
Structure guided generation of thieno[3,2-d]pyrimidin-4-amine Mycobacterium tuberculosis bd oxidase inhibitors†
Sarah M.
Hopfner
 a,
Bei Shi
Lee
b,
Nitin P.
Kalia
c,
Marvin J.
Miller
*d,
Kevin
Pethe
*b and
Garrett C.
Moraski
*a
a,
Bei Shi
Lee
b,
Nitin P.
Kalia
c,
Marvin J.
Miller
*d,
Kevin
Pethe
*b and
Garrett C.
Moraski
*a
aDepartment of Chemistry and Biochemistry, Montana State University, 103 Chemistry and Biochemistry Building, Bozeman, Montana 59717, USA
bLee Kong Chian School of Medicine, Nanyang Technological University, Experimental Medicine Building, 59 Nanyang Drive, 636921, Singapore
cSchool of Biological Sciences, Nanyang Technological University, Experimental Medicine Building, 59 Nanyang Drive, 636921, Singapore
dDepartment of Chemistry and Biochemistry, University of Notre Dame, 251 Nieuwland Science Hall, Notre Dame, Indiana 46556, USA. E-mail: mmiller1@nd.edu
First published on 12th January 2021
Abstract
Cytochrome bd oxidase (Cyt-bd) is an attractive drug target in Mycobacterium tuberculosis, especially in the context of developing a drug combination targeting energy metabolism. However, currently few synthetically assessable scaffolds target Cyt-bd. Herein, we report that thieno[3,2-d]pyrimidin-4-amines inhibit Cyt-bd, and report an initial structure–activity-relationship (SAR) of 13 compounds in three mycobacterial strains: Mycobacterium bovis BCG, Mycobacterium tuberculosis H37Rv and Mycobacterium tuberculosis clinical isolate N0145 in an established ATP depletion assay with or without the cytochrome bcc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aa3 (QcrB) inhibitor Q203. All compounds displayed activity against M. bovis BCG and the M. tuberculosis clinical isolate strain N0145 with ATP IC50 values from 6 to 54 μM in the presence of Q203 only, as expected from a Cyt-bd inhibitor. All derivatives were much less potent against M. tuberculosis H37Rv compared to N0145 (IC50's from 24 to >100 μM and 9–52 μM, respectively), an observation that may be attributed to the higher expression of the Cyt-bd-encoding genes in the laboratory-adapted M. tuberculosis H37Rv strain. N-(4-(tert-butyl)phenethyl)thieno[3,2-d]pyrimidin-4-amine (19) was the most active compound with ATP IC50 values from 6 to 18 μM against all strains in the presence of Q203, making it a good chemical probe for interrogation the function of the mycobacterial Cyt-bd under various physiological conditions.
:aa3 (QcrB) inhibitor Q203. All compounds displayed activity against M. bovis BCG and the M. tuberculosis clinical isolate strain N0145 with ATP IC50 values from 6 to 54 μM in the presence of Q203 only, as expected from a Cyt-bd inhibitor. All derivatives were much less potent against M. tuberculosis H37Rv compared to N0145 (IC50's from 24 to >100 μM and 9–52 μM, respectively), an observation that may be attributed to the higher expression of the Cyt-bd-encoding genes in the laboratory-adapted M. tuberculosis H37Rv strain. N-(4-(tert-butyl)phenethyl)thieno[3,2-d]pyrimidin-4-amine (19) was the most active compound with ATP IC50 values from 6 to 18 μM against all strains in the presence of Q203, making it a good chemical probe for interrogation the function of the mycobacterial Cyt-bd under various physiological conditions.
1. Introduction
Tuberculosis (TB) is a public health crisis. According to the 2018 World Health Organization (WHO) report, 10 million people became ill with TB, and 1.3 million died in 2017.1 TB is one of the top 10 causes of death worldwide.1 TB infection is caused by Mycobacterium tuberculosis (Mtb), a primarily aerobic bacterium. Infection is spread from person to person by aerosol transmission, usually via an infected individual's cough or sneeze. Initial symptoms are mild and often go unnoticed. Most troubling is the continued rise in drug resistant TB. It is estimated that between 483000 and 639000 people have developed TB that was resistant to rifampicin.1 Thus, there is an urgent need for the development of antimicrobials and treatment options that can swiftly eliminate Mtb infections.
Various bacteria can make adenosine triphosphate (ATP) by two methods: substrate-level phosphorylation of fermentable carbon sources or oxidative phosphorylation.2 During infection, TB demonstrates remarkable competence in its ability to adapt to environmental stresses. Of the investigated attributes, the mechanism of adapting to oxygen levels has been appreciably studied. High-density mutagenesis and deletion studies have shown that Mtb cannot sufficiently produce ATP by substrate-level phosphorylation alone, and oxidative phosphorylation is strictly required for growth.3,4
Small-molecule inhibitors have been synthesized that have various targets in the oxidative phosphorylation pathway. Clofazimine 1 (Fig. 1), a repurposed anti-leprosy drug, targets NDH-2 and probably additional respiratory cytochromes.5,6 The first line TB drug pyrazinamide incapacitates the proton motive force that maintains an electrochemical gradient across the membrane (Fig. 1).7 The diarylquinoline bedaquiline 2 targets ATP synthase8,9 whereas Q203 (3) inhibits the cytochrome bcc:aa3 complex (Fig. 1).10 Q203 belongs to the imidazopyridine class of compounds and entered phase 2 clinical trials in July of 2018.11,12 We have demonstrated that a potential clinical limitation of Q203 is that the compound is unable to inhibit oxygen respiration and is bacteriostatic due to a functional redundancy between the Cyt-bcc:aa3 and the cytochrome bd oxidase (Cyt-bd).10 Bactericidal agents are strongly desired. A neglected target in the oxidative phosphorylation pathway is the Cyt-bd, an oxidoreductase that is important for viability when the function of the Cyt-bcc:aa3 is compromised.2 The Cyt-bd is over-expressed under low oxygen conditions that coincide with the entry into a reversible non-replicating, antibiotic tolerant state.13,14
 | ||
| Fig. 1 Inhibitors of M. tuberculosis oxidative phosphorylation pathway. | ||
Knockouts and mutations of the cytochrome bcc:aa3 complex (Cyt-bcc:aa3) show an upregulation of Cyt-bd,15 making Cyt-bcc:aa3 inhibitors themselves less effective. Although non-essential, the Cyt-bd is still required to maintain oxygen respiration and ATP homeostasis in concert with the Cyt-bcc:aa3.16 In this manner, mycobacteria are protected against drug treatments by rerouting the electron flow to the Cyt-bd. Therefore, targeting cytochrome bd and adding a Cyt-bd inhibitor to a TB treatment regimen may be an effective means to kill replicating and non-replicating mycobacteria more effectively.
There are only two published Mtb Cyt-bd inhibitors, menaquinone analogue aurachin D17 (4) and ND-011992 (ref. 18) (5) (Fig. 1). Issues with aurachin D include not being able to permeate the TB cell wall effectively and having toxic off-target effects.17 However, aurachin D co-administered with Q203 shows almost identical bactericidal activity as that of Q203 within Cyt-bd knockout strains.17 As such, cytochrome bd oxidase is an attractive Mtb drug target particularly when used in combination with Cyt-bcc:aa3 inhibitors, as demonstrated by the bactericidal efficacy of the Q203/ND-011992 combination against replicating and antibiotic-tolerant non-replicating mycobacteria.18
2. Results and discussion
To discover new Cyt-bd oxidase inhibitors, we made use of a whole cell assay in Mycobacterium bovis BCG (BCG), followed by validation in M. tuberculosis H37Rv (H37Rv-Mtb) and M. tuberculosis clinical isolate N0145 (N0145-Mtb). This assay exploits the conditional essentiality of the Cyt-bd to maintain ATP homeostasis once Cyt-bcc:aa3 is selectively inhibited by Q203.18 Measurement of ATP depletion in the presence and absence of Q203 reveals whether a compound inhibits alone or synergizes with Q203. Compounds that deplete ATP in the presence of Q203 but not in its absence are putative Cyt-bd inhibitors. Next, we screened a small but diverse set of around 50 compounds selected from our long-standing antibacterial programs against M. bovis BCG to reveal active compounds like ND-011992.18 When screened in the presence of Q203, our screening revealed two thienopyrimidines—compound 6 (a thieno[2,3-d]pyrimidine-4-amine) and compound 7 (a thieno[3,2-d]pyrimidine-4-amine)—displaying divergent potency with IC50 >50 μM and 26 μM, respectively (Fig. 2). Both 6 and 7 were inactive against BCG (IC50 >50 μM when tested the absence of Q203), suggesting that these compounds work by inhibition of Cyt-bd.
 | ||
| Fig. 2 Thienopyrimidines 6 and hit 7 were identified by ATP depletion within Mycobacterium bovis BCG. | ||
When searching literature around the thienopyrimidine scaffold, we discovered an abundance of references including patent applications for use as pesticides,19 anti-cancer compounds,20,21 worm infections22 and autoimmune diseases.23 Interestingly, only one group has published on thienopyridines as anti-TB agents.24,25 This group identified a thieno[2,3-d]pyrimidine-4-amine, CWHM-728 (8, Fig. 3), through iterative screening of commercially available compounds against Mycobacterium smegmatis. CWHM-728 (8) was found to have an IC50 value of 3.2 μM against M. tuberculosis Erdman strain. Through diligent SAR studies, they developed a much more potent analogue, CWHM-1023 (9, Fig. 3), possessing an IC50 value of 0.083 μM against M. tuberculosis Erdman strain. They also determined that mutations within the QcrB gene confer resistance, suggesting Cyt-bcc:aa3 as the primarily target of these compounds.
 | ||
| Fig. 3 Thieno[2,3-d]pyrimidine-4-amines 8 and 9 identified as novel Cyt-bc1:aa3 inhibitors. | ||
The precedence of “hit to lead” development of the thieno[2,3-d]pyrimidine-4-amines combined with the structural simplicity of compound 7 makes this core an attractive scaffold to explore structure–activity-relationship (SAR) studies. Herein, we report our initial findings on thieno[3,2-d]pyrimidine-4-amines that inhibit mycobacteria Cyt-bd over Cyt-bcc:aa3 within replicating BCG, replicating Mtb-H37Rv and clinical N0145-Mtb strains.
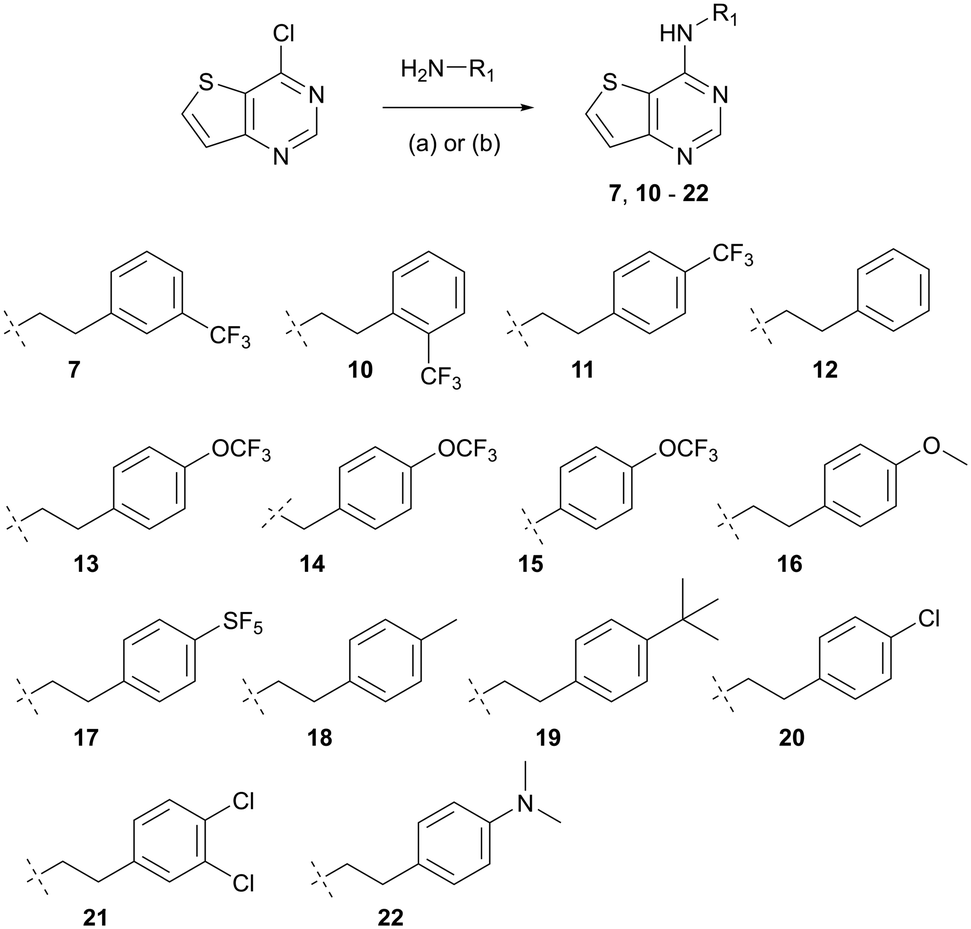
Our SAR efforts focused on probing three structural elements of hit 7: 1) positioning of aryl substitution (2′, 3′ and 4′ positions), 2) alteration of the pendant 2-(3-(trifluoromethyl)phenyl)ethan-1-amino moiety with other substitutions (i.e. H, halogen, CH3, OCF3, etc.), and 3) changing of the aliphatic chain length (i.e. anilino, benzyl, ethanyl).
Compounds were prepared in one step by classical SNAr reactions of 4-chlorothieno[3,2-d]pyrimidine, desired amines and potassium carbonate at elevated temperature (Scheme 1).26 Yields varied from 22 to 70%, after purification by recrystallization (see ESI†). We prepared thirteen analogues (10–22) that were screened in a whole-cell ATP Cyt-bd assay – in BCG with subsequent validation in H37Rv-Mtb and the clinical isolate N0145-Mtb—under replicating conditions with and without added Q203. The BCG strain was used to identify any general Cyt-bd inhibitor (when Q203 was added, “+Q203”). The laboratory adapted M. tuberculosis H37Rv strain over expresses Cyt-bd compared to clinical isolates.8 As such, strains H37Rv and N0145 were used to identify and rank compounds that target M. tuberculosis Cyt-bd (when Q203 was added). Compounds that inhibited intracellular ATP level in mycobacteria (“−Q203”) but did not show better activity when Cyt-bcc:aa3 was inhibited (“+Q203”) were presumed to not be targeting Cyt-bd, an example being the FOF1 ATP synthase inhibitor bedaquiline (BDQ)9 which is equipotent in the presence or absence of Q203. All screening data along with compound molecular mass and calculated clogP are listed in Table 1.
 | ||
| Scheme 1 Syntheses of target thieno[3,2-d]pyrimidin-4-amines (7, 10–22). Reagents and conditions: (a) K2CO3, DMSO, 100 °C, 12 h, or (b) HCl, THF:IPA (3:1), 70 °C, 12 h, yield: 22–70%. | ||
| Compound | Mol wt | clogP |
Replicating ATP IC50 (mM) | |||||
|---|---|---|---|---|---|---|---|---|
| BCG | H37Rv | N0145 | ||||||
| −Q203 | +Q203 | −Q203 | +Q203 | −Q203 | +Q203 | |||
|
IC50 values were determined by ATP depletion in the presence and absence of Q203 (see, ESI† for assay details). clogP was calculated by PerkinElmer ChemDraw Professional 16.0. Bedaquiline (BDQ) and ND-011992 were used as positive control compounds. IC50 values were determined using GraphPad Prism 9. The values reflected in the table represent the average and standard deviation, which were calculated from the IC50 values of replicates from two experimental repeats.
|
||||||||
| 7 | 323.34 | 4.91 | >50 | 25.6 ± 4.59 | >100 | 61.7 ± 7.85 | >50 | 23.4 ± 0.39 |
| 10 | 323.34 | 4.91 | >50 | 12.4 ± 3.36 | >100 | 68.3 ± 10.47 | >50 | 17.4 ± 2.10 |
| 11 | 323.34 | 4.91 | >50 | 33.1 ± 4.50 | >100 | 63.2 ± 1.55 | >50 | 24.9 ± 0.16 |
| 12 | 255.34 | 4.02 | >50 | 43.3 ± 9.44 | >100 | >100 | >50 | 51.5 ± 4.30 |
| 13 | 339.34 | 5.05 | >50 | 12.6 ± 1.10 | >100 | 24.4 ± 1.52 | >50 | 10.6 ± 0.30 |
| 14 | 325.31 | 4.40 | >50 | 38.1 ± 7.43 | >100 | 99.8 ± 19.06 | >50 | 36.1 ± 3.80 |
| 15 | 311.28 | 4.79 | >50 | 22.9 ± 3.55 | >100 | 66.7 ± 1.79 | >50 | 22.8 ± 4.34 |
| 16 | 285.36 | 3.94 | >50 | 42.8 ± 10.34 | >100 | >100 | >50 | 45.0 ± 14.63 |
| 17 | 381.39 | 5.26 | >50 | 27.1 ± 6.38 | >100 | >100 | >50 | 18.9 ± 2.88 |
| 18 | 269.36 | 4.52 | >50 | 40.4 ± 11.29 | >100 | 108.3 ± 7.80 | >50 | 37.4 ± 10.23 |
| 19 | 311.44 | 5.85 | >50 | 5.8 ± 1.06 | >100 | 18.9 ± 9.03 | >50 | 8.5 ± 2.38 |
| 20 | 289.78 | 4.74 | >50 | 30.4 ± 9.27 | >100 | 83.5 ± 0.97 | >50 | 31.8 ± 9.98 |
| 21 | 324.23 | 5.33 | >50 | 25.6 ± 8.35 | >100 | 109.3 ± 17.36 | >50 | 35.3 ± 9.49 |
| 22 | 298.41 | 4.20 | >50 | 38.9 ± 9.08 | >100 | >100 | >50 | 41.5 ± 15.31 |
| 3 (BDQ) | 555.52 | 7.25 | 0.17 ± 0.005 | 0.09 ± 0.0008 | 0.04 ± 0.004 | 0.07 ± 0.01 | 0.01 ± 0.0005 | 0.03 ± 0.003 |
| 5 (ND-011992) | 381.36 | 6.69 | >50 | 0.80 ± 0.07 | >100 | 5.8 ± 1.18 | >50 | 1.6 ± 0.69 |
Separate placement of a trifluoromethyl group at the ortho, meta, or para positions of the peripheral phenyl group (compounds 10, 7 and 11, respectively) revealed that the ortho-CF3 analogue (10) had good potency against BCG and N0145-Mtb (IC50 values of 12.4 and 15.5 μM, respectively; when Q203 was added). The ortho-CF3 (10) was slightly more potent than the screening hit meta-CF3 (7) and para-CF3 (11) compounds against those two Mtb strains. The para-CF3 compound (11) had slightly improved potency against H37Rv-Mtb but the IC50 value was high at 63 μM. Despite the slightly better potency at the ortho-position, there is greater availability of para-substituted phenyl analogues with which to probe the effect of substituents on SAR.
Considering the phenylethyl analogue (12) as electronically and sterically “neutral” we found this benchmark compound to have weak potency against the three strains (IC50 from 51 to >100 μM, when Q203 added). However, functionalization at the para position of the phenyl group significantly improved potency as the para-OCF3 compound (13) had lower IC50 values than the para-CF3 compound (11) (IC50 from 11 to 25 μM, in three strains when Q203 was added). In general, all electron withdrawing groups (SF5, Cl, di-Cl) improved potency as compared to the unsubstituted phenylethyl (12) but none were more potent than the para-OCF3 analogue (13). Electron donating groups gave diverse results as the para-methoxy (16) and para-dimethylamine (22) compounds had (weak) potency akin to the unsubstituted phenylethyl compound (12). The para-methyl analogue (18) had slightly better potency than 12 (IC50 from 41 to >100, in the presence of Q203) but not nearly as potent as the compounds with electron withdrawing groups (7, 10, 11, 13, 17, 20, and 21). Interestingly, the most potent compound was the 4-para-C(CH3)3 analogue (19) having IC50 values of 6 to 18 μM against all three stains (with Q203 added). Structurally, 19 is most like the para-SF5 analogue (17) based upon volume27 but more lipophilic (clogP of 5.85 for 19 compared to 5.26 for 17). The scope of active substituents suggests a pocket that can accommodate larger groups (SF5, t-butyl, Cl) and these larger substituents had good potency. Conformational flexibility was probed through the synthesis and screening of the 4-(trifluoromethoxy)phenylethyl (13), 4-(trifluoromethoxy)benzyl (14) and 4-(trifluoromethoxy)aniline (15) analogues. The effect of the linker between the thienopyrimidine core and peripheral phenyl group was interesting in that the compound with the rotatable benzyl side chain (14) was less active than that with aniline (15) and nearly 3-fold less active than the phenylethyl compound (13) (IC50 values of 33 to 96 μM in all three strains, when Q203 was added).
Since the increased level of Cyt-bd expression in H37Rv-Mtb implied a lower potency of the inhibitors, the extra effort of screening the clinical strain in tandem with more commonly used H37Rv-Mtb strain was justified. While it was possible that strain selectively (either BCG or Mtb) could have been revealed, instead, there was strong activity correlation between the BCG and N0145-Mtb strains for all the compounds screened. Finally, none of the compounds alone inhibited ATP, strongly suggesting that these compounds do target Cyt-bd since potency was only revealed when Cyt-bcc:aa3 was selectively inhibited by Q203. The lack of ATP inhibition against Mtb (or BCG) greatly increases the challenge to discover Cyt-bd inhibitors and establishes combination drug therapy as a viable treatment option to use with such inhibitors.
General procedure for base promoted SNAr for synthesis of compounds 7, 11, 13, 17, 19, 22
In a sealed vial, 4-chlorothieno[3,2-d]pyrimidine (100 mg, 0.57 mmol), desired amine (0.57 mmol) and K2CO3 (79 mg, 0.57 mmol) were dissolved in DMSO (4 mL). The reaction was heated to 100 °C for 12 h. The reaction mixture was concentrated to dryness and the residue was dissolved in CH2Cl2 and washed with 5% aqueous acetic acid solution (2×), water and brine. The organic phase was collected, dried over sodium sulfate, filtered, and concentrated in vacuo. Crude material obtained was purified by either silica gel column chromatography with a gradient of CH2Cl2:ethyl acetate:solvent system (0 to 80%) or recrystallized from hot isopropanol or acetonitrile to afford the product.
General procedure for acid catalyzed SNAr for synthesis of compounds 12, 14–16, 18, 20, 21
In a sealed vial, 4-chlorothieno[3,2-d]pyrimidine (100 mg, 0.57 mmol) and desired amine (0.57 mmol) were dissolved in a 3:1 tetrahydrofuran: 2-propanol solution (8 mL). Next, a drop of 12 M HCl (∼0.05 mL) was added and the solution was heated at 70 °C for 24 h. The reaction mixture was concentrated to dryness and the residue was dissolved in CH2Cl2 and washed with saturated aqueous NaHCO3 solution, water, and brine. The organic phase was collected, dried over sodium sulfate, filtered, and concentrated in vacuo. Crude material obtained was purified by either silica gel column chromatography with a gradient of CH2Cl2:ethyl acetate solvent system (0 to 80%) or recrystallized from hot isopropanol or acetonitrile to afford the product.
3. Conclusions
Herein we described our preliminary SAR assessment of the thieno[3,2-d]pyrimidin-4-amines as Cyt-bd inhibitors. This is one of a few published examples of synthetically accessible compounds that can inhibit Cyt-bd in mycobacteria. While, the IC50 values of the most potent compound (19) are merely good (6.2 μM vs. BCG and 7.3 μM vs. N0145-Mtb, when Q203 was added) this class of compounds can be used as a new tool to probe the mycobacterial oxidative phosphorylation pathway. Based upon this exploratory work, we will endeavour to design new thieno[3,2-d]pyrimidin-4-amines with improved potency and acceptable pharmacokinetics to warrant in vivo evaluation.Conflicts of interest
Hsiri Therapeutics has licensed this technology. M. J. M. and G. C. M. own equity in Hsiri. M. J. M. is CSO of Hsiri. G. C. M. and K. P. are consultants to Hsiri. Hsiri Therapeutics did not fund this study and was not involved in study design or interpretation.Acknowledgements
M. J. M. and G. C. M. acknowledge funding from the NIH R37AI054193. K. P. and G. C. M. acknowledge funding from the NIH R01AI139465. This work was supported in part by the National Research Foundation (NRF) Singapore under its NRF Competitive Research Programme (Project Award Number NRF-CRP18-2017-0).Notes and references
- Global tuberculosis report 2018, World Health Organization, Geneva, 2018, Licence: CC BY-NCSA 3.0 IGO Search PubMed.
- A. E. McDonald and G. C. Vanlerberghe, in Structural Basis of Biological Energy Generation, ed. M. F. Hohmann-Marriott, Springer, Dordrecht, 2014, vol. 39, pp. 277–293 Search PubMed.
- D. Bald, C. Villellas, P. Lu and A. Koul, mBio, 2017, 8, 11 CrossRef.
- L. G. Wayne and C. D. Sohaskey, Annu. Rev. Microbiol., 2001, 55, 139–163 CrossRef CAS.
- T. Yano, S. Kassovska-Bratinova, J. S. Teh, J. Winkler, K. Sullivan, A. Isaacs, N. M. Schechter and H. Rubin, J. Biol. Chem., 2011, 286, 10276–10287 CrossRef CAS.
- T. Beites, K. O'Brien, D. Tiwari, C. A. Engelhart, S. Walters, J. Andrews, H. J. Yang, M. L. Sutphen, D. M. Weiner, E. K. Dayao, M. Zimmerman, B. Prideaux, P. V. Desai, T. Masquelin, L. E. Via, V. Dartois, H. I. Boshoff, C. E. Barry, S. Ehrt and D. Schnappinger, Nat. Commun., 2019, 10, 12 CrossRef.
- Y. Zhang, M. M. Wade, A. Scorpio, H. Zhang and Z. H. Sun, J. Antimicrob. Chemother., 2003, 52, 790–795 CrossRef CAS.
- K. Arora, B. Ochoa-Montaño, P. S. Tsang, T. L. Blundell, S. S. Dawes, V. Mizrahi, T. Bayliss, C. J. Mackenzie, L. A. T. Cleghorn, P. C. Ray, P. G. Wyatt, E. Uh, J. Lee, C. E. Barry and H. I. Boshoff, Antimicrob. Agents Chemother., 2014, 58, 6962–6965 CrossRef.
- A. Koul, N. Dendouga, K. Vergauwen, B. Molenberghs, L. Vranckx, R. Willebrords, Z. Ristic, H. Lill, I. Dorange, J. Guillemont, D. Bald and K. Andries, Nat. Chem. Biol., 2007, 3, 323–324 CrossRef CAS.
- N. P. Kalia, E. J. Hasenoehrl, N. B. Ab Rahman, V. H. Koh, M. L. T. Ang, D. R. Sajorda, K. Hards, G. Gruber, S. Alonso, G. M. Cook, M. Berney and K. Pethe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7426–7431 CrossRef CAS.
- K. Pethe, P. Bifani, J. C. Jang, S. Kang, S. Park, S. Ahn, J. Jiricek, J. Y. Jung, H. K. Jeon, J. Cechetto, T. Christophe, H. Lee, M. Kempf, M. Jackson, A. J. Lenaerts, H. Pham, V. Jones, M. J. Seo, Y. M. Kim, M. Seo, J. J. Seo, D. Park, Y. Ko, I. Choi, R. Kim, S. Y. Kim, S. Lim, S. A. Yim, J. Nam, H. Kang, H. Kwon, C. T. Oh, Y. Cho, Y. Jang, J. Kim, A. Chua, B. H. Tan, M. B. Nanjundappa, S. P. S. Rao, W. S. Barnes, R. Wintjens, J. R. Walker, S. Alonso, S. Lee, S. Oh, T. Oh, U. Nehrbass, S. J. Han, Z. No, J. Lee, P. Brodin, S. N. Cho and K. Nam, Nat. Med., 2013, 19, 1157–1160 CrossRef CAS.
- V. R. de Jager, R. Dawson, C. van Niekerk, J. Hutchings, J. Kim, N. Vanker, L. van der Merwe, J. Choi, K. Nam and A. H. Diacon, N. Engl. J. Med., 2020, 382, 1280–1281 CrossRef.
- L. B. Shi, C. D. Sohaskey, B. D. Kana, S. Dawes, R. J. North, V. Mizrahi and M. L. Gennaro, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15629–15634 CrossRef CAS.
- J. E. Gomez and J. D. McKinney, Tuberculosis, 2004, 84, 29–44 CrossRef.
- L. G. Matsoso, B. D. Kana, P. K. Crellin, D. J. Lea-Smith, A. Pelosi, D. Powell, S. S. Dawes, H. Rubin, R. L. Coppel and V. Mizrahi, J. Bacteriol., 2005, 187, 6300–6308 CrossRef CAS.
- P. Lu, M. H. Heineke, A. Koul, K. Andries, G. M. Cook, H. Lill, R. van Spanning and D. Bald, Sci. Rep., 2015, 5, 10 Search PubMed.
- P. Lu, A. H. Asseri, M. Kremer, J. Maaskant, R. Ummels, H. Lill and D. Bald, Sci. Rep., 2018, 8, 7 CrossRef.
- B. S. Lee, K. Hards, C. A. Engelhart, E. J. Hasenoehrl, N. P. Kalia, J. S. Mackenzie, E. Sviriaeva, S. M. S. Chong, M. S. S. Manimekalai, V. H. Koh, J. Chan, J. Xu, S. Alonso, M. J. Miller, A. J. C. Steyn, G. Gruber, D. Schnappinger, M. Berney, G. M. Cook, G. C. Moraski and K. Pethe, EMBO Mol. Med., 2020, e13207 Search PubMed.
- R. G. Edie, R. E. Hackler and E. V. Krumkains, EP Pat., EP452002A2, 1991 Search PubMed.
- M. J. Munchhof and S. B. Sobolov-Jaynes, US Pat., WO9924440A1, 1999 Search PubMed.
- S. Baskaran, W. Lew, J. D. Oslob, J. C. Yoburn and Z. Min, US Pat., US2006035908A1, 2006 Search PubMed.
- R. Benayoud, W. D. Hong, P. M. O'Neil, M. J. Taylor and S. A. Ward, US Pat., US20190345157A1, 2018 Search PubMed.
- C. Beauregard, A. J. Borchardt, R. L. Davis, D. A. Gamache and J. M. Yanni, US Pat., US2010063047A1, 2010 Search PubMed.
- G. A. Harrison, A. E. M. Bridwell, M. Singh, K. Jayaraman, L. A. Weiss, R. L. Kinsella, J. S. Aneke, K. Flentie, M. E. Schene, M. Gaggioli, S. D. Solomon, S. A. Wildman, M. J. Meyers and C. L. Stallings, mSphere, 2019, 4, 14 CrossRef.
- S. Arnett, M. Meyers, M. Singh, C. Stallings, L. Weiss and S. Wildman, US Pat., WO2019018359A1, 2019 Search PubMed.
- J. M. Neri, L. N. Cavalcanti, R. M. Araujo and F. G. Menezes, Arabian J. Chem., 2020, 13, 721–739 CrossRef CAS.
- P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130–1190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, and characterisation of compounds 7, 10–22. See DOI: 10.1039/d0md00398k |
| This journal is © The Royal Society of Chemistry 2021 |