
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface oxidation for enhancing the hydrogen evolution reaction of metal nitrides: a theoretical study on vanadium nitride†
Samira
Adimi
ab,
Weiliang
Qi
 b,
Tiju
Thomas
c,
Ralph
Gebauer
*d,
Minghui
Yang
*b and
Shengping
Ruan
*a
b,
Tiju
Thomas
c,
Ralph
Gebauer
*d,
Minghui
Yang
*b and
Shengping
Ruan
*a
aState Key Laboratory on Integrated Optoelectronics and College of Electronic Science & Engineering, Jilin University, Changchun 130012, P. R. China. E-mail: ruansp@jlu.edu.cn
bSolid State functional Materials Research Laboratory, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, 1219 Zhongguan West Road, Ningbo, 315201, China. E-mail: myang@nimte.ac.cn
cDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology Madras, Adyar, Chennai 600036, Tamil Nadu, India
dThe Abdus Salam International Centre for Theoretical Physics (ICTP), Strada Costiera 11, 34151, Trieste, Italy. E-mail: rgebauer@ictp.it
First published on 1st April 2021
Abstract
The rational design of novel catalysts for energy applications has been an area of immense interest over the past decades. Recent studies have demonstrated that slight surface oxidation of nitrides can boost their catalytic activity, especially towards hydrogen adsorption. In this study, we have considered VN as a simple model nitride to simulate H adsorption on pure and oxygen polluted surfaces. First, we have evaluated the physical characteristics and stability of various facets. Diversity in the VN catalytic activity can originate from a variety of surface characteristics. Our calculations reveal the (112) and (200) surfaces as the most stable ones, while the (101), (111), and (113) surfaces are roughly in the same range of energy. The facet-dependent activity of VN toward H adsorption is then carefully discussed. A low level of oxygen contamination appears to make the VN hydrogen adsorption robust and beyond that, a thin oxide surface layer can act as an activation layer, playing a positive role in improving the catalytic performance. We anticipate this picture to be an important input for designing enhanced nitride-based catalysts with controlled oxidation of surfaces.
1. Introduction
For energy-related challenges, many material-oriented activities are in progress. For instance, there is an urgent need for developing highly active catalytic materials for processes involved in energy conversion and storage. There is an ever-increasing search for advanced rechargeable batteries with increased energy density, lower cost, long cycle life, and improved efficiency. Likewise, it is critical to develop H2 adsorbents to meet the high volumetric/gravimetric density requirements for storing hydrogen while simultaneously maintaining thermodynamic-reversibility of the adsorption, with fast kinetics.1 Water splitting and CO2 reduction with low overpotentials and fast kinetics are significant areas of investigation as well.2 Likewise, there are several efforts towards developing materials for supercapacitors to complement and improve other energy storage systems.3In energy-related materials research, transition metal nitrides have attracted considerable attention in recent years due to their potential as catalysts,4–6 hydrogen absorbents,7 and super capacitance electrodes.8 Nitrides are very attractive catalysts for some reactions, including water splitting and NH3 synthesis, because their catalytic properties are comparable to those of Pt-based metals.9
In recent years vanadium nitride (VN) has emerged as an interesting catalyst for many energy-related systems. Vanadium nitride nanowires have been used to make gas diffusion cathodes of Li–CO2 batteries.10 VN shows promise as an electrocatalyst for both oxygen (OER) and hydrogen (HER) evolution reactions in water splitting, owing to its high availability, good electrical conductivity, and corrosion resistance.11 Besides, there are several reports on hydrogen storage using VN-based nanocrystals. The reports indicate that the VN structure and its particle size are the most important factor for improving the gravimetric, thermodynamic, and kinetic properties associated with hydrogen storage.7 Interestingly VN shows remarkable characteristics like high specific capacitance and a large potential energy window, along with benefits such as a good electric conductivity and good thermal stability relevant for use as electrodes in capacitors.12,13 However, the catalytic activity, stability, and adsorptive characteristics of VN are affected by the stoichiometry and crystal structure near the surface.12,14,15 The HER, OER, and gas diffusion on cathodes are structure-sensitive reactions.16 The effect of particle size on the efficiency of hydrogen adsorbing media is inevitable.17,18 Most redox reactions take place on or near the electrode surface of the supercapacitors.19 And finally, in all these systems, the adsorption of hydrogen on the surface would be one of the main leading reactions. Therefore, a molecular-level understanding of the surface and its hydrogenation reaction remains a highly important task for the development of the aforementioned systems.
The presence of oxygen is unavoidable in nitride structures. According to some reports, oxygen incorporation with nitrides could be a deactivation factor, affecting their catalytic performance.20 Interestingly, numerous reports have also shown the positive role that oxygen could play. A controlled oxide layer on top of the vanadium nitride surface leads to a superior capacity in electrochemical capacitor application of VN, without any loss in the overall electrical conductivity.19 Furthermore, our previous work shows that the excellent OER activity of the nickel molybdenum nitride could have originated from a surface oxide-rich activation layer (SOAL).21 Recently, it has been reported that VN capacitance could be even more increased by the presence of a single form of vanadium oxides, especially VO2, on the surface.15
Experimental analysis of the surfaces is challenging and measuring the interaction strength between hydrogen and catalyst surfaces is difficult. Significant developments have been made in the use of density functional theory (DFT) for accurate simulation of the surface catalytic reactions.22 DFT offers a means to investigate the surfaces, simulate reactions, and determine the magnitude of the interactions among the reaction intermediates and the catalyst surface. The other well-known descriptors of catalytic performance, also accessible by DFT methods, are the d-band center and the geometric structure of catalyst surfaces. The d-band model is based on the electronic structure of the material and is defined as the average energy of d states when projecting the molecular orbitals onto the metal atom's d-state at the surface.23,24 An effective strategy for enhancing the catalytic activity of a surface toward the HER is tailoring its d-band center, adjusting the free energy of H adsorption (ΔGH*) close to the optimal value for the HER (i.e., zero free energy of adsorption).25 Manipulating the d-band center position has been successfully applied for improving OER and HER performance of metal nitrides previously.26 Achieving further development in the catalytic performance of nitrides could be aided through computational insights.
To the best of our knowledge, there is no prior study on VN surface structures despite the interest in VN for this wide range of catalytic applications. Investigations on the hydrogen adsorption of VN and the effects of oxidation on it are also very limited, especially from a theoretical perspective.15 The current study aims to provide a detailed and comprehensive DFT investigation of VN surfaces, hydrogen adsorption on pure and oxidized surfaces, along with offering in-depth information about the catalytic activity of this nitride.
In this study, we first consider the stability of VN facets with both low and high Miller indices by calculating the surface energies and investigating the surface physical properties. Then the H adsorption is studied on clean facets and also the surfaces with different rates of oxygen contamination. Finally, a single layer of oxygen is assumed on top of the VN surfaces and various coverages of H will be discussed below.
2. Computational method
We have used density functional theory calculations in the generalized gradient approximation,27 as implemented in the Quantum Espresso package.28 The projector augmented wave (PAW)29 and optimized norm-conserving Vanderbilt (ONCVPSP)30 pseudopotentials are utilized to describe the electron–ion interactions. The minimum energy cutoff is 45 Ry for the wavefunctions and 400 Ry for the charge density. A 6 × 6 × 1 Monkhorst–Pack k-point mesh was used for sampling the Brillouin zone.VN in the bulk structure has a halite, rock salt crystal structure in the cubic Fm-3m space group. The calculated lattice parameter is a = 4.28 Å for a fully relaxed structure. Each V atom has six equivalent bonds with the adjacent N atoms, where the bond length is 2.06 Å.
We have conducted DFT calculations for eleven cleavages on eight VN facets to evaluate the stability of the crystal facets. The surface calculations are accomplished by a slab model, while the primary unrelaxed surfaces are set up using the relaxed bulk structure. The basis of the bulk unit cell and the atomic positions are transformed such that the interesting Miller index is oriented parallel to the (001) plane.31 The slabs should be thick enough for convergence calculations, so we have considered the twelve-layer-thickness slabs. A vacuum of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Å along the c direction is sufficient for separating the periodic images. The lattice parameters of the slabs are fixed at the VN bulk value, while the atoms on the upper half of the slabs are allowed to relax until the residual force on each atom is smaller than 0.001 eV Å−1. The lower half of the slabs is frozen to simulate the bulk structure. Since we have kept atoms fixed on one side, the slab has two inequivalent surfaces. Therefore, the unrelaxed surface energy, which is the energy of the surface before any surface optimization, is also considered for finding the final surface energy of the relaxed surface.32 The surface energy (γ) is calculated using the following formula,
Å along the c direction is sufficient for separating the periodic images. The lattice parameters of the slabs are fixed at the VN bulk value, while the atoms on the upper half of the slabs are allowed to relax until the residual force on each atom is smaller than 0.001 eV Å−1. The lower half of the slabs is frozen to simulate the bulk structure. Since we have kept atoms fixed on one side, the slab has two inequivalent surfaces. Therefore, the unrelaxed surface energy, which is the energy of the surface before any surface optimization, is also considered for finding the final surface energy of the relaxed surface.32 The surface energy (γ) is calculated using the following formula,
 | (1) |
For simulating the H adsorption of the surfaces, we have adopted a procedure similar to the HER simulations.33 The HER occurs in two steps, as represented in eqn (2) and (3):
| H+ + e− + * ↔ H* | (2) |
| H* + H* ↔ H2, or H* + H+ + e− ↔ H2 | (3) |
The Gibbs free energy change is calculated as
| ΔG = ΔE − TΔS + ΔEZPE, | (4) |
 | (5) |
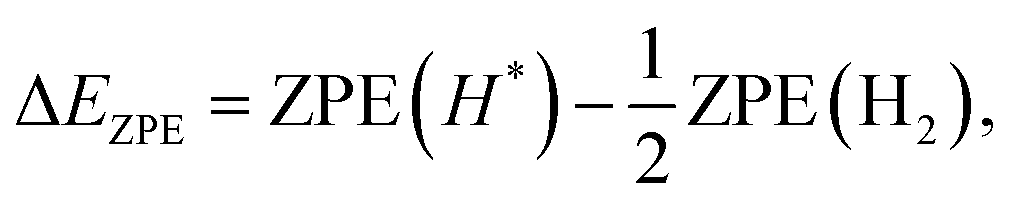 | (6) |
 | (7) |
 equal to −0.204 eV at room temperature.33 If ΔGH* has a negative value (eqn (4)), H can be easily adsorbed on the surface, while its desorption would be difficult. Therefore, the ideal HER electrocatalytic activity will belong to the material whose ΔGH* value is zero; while the energy range between −0.2 and 0.2 eV is normally considered as the criteria for showing active HER catalysts for a sample.
equal to −0.204 eV at room temperature.33 If ΔGH* has a negative value (eqn (4)), H can be easily adsorbed on the surface, while its desorption would be difficult. Therefore, the ideal HER electrocatalytic activity will belong to the material whose ΔGH* value is zero; while the energy range between −0.2 and 0.2 eV is normally considered as the criteria for showing active HER catalysts for a sample.
Oxygen doping is considered by replacing a nitrogen atom with oxygen for simulating a minor O-doping, and two out of the eight N atoms of the slab's uppermost level for simulating an oxynitride surface with 12.5% oxygen on top. Then, an oxide layer is put on the VN surface and all N atoms are replaced with O atoms. This results in a VO/VN configuration. The presence of impurities leads to a surface non-uniformity, which increases the diversity of possible hydrogen adsorption sites from a theoretical point of view. Lastly, the coverage of hydrogen atoms on the surface was raised from 1/8ML to 1/4ML and 3/8ML on pure and VO/VN surfaces.
3. Results and discussion
Surface investigation
Different synthesis methods would result in different multi-faceted samples. Finding the reactive facets and explaining their role in promoting the catalytic activity of a material at a molecular level are highly valuable. This information not only provides a guideline for uncovering the catalytic mechanisms behind, but also can lead to the design of new catalysts.The most basic step is evaluating surface stability. Eight low and high-index VN surfaces have been selected from both types of flat and step/edge facets. We have considered four previously synthesized VN surfaces, i.e. (200), (111), (220), and (311),7,15 while (001), (112), (113), and (101) surfaces will be discussed as a predictive model. Fig. S1 (ESI†) shows the lateral view of all considered slabs, before and after the structural optimization, and Table S1 (ESI†) reports the surface energies. Lower surface energy indicates a more stable surface. The stability sequence is (200)> (112) > (101), followed by (111), (311), and (113) in close stability with each other. The (220) and (001) facets have the highest surface energies and accordingly are the most unstable considered surfaces. A brief discussion on the physical properties of the surfaces is given below.
VN (001) and (220) are flat facets with rather smooth structures. A 4-fold atomic arrangement exposes on top of (001), similar to all fcc structures. Both types of V and N atoms are present on the surfaces, exposed to vacuum. After the surface optimization, there would be a slight decrease in some V–N bonding lengths, which would be compensated by increasing the length of adjacent bonds. In general, the changes due to the surface effects are negligible on both facets and as was mentioned above, these facets have the highest surface energy among all considered facets.
The (311) surface is observed on the VN sample with an oxide surface.15 Due to the geometric similarity of (113) with (311) surfaces, the geometric structure and stability of (113) are also examined in this study. At the atomic scale, two different cleavages can be considered on top of these step facets: the slabs with a V-rich layer on the topsides (referred to as (113)-I and (311)-I in Fig. S1(e) and (f), ESI†), and the slabs terminated by a layer of N atoms ((113)-II and (311)-II in Fig. S1(c) and (d), ESI†). These surfaces are good models for investigating the stability of different surface terminations, i.e., nitrogen versus vanadium. The energy considerations (Table S1, ESI†) show that V-terminated cleavages are more stable on both (113) and (311) with almost the same surface energies. To reduce the effect of surface dangling bonds, the topmost V atoms move toward the bulk level, ending to relatively smooth facets.
The VN (111) crystal plane is also experimentally observed during the HER16,35 and OER11,16 studies. Besides, VN (111) has been considered as a substrate nanosheet for improving the OER performance, providing very desirable results.11,36,37 In all fcc crystals, the (111) layers are close-packed. On VN (111), consisting of two interpenetrating fcc structures, all surface atoms have a relatively high coordination number. In accordance with (113) and (311), there would be two different possibilities on top of (111), terminated either by a V (noted by VN (111)-I) or an N layer (VN (111)-II). The geometric structures and the surface energies are reported in Fig. S1(g), (h) and Table S1 (ESI†), respectively. After the geometric relaxation, atoms in the direct vicinity of the vacuum move downward, toward the bulk layers. This leads to the shrinkage of V–N bond lengths of up to ∼9%, to neutralize the unphysical forces of the surface. The stability of this surface is close to the ones of (113) and (311). However, there is a small energy difference between the (111)-I and (111)-II cleavages, so there is a possibility for the pure nitrogen surface to be the uppermost layer. In the following, we will show that N sites are not catalytically active sites toward hydrogen, which can be a major drawback for the catalytic activity of the (111) surface.
The VN (112) surface is a completely open surface. Different colors are used in Fig. S2(a) (ESI†) to display the different heights of atoms relative to the vacuum (Z-axis). The atoms on the second and third layers are also visible through the holes of the first layer, although these atoms would not be directly accessible for an external gas molecule. Both N and V are in the vicinity of the vacuum and the difference in the height of atoms in the vicinity of the vacuum is up to 4Å before the surface relaxation. Significant changes occur on the surface due to the surface effects, so that the height and bonding lengths of the uppermost atoms are substantially decreased. This is the most energetically favorable surface, which will be discussed more in the next sections of the paper.
(200) is the second most stable surface according to our calculations (Fig. S1 and Table S1, ESI†), which is in agreement with the experimental reports, synthesizing the uniform VN (200) facets.15,36 Surface effects cause the atoms of the first layer to move downward. This increases the dependency of the V atoms to the lattice, while giving more freedom to the topmost N atoms, in a way that the final relaxed surface is slightly rippled (Fig. S1(k), ESI†). The surface reactivity can be strongly dependent on the activity of the terminating layer.
In the following, we will confine our further studies to the VN (112) and (200) surfaces, which have the lowest surface energies. This selection is not only based on these energy considerations, but also on the significant geometric changes at the surfaces, the existence of both types of N and V atoms at the surfaces, and other reasons which will be discussed below.
Gibbs free energy and H adsorption profile on clean VN surfaces
A rational design of novel catalyst materials for energy applications is essential. However, this work is challenging because of the complexity of the mechanisms involved. A large number of experimental and theoretical guidelines have already been established to find optimal catalysts.24 One of the most successful theories is known as the Sabatier principle,38 which suggests that the binding strength between a catalyst and adsorbates is the leading descriptor of the catalytic activity. In this framework, a moderate interaction leads to the best performance, while too weak or too strong interactions would limit the adsorption or desorption rate of the catalyst surface, respectively.Both VN (112) and (200) surfaces offer a variety of possible sites for adsorbing gas molecules. These sites can be considered in two general categories: (i) on-top sites, where H atoms are initially considered on top of the metal or nitrogen atoms. (ii) The hollow-sites, where H is considered above the vacant space among four atoms. We are not aware of any specific report showing the catalytic activity of VN as a function of adsorption sites.
We have considered two dissimilar positions of nitrogen for H adsorption (with two and three dangling bonds), one position on top of V, and one bridge site on the VN (112) surface. Fig. S2 (ESI†) shows the schematic view of the surface, with the H* adsorption sites marked as site L1–4. The L2 and L3 sites, locally shown in Fig. S2(b) (ESI†), are the most active sites according to the Gibbs free energy results (Fig. S2(f), ESI†). Here, the H atoms are absorbed by the V atom with a moderate V–H bond length. On the other hand, the H atom on the N site (denoted as L1) is strongly adsorbed and short H–N bonding, with an approximate length of 1.02 Å, is created on top of the surface (Fig. S2(a), ESI†). As expected, the most negative free energy, and accordingly the lowest H evolution activity, is related to this site (Fig. S2(f), ESI†).
Similarly, on the (200) surface there are three main possibilities for H adsorption, denoted as A1, A2, and A3, referring to H atoms on top of the V, N, and hollow sites, respectively (Fig. S3, ESI†). The bridge site (A3) is the most stable adsorption site, which is followed closely by A1. Their related free energies are clearly lower than the corresponding sites on the (112) surface. On the other hand, short strong H–N bonding on A2 indicates that the site won’t be active toward the HER. Both A1 and A3 sites have reached a similar final structure after surface rearrangements (Fig. S3(a) and(c), ESI†). The distance of the hydrogen-bonded V atom is considerably increased with respect to the other V atoms and also the neighboring N atoms. The lengths of H–V bonds are around ∼1.67Å on the most active sites of the clean (200) surface.
Therefore, the best adsorption sites of the clean surfaces are the topmost vanadium atoms. However, the V–H bonds are relatively strong and the corresponding free energies toward HER activity are quite negative (−0.52 eV for A3 and −0.55eV for L2 according to our results). In the following, we examine whether oxygen impurities can yield a positive effect on the catalytic performance of the surfaces.
Oxygen contamination of the VN surface
The H adsorption of VN is studied while the surfaces contain different amounts of oxygen impurities. Four contamination ratios have been studied, where 12.5%, 25%, 50%, and 100% of the N atoms on the topmost layer have been replaced by oxygen. Fig. 1, 2 and Fig. S4, S5 (ESI†), and Table 1 show the corresponding relaxed structures and H adsorption free energies.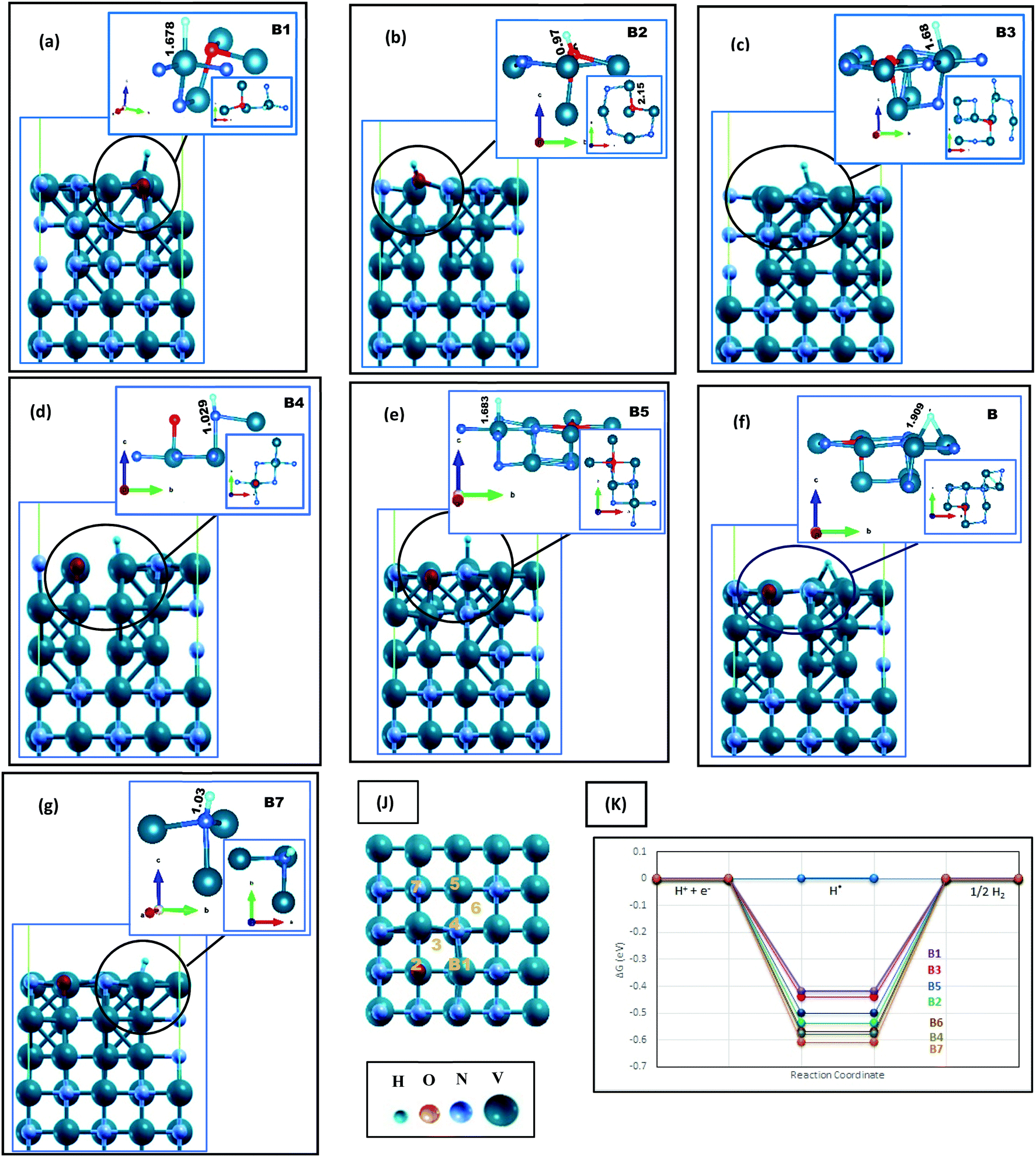 | ||
| Fig. 1 Hydrogen adsorption on O-doped VN (200) in lateral and local configurations, where H* is initially located on (a) top of V in direct vicinity of oxygen (b) top of oxygen, (c) hollow site near the oxygen, (d) top of N in direct vicinity of O, and (e–g) top of V, N and hollow space in the indirect neighboring of O, respectively. The H bonding lengths are written in Å unit on each figure. The H* adsorption sites are marked as B1–B7 represented in (k), and (J) denotes a schematic comparison between the free energies reported in Table 1. In all figures, the surface index is put in parallel to the c axis. V, N, O and H atoms are represented by aquamarine, grey, red and blue color balls, respectively. | ||
 | ||
| Fig. 2 The VO/VN configuration after H adsorption, where all the N atoms on the topmost layer are replaced with O atoms. (a–g) The local structure of adsorbed hydrogen on top of the surface, while D1–D7 sites are marked on (h) by orange color. The top and lateral views of (a–e) are shown in Fig. S6 (ESI†). The bonding lengths are reported in Å unit. (k) A comparison between the corresponding H adsorption energies reported in Table 1. | ||
| H* adsorption site | A1 | A2 | A3 |
|---|---|---|---|
| ΔGH* (eV) | −0.55 | −0.58 | −0.52 |
| H* adsorption site | B1 | B2 | B3 | B4 | B5 | B6 | B7 |
|---|---|---|---|---|---|---|---|
| ΔGH* (eV) | −0.42 | −0.54 | −0.44 | −0.58 | −0.5 | −0.56 | −0.61 |
| H* adsorption site | C1 | C2 | C3 | C4 | C5 | C6 | C7 |
|---|---|---|---|---|---|---|---|
| ΔGH*(eV) | −0.47 | −0.42 | −0.58 | −0.38 | −0.55 | −0.61 | −0.64 |
| H* adsorption site | D1 | D2 | D3 | D4 | D5 | D6 | D7 |
|---|---|---|---|---|---|---|---|
| ΔGH* (eV) | −0.32 | −0.36 | −0.38 | −0.48 | −0.54 | −0.51 | −0.56 |
| H* adsorption site | L1 | L2 | L3 | L4 |
|---|---|---|---|---|
| ΔGH* (eV) | −0.64 | −0.55 | −0.57 | −0.6 |
| H* adsorption site | M1 | M2 | M3 | M4 | M5 | M6 | M7 |
|---|---|---|---|---|---|---|---|
| ΔGH* (eV) | −0.61 | −0.63 | −0.65 | −0.6 | −0.49 | −0.52 | −0.55 |
We will first discuss the O-doped VN (112) surface. Slight surface contamination results in marginal structural changes, and the surface stability is preserved. We have considered different H adsorption sites, which can be categorized as the sites similar to that of the pure (112) surface, the sites where H are placed on-top of O, on-top of the V atom in the direct vicinity of O, or the bridge site near oxygen (denoted as M1–M7 in Fig. S4(a), ESI†). Interestingly, all three latter situations (i.e., M5, M6, and M7) lead to the same final surface configuration (Fig. S4(c), ESI†). The adsorbed H atom forms two bonds with lengths of 1.93–1.95 Å with the V atom, while it is placed 3.15 Å away from oxygen. As expected, the Gibbs free energies of M5, M6, and M7 sites are energetically very close (Fig. S4(d), ESI† and Table 1). The most active site of the O doped VN (112) surface, with a free energy amount of −0.49eV, is the V atom in the direct vicinity of oxygen.
On top of the (200) surface, each nitrogen atom has five bonds, four with the topmost V atoms and one with the V atom of the following level. On O-doped (200) surfaces, shown in Fig. 1, after replacing O with one N atom, all adjacent V atoms move away, increasing their distances up to 2.4 Å with the dopant. Besides, V atoms move toward the vacuum space, while oxygen moves clearly toward the bulk material. Finally, the only clear bonding of the oxygen (with a length of 2.01Å) is the bond with the V atom in the next layer. Thereupon, we encounter a surface with a wide variety of hydrogen uptake sites, where some possibilities are shown in Fig. 1(j) by B1–6. The local configurations around H adsorption sites are presented in Fig. 1(a)–(g). On four sites, i.e., B1, B3, B5, and B6, adsorbed hydrogen atoms are connected to vanadium. The V atom in the immediate neighborhood of O is the most active site due to the energy results reported in Table 1 and Fig. 1(k). In comparison with the clean surface, the Gibbs free energies are more favorable for the HER. Moreover, on all the aforementioned sites, the V–H bond lengths are slightly longer than the clean surface. On the other hand, H is strongly absorbed by the oxygen atom on the B2 site (Fig. 1(b)), constructing an OH unit with a length of 0.978 Å. Later, we will discuss this unit more. Both H–N bonds (Fig. 1(d) and (g)) are also short and strong, therefore, an oxygen impurity does not contribute to the better catalytic activity of these sites.
By increasing the surface contamination rate, we find that V atoms in the direct vicinity of O atoms still would be the most active sites of the surface. In Fig. S5 (ESI†), 25% of the N atoms on the outermost layer are replaced with oxygen. Not only the H adsorption energies of C1, C2, C4, and C5 are less than that of the clean (200) surface, but also the bonding length between V and H is significantly increased (Fig S6(b)–(h), ESI†). Interestingly, there is no OH unit formed on the C1 site. However, despite the longer H–V bond lengths on this site, its related free energy (∼0.47 eV) is more negative in comparison with C2 and C4 sites, with ΔGH* around −0.42eV and 0.47eV, respectively. This is due to the existence of two H–V bonds at this site, which will increase the dependency of H on the surface and thereby reduce the catalytic activity.
The efficiency of a catalyst can be better understood in terms of its electrical properties. According to the density of state (DOS) plots for the pure VN surfaces (Fig. S7(a) and S8(a), ESI†), the interaction between V and N is expectedly strong. The transferred charge between vanadium and nitrogen results in the DOS being similar to that of the Pt-group metals near the Fermi level. This similarity shows why VN has been considered as a potential candidate for catalytic activity toward evolution reactions. In doped structures, oxygen will influenceon the vanadium and nitrogen electronic states (Fig. S7(b), (c) and (b), (d), ESI†). The main peak associated with oxygen is in the energy range between −9 and −4 eV, where the creation of V–O and also N–O bonds is clear. By increasing the rate of oxygen impurities, this peak shifts toward the Fermi energy. The other important O-p DOS contribution is around the Fermi level, with −4 to 5 eV energy. The V-d and O-p orbitals are degenerate in this area, resulting in both bonding and antibonding states. However, the O-p contribution is so less than that of V-d states, so the d-character remains near the Fermi level. By increasing the rate of surface oxidation, the total density of states is even slightly increased near the Fermi level (Fig. 3(c), which promises a better catalytic activity.
 | ||
| Fig. 3 Density of state plots for (a) pure VN (200), and (b) VO/VN surfaces, where the insets represent the partial DOS plots. The Fermi level is put on zero. A comparison between the total DOSs near the Fermi level is represented in (c), and (d) denotes a comparison between the Gibbs Free energies related to the best catalytic performance of surfaces. | ||
Tailoring the d-band centers is another good way for qualifying nitrides as highly active catalysts for the HER.26 The d-band center is a physical parameter, strongly correlated with the adsorption energy of the adsorbate.33 By increasing the oxygen impurity rate, the VN d-band center shifts toward the Fermi level according to Table S2 (ESI†). The higher value of the d-band energy represents stronger adsorption.25 Thus, this shift implies a stronger H adsorption onto the VN surface and hereby the lower value of the free energy (ΔGH*).
In summary, the surface energies of VN (112) and (200) facets are in the same range, but both clean and O-doped VN (200) surfaces show a better catalytic activity toward the HER according to our results. This result could be so helpful in practice, providing a direction for laboratory synthesis. Besides, oxygen contaminations of the facets cause the enhancement of the catalytic activity of the polluted surfaces. The active sites are V atoms in the direct neighbor of oxygen. This easy surface engineering would result in modulation of the d-band center and could be a good strategy to make the hydrogen evolution robust. The aforementioned reasons persuade us to expand the oxygen coverage, placing one oxide layer on the VN (200) surface, and investigate the catalytic activity of this framework.
VO/VN configuration and catalytic activity on different H coverages
Various vanadium oxides have also been reported to be favorable for the HER, but their low inherent conductivity and insufficient cycling performance are the main issues for practical applications.39–41 By considering VO/VN configurations, the good electrical conductivity of the VN in combination with the vast variety of active hydrogenation states could boost the nitride's catalytic activity.19 Besides, we will benefit from a protective oxide layer for enhancing the stability of the material.After replacing all uppermost N atoms with oxygen, we obtain a single vanadium oxide layer on the VN (200) surface. DFT structural relaxation directs to V–O bonds with average lengths of 2.08 Å on the VO/VN surface. The possible H* adsorption sites (D1 to D7) and their lateral view are shown in Fig. 2(h). The oxygen atoms are in three non-uniform surface positions, which are marked as D1, D3, and D5 in Fig. 2. The D1 site has the lowest position among all other topmost atoms. It moves away from adjacent atoms after structural relaxations and the only remaining concrete bond is with a V atom on the following layer. The free energy values are reported in Table 1 and a schematic comparison between the best adsorption sites is shown in Fig. 3(d). The results clearly demonstrate the reduction of energy amount after oxygen doping of the surfaces, especially when there is a thin layer of oxide on top of the surface. The energy values for the VO/VN surface follow this order: D1 < D2 ≈ D3 < D4 < D6 < D5 ≈ D7, where the best energy values are −0.32 eV for D1, and −0.36 eV and −0.38 eV for D2 and D3, respectively. The value of ΔGH* for these sites shows a considerable reduction (in terms of energy amount) compared to the oxygen-free surfaces (with a value of −0.52 eV for A3 and −0.55 eV for L2 on (200) and (112) surfaces, respectively). In contrast, the difference between the ΔGH* of D4, D5, D6, and D7 sites with that of the clean (200) surface is vital (Table 1), but the V–H bonds on these sites are stretched out up to 13% (Fig. 2(b)–(d)), which shows that the sites could also be more active in comparison with the clean surface.
More interestingly, the H atom is easily adsorbed by adjacent V atoms of D1 and D5 sites (Fig. 2(a) and (c)), while the H atom on top of the D3 site is absorbed by oxygen and forms an OH unit. Oxygen in D3 has longer V–O bonds and less dependency on the surface, which could easily bound to H and freely desorbed from the surface. The metal–OH bond strength is usually a theoretical descriptor for the OER, meaning that a balanced bond strength between the metal atom and OH unit leads to optimal activity.42,43 Given that the OH unit has left the surface freely (Fig. 2(g)) and there is no strong bonding with the V atom, it does not seem like an active OER site. Although we do not calculate the binding energies of OH* on the surface and do not intend to simulate the OER, in the following paragraph we have demonstrated that higher H coverage will result in the creation of an OH unit with the appropriate dependency to the surface on site D5, which may show good OER activity of the VO/VN surface.
Finally, we have increased the coverage of hydrogen from 1/8ML to 1/4ML and 3/8ML on both VN (200) and VO/VN surfaces. We have considered different adsorbing sites of the second hydrogen on the clean surface as shown in Fig. S9(a) (ESI†). We have supposed that the first H atom is adsorbed by one of the top-most V atoms due to our previous discussions. The second H will be adsorbed by another V atom (Fig. S9(b) and (d), ESI†), or by an N atom (Fig. S9(c) and (e), ESI†), or forms a hydrogen molecule and leaves the surface (Fig. S9(f), ESI†). As this process continues and the surface hydrogen coverage increases, the V–H bonds are easily broken and the hydrogen molecules freely desorb from these sites (Fig. S9(g) and (h), ESI†). However, the N–H bonds are short and strong, and the bond breakage occurs only in high H coverage. Therefore, it is unlikely to have an H occupied surface due to the high activity of the V sites, although the strong N–H bonding will decrease the surface HER activity.
On the VO/VN surface, the second H is also adsorbed freely by another V atom, and two equal V–H bonds are formed with lengths of 1.677 Å. After the structural relaxation, a small hole is created on the surface, which is clear by a non-bonding area in ELF plots, displayed in dark blue in Fig. 4(c) and (d). Considering the small size of the H atom, the presence of this hole can create an easy path for hydrogen to penetrate into the underlying layers and enhance the HER activity.
 | ||
| Fig. 4 Hydrogen adsorption on the VO/VN surface at low H convergence. (a) and (b) The 1/4ML and 3/8ML hydrogen coverage of the surface, respectively. The corresponding Electron Localization Function (ELF) plots are reported in (c) and (d). An asterisk is placed on some sites to indicate that these atoms are linked to hydrogen. | ||
By adding the third H near the adsorbed hydrogen atoms, as expected, the hydrogen molecule detached from the surface (Fig. S10, ESI†). This indicates that the sites are fully active for the HER. However, in another interesting case, when we have placed the third hydrogen near the O atom on the D5 site, an OH unit is formed on the surface. The unit is connected to three topmost V atoms. The V–O bonding lengths are 2.0–2.2Å, and the bonds are neither too fragile nor too strong following the ELF results (Fig. 4(d)). H adsorbed on V atoms and H adsorbed on oxygen atoms has a very dissimilar chemical nature. The calculated Bader charges are reported for different H atoms in Table S3 (ESI†). Hydrogen on V atoms has a negative oxidation character, while H on O atoms has a positive oxidation character. Therefore, even though we did not consider the intermediate OH units of the OER, or simulate the OER, the activity of the VO/VN surface toward the OER has emerged.
4. Conclusions
Nitrides have been investigated as potential replacements for noble metal electrocatalysts in a wide range of applications. One of the most effective factors on their catalytic activity is the surface capability for binding or reacting with adsorbates. The surface characteristics and oxygen impurities can influence it in many ways. Motivated by this, we have calculated the H adsorption of vanadium nitride facets and determined the influences of oxygen impurities on their catalytic performance. We have studied the stability of eight VN surfaces; (001), (111), (101), (112), (200), (220), (113), and (311). The dependency of the catalytic activity on the properties of these facets is not well known, which forms one of the bases of the present study. The final nanocrystallite surface is made of a combination of these surfaces. The more stable facets, VN (200) and (112), are chosen for further inquiries of H adsorption on O-doped surfaces. We have checked many adsorption sites with different degrees of oxygen contamination of the surface. Our results demonstrate that V atoms in the direct vicinity of oxygen are the most active sites of all considered configurations. This inspired us to investigate the HER performance of VO/VN under the different H coverage. We believe that a single layer of VO acts as a surface oxide activation layer (SOAL) on top of the VN surface. Although the activity of the VO/VN surface is still far away from Pt as an HER activity indicator, it seems that this picture could be expanded to other nitrides as well for designing improved nitride-based catalysts with engineered oxidation surfaces and thereby improving HER and OER activities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to National Natural Science Foundation of China (Grant No. 12073009, 61874048) for the support to this work.References
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488(7411), 294–303, DOI:10.1038/nature11475.
- S. Chen, T. Takata and K. Domen, Particulate photocatalysts for overall water splitting, Nat. Rev. Mater., 2017, 2(10), 17050, DOI:10.1038/natrevmats.2017.50.
- P. Simon and Y. Gogotsi, Materials for electrochemical capacitors, Nat. Mater., 2008, 7(11), 845–854, DOI:10.1038/nmat2297.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Revealing nitrogen-containing species in commercial catalysts used for ammonia electrosynthesis, Nat. Catal., 2020, 3(12), 1055–1061, DOI:10.1038/s41929-020-00527-4.
- X. Meng, W. Qi, W. Kuang, S. Adimi, H. Guo, T. Thomas, S. Liu, Z. Wang and M. Yang, Chromium-titanium nitride as an efficient co-catalyst for photocatalytic hydrogen production, J. Mater. Chem. A, 2020, 8(31), 15774–15781, 10.1039/d0ta00488j.
- Y. Yuan, J. Wang, S. Adimi, H. Shen, T. Thomas, R. Ma, J. P. Attfield and M. Yang, Zirconium nitride catalysts surpass platinum for oxygen reduction, Nat. Mater., 2020, 19(3), 282–286, DOI:10.1038/s41563-019-0535-9.
- A. Goncharov, A. Guglya, A. Kalchenko, E. Solopikhina, V. Vlasov and E. Lyubchenko, Nanocrystalline Porous Hydrogen Storage Based on Vanadium and Titanium Nitrides, J. Nanotechnol., 2017, 4106067, DOI:10.1155/2017/4106067.
- A. Morel, Y. Borjon-Piron, R. L. Porto, T. Brousse and D. Bélanger, Suitable Conditions for the Use of Vanadium Nitride as an Electrode for Electrochemical Capacitor, J. Electrochem. Soc., 2016, 163(6), A1077–A1082, DOI:10.1149/2.1221606jes.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Functional carbon nitride materials—design strategies for electrochemical devices, Nat. Rev. Mater., 2017, 2(6), 17030, DOI:10.1038/natrevmats.2017.30.
- R. Pipes, J. He, A. Bhargav and A. Manthiram, Freestanding vanadium nitride nanowire membrane as an efficient, carbon-free gas diffusion cathode for Li–CO2 batteries, Energy Storage Mater., 2020, 31, 95–104, DOI:10.1016/j.ensm.2020.06.009.
- X. Peng, L. Wang, L. Hu, Y. Li, B. Gao, H. Song, C. Huang, X. Zhang, J. Fu, K. Huo and P. K. Chu, In situ segregation of cobalt nanoparticles on VN nanosheets via nitriding of Co2V2O7 nanosheets as efficient oxygen evolution reaction electrocatalysts, Nano Energy, 2017, 34, 1–7, DOI:10.1016/j.nanoen.2017.02.016.
- O. Bondarchuk, A. Morel, D. Bélanger, E. Goikolea, T. Brousse and R. Mysyk, Thin films of pure vanadium nitride: Evidence for anomalous non-faradaic capacitance, J. Power Sources, 2016, 324, 439–446, DOI:10.1016/j.jpowsour.2016.05.093.
- A. Djire, P. Pande, A. Deb, J. B. Siegel, O. T. Ajenifujah, L. He, A. E. Sleightholme, P. G. Rasmussen and L. T. Thompson, Unveiling the pseudocapacitive charge storage mechanisms of nanostructured vanadium nitrides using in-situ analyses, Nano Energy, 2019, 60, 72–81, DOI:10.1016/j.nanoen.2019.03.003.
- J.-G. Choi, J. Ha and J.-W. Hong, Synthesis and catalytic properties of vanadium interstitial compounds, Appl. Catal., A, 1998, 168(1), 47–56, DOI:10.1016/S0926-860X(97)00332-3.
- Y. Liu, L. Liu, L. Kang and F. Ran, Vanadium nitride with surface single specie oxide via vanadium-organic frameworks precursor, J. Power Sources, 2020, 450, 227687, DOI:10.1016/j.jpowsour.2019.227687.
- H. Yang, Y. Hu, D. Huang, T. Xiong, M. Li, M.-S. Balogun and Y. Tong, Efficient hydrogen and oxygen evolution electrocatalysis by cobalt and phosphorus dual-doped vanadium nitride nanowires, Mater. Today Chem., 2019, 11, 1–7, DOI:10.1016/j.mtchem.2018.10.004.
- J. O. Abe, A. P. I. Popoola, E. Ajenifuja and O. M. Popoola, Hydrogen energy, economy and storage: Review and recommendation, Int. J. Hydrogen Energy, 2019, 44(29), 15072–15086, DOI:10.1016/j.ijhydene.2019.04.068.
- S. Adimi, H. Arabi, S. R. Ghorbani and F. Pourarian, Low-index surface investigation of KAlH4: Theoretical attempt to study the surface effect on the hydrogen storage properties, 2018, pp. 8835–8845 Search PubMed.
- D. Choi, G. E. Blomgren and P. N. Kumta, Fast and Reversible Surface Redox Reaction in Nanocrystalline Vanadium Nitride Supercapacitors, Adv. Mater., 2006, 18(9), 1178–1182, DOI:10.1002/adma.200502471.
- C. Shi, A. M. Zhu, X. F. Yang and C. T. Au, On the catalytic nature of VN, Mo2N, and W2N nitrides for NO reduction with hydrogen, Appl. Catal., A, 2004, 276(1–2), 223–230, DOI:10.1016/j.apcata.2004.08.017.
- Y. Yuan, S. Adimi, X. Guo and T. Thomas, et al., A Surface-Oxide-Rich Activation Layer (SOAL) on Ni2Mo3N for a Rapid and Durable Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2020, 59(41), 18036–18041, DOI:10.1002/anie.202008116.
- M.-I. Jamesh and M. Harb, Tuning the electronic structure of the earth-abundant electrocatalysts for oxygen evolution reaction (OER) to achieve efficient alkaline water splitting – A review, J. Energy Chem., 2021, 56, 299–342, DOI:10.1016/j.jechem.2020.08.001.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Nørskov, Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5(11), 909–913, DOI:10.1038/nmat1752.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1(1), 37–46, DOI:10.1038/nchem.121.
- B. Hammer and J. K. Nørskov, Theoretical surface science and catalysis—calculations and concepts, Adv. Catal., 2000, 45, 71–129, DOI:10.1016/S0360-0564(02)45013-4.
- Z. Chen, Y. Song, J. Cai and X. Zheng, et al., Tailoring the d-Band Centers Enables Co4N Nanosheets To Be Highly Active for Hydrogen Evolution Catalysis, Angew. Chem., Int. Ed., 2018, 57(18), 5076–5080, DOI:10.1002/anie.201801834.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46(11), 6671–6687, DOI:10.1103/PhysRevB.46.6671.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- D. R. Hamann, Optimized norm-conserving Vanderbilt pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88(8), 85117, DOI:10.1103/PhysRevB.88.085117.
- W. Sun and G. Ceder, Efficient creation and convergence of surface slabs, Surf. Sci., 2013, 617, 53–59, DOI:10.1016/j.susc.2013.05.016.
- A. K. Mishra, A. Roldan and N. H. de Leeuw, CuO Surfaces and CO2 Activation: A Dispersion-Corrected DFT+U Study, J. Phys. Chem. C, 2016, 120(4), 2198–2214, DOI:10.1021/acs.jpcc.5b10431.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152(3), J23, DOI:10.1149/1.1856988.
- N. M. Marković and P. N. Ross, Surface science studies of model fuel cell electrocatalysts, Surf. Sci. Rep., 2002, 45(4), 117–229, DOI:10.1016/S0167-5729(01)00022-X.
- Z. Liang, X. Zhong, T. Li, M. Chen and G. Feng, DFT Study on the Hydrogen Evolution Reaction for Different Facets of Co2P, ChemElectroChem, 2019, 6(1), 260–267, DOI:10.1002/celc.201800601.
- P. Zhou, D. Xing, Y. Liu, Z. Wang, P. Wang, Z. Zheng, X. Qin, X. Zhang, Y. Dai and B. Huang, Accelerated electrocatalytic hydrogen evolution on non-noble metal containing trinickel nitride by introduction of vanadium nitride, J. Mater. Chem. A, 2019, 7(10), 5513–5521, 10.1039/C8TA12043A.
- H. Hajihoseini, M. Kateb, S. Ingvarsson and J. T. Gudmundsson, Effect of substrate bias on properties of HiPIMS deposited vanadium nitride films, Thin Solid Films, 2018, 663, 126–130, DOI:10.1016/j.tsf.2018.06.060.
- P. Sabatier, Catalysis in organic chemistry, D. Van Nostrand Company, 1922 Search PubMed.
- Y. Tang, X. Rui, Y. Zhang, T. M. Lim, Z. Dong, H. H. Hng, X. Chen, Q. Yan and Z. Chen, Vanadium pentoxide cathode materials for high-performance lithium-ion batteries enabled by a hierarchical nanoflower structure via an electrochemical process, J. Mater. Chem. A, 2013, 1(1), 82–88, 10.1039/c2ta00351a.
- Z. Khan, B. Senthilkumar, S. O. Park, S. Park, J. Yang, J. H. Lee, H.-K. Song, Y. Kim, S. K. Kwak and H. Ko, Carambola-shaped VO2 nanostructures: a binder-free air electrode for an aqueous Na–air battery, J. Mater. Chem. A, 2017, 5(5), 2037–2044, 10.1039/C6TA09375B.
- H. Song, C. Liu, C. Zhang and G. Cao, Self-doped V4+–V2O5 nanoflake for 2 Li-ion intercalation with enhanced rate and cycling performance, Nano Energy, 2016, 22, 1–10, DOI:10.1016/j.nanoen.2016.02.004.
- C. G. Morales-Guio, L. Liardet and X. Hu, Oxidatively Electrodeposited Thin-Film Transition Metal (Oxy)hydroxides as Oxygen Evolution Catalysts, J. Am. Chem. Soc., 2016, 138(28), 8946–8957, DOI:10.1021/jacs.6b05196.
- L. Liardet and X. Hu, Amorphous Cobalt Vanadium Oxide as a Highly Active Electrocatalyst for Oxygen Evolution, ACS Catal., 2017, 8(1), 644–650, DOI:10.1021/acscatal.7b03198.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00231g |
| This journal is © The Royal Society of Chemistry 2021 |