
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
State of charge dependent ordered and disordered phases in a Li[Ni1/3Co1/3Mn1/3]O2 cathode material†
Chi Ho
Lee‡
 a,
Byeongsun
Jun‡
a,
Seung Cheol
Lee
*c and
Sang Uck
Lee
*ab
a,
Byeongsun
Jun‡
a,
Seung Cheol
Lee
*c and
Sang Uck
Lee
*ab
aDepartment of Bionano Technology, Hanyang University, Ansan 15588, Republic of Korea. E-mail: sulee@hanyang.ac.kr
bIndo-Korea Science and Technology Center, Korea Institute of Science and Technology, Bangalore, 560065, India. E-mail: leesc@kist.re.kr
cDepartment of Applied Chemistry, Center for Bionano Intelligence Education and Research, Hanyang University, Ansan 15588, Republic of Korea
First published on 3rd May 2021
Abstract
We systematically investigated the structural phase transition of a Li[Ni1/3Co1/3Mn1/3]O2 (NCM) cathode material depending on the state of charge (SOC) using cluster expansion Monte Carlo simulation (CE-MCS) combined with density functional theory (DFT). Considering the charging/discharging process involving lithium intercalation/deintercalation, the oxidation state of transition metal (TM) cations varies with the SOC, resulting in a TM arrangement shift to a thermodynamically favorable NCM structure. Our results demonstrate that the phase transition from disordered to ordered happens at a low SOC with a high TM oxidation state, and that the phase transition is initiated by TM pop-up from the TM layer to the Li layer. Ni migration plays an especially fundamental role in the phase transition with a diffusion energy barrier comparable to that of Li ions. Furthermore, based on a thorough understanding of the structural phase transition, we propose cation dopants (Zr, Ti and V) which inhibit the Ni pop-up as an initiating step of the phase transition by enhancing the chemical bonding of Ni ions in the NCM structure, thereby preventing the phase transition from causing undesirable structural degradation and severe capacity fading in NCM. Our theoretical investigations will provide insights into the structural phase transition mechanism and the design of new cathode materials for lithium ion batteries (LIBs).
1. Introduction
The application domain for lithium ion batteries (LIBs) has rapidly progressed from portable products to large scale applications with high energy capacity requirements, such as electric vehicles (EVs) and hybrid electric vehicles (HEVs).1–6 Higher energy densities and longer cycle lives are key factors that need to be addressed in LIB applications.7,8 Recently, multi-component layered Li transition metal oxides (Li(Ni,Mn,Co)O2) have served as the most important cathode materials for electric vehicle and plug-in hybrid electric vehicle applications due to their cycling stability and large theoretical capacity compared with those of olivine or spinel materials.9,10 Following the initial commercial success of NCM 111 (LiNi1/3Co1/3Mn1/3O2), NCM cathodes have become mainstream and intensive research has improved NCM technology by steadily increasing the nickel content in each cathode generation (e.g., NCM 433, NCM 523, NCM 622 and the most recent NCM 811)11 because the Ni content is largely responsible for the cathode capacity, with Mn and Co helping with chemical and structural stability.12,13 As a matter of fact, increasing Ni content is the most effective way to enhance capacity in current state-of-the-art batteries without expanding into unexplored areas of new battery chemistries.However, a higher level of Ni incorporation causes a significant capacity fading and impedance increase, which are mainly attributed to the structural and chemical degradation of the NCM structure, even for the most stable NCM 111. Several structural phase transitions and displacement of TMs from TM layers inducing the ionic and electric impedance of batteries are known to be the crucial features plaguing the cycling performance of NCM.14–18 In addition, it has been reported that vigorous Ni migration from the TM layer to the Li layer in the NCM leads to rapid structural change. A significant number of oxygen vacancies lower the activation barrier for the TM migration, and eventually accelerate the phase transitions.19 To address the stability of NCM, multiple strategies have been investigated, such as doping with other elements/materials to improve chemical and structural stability, coating the cathode surface to passivate its reactivity, using stabilizing additives in electrolyte, or strengthening the separator safety.12,13,20,21 Recently, taking advantage of its high capacity and advanced post-processing for structural stability, NCM materials have been successfully adopted in an all-electric vehicle.
Despite the commercial success of NCM, the exact mechanism that drives the structural phase transition at the atomic level is not clear yet. Furthermore, most research facing the problem has been focused on the fully lithiated structure of NCM cathode materials, even though the oxidation state of TM cations should change depending on the state of charge (SOC) and cause a structural rearrangement of TMs to a thermodynamically favorable NCM structure. In this work, we systematically investigated the structural phase transition of NCM 111 (from now on denoted NCM) cathode material depending on the SOC using cluster expansion Monte Carlo simulation (CE-MCS) combined with density functional theory (DFT) to resolve the structural phase transition mechanism caused by the TM rearrangement. Here, we demonstrate that the structural phase transition must be accompanied by the pop-up and migration of TM cations from the TM layer into the Li layer, where the Ni cations can easily transfer to the Li layer, leading to the phase transition from the disordered to an ordered TM phase. Ni-rich local structures in the NCM structure can be anticipated, which may be the main reason for the serious capacity fading that accompanies structural phase transition. We also propose a method for effectively suppressing the migration of Ni by cation doping with elements of low electronegativity, such as Zr, Ti and V.
2. Computational methods
All calculations were performed with the Vienna Ab initio Simulation Package (VASP 5.3.5).22–25Ab initio calculations were carried out using the projector augmented wave (PAW) method26,27 with the generalized gradient approximation based on the Perdew–Burke–Ernzerhof (PBE)28,29 functional including the Hubbard U correction (GGA + U).30 The GGA + U approach has been well tested in terms of its phase stability.31 So, we used the effective interaction parameter Ueff = U − J, and the corresponding values of 6.37, 5.14 and 4.84 were used for the 3d states on Ni, Co and Mn, respectively.32 A plane-wave cutoff energy of 500 eV was used. Integration in the Brillouin zone was performed on the basis of the Monkhorst–Pack scheme33 using a k-point mesh with an interval of 0.05 Å−1 in each primitive lattice vector of the reciprocal space. Lattice constants and internal atomic positions were optimized and spin polarized until the residual forces became less than 0.04 eV Å−1. The Climbing Image-Nudged Elastic Band (CI-NEB) method is adopted to calculate the diffusion energy barriers during Li and Ni ion migration.34 Analysis of Bader charges is a widely adopted method to study charge distribution. In this work, Bader charge analysis was performed using grid-based charge density decomposition, as developed by Henkelman et al.35 The structures of and disordered phases of TM arrangements were obtained by exploring the configuration space with cluster expansion Monte Carlo (CE-MC) simulation combined with density functional theory (DFT).363. Results and discussion
3.1. SOC dependent ordered and disordered phases
Considering the charging/discharging process accompanied by deintercalation/intercalation of Li ions, the oxidation state of TM cations must change depending on the SOC, which induces the TM rearrangement into a thermodynamically stable structure at a given SOC. However, most research has been focused on the fully lithiated structure of LIB cathode materials. Here, we demonstrate that the structural phase transition of an NCM cathode material depends on the SOC using Cluster Expansion Monte Carlo (CE-MC) simulation combined with Density Functional Theory (DFT). Fig. 1(a) shows phase transitions from disordered to ordered TM arrangements in LixNi1/3Co1/3Mn1/3O2 by decreasing the Li concentration, i.e., SOC from x = 1.0 to x = 0.0 at given temperatures of 100 K, 300 K and 400 K. To quantify the degree of ordering of TMs, we constructed a local order matrix counting the number of TMs in the nearest neighbor location around each active site in the TM layers. Details are provided in the ESI,† Section A. | (1) |
 | ||
| Fig. 1 (a) Phase transitions between ordered and disordered arrangements of TMs in LixNi1/3Co1/3Mn1/3O2 (0 ≤ x ≤ 1) depending on the Li content (x = 0.0, 0.33, 0.67, and 1.0) and temperature (100 K, 300 K and 400 K). The structures at 100 K, 300 K and 400 K are obtained from Monte Carlo simulations at the given temperatures by cooling down slowly from 4000 K at 20 K intervals. (b) The ordering tendencies of TMs quantified using a local order matrix. The y-axis represents the trace of the local order matrix. | ||
Here, NTM1–TM2 indicates the number of TM2 ions in the nearest neighbor location around the TM1 ion. Therefore, the trace of the local order matrix represents the ordering tendency of cations, i.e. clustering of the same TM. A zero value of the trace means fully disordered arrangement of TMs, and the greater the trace value, the more ordered the arrangement. In the fully lithiated state of x = 1.0, all TM+3 cations are well-dispersed in NCM. The trace value stays at 0 until, at a temperature of 400 K, a disordered phase emerges, as shown in Fig. 1(b). This is in good agreement with the well-known NCM structure. However, as TM+4 increases in delithiated states of x < 1.0, the same type of TM tends to be clustered as an ordered phase by increasing the trace value. With increasing temperature, the trace value tends to increase, with the exception of the decrease in the fully delithiated x = 0.0 state. Therefore, the results imply that NCM thermodynamically prefers a different TM arrangement in each SOC, and NCM is expected to find a spontaneously stable structure during the charging/discharging process. Experimentally, an ordered Ni rich phase has been reported in previous studies.37–39
3.2. Structural phase transition mechanism
The phase transition between ordered and disordered phases of TM arrangement can be explained by the pop-up and migration of TM cations. Considering the phase transition initiated by the TM pop-up from the TM layer to the Li layer, the TM migration pathway should pass through the Li layer. To understand the underlying phase transition mechanism, we systematically studied the relative stability of the pop-up and migration of TM cations in various TM environments in the Li27[Ni12Co12Mn12]O72 structure. Details of modeling are provided in the ESI,† Section B. Fig. 2(a) shows the proposed pop-up-initiated TM migration pathway. The TM first pops up from the octahedral site of the TM layer (Oh-TM) to the tetrahedral site (Td-Li) of the Li layer, Oh-TM → Td-Li, then migrates along the Td-Li and Oh-Li sites alternately in the Li layer. We considered two different Oh-Li sites, near (Oh-Li_near) and far (Oh-Li_far), to describe the TM diffusion through the Li layer. Interestingly, the relative stability of TMs in the pop-up-initiated TM migration pathway shows a strong dependence on the type of pop-up TM element and the configurations of the adjacent TMs around the pop-up TM. Fig. 2(b)–(d) show that Mn is thermodynamically unfavored in the pop-up-initiated TM migration pathway compared to Ni and Co. In addition, the TMs surrounded by different types of TM elements, Ni@Mn3Co3 (red), Co@Mn3Ni3 (green) and Mn@Co3Ni3 (blue), have the energetic advantage in TM migration over TMs surrounded by the same type of TM element. Looking at the first TM pop-up stage, Co is the most thermodynamically favored regardless of the configurations of the adjacent TMs. However, considering further migration of the popped-up TM, only Ni@Mn3Co3 (red) has a downhill energy profile with steadily increasing stability along the TM migration pathway, Oh-TM → Td-Li → Oh-Li_near → Oh-Li_far. Therefore, among the three TMs considered, Ni, Co and Mn, Ni is most favored to migrate through the Li layer and to initiate the phase transition from the disordered to an ordered TM arrangement. | ||
| Fig. 2 (a) Pop-up and migration pathway of TM (denoted by yellow circles) in the Li27[Ni12Co12Mn12]O72 structure and the relative stability (eV) of (b) Ni, (c) Co, and (d) Mn (denoted by white circles) along the pop-up-initiated TM migration pathway in different TM environments. | ||
Then, we also investigated the kinetic properties in the pop-up-initiated TM migration pathway by calculating the diffusion energy barriers of the pop-up and migration of TM cations, especially those of Ni. In terms of reaction kinetics, the ion transport mechanism in the layered structure follows a vacancy diffusion, and the diffusivity shows an exponential dependence on the diffusion energy barrier along a migration pathway.40,41Fig. 3 shows the calculated diffusion energy barriers of Ni moving over the different types of TM in the NCM 111 structure with no TM vacancy compared to the diffusion of Li. The energy barriers of Li diffusion through the Oh-Li → Td-Li → Oh-Li pathway are calculated to be almost 0.7 eV regardless of the type of TM on the diffusion path. The analogous energy barriers for Ni are 1.5 eV, about twice those of Li. However, empty octahedral Ni sites (Oh-vNi) are always produced by Ni pop-ups and can have a significant effect on Ni diffusion through the Li layer. To verify the effect of Oh-vNi on the diffusion kinetics, we further studied the Ni diffusion energy barriers with Oh-vNi along the Oh-TM → Td-Li → Oh-Li_near diffusion pathway, as shown in Fig. 3(c). The diffusion energy barrier of the first Ni pop-up stage is considerably lower than that of Li migration. The Ni pop-up can apparently occur easily within the LIB working voltage. Moreover, the diffusion energy barrier of Ni migration with Oh-vNi is almost 0.9 eV, comparable to the Li diffusion. The favorable pop-up and migration of Ni can be convincingly attributed to the structural phase transition.
 | ||
| Fig. 3 Diffusion energy barriers of (a) Li and (b) Ni moving over the different types of TM in the NCM 111 structure with no TM vacancy. (c) Diffusion energy barriers of Ni pop-up and migration from Oh-TM to Oh-Li_near via Td-Li considering the vacant Oh-TM site caused by Ni pop-up. The insets indicate Ni migration. (Green, blue, purple and gray are Ni, Co, Mn and Li ions, respectively.) | ||
3.3. Suppression of structural phase transition by doping
Based on the understanding of the structural phase transition mechanism in the NCM structure, we looked for a way to suppress the Ni pop-up-initiated phase transition. Our simple strategy is enhancing the Ni–O chemical bond strength to keep the Ni on the Oh-TM site of the TM layer, blocking Ni pop-up to the Li layer and subsequent migration. Because the ionic bond strength of Ni–O can be improved by increasing the ionic charge, we employed doping with low electronegativity elements (Zr, Ti, and V42–44) to increase the negative charges of oxygen around Ni in NCM. In particular, we substituted Zr, Ti, or V for Co in Ni@Mn3Co3, the environment where Ni pop-up is the most favorable considering a 2.94 mol% doping ratio similar to the experiments,45–48 as shown in Fig. 4(a). Details of doping are provided in the ESI,† Section C. As expected, the charge analysis in Fig. 4(b) shows that the doping Zr, Ti and V have relatively positive charges compared to Co and induce increased relatively negative charges of oxygen. In addition, Fig. 4(c) shows that the thermodynamic stabilities of Ni at Td-Li, Oh-Li_near and Oh-Li_far of Li27[Ni12Mn12Co11X]O72 (X = Zr, Ti, or V) are evaluated as unstable compared to undoped Li27[Ni12Mn12Co12]O72. Even though Ni pop-up can occur with thermodynamic stability, the kinetically higher Ni diffusion energy barrier (around 1.8 eV for the Zr and Ti doped structures) prevents further migration of Ni from Td-Li to Oh-Li_near, Fig. 4(d). The chemical doping with the electronegatively weak element Zr, Ti, or V can modulate the local electronic structure enhancing the negative charge of oxygen around Ni and the Ni–O chemical bond to restrain Ni from pop-up and migration. Therefore, we propose Zr, Ti, or V dopant, especially Zr, for suppressing the Ni pop-up-initiated phase transition from causing undesirable structural degradation and severe capacity fading in NCM cathode materials.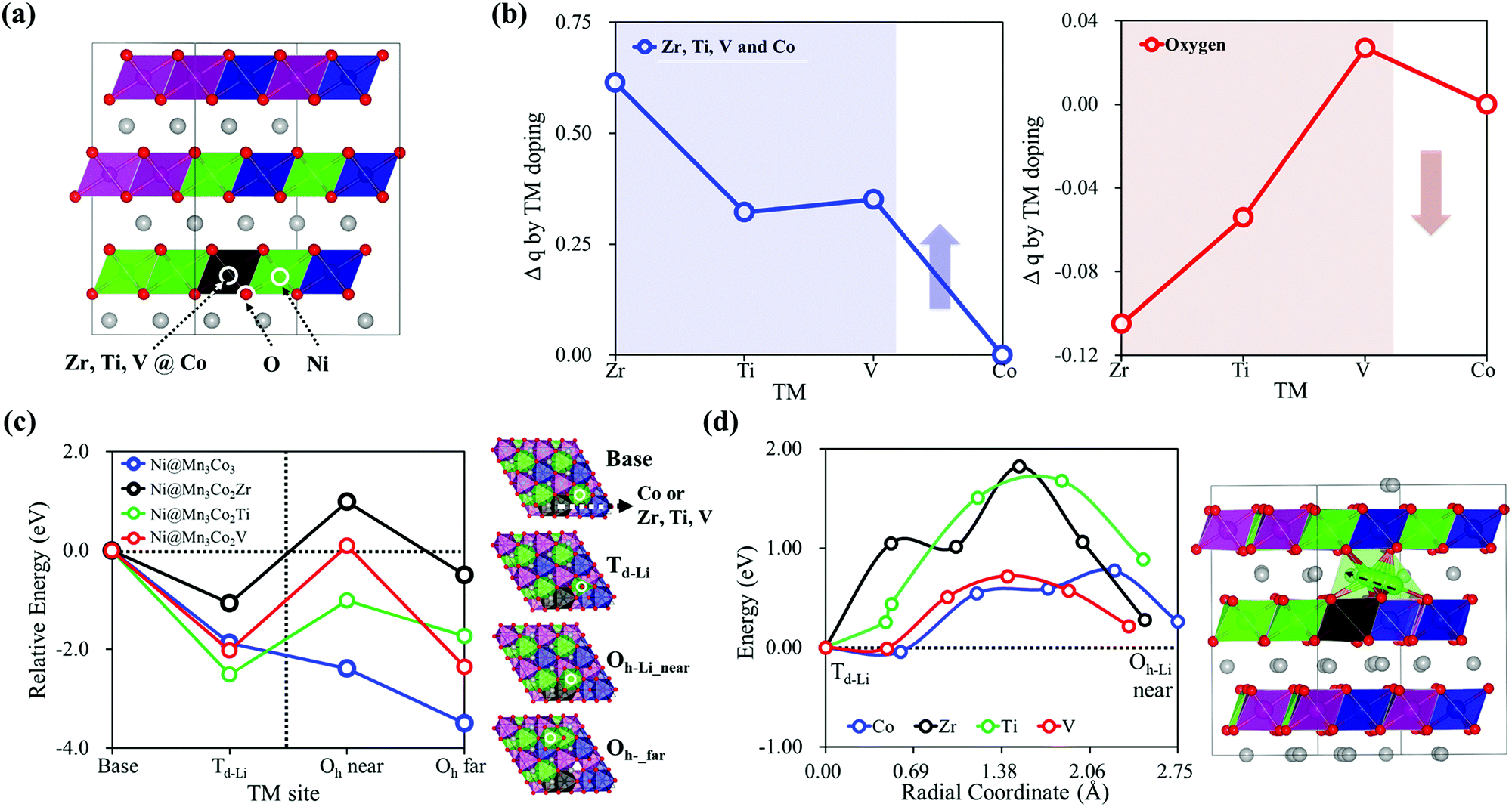 | ||
| Fig. 4 (a) Electronegatively weak Zr, Ti, or V doped structure, where the dopant replaces Co in the Ni@Mn3Co3 local structure of Li27[Ni12Co12Mn12]O72. (b) Relative charge states of Zr, Ti, and V dopants and Co, and oxygen near Ni in Li27[Ni12Co11Mn12TM]O72 (TM = Zr, Ti or V) compared to the charges in undoped Li27[Ni12Co12Mn12]O72. (c) Relative stability of Ni along the pop-up-initiated Ni migration pathway in different local structures, Ni@Mn3Co3 and Ni@Mn3Co2TM (TM = Zr, Ti or V). (d) Ni diffusion energy barriers from Td-Li to Oh-Li_near. | ||
4. Conclusions
In this work, we systematically investigated that the TM arrangement of Li[Ni1/3Co1/3Mn1/3]O2 (NCM) cathode materials can be transformed to a thermodynamically favorable phase for the corresponding state of charge (SOC). Our results unveil that at a given SOC the structural phase transition from the disordered to ordered TM phase can occur by TM pop-up to the Li layer and migration. Ni surrounded by Mn3Co3 is thermodynamically most advantageous for TM pop-up-initiated phase transition with a downhill energy profile of continuously improved stability along the TM migration pathway, Oh-TM → Td-Li → Oh-Li_near → Oh-Li_far. In addition, by evaluating the Ni diffusion energy barrier, we proved that Ni migration can easily occur and that it is kinetically comparable to Li diffusion. Therefore, Ni pop-up-initiated phase transition causes capacity fading due to the formation of a Ni enriched surface structure and surface degradation during the charging process. Based on the understanding of the structural phase transition mechanism in the NCM structure, we proposed Zr-doping to prevent the phase transition by suppressing the Ni pop-up and migration. Zr-doping has the effect of enhancing the Ni–O chemical bond strength to keep the Ni on the Oh-TM site of the TM layer, suppressing the Ni pop-up and migration. Our theoretical investigations provide further understanding of the structural phase transition in the NCM cathode material and can be used to promote the development of new cathode materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2021R1A2B5B01002879) and the Supercomputing Center/Korea Institute of Science and Technology Information with supercomputing resources including technical support (KSC-2019-CRE-0192).References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef.
- H. Jiang, H. X. Zhang, Y. Fu, S. J. Guo, Y. J. Hu, L. Zhang, Y. Liu, H. L. Liu and C. Z. Li, ACS Nano, 2016, 10, 1648–1654 CrossRef PubMed.
- S. M. Kim, J. D. Chang and J. H. Lim, Adv. Mater. Sci. Eng., 2017, 1–1, DOI:10.1155/2017/5792427.
- Y. K. Sun, M. J. Lee, C. S. Yoon, J. Hassoun, K. Amine and B. Scrosati, Adv. Mater., 2012, 24, 1192–1196 CrossRef PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef PubMed.
- D. L. Ye, K. Ozawa, B. Wang, D. Hulicova-Jurcakova, J. Zou, C. H. Sun and L. Z. Wang, Nano Energy, 2014, 6, 92–102 CrossRef.
- K. S. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef.
- A. Rougier, P. Gravereau and C. Delmas, J. Electrochem. Soc., 1996, 143, 1168–1175 CrossRef.
- F. Lin, I. M. Markus, D. Nordlund, T. C. Weng, M. D. Asta, H. L. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5 Search PubMed.
- A. Manthiram, J. C. Knight, S. T. Myung, S. M. Oh and Y. K. Sun, Adv. Energy Mater., 2016, 6 Search PubMed.
- P. Y. Hou, J. M. Yin, M. Ding, J. Z. Huang and X. J. Xu, Small, 2017, 13 Search PubMed.
- J. Kim, H. Lee, H. Cha, M. Yoon, M. Park and J. Cho, Adv. Energy Mater., 2018, 8 Search PubMed.
- N. Y. Kim, T. Yim, J. H. Song, J. S. Yu and Z. Lee, J. Power Sources, 2016, 307, 641–648 CrossRef.
- M. Lang, M. S. D. Darma, K. Kleiner, L. Riekehr, L. Mereacre, M. A. Perez, V. Liebau and H. Ehrenberg, J. Power Sources, 2016, 326, 397–409 CrossRef.
- G. Y. Li, Z. J. Zhang, Z. L. Huang, C. K. Yang, Z. C. Zuo and H. H. Zhou, J. Solid State Electrochem., 2017, 21, 673–682 CrossRef.
- R. Xu and K. J. Zhao, J. Electrochem. Energy Convers. Storage, 2016, 13 Search PubMed.
- S. L. Zhang, K. J. Zhao, T. Zhu and J. Li, Prog. Mater. Sci., 2017, 89, 479–521 CrossRef.
- S. M. Bak, E. Y. Hu, Y. N. Zhou, X. Q. Yu, S. D. Senanayake, S. J. Cho, K. B. Kim, K. Y. Chung, X. Q. Yang and K. W. Nam, ACS Appl. Mater. Interfaces, 2014, 6, 22594–22601 CrossRef PubMed.
- A. L. Lipson, J. L. Durham, M. LeResche, I. Abu-Baker, M. J. Murphy, T. T. Fister, L. X. Wang, F. Zhou, L. Liu, K. Kim and D. Johnson, ACS Appl. Mater. Interfaces, 2020, 12, 18512–18518 CrossRef PubMed.
- S. T. Myung, F. Maglia, K. J. Park, C. S. Yoon, P. Lamp, S. J. Kim and Y. K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef PubMed.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef.
- F. Zhou, T. Maxisch and G. Ceder, Phys. Rev. Lett., 2006, 97 Search PubMed.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Chandran, Comput. Mater. Sci., 2015, 108, 192–204 CrossRef.
- D. P. Abraham, R. D. Twesten, M. Balasubramanian, J. Kropf, D. Fischer, J. McBreen, I. Petrov and K. Amine, J. Electrochem. Soc., 2003, 150, A1450–A1456 CrossRef.
- D. P. Abraham, R. D. Twesten, M. Balasubramanian, I. Petrov, J. McBreen and K. Amine, Electrochem. Commun., 2002, 4, 620–625 CrossRef.
- S. Muto, Y. Sasano, K. Tatsumi, T. Sasaki, K. Horibuchi, Y. Takeuchi and Y. Ukyo, J. Electrochem. Soc., 2009, 156, A371–A377 CrossRef.
- F. T. Kong, R. C. Longo, M. S. Park, J. Yoon, D. H. Yeon, J. H. Park, W. H. Wang, K. C. Santosh, S. G. Doo and K. Cho, J. Mater. Chem. A, 2015, 3, 8489–8500 RSC.
- Y. Wei, J. X. Zheng, S. H. Cui, X. H. Song, Y. T. Su, W. J. Deng, Z. Z. Wu, X. W. Wang, W. D. Wang, M. M. Rao, Y. Lin, C. M. Wang, K. Amine and F. Pan, J. Am. Chem. Soc., 2015, 137, 8364–8367 CrossRef PubMed.
- L. C. Allen, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef.
- J. B. Mann, T. L. Meek and L. C. Allen, J. Am. Chem. Soc., 2000, 122, 2780–2783 CrossRef.
- J. B. Mann, T. L. Meek, E. T. Knight, J. F. Capitani and L. C. Allen, J. Am. Chem. Soc., 2000, 122, 5132–5137 CrossRef.
- J. Choi, S. Y. Lee, S. Yoon, K. H. Kim, M. Kim and S. H. Hong, ChemSusChem, 2019, 12, 2439–2446 Search PubMed.
- S. Y. Liu, Z. Y. Dang, D. Liu, C. C. Zhang, T. Huang and A. S. Yu, J. Power Sources, 2018, 396, 288–296 CrossRef.
- Y. Lu, M. Pang, S. L. Shi, Q. Ye, Z. J. Tian and T. Wang, Sci. Rep., 2018, 8 CrossRef.
- K. Park, D. J. Ham, S. Y. Park, J. Jang, D. H. Yeon, S. Moon and S. J. Ahn, RSC Adv., 2020, 10, 26756–26764 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00289a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |