
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ-Conjugated stannole copolymers synthesised by a tin-selective Stille cross-coupling reaction†‡
Isabel-Maria
Ramirez y Medina
 ab,
Markus
Rohdenburg
cd,
Pascal
Rusch
ef,
Daniel
Duvinage
bg,
Nadja C.
Bigall
ef and
Anne
Staubitz
*ab
ab,
Markus
Rohdenburg
cd,
Pascal
Rusch
ef,
Daniel
Duvinage
bg,
Nadja C.
Bigall
ef and
Anne
Staubitz
*ab
aInstitute for Organic and Analytical Chemistry, University of Bremen, Leobener Str. 7, 28359 Bremen, Germany. E-mail: staubitz@uni-bremen.de
bMAPEX Center for Materials and Processes, University of Bremen, Bibliothekstr. 1, 28359 Bremen, Germany
cUniversity of Bremen, Institute for Applied and Physical Chemistry, Leobener Str. 5, 28359 Bremen, Germany
dUniversity of Leipzig, Wilhelm-Ostwald-Institute for Physical and Theoretical Chemistry, Linnéstr. 2, 04103 Leipzig, Germany
eInstitute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Callinstr. 3A, 30167 Hannover, Germany
fCluster of Excellence PhoenixD (Photonics, Optics, and Engineering – Innovation Across Disciplines), Hannover, Germany
gInstitute of Inorganic Chemistry and Crystallography, University of Bremen, Leobener Str. 7, 28359 Bremen, Germany
First published on 27th March 2021
Abstract
The synthesis of four well-defined conjugated polymers TStTT1–4 containing unusual heterocycle units in the main chain, namely stannole units as building blocks, is reported. The stannole–thiophenyl copolymers were generated by tin-selective Stille coupling reactions in nearly quantitative yields of 94% to 98%. NMR data show that the tin atoms in the rings remain unaffected. Weight-average molecular weights (Mw) were high (4900–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 Da and 9600–21900 Da); and molecular weight distributions (Mw/Mn) were between 1.9 and 2.3. The new materials are strongly absorbing and appear blue-black to purple-black. All iodothiophenyl–stannole monomers St1–4 and the resulting bisthiophenyl–stannole copolymers TStTT1–4 were investigated with respect to their optoelectronic properties. The absorption maxima of the polymers are strongly bathochromically shifted compared to their monomers by about 76 nm to 126 nm in chloroform. Density functional theory calculations support our experimental results of the single stannoles St1–4 showing small HOMO–LUMO energy gaps of 3.17–3.24 eV. The optical band gaps of the polymers are much more decreased and were determined to be only 1.61–1.79 eV. Furthermore, both the molecular structures of stannoles St2 and St3 from single crystal X-ray analyses and the results of the geometry optimisation by DFT confirm the high planarity of the molecules backbone leading to efficient conjugation within the molecule.
900 Da and 9600–21900 Da); and molecular weight distributions (Mw/Mn) were between 1.9 and 2.3. The new materials are strongly absorbing and appear blue-black to purple-black. All iodothiophenyl–stannole monomers St1–4 and the resulting bisthiophenyl–stannole copolymers TStTT1–4 were investigated with respect to their optoelectronic properties. The absorption maxima of the polymers are strongly bathochromically shifted compared to their monomers by about 76 nm to 126 nm in chloroform. Density functional theory calculations support our experimental results of the single stannoles St1–4 showing small HOMO–LUMO energy gaps of 3.17–3.24 eV. The optical band gaps of the polymers are much more decreased and were determined to be only 1.61–1.79 eV. Furthermore, both the molecular structures of stannoles St2 and St3 from single crystal X-ray analyses and the results of the geometry optimisation by DFT confirm the high planarity of the molecules backbone leading to efficient conjugation within the molecule.
1 Introduction
The development of polymers containing group 14 (Si, Ge, Sn, Pb) metalloles or their ring-fused analogues as repeating units has enjoyed increased interest for several decades due to their unique properties resulting in a potential as materials in applications.1 Compared to other structural motifs, their major benefit is a low HOMO–LUMO gap in single molecules and a low band gap in polymers resulting from the incorporation of the element = Si, Ge, Sn, Pb. Contrary to generally used principles to attain these low energy gaps, i.e. push–pull systems, quinoid systems2 or a longer effective conjugation length in polymers, there is an entirely different mechanism at the root of this phenomenon.3The exchange of the methylene carbon atom in cyclopentadienes by other group 14 “ER2” moieties drastically changes the electronic structure. Compared to cyclopentadiene, the siloles, germoles, stannoles and plumboles have an orbital lobe on the heteroatom (σ*-orbital), which interacts with the π*-orbital of the diene unit producing a strong, efficient σ*–π*-conjugation, also called cross-hyperconjugation.4 This leads to a lowering of the lowest unoccupied molecular orbital (LUMO) energy level, while the highest occupied molecular orbital (HOMO) is nearly unaffected. Thus, a much narrower HOMO–LUMO energy gap results compared to cyclopentadiene and its derivatives.3a,b,5 Tamao and co-workers discovered this phenomenon to be the origin of the unusual optical properties of the group 14 heteroles by computational and spectroscopic investigations in 1996.3a
The combination of cross-hyperconjugation with π-conjugation by mixing group 14 metalloles with thiophenes to build polymers, leads to very low band gap materials making them highly attractive for potential applications.1n,6 While polymers with siloles as building blocks have already been used in applications (e.g. OLEDs, OSCs, OFETs, chemical/biological sensing), germoles are less common.1f–i,l–n Polymers with plumbole units in the main chain have not been reported yet. For stannoles, the main body of work has focussed on ring-fused compounds (i.e. stannafluorenes), but they are rather different from non-fused stannoles.1o,7 The only application for non-ring fused stannoles that has been reported so far was fluoride sensing by Tomita and co-workers.1n,8
Although stannoles are supposed to have the same beneficial properties as siloles or germoles, there is only a small amount of literature about polymers with stannole units; this might be due to the difficulty in their synthesis.8,9 These attractive materials can be accessed via different synthetic strategies. On the one hand, polymerisation of the readily prepared organometallic building block is an often-used tool,1d,e,g,9 but also the synthesis of precursor polymers and subsequent reaction with a reagent of the desired main group element is a well-known strategy to furnish polymers containing group 14 metalloles.8,10
To the best of our knowledge, to date only five polymers with non-annulated stannoles in the main chain were reported: one by Staubitz and co-workers in 2014,9 synthesised by tin-selective Stille coupling and in total four by Tomita and co-workers in 20098b and 20198a created by post-element transformation of organotitanium polymers as precursors (Fig. 1).
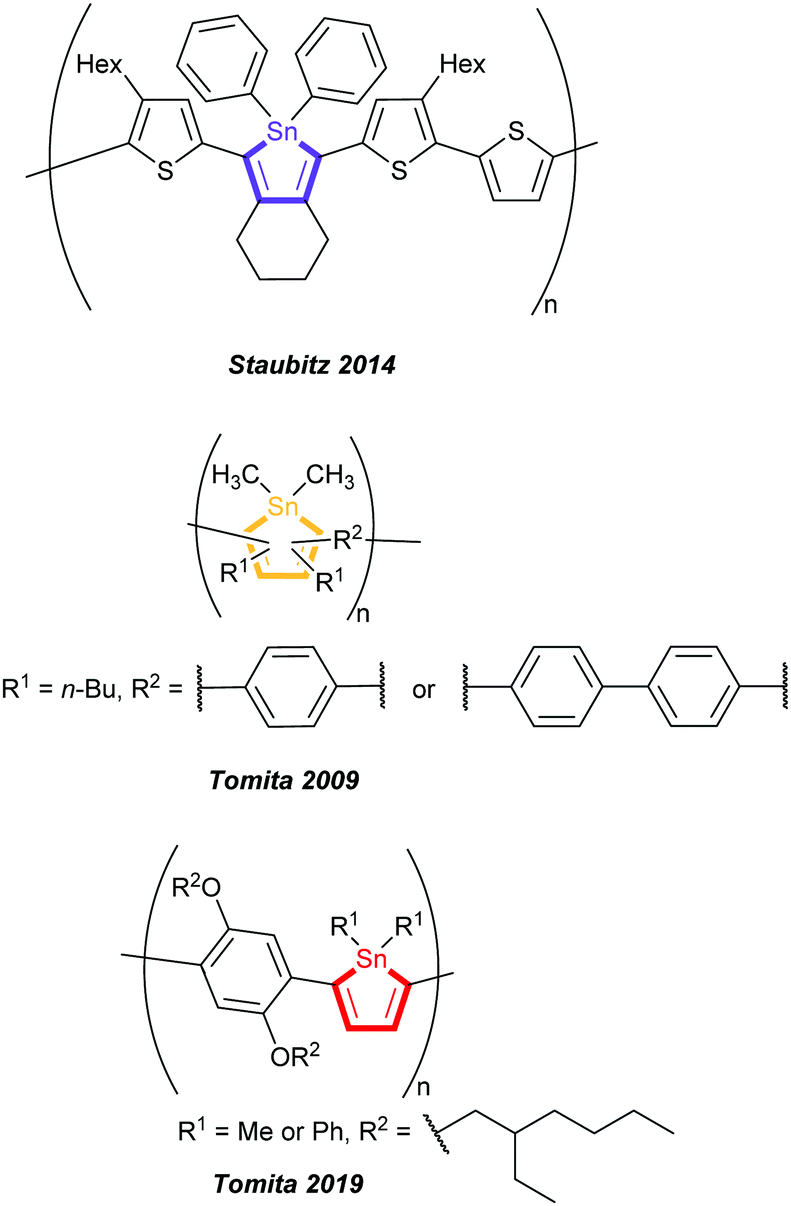 | ||
| Fig. 1 Previously reported stannole containing polymers having non-annulated stannole units in the main chain by Staubitz and coworkers and Tomita and coworkers.8,9 | ||
Here, we report four iodothiophenyl–stannoles St1–4 (Scheme 2), which were used as monomers in a polycondensation reaction. We describe their synthesis, computed molecular geometries, crystal structures, the frontier molecular orbitals (FMO's) and their energies, and the optical properties. The tin-selective polymerisation by Stille cross-coupling led to the four corresponding polymers TStTT1–4 in high yields, which were studied by gel-permeation chromatography (GPC), MALDI, thermogravimetric analysis (TGA), absorption and emission measurements.
2 Results and discussion
2.1 Syntheses
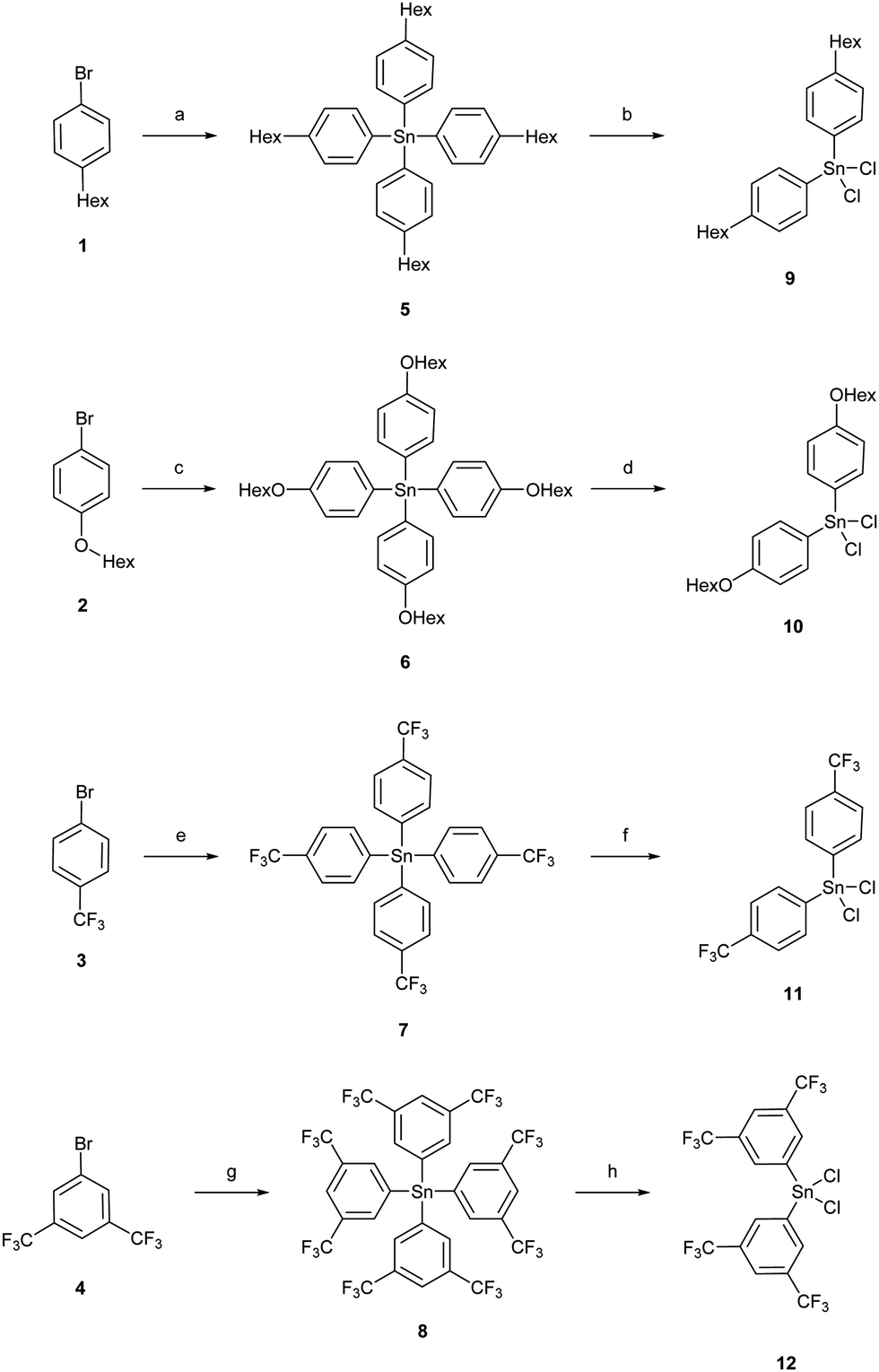 | ||
| Scheme 1 Reactions to tetraorganostannanes and diorgano-dichlorostannanes. (a) 1. t-BuLi, THF, −78 °C, 5 min, 2. SnCl4, −78 °C, 40 min, −78 °C to 20 °C, 16 h, 87%; (b) SnCl4, 9 × 10−3 mbar, 180 °C, 10 h, 83%; (c) t-BuLi, THF, −78 °C, 5 min, 2. SnCl4, −78 °C, 30 min, −78 °C to 20 °C, 16 h, 89%; (d) SnCl4, 9 × 10−3 mbar, 230 °C, 10 h, 89%; (e) n-BuLi, THF, −78 °C, 2.5 h, 2. SnCl4, −78 °C, 1 h, −78 °C to 22 °C, 16 h, 67%; (f) SnCl4, 1.0 × 10−2 mbar, 180 °C, 48 h, 66%; (g) n-BuLi, Et2O, −78 °C, 3 h, 2. SnCl4, −78 °C, 30 min, −78 °C to 22 °C, 16 h, 70%; (h) SnCl4, 3.0 × 10−2 mbar, 180 °C, 48 h, 70%. | ||
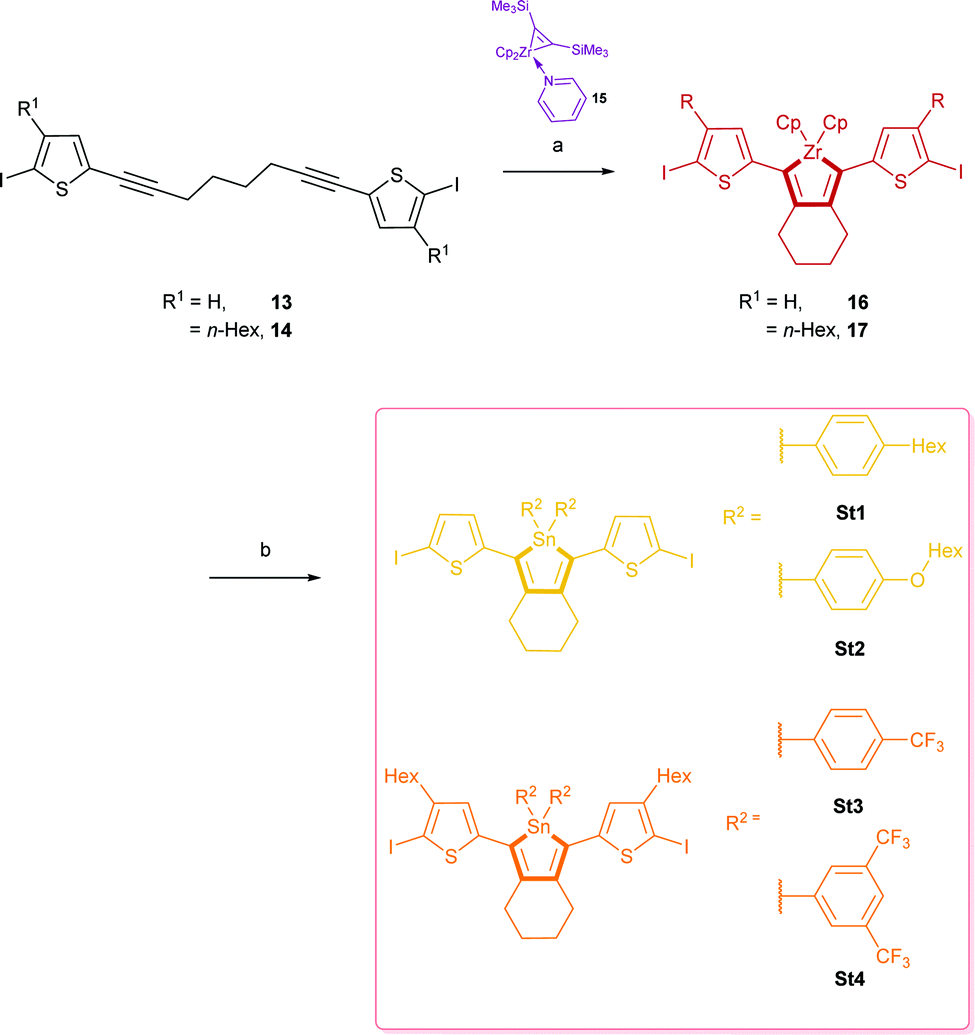 | ||
| Scheme 2 Reaction of diynes 11, 12 with Rosenthal's zirconocene to zirconacyclopentadienes 14, 15 and further transformation to stannoles St1–4. (a) Toluene, 22 °C, 1 h; (b) diorgano-dichlorostannanes 4/6/8/10, Cu(I)Cl, toluene or THF, 22 °C, 6 h, St1: 64%, St2: 68%, St3: 75%, St4: 32%. | ||
The di-(aryl)-dichlorostannanes 9, 10, 11, and 12 were obtained in a Kocheshkov reaction of the tetra-organostannanes with tin(IV)chloride in yields of 83%, 89%, 66% and 70%, respectively (Scheme 1). Di-[3,5-bis(trifluoromethyl)phenyl]-dichloro-stannane (12) could not be obtained as completely pure material by fractionated sublimation; the by-product was tri-[3,5-bis(trifluoromethyl)phenyl]-chlorostannane. However, the stannane could be used for the subsequent synthetic pathway.11,12
1,8-Bis(5-iodo-thiophen-2-yl)octa-1,7-diyne (13) and 1,8-bis(5-iodo-4-hexylthiophen-2-yl)octa-1,7-diyne (14) were synthesised by an electrophile-selective Sonogashira coupling of one equivalent 1,7-octadiyne and two equivalents functionalised thiophenes followed by a bromo-iodo exchange according to published procedures.13
Reductive coupling of the respective diynes 13 or 14 with the active “Cp2Zr(II)” species generated from Rosenthal's zirconocene (15)13b,14 led to zirconacyclopentadienes 16 and 17 with quantitative conversion (measured by 1H NMR).
These precursor molecules were not isolated but converted in situ into the desired stannole monomers St1–4 (32–75%) by reaction with the respective di-organo-dichlorostannanes with an in-situ transmetalation step with Cu(I)Cl (Scheme 2).
The 119Sn{1H} NMR spectra of St1–4 showed a signal at −74.9 ppm (St1), −70.2 ppm (St2), −83.2 ppm (St3) and −82.7 ppm (St4) respectively.
Yellow and orange-yellow single crystals of stannoles St2 and St3 could be obtained from a saturated solution of dichloromethane/n-hexane for X-ray analyses. St1 and St4 did not crystallise.
2.2 Structures in the solid state and computational study of monomers St1–4
Single crystal X-ray crystallography of St2 and St3 confirms their molecular structures. Both are depicted in Fig. 2, selected bond lengths, bond angles and torsion angles are summarised in Table S5 in the ESI.‡ | ||
| Fig. 2 Molecular structures of St2 and St3 showing 50% probability ellipsoids and the crystallographic numbering scheme. | ||
The Sn–C bond lengths (Sn1–C1 2.140(2) Å St2, 2.127(3) Å St3) within the ring are comparable to literature values and are longer than reported Ge–C and Si–C bonds in the germoles (1.95 Å) and siloles (1.88 Å). Also, the small C–Sn–C angles (83.0(2)° St2 and 84.23(7)° St3) are consistent with the literature.3b,c The stannole ring itself is nearly perfectly planar (−3.6(3)° St2 and 5.2(2)° St3), but the thiophenyl substituents are slightly twisted to the central ring with torsion angles (Sn1–C1–C10–C11) of −14.5(3)° (St2) and −10.5(3)° (St3), respectively.
The high coplanarity of the overall molecules’ backbones leads to efficient conjugation, which is an important characteristic for the polymers later on. The phenyl-groups on the Sn atom are twisted out of the plane (Fig. 2 and Table S5 in the ESI‡), but do not affect the conjugation.
The calculated molecular structures and absorption spectra are presented in the ESI‡ (Fig. S58–S69). The distribution of the frontier molecular orbitals (FMOs), the HOMO and LUMO energy levels (eV) and HOMO–LUMO energy gaps (eV) are shown in Fig. 3. The energy minimised conformational geometries of St2 and St3 are in agreement with the X-ray analysis with the exception of the orientation of the hexyl chains, likely due to crystal packing effects (Fig. 2 and Fig. 3).
 | ||
| Fig. 3 FMOs of St1–4, HOMO and LUMO energy levels (eV) and HOMO–LUMO energy gaps (eV). | ||
Theoretical torsion angles of all four monomers St1–4 reveal that not only the stannole ring itself is planar (Sn1–C1–C2–C3 0.4 to −2.1°) but also the thiophenyl–stannole–thiophenyl systems are almost coplanar (Sn1–C1–C10–C11 8.5 to 10.0°) (Table S10 in the ESI‡). Isosurface representations of the FMOs show that the HOMOs and LUMOs are delocalised over the whole molecules’ backbones. The LUMOs of all show a contribution of the Sn, which indicates a σ*π*-conjugation within the stannole ring (Fig. 3).
The HOMO–LUMO energy gap of St1 and St2 is equal (3.24 eV); but the respective HOMO and LUMO energy levels of St2 are slightly increased compared to St1 due to the change from hexylphenyl groups (St1) to hexyloxyphenyl substituents (St2) on the Sn (1,1-position) destabilising the FMOs. Going from St1–2 to St3 and St4, the HOMO and LUMO energy levels are decreased by ca. 0.3–0.4 eV due to the electron-withdrawing groups on the Sn (1,1-position) stabilising the FMOs. Furthermore, the HOMO–LUMO energy gaps are smaller than the ones of St1–2 with 3.19 eV (St3) and 3.17 eV (St4), respectively. These results are in agreement with the findings from a previous study in which we investigated the influence of different substituents, attached on the Sn in the 1,1-position, on the HOMO and LUMO energy levels.3c
2.3 Optical properties of St1–4
The absorption and emission of St1–4 were recorded in chloroform, toluene, and tetrahydrofuran at 295 K. Excitation and emission were also measured in the solid state. All spectra can be found in the ESI.‡Fig. 4 displays the absorption and emission spectra of St1–4 in chloroform (Fig. 4a and b), the temperature-dependence of the emission of St3 (Fig. 4c) and solid-state excitation/emission spectra of St3 (Fig. 4d). Tables 1 and 2 summarise all experimental and predicted optical data. | ||
| Fig. 4 (a) Absorption spectra of stannoles St1–4 in chloroform at 295 K; (b) emission spectra of stannoles St1–4 in chloroform at 295 K; (c) emission spectra of St3 in 2-methyl-tetrahydrofuran from 100 K to 280 K, irradiated at 445 nm and a photography of the irradiated frozen solution at 100 K; (d) excitation (measured at 585 nm) and emission (irradiated at 507 nm) spectra of St3 in the solid state at 295 K and a photography of irradiated (366 nm) crystals. | ||
| Monomers | Solvent | λ abs,expt/nm (ε/L mol−1 cm−1) | λ abs,calc/nm | λ em,expt/nm | Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 /cm−1 |
Φ F/% |
|---|---|---|---|---|---|---|
| St1 | CHCl3 | 434 (22600) |
451 | 522 | 3884 | 0.57 |
| PhMe | 438 | 522 | 3674 | 0.95 | ||
| THF | 434 | 519 | 3774 | 0.50 | ||
| St2 | CHCl3 | 434 (22400) |
450 | 523 | 3921 | 0.42 |
| PhMe | 437 | 523 | 3763 | 0.87 | ||
| THF | 434 | 520 | 3811 | 0.38 | ||
| St3 | CHCl3 | 443 (24500) |
463 | 534 | 3847 | 1.32 |
| PhMe | 446 | 535 | 3730 | 2.72 | ||
| THF | 443 | 533 | 3812 | 1.93 | ||
| St4 | CHCl3 | 446 (17300) |
469 | 543 | 4005 | 3.51 |
| PhMe | 451 | 545 | 3824 | 6.75 | ||
| THF | 445 | 543 | 4055 | 2.45 | ||
| Monomer | λ exc/nm | λ em/nm | Δ/cm−1 |
Φ F/% | Φ F/% (thin film) |
|---|---|---|---|---|---|
| St1 | 373 | 537 | 8188 | 0.22 | 0.77 |
| St2 | 355 | 530 | 9302 | 0.21 | 0.29 |
| St3 | 368 | 583 | 10021 |
8.15 | 3.71 |
| St4 | 344 | 561 | 11244 |
9.36 | 8.89 |
The absorption maxima of the stannoles St1, St2, St3 and St4 were found at 438 nm, 437 nm, 446 nm and 451 nm in toluene at 295 K, respectively. They are consistent with the small HOMO–LUMO energy gaps and the efficient conjugation within the molecule due to the high planarity and the mixed conjugation. All also show the typical vibrational fine-structures that were already observed for other stannoles.3c The absorption maxima show a slight bathochromic shift changing the solvent from tetrahydrofuran or chloroform to toluene. A second absorption maximum is located in the short wavelength area <280 nm (Fig. 4a and Table 1).
Computed absorption spectra by TD-DFT also display transitions in the short and long wavelength area giving two maxima for each compound (see Fig. S60, S63, S66 and S69 in the ESI‡). The more interesting maxima at 450 nm (St2) to 469 nm (St4) arise from direct HOMO–LUMO transitions of π–π* character. All computed values are systematically bathochromically shifted by ca. 13 nm to 18 nm from the experimental ones in toluene due to the consideration of single molecules in the gas phase without interactions between the molecules or with solvent molecules (Table 1). High extinction coefficients between 17400 L mol−1 cm−1 (St4) and 24500 L mol−1 cm−1 (St3) demonstrate strongly absorbing compounds, which are likely to also yield strongly absorbing polymers.
Emission spectra revealed maxima at 525 nm (St1, toluene) to 545 nm (St4, toluene), which are hardly affected by the solvent. Stokes shifts range between 3674 cm−1 (St1, toluene) and 4055 cm−1 (St4, tetrahydrofuran), and both absorption and emission bands are not completely separated. The fluorescence quantum yields ΦF increased in the solvent order tetrahydrofuran < chloroform < toluene, which is often observed when the polarity of the medium is changed. Furthermore, St3 and St4 showed much more intense emission with ΦF of up to 2.72% and 6.75% compared to St1 and St2 with ΦF of 0.95% and 0.87%. Compared to ΦF values for non-fused stannoles in solution at 295 K found in the literature, these are high quantum yields, observed for this class of system for the first time.3b,13a,15
The emission maxima in the solid state at 530 nm (St2) to 583 nm (St3) are red-shifted as compared to the solutions due to the higher extent of planarity and conjugation in the bulk (Table 2). While St1 and St2 showed slightly lower quantum yields at 295 K in the solid state and similar numbers in thin film, the emission for St3 and St4 was enhanced. The ΦF increased to 8.15% (St3) and 9.36% (St4) in the solid and to 3.71% (St3) and 8.89% (St4) in thin film. In a recent publication we investigated the phenomenon of Aggregation Induced Emission (AIE) in a series of six stannoles. Although all of those showed enhanced emission in the solid state or thin film, this could be not observed for St1 and St2; but Aggregation Induced Emission Enhancement (AIEE) could be seen for St3 and St4 (Table 2).13a
Finally, the temperature dependency of the emission of the four monomers was studied. The photoluminescence was measured in 2-methyltetrahydrofuran from 100 K to 280 K in 10 K steps.
Compared to 280 K, the emission intensity much enhanced. Formally, it is multiplied by a factor of 122 (St1), 237 (St2), 71 (St3), and 61 (St4) at 100 K, although one has to take into consideration that the low fluorescence quantum yields at 280 K for St1 and St2 cannot be measured with high accuracy. All showed intense green emission at 100 K (see Fig. 4, Fig. S12–S15, S22–S25, S34–S37, S44–S47 in the ESI‡). The wavelength of the absorption maximum is only marginally affected by the temperature and slightly hypsochromically shifted. The reason for the enhanced emission in the cold and in the solid state is likely to be restricted intramolecular rotation (RIR), a typical phenomenon for such structure motifs consisting of a stator (stannole ring) and several rotors (free rotatable substituents). The non-radiative pathways are reduced and the radiative become dominant leading to stronger photoluminescence.13a,15a,16
In total, the small HOMO–LUMO energy gaps (3.17–3.24 eV), the high planarity and large π-conjugation within the molecule and the strong absorption in the long wavelength area (434–451 nm), which is also bathochromically shifted about 81 nm to 98 nm compared to similar pure thiophene systems, e.g. terthiophene,17 makes these molecules highly efficient as monomers for potential semiconducting polymers.
2.4 Polymers, analysis of molecular weight and thermal stability
Polycondensation reactions under Stille conditions in mixtures of toluene/dimethylformamide for 3 days led to well-defined blue-black and purple-black polymers TStTT1–4 in nearly quantitative yields of 94–98%. During the polymerisation, the colour of the mixture changed from orange-yellow to red and then to dark blue (TStTT1–2) or dark purple (TStTT3–4) indicating the formation of long chains and an effective conjugation. The respective stannoles St1–4 reacted tin-selectively with 2,5-bis(trimethylstannyl)thiophene (18), while the Sn within the stannole ring remained stable (Scheme 3). The 119Sn{1H} NMR spectra showed signals at −76.4 ppm (TStTT1), −71.8 ppm (TStTT2), −83.9 ppm (TStTT3), and −84.2 ppm (TStTT4), which are nearly the same as found for the corresponding monomers. All polymers were moisture and air stable, but they were attacked by acids or traces of acids in solvents. A decomposition of the polymers in chloroform could be observed by eye (colour change to yellow) over approximately a day due to traces of hydrochloric acid. | ||
| Scheme 3 Polycondensation reactions by Stille cross-coupling of stannoles St1–4 with 2,5-bis(trimethylstannyl)thiophene (18) to furnish polymers TStTT1–4. (a) [Pd(t-Bu3P)2], DMF/toluene, reflux, 3 d, TStTT1: 97%, TStTT2: 96%; (b) [Pd(PPh3)4], DMF/toluene, reflux, 3 d, TStTT3: 98%, TStTT4: 94%. | ||
For polycondensation reactions, it is not only essential to mix two monomers in exactly the desired ratio to obtain long polymer chains; it is also important to have a quantitative conversion for the condensation step. Therefore, the polymerisation was carefully optimised. The catalyst is perhaps the most decisive parameter to the success of a Stille cross-coupling reaction. Therefore, four different palladium catalysts ([Pd(PPh3)4], [PdCl2(PPh3)2], [Pd(t-Bu3P)2], [Pd(dba)2]) and three different solvents (tetrahydrofuran, toluene, toluene/dimethyl-formamide) were investigated.
GPC and UV-Vis spectroscopy of the products showed that the best catalysts were [Pd(t-Bu3P)2] for the coupling of St2 and [Pd(PPh3)4] for the reaction of St3 and a mixture of toluene (2 equivalents) and dimethylformamide (1 equivalent) as the solvent (see experimental procedures and Table S3 in the ESI‡). Pure toluene as solvent led to better results than tetrahydrofuran, but it was not as good as the combination with dimethylformamide. In general, the palladium catalysts [PdCl2(PPh3)2] and [Pd(dba)2] furnished shorter polymers. The optimised reaction conditions of St2/St3 were then adopted for St1 and St4.
MALDI measurements confirm the repeating unit of each polymers as [Thiophenyl–Stannole–Thiophenyl–Thiophenyl]. However, end group analysis by MALDI was not possible. End groups can be –I, –SnMe3 or –H from the work-up procedure (see Fig. S76, S79, S82 and S85 in the ESI‡).
Gel permeation chromatography results (GPC, conventional calibration with polystyrene standards) reveal number-average molecular weights (Mn) and weight-average molecular weights (Mw) from 4900 Da to 10900 Da and 9600 Da to 21900 Da, and molecular weight distributions (Mw/Mn = polydispersity index = PDI) between 1.9 and 2.3 (Table 3). Compared to TStTT1–2, the Mn and Mw were estimated to be approximately twice as high for TStTT3–4. This is most probably due to the different attachment positions of the n-hexyl-chains: Polymers with n-hexyl-chains at the thiophenyl unit in the main chain showed much better solubility than the ones with n-hexylphenyl-groups at the Sn atom of the stannole-unit.
| Polymer | M n [Da] | M w [Da] | PDI | T 1d [°C] |
|---|---|---|---|---|
| TStTT1 | 4900 | 11200 |
2.3 | 264 |
| TStTT2 | 5100 | 9600 | 1.9 | 270 |
| TStTT3 | 10900 |
21900 |
2.0 | 262 |
| TStTT4 | 10300 |
21500 |
2.1 | 245 |
While TStTT3 and TStTT4 were soluble in common organic solvents (chloroform, dichloromethane, tetrahydrofuran, 2-methyltetrahdydrofuran, toluene) at 22 °C, TStTT1 and TStTT2 were not completely soluble. Therefore, GPC samples were placed into the ultrasonic bath for about 10 minutes and then filtered with PTFE filters prior measurement. However, small dark blue-black particles of TStTT1–2 were visible on the PTFE filters indicating that longer polymer chains of these were insoluble and therefore not detected by GPC. A further reason for the shorter polymer chains of TStTT1–2 is that because of the lower solubility of polymers with a certain chain length, the polymers precipitated in the reaction mixture and could not further react.
In total, the values for Mn and Mw of two polymers were higher and all PDI values smaller than the first stannole-containing polymer published in 2014 (Mn = 6,800 Da, Mw = 17000 Da, PDI = 2.5).9 Although the other two polymers were not completely soluble and therefore most probably the detected Mn and Mw were lower than the actual molecular weights, the values are high.
The thermal stability of the materials was measured under a nitrogen flow by thermogravimetric analysis (TGA) from 25 °C to 600 °C. The most unstable polymer was TStTT4 with a T1d = 245 °C and the most stable polymer was TStTT2 with a T1d = 270 °C. All showed several decomposition steps (Td) up to 600 °C. TGA spectra with mass losses (% and mg) can be found in the ESI,‡ Fig. S86–S89.
2.5 Optical properties of polymers TStTT1–4
The absorption spectra of the polymers TStTT1–4 were measured in chloroform at 295 K and reveal broad maxima at 522 nm to 560 nm, which are strongly red-shifted by 76 nm to 126 nm compared to their monomers (Tables 1, 4 and Fig. 7a). Contrary to the UV-Vis results of the monomers, the absorption maximum of TStTT1 and TStTT2 is bathochromically shifted about 24 nm to 38 nm in comparison to TStTT3 and TStTT4. The reason for this is most probably a longer effective conjugation length within polymers TStTT1–2 than in TStTT3–4 due to the difference in the position of the n-hexyl-chains and therefore a higher degree of planarity. The previous mentioned disadvantage of the lower solubility of these two polymers might be solved by the establishment of soluble groups in the 3,4-positions of the stannole ring as already done by Tamao and coworkers or by the introduction of better solubilising groups in the 1,1-position. This will combine the good solubility of TStTT3–4 with the better optoelectronic properties of TStTT1–2.1k,17| Polymer | λ abs/nm (CHCl3) | ε/L mol−1 cm−1 | λ abs/nm (thin film) | λ em/nm | Δ/cm−1 |
Φ F/% |
|---|---|---|---|---|---|---|
| TStTT1 | 556 | 35600 |
556 | 717 | 4038 | 0.26 |
| TStTT2 | 560 | 28600 |
595 | 716 | 3891 | <0.1 |
| TStTT3 | 532 | 32000 |
568 | 654 | 3506 | 0.32 |
| TStTT4 | 522 | 26400 |
550 | 655 | 3890 | <0.1 |
The extinction coefficient is maximal for TStTT1 (35600 L mol−1 cm−1) and in general, all are much higher for the polymers than for the monomers, giving well-absorbing materials (Table 4). Furthermore, the four polymers TStTT1–4, combinations of thiophenyl and stannole monomers, show a high red-shift in the absorption towards pure poly(3-hexylthiophene) (λabs = 435 nm) demonstrating the influence of the group 14 element on the conjugation within the polymer and on the properties (Table 4).17
When the absorption spectra were measured from thin films, which were prepared on a microscope slide by using a spin-coater, the broad absorption maxima were again bathochromically shifted for TStTT2–4 by about 28 nm to 36 nm. The absorption maximum of TStTT1 was not shifted, only the absorption spectrum itself was more broadened. The optical band gaps were determined, using the λonset of the absorption maximum, to be 1.85 eV (TStTT1), 1.84 eV (TStTT2), 1.93 eV (TStTT3), and 1.95 eV (TStTT4) from the spectra in solution and 1.75 eV (TStTT1), 1.61 eV (TStTT2), 1.72 eV (TStTT3), and 1.79 eV (TStTT4), respectively, in thin film. These values demonstrate extremely low-band gap materials.
Compared to the phenyl–stannole copolymers published in 2019 (λabs,chloroform = 457–491 nm, λabs,film = 454–503 nm, Eg,opt (thin film) 1.99–2.05 eV), the absorption maxima of TStTT1–4 are substantially bathochromically shifted and the optical band gaps smaller due to the high planarity of the polymers backbone and therefore the more efficient conjugation;8a while the results (λabs,chloroform = 536 nm, λabs,film = 585 nm, Eg,opt (thin film) = 1.70 eV) of the thiophenyl–stannole copolymer reported in 2014 are alike to our findings (Table 4).13c
Furthermore, the influence of the Sn becomes clear by comparing the optical band gaps of TStTT1–4 (Eg,opt = 1.84–1.95 eV in chloroform, Eg,opt = 1.61–1.79 eV in thin film) with those of common polymers, such as polythiophene (1.8–2.21 eV), polypyrrole (2.9–3.2 eV) or polyfuran (1.94–2.7 eV).18
The phenylated phosphole and arsole polymers (heavier gr. 15 metalloles) by Tomita and coworkers in 2015 and 2016, respectively, also showed a low band gap of Eg,opt = 2.00 eV (λabs,chloroform = 522 nm, λabs,film = 525 nm) and Eg,opt = 2.00 eV (λabs,chloroform = 517 nm, λabs,film = 517 nm) due to σ*–π*-conjugation as in the gr. 14 metalloles.19 However, the related bismole polymer, which contains the heaviest group 15 congener, only had an absorption maximum of λabs,chloroform = 311 nm.20
In 2013, Rivard and coworkers published a series of thiophenyl copolymers very similar to ours (butadiene–thiophenyl and heterole–thiophenyl hybrid polymers with E = S, Se, Te), which were synthesised by Suzuki–Miyaura cross-coupling reaction (Fig. 5).21
 | ||
| Fig. 5 Hybrid polymers with butadiene, thiophene, selenophene, or tellurophene units. | ||
Surprisingly, their butadiene-thiophenyl hybrid polymer without the heteroelements S, Se or Te bridging this unit showed the lowest band gap and most red-shifted absorption maximum (Eg,opt = 2.36 eV, λabs,THF = 430 nm) in THF; while the band gap and the absorption maxima for the other copolymers were Eg,opt = 2.75 eV, λabs,THF = 404 nm (thiophene), Eg,opt = 2.70 eV, λabs,THF = 408 nm (selenophene) and Eg,opt = 2.64 eV, λabs,THF = 420 nm (tellurophene).21 Compared to these values, the absorption maxima (λabs,chloroform = 522–560 nm) of the polymers TStTT1–4 are considerably more red-shifted and the band gaps smaller (Eg,opt = 1.84–1.95 eV in chloroform). Group 14 metalloles (Si, Ge, Sn, Pb) are non-aromatic compounds in comparison to group 16 heterocycles (O, S, Se, Te); however, the σ*–π*-conjugation in combination with the π-conjugation from thiophene appears to be efficient in lowering the HOMO–LUMO gap.1e An exact quantification of this effect is, however, not possible, because the group 14 element changes not only the frontier molecular orbitals, but also the geometry. To be able to obtain qualitative information on the importance of the σ*–π*-conjugation, a comparative calculation was performed of both a pentamer of a thiophene–cyclopentadiene–thiophene triad and the corresponding thiophene–stannole–thiophene triad (Fig. 6, for the corresponding images of the HOMO and LUMO orbitals see Fig. S70–S73 in the ESI‡).
 | ||
| Fig. 6 Model compounds of a thiophenyl–stannole and thiophenyl–cyclopentadiene co-oligomer for DFT calculations. | ||
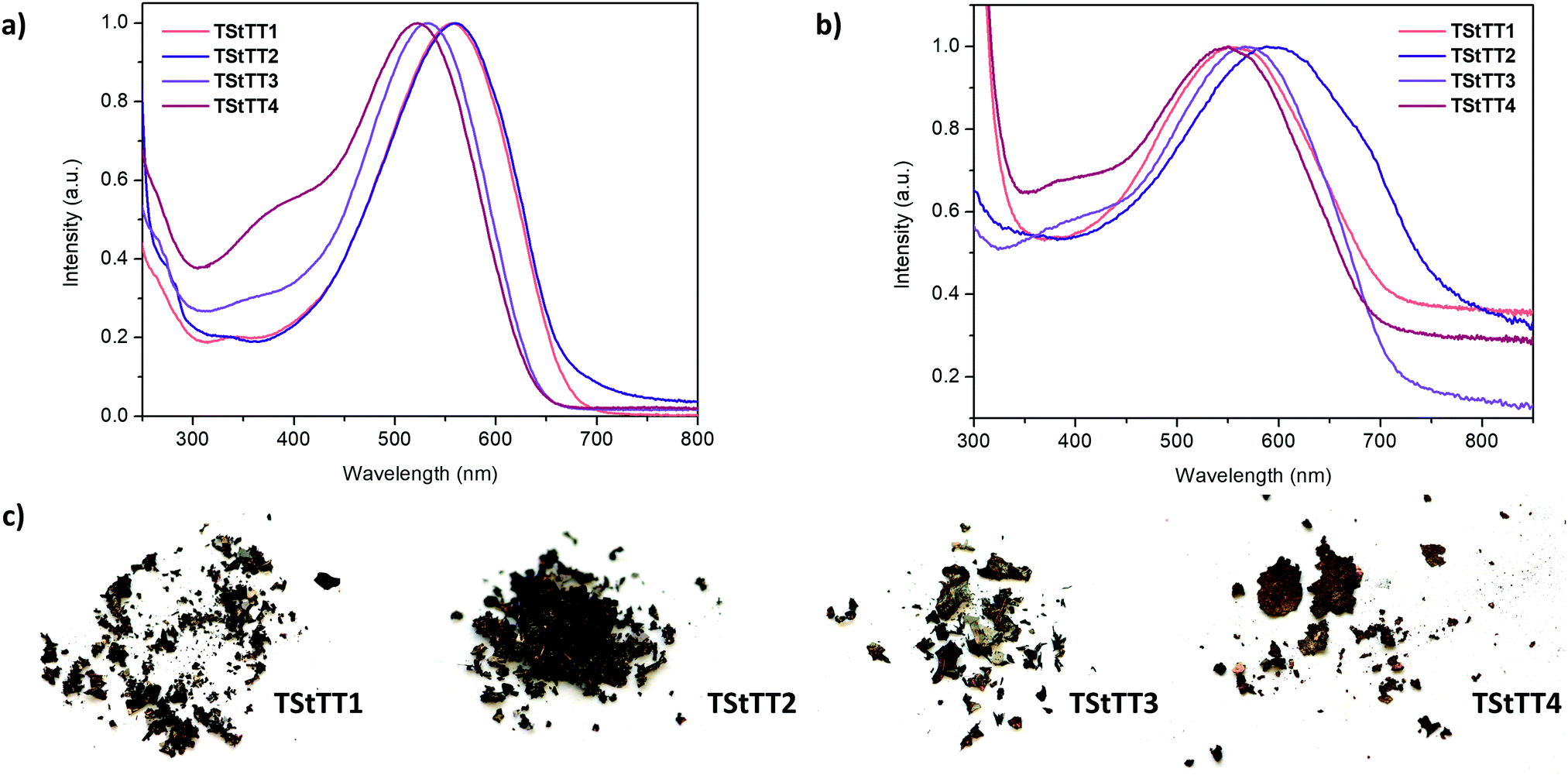 | ||
| Fig. 7 (a) UV-Vis spectra of all polymers in chloroform at 295 K; (b) UV-Vis spectra of thin films of TStTT1–4; (c) photographic images of all four polymers. | ||
For the carbon analogue, the calculated energy gap was 2.24 eV whereas for the Sn analogue it was 2.10 eV. This difference appears small at first glance; however, it appears that in the carbon analogue there is also a σ*-contribution, which is absent in the monomer. Therefore, one might tentatively conclude that a butadiene motif is not sufficiently analogous to make comparative conclusions concerning the σ*–π*-conjugation or absence thereof. Secondly one might conclude that the carbon congeners of the presented polymers might be interesting targets to explore, because this effect appears to occur only in a polymer. The cyclopentadiene motif has only rarely been used in polymers, but the possibility of σ*–π*-conjugation has not yet been explored.1e,3a,4a
Looking at the emission properties, the fluorescence turned extremely weak to non-existent with quantum yields of <0.1% to 0.32% (Table 4, see Fig. S50–S53 in the ESI‡). These results are unsurprising, because the emission maxima λem = 654–717 nm are far red-shifted and because of the heavy-metal effect of all the Sn atoms. However, the values are quite promising compared to the tellurophene copolymer (by Rivard and coworkers) showing no fluorescence at all.21 However the BPin–tellurophene monomer and a other tellurophenes exhibited promising properties in the past, namely strong aggregation induced emission and phosphorescence.22 Furthermore, for the phenylated arsole and bismole polymers emission maxima at λem = 600 nm (ΦF = 5%) and 440 nm (ΦF = 13%) were published, which are high for compounds with these heavy elements.19b,20
3 Conclusions
In conclusion, four new polymers TStTT1–4 with mixed conjugation (cross-hyperconjugation and π-conjugation) were furnished in high yields of 94–98% by tin-selective Stille coupling without decomposition of the stannole ring itself. All materials showed broad strong absorption maxima in solution between 522–560 nm and in thin films at 550–595 nm with high molar extinction coefficients up to 35571 L mol−1 cm−1. The combination of the long effective conjugation within the polymers, the high planarity of the backbones of each repeating unit, the low-band gap (1.61–1.79 eV), and the unique optoelectronic properties makes these materials to be promising candidates for applications in organic electronics. The position of solubilising chains (i.e. on thiophene or Sn) or the fluorinated electron-withdrawing groups affect the final properties of the polymer. Although one must be careful not to overinterpret the data, certain plausible points can be made: One fact is that electron withdrawing groups at the Sn atom decrease the HOMO–LUMO gap; therefore, such groups were used in two polymers TStTT3–4. On the other hand, it was important to understand the effect on the polymers’ properties, if the solubilising chains were not attached at the π-conjugated backbone. Somewhat surprisingly, the polymers TStTT1–2 showed bathochromically shifted absorption maxima than TStTT3–4, although the electronic effects of the substituents on Sn would have led us to predict otherwise. Therefore, it seems that the effective conjugation of the first two polymers TStTT1–2 is larger than of TStTT3–4, most probably due to a higher degree of planarity. This in turn is most likely the effect of not having any additional solubilising alkyl chains on the conjugated backbone of the polymer. However, aside from this electronic or stereoelectronic effect, the position of the hexyl-chains of TStTT1/2 also affected the solubility making them much less soluble than TStTT3–4. Therefore, it is necessary to install the solubilising groups either in the 1,1-position or maybe in the 3,4-positions on the stannole ring itself and not on the backbone of the polymer, as in TStTT3–4, to achieve the larger effective conjugation within the polymer and with this, better optoelectronic properties. However, n-hexyl-groups are not good enough; either the chains have to be elongated, i.e. dodecyl, or branched groups, i.e. tert-butyldimethylsiloxy, might be introduced to increase the solubility of the polymer. Finally, the positive influence of the electron-withdrawing CF3-groups on the molecules optoelectronic properties was significant for the monomers St3–4, but could not be verified for the corresponding polymers TStTT3–4, most probably due to the less effective conjugation.
4 Experimental section
4.1 Materials and methods
All reactions were carried out using standard Schlenk techniques under a dry, inert nitrogen or argon atmosphere or in a nitrogen filled glove box (GB) from Inert unless noted otherwise. Commercially available chemicals were used without further purification unless noted otherwise. Anhydrous solvents were taken from the solvent purification system(SPS) from Inert and were degassed by three freeze–pump–thaw cycles.1H, 13C, 19F, and 119Sn{1H} NMR spectra were recorded on a Bruker Avance Neo 600 or Bruker DRX 500 at 23 °C. All 1H NMR and 13C{1H} NMR spectra were referenced against the solvent residual proton signals (1H), or the solvent itself (13C). The reference for the 19F and 119Sn{1H} NMR spectra was calculated based on the 1H NMR spectrum of tetramethylsilane (TMS). High resolution (HR) EI mass spectra were recorded on the double focusing mass spectrometer MAT 95XL from FINNIGAN MAT. HR-APCI mass spectra were recorded on a Bruker Impact II. MALDI mass spectra were recorded on a AutoflexMax from Bruker Daltonik, Bremen using DCTB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile) (20 mg mL−1) as the matrix. Gel permeation chromatography (GPC) was performed on a GPC – PSS/Agilent SECurity 1260 System. The columns were heated at 35 °C with a column thermostat SECurity TCC6000. Conventional calibration using polystyrene standards (PS) was conducted to calibrate the system. Chloroform (HPLC grade without stabiliser) was used as an eluent. IR spectra were recorded on a Nicolet Thermo IS10 SCIENTIFIC spectrometer with a diamond ATR unit. The resolution was 4 cm−1. Relative intensities of the IR bands were described by s = strong (0–33% T), m = medium (34–66% T) or w = weak (67–100% T). All melting points were measured with a Büchi Melting Point M-560 apparatus. UV-Vis spectra were recorded with a resolution of 0.1 nm on a UV-2700 spectrometer from Shimadzu. Emission spectra were recorded on an Edinburgh Instruments FLS 1000 photoluminescence spectrometer. For temperature dependent measurements this spectrometer was equipped with an Oxford Instruments OptistatCF cryostat cooled with liquid nitrogen. Absolute quantum yields were measured with an Edinburgh Instruments integrating sphere. All emission spectra are corrected spectra. Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TGA instrument using aluminium crucibles under N2 at a flow rate of 20 mL min−1 and a heating rate of 10 K min−1.
4.2 Crystallography
Intensity data of ST2 and ST3 were collected on a Bruker Venture D8 diffractometer at 100 K with Mo-Kα (0.7107 Å) radiation. All structures were solved by direct methods and refined based on F2 by use of the SHELX23 program package as implemented in OLex2 1.2.24 All non-hydrogen atoms were refined using anisotropic displacement parameters. Hydrogen atoms attached to carbon atoms were included in geometrically calculated positions using a riding model. Crystal and refinement data are collected in Table S4 in the ESI.‡ Figures were created using the programme Diamond (Diamond – Crystal and Molecular Structure Visualization, Crystal Impact – Dr H. Putz & Dr K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany http://www.crystalimpact.com/diamond).4.3 Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations
Optimised equilibrium geometries were calculated using DFT with the Gaussian 16, revision A.0325 quantum software package for a single molecule in the gas phase using the PBE1PBE/6-311++G(2d,2p)26 level of theory including empirical dispersion corrections according to Grimme's D327 method involving Becke–Johnson damping (GD3BJ). For the Sn atom, we employed the Stuttgart/Dresden (SDD) pseudo potential.28 Absorption data was calculated using time-dependent DFT (TD-DFT) level on the optimised ground state geometries with the same functional and basis set as described above, i.e., TD-PBE1PBE-GD3BJ/6-311++G(2d,2p)//PBE1PBE-GD3BJ/6-311++G(2d,2p) employing SDD pseudo potentials for Sn.26–284.4 Synthetic procedures
Detailed procedures including the analytical data of all compounds can be found as a part of the ESI.‡
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computations for this work were done with resources of Leipzig University Computing Center (M. R.). N. C. B. and P. R. thank the DFG for partial funding under Germany's Excellence Strategy within the Cluster of Excellence PhoenixD (EXC 2122, Project ID 390833453) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 714429). We thank Fangshun Yang (Leibniz IOM) and Katharina Richter (IFAM Bremen) for their support with the MALDI results.Notes and references
- (a) J. Chen, Z. Xie, J. W. Y. Lam, C. C. W. Law and B. Z. Tang, Macromolecules, 2003, 36, 1108–1117 CrossRef CAS; (b) S. J. Toal, D. Magde and W. C. Trogler, Chem. Commun., 2005, 5465–5467 RSC; (c) S. J. Toal, J. C. Sanchez, R. E. Dugan and W. C. Trogler, J. Forensic Sci., 2007, 52, 79–83 CrossRef CAS PubMed; (d) B. L. Lucht, M. A. Buretea and T. D. Tilley, Organometallics, 2000, 19, 3469–3475 CrossRef CAS; (e) M. Hissler, P. W. Dyer and R. Réau, Coord. Chem. Rev., 2003, 244, 1–44 CrossRef CAS; (f) H. Usta, G. Lu, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 9034–9035 CrossRef CAS PubMed; (g) J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714–1742 CrossRef CAS; (h) J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS; (i) G. Lu, H. Usta, C. Risko, L. Wang, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 7670–7685 CrossRef CAS; (j) K. Murakami, Y. Ooyama, H. Higashimura and J. Ohshita, Organometallics, 2016, 35, 20–26 CrossRef CAS; (k) K. Tamao, S. Yamaguchi, Y. Ito, Y. Matsuzaki, T. Yamabe, M. Fukushima and S. Mori, Macromolecules, 1995, 28, 8668–8675 CrossRef CAS; (l) D. Gendron, P.-O. Morin, P. Berrouard, N. Allard, B. R. Aïch, C. N. Garon, Y. Tao and M. Leclerc, Macromolecules, 2011, 44, 7188–7193 CrossRef CAS; (m) X. Guo, N. Zhou, S. J. Lou, J. W. Hennek, R. Ponce Ortiz, M. R. Butler, P.-L. T. Boudreault, J. Strzalka, P.-O. Morin, M. Leclerc, J. T. López Navarrete, M. A. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 18427–18439 CrossRef CAS PubMed; (n) S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC; (o) C. Gu, D. Zhu, M. Qiu, L. Han, S. Wen, Y. Li and R. Yang, New J. Chem., 2016, 40, 7787–7794 RSC.
- J. Casado, M. Z. Zgierski, P. C. Ewbank, M. W. Burand, D. E. Janzen, K. R. Mann, T. M. Pappenfus, A. Berlin, E. Pérez-Inestrosa, R. P. Ortiz and J. T. López Navarrete, J. Am. Chem. Soc., 2006, 128, 10134–10144 CrossRef CAS PubMed.
- (a) S. Yamaguchi and K. Tamao, Bull. Chem. Soc. Jpn., 1996, 69, 2327–2334 CrossRef CAS; (b) S. Yamaguchi, Y. Itami and K. Tamao, Organometallics, 1998, 17, 4910–4916 CrossRef CAS; (c) I.-M. Ramirez y Medina, M. Rohdenburg, F. Mostaghimi, S. Grabowsky, P. Swiderek, J. Beckmann, J. Hoffmann, V. Dorcet, M. Hissler and A. Staubitz, Inorg. Chem., 2018, 57, 12562–12575 CrossRef CAS PubMed; (d) A. Moliton and R. C. Hiorns, Polym. Int., 2004, 53, 1397–1412 CrossRef CAS.
- (a) K. Jorner, R. Emanuelsson, C. Dahlstrand, H. Tong, A. V. Denisova and H. Ottosson, Chem. – Eur. J., 2014, 20, 9295–9303 CrossRef CAS PubMed; (b) A. Denisova, J. Tibbelin, R. Emanuelsson and H. Ottosson, Molecules, 2017, 22, 370 CrossRef PubMed.
- (a) M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura and S. Nagase, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 1068–1076 CrossRef CAS; (b) T. Kawamura, M. Abe, M. Saito and M. Hada, J. Comput. Chem., 2014, 35, 847–853 CrossRef CAS PubMed.
- A. Pöcheim, G. A. Özpınar, T. Müller, J. Baumgartner and C. Marschner, Chem. – Eur. J., 2020, 26, 17252–17260 CrossRef PubMed.
- (a) S. Urrego-Riveros, I. M. Ramirez y Medina, J. Hoffmann, A. Heitmann and A. Staubitz, Chem. – Eur. J., 2018, 24, 5680–5696 CrossRef CAS PubMed; (b) I.-M. Ramirez y Medina, W. Kipke, J. Makow and A. Staubitz, Sci. Synth., Knowl. Updates, 2020, 2, 31 Search PubMed.
- (a) Y. Matsumura, M. Sugihara, S. E. Tan, T. Sato, K. Hayashi, H. Nishiyama, W. M. Zhou, S. Inagi and I. Tomita, Macromol. Rapid Commun., 2019, 40, 1800929 CrossRef PubMed; (b) W.-M. Zhou and I. Tomita, J. Inorg. Organomet. Polym., 2009, 19, 113–117 CrossRef CAS.
- J. Linshoeft, E. J. Baum, A. Hussain, P. J. Gates, C. Näther and A. Staubitz, Angew. Chem., Int. Ed., 2014, 53, 12916–12920 CrossRef CAS PubMed.
- F. Zheng, S.-E. Tan, Y. Yanamoto, N. Shida, H. Nishiyama, S. Inagi and I. Tomita, NPG Asia Mater., 2020, 12, 41 CrossRef CAS.
- V. Y. Lu and T. D. Tilley, Macromolecules, 2000, 33, 2403–2412 CrossRef CAS.
- T. B. D. Miles, A. Lough and D. Foucher, J. Inorg. Organomet. Polym., 2010, 20, 544–553 CrossRef.
- (a) I.-M. Ramirez y Medina, M. Rohdenburg, E. Lork and A. Staubitz, Chem. Commun., 2020, 56, 9775–9778 RSC; (b) S. Urrego-Riveros, I.-M. Ramirez y Medina, D. Duvinage, E. Lork, F. D. Sönnichsen and A. Staubitz, Chem. – Eur. J., 2019, 25, 13318–13328 CrossRef CAS PubMed; (c) J. Linshoeft, E. J. Baum, A. Hussain, P. J. Gates, C. Näther and A. Staubitz, Angew. Chem., Int. Ed., 2014, 53, 12916–12920 CrossRef CAS PubMed.
- (a) U. Rosenthal, A. Ohff, W. Baumann, A. Tillack, H. Görls, V. V. Burlakov and V. B. Shur, Z. Anorg. Allg. Chem., 1995, 621, 77–83 CrossRef CAS; (b) J. R. Nitschke, S. Zürcher and T. D. Tilley, J. Am. Chem. Soc., 2000, 122, 10345–10352 CrossRef CAS.
- (a) H. J. Tracy, J. L. Mullin, W. T. Klooster, J. A. Martin, J. Haug, S. Wallace, I. Rudloe and K. Watts, Inorg. Chem., 2005, 44, 2003–2011 CrossRef CAS PubMed; (b) J. L. Mullin and H. J. Tracy, Aggregation-Induced Emission: Fundamentals and Applications, John Wiley and Sons Ltd, 2013, vol. 1 and 2, ch. 2, pp. 39–60 DOI:10.1002/9781118735183.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- K. Tamao, S. Yamaguchi, M. Shiozaki, Y. Nakagawa and Y. Ito, J. Am. Chem. Soc., 1992, 114, 5867–5869 CrossRef CAS.
- M. S. Senevirathne, A. Nanayakkara and G. K. R. Senadeera, J. Natl. Sci. Found. Sri Lanka, 2011, 39, 183–185 CrossRef CAS.
- (a) Y. Matsumura, M. Ueda, K. Fukuda, K. Fukui, I. Takase, H. Nishiyama, S. Inagi and I. Tomita, ACS Macro Lett., 2015, 4, 124–127 CrossRef CAS; (b) Y. Matsumura, M. Ishidoshiro, Y. Irie, H. Imoto, K. Naka, K. Tanaka, S. Inagi and I. Tomita, Angew. Chem., Int. Ed., 2016, 55, 15040–15043 CrossRef CAS PubMed; (c) H. Imoto and K. Naka, Chem. – Eur. J., 2019, 25, 1883–1894 CrossRef CAS PubMed.
- Y. Morisaki, K. Ohashi, H.-S. Na and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4857–4863 CrossRef CAS.
- G. He, L. Kang, W. Torres Delgado, O. Shynkaruk, M. J. Ferguson, R. McDonald and E. Rivard, J. Am. Chem. Soc., 2013, 135, 5360–5363 CrossRef CAS PubMed.
- (a) E. Rivard, Chem. Rec., 2020, 20, 640–648 CrossRef CAS PubMed; (b) G. He, W. Torres Delgado, D. J. Schatz, C. Merten, A. Mohammadpour, L. Mayr, M. J. Ferguson, R. McDonald, A. Brown, K. Shankar and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 4587–4591 CrossRef CAS PubMed.
- (a) G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, H. P. H. X. Li, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 16 Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (c) R. C. Binning Jr. and L. A. Curtiss, J. Comput. Chem., 1990, 11, 1206–1216 CrossRef; (d) M. P. McGrath and L. Radom, J. Chem. Phys., 1991, 94, 511–516 CrossRef CAS; (e) L. A. Curtiss, M. P. McGrath, J. Blaudeau, N. E. Davis, R. C. Binning and L. Radom, J. Chem. Phys., 1995, 103, 6104–6113 CrossRef CAS; (f) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) G. Igel-Mann, H. Stoll and H. Preuss, Mol. Phys., 1988, 65, 1321–1328 CrossRef CAS; (b) A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
Footnotes |
| † This article was submitted on the occasion of the 85th birthday of Ei-ichi Negishi, the discoverer of the Negishi coupling and Negishi's zirconocene. |
| ‡ Electronic supplementary information (ESI) available: Chemicals and methods, experimental procedures, analytical data, crystal data, UV-Vis, emission, GPC, MALDI, TGA, and NMR spectra. CCDC 2052177 and 2052178. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00104c |
| This journal is © The Royal Society of Chemistry 2021 |