
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-purpose heterogeneous catalyst material from an amorphous cobalt metal–organic framework†
Kefeng
Ping‡
a,
Mahboob
Alam‡
a,
Sean Ray
Kahnert§
a,
Rohit
Bhadoria
a,
Arvo
Mere
b,
Valdek
Mikli
b,
Maike
Käärik
c,
Jaan
Aruväli
d,
Päärn
Paiste
d,
Arvo
Kikas
e,
Vambola
Kisand
e,
Ivar
Järving
a,
Jaan
Leis
c,
Nadežda
Kongi
 *c and
Pavel
Starkov
*a
*c and
Pavel
Starkov
*a
aDepartment of Chemistry and Biotechnology, Tallinn University of Technology, Tallinn 12618, Estonia. E-mail: pavel.starkov@taltech.ee
bDepartment of Materials and Environmental Technology, Tallinn University of Technology, Tallinn 19086, Estonia
cInstitute of Chemistry, University of Tartu, Tartu 50411, Estonia. E-mail: nadezda.kongi@ut.ee
dInstitute of Ecology and Earth Sciences, University of Tartu, Tartu 50411, Estonia
eInstitute of Physics, University of Tartu, Tartu 50411, Estonia
First published on 17th May 2021
Abstract
Sustainable technologies rely on the development of universal catalyst materials. While a lot of the attention has been given to improving the performance of one single catalyst material for one specific application, there is still a need to find ways to develop catalysts that can simultaneously be utilized for several chemo- and electrocatalytic processes. In this work, we have surveyed a series of novel, cobalt-based catalyst materials derived from an amorphous MOF in an array of diverged applications. Specifically, we have focused on organic transformations such as oxidative of alkylarenes and benzylic homocoupling reactions as well as several electrocatalytic processes, which directly relate to energy conversion and storage devices, such as oxygen reduction (ORR), oxygen evolution (OER) and hydrogen evolution (HER) reactions. We have observed that only one material, TAL-2-900, delivered the optimal solution. The stability and recyclability of this unique multifunctional material has been examined.
1. Introduction
The notion of M–N–C catalysts, where M is typically Fe, Co, Mn or Ni, is often reserved for the catalysts developed for applications in energy conversion and storage devices.1–3 However, recent reports have also emphasized the potential of this type of materials in facilitating heterogeneous organic transformations.4–6There is an accountable divergence in the ways that the M–N–C materials are prepared, and they depend on the ultimate application prerogative.1–12 When used in the electrochemical settings, the underlying materials are typically acid-etched to make sure that the additional influence of related nanoparticles (metal and/or that of metal oxides, carbides, nitrides, sulfides and phosphides) deposited directly at the surface of the catalyst is eliminated. In heterogeneous catalysis applications, however, the contribution of these species to the overall chemical reactivity – while being well acknowledged and often relied upon – is not readily distinguished from the instances of single-atom catalysis9,10 and the role that nanocrystals protected by graphitic carbon layers might play.
Metal–organic frameworks (MOFs) are one of the most attractive building blocks for the formation of active M–N–C materials.12 Whereas only a handful of crystalline MOFs can directly be used as electrocatalysts,13–15 a vast majority of MOFs still require an additional carbonization step to allow for increased stability and conductivity as well as to introduce a variety of catalytically active species (M–Nx).12 During this process, additional doping with heteroatoms and metals7,8 or the use of alternative supports16,17 is often advantageous.
While crystalline MOFs are widely used for carbonization,7–9 their innate crystallinity may not always be a critical factor to achieve high catalytic activities (aside from contributing to the topology). All the catalyst materials reported to date are based on monometallic (e.g. MIL-101, ZIF-67, MOF-74-Zn, Prussian blue analogues)11 and mixed ligand/mixed metal (e.g. the MIL-101/ZIF-67 composite18 and ZnCo-MOFs19) MOFs are highly crystalline. During the pyrolysis, however, MOFs undergo drastic molecular rearrangements; hence, amorphous MOFs can be used as the precursors.20,21 To this end, we recently assessed iron-based catalysts derived from polycrystalline metal–organic framework precursors (TAL-1-900, TAL-6-900, TAL-7-900),22,23 which were based on carbon-rich24 organic linkers. Even the slightest variations around the benzimidazole core have led to alterations in the performance of the materials as bifunctional oxygen electrocatalysts22 and in heterogeneous catalysis.23
Herein, we report a unique amorphous cobalt-based MOF-derived multifunctional material, which acts as an efficient trifunctional electrocatalyst25,26 and as a robust heterogeneous catalyst.
2. Results and discussion
2.1 Morphological and physical characterizations
We prepared a series of catalysts from an amorphous metal–organic framework precursor TAL-2 by carbonizing it at different temperatures (Fig. 1). Importantly, we have treated these carbonized materials with 0.5 M HNO3 to remove the traces of cobalt(0) and cobalt oxides from the surface of the materials. Regardless of this treatment, the final porous M–N–C materials still contained cobalt(0) nanoparticles, which were protected by the graphitic layers as is evident from the high resolution transmission electron microscopy (HRTEM; Fig. 1B–D and Fig. S1, S2, ESI†) micrographs. Increasing the temperature of carbonization did not lead to a substantial change in the N2 adsorption/desorption isotherms or the distribution of pore sizes, which effectively remained in the 3–4 nm range (Fig. 1E and F). However, as is seen in the powder X-ray diffraction (PXRD; Fig. 1G) patterns and the microwave plasma-atomic emission spectroscopy (MP-AES; Table S1, ESI†) datasets, the graphitic carbon and cobalt(0) content was significantly higher in the samples that have been treated at 900 and 1000 °C. The X-ray photoelectron spectroscopy (XPS) data indicated that carbonization at higher temperatures both lowered the nitrogen content and altered the distribution of nitrogen species at the surface (Fig. S3 and Tables S1–S5, ESI†).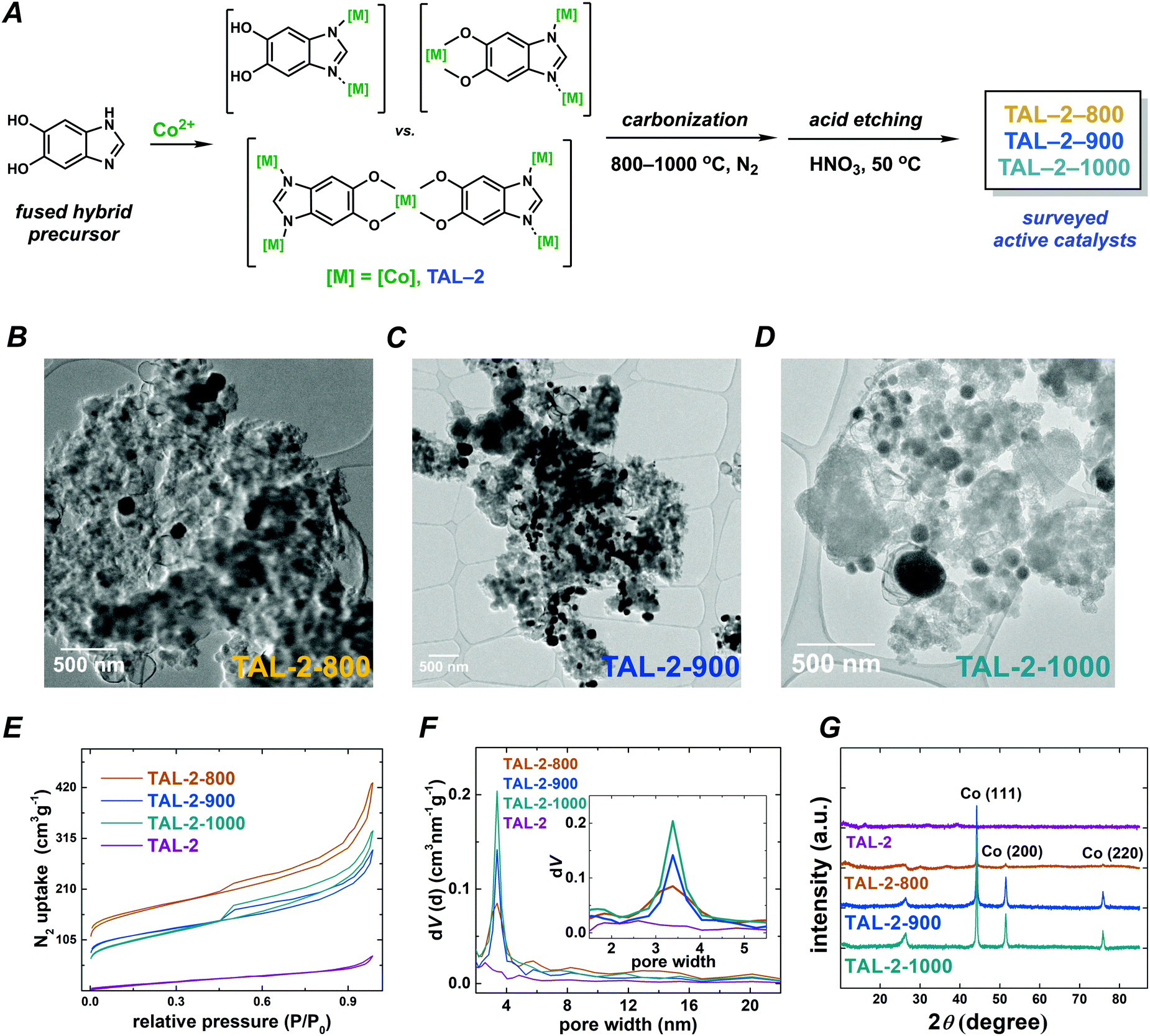 | ||
| Fig. 1 (A) Preparation of TAL-2 derived catalysts materials. HRTEM micrographs of (B) TAL-2-800, (C) TAL-2-900 and (D) TAL-2-1000 catalysts. Physical characterization of the materials by (E) N2 uptake, and (F) pore size distribution and (G) PXRD patterns. | ||
2.2 Oxidative organic transformations
The de novo Co–N–C catalysts were screened against oxidative transformations with tert-butyl peroxide (TBHP) as terminal oxidant at a considerably low catalyst loadings, namely, that of 5 mg mmol−1. While TBHP-driven oxidations of arylmethanes have been reported to be catalyzed by Fe–N–C materials effectively,23,27–29 the cobalt corresponding Co–N–C catalysts were not.30 Typically, Co–N–C catalysts are used for reduction reactions (nitroarenes,31,32N-heterocycles,33 and hydrocarbons34), reductive aminations35 and alkylations,36,37 while the oxidative esterification of benzyl alcohols were done under dioxygen.38–40Overall, the cobalt materials outperformed the three previously reported related Fe–N–C systems (TAL-1-900, TAL-6-900, TAL-7-900),22,23 and importantly, they sustained their activity over a larger number of cycles (Fig. 2A and B; Tables S6 and S7, ESI†). TAL-2-900 and TAL-2-1000 were more active because they contained metallic Co nanoparticles, which were protected by graphitic carbon layers, as confirmed by PXRD and HRTEM.
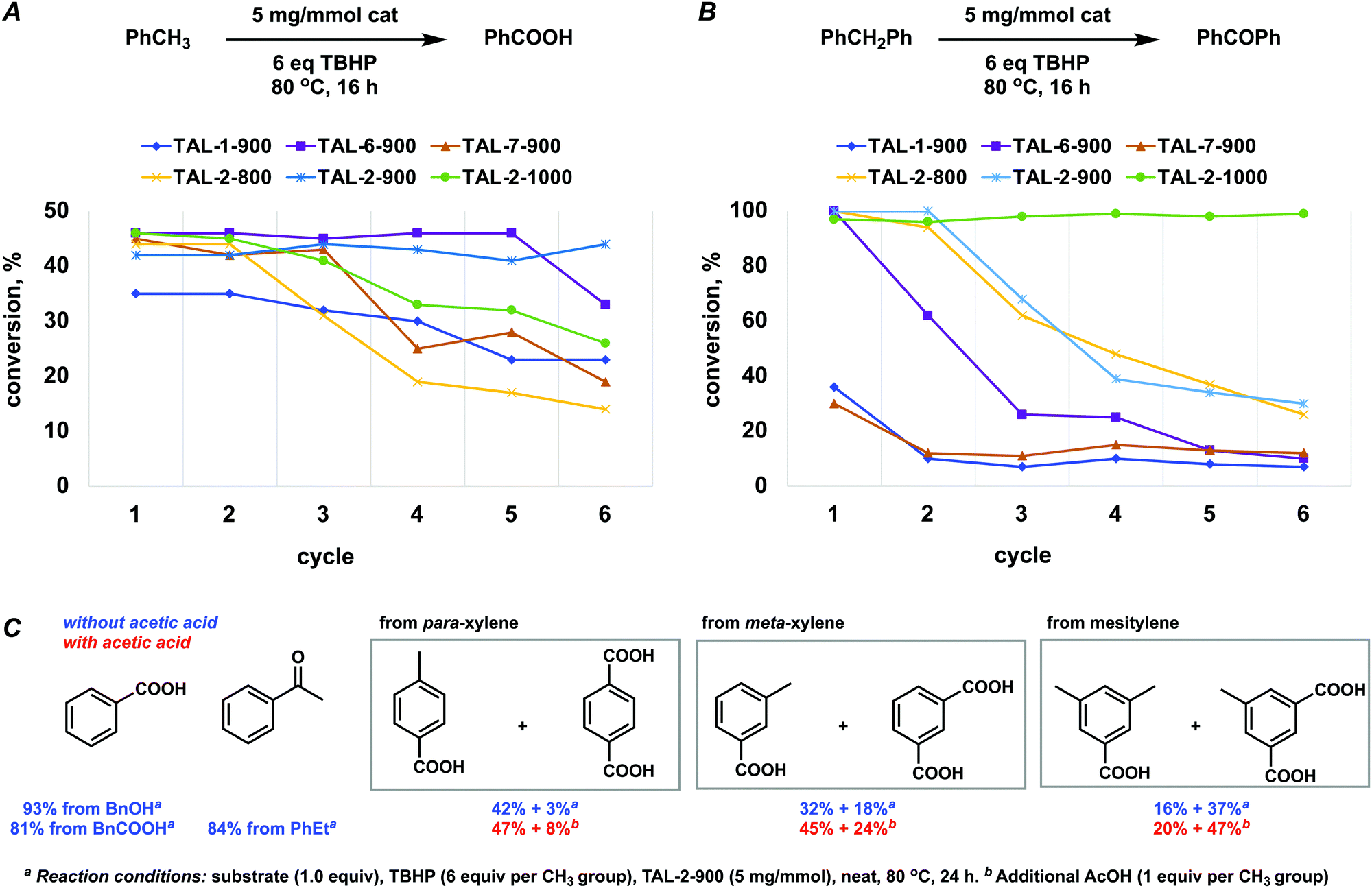 | ||
| Fig. 2 Use of TAL-derived catalysts in oxidative transformations. (A) Catalyst recyclability during oxidation of toluene into benzoic acid. (B) Catalyst recyclability during oxidation of diphenylmethane into benzophenone (conversion were determined by NMR using 1,3,5-trimethoxybenzene as internal standard). (C) Substrate scope for TAL-2-900-catalyzed oxidations (isolated yields). | ||
Interestingly, TAL-2-900 was a more robust catalyst for converting toluene to benzoic acid, while TAL-2-1000 was better at oxidizing diphenylmethane to benzophenone. This indicates that small differences in the active sites (e.g. the total content of pyridinic nitrogen) at the surface of the catalyst play an important role in catalyst stability. A further set of substrates was subjected to oxidation (Fig. 2C). Addition of acetic acid slightly improved the yields in the cases where polymethylated arenes were converted to the corresponding carboxylic acids.
2.3 Benzylic homocoupling
Knowing that radical pathways were involved in oxidative transformation reactions, has encouraged us to extend the use of TAL-2-900 as a heterogeneous catalyst for carbon–carbon bond forming reactions via activation at the benzylic positions. While M–N–C materials are widely explored in oxidative and reductive processes,4–6 there is only a handful of examples of their use in other organic transformations. For instance, Zhang et al. developed an oxidative coupling between primary and secondary alcohols.41 This reaction was shown to proceed via formation of intermediary aldehydes and ketones, respectively, which undergo aldol condensation. A reductive C–H alkylation of quinolines by aldehydes using cobalt-based catalysts was also demonstrated.42,43We were interested to see whether the M–N–C catalysts could be compatible with organometallic reagents; hence, we explored C(sp3)–C(sp3) homocoupling reaction of benzyl bromides in the presence of a Grignard reagent. This reaction traditionally requires stoichiometric metallic lithium,44 magnesium,45 copper46 or nickel.47 Recently, a rhodium-catalyzed version employing dimethylzinc was demonstrated,48 and several photocatalyzed conditions were developed.49,50 Gratifyingly, we observed the desired homocoupling products with cobalt based catalyst TAL-2-900 but not the iron version (TAL-1-900) (Scheme 1). The reactions were carried out using methylmagnesium bromide at ambient temperature to give corresponding bibenzyls in good to excellent yields, while aryliodides and bromides were also well tolerated. After standard aqueous workup, the spent catalyst had a significantly reduced activity. Specifically, on the next cycle, the isolated yield of homocoupled product has dropped from 93% to 38%. Alternatively, the THF solution containing the product was removed by syringe under anhydrous conditions and the reaction vessel was recharged with THF, benzyl bromide and MeMgBr. This modification gave the desired product with an improved yield on the second run (93%).
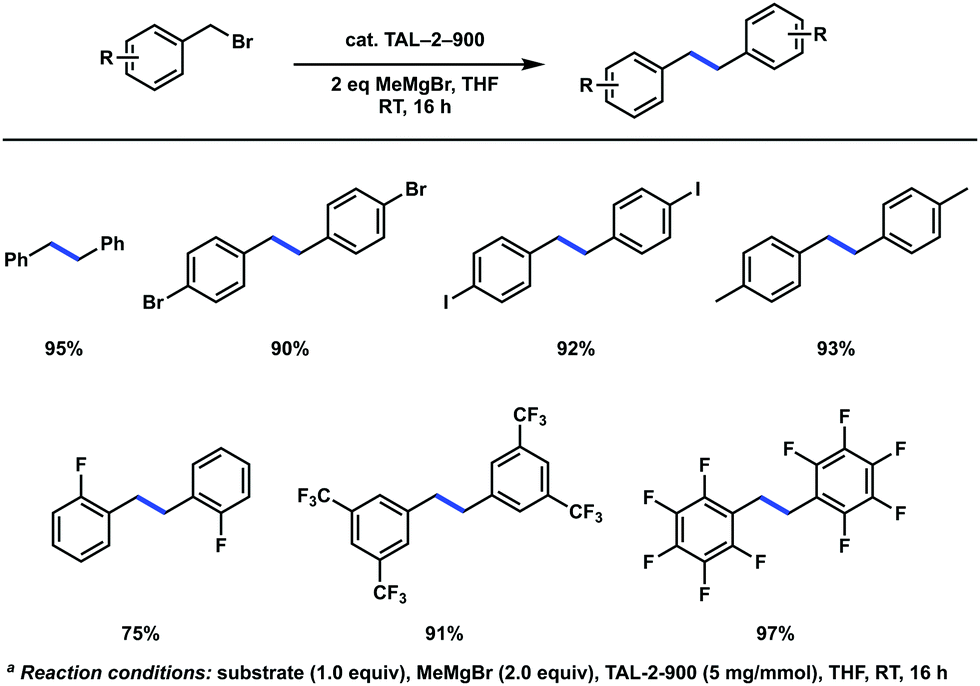 | ||
| Scheme 1 TAL-2-900 catalyzed C(sp3)–C(sp3) homocoupling reactionsa. | ||
2.4 Electrocatalytic transformations
After having identified the most promising catalyst for the organic transformations, we wished to screen the TAL-2 catalyst series as trifunctional electrocatalyst materials across oxygen reduction (ORR), oxygen evolution (OER) and hydrogen evolution (HER) reactions (Fig. 3). Potentially, these datasets should give insights into whether there is a link between the performance of the individual catalysts across electrocatalytic and chemocatalytic interconversions.51,52 Out of the three catalysts, TAL-2-900, once again, showed a superior activity profiles in all of the electrocatalytic reactions.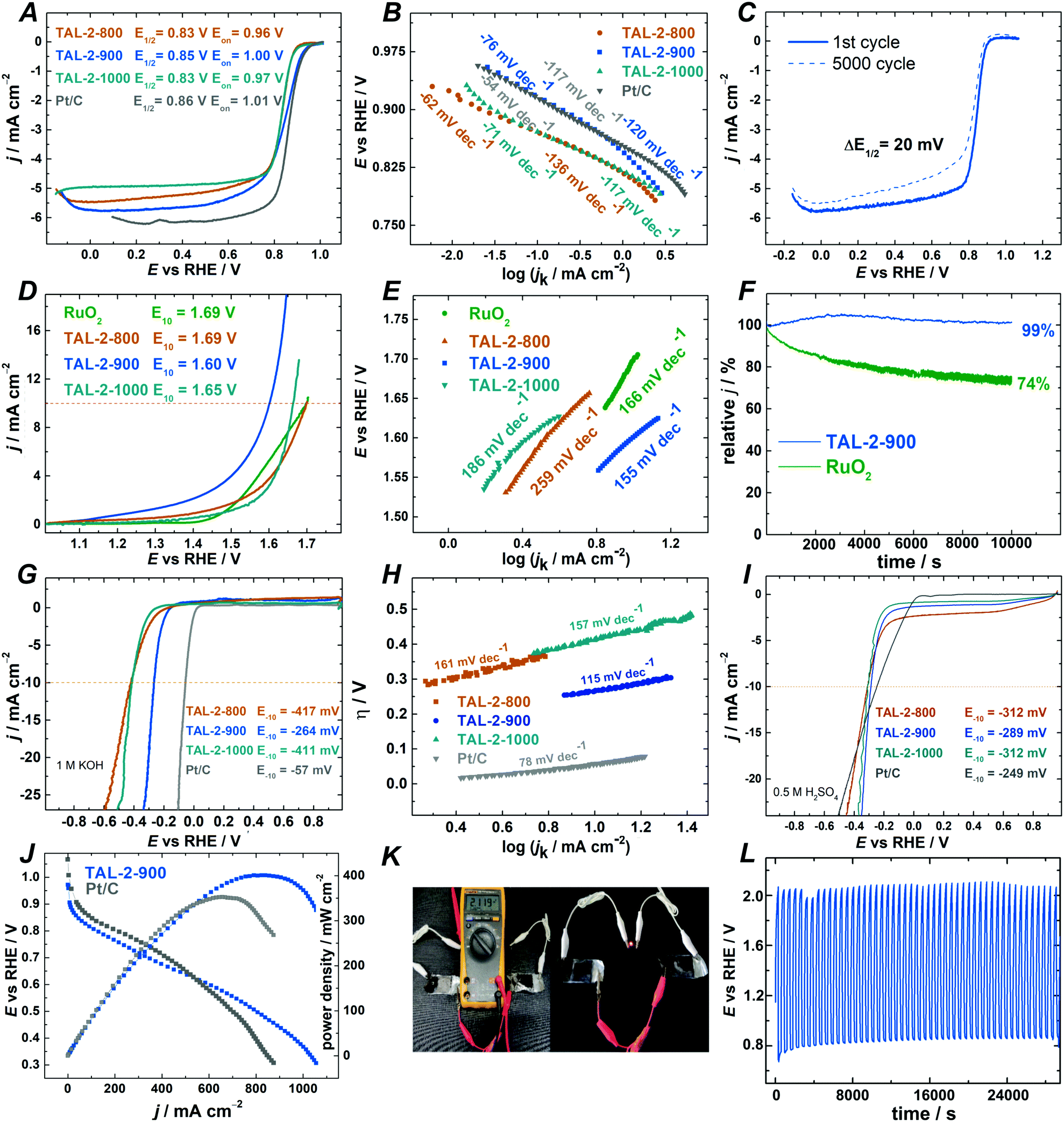 | ||
Fig. 3 Electrochemical characterization of TAL-2 derived catalyst materials. (A) ORR polarization curves for TAL and Pt/C-modified GC electrodes at 1600 rpm; ν = 10 mV s−1. (B) Tafel plots for ORR on TAL and Pt/C catalysts. (C) Electrochemical ORR stability test for TAL-2-900 (mid-range: 0.6–1.0 V). (D) OER polarization curves (under argon); ν = 10 mV s−1. (E) OER Tafel plots. (F) OER chronoamperometric stability of TAL-2-900 and RuO2 in 0.1 M KOH over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 seconds (1.6 V). (G) HER polarization curves for TAL and Pt/C electrocatalysts in 1 M KOH; ν = 10 mV s−1. (H) HER Tafel plots for TAL and Pt/C electrocatalysts in 1 M KOH. (I) HER polarization curves for TAL and Pt/C electrocatalysts in 0.5 M H2SO4; ν = 10 mV s−1. (J) Alkaline single fuel cell test of TAL-2-900 and Pt/C. (K) Zinc–air battery testing with TAL-2-900, shows the voltage produced by two batteries, LED light using the same batteries, and (L) galvanostatic cycling of one battery for 8 h. 000 seconds (1.6 V). (G) HER polarization curves for TAL and Pt/C electrocatalysts in 1 M KOH; ν = 10 mV s−1. (H) HER Tafel plots for TAL and Pt/C electrocatalysts in 1 M KOH. (I) HER polarization curves for TAL and Pt/C electrocatalysts in 0.5 M H2SO4; ν = 10 mV s−1. (J) Alkaline single fuel cell test of TAL-2-900 and Pt/C. (K) Zinc–air battery testing with TAL-2-900, shows the voltage produced by two batteries, LED light using the same batteries, and (L) galvanostatic cycling of one battery for 8 h. | ||
To learn the effect of different carbonization temperatures on electrocatalytic ORR performance, TAL-2 based catalysts were tested by rotating disc electrode (RDE) and rotating ring-disc electrode (RRDE) techniques in 0.1 M KOH electrolyte. Oxygen reduction polarization curves (Fig. 3A) confirmed that TAL-2 based catalyst carbonized at 900 °C had the highest ORR electrocatalytic activity with an onset potential (Eon) of 1.00 V, a half-wave potential (E1/2) of 0.85 V and a diffusion-limiting current density of −5.78 mA cm−2. The kinetic parameters of TAL-2-900 were on par with the ones obtained for commercial Pt/C catalyst (Eon = 1.01 V; E1/2 = 0.86; jd = −6.16 mA cm−2) and other promising non-noble metal ORR catalysts (Table S9, ESI†). Despite the fact that BET surface area of TAL-2-900 (421 m2 g−1) is lower than that obtained for TAL-2-800 (589 m2 g−1) (Table S11, ESI†), the superior ORR performance of TAL-2-900 indicates higher density of electrochemically accessible active sites for ORR.
To shed light on the enhancement of the ORR activity, electrochemically active surface area (ECSA) of TAL-2 derived catalysts was estimated by collecting the electrochemical double-layer capacitance (Cdl) from CV curves (Table S10, ESI†). The ECSA values have decreased for the samples that were obtained by increasing the carbonization temperatures. Hence, the superior electrocatalytic activity of the TAL-2-900 sample may be explained by a higher amount of ORR active sites, specifically in the form of cobalt(0) nanoparticles.
The Koutecky–Levich (K–L) plots were constructed using data derived from RDE (Fig. S4, ESI†) and the calculated number of electrons transferred per oxygen molecule n was in all the cases approximately four. RRDE studies confirmed that oxygen reduction proceeds via a two-step 2 × 2 H2O2 mechanism (Fig. S5B and C, ESI†). The Tafel plot analysis demonstrated that the TAL-2-900 material had the highest slope value (−76 mV decade−1) against all the other samples that were surveyed in this study (Fig. 3B). Similarly to Pt/C, this means that the rate determining step for ORR is the first electron transfer step, whereby O2(ads) is reduced to O2(ads)−. A continuous potential cycling in the range between 0.6 and 1.0 V was used to assess the long-term stability of TAL-2-900, with E1/2 shifting only by 20 mV after 5,000 cycles (Fig. 3C). The RDE studies have confirmed that TAL-2-900 was a highly stable active ORR electrocatalyst in 0.1 M KOH.
The electrocatalytic activity towards ORR of the TAL-2-900 material has also been tested under acidic conditions (0.5 M H2SO4; Fig. S7, ESI†). As in the case of alkaline media, TAL-2-900 has shown higher onset and half-wave potentials (0.82 V and 0.73 V vs. RHE) than those for TAL-2-800 and TAL-2-1000. Notably, TAL-2-900 remained stable after 5000 cycles with E1/2 having decreased only by 20 mV (Fig. S7D, ESI†).
The OER performance of the TAL-2 derived catalysts was assessed and iR-corrected OER polarization curves are shown in Fig. 3D. The benchmark current density of 10 mA cm−2 was achieved at 1.60 V for TAL-2-900 material, which is superior to that of ruthenium oxide (1.69 V). The Tafel slopes obtained for the OER electrocatalytic activity among the series, confirm that TAL-2-900 functions similarly to the RuO2 systems. Under the long-term OER operation, this catalyst material is stable (Fig. 3F). The overall oxygen bifunctional electroactivity (ΔE) value for TAL-2-900 is 0.75 V, which makes it much lower than the rest of the M–N–C samples, whilst haven been screened at the similar loadings (Table S10, ESI†).
The electrocatalytic HER activity of the best performing catalyst was compared with the commercial Pt/C in 1 M KOH (Fig. 3G and H) at the current density of −10 mA cm−2. Under these conditions, the overpotential with TAL-2-900 as a catalyst was achieved at −264 mV vs. RHE with a low Tafel slope value of 115 mV dec−1. The HER overpotential values η for TAL-2-800 and TAL-2-1000 were 417 and 411 mV, respectively, indicating that the carbonization step at 900 °C was the optimal path to improve the electroactivity toward HER. In acidic media, the overpotential (η) was the smallest for TAL-2-900, while its overpotential was comparable to the values obtained for other Pt-free HER catalysts (Fig. 3I and Table S9, ESI†).
