
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-colour photoswitching in photoresponsive inorganic thin films†
Elin
Sundin
,
Fredrik
Johansson
,
Valeria
Saavedra Becerril
,
Joachim
Wallenstein
,
August
Gasslander
,
Jerker
Mårtensson
 and
Maria
Abrahamsson
*
and
Maria
Abrahamsson
*
Department of Chemistry and Chemical Engineering, Chalmers University of Technology, Gothenburg, Sweden. E-mail: abmaria@chalmers.se
First published on 4th March 2021
Abstract
Herein we report the first example of an inorganic photochromic material that allows for spatial addressability. We designed a photoresponsive thin film based on a ruthenium sulfoxide complex, [Ru(deeb)2PySO-iPr]2+, which was immobilized onto a mesoporous ZrO2 surface. The resulting material allows for all-optical detection and shows reversible, selective, photochromic behavior that can be cycled back and forth. The photosiomerization quantum yield is lower on the film than in solution which is attributed to a combination of the immobilization and steric hindrance by molecules in close proximity as evident from a surface coverage dependence of the photoisomerization quantum yield. The results reported here provide an important step towards inorganic photoswitchable materials.
Introduction
Photochromic molecules are compounds that isomerize and change colour upon photoexcitation. The colour change typically represents a metastable state that reverts to the ground state thermally and/or through triggering with light.1,2 Photochromic materials are promising for applications where photoresponsivity is beneficial such as logic gates, molecular machines and memory storage.3–6 For such applications, selective conversion between the isomers by irradiation with light of different wavelengths is required, as is control over the precise timing of these events. Several examples of organic molecules with light-induced reversible photoisomerization exists, including but not limited to azobenzene,7 diarylethene,8 and spiropyran,9 while, as noted by Rack and co-workers, two-colour reversible switching is rarely observed in metal complexes11,38 and only a few reports exist in the literature.11–15For practical applicability, the materials used must retain function in the solid state and at ambient conditions, something that is not straightforward since photochromic molecules in the solid state typically display a much lower isomerization ability compared to the same molecules in fluid solution.16 Moreover, for applications such as logic gates and molecular memory storage, spatial addressability is required. Thus, attaching the photochromic molecules onto solid surfaces becomes necessary. Attaching organic photoswitches directly onto surfaces however usually results in a loss of the photoisomerization ability17,18 and both interactions with the surface as well as steric hindrance between neighbouring molecules needs to be inhibited to retain the photoisomerization on the surface.17 This has been achieved for organic molecules on metallic surfaces by adding spacer groups17–19 or incorporating the molecules in different matrixes.20,21 By these modifications, organic photoswitches have been possible to photoisomerize on surfaces and nanoparticles.17–31 These approaches are however generally restricted to monolayers on flat surfaces which limits the surface coverage, this was however recently circumvented by incorporating molecules into a porous network on the surface.21 By using a mesoporous semiconductor substrate instead of a flat surface, the molecular density directly on the surface can be dramatically increased since several molecules can be attached in close proximity on the porous surface.32 Moreover, using metal complexes instead of organic molecules could also come with additional benefits such that their properties can be tuned with relative ease. By varying the ligands in metal complexes, both the bulkiness and binding to the surface as well as other properties such as light absorption and redox properties can be easily tuned synthetically, and the ligand can be used for direct immobilization to the surface.
Ruthenium(II) complexes with sulfoxide ligands are a well-known type of photochromic molecules which undergo S-to-O linkage isomerization upon metal-to-ligand charge transfer (MLCT) excitation in weakly basic liquid solutions.10,11,33–41 The quantum yield for the photoinduced S-to-O isomerization in these complexes varies depending on the structure of both the sulfoxide ligand and the ancillary ligands. Generally, a more electron withdrawing sulfoxide ligand results in a larger quantum yield36 and the same appears to be true for the ancillary ligands.39 For most Ru-sulfoxide complexes, the O-bonded isomer is insensitive to light and the thermodynamically stable S-bonded isomer is regenerated thermally.34,38 A few years ago however, the Rack-group observed two-colour reversible switching in a RuII-sulfoxide complex, [Ru(bpy)2PySO-iPr]2+, where PySO-iPr is 2-((isopropylsulfinyl)methyl)pyridine and bpy is 2,2′-bipyridine. When the complex was irradiated with UV-light, the S-form isomerized to the O-bonded form, and subsequent irradiation with visible light triggered the isomerization back to the S-bonded form.11,38,42 The light-triggered reversibility highlights an enhanced potential for these complexes in applications such as molecular machines, and a few examples of reversible photoinduced switching between the isomers have now been reported.11,43,44 No examples of photoisomerizable solid state materials based on ruthenium sulfoxide complexes on solid substrates have been reported so far.
Here, as a step towards a functional photochromic inorganic material with wavelength selective spatial addressability, we have synthesized a ruthenium sulfoxide complex, [Ru(deeb)2PySO-iPr]2+, with the previously studied PySO-iPr ligand combined with deeb ligands (deeb is 4,4′-diethyl ester 2,2′-bipyridine) that allows for anchoring of the complex to mesoporous metal oxide surfaces via diethyl ester groups (see Scheme 1) and studied how the immobilization affects the photoisomerization. Using mesoporous semiconductor surfaces as the substrate comes with several advantages compared to other previously studied surfaces; firstly, as previously mentioned, it is an ideal scaffold to achieve a high surface coverage.32 Secondly, the surface is semi-transparent which allows for optical detection of the photo-switching.23 Thirdly, it allows for use of a variety of binding groups.45 Mesoporous ZrO2 films were chosen as the substrate since no electronic interaction (e.g. electron injection) between the excited complex and the mesoporous surface is expected,46 hence the photoisomerization of the Ru(deeb)2PySO-iPr/ZrO2 material can be exclusively monitored by optical spectroscopic methods.
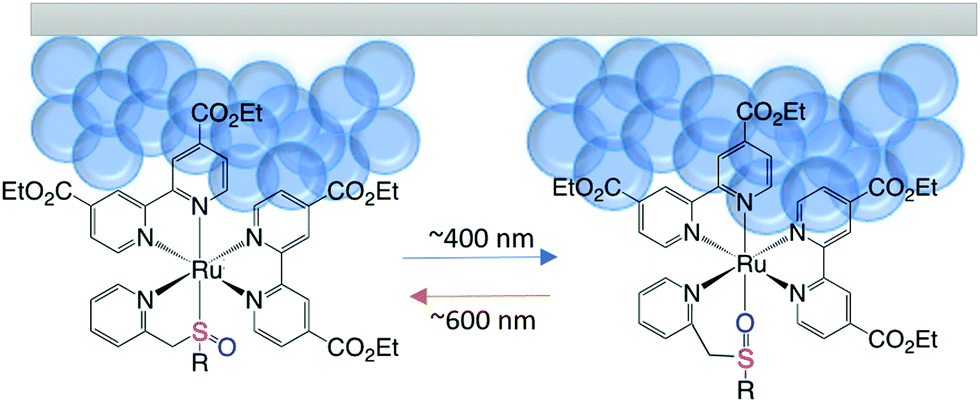 | ||
| Scheme 1 Schematic illustration showing the reversible photoisomerization of Ru(deeb)2PySO-R, (R = iPr) attached to a mesoporous surface. | ||
Experimental
Description of the synthesis can be found in the ESI.† All samples were prepared under red light and stored in the dark to prevent any photoisomerization prior to the measurements. The samples were stored in powder form and solutions were typically freshly prepared. ZrO2 paste were prepared following procedures in the literature.47 Semitransparent ZrO2 films were prepared by doctor blading the ZrO2 colloidal paste with Scotch tape on glass substrates (Ted Pella, Inc.) that had been cleaned by ultrasonication with 2% RBS detergent solution and acetone. The films were progressively heated and sintered (450 °C for 30 min) in an oven in air. The ZrO2 films were sensitized by immersing them in dichloromethane (DCM) solutions of the compound for at least 8 hours. The surface coverage, Γ (mol cm−2), of the films were calculated using Γ = A/(ε × 1000), where ε was assumed to be the same as in liquid solution.UV-vis absorption spectra were recorded using a Varian Cary 50 Bio spectrophotometer. In the photoisomerization photolysis experiments, the absorption spectra were continuously measured (0.1–10 min between each spectrum depending on the measurement; to record the S-to-O photoisomerization 0.1–1 min between the spectra were used depending on the measurement and the kinetics of interest and for the O-to-S photoisomerization 1–5 min was used between each scan) while a LED lamp of different wavelengths; 365 nm (LZ1-00UV00), 385 (LZ1-00UB00), 405 (LZ1-00UB00), 523 (LZ1-00G100), 590 (LZ1-00A100), 623 nm (LZ1-00R100) or a Xe-arc lamp (250 W) irradiated the sample (0.5–2 mW at the sample). The photo-isomerization quantum yields were estimated in a similar fashion to previously published methods.11,48 A detailed description of the quantum yield calculations can be found in the ESI.†
Results and discussion
The synthetic procedure for [Ru(deeb)2PySO-iPr][PF6]2 is based on a modified protocol used by the Rack-group to prepare [Ru(bpy)2PySO-iPr][PF6]2.11 The synthesis and the NMR data of the required ligands, the intermediate cis-dichloro-Ru complex and the target Ru-complex are presented in the ESI,† as is HRMS data for the final compound. Room temperature photoluminescence measurements reveals that the complex is non-emissive, consistent with previously studied ruthenium sulfoxide complexes. The complex attaches successfully to mesoporous ZrO2 thin films following overnight immersion in a DCM solution of the complex, yielding the Ru(deeb)2PySO-iPr/ZrO2 solid films. The surface coverage of the films is controlled by using different concentrations of the complex in the DCM solutions. The obtained surface coverages varies between ∼10 nmol cm−2 and ∼70 nmol cm−2, which are ∼200 times higher than for self-assembled monolayers on flat surfaces and similar to reported surface coverages in a porous nanowire network.21The UV-vis absorption spectrum of the thermodynamically stable S-bonded isomer in DCM solution is shown in Fig. 1a (red trace). The expected MLCT band38,49 is centred at 411 nm (ε ∼ 7500 M−1 cm−1) and appears at the same position in propylene carbonate (PC) and acetonitrile (MeCN). This peak is red-shifted ca. ∼20 nm compared to the previously studied [Ru(bpy)2PySO-iPr]2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and this red-shift is attributed to the electron withdrawing nature of the deeb-ligands, in agreement with observations for other heteroleptic Ru-complexes.49,50 The UV-vis absorption spectrum of Ru(deeb)2PySO-iPr/ZrO2 recorded in air, Fig. 1b (red trace), shows a broadened and slightly blue-shifted (405 nm) MLCT band. The broadening is common for molecules attached to semiconductor surfaces32 and the blue-shift could possibly be related to a lack of solvent stabilization since there is no solvent surrounding the complex.
