
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ formation of core–shell nanoparticles in epoxy resin via reversible addition–fragmentation chain transfer dispersion polymerization†
Eun Ho
Lee‡
a,
Kyoungho
Kim‡
a,
Gyeongdong
Yeom
a,
Bongkuk
Seo
b,
Wonjoo
Lee
b,
Youngchang
Yu
*b,
Aruna Kumar
Mohanty
 *a and
Hyun-Jong
Paik
*a
*a and
Hyun-Jong
Paik
*a
aDepartment of Polymer Science and Engineering, Pusan National University, Busan 46241, Korea. E-mail: akmohanty07@pusan.ac.kr; hpaik@pusan.ac.kr
bCenter for Advanced Specialty Chemicals, Korea Research Institute of Chemical Technology, Ulsan, 44412, Republic of Korea. E-mail: ycyu@krict.re.kr
First published on 2nd November 2021
Abstract
We report for the first time in situ reversible addition–fragmentation chain transfer (RAFT) dispersion polymerization in an epoxy resin mixture for toughening of epoxy. Good dispersion of latex particles in the epoxy resin by the selection of suitable monomers and block copolymerization eliminated the need for tedious re-dispersion of preformed particles.
Epoxy polymers are widely used in adhesives and structural engineering applications due to their high Young's moduli and failure strengths.1–3 However, a highly crosslinked epoxy structure after curing with a hardener often results in undesired brittleness. Hence, liquid rubber or core–shell rubber particles are typically used to increase the toughness of epoxy resins.4–8 The dispersed rubber phases can effectively dissipate the impact energy, thereby producing a strong toughening effect on the epoxy polymers by preventing crack initiation and growth.7–9 In a simple technique of blending with liquid rubber, curing conditions mainly determine the morphology of the rubber phase, which often with a poor state of dispersion leads to a deterioration of the thermomechanical properties.10–12 Meanwhile, dispersion of pre-formed well-defined core–shell latex particles provides a promising alternative in terms of morphology control and a separate examination of the toughening effect.1,13,14
Core–shell rubber particles are usually dispersed in epoxy resins by two methods. The first method utilizes coagulated latex particles of heterogeneous polymerization for re-dispersion in the epoxy resin.15,16 However, dispersion of coagulated latex particles in epoxy resins is difficult (if not impossible) even by applying a strong mechanical shear force. The second method involves the dispersion transfer of latex particles to a solvent(s), followed by their uniform re-dispersion in the epoxy resin and the removal of volatile components.16 This method represents a complex multi-step process and requires the selection of a suitable solvent depending on the type of rubber particles. Thus, there is a need for an alternative method that removes the hardship of re-dispersion and saves time and valuable resources.
Spherical core–shell particles are used in various applications, such as thermosets, coatings, adhesives, sealants, and biomedical devices.15,17–19 Heterogeneous (such as emulsion or dispersion) polymerization is usually used to design and synthesize well-defined particles with desired cores, shell functionalities, sizes, and shell thicknesses.15,20–22 In recent years, heterogeneous reversible addition–fragmentation chain transfer (RAFT) polymerization that forms dispersions and (mini)emulsions has developed as a promising technique due to its ability to synthesize a wide variety of nano-objects (such as spheres, worms, and vesicles) via polymerization-induced self-assembly (PISA).23–26 Besides the usual aqueous continuous phase, PISA based on RAFT dispersion polymerization in non-conventional continuous phases, such as alcoholic, and aqueous–alcoholic ones as well as in supercritical CO2, alkanes, and silicone oil indicated their tolerance towards various solvent systems.25,27–30
RAFT polymerization is well-known for the preparation of polymers with predetermined molecular weights (MWs) and narrow molecular weight distributions (MWDs).31–33 It proceeds by achieving an equilibrium between propagating radicals and dormant species through a degenerative chain-transfer reaction. Thiocarbonylthio compounds with the generic formula R–S–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–Z are typically used as the active reversible chain transfer agents (RAFT agents), which allow the rapid establishment of equilibrium and production of polymers with narrow MWDs.34,35 Controlled polymerization is possible in the RAFT technique because the majority of polymer chains exist as the dormant species with thiocarbonylthio moieties (living chains).36,37 Nonetheless, a small amount of dead chains without the thiocarbonylthio moiety are also formed through the termination reactions of propagating radicals. Due to the living nature, macro-RAFT agents can also participate in chain extension reactions into diverse polymer architectures, including well-defined block copolymers.38,39 In the RAFT PISA process of block copolymer synthesis, a solvophilic macro-RAFT agent is chain-extended by a solvophobic monomer to form soluble block copolymers at the beginning of the polymerization process. As the conversion increases, the growing block copolymers become insoluble in the continuous phase at a critical MW of the solvophobic moiety, leading to the in situ formation of self-assembled nano-objects.40
S)–Z are typically used as the active reversible chain transfer agents (RAFT agents), which allow the rapid establishment of equilibrium and production of polymers with narrow MWDs.34,35 Controlled polymerization is possible in the RAFT technique because the majority of polymer chains exist as the dormant species with thiocarbonylthio moieties (living chains).36,37 Nonetheless, a small amount of dead chains without the thiocarbonylthio moiety are also formed through the termination reactions of propagating radicals. Due to the living nature, macro-RAFT agents can also participate in chain extension reactions into diverse polymer architectures, including well-defined block copolymers.38,39 In the RAFT PISA process of block copolymer synthesis, a solvophilic macro-RAFT agent is chain-extended by a solvophobic monomer to form soluble block copolymers at the beginning of the polymerization process. As the conversion increases, the growing block copolymers become insoluble in the continuous phase at a critical MW of the solvophobic moiety, leading to the in situ formation of self-assembled nano-objects.40
Here, we report a new synthetic method of in situ RAFT dispersion polymerization in a mixture of ethanol/water/epoxy for the good dispersion of core–shell rubber particles in the epoxy resin (Scheme 1). Poly(methyl methacrylate-co-glycidyl methacrylate) (P(MMA-co-GMA)) and poly(2-hydroxypropyl methacrylate) (PHPMA) were synthesized for their utilization as the macro-RAFT agent and steric stabilizer forming the shell in a RAFT dispersion polymerization. Chain extension of macro-RAFT agents with a solvophobic monomer, 2-ethylhexyl methacrylate (EHMA) or hexyl acrylate (HA), was then performed in a RAFT dispersion polymerization to form the rubbery core. This work confirms the effect of the selection of macro-RAFT agents and block extension on polymerization kinetics, control over MW and dispersity (Đ), particle size, and impact strength of the particle dispersed epoxy system.
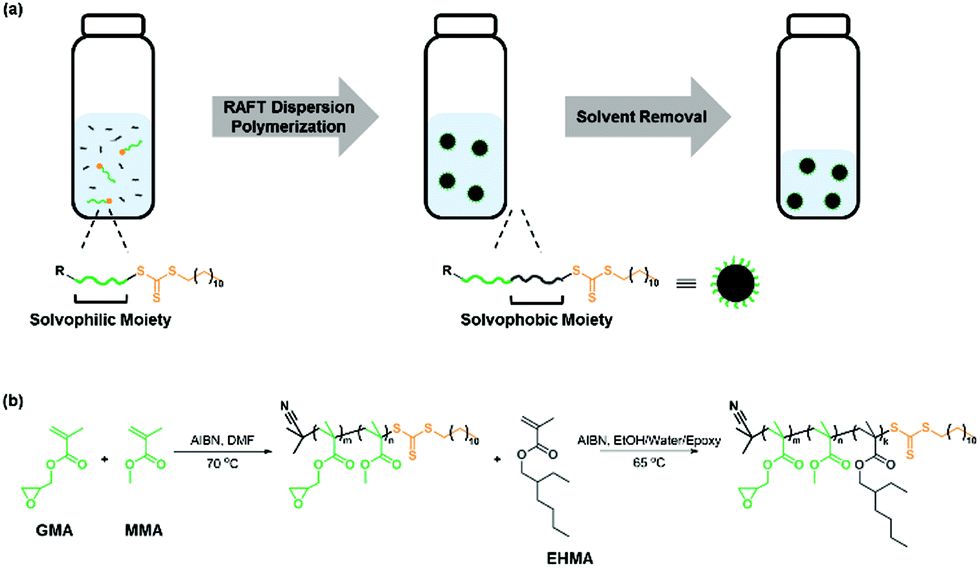 | ||
Scheme 1 (a) RAFT dispersion polymerization and preparation of the core–shell particle dispersed epoxy resin. (b) Synthesis of P(MMA-co-GMA) as a macro-RAFT agent with [RAFT agent]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[GMA]0:[MMA]0:[initiator]0 = 1:68:23:0.1 in DMF at 70 °C, followed by the RAFT dispersion polymerization of EHMA with [P(MMA-co-GMA)]0:[EHMA]0:[AIBN]0 = 1:400:0.3 in the EtOH/water/epoxy mixture at 65 °C. :[GMA]0:[MMA]0:[initiator]0 = 1:68:23:0.1 in DMF at 70 °C, followed by the RAFT dispersion polymerization of EHMA with [P(MMA-co-GMA)]0:[EHMA]0:[AIBN]0 = 1:400:0.3 in the EtOH/water/epoxy mixture at 65 °C. | ||
The selection of a suitable RAFT agent (R–Z) is essential for the synthesis of well-defined polymers. Previously, RAFT agents bearing a trithiocarbonate moiety with an alkyl chain (Z group) and an isobutyronitrile moiety (R group) were successfully used for the polymerization of (meth)acrylic monomers.33,41 PHPMA and P(MMA-co-GMA) have been widely reported as steric stabilizers in alcoholic media and epoxy resins, respectively.42–44 Hence, two macro-RAFT agents (P(MMA-co-GMA) and PHPMA) were initially prepared by RAFT solution polymerization using 2-cyano-2-propyl dodecyl trithiocarbonate as the RAFT agent and azobisisobutyronitrile (AIBN) as the thermal initiator. P(MMA-co-GMA) was prepared by heating a mixture of [RAFT agent]0:[MMA]0:[GMA]0:[AIBN]0 = 1:75:25:0.1 in N,N-dimethylformamid (DMF) at 70 °C. The polymerization conversion reached 95% at 12 h, as determined by 1H NMR spectroscopy. The theoretical number fraction of living chains (livingness, L = 93%) during polymerization was calculated using the equation (see the ESI† and Table S1):36,39
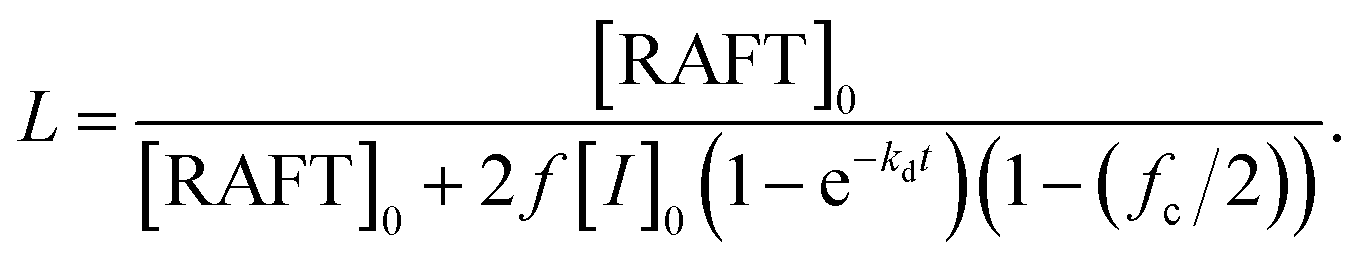
After precipitation in methanol, the number-average MW (Mn) and dispersity (Đ = Mw/Mn) of P(MMA-co-GMA) measured by SEC were 11.5 kg mol−1 and 1.13, respectively. The Mn,SEC of P(MMA-co-GMA) matched well with the theoretical Mn value (11.2 kg mol−1). The SEC chromatogram and 1H NMR spectrum of P(MMA-co-GMA) are shown in Fig. S1(a) and (b) (ESI†), respectively. The degrees of polymerization (DP) of MMA and GMA in P(MMA-co-GMA) were 71 and 27, respectively. PHPMA was also prepared by heating a mixture of [RAFT agent]0:[HPMA]0:[initiator]0 = 1:50:0.1 in DMF at 70 °C. The polymerization conversion reached 96% at 12 h with Mn = 11.3 kg mol−1 (DP = 75) and Đ = 1.14. The Mn,SEC of PHPMA was higher than the theoretical Mn value of 7.30 kg mol−1 because of the small amount of dimethacrylate impurity in the HPMA monomer.45 Further, this might be due to the loss of low-MW PHPMA during the repeated precipitation process. The SEC chromatogram and 1H NMR spectrum of PHPMA are shown in Fig. S1(c) and (d) (ESI†), respectively.
RAFT dispersion polymerizations were performed by heating the synthesized macro-RAFT agent, a monomer, HA or EHMA, and AIBN in an epoxy resin (bisphenol A diglycidyl ether), ethanol, and water mixture at 65 °C. The chain growth leads to the formation of core–shell nanoparticles with a rubbery core of PHA or PEHMA and a glassy shell of P(MMA-co-GMA) or PHPMA for toughening of the epoxy matrix. The detailed synthetic procedure is described in the ESI.†
The dispersion polymerization of EHMA was performed using a mixture of [P(MMA-co-GMA)]0:[EHMA]0:[AIBN]0 = 1:400:0.3. P(MMA-co-GMA) was utilized as both the chain transfer agent and steric stabilizer. During polymerization, samples were taken periodically for the 1H NMR spectroscopy measurement to monitor the conversion of EHMA by comparing it with epoxy (Fig. S2 and S3, ESI†). The monomer conversion was 95% after 24 h of reaction. A semi-logarithmic plot of the monomer concentration versus time is presented in Fig. 1(a). It shows that the rate of polymerization significantly increased after 4 h, with the homogeneous reaction medium turning opaque. This indicates the formation of micelles (reaction loci), responsible for an increase in the rate of polymerization at a high local EHMA concentration. The DLS analysis also shows the presence of nanometer-scale polymer particles, which further confirmed the establishment of a compartmentalized system (micelles) that increased the rate of polymerization due to the segregation of propagating radicals. Fig. 2(a) displays the narrow particle size distribution (37–53 nm, polydispersity index: 0.277) of the dispersion polymerization. The results of the dispersion polymerizations including the chain extensions of PHPMA and P(MMA-co-GMA) with HA are summarized in Table 1 (as well as in Fig. S4 and S5, ESI†). Finally, the solvents, ethanol and water in the latex dispersion, were distilled out under reduced pressure to produce an epoxy resin with dispersed polymer particles. The chemical structure of P(MMA-co-GMA)-b-PEHMA was confirmed by 1H NMR spectroscopy (Fig. S2, ESI†). The peaks in the spectrum corresponded well with the magnetically different protons of the resulting polymers and epoxy resin in the mixture. The 1H NMR spectra of PHPMA-b-PHA and P(MMA-co-GMA)-b-PHA are also shown in Fig. S4(c) and S5(c) (ESI†), respectively.
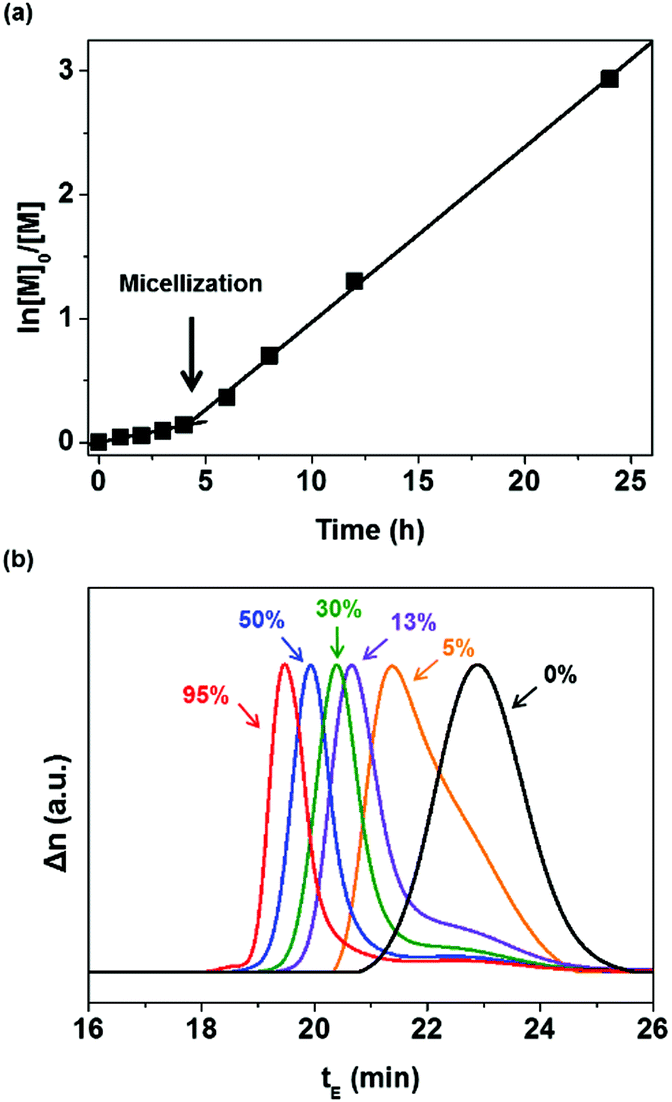 | ||
| Fig. 1 Kinetic analysis of the RAFT dispersion polymerization of EHMA using P(MMA-co-GMA). (a) Plot of ln([M]0/[M]) versus time. (b) SEC chromatograms recorded at different conversions between 0% and 95%. | ||
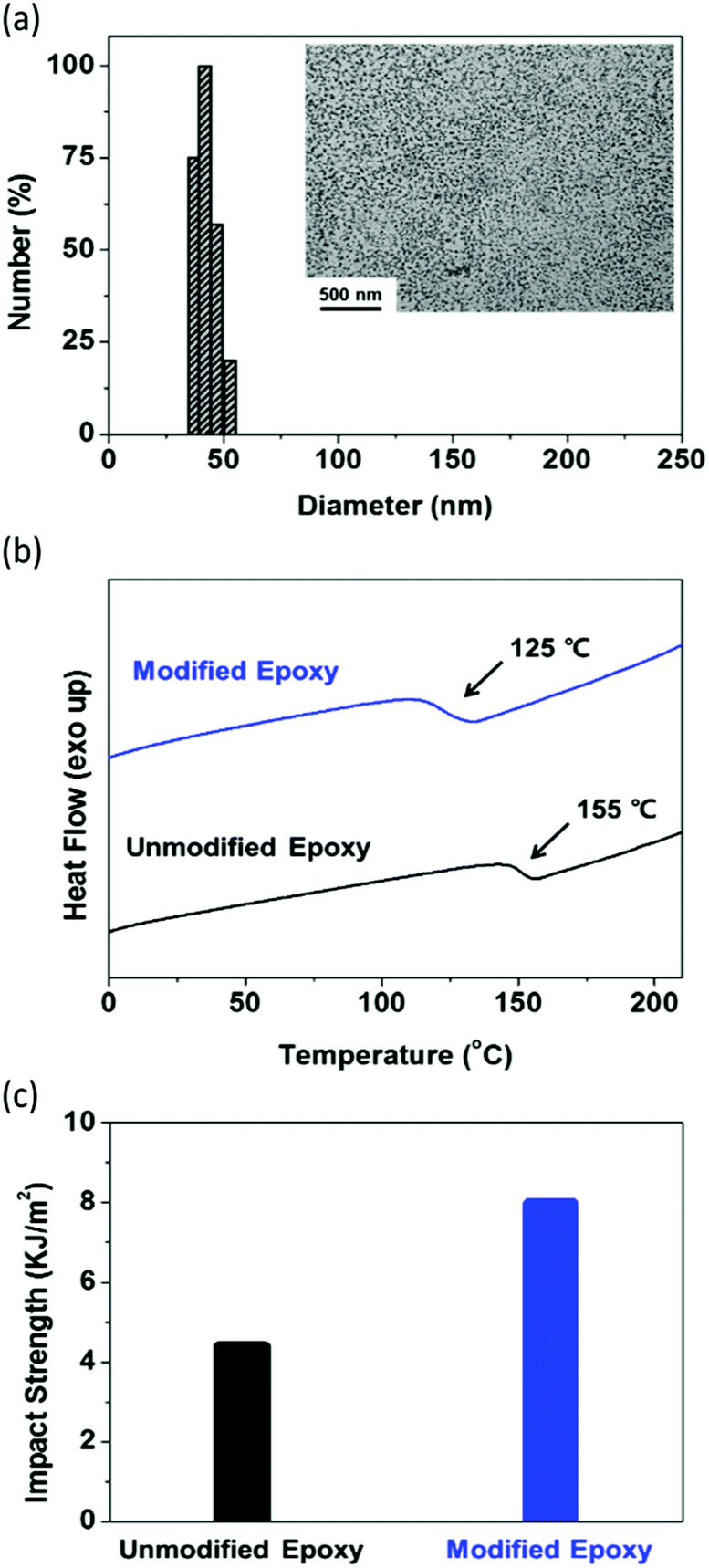 | ||
| Fig. 2 (a) DLS number-based particle size distribution of the P(MMA-co-GMA)-b-PEHMA copolymer prepared by RAFT dispersion polymerization. Inset: TEM image of the P(MMA-co-GMA)-b-PEHMA particles dispersed in the epoxy resin. The scale bar is 500 nm. (b) DSC curves of the unmodified epoxy and the modified epoxy containing the P(MMA-co-GMA)-b-PEHMA particles. (c) Impact strengths of the unmodified epoxy and the modified epoxy containing the P(MMA-co-GMA)-b-PEHMA particles. | ||
| M n,SEC (kg mol−1) | Đ | Conversionb (%) | Diameterc (nm) | |
|---|---|---|---|---|
| a M n and Đ were determined in SEC using poly(methyl methacrylate) calibration standards. b Monomer conversion was calculated by 1H NMR spectroscopy. c The particle size in the solvent mixture (epoxy/ethanol/water) was measured by DLS. | ||||
| PHPMA | 11.3 | 1.14 | 95.8 | — |
| P(MMA-co-GMA) | 11.5 | 1.13 | 94.5 | — |
| PHPMA-b-PHA | 21.4 | 1.64 | 72.6 | 51.0 |
| P(MMA-co-GMA)-b-PHA | 29.8 | 1.78 | 73.5 | 60.0 |
| P(MMA-co-GMA)-b-PEHMA | 55.5 | 1.41 | 94.6 | 43.0 |
The MW of the synthesized polymer was measured using SEC by mixing it with a small amount of THF. In Fig. 1(b), the SEC chromatograms show the molecular weight transition (with a shorter elution time, tE) due to the chain addition reaction of EHMA. The resulting P(MMA-co-GMA)-b-PEHMA polymer exhibited Mn,exp = 55.5 kg mol−1 and Đ = 1.41. In the SEC trace of the P(MMA-co-GMA)-b-PEHMA polymer, most of the P(MMA-co-GMA) chains participated in the chain extension reaction, while a less intense broad peak remained in the lower MW region corresponding to the 12% of unreacted P(MMA-co-GMA). This is in good agreement with the calculated amount of the dead chains formed during synthesis (L ∼93%). The small deviation is due to the production of low-MW polymers through radical initiation and chain termination.46 In contrast to the polymerization reaction of EHMA, the dispersion polymerization of HA with PHPMA or P(MMA-co-GMA) resulted in SEC chromatograms showing bimodal distributions representing a mixture of the chain-extended polymers and unreacted macro-RAFT agents (Fig. S4 and S5, ESI†). It is related to the initial rate of reaction in the continuous phase and the subsequent ability to form a micellar system.
Since the propagation rate constants of EHMA are smaller than those of HA, the initial chain extension reaction (in the continuous phase) proceeds slowly in the presence of EHMA.47 Thus, this slow chain growth is responsible for the induction period before the formation of micelles during the dispersion polymerization of EHMA. However, after the attainment of the critical MW in the continuous phase, micelles with uniform size are formed via the self-assembly of PEHMA blocks. Thus, a perfect phase separated system results in particles with cores consisting of PEHMA blocks that increase the interaction degree/solubility of the EHMA monomer, thereby promoting rapid chain growth with an almost full conversion of macro-RAFT chains and the highest monomer conversion (∼95%). In the case of HA, even though the higher propagation rate in the continuous phase results in a rapid formation of phase-separated (heterogeneous) system, it does not result in uniform self-assembly into nanoparticles. Hence, the lack of a perfect compartmental system decreases the chance of 100% conversion of macro-RAFT chains during the dispersion polymerization of PHA. For this reason, the dispersion polymerization reaction also did not result in a higher monomer conversion (∼75%). These results indicate that only the RAFT dispersion polymerization of EHMA involving P(MMA-co-GMA) proceeded with typical characteristics of controlled radical polymerization.
The digital photographs of the samples (P(MMA-co-GMA)-b-PEHMA, P(MMA-co-GMA)-b-PHA, and PHPMA-b-PHA in epoxy) after the removal of ethanol and water are shown in Fig. S6 (ESI†). In the figure, PHPMA caused the aggregation of polymer particles in the epoxy matrix, while P(MMA-co-GMA) exhibited good dispersibility without aggregation. Although both P(MMA-co-GMA) and PHPMA are soluble in the ethanol/water/epoxy mixture, the relatively high apparent solubility of P(MMA-co-GMA) in epoxy indicates its superior compatibility and steric stabilization properties as the particle shell material as compared to particles with PHPMA shells. To check the dispersion of the particles in the epoxy resin, the amine cured epoxy sample was measured by TEM. The sample was prepared by mixing equal amounts of the curing agent and diethylenetriamine with the epoxy resin containing dispersed particles, and curing by hot-pressing at 120 °C. The TEM image (inset of Fig. 2(a)) shows that the P(MMA-co-GMA)-b-PEHMA particles are uniformly dispersed in the epoxy resin. After considering all the above results, only the epoxy resin with uniformly dispersed well-defined P(MMA-co-GMA)-b-PEHMA particles was investigated for other physical properties.
The glass transition temperature (Tg) and mechanical properties of the cured epoxy resin containing dispersed P(MMA-co-GMA)-b-PEHMA particles were measured and compared with those of the unmodified cured epoxy (details of the testing procedure are provided in the ESI†). In the DSC plot in Fig. 2(b), the modified epoxy exhibits a lower Tg of 125 °C than the unmodified epoxy (Tg = 155 °C). This indicates the plasticization effect of P(MMA-co-GMA)-b-PEHMA particles on the epoxy matrix. Due to the same reason, the tensile strength of the modified epoxy is also smaller than that of the unmodified epoxy (for the unmodified epoxy, tensile strength (TS) = 75.4 MPa; for the particle modified epoxy, TS = 41.9 MPa).48,49 However, this was expected to increase the toughness of the epoxy resin. Therefore, the impact strengths of the modified and unmodified epoxy resins were measured by performing Izod impact tests to investigate the effect of core–shell rubber particles. In Fig. 2(c), the epoxy resin with well-dispersed P(MMA-co-GMA)-b-PEHMA particles exhibits a higher impact strength of 7.99 MPa than the unmodified epoxy (4.39 MPa). This proves that the uniformly well-dispersed P(MMA-co-GMA)-b-PEHMA rubber particles increased the toughness of the epoxy polymer.
Conclusions
Core–shell rubber particles were synthesized as a dispersed phase in the epoxy matrix via the simple RAFT dispersion polymerization in the epoxy/ethanol/water mixture. For this purpose, P(MMA-co-GMA) and PHPMA macro-RAFT agents were prepared and employed as the reversible chain transfer agents and steric stabilizers in the dispersion polymerization of EHMA or HA. The RAFT dispersion polymerization of EHMA with P(MMA-co-GMA) led to a successful uniform chain extension, while the RAFT dispersion polymerization of HA resulted in a mixture with the residual unreacted macro-RAFT agents (P(MMA-co-GMA) or PHPMA). The obtained block copolymer P(MMA-co-GMA)-b-PEHMA was characterized by SEC, 1H NMR spectroscopy, DLS, and TEM. The formed P(MMA-co-GMA)-b-PEHMA polymer particles had a size of 45 nm, a Mn,exp of 55.5 kg mol−1, and a Đ of 1.41; moreover, they produced a strong plasticization effect on the epoxy resin and considerably increased its impact strength. Thus, we believe that this RAFT dispersion polymerization in the ethanol/water/epoxy mixture can be potentially useful for the future design and synthesis of various polymer particles in a different reaction medium.Author contributions
E. H. Lee: conceptualization, investigation, writing – original draft. K. Kim: conceptualization, investigation, writing – original draft. G. Yeom: investigation. B. Seo: funding acquisition. W. Lee: investigation. Y. Yu: writing – review & editing, supervision. A. K. Mohanty: conceptualization, writing – review & editing. H. Paik: conceptualization, writing – review & editing, supervision, funding acquisition. E. H. Lee and K. Kim contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2021R1A2B5B01002081), and the Industrial Technology Innovation Program (20010566) funded by the Ministry of Trade, Industry and Energy (MOTIE) of Korea. This work was also supported by the Korea Research Institute of Chemical Technology (grant number SS2141-10).Notes and references
- J. Hodgkin, G. P. Simon and R. J. Varley, Polym. Adv. Technol., 1998, 9, 3–10 CrossRef CAS.
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- C. May, Epoxy resins: chemistry and technology, Routledge, 2018 Search PubMed.
- N. Chikhi, S. Fellahi and M. Bakar, Eur. Polym. J., 2002, 38, 251–264 CrossRef CAS.
- G. Giannakopoulos, K. Masania and A. Taylor, J. Mater. Sci., 2011, 46, 327–338 CrossRef CAS.
- A. Kinloch, S. Shaw, D. Tod and D. Hunston, Polymer, 1983, 24, 1341–1354 CrossRef CAS.
- J. Wang, Z. Xue, Y. Li, G. Li, Y. Wang, W.-H. Zhong and X. Yang, Polymer, 2018, 140, 39–46 CrossRef CAS.
- N. Ning, W. Liu, Q. Hu, L. Zhang, Q. Jiang, Y. Qiu and Y. Wei, Compos. Sci. Technol., 2020, 199, 108364 CrossRef CAS.
- W. L. Tsang and A. C. Taylor, J. Mater. Sci., 2019, 54, 13938–13958 CrossRef CAS.
- M. Abadyan, V. Khademi, R. Bagheri, H. Haddadpour, M. Kouchakzadeh and M. Farsadi, Mater. Des., 2009, 30, 1976–1984 CrossRef CAS.
- S. Kunz, J. Sayre and R. Assink, Polymer, 1982, 23, 1897–1906 CrossRef CAS.
- A. C. Meeks, Polymer, 1974, 15, 675–681 CrossRef CAS.
- J. Chen, A. Kinloch, S. Sprenger and A. Taylor, Polymer, 2013, 54, 4276–4289 CrossRef CAS.
- A. Keller, H. M. Chong, A. C. Taylor, C. Dransfeld and K. Masania, Compos. Sci. Technol., 2017, 147, 78–88 CrossRef CAS.
- R. He, X. Zhan, Q. Zhang and F. Chen, RSC Adv., 2016, 6, 35621–35627 RSC.
- K. Yamaguchi, M. Ueno and M. Miyamoto, US Pat., US8222324B2, 2012 Search PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- F. D’Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef PubMed.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- L. Becu-Longuet, A. Bonnet, C. Pichot, H. Sautereau and A. Maazouz, J. Appl. Polym. Sci., 1999, 72, 849–858 CrossRef CAS.
- S. Kawaguchi and K. Ito, Polymer Particles, Springer, 2005, pp. 299–328 Search PubMed.
- B. Peng, E. van der Wee, A. Imhof and A. van Blaaderen, Langmuir, 2012, 28, 6776–6785 CrossRef CAS PubMed.
- I. Chaduc, A. s. Crepet, O. Boyron, B. Charleux, F. D’Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- M. J. Rymaruk, S. J. Hunter, C. T. O’Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 2822–2832 CrossRef CAS.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- L. P. Ratcliffe, B. E. McKenzie, G. L. M. Le Bouëdec, C. N. Williams, S. L. Brown and S. P. Armes, Macromolecules, 2015, 48, 8594–8607 CrossRef CAS.
- A. Xu, Q. Lu, Z. Huo, J. Ma, B. Geng, U. Azhar, L. Zhang and S. Zhang, RSC Adv., 2017, 7, 51612–51620 RSC.
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- M. Destarac, Polym. Rev., 2011, 51, 163–187 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. B. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- K. Kim, J. Ahn, M. Park, H. Lee, Y. J. Kim, T. Chang, H. B. Jeon and H.-J. Paik, Macromolecules, 2019, 52, 7448–7455 CrossRef CAS.
- Y. Chong, T. P. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071–2074 CrossRef CAS.
- P. B. Zetterlund, G. Gody and S. Perrier, Macromol. Theory Simul., 2014, 23, 331–339 CrossRef CAS.
- G. N. Smith, S. L. Canning, M. J. Derry, O. O. Mykhaylyk, S. E. Norman and S. P. Armes, Polym. Chem., 2020, 11, 2605–2614 RSC.
- M. Park, K. Kim, A. K. Mohanty, H. Y. Cho, H. Lee, Y. Kang, B. Seo, W. Lee, H. B. Jeon and H. J. Paik, Macromol. Rapid Commun., 2020, 41, 2000399 CrossRef CAS PubMed.
- J. M. Dean, N. E. Verghese, H. Q. Pham and F. S. Bates, Macromolecules, 2003, 36, 9267–9270 CrossRef CAS.
- K. Wang, Y. Wang and W. Zhang, Polym. Chem., 2017, 8, 6407–6415 RSC.
- L. Ruiz-Perez, G. J. Royston, J. P. A. Fairclough and A. J. Ryan, Polymer, 2008, 49, 4475–4488 CrossRef CAS.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27, 191–254 CrossRef CAS.
- B. Meng, J. Deng, Q. Liu, Z. Wu and W. Yang, Eur. Polym. J., 2012, 48, 127–135 CrossRef CAS.
- T. H. Kim, M. Kim, W. Lee, H.-G. Kim, C.-S. Lim and B. Seo, Coatings, 2019, 9 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, polymer preparation and characterization procedures. See DOI: 10.1039/d1ma00850a |
| ‡ These authors contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2021 |