polymer brushes with temperature in an aqueous environment. Firstly, the comb copolymer, PMOXA-r-GMA was synthesized via cationic ring opening polymerization (CROP) of MOXA followed by its random copolymerization with GMA according to our previous work;28,31,44 and poly(glycidyl methacrylate)-block-poly(N-isopropyl acrylamide)-block-polystyrene (PGMA-b-PNIPAM-b-PSt) was synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization of GMA, N-isopropyl acrylamide (NIPAM), and styrene (St) step by step. Then the mixed brushes based on PMOXA and PNIPAM-b-PSt were fabricated by spin coating the mixture of PMOXA-r-GMA and PGMA-b-PNIPAM-b-PSt solutions onto silicon or glass substrates followed by an annealing protocol. A strategy to design mixed brushes is shown in Scheme 1. The hydrophilic character of the mixed brush is provided by highly hydrophilic PMOXA, which is end grafted to the surface by using GMA units existing in PMOXA-r-GMA as an anchor. PGMA-b-PNIPAM-b-PSt was grafted to the surface by using a PGMA block as the anchor, where the PNIPAM block tethered to PGMA anchor is a thermoresponsive polymer chain and serves as a macromolecular actuator to bring the highly hydrophobic PSt block to the surface upon the external temperature change. In PMOXA-r-GMA/PGMA-b-PNIPAM-b-PSt mixed brushes, the crosslinking of GMA segments next to the surface could strengthen the anchoring to the surface. In an environment where the tethered PNIPAM blocks are swollen (environmental temperature lower than the LCST of PNIPAM), the PSt hydrophobic domain is exposed to the surface. Such a surface has strong interactions with hydrophobic entities. When the PNIPAM block is not swollen (environmental temperature higher than the LCST of PNIPAM), the hydrophobic domain of the brush is hidden inside the hydrophilic PMOXA grafted polymer layer. Therefore, the hydrophobic/hydrophilic behavior of mixed polymer brushes could be tuned by moving the hydrophobic domains up or down with respect to the top surface. Prepared PMOXA-r-GMA/PGMA-b-PNIPAM-b-PSt coatings were characterized by using X-ray photoelectron spectroscopy (XPS), variable angle spectroscopic ellipsometry (VASE), atomic force microscopy (AFM), and static water contact angle (CA) measurements in order to assess their composition, morphology, thickness, and hydrophilicity respectively. The fluorescein isothiocyanate-labeled protein assay and VASE were used for qualitative and quantitative evaluation of the adsorption of protein on mixed brush coatings respectively through varying the temperature.
 | ||
| Scheme 1 Strategy of design for PMOXA-r-GMA and PGMA-b-PNIPAM-b-PSt mixed polymer brushes. | ||
Experimental
Materials
The water used in all experiments was deionized water. N,N-Dimethylformamide (DMF, Sinopharm Chemical Reagents, Shanghai, China) was distilled under reduced pressure before use. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized from methanol. Glycidyl methacrylate (GMA, 97%, Aladdin, Shanghai, China) and styrene (St, Sinopharm Chemical Reagents, Shanghai, China) were respectively passed through an activated basic alumina column to remove the inhibitor before use. N-Isopropyl acrylamide (NIPAM, Aladdin, Shanghai, China) was recrystallized from hexane. S-1-Dodecyl-S′-(α,α′-dimethyl-α′′-acetic acid) trithiocarbonate (DDMAT) was synthesized according to the previously published procedure.42 Fluorescein isothiocyanate-labeled bovine serum albumin (FITC-BSA), fibrinogen (FITC-Fib), and lysozyme (FITC-Lys) were prepared by mixing fluorescein isothiocyanate (FITC, Sigma-Aldrich, USA) with bovine serum albumin (BSA, pI ∼ 4.8, Mw ∼ 68 kDa, Sigma-Aldrich, USA), fibrinogen (Fib, pI ∼ 5.8, Mw ∼ 300 kDa, Sigma-Aldrich, USA) and lysozyme (Lys, pI ∼ 10.8, Mw ∼ 15 kDa, Sigma-Aldrich, USA) respectively at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in Na2CO3–NaHCO3 buffer for 2 h, followed by thorough dialysis in pH 7.4 phosphate-buffered saline (PBS, 10 mM) for three days.31 Glass wafers and microscope glass slides for FITC-immunoassay were bought from Shanghai Jinglun Industry Glass Co. (Shanghai, China). Silicon(111) wafers with a natural oxidized layer were received from Zhejiang Crystal Photoelectric Technology Co. (Zhejiang, China). Other reagents were all obtained from Sinopharm Chemical Reagents (Shanghai, China).
:1 in Na2CO3–NaHCO3 buffer for 2 h, followed by thorough dialysis in pH 7.4 phosphate-buffered saline (PBS, 10 mM) for three days.31 Glass wafers and microscope glass slides for FITC-immunoassay were bought from Shanghai Jinglun Industry Glass Co. (Shanghai, China). Silicon(111) wafers with a natural oxidized layer were received from Zhejiang Crystal Photoelectric Technology Co. (Zhejiang, China). Other reagents were all obtained from Sinopharm Chemical Reagents (Shanghai, China).
Synthesis of polymers
:1. Relative number average molar mass (Mn) of resultant PMOXA-MAA and molar ratio of PMOXA-MAA to GMA were calculated by 1H NMR. The synthesized copolymer PMOXA-r-GMA was called PGM for convenience.
Surface modification
Silicon/glass wafers were cut into 1 × 1 cm2 pieces and cleaned by sonication in ethanol (15 min), and then in deionized water (15 min). Subsequently, the wafers were cleaned in a piranha solution (7:3 v/v mixture of H2SO4 (95–98%) and H2O2 (30%)) for 60 min, rinsed extensively with water, ethanol, and then dried under a nitrogen atmosphere before use. Solutions of polymers with PGNS(PGNS1k, PGNS6k, and PGNS8k)/PGM mass ratio of 10/0 (pure PGNS), 0/10 (pure PGM), 5/5, were prepared by mixing stock chloroform solutions of PGM and PGNS at 10 mg mL−1. Spin coating of copolymer solutions (150 μL for 1 × 1 cm2) was performed on all above substrates for 20 s at 2000 rpm under vacuum by an EZ4 SPIN COATER (LEBO SCEINCE, China). The coated substrates were subsequently annealed for 12 h at 110 °C. After annealing, substrates were allowed to cool to room temperature and rinsed with chloroform, alcohol, and deionized water to remove unattached polymer in succession and then dried with nitrogen. To compare with the mixed brush PGNS/PGM coating, a PGN/PGM brush coating was also prepared by using the same annealing protocol as above with a PGN/PGM mass ratio of 10/0 (pure PGN), 5/5 (PGN/PGM), based on the stock chloroform solutions of PGN and PGM at 10 mg mL−1.
Characterizations
Switchable properties
| Δm = ρ × Δh × S | (1) |
Results and discussion
Preparation of polymer
PMOXA-r-GMA (PGM) was synthesized via CROP of MOXA followed by its random copolymerization with GMA according to our previous work.31 The 1H NMR result (Fig. S1 in the ESI†) showed that Mn of PMOXA chain on PGM is about 4900 g mol−1, and contour chain length of PMOXA side chains on PGM is about 20.1 nm (Table S1 in the ESI†).PGMA-b-PNIPAM-b-PSt with a different design Mn of PSt block (PGNS1k, PGNS6k and PGNS8k) was synthesized by RAFT polymerization of St with the PGMA-b-PNIPAM-macro-RAFT agent (PGN). As shown in Fig. 1a, the resonance peaks a (δ ∼2.6 ppm, 2.8 ppm) and b (δ ∼ 3.5 ppm) were assigned to the protons of methylene and methine of the GMA epoxide group individually, while the peak c (δ ∼ 3.8 ppm, 4.3 ppm) was assigned to the methylene connected to the epoxide groups. The signals of proton from the isopropyl group of NIPAM were detected at the resonance peaks of i (δ ∼ 4.0 ppm), and j (δ ∼ 1.2 ppm). These significant proton signals confirm the successful synthesis of PGN. The Mn of the PGMA block in PGN is about 770 g mol−1, determined by calculating relative peak areas corresponding to the peak n from DDMAT (δ ∼ 0.8–0.9 ppm) and peak c (δ ∼ 3.8 ppm, 4.3 ppm); the Mn of PNIPAM block in PGN is about 7900 g mol−1 determined by calculating the relative peak areas corresponding to the peak n from DDMAT RAFT agent's end CH3 (δ ∼ 0.8–0.9 ppm) and peak i (δ ∼ 4.0 ppm). The theoretical chain length of the PNIPAM block is 20.3 nm (Table S1 in the ESI†). Fig. 1b shows the 1H NMR of PGNS with varying Mn of the PSt block. In addition to all other corresponding peaks of PGN, a new significant peak was detected at 6.7–7.2 ppm, assigned to the phenyl group of styrene, confirming the successful synthesis of PGNS. The intensity of this peak (δ ∼ 6.7–7.2 ppm) increases by increasing the designed Mn of the PSt block from 1k to 8k respectively. The Mn of the PSt block of PGNS1k, PGNS6k and PGNS8k are 1232, 5897, and 7925 g mol−1 respectively, determined by the relative peak areas of the DDMAT's end methyl peak (δ ∼ 0.8–0.9 ppm) and phenyl group signal of styrene (δ ∼ 6.7–7.2 ppm), close to the designed Mn of PSt block. The contour chain length of PGNS1k, PGNS6k and PGNS8k are 23.2, 37.5 and 43.2 nm, respectively, while the contour chain lengths of the PSt block of PGNS1k, PGNS6k and PGNS8k are 2.9, 17.2and 22.9 nm respectively. The detailed chain length calculation is shown in Table S1 of the ESI.†
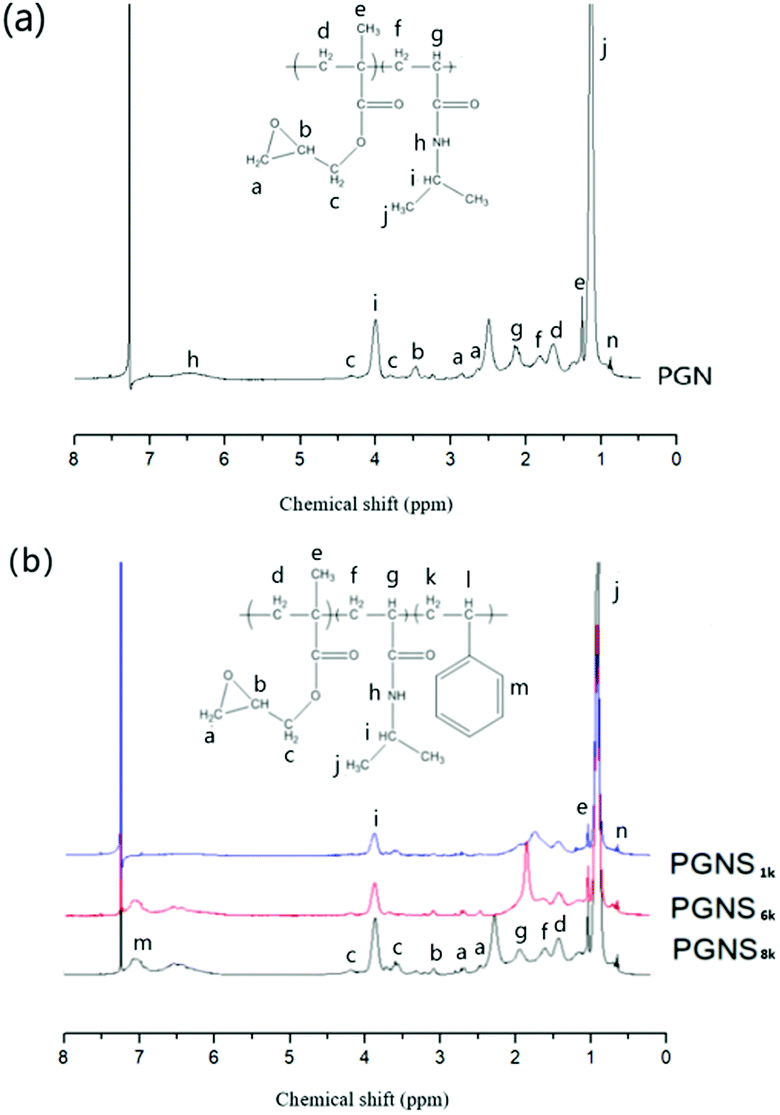 | ||
| Fig. 1 1H NMR spectra of (a) PGN, and (b) PGNS1k, PGNS6k, and PGNS8k in CDCl3. | ||
Characterization of polymer modified surfaces
The XPS measurements were used to determine the surface composition of the polymer coatings formed on the silicon wafers. The XPS wide scan spectra and high-resolution C1s of bare, PGN, PGM, PGNS1k, PGNS6k, PGNS8k, PGN/PGM, PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM modified silicon wafers are shown in Fig. S3 and S4 of the ESI,† and the corresponding atomic percentage of elements on bare and modified substrates is listed in Table 1. Compared to the bare silicon surface, clear changes in carbon (C1s, 287 eV), nitrogen (N1s, 400.5 eV), oxygen (O1s, 533 eV), and silicon (154 eV for Si2s and 103 eV for Si2p) signals were observed in modified silicon surfaces. For instance, in the PGM modified substrate, increments in carbon (C1s, 287 eV), nitrogen (N1s, 400.5 eV), and oxygen signals (O1s, 533 eV), with corresponding decreases in silicon (154 eV for Si2s and 103 eV for Si2p) signals confirmed the successful immobilization of PGM on the substrate as reported previously.31 The intensities of the carbon (C1s, 287 eV) signal increase with increasing the molar mass of the PSt block from PGN to PGNS8k respectively. For the mixed polymer brushes of PGN/PGM, PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM (mass ratio of PGN (PGNS) and PGM was 5/5, the details for determination of the ratio of PGNS and PGM are described in the ESI†), the intensity of the carbon (C1s, 287 eV) and nitrogen (N1s, 400.5 eV) signal between the intensity of pure PGM and pure PGN or corresponding PGNS modified silicon wafers, suggested the presence of both polymers on the silicon wafer. The area of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N peak (288.6 eV) from the amide bond of the PNIPAM block or PMOXA part (Fig. S4 of the ESI†) decreased with the increment of the PSt block from PGN/PGM to PGNS8k/PGM (Table S2 of the ESI†), further indicating the presence of both PGM and PGN(PGNS) copolymers on the PGN(PGNS)/PGM modified surface.
C–N peak (288.6 eV) from the amide bond of the PNIPAM block or PMOXA part (Fig. S4 of the ESI†) decreased with the increment of the PSt block from PGN/PGM to PGNS8k/PGM (Table S2 of the ESI†), further indicating the presence of both PGM and PGN(PGNS) copolymers on the PGN(PGNS)/PGM modified surface.
| Samples | Element mole percent (atom%) | f PGNS(PGN)/fPGMb | Thicknessc (nm) | Γ (chain per nm2) | Water contact anglee (°) | |||
|---|---|---|---|---|---|---|---|---|
| C1s | N1s | O1s | Si2s | |||||
| a Feed mass ratio of PGNS (or PGN) to PGM in mixed polymer brushes. b Mass ratio of PGNS (or PGN) to PGM in mixed polymer brushes calculated according to eqn (S4) and (S5), ESI. c Data obtained in dry film and expressed as mean ± SD (n = 3). d The grafting density Γ of polymer brushes expressed by the chain number per square nanometer calculated according to eqn (S6)–(S13) (ESI). The grafting density ratio of PGNS (or PGN) to PGM in mixed polymer brushes are given in parentheses calculated according to eqn (S9)–(S12) (ESI). e Data measured at room temperature and expressed as mean ± SD (n = 3). | ||||||||
| Bare | 27.15 | 1.20 | 29.01 | 42.64 | 5.2 ± 1.6 | |||
| PGM | 40.46 | 5.18 | 25.32 | 29.04 | 2.0 ± 0.1 | 0.24 | 9.4 ± 1.8 | |
| PGN | 46.06 | 5.70 | 23.72 | 24.51 | 2.6 ± 0.1 | 0.20 | 30.6 ± 1.9 | |
| PGNS1k | 46.72 | 4.80 | 26.88 | 21.59 | 2.6 ± 0.3 | 0.17 | 31.8 ± 1.6 | |
| PGNS6k | 53.68 | 1.54 | 23.64 | 22.14 | 2.5 ± 0.2 | 0.11 | 43.5 ± 1.4 | |
| PGNS8k | 57.04 | 1.82 | 22.64 | 18.50 | 2.2 ± 0.1 | 0.08 | 50.1 ± 0.7 | |
| PGN/PGM (5/5)a | 42.37 | 5.31 | 24.91 | 26.41 | 3.8/6.2 | 2.5 ± 0.2 | 0.23(0.07/0.16) | 20.3 ± 1.2 |
| PGNS1k/PGM (5/5)a | 45.89 | 4.87 | 23.07 | 26.17 | 3.9/6.1 | 3.0 ± 0.1 | 0.23(0.08/0.15) | 28.9 ± 2.1 |
| PGNS6k/PGM (5/5)a | 46.42 | 2.62 | 24.31 | 23.65 | 4.3/5.7 | 2.9 ± 0.2 | 0.18(0.03/0.15) | 29.1 ± 1.1 |
| PGNS8k/PGM (5/5)a | 52.10 | 1.95 | 24.76 | 21.19 | 4.2/5.8 | 2.1 ± 0.1 | 0.12(0.02/0.10) | 45.8 ± 1.4 |
The relative mass fraction of PGN, PGM, and PGNS (fPGN, fPGM, fPGNS) in the mixed polymer brushes of PGN/PGM, PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM were calculated by using eqn (S4) and (S5) in the ESI† based on XPS results, and the results are presented in Table 1. The results demonstrated that the resultant mass fraction of PGN, PGM, and PGNS on the mixed polymer brush modified surfaces are close to the feed mass ratio (5/5) (resultant mass fraction based on the XPS results for other feed mass ratios of PGNS6k/PGM is described in Table S2, ESI†), implying that the surface composition could be controlled by the concentration ratio of polymers in the mixed polymer solutions which were used to fabricate mixed polymer brush coatings.
Furthermore, the dry thickness of polymer modified surfaces were determined at room temperature and the results are given in Table 1. The results showed that the dry thickness of all the polymer modified surfaces are in the range of 2–3 nm and it is higher than that of the bare silicon wafer (∼0 nm). The grafting density of polymers on these samples could be estimated by using eqn (S6)–(S13) of the ESI† based on ellipsometry thickness. As shown in Table 1, the grafting density of the PGM is 0.24 chain per nm2. From PGN to PGNS8k the grafting density decreased from 0.20 chain per nm2 to 0.08 chain per nm2, suggesting that the polymer brushes with the larger PSt block have the lowest graft density on the substrate. From PGN/PGM to PGNS8k/PGM the grafting density decrease from 0.23 to 0.12 chain per nm2, the grafting density of PGM also decrease from 0.16 to 0.10 chain per nm2 with an increment of PSt block's molar mass, because of the steric hindrance of the PSt block.
The WCA of bare and polymer modified silicon wafers were also measured at room temperature and the results are depicted in Table 1. The value of WCA on bare silicon wafer is 5.2° representing its hydrophilic nature after dealing with piranha solution. The value of the PGM modified silicon wafer is 9.4°, confirming the hydrophilic properties of PMOXA chains as reported previously.31 While for PGN modified surfaces, the WCA increased sharply to 30.6°, because PNIPAM is more hydrophobic than PMOXA at room temperature.39 The WCAs of the PGNS modified wafters are higher than the PGN modified wafers, due to the presence of a PSt block with hydrophobic nature, and the WCA increased from 31.8°, 43.5° to 50.1° with increasing the molar mass of the PSt block from PGNS1k, PGNS6k, to PGNS8k respectively. For the modified silicon wafer with PGN/PGM brushes, WCA (20.3°) was observed to be in between that of the pure PGM and pure PGN modified wafer. For modified silicon wafers with PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM, WCA values decreased compared with their corresponding pure PGNS brushes, and WCA values increased from 28.9°, 29.1° to 45.8° with increasing the molar mass of PSt block from PGNS1k, PGNS6k, to PGNS8k respectively. Taking into account of XPS, thickness, and WCA data, it is confirmed that a silicon substrate could be modified successfully by PGM, PGN, PGNS and mixed polymer brushes of PGN/PGM, and PGNS/PGM with good surface coverage and controlled surface composition using an annealing protocol.
To further examine the grafting of polymers on the silicon substrates, surface morphology was investigated by using AFM with tapping mode and the results are illustrated in Fig. 2. The arithmetical mean deviation of the roughness profile https://www.baidu.com/javascript; (Ra) value increased from 0.294 nm of bare silicon to 5.05 nm of modified silicon wafers. The higher roughness values of the polymer modified surfaces comparing with bare substrate clearly indicated the successfully grafting of the polymers on the silicon surfaces. Compared with PGN (1.86 nm) or PGN/PGM (2.54 nm), the Ra of PGNS or PGNS/PGM increased with increasing the molar mass of PSt block, implying that the hydrophobic segregated PSt domain would reach to the top exterior boundary of the brush with increasing the molar mass of the PSt block. These results are consistent with the WCA data obtained at room temperature, in which WCA values increased with increasing the molar mass of the PSt block from PGNS1k to PGNS8k in either the PGNS brush or PGNS/PGM mixed brushes coating.
 | ||
| Fig. 2 AFM images of bare, PGN, PGM, PGNS1k, PGNS6k, PGNS8k and PGNS1k/PGM, PGNS6k/PGM, PGNS8k/PGM modified silicon wafer. The scan size was 2 × 2 μm2, scale bar was 200 nm, and the vertical scale was 20 nm. | ||
Switching of hydrophilic/hydrophobic properties
The WCA measurement is the basic method to investigate the surface hydrophilicity that is commonly used to examine the antifouling properties of the material surfaces. Fig. 3a shows polymer modified silicon surfaces at 0 °C and 38 °C, respectively. The WCA of PGM modified silicon surfaces exhibited negligible transition from 0 °C to 38 °C, anticipating the temperature independent nature of PMOXA. Also, very low WCA values (around 10° both at 0 °C and 38 °C) represent the strong hydrophilic property of PMOXA. The WCA of PGN modified silicon surfaces jumped from 20° to 40° by increasing the temperature from 0 °C to 38 °C. The difference in WCA (ΔWCA) between 0 °C and 38 °C represents the thermoresponsive behavior of PNIPAM. Below the LCST of PNIPAM (0 °C), PNIPAM chains would swell and adsorb water, causing the lower WCA of the PGN modified surface; while above the LCST of PNIPAM (38 °C), the PNIPAM chains collapse and undergo water repellent properties, thus an increment in the WCA was observed in the PGN modified surface.50–52 As compared to the PGN modified surface, increment in the WCA was observed for the PGNS modified surface both at 0 °C and 38 °C, and the values of WCA increased with increasing the molar mass of the PSt block from PGNS1k to PGNS8k. Moreover, ΔWCA from 0 °C to 38 °C is less than 5°. These results indicated that the addition of a PSt block in the PGNS modified surface could improve the hydrophobicity of the surface both at 0 °C and 38 °C. For the mixed polymer brushes of PGN/PGM, WCA varies from 16° to 22° by increasing the temperature from 0 °C and 38 °C, representing its thermoresponsive abilities but the extent of responsiveness is lower than the pure PGN modified surface due to the presence of PMOXA side chains in PGM. Where the mixed polymer brushes of PGNS/PGM are concerned, a very interesting behavior of WCA was observed. For instance, the WCA of PGNS1k/PGM remains approximately the same both at 0 °C and 38 °C and its value is about 28°. For PGNS6k/PGM, the WCA increased to 43° at 0 °C and reduced to 30° at 38 °C suggesting its shifting from a hydrophobic to hydrophilic nature below and above the LCST of PNIPAM respectively. To PGNS8k/PGM, very little difference in WCA (42° at 0 °C, 44° at 38 °C) was observed as the temperature was moved from 0 °C to 38 °C.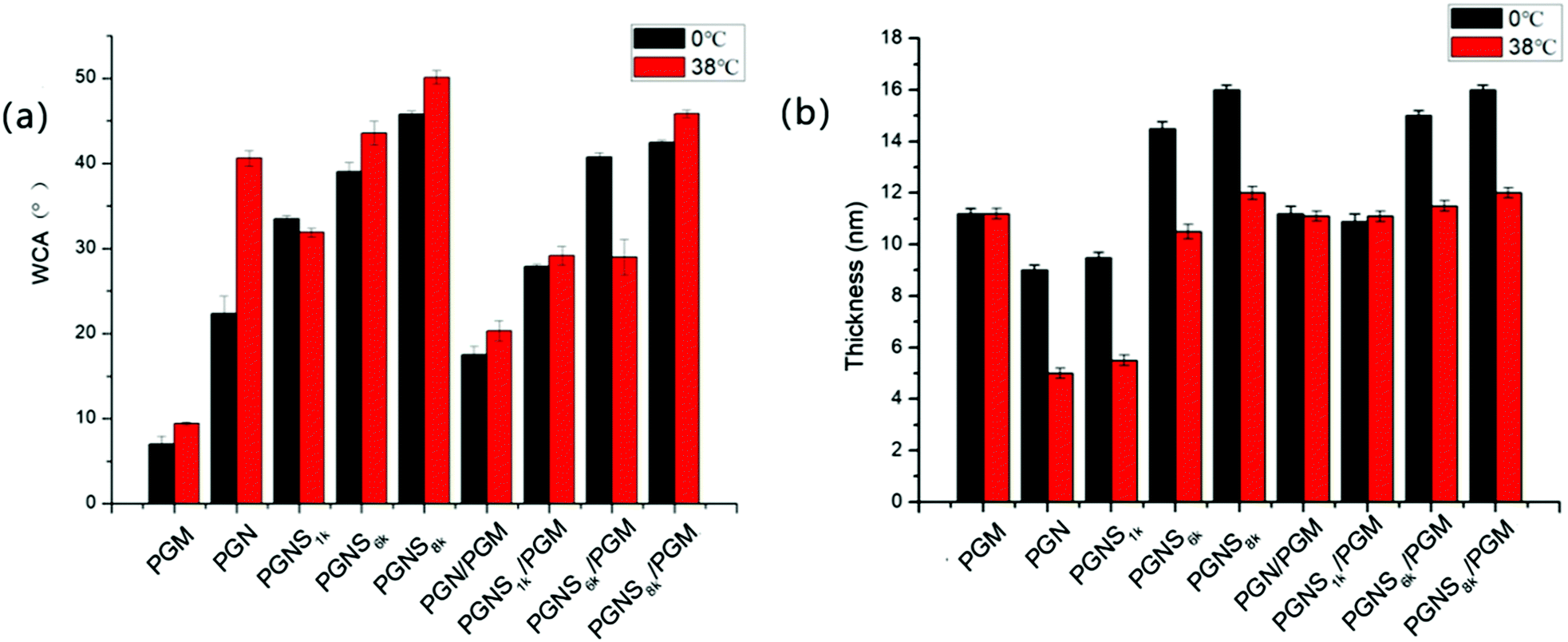 | ||
| Fig. 3 WCA value (a) and hydrated thickness (b) at 0 °C and 38 °C for PGN, PGM, PGNS1k, PGNS6k, PGNS8k and PGN/PGM, PGNS1k/PGM, PGNS6k/PGM, PGNS8k/PGM modified silicon wafer. Data are expressed as mean ± SD (n = 3). | ||
To further evaluate the switchable properties of the polymer modified surfaces, the hydrated thickness was measured after wafers were immersed in the solution of 0 °C and 38 °C, respectively, and they were subsequently lyophilized, and the results are depicted in Fig. 3b. It was observed that the average hydrated thickness of PGM was not changed by varying the temperature from 0 °C to 38 °C. In contrast, the hydrated thickness of PGN varied significantly with increasing the temperature from 0 °C to 38 °C. For instance, at 0 °C, the average hydrated thickness of PGN was 9 nm which was reduced to 4 nm by increasing the temperature to 38 °C. The difference in thickness is due mainly to swelling of PNIPAM chains below the LCST (0 °C) and deswelling above the LCST (38 °C). For PGNS modified surfaces, the average hydrated thickness increased both at 0 °C and 38 °C respectively by increasing the molar mass of the PSt block in PGNS from PGNS1k to PGNS8k. Moreover, regardless of the molar mass of PSt, the average hydrated thickness of PGNS with the same PSt block was higher at 0 °C and lower at 38 °C. This might be due to the swelling of PNIPAM chains in PGNS at 0 °C and deswelling of PNIPAM chains in PGNS at 38 °C respectively. For the mixed polymer brushes of PGN/PGM, the hydrated thickness increased as compared to the pure PGN brush both at 0 °C and 38 °C, and remained almost the same as that of the PGM brush. The hydrated thickness of the PGNS1k/PGM coating remains constant both at 0 °C and 38 °C, and also the value of hydrated thickness was almost the same as that of PGN/PGM, illustrating little contribution of the PSt block with low molar mass. For PGNS6k/PGM, and PGNS8k/PGM modified silicon wafers, the hydrated thickness increases sharply to 15 nm and 16.5 nm respectively at 0 °C, while the values of hydrated thickness decrease at 38 °C both for PGNS6k/PGM and PGNS8k/PGM. The pure PGM brush modified silicon wafer remains hydrophilic irrespective to the change of temperature due to the presence of PMOXA chains in PGM. On the other hand, the pure PGN brush shows swelling when T < LCST of PNIPAM and obtains a hydrophilic nature; by increasing the temperature (T > LCST), PNIPAM chains collapse and show strong hydrophobic behavior, representing the dominant role of PNIPAM in PGN. After being modified with PGNS, the substrates displayed a more hydrophobic nature compared with pure PGN modified substrates, and this hydrophobic property varied a little with the temperature and increased with increasing the molar mass of the PSt block (Fig. 3a); meanwhile the increment in brush thickness with the molar mass of the PSt block and the switchable behavior in thickness upon temperature change were observed (Fig. 3b). These results suggested that the hydrophobicity of PGNS modified substrates was determined by the PSt block, and the switchable behavior in thickness was determined by the PNIPAM block. Although the contour chain length of PMOXA (20.1 nm) in PGM is similar to that of PNIPAM (20.3 nm) in PGN (Table S1 of the ESI†), the hydrated thickness of the PGM modified substrate is higher than that of the PGN modified substrate at both 0 °C and 38 °C (Fig. 3b), and the PGM modified substrate is more hydrophilic than the PGN modified substrate at both 0 °C and 38 °C (Fig. 3a). Therefore, for PGN/PGM mixed polymer brushes coating, the hydrated thickness and hydrophilicity are determined by the PGM part owing to its position in the mixed brush exterior, and is kept almost the same at both 0 °C and 38 °C as shown in Scheme 2. For the PGNS/PGM mixed polymer brushes coating, though the total contour chain length of PNIPAM and PSt block in all PGNS is longer than that of PMOXA in the PGM part (23.2 nm for PGNS1k, 37.5 nm for PGNS6k, and 43.2 nm for PGNS6k, Table S1 in the ESI†), the hydrophilicity/hydrophobicity of modified substrates are different. For PGNS1k/PGM mixed brushes, the total contour chain length of PNIPAM and PSt block in PGNS1k is a little longer than that of PMOXA in PGM, but the hydrated thickness of pure PGM is much higher than pure PGNS1k at both 0 °C and 38 °C, leading to the dominant role of PGM in mixed brushes; hence the hydrated thickness and hydrophilicity are determined by the PGM part, and kept almost the same at both 0 °C and 38 °C as shown in Scheme 2. For PGNS6k/PGM mixed brushes, the total contour chain length of PNIPAM and the PSt block in PGNS6k increased greatly with increasing the molar mass of the PSt block, meanwhile, the hydrated thickness of pure PGNS6k is much higher than that of PGM at 0 °C, and lower than that of PGM at 38 °C (Fig. 3b), suggesting that PGNS6k would dominate the modified substrate at 0 °C representing the hydrophobicity of the PSt block, and PGM would dominate the modified substrate at 38 °C representing hydrophilicity (Fig. 3a) as shown in Scheme 2. By further increases the molar mass of PSt in the mixed polymer brushes (PGNS8k/PGM), the contour chain length of the PSt block increased greatly, and the total contour chain length of PNIPAM and the PSt block in PGNS8k is much longer than that of PMOXA in PGM; moreover, both the hydrated thickness of pure PGNS8k are higher than PGM at 0 °C and 38 °C (Fig. 3b) due to the high molar mass of the PSt block, implying that PGNS8k would control the wettability of the PGNS8k/PGM modified surface; consequently the PGNS8k/PGM modified substrate remains hydrophobic both at 0 °C and 38 °C as shown in Scheme 2. The polymer brush which had a higher hydrated thickness would occupy the top of the mixed brushes modified surface, therefore the hydrophilic/hydrophobic property of the surface could be controlled by this exposed part. For PGNS/PGM mixed polymer brushes coating, the overall hydrophilic character of the mixed brush is provided by the highly hydrophilic PMOXA in the PGM. The PNIPAM block is an inherently responsive polymer chain and serves as a macromolecular trigger to bring the PSt hydrophobic part to the surface upon the change of external temperature above or below the LCST of PNIPAM. The PSt block is a hydrophobic probe. When PNIPAM is not swollen (e.g. at 38 °C higher than the LCST of PNIPAM), the hydrophobic probe with appropriate Mn of PSt block (PGNS6k) would be hidden inside the hydrophilic PGM layer; when PNIPAM blocks are more extended (e.g. at 0 °C lower than the LCST of PNIPAM), the hydrophobic probe of PSt would be exposed to the surface. Consequently, with PGNS6k/PGM mixed brushes, by tuning the environmental temperature from 38 °C to 0 °C, the surface behavior could be changed from hydrophilic to hydrophobic.
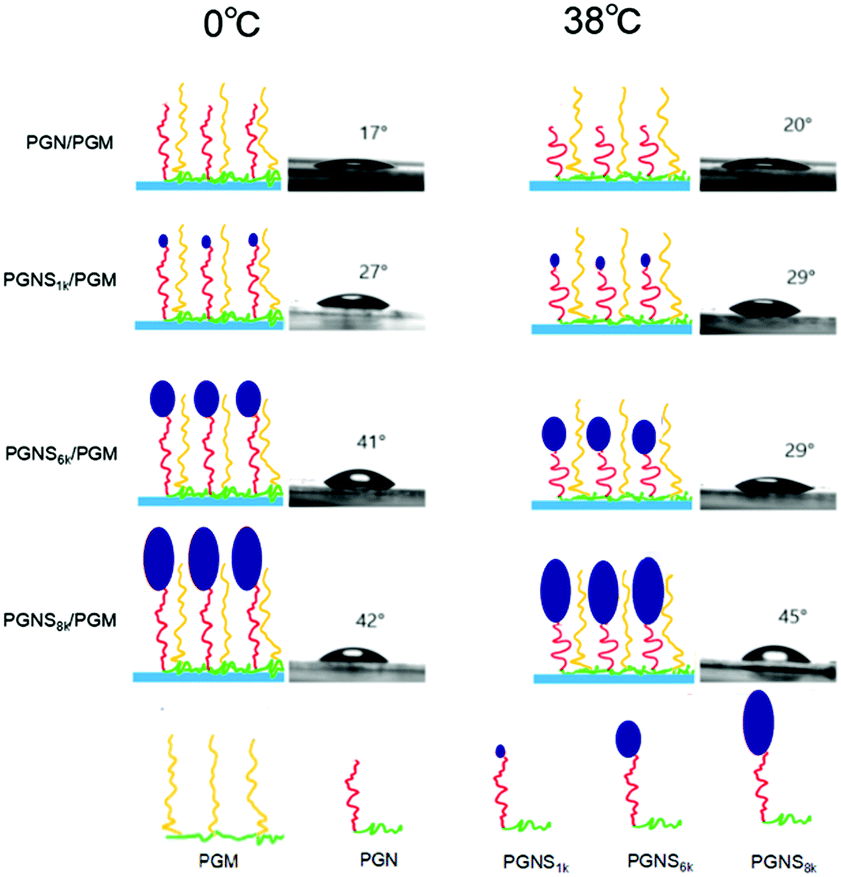 | ||
| Scheme 2 Illustration of the thermoresponsive behavior of PGN/PGM, PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM mixed polymer brushes coating. | ||
Adsorption of protein
The effect of switching between hydrophilic and hydrophobic properties of the developed system on its adsorption of protein was investigated. To qualitatively assess the protein adsorption, a fluorescent test was performed using FITC-BSA, FITC-Fib and FITC-Lys as the model proteins, respectively. Fig. 4 shows the surface images by fluorescence microscopy after the adsorption of proteins at 0 °C and 38 °C, respectively. The bar graphs are the relative quantitative analysis of the fluorescence intensities of the corresponding fluorescence images in Fig. 4. Herein the green fluorescence illustrates the adsorption of protein. For PGM modified surfaces, no adsorption was detected for all three types of protein (BSA, Fib, and Lys) at either 0 °C or 38 °C, representing the strong anti-fouling property of PMOXA. For PGN modified polymer brushes, strong adsorption of proteins with the fluorescence intensity of 40% for BSA, 52% for Fib, 50% for Lys was observed at 38 °C, but by changing the temperature to 0 °C, very little adsorbed protein was observed (fluorescence intensity was only 1% for BSA, 11% for Fib, 19% for Lys) because of the protein-resistant ability of the hydrated layer of swollen PNIPAM in PGN.50–52 After the block copolymerization of PGN with PSt (PGNS), the adsorption of three proteins on PGNS modified substrates was observed at 0 °C, while by shifting the temperature to 38 °C, the protein adsorption amount was nearly the same as at 0 °C, and the amounts of protein adsorbed on PGNS modified substrates increased with increasing the molar mass of PSt both at 0 °C and 38 °C. In the mixed polymer brushes of PGN/PGM, no evident fluorescence was observed (fluorescence intensity was nearly 3% for BSA, 3% for Fib, 1% for Lys) both at 0 °C and at 38 °C due to strong protein repellent property of exposed PGM. As long as mixed polymer brushes of PGNS/PGM were concerned, an almost negligible amount of adsorption of these three proteins was observed for PGNS1k/PGM, both at 0 °C and at 38 °C. For PGNS6k/PGM, strong adsorption of protein was observed at 0 °C and its fluorescence intensity was 65% for BSA, 78% for Fib, 77% for Lys; while inconspicuous adsorption of protein took place by changing the temperature to 38 °C (fluorescence intensity was 8% for BSA, 9% for Fib, 10% for Lys). These results further demonstrated that the exposed PSt block was more hydrophobic at 0 °C which would result in strong protein adsorption and exposed PGM that was more hydrophilic at 38 °C would display a strong protein repellent property for PGNS6k/PGM mixed brushes as shown in Scheme 2. However, for PGNS8k/PGM, where the molar mass of the PSt block was at its maximum, the same intensive fluorescence on PGNS8k/PGM was observed at either 0 °C or 38 °C for each protein (67% for BSA, 84% for Fib, 95% for Lys), due to the predominant role of the exposed PSt block irrespective of temperature as shown in Scheme 2. In brief, the adsorption degree of proteins on PGNS/PGM mixed brushes, regardless of acidic or basic protein (BSA/Lys), large or small size protein (Fib/Lys) could be adjusted through the hydrophobic/hydrophilic interaction between protein and the exposed part of mixed brushes by varying the temperature of the environment.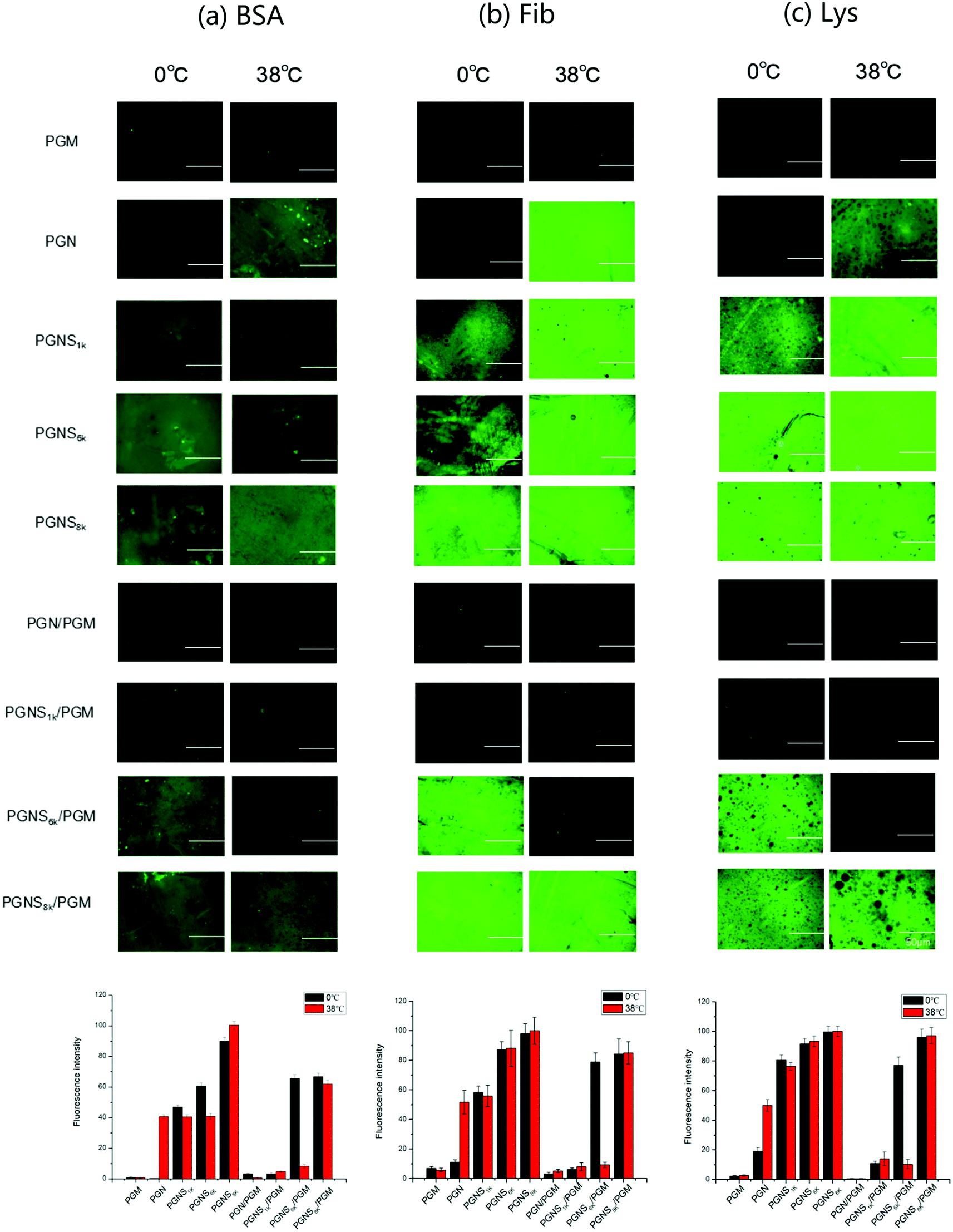 | ||
| Fig. 4 Fluorescence microscopy images and fluorescence intensity bar graph of the (a) FITC-BSA, (b) FITC-Fib and (c) FITC-Lys adsorption on PGM, PGN, PGN/PGM, PGNS1k, PGNS6k, PGNS8k, PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM modified glass wafer at 0 °C and 38 °C. Scale bar is 50 μm. The fluorescence intensity of PGNS8k at 38 °C was set as 100% for each protein. Data are expressed as mean ± SD (n = 3). | ||
The quantification of the adsorption of protein was performed by using VASE. To verify the reliability of this method, after measuring the dry thickness of polymer modified wafers, the same wafers were immersed in PBS solution (without protein) for 2 h at 0 °C or 38 °C, then its thickness was remeasured after being dried under nitrogen at 25 °C. The results showed no considerable difference in thickness, suggesting the validity of this method. The details of the experiment and the corresponding results are shown in the ESI.† The ellipsometry thickness of polymer modified silicon wafers before and after BSA, Fib and Lys adsorption at 0 °C and 38 °C are shown in Fig. S8 of the ESI,† and the amounts of protein adsorption calculated by eqn (1) were shown in Fig. 5. As shown in Fig. 5, the amounts of protein adsorption on the PGM modified surfaces remain nearly the same at either 0 °C or 38 °C, representing the strong protein repellent properties of PMOXA. The amounts of protein adsorbed on PGN at 0 °C were lower than that adsorbed at 38 °C, indicating that the hydrophobic interaction between PGN and proteins could be adjusted through the thermoresponsive ability of PNIPAM. Similar to fluorescence microscopy results, the amounts of protein adsorbed on PGNS modified wafers increased with increasing the molar mass of PSt block at either 0 °C or 38 °C. In the mixed polymer brushes of PGN/PGM, a little adsorption of protein (nearly 10 ng cm−2 for BSA, 30 ng cm−2 for Fib, 10 ng cm−2 for Lys) was observed both at 0 °C and 38 °C. While in the mixed polymer brushes of PGNS1k/PGM, a small amount of adsorption (nearly 10 ng cm−2 for BSA, 60 ng cm−2 for Fib, 50 ng cm−2 for Lys) took place both at 0 °C and at 38 °C. However, by increasing the molar mass of PSt in the mixed polymer brushes (PGNS6k/PGM), a large amount of protein adsorption (207 ng cm−2 for BSA, 700 ng cm−2 for Fib, 408 ng cm−2 for Lys) was observed at 0 °C, but by changing the temperature to 38 °C, the amounts of protein adsorbed on PGNS6k/PGM decreased sharply (11 ng cm−2 for BSA, 83 ng cm−2 for Fib, 77 ng cm−2 for Lys), representing its strong temperature responsive antifouling properties. By further increasing the molar mass of PSt, considerable amounts of protein adsorbed on PGNS8k/PGM were observed at either 0 °C or 38 °C, and the amounts of protein adsorbed on PGNS8k/PGM are of the same order of magnitude at both 0 °C and 38 °C as shown in Fig. 5. These results were fairly consistent with the observations obtained from fluorescence microscopy. Finally, the PGNS6k/PGM shows the most attractive thermally responsive property and excellent stability (Fig. S9 and S10, ESI†), having important implication in developing chemically modified surfaces.
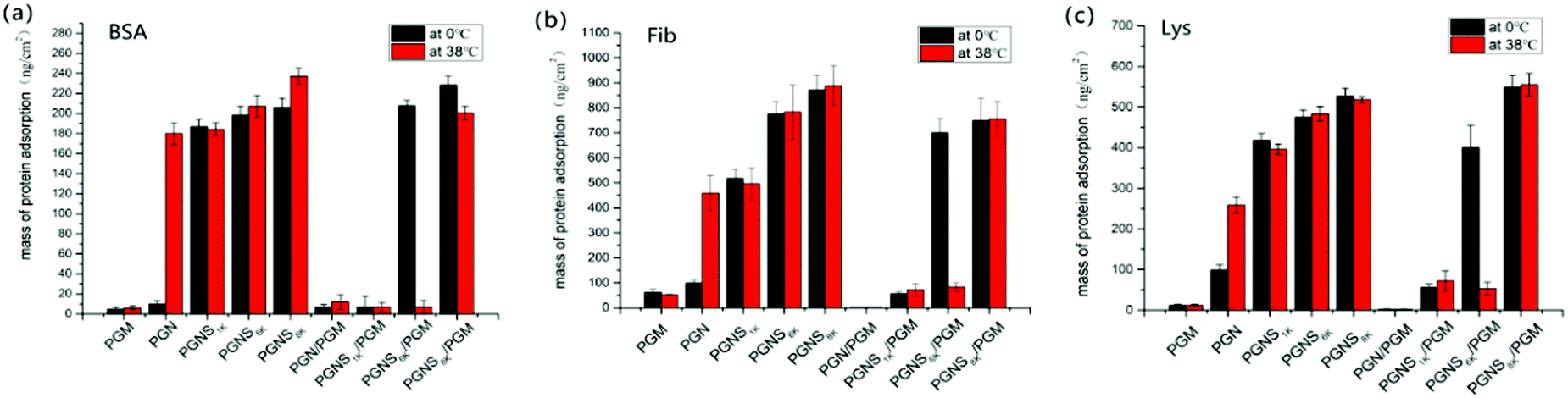 | ||
| Fig. 5 The adsorption amount of (a) BSA, (b) Fib and (c) Lys on PGM, PGN, PGNS1k, PGNS6k, PGNS8k, PGN/PGM, PGNS1k/PGM, PGNS6k/PGM, and PGNS8k/PGM modified silicon wafers at 0 °C and 38 °C, respectively, obtained by using VASE. Data are expressed as mean ± SD (n = 3). | ||
Conclusions
In this study, a mixed polymer brushes coating with thermoresponsive ability was fabricated by spin coating a mixture of PMOXA-r-GMA and PGMA-b-PNIPAM-b-PSt solutions onto silicon or glass substrates followed by an annealing protocol via GMA units as the anchor part. Under the selected temperature (below or above the LCST of PNIPAM), the hydrophilic/hydrophobic behavior of mixed brushes in an aqueous environment was mainly governed by the exterior part through varying the molar mass (i.e. chain length) of the PSt block. When the environment temperature remained below the LCST of PNIPAM, the PNIPAM block was swollen. The hydrated thickness of the PGMA-b-PNIPAM-b-PSt brush increased with increasing the molar mass of PSt. When the hydrated thickness of PGMA-b-PNIPAM-b-PSt with lower molar mass of PSt block (PGNS1k) was lower than that of PMOXA-r-GMA, the surface showed hydrophilic behavior; when the hydrated thickness of PGMA-b-PNIPAM-b-PSt with higher molar mass of PSt block (PGNS6k and PGNS8k) was higher than that of PMOXA-r-GMA, the surface showed hydrophobic behavior. When the environment temperature remained above the LCST of PNIPAM, the PNIPAM block collapsed. The surface only displayed the hydrophobic behavior when the hydrated thickness of PGMA-b-PNIPAM-b-PSt with the maximum molar mass of the PSt block (PGNS8k). The reversible transition between hydrophilic and hydrophobic behavior was realized for PGNS6k/PGM mixed brushes in an aqueous environment upon temperature change (lower or higher than LCST of PNIPAM) through the reversibly exposing hydrophobic fragment at its hydrophilic surface. Moreover, switching of the exterior part for PGNS6k/PGM mixed brushes could be used to adjust the adsorption of proteins by tuning the environmental temperature (lower or higher than the LCST of PNIPAM). This strategy of reversibly exposing a fragment will lead to potential applications in coating design.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21674102).References
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2006, 17, 617–621 Search PubMed.
- Z. Lei, J. Gao, X. Liu, D. Liu and Z. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 10174–10182 CrossRef CAS.
- H. Chen, J. Yang, S. Xiao, R. Hu, S. M. Bhaway, B. D. Vogt, M. Zhang, Q. Chen, J. Ma, Y. Chang, L. Li and J. Zheng, Acta Biomater., 2016, 40, 62–69 CrossRef CAS PubMed.
- V. Bugatti, A. Sorrentino and G. Gorrasi, Eur. Polym. J., 2017, 93, 495–506 CrossRef CAS.
- N. M. S. Bettahalli, I. T. M. Arkesteijn, M. Wessling, A. A. Poot and D. Stamatialis, Acta Biomater., 2013, 9, 6928–6935 CrossRef CAS PubMed.
- S. Jana, B. J. Tefft, D. B. Spoon and R. D. Simari, Acta Biomater., 2014, 10, 2877–2893 CrossRef CAS PubMed.
- H. Chen, M. Zhang, J. Yang, R. Hu, Q. Chen, Y. Chang and J. Zheng, Langmuir, 2014, 30, 10398–10409 CrossRef CAS PubMed.
- H. Yuan, J. Xue, B. Qian, H. Chen, Y. Zhu and M. Lan, Appl. Surf. Sci., 2017, 394, 403–413 CrossRef CAS.
- J. Li, M. Li, J. Miao, J. Wang, X. Shao and Q. Zhang, Appl. Surf. Sci., 2012, 258, 6398–6405 CrossRef CAS.
- C. Alexander and K. M. Shakesheff, Adv. Mater., 2006, 18, 3321–3328 CrossRef CAS.
- B. Tripathi, N. Dubey, S. Choudhury, F. Simona and M. Stamm, J. Mater. Chem. B, 2013, 1, 3397–3409 RSC.
- Y. Wang, J. Wu, D. Zhang, F. Chen, P. Fan, M. Zhong, S. Xiao, Y. Chang, X. Gong, J. Yang and J. Zheng, J. Mater. Chem. B, 2019, 7, 5762–5774 RSC.
- A. Kikuchi and T. Okano, J. Controlled Release, 2005, 101, 69–84 CrossRef CAS PubMed.
- I. C. Alard, J. Soubhye and G. Berger, Polym. Chem., 2017, 8, 2450–2456 RSC.
- M. F. Delcroix, G. L. Huet, T. Conard, S. Demoustier-Champagne, F. E. Du Prez, J. Landoulsi and C. C. Dupont-Gillain, Biomacromolecules, 2013, 14, 215–225 CrossRef CAS PubMed.
- M. F. Delcroix, S. Demoustier-Champagne and C. C. Dupont-Gillain, Langmuir, 2014, 30, 268–277 CrossRef CAS PubMed.
- D. Aulich, O. Hoy, I. Luzinov, K. J. Eichhorn, M. Stamm, M. Gensch, U. Schade, N. Esser and K. Hinrichs, Phys. Status Solidi C, 2010, 7, 197–199 CrossRef CAS.
- J. Draper, I. Luzinov, S. Minko, I. Tokarev and M. Stamm, Langmuir, 2004, 20, 4064–4075 CrossRef CAS PubMed.
- S. Minko, S. Patil, V. Datsyuk, F. Simon, K.-J. Eichhorn, M. Motornov, D. Usov, I. Tokarev and M. Stamm, Langmuir, 2002, 18, 289–296 CrossRef CAS.
- S. Minko, D. Usov, E. Goreshnik and M. Stamm, Macromol. Rapid Commun., 2001, 22, 206–211 CrossRef CAS.
- A. Sidorenko, S. Minko, K. Schenk-Meuser, H. Duschner and M. Stamm, Langmuir, 1999, 15, 8349–8355 CrossRef CAS.
- L. Ionov and S. Minko, ACS Appl. Mater. Interfaces, 2012, 4, 483–489 CrossRef CAS PubMed.
- O. Hoy, B. Zdyrko, R. Lupitskyy, R. Sheparovych, D. Aulich, J. Wang, E. Bittrich, K. Eichhorn, P. Uhlmann, K. Hinrichs, M. Muller, M. Stamm, S. Minko and I. Luzinov, Adv. Funct. Mater., 2010, 20, 2240–2247 CrossRef CAS.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- L. Tan, L. Bai, H. Zhu, C. Zhang, L. Chen and Y. Wang, J. Mater. Sci., 2015, 50, 4898–4913 CrossRef CAS.
- H. Du, C. Zhang, K. Mao and Y. Wang, Talanta, 2017, 170, 275–285 CrossRef CAS PubMed.
- C. Pan, L. Chen, S. Liu, Y. Zhang, C. Zhang, H. Zhu and Y. Wang, J. Mater. Sci., 2016, 51, 2427–2442 CrossRef CAS.
- F. Mumtaz, C. Chen, H. Zhu, C. Pan and Y. Wang, Appl. Surf. Sci., 2018, 439, 148–159 CrossRef CAS.
- H. Zhu, F. Mumtaz, C. Zhang, L. Tan, S. Liu, Y. Zhang, C. Pan and Y. Wang, Appl. Surf. Sci., 2017, 426, 817–826 CrossRef CAS.
- A. A. Cavallaro, M. N. Macgregor-Ramiasa and K. Vasilev, ACS Appl. Mater. Interfaces, 2016, 8, 6354–6362 CrossRef CAS PubMed.
- L. Bai, L. Tan, L. Chen, S. Liu and Y. Wang, J. Mater. Chem. B, 2014, 2, 7785–7794 RSC.
- H. Zou, S. Lin, Y. Tu, G. Liu, J. Hu, F. Li, L. Miao, G. Zhang, H. Luo, F. Liu, C. Hou and M. Hu, J. Mater. Chem. A, 2013, 1, 11246–11260 RSC.
- E. Soto-Cantu, B. S. Lokitz, J. P. Hinestrosa, C. Deodhar, J. M. Messman, J. F. Ankner and S. M. Kilbey II, Langmuir, 2011, 27, 5986–5996 CrossRef CAS PubMed.
- B. S. Lokitz, J. Wei, J. P. Hinestrosa, I. Ivanov, J. F. Browning, J. F. Ankner, S. M. Kilbey and J. M. Messman, Macromolecules, 2012, 45, 6438–6449 CrossRef CAS.
- B. Wang, Z. Ye, Q. Xu, H. Liu, Q. Lin, H. Chen and K. Nan, Biomater. Sci., 2016, 4, 1731–1741 RSC.
- A. Krieg, E. Arici, N. Windhab, J. H. Schattka, S. Schubert and U. S. Schubert, ACS Comb. Sci., 2014, 16, 386–392 CrossRef CAS PubMed.
- W. Ji, N. Li, D. Chen, X. Qi, W. Sha, Y. Jiao, Q. Xu and J. Lu, J. Mater. Chem. B, 2013, 1, 5942–5949 RSC.
- H. Omidian, J. G. Rocca and K. Park, Advances in superporous hydrogels, J. Controlled Release, 2005, 102, 3–12 CrossRef CAS PubMed.
- H. K. Ju, S. Y. Kim, S. T. Kim and Y. M. Lee, J. Appl. Polym. Sci., 2002, 83, 1128–1139 CrossRef CAS.
- T. Baltes, F. Garret-Flaudy and R. Freitag, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2977–2989 CrossRef CAS.
- M. Heskins, J. E. Guillet and E. J. James, J. Macromol. Sci., 1968, A2, 1441–1445 Search PubMed.
- Y. H. Bae, T. Okano and S. W. Kim, J. Polym. Sci.: Polym. Phys., 1990, 28, 923–936 CrossRef CAS.
- H. Kuroki, I. Tokarev, D. Nykypanchuk, E. Zhulina and S. Minko, Adv. Funct. Mater., 2013, 23, 4593–4600 CrossRef CAS.
- F. Mumtaz, C. Chen, H. Zhu, M. Atif and Y. Wang, Chinese J. Polym. Sci., 2018, 36, 1328–1341 CrossRef CAS.
- O. Borozenko, C. Ou, W. G. Skene and S. Giasson, Polym. Chem., 2014, 5, 2242–2252 RSC.
- S. Colak and G. N. Tew, Langmuir, 2011, 28, 666–675 CrossRef PubMed.
- P. A. Cuypers, J. W. Corsel, M. P. Janssen, J. Kop, W. T. Hermens and H. C. Hemker, J. Biol. Chem., 1983, 258, 2426–2431 CrossRef CAS.
- H. Elwing, Biomaterials, 1998, 19, 397–406 CrossRef CAS PubMed.
- S. Sharma, R. W. Johnson and T. A. Desai, Langmuir, 2004, 20, 348–356 CrossRef CAS PubMed.
- S. Burkert, E. Bittrich, M. Kuntzsch, M. Muller, K. J. Eichhorn, C. Bellmann, P. Uhlmann and M. Stamm, Langmuir, 2010, 26, 1786–1795 CrossRef CAS PubMed.
- S. Chen, L. Li and C. Zhao, Polymer, 2010, 23, 5283–5293 CrossRef.
- C. Xur, N. Yonet-Yanyeri, N. Brouette, M. Sferrazza, P. V. Braaun and D. E. Leckband, Langmuir, 2011, 27, 8810–8818 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00932f |
| This journal is © The Royal Society of Chemistry 2021 |