Finally, the performance of TAL-2-900 as catalyst material was evaluated in alkaline electrolyte membrane fuel cell and zinc–air battery. Alkaline membrane fuel cell test yielded current density up to 1100 mA cm−2 (Fig. 3J). Loading of the catalyst was kept at 2 mg cm−2 on GDL membrane modified with direct suspension pipetting. HMT-PMBI (hydroxide conducting membrane) was used as polymer electrolyte in both tests for Pt/C and TAL-2-900. Under similar conditions, TAL-2-900 showed 20% better activity than Pt/C. The value of power density for TAL-2-900 is >400 mW cm−2 leaving behind Pt/C 350 mW cm−2. Similar results were recently reported using the same anion exchange membrane and an alternative Co–N–C material as a cathode catalyst.53 Durability tests of poly(vinyl alcohol)-based solid-state zinc–air battery assembled using TAL-2-900 as an electrode material were performed by cycling at constant current density of 5.0 mA cm−2 (Fig. 3K). As expected from the ORR/OER data, TAL-2-900 catalyst-driven Zn–air battery delivered long cycle life over 8 h (Fig. 3L).
3. Conclusions
In conclusion, we have introduced a new materials preparation strategy by involving amorphous not crystalline MOFs, which were obtained from dihydroxybenzimidazole as a carbonaceous linker. Upon carbonization, it delivered a unique Co–N–C material, which simultaneously served as a heterogeneous catalyst for several organic transformations (incl. oxidation and homocoupling reactions) as well as an electrocatalyst material for ORR, OER and HER processes. This is the first example of an M–N–C catalyst being used for C(sp3)–C(sp3) carbon–carbon bond formation, and it was carried out in the presence of an organometallic reagent.Author contributions
Kefeng Ping, Mahboob Alam: investigation, methodology, formal analysis, validation, writing – original draft; writing – review and editing; Sean Ray Kahnert, Rohit Bhadoria, Arvo Mere, Valdek Mikli, Maike Käärik, Jaan Aruväli, Päärn Paiste, Arvo Kikas, Vambola Kisand, Ivar Järving, Jaan Leis: investigation, methodology, formal analysis, writing – review and editing; Nadežda Kongi, Pavel Starkov: conceptualization, supervision, writing - original draft, writing – review and editing, funding acquisition.Conflicts of interest
K. P., N. K. and P. S. are inventors on the PCT Appl. filed by the University of Tartu and Tallinn University of Technology.Acknowledgements
This research was supported by Estonian Research Council (PSG250 and PUT1290) and TalTech Young Investigator Grant B62. We thank Prof. Steven Holdcroft (Simon Fraser University) for providing the HMT-PMBI membrane. The measurement facilities were supported by Institutional Research Grants IUT34-14 and TK141 (Advanced materials and high-technology devices for energy recuperation systems). K. P. acknowledges Estonian Smart Specialization PhD Fellowship. P. S. thanks COST Action CA18224 members (https://greenering.eu) for fruitful discussions.Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
.
- J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 15298–15312 CrossRef CAS PubMed
- A. Sarapuu, E. Kibena-Põldsepp, M. Borghei and K. Tammeveski, J. Mater. Chem. A, 2018, 6, 776–804 RSC
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS
- H. B. Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS
- H. Yan, C. Su and J. He, J. Mater. Chem. A, 2018, 6, 8793–8814 RSC
- S. Mitchell, E. Vorobyeva and J. Pérez–Ramírez, Angew. Chem., Int. Ed., 2018, 57, 15316–15329 CrossRef CAS PubMed
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC
- X. F. Lu, B. Y. Xia, S.-Q. Zang and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 4634–4650 CrossRef CAS PubMed
- E. M. Miner, L. Wang and M. Dincă, Chem. Sci., 2018, 9, 6286–6291 RSC
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS PubMed
- S. S. Shinde, C. H. Lee, J.-Y. Jung, N. K. Wagh, S.-H. Kim, D.-H. Kim, C. Lin, S. U. Lee and J.-H. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC
- M. Aygün, T. W. Chamberlain, M. C. Gimenez–Lopez and A. N. Khlobystov, Adv. Funct. Mater., 2018, 28, 1802869 CrossRef
- X.-J. Bai, X.-Y. Lu, R. Ju, H. Chen, L. Shao, X. Zhai, Y.-N. Li, F.-Q. Fan, Y. Fu and W. Qi, Angew. Chem., Int. Ed., 2021, 60, 701–705 CrossRef CAS PubMed
- S. L. Zhang, B. Y. Guan and X. W. Lou, Small, 2019, 15, 1805324 CrossRef PubMed
- X. P. Han, X. F. Ling, Y. Wang, T. Y. Ma, C. Zhong, W. B. Hu and Y. D. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47(5), 1555–1562 CrossRef CAS PubMed
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef
- K. Ping, A. Braschinsky, M. Alam, R. Bhadoria, V. Mikli, A. Mere, J. Aruväli, P. Paiste, S. Vlassov, M. Kook, M. Rähn, V. Sammelselg, K. Tammeveski, N. Kongi and P. Starkov, ACS Appl. Energy Mater., 2020, 3, 152–157 CrossRef CAS
- K. Ping, M. Alam, M. Käärik, J. Leis, N. Kongi, I. Järving and P. Starkov, Synlett, 2019, 1536–1540 CAS
- D. Kong, Y. Gao, Z. Xiao, X. Xu, X. Li and L. Zhi, Adv. Mater., 2019, 31, 201804973 Search PubMed
- S. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2019, 7, 18674–18707 RSC
- W. Zhang, L. Cui and J. Liu, J. Alloys Compd., 2020, 821, 153542 CrossRef CAS
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed
- T. Song, Z. Ma, Y. Yuan, J. Xiao, Y. Yang and P. Ren, ACS Catal., 2020, 10, 4617–4629 CrossRef CAS
- M. Nakanishi and C. Bolm, Adv. Synth. Catal., 2007, 349, 861–864 CrossRef CAS
- Y. Li, S. Jie, K. Li and Z. Liu, New J. Chem., 2018, 42, 12677–12683 RSC
- P. Zhou, L. Jiang, F. Wang, K. Deng, K. Lv and Z. Zhang, Sci. Adv., 2017, 3, e1601945 CrossRef PubMed
- S. Yang, L. Peng, D. T. Sun, E. Oveisi, S. Bulut and W. L. Queen, ChemSusChem, 2018, 11, 3131–3138 CrossRef CAS PubMed
- F. Chen, A. Surkus, L. He, M. Pohl, J. Radnik, C. Topf, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 11718–11724 CrossRef CAS PubMed
- F. Chen, C. Kreyenschulte, J. Radnik, H. Lund, A. Surkus, K. Junge and M. Beller, ACS Catal., 2017, 7, 1526–1532 CrossRef CAS
- R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M. M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed
- R. Xie, F. Xie, C. J. Zhou, H. F. Jiang and M. Zhang, J. Catal., 2019, 377, 449–454 CrossRef CAS
- F. Xie, R. Xie, J. X. Zhang, H. F. Jiang, L. Du and M. Zhang, ACS Catal., 2017, 7, 4780–4785 CrossRef CAS
- H. Luo, L. Wang, S. Shang, G. Li, Y. Lv, S. Gao and W. Dai, Angew. Chem., Int. Ed., 2020, 59, 19268–19274 CrossRef CAS PubMed
- Y. Zhou, Y. Chen, L. Cao, J. Lu and H. Jiang, Chem. Commun., 2015, 151, 8292–8295 RSC
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS
- L. Zhang, A. Wang, W. Wang, Y. Huang, X. Liu, S. Miao, J. Liu and T. Zhang, ACS Catal., 2015, 5, 6563–6572 CrossRef CAS
- F. Xie, R. Xie, J. X. Zhang, H. F. Jiang, L. Du and M. Zhang, ACS Catal., 2017, 7, 4780–4785 CrossRef CAS
- R. Xie, F. Xie, C. J. Zhou, H. F. Jiang and M. Zhang, J. Catal., 2019, 377, 449–454 CrossRef CAS
- H. Gilman and R. G. Gorsich, J. Am. Chem. Soc., 1955, 77, 3134–3135 CrossRef CAS
- R. A. Aitken, P. K. G. Hodgson, J. J. Morrison and A. O. J. Oyewale, J. Chem. Soc., Perkin Trans. 1, 2002, 3, 402–415 RSC
- F. O. Ginah, T. A. Donovan Jr., S. D. Suchan, D. R. Pfennig and G. W. Ebert, J. Org. Chem., 1990, 55, 584–589 CrossRef CAS
- S.-i. Inaba, H. Matsumoto and R. D. Rieke, Tetrahedron Lett., 1982, 23, 4215–4216 CrossRef CAS
- K. Sato, Y. Inoue, T. Mori, A. Sakaue, A. Tarui, M. Omote, I. Kumadaki and A. Ando, Org. Lett., 2014, 16, 3756–3759 CrossRef CAS PubMed
- V. V. Levin, D. P. Agababyan, M. I. Struchkova and A. D. Dilman, Synthesis, 2018, 2930–2935 CAS
- G. Park, S. Y. Yi, J. Jung, E. J. Cho and Y. You, Chem. – Eur. J., 2016, 22, 17790–17799 CrossRef CAS PubMed
- R. Paul, F. Du, L. Dai, Y. Ding, Z. L. Wang, F. Wei and A. Roy, Adv. Mater., 2019, 31, 1805598 CrossRef PubMed
- S. Ghosh and R. N. Basu, Nanoscale, 2018, 10, 11241–11280 RSC
- J. Lilloja, E. Kibena-Põldsepp, A. Sarapuu, M. Kodali, Y. Chen, T. Asset, M. Käärik, M. Merisalu, P. Paiste, J. Aruväli, A. Treshchalov, M. Rähn, J. Leis, V. Sammelselg, S. Holdcroft, P. Atanassov and K. Tammeveski, ACS Appl. Energy Mater., 2020, 3, 5375–5384 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00414j |
| ‡ These authors contributed equally. |
| § Present address: Institute of Organic Chemistry, Clausthal University of Technology, Clausthal-Zellerfeld 38678, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |