
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable cycling via absolute intercalation in graphite-based lithium-ion battery incorporated by solidified ether-based polymer electrolyte†
Hyunjin
Kim
a,
Do Youb
Kim
 a,
Jungdon
Suk
a,
Yongku
Kang
*a,
Jin Bae
Lee
b,
Hae Jin
Kim
b and
Dong Wook
Kim
*a
a,
Jungdon
Suk
a,
Yongku
Kang
*a,
Jin Bae
Lee
b,
Hae Jin
Kim
b and
Dong Wook
Kim
*a
aEnergy Materials Research Center, Advanced Materials Division, Korea Research Institute of Chemical Technology, Daejeon 34114, Republic of Korea. E-mail: ykang@krict.re.kr; dongwook@krict.re.kr
bResearch Center for Materials Analysis, Korea Basic Science Institute, Daejeon, 34133, Republic of Korea
First published on 27th April 2021
Abstract
Current lithium-ion batteries are vulnerable to fire accidents and explosions because liquid electrolytes have a low flash point and poor thermal stability. This intrinsic problem has led to an ever-growing interest in solid-state polymer electrolytes with high thermal stability. In this study, a solidified polyether-based polymer electrolyte is incorporated into a graphite/LiFePO4 full-cell battery. A liquid precursor, which is prepared by mixing the bisphenol A ethoxylate diacrylate (BisA) crosslinker and the poly(ethylene glycol) dimethyl ether (PEGDME) plasticizer, first wets the anode and cathode, and is then solidified by in situ thermal crosslinking to produce a solid polymer electrolyte. BisA forms a rigid crosslinked network and PEGDME conducts lithium ions within the network. Analysis results, including in situ X-ray diffraction, show that PEGDME in the polymer electrolyte is co-intercalated with lithium ions into the gallery of the graphite electrode, which causes electrode exfoliation and severe capacity fading. Fluoroethylene carbonate is highly effective to prevent the co-intercalation of lithium–PEGDME complex ions into the graphite, via the formation of a solid electrolyte interphase layer, which leads to the ‘absolute intercalation’ of lithium ions. Consequently, the graphite/LiFePO4 full-cell battery based on the solid polymer electrolyte runs stably at a coulombic efficiency higher than 99% for most cycles and the residual capacity of the cell reaches 80% after 100 cycles.
Introduction
As the major market for rechargeable batteries has shifted from portable devices to electric vehicles (EV) and energy storage systems (ESS), the safety of batteries has become a critical issue.1,2 The batteries of EV and ESS, which require a significant high capacity, have caused an increasing number of fatal accidents over the past years.3–8 The main culprit for battery fires is the liquid electrolyte, which consists of an organic carbonate solvent.9–11 Because solvents are associated with a high risk of ignition due to their volatility and potential leakage, solid electrolytes are considered a promising alternative to solve this problem.12–14 Solid electrolytes can be divided into inorganic and poly(ethylene oxide) (PEO)-based polymer electrolytes.13,14 Although inorganic electrolytes have a high thermal stability and dimensional stability, their rigidity and complex fabrication process hinder their adoption in the roll-to-roll mass-production of batteries.15–17 PEO-based polymer electrolytes are solid but flexible, and they can be compatible with current battery fabrication processes.18 However, polymer electrolytes show a poor performance at room temperature due to their low ionic conductivity and high interfacial resistance with electrodes.19–22 Many attempts have been made to increase the ionic conductivity of polymer electrolytes, for example, by adding ceramic fillers or mixing with other materials.19–22 In our previous studies, we presented a crosslinked polymer network system for a solid polymer electrolyte,23 in which a crosslinker formed a rigid crosslinked network and a plasticizer conducted lithium ions within the network. This system showed reasonably high ionic conductivity and low interfacial resistance between the electrode and electrolyte. So far, most studies with polymer electrolytes have used lithium as the anode to fabricate batteries due to its high theoretical capacity.3–6 However, lithium is currently not suitable for such batteries due to its high reactivity and dendrite problems.24,25 To realize a safe polymer electrolyte battery, a more stable anode is required. Graphite serves as a typical anode for commercialized lithium-ion batteries owing to its high stability, moderately large capacity, and low cost.26–28 In spite of such advantages, graphite has rarely been studied for use in polymer electrolyte batteries.Herein, we report the stable cycling of a graphite-based full-cell battery with a polymer electrolyte. Bisphenol A ethoxylate diacrylate (BisA) and poly(ethylene glycol) dimethyl ether (PEGDME) were used as the crosslinker and plasticizer, respectively. A liquid-state precursor, which was prepared by mixing BisA and PEGDME, first wetted the anode and cathode, and was then solidified by in situ thermal crosslinking between the electrodes. However, when the polymer electrolyte was used with the graphite anode, severe capacity degradation was unexpectedly observed during the charge and discharge cycles. In situ X-ray diffraction (XRD) analysis suggested that the PEGDME in the polymer electrolyte was co-intercalated with lithium ions into the gallery of the graphite electrode. Such co-intercalation of lithium–PEGDME complex ions likely caused the electrode exfoliation and extraordinary capacity fading. This co-intercalation and severe capacity fading were effectively prevented by the addition of fluoroethylene carbonate (FEC), and the resulting “absolute intercalation” enabled stable cycling of the graphite/LiFePO4 polymer electrolyte battery.
Result and discussions
A liquid-state precursor was prepared by mixing BisA and PEGDME, and it was solidified by thermal crosslinking at 90 °C to produce a solid polymer electrolyte. The crosslinked BisA provided a three-dimensional network in which to confine PEGDME, and through which lithium ions were conducted. The molecular weight of PEGDME had a dominant effect on the ionic conductivity and stability of the polymer electrolyte.23 With an exceedingly high molecular weight of PEGDME, the segmental motion of the molecules was highly retarded at room temperature and ionic conduction did not occur properly.18–21 By contrast, PEGDME with an exceedingly low molecular weight was volatile and deteriorated the stability of the polymer electrolyte.18 In this work, we prepared a polymer electrolyte by using a PEGDME with a molecular weight of 500. When the BisA–PEGDME polymer electrolyte was used with graphite, the capacity of the lithium/graphite half-cell was severely faded during the lithiation/delithiation cycles, as shown in Fig. S1 (ESI†). After 10 cycles, the residual capacity was only approximately 50%. The capacity fading occurred due to the co-intercalation of PEGDME, as will be discussed later. As reported in a previous study, ether-based electrolytes such as diethylene glycol dimethyl ether (DEGDME) have a strong tendency for co-intercalation with lithium ions into the graphite gallery.29,30 Moreover, despite the significantly lower capacity obtained from this co-intercalation compared with usual intercalation in graphite, DEGDME show reversible co-intercalation for long discharge/charge cycling without any capacity fading.29 Our study indicates that when PEGDME (Mn = 500) was used as an electrolyte, co-intercalation of Li–PEGDME complex ions and intercalation of lithium ions into graphite occurred simultaneously. In addition, PEGDME co-intercalation caused electrode exfoliation and severe capacity fading as the cycling progressed, which is in significant contrast with the results obtained for DEGDME. DEGDME showed only co-intercalation in graphite, which allowed stable cycling without capacity fading.30 We have found that it was possible to prevent the co-intercalation and loss of capacity through the solid electrolyte interphase (SEI) layer, which was formed upon addition of FEC. Fig. 1a illustrates the co-intercalation of Li–PEGDME complex ions and the intercalation of lithium ions into the gallery of the graphite electrode. The additive FEC, which is known to form a stable SEI layer on graphite electrodes, was used.31–34 When the SEI layer was formed, it was possible to conduct only lithium ions and prevent the capacity loss caused by the co-intercalation of PEGDME, as shown in Fig. 1b.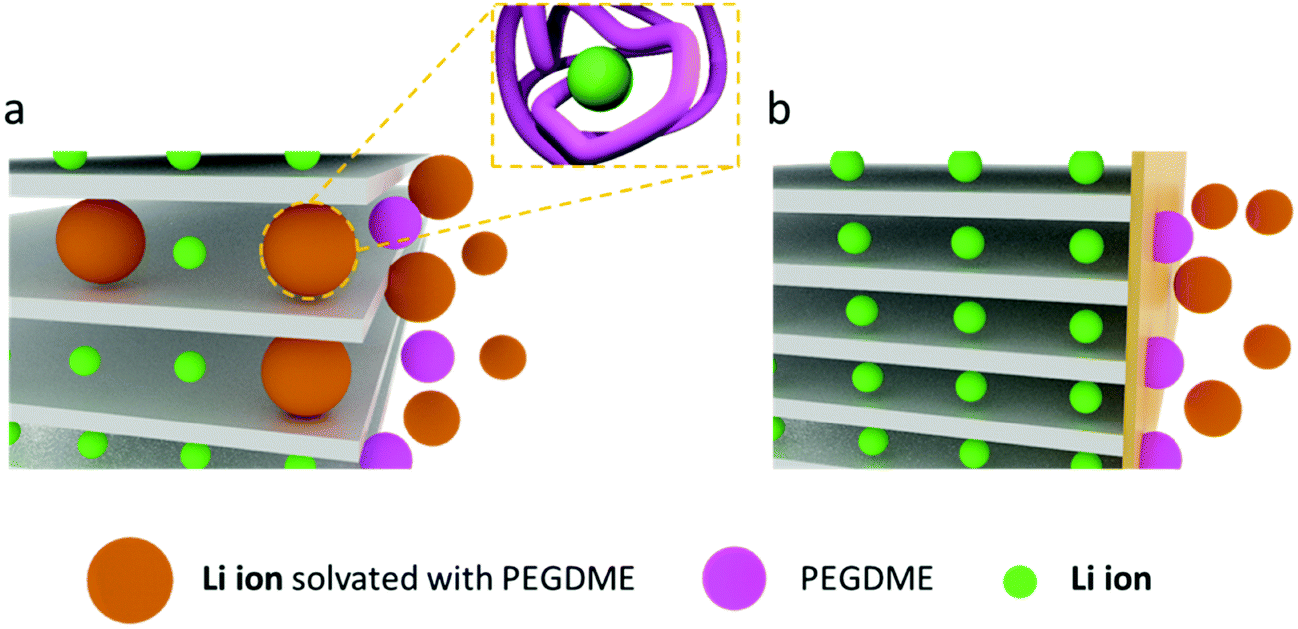 | ||
| Fig. 1 Schematics of (a) intercalation of lithium ions coexisting with co-intercalation of Li–PEGDME complex ions in a graphite electrode, and (b) absolute intercalation of lithium ions in graphite with an SEI layer (formed upon use of FEC). | ||
Transmission electron microscopy (TEM) measurements were performed to confirm the formation of the SEI layer on the graphite electrode by FEC. For that, a lithium/graphite half-cell with PEGDME electrolyte was fabricated, and the cell was run for one lithiation/delithiation cycle. The pristine graphite surface image is shown in Fig. S2 (ESI†). After the first cycle of the cell without FEC, the morphology of the graphite surface did not change significantly compared with the pristine form, as shown in Fig. 2a. These results are in good agreement with a previous report.29 In energy dispersive X-ray spectroscopy (EDS) analysis, F, O, and S elements were observed due to the lithium salt (lithium bis(trifluoromethane sulfonyl)imide, LiTFSI) along with some of the remaining PEGDME electrolyte. However, nothing was observed outside the graphite boundary (Fig. 2c). When the FEC additive was used, an amorphous layer was observed outside the graphite surface (which is surrounded by white dashed lines in Fig. 2b). The layer was likely formed due to the addition of FEC, as reported by other literature reports.31–34 According to EDS analysis (Fig. 2d), this region is rich in F and O elements. The TEM images show the formation of a layer on the graphite surface upon use of the additive.
 | ||
| Fig. 2 Transmission electron microscopy (TEM) image of graphite (a) without and (b) with FEC after the first cycle. EDS mapping images of graphite (c) without and (d) with FEC after the first cycle. X-ray photoelectron spectroscopy (XPS) spectra of the graphite electrode: (e) pristine, (f) without FEC after the first cycle, and (g) with FEC after the first cycle. A lithium/graphite half-cell with PEGDME electrolyte was fabricated, and the cell was run for one lithiation/delithiation cycle. | ||
X-Ray photoelectron spectroscopy (XPS) was also performed for a more comprehensive analysis of the layer. Fig. 2e shows the pristine graphite electrode. A strong peak appears at 688.1 eV due to the polyvinylidene fluoride (PVdF) binder in the F1s spectrum, and there were no apparent peaks in Li1s.35 The graphite surface without the additive was identical to that of pristine graphite, but the Li1s spectrum presented a peak at 56.6 eV, which was attributed to lithium salts (Fig. 2f).36 The F1s spectrum due to LiTFSI was overlapped with the one due to PVDF (Fig. 2f).35 When the FEC additive was used, peaks due to LiF appeared at 685.2 eV in the F1s spectrum and at 56.4 eV in the Li1s spectrum (in fact, the Li1s peak due to LiF overlapped with the one due to LiTFSI), as shown in Fig. 2g.5,6,36 Overall, the TEM, EDS, and XPS data confirmed that an SEI layer was formed on the graphite surface upon addition of FEC, and that the SEI layer contained LiF.
The lithium/graphite half-cells containing the PEGDME electrolyte with or without FEC were cycled with the change of d-spacing in graphite observed by in situ XRD. Fig. 3a shows the potential profile and the corresponding in situ XRD 3D graph (side view) during lithiation and delithiation in the cell without FEC, and Fig. 3d presents the results of the cell with FEC. The top view images for the in situ XRD 3D data are also displayed in Fig. S3 (ESI†). Fig. 3b, c and e, f show the XRD data by rescaling Fig. 3a and d, respectively, to a lower angle of 2θ = 15°. As the lithiation progressed in the cell with FEC, the XRD peak at 26.5° gradually moved to 25.2° and finally converged to 23.9° at the end of lithiation (Fig. 3d). As shown in Fig. S4 (ESI†) and other literature,37 the XRD peak at 26.5° was attributed to the (002) plane of pristine graphite.37 Intercalation of lithium ions into the graphite should result in expansion of the d-spacing of the graphite. Delithiation occurred in a mirror-symmetric manner with lithiation. Such XRD data are well matched with those of a previous paper on the change of d-spacing in the graphite, which are intercalated and deintercalated by lithium ions during cycling.37,38
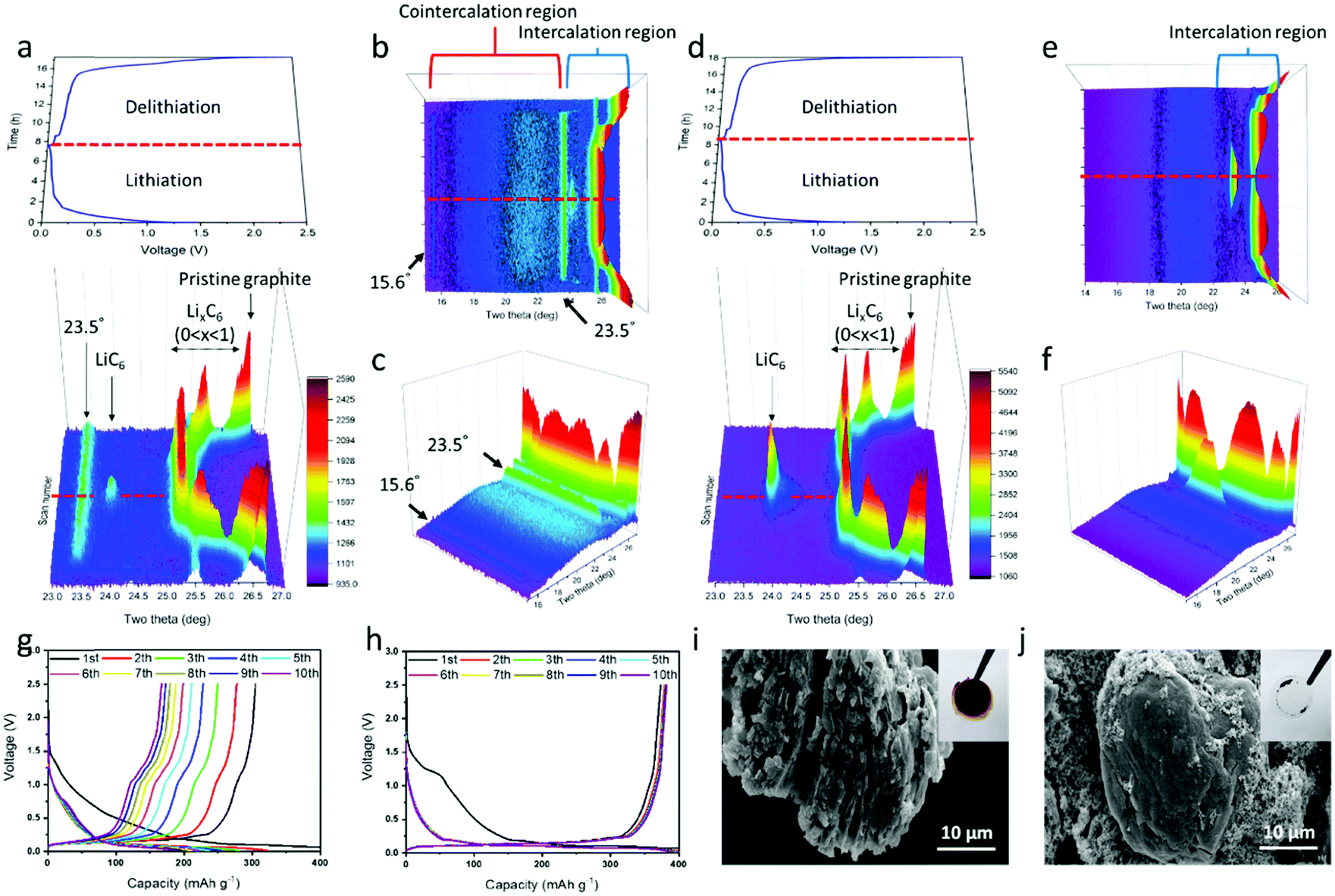 | ||
| Fig. 3 Potential profiles and in situ XRD patterns of lithium/graphite half-cell containing PEGDME electrolyte (a–c) without and (d–f) with FEC during lithiation and delithiation. Red dotted lines of (a, b, d and e) indicate the point where lithiation finishes and delithiation starts. LiC6 represents the fully lithiated graphite. Potential profiles of lithium/graphite half-cell containing PEGDME electrolyte (g) without and (h) with FEC for 10 cycles at the 0.1C-rate. SEM images of graphite electrode (i) without and (j) with FEC after 10 cycles. The inset pictures in (i) and (j) show the separator membranes after the cell had been disassembled. | ||
The cell without the additive presented a different behavior, with new prominent XRD peaks at 15.6° and 23.5° together with the peaks from 25.2° to 23.9° due to the intercalation of lithium ions (Fig. 3a–c). As reported by a previous study on the co-intercalation of DEGDME,29,30,39 the new peaks at 15.6° and 23.5° likely occurred due to the co-intercalation of PEGDME. Fig. 3a–c strongly suggest the coexistence of intercalation by lithium ions and co-intercalation by Li–PEGDME complex ions, as illustrated in Fig. 1a. When the in situ XRD data are compared with the corresponding potential profile in Fig. 3a–c, the co-intercalation-related peaks at 15.6° and 23.5° appear at approximately 0.5 V, whereas the intercalation-related peaks start to occur at approximately 0.2 V.29,30,39 The position and intensity of the co-intercalation-related peaks were nearly unchanged during the remaining lithiation process. This indicates that the co-intercalation of Li–PEGDME preferentially occurred at the initial stage of the lithiation process, and the intercalation of lithium ions predominantly proceeded in the remaining stage. Solvated lithium ions in electrolyte require more energy in order to be released from the solvent before intercalation into graphite, but co-intercalation may occur rapidly at relatively low energy because the lithium ions do not need to be released from the solvent.29 Consequently, when the lithiation process started, co-intercalation first occurred at a high potential of approximately 0.5 V, and then lithium ions separated from the solvent were dominantly intercalated at a lower potential of approximately 0.2 V. We also confirmed the absence of co-intercalation of PEGDME with the assistance of FEC by infrared (IR) spectroscopy. As shown in Fig. S5 (ESI†), discharged graphite with the assistance of FEC did not show any distinguishable peaks, which were same with those of the pristine graphite powder. However, the graphite without FEC showed evident peaks at 2860 cm−1 due to the C–H stretching vibration, suggesting that PEGDME was present in the graphite.
The adverse effects of PEGDME co-intercalation can be observed in Fig. 3g and i. Fig. 3g displays the severe capacity fading in the Li/PEGDME/graphite half-cell without FEC throughout the cycles. After 10 cycles, the residual capacity decreased to less than 50%. Fig. 3i shows an SEM image in which the graphite electrode in the cell without FEC was severely damaged by exfoliation after 10 cycles (for comparison, see the SEM image of the pristine graphite electrode in Fig. S6, ESI†). It can be conjectured that the co-intercalation of PEGDME caused the exfoliation of graphite particles, thereby resulting in capacity fading.29 Fig. S7a (ESI†) shows that the intensity of the XRD peak at 26.5° decreased as the cycling advanced. This indicates that the graphite electrode continued to be exfoliated by the co-intercalation. Such exfoliation also caused the delamination of graphite particles from the current collector. When the cell was disassembled, most of the graphite particles were easily detached from the current collector and attached to the separator membrane, as shown in the inset of Fig. 3i. This indicates that such exfoliation happened in most graphite particles. By contrast, the cell with FEC did not present the co-intercalation-related XRD peaks (15.6° and 23.5°) and showed only Li ion intercalation-related peaks (Fig. 3d–f). The cell with FEC demonstrated a stable cycling profile without significant capacity fading for 10 cycles, as shown in Fig. 3h. Moreover, no traces of exfoliation were observed in the graphite electrode after cycling (Fig. 3j). Furthermore, only a part of the graphite was attached to the membrane, and most particles were firmly attached to the current collector after the cell was disassembled (inset of Fig. 3j). In addition, the graphite XRD intensity of the cell with FEC remained almost constant (Fig. S7b, ESI†), thus indicating that the structure of graphite remained intact throughout cycling. As shown in Fig. 2, an SEI layer was formed on the graphite surface by FEC, and that layer was crucial to prevent the co-intercalation of PEGDME and suppress capacity fading, thereby resulting in stable reversible cycling. It should be noted that a small bump at approximately 1.2 V in the potential profile of the first lithiation (Fig. 3h) occurred due to the electrochemical reaction of FEC to form the SEI layer.
After verifying the effect of FEC in the graphite electrode with the PEGDME electrolyte, we attempted to use the additive for the cell with a solidified polymer electrolyte from BisA and PEGDME. Fig. 4a shows the fabricated free-standing BisA–PEGDME polymer electrolyte, which is a semi-transparent solid film with high flexibility. Its ionic conductivity was measured as 5.9 × 10−4 S cm−1 at room temperature, and its electrochemical stability window was widened up to 4.7 V, as depicted in the graphs of Fig. 4b and c, respectively. As shown in Fig. S1 (ESI†), the lithium/graphite half-cell with the BisA–PEGDME polymer electrolyte exhibited severe capacity fading, which is a similar behavior to that of the cell with the PEGDME electrolyte (Fig. 3g). Fig. 4d shows that the capacity fading was highly suppressed by the addition of FEC, as in the cell that was incorporated with PEGDME electrolyte and FEC in Fig. 3h. The in situ XRD data in Fig. 4e show only Li ion intercalation-related peaks without any co-intercalation-related peaks at 23.5°. That is, FEC had a similar function in the polymer electrolyte cell, which resulted in “absolute intercalation” by lithium ions in graphite, as illustrated in Fig. 1b.
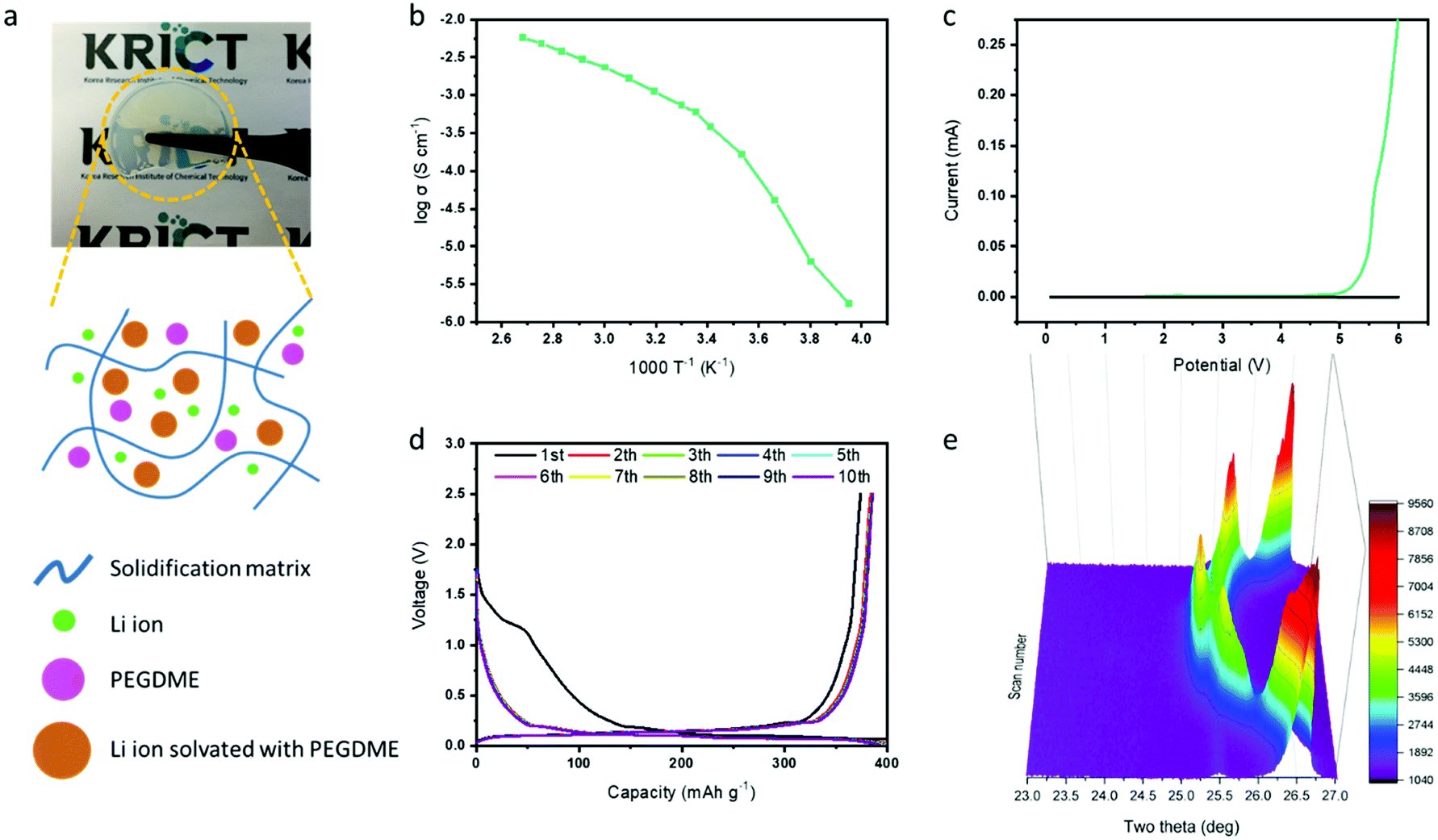 | ||
| Fig. 4 (a) Photo image of the free-standing BisA–PEGDME polymer electrolyte film and a schematic illustration of the crosslinked polymer electrolyte structure. (b) Ionic conductivity of the polymer electrolyte at various temperatures, and (c) its linear sweep voltammetry (LSV) curve at a scan speed of 1 mV s−1. (d) Potential profiles for ten cycles at the 0.1 C-rate, and (e) in situ XRD data for one lithiation/delithiation cycle of the lithium/graphite half-cell with the BisA–PEGDME polymer electrolyte including the FEC additive. | ||
A polymer electrolyte full-cell with graphite as the anode and LiFePO4 as the cathode was also prepared to confirm the applicability in the actual battery system. LiFePO4 was chosen as a cathode because it has a highly stable structure and moderately high capacity.5,6,40Fig. 5a shows the charging and discharging profiles at different C-rates in the graphite/LiFePO4 full-cell with the BisA–PEGDME polymer electrolyte including FEC. The graphite/LiFePO4 full-cell presented a capacity of 114 mA h g−1 at 0.1C and 104 mA h g−1 at 0.2C. The long-term cycling stability was also tested at 0.5C for 100 cycles, as shown in Fig. 5b. Charge–discharge profiles for every 20 cycles are shown in Fig. S8 (ESI†). Except for the initial several cycles, the cell runs stably at a coulombic efficiency higher than 99% for most cycles. Although the severe capacity fading of the full-cell with the polymer electrolyte was efficiently prevented by the addition of FEC, the cell exhibited a tendency of slow capacity decrease with long-term cycling. After 100 cycles, the residual capacity of the cell reached approximately 80% of the initial capacity. Considering the results for the Li/LiFePO4 or Li/graphite half-cell (Fig. S9, ESI†), it can be concluded that the capacity decrease of the full-cell during the cycling was possibly related to the anode side. Finally, a pouch cell (30 mm × 42 mm) of the graphite/LiFePO4 full-cell with polymer electrolyte was also prepared. The pouch cell with the polymer electrolyte performed well even when the cell was bent in various directions, as shown in Fig. 5c.
 | ||
| Fig. 5 (a) Charge–discharge profile of the graphite/LiFePO4 full-cell with the BisA–PEGDME polymer electrolyte including FEC at various C-rates. (b) Cycle stability of graphite/LiFePO4 full-cell at 0.5 C. (c) Flexibility test of the graphite/LiFePO4 pouch cell with the BisA–PEGDME polymer electrolyte. | ||
Conclusions
This study presented a solidified polyether-based polymer electrolyte used in a graphite/LiFePO4 full-cell battery. A simple in situ thermal crosslinking of bisphenol A ethoxylate diacrylate (BisA) and poly(ethylene glycol) dimethyl ether (PEGDME) was used to produce a solidified polymer electrolyte, which was sandwiched between the anode and cathode. Real-time monitoring of the change in the graphite gallery by in situ XRD analysis demonstrated the coexistence of co-intercalation by Li–PEGDME complex ions and intercalation by lithium ions into the gallery during the lithiation process. Such co-intercalation induced exfoliation of the graphite and a severe capacity fading after repeated cycles. We successfully suppressed the co-intercalation by introducing an SEI layer formed by the addition of the FEC additive. The resulting “absolute intercalation” led to stable cycling of the graphite/LiFePO4 full-cell battery with polymer electrolyte. Except for the initial several cycles, the cell could run stably at a coulombic efficiency higher than 99% for most cycles, and it could retain 80% of the initial capacity after 100 cycles. To further enhance the reversibility of the polymer electrolyte full-cell battery, other strategies should be developed, for example, using a polymer electrolyte more compatible with the graphite electrode and a more efficient additive to stabilize the interface between the electrolyte and electrode. We also demonstrated a flexible pouch-type cell with the polymer electrolyte, which worked well even when the cell was bent in various directions.Experimental section
Materials
Poly(ethylene glycol) dimethyl ether (PEGDME, average Mn = 500) and bisphenol A ethoxylate diacrylate (BisA, average Mn = 688, EO/phenol = 4) used for the solid polymer electrolytes were purchased from Sigma-Aldrich and were thoroughly dried under vacuum and by molecular sieves before use. The moisture level was controlled to be less than 20 ppm, which was measured by Karl Fischer titration (C30, Mettler Toledo). Lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and FEC were obtained from Sigma-Aldrich and dried under vacuum oven before use. t-Butyl peroxypivalate (Seki Arkema Co., Japan) was purchased and used as received. Graphite (SAG-R, MTI Korea) and LiFePO4 (LFP, Hanwha Chemical Co.) were purchased for the anode and cathode materials, respectively. Polyvinylidene fluoride (PVdF, Kynar Flex® 2851) was provided by Arkema.Preparation of solid polymer electrolytes
A liquid-state precursor was prepared by dissolving LiTFSI (0.287 g) in PEGDME (0.8 g) and BisA (0.2 g). FEC was added at 5.0 wt% with respect to the mixture of PEGDME and BisA. t-Butyl peroxypivalate was used at 2.0 wt% of BisA as a thermal initiator. The precursor was then solidified by heating at 90 °C to produce a semi-transparent free-standing solid polymer electrolyte film. The ionic conductivity and electrochemical stability of the polymer electrolytes were measured via electrochemical impedance analysis and linear sweep voltammetry, as described in our previous paper.23Preparation of graphite/Li half-cells
A slurry was prepared by mixing graphite, PVdF binder, and super P at a mass ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in N-methyl-2-pyrrolidone (NMP). The slurry was coated on a copper current collector and dried. Lithium metal (Honjo Metal Co., Japan) was used as the counter electrode with copper foil as the current collector. The loading density of graphite was approximately 2.5 mg cm−2. An electrospun membrane (thickness: 30 μm), which was purchased from Amogreentech Co. (model: Nano Membrane), was used as a supporting separator membrane of the polymer electrolyte. The membrane was wetted with the liquid precursor and then placed between the positive and negative electrodes. After the electrodes were fully soaked with the liquid precursor, the thermal crosslinking reaction was then carried out to produce the crosslinked solid polymer electrolyte by placing the cell in an oven at 90 °C. Such in situ solidification was very important for resulting in conformal contact between the solid polymer electrolyte and the electrode, and hence decreasing the interfacial resistance. The complete crosslinking was monitored using a Fourier transform-infrared (FT-IR) spectrometer (Nicolet 6700, Thermo Fisher Scientific Inc.).
:1:1 in N-methyl-2-pyrrolidone (NMP). The slurry was coated on a copper current collector and dried. Lithium metal (Honjo Metal Co., Japan) was used as the counter electrode with copper foil as the current collector. The loading density of graphite was approximately 2.5 mg cm−2. An electrospun membrane (thickness: 30 μm), which was purchased from Amogreentech Co. (model: Nano Membrane), was used as a supporting separator membrane of the polymer electrolyte. The membrane was wetted with the liquid precursor and then placed between the positive and negative electrodes. After the electrodes were fully soaked with the liquid precursor, the thermal crosslinking reaction was then carried out to produce the crosslinked solid polymer electrolyte by placing the cell in an oven at 90 °C. Such in situ solidification was very important for resulting in conformal contact between the solid polymer electrolyte and the electrode, and hence decreasing the interfacial resistance. The complete crosslinking was monitored using a Fourier transform-infrared (FT-IR) spectrometer (Nicolet 6700, Thermo Fisher Scientific Inc.).
A graphite/Li half-cell with PEGDME electrolyte was also prepared for comparison. The half-cell has used the same graphite and lithium foil electrode, but PEGDME is used as the electrolyte.
Preparation of graphite/LiFePO4 full-cells
The anode slurry was prepared using graphite, PVdF binder, and super P at a mass ratio of 8:1:1 in NMP. The slurry was coated on copper foil and dried. The cathode electrode was prepared by a similar method to the anode by using LFP as the cathode material and aluminum foil as the current collector. The loading densities of graphite and LFP were typically 2.7 mg cm−2 and 7.0 mg cm−2, respectively. Solid polymer electrolyte was prepared by the in situ crosslinking method, which was the same as for the half-cell.
Coin cells (CR2032) were assembled to evaluate electrochemical properties. The charge/discharge cycling measurements for 100 cycles were performed at room temperature and at 0.5C. The cut-off voltage was 2.5–4.2 V. Cycle tests were performed using TOSCAT-3100 apparatus (TOYO SYSTEM).
Characterization
A binder-free graphite electrode was used for the transmission electron microscopy (TEM) measurements. Lithium metal and graphite on Cu foil were assembled in a coin cell. After the SEI layer was formed, the cell was disassembled and the graphite was washed with 1,2-dimethoxyethane. The washed graphite was dried for 2 days at room temperature. The graphite was dispersed in 1,2-dimethoxyethane and then placed on a lacey grid. All the processes were performed in an Ar-filled grove box.In situ X-ray diffraction (XRD) measurements were carried out by X-ray diffractometer (Empyrean, Malvern Panalytical Co.) using Cu Kα radiation (0.154 nm) at 40 kV and 30 mA in a coin cell configuration. The X-ray holder used a beryllium window allowing X-ray penetration. Two theta scans were conducted from 10 to 60°. Electrochemical measurements were performed using WBCS-3000 (Wonatech).
The X-ray photoelectron spectroscopy (XPS) instrument was an Axis-Supra (Kratos Co.) with an Al monochromatic X-ray source (1486.7 eV). An Ar-filled glove box was attached to the XPS instrument, preventing the surface of the electrode from being exposed to air.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the R&D Convergence Program of the National Research Council of Science and Technology (CAP-15-02-KBSI), Republic of Korea and by a project no. SS2122-20 (A Study on the Convergence Materials for Off-Grid Energy Conversion/Storage Integrated Devices) of Korea Research Institute of Chemical Technology (KRICT).Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Armand, Nature, 2001, 414, 359–367 CrossRef PubMed.
- B. Liu, J. G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef CAS.
- H. Kim, Y. J. Gong, J. Yoo and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15540–15545 RSC.
- H. Kim, Y. S. Kim and J. Yoo, Sustainable Energy Fuels, 2020, 4, 522–527 RSC.
- H. Kim, Y. S. Kim and J. Yoo, Sustainable Energy Fuels, 2020, 4, 3282–3287 RSC.
- Q. Wang, J. Sun, X. Yao and C. Chen, J. Loss Prev. Process Ind., 2006, 19, 561–569 CrossRef.
- D. Lisbona and T. Snee, Process Saf. Environ. Prot., 2011, 89, 434–442 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama and A. Yamada, Nat. Energy, 2018, 3, 22–29 CrossRef CAS.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- L. Hu and K. Xu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3205–3206 CrossRef CAS PubMed.
- K. K. Fu, Y. Gong, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey, J. Dai, Y. Chen, Y. Mo, E. Wachsman and L. Hu, Sci. Adv., 2017, 3, 1–12 Search PubMed.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- P. J. Lian, B. S. Zhao, L. Q. Zhang, N. Xu, M. T. Wu and X. P. Gao, J. Mater. Chem. A, 2019, 7, 20540–20557 RSC.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- Z. Ding, J. Li, J. Li and C. An, J. Electrochem. Soc., 2020, 167, 070541 CrossRef.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- X. Li, Y. Zhao, L. Cheng, M. Yan, X. Zheng, Z. Gao and Z. Jiang, J. Solid State Electrochem., 2005, 9, 609–615 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nat. Lett., 1998, 394, 456–458 CrossRef CAS.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 2–5 CrossRef PubMed.
- X. Chen, W. He, L. X. Ding, S. Wang and H. Wang, Energy Environ. Sci., 2019, 12, 938–944 RSC.
- J. Suk, Y. H. Lee, D. Y. Kim, D. W. Kim, S. Y. Cho, J. M. Kim and Y. Kang, J. Power Sources, 2016, 334, 154–161 CrossRef CAS.
- K. Liu, A. Pei, H. R. Lee, B. Kong, N. Liu, D. Lin, Y. Liu, C. Liu, P. Chun Hsu, Z. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 4815–4820 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- J. Ming, Z. Cao, W. Wahyudi, M. Li, P. Kumar, Y. Wu, J. Y. Hwang, M. N. Hedhili, L. Cavallo, Y. K. Sun and L. J. Li, ACS Energy Lett., 2018, 3, 335–340 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- H. Kim, K. Lim, G. Yoon, J. H. Park, K. Ku, H. D. Lim, Y. E. Sung and K. Kang, Adv. Energy Mater., 2017, 7, 1–10 Search PubMed.
- H. Kim, G. Yoon, K. Lim and K. Kang, Chem. Commun., 2016, 52, 12618–12621 RSC.
- E. Markevich, G. Salitra and D. Aurbach, ACS Energy Lett., 2017, 2, 1337–1345 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- T. Hou, G. Yang, N. N. Rajput, J. Self, S. W. Park, J. Nanda and K. A. Persson, Nano Energy, 2019, 64, 103881 CrossRef CAS.
- Y. Chu, Y. Shen, F. Guo, X. Zhao, Q. Dong, Q. Zhang, W. Li, H. Chen, Z. Luo and L. Chen, Advanced Characterizations of Solid Electrolyte Interphases in Lithium-Ion Batteries, Springer, Singapore, 2020, vol. 3 Search PubMed.
- B. Flamme, J. Światowska, M. Haddad, P. Phansavath, V. Ratovelomanana-Vidal and A. Chagnes, J. Electrochem. Soc., 2020, 167, 070508 CrossRef CAS.
- D. Ensling, M. Stjerndahl, A. Nytén, T. Gustafsson and J. O. Thomas, J. Mater. Chem., 2009, 19, 82–88 RSC.
- C. J. Pan, C. Yuan, G. Zhu, Q. Zhang, C. J. Huang, M. C. Lin, M. Angell, B. J. Hwang, P. Kaghazchi and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5670–5675 CrossRef CAS PubMed.
- J. Park, S. S. Park and Y. S. Won, Electrochim. Acta, 2013, 107, 467–472 CrossRef CAS.
- J. Zhang, D. W. Wang, W. Lv, L. Qin, S. Niu, S. Zhang, T. Cao, F. Kang and Q. H. Yang, Adv. Energy Mater., 2018, 8, 1–14 CrossRef.
- L. X. Yuan, Z. H. Wang, W. X. Zhang, X. L. Hu, J. T. Chen, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00946f |
| This journal is © The Royal Society of Chemistry 2021 |