
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoparticle-infused-biodegradable-microneedles as drug-delivery systems: preparation and characterisation†
Rachel E.
Sully
 a,
Hemda
Garelick
b,
Eriketi Z.
Loizidou
b,
Adrian G.
Podoleanu
*c and
Vladimir
Gubala
*a
a,
Hemda
Garelick
b,
Eriketi Z.
Loizidou
b,
Adrian G.
Podoleanu
*c and
Vladimir
Gubala
*a
aMedway School of Pharmacy, Universities of Greenwich and Kent, Anson Building, Central Avenue, Chatham ME4 4TB, UK. E-mail: V.Gubala@kent.ac.uk
bDepartment of Natural Sciences, Middlesex University, The Burroughs, Hendon, London NW4 4BT, UK
cSchool of Physical Sciences, University of Kent, Ingram Building, Parkwood Road, Canterbury CT2 7NH, UK
First published on 8th July 2021
Abstract
For almost two decades, scientists have been exploring the use of nanoparticles as drug vesicles capable of protecting their cargo and deliver it to the target site while evading detection by the body. However, their translation to clinical use has been slower than expected. To a large degree, this is due to the difficulty to formulate the nanomaterial into a usable form, in which they retain their unique, size-dependent properties without aggregating into a bulk material. In this work, we describe a simple methodology for synthesising novel biodegradable microneedle systems infused with silica nanoparticles (SiNP). SiNP were doped with small library of model anti-cancer drugs or drug surrogates before being characterised and encapsulated into biodegradable microneedles. Detailed preparation and characterisation methods for both the nanoparticles and the microneedles-infused with nanoparticles are presented here. We demonstrated the distribution of the nanoparticles within the microneedle matrix in a uniform, un-aggregated form, which enabled the release of the nanoparticles in a sustained manner. Formulating nanomaterial into biodegradable, hydrogel-like microneedles was shown to be effective in preserving their colloidal properties, whilst simultaneously enabling the transdermal delivery of the nanomaterial into the body. Although the concepts of nanoparticles and biodegradable microneedles have been researched individually, the combination of the two, to the best of our knowledge, offers a new pathway to nanomedicine-related applications.
Introduction
Nanoparticles (NPs) are minute objects between 1–100 nm in size with three major properties – they are highly mobile in the free state; have a very large surface area-to-volume ratio; and may exhibit quantum effects, which influences their electronic characteristics.1–3 These properties may help them escape the natural defence mechanisms of the human body.4 There are many types of nanoparticles which are made from biocompatible and biodegradable materials,5 including polymeric nanoparticles, metallic nanoparticles, magnetic nanoparticles, inorganic nanoparticles, organic nanoparticles, etc. They have been shown to present great potential for transportation of drugs.6–9Using nanoparticles as drug vesicles provides many advantages, such as good biocompatibility; protection of the drug molecules; and specific targeting, to name a few.10,11 The efficiency of drug-delivery systems is related to their size. Nanoparticles are small in size with a large surface area, meaning the drug is closer to the surface and therefore released faster. If they are too large in size, i.e. over 200 nm, the lymphatic system is activated and the nanoparticles are cleared from the body quicker.10,12
Another characteristic of nanoparticles that influences their clearance from the body is their surface properties. Targeting ligands, surface curvature and surface reactivity can be carefully designed to prevent aggregation, to increase stability and to influence receptor binding to the nanoparticles.10 The more hydrophobic the surface, the more likely the nanoparticle is to be cleared from the body. To overcome clearance from the body,10,12 their surfaces can be coated with polymers, however this does not overcome aggregation. Coating with capping agents or altering the zeta (ζ) potential (surface charge) have been reported to tackle the aggregation problem with more success.10,12
Drug release from nanoparticles is achieved either by erosion of the matrix, releasing drugs from their interior, producing a rapid burst of drug release followed by a sustained release; or by release of drugs bound to the exterior surface of nanoparticles.5,10,12,13 Once in the blood system, nanoparticles migrate to their desired target by active or passive targeting. Active targeting involves conjugating the nanoparticles to a specific tissue or cell ligand. Whereas, passive targeting involves migrating to the specific tissue through leaky junctions.10
For nanoparticles to be ideal drug-delivery systems, they should be able to reach their target, be recognised, bind and deliver their load to the specific tissue whilst minimising or avoiding drug induced damage to healthy tissues.
An excellent example of the earlier studies of nanoparticles used as drug-delivery systems showed the entrapment of anti-cancer drugs into silica nanoparticles (SiNP) of size 60–70 nm and wall thickness of 10 nm.14 The release of these drugs was studied over 12 hours and showed an initial large release within the first 20 minutes followed by a slow sustained release over the following 10 hours. From these findings it was concluded that hollow porous SiNP could be used successfully as drug-delivery systems.
Some examples of polymeric nanoparticles as drug-delivery systems were applied for treatment of HIV/AIDS,10 malaria,11 and tuberculosis;5 delivery of insulin12 or of psychotic drugs;11 hormone treatments;11 and many more.11
Although nanoparticles have been shown to be good drug carriers, their formulation remains to be a major challenge. They tend to aggregate when in solution due to the effects of Ostwald Ripening and Brownian motion, where smaller particles move, dissolve and deposit onto larger particles to form a thermodynamically stable state.15 Previous research presented by Giovannini et al.15 showed that entrapping nanoparticles in hydrogels could represent a very good storage environment for SiNP, as the dense, hygroscopic matrix of a hydrogel can slow down any movements of nanoparticles and inhibit any reactions of the silica matrix, thus effectively preventing undesired aggregation.15
Giovannini et al.15 studied the long-term stability of SiNP and gold nanoparticles (AuNP) by storing them in both aqueous solutions and hydrogels for several weeks. The carefully engineered nanoparticles stored in solutions displayed visible aggregates from as early as one week. However, the nanoparticles in hydrogels were stable and without aggregates for many weeks or months (as illustrated in Fig. 1). Because the studied hydrogel was responsive to mechanical forces, the release of the nanoparticles from the hydrogel was facilitated by shaking/vortexing of the nanoparticle–hydrogel formulation, which turned the gel back into an injectable solution. When this solution was injected into an animal model (i.e. chicken embryos), we reported on the presence of aggregation, the cytotoxicity and delivery effects. While this was an elegant resolution to nanoparticle aggregation, formulation and storage, our next attempt was to formulate the nanoparticles into a gel that could have the potential to release and deliver the nanoparticles directly, without additional manipulation and changing the phase from gel to a solution.
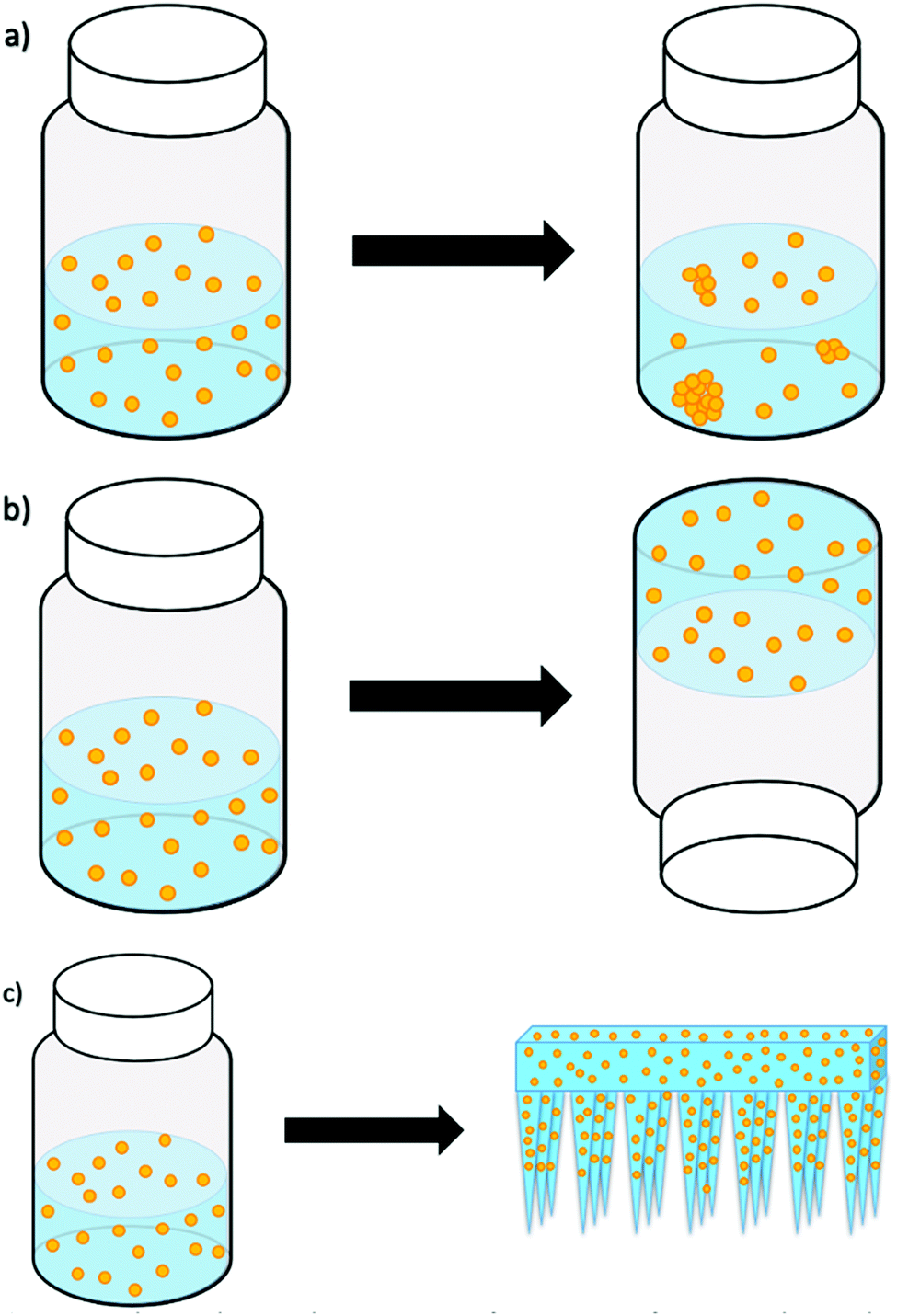 | ||
| Fig. 1 Scheme showing the prevention of aggregation of nanoparticles in gels – (a) nanoparticles in solution; (b) nanoparticles in self-supporting gels; (c) nanoparticles in biodegradable microneedle arrays. | ||
Therefore, an alternative strategy, exploiting the use of biodegradable microneedles (MN), as a novel delivery system for nanoparticles is being introduced in this work. A microneedle–nanoparticle-gel formulation would enable the delivery of nanoparticles directly (i.e. without the need to change the gel into an injectable solution), while it would be possible to store the microneedle–nanoparticle-gel formulation for longer time without risking nanoparticle aggregation. Although there has been extensive research into the use of nanoparticles as drug vesicles, the field of microneedles for transdermal drug-delivery is still in its infancy and there is very little research into the incorporation of nanoparticles into dissolvable microneedles.
Microneedles have been defined as ‘a canula which is either solid or hollow with an approximate needle length ranging between 50–900 μm and an external diameter of around 300 μm’.16 The length and shape of the needle is carefully engineered so that it painlessly penetrates the stratum corneum and deposits the cargo in the epidermis/dermis interface. There are many different types of microneedles including solid microneedles, coated microneedles, hollow microneedles, polymer microneedles, biodegradable/dissolvable microneedles and hydrogel forming microneedles,17 and the potential of microneedles for drug delivery applications have been extensively researched.18–23
In this work, we prepared SiNP infused with various model drugs (e.g., a small library of known anti-cancer agents), drug surrogate (e.g., fluorescein isothiocyanate (FITC)) and formulated the nanoparticles into biodegradable microneedle gels for transdermal delivery. The materials used for microneedles in this project was biodegradable carboxymethyl cellulose (CMC), mixed with readily available carbohydrates (e.g., maltose, sucrose, or trehalose). We hypothesised that from a long-term perspective, incorporating the nanoparticles into a biodegradable polymer not only prevents their aggregation, but also allows them to be sustainably released into the epidermis, surpassing the stratum corneum barrier. CMC has been used as it has been shown to form an anti-aggregation layer,24 which makes it more useful as a matrix for nanoparticle encapsulation. In addition, microneedles formed from CMC/sugar are not too brittle and do not break upon insertion into the skin,25 as well as the material being inexpensive and available even for a potential scale-up production. The SiNP used in this research were synthesised by a microemulsion method.26 The fabricated nanoparticles: pristine SiNP; fluorescein isothiocyanate dye-doped SiNP (FITC-NP); and anti-cancer drug-doped SiNP (Drug-NP), were characterised using various techniques including Dynamic Light Scattering (DLS), Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM).
The nanoparticles were incorporated into the microneedle arrays at varying concentrations, following the synthesis described by Loizidou et al.25 The nanoparticle-microneedles were characterised using SEM and imaged by darkfield and confocal microscopy. Their degradation time and storage stability were also studied. The optimisation of the microneedle preparation is also discussed within this paper.
Results and discussion
Synthesis of silica nanoparticles
The synthetic route used to synthesise the SiNP is known as the microemulsion method and involves dispersing two or more immiscible liquids as droplets by stirring. Ammonium hydroxide is used as a catalyst and ethanol is used to break the microemulsion.26 The products of this synthetic route appeared to show very uniform samples (PdI < 0.1) of neatly spherical particles.DLS was used to study both the pristine SiNP and the FITC-NP, giving average sizes of 109.41 ± 3.21 nm and 96.09 ± 8.33 nm respectively (Tables S1–S2, ESI†). These sizes were compared to those observed by TEM, which were 73.20 ± 6.23 nm for the pristine SiNP and 84.17 ± 1.18 nm for FITC-NP (Fig. 2a and b). Often there can be a discrepancy between the size data by DLS and TEM due to the DLS measuring the hydrodynamic radius of the nanoparticles in a solution, whereas TEM measures their diameter in vacuum. Depending on what type of nanoparticles are being observed, as well as their synthetic route, their hydrodynamic radius varies proportionally with the quantity of water molecules that their surface attracts.
 | ||
| Fig. 2 TEM images of nanoparticles (scale bar represents 200 nm for all images) – (a) pristine silica nanoparticles; (b) FITC-doped silica nanoparticles; (c) doxorubicin-doped silica nanoparticles; (d) temozolomide-doped silica nanoparticles; (e) 5-fluorouracil-doped silica nanoparticles; (f) paclitaxel-doped. | ||
The surface charge of both types of nanoparticles were also measured using DLS. Pristine SiNP were slightly less negative than the FITC-NP, showing an average ζ-potential of −30.178 ± 5.30 mV compared to −37.37 ± 5.03 mV (Tables S1–S2, ESI†). Nanoparticles with such negative charge tend to be more stable in buffered solutions, such as phosphate buffered saline (PBS). We have previously observed that the colloidal stability of silica nanoparticles stored in aqueous media, such as PBS or cell culture media ranges typically between 1–4 h.27
Degradation of silica nanoparticles
SiNP are produced by hydrolysis of –Si–OH groups. This reaction is reversible, meaning that in aqueous solution, SiNP can also disintegrate into smaller fragments. This, in fact, is a useful feature, the slowly degrading silica matrix is able to release any cargo into the environment, while at the same time, when the SiNP degrades into fragments smaller than 5 nm, the nanoparticle can be renally cleared from the body. In this work, the degradation of the nanoparticles was studied in both PBS and in an aqueous solution containing bovine serum albumin (BSA). FITC-NP enabled us to track the nanoparticle degradation by monitoring the changes of the fluorescence signal over time. The fluorescence intensity of the supernatant, is proportional to the concentration of FITC molecules released from the silica matrix into the solution, thus indicating nanoparticle degradation. This process was measured by fluorescence microscopy at regular intervals over a period of 72 hours. The plotted data can be seen in the Fig. S1 and S2 (ESI†). The nanoparticles began to degrade between 8 and 24 hours in PBS but the process was slower in a BSA solution, where an observable degradation was recorded between 48 and 72 hours. This is presumably due to BSA being adsorbed onto the FITC-NP surface, slowing down the release of FITC molecules into the solution. In both cases, the degradation half-life suggests that nanoparticles may possess the capacity to migrate and target the desired tumour cells before they begin to degrade and release their cargo.26Synthesis of anti-cancer drug-doped silica nanoparticles
In the degradation experiment, FITC was used as a drug-surrogate in the preparation if the nanoparticles, as it was relatively easy to visualise the FITC-NP by fluorescence. Encapsulation of FITC within SiNP is well studied and the synthesis is robust and reproducible. However, encapsulating the SiNP with anti- cancer drugs is not as straightforward as it seems. Four anti-cancer drugs were chosen to be used in the synthesis of nanoparticles. Doxorubicin (DOX), temozolomide (TMZ), 5-fluorouracil (5-FU) and paclitaxel (PAC) were selected because they are all routinely prescribed for patients with cancer and because their relatively subtle structural differences allowed us to understand the influence of charge and or functional groups (i.e. hydrophobicity/hydrophilicity) on the size, shape and charge of the resulting nanoparticles. The nanoparticles were doped using solutions with drug at 1-5% (w/w) concentrations, which equate to critically relevant concentrations stated in literature,28 and the changes in size and surface charge between the different samples were observed using DLS and TEM. Average sizes ranged from ∼162–184 nm by DLS (Tables S4–S7, ESI†) and ∼39–56 nm by TEM (Fig. 2 and Table S8, ESI†). The discrepancy between these results implies that the drug–NP have large hydrodynamic radii, suggesting that they could potentially form a large protein corona when in plasma. A protein corona is a phenomenon that occurs when nanoparticles come into contact with protein-rich media such as blood or plasma. The nanoparticles become covered in proteins which significantly impacts on their size, charge, aggregation and how quickly they are cleared from the systemic circulation.2 The average surface charges (Tables S4–S8, ESI†) for the drug–NP was −32 mV, which is similar to that of the pristine SiNP (−30 mV), suggesting that incorporating molecules into nanoparticles can have some influence on their surface properties.The concentration of the drug, in μg of the drug per 1 mL of nanoparticles, expressed as a loading efficiency, was measured by UV/Vis absorbance. Calibration curves (Fig. S3–S6, ESI†) for each anti-cancer drug in ethanol were plotted and used to extrapolate the mass of the drugs within the nanoparticles, which was then used to calculate the loading efficiency of the nanoparticles using equation 1.
 | (1) |
The results are displayed in Fig. 3. The data shows trends between the change in initial mass of the drug used in the synthesis and the concentration of the drug that becomes encapsulated inside the drug–NP. DOX shows no specific trend and very little drug encapsulation, which could be due to the large size of the drug molecule itself. Whereas, the smaller drug molecules, TMZ and 5-FU, show good encapsulation efficiency. PAC shows good encapsulation for the initial 1% and 2% (w/w) concentration, however above these percentages, the encapsulation concentration decreases again, this may be due to the extremely large size of paclitaxel molecules leading to lower encapsulation due to limited space within the SiNP core. Previous investigations into doping nanoparticles with paclitaxel29 have also discussed the effect of steric hindrance, which is defined as the steric effect arising from crowding of substituents,30 causing maximum drug loading. For the drug–NP to be synthesised reproducibly and with predictable physico-chemical properties, a fine balance between the organic phase, aqueous phase and the surfactant must be maintained. The presented data indicate that this balance can be influenced by the initial mass of the drug used in the synthesis, as well as by the intrinsic properties of the drug itself. While all the tested drugs have limited water solubility, TMZ and 5-FU are structurally more similar than the bulkier DOX and PAC. The encapsulation efficiencies for the smaller TM and 5-FU were also following a more similar trend, when compared to the seemingly random trends for DOX and PAC. The drug–NP synthesis for all four drugs was performed with multiple replicates, at each initial mass concentration of the drug. The fact that the standard deviation for each sample (expressed as error bars in Fig. 3) is generally high, simply illustrates that while it is possible to prepare nanoparticles doped with the anti-cancer drugs as shown in this study, the range of the clinically relevant drug concentrations is relatively broad and does not follow a very clear, pre-determined pattern. However, examples from the literature showed that it is possible to fine-tune this microemulsion process to enable encapsulation of poorly soluble drugs using organically modified silica matrices.31
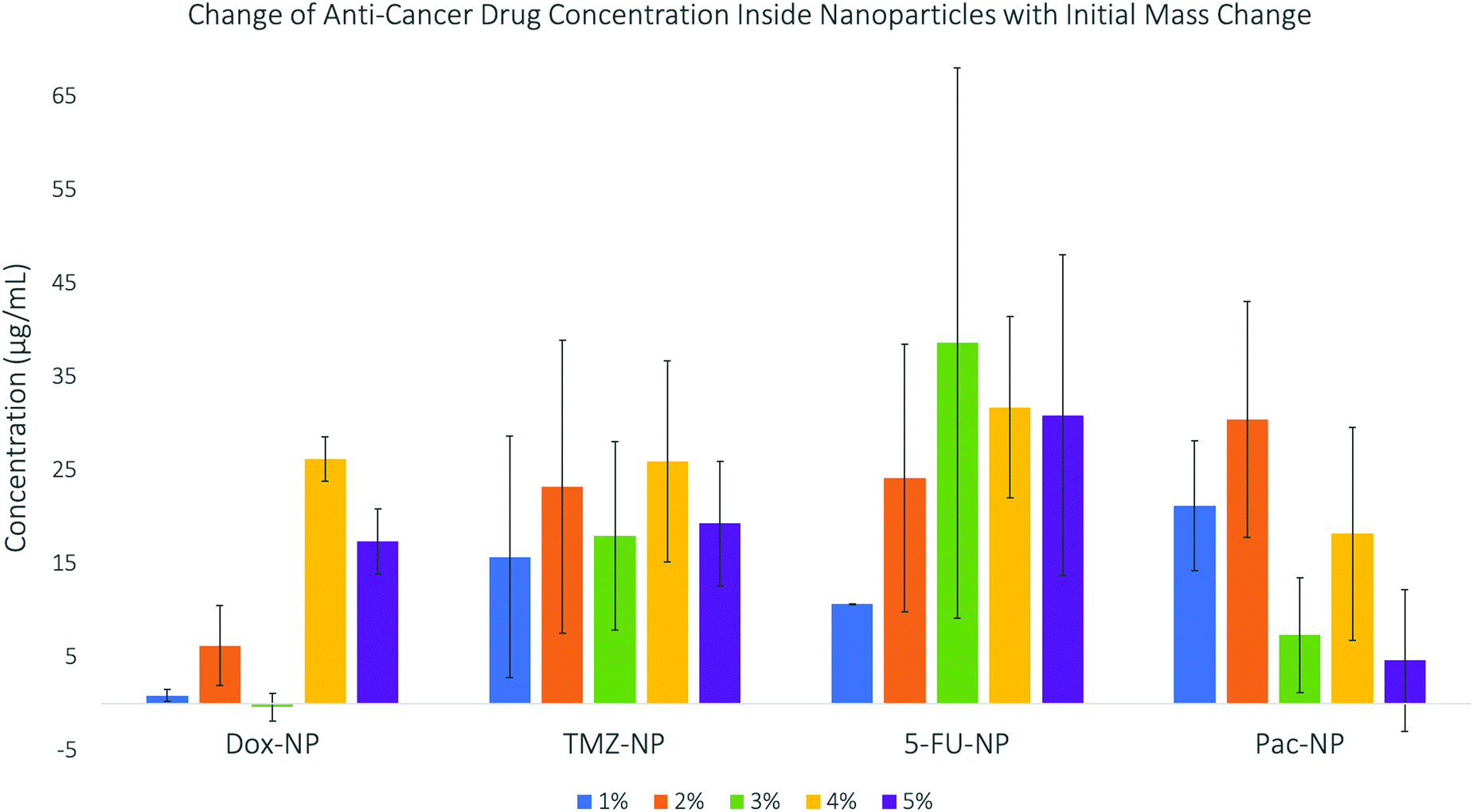 | ||
| Fig. 3 Concentrations of anti-cancer drugs encapsulated into silica nanoparticles – this data shows a general increase in concentration as initial mass increases until a maximum. | ||
Synthesis of microneedle arrays
Microneedle arrays from Carboxymethylcellulose (CMC), combined with either Trehalose (TRD), Sucrose (SUC) or Maltose (MAL), each one at 50% (w/w) concentration were successfully synthesised with and without nanoparticles incorporated into them. Their stability was studied over 30 days at room temperature and 4 °C both quantitatively and qualitatively. No change in mass or appearance was observed over a long period of time, thus suggesting that the arrays are stable, presumably suitable for medicinal use or for transport if needed for any other research purposes.The standard procedure to prepare microneedle arrays with nanoparticles incorporated inside them requires a 72 hour-long evaporation step at atmospheric pressure, during which the CMC/TRD (or CMC/SUC or CMC/MAL) solution solidifies into a hydrogel matrix. We found out that most of the nanoparticles could aggregate within the 72 hours (unpublished data) before the material solidifies into a hydrogel. Therefore, we have modified the standard procedure to avoid or minimise the risk of nanoparticle aggregation. To this end, we have fabricated the nanoparticle-microneedles at decreased pressures in order to speed up the evaporation process, thus keeping the nanoparticles in solution for as short time as possible. The pressures used were over the range of 100–400 mBar at 100 mBar intervals. The low pressure drastically accelerated the evaporation time to 3–4 hours instead of 72 hours.
Initially, we attempted to prepare the microneedles in a vacuum oven, which would enable us to control the temperature as well. However, the microneedles are formed on a silicone mould, which needs to be evacuated before the CMC/sugar/nanoparticle solution is poured over it. Placing the mould in vacuum ensures that the solution fills completely all the needle-cavities and forms a very uniform, microneedle array when the solution solidifies and is peeled off the mould. Because the vacuum oven had to be opened and exposed to an atmospheric pressure to allow the solution to be deposited over the mould, we observed the formation of bubbles in the microneedle array. The set up was therefore changed. A simple, wide-neck vacuum flask with rubber septum was employed to hold the mould. Once the flask with the mould was evacuated, the solution was poured over the mould via septum using a syringe, while maintaining the vacuum inside the flask. Although this might appear trivial, the optimisation of the protocol was critical for us to produce high quality microneedle arrays with infused nanoparticles, in a reproducible and robust way (Fig. 4).
 | ||
| Fig. 4 CMC-MAL microneedle array structure consisting of 324 needles of height 750 μm, base diameter 200 μm, tip diameter 10 μm and centre-to-centre spacing of 600 μm. | ||
Characterisation of microneedle arrays
With the optimised vacuum protocol, FITC-NP were incorporated into the microneedle arrays in order to characterise the FITC-NP-microneedles and observe the distribution of the nanoparticles inside of the microneedle arrays. Various microscopy techniques were used to image and locate the nanoparticles, including darkfield microscopy, fluorescence microscopy, confocal microscopy and back-scattered SEM. Darkfield microscopy provided the overall fluorescence image of the arrays. While the individual microneedles were clearly visible, which offered very useful information about their size and shape, the resolution of this technique did not allow to identify the exact position of the nanoparticles inside the needles. Both fluorescence microscopy and confocal microscopy were used to form z-stack 3D images which again showed very clearly the shape and size of the individual microneedles; however, the fluorescence intensity maps could not pinpoint the spatial distribution of the nanoparticles inside the microneedles (Fig. 5i and ii). Therefore, we have prepared another set of microneedles with incorporated commercial gold nanoparticles (with diameter and surface charge similar to that of FITC-NP) and observed them using back-scattered SEM. The images presented in Fig. S11–S13 (ESI†) demonstrated mostly unaggregated gold nanoparticles, uniformly distributed within the microneedle matrix. The mechanical properties of the microneedle arrays without nanoparticles have been extensively studied in a previous work.32 That work involved a combination of experimental and finite element analyses which showed that microneedles made of CMC/MAL are superior to those made of CMC/TRD and CMC/SUC in terms of mechanical strength. Buckling was predicted to be the main mode of microneedle failure and the order of buckling was positively correlated to the Young's modulus values of the sugar constituents of each microneedle. Furthermore, in the same study it was identified that small changes (i.e., less than 10% change) to the sugar formulation are unlikely to affect the overall mechanical properties of the microneedles. Similar results have been observed in this project when investigating the effect of nanoparticle presence on gelation time and morphology. SEM imaging showed no changes in morphology between biodegradable gels fabricated with or without silica nanoparticles at varying pressures. Minimal differences in gelation time between gels with and without nanoparticles encapsulated confirm that the presence of nanoparticles does not alter the mechanical properties of the microneedle arrays. This data can be found in Fig. S25 and Table S11 (ESI†). | ||
| Fig. 5 (i) Confocal microscopy image of FITC-NP encapsulated into CMC-MAL array; (ii) backscattered SEM images of CMC-MAL microneedle array with AuNP incorporated into the matrix – (a) microneedle array with AuNP; (b) single needle of microneedle array with AuNPs incorporated showing an isolated cluster of AuNP on the tip of the needle; (c) backing plate of microneedle array with AuNP showing some aggregated AuNP and some single AuNPs distributed; (iii) optical coherence tomography image of biodegradable microneedle array in mouse skin demonstrating the ability of these arrays to penetrate skin without bending or breaking. Yellow colour outlines the position of the individual microneedles. | ||
The ability of the prepared microneedle arrays to penetrate through the skin layers was qualitatively demonstrated on an ex vivo model, using skin of 6 weeks old nude mice. 1 × 1 cm square piece of mouse skin was cut and a CMC-TRD microneedle array was gently pressed into the skin. The sample was then mounted in the sample holder and images taken by a non-invasive technique for cross-sectional tissue imaging, called Optical Coherence Tomography (OCT). OCT uses light in the near-infrared spectral range, with a penetration depth of several hundred micrometres in tissue. The backscattered light is measured to reconstruct the depth profile of the sample, as clearly demonstrated on Fig. 5iii. This image clearly shows a penetration of the microneedle array through the stratum corneum and into the epidermis/dermis interface layer of the nude mice skin. The microneedles did not appear to be bending or breaking upon the contact with the skin, which is a good indication of the applicability of the studied concept to deliver nanomaterial via a transdermal route.
Aggregation of nanoparticles in solution and in microneedle arrays
Nanoparticle aggregation in solution is one of the major obstacles when translating the results from laboratory bench into a commercial product. A classic example of nanoparticle aggregation, which typically results in precipitation of the nanomaterial is shown in Fig. S14 (ESI†). The aggregation timeframe depends on the type of nanomaterial and its intrinsic properties such as the size, surface chemistry and surface charge. Nonetheless, silica-based nanomaterial tends to aggregate in aqueous solutions within a few hours. If SiNP are stored in high salt solution, the aggregation typically occurs within a few minutes. To compare the effect of solution vs microneedle gel, we have formulated FITC-NP both into a brine solution and a brine-hydrogel (i.e. both formulations inserted in saturated NaCl solution). When FITC-NP were placed in the brine solution, we were able to track the aggregation process by fluorescence, measuring the presence of nanoparticles from the top layer of the solution. Over time, the FITC-NP aggregated and precipitated to the bottom of the vial. We measured the fluorescence in the top part of the vial. As demonstrated in Fig. S15 (ESI†), the nanoparticle fluorescence kept decreasing over time, indicating fast aggregation and nanoparticle precipitation. For comparison, we have also formulated the FITC-NP into a hydrogel and immersed it into a brine solution (Fig. S16, ESI†). We tracked the movement and precipitation of the FITC-NP by image analysis software. Fig. S17 (ESI†) shows that the position of FITC-NP did not dramatically change over the same period of time. The hydrogel prevented the FITC-NP from moving inside the matrix, thus inhibited the nanoparticle aggregation. In line with our previously published data,4 we have confirmed that the FITC-SiNP were stable in the hydrogel matrix for at least 4 hours (Fig. S18 and S19, ESI†). This is in stark contrast with the same nanoparticles stored in brine solution, where a visible aggregation was observed in just 15 minutes.Degradation of microneedle arrays
The prerequisite for the dissolvable microneedles to be used as efficient formulation to deliver drugs into a body is that the individual needles, once inserted into the epidermis, degrade over time and release the nanoparticles with their cargo. The degradation of the different FITC-NP-microneedle arrays was performed over 72 hours in either a solution of PBS or a solution containing tyrosinase, an enzyme that is present in our skin, normally responsible for skin pigmentation abnormalities. Tyrosinase is however also known to play role in oxidation of and degradation of various carbohydrates33–35 which is why we aimed to employ it to understand its function in the degradation of the sugar-based, dissolvable microneedles.The results summarised in Fig. S20 (ESI†) were surprising. The FITC-NP–microneedle arrays immersed in PBS released their FITC-NP faster than those immersed in the solution with tyrosinase. The results were normalised to 10 mg of the material due to the difference in the microneedle array mass at the beginning of the experiment. The control data (Fig. S21, ESI†) also showed that the FITC-NP immersed in PBS (without the microneedles) degraded faster than FITC-NP immersed in the tyrosinase solution within the first 24 hours. Therefore, we continued with the degradation study of the FITC-NP-microneedle arrays in PBS only.
The degradation of microneedles, measured as the release of the FITC molecules from the FITC-NP–hydrogel matrix, was done over 4 hours (Fig. 6). As before, the results were again normalised to 10 mg of the material. The results showed that the FITC-NP in microneedle arrays degraded slower than those in the solutions. The sugar solutions also showed a greater intensity throughout the experiment than the FITC-NP only in PBS. Therefore, the calibration curves of FITC alone and FITC-NP were studied to investigate if their concentrations effected the fluorescence intensity (Fig. S22–S24, ESI†). The release of FITC from the FITC-NP was also studied to understand whether the maximum amount of FITC had been released from the nanoparticles over the four hours of degradation.
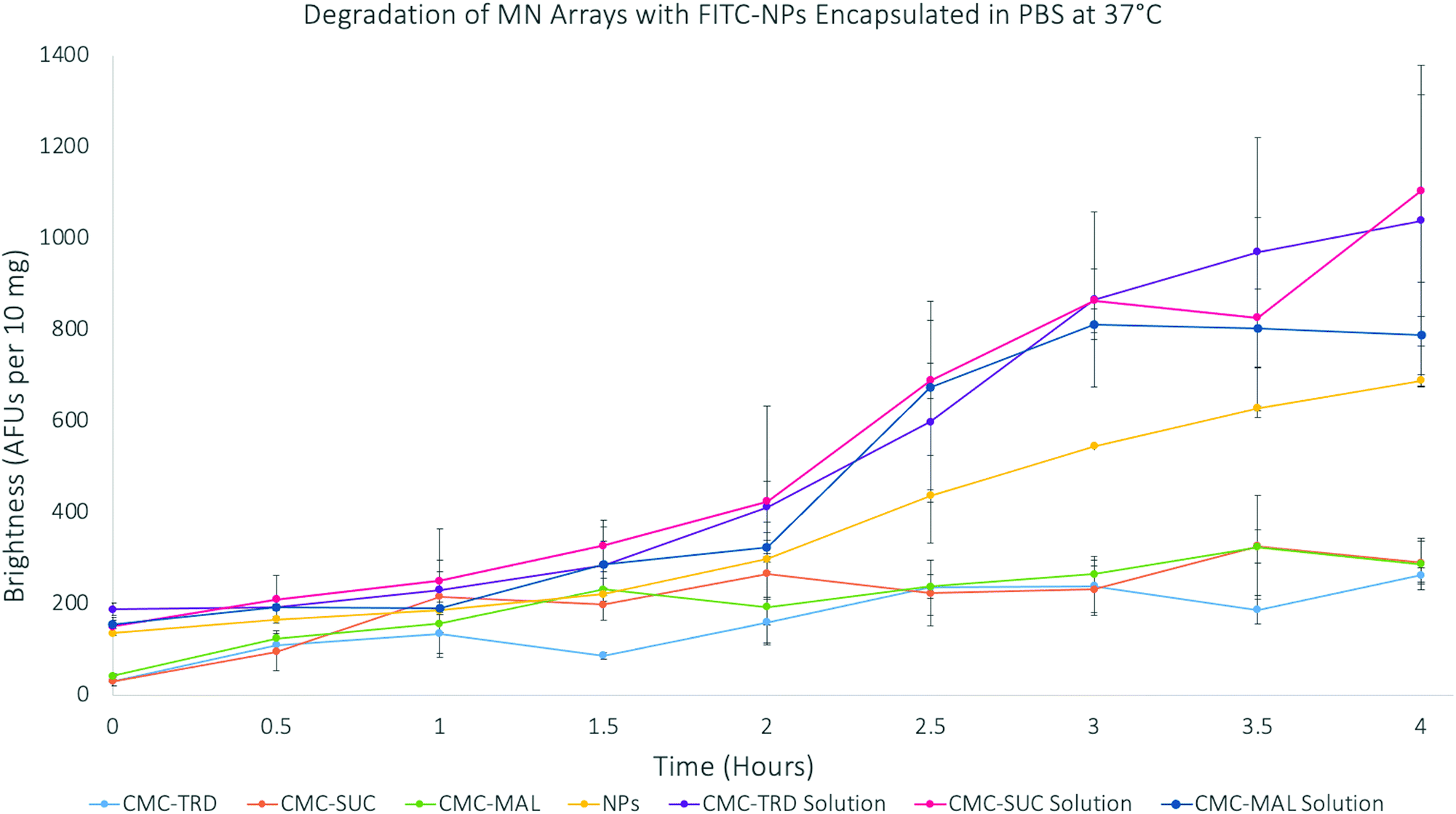 | ||
| Fig. 6 Graph showing the degradation of microneedle arrays with FITC-SiNP encapsulated at 37 °C over 4 hours. | ||
The calibration curve of FITC in PBS and ethanol (90% v/v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10% v/v) showed the fluorescence intensity of FITC is directly proportional to the concentration of FITC. However, the calibration curve for FITC-NP in PBS showed the fluorescence intensity initially decreased at lower concentrations before proportionally increasing with higher concentrations. This can be attributed to a homo-FRET effect of two adjacent FITC molecules (i.e. self-quenching). The self-quenching phenomenon is caused by the re-emission of absorbed energy from molecules in the excited state. As self-quenching occurs, these molecules dissipate energy between the ground state and excited state, resulting in a state with reduced fluorescence intensity in comparison to molecules that are not self-quenching. There are many ways to overcome the self-quenching effect of dye molecules, including the optimisation of dye molecules concentration within the nanoparticles.36
:10% v/v) showed the fluorescence intensity of FITC is directly proportional to the concentration of FITC. However, the calibration curve for FITC-NP in PBS showed the fluorescence intensity initially decreased at lower concentrations before proportionally increasing with higher concentrations. This can be attributed to a homo-FRET effect of two adjacent FITC molecules (i.e. self-quenching). The self-quenching phenomenon is caused by the re-emission of absorbed energy from molecules in the excited state. As self-quenching occurs, these molecules dissipate energy between the ground state and excited state, resulting in a state with reduced fluorescence intensity in comparison to molecules that are not self-quenching. There are many ways to overcome the self-quenching effect of dye molecules, including the optimisation of dye molecules concentration within the nanoparticles.36
The results of the FITC release from FITC-NP were used to calculate the percentage release of FITC from the microneedle arrays. This study showed that the results from the 4 hour degradation study for the microneedle arrays support that only the FITC-NP (39.88% from CMC-TRD arrays; 42.38% from CMC-SUC arrays; and 30.27% from CMC-MAL arrays) were released from the microneedle arrays, due to the very low percentage release of FITC (26.44%), and that the FITC-NP were not degraded themselves. This supports the conclusion that the microneedle arrays release the FITC-NP in a sustained manner, showing a very gradual release between 0–2 hours and then more significant release between 2–4 hours, without affecting the nanoparticle vesicle itself. This holds promise that ligand-coated nanoparticles could be released from the microneedle arrays and target the cancer cells before the nanoparticles degrade.
Conclusions
The work presented here demonstrated a useful step-by-step strategy to produce biodegradable, dissolvable microneedle arrays, infused with SiNP. We showed that SiNP can be successfully prepared to hold a variety of anti-cancer drug molecules, albeit the synthetic procedure for DOX and TMZ should be further optimised. The aggregation studies on FITC-NP and AuNP in different microneedle matrix, showed no observable aggregation of nanoparticles in the microneedle arrays when imaged by confocal microscopy and SEM respectively. The degradation of the microneedle arrays displayed a sustained release of FITC-NP, without disintegration of the nanoparticle and thus without the premature release of the cargo. Nanoparticles as drug delivery concepts have been a subject of intense studies for the past 20 years, yet without significant commercial and/or clinical success. Formulation of the material that is intended to be used as the next generation of nanomedicine still remains a challenge. We believe that the work presented in this article represents a significant step towards the development of painless drug delivery systems, in which the carefully designed nanomaterial can be stored and applied at any time, while retaining its unique, size-dependant physico-chemical and biological properties.Experimental section
Materials
The following chemicals were purchased from Sigma Aldrich: fluorescein isothiocyanate (FITC) (90%); (3-aminopropyl)trimethoxysilane (APTMS) (97%); methanol (anhydrous, 99.8%); tetraethyl orthosilicate (TEOS) (trace metal basis, 99.999%); ammonium hydroxide (28% NH3 in water, 99.99%); 1-hexanol (anhydrous, 99%); cyclohexane (anhydrous, 99.5%); Triton™ X-100 (laboratory grade); 3-(trihydroxysilyl)propyl methylphosphonate, monosodium salt solution (THPMP) (50 wt% in H2O); phosphate buffered saline (PBS) tablets; bovine serum albumin (BSA) (pH 7, 98%); 5-fluorouracil (99%); carboxymethylcellulose sodium salt (CMC) (medium viscosity); D-(+)-maltose monohydrate (MAL) (from potato, 99%); D-(+)-trehalose dehydrate (TRD) (from saccharomyces cerevisiae, 99%); sucrose (SUC) (BioXtra, 99.5%); N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) (purum, ≥98.0%); poly(amidoamine) (PAMAM) dendrimer (ethylenediamine core, generation 3.5 solution, 10 wt% in methanol); 2-(N-morpholino)ethanesulfonic acid (MES) (low moisture content, ≥99%).The following chemicals were purchased from Fisher Scientific: ethanol (absolute, 99.8%); doxorubicin hydrochloride (BioReagent); paclitaxel (95%); Alexa Flour 647 Goat Anti-Rabbit IgG (H + L) (2 mg mL−1); hydrochloric acid (32% extra pure, d = 1.16).
The following chemicals were purchased from Tokyo Chemical Industries (TCI): temozolomide (98%); N-hydroxysulfosuccinimide sodium salt (NHS) (>98.0%).
The animal models were purchased from Charles River as CD-1 Nude Mice Homozygous Nu/Nu Strain 086 at 6 weeks of age.
The following instruments were used for characterisation:
• Dynamic light scattering (DLS) – Malvern Panalytical Zetasizer Nano Series Nano-ZS.
• Transmission electron microscopy (TEM) – JEOL 1230 Gatan One View.
• Tecan Infinite M200Pro Plate Reader.
• WPA Biowave II UV-Vis Spectrometer.
• Scanning electron microscopy (SEM) – Hitachi High Technologies SU8030 (Tokyo, Japan).
• Olympus BX40 with Transmitted Darkfield Condenser.
• ThorLabs Ganymede™ Series Spectral Domain OCT Imaging System.
Experimental
Synthesis of silica nanoparticles
This synthetic method was adapted from Moore et al.26 and was followed to prepare pristine SiNP, FITC-doped SiNP and anti-cancer drug-doped SiNP. Briefly, APTMS (5.6 μL; 0.0321 mmol) was stirred in 1-hexanol (2 mL) for 2 hours. For the preparation of the FITC-doped SiNP or anti-cancer drug-doped SiNP the FITC or one of four anti-cancer drugs at different w/w% (see Table S3, ESI†) respectively, were also added to this solution before stirring. After 2 hours, 0.666 mL of this solution was mixed with cyclohexane (7.5 mL), 1-hexanol (1.133 mL), Triton X-100 (1.77 mL; 2.93 mmol) and deionised (DI) water (0.48 mL) under vigorous stirring. Immediately after, TEOS (100 μL; 0.451 mol) was added, followed by ammonium hydroxide (40 μL; 28% (v/v)) 30 minutes later. This mixture was left to stir for 24 hours before subsequently adding TEOS (50 μL; 0.226 mol). After 30 minutes, THPMP (40 μL; 0.2103 mmol) was added and 5 minutes later APTMS (10 μL; 0.0573 mmol) before being left to stir for a further 24 hours. The microemulsion was then broken through the addition of ethanol (30 mL) before purifying the nanoparticles with ethanol (3 × 30 mL) by centrifugation (7800 rpm for 20 mins). The size and ζ-potential of the particles were determined by DLS. The synthesised nanoparticles were observed using TEM with an accelerating voltage of 80 kV and a beam amplitude of 15.4 μA.Degradation of FITC-doped silica nanoparticles
Nanoparticle solutions of concentration 1 mg mL−1 were placed in an Eppendorf tube and centrifuged (14000 rpm for 5 mins). The ethanol was removed and replaced with 1 mL of either PBS (pH = 7.4), BSA (pH = 6.3) or tyrosinase. 100 μL of the surfactant was removed and measured under fluorescence. The remaining solution was placed in an incubator at 37 °C taking fluorescence measurements of the supernatant at regular intervals.
Absorbance of anti-cancer drug-doped nanoparticles
The absorbance of the different drug-doped nanoparticles was observed using the Biowave II UV-Vis Spectrometer. The different nanoparticles, all at a concentration of 0.1 mg mL−1, were dispersed in ethanol before running absorbance scans from 200–900 nm. TEOS was also combined with ethanol and run as a control. Each drug alone was dissolved in ethanol at concentrations of 0.1–1 mg mL−1 and their absorbance determined to plot a calibration curve. The concentrations of anti-cancer drugs within the nanoparticles were calculated from the calibration curves displayed in Fig. S3–S6 (ESI†).Synthesis of microneedle arrays
The protocol for synthesising the microneedle arrays was from Loizidou et al.25 and was used to prepare plain microneedle arrays and nanoparticle-infused microneedle arrays. Briefly, microneedle arrays consisting of CMC (50 w/w%) and either TRD (50 w/w%), SUC (50 w/w%) or MAL (50 w/w%) were prepared by dissolving in DI water under stirring. For the nanoparticle-infused microneedle arrays, either FITC-NP or Dox-NPs were added at a concentration of 0.1 mg mL−1 during stirring. 200 μL of the gel solutions were then injected onto an inverted cone shaped silicone micromould under vacuum. The micromould array consisted of 324 needles of height 750 μm, base diameter 200 μm and centre-to-centre spacing of 600 μm. Once the sugar solutions had been applied, the vacuum was released and the micromoulds were left to dehydrate in a fumehood at room temperature for three days.Synthesis of microneedle arrays at various pressures
Microneedle arrays consisting of CMC (50 w/w%) and either TRD (50 w/w%), SUC (50 w/w%) or MAL (50 w/w%) were prepared by injecting 200 μL of the gel solutions (of CMC and sugars in DI water) onto an inverted cone shaped silicone micromould. They were then placed in a vacuum oven at 33 °C at various pressures of 100 mBar, 200 mBar, 300 mBar or 400 mBar to dehydrate. This was also carried out using gel solutions with nanoparticles-infused in them.Optical coherence tomography imaging of microneedle arrays
Microneedle arrays were imaged in situ using optical coherence tomography. The mouse skin was prepared by euthanising CD-1 Nude Mice Homozygous Nu/Nu Strain 086 at 6 weeks of age (purchased from Charles River) and removing their skin from the main body of the mice. The parameters of the ThorLabs imaging system are as follows: superluminescent diode source at 930 nm wavelength with a 100 nm tuning range; an axial resolution of 5.5 μm.The sample was prepared for imaging by cutting a 1 × 1 cm square piece of mouse skin and pressing the CMC-TRD microneedle array into the skin. The sample was then mounted in the sample holder and images taken.
Degradation of microneedle arrays with FITC-NP encapsulated in PBS and tyrosinase
Microneedle arrays synthesised from each sugar with FITC-NP encapsulated were placed into the wells of a 24-well plate. These were covered with PBS only or PBS with tyrosinase and incubated at 37 °C for 72 hours. 50 μL aliquots were taken at regular time intervals and the fluorescence intensity measured (λexc = 488 nm and λemm = 525 nm). Controls of FITC-NP in PBS; FITC-NP in PBS & tyrosinase; and tyrosinase in PBS were also studied.Degradation of microneedle arrays with FITC-NP encapsulated in PBS
Microneedle arrays synthesised from each sugar with FITC-NP encapsulated were placed into the wells of a 24-well plate. These were covered with PBS only and incubated at 37 °C for 4 hours. 50 μL aliquots were taken at half hour time intervals and the fluorescence intensity measured (λexc = 488 nm and λemm = 525 nm). Controls of FITC-NP and the sugar solutions before gelation were also studied.Calibration curves of FITC and FITC-NP
Various concentrations of FITC and FITC-NP were dissolved in PBS and ethanol (90% v/v:10% v/v) or PBS only. The fluorescence intensity for each concentration was measured at λexc = 488 nm and λemm = 525 nm. These intensities were plotted against the concentration to give calibration curves.
FITC release from FITC-NPs
FITC-NP (100 μL; 0.1 mg mL−1) were stirred with 10 μL of 10% sodium carbonate solution (in DI water) for one hour at 1200 rpm. The pH was then adjusted back to pH 7.4 with hydrochloric acid, before measuring the fluorescence intensity (λexc = 488 nm and λemm = 525 nm) of the released FITC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge and thank Medway School of Pharmacy, Universities of Kent and Greenwich for providing funding for this project and Middlesex University for use of their facilities and materials. We would also like to acknowledge Mr Ian Brown at School of Biosciences at the University of Kent for imaging and training on the TEM; and Mr Andrew Hurt & Dr Hiezl Zoltan from the Faculty of Engineering and Science at the University of Greenwich for training on the SEM. We would also like to thank Professor Stephen Dilworth at the Department of Natural Sciences at Middlesex University for training on the confocal microscope; and Dr Leonardo P. Munoz from the Department of Natural Sciences at Middlesex University for allowing us to use the OCT imaging system and training on this system.References
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435–451 CrossRef CAS.
- T. Daniels-Race, Nanolithography, Elsevier, 2014, pp. 399–423 Search PubMed.
- TWI Ltd, What are Nanoparticles? Definition, size, uses and properties, https://www.twi-global.com/technical-knowledge/faqs/what-are-nanoparticles, (accessed 16 November 2020).
- D. Boraschi, Nanoparticles and the Immune System, Elsevier, 2014, pp. 1–7 Search PubMed.
- S. Gelperina, K. Kisich, M. D. Iseman and L. Heifets, Am. J. Respir. Crit. Care Med., 2005, 172, 1487–1490 CrossRef PubMed.
- I. Slowing, J. Viveroescoto, C. Wu and V. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, B. G. Trewyn and V. S. Y. Lin, Small, 2010, 6, 1952–1967 CrossRef CAS PubMed.
- T.-H. Chung, S.-H. Wu, M. Yao, C.-W. Lu, Y.-S. Lin, Y. Hung, C.-Y. Mou, Y.-C. Chen and D.-M. Huang, Biomaterials, 2007, 28, 2959–2966 CrossRef CAS PubMed.
- J. Lu, M. Liong, Z. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS PubMed.
- S. A. A. Rizvi and A. M. Saleh, Saudi Pharm. J., 2018, 26, 64–70 CrossRef PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- M. L. Hans and A. M. Lowman, Curr. Opin. Solid State Mater. Sci., 2002, 6, 319–327 CrossRef CAS.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- J.-F. Chen, H.-M. Ding, J.-X. Wang and L. Shao, Biomaterials, 2004, 25, 723–727 CrossRef CAS PubMed.
- G. Giovannini, F. Kunc, C. C. Piras, O. Stranik, A. A. Edwards, A. J. Hall and V. Gubala, RSC Adv., 2017, 7, 19924–19933 RSC.
- M. Ogundele and H. Okafor, J. Pharm. Res. Int, 2017, 18, 1–14 CrossRef.
- Y. Hao, W. Li, X. Zhou, F. Yang and Z. Qian, J. Biomed. Nanotechnol., 2017, 13, 1581–1597 CrossRef CAS PubMed.
- D. Liu, Y. Zhang, G. Jiang, W. Yu, B. Xu and J. Zhu, ACS Biomater. Sci. Eng., 2018, 4, 1687–1695 CAS.
- T. Liu, G. Jiang, G. Song, J. Zhu and Y. Yang, Biomed. Microdevices, 2020, 22, 12 CrossRef PubMed.
- G. Song, G. Jiang, T. Liu, X. Zhang, Z. Zeng, R. Wang, P. Li and Y. Yang, ACS Biomater. Sci. Eng., 2020, 6, 4116–4125 CrossRef CAS PubMed.
- B. Xu, Q. Cao, Y. Zhang, W. Yu, J. Zhu, D. Liu and G. Jiang, ACS Biomater. Sci. Eng., 2018, 4, 2473–2483 CrossRef CAS PubMed.
- Y. Zhang, G. Jiang, W. Yu, D. Liu and B. Xu, Mater. Sci. Eng., C, 2018, 85, 18–26 CrossRef CAS PubMed.
- Y. Zhang, D. Wang, M. Gao, B. Xu, J. Zhu, W. Yu, D. Liu and G. Jiang, ACS Biomater. Sci. Eng., 2018, 4, 2879–2888 CrossRef CAS PubMed.
- G. Aguilera, C. C. Berry, R. M. West, E. Gonzalez-Monterrubio, A. Angulo-Molina, Ó. Arias-Carrión and M. Á. Méndez-Rojas, Nanoscale Adv., 2019, 1, 671–685 RSC.
- E. Z. Loizidou, N. A. Williams, D. A. Barrow, M. J. Eaton, J. McCrory, S. L. Evans and C. J. Allender, Eur. J. Pharm. Biopharm., 2015, 89, 224–231 CrossRef CAS PubMed.
- C. J. Moore, H. Montón, R. O’Kennedy, D. E. Williams, C. Nogués, C. Crean and V. Gubala, J. Mater. Chem. B, 2015, 3, 2043–2055 RSC.
- C. J. Moore, G. Giovannini, F. Kunc, A. J. Hall and V. Gubala, J. Mater. Chem. B, 2017, 5, 5564–5572 RSC.
- D. R. Liston and M. Davis, Clin. Cancer Res., 2017, 23, 3489–3498 CrossRef CAS PubMed.
- M.-Y. Hua, H.-W. Yang, C.-K. Chuang, R.-Y. Tsai, W.-J. Chen, K.-L. Chuang, Y.-H. Chang, H.-C. Chuang and S.-T. Pang, Biomaterials, 2010, 31, 7355–7363 CrossRef CAS PubMed.
- A. D. McNaught and A. Wilkinson, The IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2019 Search PubMed.
- F. Yin, B. Zhang, S. Zeng, G. Lin, J. Tian, C. Yang, K. Wang, G. Xu and K.-T. Yong, J. Mater. Chem. B, 2015, 3, 6081–6093 RSC.
- E. Z. Loizidou, N. A. Williams, D. A. Barrow, M. J. Eaton, J. McCrory, S. L. Evans and C. J. Allender, Eur. J. Pharm. Biopharm., 2015, 89, 224–231 CrossRef CAS PubMed.
- E. Neeley, G. Fritch, A. Fuller, J. Wolfe, J. Wright and W. Flurkey, Int. J. Mol. Sci., 2009, 10, 3811–3823 CrossRef CAS PubMed.
- S. Zolghadri, A. Bahrami, M. T. Hassan Khan, J. Munoz-Munoz, F. Garcia-Molina, F. Garcia-Canovas and A. A. Saboury, J. Enzyme Inhib. Med. Chem., 2019, 34, 279–309 CrossRef CAS PubMed.
- K. Iozumi, G. E. Hoganson, R. Pennella, M. A. Everett and B. B. Fuller, J. Invest. Dermatol., 1993, 100, 806–811 CrossRef CAS PubMed.
- S. Santra, K. Wang, R. Tapec and W. Tan, J. Biomed. Opt., 2001, 6, 160 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00135c |
| This journal is © The Royal Society of Chemistry 2021 |