11 and this red-shift is attributed to the electron withdrawing nature of the deeb-ligands, in agreement with observations for other heteroleptic Ru-complexes.49,50 The UV-vis absorption spectrum of Ru(deeb)2PySO-iPr/ZrO2 recorded in air, Fig. 1b (red trace), shows a broadened and slightly blue-shifted (405 nm) MLCT band. The broadening is common for molecules attached to semiconductor surfaces32 and the blue-shift could possibly be related to a lack of solvent stabilization since there is no solvent surrounding the complex.
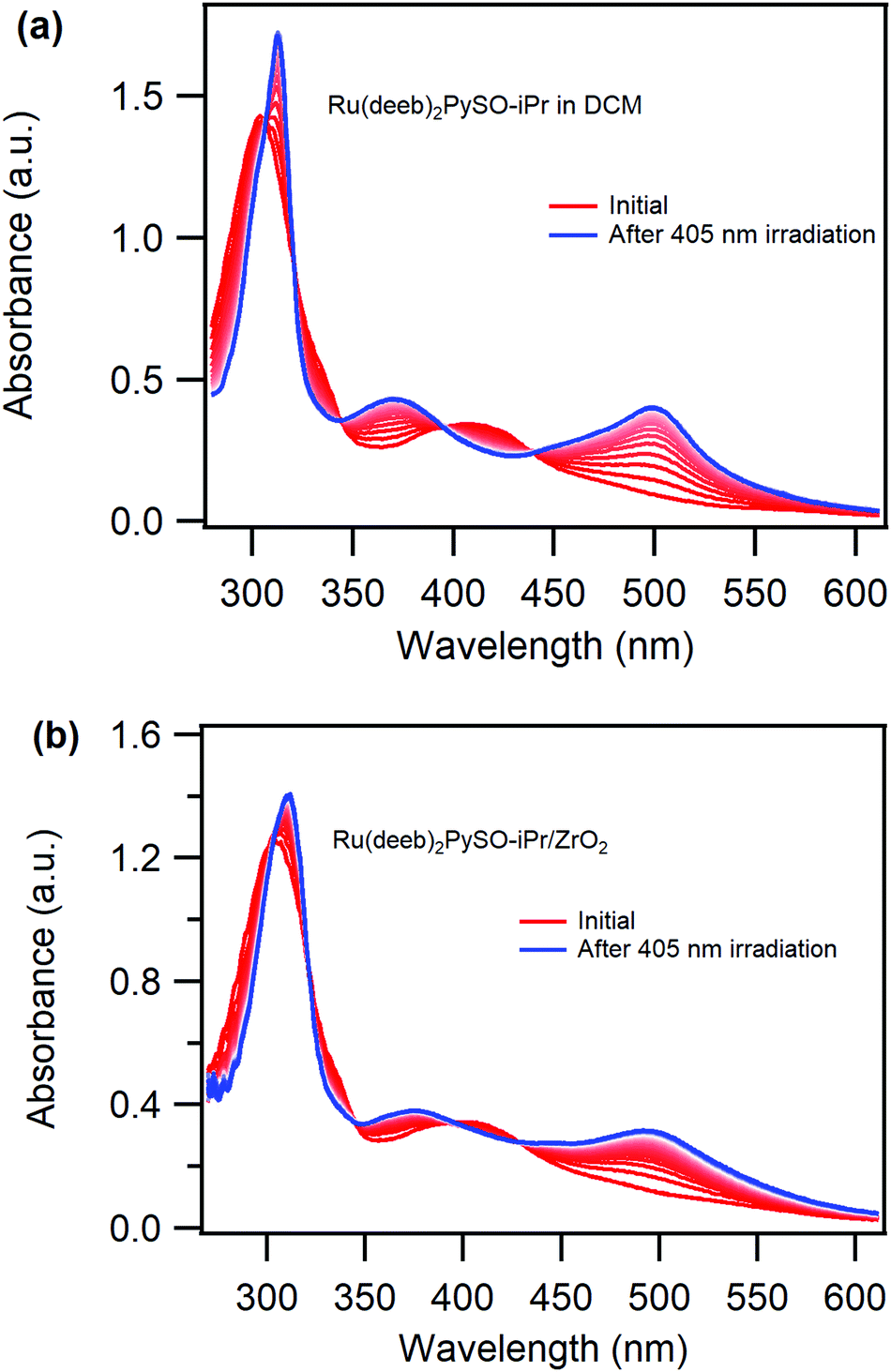 | ||
| Fig. 1 Absorption spectrum of [Ru(deeb)2PySO-iPr]2+ (initial spectrum, red line) and absorption changes over time (from red to blue spectra, 30 s between each scan) following continuous irradiation with 405 nm light in (a) DCM liquid solution and (b) attached to a ZrO2 thin film (∼50 nmol cm−2) in air. | ||
Irradiating the MLCT band (λ ∼ 400 nm) of the S-bonded isomer results in growth of two new absorption peaks, centred at 375 nm and 500 nm respectively and three isosbestic points which are maintained through the course of the experiment both in DCM (Fig. 1a) and PC (Fig. S9, ESI†) solution as well as when attached to ZrO2 in air (slightly blue-shifted, Fig. 1b). The spectral changes are attributed to formation of the O-bonded isomer, in agreement with previously reported S-to-O isomerization for ruthenium sulfoxide complexes11,36,37,39,44 demonstrating that the expected S-to-O photoisomerization in solution is successful also in solid state when the complex is attached to ZrO2. This is the first observation of photoinduced isomerization of a Ru-sulfoxide complex attached to a film. The photoinduced S-to-O isomerization of the Ru(deeb)2PySO-iPr/ZrO2 material proceeds in a similar fashion as in solution despite the relatively high surface coverage and the lack of a solvent surrounding the complex. Moreover, the photoisomerization proceeds with maintained isosbestic points which suggests that no degradation occurs. A somewhat lower photoisomerization ability on the surface is however apparent from comparison between the final spectra of the complex in liquid solution (Fig. 1a blue trace) and of the Ru(deeb)2PySO-iPr/ZrO2 material (Fig. 1b blue trace) which reveals slightly muted spectral changes on the film. The photoisomerization on the surface does not appear to proceed until completion despite being irradiated until no further spectral changes was observed (see also normalized spectra in Fig. S11, ESI†). Thus, the photoisomerization is somewhat hampered on the surface compared to in non-restricted solution environments and it appears as if some molecules on the surface cannot undergo the photoisomerization, likely due to steric hindrance from either nearby molecules or a restricted attachment to the surface.
The quantum yield of photoisomerization was estimated following previously published methods11,48 (see ESI† for details), and revealed a higher quantum yield in liquid solution (33% in DCM solution and 29% in PC solution) compared to attached to the films (∼4–7% depending on the surface coverage). This is consistent with the observed muted spectral changes on the film and could either be a result of the immobilization itself or originate from steric hindrance due to the close proximity between molecules. Steric hindrance from neighbouring molecules would likely result in a concentration dependence such that a lower surface coverage results in a larger photoisomerization ability, and indeed, the quantum yield of the photoisomerization displays such a dependence. A ZrO2 film with a surface coverage of 60 nmol cm−2 results in an estimated quantum yield of ∼4% and a film with a surface coverage of 10 nmol cm−2 in an estimated quantum yield of ∼7%. This surface coverage dependence suggests that the photoisomerization is hindered to some degree by neighbouring molecules. However, since both high and low surface coverage results in a significantly lower quantum yield of photoisomerization compared to in the two solvents studied, the photoisomerization efficiency appears to be more affected by being attached to the surface.
The effect from the surface on the photoisomerization could either be a result of interactions between the molecules and the surface or by a decreased flexibility of the molecules when they are attached to the surface. A decreased flexibility of the molecules on the surface would likely result in obstruction of the molecular rearrangement. The solvent dependence of the quantum yield in solution, with a higher quantum yield in DCM (33%) compared to in PC (29%), could possibly be explained in a similar way and be related with the viscosity of the solvent. This solvent dependence is consistent with previously reported solvent dependence of the quantum yield for the bpy-version, which was reported to 11% in PC solution and to 17% in dichloroethane (DCE) solution, as well as with the ultrafast kinetics of the photoisomerization.11,42 The solvent dependence for the bpy-version was attributed to the viscosity of the solvent and that a higher viscosity obstructs the necessary molecular rearrangements and coupling between the excited state and ground state potential energy surfaces.42 It is likely that the solvent has a similar effect on the molecular rearrangement of our complex, given the similar structures and it is expected that attaching the molecules onto a surface disturbs the molecular rearrangements even more.
To further improve the photoisomerization ability on the film, chemical modification of the molecule is likely necessary. Using only one ancillary ligand with anchoring groups for surface binding could possibly enhance the photoisomerization, since this way of binding to the surface for ruthenium dyes in dye sensitized solar cells have been suggested to result in more flexibility.51 Another possible modification would be to increase the photoisomerization per area on the film using a bi-sulfoxide ligand with two sulfoxide groups, similar to previously described molecules.44,52–54 This may however result in more steric hindrance and a loss of the reversibility of the photoisomerization. Importantly, the quantum yield of photoisomerization is high enough to achieve the desired spatial addressability for the Ru(deeb)2PySO-iPr/ZrO2 material, as is evident from Fig. 2 where irradiation through a star-shaped pattern resulted in distinct colour differences visible to the naked eye.
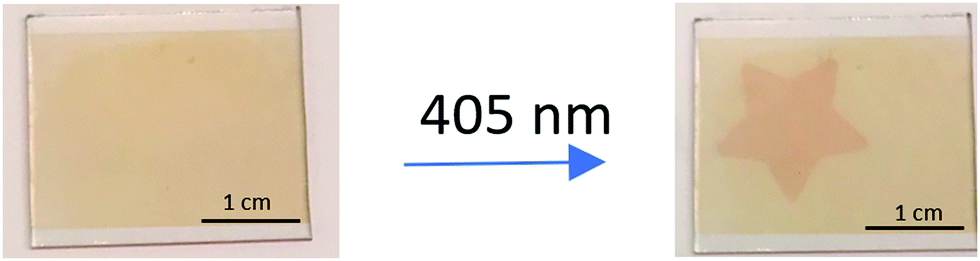 | ||
| Fig. 2 Photograph of a S-bonded Ru(deeb)2PySO-iPr/ZrO2 film before (left) and after (right) being illuminated with 405 nm light through a star-shaped scaffold. | ||
We note that the quantum yield for the S-to-O photoisomerization in solution is remarkably high for our complex compared to the bpy-version. This is attributed to the more electron withdrawing nature of the deeb-ligands compared to bpy-ligands. The electron withdrawing ligands could stabilize the O-bonded isomer in analogy to the fact that the O-bonded isomer also can be obtained by oxidation of the metal centre in many ruthenium sulfoxide complexes. Thus, modifying the ancillary ligand appears to be a straightforward way of tuning the S-to-O photoisomerization quantum yield. The high quantum yield for the S-to-O photoisomerization for our complex is also reflected in the photo-stationary state achieved from white-light irradiation which is largely shifted towards the O-bonded isomer (Fig. S12, ESI†) compared to for the bpy-version.11
Reversible photoisomerization is observed by irradiating the complex with visible light, both in liquid solution and attached to ZrO2. MLCT excitation of the O-bonded isomer results in a decrease of the absorption peaks at 375 and 500 nm and a regrowth of the absorption peak at 411 nm. Irradiation close to the maximum of the peak of the O-bonded isomer results in a photo-stationary state that still contains a high concentration of the O-bonded isomer (Fig. S13, ESI†), reflecting a lower O-to-S than S-to-O quantum yield of photoisomerization. However, selective excitation of the O-bonded isomer in solution with ∼600 nm light results in complete O-to-S photoisomerization, Fig. 3a. Importantly, the photoinduced back isomerization goes almost to completion also for the Ru(deeb)2PySO-iPr/ZrO2 material when it is irradiated with ∼600 nm light, Fig. 3b, albeit with a lower quantum yield than in solution. On the surface the quantum yield was estimated to ∼0.4% (∼10 nmol cm−2), and in DCM and PC liquid solution to 2.5% and 1.4% respectively. These values are lower than for the bpy-version in solution,11 which further supports the notion that the electron withdrawing deeb-ligands results in a stabilization of the O-bonded isomer. Cycling between the isomers is possible both in solution as well as on ZrO2 films (Fig. S14–S17, ESI†) using wavelengths of ∼400 and ∼600 nm, although on ZrO2 not all molecules can be photoisomerized, as previously mentioned, which is apparent from the difference between the initial absorbance and the absorbance in the following cycles (Fig. S16, ESI†). The reversible and cyclable photoisomerization of the Ru(deeb)2PySO-iPr/ZrO2 film indicates that designing photoresponsive materials by attaching photochromic molecules onto mesoporous semiconductors surfaces is a promising approach.
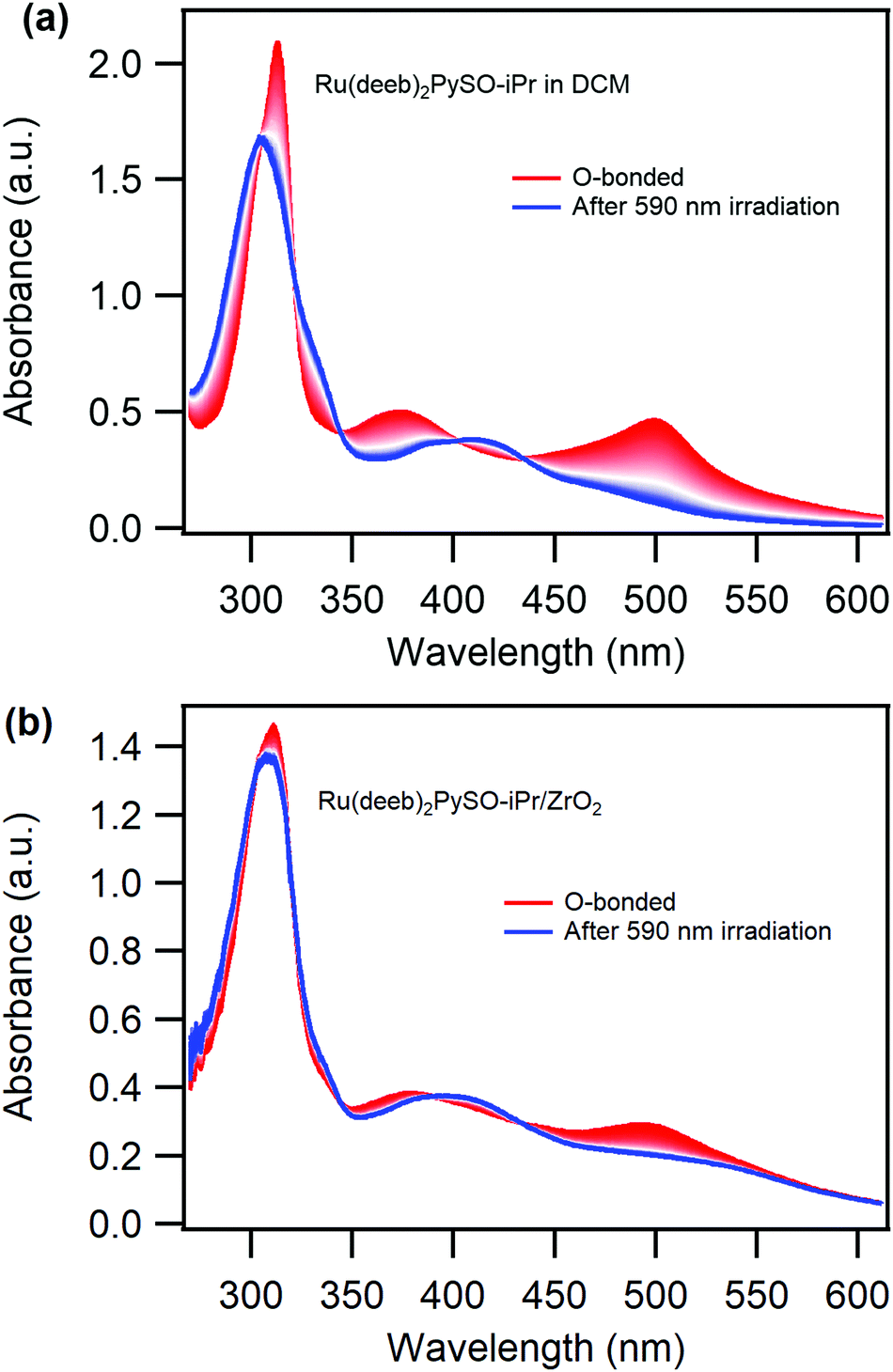 | ||
| Fig. 3 Absorption changes over time (from red to blue spectra, 2 min between each scan) of initially O-bonded [Ru(deeb)2PySO-iPr]2+, (a) in DCM solution, and (b) attached to a ZrO2 thin film (∼50 nmol cm−2) continuously irradiated with 590 nm light. | ||
Importantly, the O-bonded isomer of the complex is thermally trapped for a considerable amount of time, both in liquid solution and when immobilized on the ZrO2 film. Leaving it in room temperature in the dark subsequent to S-to-O photoisomerization results in thermal reversion to the S-bonded isomer over the course of several hours, Fig. 4, in a similar fashion as previously reported for other ruthenium sulfoxide complexes.40
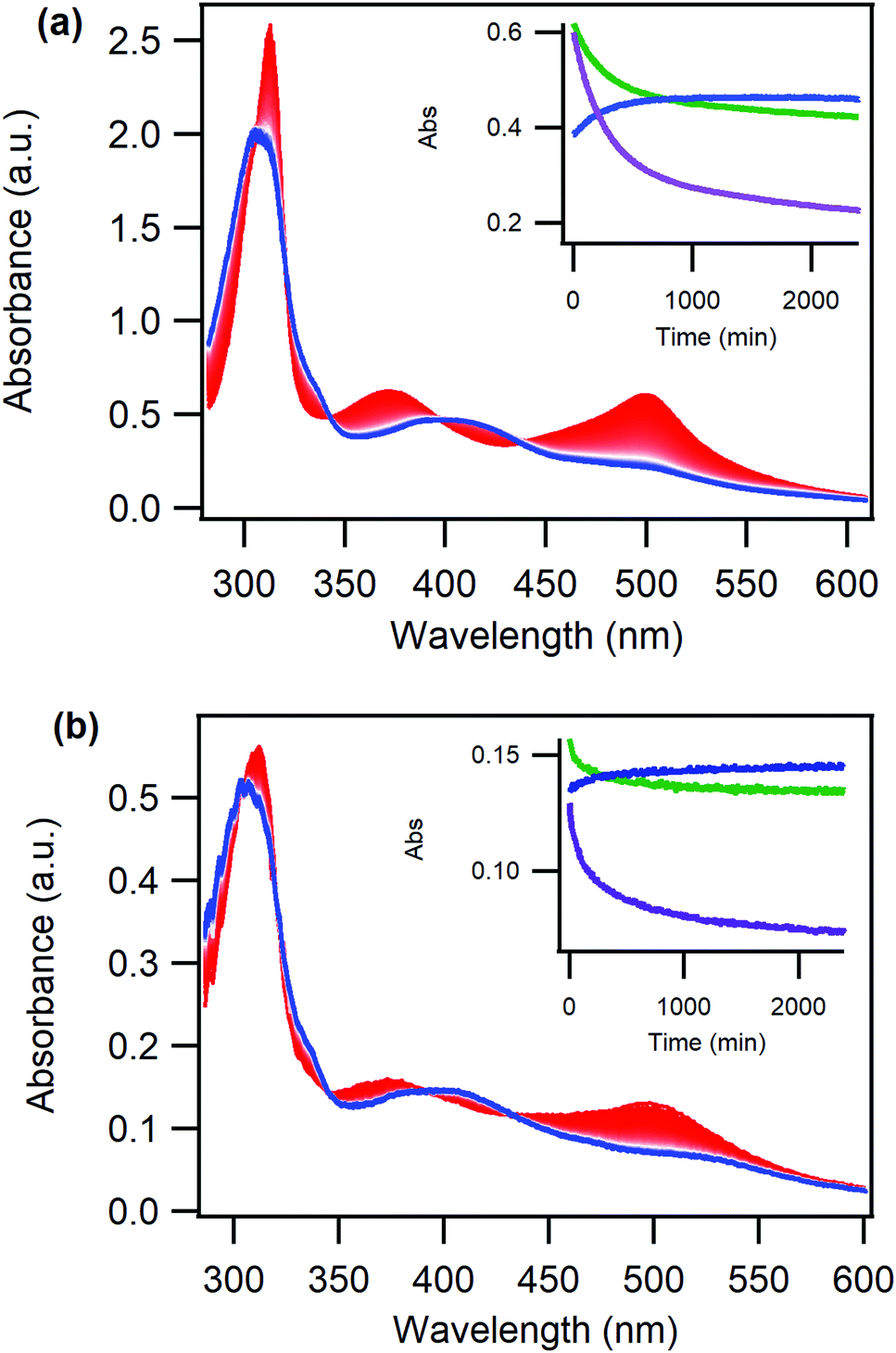 | ||
| Fig. 4 Absorption changes over time in the dark (from red to blue spectra) of (a) a solution of initially O-bonded [Ru(deeb)2PySO-iPr]2+ in DCM, inset shows single wavelength kinetics at 375 nm (blue), 411 nm (green) and 500 nm (purple) and (b) an initially O-bonded Ru(deeb)2PySO-iPr/ZrO2 film in air, inset shows single wavelength kinetics at 370 nm (blue), 405 nm (green) and 500 nm (purple). | ||
The rate of thermal reversion was determined through fitting of the absorption vs time data at 500 nm to a biexponential model, Table 1. Biexponential kinetics of thermal reversion for ruthenium sulfoxide complexes have previously been assigned to a molecular rearrangement (fast component) and the isomerization (slow component).55
| Sample | k 1,thermal (s−1) | k 2,thermal (s−1) |
|---|---|---|
| PC solution | 2.13 × 10−4 (50%) | 2.24 × 10−5 (50%) |
| DCM solution | 6.90 × 10−5 (65%) | 7.81 × 10−6 (35%) |
| ZrO2 | 1.40 × 10−4 (53%) | 1.40 × 10−5 (47%) |
Interestingly, the kinetics of the thermal reversion is similar for the Ru(deeb)2PySO-iPr/ZrO2 material and for the complex in liquid PC solution, and these rates are also in agreement with previously reported thermal reversion for other ruthenium sulfoxide complexes.36,37 Hence, the thermal reversion proceeds in a comparable way for the Ru(deeb)2PySO-iPr/ZrO2 material as it does for the complex in liquid solution, despite the restricted movement of the molecules. The similar kinetics suggests that the mechanism for thermal back-isomerization, where the excited state potential surface is not involved, does not require the same amount of rearrangement of the molecule as the photoinduced process. It is interesting to note that the thermal reversion is faster for the Ru(deeb)2PySO-iPr/ZrO2 material compared to in DCM liquid solution, the reason for this is not clear but further reflects the different mechanisms for thermal and photoinduced isomerization.
Conclusions
In summary, we report for the first time a solid-state photochromic material based on a ruthenium sulfoxide complex immobilized onto a mesoporous metal oxide surface. The resulting film has a high surface coverage, displays reversible two-colour switching and provides spatial addressability of the photo-switching process, paving the way for various photonic applications. Importantly, the process proceeds in air at ambient conditions. The quantum yield of the S-to-O photoisomerization is remarkably high for the complex in liquid solution, attributed to the electron withdrawing nature of the ancillary ligands. The quantum yield is however reduced on the film, likely due to a restricted ability of conformational movement/changes of the molecules. To further improve the photoisomerization of the film, modification of the complex and the binding to the surface is likely necessary. The results presented herein suggests that attaching photochromic molecules onto mesoporous surfaces is a promising approach to make photo-responsive films that can find use in applications such as molecular memory storage and logic gates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge August Runemark and Benedikt Bagemihl for preliminary synthetic studies, Dr. Andreas Ekebergh is acknowledged for preparing ZrO2 paste and the Chalmers Mass Spectrometry Infrastructure (CMSI) is acknowledged for mass spectrometric analysis. The Swedish Energy Agency is acknowledged for financial support.Notes and references
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672–687 RSC.
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef PubMed.
- R. Ballardini, A. Credi, M. Teresa Gandolfi, F. Marchioni, S. Silvi and M. Venturi, Photochem. Photobiol. Sci., 2007, 6, 345–356 CrossRef PubMed.
- V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- S. Bonnet and J.-P. Collin, Chem. Soc. Rev., 2008, 37, 1207–1217 RSC.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- H. Tian and S. Yang, Chem. Soc. Rev., 2004, 33, 85–97 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- A. W. King, L. Wang and J. J. Rack, Acc. Chem. Res., 2015, 48, 1115–1122 CrossRef CAS PubMed.
- B. A. McClure and J. J. Rack, Angew. Chem., Int. Ed., 2009, 48, 8556–8558 CrossRef CAS.
- T. E. Bitterwolf, Coord. Chem. Rev., 2006, 250, 1196–1207 CrossRef CAS.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef CAS PubMed.
- D. Schaniel, M. Imlau, T. Weisemoeller, T. Woike, K. W. Krämer and H. U. Güdel, Adv. Mater., 2007, 19, 723–726 CrossRef CAS.
- T. T. To, E. J. Heilweil, C. B. Duke, K. R. Ruddick, C. E. Webster and T. J. Burkey, J. Phys. Chem. A, 2009, 113, 2666–2676 CrossRef CAS PubMed.
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. G. D. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- P. Tegeder, J. Phys.: Condens. Matter, 2012, 24, 394001 CrossRef.
- M. J. Comstock, N. Levy, A. Kirakosian, J. Cho, F. Lauterwasser, J. H. Harvey, D. A. Strubbe, J. M. J. Fréchet, D. Trauner, S. G. Louie and M. F. Crommie, Phys. Rev. Lett., 2007, 99, 038301 CrossRef PubMed.
- M. Ito, T. X. Wei, P.-L. Chen, H. Akiyama, M. Matsumoto, K. Tamada and Y. Yamamoto, J. Mater. Chem., 2005, 15, 478–483 RSC.
- A. S. Kumar, T. Ye, T. Takami, B.-C. Yu, A. K. Flatt, J. M. Tour and P. S. Weiss, Nano Lett., 2008, 8, 1644–1648 CrossRef CAS.
- Z. Chu and R. Klajn, Nano Lett., 2019, 19, 7106–7111 CrossRef CAS.
- C. Bronner, M. Schulze, S. Hagen and P. Tegeder, New J. Phys., 2012, 14, 043023 CrossRef.
- W. R. Browne and B. L. Feringa, Annu. Rev. Phys. Chem., 2009, 60, 407–428 CrossRef CAS.
- V. Dryza and E. J. Bieske, J. Phys. Chem. C, 2015, 119, 14076–14084 CrossRef CAS.
- J. Iwicki, E. Ludwig, M. Kalläne, J. Buck, F. Köhler, R. Herges, L. Kipp and K. Rossnagel, Appl. Phys. Lett., 2010, 97, 063112 CrossRef.
- A. Khayyami and M. Karppinen, Chem. Mater., 2018, 30, 5904–5911 CrossRef CAS.
- E. Ludwig, T. Strunskus, S. Hellmann, A. Nefedov, C. Wöll, L. Kipp and K. Rossnagel, Phys. Chem. Chem. Phys., 2013, 15, 20272–20280 RSC.
- F. Schreiber, J. Phys.: Condens. Matter, 2004, 16, R881–R900 CrossRef CAS.
- S. Yasuda, T. Nakamura, M. Matsumoto and H. Shigekawa, J. Am. Chem. Soc., 2003, 125, 16430–16433 CrossRef CAS PubMed.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS PubMed.
- T. R. Rusch, M. Hammerich, R. Herges and O. M. Magnussen, Chem. Commun., 2019, 55, 9511–9514 RSC.
- J. C. Wang, S. P. Hill, T. Dilbeck, O. O. Ogunsolu, T. Banerjee and K. Hanson, Chem. Soc. Rev., 2018, 47, 104–148 RSC.
- A. A. Cordones, J. H. Lee, K. Hong, H. Cho, K. Garg, M. Boggio-Pasqua, J. J. Rack, N. Huse, R. W. Schoenlein and T. K. Kim, Nat. Commun., 2018, 9, 1989 CrossRef.
- K. Garg, J. T. Engle, C. J. Ziegler and J. J. Rack, Chem. – Eur. J., 2013, 19, 11686–11695 CrossRef CAS PubMed.
- A. J. Göttle, I. M. Dixon, F. Alary, J.-L. Heully and M. Boggio-Pasqua, J. Am. Chem. Soc., 2011, 133, 9172–9174 CrossRef.
- B. A. McClure, E. R. Abrams and J. J. Rack, J. Am. Chem. Soc., 2010, 132, 5428–5436 CrossRef CAS.
- B. A. McClure, N. V. Mockus, D. P. Butcher, D. A. Lutterman, C. Turro, J. L. Petersen and J. J. Rack, Inorg. Chem., 2009, 48, 8084–8091 CrossRef CAS PubMed.
- B. A. McClure and J. J. Rack, Eur. J. Inorg. Chem., 2010, 3895–3904 CrossRef CAS.
- J. J. Rack, Coord. Chem. Rev., 2009, 253, 78–85 CrossRef CAS.
- J. J. Rack and N. V. Mockus, Inorg. Chem., 2003, 42, 5792–5794 CrossRef CAS.
- A. Yeh, N. Scott and H. Taube, Inorg. Chem., 1982, 21, 2542–2545 CrossRef CAS.
- A. W. King, B. A. McClure, Y. Jin and J. J. Rack, J. Phys. Chem. A, 2014, 118, 10425–10432 CrossRef CAS.
- Y. Jin, S. I. M. Paris and J. J. Rack, Adv. Mater., 2011, 23, 4312–4317 CrossRef CAS PubMed.
- N. V. Mockus, D. Rabinovich, J. L. Petersen and J. J. Rack, Angew. Chem., Int. Ed., 2008, 47, 1458–1461 CrossRef CAS.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS.
- M. A. Butler and D. S. Ginley, J. Electrochem. Soc., 1978, 125, 228–232 CrossRef CAS.
- T. A. Heimer, S. T. D'Arcangelis, F. Farzad, J. M. Stipkala and G. J. Meyer, Inorg. Chem., 1996, 35, 5319–5324 CrossRef CAS.
- K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 41145 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- B. H. Farnum, J. J. Jou and G. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15628 CrossRef CAS PubMed.
- K. Kilså, E. I. Mayo, B. S. Brunschwig, H. B. Gray, N. S. Lewis and J. R. Winkler, J. Phys. Chem. B, 2004, 108, 15640–15651 CrossRef.
- K. Garg, A. W. King and J. J. Rack, J. Am. Chem. Soc., 2014, 136, 1856–1863 CrossRef CAS PubMed.
- A. W. King, Y. Jin, J. T. Engle, C. J. Ziegler and J. J. Rack, Inorg. Chem., 2013, 52, 2086–2093 CrossRef CAS.
- M. Y. Livshits, L. Wang, S. B. Vittardi, S. Ruetzel, A. King, T. Brixner and J. J. Rack, Chem. Sci., 2020, 11, 5797–5807 RSC.
- T. A. Grusenmeyer, B. A. McClure, C. J. Ziegler and J. J. Rack, Inorg. Chem., 2010, 49, 4466–4470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Description of the synthesis, additional isomerization experiments and quantum yield calculations. See DOI: 10.1039/d1ma00013f |
| This journal is © The Royal Society of Chemistry 2021 |