
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic RGD and GHK peptides synergistically enhance liposomal delivery into cancer and endothelial cells†
Mohamed
Zoughaib
 a,
Rais V.
Pavlov
b,
Gulnara A.
Gaynanova
b,
Ruslan
Garifullin
ac,
Vladimir G.
Evtugyn
d and
Timur I.
Abdullin
*a
a,
Rais V.
Pavlov
b,
Gulnara A.
Gaynanova
b,
Ruslan
Garifullin
ac,
Vladimir G.
Evtugyn
d and
Timur I.
Abdullin
*a
aInstitute of Fundamental Medicine and Biology, Kazan (Volga Region) Federal University, 18 Kremlyovskaya St., 420008 Kazan, Russia. E-mail: timur.abdullin@kpfu.ru; tabdulli@gmail.com
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, 8 Arbuzov St., 420088 Kazan, Russia
cInstitute of Materials Science and Nanotechnology, Bilkent University, 06800 Ankara, Turkey
dInterdisciplinary Center for Analytical Microscopy, Kazan (Volga Region) Federal University, 18 Kremlyovskaya St., 420008 Kazan, Russia
First published on 28th September 2021
Abstract
This study reveals enhanced cancer-targeting properties of a peptide composition consisting of RGD and GHK, recognized as an important cell adhesion factor and pleiotropic modulator of cellular functions, respectively. C12-GGRGD-NH2 and C12-GGGHK-NH2 amphiphilic peptides comprising a lauric acid moiety capable of insertion into the liposomal membrane were synthesized. Composite liposomes made of phosphatidylcholine, cationic DOTAP and the peptide(s) were used at a pre-optimized PC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOTAP ratio of 35:1 and relative peptide content of 4 mol%. The RGD/GHK dual targeting system exhibited a profound synergistic effect on the cellular uptake of the liposomal formulation in integrin-overexpressing cancer and endothelial cells. Effective liposome activation via in situ association of the amphiphilc peptide(s) with the liposomal membrane was carried out. Dual peptide-modified liposomes loaded with doxorubicin or paclitaxel induced enhanced cytotoxicity accompanied by oxidative stress and mitochondria depolarization in the target cells. The study shows joint potential of RGD and GHK tripeptides as a targeting system in anticancer/antiangiogenic therapy and provides a methodology for screening of combinatorial effects of bioactive peptides displayed on the liposome surface. Peptide-modified liposomes were employed to reveal GHK–heparin binding, suggesting a potential complementary role of glycosaminoglycans in RGD/GHK-mediated liposomal delivery.
:DOTAP ratio of 35:1 and relative peptide content of 4 mol%. The RGD/GHK dual targeting system exhibited a profound synergistic effect on the cellular uptake of the liposomal formulation in integrin-overexpressing cancer and endothelial cells. Effective liposome activation via in situ association of the amphiphilc peptide(s) with the liposomal membrane was carried out. Dual peptide-modified liposomes loaded with doxorubicin or paclitaxel induced enhanced cytotoxicity accompanied by oxidative stress and mitochondria depolarization in the target cells. The study shows joint potential of RGD and GHK tripeptides as a targeting system in anticancer/antiangiogenic therapy and provides a methodology for screening of combinatorial effects of bioactive peptides displayed on the liposome surface. Peptide-modified liposomes were employed to reveal GHK–heparin binding, suggesting a potential complementary role of glycosaminoglycans in RGD/GHK-mediated liposomal delivery.
Introduction
The efficacy of modern chemotherapy of cancer diseases relies on an appropriate balance between the ability to induce cancer-targeted and non-specific toxic effects. Despite the progress achieved in anticancer drug discovery, existing treatments are often not sufficiently effective and/or poorly selective against cancer cells/tissues, especially in the case of multi-drug resistant (MDR) tumors requiring administration of increased drug doses. Different strategies to improve the selectivity of chemotherapy have been introduced, ranging from rational design of small inhibitors of functional/signaling biomacromolecules1 to carrier formulation of established drugs aiming at increasing their circulation half-life and delivery to tumors via different mechanisms.2,3Modification of both plain and formulated drugs with targeting ligands is an attractive approach to enhance delivery into cancer cells/tissues, reduce therapeutic doses/side effects and improve treatment of MDR tumors.4,5 Oligopeptides are one of the most powerful molecular tools for tumor targeting by virtue of their natural specificity and potential safety in addition to higher availability and stability compared to full-length proteins such as antibodies. Molecular biology and in silico techniques can be used to identify cancer-specific peptide sequences,6 whereas solid-phase peptide synthesis (SPPS) methods provide effective production as well as on-resin derivatization of the sequence of interest for drug development purposes.7
The state of the art in cancer-specific oligopeptides and their applications is considered in recent reviews.6,8,9 Distinct tumor targeting can be provided by the simplest peptide sequences from extracellular matrix (ECM) components carrying different cell-adhesive peptide motifs. Overexpression of adhesion receptors in various solid tumor cells and tumor-associated endothelial cells underlies their targeting by ECM-derived oligopeptides. For instance, the RGD peptide, essentially found in fibronectin and vitronectin, has been extensively studied as a malignant tumor-specific ligand capable of binding to integrins such as αvβ3, αvβ1, αvβ5, α5β1, and α4β1 involved in cancer invasion, angiogenesis and metastasis.10,11 Another fibronectin motif, NGR, is recognized by CD13/aminopeptidase N (APN) receptor isoforms overexpressed in newly formed tumor vasculature.12 The YIGSR peptide from the laminin β1 chain can be specifically targeted to the 67 kDa laminin receptor overexpressed in metastatic cancer cells.13
An important issue that remains poorly addressed is the exploration of combinatorial effects of oligopeptide ligands on drug delivery into cancer cells. We have shown recently that affinity immobilized RGD and GHK peptides in a cryogel ECM model provide synergistic interactions with mammalian cells including human umbilical vein endothelial cells (HUVECs), profoundly increasing cell proliferation and differentiation over RGD alone.14 GHK is recognized as a tripeptide motif abundant in ECM proteins such as collagen α2(I) and α2(V) chains, SPARC glycoprotein and thrombospondin-1.15,16 GHK can be released as a matrikine with pleotropic growth factor-like activities; however, it does not seem to have distinct molecular targets.16
Considering the above data, it was of particular interest to establish whether the combination of the RGD and GHK peptides can increase the anticancer effects of drug formulations against tumor cells of epithelium and endothelium origin. For this purpose, liposomes were used as powerful drug carriers, which improve systemic and localized drug delivery17 and can be modified with peptides in a controllable manner to assess their cell-targeting properties. Liposomal formulations are particularly endowed with beneficial characteristics including decreased toxicity and increased bioavailability as well as compatibility with various drugs/labels and ligands with stimuli-responsive, stealth and biospecific properties.18,19 In addition to covalent conjugation, functional ligands can be attached to the liposomes via hydrophobic/supramolecular interactions.
The latter principle was exploited in our previous studies of cationic surfactant-modified liposomes with improved electrostatic interactions with cellular membranes and organelles20–22 and was extended in this work to activate cationic liposomes with the targeting peptides. The RGD and GHK peptides were synthesized in the form of amphiphilic conjugates of lauric acid, which were introduced to the liposomal membrane by addition to the organic lipid phase or in situ surface association with the liposomes in solution. Synergistically enhanced delivery of chemotherapeutic drug-loaded liposomes modified with the RGD/GHK composition into cancer and endothelial cells was reported for the first time.
Experimental
Materials and reagents
Soybean phosphatidylcholine (PC, 98%), 1,2-dioleoyl-3-trimethylammonium propane (DOTAP, 98%) (both are a gift from Lipoid GmbH, Germany), cholesterol (CHOL, 95%, Acros Organics), rhodamine B (RhB, ≥95%) and paclitaxel (PTX, ≥95%) (Sigma-Aldrich), doxorubicin hydrochloride (DOX, ≥98%, Alfa Aesar), Fmoc-Gly-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Asp(OtBu)-OH, Fmoc-His(Trt)-OH, Fmoc-Lys(Boc)-OH, lauric acid (C12, 99%), Fmoc-Rink amide resin, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, N,N-diisopropylethylamine, triisopropylsilane, trifluoroacetic acid, N,N-dimethylformamide, dichloromethane (Novabiochem, Merck), and HPLC grade acetonitrile (Merck) were used. The highly oriented pyrolytic graphite (HOPG, ZYA grade) substrate was purchased from Tips Nano.Cell culture media and reagents (Paneco, Russia), LysoTracker Green (Thermo Fisher Scientific), Hoechst 33258 (Acros Organics), 2′,7′-dichlorodihydrofluorescin diacetate (DCFDA), tetramethylrhodamine ethyl ester (TMRE), heparin sodium salt from porcine intestinal mucosa and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) (Sigma-Aldrich) were used. Aqueous solutions were prepared with deionized water (18.2 MΩ cm) from Direct-Q 5 UV equipment (Millipore).
Peptide synthesis
C12-GGRGD-NH2 and C12-GGGHK-NH2 as well as the free RGD-NH2 and GHK-NH2 (competitive inhibitors) peptide sequences with a C-terminal amide group were synthesized by the Fmoc solid-phase peptide synthesis (SPPS) method as detailed previously.23 Briefly, Fmoc-Rink amide resin was swollen in N,N-dimethylformamide (DMF) and deprotected using 20% (v/v) piperidine in DMF. Subsequent coupling and deprotection cycles were carried out using 2 equiv. of Fmoc-protected amino acids, 1.98 equiv. of 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 3 equiv. of N,N-diisopropylethylamine (DIPEA) in DMF. Finally, the peptides were released from the resin in a cleavage mixture (95% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% triisopropylsilane (TIPS)) and collected with dichloromethane (DCM), which was removed alongside TFA on a rotary evaporator. Following trituration with cold diethyl ether, the peptides were separated through centrifugation, and finally lyophilized from water. The peptides were purified by preparative HPLC and analyzed by the LC-MS technique.Determination of the critical micelle concentration (CMC)
The CMC of the amphiphilic peptides was assessed using a curcumin fluorescent probe in 96-well plate format24 with some modifications. Briefly, a 20 μL aliquot of curcumin was added to 80 μL of serially diluted peptides in 50 mM HEPES (pH = 7.4) to obtain final concentrations of 10 μM (curcumin) and 1–1000 μM (peptides). The fluorescence intensity of micelle-bound curcumin was measured on an Infinite M200 PRO microplate analyzer at a λex/λem of 423/498 nm. CMC values were determined from the inflection point on peptide concentration vs. fluorescence signal curves.24Preparation of liposomal formulations
:1 (PC:CHOL); DOTAP liposomes 4:1 (DOTAP:CHOL); PC/DOTAP liposomes 35:1:9 (PC:DOTAP:CHOL).
Modification with peptides and drug loading
The liposomes were modified with amphiphilic peptides by means of mixing the peptide(s) with lipids in organic solution followed by thin film formation (method I). Alternatively, the peptides in aqueous solution were added to the plain liposomes and incubated for 2 h at 37 °C to allow in situ association of the peptides with the liposome surface via insertion of a lauroyl group into the lipid bilayer (method II). The amount of peptide(s) in the modified liposomes was presented relative to the amount of PC + DOTAP lipids. The corresponding molar ratio of the lipid and peptide components varied in the range from 125:1 to 5:1.
To obtain rhodamine B (RhB)- or doxorubicin (DOX)-loaded liposomes, the thin lipid (lipid–peptide) films were hydrated with a 0.5 mg mL−1 solution of the compounds in deionized water. Paclitaxel (PTX) was pre-distributed in the film by dissolving the drug in the organic phase. The PTX concentration in the resulting liposome suspension amounted to 0.34 mg mL−1.
Analysis of modification/loading of liposomes
The encapsulation efficiency (EE) and loading capacity (LC) of the RhB and DOX-loaded liposomes were determined as the ratio of the entrapped drug amount, respectively, to the total drug or drug and lipids amounts by centrifugal filtration using Amicon Ultra 100 kDa cut-off concentrators (Merck Millipore). RhB or DOX were quantified in the filtrate using extinction coefficients of 94000 M−1 cm−1 (λ = 555 nm) for RhB and 10000 M−1 cm−1 (λ = 482 nm) for DOX.
Similarly, peptide-modified liposomes were separated by centrifugal filtration using Millipore Amicon Ultra 0.5 mL filters with a 100 kDa threshold at 6700g (10000 rpm Eppendorf MiniSpin), and the filtrate was analyzed for the detection of free peptides in extra-liposomal solution by reverse-phase HPLC. A Dionex UltiMate 3000 HPLC system (Thermo Scientific) equipped with a UV-Vis detector and Kromasil C18 column was used. Gradient elution with a water/acetonitrile mobile phase was applied at a constant flow rate of 0.5 mL min−1. The peptide concentration was determined from the linear calibration graph (0.1–156 μM) by the peak area.
The amount of liposome-bound PTX was determined as described previously25 with some modifications. The liposomes were centrifuged at 27000g for 2 h (4 °C), mixed with 500 μL of acetonitrile and vortexed to solubilize the drug. An isocratic elution with acetonitrile/water (60:40) was applied at a flow rate of 0.5 mL min−1. The PTX concentration was calculated from the linear calibration graph (0.02–1.62 mM).
Dynamic light scattering (DLS)
The colloidal properties of the liposomes were analyzed by the DLS technique on a Zetasizer NanoZS analyzer (Malvern Instruments) in a transparent zeta-cuvette at 25 °C with a backscattering angle of 173°, and using a 633 nm He–Ne laser. The samples were diluted to a final lipid concentration of about 1 mM to ensure optimal count rates.For studying peptide–heparin interactions, DOTAP-free PC liposomes were used. The liposomes were modified with the peptides by method II at a lipid:peptide ratio of 25:1 and incubated in the presence of heparin (25.4 μg mL−1) for 15 min prior to measurements.
The mean hydrodynamic diameter and zeta potential of the liposomes were analyzed in MilliQ water or 50 mM HEPES buffer (pH = 7.4). The data were processed using Dispersion Technology Software 6.2 (Malvern Instruments).
Visualization of the liposomes
For transmission electron microscopy (TEM) analysis, an aliquot (3–5 μL) of the liposomes diluted in MilliQ water was distributed on 3 mm formvar-carbon coated copper grids and dried at room temperature. The samples were analyzed on a Hitachi HT7700 Exalens microscope at an accelerating voltage of 100 kV in high contrast mode.Atomic force microscopy (AFM) measurements were made on freshly prepared surfaces of highly oriented pyrolytic graphite (HOPG) and mica sheets using a Bruker Dimension FastScan microscope (Bruker). AFM images were obtained in PeakForceQNM (quantitative nanomechanical mapping) mode with the use of standard silicon cantilevers ScanAsyst-Air (Bruker) having curvature 2 nm and stiffness 0.4 N m−1.
Cell culture
Primary human umbilical vein endothelial cells (HUVECs) were grown in DMEM/F-12 supplemented with 20% fetal bovine serum (FBS), penicillin (100 U mL−1)/streptomycin (100 μg mL−1), L-glutamine (2 mM), sodium pyruvate (2 mM), heparin (100 μg mL−1) and endothelial cell growth supplements (ECGS) in a temperature- and humidity-controlled incubator at 37 °C. The experiments with HUVECs were conducted between passages 3 and 6. Human skin fibroblasts (HSF, passage 5) and human prostate carcinoma PC-3 cells (ATCC) were cultured in α-MEM culture medium supplemented with 10% FBS, penicillin (100 U mL−1)/streptomycin (100 μg mL−1) and L-glutamine (2 mM) under standard conditions (temperature 37 °C, humidified air atmosphere with 5% CO2). The culture medium was refreshed every 2 days.MTT assay
The cytotoxicity of plain PC, DOTAP and PC/DOTAP liposomes in a concentration range of lipids from 2 μM to 5 mM was assessed on HSF and PC-3 cells (72 h). The cytotoxicity of liposomal or free DOX (0.05–100 μM) and PTX (0.5–1000 nM) was studied on PC-3 and HUVECs, respectively (24 h). The cells were seeded in 96-well cell culture plates at a density of 5 × 103 cells per well and allowed to grow for 24 h followed by the addition of serially diluted formulations. Half-maximal inhibitory concentrations (IC50) were determined using the MTT indicator according to the conventional procedure and calculated using GraphPad Prism 5.0 software. Data were presented as mean ± SD (n = 3).Cellular uptake study
PC-3 cells and HUVECs were plated at a density of 2 × 105 cells per well in a 12-well plate. Following 24 h culturing, the cells were exposed to free RhB (0.8 μM) or its liposomal formulations for 2 h in serum-free medium. Additionally, the cells were treated with the formulations for 2 h in the presence of different effectors, namely, MnCl2 (1 mM), free RGD-NH2 or GHK-NH2 peptides (both 1 mM), or an ATP-mediated endocytosis inhibitor cocktail (0.65 mg mL−1 NaN3, 1 mg mL−1 NaF), which were added to the culture medium 30 min prior to the liposomes. The treated cells were washed with PBS three times, harvested by trypsinization, and analyzed on a Guava EasyCyte flow cytometer (Millipore). The percentage increment of the fluorescence intensity (FI) signals generated by the peptide-targeted liposomes was calculated relative to the control non-targeted liposomes using the formula: (FIpeptide − FIctrl)/FIctrl × 100%.Intracellular trafficking
To assess the intracellular penetration and localization of the RhB-loaded liposomes, HUVECs and PC-3 cells were cultured on glass coverslips placed in a 6-well cell culture plate at a density of 2 × 105 cells per well for 24 h. Then the cells were incubated with the liposomes for 30 or 120 min, rinsed with PBS three times and subsequently stained with LysoTracker Green (50 nM, 40 min) and Hoechst 33258 (2 μg mL−1, 10 min) for visualization of the lysosomes and nuclei, respectively, using an LSM 780 laser scanning confocal microscope (Carl Zeiss).Detection of intracellular DOX and ROS and the mitochondrial potential
HUVECs and PC-3 cells pre-cultured in a 12-well plate at a density of 2 × 105 cells per well were respectively treated with 1 μM DOX and 0.5 μM PTX or their formulations with PC/DOTAP liposomes for 2 h. To detect reactive oxygen species (ROS), the cells were stained with 20 μM DCFDA for 30 min at 37 °C. HUVECs were additionally labeled with 200 nM TMRE for 30 min to assess the mitochondrial potential. Simultaneous analysis of ROS and DOX in PC-3 as well as ROS and the mitochondrial potential in HUVECs was performed on a Guava EasyCyte flow cytometer.Statistical analysis
Data were presented as means with standard deviations (SD). Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Tukey's Multiple Comparison post-test (*p < 0.05, **p < 0.01, ***p < 0.001).Results
Synthesis and characterization of the peptides
The amphiphilic peptides C12-GGRGD-NH2 and C12-GGGHK-NH2 (Fig. 1) synthesized by the Fmoc SPPS method consisted of RGD and GHK bioactive motifs, a diglycyl spacer and a lauric acid (C12) residue. C12 was selected to impart membranotropic properties26,27 while ensuring better aqueous solubility compared with higher fatty acid homologues. A glycine-based spacer was previously introduced to self-assembling bioactive peptide amphiphiles owing to its low intermolecular H-bonding and β-sheet propensity.28,29 | ||
| Fig. 1 Structural formulas and mass spectra of the C12-GGRGD-NH2 and C12-GGGHK-NH2 peptides. | ||
An additional aminohexanoic acid spacer used in related peptide sequences for biomaterials modification23 was avoided here as it could adversely affect the hydrophilic–lipophilic balance of the peptide conjugates and impair their interaction with the lipid bilayer. The identity of the peptides, hereafter designated as RGD (C12-GGRGD-NH2) and GHK (C12-GGGHK-NH2), was confirmed by the LC-MS technique (Fig. 1).
The micelle-forming properties of the peptides were studied using a curcumin probe24 with more convenient fluorescence parameters (λex/λem = 423/498 nm) for the microplate format than a pyrene probe (λex/λem = 337/373(I1) nm). The working concentration of curcumin was reduced to 10 μM instead of a higher reported concentration24 to avoid deviation from the linear concentration range. The calculated CMC values of RGD and GHK were 61 ± 2 and 228 ± 16 μM, respectively (Fig. S1, ESI†). The increased micelle-forming ability of RGD can be explained by its lower expected net charge (0), which leads to decreased mutual electrostatic repulsion of its headgroups compared with GHK (+2) (given their amide form) and thus facilitates hydrophobic association of the C12 moiety. RGD and GHK in an equimolar mixture had an intermediate CMC value of 174 ± 10 μM. RGD/GHK mixed micelles induced ca. 3-fold higher fluorescence of the probe than the single-peptide systems attributed to additional electrostatic interactions of the peptide moieties, which however does not significantly promote the unimer–micelle transition compared with RGD alone (Fig. S1, ESI†).
Optimization of the liposomal composition
To optimize the targeted liposomal formulation, the effect of the lipid and peptide composition on the cellular uptake of PC, DOTAP and PC/DOTAP liposomes was initially studied. RGD-modified liposomes were prepared from mixed lipid–peptide films (method I). The targeting ability of the RhB-loaded liposomes was assessed using PC-3 prostate cancer cells (Fig. 2), overexpressing integrins α5β1 and αvβ3.30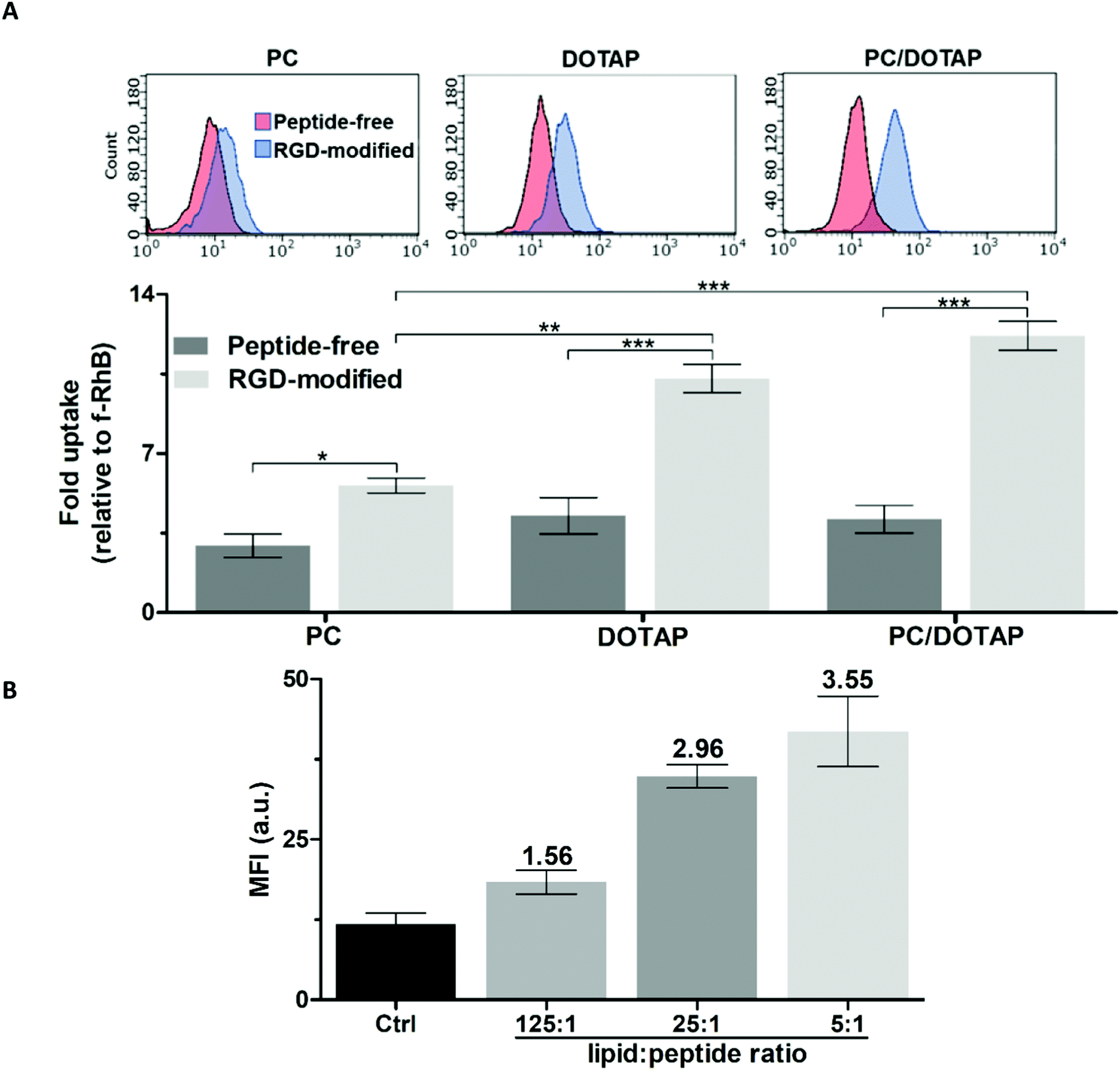 | ||
| Fig. 2 Accumulation of RhB-loaded liposomes in PC-3 cells according to flow cytometry (2 h incubation). (A) Effect of the lipid composition and modification with RGD (the molar lipid–peptide ratio was 25:1). (B) Effect of the RGD–lipid ratio in PC/DOTAP liposomes. Mean fluorescence intensity values (MFI) are shown. | ||
The liposomes formed purely from DOTAP exhibited 1.5-fold better penetration into the cells compared with the equivalent PC liposomes (Fig. 2A) apparently due to facilitation of liposome–plasma membrane interactions by the quaternary amine group of DOTAP.31 RGD noticeably enhanced the cellular accumulation of both the PC and DOTAP liposomes by 1.9 and 2.4 times, respectively. Doping of the PC liposomes with as low as ∼3 mol% DOTAP was sufficient to achieve a positive zeta potential (ZP) over +40 mV (data not shown), which should favor the cellular uptake32,33 and shelf life34 of the liposomes. The PC/DOTAP (35:1) liposomes were characterized by similar targetability to that of the DOTAP liposomes both in the case of the peptide-free and RGD-modified formulations (Fig. 2A). Therefore, the cell penetration of the PC liposomes is improved by minimal DOTAP additive increasing their cationic charge.
In addition, the plain PC/DOTAP liposomes were, on average, 1.6 times less cytotoxic than DOTAP with corresponding IC50 values (mM) as follows: 0.59 ± 0.04 (PC/DOTAP) and 0.39 ± 0.01 (DOTAP) for PC-3 cells, and 0.38 ± 0.05 (PC/DOTAP) and 0.23 ± 0.04 (DOTAP) for human skin fibroblasts (HSF) (Fig. S2A and B, ESI†). Considering the increased cost and cytotoxicity of the DOTAP lipid, the PC/DOTAP (35:1) liposomes are preferable for drug delivery applications.
The cellular uptake of the PC/DOTAP liposomes increased in proportion to the relative content of RGD (Fig. 2B and Fig. S3, ESI†). In the range of molar lipid–peptide ratio up to 25:1 the RhB fluorescence in the cells linearly increased (r2 = 0.98), whereas a further 5-fold increment of the peptide content insignificantly enhanced the signal. Therefore, a 25:1 lipid–peptide ratio was selected to prepare the targeted liposomes.
Characterization of the peptide-modified liposomes
The optimized liposomal formulations were characterized by the DLS, AFM and TEM techniques. The plain PC/DOTAP liposomes formed a monodisperse positively charged system with a hydrodynamic diameter (DH) of 117 nm and ZP of +55 mV. The amphiphilic peptides insignificantly altered the characteristics of the as-prepared liposomes in deionized water (Table S1, ESI,† and Fig. 3), which therefore preserved their colloidal properties after modification. | ||
| Fig. 3 Representative size distribution of the as-prepared plain and peptide-modified PC/DOTAP liposomes in deionized water (upper panel). TEM images of the corresponding liposomes attached to the formvar-carbon substrate (lower panel). | ||
According to AFM, on the surface of highly hydrophobic HOPG the dried liposomes (both plain and peptide-modified) were found to be disrupted, apparently as a result of hydrophobic interactions with the substrate. On a mica substrate, round structures of different diameter were detected (Fig. S3, ESI†). These were generally attributed to individual liposomal particles, which however were not expected to have relevant morphology due to repulsion forces between the lipids and highly hydrophilic mica surface.
According to TEM, the plain liposomes appeared as spread roundish structures with somewhat variable morphology but a typical diameter of 104 ± 18 nm, which corresponds to individual vesicles. Some liposomes tended to aggregate and form bigger partially fused structures presumably as a result of inter-particle interactions upon liquid evaporation. The individual peptide-modified liposomes were also visualized but relatively rarely detected (Fig. 3). Thus, the moderately hydrophobic formvar surface seems to better support attachment of the liposomes, allowing for more informative visualization by TEM. However, the resultant morphology of the liposomes could be variably affected by different factors (e.g., liposomal cargo/composition, drying conditions, vacuum and electron ionization in the TEM chamber, etc.).
Additional DLS analysis of the liposomes in HEPES buffer (pH = 7.4) allowed for more sensitive detection of the peptide component. Liposome modification with the peptides by method I neither changed the size nor disturbed the homogeneity of the system (Table 1). However, RGD and GHK in accordance with their net charge induced, respectively, a moderate but significant (p < 0.05) decrease and increase in the liposome ZP. This confirms immobilization of the peptide component at the liposome–solution interface apparently due to the insertion of the C12 tail into the lipid bilayer.
| Formulationa | D H (nm) | ZP (mV) | PdI |
|---|---|---|---|
|
a 50 mM HEPES buffer (pH = 7.4), lipid concentration 1 mM, lipid–peptide ratio 25:1. For method II the liposomes and peptides were incubated for 2 h.
|
|||
| Plain | 101 ± 1 | 18.9 ± 0.6 | 0.052 ± 0.005 |
| RGD | 102 ± 1 | 16.2 ± 0.6 | 0.069 ± 0.007 |
| GHK | 101 ± 2 | 23.1 ± 0.3 | 0.038 ± 0.016 |
| RGD/GHK | 99 ± 3 | 16.6 ± 0.4 | 0.086 ± 0.027 |
| RGD | 120 ± 2 | 11.6 ± 0.7 | 0.119 ± 0.019 |
| GHK | 117 ± 3 | 27.4 ± 0.6 | 0.113 ± 0.018 |
| RGD/GHK | 114 ± 1 | 23.7 ± 0.5 | 0.110 ± 0.017 |
The liposomes modified by method II were characterized by noticeably higher DH (on average by 16.6%) and polydispersity index (PdI), remaining quasi-monodisperse, as well as more distinct ZP values (Table 1 and Fig. S5, ESI†). The increased effects of in situ associated peptides can be explained by their predominant localization at the outer surface of the liposomal membrane, whereas with method I they should be equally distributed at both membrane surfaces. According to HPLC analysis, no peptides were detected in the extraliposomal solution for the peptide-modified liposomes prepared by both methods. This shows that the concentration of unbound peptides is below their established detection limit of 0.1 μM, which corresponds to less than 1% of the amount of peptides used for the liposome preparation/modification (for the optimized formulations with a 25:1 lipid:peptide ratio), and therefore at least 99% of the added peptide molecules should be associated with the liposomal surface.
Evaluation of the in vitro cellular uptake of the peptide-modified liposomes
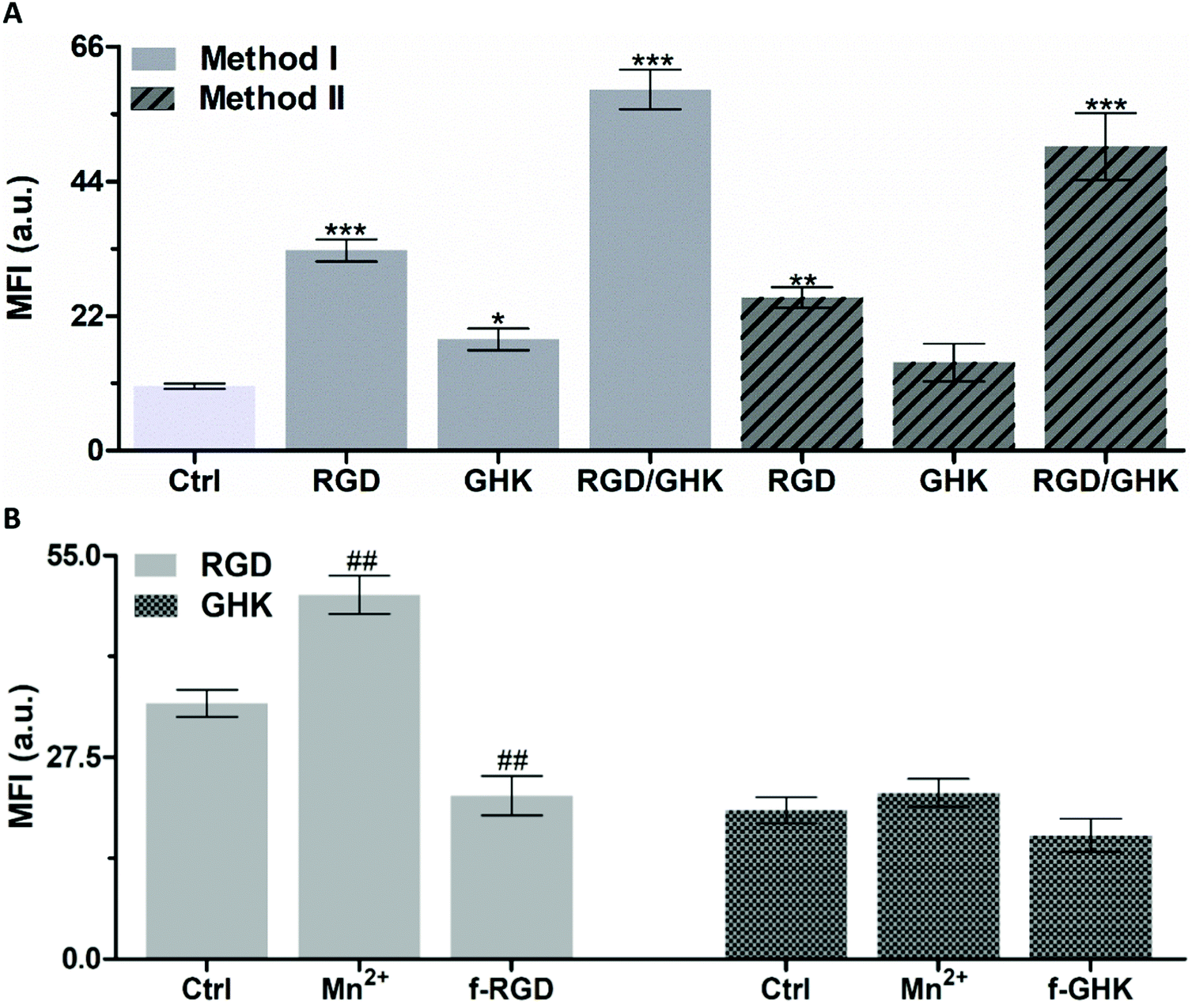 | ||
| Fig. 4 (A) Accumulation of RhB-loaded PC/DOTAP liposomes prepared from lipid–peptide films (method I) and in situ modified with peptides (method II) in PC-3 cells (2 h incubation). *p < 0.05, **p < 0.01, ***p < 0.001 vs. non-targeted liposomes (Ctrl). (B) Effect of manganese chloride (1 mM) and the free non-conjugated peptides (f-RGD, f-GHK, 2 mM) on the cellular uptake of the peptide-modified liposomes. The molar lipid–peptide ratio was 25:1. ##p < 0.01 vs. RGD-targeted liposomes (Ctrl). | ||
As additional proof of the specificity of the RGD-mediated cellular uptake of the liposomes, free non-conjugated RGD was used as a competitive inhibitor of interaction of formulated RGD with integrins35,37 (Fig. 4B). The results demonstrate that in Mn2+-supplemented medium, the signal of intracellular RhB was increased by 42.5%, whereas in the presence of free RGD-NH2 it was decreased by 36.1% (p < 0.01).
Although the exact mechanism of the interaction of GHK with the cell surface remains unknown, it may potentially involve low-affinity binding to some membrane receptors, thus providing less efficient accumulation of GHK-modified liposomes compared with the RGD based counterpart (by ca. 1.7 times, Fig. 4A). The interaction of GHK with integrins was reported;38 however, GHK-mediated uptake of the liposomes was insignificantly affected by the integrin-binding modulators (Fig. 4B), assuming other potential membrane target(s) for the GHK ligand.
It was found that the combination of RGD and GHK provided a clear synergistic enhancement of liposomal delivery (461%) exceeding the additive effect of the individual peptides (284%) (both relating to non-targeted liposomes).
Furthermore, the in situ modified liposomes (method II) showed close individual and joint effects of RGD and GHK on the cellular uptake to those of the counterpart lipid–peptide liposomes (method I) (Fig. 4A). This suggests that the inclusion of C12-conjugated peptides into the lipid phase upon liposome preparation (method I) and the association of peptides with the surface of pre-formed liposomes (method II) ensure similar liposome activation for targeting purposes.
Distribution of liposomal RhB in PC-3 cells
LSCM analysis confirmed increased uptake of the peptide-modified liposomes by PC-3 cells (RGD ≪ RGD/GHK) over the control non-targeted liposomes (Fig. 5). The intracellular accumulation of RhB was in proportion to the time of cell exposure to all liposomal formulations during 2 h. At 30 min exposure, RhB fluorescence was partially co-localized with the lysosomes according to probing with LysoTracker Green and was detected in some other structures in the cytoplasm (Fig. 5A).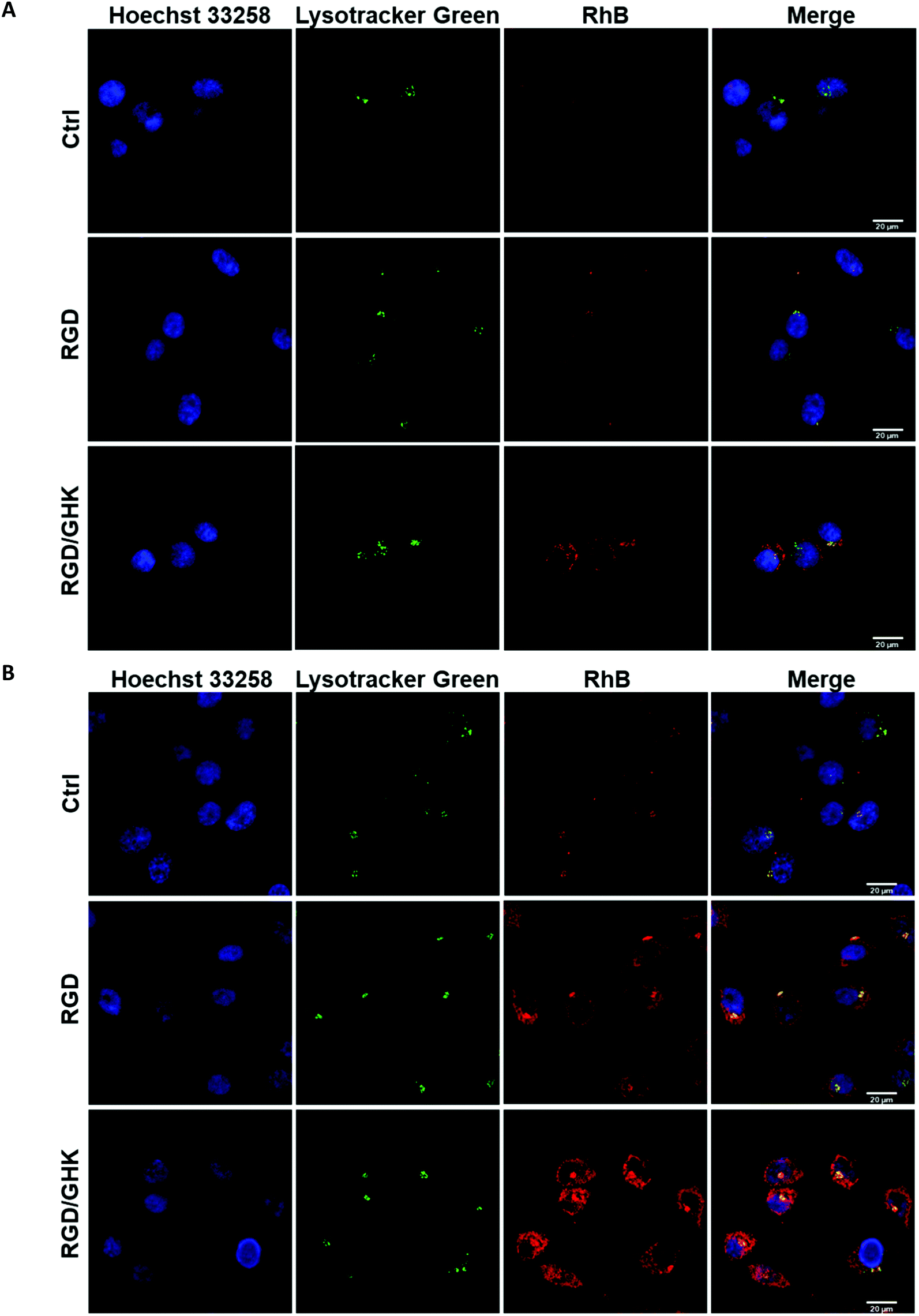 | ||
| Fig. 5 LSCM images of PC-3 cells exposed to RhB-loaded PC/DOTAP liposomes for 30 min (A) and 120 min (B). The liposomes were modified with the peptides by method I. The cells were stained with Lysotracker Green for lysosomes and Hoechst 33258 for nuclei. | ||
At 120 min exposure, RhB was mainly distributed in the compartments (Fig. 5B) attributed to mitochondria previously shown to accumulate this dye.39 These data suggest that the liposomes may enter the cells involving receptor-mediated endocytosis and lysosomal uptake as well as presumably direct fusion with the plasma membrane.
Liposome uptake by HUVECs
A similar delivery profile of the liposomal formulations was observed for HUVECs, actively proliferating fetal endothelial cells expressing αvβ3 integrins.40 In comparison with PC-3 cells, HUVECs were somewhat more sensitive to RGD and GHK, which promoted cellular uptake of the liposomes by 396 and 96%, respectively (Fig. 6A). The RGD/GHK composition further increased the sum of the effects of the individual peptides by 156%. The flow cytometry analysis correlated with LSCM data on increased intracellular accumulation of RhB formulated in RGD/GHK-modified liposomes (Fig. 6B). | ||
| Fig. 6 (A) Accumulation of RhB-loaded PC/DOTAP liposomes in HUVECs (2 h incubation). (B) LSCM images of treated HUVECs stained with Lysotracker Green and Hoechst 33258. The liposomes were modified with the peptides by method I. | ||
Inhibition of active transport by NaN3/NaF41 somewhat decreased the uptake by HUVECs in the following order: control (6.3%) < GHK (7.3%) < RGD/GHK (10.1%) < RGD (20.3%) (Fig. S6, ESI†). Therefore, although receptor-mediated endocytosis may contribute to liposome delivery depending on the type of peptide (RGD > GHK, p < 0.01), their direct fusion seems to mainly determine the transport. Together, the results demonstrate that co-presenting RGD/GHK on PC/DOTAP liposomes provides enhanced delivery both into cancer and endothelial cells expressing specific integrins.
Study of targeted liposomal formulations of anticancer drugs
The in vitro drug delivery properties of the dual-targeted RGD/GHK liposomes were assessed using doxorubicin (DOX) and paclitaxel (PTX) as well-established anticancer/antiangiogenic drugs.42,43 DOX was encapsulated into the liposomes from the aqueous phase, whereas PTX was pre-distributed in the lipid phase. The EE of the drugs was determined by spectrophotometric and HPLC analyses, respectively. The peptide-free PC/DOTAP liposomes encapsulated 45 ± 1% and 73.9 ± 2.7% DOX and PTX amounts, respectively, demonstrating better ability to retain membrane-bound PTX molecules. The modification with RGD and GHK alone did not alter the EE for both drugs; RGD/GHK together somewhat increased the EE for DOX (55 ± 3%). The drug loading was not accompanied by a noticeable change in the DLS characteristics of the as-prepared liposomes (Table S1, ESI†).Effects on PC-3 cells
IC50 values of free and encapsulated DOX toward PC-3 cells were determined using the MTT assay (24 h). All liposomal formulations increased the DOX cytotoxicity as follows: control < GHK < RGD < RGD/GHK (Fig. 7A). This dependence correlated with the intracellular accumulation of DOX according to flow cytometry analysis (2 h) (Fig. 7B). The DOX uptake was accompanied by the induction of oxidative stress as revealed by increased fluorescence of DCFDA, the cytoplasm probe for ROS, in agreement with the established prooxidant activity of anthracycline antibiotics.44,45 The results show that the dual-targeted liposomes provide a considerable ca. 4- and 2-fold decrease of the IC50 values over the free drug and RGD-modified liposomes, respectively, supporting increased tumor-targeting potential of the RGD/GHK composition (Fig. 7A). | ||
| Fig. 7 (A) IC50 values of free DOX and DOX-loaded PC/DOTAP liposomes for PC-3 cells (MTT assay, 24 h). ***p < 0.001 vs. free DOX, ##p < 0.01, and ###p < 0.001 vs. DOX-loaded non-targeted liposomes. (B) MFI of DOX (left Y-axis) and DCFDA (right Y-axis) in PC-3 cells treated with free and encapsulated DOX (1 μM) for 2 h according to flow cytometry. ***p < 0.001 vs. free DOX. | ||
Effects on HUVECs
The liposomal formulations also promoted PTX cytotoxicity toward HUVECs by 1.2–3.0 times with corresponding IC50 values (nM) of 10.8 ± 1.3 (non-targeted liposomes), 7.4 ± 0.7 (GHK), 6.0 ± 0.6 (RGD) and 4.3 ± 0.6 (RGD/GHK) (Fig. 8A). These results demonstrate that the RGD/GHK composition ensures enhanced cytotoxic activity of liposomal PTX against endothelial cells. Probing with DCFDA showed that the PTX-loaded liposomes modified with the peptide composition also had a dramatically increased ability to induce oxidative stress in HUVECs over the other formulations (Fig. 8B). This effect was observed along with a proportional decrease in the transmembrane potential of mitochondria according to TMRE fluorescence (Fig. 8B). The latter depolarizing effect apparently reflects earlier mitochondria-damaging and proapoptotic action due to enhanced peptide-mediated liposomal delivery of PTX into the cells.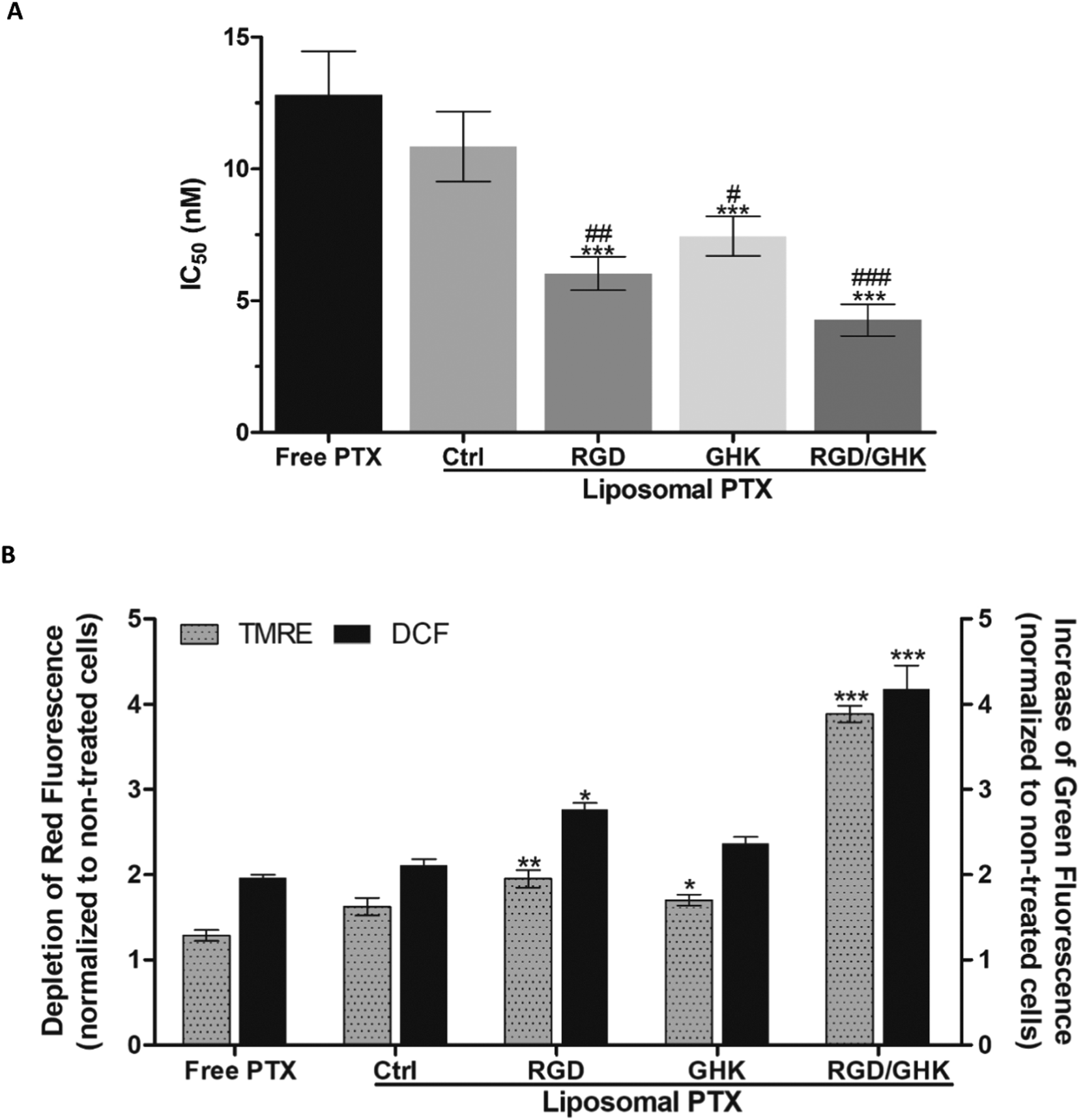 | ||
| Fig. 8 (A) IC50 values of free PTX and PTX-loaded PC/DOTAP liposomes for HUVECs (MTT assay, 24 h). **p < 0.01, ***p < 0.001 vs. free PTX, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. non-targeted liposomes (Ctrl). (B) Fold changes of the TMRE (left Y-axis) and DCFDA (right Y-axis) fluorescence intensities in HUVECs treated with free and encapsulated PTX (0.5 μM) for 2 h according to flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001 vs. free PTX. | ||
Discussion
PC/DOTAP (35:1) liposomes were used to develop peptide-targeted liposomal formulations. Different optimal ratios of cationic and neutral lipids were earlier reported for the liposomes.19,46 The DOTAP additive (≥10 mol%) was shown to reduce the size distribution of PC liposomes and improve PTX incorporation into the liposomal membrane.47 Based on our results, increased DOTAP content does not seem reasonable as it should increase the cytotoxicity of the formulations (Fig. S2, ESI†) in addition to its high cost.48
Non-covalent introduction of fatty acid-conjugated peptides to the liposomal membrane was advantageous for studying targeting peptides, considering that conventional covalent methods for liposome modification are characterized by relative laboriousness, variable yield, side reactions and by-products.49 Therefore, RGD and GHK based sequences (Fig. 1) were synthesized in the form of conjugates with lauric acid with proven ability to associate with and be inserted into lipid bilayers.26,27 Even shorter hexanoyl and decanoyl groups were shown to anchor in PC based liposomal membranes, allowing oligopeptide molecules to be presented at the outer liposome surface.50
We compared two methods for liposome modification with the amphiphilic peptides based on the preparation of lipid–peptide films (method I) and post-insertion of the peptides into the liposomal membrane (method II). Although the latter method is less conventional, it has proved suitable for the immobilization of antibody fragments and peptides on liposomes.51
The synthesized peptides had well-defined micelle-forming properties with comparable CMC values to those previously reported for short C12-conjugated peptides such as C12-KKGRGDS (CMC = 95 μM).52 To provide appropriate ionization of peptide groups, the CMC was determined in HEPES buffer (pH = 7.4), which was also advantageous for ZP measurements. Interestingly, in HEPES buffer the immobilized peptides induced more distinct changes in the ZP of the liposomes, allowing for better characterization of the liposome–solution interface (Table 1). These data are in agreement with reported ZP modulation of liposomes by the RGD peptide in PBS buffer solution at pH 7.4.53 DLS and HPLC together showed effective redistribution of the amphiphilic peptides from solution to the liposome surface, supporting the feasibility of liposome modification with the peptides by co-incubation in situ (method II).
Along with the lipid composition, the optimal peptide content in the liposomes was found to be ca. 4 mol% of the lipid amount. This peptide content, as shown for RGD, ensured almost saturated intracellular accumulation of the liposomes; at 20 mol% content the cellular uptake increased slightly and became more variable (Fig. 2B).
Using the optimized liposomal formulation, the cell-penetrating ability of the RGD, GHK and RGD/GHK systems was accurately compared. Most of the previous studies were focused on cell-targeting systems based on RGD peptide derivatives;10,37 no data exist on such a characterization of the GHK peptide and its composition with the RGD peptide. Epithelial cancer cells (PC-3) and primary endothelial cells (HUVECs) both overexpressing membrane integrins30,40 were used as relevant cells for cancer targeting.
Our results for the first time show that the dual modification with RGD and GHK allows for a profound synergistic increase of liposomal delivery to both types of cells (Fig. 4–6). Taking into account the cationic nature of PC/DOTAP liposomes, the enhancing effect of the GHK peptide in the GHK/RGD composition was attributed to its specific binding to certain plasma membrane component(s) rather than electrostatic interactions of the liposomes with the membrane (mediated by the positive charge of GHK). The fact that the revealed synergy was observed against a weak effect of GHK alone might suggest multiple low-affinity interactions of the peptide with cell membrane targets to complement high-affinity integrin–RGD binding.
Potential interaction of GHK with α6 and β1 integrins in mesenchymal stem cells was shown.38 Based on the previously reported growth factor-like activities of free and material-conjugated GHK peptides,14,23,54,55 their affinity to some membrane receptors of polypeptide growth factors can be envisaged. GHK may activate the angiotensin II AT1 receptor56 involved in the regulation of tumor growth and induction of tumor-associated angiogenesis.57 Moreover, the GHK motif within SPARC113 and SPARC118 was shown to stimulate endothelial cell angiogenic differentiation and proliferation.58
Furthermore, we hypothesized that the effect of the RGD/GHK composition could be mediated by the GHK interaction with anionic glycosaminoglycans presented on the surface of mammalian cells playing important roles in cell–matrix interactions and regulation of various cellular functions including activation of growth-factor receptors (e.g., heparin-binding EGFR, FGFR, and others).
Based on this assumption, the peptide-modified liposomes were additionally employed to assess binding of GHK and heparin. To prevent non-specific electrostatic interactions with anionic heparin, DOTAP-free PC liposomes were used to ensure a negative charge of both the control and peptide-modified formulations under experimental conditions. Interestingly, heparin was found to strongly increase the negative charge of the GHK-modified liposomes in contrast to the unmodified ones, indicating GHK-mediated attachment of the glycosaminoglycan to the liposomal surface. The additional RGD peptide in the composition with GHK did not inhibit this binding (Fig. 9A). Interaction of the liposomes with the peptides and heparin modulated the liposome size without inducing any decrease in homogeneity (Fig. 9B). These data support the interaction of GHK with heparin in accordance with an earlier spectroscopic study59 and also the idea that GHK-modified liposomes are potentially capable of binding to membrane glycosaminoglycans. Moreover, these results for the first time suggest in situ peptide-modified liposomes as a useful tool to characterize biorelevant interactions of peptide ligands.
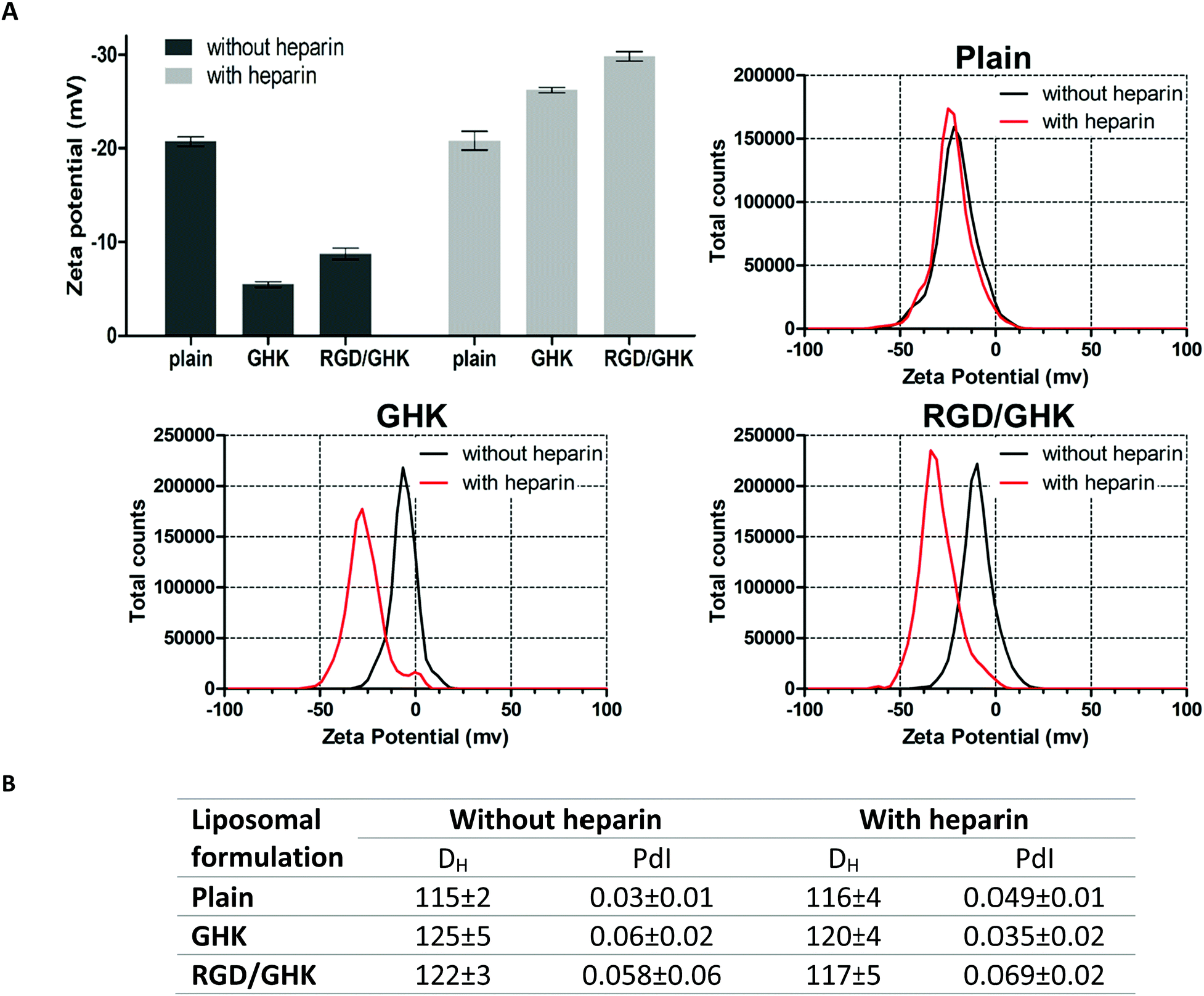 | ||
| Fig. 9 DLS characteristics of peptide-modified PC liposomes prepared by method II in the absence or presence of heparin (25.4 μg mL−1). (A) Mean values and corresponding distributions of the zeta potential. (B) Mean hydrodynamic diameter and polydispersity index of the formulations. 50 mM HEPES buffer (pH = 7.4), lipid concentration 1 mM, lipid–peptide ratio 25:1. | ||
The reported mechanisms of internalization of soft liposomes may involve both fusion with the plasma membrane, which is promoted by fusogenic lipids like DOTAP,41 and an endocytosis process with the participation of cell-adhesive peptides like RGD.60 Our data suggest that peptide-mediated delivery of the liposomes is mainly determined by their interaction/fusion with the cellular membrane rather than being limited to receptor-mediated uptake. This is supported by both the low effect of active transport inhibitors and the low degree of co-localization of the liposomal dye with lysosomes.
Recently, in a peptide-modified cryogel ECM model, GHK did not significantly promote primary adhesion and proliferation of mammalian cells, though when combined with RGD it ensured synergistic regeneration-related effects in vitro also attributed to specific interactions of the peptides with the cell surface.14,23 This finding together with some established relationships between regenerative and oncogenic processes61 served as the basis for studying the cancer-targeting properties of the RGD/GHK composition.
Previously, increased targeted delivery of liposomes into glioma cells was also achieved by a combination of cyclic RGD variants with transferrin62 or C-type natriuretic peptide-22.63 Both of these ligands are of more complex sequences than GHK, and their immobilization required covalent pre-conjugation steps. According to our data, the combination of the simplest RGD and GHK motif-based amphiphilic peptides allows effective single-step activation of the liposomes to boost their intracellular delivery. The cancer-targeting potential of the composition was additionally assessed in vitro using pre-characterized DOX and PTX-loaded liposomes as formulations against different solid tumors64 and tumor vessels,43 respectively. The capability of peptide-targeted liposomes to efficiently encapsulate both hydrophilic and hydrophobic anticancer agents and deliver them to the target cells was demonstrated. The presented peptides did not negatively affect the drug EE of the liposomes and, to the contrary, increased the EE presumably due to the stabilizing effect on the lipid bilayer and/or additional interactions with encapsulated drugs.
The encapsulation of DOX and PTX within RGD/GHK-modified liposomes was accompanied, respectively, by a 4- and 3-fold increase in drug cytotoxicity against PC-3 cells (Fig. 7A) and HUVECs (Fig. 8A). This effect considerably exceeded that of other targeted formulations and was associated with enhanced intracellular accumulation of DOX, a decrease in mitochondrial potential by PTX and oxidative stress induced by both drugs (Fig. 7B and 8B). ROS generation and mitochondria depolarization are key cellular processes involved in pro-apoptotic activity of anticancer drugs.45,65 Although the cancerous phenotype often relies on an increased ROS level, the induction of an intense prooxidant effect is generally required to kill cancer cells.66,67 ROS overproduction is known to underlie the cytotoxicity of DOX in addition to its direct DNA-damaging ability.66
The early-detected effects of the formulated drugs, in addition to intense intracellular fluorescence of RhB and DOX (Fig. 5, 6 and 7B), which is not expected for fluorophores if they were still retained in the liposomes,68 suggest fast intracellular release of the drugs. These results further support that combined targeting effects of RGD and GHK peptides are of particular interest for the development of improved anticancer liposomal formulations carrying drugs with different properties and modes of encapsulation.
Conclusions
Our study identifies the dual composition of synthesized RGD and GHK peptides (Fig. 1) as a novel promising targeting system. Elucidation of the exact mechanisms of this composition should involve establishment of the targets for the GHK peptide along with further optimization of the RGD- and GHK-containing sequences. This requires a separate study as no specific targets for the GHK peptide have been proposed in spite of its multiple bioactivities, and pharmacological and cosmeceutical significance.15,16 Along with demonstrating the targeting potential of the RGD/GHK composition, our study provides a required methodology for studying combinatorial effects of short peptide ligands on the surface of liposomal formulations, including the synthesis of amphiphilic peptide sequences and their controllable immobilization on the liposomes. Two immobilization procedures, namely, mixing lipids and peptides upon liposome preparation (method I) or post-insertion of peptide molecules into the liposomal membrane (method II), showed comparable in vitro delivery profiles (Fig. 4A). We believe that the latter method is of particular interest for the detection of biorelevant peptide-mediated interactions as well as screening of targeted peptide systems based on the combination of different peptide signals, and this can be performed using both synthetic liposomes and potentially natural vesicles derived from living cells.69 Furthermore, our results pave the way for the development of improved liposomal formulations capable of carrying both hydrophilic and hydrophobic anticancer drugs along with multiple membrane-bound peptides with synergistic therapeutic effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work of M. Z., R. G., and T. I. A. was supported by Russian Foundation for Basic Research (project 19-03-01010) (in vitro study of peptide-modified liposomal formulations) and Russian Science Foundation (project 20-73-10105) (design, synthesis, structure verification of amphiphilic targeting peptides). R. V. P. and G. A. G. are grateful for financial support from the government assignment for FRC Kazan Scientific Center of RAS in part of the sample preparation for morphological analysis and further experimentation. The authors thank S. V. Fedosimova and A. M. Rogov (Interdisciplinary Center for Analytical Microscopy of Kazan Federal University) for LSCM and AFM analysis as well as V. A. Nikolaeva and M. I. Kamalov (Laboratory of Bioactive Polymers and Peptides, Kazan Federal University) for helpful experimentation during manuscript revision. This research is part of Kazan Federal University Strategic Academic Leadership Program.References
- Y. Yang, X. M. Li, T. Wang, Q. Q. Guo, T. Xi and L. F. Zheng, J. Hematol. Oncol., 2020, 13, 60, DOI:10.1186/S13045-020-00901-6.
- P. Lee, R. Z. Zhang, V. Li, X. L. Liu, R. W. Y. Sun, C. M. Che and K. K. Y. Wong, Int. J. Nanomed., 2012, 7, 731–737, DOI:10.2147/Ijn.S28783.
- W. C. Zamboni, Oncologist, 2008, 13, 248–260, DOI:10.1634/theoncologist.2007-0180.
- M. Srinivasarao, C. V. Galliford and P. S. Low, Nat. Rev. Drug Discovery, 2015, 14, 203–219, DOI:10.1038/nrd4519.
- U. K. Marelli, F. Rechenmacher, T. R. Sobahi, C. Mas-Moruno and H. Kessler, Front. Oncol., 2013, 3, 222, DOI:10.3389/fonc.2013.00222.
- A. Tyagi, P. Kapoor, R. Kumar, K. Chaudhary, A. Gautam and G. P. Raghava, Sci. Rep., 2013, 3, 2984, DOI:10.1038/srep02984.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672, 10.1039/c4ra02458c.
- Z. Jiang, J. Guan, J. Qian and C. Zhan, Biomater. Sci., 2019, 7, 461–471, 10.1039/c8bm01340c.
- A. Accardo and G. Morelli, Biopolymers, 2015, 104, 462–479, DOI:10.1002/bip.22678.
- S. Naik, D. Patel, K. Chuttani, A. K. Mishra and A. Misra, Nanomedicine, 2012, 8, 951–962, DOI:10.1016/j.nano.2011.11.008.
- K. Chen and X. Chen, Theranostics, 2011, 1, 189–200, DOI:10.7150/thno/v01p0189.
- K. N. Enyedi, S. Toth, G. Szakacs and G. Mezo, PLoS One, 2017, 12, e0178632, DOI:10.1371/journal.pone.0178632.
- G. Sarfati, T. Dvir, M. Elkabets, R. N. Apte and S. Cohen, Biomaterials, 2011, 32, 152–161, DOI:10.1016/j.biomaterials.2010.09.014.
- M. Zoughaib, D. Luong, R. Garifullin, D. Z. Gatina, S. V. Fedosimova and T. I. Abdullin, Mater. Sci. Eng., C, 2021, 120, 111660, DOI:10.1016/j.msec.2020.111660.
- L. Pickart, J. Biomater. Sci., Polym. Ed., 2008, 19, 969–988, DOI:10.1163/156856208784909435.
- F. X. Maquart and J. C. Monboisse, Pathol. Biol., 2014, 62, 91–95, DOI:10.1016/j.patbio.2014.02.007.
- R. Kashapov, G. Gaynanova, D. Gabdrakhmanov, D. Kuznetsov, R. Pavlov, K. Petrov, L. Zakharova and O. Sinyashin, Int. J. Mol. Sci., 2020, 21, 6961, DOI:10.3390/ijms21186961.
- M. Riaz, M. Riaz, X. Zhang, C. Lin, K. Wong, X. Chen, G. Zhang, A. Lu and Z. Yang, Int. J. Mol. Sci., 2018, 19, 195, DOI:10.3390/ijms19010195.
- S. Lee, S. Y. Lee, S. Park, J. H. Ryu, J. H. Na, H. Koo, K. E. Lee, H. Jeon, I. C. Kwon, K. Kim and S. Y. Jeong, Macromol. Biosci., 2012, 12, 849–856, DOI:10.1002/mabi.201200001.
- D. A. Kuznetsova, L. A. Vasileva, G. A. Gaynanova, R. V. Pavlov, A. S. Sapunova, A. D. Voloshina, G. V. Sibgatullina, D. V. Samigullin, K. A. Petrov, L. Y. Zakharova and O. G. Sinyashin, J. Mol. Liq., 2021, 330, 115703, DOI:10.1016/j.molliq.2021.115703.
- D. A. Kuznetsova, G. A. Gaynanova, L. A. Vasileva, G. V. Sibgatullina, D. V. Samigullin, A. S. Sapunova, A. D. Voloshina, I. V. Galkina, K. A. Petrov and L. Y. Zakharova, J. Mater. Chem. B, 2019, 7, 7351–7362, 10.1039/c9tb01853k.
- R. V. Pavlov, G. A. Gaynanova, D. A. Kuznetsova, L. A. Vasileva, I. V. Zueva, A. S. Sapunova, D. N. Buzyurova, V. M. Babaev, A. D. Voloshina, S. S. Lukashenko, I. K. Rizvanov, K. A. Petrov, L. Y. Zakharova and O. G. Sinyashin, Int. J. Pharm., 2020, 587, 119640, DOI:10.1016/j.ijpharm.2020.119640.
- T. D. Luong, M. Zoughaib, R. Garifullin, S. Kuznetsova, M. O. Guler and T. I. Abdullin, ACS Appl. Bio Mater., 2020, 3, 1116–1128, DOI:10.1021/acsabm.9b01059.
- S. Mondal and S. Ghosh, J. Photochem. Photobiol., B, 2012, 115, 9–15, DOI:10.1016/j.jphotobiol.2012.06.004.
- S. Jain, D. Kumar, N. K. Swarnakar and K. Thanki, Biomaterials, 2012, 33, 6758–6768, DOI:10.1016/j.biomaterials.2012.05.026.
- L. Farkuh, P. T. Hennies, C. Nunes, S. Reis, L. Barreiros, M. A. Segundo, P. L. Oseliero Filho, C. L. P. Oliveira, A. Cassago, R. V. Portugal, R. A. Muramoto, G. P. B. Carretero, S. Schreier, H. Chaimovich and I. M. Cuccovia, Heliyon, 2019, 5, e02648, DOI:10.1016/j.heliyon.2019.e02648.
- P. Hoyrup, J. Davidsen and K. Jorgensen, J. Phys. Chem. B, 2001, 105, 2649–2657, DOI:10.1021/jp003631o.
- R. Garifullin, O. Ustahuseyin, A. Celebioglu, G. Cinar, T. Uyar and M. O. Guler, RSC Adv., 2013, 3, 24215–24221, 10.1039/C3RA43930E.
- G. Gulseren, G. Tansik, R. Garifullin, A. B. Tekinay and M. O. Guler, Macromol. Biosci., 2019, 19, 1800080, DOI:10.1002/mabi.201800080.
- A. Stachurska, J. Elbanowski and H. M. Kowalczynska, Cell Biol. Int., 2012, 36, 883–892, DOI:10.1042/Cbi20110522.
- R. Kolasinac, C. Kleusch, T. Braun, R. Merkel and A. Csiszar, Int. J. Mol. Sci., 2018, 19, 346, DOI:10.3390/ijms19020346.
- J. Wu, A. Lee, Y. Lu and R. J. Lee, Int. J. Pharm., 2007, 337, 329–335, DOI:10.1016/j.ijpharm.2007.01.003.
- J. Jimenez-Lopez, I. Bravo-Caparros, L. Cabeza, F. R. Nieto, R. Ortiz, G. Perazzoli, E. Fernandez-Segura, F. J. Canizares, J. M. Baeyens, C. Melguizo and J. Prados, Biomed. Pharmacother., 2021, 133, 111059, DOI:10.1016/j.biopha.2020.111059.
- D. A. Kuznetsova, D. R. Gabdrakhmanov, S. S. Lukashenko, L. R. Ahtamyanova, I. R. Nizameev, M. K. Kadirov and L. Y. Zakharova, Colloids Surf., B, 2019, 178, 352–357, DOI:10.1016/j.colsurfb.2019.03.025.
- L. Y. Wang, D. Pan, Q. Yan and Y. H. Song, Protein Sci., 2017, 26, 1124–1137, DOI:10.1002/pro.3163.
- F. Ye, C. Kim and M. H. Ginsberg, Blood, 2012, 119, 26–33, DOI:10.1182/blood-2011-04-292128.
- Z. Song, Y. Lin, X. Zhang, C. Feng, Y. Lu, Y. Gao and C. Dong, Int. J. Nanomed., 2017, 12, 1941–1958, DOI:10.2147/IJN.S125573.
- M. E. Klontzas, S. Reakasame, R. Silva, J. C. F. Morais, S. Vernardis, R. J. MacFarlane, M. Heliotis, E. Tsiridis, N. Panoskaltsis, A. R. Boccaccini and A. Mantalaris, Acta Biomater., 2019, 88, 224–240, DOI:10.1016/j.actbio.2019.02.017.
- P. Reungpatthanaphong, S. Dechsupa, J. Meesungnoen, C. Loetchutinat and S. Mankhetkorn, J. Biochem. Biophys. Methods, 2003, 57, 1–16, DOI:10.1016/S0165-022x(03)00032-0.
- D. G. Vartak, B. S. Lee and R. A. Gemeinhart, Mol. Pharmaceutics, 2009, 6, 1856–1867, DOI:10.1021/mp900152t.
- M. Farid, T. Faber, D. Dietrich and A. Lamprecht, Colloids Surf., B, 2020, 194, 111193, DOI:10.1016/j.colsurfb.2020.111193.
- M. H. Zoughaib, D. T. Luong, Z. Y. Siraeva, A. A. Yergeshov, T. I. Salikhova, S. V. Kuznetsova, R. G. Kiyamova and T. I. Abdullin, Bull. Exp. Biol. Med., 2019, 167, 590–598, DOI:10.1007/s10517-019-04577-y.
- F. Tan, X. H. Mo, J. Zhao, H. Liang, Z. J. Chen and X. L. Wang, J. Nanopart. Res., 2017, 19, 51, DOI:10.1007/s11051-016-3721-6.
- A. Mordente, E. Meucci, A. Silvestrini, G. E. Martorana and B. Giardina, Curr. Med. Chem., 2009, 16, 1656–1672, DOI:10.2174/092986709788186228.
- H. He, L. Wang, Y. Qiao, Q. Zhou, H. Li, S. Chen, D. Yin, Q. Huang and M. He, Front. Pharmacol., 2019, 10, 1531, DOI:10.3389/fphar.2019.01531.
- Z. Drulis-Kawa, J. Gubernator, A. Dorotkiewicz-Jach, W. Doroszkiewicz and A. Kozubek, Int. J. Pharm., 2006, 315, 59–66, DOI:10.1016/j.ijpharm.2006.02.017.
- R. B. Campbell, S. V. Balasubramanian and R. M. Straubinger, J. Pharm. Sci., 2001, 90, 1091–1105, DOI:10.1002/jps.1063.
- S. Y. Kim, S. J. Lee, J. K. Kim, H. G. Choi and S. J. Lim, Int. J. Nanomed., 2017, 12, 7323–7335, DOI:10.2147/IJN.S146785.
- P. Marques-Gallego and A. I. de Kroon, BioMed Res. Int., 2014, 2014, 129458, DOI:10.1155/2014/129458.
- N. Alminana, M. A. Alsina and F. Reig, Colloids Surf., B, 2007, 57, 243–249, DOI:10.1016/j.colsurfb.2007.02.006.
- T. M. Allen, P. Sapra and E. Moase, Cell. Mol. Biol. Lett., 2002, 7, 889–894 CAS.
- J. Liang, W. Wu, D. Lai, J. Li and C. Fang, J. Biomater. Sci., Polym. Ed., 2015, 26, 369–383, DOI:10.1080/09205063.2015.1012034.
- M. Chang, S. Lu, F. Zhang, T. Zuo, Y. Guan, T. Wei, W. Shao and G. Lin, Colloids Surf., B, 2015, 129, 175–182, DOI:10.1016/j.colsurfb.2015.03.046.
- P. J. Huang, Y. C. Huang, M. F. Su, T. Y. Yang, J. R. Huang and C. P. Jiang, Photomed. Laser Surg., 2007, 25, 183–190, DOI:10.1089/pho.2007.2062.
- X. M. Zhou, G. L. Wang, X. B. Wang, L. Liu, Q. Zhang, Y. Yin, Q. Y. Wang, J. Kang and G. Hou, Front. Pharmacol., 2017, 8, 904, DOI:10.3389/fphar.2017.00904.
- J. A. Garcia-Sainz and J. A. Olivares-Reyes, Peptides, 1995, 16, 1203–1207, DOI:10.1016/0196-9781(95)02015-o.
- N. Imai, T. Hashimoto, M. Kihara, S. Yoshida, I. Kawana, T. Yazawa, H. Kitamura and S. Umemura, Lab. Invest., 2007, 87, 189–198, DOI:10.1038/labinvest.3700504.
- I. T. Tai and M. J. Tang, Drug Resist. Updates, 2008, 11, 231–246, DOI:10.1016/j.drup.2008.08.005.
- D. L. Rabenstein, J. M. Robert and S. Hari, FEBS Lett., 1995, 376, 216–220, DOI:10.1016/0014-5793(95)01286-5.
- M. Amin, M. Mansourian, G. A. Koning, A. Badiee, M. R. Jaafari and T. L. M. Ten Hagen, J. Controlled Release, 2015, 220, 308–315, DOI:10.1016/j.jconrel.2015.10.039.
- L. A. Rocha, D. A. Learmonth, R. A. Sousa and A. J. Salgado, Biotechnol. Adv., 2018, 36, 208–227, DOI:10.1016/j.biotechadv.2017.11.004.
- L. Qin, C. Z. Wang, H. J. Fan, C. J. Zhang, H. W. Zhang, M. H. Lv and S. D. Cui, Oncol. Lett., 2014, 8, 2000–2006, DOI:10.3892/ol.2014.2449.
- C. Chen, Z. Duan, Y. Yuan, R. Li, L. Pang, J. Liang, X. Xu and J. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 5864–5873, DOI:10.1021/acsami.6b15831.
- A. Accardo, S. Mannucci, E. Nicolato, F. Vurro, C. Diaferia, P. Bontempi, P. Marzola and G. Morelli, Drug Delivery Transl. Res., 2019, 9, 215–226, DOI:10.1007/s13346-018-00606-x.
- H. Fu, L. Huang, C. Xu, J. Zhang, D. Li, L. Ding, L. Liu, Y. Dong, W. Wang and Y. Duan, Nanomedicine, 2019, 21, 102062, DOI:10.1016/j.nano.2019.102062.
- Z. Farhane, F. Bonnier, M. A. Maher, J. Bryant, A. Casey and H. J. Byrne, J. Biophotonics, 2017, 10, 151–165, DOI:10.1002/jbio.201600019.
- Y. Mai, J. J. Yu, B. Bartholdy, Z. Y. Xu-Monette, E. E. Knapp, F. Yuan, H. Chen, B. B. Ding, Z. Yao, B. Das, Y. Zou, K. H. Young, S. Parekh and B. H. Ye, Blood, 2016, 128, 2797–2807, DOI:10.1182/blood-2016-03-705814.
- X. B. Xiong, Y. Huang, W. L. Lu, X. Zhang, H. Zhang, T. Nagai and Q. Zhang, J. Pharm. Sci., 2005, 94, 1782–1793, DOI:10.1002/jps.20397.
- T. Tian, H. X. Zhang, C. P. He, S. Fan, Y. L. Zhu, C. Qi, N. P. Huang, Z. D. Xiao, Z. H. Lu, B. A. Tannous and J. Gao, Biomaterials, 2018, 150, 137–149, DOI:10.1016/j.biomaterials.2017.10.012.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00498k |
| This journal is © The Royal Society of Chemistry 2021 |