
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile intra- and intermolecular charge transfer control for efficient mechanofluorochromic material†
Min-Ji
Kim
,
Mina
Ahn
and
Kyung-Ryang
Wee
 *
*
Department of Chemistry and Institute of Natural Science, Daegu University, Gyeongsan 38453, Republic of Korea. E-mail: krwee@daegu.ac.kr
First published on 10th June 2021
Abstract
A series of donor–acceptor–donor (D–A–D) pyrene (Py) 2,7-position-based compounds (CN, F, H, Me, and OMe) were designed and synthesized to demonstrate facile intra- and intermolecular charge transfer control by an electron push–pull effect for an efficient mechanofluorochromic (MFC) material. An examination of the photophysical properties of CN–OMe revealed that the locally excited (LE) and intramolecular charge transfer (ICT) state of the compounds were modulated finely by the electron push–pull substituent effect. In addition, it is noteworthy that the CN emission originated only from the LE state despite the D–A–D molecular system. Moreover, we confirmed that the selective LE and/or ICT state modulation affects intra- and intermolecular charge transfer control in the solid-state emission. In particular, this intra- and intermolecular charge transfer control is directly dominated by MFC phenomena, which means that the molecular electron push–pull substituent effect is dependent on MFC behavior. As a result, this study shows that as the electron-withdrawing group ability increases, the intramolecular interactions become insufficient and induce strong intermolecular D–A interactions for stabilizing molecules, leading to high MFC efficiency that is reversibly repeated several times.
Introduction
Mechanofluorochromic (MFC) organic materials based on an electron donor (D) and electron acceptor (A) are attracting considerable attention because of their unique optical characteristics and various applications such as mechanical sensors, deformation detectors, security inks, memory chips, and optical data storage devices.1–6 MFC organic materials can convert the emission wavelength under external physical stimuli (grinding, pressing, crushing, or rubbing) in the solid-state and can return to their original state by annealing or solvent fuming; this phenomenon can be switched repeatedly. MFC organic materials depend greatly on the transformation of the molecular packing mode, which can occur between the crystalline state and amorphous state or between two different crystalline states. A complicated intermolecular interaction affects the molecular packing mode, such as π–π, hydrogen bonding, and dipole–dipole interactions, making it difficult to predict the properties of MFC organic materials.7–10 Hence, although the development of an excellent MFC organic material is meaningful, there is no clear strategy for the rational molecular design of MFC organic materials. Recently, it was reported that the twisted conformation,11–14 the alkyl chain length,15–17 and the D–A structure9,18–20 directly affect the performance of MFC organic materials. However, a prototypical strategy is required to design new MFC organic materials (e.g., simple, planar, and D–A–D structures) that meet the basic concepts mentioned above and are not limited to these influencing factors. Since the solid-state optical properties largely depend on the molecular packing mode, the properties of the MFC organic material can be predicted if the molecular packing mode can be easily controlled. Therefore, in order to control the molecular packing mode for designing new MFC organic materials, it is necessary to understand the correlation between intra- and intermolecular interactions.This paper presents a novel strategy for the facile intra- and intermolecular control using triphenylamine (TPA) and pyrene (Py) units. TPA is a very popular building block for constructing optoelectronic materials because of its excellent electron donor and hole transport properties.21–24 The nonplanar molecular structure of TPA can induce MFC properties (i.e., facilitate rearrangement of the crystal packing structure upon the application of mechanical stimuli).25–28 As a polycyclic aromatic hydrocarbon, Py is used widely as a classic flat molecule to facilitate a high quantum yield, long fluorescence lifetime, high thermal stability, and high charge carrier mobility.29–32 The photophysical properties of Py depend on the substitution positions in the Py ring, including the active sites at the 1-, 3-, 6-, and 8-positions;33–38 the K-region sites at the 4-, 5-, 9-, and 10-positions;39–41 and the inactive sites at the 2-, and 7-positions.31,42–44 Among the various positions of Py, the 2,7-positions were selected as the target positions for three reasons. (i) when constructing a planar molecular structure with MFC properties, substitution at the 2,7-positions of Py improves the planarity of the molecular structure. (ii) Substitution at the 2,7-positions of Py is synthetically challenging because of the presence of the nodal plane in the HOMO and LUMO,45,46 so there are few reports on the structure–property relationship and MFC properties of Py derivatives functionalized at 2,7-positions. (iii) Because Py possesses a large planar π-conjugated aromatic system, it has strong π–π stacking, causing the quenched emission in the solid state,32,47 but the problem can be overcome, and various fluorescence states can be induced through the appropriate combination of TPA and Py. Cyano-substituted organic molecules have been found to exhibit obvious emission color changes under mechanical stimuli.48–52 On the other hand, further understanding of the interactions between the MFC properties and single-molecule photophysical properties for cyano-substituted organic molecules could be key in the application of an efficient MFC material. Therefore, the correlation between the electron push–pull substituent effect and MFC properties was also investigated by expanding the substituted R group (R = CN, F, H, Me, and OMe) at the para-position of TPA.
In this study, to prove the electron push–pull substituent effect on the single molecular photophysical properties and MFC properties, Py 2,7-position-based D–A–D compounds were designed and synthesized. Interestingly, femtosecond transient absorption (fs-TA) spectroscopic results showed that the emission origin of Py 2,7-position-based D–A–D compounds was controlled by the electron push–pull substituent effect. X-ray crystallographic analysis revealed these photophysical properties and MFC properties to be closely related. These results are helpful for better understanding the influence of the electron push–pull substituent effect on the MFC phenomenon and provide structural insight into the design of new Py 2,7-position D–A–D compound-based MFC materials.
