
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable oxygen evolution catalysis – electrochemical generation of mössbauerite via corrosion engineering of steel†‡
Sebastian
Weiß
ab,
A. V.
Radha
b,
Michael
Ertl
b,
Catherine
McCammon
 c and
Josef
Breu
*ab
c and
Josef
Breu
*ab
aBavarian Center for Battery Technology (BayBatt), University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany
bDepartment of Chemistry, University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany. E-mail: josef.breu@uni-bayreuth.de
cBayerisches Geoinstitut, University of Bayreuth, Universitätsstr. 30, 95447 Bayreuth, Germany
First published on 13th July 2021
Abstract
A versatile electrochemical corrosion process was used to generate green rust (GR) on a steel substrate and to transform it into the trivalent iron-only oxygen evolution catalyst mössbauerite (GR*) upon a subsequent oxidation process. Besides being scalable, cheap, and time-efficient, the demonstrated procedure is based on earth-abundant and non-hazardous materials only. Oxidation of mixed-valence Fe2+/Fe3+ GR to GR* was achieved by chemical and electrochemical processes. The latter induces less grafting of interlayer CO32−, leading to less contraction of the interlayer space and a reduction in the observable stacking disorder. The direct electrodeposition of the catalyst on a conductive substrate enabled the first systematic study of the impact of grafting on the performance of GR* in the oxygen evolution reaction (OER). The materials subjected to chemical (GR*ChOx) and electrochemical oxidation (GR*ElOx) allowed obtaining a current density of 100 mA cm−2 at 1.82 V and 1.84 V, respectively, thus improving the electrocatalytic capabilities and outperforming the bare steel substrate operated in 1 M KOH solutions.
Introduction
Resolving energy and environmental issues has been receiving enormous attention. It can be regarded as the critical challenge of the 21st century.1,2 Most of the proposed strategies focus on renewable energies to curb CO2 emissions by gradually turning away from conventional fossil energy sources. Due to the intermittent nature of these energy sources, suitable storage methods are imperative to provide a continuous energy supply for grid management.3 For instance, energy can be stored in the form of chemical bonds, e.g., hydrogen. However, the bottleneck for establishing a hydrogen-based storage cycle is linked to the oxidative half-reaction, called the oxygen evolution reaction (OER).4 Moreover, for green energy production, environmentally friendly, earth-abundant, non-hazardous, and affordable elements allowing for technically benign processing are preferred. Such materials with efficient and durable electrocatalytic properties will play a pivotal role in future developments in this field.5Ideally, cheap and scalable anodes for the OER are highly sought-after since noble metal oxides are considered unsuitable due to dissolution, high production price, and catalyst material maintenance.6 Being affordable and easy to manufacture, iron-based electrodes represent a viable option. Unfortunately, under OER operating conditions, the Pourbaix diagram of iron implies the dissolution of the electrode through the formation of ferrate (FeO42−) anions.7 The dissolved ferrate is not stable in aqueous environments at pH 14 and will thus oxidize water to produce oxygen.8 Concomitantly, with the reduction of the ferrate, undesirable corrosion products of iron are formed on the steel surface.9 The formation of this passivation layer may impede further corrosion but may also impair the performance of the electrode due to the low conductivity and/or lacking activity of the formed products.10
Using stainless steel, this uncontrolled rusting of the electrode can be mitigated. Among the other transition metal additives, stainless steels typically contain at least 12 wt% of chromium to reduce susceptibility to corrosion.11 Under ambient conditions, a thin chromium oxide layer passivates the surface. This, however, has been reported to contribute little or even reduce the OER activity of the stainless steel.12,13 The Pourbaix diagram of Cr, moreover, indicates that the formation of CrO42− is thermodynamically favored under OER operating conditions.14 Indeed, previous studies have shown Cr leaching into the electrolyte in the long term.13,15,16 The dissolution of chromium under sufficiently high anodic potentials has even been utilized to obtain thin Ni–Fe oxide layers representing a robust OER catalyst.17 However, Cr(VI) is known to be carcinogenic and must not be released into the environment but rather needs to be completely removed upon exchanging the electrolyte.18,19 Nevertheless, these and other13,20,21 studies clearly have proven the efficiency of oxidic layers on Fe-based substrates for OER catalysis. The steel types applied, however, often contain environmentally unfriendly or even toxic elements like Co, Cr, and Ni while processing often is time-consuming and requires harsh conditions.
Layered double hydroxides (LDHs) are regarded as one of the most promising classes of OER catalysts22 owing to the fact that they exhibit compositional flexibility and allow for tuning of the morphology. When optimizing the electrocatalytic properties, these features allow for the optimization of electronic structures while providing high specific surface area at the same time.22 Generally, the composition of carbonate-LDHs can be described as M6−x2+Mx3+(OH)12(CO3)x/2·nH2O with 0 ≤ x ≤ 2. Previously, we have shown that the trivalent Fe-only (oxy)hydroxide mössbauerite (GR*; FeIII6(O)4(OH)8(CO3)·3H2O) is an n-type semiconductor and exhibits electrocatalytic activity promoting the OER.23 The catalyst is obtained by the quick oxidation of the mixed-valence mineral green rust (GR; FeII4FeIII2(OH)12(CO3)·3H2O). In terms of structure, GR consists of positively charged brucite-like layers containing di-/trivalent cations, interlayered anions, and structural water.24 Under ambient oxygen fugacity, GR is susceptible to oxidation and can form various ferric oxyhydroxides like goethite, feroxyhyte, and lepidocrocite.25,26 However, fast chemical oxidation using H2O2 leads to the formation of GR*.23
Herein, we report the direct electrochemical coating of environmentally friendly, all-iron GR on a steel surface and its subsequent conversion into the efficient OER catalyst GR* (Fig. 1). Anodes are fabricated from inexpensive steel via an electrochemical corrosion process producing GR, which is subsequently transformed into the active OER catalyst GR*. Two alternative routes for the conversion were explored, which influence the grafting behavior of the interlayer carbonate: either via chemical oxidation (GR*ChOx) using H2O2 or via an electro-oxidation process (GR*ElOx). The variation in the structure and the electrochemical performance of the oxidized products obtained by these two methods are compared.
 | ||
| Fig. 1 Schematic comparison of the current–voltage curve for the OER of an unmodified bare steel substrate (a) and the same substrate coated with the all-iron electrocatalyst GR* via a corrosion engineering process (b). | ||
Experimental
Materials
All solutions used to synthesize the catalysts were prepared with Ar-saturated Milli-Q water (18.2 MΩ cm). Hydrogen peroxide solution (30%) and ethanol absolute were supplied by VWR Chemicals. Potassium hydroxide was ordered from Bernd Kraft GmbH and sodium bicarbonate from Grüssing GmbH. Substrates were prepared using custom-cut steel plates (DC 01 – DIN EN 10130, Maschinenbau Feld GmbH). The nail varnish (shine last & go! gel nail polish; absolute pure) was bought from Cosnova GmbH.Electrodeposition of GR and its conversion into GR*
Steel plates were cut to square-sized pieces of 1.4 cm length and any potentially pre-formed corrosion products were removed using grade P400 grinding paper and were further polished with 0.3 μm Al2O3 powder. Then, they were cleaned by ultrasonication with ethanol for 5 minutes. Finally, each electrode was rinsed with water and ethanol to remove the remaining loose metal particles and organic contaminants. Electrical contact was provided by using a PTFE-insulated Ag-wire and conductive silver adhesive. UV-curing glue was used to seal any exposed silver. According to a previously published procedure,27 the plates were placed in a 0.4 M NaHCO3 solution with the pH adjusted to 9.6 by the dropwise addition of 10 M NaOH solution. Electrodeposition and electro-oxidation were conducted using a CH-Instruments potentiostat 710E in a 3-electrode setup. A Fisher-type double net Pt/Ir electrode (∅ 3 cm) and a HydroFlex® hydrogen reference electrode from Gaskatel were used as the counter electrode and reference electrode, respectively. The plates were cycled from 0 to 0.2 V vs. RHE for 90 cycles with a scan speed of 10 mV s−1. After completion, a greenish deposit was visible on the surface of the substrate. As a control experiment, the substrate was immersed in the given solution without any applied potential. Here, no formation of a deposit was observed after 12 weeks.For the complete oxidation of the mixed-valence GR to GR*, two different methods were employed. For chemical oxidation, the sample was immersed in 40 mL of a 25 mM aqueous solution of H2O2. Electrochemical oxidation was achieved by simply increasing the voltage from 1.10 to 1.47 V vs. RHE (typically 5–10 cycles) and subsequently to the electrodeposition of GR. Scanning experiments were performed until no notable current was detectable. Both procedures yielded orange-brown deposits on the steel substrate plates and were then applied as anodes.
Structural characterization of the materials
To allow for quick measurements to minimize oxidation, powder X-ray diffraction (PXRD) traces for GR were collected using a Bragg–Brentano-type diffractometer (Empyrean) with a Ni-filtered Cu Kα radiation source (λ = 1.540598 Å) equipped with a PIXcel detector. To further reduce fluorescence, PXRD patterns of air-stable GR*-samples were collected using a STOE STADI-P diffractometer with a Ag Kα1 radiation source (λ = 0.5594075 Å) in transmission geometry equipped with four MYTHEN2 R 1 K detectors. Data collection was performed in the static mode for one hour, with samples loaded in 0.5 mm glass capillaries. However, to assist comparability, the diffraction traces acquired with Ag Kα1 radiation (Fig. 2b) were recalculated assuming Cu Kα radiation. The infrared spectra of the powder samples were recorded using a JASCO FT-IR 6100 spectrometer (400–4000 cm−1 range, 1 cm−1 resolution). Transmission electron microscopy (TEM) images were obtained using a Zeiss/LEO EM922 Omega transmission electron microscope. Scanning electron microscopy (SEM) images were acquired using a Zeiss/LEO 1530 system. For Mössbauer spectroscopy, a small amount of the powder sample was mixed with transparent nail varnish and spread over a region of 1 mm diameter on a plastic foil. The dimensionless effective sample thickness was estimated to be 12 (∼30 mg Fe cm−2), which is close to the optimum thickness for this composition.28 The Mössbauer spectra were recorded at room temperature (293 K) in the transmission mode using a constant acceleration Mössbauer spectrometer with a nominal 370 MBq 57Co high specific activity source in a 12-μm-thick Rh matrix.29,30 The velocity scale was calibrated relative to a 25-μm-thick α-Fe foil using the positions certified for the (former) National Bureau of Standards, standard reference material no. 1541; line widths of 0.36 mm s−1 for the outer lines of α-Fe were obtained at room temperature. The spectrum was collected for one day. The Mössbauer spectra were fitted with MossA software31 using the full transmission integral. Data were fitted into doublets with pseudo-Voigt lineshape and conventional constraints (doublet components constrained to have equal widths and areas).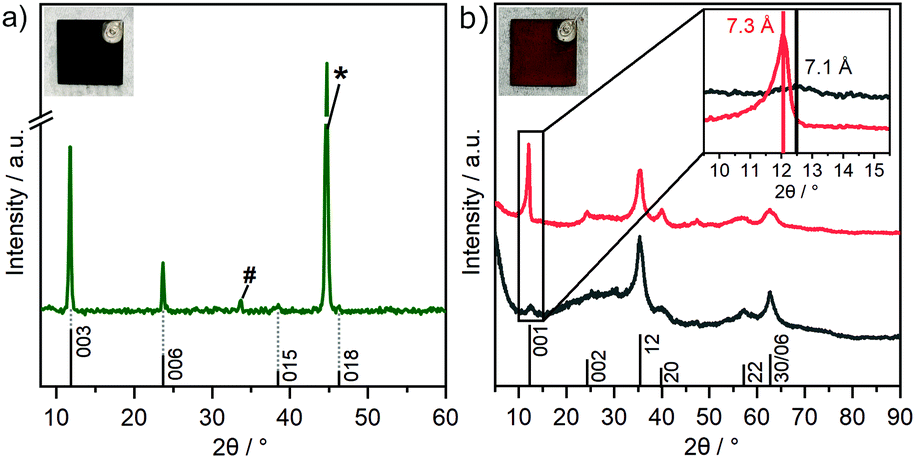 | ||
| Fig. 2 PXRD patterns of (a) GR on a steel substrate as obtained through an electrochemical corrosion engineering process (*: substrate, #: partially oxidized product formed during measurements) and (b) scraped off GR* powder as obtained through electro-oxidation (GR*ElOx; red) and chemical oxidation (GR*ChOx; gray) of GR. Photographs of GR and GR*ElOx are shown in the insets. | ||
Electrochemical characterization
Electrochemical characterization was performed using an Autolab potentiostat/galvanostat PGSTAT204. Counter and reference electrodes were the same as those used for the preparation of the material. If not noted otherwise, all electrocatalytic performance measurements were performed in O2 saturated 1 M KOH solutions. The electrolyte for all measurements was purified using the cation-exchange resin Chelex® 100 (Bio-Rad Laboratories) following the procedure recommended by the producer.32 For electrochemical measurements, the exposed surface area was reduced to 1 cm2 by sealing the rest of the plate with hot melt adhesives. The obtained potentials were corrected by 85% of the uncompensated resistance (iR) determined via current interrupt. Polarization curves were acquired by performing linear sweep voltammetry (LSV) in the range of 1.0–2.5 V with 5 mV s−1. The overpotential (η) is given as the voltage potential difference of 1.23 V. Stability measurements were performed galvanostatically by applying a current of 10 mA, corresponding to a current density of 10 mA cm−2, for 24 h. Galvanostatic stability experiments were carried out without iR compensation.The determination of the electrochemically active surface area (ECSA) was performed by cyclic voltammetry (CV) measurements in an Ar-saturated 1 M KOH at various scan rates (10, 20, 40, …, 100 mV s−1) in the potential window between 0.210 V and 0.310 V vs. RHE where there was no faradaic current response. The double-layer capacitance (Cdl) was estimated by plotting Δj = ja − jc at 0.260 V as a function of the scan rate ν (eqn (1)).33
| Cdl = 0.5·(d(Δj)/dν) | (1) |
The ECSA was calculated by dividing Cdl by the specific capacitance Cs of a flat surface. A value of Cs = 0.04 mF cm−2 was reported as an average value for different metal electrodes in an alkaline environment.34
Results and discussion
Various strategies have been employed to synthesize GR. These include FeII/FeIII co-precipitation,23 oxidation of ferrous hydroxide,35 bacterial reduction of ferric compounds,36 and electrosynthesis.37 Formation of carbonate interlayered GR has been followed by Génin et al. by tracking the Eh during the reaction.38 They further constructed the corresponding Pourbaix diagram, which identifies the stability field of GR with variation in pH and Eh.38 Based on previous investigations, a pH of 9.6 was considered most suitable to enable a successful deposition.27 For this pH, the Eh range of GR can be identified to be located between 0 V and 0.15 V. For cyclic voltammetry, the window was chosen from 0 V to 0.2 V to account for possible overpotentials. After cycling, a greenish deposit was formed on the steel electrode. The intensity of the wave peak recorded during deposition increased steadily, indicating the thickening of the deposited material layer with time (Fig. S1, ESI‡). The obtained product was characterized utilizing XRD directly on the steel substrate to minimize unintended oxidation by air (Fig. 2a).The diffraction pattern of GR is in accordance with previous reports on the material (PDF #00-046-0098).23,39,40 The d-spacing of 7.5 Å for the first reflection is characteristic for GR containing carbonate ions in the interlayer space and is usually assigned as the 003 plane of the 3R1 polymorph.23,40 The coefficient of variation of the average of the basal spacing as calculated from individual 00l reflections was found to be small for GR (0.12%).
This indicates little variance in the stacking distance of individual interlayers.39 As previously found, the remaining, weak in-plane reflections observed are in line with the 3R1 polymorph.23,40 A prominent reflection at 44.7° is caused by the underlying substrate and can be assigned to α-Fe(110) (PDF #00-006-0696).
Because of the oxygen sensitivity of GR,25 a controlled oxidation process is necessary to obtain phase pure GR*.23 Previously, we have shown that this can be achieved by subjecting GR to a H2O2 treatment as it allows rapid oxidation. Applying this chemical oxidation (GR*ChOx) to the GR deposited on the steel electrode led to an immediate color change from green to orange-brown. Alternatively, the previously deposited GR on the steel substrate was subjected to electrochemical oxidation (GR*ElOx) via cyclic voltammetry in the range of 1.10–1.47 V vs. RHE. A similar color change to orange-brown was observed. A suitable potential range to facilitate the oxidation was identified using cyclic voltammetry over a broad range from 0.60 to 1.57 V (Fig. S2, ESI‡). Because both GR* samples are air-stable, the obtained products were scraped off the plate before being characterized using XRD (Fig. 2b).
The powder XRD patterns of both GR* samples exhibit very few asymmetric and rather broad reflections. The featureless diffraction pattern is typical for GR*.23,39,41 The diffraction pattern can be fully explained with a turbostratic disorder and concomitant interstratification of domains with grafted carbonate and domains with intercalated carbonate. In any case, side phases are not indicated in the diffraction pattern. This is in contrast with previous reports, where GR was oxidized in an ambient atmosphere or by bubbling oxygen through the suspension where mixtures of GR* with goethite are obtained instead of phase pure GR*.27,42 Both the strategies presented herein, however, yield the desired product with no side phases. The first reflex of GR* appears at 7.1 Å (GR*ChOx) and 7.3 Å (GR*ElOx) and has been related to the basal spacing.41 However, an integral 00l series is missing, indicating heavy random interstratification. The apparent basal spacing of both samples is lower as compared to that of the mixed-valence parent GR (7.5 Å).40 In GR*ElOx, the (001) reflection is rather sharp and more intense as compared to that in GR*ChOx. Moreover, even the (002) reflection (d = 3.7 Å) is visible while being submerged into the background when the oxidation is performed chemically.
We have previously shown that the oxidation of mixed-valence Fe-LDHs triggers partial grafting (i.e., chemical bonding) of interlayer carbonate to the brucite-like layers.39 Density functional theory (DFT) calculations have shown that the completely grafted structure shows a basal spacing, which is 0.3 Å smaller than that for the non-grafted LDH.39 A similar trend was found in CoFe-LDHs, where it was found that a higher Fe-content promoted more grafting.43 In the presence of H2O2, the grafting was shown by DFT to proceed exothermically, which constitutes the driving force for the grafting process.39 Grafting can also be qualitatively supported by FT-IR, as discussed below. Thus, the variations in the apparent basal spacings found for GR*ChOx and GR*ElOx are attributed to a random interstratification of grafted and intercalated domains. The higher intensity and higher d-value observed for GR*ElOx indicate significantly fewer grafted domains being produced by electrochemical oxidation as compared to chemical oxidation (GR*ChOx). Employing electro-oxidation (i.e., in the absence of H2O2), the driving force for grafting might be smaller as compared to chemical oxidation and consequently, more intercalated (i.e. ungrafted) domains contribute to random interstratification leading to an overall higher observed basal spacing of 7.3 Å.
To corroborate the varying grafting probability with the oxidation method, FT-IR spectra were recorded (Fig. 3). Based on the symmetry of the carbonate ion, it is possible to experimentally distinguish between non-grafted interlayer carbonate and carbonate ions that are grafted to the brucite layers. The former features a D3h symmetry, whereas the latter shows symmetry reduction to C2v. As shown in the previous work, the ν3 vibration band of CO32− at 1350 cm−1 may be used to quantify grafting. Symmetry reduction due to grafting manifests as an energetic splitting of this band to a second one observable at 1485 cm−1, which is characteristic of the grafted carbonate species.39 Irrespective of the oxidation method, these two bands are present. The relative intensity of these two bands furthermore allows for the assessment of the extent of grafted and non-grafted species qualitatively. Both bands, assigned to grafted and ungrafted carbonate species, exhibit roughly similar intensity for GR*ChOx. For GR*ElOx, however, the band's relative intensity at 1485 cm−1 is notably decreased. Thus, it can be concluded that grafting of carbonate species is less prevalent when performing electro-oxidation. This phenomenon is in line with the above interpretation of the XRD patterns (Fig. 2b). It suggests that oxidation with a potent chemical oxidation agent, such as H2O2, provides a stronger driving force for grafting as compared to electro-oxidation.
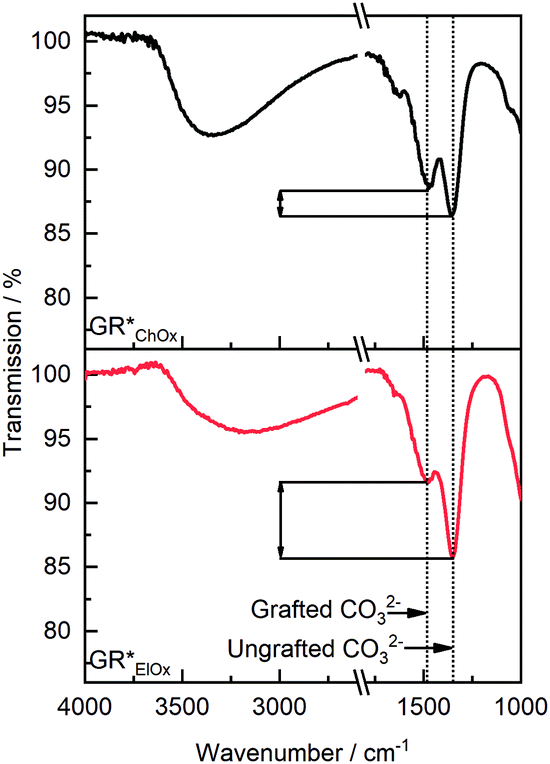 | ||
| Fig. 3 Normalized FT-IR spectra of GR*ChOx (gray) and GR*ElOx (red). Carbonate peak splitting is found in both samples. Based on the relative intensity of grafted and ungrafted carbonate peaks, a lower proportion of grafted CO32− can be obtained for GR*ElOx. | ||
Besides basal reflections, only asymmetric, non-uniformly broadened reflections are observed in the diffraction patterns of both GR* samples. As previously shown,39 they originate from two-dimensional diffraction by the brucite layers being stacked with the random phase (turbostratic disorder) leading to hk-bands.
The SEM images of both GR* samples (Fig. S3a–d, ESI‡) confirm the coating on the substrate. The micrographs of the bare steel substrate merely show some surface roughness due to polishing (Fig. S3e and f, ESI‡). For both chemically and electrochemically oxidized samples, the deposited layer features a plate-like morphology typically observed for LDHs. The steel substrate is completely coated and the coating appears to be uniform. The platy crystals of both GR* samples feature dimensions ranging from 0.4 μm to 4.5 μm in diameter and 40–80 nm in thickness. Large platelets are found to be intergrown, with smaller platelets providing an increased number of edges and corner sites, contributing to the OER activity.22 A similar appearance of both samples indicates the topotactic transformation of GR to GR* and thus the morphology is determined already in the first step, the electrochemical deposition of GR, with little to no morphology change occurring upon the formation of GR*.44 This conclusion is supported by the SEM micrographs of GR acquired prior to oxidation (Fig. S4, ESI‡).
The Mössbauer spectra of chemically and electrochemically oxidized samples are also very similar (Fig. S5, ESI‡). The 57Fe spectra were acquired at room temperature consisting of a single, broad doublet with a mean quadrupole splitting of 0.67 mm s−1 and 0.75 mm s−1 and an isomer shift of 0.35 mm s−1 and 0.36 mm s−1 with respect to metallic iron. These values agree well with those expected for GR*.40,41 Complete oxidation from Fe2+ to Fe3+ can be achieved for both samples as no residual ferrous doublet can be observed in the acquired spectra. Not surprisingly, Mössbauer spectroscopy is insensitive to the degree of grafting.
Both GR* samples were tested for OER catalysis. The electrochemically active surface area (ECSA) is calculated (Fig. 4a) by performing CV in a non-faradaic regime at different scan rates (Fig. S6, ESI‡). Due to the influence of the setup and the method of determining the absolute ECSA,12 we only use the ECSA as an activity parameter to compare the samples obtained within this study. For this purpose, the relative ECSA of the untreated steel plate is compared with those of GR*ChOx and GR*ElOx.
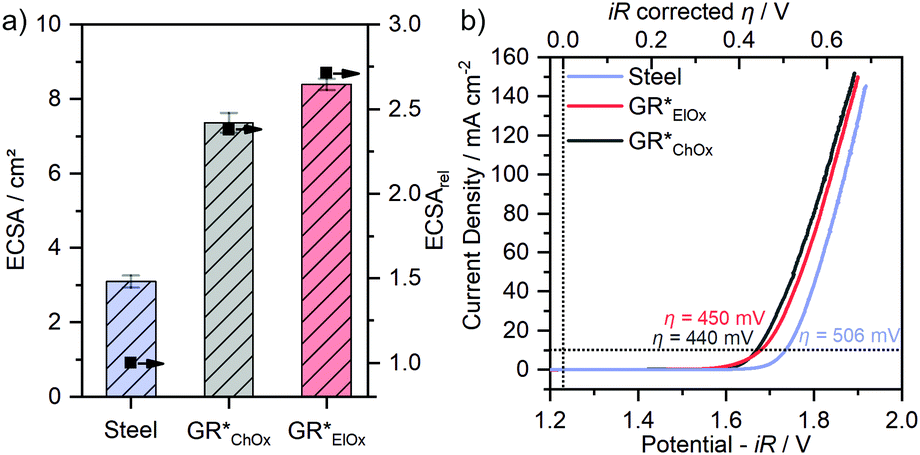 | ||
| Fig. 4 (a) Electrochemically active surface area with normalized relative ECSA and (b) linear sweep voltammograms for all three samples: the pristine steel plate, GR*ElOx, and GR*ChOx. LSVs were recorded with a scan rate of 5 mV s−1 in O2-saturated purified 1 M KOH solutions. | ||
Compared to a non-coated sample, the controlled corrosion producing GR and then GR* leads to an approximately 2.4- and 2.7-fold increase in the ECSA for GR*ChOx and GR*ElOx, respectively. Regarding the different oxidation methods, GR*ElOx has a slightly higher relative ECSA.
LSV experiments are performed and compared to the overpotential (η) of the uncoated substrate (Fig. 4b). Comparing the two GR* samples with varying degrees of grafting furthermore will allow for the first-time conclusion regarding the effect of grafting on the OER performance.
GR*ChOx and GR*ElOx showed overpotentials of η = 440 mV and η = 450 mV, respectively, at a current density of 10 mA cm−2 while for the uncoated bare steel plate, η = 506 mV is necessary. A current density of 100 mA cm−2 was reached at the overpotentials of 598 mV for GR*ChOx and 610 mV for GR*ElOx. The absence of any notable pre-peaks is consistent with the expectations and conclusions from the Mössbauer spectroscopy measurements, as both electrocatalysts have been fully oxidized to the all-ferric oxidation state.
Compared to the uncoated steel plate, both GR* catalyst materials show a clear OER activity improvement. The overpotentials at 10 mA cm−2 are in the range reported for mössbauerite synthesized at room temperature (η = 540 mV) and at a temperature of 50 °C (η = 400 mV). The offset may be attributed to the different synthesis methods and the rotating disc electrode setup being used to determine the latter values.
Despite the slightly higher ECSA of GR*ElOx compared to GR*ChOx, the latter exhibits a higher activity over the probed potential range. Normalization of the polarization curves to the ECSA (Fig. S7, ESI‡) further confirms the higher intrinsic activity of GR*ChOx compared to GR*ElOx. We attribute this to the higher degree of carbonate grafting of GR*ChOx which can be regarded as a formal replacement of OH− with CO32−, modulating the electronic structure and thus the redox activity of the metal center. According to the HSAB principle, the softer base OH− donates electron density to the adjacent metal centers more readily than the harder base CO32−.45 Therefore, a lower electron density is expected at the electrocatalytically active transition-metal center for GR*ChOx as compared to GR*ElOx. Investigations on NiFe-LDHs have shown that such reduced electron density, charge delocalization and the incorporation of electron-withdrawing anions or cations can be beneficial for the facilitation of higher-valent electrophilic transition metal ion sites. At these sites, the nucleophilic attack of hydroxyl and water molecules is promoted, leading to improved adsorption of reaction intermediates during the OER.46,47
In order to investigate the stability and versatility of the catalyst during continuous operation, chronopotentiometry experiments were conducted. Here, the galvanostatic performance was investigated by applying a current corresponding to a current density of 10 mA cm−2 for 24 h in 1 M KOH (Fig. S8, ESI‡). Over this time, the necessary overpotential to maintain this current was recorded. Initially, GR*ElOx and GR*ChOx allow achieving the current density of 10 mA cm−2 at lower potentials than the uncoated substrate. Throughout the experiment, an increase of the potential was recorded for GR*ElOx (+21 mV), GR*ChOx (+30 mV), and the uncoated bare steel substrate (+26 mV).
The decrease in the performance of both GR* samples may be related to various effects such as the physical detachment of the catalyst material under operating conditions or degradation resulting in the loss of activity and active sites.23 Interestingly, the potential of GR*ChOx increased the most during the first 2 hours of the experiment (inset Fig. S8, ESI‡). Most likely, this is caused by the energy released during harsh chemical oxidation, which might result in a less intimate contact between the substrate and the catalyst. Thus, GR*ChOx is more prone to a loss of electrical contact during operation. Due to the prior electrochemical treatment, GR*ElOx shows an initial performance increase which could be related to the easier formation of percolation paths and active sites. In GR*ChOx, these improving changes are delayed and masked by possible detrimental alterations discussed earlier.
As expected, the performance of the uncoated steel also decreases, likely due to the formation of inactive corrosion products blocking the access to more electroactive centres. The formation of such products was also evidenced by the brownish-black staining of the surface after the experiment.
To gain insight into the alteration of catalysts during catalysis, post-catalysis SEM studies of the employed electrodes were performed. For the bare steel substrate, the results evidence the formation of a porous surface layer (Fig. S9a and b, ESI‡). This indicates the oxidation of Fe0 to soluble Fe-species as predicted by the Pourbaix diagram discussed earlier. Alteration of the electrode under the current load was also evident by the previously described brown-black staining. The post-catalysis XRD measurements (Fig. S10, ESI‡) indicate the presence of Fe1−xO and Fe3O4 deposits formed via reprecipitation. Presumably, these deposits are responsible for the deterioration of catalytic performance during the chronopotentiometry measurements.
For GR*ChOx and GR*ElOx catalysts, the platelet-like morphology was generally preserved, although a loss of the catalyst material (−23%, determined gravimetrically) was observed during the stability experiment. The SEM analysis moreover indicates the degradation of the hexagonal platelet shapes to more rounded corners and the secondary deposits covering the platelets after 24 h (Fig. S9c–f, ESI‡). Based on our previous study examining the stability of mössbauerite in a setup using a rotating disc electrode, we conclude that during electrolysis, small amounts of the catalyst can be dissolved into the electrolyte and are redeposited as amorphous iron (oxy)hydroxides.23 The post-catalysis XRD measurements (Fig. S10, ESI‡) do not show any crystalline phase other than GR* and steel.
Conclusions
Manufacturing catalyst-coated electrodes through a controlled, cheap, and time-efficient corrosion engineering process of steel presents a viable way to obtain electrodes for the oxygen evolution reaction. A carbon steel substrate was used to deposit the known iron-only electrocatalyst GR* via its precursor GR. Clearly, the choice of oxidation method, i.e., chemical or electrochemical oxidation, has a notable influence on the structural constitution and performance of the catalyst. Chemical oxidation gives a higher degree of grafting, which appears to be advantageous for the catalytic performance.Based on these results, further developments will aim towards maximizing the catalyst's coverable surface area, e.g., by deposition on steel wool or iron foam. Of course, concomitant doping can further improve the performance as well.
Author contributions
Sebastian Weiß: investigation, formal analysis, writing – original draft, and writing – review & editing; A. V. Radha: investigation; Catherine McCammon: investigation, formal analysis, and resources; Michael Ertl: investigation; Josef Breu: resources, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for financial support from the Deutsche Forschungsgemeinschaft (DFG) within the collaborative research center 840 “From particulate nanosystems to mesotechnology” (79971943; SFB 840). They thank Mirijam Zobel for the measurement time with her powder X-ray diffractometer. The authors thank O. Khoruzhenko for the graphic design. They further thank R. Timmins and C. Habel for their advice and proofreading the manuscript. They appreciate the support from the Keylab for Optical and Electron Microscopy of the Bavarian Polymer Institute (BPI). They thank Marco Schwarzmann for the corresponding measurements. S. W. acknowledges the support from the Elite Network of Bavaria and thanks the graduate school of the Bavarian Center for Battery Technology (BayBatt) for ongoing support.Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. Yin, A. Molini and A. Porporato, Nat. Commun., 2020, 11, 4781 CrossRef CAS PubMed.
- D. Zhao, Y. Pi, Q. Shao, Y. Feng, Y. Zhang and X. Huang, ACS Nano, 2018, 12, 6245–6251 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- L. Wu, L. Yu, X. Xiao, F. Zhang, S. Song, S. Chen and Z. Ren, Research, 2020, 2020, 17 Search PubMed.
- R. Tolouei, J. Harrison, C. Paternoster, S. Turgeon, P. Chevallier and D. Mantovani, Phys. Chem. Chem. Phys., 2016, 18, 19637–19646 RSC.
- D. G. Lee and H. Gai, Can. J. Chem., 1993, 71, 1394–1400 CrossRef CAS.
- J. M. Schreyer and L. T. Ockerman, Anal. Chem., 1951, 23, 1312–1314 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, Wiley, 2006 Search PubMed.
- S. Papavinasam, in Corrosion Control in the Oil and Gas Industry, ed. S. Papavinasam, Gulf Professional Publishing, Boston, 2014, pp. 133–177 Search PubMed.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- H. Zhong, J. Wang, F. Meng and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9937–9941 CrossRef CAS PubMed.
- B. A. Marinho, R. O. Cristóvão, R. A. R. Boaventura and V. J. P. Vilar, Environ. Sci. Pollut. Res., 2019, 26, 2203–2227 CrossRef CAS PubMed.
- F. Guo, Y. Wu, H. Chen, Y. Liu, L. Yang, X. Ai and X. Zou, Energy Environ. Sci., 2019, 12, 684–692 RSC.
- X. Liu, B. You and Y. Sun, ACS Sustainable Chem. Eng., 2017, 5, 4778–4784 CrossRef CAS.
- H. Schäfer, S. Sadaf, L. Walder, K. Kuepper, S. Dinklage, J. Wollschläger, L. Schneider, M. Steinhart, J. Hardege and D. Daum, Energy Environ. Sci., 2015, 8, 2685–2697 RSC.
- H. Sun, J. Brocato and M. Costa, Curr. Environ. Health Rep., 2015, 2, 295–303 CrossRef CAS PubMed.
- S. Mitra, A. Sarkar and S. Sen, Nanotechnol. Environ. Eng., 2017, 2, 11 CrossRef.
- Y. Liu, X. Liang, L. Gu, Y. Zhang, G.-D. Li, X. Zou and J.-S. Chen, Nat. Commun., 2018, 9, 2609 CrossRef PubMed.
- H. Schäfer, S. M. Beladi-Mousavi, L. Walder, J. Wollschläger, O. Kuschel, S. Ichilmann, S. Sadaf, M. Steinhart, K. Küpper and L. Schneider, ACS Catal., 2015, 5, 2671–2680 CrossRef.
- Y. Wang, D. Yan, S. El Hankari, Y. Zou and S. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef PubMed.
- M. Ertl, C. Andronescu, J. Moir, M. Zobel, F. E. Wagner, S. Barwe, G. Ozin, W. Schuhmann and J. Breu, Chem. – Eur. J., 2018, 24, 9004–9008 CrossRef CAS PubMed.
- M. Usman, J. M. Byrne, A. Chaudhary, S. Orsetti, K. Hanna, C. Ruby, A. Kappler and S. B. Haderlein, Chem. Rev., 2018, 118, 3251–3304 CrossRef CAS PubMed.
- U. Schwertmann and H. Fechter, Clay Miner., 1994, 29, 87–92 CrossRef CAS.
- M. Abdelmoula, P. Refait, S. H. Drissi, J. P. Mihe and J. M. R. Génin, Corros. Sci., 1996, 38, 623–633 CrossRef CAS.
- L. Legrand, L. Mazerolles and A. Chaussé, Geochim. Cosmochim. Acta, 2004, 68, 3497–3507 CrossRef CAS.
- G. J. Long, T. Cranshaw and G. Longworth, Moessbauer Eff. Ref. Data J., 1983, 6, 42–49 Search PubMed.
- C. McCammon, V. Chaskar and G. Richards, Meas. Sci. Technol., 1991, 2, 657 CrossRef CAS.
- C. A. McCammon, Hyperfine Interact., 1994, 92, 1235–1239 CrossRef CAS.
- C. Prescher, C. McCammon and L. Dubrovinsky, J. Appl. Crystallogr., 2012, 45, 329–331 CrossRef CAS.
- Bio-Rad Laboratories, Chelex(R) 100 and Chelex 20 Chelating Ion Exchange Resin – Instruction Manual,https://www.bio-rad.com/webroot/web/pdf/lsr/literature/LIT200.pdf, 2021.
- H. Deng, C. Zhang, Y. Xie, T. Tumlin, L. Giri, S. P. Karna and J. Lin, J. Mater. Chem. A, 2016, 4, 6824–6830 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- P. Refait and J.-M. Génin, Corros. Sci., 1993, 34, 797–819 CrossRef CAS.
- G. Ona-Nguema, M. Abdelmoula, F. Jorand, O. Benali, A. Géhin, J. C. Block and J. M. R. Génin, Hyperfine Interact., 2002, 139, 231–237 CrossRef.
- L. Legrand, S. Savoye, A. Chausse and R. Messina, Electrochim. Acta, 2000, 46, 111–117 CrossRef CAS.
- J.-M. R. Génin, C. Ruby, A. Géhin and P. Refait, C. R. Geosci., 2006, 338, 433–446 CrossRef.
- P. Lyu, M. Ertl, C. J. Heard, L. Grajciar, A. V. Radha, T. Martin, J. Breu and P. Nachtigall, J. Phys. Chem. C, 2019, 123, 25157–25165 CrossRef CAS.
- J. M. R. Génin, A. Christy, E. Kuzmann, S. Mills and C. Ruby, Hyperfine Interact., 2014, 226, 459–482 CrossRef.
- J. M. R. Génin, S. J. Mills, A. G. Christy, O. Guérin, A. J. Herbillon, E. Kuzmann, G. Ona-Nguema, C. Ruby and C. Upadhyay, Mineral. Mag., 2014, 78, 447–465 CrossRef.
- C. Ruby, M. Abdelmoula, S. Naille, A. Renard, V. Khare, G. Ona-Nguema, G. Morin and J.-M. R. Génin, Geochim. Cosmochim. Acta, 2010, 74, 953–966 CrossRef CAS.
- S. Weiß, M. Ertl, S. D. Varhade, A. V. Radha, W. Schuhmann, J. Breu and C. Andronescu, Electrochim. Acta, 2020, 350, 136256 CrossRef.
- J.-M. R. Génin, C. Ruby and C. Upadhyay, Solid State Sci., 2006, 8, 1330–1343 CrossRef.
- H. Xu, D. C. Xu and Y. Wang, ACS Omega, 2017, 2, 7185–7193 CrossRef CAS PubMed.
- L. Fan, P. Zhang, B. Zhang, Q. Daniel, B. J. J. Timmer, F. Zhang and L. Sun, ACS Energy Lett., 2018, 3, 2865–2874 CrossRef CAS.
- Y.-Y. Dong, D.-D. Ma, X.-T. Wu and Q.-L. Zhu, Inorg. Chem. Front., 2020, 7, 270–276 RSC.
Footnotes |
| † Dedicated to Prof. Dr Wolfgang Schuhmann on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: CV, SEM, Mössbauer spectroscopy, CP, and post-catalysis SEM/XRD. See DOI: 10.1039/d1ma00381j |
| This journal is © The Royal Society of Chemistry 2021 |