
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerformance and application of carbon-based electrocatalysts in direct methanol fuel cell
Bhagyalakhi
Baruah
ab and
Pritam
Deb
 *a
*a
aDepartment of Physics, Tezpur University (Central University), Tezpur 784028, Assam, India. E-mail: pdeb@tezu.ernet.in
bDepartment of Physics, Duliajan College, 786602, Assam, India
First published on 12th July 2021
Abstract
The growing energy demand with rapid consumption of fossil fuels and continuous rise in environmental issues have led to the development of an alternative energy conversion system, direct methanol fuel cell (DMFC). The high energy density, easy transportability of methanol and compact design, lightweight are some of the advantages of DMFC over other types of fuel cells. However, the high cost of platinum (Pt) catalyst and the poisoning of Pt electrode, sluggish reaction kinetics as well as methanol crossover are the major limitations of DMFC. Currently, carbon materials have drawn tremendous attention from researchers as catalyst support in DMFC due to remarkable properties, such as good electronic conductivity, better stability and environment friendliness that have an immense influence on fuel cell performance. In this regard, recently, advancements in carbon-based materials, including mesoporous carbon, carbon black, carbon quantum dots, carbon nanotubes, carbon nanofibre, graphene and other carbon forms such as carbon nanosheet, nanohorns, nanosphere as electrocatalyst support have been reported. The influence of doping with heteroatoms, functionalization, presence of defects and porous structure of carbon on the electrocatalytic performance has also been discussed in this report. Finally, challenges related to new generation carbon material-based electrocatalysts and future perspectives are discussed.
1. Introduction
In recent years, the rapid consumption of fossil fuels with the growing need of human beings has accelerated the development of alternative energy sources over conventional ones. Furthermore, severe harmful impacts of conventional combustion-based power generation technologies are climate change, global warming, ozone depletion, acidic rains, etc. Therefore, in recent times, people have shown much interest in the development of sustainable and renewable green energy sources to suppress the emissions of toxic gases and decrease environmental pollution. Among the various green power technologies, fuel cell-based technologies have several benefits, including clean energy generation, high quality of the generated energy, electrical adaptability to the load and silent functioning.1,2 Fuel cell technology can also overcome the issues including power fluctuation and intermittent nature related to the other green renewable energy sources including solar and wind systems.3 Moreover, the maximum attainable theoretical efficiency of the fuel cell is not limited by the Carnot efficiency and it can reach higher than 80% in cogeneration of heat and power (CHP).4In fuel cells, chemical energy gets converted into electrical energy via a chemical reaction. Fuel cell differs from the battery in the sense that, batteries are closed systems, where reactants are stored inside the cell compartment. On the other hand, fuel cells are open systems, where fuels (hydrocarbon, hydrogen, alcohol) are constantly fed from outside the cell compartment.5 Moreover, recharging a battery takes much more time than refueling an external fuel. Therefore, fuel cells can be considered as a suitable alternative to conventional batteries. Fuel cells do not store charges but directly deliver electricity obtained from the chemical reaction of the reactants. The main advantages of fuel cells over conventional batteries are increased operation time, extended durability, compact load, and ease of recharging.6 Fuel cells have been considered as a promising renewable energy source with diverse applications in portable electronics such as cellular phones, laptops, notepad, battery chargers, electronic appliances, stationary items, electric vehicles, military equipment and remote controlled gadgets.7,8 These can be also used in the residential, industrial, and commercial purpose for the generation of electricity and heat.9
Hydrogen is the most common fuel that has been used in fuel cells with many advantages such as environmental friendliness and high power density. Moreover, the fast reaction kinetics in hydrogen fuel cell results in excellent cell performance. However, the major limitations are difficulty in storage, transportation and handling of hydrogen. To overcome these issues, liquid fuels including methanol, ethanol, propanol, ethylene glycol, formic acid have been used to draw hydrogen for fuel cell applications. In this process, liquid fuel is converted into hydrogen by using a fuel reformer and therefore the overall cost of the cell increases.10 However, liquid fuels such as methanol, ethanol, ethylene glycol, glycerol and formic acid can be directly fed into the cell compartment without using a reformer system in the direct alcohol fuel cell.11–14 Among different alcohols, methanol attracts much attention since it is liquid at room temperature having a high energy density (5.04 kW h L−1) and is easy to store and transport. It can also be processed from various carbon-based components such as biomass, coal, wood and natural gas.15–18 Direct methanol fuel cell (DMFC) is based on proton exchange membrane (PEM) technology, where a polymer electrolyte membrane is used. DMFCs have gained much attention from researchers due to their simple design, low temperature (60–120 °C) of operation, cost-effective, easy portability and lightweight. Moreover, the lifetime of DMFC is also longer than that of conventional lithium-ion batteries and also refueling can be done by simply replacing the cartridge of the fuel. Nevertheless, there are some challenges related to DMFC such as methanol crossover, sluggish reaction kinetics and higher fabrication cost due to the use of platinum (Pt) catalyst and Nafion membrane. Pt catalysts are the commonly used catalyst for both methanol oxidation and oxygen reduction reactions in DMFC due to excellent electrocatalytic performance.19–22 Although, the high price and global scarcity of Pt impede the commercialization of DMFC. Moreover, the intermediates, formed during MOR, get adsorbed easily over pure Pt catalysts causing CO poisoning that deteriorates the electrocatalytic activity. In order to overcome these limitations and to increase the electrocatalytic activity, Pt loading is needed to be minimized by incorporating a support material over which metal nanostructures are uniformly distributed. There are some important characteristics that should be fulfilled by the catalyst support materials: (i) large surface area and sufficient interaction between metal nanostructures and support material to obtain a highly stable dispersion; (ii) good electrochemical stability and (iii) high electrical conductivity to enhance electron transfer rate during the electrochemical reaction.
Carbon materials have been frequently used as support to electrocatalysts in DMFC because of their high conductivity, large surface area, and stability in both acidic and alkaline media.23–26 The use of carbon materials greatly influences the size, morphology and distribution of particles, as well as properties including dispersion and stability of the supported metal.27,28 Moreover, the carbon materials can improve the conductivity of the catalyst layer, mass transportation and stability of the metal-based catalyst. Therefore, several carbon-based materials have been developed in various structural forms such as particles, tubes, fibers, and sheets.29–32
The prime goal of the present report is to discuss the recent advancement of new generation carbon-based materials and their composites as a support to Pt, Pd, Ru, Au and other non-noble metal-based catalysts in DMFC. Characteristics of different forms of carbon materials and their applications in DMFC have been discussed in detail. The challenges related to carbon-based electrocatalysts and future prospects for the development of efficient electrocatalysts have been described. The critical analysis of recent literature on the currently developed carbon-based electrocatalysts for their future utilization in DMFC has been presented in this report.
2. Operating principle of DMFC
The basic operating principle of DMFC is similar to PEMFC; however, the main difference is the supplied fuel. The main components of DMFC are the two electrodes- anode and cathode and a proton-conducting electrolyte membrane sandwiched between the two electrodes. A schematic illustration of DMFC is shown in Fig. 1, showing the reaction mechanisms. In DMFC, methanol is directly fed into the anode that reacts with water molecules to oxidize into carbon dioxide and to form six protons and six electrons.33,34 The migration of protons occurs from the anode to the cathode via the electrolyte membrane, while the electrons move through the external circuit to reach the cathode. At the cathode, the electrons and protons combine with oxygen and form water molecules. The half-cell reactions in DMFC is shown below:34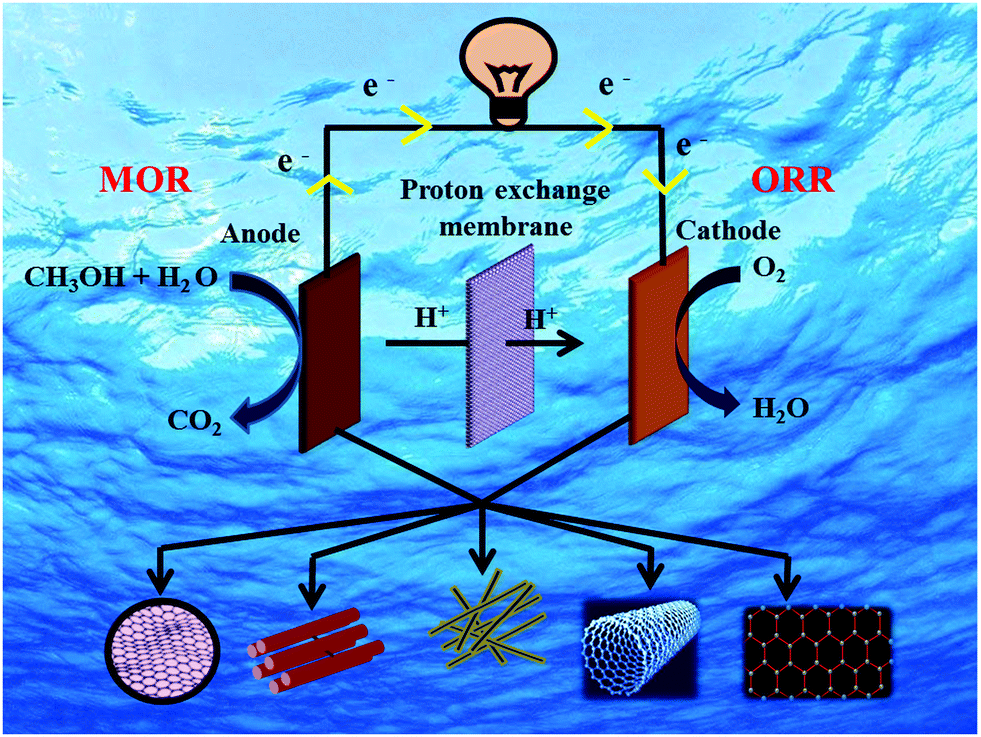 | ||
| Fig. 1 Schematic illustration showing the reaction mechanisms in DMFC. | ||
At anode:
| CH3OH + H2O → CO2 + 6H+ + 6e− E0anode = 0.02 V (vs. SHE) (oxidation) | (1) |
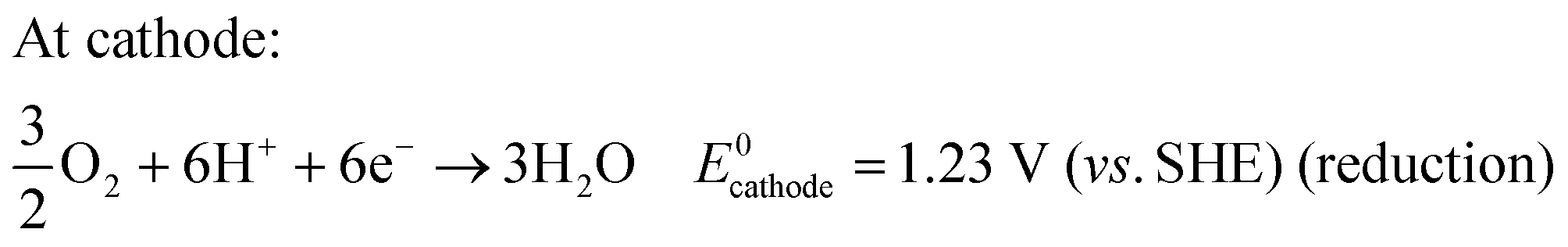 | (2) |
The overall cell reaction can be written as:
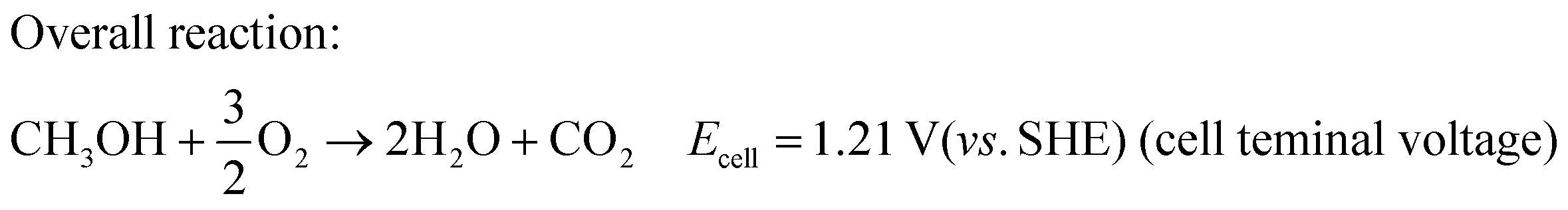 | (3) |
The formation of excess electrons at the anode as compared to the cathode causes a potential difference between the anode and the cathode, so that the current is driven through the external electrical circuit. Hence, the fuel cell can be considered as a power source. From the overall reaction in DMFC (eqn (3)), the maximum attainable voltage can be 1.21 V (vs. SHE) with a theoretical efficiency of 96.5%. However, one cannot achieve this theoretical voltage and efficiency in DMFC due to sluggish reaction kinetics of methanol oxidation forming poisonous CO and also various kinds of losses such as activation loss, ohmic loss, and concentration loss and methanol crossover in the electrolyte.35
2.1 Methanol oxidation reaction (MOR) mechanism
The MOR mechanism was explained earlier34 as follows: (i) the adsorption of methanol molecules over the catalyst surface by dehydrogenation or deprotonation, (ii) dissociation of methanol units due to activation of the C–H bond, (iii) adsorption and activation of water molecules to release oxygen (O2) molecule and (iv) formation of CO2 due to addition of O2 to the adsorbed intermediates over the surface of the catalyst. The methanol oxidation mechanism in acidic media over Pt surface is depicted in Fig. 2a.36,37 The methanol oxidation mechanism results in the formation of carbonaceous intermediates such as formic acid, formaldehyde and carbon monoxide that are strongly adsorbed onto the surface of Pt catalyst. Hence, the performance of DMFC lowers due to a decrease in electroactive sites over the catalyst surface. Bagotzky et al.38 and Parsons et al.34 suggested that the MOR can be explained by a dual path mechanism in acidic media. Among these two mechanisms, indirect one is involved in the formation of CO from methanol, which further gets oxidized into CO2 (a path I in Fig. 2a), while in direct mechanism (paths II and III in Fig. 2a), methanol molecules are directly oxidized into CO2 without forming CO.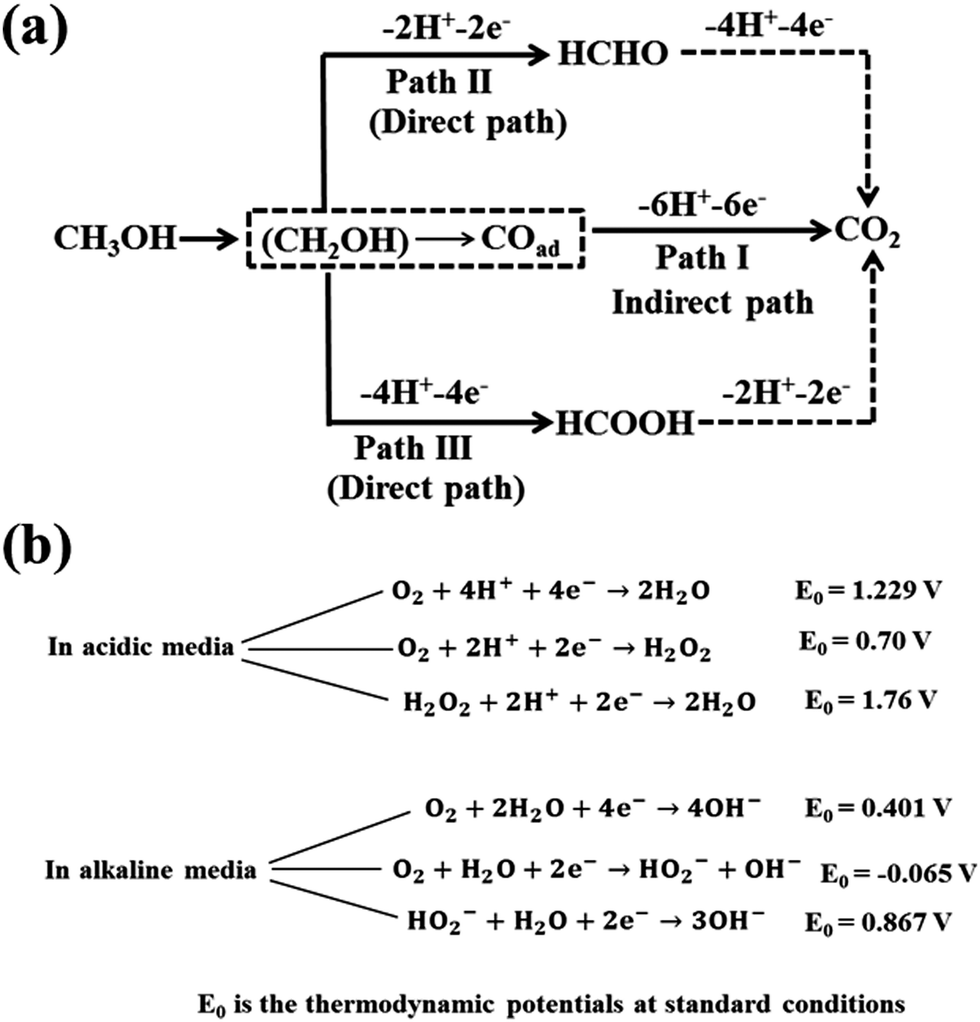 | ||
| Fig. 2 Schematic diagrams for (a) methanol oxidation reaction (MOR) in acidic media and (b) oxygen reduction reaction (ORR) mechanisms in both acidic and alkaline media. | ||
The methanol oxidation mechanism in alkaline media is discussed below:39
| Pt + OH− → Pt − (OH)ads + e− | (4) |
| Pt + (CH3OH)sol → Pt − (CH3OH)ads | (5) |
| Pt − (CH3OH)ads + Pt − (OH)ads → Pt − (CH3O)ads + Pt + H2O | (6) |
| Pt − (CH3O)ads + Pt − (OH)ads → Pt − (CH2O)ads + Pt + H2O | (7) |
| Pt − (CH2O)ads + Pt − (OH)ads → Pt − (CHO)ads + Pt + H2O | (8) |
| Pt − (CHO)ads + Pt − (OH)ads → Pt − (CO)ads + Pt + H2O | (9) |
| Pt − (CO)ads + Pt − (OH)ads → Pt − (COOH)ads + Pt | (10) |
| Pt − (COOH)ads + OH− → Pt − (OH)ads + HCOO− | (11) |
| Pt − (COOH)ads + Pt − (OH)ads + 2OH− → 2Pt + CO32− + 2H2O | (12) |
In alkaline media, there is simultaneous adsorption of methanol and hydroxide ions over the catalyst surface at similar potentials during methanol oxidation, which further combines to form active intermediates such as formaldehyde, formic acid, carbon monoxide in methanol solution. Thus, carbonate (CO32−) ions and water are formed as a result of the complete oxidation of methanol.
Electrocatalysis of methanol in alkaline media is more facile than that in acidic media since the poisoning intermediates formed during the oxidation of methanol are loosely bound to the electrode surface in alkaline media.
2.2 Oxygen reduction reaction (ORR) mechanism
ORR is a significant electrochemical reaction in energy conversion devices including batteries and fuel cells. The ORR mechanism is complex with many intermediate steps, including the adsorption of O2 at the surface of the cathode catalyst, transfer of multiple electrons, proton and oxygen-containing groups (OH, HO2−, O2), weakening and cleavage of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, oxide or peroxide formation and removal of surface hydroxide.40,41 Therefore, nature of the cathode catalyst and pH of the electrolyte play a pivotal role in ORR mechanism. The reaction pathways of the ORR mechanism in both acidic and alkaline media are similar. However, alkaline media are preferred over acidic media for ORR due to the enhancement in kinetics, i.e., the reaction occurs at a more positive potential and high charge transfer.42 In addition, the alkaline electrolyte is less corrosive compared to the acidic one thereby increasing the use of non-noble metal catalysts for ORR. In both alkaline and acidic media, ORR can proceed through two pathways, such as direct path involving the transfer of four electrons and indirect path involving two-electron transfer process, as illustrated in Fig. 2b. In the two-electron path, O2 molecules are reduced to H2O2 in the acidic solution and HO2− in the alkaline solution by accepting two electrons and then further reduced to H2O and OH− in acidic and alkaline solutions, respectively, by adsorbing another two electrons. The four-electron pathway is more preferable since there is no formation of peroxide and the faradaic efficiency is high. However, many intermediate steps are involved with the direct path, including dissociation of oxygen molecules at the surface and recombination with hydrogen ions to produce water. The two-electron process is not favourable because of low energy efficiency and also degradation of the cathode and proton exchange membrane caused by the formation of HO2− and H2O2.
O bond, oxide or peroxide formation and removal of surface hydroxide.40,41 Therefore, nature of the cathode catalyst and pH of the electrolyte play a pivotal role in ORR mechanism. The reaction pathways of the ORR mechanism in both acidic and alkaline media are similar. However, alkaline media are preferred over acidic media for ORR due to the enhancement in kinetics, i.e., the reaction occurs at a more positive potential and high charge transfer.42 In addition, the alkaline electrolyte is less corrosive compared to the acidic one thereby increasing the use of non-noble metal catalysts for ORR. In both alkaline and acidic media, ORR can proceed through two pathways, such as direct path involving the transfer of four electrons and indirect path involving two-electron transfer process, as illustrated in Fig. 2b. In the two-electron path, O2 molecules are reduced to H2O2 in the acidic solution and HO2− in the alkaline solution by accepting two electrons and then further reduced to H2O and OH− in acidic and alkaline solutions, respectively, by adsorbing another two electrons. The four-electron pathway is more preferable since there is no formation of peroxide and the faradaic efficiency is high. However, many intermediate steps are involved with the direct path, including dissociation of oxygen molecules at the surface and recombination with hydrogen ions to produce water. The two-electron process is not favourable because of low energy efficiency and also degradation of the cathode and proton exchange membrane caused by the formation of HO2− and H2O2.
3. Carbon-based materials as catalyst support
One important aspect of the development of DMFC is the proper choice of the support material since it can increase the dispersion of metal nanostructures and also makes the distribution of metal nanostructures uniform over the catalyst support. The widespread use of carbon-based materials in supercapacitors,43,44 batteries,45,46 fuel cells,47,48 as MRI contrast agents,49,50 sensors,51,52 and in hydrogen53,54 and oxygen evolution reactions55,56 is due to several excellent characteristics, such as large abundance, available in variable forms, high electrical conductivity, inert surface, and good resistance to corrosion in both acidic and alkaline media. The comparison of different carbon materials in terms of the synthesis process, surface area, solubility and electrical conductivity is listed in Table 1. The uniform distribution of catalysts over carbon support promotes the electrocatalytic performance of the catalyst. The porous architecture of carbon materials also plays an important role in the performance of DMFC. The role of different carbon materials as electrocatalyst support for both anode and cathode catalysts is discussed in the following sections:| Carbon material | Synthesis process | Surface area | Solubility | Electrical conductivity |
|---|---|---|---|---|
| Carbon quantum dot | Arc discharge, laser ablation, acidic oxidation, combustion method, microwave irradiation, Solvothermal/hydrothermal preparation | Large | Highly soluble | Medium |
| Carbon black | Incomplete combustion, thermal decomposition, oil furnace method | ∼800 m2 g−1 | Insoluble | High |
| Mesoporous carbon | Hard template and soft template method | Large | Poor | High |
| Carbon nanofibre | Chemical vapour deposition, electrospinning method | 300–700 m2 g−1 | Insoluble | High |
| Carbon nanotube | Electric arc discharge, laser ablation, chemical vapour deposition | 1315 m2 g−1 | Insoluble | Very high |
| Graphene | Chemical vapour deposition, mechanical exfoliation, pyrolysis, chemical synthesis, electrochemical process | 2630 m2 g−1 | Insoluble | Excellent (106 S cm−1) |
| Single walled carbon nanohorn | CO2 laser ablation, arc discharge method | ∼1720 m2 g−1 | Poor | High |
| Carbon nano-onion | Detonation method, arc discharge, chemical vapour deposition, ion implantation, electron beam irradiation, pyrolysis | 200–600 m2 g−1 | Poor | Medium |
| Hollow carbon nanosphere | Soft template and hard template method, emulsion process | Large | Poor | Medium |
| Carbon nanocage | Hard template, soft template and template free method | Large | Poor | High |
| Carbon aerogel | Sol–gel, chemical vapour deposition method | 400–1000 m2 g−1 | Poor | Medium |
3.1 Monophasic carbon material as catalyst support
The use of lignin-based carbon dots supports Pt catalysts (Pt/Lg-CDs) in DMFC shows a rise in the oxidation peak current density from 0.65 mA cm−2 Pt to 0.76 mA cm−2 Pt compared to PtRu/C.57 The enhancement in the catalytic activity of Pt/Lg-CDs-800 catalyst can be attributed to lignin-based carbon dots and summarized as follows: (i) the abundance of oxygenated functional groups on the carbon dot offers excellent water solubility, hence the synthesis of Pt occurs in the homogenous system. Thus, the aggregation of Pt is lowered to a certain extent thereby increasing its utilization rate; (ii) the presence of a large number of functional groups enriches the interaction between Pt catalysts and carbon dots resulting in long-term stability of the catalyst; and (iii) incorporation of carbon dots increases the number of –C–OH– groups, which can promote the conversion of oxidation intermediates into CO2. Pt nanoflowers supported on CQDs show a considerably increased electrocatalytic performance, long-term stability, lower onset potential and higher CO tolerance.58 The Pt-CQDs hybrid catalyst exhibits If and If/Ib values that are 5.28 and 2.04 times larger than those of pure Pt catalyst. The reasons behind the superior methanol oxidation activity of Pt-CQDs can be rationalized as follows: (i) the presence of oxygenated groups in CQD improves the catalytic active sites over the surface of the electrode, thus providing a larger electroactive surface area of the catalyst during the oxidation process, (ii) maximum utilization of 3D Pt nanoflowers and thus favouring fast diffusion of electrolyte on Pt surface, (iii) the larger proportion, i.e., 83% of Pt0+ in Pt-CQD catalyst is less corrosive. The comparison of methanol oxidation activity between Pd-NPs/RCQDs/GCE and Pd-NPs/GCE reveals a more negative oxidation peak potential and a fivefold higher oxidation peak current density for Pd-NPs/RCQD/GCE.59 The presence of CQDs in the catalyst improves the conductivity of electrodes and also offers a large surface area for catalysis. Moreover, the oxygenated groups on CQDs oxidize poisonous CO to CO2 and thereby inhibiting surface poisoning. Thus, there is a considerable increase in CO tolerance of Pd-NPs/RCQD/GCE than that of Pd-NPs/GCE.
Ni nanoparticles decorated CQD-modified glassy carbon electrode (Ni-NPs/RCQD/GCE) produced a higher methanol oxidation peak current density of 32 mA cm−2 at a lower voltage of 0.56 V (vs. Ag/AgCl) compared to Ni-NPs/GCE.60 The enhanced activity of Ni-NPs/RCQD/GCE is due to the increased electroactive area offered by CQDs and strong interaction between Ni-NPs and CQDs, favouring the rapid methanol oxidation process and enhanced electron transfer kinetics. The confinement effect of CQDs prevents the agglomeration of Ni-NPs that result into uniform morphology and thus increasing the stability of the catalyst during the oxidation process. In another work, CDs have been produced from a C source of g-C3N4 and then in situ incorporated into Co3O4 for the formation of CDs/Co3O4 composite.61 The as-synthesized composite has been used as a cathode electrocatalyst for ORR and a peak current density of 0.85 mA cm−2 has been obtained. The incorporation of CDs into the catalyst facilitates the electron transfer kinetics as well as increases the conductivity of Co3O4. ORR proceeds via four-electron transfer process in the case of CDs/Co3O4 composite.
N-doped carbon dots encapsulated ionic liquid, poly(imidazolium) modified glassy carbon electrode has been prepared as a cathode catalyst and a high selectivity for the reduction of oxygen into water (97%) at −0.2 V (vs. RHE) has been achieved.62 The ORR at the as-prepared hybrid electrode proceeds via a four-electron path, with H2O as the main product due to the presence of poly(ionic liquid). The synergistic contributions from the poly(ionic liquid) and CDs are the reasons behind the change in the electron transfer path of ORR in CDs after deposition on poly(ionic liquid). The integration of poly(ionic liquid) and CDs may give rise to new active sites for catalysis that facilitate the four-electron pathway for ORR. There is a strong electrostatic attraction between the positive charge of the imidazolium ring and oxygen, nitrogen-containing functional groups of CDs. This facilitates oxygen adsorption and the reduction becomes easier. It has been found that sandwich-like electrode of N doped CDs decorated on tungsten nitride nanosheets (WN NSs) hybrid catalyst (N-CDs@WN) displays excellent ORR performance via a four-electron path and exhibits a long term stability and high resistance to methanol crossover.63 The improved ORR activity of the N-CDs@WN catalyst can be attributed to the following factors: (i) zero-dimensional CDs and two-dimensional WN nanosheets form the sandwich-like structure that leads to the formation of large mesopores resulting in catalytic sites, which are easily accessible to the electrolyte solution and (ii) the conductive channels in the hybrid catalyst favour electron transfer to the WN surface.
Pd nanoparticles supported on graphitic carbon nitride and carbon black (g-C3N4/carbon black) have been fabricated via wet chemistry route.70 The optimized Pd/g-C3N4/carbon black catalyst shows a high peak current density of 1720 mA mg Pd−1 for methanol oxidation in an alkaline solution. The high electrocatalytic activity of Pd/g-C3N4/carbon black catalyst can be attributed to three important factors: (i) high surface area provided by carbon black in the hybrid catalyst for easy accessibility of the electrolyte and simultaneously maintaining high electrical conductivity causing fast electron transfer during electrochemical reactions; (ii) the strong interaction between Pd and the substrate is due to the presence of g-C3N4, which also inhibits electrochemical corrosion by protecting the defect sites of carbon black; (iii) good dispersion of small-sized Pd particles over g-C3N4/carbon black plays a major role in the enhancement of electrocatalytic activity toward methanol oxidation.
In DMFC, the methanol crossover effect can be reduced by increasing the catalyst loading at the cathode. The high loading of Pt catalyst makes the cathode layer thick, causing mass transfer limitation. Carbon blacks with good mesoporous distribution possess gas diffusion capability and water handling ability and these can be considered as a promising alternative to porous materials. Therefore, the porosity and thickness of the cathode catalyst layer can be controlled by adding an appropriate quantity of carbon black to Pt catalyst via the mechanical mixing method. A comparison of electrocatalytic activities in DMFC has been performed between the Pt black cathode catalysts supported on Vulcan XC-72R, Ketjen Black EC 300J and Black Pearls 2000 carbon blacks.71 The structure of pores on the cathode catalyst is dependent upon the type of carbon black used as an additive. The presence of a large number of pores with considerable size in the cathode catalyst is desirable to decrease oxygen transport resistance and enhance the removal of water when the airflow rate is low in DMFC. Cathode catalysts with Ketjen Black EC 300J exhibit a maximum power density of 48.9 mW cm−2, while the Vulcan XC-72R and Black Pearls 2000 added cathode catalyst layers show power densities of 26.4 mW cm−2 and 31.3 mW cm−2, respectively at 60 °C and an air-breathing mode. Compared to these three carbon blacks, Ketjen Black EC 300J has shown superior performance as a DMFC cathode catalyst due to the largest mesoporous area that increases the transport of oxygen and removal of water in the cathode catalyst at a low airflow rate. A catalyst comprising of NiCo alloy, layered perovskite oxide Pr0.95Ba0.95Mn1.8O5+δ and a varying amount of Ketjen Black EC 300J (EC) has been prepared by a one-pot pyrolysis method.72 The insertion of an appropriate amount of EC to the catalyst material can enhance electrocatalytic activity because of high specific surface area, good dispersion of Pr0.95Ba0.95Mn1.8O5+δ and NiCo alloy on the EC support, high conductivity and synergistic interaction among EC, Pr0.95Ba0.95Mn1.8O5+δ and NiCo alloy. However, a large amount of EC may reduce the ORR activity of the electrocatalyst due to a decrease in the number of reaction sites.
A cathode catalyst consisting of polypyrrole (PPY) and carbon black (CB) with cobalt (Co) at various compositions has been developed.73 The strong attraction between the aromatic ring of PPY and the graphitic character of carbon black allows easy deposition of N on carbon black. ORR performance and selectivity are influenced by the composition of Co-PPY-CB electrocatalysts. The catalyst composition of CB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPY with weight ratio 2 and PY:Co with molar ratio 4 reveals good results. The lower value of CB:PPY ratio than 2 indicates a larger amount of N causing a decrease in ORR kinetics. This is due to the oversaturation of smaller pores on the surface of carbon black by decomposition products of PPY, thereby decreasing the porosity of the surface. The presence of many micropores (<2 nm) in Vulcan XC-72 minimizes the utilization of catalysts since large solvated ions are unable to penetrate inside the micropores. Therefore, apart from the surface area, the pore size also plays a significant role in the electrocatalytic activity of the catalyst and has to be properly optimized to obtain improved performance. Moreover, the catalyst nanoparticles trapped within the deep micropores and the presence of organo-sulfur impurities of carbon black make them inaccessible to reactant molecules. The corrosion of the carbon support occurs due to thermochemical instability, which finally disintegrates the catalyst layer.
:PPY with weight ratio 2 and PY:Co with molar ratio 4 reveals good results. The lower value of CB:PPY ratio than 2 indicates a larger amount of N causing a decrease in ORR kinetics. This is due to the oversaturation of smaller pores on the surface of carbon black by decomposition products of PPY, thereby decreasing the porosity of the surface. The presence of many micropores (<2 nm) in Vulcan XC-72 minimizes the utilization of catalysts since large solvated ions are unable to penetrate inside the micropores. Therefore, apart from the surface area, the pore size also plays a significant role in the electrocatalytic activity of the catalyst and has to be properly optimized to obtain improved performance. Moreover, the catalyst nanoparticles trapped within the deep micropores and the presence of organo-sulfur impurities of carbon black make them inaccessible to reactant molecules. The corrosion of the carbon support occurs due to thermochemical instability, which finally disintegrates the catalyst layer.
The utilization of ordered mesoporous carbon/tungsten carbide (OMC/WC) catalyst with pore size 3.86 nm shows electrocatalytic activity that is almost comparable to that of the platinum disk electrode.85 OMC present in the catalyst acts both as the support and source of carbon. The use of OMC increases the specific surface area of materials and also the formation of carbon deposits over the catalytic site can be avoided. From the in situ FTIR spectroscopy studies as depicted in Fig. 3, it is observed that the intermediates formed during MOR are easily desorbed and oxidized; showing that OMC/WC catalyst is resistant to CO poisoning.
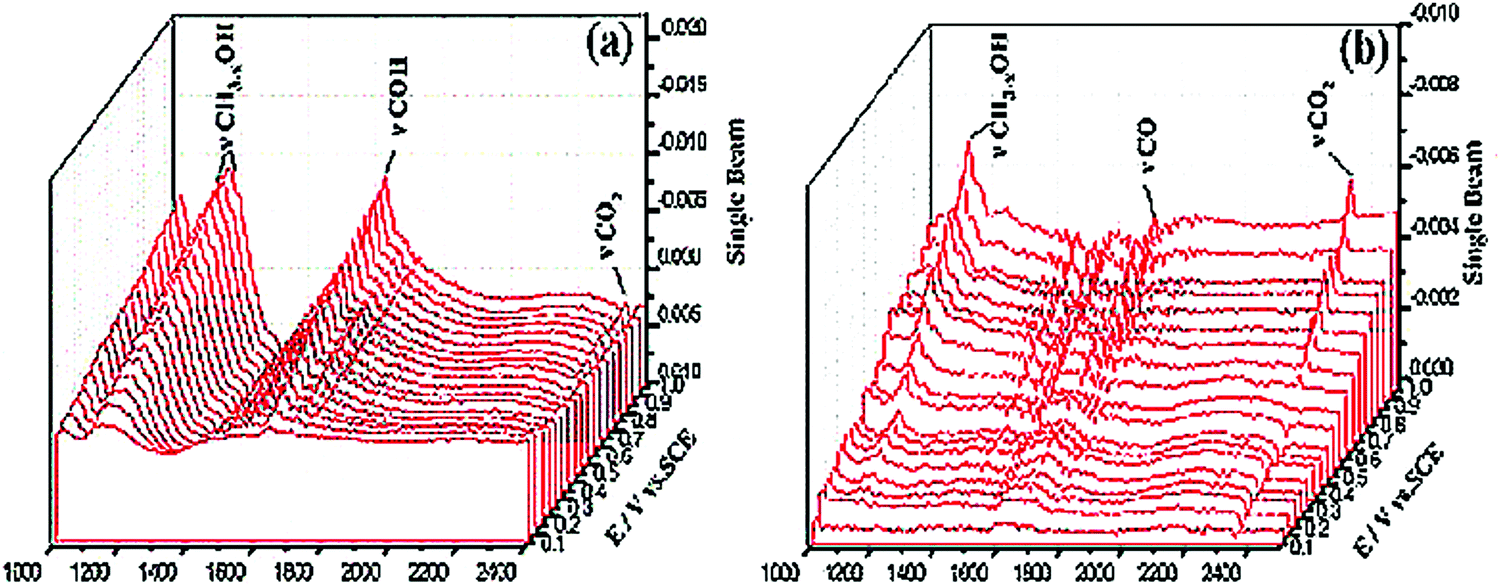 | ||
| Fig. 3 In situ MS-FTIR spectra recorded during methanol oxidation in the acidic electrolyte on (a) OMC/WCs electrode and (b) A platinum disk electrode within the range from 0.1 to 1.0 V (vs. SCE). Reproduced with permission.85 Copyright 2013, Elsevier. | ||
CMK-3 is one of the mesoporous carbon materials that has drawn much attention from researchers, which can be obtained by the replication of mesoporous silica, SBA-15. CMK-3, which have highly ordered two-dimensional structures with outstanding textural properties. Therefore, this mesoporous carbon has been used to deposit noble metal particles for their application as a catalyst material in DMFC.
The electrocatalytic activity of PdNi nanoparticles supported on defective CMK-3 carbon (CMK-3-D) with different molar ratios of Pd and Ni for both MOR and ORR has been studied.86 The optimized PdNi/CMK-3-D displays improved performance for the oxidation of methanol with a larger peak current of methanol oxidation, a more negative onset voltage, and higher If/Ib ratio due to the greater number of reactive sites for MOR from the synergistic contribution of Pd and Ni. PdNi/CMK-3-D also shows superior ORR activity comparable to Pt/C. The electronic structure of catalysts for improved catalytic activity is influenced by the presence of defects. Moreover, carbon defects provide numerous electroactive sites for the anchoring of metals. The defective mesoporous carbon can weaken the adsorption of oxygen over the catalyst surface and thus reduce the energy barrier for the oxygen reduction reaction. The mesoporous carbon provides sufficient substrate for attachment of nanoparticles and also has water handling ability for removal of water produced at the cathode. However, the hard template method used for the synthesis of CMK type OMCs has limitations as it requires extra steps for the removal of the hard template making it time-consuming and costly. In addition, the structure of pores may also get affected during the removal of the hard template.
An evaporation-induced self-assembly strategy also termed as a soft template method87 was proposed for the synthesis of OMC. The FDU materials such as FDU-14 (3-D cubic), FDU-15 (2-D hexagonal) and FDU-16 (3-D bicontinuous) are the OMCs prepared using the soft template method. Fig. 4a depicts FDU-15, which is an inverted replica of CMK-3 (Fig. 4b). Furthermore, the influence of pore size in OMC FDU-15 on the electrocatalytic activity of the cathode has been investigated.88 The pore volume and specific surface area of FDU-15 become larger with an increase in pore size, although the conductivity decreases. The large surface area with a significant pore volume of FDU-15 offers numerous active sites for the deposition of Pt catalyst. Although, the particle size of Pt reduces with an increase in pore size of FDU-15. The Pt catalyst supported on FDU-15 having pore size 6.5 nm shows superior ORR activity with a kinetic current density of 276 mA mgPt−1 at a specific surface area of 70.2 m2 g−1 and a peak power density of 0.141 W cm−2.
 | ||
| Fig. 4 Schematic representations of (a) FDU-15 and (b) CMK-3. Reproduced with permission.88 Copyright 2017, Elsevier. (c) CV and LSV (inset) curves of the catalysts in 0.5 M H2SO4 solution with 1.0 M methanol at a sweep rate of 50 mV s−1; (d) CO stripping voltammograms of all the catalysts in 0.5 M H2SO4 solution at a sweep rate of 50 mV s−1; (e) Chronoamperometric response of the catalysts at 0.7 V (vs. Ag/AgCl). Reproduced with permission.95 Copyright 2014, Elsevier. | ||
Doping of heteroatoms, such as Fe,89 N,90 P,91 S,48 into carbon materials boosts the electrocatalytic performance of the catalysts. The bonding of heteroatoms with carbon materials can generate various defect sites owing to variation in the bond length, atomic size and electronegativity. The non-uniform distribution of charge has a significant impact on the electrocatalytic properties. A non-precious metal catalyst of iron-nitrogen doped perfectly ordered mesoporous carbon material (Fe–N/C), followed by NH3 activation was investigated for ORR.92 According to literature,93,94 increase in the concentration of pyridinic N promotes the creation of a more electroactive sites for ORR. The amount of pyridinic N in carbon materials can be increased using carbon precursors with N content such as polyaniline and melamine or by NH3 activation. A significant amount of pyridinic N can be incorporated by preserving the ordered mesoporous structure of carbon with a large specific surface area and increased pore volume through post-treatment of NH3 activation. Moreover, porous structures have the ability to enhance the transport of ORR-related species and also improve the availability of electroactive sites for catalysis. The ORR activities are largely promoted by the NH3–Fe0.25–N/C catalyst with onset voltages of 1.018 and 0.935 V (vs. RHE) and limited diffusion current densities of 5.98 and 5.60 mA cm−2 in alkaline and acidic media, respectively. In both alkaline and acidic solutions, NH3–Fe0.25–N/C catalyst displays superior durability and methanol tolerance as compared to 20 wt% Pt/C. The electroactive species for ORR, such as iron carbides and iron-nitrogen coordinates are formed upon the combination of doped Fe with carbon and nitrogen, respectively. Phosphorous (P) is one of the elements in the N-group with higher atomic radius and greater electron donating capability that make it a suitable dopant atom in the carbon framework. The size of Pt nanoparticles decreases upon the introduction of P and also enhances the distribution of particles on carbon materials. In addition, the incorporation of P can improve the content of oxygenated groups that increases the tolerance against CO poisoning as a result of MOR. The synthesis of P doped OMC (POMC) with varying P content, followed by the deposition of Pt catalyst over POMC framework has been reported.95 The Pt/P7OMC catalyst exhibits larger specific surface area, superior mass current density, lower onset potential (Fig. 4c), higher tolerance against CO poisoning (Fig. 4d) and better stability (Fig. 4e) towards methanol oxidation in acidic solution as compared to commercial Pt/C, Pt/OMCs, PtRu/XC. The superior electrocatalytic performance of Pt/P7OMC can be attributed to the following factors: (i) the periodicity of mesopores, large electroactive surface area and enlarged pores may boost the rate of electron and mass transport, (ii) incorporation of P to OMC increases the number of oxygen containing functional groups that can react with the intermediates such as CO. Thus large numbers of active sites are accessible due to removal of CO from the surface of electrode and hence the electrode becomes CO tolerant.
 | ||
| Fig. 5 Schematic representations of three different types of carbon nanofibres (CNFs). Reproduced with permission.96 Copyright 2017, Elsevier. | ||
The electrocatalytic activity for ORR has been investigated at Pt nanoparticles supported on fishbone-type CNFs.97 The characteristics of CNFs including textural properties such as specific surface area, porosity or degree of graphitization of CNFs have been modified by variation of synthesis temperature, which in turn increases the electrical conduction of CNFs. According to half-cell studies, CNFs with a high degree of graphitization exhibit improved ORR activity in spite of having low surface area and pore volume. In another study, the growth of CNF on graphite paper with controlled morphology by plasma-enhanced chemical vapour deposition, followed by the electrodeposition of Pt over it has been reported.98 The direct growth of CNF on graphite paper can act as a single layer having both diffusive and catalyst layer functions. The orientation of graphene layers with respect to the growth axis is influenced by growth temperature and pressure, which leads to the formation of platelet and ribbon-like CNFs. The electrocatalytic activity of the as-prepared electrodes depends on the structure and morphology of nanofibers. The platelet CNF supported Pt (Pt/pCNF) showed superior electrocatalytic performance and better CO tolerance. The uniform dispersion, smaller size and lower coalescence of Pt particles over platelet CNF are the reasons behind higher electrocatalytic activity of Pt/pCNF electrodes. The better CO tolerance of Pt/pCNF is attributed to the following reasons: (i) the presence of oxygenated groups on the surface of pCNFs help in the removal of CO by promoting the oxidation of CO to CO2 and (ii) the morphology of pCNF allowing a stronger anchorage with Pt nanoparticles even after methanol oxidation reaction cycle.
Metal catalysts supported on CNFs are synthesized by electrospinning a solution containing transition metal acetate precursors and thermally less stable polymer, e.g., poly(vinyl alcohol) (PVA), polyvinylpyrrolidone (PVP); followed by calcination under an inert atmosphere. In this process, the metal atoms are formed due to the decomposition of transition metal precursors and enclosed within CNF. The metallic nanoparticles are usually embedded within a thin layer of carbon that acts as a protective layer for the metallic surface. In addition, the adsorption of fuel increases over the surface of the catalyst due to the presence of the carbon layer.99 A bimetallic catalyst Pt@Ni supported on CNFs has been developed, where the porous Ni/CNF was synthesized via electrospinning, followed by a carbonization process.100 The presence of micropores and large specific surface area of Ni/CNFs enhance the adsorption of Pt nanoparticles via chemical reduction that improves the catalytic behaviour of the catalysts for methanol oxidation and oxygen reduction reactions. The combination of CNFs with electrochemically active Ni and Pt offers a considerably large surface area and exposed electroactive sites for catalytic reactions. The stability of Pt deposited Ni/CNFs was enhanced with an increase in the content of Ni in the catalysts, owing to the transfer of electrons between Pt and Ni, contributing to improved oxidation of CO produced during MOR. The influence of nitrogen content on the MOR activity at Ni/N-CNFs catalyst supported on a graphite disk has been investigated.101 The incorporation of nanofibers on graphite disk shows significant improvement in electrocatalytic activity since the underneath nanofibers merge with the graphite support that lowers the interfacial resistance. Ni/N (5.0 wt%)-CNFs/graphite catalyst exhibits a current density of 270.4 mA cm−2, while unsupported fibers reveal a current density of 80.5 mA cm−2 at a sweep rate of 50 mV s−1 in 0.5 M methanol in 1 M KOH solution. The current density of methanol oxidation increases with an increase in the nitrogen content owing to the presence of lone pair of electrons in nitrogen. N doping boosts the immobilization of Ni NPs onto CNFs and also improves the binding between the catalyst and graphite disk without using Nafion. In a recent study, the synthesis of carbon nanofibers/poly(para-phenylenediamine) hybrid on the carbon paste electrode (CPE/CNF/PpPD), followed by electrodeposition of nickel particles (NiPs) has been reported.102 The CPE/CNF/PpPD/NiPs electrode exhibits superior electrocatalytic activity toward MOR with a peak current density of 38.1 mA cm−2, better stability and durability than that of CPE/CNF/NiPs. The presence of hybrid structure CNF/PpPD in the catalyst increases the electroactive surface area that in turn promotes methanol adsorption rate over the surface of the electrode. A significant decrease in charge transfer resistance and increment in the electron transfer rate is attributed to the synergistic effect of CNFs and PpPD that contributes to the superior electrical conduction and high specific surface area.
Fe and N co-containing porous CNF (Fe-N/N-C pNF) has been synthesized by electrospinning technique, followed by heat treatment to avoid agglomeration during the fabrication of the catalyst.103 The polymethyl methacrylate (PMMA) has been added to polyacrylonitrile (PAN) in the precursor solution for the formation of pores in N-CNF. The polymer is stabilized at a temperature of 280 °C to inhibit the aggregation and entanglement of nanofibres with each other during carbonization. Complete decomposition of PMMA after carbonization releases lots of small molecules from nanofibres increasing surface roughness and number of pores. Then, the removal of extra iron-containing particles over the surface of nanofibre via HF chemical etching also produces numerous pores and thus enhances the surface area. Finally, the heat treatment in NH3 atmosphere generates more Fe–Nx moieties, which are electroactive for ORR. Hence, Fe–N/N–C pNF prepared with a large concentration of Fe-Nx reveals higher ORR activity than that of Fe/N–C pNF. Electroless deposition of metal nanoparticles over Cu microsphere dispersed porous N-doped CNF (Cu/p-NCNFs) catalyst for methanol oxidation has been reported.104 The PtCu/p-NCNFs with an alloy size around 3.5 nm display 4.1 times larger specific peak current density, lower onset potential of a forward peak, better cycling stability, long-term durability and higher CO oxidation ability than that of commercial Pt/C catalyst. The reasons behind the superior electrocatalytic performance and stability of PtCu/p-NCNFs is discussed as follows: (i) p-NCNFs can strongly stabilize PtCu NPs via the interaction between metal and support to reduce agglomeration providing a short path for mass transport and a higher conductivity for charge transfer; (ii) availability of a large number of electroactive sites due to good dispersion of PtCu NPs and thereby increasing the electrocatalytic activity; and (iii) alteration in the electronic state of Pt (4d) due to synergic interaction between Pt, Cu and p-NCNFs that reduces binding energy of Pt and also weakens the adsorption of poisonous CO over the PtCu/p-NCNF surface, and thereby increasing the electrocatalytic activity and tolerance against CO poisoning, respectively.
Electrochemical characterizations have shown that PtAu/CNTs and PtPd/CNTs catalysts with 1:1 ratio exhibit remarkable methanol oxidation activity that results into lower onset voltages, smaller charge transfer resistances, larger oxidation peak current densities with better stability and tolerance against CO poisoning as compared to commercial PtRu/C.107 The excellent electrocatalytic activity of PtxMy/CNT catalysts can be ascribed to the following reasons as summarized below: (i) good dispersion of PtxMy nanoparticles on the functionalized CNTs resulting into a large accessible electroactive sites that increase the peak current density of methanol oxidation; (ii) functionalized CNTs act as good catalyst support providing channels for the transfer of electrons among CNTs, PtxMy nanoparticles and electrolytes; and (ii) a synergetic effect of M and Pt sites for the enhancement of tolerance against CO poisoning as M is more easily oxidizable compared to Pt. The electrocatalytic activity towards ORR has been investigated at a 3-D network containing MnO2 nanowires and CNTs as shown in Fig. 6.108 The MnO2/CNT hybrid catalyst exhibits ORR activity higher than commercial Pt/C with a reduction peak current density of 1.77 mA cm−2 at −0.36 V (vs. Ag/AgCl) in an alkaline solution. MnO2/CNT composite is much stable with 79% of the initial current density than Pt/C with 51% of initial current density after 20000 s. The significant electrocatalytic activity of MnO2/CNT is attributed to the following reasons: (i) presence of conductive CNTs facilitates charge transfer and also increases the conductivity of the composite; (ii) a large number of electroactive sites on MnO2 nanowires and (ii) high methanol tolerance and large stability of the 3D network formed by MnO2 nanowires and CNTs.
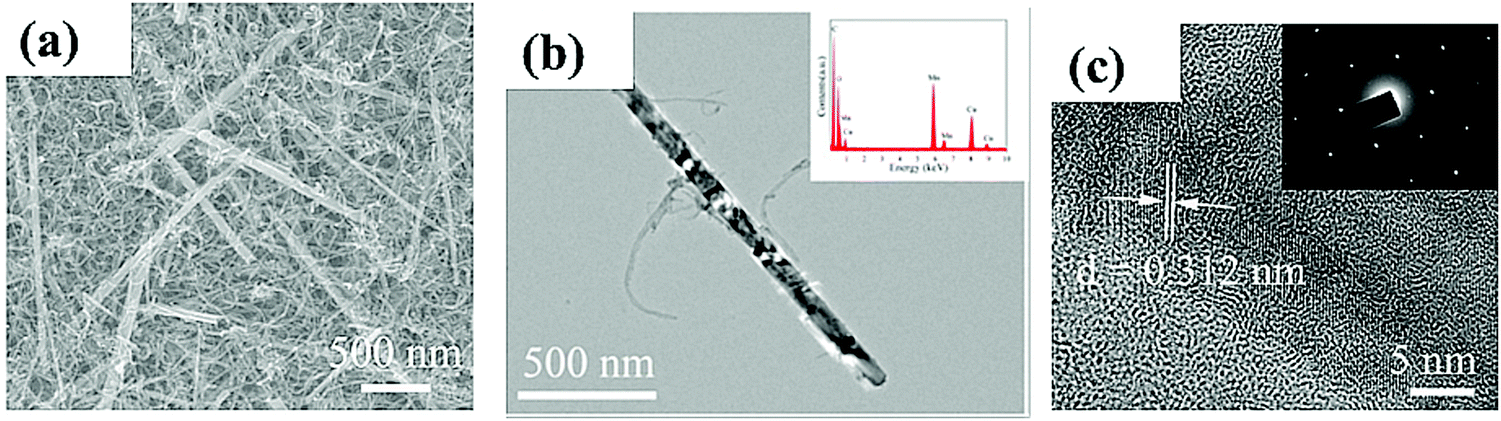 | ||
| Fig. 6 (a) SEM; (b) TEM and (c) HRTEM images of MnO2/CNTs nanohybrid. The inset of (b) shows EDS and the inset of (c) depicts SAED pattern of MnO2/CNTs. Reproduced with permission.108 Copyright 2019, Elsevier. | ||
In another work, a multi-component hybrid of MoS2QDs@Ti3C2TxQDs@MWCNTs was reported as a bifunctional catalyst for MOR and ORR.109 The MoS2QDs@Ti3C2TxQDs@MWCNTs-2 hybrid with MoS2 to Ti3C2Tx ratio 200:1 showed superior ORR performance with a Tafel slope 90 mV dec−1 and half-wave potential of 0.75 V (vs. RHE), which are comparable to the commercial Pt/C (20%) catalyst. In addition, the MoS2QDs@Ti3C2TxQDs@MWCNTs-2 exhibits good methanol oxidation activity with a peak oxidation current density of 160 A g−1 at 2.2 V (vs. RHE) in alkaline media. The superior electrocatalytic activity of catalysts can be ascribed to the presence of functional groups and uniform structure of nanotubes due to coupling between them. The effect of the addition of MWCNTs (15, 25, 35, and 45 wt%) into NiCo/C–N (synthesized via direct carbonization of Ni/Co salts doped polyaniline (PANI) composites at a temperature of 800 °C under N2 atmosphere), on the catalytic activities of methanol and ethanol oxidation, has been demonstrated.110 Ni3Co1/C–N/CNT catalyst with 35 wt% of MWCNT content exhibits high electrocatalytic activity with peak current densities of 213 mA cm−2 and 181 mA cm−2 towards 1 M methanol and 1 M ethanol, respectively. The incorporation of 35 wt% MWCNTs increases the rate of charge transfer between the catalyst and electrolyte solution, and also assists in good dispersion of Ni3Co1/C–N nanoparticles, however, a further increase in the amount of MWCNT (45 wt%) reduces the number of electroactive sites and thereby decreases the electrocatalytic activity of the catalyst. The ORR activity of Fe-Ni alloy nanoparticles encapsulated in oxidized CNTs (FeNi-OCNT) has been studied by varying the molar ratio of Fe/Ni.111 The physicochemical characteristics of FeNi-OCNTs can be tuned by changing the molar ratio of Fe/Ni. CNTs with higher iron content are shorter and thicker as compared to those with low molar ratio of Fe/Ni. However, a high amount of Ni results in longer, thinner and homogeneous CNTs and this can be attributed to the good dispersion of small-sized metal particles because of the strong interaction between Ni particles and CNTs. The FeNi12-OCNT catalyst with a Fe/Ni molar ratio of 1:2 of Fe/Ni displays excellent catalytic activity towards ORR that is almost close to a commercial 20% Pt/C catalyst. The onset potential of FeNi12-OCNT is lower by 30 mV and half-wave potential is lower by 20 mV in comparison to commercial 20% Pt/C catalyst. FeNi12-OCNT exhibits long term stability and methanol tolerance owing to the distinctive structure of the as-prepared CNT based catalyst, in which OCNT wrap the FeNi alloy particles and protect them from agglomeration and corrosion. The remarkable electrochemical activity can be ascribed to the presence of Fe–Ni alloy particles.
The comparison of MOR activity for CB, CNT and N-CNT-supported PtRu nanoparticles has been reported.112 Small-sized metal clusters are formed due to the interaction between metal particles and support materials and also the presence of a large number of electroactive sites on the catalyst surface leads to superior electrochemical performance. The methanol oxidation activity of PtRu/N-CNT is 3 and 1.9 times higher than that of PtRu/CB and PtRu/CNT, respectively. The size of PtRu nanoparticles supported on N-CNTs is smaller with a higher surface area and also the diameter of CNT increases as a result of N doping. Co-doping of transition metal and nitrogen on CNTs results in higher electrocatalytic activity since the curved surface improves the bonding between atoms inside CNTs and other species. A recent study revealed an excellent electrocatalytic activity of Fe, N-CNT/FeNP catalyst towards ORR with an onset potential of 0.95 V (vs. RHE) due to the presence of uniformly distributed active species such as FeCxNy or FeNx and Fe, CFe15.1 and Fe4N nanoparticles encapsulated in Fe, N-CNTs.113 In addition, the Fe, N-CNT/FeNP catalyst exhibits higher stability and superior resistance towards methanol crossover than that the commercial Pt/C catalyst. Moreover, the heavy doping of Fe and N on CNTs creates a large number of defects, contributing to the enhancement of electrocatalytic activity.
According to literature, doping of heteroatoms including N,125 B,126 P127 and S128 into graphene can generate a huge number of defects that can act as anchoring sites of small-sized catalyst particles with improved dispersion. Moreover, the adsorption of oxygen also increases with doping of heteroatoms into graphene, where the charge density of C atoms can be reallocated, which further enhances the electrocatalytic performance. The doping of N into graphene: (i) improves its electrical conductivity due to higher electronegativity of N than that of C, favouring the transfer of electrons from C to N atoms; (ii) strengthens the binding energy between catalyst and graphene that inhibits the aggregation and the dissolution of catalyst particles from the substrate into the solution; (iii) enhances the durability of the catalyst and support by improving the bonding between them; (iv) controls the nucleation and growth of catalyst particles over the surface of graphene; and (v) improves the dispersion of small-sized catalyst particles due to the increased electron density and electron-donating behaviour. An enhancement in the defect level has been obtained upon functionalization of graphene by Fe3O4 and co-doping with S and N.129 A large number of defects in graphene affects the growth of Pt nanoparticles, thereby forming worm-like morphology of Pt on Fe3O4-SNG. Pt/Fe3O4-SNG has a high electroactive surface area of 162 m2 g−1 and shows a peak current density of 1129 mgPt−1 for methanol oxidation. The Pt/Fe3O4-SNG shows improved methanol oxidation activities than commercial Pt/C owing to the strong interaction between Pt and Fe3O4-SNG support, which boosts the removal of poisonous intermediates formed as a result of methanol oxidation and thus increasing the stability of the catalyst. The synthesis of N, S co-doped graphene supported Fe3C (NS-GR/Fe3C) results in a superior ORR activity with a half-wave potential of 0.859 V (vs. RHE), high methanol tolerance and good cycling stability in the alkaline solution.130 The optimized NS-GR/Fe3C electrode exhibits superior ORR activity. The electrocatalytic performance is increased significantly due to the doping of the S atom and incorporation of Fe3C on N-doped graphene. The electronic properties of graphene are altered due to doping with N and S and the electron acceptor or donor levels are activated depending on the nature of bonding of dopant atoms that results in the formation of new reactive sites for catalysis of ORR. A quaternary alloy catalyst supported on N-doped graphene sheets (PtRuFeCo/NG) exhibits an enhanced ORR activity with a four-electron transfer, which is attributed to N-doping.131 The addition of N atoms generates electron spin density and also the distribution of atomic charge on graphene that assists in bond weakening and the O–O bond dissociation via the transfer of electrons to the adsorbed oxygen. The existence of pyridinic, pyrollic and graphitic nitrogen facilitates the decomposition of H2O2 molecule to the O atom during ORR and leads to the stabilization of ORR active sites in N-doped graphene.
It has been reported that rGO-supported bimetallic Pt–Pd alloy nanoparticles synthesized by the surfactant-free one-pot hydrothermal-assisted chemical reduction process (Fig. 7a) exhibit a higher electroactive surface area of 0.91 cm2 with an enhanced forward current density of 59.6 mA cm−2, much negative onset potential of 0.34 V (vs. Ag/AgCl) and better stability in comparison to RGO/Pt and RGO/Pd catalysts.132 SEM images of RGO/Pt-Pd catalysts are shown in Fig. 7b, c, d and e. The high electrocatalytic activity of RGO/Pt-Pd catalysts is attributed to the large surface area and synergistic effect of well-dispersed binary Pt–Pd nanoparticles over RGO. The presence of RGO inhibits aggregation of alloy nanoparticles during methanol oxidation and thus increases the durability of the catalyst.
 | ||
| Fig. 7 (a) Schematic illustration for the synthesis of RGO/Pt-Pd catalyst; FE-SEM micrographs of (b–e) RGO/Pt-Pd catalyst at different magnifying scales. Reproduced with permission.132 Copyright 2019, Elsevier. | ||
A bimetallic Pd@Au (PdCore–AuShell) catalyst supported on RGO (Pd@Au/RGO) displays a superior electrocatalytic activity with 6.24 mA cm−2 at −0.8 V (vs. Ag/AgCl), which is higher as compared to Pt/C.133 The enhanced ORR performance of Pd@Au/RGO originates from the modification of the electronic states of Pd by the surface of Au atoms allowing the exchange of electrons between O and Pd and enhances the adsorption of oxygen and thereby improving the oxygen reduction. Moreover, the higher interaction between Au and Pd lowers the adsorption of hydroxyl groups on the active sites of the catalyst favouring the oxygen reduction kinetics. In addition, RGO support prevents the agglomeration of the Pd@Au catalyst, thereby increasing the surface area for the adsorption of oxygen molecules.
A nitrogen-rich carbon nano-onion (N-CNO) was synthesized using a biological resource, collagen for catalytic reduction of oxygen.136 The N-CNO, containing a nitrogen content of 7.5%, exhibits ORR activity almost comparable to that of the Pt/C catalyst. Moreover, the synthesized catalyst also has better methanol tolerance and higher durability compared to Pt/C in alkaline solution. The enhanced electrocatalytic activity of the catalyst is due to a large number of pyridinic-N content, which not only act as catalytic sites but also provide a positive charge on nearby carbon atoms, favouring adsorption of oxygen molecules and attraction of electrons from the anode. In addition, the high mesoporous surface of N-CNO promotes electrocatalytic performance by offering multiple channels for mass diffusion during ORR. A cathode catalyst based on Fe supported on N-doped carbon nanospheres (Fe–N–C HNSs) catalyst was developed and a superior ORR activity, better stability and higher methanol tolerance were obtained compared to the commercial Pt/C.137 The excellent ORR activity of Fe–N–C HNSs can be attributed to the following factors: (i) exposure of the catalytic active sites of the hollow carbon nanosphere with an open framework for full utilization of the active components. Moreover, the interconnected spherical structure also facilitates the mass transport process during oxygen reduction reactions. Furthermore, the bimodal mesopores present within the carbon nanosphere can also increase the area of the interfacial region of the electrode and electrolyte and improve the diffusion of oxygen molecules, (ii) highly graphitized Fe–N–C HNSs can improve the conductivity and increase the electron transfer rate, (iii) the strong interaction between Fe and carbon nanosphere facilitates the uniform distribution of electroactive sites and also enhances the stability, (iv) the presence of a large amount of pyridinic-N and graphitic-N in N-doped C HNS facilitates the adsorption of oxygen molecules. This is due to a change in the electronic structure of carbon atoms upon N-doping. As reported earlier,138 optimized Co, N-doped carbon nanocages (Co, N-CNC) catalyst (Co, N-CNC) exhibits good ORR performance with an onset voltage of 0.924 V (vs. RHE) and electron transfer number of around 3.4. Moreover, the stability and resistance to methanol crossover are higher than those on commercial 40% Pt/C. The superior ORR activity of the optimized Co, N-CNC is due to the unique nanocage structure, over which N and Co are distributed homogeneously.
In a recent study, carbon aerogel was prepared by freeze-drying and carbonization of bacterial cellulose hydrogel under a nitrogen environment, and then NiS particles were loaded over it.139 The optimized NiS supported on carbon aerogel displayed a maximum oxidation current density of 43 mA cm−2 at 0.8 V (vs. SCE) with current retention of 92% of the first cycle after continuous 1000 cycles in an alkaline solution. Besides the above, other carbon forms such as carbon xerogel,140 carbon nanocoil,141 carbon nanocapsule,142 and carbon paper143 have also been used as catalyst supports and display excellent electrocatalytic performance.
3.2 Multiphase carbon materials as catalyst support
From the above discussions, it is clear that each carbon form has some merits and demerits as catalyst support in DMFC. Hence, the integration of a metal-based catalyst with single carbon material may not result in very satisfactory electrocatalytic activity. In this context, combining multiple-carbon materials as a support to catalysts can enhance the catalytic performance by exploiting the respective advantages of each carbon material.A cathodic catalyst of nitrogen-doped graphite encapsulated Fe/Fe3C and carbon black (Fe/Fe3C@NC/CB) has been developed.144 In both acidic and alkaline solutions, the Fe/Fe3C@NC/CB catalyst shows almost comparable ORR activity to the commercial Pt/C catalyst with better methanol tolerance and durability as compared to Pt/C. The enhanced electrocatalytic activity of Fe/Fe3C@NC/CB catalyst is ascribed to the presence of nitrogen-rich layer over Fe/Fe3C nanoparticles as a result of the chemical reaction with urea during pyrolysis that leads to the four-electron pathway as same as that of the Pt/C electrocatalyst. The schematic of the ORR mechanism at Fe/Fe3C@NC/CB catalyst is depicted in Fig. 8.
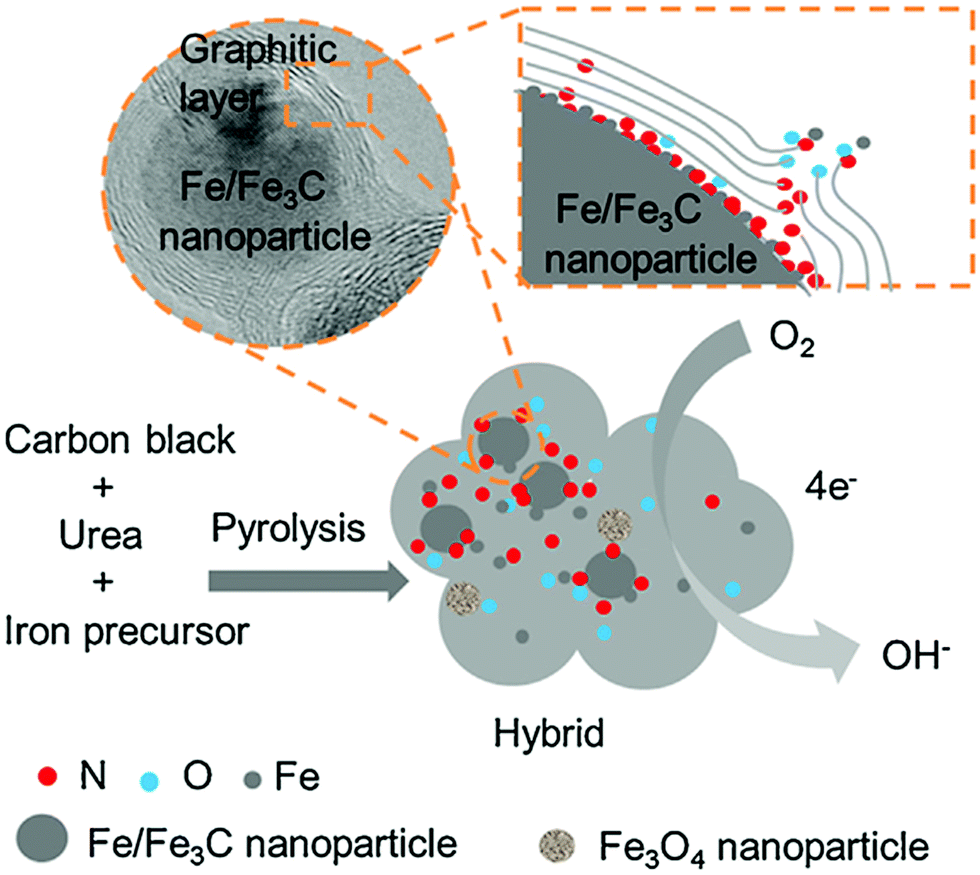 | ||
| Fig. 8 Schematic diagram and oxygen reduction reaction at the Fe/Fe3C@NC/CB catalyst. Reproduced with permission.144 Copyright 2019, Elsevier. | ||
A hollow porous carbon sphere@cobalt phosphide/nitrogen-doped carbon (HPCS@CoP/NC) structure has been designed.145 Pt nanoparticles have been deposited onto HPCS@CoP/NC support using microwave-assisted polyol strategy. Electrochemical studies verified that the mass activity of Pt-HPCS@CoP/NC catalyst is much higher and the onset potential is more negative (688 mA mgPt−1 and 0.339 V (vs. SCE)) upon oxidation of methanol as compared to commercial Pt–C (393 mA mgPt−1 and 0.412 V (vs. SCE)), Pt–CoP/NC (116 mA mgPt−1 and 0.430 V (vs. SCE)) and Pt-HPCS/NC (157 mA mgPt−1 and 0.422 V (vs. SCE)). The excellent electrocatalytic activity, good tolerance against CO, and better durability of Pt-HPCS@CoP/NC catalyst towards methanol oxidation can be attributed to the following factors: (i) the hollow porous architecture with good conductivity, allowing easy transfer of electrons and reactant species and thus boosting the reaction kinetics at the catalytic sites and (ii) the co-catalytic effect due to considerable electronic interaction between CoP and the Pt catalyst.
In recent years, graphene has been considered a major research topic in the area of fuel cells. However, the deterioration of electrical conductivity and mechanical stability of graphene has been observed upon restacking of exfoliated sheets of graphene due to strong van der Waals force and the presence of π–π interactions between the graphene sheets. Therefore, it becomes necessary to suppress the aggregation of graphene layers by incorporating another carbon material into the catalytic system. To attain this goal, PtCo-graphene/carbon dots/graphene (PtCo/GCG) catalyst modified glassy carbon electrode has been fabricated by electrodeposition of PtCo nanoparticles on the sandwich-structured GCG as illustrated in Fig. 9.146 The insertion of CDs between graphene sheets has the following advantages: (i) the incorporated CDs can widen the distance between graphene sheets, thus facilitating the diffusion of reactants; (ii) the electrical conduction between different layers of graphene can be considerably increased by embedding the conductive CDs as a “bridge”; (iii) the optimized graphene layers by incorporating CDs can enhance the distribution of PtCo nanoparticles that would maximize the use of Pt and thus improving the catalytic activity towards methanol oxidation; and (iv) oxygenated groups including carboxyl, hydroxyl, carbonyl present on CDs favour the adsorption of intermediate products.
 | ||
| Fig. 9 Schematic representation of the synthesis procedure of PtCo/GCG catalyst. Reproduced with permission.146 Copyright 2017, Elsevier. | ||
According to recent reports,147–149 the combination of graphene and CNT support reveals good electrocatalytic performance towards both MOR and ORR in DMFC. One such example is the incorporation of pure low defect CNTs into the graphene network that can offer high resistance to structural disintegration in hybrid material.150 Moreover, the excellent mechanical properties of CNTs can enhance the stability of graphene-based composites and boost the cyclic performance of catalytic materials by inhibiting the aggregation of graphene sheets. The nitrogen-doped graphene and low-defect carbon nanotube (NG-LCNT) supported Pd nanocrystals (Pd/NG-LCNT) possess extraordinary catalytic activity toward the oxidation of methanol, including high electroactive surface area, large mass activity, and considerable lifetime due to the three-dimensional interconnected porous network, proper optimization of electronic structure and low defect density in CNTs. The higher stability of Pd/NG-LCNT catalyst can be ascribed to the presence of N atoms in the framework that can be useful for anchoring of Pd nanocrystals on the NG-LCNT support and also assisting in the dissociation of water molecules to form numerous hydroxyl species to accelerate the elimination of poisonous CO absorbed on the catalyst surface and thus increasing the lifespan of the catalyst. A free-standing three-dimensional interconnected embossed graphene (e-RGO)-SWCNTs paper has been reported, where e-RGO has been synthesized using monodispersed polystyrene (PS) spheres as hard templates to prevent the aggregation of RGO sheets, and then SWCNTs have been introduced for the penetration of electrolyte into the interior of the porous electrode surface.151 By integrating the distinctive structures and characteristics of two carbon nanomaterials, the paper-based new carbon structure offers a high electroactive surface area, enhanced electrical conductivity that results in excellent electrochemical activity. In addition, SWCNT can be considered as the distinctive “superhighways” for the fast diffusion of electrons in the catalyst. The as-obtained e-RGO-SWCNT paper-supported Pt nanoparticles show superior electrocatalytic performance and better stability towards methanol electro-oxidation, compared to those of Pt/e-RGO and Pt/RGO catalysts. Recently, a mixture of pristine graphite and CNTs has been used as a carbon precursor for the synthesis of N, S-co-doped CNTs/graphene/MnS (NSCNT/NSG/MnS) ternary composite by modified Hummers' method, followed by pyrolysis process as shown in Fig. 10a.152 The morphological structures of the catalysts are shown in Fig. 10b and c. The as-obtained NSCNT/NSG/MnS composite has a high specific surface area, a large amount of N doping and distinctive three-dimensional structures. The MnS nanoparticles anchored into the NSCNT/NSG matrix exhibit reduced size and good dispersion that leads to a large number of exposed active sites for ORR. Furthermore, co-doping of N and S into the NSCNT/NSG matrix can enhance the electrical conductivity and electrocatalytic performance towards ORR. The NSCNT/NSG/MnS catalyst displays an almost comparable ORR activity (positive shifting of onset potential to 1.00 V (vs. RHE); limiting current density reaches up to 4.93 mA cm−2; electron transfer number in ORR is approximately four) to commercial Pt/C catalyst.
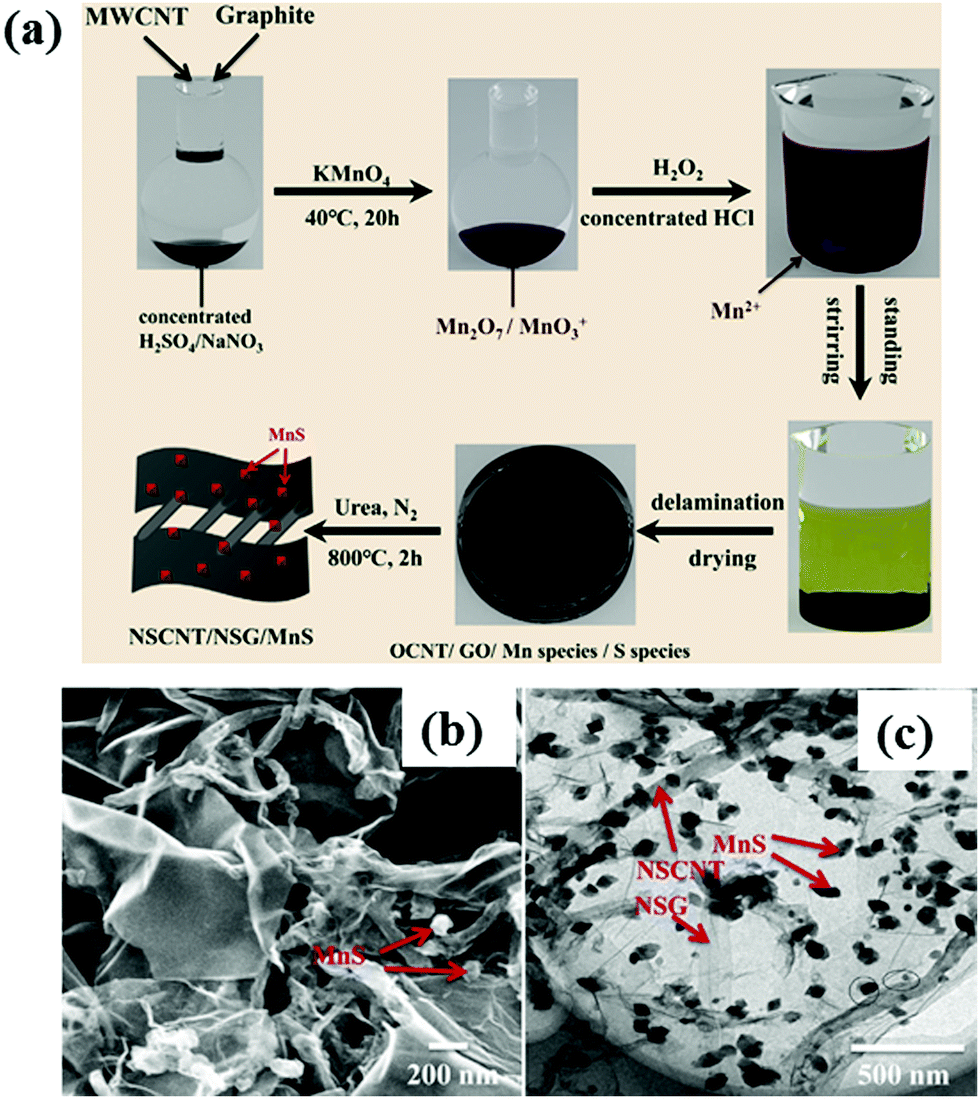 | ||
| Fig. 10 (a) Schematic illustration of the synthesis of 3D NSCNT/NSG/MnS nanohybrid; (b) SEM and (c) TEM micrographs of NSCNT/NSG/MnS catalyst. Reproduced with permission.152 Copyright 2020, Elsevier. | ||
4. Conclusion and future outlook
The development of highly conductive, efficient and cost-effective catalyst support is a major research interest for the commercialization of DMFC to meet the requirement of the growing need of human beings. Carbon-based materials are one of the most widely used catalyst supports due to easy preparation, eco-friendliness, low price and large surface area. The use of carbon support can reduce the amount of catalyst loading and thus lowering the manufacturing cost of electrodes in DMFC. Hydrophilic carbon support is required for catalytic reactions of methanol oxidation, while a hydrophobic carbon material is needed for the fast removal of products, i.e., water from the reactive sites and easy accessibility of catalytic sites for oxygen reduction reactions. This can be attained by modifying the carbon support and integrating with Pt or other metal-based catalysts. This report mainly focuses on the recent advancement of smart carbon-based materials and their composites as support for both anodic and cathodic electrocatalysts in DMFC. The carbon-based support discussed in this report is carbon black, mesoporous carbon, carbon nanofibres, carbon nanotubes, graphene, other carbon nanoforms and multiple carbon materials-based supports have been taken into consideration for a detailed discussion. Table 2 and Table 3 summarize the electrocatalytic activity of carbon-based cathode and anode catalysts in DMFC, respectively. This report also discusses the effect of doping of heteroatoms such as N, S, P and the presence of defects in carbon materials on the electrocatalytic performance of the supported catalyst. The incorporation of heteroatoms and preparation of defective carbon materials favour the creation of more number of catalytic sites and thereby promoting the electrocatalytic activity. Furthermore, the synergistic contributions of doping and defect also reduce the activation barrier and overvoltage of the reaction, thus making the electrocatalytic activity of carbon-supported catalysts comparable to f Pt/C. However, carbon corrosion during oxygen reduction and easy poisoning of Pt catalysts by CO lowers the stability and durability of the catalyst. Therefore, graphitization of carbon and decoration with metal oxides, sulfides, and carbides has been done to enhance stability. Another issue is the control of oxygen-containing functional groups on a carbon support, which has a considerable impact on the size and structure of Pt catalyst. Hence, the development of new generation carbon materials with a well-defined framework and porous structure has been greatly emphasized in recent times. Among the various carbon materials, graphene is the most widely used carbon form with a large surface area that provides homogenous dispersion of catalysts. The excellent electrical conductivity, high thermal and chemical stability of graphene make it suitable carbon support for electrocatalysis. However, restacking of graphite layers decreases the efficiency of the catalyst. Recently, the development of graphene heterostructures has become an emerging area of research for catalytic applications, where catalyst materials such as noble metal particles and metal oxides are sandwiched between two graphene layers. The availability of a large number of active sites at the interface of heterostructure favours the deposition of noble metals without agglomeration. Hence, the loading of noble metals can be reduced using graphene heterostructures. However, the large-scale production of pure graphene structures for commercial applications is still a challenge. In this context, organic polymers have also been used as precursors for the synthesis of porous carbon support. It is expected that carbon support with a well-defined structure, highly porous surface and large surface area, considerable anti-corrosion property and high electrical conductivity would be available in the near future.| Catalyst | Electrolyte | Reference electrode | Onset potential (Eonset, ORR) (V) | Half-wave potential (V) | Limiting current density (mA cm−2) | Electron transfer number (n) | Ref. |
|---|---|---|---|---|---|---|---|
| CuCo@NC | 0.1 M KOH | RHE | 0.96 | 0.884 | 5.5 | 3.96 | 153 |
| NiCo@NpC-12% | 0.1 M KOH | RHE | 0.879 | 0.780 | 5.120 | 3.98 | 154 |
| NPC-1000 | 0.1 M KOH | RHE | 1.02 | 0.902 | −5.85 | ∼ 4.0 | 155 |
| Fe-SNC | 0.1 M KOH | Ag/AgCl | 0.015 | −0.130 | 7.20 | 3.9–4.0 | 156 |
| NHCS-1000 | 0.1 M KOH | SCE | −0.08 | −0.215 | 5.94 | 3.99 | 157 |
| N, S-CN | 0.1 M KOH | Ag/AgCl | −0.05 | −0.20 | 16 | 3.98 | 158 |
| N-CNS-120 | 0.1 M KOH | Ag/AgCl | −0.076 | −0.210 | 5.79 | 3.9–4.0 | 159 |
| TiN/Fe–N-CNS | 0.1 M KOH | RHE | 1.03 | 0.87 | 4.43 | ∼4.0 | 160 |
| NiCoP/NSP-HPCNS | 0.1 M KOH | RHE | 0.92 | 0.84 | 6.0 | 4.0 | 32 |
| NiCo2O4/HCS-320-V | 0.1 M KOH | RHE | 0.90 | 0.78 | 5.8 | ∼4.0 | 161 |
| NSC | 0.1 M KOH | RHE | 0.94 | 0.80 | 5.3 | 3.82 | 162 |
| Tri(Fe/N/F)-doped MCNFs | 0.1 M KOH | RHE | 0.90 | 0.822 | −5.23 | 4.0 | 163 |
| FeNB/C-800 | 0.1 M KOH | RHE | 0.97 | 0.81 | 5.59 | 3.97 | 164 |
| Co@NCNT-800 | 0.1 M KOH | RHE | 0.86 | 0.78 | 5.46 | 3.78 | 165 |
| Fe-N-CNTAs-5-900 | 0.1 M KOH | RHE | 0.97 | 0.88 | 131.4 | ∼4.0 | 166 |
| (Co,Fe)–CN-1/RGO | 0.5 M H2SO4 | RHE | 0.875 | 0.716 | 5.32 | 3.97 | 167 |
| BDC/rGO-HT-NH3 | 0.1 M KOH | RHE | 0.91 | 0.75 | 2.39 | 3.53 | 168 |
| NSCNT/NSG/MnS | 0.1 M KOH | RHE | 1.00 | 0.74 | 4.93 | 3.97 | 152 |
| MoSe2/NG-4 | 0.1 M KOH | RHE | 0.92 | 0.78 | 4.02 | ∼4.0 | 169 |
| 3D BNG-Co | 0.1 M KOH | Ag/AgCl | −0.02 | −0.3 | 5.6 | ∼4.0 | 170 |
| Catalyst | Experimental condition | Reference electrode | Electrochemical surface area/BET surface area (m2 g−1) | Onset voltage (V) | Specific/mass activity | Ref. |
|---|---|---|---|---|---|---|
| PtPdCr/C | 0.1 M HClO4 + 0.5 M CH3OH, 10 mV s−1 | Ag/AgCl | 65 | — | 969 mA mgPt−1 | 171 |
| NiMoO4/C | 1.0 M KOH + 2 M CH3OH, 50 mV s−1 | Hg/HgO | 36.5 | 0.45 | 49 mA cm−2 | 172 |
| Pt-CoOOH-CDs/C | 1.0 M KOH + 1.0 M CH3OH, 50 mV s−1 | SCE | 63.64 | −0.60 | 856.3 mA mgPt−1 | 173 |
| CB-CNT/CeO2/Pt | 0.5 M H2SO4 + 0.5 M CH3OH, 50 mV s−1 | Ag/AgCl | 5512.19 | 0.45 | 740.14 mA mg−1 | 174 |
| Pt/Co@NCNT-NG-900 | 0.5 M H2SO4 + 1.0 M, CH3OH, 50 mV s−1 | SCE | 55.8 | 0.571 | 791 mA mgPt−1 | 175 |
| Pt/Ni/CNTs | 0.5 M H2SO4 + 1.0 M, CH3OH | Ag/AgCl | 65.8 | — | 385 mA mg−1 | 176 |
| PtCu-CeO2/CNTs | 0.5 M H2SO4 + 1.0 M, CH3OH, 50 mV s−1 | Ag/AgCl | 66.38 | 0.441 | 1.28 A mg−1 | 177 |
| Pt/Cu/rGO@CNTs | 0.5 M H2SO4 + 1.0 M, CH3OH, 50 mV s−1 | RHE | 66.0 | — | 587 A g−1 | 178 |
| Pt1Cu0.25 ANCs/MWCNTs | 0.5 M H2SO4 + 0.5 M, CH3OH, 50 mV s−1 | SCE | 70.9 | — | 1247.3 mA mgPt−1 | 179 |
| Pt-RuO2/DAAQ-MWCNTs | 0.5 M H2SO4 + 1.0 M, CH3OH, 50 mV s−1 | SCE | 79.8 | 0.11 | 500 mA mgPt−1 | 180 |
| Pt NWs/NL-CNS | 0.5 M H2SO4 + 0.5 M, CH3OH, 50 mV s−1 | SCE | 115.9 | — | 1949 mA mg−1 | 181 |
| Pd NPs inside S-CMS | 1.0 M NaOH + 1.0 M CH3OH, 50 mV s−1 | Ag/AgCl | 32.09 | −0.31 | 250.9 mA mg−1 | 182 |
| Pt/MNCS | 1.0 M NaOH + 1.0 M CH3OH, 50 mV s−1 | Ag/AgCl | 89.2 | −0.53 | 1007 mA mg−1 | 183 |
| Pt/LP -TiO2/CFP | 0.5 M H2SO4 + 1.0 M, CH3OH, 50 mV s−1 | SCE | 230.04 | 0.14 | 1182 mA mgPt−1 | 184 |
| NiCO2O4/CX | 1.0 M KOH + 0.5 M CH3OH, 50 mV s−1 | Ag/AgCl | 64.0 | 0.29 | 98 mA cm−2 | 185 |
| Pd-Co/NG | 1.0 M KOH + 1.0 M CH3OH, 50 mV s−1 | SCE | 44.2 | −0.6 | 21.33 mA cm−2 | 186 |
| Pt NW/N-LDG | 0.5 M H2SO4 + 1.0 M, CH3OH, 20 mV s−1 | RHE | 60.9 | — | 1283 mA mg−1 | 187 |
| Rh-NSs/RGO | 1.0 M KOH + 1.0 M CH3OH, 50 mV s−1 | RHE | 48.66 | 0.49 | 264 A gRh−1 | 188 |
| Pt3Ni-C/rGO | 0.1 M HClO4 + 1.0 M CH3OH, 50 mV s−1 | RHE | 52.7 | 0.65 | 360.4 mA mg−1 | 189 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
BB is thankful to Tezpur University for providing financial and research support. P. Deb would like to acknowledge the N-BIOS award project, Department of Biotechnology, Government of India.Notes and references
- M. Hu, G. Triulzi and M. Sharifzadeh, in Design and Operation of Solid Oxide Fuel Cells, Elsevier, 2020, pp. 3–41 Search PubMed.
- S. Basu, in Recent Trends in Fuel Cell Science and Technology, Springer, 2007, pp. 356–365 Search PubMed.
- L. Yao, B. Yang, H. Cui, J. Zhuang, J. Ye and J. Xue, J. Mod. Power Syst. Clean Energy, 2016, 4, 519–528 CrossRef.
- A. E. Lutz, R. S. Larson and J. O. Keller, Int. J. Hydrogen Energy, 2002, 27, 1103–1111 CrossRef CAS.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- L. Carrette, K. Friedrich and U. Stimming, Fuel Cells, 2001, 1, 5–39 CrossRef CAS.
- K. Joon, J. Power Sources, 1998, 71, 12–18 CrossRef CAS.
- T. Wilberforce, A. Alaswad, A. Palumbo, M. Dassisti and A.-G. Olabi, Int. J. Hydrogen Energy, 2016, 41, 16509–16522 CrossRef CAS.
- H. Dohle, J. Mergel and D. Stolten, J. Power Sources, 2002, 111, 268–282 CrossRef CAS.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J.-M. Léger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- G. Zhang, Z. Yang, W. Zhang and Y. Wang, J. Mater. Chem. A, 2017, 5, 1481–1487 RSC.
- J. L. Tan, A. M. De Jesus, S. L. Chua, J. Sanetuntikul, S. Shanmugam, B. J. V. Tongol and H. Kim, Appl. Catal., A, 2017, 531, 29–35 CrossRef CAS.
- H. Xu, B. Yan, J. Wang, K. Zhang, S. Li, Z. Xiong, C. Wang, Y. Shiraishi, Y. Du and P. Yang, J. Mater. Chem. A, 2017, 5, 15932–15939 RSC.
- Y. Chen, Y. Yang, G. Fu, L. Xu, D. Sun, J.-M. Lee and Y. Tang, J. Mater. Chem. A, 2018, 6, 10632–10638 RSC.
- D. Güllü and A. Demirbaş, Energy Convers. Manage., 2001, 42, 1349–1356 CrossRef.
- T. Chmielniak and M. Sciazko, Appl. Energy, 2003, 74, 393–403 CrossRef CAS.
- S. Leduc, D. Schwab, E. Dotzauer, E. Schmid and M. Obersteiner, Int. J. Energy Res., 2008, 32, 1080–1091 CrossRef CAS.
- M. Steinberg, Int. J. Hydrogen Energy, 1998, 23, 419–425 CrossRef CAS.
- G. Chen, Z. Dai, L. Sun, L. Zhang, S. Liu, H. Bao, J. Bi, S. Yang and F. Ma, J. Mater. Chem. A, 2019, 7, 6562–6571 RSC.
- F. Zhao, J. Ye, Q. Yuan, X. Yang and Z. Zhou, J. Mater. Chem. A, 2020 Search PubMed.
- C. Zhang, Y. Dai, H. Chen, Y. Ma, B. Jing, Z. Cai, Y. Duan, B. Tang and J. Zou, J. Mater. Chem. A, 2018, 6, 22636–22644 RSC.
- Y. Wang, K. Yin, J. Zhang, C. Si, X. Chen, L. Lv, W. Ma, H. Gao and Z. Zhang, J. Mater. Chem. A, 2016, 4, 14657–14668 RSC.
- H. Huang, Y. Wei, B. Shen, Y. Zhang, H. He, Q. Jiang, L. Yang, A. K. Nanjundan, J. Na and X. Xu, Adv. Mater. Interfaces, 2020, 2000142 CrossRef CAS.
- Y. P. Zhu, Y. Liu, Y. P. Liu, T. Z. Ren, T. Chen and Z. Y. Yuan, ChemCatChem, 2015, 7, 2903–2909 CrossRef CAS.
- L. Dai, D. W. Chang, J. B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- H. Tian, N. Wang, F. Xu, P. Zhang, D. Hou, Y. Mai and X. Feng, J. Mater. Chem. A, 2018, 6, 10354–10360 RSC.
- H. Huang and X. Wang, J. Mater. Chem. A, 2014, 2, 6266–6291 RSC.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS.
- P. Wang, B. Qiao, Y. Du, Y. Li, X. Zhou, Z. Dai and J. Bao, J. Phys. Chem. C, 2015, 119, 21336–21344 CrossRef CAS.
- C.-Y. Su, B.-H. Liu, T.-J. Lin, Y.-M. Chi, C.-C. Kei, K.-W. Wang and T.-P. Perng, J. Mater. Chem. A, 2015, 3, 18983–18990 RSC.
- X. Mu, Z. Xu, Y. Ma, Y. Xie, H. Mi and J. Ma, Electrochim. Acta, 2017, 253, 171–177 CrossRef CAS.
- H.-J. Niu, A.-J. Wang, L. Zhang, J.-J. Guo and J.-J. Feng, Mater. Chem. Front., 2019, 3, 1849–1858 RSC.
- M. Baldauf and W. Preidel, J. Power Sources, 1999, 84, 161–166 CrossRef CAS.
- R. Parsons and T. VanderNoot, J. Electroanal. Chem. Interfacial Electrochem., 1988, 257, 9–45 CrossRef CAS.
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef CAS.
- E. Batista, H. Hoster and T. Iwasita, J. Electroanal. Chem., 2003, 554, 265–271 CrossRef.
- E. Batista, G. Malpass, A. Motheo and T. Iwasita, J. Electroanal. Chem., 2004, 571, 273–282 CrossRef CAS.
- V. Bagotzky, Y. B. Vassiliev and O. Khazova, J. Electroanal. Chem. Interfacial Electrochem., 1977, 81, 229–238 CrossRef.
- S. Fritts and R. Sen, Assessment of methanol electro-oxidation for direct methanol-air fuel cells, Pacific Northwest Lab., Richland, WA (USA), 1988 Search PubMed.
- B. Wang, X. Cui, J. Huang, R. Cao and Q. Zhang, Chin. Chem. Lett., 2018, 29, 1757–1767 CrossRef CAS.
- X. Huang, Y. Wang, W. Li and Y. Hou, Sci. China: Chem., 2017, 60, 1494–1507 CrossRef CAS.
- K. I. Ozoemena, RSC Adv., 2016, 6, 89523–89550 RSC.
- K. Bhattacharya and P. Deb, Dalton Trans., 2015, 44, 9221–9229 RSC.
- Z. Bi, Q. Kong, Y. Cao, G. Sun, F. Su, X. Wei, X. Li, A. Ahmad, L. Xie and C.-M. Chen, J. Mater. Chem. A, 2019, 7, 16028–16045 RSC.
- M. Zheng, Y. Chi, Q. Hu, H. Tang, X. Jiang, L. Zhang, S. Zhang, H. Pang and Q. Xu, J. Mater. Chem. A, 2019, 7, 17204–17241 RSC.
- J. Wang, P. Nie, B. Ding, S. Dong, X. Hao, H. Dou and X. Zhang, J. Mater. Chem. A, 2017, 5, 2411–2428 RSC.
- S. M. Unni, S. Ramadas, R. Illathvalappil, S. N. Bhange and S. Kurungot, J. Mater. Chem. A, 2015, 3, 4361–4367 RSC.
- J.-J. Fan, Y.-J. Fan, R.-X. Wang, S. Xiang, H.-G. Tang and S.-G. Sun, J. Mater. Chem. A, 2017, 5, 19467–19475 RSC.
- K. Deka, A. Guleria, D. Kumar, J. Biswas, S. Lodha, S. D. Kaushik, S. Dasgupta and P. Deb, Curr. Appl. Phys., 2020, 20, 89–95 CrossRef.
- O. N. Metelkina, R. W. Lodge, P. G. Rudakovskaya, V. M. Gerasimov, C. H. Lucas, I. S. Grebennikov, I. V. Shchetinin, A. G. Savchenko, G. E. Pavlovskaya and G. A. Rance, J. Mater. Chem. C, 2017, 5, 2167–2174 RSC.
- N. A. Travlou, M. Seredych, E. Rodríguez-Castellón and T. J. Bandosz, J. Mater. Chem. A, 2015, 3, 3821–3831 RSC.
- S. W. Lee, W. Lee, Y. Hong, G. Lee and D. S. Yoon, Sens. Actuators, B, 2018, 255, 1788–1804 CrossRef CAS.
- S. K. Behera, P. Deb and A. Ghosh, ChemistrySelect, 2017, 2, 3657–3667 CrossRef CAS.
- G. Dong, M. Fang, H. Wang, S. Yip, H.-Y. Cheung, F. Wang, C.-Y. Wong, S. T. Chu and J. C. Ho, J. Mater. Chem. A, 2015, 3, 13080–13086 RSC.
- X. Lin, X. Li, F. Li, Y. Fang, M. Tian, X. An, Y. Fu, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 6505–6512 RSC.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, J. Sainio, H. Jiang, E. I. Kauppinen and K. Laasonen, J. Mater. Chem. A, 2016, 4, 5216–5222 RSC.
- X. Li, Y. Lv and D. Pan, Colloids Surf., A, 2019, 569, 110–118 CrossRef CAS.
- R. Sha, S. S. Jones and S. Badhulika, Surf. Coat. Technol., 2019, 360, 400–408 CrossRef CAS.
- H. Javan, E. Asghari and H. Ashassi-Sorkhabi, Synth. Met., 2019, 254, 153–163 CrossRef CAS.
- H. Javan, E. Asghari, H. Ashassi-Sorkhabi and M. Moradi-Haghighi, Chem. Eng. Sci., 2020, 217, 115534 CrossRef CAS.
- X. Zhang, X. Cheng, F. Chen, L. Xu, Y. Wang, J. Qian, Z. Wu and Q. Zhang, Chem. Phys. Lett., 2020, 740, 137058 CrossRef CAS.
- T.-N. Pham-Truong, C. Ranjan, H. Randriamahazaka and J. Ghilane, Catal. Today, 2019, 335, 381–387 CrossRef CAS.
- J. Zhang, J. Chen, Y. Luo, Y. Chen, X. Wei, G. Wang and R. Wang, Appl. Surf. Sci., 2019, 466, 911–919 CrossRef CAS.
- H. Yang, D. Lee, K. Park and W. Kim, Energy, 2015, 89, 500–510 CrossRef CAS.
- C. Hu and X. Wang, Int. J. Hydrogen Energy, 2015, 40, 12382–12391 CrossRef CAS.
- A. G. Oshchepkov, A. Bonnefont, S. N. Pronkin, O. V. Cherstiouk, C. Ulhaq-Bouillet, V. Papaefthimiou, V. N. Parmon and E. R. Savinova, J. Power Sources, 2018, 402, 447–452 CrossRef CAS.
- R. A. Hameed and S. S. Medany, Int. J. Hydrogen Energy, 2019, 44, 3636–3648 CrossRef.
- F. Fathirad, A. Mostafavi and D. Afzali, Int. J. Hydrogen Energy, 2017, 42, 3215–3221 CrossRef CAS.
- X. Chang, F. Dong, S. Yang, Z. Tang and F. Zha, Int. J. Hydrogen Energy, 2019, 44, 21559–21568 CrossRef CAS.
- H. Qian, H. Huang and X. Wang, J. Power Sources, 2015, 275, 734–741 CrossRef CAS.
- G. Wang, G. Sun, Q. Wang, S. Wang, H. Sun and Q. Xin, Int. J. Hydrogen Energy, 2010, 35, 11245–11253 CrossRef CAS.
- F. Li, Y. Yin, W. Li, C. He, Q. Zhang, J. Liu and L. Fan, Int. J. Hydrogen Energy, 2018, 43, 12637–12645 CrossRef CAS.
- D. Nguyen-Thanh, A. I. Frenkel, J. Wang, S. O’Brien and D. L. Akins, Appl. Catal., B, 2011, 105, 50–60 CrossRef CAS.
- S. Chatterjee and K. Sengupta, in Direct Methanol Fuel Cell Technology, Elsevier, 2020, pp. 135–176 Search PubMed.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- B. Tian, S. Che, Z. Liu, X. Liu, W. Fan, T. Tatsumi, O. Terasaki and D. Zhao, Chem. Commun., 2003, 2726–2727 RSC.
- M. Kruk, M. Jaroniec, R. Ryoo and S. H. Joo, J. Phys. Chem. B, 2000, 104, 7960–7968 CrossRef CAS.
- N. Krishna and P. Selvam, Adv. Porous Mater., 2014, 2, 106–112 CrossRef.
- Y. E. Song, S. Lee, M. Kim, J.-G. Na, J. Lee, J. Lee and J. R. Kim, J. Power Sources, 2020, 451, 227816 CrossRef CAS.
- G. Hu, F. Nitze, X. Jia, T. Sharifi, H. R. Barzegar, E. Gracia-Espino and T. Wågberg, RSC Adv., 2014, 4, 676–682 RSC.
- Y. Fang, Y. Lv, F. Gong, A. A. Elzatahry, G. Zheng and D. Zhao, Adv. Mater., 2016, 28, 9385–9390 CrossRef CAS PubMed.
- D. Saikia, J. R. Deka, C.-J. Chou, C.-H. Lin, Y.-C. Yang and H.-M. Kao, ACS Appl. Energy Mater., 2019, 2, 1121–1133 CrossRef CAS.
- T. Panja, D. Bhattacharjya and J.-S. Yu, J. Mater. Chem. A, 2015, 3, 18001–18009 RSC.
- D. Zhang, L. Zheng, Y. Ma, L. Lei, Q. Li, Y. Li, H. Luo, H. Feng and Y. Hao, ACS Appl. Mater. Interfaces, 2014, 6, 2657–2665 CrossRef CAS PubMed.
- H. Zheng, Z. Chen and Y. Li, Electrochim. Acta, 2013, 108, 486–490 CrossRef CAS.
- G. Li, B. Shi, Y. Gong, Y. Zhang, X. Wang, M. Guo and X. Lyu, Mater. Chem. Phys., 2020, 243, 122570 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng and Z. Wu, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- G. Zhao, T. Zhao, J. Xu, Z. Lin and X. Yan, Int. J. Hydrogen Energy, 2017, 42, 3325–3334 CrossRef CAS.
- G. A. Ferrero, K. Preuss, A. Marinovic, A. B. Jorge, N. Mansor, D. J. Brett, A. B. Fuertes, M. Sevilla and M.-M. Titirici, ACS Nano, 2016, 10, 5922–5932 CrossRef CAS PubMed.
- J. Wang, L. Li, X. Chen, Y. Lu and W. Yang, J. Mater. Chem. A, 2016, 4, 11342–11350 RSC.
- Z. Liu, Q. Shi, F. Peng, H. Wang, R. Zhang and H. Yu, Electrochem. Commun., 2012, 16, 73–76 CrossRef CAS.
- H. Tan, Y. Li, X. Jiang, J. Tang, Z. Wang, H. Qian, P. Mei, V. Malgras, Y. Bando and Y. Yamauchi, Nano Energy, 2017, 36, 286–294 CrossRef CAS.
- D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127–15129 CrossRef CAS PubMed.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS.
- P. Song, L. Zhu, X. Bo, A. Wang, G. Wang and L. Guo, Electrochim. Acta, 2014, 127, 307–314 CrossRef CAS.
- P. You and S. Kamarudin, Chem. Eng. J., 2017, 309, 489–502 CrossRef CAS.
- D. Sebastián, M. Lázaro, I. Suelves, R. Moliner, V. Baglio, A. Stassi and A. Aricò, Int. J. Hydrogen Energy, 2012, 37, 6253–6260 CrossRef.
- E. Salernitano, L. Giorgi and T. D. Makris, Int. J. Hydrogen Energy, 2014, 39, 15005–15016 CrossRef CAS.
- A. Yousef, R. M. Brooks, M. El-Halwany, M. A. Abdelkareem, J. A. Khamaj, M. H. EL-Newehy, N. Barakat and H. Y. Kim, Int. J. Electrochem. Sci., 2015, 10, 7025–7032 CAS.
- J. Chen, Q. Niu, G. Chen, J. Nie and G. Ma, J. Phys. Chem. C, 2017, 121, 1463–1471 CrossRef CAS.
- B. M. Thamer, M. H. El-Newehy, N. A. Barakat, M. A. Abdelkareem, S. S. Al-Deyab and H. Y. Kim, Int. J. Hydrogen Energy, 2015, 40, 14845–14856 CrossRef CAS.
- M. Elbasri, H. Perrot, O. Sel, K. Lafdi and M. El Rhazi, Int. J. Hydrogen Energy, 2019, 44, 24534–24545 CrossRef.
- M. Sun, Z. Xie, Z. Li, X. Deng, Q. Huang and Z. Li, Int. J. Hydrogen Energy, 2019, 44, 24617–24627 CrossRef CAS.
- J. J. Chen, J. B. Tan, C. F. Li, L. F. Gu, X. F. Lu and G. R. Li, J. Phys. Chem. C, 2020, 124, 13036–13044 CrossRef CAS.
- S. Kumar, M. Nehra, D. Kedia, N. Dilbaghi, K. Tankeshwar and K.-H. Kim, Prog. Energy Combust. Sci., 2018, 64, 219–253 CrossRef.
- M. A. Abdelkareem, E. T. Sayed, H. O. Mohamed, M. Obaid, H. Rezk and K.-J. Chae, Prog. Energy Combust. Sci., 2020, 77, 100805 CrossRef.
- S. Themsirimongkon, T. Sarakonsri, S. Lapanantnoppakhun, J. Jakmunee and S. Saipanya, Int. J. Hydrogen Energy, 2019, 44, 30719–30731 CrossRef CAS.
- Y. Dong, Y. Xue, W. Gu, Z. Yang and G. Xu, J. Electroanal. Chem., 2019, 837, 55–59 CrossRef CAS.
- X. Yang, Q. Jia, F. Duan, B. Hu, M. Wang, L. He, Y. Song and Z. Zhang, Appl. Surf. Sci., 2019, 464, 78–87 CrossRef CAS.
- Z. Deng, Q. Yi, Y. Zhang and H. Nie, J. Electroanal. Chem., 2017, 803, 95–103 CrossRef CAS.
- N. Cai, H. Yang, X. Zhang, S. Xia, D. Yao, P. Bartocci, F. Fantozzi, Y. Chen, H. Chen and P. T. Williams, Waste Manage., 2020, 109, 119–126 CrossRef CAS PubMed.
- H. F. Fard, M. Khodaverdi, F. Pourfayaz and M. H. Ahmadi, Int. J. Hydrogen Energy, 2020, 45, 25307–25316 CrossRef.
- Y. Zhang, R. Jiang, Z. Wang, Y. Xue, J. Sun and Y. Guo, J. Colloid Interface Sci., 2020, 579, 391–400 CrossRef CAS PubMed.
- A. Ali and P. K. Shen, J. Mater. Chem. A, 2019, 7, 22189–22217 RSC.
- D. Li, L. Zhang, H. Chen, J. Wang, L.-X. Ding, S. Wang, P. J. Ashman and H. Wang, J. Mater. Chem. A, 2016, 4, 8630–8635 RSC.
- J. Azadmanjiri, V. K. Srivastava, P. Kumar, M. Nikzad, J. Wang and A. Yu, J. Mater. Chem. A, 2018, 6, 702–734 RSC.
- N. Li, T. Lv, Y. Yao, H. Li, K. Liu and T. Chen, J. Mater. Chem. A, 2017, 5, 3267–3273 RSC.
- H. Huang, Q. Chen, M. He, X. Sun and X. Wang, J. Power Sources, 2013, 239, 189–195 CrossRef CAS.
- W. Liu, M. Shi, X. Lang, Y. Chu, Z. Chen, D. Zhao, W. Lin and C. Hardacre, Electrochim. Acta, 2013, 114, 133–141 CrossRef.
- J. Yang, G. Zhu, Y. Liu, J. Xia, Z. Ji, X. Shen and S. Wu, Adv. Funct. Mater., 2016, 26, 4712–4721 CrossRef CAS.
- C. Zhai, M. Zhu, D. Bin, F. Ren, C. Wang, P. Yang and Y. Du, J. Power Sources, 2015, 275, 483–488 CrossRef CAS.
- K.-H. Luo, C.-K. Cheng, J.-Y. Lin, C.-H. Huang, T.-K. Yeh and C.-K. Hsieh, Surf. Coat. Technol., 2020, 125850 CrossRef CAS.
- J. Gautam, D. T. Tran, T. I. Singh, N. H. Kim and J. H. Lee, J. Power Sources, 2019, 427, 91–100 CrossRef CAS.
- B. Baruah and A. Kumar, Synth. Met., 2018, 245, 74–86 CrossRef CAS.
- D. Liu, L. Li and T. You, J. Colloid Interface Sci., 2017, 487, 330–335 CrossRef CAS PubMed.
- A. K. Nair, N. Kalarikkal, S. Thomas, M. Kala, V. Sahajwalla, R. K. Joshi and S. Alwarappan, Sci. Rep., 2016, 6, 37731 CrossRef CAS PubMed.
- M. An, C. Du, L. Du, Y. Sun, Y. Wang, C. Chen, G. Han, G. Yin and Y. Gao, Chem. Phys. Lett., 2017, 687, 1–8 CrossRef CAS.
- D. Higgins, M. A. Hoque, M. H. Seo, R. Wang, F. Hassan, J. Y. Choi, M. Pritzker, A. Yu, J. Zhang and Z. Chen, Adv. Funct. Mater., 2014, 24, 4325–4336 CrossRef CAS.
- J. Zhong, L. Wu, J. Lan, M. Waqas, M. Sun, Y. Fan, W. Chen, L. Liu and J. Yang, Int. J. Hydrogen Energy, 2020, 45, 22929–22937 CrossRef CAS.
- K. K. Rani, C. Karuppiah, S.-F. Wang, S. O. Alaswad, P. Sireesha, R. Devasenathipathy, R. Jose and C.-C. Yang, Ultrason. Sonochem., 2020, 105111 CrossRef PubMed.
- M. Rethinasabapathy, S.-M. Kang, Y. Haldorai, N. Jonna, M. Jankiraman, G.-W. Lee, S.-C. Jang, B. Natesan, C. Roh and Y. S. Huh, J. Ind. Eng. Chem., 2019, 69, 285–294 CrossRef CAS.
- M. F. R. Hanifah, J. Jaafar, M. Othman, A. Ismail, M. Rahman, N. Yusof and F. Aziz, J. Alloys Compd., 2019, 793, 232–246 CrossRef CAS.
- P. Raghavendra, G. V. Reddy, R. Sivasubramanian, P. S. Chandana and L. S. Sarma, Int. J. Hydrogen Energy, 2018, 43, 4125–4135 CrossRef CAS.
- Y. Lv and X. Li, Int. J. Hydrogen Energy, 2020, 45, 12766–12776 CrossRef CAS.
- X. Guo, L. Yang, B. Shen, Y. Wei, Y. Yang, C. Yang, Q. Jiang, H. He and H. Huang, Mater. Chem. Phys., 2020, 123167 CrossRef CAS.
- K. Chatterjee, M. Ashokkumar, H. Gullapalli, Y. Gong, R. Vajtai, P. Thanikaivelan and P. M. Ajayan, Carbon, 2018, 130, 645–651 CrossRef CAS.
- Y. Chen, Z. Li, Y. Zhu, D. Sun, X. Liu, L. Xu and Y. Tang, Adv. Mater., 2019, 31, 1806312 CrossRef PubMed.
- J. Wang, S. Yang, W. Song, J. Zhao, Z. Lu, Q. Zhai and L. Jiang, Int. J. Hydrogen Energy, 2020, 45, 6318–6327 CrossRef CAS.
- G. Y. Hou, Z. Y. Lyu, Y. P. Tang, H. Z. Cao and G. Q. Zheng, Int. J. Hydrogen Energy, 2020, 45, 16049–16059 CrossRef CAS.
- C. Alegre, M. E. Gálvez, E. Baquedano, R. Moliner, E. Pastor and M. J. S. Lázaro, J. Phys. Chem. C, 2013, 117, 13045–13058 CrossRef CAS.
- M. Sevilla, C. Sanchís, T. Valdés-Solís, E. Morallón and A. Fuertes, Electrochim. Acta, 2009, 54, 2234–2238 CrossRef CAS.
- L. Zhao, L. Wang, P. Yu, D. Zhao, C. Tian, H. Feng, J. Ma and H. Fu, Chem. Commun., 2015, 51, 12399–12402 RSC.
- S. H. Ahn, I. Choi, O. J. Kwon and J. J. Kim, Chem. Eng. J., 2012, 181, 276–280 CrossRef.
- J. Zhu, Z. Xiong, J. Zheng, Z. Luo, G. Zhu, C. Xiao, Z. Meng, Y. Li and K. Luo, J. Mater. Sci. Technol., 2019, 35, 2543–2551 CrossRef.
- H. Zhang, L. Ma, M. Gan, F. Xie, H. He, L. Hu and M. Jiang, New J. Chem., 2019, 43, 10784–10791 RSC.
- S. Yang, F. Zhang, C. Gao, J. Xia, L. Lu and Z. Wang, J. Electroanal. Chem., 2017, 802, 27–32 CrossRef CAS.
- S.-S. Li, H.-P. Cong, P. Wang and S.-H. Yu, Nanoscale, 2014, 6, 7534–7541 RSC.
- Y. Zhou, X. Hu, S. Guo, C. Yu, S. Zhong and X. Liu, Electrochim. Acta, 2018, 264, 12–19 CrossRef CAS.
- Y. Ma, Q. Wang, Y. Miao, Y. Lin and R. Li, Appl. Surf. Sci., 2018, 450, 413–421 CrossRef CAS.
- J. Ren, J. Zhang, C. Yang, Y. Yang, Y. Zhang, F. Yang, R. Ma, L. Yang, H. He and H. Huang, Mater. Today Energy, 2020, 16, 100409 CrossRef.
- Q. Zhang, F. Yue, L. Xu, C. Yao, R. D. Priestley and S. Hou, Appl. Catal., B, 2019, 257, 117886 CrossRef CAS.
- Y. Chen, C. Xu, Z. Hou, M. Zhou, B. He, W. Wang, W. Ren, Y. Liu, L. Chen and W. Xu, Mater. Today Energy, 2020, 16, 100402 CrossRef.
- M. Kuang, Q. Wang, P. Han and G. Zheng, Adv. Energy Mater., 2017, 7, 1700193 CrossRef.
- D. Wang, Q. Wang, S. Jiang, K. Dong, Z. Wang, S. Luo, Y. Liu, Y. Zhang, Q. Wang and T. Yi, Int. J. Hydrogen Energy, 2020, 45, 22797–22807 CrossRef CAS.
- L. Ye, G. Chai and Z. Wen, Adv. Funct. Mater., 2017, 27, 1606190 CrossRef.
- M. H. Naveen, K. Shim, M. S. A. Hossain, J. H. Kim and Y. B. Shim, Adv. Energy Mater., 2017, 7, 1602002 CrossRef.
- T. Zhou, Y. Zhou, R. Ma, Z. Zhou, G. Liu, Q. Liu, Y. Zhu and J. Wang, Carbon, 2017, 114, 177–186 CrossRef CAS.
- K. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2016, 19, 373–381 CrossRef CAS.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. Waterhouse, Y. Zhao, C. Zhou, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- Y. Liu, Y. Shen, S. Zhu and D. Li, Chem. Eng. J., 2020, 400, 125968 CrossRef CAS.
- H. Yuan, J. Li, W. Yang, Z. Zhuang, Y. Zhao, L. He, L. Xu, X. Liao, R. Zhu and L. Mai, ACS Appl. Mater. Interfaces, 2018, 10, 16410–16417 CrossRef CAS PubMed.
- A. M. B. Honorato, M. Khalid, Q. Dai and L. A. Pessan, Synth. Met., 2020, 264, 116383 CrossRef.
- Y.-G. Lee and H.-J. Ahn, Appl. Surf. Sci., 2019, 487, 389–397 CrossRef CAS.
- Y. Wang, Y. Liu, H. Yang, Y. Liu, K.-H. Wu and G. Yang, J. Colloid Interface Sci., 2020, 579, 637–644 CrossRef CAS PubMed.
- F. Liu, X. Zhang, X. Zhang, L. Wang, M. Liu and J. Zhang, J. Colloid Interface Sci., 2020, 581, 523–532 CrossRef PubMed.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, M. H. Engelhard, X. Li, D. Xiao, D. Li and L. Estevez, Small, 2017, 13, 1603407 CrossRef PubMed.
- W.-K. Jo, S. Moru, D.-E. Lee and S. Tonda, Appl. Surf. Sci., 2020, 531, 147367 CrossRef CAS.
- Y. Sun, R. Zhong, H. Zhang, T. Huang, J. Yu, H. Fang, D. Liang and Z. Guo, Int. J. Hydrogen Energy, 2019, 44, 21790–21802 CrossRef CAS.
- D. Zheng, P. Cheng, Q. Yao, Y. Fang, M. Yang, L. Zhu and L. Zhang, J. Alloys Compd., 2020, 848, 156588 CrossRef CAS.
- R. Yuan, Y. Xu, Y. Wang, F. You, W. Chen, C. Ding, D. Jiang and K. Wang, J. Electroanal. Chem., 2020, 877, 114555 CrossRef CAS.
- K. Peng, N. Bhuvanendran, S. Ravichandran, W. Zhang, Q. Ma, L. Xing, Q. Xu, L. Khotseng and H. Su, Int. J. Hydrogen Energy, 2020, 45, 22752–22760 CrossRef CAS.
- P. R. Jothi, S. Kannan and G. Velayutham, J. Power Sources, 2015, 277, 350–359 CrossRef CAS.
- W. Tu, Y. Sun, D. Wu, H. Wang, H. Huang, M. Shao, Y. Liu and Z. Kang, Mater. Chem. Phys., 2019, 225, 64–71 CrossRef CAS.
- N. Pongpichayakul, S. Themsirimongkon, S. Maturost, K. Wangkawong, L. Fang, B. Inceesungvorn, P. Waenkaew and S. Saipanya, Int. J. Hydrogen Energy, 2021, 46, 2905–2916 CrossRef CAS.
- J. Ding, L. Ma, M. Gan, W. Zhan, C. Zhou, D. Wei, S. Han, J. Shen, F. Xie and X. Zhong, Int. J. Hydrogen Energy, 2019, 44, 30388–30400 CrossRef CAS.
- Y. Tan, Y. Zhang, X. Wang, L. Zeng, F. Luo and A. Liu, J. Electroanal. Chem., 2020, 856, 113739 CrossRef CAS.
- C. Liu, L. Zhang, L. Sun, W. Wang and Z. Chen, Int. J. Hydrogen Energy, 2020, 45, 8558–8567 CrossRef CAS.
- M. Guo, Q. Tu, L. Wang, Y. Tang, H. Song, J. Zhou, Z. Zhang, Y. Wang and C. Liu, Int. J. Hydrogen Energy, 2019, 44, 6886–6895 CrossRef CAS.
- J. Zhong, L. Li, M. Waqas, X. Wang, Y. Fan, J. Qi, B. Yang, C. Rong, W. Chen and S. Sun, Electrochim. Acta, 2019, 322, 134677 CrossRef CAS.
- R. Liu, Z. Luo, Q. Wei and X. Zhou, Mater. Des., 2016, 94, 132–138 CrossRef CAS.
- H. Huang, Y. Wei, Y. Yang, M. Yan, H. He, Q. Jiang, X. Yang and J. Zhu, J. Energy Chem., 2021, 57, 601–609 CrossRef.
- X. Niu, Q. Xiong, J. Pan, X. Li, W. Zhang, F. Qiu and Y. Yan, Fuel, 2017, 190, 174–181 CrossRef CAS.
- Y. Zhang, Y. Liu, W. Liu, X. Li and L. Mao, Appl. Surf. Sci., 2017, 407, 64–71 CrossRef CAS.
- J. Deng, J. Zhang, J. Chen, Y. Luo, Y. Chen, Y. Xue, G. Wang and R. Wang, J. Electroanal. Chem., 2020, 874, 114468 CrossRef CAS.
- M. M. El-Deeb, W. M. El Rouby, A. Abdelwahab and A. A. Farghali, Electrochim. Acta, 2018, 259, 77–85 CrossRef CAS.
- R. Kiyani, S. Rowshanzamir and M. J. Parnian, Energy, 2016, 113, 1162–1173 CrossRef CAS.
- H. Huang, L. Ma, C. S. Tiwary, Q. Jiang, K. Yin, W. Zhou and P. M. Ajayan, Small, 2017, 13, 1603013 CrossRef PubMed.
- Y. Kang, Q. Xue, P. Jin, J. Jiang, J. Zeng and Y. Chen, ACS Sustainable Chem. Eng., 2017, 5, 10156–10162 CrossRef CAS.
- W. Niu, L. Li, X. Liu, W. Zhou, W. Li, J. Lu and S. Chen, Int. J. Hydrogen Energy, 2015, 40, 5106–5114 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |