
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale, automated, high throughput synthesis and screening for the accelerated discovery of protein modifiers†
Kai
Gao‡
,
Shabnam
Shaabani‡
 ,
Ruixue
Xu‡
,
Tryfon
Zarganes-Tzitzikas
,
Li
Gao
,
Maryam
Ahmadianmoghaddam
,
Matthew R.
Groves
and
Alexander
Dömling
*
,
Ruixue
Xu‡
,
Tryfon
Zarganes-Tzitzikas
,
Li
Gao
,
Maryam
Ahmadianmoghaddam
,
Matthew R.
Groves
and
Alexander
Dömling
*
Pharmacy Department, Drug Design group, University of Groningen, A. Deusinglaan 1, 9700 AD Groningen, The Netherlands. E-mail: a.s.s.domling@rug.nl
First published on 5th May 2021
Abstract
Hit finding in early drug discovery is often based on high throughput screening (HTS) of existing and historical compound libraries, which can limit chemical diversity, is time-consuming, very costly, and environmentally not sustainable. On-the-fly compound synthesis and in situ screening in a highly miniaturized and automated format has the potential to greatly reduce the medicinal chemistry environmental footprint. Here, we used acoustic dispensing technology to synthesize a library in a 1536 well format based on the Groebcke–Blackburn–Bienaymé reaction (GBB-3CR) on a nanomole scale. The unpurified library was screened by differential scanning fluorimetry (DSF) and cross-validated using microscale thermophoresis (MST) against the oncogenic protein–protein interaction menin–MLL. Several GBB reaction products were found as μM menin binder, and the structural basis of the interactions with menin was elucidated by co-crystal structure analysis. Miniaturization and automation of the organic synthesis and screening process can lead to an acceleration in the early drug discovery process, which is an alternative to classical HTS and a step towards the paradigm of continuous manufacturing.
Introduction
Growing costs and timelines in drug discovery present a significant challenge to pharmaceutical R&D to deliver the best possible medicine to the patient.1 Current early drug discovery is based on HTS of large and existing libraries followed by property optimization i.e., multiple design-make-test-analyze cycles (DMTA).2 Synthesis in early drug discovery is neither optimal nor green.3 Conservative estimates of waste produced per year in drug discovery are as high as 2 million kilograms with up to 1.5 million kilograms of additional waste produced per year in preclinical studies.4 Amongst the 12 principles of green chemistry, early industrial drug discovery and development is particular badly performing with the principle of prevention which greatly overshadows all other principles.5 Reactions in early discovery are typically run on a near mmol scale for thousands of derivatives during an optimization campaign, while with new HTS technologies, material requirements are vanishingly small. For example, 1 μg of an average compound (MW ∼400 Dalton) can be used in ∼1500 HTS campaigns as well as in early pharmacokinetic profiling. Why is it then that synthesis is still mostly performed at an unnecessary large milligram scale?New enabling technologies, laboratory automation and high-throughput experimentation (HTE) aim towards the paradigm of continuous manufacturing and to accelerate early preclinical discovery and development.6 For example, HTE is regularly used to optimize reaction conditions and expand the scope of known reactions.7 The combination of acoustic mist ionization and time-of-flight mass spectrometry finds already sporadic use in HTS approaches to triage assays and in metabolic profiling.8 Acoustic dispensing is used for the fast, mild and effective crystal soaking for the formation of ligand–protein complexes.9 Although novel technologies are constantly surfacing, they have to be validated, implemented and integrated with academic and pharmaceutical R&D activities to obtain a better understanding of the molecular basis of diseases and to ultimately accelerate drug discovery. Forefront developments aiming to automate and accelerate drug discovery are underway, e.g. a specialized platform for innovative research exploration.10
We are working on the concept of artificial intelligence-driven (AI) autonomous early drug discovery towards the paradigm of continuous manufacturing. We have recently shown miniaturized and automated synthesis of large and diverse small molecular weight libraries, including isoquinolines, indole polycycles, quinoxalines, boronic acids, and iminopyrrolidines that resulted in useful, scalable synthetic protocols.10,11 Here, we present the miniaturized, automated, on-the-fly synthesis, quality control, and biophysical screening of a large unpurified library on a nanomole scale to rapidly discover novel binders to the menin protein playing a role in the oncogenic protein–protein interaction menin–MLL. The discovered hits were biophysically characterized and culminated in an experimentally derived molecular model of a hit compound bound to the menin receptor provided by X-ray structure analysis.
Chromosomal rearrangements of the MLL gene12 are associated with patients with acute myeloid (AML), acute lymphoblastic (ALL) leukemias13 and in therapy related leukemias or myelodysplastic syndrome (MDS).14 As a result of these chromosomal rearrangements a gene fusion product may be formed with one of over 60 distinct protein partners, including AF4, AF9, ENL, AF6, ELL and AF10.15 Disruption of MLL activity by this gene translocation has previously been shown to upregulate genes critical in leukemogenesis (HOXA9 and MEIS1).16 The onogenic function of these MLL-fusion proteins17 is dependent upon the direct interaction of the N-terminus of MLL with menin, a 67 kDa ubiquitously protein that is located in the nucleus.18 Antagonizing menin binding to the MLL fusion proteins has been shown to abolish their oncogenic properties in vitro and in vivo and thus represents an attractive drug target in hematology (Fig. 1B).19 Several groups have developed potent small molecules using structure-based drug design methods and involving many optimization cycles.20 Structural studies demonstrated that menin is composed of 3 tetratricopeptide (TPR) motifs that generate a large central cavity of ∼5000 Å3 that is lined with hydrophobic and acidic amino acids.21 Such cavities are typically attractive “druggable” sites and mutation and structural studies demonstrated that this cavity was responsible for the menin:MLL interaction.22 As a result many groups have focused on this cavity of menin as a target for the development of novel therapies in leukemia treatment.
 | ||
| Fig. 1 Chemistry, known menin inhibitors, and overview of the overall workflow. (A) The reaction scheme of the GBB-3CR; (B) previously described highly optimized menin–MLL antagonists; (C) a 384-well destination/reaction plate performed on the 500 nmol scale and charged by an Echo 555; (D) MS analytics of the crude reaction wells; (E) differential scanning fluorimetry (DSF) screening of the crude reaction wells for menin binding; (F) resynthesis of discovered hits on a mmol scale; (G) determination of accurate Kd by microscale thermophoresis (MST); (H) determination of the 3D ligand–receptor interaction by co-crystal structure analysis. | ||
Our workflow towards discovering menin–MLL binders consisted of the acoustic dispensing ejection-enabled (ADE) autonomous synthesis and analysis of a random library of 1536 heterocycles on a nanomole scale (Fig. 1A and C–H). The compounds were then screened as crude reaction mixtures for menin affinity by a high throughput differential scanning fluorimetry/thermal shift analysis (DSF/TSA)-based assay. Identified hits were resynthesized, purified and further characterized by microscale thermophoresis (MST), and our best performing compound was co-crystallized with the menin receptor to confirm binding and understand mode-of-action.
Results
On-the-fly nano-scale high throughput synthesis of a heterocyclic library
As suitable chemistry for the project, we choose the Groebcke–Blackburn–Bienaymé three-component reaction (GBB-3CR, Fig. 1A), since the resulting heterocycles are drug-like, reminiscent to approved drugs and have been shown to yield multiple biologically active compounds previously.23 The GBB-3CR incorporates three building blocks: isocyanide, aldehyde and heterocyclic 5- or 6-membered aromatic amidine. The reaction can be performed under mild conditions in protic solvents, and several Lewis acid catalysts have been described.24We used 71 isocyanides, 53 aldehydes, and 38 cyclic amidines (Fig. 2). The starting materials were chosen to represent multiple physicochemical properties and spanning an interesting chemical screening space. Specifically, we integrated multiple functional groups such as aliphatic and aromatic hydroxyl groups, amines, heterocycles, (pseudo)halogens, and carbonyls to enhance the chances of creating specific small molecule–receptor interactions. Many of the building blocks used herein were previously unknown or are unprecedented in the GBB-3CR.23 For example, amongst the cyclic amidine components (A), sterically hindered A10, hydrophilic A12, bifunctional A14 (–CHO), iodo building block A16, benzoxazole A29, and tetrazole A37. Unique aldehyde components (B) include, for example, hindered benzaldehyde B17, bifunctional B46 (–COOH), α,β-unsaturated B47 and B52, and acetal protected B50. Moreover, the majority of the isocyanides (C) used were previously unknown in the GBB-3CR, including amino acid derivatives C16, C20, C25, C29, acrylamide derivative C32 (as potential covalent inhibitors), or azide derivative C56 (for orthogonal tagging of protein targets) just to name a few. To avoid chemical space bias by synthesizing combinatorial libraries, we used a script that randomly combines building blocks in the reaction wells (ESI†-2.3.4). The theoretical chemical space of the above building blocks is 142![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 994 (71 × 53 × 38); however, we synthesized only a small subspace of 1536 GBB-3CR products.
994 (71 × 53 × 38); however, we synthesized only a small subspace of 1536 GBB-3CR products.
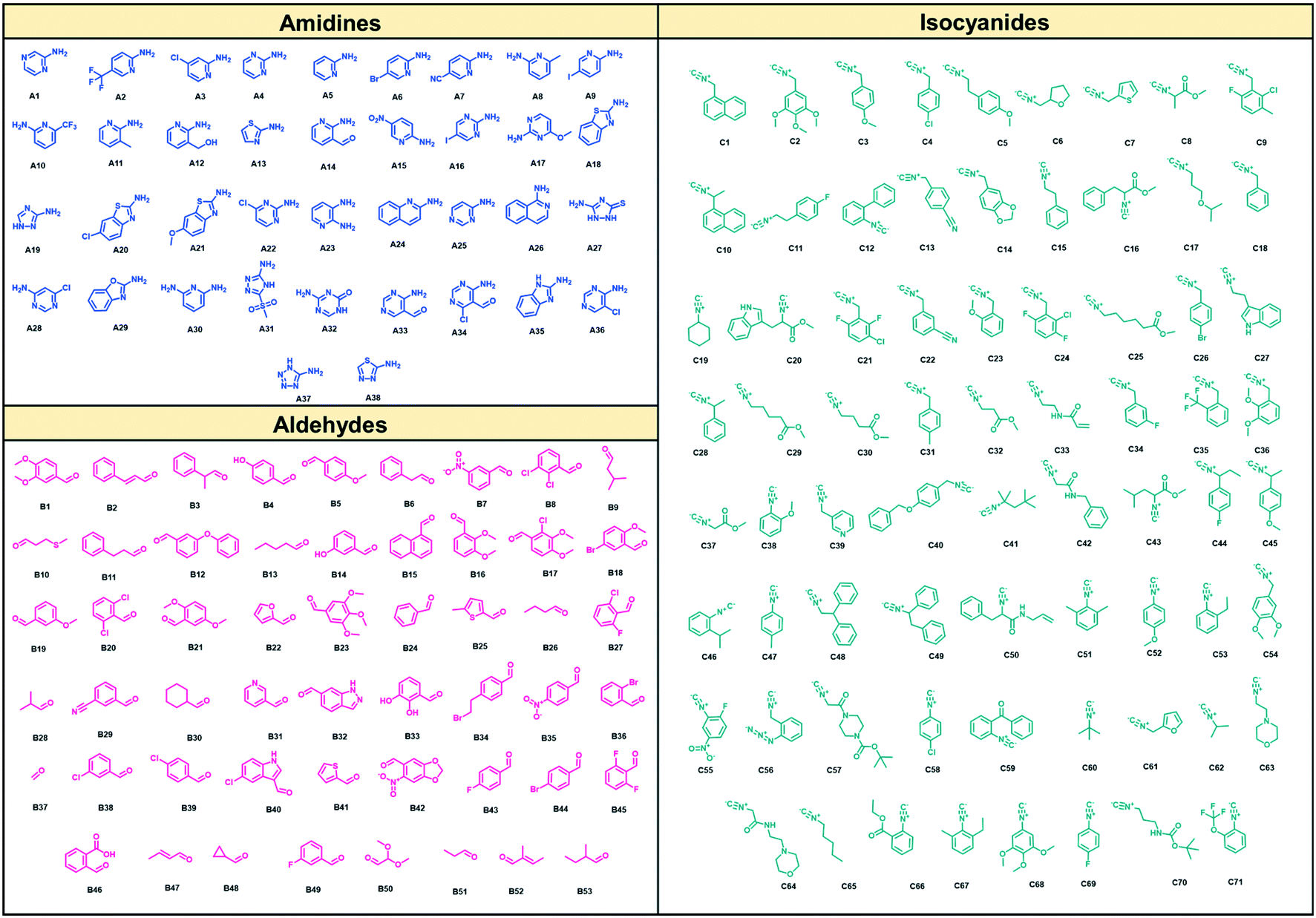 | ||
| Fig. 2 Building block chemical space, cyclic aromatic amidines, aldehydes, and isocyanides, used. | ||
To assemble the library on a nanomole scale, we used an Echo 555 acoustic dispensing instrument.10,11b Briefly, acoustic dispensing is a contact-less, fast, precise, and accurate fluid transfer technology using ultrasound.25 A transducer below a source plate containing the building blocks stock solutions emits focused sound energy repeatedly to the meniscus of the fluid to be transferred. A stream of 2.5 nL droplets is ejected into the wells of an inverted destination microplate. By combining different building blocks in the destination plate, reactions can take place in a fast and miniaturized fashion. Each well was filled with 500 nanomole of reagents yielding a total volume of 3.1 μL. Thus, the filling of a 1536-well plate takes ∼10 h employing the Echo 555. Importantly, standard plate formats can be utilized in acoustic dispensing, such as 96-, 384-, 1536- or 3456-well plates that allow for facile integration in an established laboratory environment. A number of different solvents were tested and can be transferred on the Echo 555 platform: DMSO, DMF, water, ethylene glycol, 2-methoxyethanol, and N-methylpyrrolidone. The polar protic solvent ethylene glycol and 2-methoxyethanol were chosen for the nano-scale GBB reaction.
Next, we analyzed the reactions using direct mass spectrometry (Fig. 3A). After 24 h of reaction, each well was diluted with 100 μL of ethylene glycol and the reaction mixtures were injected directly into the mass spectrometer. We categorized the reaction success according to three different classes. Reactions showing as the main peak (M + H)+, (M + Na)+, (M + K)+, showing a peak corresponding to the desired products but not as the highest peak or not showing the desired product peaks at all, were classified as green, yellow and blue, respectively. Analysis of 1536 wells yielded 323 green, 281 yellow, and 932 blue reactions (Fig. 3 and S4–S7†).