Results and discussion
Synthesis
Scheme 1 shows the synthetic procedure for a series of pyrene-based D–A–D compounds. First, direct borylation of pyrene at the 2 and 7 positions was performed in situ via a reaction of [{Ir(μ-OMe)cod}2] with 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) according to a previous report.53 As an electron donor, the functional R group (R = CN, F, H, Me, and OMe) at the para position and bromo-substituted triphenylamine derivatives were prepared using previously reported procedures.54 Pyrene-based D–A–D compounds were then synthesized by the palladium-catalyzed Suzuki–Miyaura coupling reaction using 2,7-bis(Bpin)pyrene (pin = OCMe2CMe2O) with two molar ratios of bromo-substituted p-(R)-triphenylamines (R = CN, F, H, Me, and OMe). All compounds were purified by silica gel column chromatography with yields ranging from 16 to 53%. The molecular structures of all products were determined by 1H- and 13C{1H}-nuclear magnetic resonance (NMR) spectroscopy, elemental analysis, and mass spectrometry (Fig. S1–S11, ESI†). Finally, the structures of CN, F, and Me were authenticated by single-crystal X-ray crystallography (Fig. S12–S14, ESI†). The Experimental section and ESI† provide details of the synthetic procedures and characterization data.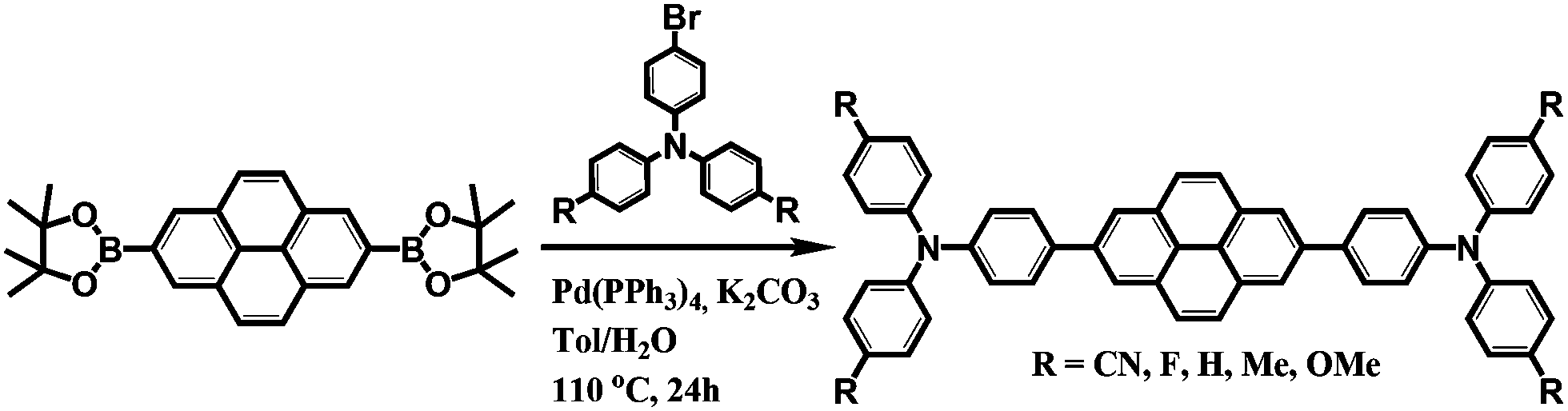 | ||
| Scheme 1 Synthesis of pyrene 2,7-positions based D–A–D compounds. | ||
Theoretical characterization and electrochemical properties
To consider the optical properties of pyrene derivatives, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed at the B3LYP 6-31G (d,p) level. In all the compounds studied, the highest occupied molecular orbital (HOMO) was largely distributed on the TPA moiety, and the lowest unoccupied molecular orbital (LUMO) was strongly localized on the Py moiety (Fig. S34 and S35–S39, ESI†). These changes in the orbital distributions of the HOMOs and LUMOs showed that charge transfer occurs from the TPA to Py in the excited state. The experimental HOMO energy levels could be determined from the oxidation onset potentials from the cyclic voltammetry (CV) curves (Fig. S15, ESI†), and the HOMO energy levels of OMe–CN were estimated to be ca. −5.44 to −5.92 eV. Because no cathodic reduction processes were measured, the experimental LUMO energy levels were estimated to be ca. −2.48 to −2.70 eV from the optical band gaps calculated from the edges of the absorption spectra and HOMO energy levels. The calculated HOMO energy levels ranged from −4.55 to −5.77 eV, and the calculated LUMO energy levels ranged from −1.39 to −2.01 eV (Table S4, ESI†). The calculated values were higher than the experimental values for all compounds, but the tendencies were similar. These results show that an increase in the electron-donating ability destabilizes the HOMO more than the LUMO, thereby narrowing the bandgap and producing a red-shift in the absorption spectrum from CN to OMe. The electron push–pull effect of the para-substituents of triphenylamine on the HOMO and LUMO levels is also explained as a function of the Hammett substituent constants (σp).55 Fig. S16 (ESI†) shows that, the HOMO and LUMO energy levels change according to the σp values with a good linear correlation and the slope of the HOMO was larger than that of the LUMO. These results show that the substituent effect has a greater impact on the HOMO levels than the LUMO levels, resulting in a tuning of the bandgap. For all compounds, the lowest-energy singlet transition involved the LUMO ← HOMO transitions, all of which were ICT transitions from the TPA to Py. This transition shifting theoretically followed the order, CN (381 nm), F (411 nm), H (415 nm), Me (425 nm), and OMe (442 nm), which is consistent with the electron push–pull substituent effect (Fig. S40–S44 and Table S16–S20, ESI†).Photophysical properties
Fig. 1 shows the steady-state UV-visible absorption and emission spectra of CN–OMe, and the spectral parameters are summarized in Table 1. In the UV-visible absorption spectra, all pyrene-based D–A–D compounds showed relatively weak absorption band in the range of 250–300 nm, which was attributed to a locally excited (LE) transition of the TPA and Py moieties. All compounds exhibited a strong absorption band with a vibrational progression corresponding to the S2 ← S0 excitation at approximately 345 nm, which is characteristic of the S2 ← S0 excitation (LE transition) of pyrene itself, called a “pyrene-like” transition (Fig. S17, ESI†).45 In addition, all compounds produced a weak absorption shoulder corresponding to the S1 ← S0 excitation at ∼362 nm, which was attributed to intramolecular charge transfer (ICT) transition from the para-substituted TPA to the Py moiety. In particular, the ICT transition in the absorption spectra showed a gradual red-shift in the order CN < F < H < Me < OMe, and the absorption intensity of the CT band also increased gradually. This indicates a strong electronic coupling between the para-substituted TPA and Py, as the electron-donating ability increases in the ground state. In addition, despite Py substitution at the 2,7-positions, the result agreed well with the substituent effect in the ground state. In the emission spectra, the emission maxima of the pyrene-based D–A–D compounds ranged from 421 to 522 nm in CH2Cl2 solutions with a systematic bathochromic shift in the order CN < F < H < Me < OMe (Fig. 1b), indicating that the energy band gap was fine-tuned between the ground and excited states by the electron push–pull substituents. Fluorescence quantum yield (Φf) measurements were conducted in CH2Cl2 to investigate the properties of the excited state for the compounds CN–OMe. The quantum yields for the compounds were in the range of 0.1–0.06, all being lower than that of pyrene (Φf = 0.28, in CH2Cl2),56 because of the deactivation pathways from the excited state, caused by the weak π-conjugated of pyrene 2,7-positions and the rotational motion of the phenyl rings of the donor moiety. Fluorescence lifetime measurements were performed using the time-correlated single-photon counting detection (TCSPC) method (Fig. S29–S31, and Table S8, ESI†), and the fluorescence lifetimes increased with the increasing electron-withdrawing ability of the substituents. These results can be mainly by the radiative and nonradiative decay rate constants (krad and knr), which were calculated from the Φf and τF values (Table 1). The krad values of all compounds were similar, while the knr value increased with increasing electron-donating ability, which means that the knr value is controlled by the electron effect of the substituents. In addition, the knr value of all compounds was larger than krad, indicating that the weak π-conjugated structure of the 2,7-positions of pyrene favors nonradiative decay. To examine the properties in the solid state, thin-film absorption and emission spectra were also measured (Fig. S18, ESI†). The thin-film absorption and emission spectra of CN–OMe exhibited red-shifts compared to the corresponding spectra in an n-hexane solution, suggesting that pyrene-based D–A–D compounds formed J-aggregates.57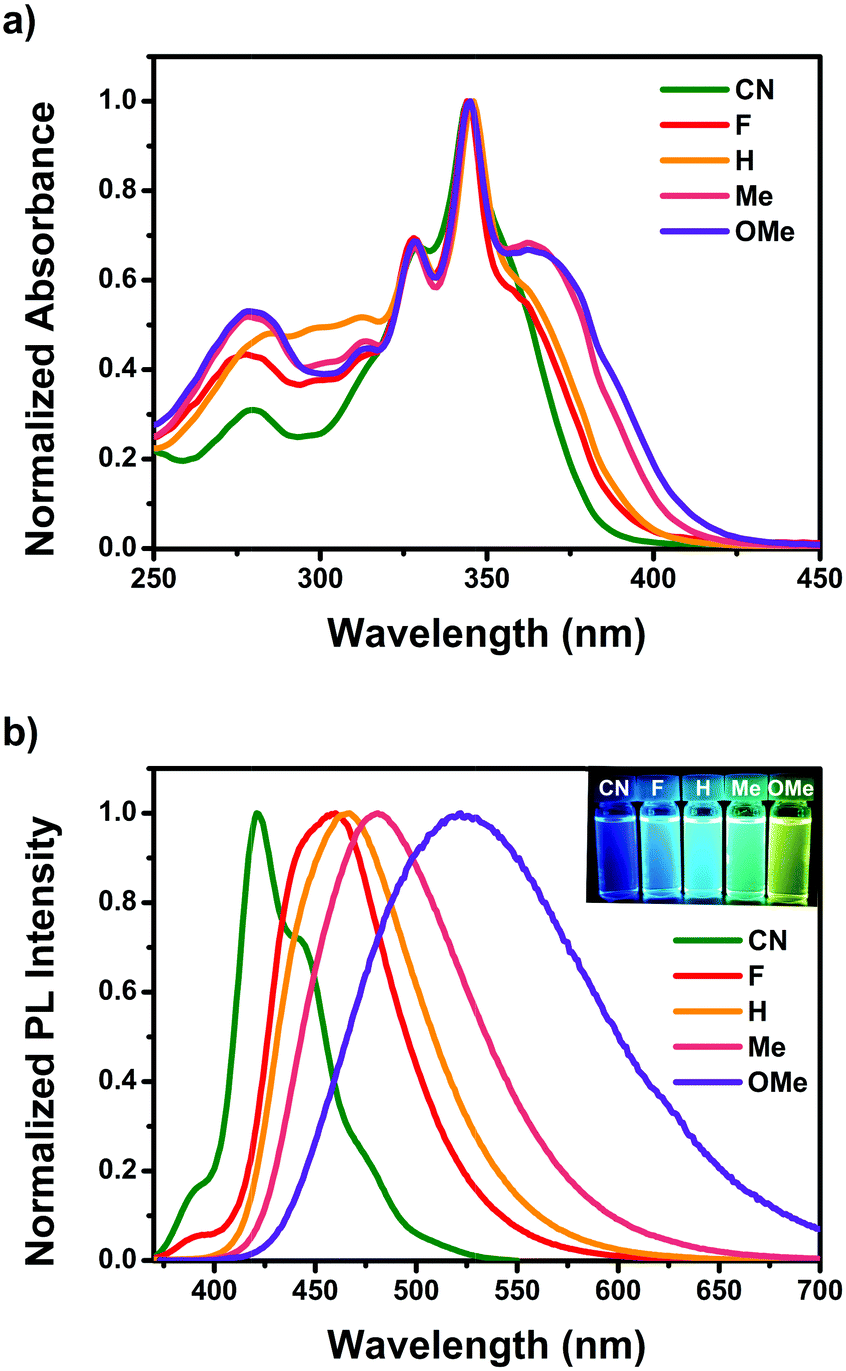 | ||
| Fig. 1 (a) Optical absorption and (b) emission spectra of CN–OMe in CH2Cl2. Insets: Photographs of CN–OMe in CH2Cl2 solution under UV irradiation at 365 nm. | ||
| λ abs (nm) | λ em (nm) | Stokes Shift (cm−1) | Φ f | τ F (ns) | k rad × 107 (s−1) | k nr × 107 (s−1) | k rad/knr | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Sola | Filmb | Sola | Filmb | |||||||
| a Measured in CH2Cl2 at room temperature (RT). b Measured from a drop-casted film on glass. Double exponential factors scaled to 100 and given in parentheses. c Fluorescence quantum yields, with 9,10-diphenylanthracene (Φf = 0.95, ethanol) as the standard, in CH2Cl2 at RT. d Fluorescence lifetime measured in CH2Cl2. e Values of krad and knr were calculated using krad = Φf/τF and knr = (1/τF) − krad, respectively. | ||||||||||
| CN | 357 | 376 | 421 | 453 | 4258 | 0.10 | 20.3 | 0.50 | 4.43 | 0.11 |
| F | 360 | 358 | 460 | 465 | 6039 | 0.11 | 8.8 (31), 19.6 (69) | 1.24, 0.56 | 10.13, 4.56 | 0.12, 0.12 |
| H | 362 | 357 | 467 | 467 | 6211 | 0.11 | 7.2 (29), 17.8 (71) | 1.52, 0.61 | 12.41, 5.02 | 0.12, 0.12 |
| Me | 363 | 352 | 481 | 468 | 6758 | 0.092 | 9.2 (37), 15.6 (63) | 0.99, 0.59 | 9.82, 5.81 | 0.10, 0.10 |
| OMe | 365 | 348 | 522 | 475 | 8240 | 0.060 | 8.6 (35), 16.1 (65) | 0.70, 0.37 | 10.98, 5.84 | 0.06, 0.06 |
As shown in Fig. S26 (ESI†), the emission spectra of F–OMe showed a hypsochromic shift in methanol, a strong polar solvent, but CN did not show this phenomenon due to the protic solvent. To investigate their intermolecular interaction by the protic solvent, the aggregation-induced PL spectra of pyrene 2,7-positions based D–A–D compounds were measured (Fig. S19, ESI†) in THF/water mixtures with different water volume fractions (fw). As the fw increased from 10 to 60%, the emission spectra of F–OMe exhibited a gradually reducing PL intensity and a red-shifted emission peak (465, 471, 486, and 556 nm at fw = 60%, respectively), which was attributed to the stabilized CT states resulting from an increase in the polarity of the solvent mixture. As the fw increased from 70 to 90%, the emission spectra of F–OMe showed a more pronounced vibronic structure, and blue-shifted emission peaks were observed at approximately 450 nm. These results were attributed to the formation of aggregates in the THF/water mixtures with fw > 60%. Generally, as the water volume fractions increase, aggregates are formed, resulting in a bathochromic shift,58 so a blue-shift in the aggregated state is uncommon. In particular, in the emission spectrum of CN, when the fw was increased from 10 to 60%, showed neither a red-shift due to CT state stabilization nor an unusual blue-shift due to the formation of aggregates when the fw > 60% were also observed. Therefore, from the observations, despite the D–A–D molecule, CN is unaffected by a protic solvent, suggesting that CN does not have an intermolecular interaction in the solution state.
Mechanofluorochromic properties
As shown in Fig. 2a and b (Fig. S20 and Table S6, ESI†), the pristine powder of CN–Me showed emission peaks/shoulders at 432/446, 437/450, 450, and 449 nm, respectively. Upon grinding, the emission profiles exhibited a bathochromic shift with emission peaks/shoulders at 440/456, 444, 466, and 453 nm for CN, F, H, and Me, respectively, mostly accompanied by a broader emission band than that of the pristine powder. These results imply that the internal space between molecules is collapsed by mechanical stimulation, and the packing morphology is changed, resulting in stabilization of the CT state. After exposure to acetone vapor, the emission spectrum of CN showed peak/shoulder at 430/448 nm, which means that the emission of ground powder has been restored to its original condition, indicating the reversibility of mechanofluorochromism. Although not as much as CN, the emission band of F almost returned to its original shape by acetone fuming with an emission peak/shoulder at 430/448 nm. On the other hand, the ground powder of H did not perfectly revert to the pristine unground state emission after fuming with acetone vapor and showed an emission maximum at 458 nm, which was red-shifted compared to that of the pristine powder. Similarly, when the ground powder of Me was fumed with acetone vapor, the emission maximum (450 nm) was similar to that of the pristine powder, but a broader emission band and large FWHM were observed compared to the pristine powder. This means that the ground powder of Me does not perfectly return to its original state. In the case of a strong electron-donating group OMe, OMe showed an emission peak at 481 nm. Moreover, the emission spectrum of OMe remained unchanged even during the grinding and acetone fuming procedures, suggesting that OMe does not possess MFC properties. Furthermore, as the electron-withdrawing ability increased, the emission color indicated that the transition between two different states could be repeated reversibly many times without a fatigue response by the repeated grinding and acetone fuming treatments (Fig. 2c, d and Fig. S21, ESI†). Based on these results, the MFC properties of the Py 2,7-position-based D–A–D compounds showed excellent reversibility of the mechanofluorochromic processes as the electron-withdrawing ability was increased. Hence, the mechanofluorochromism was well controlled by the electron push–pull substituent effect. | ||
| Fig. 2 Emission spectra of (a) CN, and (b) OMe in different solid-state: pristine, ground, and fumed powder. Reversible switching of emission of pristine powder (c) CN, and (d) OMe by repeating grinding-acetone fuming cycles. (e) Fluorescent images of filter papers loaded with CN–OMe were written with the letter ‘D’, fumed with acetone vapor, and rewritten with the letter ‘U’ under a 365 nm UV lamp. | ||
Based on the MFC properties of the Py 2,7-position-based D–A–D molecules, the mechanofluorochromism in potential applications for mechano sensors and security inks was compared visually. The powder of CN–OMe was dispersed on the filter paper as a thin-film, and upon excitation by UV lamp (365 nm), exhibited light violet emission for CN, blue emission for F, sky blue emission for H, light blue for Me, and blue-green emission for OMe (Fig. 2e). The letter “D” was written on the thin film with a spatula as a pen. For CN–Me, when viewed with the naked eye, the letter emitted bluish fluorescence compared to the background color, highlighting the red-shift of the PL spectra due to grinding. In contrast, for OMe, the letter emitted blue-green fluorescence similar to the background color. As the electron-withdrawing ability increased, i.e., going from Me to CN, the letter “D” was erased quickly and easily using acetone vapor as an eraser. In the case of OMe, there was no change, such as that the letter “D” was erased. The letter “U” was rewritten on the thin-film and was observed distinctly as the electron-withdrawing ability was increased, which was affected by the degree of erasing the letter “D”. These results visually show that the mechanofluorochromism increases with increasing electron-withdrawing ability, indicating a proportional correlation.
The absorption and emission spectra of the poly(methyl methacrylate) (PMMA) thin film were measured to determine if the origin of the MFC behavior of the Py 2,7-position-based D–A–D compounds was due to a rigid single molecular in the solid state or an intermolecular interaction (Fig. S22 and S23, ESI†). The absorption and emission spectra of the 1 wt% doped PMMA films showed a red-shift as the electron-donating ability increased, which was in agreement with the electron push–pull substituent effect. PMMA film emission maxima of CN–OMe were observed at 419, 430, 430, 437, and 448 nm with a broader emission band than in a n-hexane solution. In particular, the PMMA film emission spectra of CN–OMe were similar to that of the n-hexane solution with single molecular properties than that of the pristine powder, meaning that the MFC behavior originates from intermolecular interactions.
Powder X-ray diffraction (PXRD) of the pristine state and the single-crystal X-ray diffraction were conducted for CN, F, and Me to gain insight into the MFC behavior of the powder state. As shown in Fig. S24 (ESI†), the PXRD patterns of the pristine powder samples showed sharp and intense diffraction peaks in the range of 5°–50°, suggesting that these compounds are the microcrystalline structure in the pristine powder state. The PXRD patterns of the pristine powder of the three compounds were similar to single-crystal X-ray diffraction powder patterns. In particular, for F, the PXRD pattern of the pristine powder corresponded almost perfectly to the single-crystal X-ray diffraction powder patterns. Thus, the same molecular packing was present in the crystals and pristine powders, which means that the single crystal X-ray structure provides more insight into the MFC behavior.
For X-ray crystallographic analysis, a single crystal of CN, F, and Me was grown by slow evaporation in a CH2Cl2/n-hexane or CHCl3/n-hexane mixture at room temperature. As expected, the X-ray structure and molecular packing of the crystals of these three compounds were different, and their crystal data (Fig. 3; Tables S1–S3, ESI†) list the collection parameters. CN molecules consist only of a strong donor–acceptor contact packing structure, forming two dimeric structures with a parallel arrangement (Fig. 3a and b). In one dimeric form, the Py moiety forms short contacts with the TPA moiety: intermolecular C⋯H (2.89 Å), and C⋯H (2.87 Å). Similarly, in the other dimeric form, intermolecular donor and acceptor moieties interact via the C⋯H interaction with a distance of 3.12 Å. In contrast, the F and Me molecules are formed in two different dimeric structures with a weaker donor–acceptor contact packing structure than that of CN. In the case of F, although one dimeric structure is arranged in a parallel packing mode, unlike CN, the contact between Py and TPA with a distance of 3.12 Å is longer than the contact between the phenyl groups of the TPA moiety with a distance of 2.87 Å (Fig. 3c (top)). In the other dimeric structures (Fig. 3c (bottom)), F molecules are formed in a twisted arrangement, and the intermolecular donor-donor interaction (C⋯H contacts of 2.72 and 2.82 Å) is more dominant than the intermolecular donor–acceptor interaction (C⋯H contact of 3.70 Å). In the case of Me (Fig. 3e (top)), one dimeric form has an intermolecular donor–donor interaction and acceptor–acceptor interaction due to the bulky electron-donating methyl group without an interaction between the Py and the TPA moiety: C⋯H (2.74 Å), C⋯H (3.25 Å), and C⋯H (2.66 Å). The other dimeric structure of Me is formed in a twisted packing mode to enhance the intermolecular donor–acceptor interaction (Fig. 3e (bottom)), stabilizing the bulky molecular structures; intermolecular Py–TPA interaction (C⋯H with a distance of 2.61 Å). Therefore, these results show that as the electron-withdrawing ability increases, the intermolecular donor–acceptor interactions also increase, leading to high mechanofluorochromic efficiency that can be repeated reversibly many times. Also, these intermolecular donor–acceptor interactions are related to the torsion angle of the donor–acceptor connected moiety.
 | ||
| Fig. 3 Molecular packing of two dimeric structures of (a) CN, (c) F, and (e) Me in single crystals. The stacking images in the crystal (b) CN, (d) F, and (f) Me. (g) Schematic diagram of the different packing structures. | ||
In the crystal packing structure, the molecules of the three compounds were not a coplanar conformation due to the nonplanar shape of the donor moiety. Upon mechanical stimulation, these twisted structures can be transformed to a more planar structure, causing morphological changes and leading to a bathochromic shift of the MFC properties.59,60 The smaller the torsion angles of the donor–acceptor connected moiety, the closer the intermolecular distance, which further promotes the intermolecular donor–acceptor interaction. From this point of view, the torsion angles between the central Py plane and the TPA increased in the order CN (30.0°) < F (38.8°) < Me (40.4°), based on the largest value (Fig. 3). Me had the largest torsion angle of the three compounds due to the introduction of a relatively bulky electron-donating methyl group. Interestingly, within the electron-withdrawing group, F had a larger torsion angle than the bulky CN. These results suggest that it is related to the single-molecule optical properties according to the electron push–pull substituent effect.
Influence of single-molecule optical properties on mechanofluorochromism
Photophysical spectra for the pyrene-based D–A–D compounds were initially measured in various solvent environments to investigate single-molecular intrinsic characteristics. The absorption spectra of all compounds showed a weak solvent polarity dependency (Fig. S25 and Table S7, ESI†). In contrast, the emission spectra of F–OMe exhibited a markedly bathochromic shift, and a gradually broadened and gradually vanished vibronic resolution with increasing solvent polarity from n-hexane to acetonitrile, indicating the presence of an ICT state, which is sensitive to the surroundings, in the excited state (Fig. S26, ESI†). The F–OMe exhibited a large solvatochromic shift (47, 55, 110, and 189 nm) in the emission maxima between n-hexane and acetonitrile, as the electron-donating ability of the substituents increased. Also, in a high-polarity solvent ACN, the decays of the fluorescence profiles of F–Me are a double exponential, whereas the decay of fluorescence profiles of OMe are a single exponential (Fig. S31, ESI†). Based on these results, the double exponential fluorescence decay of F–Me can be attributed to the LE and ICT. And, the single exponential fluorescence decay of OMe can be attributed to the favorable and fast ICT by the strong electron-donating group and stabilized ICT state in ACN. On the other hand, with increasing solvent polarity, the emission spectrum of CN did not show a bathochromic shift and had a distinguished vibronic structure. In addition, CN exhibited identical single exponential fluorescence decay, despite the changing polar environment. Thus, the fluorescence decay of CN can be attributed to the typical LE state character insensitive to the changing of the polar environment.To clarify the solvation effects, this study analyzed the dipole moments between the ground and excited states using a Lippert–Mataga plot (Fig. S27, S28 and Table S8, ESI†).61,62 As shown in Fig. S27b, ESI,† the plot of F–OMe did not obey the linear relationship predicted by the Lippert–Mataga equation well and showed two sets of linearity indicative of two different excited states: one for high polarity solvents and the other for low polarity solvents. As the solvent polarity increases, the ICT state begins to stabilize, leading to a significantly lower energy barrier between the LE and ICT states. As a result, F–OMe, which favorably transitioned from the LE to the ICT state, has strong ICT character in a high polarity solvent; the excited state dipole moments of F–OMe were 45.5, 41.4, 94.8, and 108.3 D in high polarity solvents. As the solvent polarity was decreased, the energy level of the ICT state began to increase, which significantly expanded the energy barrier between the LE and ICT states. Accordingly, the fraction of the transition from the LE to ICT state decreased. This means that in low polarity solvents, the fluorescence of F–OMe originates mainly from the LE state; the excited state dipole moments of F–OMe (9.0, 10.1, 46.0, and 45.2 D) in low polar solvents. On the other hand, the Lippert–Mataga plot of CN obtained one type of linear relationship regardless of the solvent polarity. Thus, the slope of the approximate line was almost zero, and ground state dipole moment was calculated to be 0.0 D, indicating a typical LE state despite the D–A–D system. As a result, the slope trend of the Lippert–Mataga plot in both the high and low polarity solvent regions follows the order CN < F, H < Me < OMe, suggesting that the transition from LE to ICT state is controlled by the substituent effect.
Femtosecond transient absorption (fs-TA) measurements were conducted to obtain more information on the excited state properties for the pyrene-based D–A–D compounds. And the origin of the MFC behavior is further evidenced by the excited state properties. The fs-TA spectra of CN–OMe in the medium polarity solvent CH2Cl2 were obtained by laser excitation at 340 nm (Fig. 4 and Fig. S32, S33, ESI†). All compounds exhibited a transient absorption band at ∼550 nm during the first hundreds of femtoseconds, which was attributed to the Franck–Condon (FC) state. Subsequently, the TA band was observed at approximately 633 nm (CN), 610 nm (F), 610 nm (H), 619 nm (Me), and 573 nm (OMe), which were ascribed to the LE state of 1(CN–OMe)*. Except for CN, as the time delay increased, for F–OMe, the TA band around 715 nm increased concomitantly with decay of the TA band at ∼610 nm, which decayed with time constants of 0.9 ± 0.04, 1.8 ± 0.08, 1.5 ± 0.1, and 2.0 ± 0.1 ps, respectively. The increasing TA band at approximately 715 nm was attributed to the ICT state of 1(F–OMe)* with rise time constants of 2.3 ± 0.3, 0.06 ± 0.02, 0.1 ± 0.08, and 3.9 ± 0.4 ps, respectively. In particular, in the case of H and Me, the TA band at approximately 715 nm exhibited decay profiles at a time delay of 2–2.6 ps. This suggests that compounds H and Me are relatively less polarized than F and OMe by the intermediate electron push–pull substituent effect, resulting in the rapid stabilization of the ICT state. For CN, the TA band at approximately 633 nm increased together with a rise time constant of 1.6 ± 0.4 ps after the initial FC state with a decay time constant of 0.7 ± 0.06 ps, and no TA band at approximately 715 nm was observed. This means that the emissive state of CN originates from only the LE state despite the D–A–D system. These results show that the LE and ICT state of the 2,7-positions pyrene-based D–A–D compounds were modulated finely by the electron push–pull substituent effect. Therefore, the evolution of the excited state of the Py 2,7-position-based D–A–D compounds in a high polarity solvent, such as ACN, can be summarized as follows, as shown in Fig. 4c.
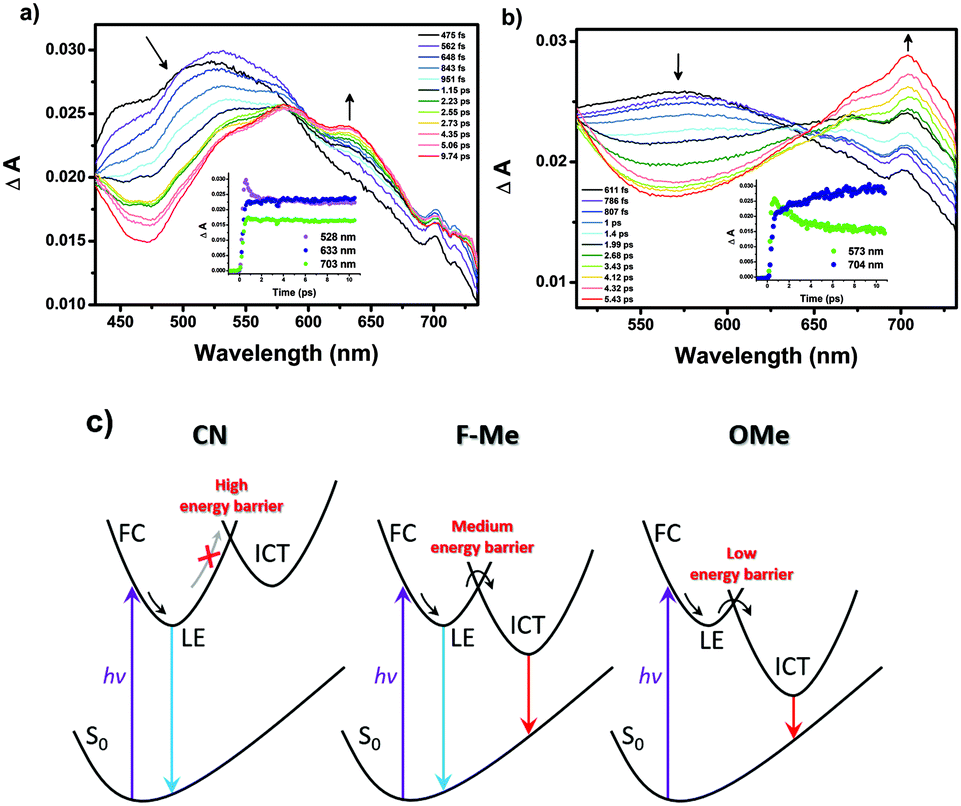 | ||
| Fig. 4 Chirping corrected transient absorption spectra of (a) CN, and (b) OMe in CH2Cl2. The excitation wavelength was 340 nm. Inset: Decay and rise profiles were monitored at selected wavelengths. (c) Schematic potential curves for CN–OMe in a high polarity solvent. | ||
Based on the results obtained, the MFC properties and molecular packing mode are related to the optical properties of Py 2,7-position-based D–A–D compounds according to the electron push–pull substituent effect. As the electron-withdrawing ability increases, the intramolecular charge transfer of the Py 2,7-position-based D–A–D compounds decreases, and consequently the intramolecular interaction becomes insufficient (Fig. 5). Insufficient intramolecular interactions led to strong intermolecular interactions, causing small torsion angles by the strong intermolecular interactions. This smaller torsion angle appears as interactions between the donor and acceptor and facilitates intermolecular short contact stacking. In other words, it induces strong intermolecular donor–acceptor charge transfer for molecular stabilization. These strong intermolecular donor–acceptor interactions cause strong molecular rearrangements when the pristine state is ground and then fumed. It leads to high MFC efficiency, which can be reversibly repeated many times. Thus, the MFC efficiency in the Py 2,7-position-based D–A–D compounds is controlled by the electron push–pull substituent effect. The relationship between the photophysical properties according to the electron push–pull substituent effect of the compounds and the MFC properties can be used to build a predictive method for the properties of the MFC organic material at the molecular level.
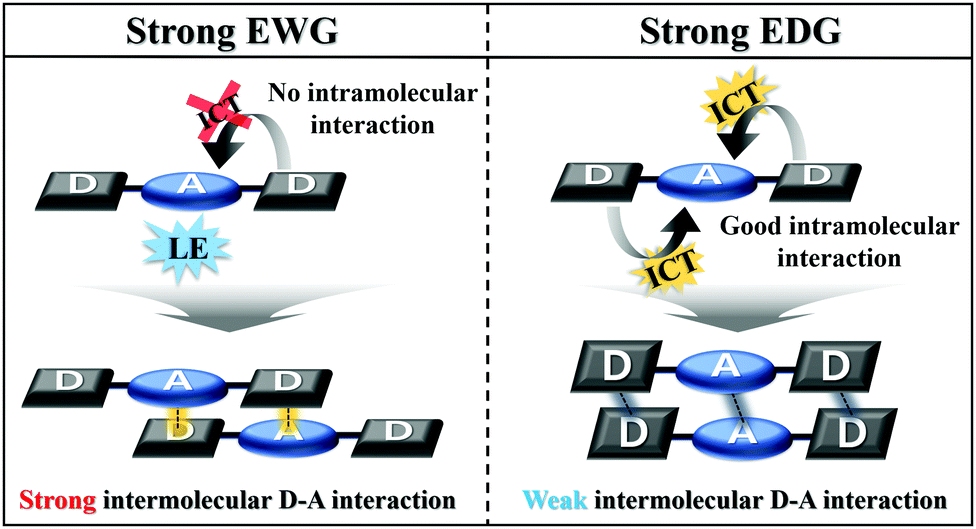 | ||
| Fig. 5 Schematic explanation of the correlation between single-molecule optical properties and molecular packing mode. | ||
Conclusions
Py 2,7-position-based D–A–D compounds were designed and synthesized to understand the correlation of single-molecule photophysical properties (intramolecular interaction) and MFC properties (intermolecular interaction) according to electron push–pull substituent effect. The MFC properties of the Py 2,7-position-based D–A–D compounds showed the excellent reversibility of the MFC processes as the electron-withdrawing ability was increased. These MFC behavior by the substituent effect are closely related to the photophysical properties. For the photophysical properties of the compounds, the ICT state is not simply controlled, but the energy levels of the two different excited states are regulated by the substituent effect. Femtosecond transient absorption measurements revealed the excited state properties according to the electron push–pull substituent effect: pure LE state of CN and transition from LE to ICT state of F–OMe. Based on these results, it was shown that as the electron-withdrawing ability increases, the intramolecular charge transfer decreases, and the intramolecular interactions become insufficient, resulting in strong intermolecular donor–acceptor interactions. Therefore, the MFC behavior increases as the intermolecular donor–acceptor interactions are improved. Overall, the solid-state optical properties and molecular packing mode of organic molecules can be tuned by a simple alteration of the functional R group. These results provide a molecular design strategy for highly efficient new MFC organic materials. Further studies on color-tunable pyrene-based MFC organic materials are ongoing.Experimental
General information
Based on standard Schlenk techniques, all of the synthesis experimental procedures were performed under a dry argon condition. Reagents and solvents were purchased from commercial sources and used as received without further purification, unless otherwise stated. All reactions were monitored with thin layer chromatography (TLC) using commercial TLC plates (Merck Co.). Silica gel column chromatography was performed on silica gel 60 G (230–400 mesh ASTM, Merck Co.). The synthesized compounds were characterized by 1H-NMR or 13C{1H}-NMR, and elemental analysis. The 1H and proton-decoupled 13C spectra were recorded on a Bruker500 spectrometer operating at 500 and 125 MHz, respectively, and all proton and carbon chemical shifts were measured relative to internal residual chloroform (99.5% CDCl3) from the lock solvent. The elemental analyses (C, H, N, O) were performed using Thermo Fisher Scientific Flash 2000 series analyzer. The GC-MS analysis was performed using a highly sensitive Gas Chromatograph/Mass Selective Detector spectrometer (Agilent, 7890B-5977B GC/MSD). The 2,7-bis(Bpin)pyrene53 and bromo-substituted p-(R)-triphenylamine54 were prepared based on the previously published method. The crystal structure was determined by single-crystal X-ray diffractometer at the Western Seoul Center of Korea Basic Science Institute.Synthesis of 2,7-bis(p-(R)-triphenylamino)pyrene
A mixture of 2,7-bis(Bpin)pyrene (0.5 g, 1.1 mmol), bromo-substituted p-(R)-triphenylamine (2.42 mmol, R = CN, F, H, Me, OMe), Pd(PPh3)4 (10 mol%), and K2CO3 (1.37 g, 9.9 mmol) in toluene/H2O (40 mL/10 mL) was refluxed under argon at 110 °C for overnight. After cooling to room temperature, deionized water (50 ml) was added and the organic layer was collected. The water layer was washed using methylene chloride (×3) to extract remaining organics. After combined organic extracts, the organic layer was dried over anhydrous MgSO4 and filtered. Solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography.2,7-Bis[4′-[bis(4′′-cyanophenyl)amino]phenyl]pyrene (CN)
Eluent: ethyl acetate/n-hexane = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Pale brown powder (0.46 g, yield: 53%). 1H-NMR (500 MHz, CDCl3, ppm) δ 8.43 (s, 1H), 8.20 (s, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 9.0 Hz, 2H), 7.34 (d, J = 8.5 Hz, 1H), 7.25 (d, J = 9.0 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 150.15, 144.52, 139.45, 137.74, 133.68, 131.68, 129.77, 128.10, 127.22, 123.89, 123.73, 123.18, 118.89, 106.17. GC-MS (m/z) calcd. for C56H32N6: 788.27, found: 788.4 [M]+. Anal. calcd. for C56H32N6: C 85.26, H 4.09, N 10.65. Found: C 85.71, H 3.88, N, 10.34.
:2. Pale brown powder (0.46 g, yield: 53%). 1H-NMR (500 MHz, CDCl3, ppm) δ 8.43 (s, 1H), 8.20 (s, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 9.0 Hz, 2H), 7.34 (d, J = 8.5 Hz, 1H), 7.25 (d, J = 9.0 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 150.15, 144.52, 139.45, 137.74, 133.68, 131.68, 129.77, 128.10, 127.22, 123.89, 123.73, 123.18, 118.89, 106.17. GC-MS (m/z) calcd. for C56H32N6: 788.27, found: 788.4 [M]+. Anal. calcd. for C56H32N6: C 85.26, H 4.09, N 10.65. Found: C 85.71, H 3.88, N, 10.34.
2,7-Bis[4′-[bis(4′′-fluorophenyl)amino]phenyl]pyrene (F)
Eluent: CH2Cl2/n-hexane = 1:7. Pale yellow powder (0.22 g, yield: 26%). 1H-NMR (500 MHz, CDCl3, ppm) δ 8.38 (s, 1H), 8.14 (s, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.20–7.15 (m, 3H), 7.04 (t, J = 8.0 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 147.53, 143.73, 135.06, 131.52, 128.73, 127.91, 126.25, 126.18, 123.62, 123.37, 122.85, 116.36, 116.18. GC-MS (m/z) calcd. for C52H32F4N2: 760.25, found: 760.3 [M]+. Anal. calcd. for C52H32F4N2: C 82.09, H 4.24, N 3.68. Found: C 81.96, H 4.21, N 3.52.
2,7-Bis(triphenylamino)pyrene (H)
Eluent: CH2Cl2/n-hexane = 1:5. Pale yellow powder (0.2 g, yield: 26%). 1H-NMR (500 MHz, CDCl3, ppm) δ 8.40 (s, 1H), 8.15 (s, 1H), 7.80 (d, J = 9.0 Hz, 1H), 7.33 (t, J = 8.5 Hz, 2H), 7.29–7.28 (m, 1H), 7.22 (d, J = 8.5 Hz, 2H), 7.09 (t, J = 7.0 Hz, 1H). 13C{1H} (125 MHz, CDCl3, ppm) δ 147.71, 131.51, 129.34, 128.65, 127.90, 124.52, 124.12, 123.41, 123.04. GC-MS (m/z) calcd. for C52H36N2: 688.29, found: 688.4 [M]+. Anal. calcd. for C52H36N2: C 90.67, H 5.27, N 4.07. Found: C 90.22, H 5.17, N 3.96.
2,7-Bis[4′-[bis(4′′-methylphenyl)amino]phenyl]pyrene (Me)
Eluent: CH2Cl2/n-hexane = 1:5. Pale yellow powder (0.39 g, yield: 48%). 1H-NMR (500 MHz, CDCl3, ppm) δ 8.28 (s, 1H), 8.03 (s, 1H), 7.66 (d, J = 8.5 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 7.05-7.01 (m, 4H). 13C{1H} (125 MHz, CDCl3, ppm) δ 145.26, 134.38, 132.71, 131.47 129.97, 128.47, 127.85, 124.78, 123.32, 122.90, 20.87. GC-MS (m/z) calcd for C56H44N2: 744.35, found: 744.4 [M]+. Anal. calcd. for C56H44N2: C 90.29, H 5.95, N 3.76. Found: C 90.22, H 5.81, N 3.59.
2,7-Bis[4′-[bis(4′′-methoxyphenyl)amino]phenyl]pyrene (OMe)
Eluent: CH2Cl2/n-hexane = 1:1. Yellow powder (0.14 g, yield: 16%). 1H-NMR (500 MHz, CDCl3, ppm) δ 8.36 (s, 1H), 8.12 (s, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.18–7.12 (m, 3H), 6.90 (d, J = 9.0 Hz, 2H). 13C{1H} (125 MHz, CDCl3, ppm) δ 131.45, 127.83, 126.68, 123.20, 121.23, 114.77, 55.54. GC-MS (m/z) calcd. for C56H44N2O4: 808.33, found: 808.5 [M]+. Anal. calcd. for C56H44N2O4: C 83.14, H 5.48, N 3.46, O 7.91. Found: C 83.09, H 5.41, N 3.40, O 7.88.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2020R1C1C1009007).Notes and references
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- B. R. Crenshaw and C. Weder, Chem. Mater., 2003, 15, 4717–4724 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez and S. R. White, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- Q. Zhu, K. Van Vliet, N. Holten-Andersen and A. Miserez, Adv. Funct. Mater., 2019, 29, 1808191 CrossRef.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed.
- J. Wu, Y. Cheng, J. Lan, D. Wu, S. Qian, L. Yan, Z. He, X. Li, K. Wang and B. Zou, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed.
- X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC.
- D. Xu, J. Hao, H. Gao, Y. Wang, Y. Wang, X. Liu, A. Han and C. Zhang, Dyes Pigm., 2018, 150, 293–300 CrossRef CAS.
- H. Zhu, J. Huang, L. Kong, Y. Tian and J. Yang, Dyes Pigm., 2018, 151, 140–148 CrossRef CAS.
- J. Jia and H. Zhao, New J. Chem., 2019, 43, 2231–2237 RSC.
- Z.-H. Guo, Z.-X. Jin, J.-Y. Wang and J. Pei, Chem. Commun., 2014, 50, 6088–6090 RSC.
- S. Xue, X. Qiu, Q. Sun and W. Yang, J. Mater. Chem. C, 2016, 4, 1568–1578 RSC.
- X. Zhang, Z. Chi, B. Xu, L. Jiang, X. Zhou, Y. Zhang, S. Liu and J. Xu, Chem. Commun., 2012, 48, 10895–10897 RSC.
- J. Jia and Y. Wu, Dyes Pigm., 2017, 147, 537–543 CrossRef CAS.
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed.
- X. Zhang, Z. Ma, Y. Yang, X. Zhang, X. Jia and Y. Wei, J. Mater. Chem. C, 2014, 2, 8932–8938 RSC.
- J. Sun, X. Lv, P. Wang, Y. Zhang, Y. Dai, Q. Wu, M. Ouyang and C. Zhang, J. Mater. Chem. C, 2014, 2, 5365–5371 RSC.
- N. Metri, X. Sallenave, C. D. Plesse, L. Beouch, P.-H. Aubert, F. Goubard, C. Chevrot and G. Sini, J. Phys. Chem. C, 2012, 116, 3765–3772 CrossRef CAS.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li and P. Lu, Chem. Sci., 2015, 6, 3797–3804 RSC.
- J. Zhang, D. Deng, C. He, Y. He, M. Zhang, Z.-G. Zhang, Z. Zhang and Y. Li, Chem. Mater., 2011, 23, 817–822 CrossRef CAS.
- J. Jia and H. Zhao, Tetrahedron Lett., 2019, 60, 252–259 CrossRef CAS.
- P. Gayathri, M. Pannipara, A. G. Al-Sehemi and S. P. Anthony, New J. Chem., 2020, 44, 8680–8696 RSC.
- T. Ishi-i, H. Tanaka, R. Youfu, N. Aizawa, T. Yasuda, S.-I. Kato and T. Matsumoto, New J. Chem., 2019, 43, 4998–5010 RSC.
- P. Josse, M. Allain, J. P. Calupitan, Y. Jiang, C. Cabanetos and J. Roncali, Adv. Opt. Mater., 2020, 8, 2000420 CrossRef CAS.
- Y. Jiang, D. Gindre, M. Allain, P. Liu, C. Cabanetos and J. Roncali, Adv. Mater., 2015, 27, 4285–4289 CrossRef CAS PubMed.
- T. M. Figueira-Duarte and K. Mullen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CrossRef CAS.
- J. Yang, L. Li, Y. Yu, Z. Ren, Q. Peng, S. Ye, Q. Li and Z. Li, Mater. Chem. Front., 2017, 1, 91–99 RSC.
- X. Yang, Z. Zhao, H. Ran, J. Zhang, L. Chen, R. Han, X. Duan, H. Sun and J.-Y. Hu, Dyes Pigm., 2020, 173, 107881 CrossRef CAS.
- Y. Niko, S. Kawauchi and G. I. Konishi, Chem. – Eur. J., 2013, 19, 9760–9765 CrossRef CAS.
- X. Feng, H. Tomiyasu, J.-Y. Hu, X. Wei, C. Redshaw, M. R. Elsegood, L. Horsburgh, S. J. Teat and T. Yamato, J. Org. Chem., 2015, 80, 10973–10978 CrossRef CAS PubMed.
- Y. Niko, S. Sasaki, K. Narushima, D. K. Sharma, M. Vacha and G. I. Konishi, J. Org. Chem., 2015, 80, 10794–10805 CrossRef CAS PubMed.
- P. Sonar, M. S. Soh, Y. H. Cheng, J. T. Henssler and A. Sellinger, Org. Lett., 2010, 12, 3292–3295 CrossRef CAS.
- K.-R. Wee, H.-C. Ahn, H.-J. Son, W.-S. Han, J.-E. Kim, D. W. Cho and S. O. Kang, J. Org. Chem., 2009, 74, 8472–8475 CrossRef CAS PubMed.
- A. Karuppusamy, T. Vandana and P. Kannan, J. Photochem. Photobiol., A, 2017, 345, 11–20 CrossRef CAS.
- S. N. Keller, N. L. Veltri and T. C. Sutherland, Org. Lett., 2013, 15, 4798–4801 CrossRef CAS.
- L. Zöphel, V. Enkelmann and K. Müllen, Org. Lett., 2013, 15, 804–807 CrossRef.
- Z. H. Wu, Z. T. Huang, R. X. Guo, C. L. Sun, L. C. Chen, B. Sun, Z. F. Shi, X. Shao, H. Li and H. L. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13031–13035 CrossRef CAS.
- L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS PubMed.
- L. Ji, A. Lorbach, R. M. Edkins and T. B. Marder, J. Org. Chem., 2015, 80, 5658–5665 CrossRef CAS PubMed.
- S.-S. Li, K.-J. Jiang, C.-C. Yu, J.-H. Huang, L.-M. Yang and Y.-L. Song, New J. Chem., 2014, 38, 4404–4408 RSC.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Palsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS.
- J. Merz, J. Fink, A. Friedrich, I. Krummenacher, H. H. Al Mamari, S. Lorenzen, M. Haehnel, A. Eichhorn, M. Moos and M. Holzapfel, Chem. – Eur. J., 2017, 23, 13164–13180 CrossRef CAS PubMed.
- H. Zhang, H. Zhang, G. Huang and B. S. Li, Dyes Pigm., 2021, 185, 108947 CrossRef CAS.
- Y. Zhang, J. Sun, G. Bian, Y. Chen, M. Ouyang, B. Hu and C. Zhang, Photochem. Photobiol. Sci., 2012, 11, 1414–1421 CrossRef CAS PubMed.
- X. Y. Wang, J. Zhang, J. Yin and S. H. Liu, Chem. – Asian J., 2019, 14, 2903–2910 CAS.
- P. Xue, J. Ding, Y. Shen, H. Gao and J. Zhao, Dyes Pigm., 2017, 145, 12–20 CrossRef CAS.
- P. Xue, B. Yao, Y. Shen and H. Gao, J. Mater. Chem. C, 2017, 5, 11496–11503 RSC.
- Y. Wang, D. Xu, H. Gao, Y. Wang, X. Liu, A. Han, C. Zhang and L. Zang, J. Phys. Chem. C, 2018, 122, 2297–2306 CrossRef CAS.
- A. G. Crawford, Z. Liu, I. A. Mkhalid, M. H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov and J. A. Howard, Chem. – Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- D. Lumpi, B. Holzer, J. Bintinger, E. Horkel, S. Waid, H. D. Wanzenböck, M. Marchetti-Deschmann, C. Hametner, E. Bertagnolli and I. Kymissis, New J. Chem., 2015, 39, 1840–1851 RSC.
- C. Hansch, A. Leo and R. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- Y. Qiao, J. Zhang, W. Xu and D. Zhu, Tetrahedron, 2011, 67, 3395–3405 CrossRef CAS.
- M. Kasha, Radiat. Res., 1963, 20, 55–70 CrossRef CAS.
- M. M. Islam, Z. Hu, Q. Wang, C. Redshaw and X. Feng, Mater. Chem. Front., 2019, 3, 762–781 RSC.
- W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Lam, X. Y. Shen, C. Feng, H. H. Y. Sung, Y. Lu and I. D. Williams, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed.
- Y. Gong, Y. Zhang, W. Z. Yuan, J. Z. Sun and Y. Zhang, J. Phys. Chem. C, 2014, 118, 10998–11005 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- M. Ahn, M.-J. Kim, D. W. Cho and K.-R. Wee, J. Org. Chem., 2021, 86, 403–413 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra; GC-MS data; X-ray crystallographic data of CN, F, and Me; PXRD of CN, F, and Me; absorption and emission spectra of TPA donor and pyrene acceptor; absorption and emission spectra in various solvents and solid state; emission lifetime profile; Lippert–Mataga plots and dipole moment values; femtosecond transient absorption spectroscopic data; CV; HOMO and LUMO energy; DFT/TD-DFT calculation results. CCDC 2060789 (CN), 2060790 (F), 2060791 (Me). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ma00407g |
| This journal is © The Royal Society of Chemistry 2021 |