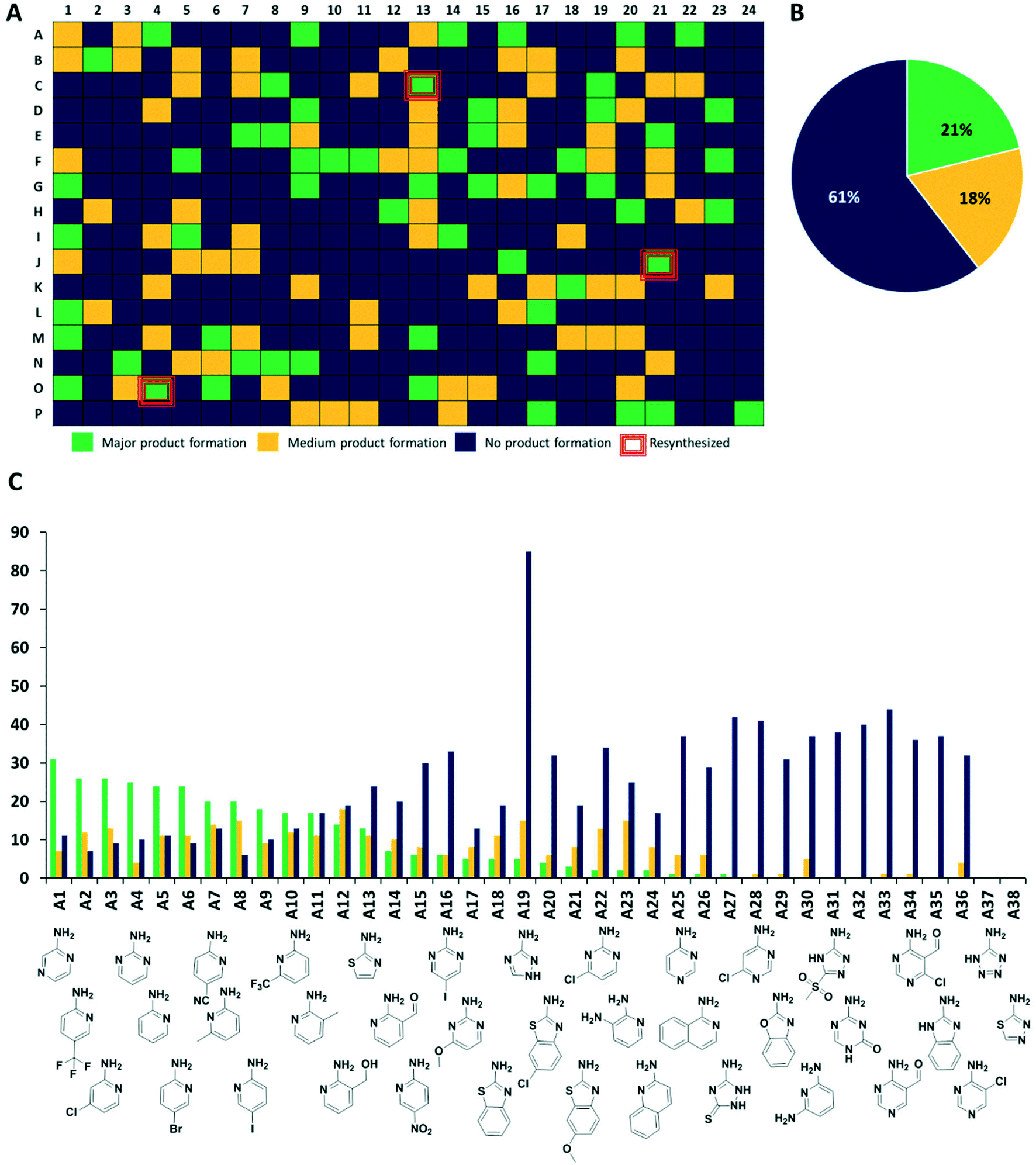 | ||
| Fig. 3 Quality control of the nanomole scale syntheses. (A) Heat map of destination plate I; (B) pie chart of the MS analytic of the crude reaction mixtures of 1536 wells; (C) success-analysis of the cyclic amidine building blocks. | ||
Scalability: from nano to millimole
The synthesis of organic compounds is not always scalable and can pose considerable issues for optimization. This is mostly observed for up- but also for down-scaling. To validate the chemistries and crude analytics, we randomly picked 22 GBB-3CR products, which were previously unknown for resynthesis from the green and yellow categories (Fig. 4). As standard conditions for the reaction, we chose room temperature, 1 M reagents in methanol, and 10 mol% of Sc(OTf)3 as a Lewis acid catalyst on a 1 mmol scale, similar to the miniaturized format.23,26 To avoid the complicated workup of non-volatile solvent, ethylene glycol was replaced by methanol. The GBB-3CR products were obtained in yields of 25–92% (123–462 mg), with an average of 61%.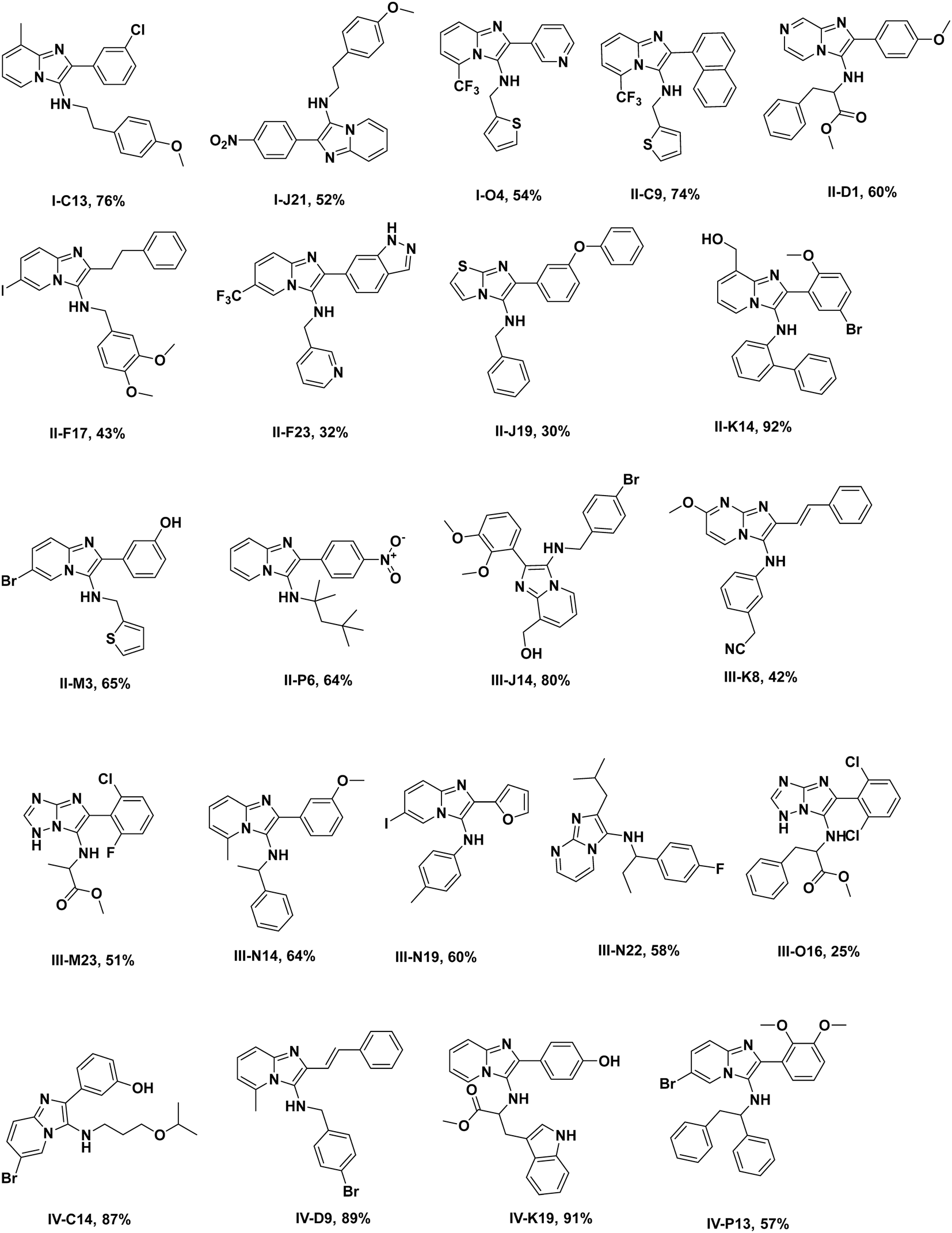 | ||
| Fig. 4 Resynthesized and purified molecules (green and yellow) on a 1 mmol scale, their structural diversity, and determined yields. | ||
HTS by MST
Time and resource-consuming and operationally complex compound purification of screening libraries of new synthetic compounds is a major bottleneck in early drug discovery.27 To test if unpurified reaction mixtures can give useful screening results, we next screened the raw, unpurified GBB-3CR plates for binding to the protein target menin. We used DSF as a fast screening method followed by MST as a cross-validation technique.28The crude reaction mixtures were diluted with DMSO as an initial stock, then further mixed with protein solution at a ratio of 1:9 and screened against menin by DSF at a throughput of ∼190 wells per h (Scheme S1†). Thus a 1536 well plate can be measured in less than 1 day. Hits were then resynthesized, purified, and MST was utilized to cross-validate the binding affinity of the hits. The MST technique relies on the thermophoretic motion of the molecule in a temperature gradient induced by a high precision IR-laser beam.29 Changes in molecular diffusion resulting from binding events provide information on affinity. The validated hits showed binding affinity to the target protein in the μM range (Fig. 5F and S8†).
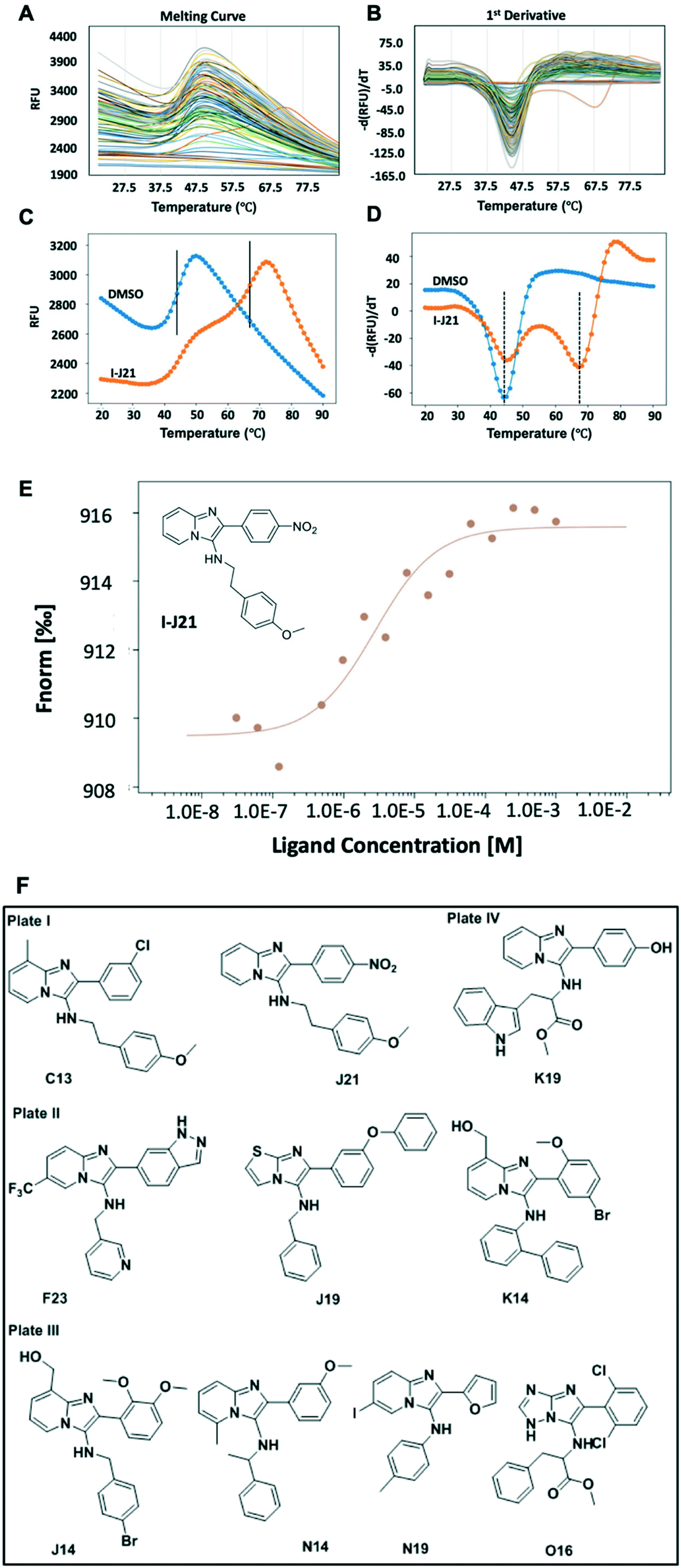 | ||
| Fig. 5 Screening results. (A) Overview of the DSF fluorescence changes against the temperature of one GBB plate including 95 reaction mixtures; (B) first deviation of the fluorescence, the valley indicates the Tm, hits were selected based on the ΔTm compared to the control; (C) compound I-J21 result (orange) compared to the control of DMSO (blue), there clearly show a second peak indicating the stabilizing effect of the compound; (D) the first deviation of I-J21 (orange) compared to DMSO (blue), the dual peaks show two states of protein during denaturing, one indicates the Tm close to the control, the second shows compound helps part of the protein even stable to 67 °C; (E) dose–response binding curve of purified I-J21 to menin; (F) structures of primary hits. | ||
Generally, as ligand binding enhances the thermal stability of target protein, DSF would be the ideal way in which to distinguish the minor difference caused by the presence and absence of ligands.28 As a rapid readout, the first derivative (Fig. 5B) of the experimental fluorescence (Fig. 5A) provides a clear estimate of the Tm, indicated by the minima of the 1st derivative curve. Amongst one of the 96-well plates, while most of the mixtures remain close to the DMSO control, a compound labeled with its position code I-J21 showed a distinct pattern of two different dual peaks in the first derivative (Fig. 5B, orange curve). Comparing with the blank control, it dramatically elevated the Tm from 44 °C to 67 °C (Fig. 5C), even though a fraction of the protein remained the same Tm as in the DMSO control, as revealed in the first derivative curve (Fig. 5D). This biphasic melting phenomenon in thermal shift assay has been described before as an indicator of the presence of two populations caused by under titration of compound binding.30 In a calorimetric study, calcium at various concentration induced bovine α-lactalbumin undergoing a two-state transition, the excess calcium shifts the temperature from 27 °C to 63 °C, coming with a single peak split into dual-peak state.31 The same behavior has also been observed in ligands screening against the human estrogen receptor α ligand-binding domain (ERαLBD). Three compounds displayed significant secondary melting points, which are already known ERα agonists or antagonists with sub-nanomolar EC50.32 Inspired by this obvious hint, we resynthesized and tested the pure compound I-J21, for which the MST subsequently determined a 2.8 μM affinity to menin. Furthermore, I-C13 and III-O16 hits were also confirmed by MST (Fig. S8†).
Structural basis of the interaction of compounds binding to menin
Aiming to understand the structural basis of the interaction between the ligands and menin, we either soaked or co-crystallized menin with the most potent hit compound we found, I-J21. We were able to get the structure by soaking the crystal into a compound solution (Table S1†). Due to previously described properties of the protein construct, we found 3 unstructured loops inside the sequence (residues 54–73, residues 387–397 and residues 520–547), and these flexible segments likely lead the limited resolution of the crystals, which diffracted to a resolution of 3.1 Å. However, the electron density of the GBB-3CR product could be clearly located (PDB: 6S2K) (Fig. 6). The I-J21 molecule binds on top of helix opposite to the deep part of the binding pocket. It undergoes mainly hydrogen bonding interactions with Glu366 and Arg330 through the anisole and nitro group, respectively. The core imidazopyridine heterocycle fits well into the pocket formed by the orthogonal oriented Tyr321 and Tyr325 (Fig. 6B), and performs a T-shaped pi–pi interaction with Tyr 321 as well as with Glu365. The nitro group forms cation–dipole interactions with Arg330. Multiple van der Waals interactions can be observed to Val373, Val369, Glu368, and Arg332. Unfortunately, compound III-O16 could not be co-crystallized with menin; however, a docking model is shown in Fig. 6C. Based on molecular modeling III-O16 is proposed to bind in a deep pocket in menin in between the three α-helices. The core imidazo-1,2,4-triazol of the GBB scaffold is involved in a trifurcated hydrogen bonding network to the Tyr325–OH, Tyr278–OH, and Cys241–SH. Additionally, the methoxyester carbonyl makes a hydrogen bond to the Tyr325. The phenylalanine moiety in the ligand undergoes multiple stacking and T-shaped pi–pi interactions with Tyr321 and Tyr325. Finally, the 2,6-dichlorphenyl moiety is embedded in a narrow pocket and undergoes multiple hydrophobic interactions to Leu179.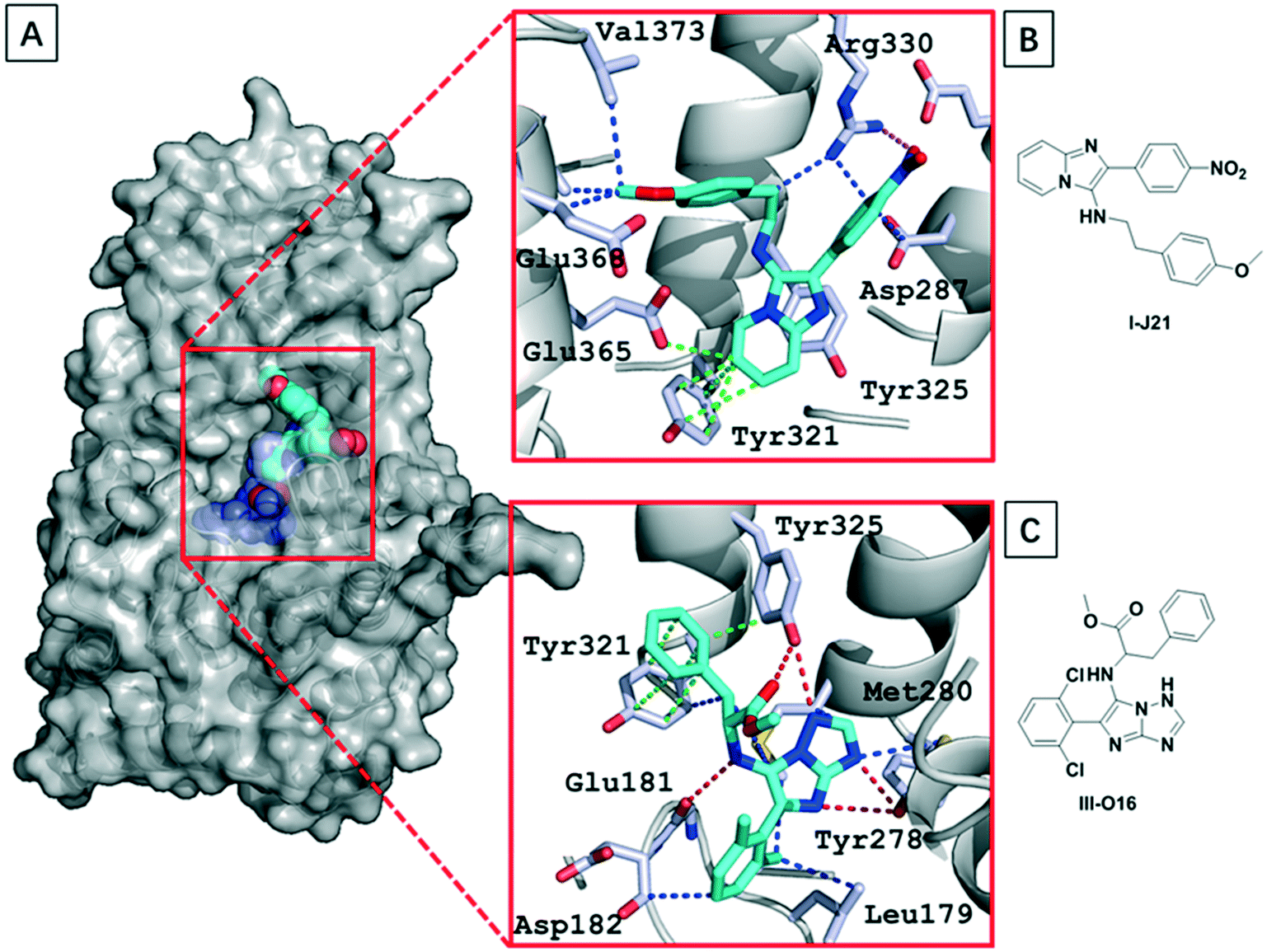 | ||
| Fig. 6 Structural basis of GBB-3CR compounds binding to menin. (A) The co-crystal structure of menin with I-J21 (PDB: 6S2K) and modeled III-O16 (menin is shown in surface representation, I-J21, and III-O16 as spheres); (B) and (C) details of the interaction between I-J21; (B) and III-O16; (C) residues in the receptor (the ligands are shown as cyan sticks, nitrogen and oxygen are shown in blue and red, respectively; hydrogen bondings, hydrogen bonding-pi interactions, pi–pi interactions, and van der Waals interactions are shown as red, blue, orange and green dotted lines, respectively). | ||
Discussion
Synthetic chemistry, as a central science, ultimately serves to discover novel matter to advance humanity.Here, we describe for the first time the automated synthesis of a library of 1536 compounds of heterocycles on a nanomole scale using fast and precise acoustic dispensing technology, followed by an equally fast biophysical screening of the crude library against a protein of therapeutic use. The reader should be very clear that while our approach allows an unprecedented speed of screening – due to the omission of a purification step – there is a significant price to be paid as many designed compounds are inefficiently synthesized. We believe that this issue can be addressed by overpopulating the reaction space. Our present and previous studies demonstrated that large compound libraries could be produced unattended in an automatic and very fast fashion on a nanomole scale, including quality control using mass spectrometry.10,11b,e Screening of the library for the protein–protein interaction menin–MLL using differential scanning fluorometry and microscale thermophoresis yielded several hits. Binding of compound I-J21 was confirmed through crystallization with menin. Although, our biophysical assays are not suitable to proof functional antagonisms, it has to be noted that I-J21 bind in the center of the menin–MLL protein–protein interaction. The progress of our work consists of the automated, miniaturized, and accelerated library synthesis and the screening of the unpurified compounds in the same format yielding in rapid, high-quality hit finding. Unlike the traditional screening process using prepared stored (often historical) libraries, we freshly synthesized a large 1536 compound library ‘on-the-fly’ and screened it using fast DSF and MST. Although the screening hits are far from optimized compounds, our approach shows an alternative process to classical HTS and triaging, followed by lengthy manual medicinal chemistry. Perhaps this presents a stepping stone for a future more sustainable continuous manufacturing and has the potential to greatly reduce the ecological footprint of early drug discovery.
The advantages of our process in the sense of green metrics are multiple. The miniaturized synthesis in our example on a 1536-well format leads to a great reduction of compound mass and waste (reduced E-factor), thus addressing the green principle avoidance. In the classical process of property optimization, compounds are often synthesized on a 0.5 mmol scale and purified, thus leading to the usage of multi kg amounts of precious reagents and solvents and chromatography material. The automation aspect of the synthesis leads to greatly increased safety and less exposure to hazardous fumes. The in situ quality control and biophysical screening of unpurified compounds not only removes the necessity of mass intensive purification but also adds to the green principles of real-time analysis for pollution prevention and inherently safer chemistry for accident prevention. An additional benefit of the herein used multicomponent reaction GBB-3CR is the inherent atom economy.23,33 The well-established convergency of MCRs (three or more building blocks assembly leading to a complex product) leads to a reduced environmental burden and to improved process efficiency through fewer operations. Clearly, all these factors not only contribute to the greenness of the process but also greatly reduced the costs. Based on the expected affinity we could reasonably expect of the fragments (at best ∼10 μM) we decided that a low value of thermal stability increase (0.5°) would reduce our false negative rates – allowing us to identify these initial weakly binding hits. We believe that the subsequent cross-validation by MST allows us to filter out the false positives, but choice of appropriate filters will likely differ with different targets. Equipment costs for the biophysical data collection are high (MST > 100kE, DSF > 10kE), consumable costs are low and such equipment can provide data on many targets. While we have previously published advances in the use of nanosynthesis coupled with direct biophysical screening,11e the herein exemplified miniaturized and accelerated synthesis, analytics, and screening provides the basis to come one step closer to the ultimate goal, a closed loop of automated and accelerated early drug discovery in the sense of continuous manufacturing.5
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Labcyte Inc. for lending an Echo 555 instrument to perform the herein described chemistry and Dominik Zahr for technical support. This work was supported by the National Institute of Health (2R01GM097082-05), the Qatar National Research Foundation (NPRP6-065-3-012), KWF Kankerbestrijding grant (grant agreement No. 10504) and the European Union (ITN “Accelerated Early stage drug dIScovery” AEGIS, grant agreement No. 675555; European Lead Factory (IMI) grant agreement number 115489; COFUNDs ALERT (grant agreement No. 665250) and Prominent (grant agreement No. 754425)). K. G., R. X. and L. G. are grateful for the CSC Fellowships from the Chinese government.References
-
(a) S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203 CrossRef CAS PubMed
; (b) J. A. DiMasi, H. G. Grabowski and R. W. Hansen, J. Health Econ., 2016, 47, 20–33 CrossRef PubMed
- A. T. Plowright, C. Johnstone, J. Kihlberg, J. Pettersson, G. Robb and R. A. Thompson, Drug Discovery Today, 2012, 17, 56–62 CrossRef CAS PubMed
- H. Wong and T. Cernak, Curr. Opin. Green Sustain. Chem., 2018, 11, 91–98 CrossRef
-
H. F. Sneddon, in Green Chemistry Strategies for Drug Discovery, The Royal Society of Chemistry, 2015, pp. 13–38, 10.1039/9781782622659-00013
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC
-
(a) W. Czechtizky, J. Dedio, B. Desai, K. Dixon, E. Farrant, Q. Feng, T. Morgan, D. M. Parry, M. K. Ramjee, C. N. Selway, T. Schmidt, G. J. Tarver and A. G. Wright, ACS Med. Chem. Lett., 2013, 4, 768–772 CrossRef CAS PubMed
-
(a) K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed
- I. Sinclair, G. Davies and H. Semple, Expert Opin. Drug Discovery, 2019, 14, 609–617 CrossRef CAS PubMed
- P. M. Collins, J. T. Ng, R. Talon, K. Nekrosiute, T. Krojer, A. Douangamath, J. Brandao-Neto, N. Wright, N. M. Pearce and F. von Delft, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 246–255 CrossRef CAS PubMed
- S. Shaabani, R. Xu, M. Ahmadianmoghaddam, L. Gao, M. Stahorsky, J. Olechno, R. Ellson, M. Kossenjans, V. Helan and A. Domling, Green Chem., 2019, 21, 225–232 RSC
-
(a) Y. Wang, P. Patil, K. Kurpiewska, J. Kalinowska-Tluscik and A. Domling, Org. Lett., 2019, 21, 3533–3537 CrossRef CAS PubMed
-
(a) Y.-X. Chen, J. Yan, K. Keeshan, A. T. Tubbs, H. Wang, A. Silva, E. J. Brown, J. L. Hess, W. S. Pear and X. Hua, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1018–1023 CrossRef CAS PubMed
-
(a) R. K. Slany, Haematologica, 2009, 94, 984–993 CrossRef CAS PubMed
- J. D. Rowley and H. J. Olney, Genes, Chromosomes Cancer, 2002, 33, 331–345 CrossRef PubMed
-
(a) R. Popovic and N. J. Zeleznik-Le, J. Cell. Biochem., 2005, 95, 234–242 CrossRef CAS PubMed
- B. B. Zeisig, T. Milne, M.-P. García-Cuéllar, S. Schreiner, M.-E. Martin, U. Fuchs, A. Borkhardt, S. K. Chanda, J. Walker, R. Soden, J. L. Hess and R. K. Slany, Mol. Cell. Biol., 2004, 24, 617 CrossRef CAS PubMed
- R. K. Slany, Hematol. Oncol., 2005, 23, 1–9 CrossRef CAS PubMed
-
(a) S. C. Guru, P. K. Goldsmith, A. L. Burns, S. J. Marx, A. M. Spiegel, F. S. Collins and S. C. Chandrasekharappa, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1630 CrossRef CAS PubMed
- T. Cierpicki and J. Grembecka, Future Med. Chem., 2014, 6, 447–462 CrossRef CAS PubMed
-
(a) A. Shi, M. J. Murai, S. He, G. Lund, T. Hartley, T. Purohit, G. Reddy, M. Chruszcz, J. Grembecka and T. Cierpicki, Blood, 2012, 120, 4461–4469 CrossRef CAS PubMed
- A. Shi, M. J. Murai, S. He, G. Lund, T. Hartley, T. Purohit, G. Reddy, M. Chruszcz, J. Grembecka and T. Cierpicki, Blood, 2012, 120, 4461–4469 CrossRef CAS PubMed
- J. Grembecka, S. He, A. Shi, T. Purohit, A. G. Muntean, R. J. Sorenson, H. D. Showalter, M. J. Murai, A. M. Belcher, T. Hartley, J. L. Hess and T. Cierpicki, Nat. Chem. Biol., 2012, 8, 277–284 CrossRef CAS PubMed
- A. Boltjes and A. Dömling, Eur. J. Org. Chem., 2019, 7007–7049 CrossRef CAS PubMed
-
(a) K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 1998, 661–663 CrossRef
- R. Ellson, Drug Discovery Today, 2002, 7, S32–S34 CrossRef
- S. Shaaban and B. F. Abdel-Wahab, Mol. Diversity, 2016, 20, 233–254 CrossRef CAS PubMed
- A. Espada, A. Marin and C. Anta, J. Chromatogr. A, 2004, 1030, 43–51 CrossRef CAS PubMed
- F. H. Niesen, H. Berglund and M. Vedadi, Nat. Protoc., 2007, 2, 2212–2221 CrossRef CAS PubMed
- C. J. Wienken, P. Baaske, U. Rothbauer, D. Braun and S. Duhr, Nat. Commun., 2010, 1, 100 CrossRef PubMed
-
(a) M. Vivoli, H. R. Novak, J. A. Littlechild and N. J. Harmer, J. Visualized Exp., 2014, e51809 Search PubMed
- T. Hendrix, Y. V. Griko and P. L. Privalov, Biophys. Chem., 2000, 84, 27–34 CrossRef CAS PubMed
- K. DeSantis, A. Reed, R. Rahhal and J. Reinking, Nucl. Recept. Signaling, 2012, 10, 1–5 Search PubMed
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00087j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |