
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonobodies as tool biologics for accelerating target validation and druggable site discovery
Padma
Akkapeddi†
 a,
Kai Wen
Teng†‡
a and
Shohei
Koide
*ab
a,
Kai Wen
Teng†‡
a and
Shohei
Koide
*ab
aPerlmutter Cancer Center, New York University Langone Medical Center, New York, NY, USA. E-mail: Shohei.Koide@nyulangone.org
bDepartment of Biochemistry and Molecular Pharmacology, New York University School of Medicine, New York, NY, USA
First published on 13th September 2021
Abstract
Despite increased investment and technological advancement, new drug approvals have not proportionally increased. Low drug approval rates, particularly for new targets, are linked to insufficient target validation at early stages. Thus, there remains a strong need for effective target validation techniques. Here, we review the use of synthetic binding proteins as tools for drug target validation, with focus on the monobody platform among several advanced synthetic binding protein platforms. Monobodies with high affinity and high selectivity can be rapidly developed against challenging targets, such as KRAS mutants, using protein engineering technologies. They have strong tendency to bind to functional sites and thus serve as drug-like molecules, and they can serve as targeting ligands for constructing bio-PROTACs. Genetically encoded monobodies are effective “tool biologics” for validating intracellular targets. They promote crystallization and help reveal the atomic structures of the monobody-target interface, which can inform drug design. Using case studies, we illustrate the potential of the monobody technology in accelerating target validation and small-molecule drug discovery.
 Padma Akkapeddi | Padma Akkapeddi completed her PhD in Medical Biochemistry in 2019, from Instituto de Medicina Molecular, University of Lisbon, Portugal as a Marie Sklodowska-Curie ITN fellow, under the supervision of Dr. Gonçalo Bernardes and Prof. João Barata. She is currently a postdoctoral fellow in Prof. Shohei Koide's lab at Perlmutter Cancer Center, NYU Langone Health, developing highly selective and functional synthetic proteins against oncogenic KRAS mutants and exploring the impact of mutant-selective inhibition on RAS driven cancers. Her major scientific interest includes structure guided protein engineering to facilitate targeted delivery of biologics to the brain and towards intracellular targets. |
 Kai Wen Teng | Kai Wen Teng received his PhD in Biophysics from the University of Illinois at Urbana Champaign. After graduation, he pursued postdoctoral training in protein engineering and biologics design under the guidance of Dr. Shohei Koide at NYU Langone Health. For his postdoctoral project, Kai Wen focused on engineering synthetic binders that selectively inhibit and degrade KRAS mutants. |
 Shohei Koide | Shohei Koide, PhD is a synthetic protein scientist. His research integrates mechanistic biochemistry and directed evolution to create highly functional but still simple proteins. He is the inventor of the FN3 Monobody technology, and he has made important contributions to synthetic antibody technologies and to the application of synthetic binding proteins to biology, chemistry and medicine. He is Professor of Biochemistry and Molecular Pharmacology at NYU School of Medicine. Previously he was Professor at the University of Chicago and at the University of Rochester School of Medicine and Dentistry. |
Introduction
Drug development is an expensive, time consuming yet important endeavor to address an unmet medical need. Drug discovery starts with the initial research, often in academia1,2 that generates a hypothesis that modulation, i.e., inhibition or activation, of a biological function leads to a therapeutic effect. The outcome of the initial research leads to the identification of a potential drug target, i.e., a macromolecule involved in the biological function. Still further validation is required before the project proceeds to target-based lead discovery. Target-based drug discovery has become the dominant paradigm, but, despite the advances in the mechanistic understanding of the disease biology and in target-based drug discovery, it remains challenging to bring a new drug into the market.3,4The overall failure rate in drug development is >96%, including a 90% failure rate during clinical development5–10 and the failure rates are the highest for drugs against previously ‘undrugged’ targets. A number of factors contribute to this high rate of failure, including a lack of preclinical experiments in cells, tissues, and animal models to support drug target validation. Insufficient validation of drug targets at an early stage has been linked to costly clinical failures11 and low drug approval rates.6,12 Effective target validation as well as early proof-of-concept studies have been predicted to reduce the phase II clinical trial failures by ∼24%, thereby lowering the cost of development of new molecular entities by ∼30%.5 Consequently, there is a clear unmet need for robust technologies for drug target validation.
The discovery and development of first-in-class drugs often begins with identification of a new drug target (Fig. 1a). Fortunately, there are many powerful technologies for target identification. Omics technologies have brought about unprecedented capacities to screen biological samples at the levels of gene, transcript, protein and their interaction network in a high-throughput manner.13 Although robust and efficient, an omics study often results in a vast number of hits for a disease of interest. Efficient prioritization of hits at an early stage data is a key to a successful drug discovery project.
 | ||
| Fig. 1 (a) Schematic representation of major steps in the drug discovery process highlighting the time it takes in the conventional approach and how monobody assisted approaches could facilitate target validation. We propose that an addition of a target validation step with synthetic binding proteins such as monobodies helps address the efficacy question early in the project. (b) The three-dimensional structures of common protein scaffolds for developing binding proteins. The yellow spheres indicate positions that are diversified in combinatorial libraries and thus likely to be involved in target binding. (c) Schematic representation of main processes of target validation utilizing monobodies. | ||
In this article, we define drug target validation as the evaluation of the following question: whether pharmacological modulation of the target can provide therapeutic benefit with an acceptable safety window (i.e., a substantial therapeutic window). A wide spectrum of validation methods involves genetic modulation of a target of interest or biochemical perturbation, where a reagent binds to a target and modulates its function, in cell and animal models. Ideally, these perturbations should have a mode of action close to that of the ultimate drug so that the outcomes of perturbation studies have high predictive power for the efficacy and adverse effects of the drug in patients.
Genetic manipulation is a powerful technique for target validation and it leverages the ease of designing and producing nucleic-acid reagents for selective targeting of a region of interest and of performing large-scale experiments.14 Examples include loss-of-function experiments utilizing genetic knockout and knockdown approaches ultimately resulting in reductions of protein levels. In addition to the conventional RNA interference, recent genome editing methods, particularly CRISPR/Cas9, have accelerated and expanded these approaches.15–25 New CRISPR/Cas9 variations can also increase the protein abundance, in addition to reducing the protein abundance and causing a loss of function. However, these approaches alter the abundance of the target, which is not equivalent to functional inhibition conferred by the binding of a drug to a specific site within the target (see below for an important exception with degrader drugs). Genetically modifying a target, such as site-directed mutations, could offer higher-resolution information than altering the target abundance, but substantially more prior knowledge is required to design such mutations and still mutations rarely mimic the action of small molecule therapeutics. In addition, it is difficult to predict the final biological outcome due to post-transcriptional and post-translational modifications using genetic approaches. For example, many genes produce multiple isoforms that a single genetic manipulation can perturb and hence it is difficult to determine which isoforms are valid drug targets. Genetic modifications are often irreversible, and it does not accurately mimic temporal modulation of biological functions by a drug. Taken together, whereas genetic modification is powerful as the initial screening tools, higher-resolution tools that selectively target a particular region (e.g., a domain and a site) within a target molecule are needed for high-confidence target validation.
Small molecule chemical probes, compounds that bind to the target of interest with sufficient selectivity and potency, are powerful tools for target validation, including the feasibility of perturbing the target to modulate downstream consequences and predicting adverse effects. Chemical probes are developed using techniques such as high-throughput screening (HTS), fragment-based drug discovery (FBDD) and newer technologies. HTS has remained a pillar of small molecule drug discovery with relatively high success rates of delivering hits that make it to the clinic.64–66 It generally takes a long time to develop a tool compound with sufficient selectivity and affinity for in-cell and in-animal validation. Essentially one needs to develop a compound that is almost a drug to be able to validate a target, which requires substantial investment of time and resources (Fig. 1a).
As a variation of small molecule probes, proteolysis targeting chimeras (PROTACs) are increasingly being developed as drugs to degrade protein of interest and also used as chemical tools to validate therapeutic targets.67,68 PROTACs are heterobifunctional compounds that recruit the E3 ubiquitin ligase machinery to the target protein, resulting in their ubiquitination and subsequent proteasomal degradation. PROTACs function in an event-driven fashion compared to the traditional inhibitors that function via the occupancy-driven paradigm and as a result PROTACs overcome the challenge of maintaining high target engagement for effective inhibition. The mode of action of PROTACs is conceptually related to the reduction of protein abundance using genetic manipulation, which increases the relevance of genetic manipulation in target validation. However, PROTACs still require a highly selective ligand for target engagement and thus suffers from the same bottleneck as in chemical probe development.
In this review, we describe an emerging approach in which synthetic binding proteins are used as “tool biologics” for target identification and validation, with a particular emphasis on the monobody technology (Fig. 1). These molecules bind to a specific surface of a target of interest and biochemically perturb its function, thus mimicking the mode of action of small molecule drugs. Using advanced protein engineering technologies, synthetic binding proteins with exquisite selectivity and high potency can be rapidly developed. Genetically encoded synthetic binding proteins can be delivered to the cell and organ of interest and their levels can be temporally controlled with the convenience and efficiency of genetic manipulation. Over the last three decades, a number of studies have demonstrated the potential of this concept (Table 1). We propose that synthetic binding proteins, such as monobodies, are powerful technologies that bridge the gap between genetic modification and chemical probes and hence they will have substantial impact on drug discovery (Fig. 1a).
| Binding protein platform | Target |
|---|---|
| Peptide-aptamer | CDK2 (ref. 26) |
| HRAS27 | |
| Rho-GEF28 | |
| Affibody | HRAS/Raf-1 (ref. 29–31) |
| Nanobody | PKCε32 |
| F-Actin33 | |
| α-Synuclein34 | |
| p53 (ref. 35) | |
| GPCR36–38 | |
| scFv (Intrabody) | RAS39 |
| HHV-8/IL-6 (ref. 40) | |
| pYSTAT3 (ref. 41) | |
| HP-1β42 | |
| BCR-ABL43 | |
| DARPin | ERK44 |
| Tubulin caps45 | |
| KRAS46–48 | |
| c-Jun N-terminal kinase49,50 | |
| Monobody | Abl-Kinase51,52 |
| SHP2 (ref. 53) | |
| PRDM14-MTGR1 (ref. 54) | |
| sNASP55 | |
| HRAS and KRAS56 | |
| WDR5 (ref. 57) | |
| ICMT58 | |
| Aurora kinase A59 | |
| MLKL60,61 | |
| STAT3 (ref. 62) | |
| KRAS(G12C) and KRAS(G12V)63 |
Monobodies and other synthetic binding proteins
Synthetic binding proteins are proteins that are tailored to bind to a target of interest. Development of synthetic binding proteins has been inspired by the capability of the immune system to generate antibodies against virtually any antigen and successes of monoclonal antibodies as transformative therapeutics. It has also been motivated by the desire to overcome shortcomings of the immunoglobulin framework of the conventional antibodies including its complex molecular architecture and the dependence of its folding on the formation of multiple disulfide bonds. Advances in the knowledge of the mechanisms governing protein–protein interaction and in protein engineering technology over the last three decades have enabled the generation of synthetic binding proteins whose affinity and selectivity match those of therapeutic antibodies.69–77Synthetic binding proteins are mostly generated by varying portions of a stable protein scaffold (Fig. 1b).77 Using molecular display technologies, such as phage display, yeast display, ribosome display and mRNA display, and tailored oligonucleotide synthesis methods such as trimer nucleotides and massively parallel synthesis, one can construct a large combinatorial library of synthetic binding proteins in which typically 10–20 residues are diversified.78–82 Modern libraries contain 1010–1013 independent sequences, and the effective library size can be readily expanded by the incorporation of genetic recombination and random mutagenesis steps. Clones that bind to the target of interest with desired affinity and specificity are identified by subjecting a library to selection and screening. Many potent and exquisitely selective binding proteins have been rapidly developed, typically within a few months, using multiple scaffold systems, underscoring the fundamental ability of larger interfaces afforded by a scaffold protein to achieve high functionality.
Peptide aptamers are short peptides embedded within a small protein scaffold. They can be viewed as pioneers of synthetic binding proteins particularly for intracellular applications.26,83,84 As one might expect, their simple architecture does not often lead to high affinity or high specificity and the flexible linkage between the peptide segment and the scaffold makes it extremely challenging to determine the crystal structures of the complexes. Consequently, peptide aptamers have largely been superseded by systems discussed below.
Among numerous synthetic binding protein platforms, the most established systems include designed ankyrin repeat proteins (DARPins),72,85–87 monobodies,88,89 anticalins,90–92 and affibodies.93–95 Numerous binding proteins, or “binders”, have been derived from “natural” scaffolds of antibody fragments such as single-chain variable fragment (scFv) and nanobodies.96–101 The distinction between synthetic and natural binders is subtle in that the latter originate from natural immune repertoires rather than synthetic libraries. Similar technologies are used for both types in clone identification, characterization and production. Successes of these and other scaffold systems in generating potent binding proteins demonstrate that the field has established robust knowledge and technologies for synthetic binding protein development. For this review we will not elaborate further on these and many other binding protein systems, because there are already many excellent reviews on this topic.72,92,95,100,102 As one might expect, the major interest of synthetic binding protein developers is in therapeutics development, and drug target validation is a relatively unexplored application of synthetic binding proteins.
Monobodies are the founding system of synthetic binding proteins based on a fibronectin type III (FN3) domain that has the immunoglobulin (Ig) fold.103 A typical monobody development process involves enriching clones that bind to the target of interest and optionally negative selection for eliminating clones that cross-react with an undesirable molecule (e.g., a homologous protein).104 In the end, the lead clones are chosen from many candidates in terms of affinity, specificity and biophysical properties. Ultimately monobodies are expressed in E. coli for biochemical and structural studies, and alternatively converted into genetically encoded reagents, i.e., expression vectors, for in-cell and in vivo studies (Fig. 1c).57,62 FN3 is monomeric and stable but unlike conventional Ig domains it has no disulfide bonds.103 The lack of disulfide bonds makes it straightforward to express fully functional monobodies under the reducing environment within the cell. Along with monobodies, scaffolds such as DARPins and affibodies also do not have disulfide bonds, and have been used for targeting intracellular proteins of interest.29–31,44,46,47,50,93,105–107 The absence of a disulfide bond is clearly desirable but not an absolute requirement for intracellular applications. The so-called intrabodies have been constructed using the scFv,108,109 VL,110 and nanobody111,112 frameworks that retain functionality in the intracellular reducing environment.108,113 These examples (Table 1) clearly show that, in principle, these and other scaffold systems can be used to generate binders suitable for intracellular applications.
The robust scaffold of monobodies also makes it compatible with diverse fusion partners (e.g., epitope tags and fluorescent proteins) and with virtually all molecular display technologies.88,104,114 Monobodies have strong tendency to bind to a functional surface on the target protein,88 and consequently, monobodies identified simply based on target binding are usually modulators of biological function (Fig. 2). There are distinct types of monobody library designs developed to date, one that diversifies residues in loops at one end of the scaffold and the other that diversifies residues on a beta-sheet in addition to loop residues (Fig. 1b). These libraries present diversified positions with distinct surface topography and thus have distinct preference of binding towards surfaces of the target. Monobodies from the “loop” library prefer concave surfaces, whereas those from the “side” library prefer flatter and convex surfaces.88 Together, monobodies binding to diverse surfaces of a target can be developed88,89 resulting in identification of potential druggable sites with diverse topography (Fig. 2).
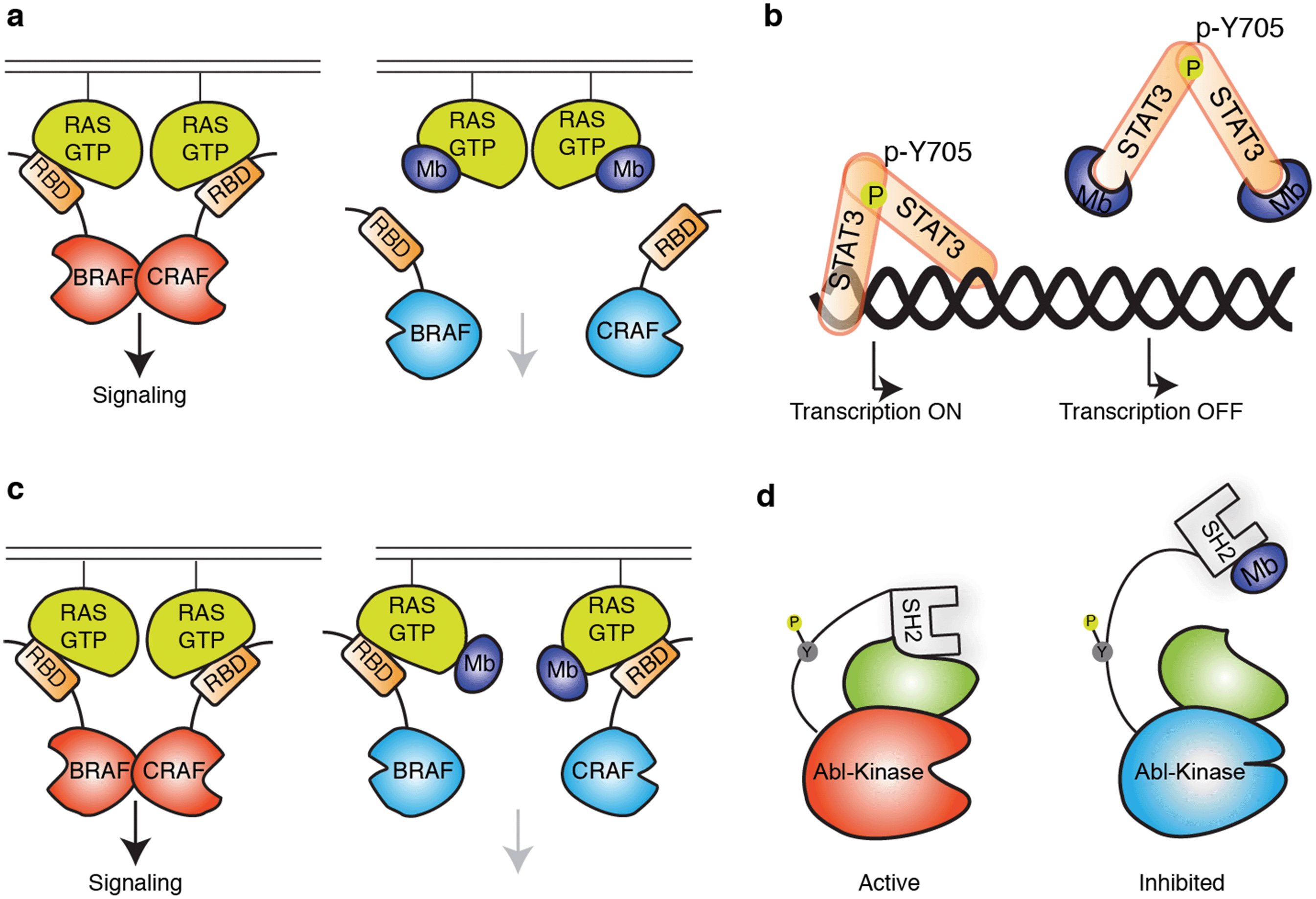 | ||
| Fig. 2 Schematics showing modes of regulation of target functions using synthetic binding proteins. (a and b) Competitive disruption of protein–ligand interaction with a monobody (Mb). (a) Competitive inhibition of the interaction between the active form of RAS mutant and the RAS-binding domain (RBD), which in turn inhibits the downstream signalling.63 (b) Inhibition of transcriptional activity of STAT3 by monobody. Monobody binds to STAT3 thus decreasing STAT3 binding to DNA.62 (c and d) Allosteric regulation of the target protein. (c) Monobody binding to a self-association interface of RAS, disrupting the RAS–RAF hetero-tetramer and inhibiting RAS-mediated activation of RAF.56 (d) Monobody binding to the SH2 domain of ABL kinase disrupts the intramolecular, domain–domain interaction, leading to the inhibition of kinase activity.51 | ||
Complementing synthetic proteins, cyclic peptides, usually composed of 5 to 14 amino acids, are alternative synthetic binding modalities. Details on application of cyclic peptides in drug discovery is beyond the scope of this article and the reader is directed to many excellent review articles on this topic.115–117 The moderate size and diverse functional groups of the cyclic peptides ensure the contact is large enough to provide high selectivity, added to that the cyclization helps resist degradation by proteases in blood.115 Similar to synthetic binding proteins, it is often challenging to deliver cyclic peptides into the cytoplasm of the cells,118 but, unlike synthetic binding proteins, most cyclic peptides cannot be produced genetically, further limiting their utility for the validation of intracellular targets.
For the remainder of this review we present applications of monobodies as “tool biologics” towards drug target validation.
The use of monobody technology for facilitating drug discovery
The monobody technology enables the rapid generation of highly specific and potent inhibitors against difficult targets. Therefore, it can provide valuable information during each step of the drug discovery process, complementing the drug discovery effort (Fig. 1a). In this section we will illustrate how the use of monobody technology has facilitated various steps of drug discovery against challenging targets.Monobody in drug target validation
Many potential drug targets that were discovered in basic research are not validated due to a lack of suitable tool compounds that can act as target-specific inhibitors. The monobody technology can help bridge this gap by offering a quick path for generating selective inhibitors for a given target and validate the target as druggable by functional readouts. Furthermore, the levels of specificity and selectivity that the monobodies can achieve have allowed it to be used as a tool for validating targets even in a situation where the biology of the target is poorly understood. Other synthetic binding protein platforms could, in principle, be employed in a similar manner.In one example, the monobody technology helped advance the understanding of a poorly characterized and unvalidated drug target. Members of G-protein coupled receptor (GPCR) super-family are important drug targets because of their roles in development and signaling regulation in both healthy and diseased cells.119,120 In particular, adhesion GPCRs (aGPCRs) are difficult to target due to their complex structures and relatively poorly characterized biological roles.121 A substantial fraction of pharmaceutical agents currently on the market target GPCR. However, to date, there has yet to be a clinically approved drug that targets an aGPCR. Only recently have small molecule ligands for aGPCRs been identified122–124 and tool reagents such as antibodies against aGPCRs are limited. In addition to the seven-transmembrane helix domain characteristic of GPCRs, aGPCRs have a large, multi-domain extracellular region (ECR) whose roles in signaling remain poorly understood. The lack of well-characterized, soluble ligands to aGPCR makes it difficult to evaluate whether it is a viable drug target, which calls for the ongoing development of research tools that can dissect the functional consequences of targeting aGPCR and its individual extracellular domains.
A series of monobodies to an aGPCR (GPR56/ADGRG1) of mouse and human were generated using the monobody technology targeting different parts of the ECR (Fig. 3a).125 A monobody was used to facilitate the determination of the crystal structure of the mouse GPR56 ECR, which revealed two domains: a novel N-terminal pentraxin and laminin/neurexin/sex hormone-binding globulin-like (PLL) domain and an unusually short G protein-coupled receptor autoproteolysis-inducing (GAIN) domain. Using the knowledge of the domain architecture, more monobodies targeting the PLL domain, the GAIN domain, and an epitope that included both domains were developed. These monobodies became valuable reagents for detecting splice variants of aGPCRs. Importantly, this panel of distinct monobodies enabled the authors to elucidate signaling implications for targeting specific extracellular domains of GPR56. A monobody that interacted with both the PLL and GAIN domains resulted in a decrease in GPR56 signaling, leading to a model in which a rigid ECR may decrease basal activity of the receptor. In contrast, some PLL domain-binding monobodies acted as agonists. Lastly, none of the GAIN-domain-binding monobodies caused significant changes in GPR56 signaling. These studies have established that aGPCR signaling can be modulated with a soluble ligand targeting the ECR and that targeting of specific extracellular domains in an aGPCR can lead to different downstream signaling outcomes. As aGPCR ECRs are extracellularly accessible, these monobodies are potentially new therapeutics.
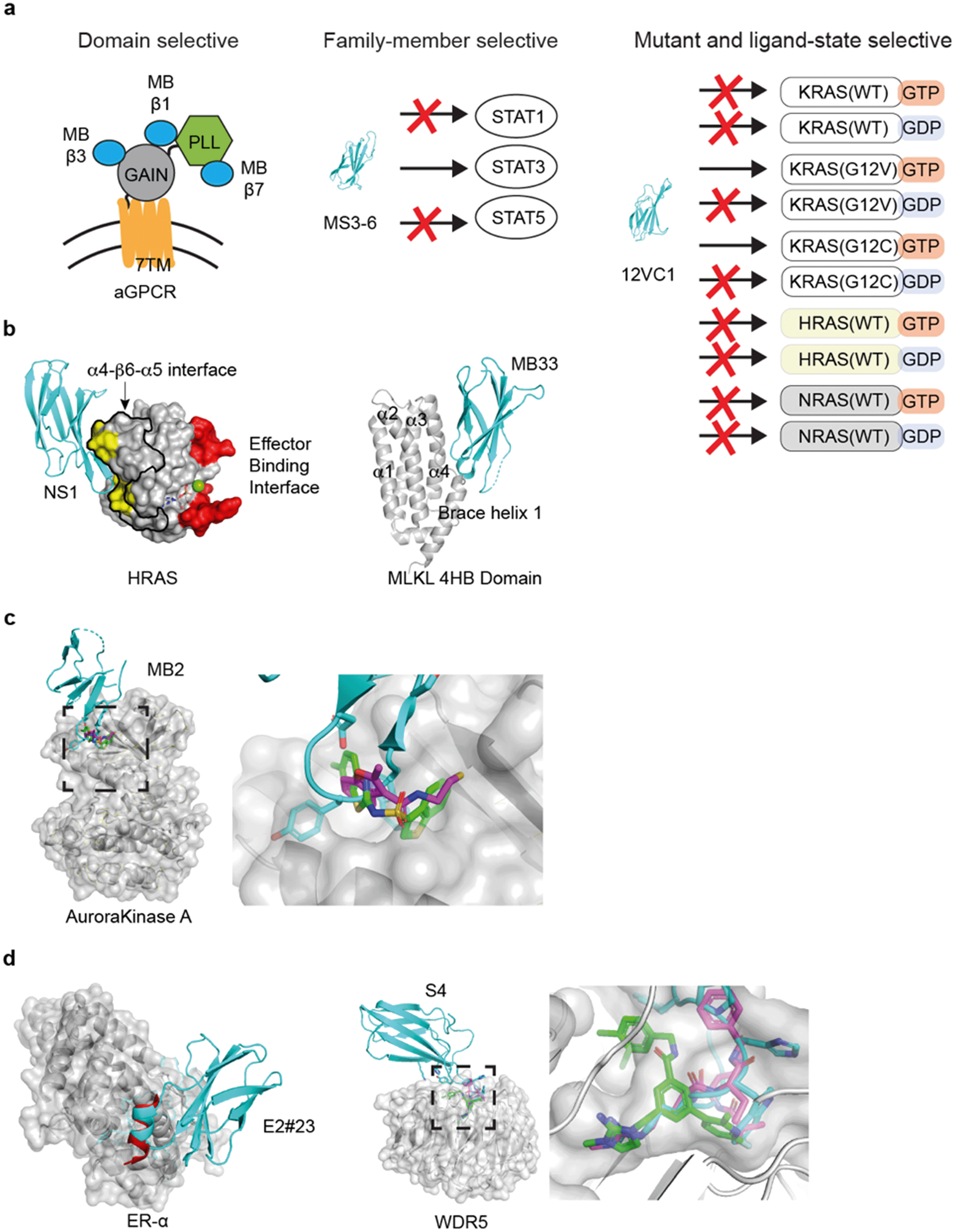 | ||
| Fig. 3 Applications of the monobody technology for facilitating drug discovery. (a) Exquisite selectivity of monobodies facilitates drug target validation and discovery of druggable sites. A panel of monobodies (teal) was developed for inhibiting aGPCR and dissecting the functional role of individual extracellular domains (left). A monobody selectively recognizes STAT3 over other STAT family members. Monobody 12VC1 selectively inhibits RAS mutants, G12C and G12V, over other WT RAS isoforms. (b) Monobody-assisted discovery of new targetable interfaces for inhibition (PDB ID: 5E95).56 The NS1 monobody inhibited RAS by binding (yellow) to the α4–β6–α5 interface (enclosed with the black boundary) that is important for RAS: RAS association, which is away from the effector binding interface (red). Structure of monobody Mb33 bound to MLKL 4HB domain (PDB ID: 6UX8).60 (c) Monobody MB2 (PDB ID: 6C83, teal)59 allosterically inhibited AurA by targeting the pocket. The equivalent pocket of a homologous kinase, PDK1, is targeted by small molecules inhibitors (PDB ID: 3ORX, magenta, PDB ID: 4RQK, green)146,147 (d) A peptide inhibitor (PDB ID: 5WGQ, red)148 that binds to ER-α closely resembles the FG loop of a monobody (PDB ID: 2OCF, teal).149 Small molecule (PDB ID: 6E23, green)150 and peptide inhibitor (PDB ID: 5VFC magenta)151 that targets WDR5 closely resembles the FG loop of a WDR5-binding monobody (PDB ID: 6BYN, teal).57 | ||
One of the major class of undruggable proteome is the transcription factors.126,127 Although numerous studies have highlighted the role of transcriptional regulation in human disease, it has been challenging to validate transcription factors as drug targets. A loss of function upon genetic silencing of transcription factors may not be sufficient for validating them as drug targets. Such an outcome may be due to the disruption of a protein complex, which may not be recapitulated with a small molecule drug. Furthermore, some transcription factors belong to a family of proteins with high sequence homology, making it challenging to selectively target one particular family member for target validation. Many monobodies with exceptional selectivity have been developed, including a monobody that is selective to a single SH2 domain among >100 human SH2 domains.53 Furthermore, monobody is an ideal reagent for validating nuclear targets because of its compact size that allows it to pass through the nuclear pores and reach its target inside the nucleus.
STAT3 is an example of a transcription factors that have so far been undruggable,128 and activation of STAT3 is frequently found in solid tumors and hematopoietic diseases.129,130 The main challenges in targeting STAT3 include the absence of an obvious pocket for inhibiting STAT3 monomer and the high sequence similarity among STAT-family member proteins. Although there has been reports of inhibitors that prevent the dimerization of STAT3, which is required for activation, the STAT3 monomer has been reported to have residual signaling activity, suggesting the need to inhibit the monomer.131 Recently, a series of monobodies collectively termed MS3 that bind STAT3 were developed (Fig. 3a).62 They bind to STAT3 with affinity in the mid-nanomolar range, and their high selectivity for STAT3 over other STAT-family member proteins were confirmed by affinity capture followed by mass spectrometry. Expression of MS3 monobodies in cells inhibited the transcription of STAT3 target genes. In particular, one of these monobodies, MS3–6, binds to the coiled-coil domain of STAT3 and inhibits STAT3 signaling through multiple fronts. First, it allosterically inhibited the binding of STAT3 to DNA by stabilizing a coiled-coil conformation of STAT3 that is incompatible with its binding to DNA (Fig. 2b). Second, it binds to a region of STAT3 proximal to the nuclear localization sequence (NLS), which inhibited translocation of STAT3 into the nucleus. Third, it reduces IL-22 signaling by perturbing binding of STAT3 to the IL-22 receptor. Taken together, this study has generated a powerful tool to study the functional consequences of inhibiting STAT3, and demonstrated that STAT3 can be inhibited by a variety of mechanisms.
In another study, a monobody termed S4 was developed to bind to WDR5,57 a nuclear protein that is a part of the mixed lineage leukemia 1 (MLL1) transferase complex.132 A crystal structure revealed that monobody S4, just like the small molecule inhibitors, binds to the central cavity (WIN site) of WDR5 in which WIN-motif of MLL family protein binds and disrupts the formation of MLL1–WDR5 complex. MLL1 plays an important role in MLL1-rearrange leukemia. However, the efficacy of current WDR5 inhibitors has only been demonstrated in cells but not in animal models due to their low bio-availability. Leukemia cell lines with genetically encoded S4 under a DHFR-degron based inducible system was generated for studying the functional impact of WDR5 inhibition. In this system, the intracellular concentration of monobody is controlled with trimethoprim (TMP), which stabilizes the DHFR-monobody fusion that is otherwise rapidly degraded. In the absence of TMP, DFHR-monobody was degraded. The cells were then engrafted in mice to examine whether the approach of inhibiting WDR5 is effective in vivo. Expression of S4 potently decreases Hox4A mRNA level, a signature of MLL1’s transcriptional activity. Cellular effects of S4 were similar to those of small molecule inhibitors, supporting the use of monobodies for target validation. In a mouse model, the expression of monobody S4 significantly improved the survival of mouse engrafted with leukemia cells, establishing the on-tissue efficacy of targeting the WIN site of WDR5 and validating the site as a therapeutic target. Through these examples, we can perceive monobody being a useful tool for validating difficult to drug targets, and a significant improvement over genetically based verification.
Discovering new druggable sites and new modes of inhibition
A monobody selection campaign often yields clones that bind to multiple, different epitopes within a target. Because monobodies are good crystallization chaperones,88,89,133 there is a high probability that several different structures of monobodies bound to a drug target can be readily determined, which can reveal new potential druggable sites. The monobody bound structures also reveal druggable conformations of a target, establishing the feasibility of targeting an epitope with a non-covalent ligand.RAS has been known as a potential drug target for more than 30 years.134,135 RAS mutation is a key driver in estimated 19% of human cancer,136 and currently there are no clinical-approved inhibitors directly targeting RAS. RAS has long earned its reputation as an undruggable target due to its lack of surface pocket and its sole ligand binding pocket has picomolar affinity to nucleotides. Over the last decade, there have been significant breakthroughs in targeting the G12C mutation of RAS by using covalent inhibitor,137–139 and multiple drugs are being evaluated for the treatment of lung and colorectal cancers in clinical trials. These breakthroughs have validated RAS as a druggable target and invigorated RAS-targeted drug discovery. However, the G12C mutation is a small subset of oncogenic mutations found in RAS, therefore it calls for new approaches to non-covalently inhibit RAS function.
The feasibility of targeting RAS directly with a non-covalent inhibitor has been a major unanswered question in the field of RAS drug discovery. Recently, the monobody technology has been used to address this question on several fronts.56,63 A monobody termed 12VC1 was developed that selectively target the oncogenic mutant forms of RAS, G12V and G12C, in the active, GTP-bound state, and it does not bind to any of the wildtype RAS isoforms in either active or inactive states (Fig. 3a). Binding of 12VC1 to the active RAS mutants competitively inhibits RAS from interacting with its downstream effector RAFs (Fig. 2a). Fusion constructs of 12VC1 with fluorescent proteins readily enabled the confirmation of its selective engagement with the RAS mutants in cells.63 The unprecedented level of selectivity of 12VC1 demonstrated that RAS mutant can be selectively targeted in a non-covalent manner. When expressed as an intracellular biologic, 12VC1 is effective in inhibiting RAS mediated signaling and proliferation in RAS-driven cancer cell lines and a mouse xenograft model. This monobody has established the feasibility of selectively inhibiting a RAS mutant using non-covalent means, as well as validating active mutants of RAS as a drug target, which significantly de-risk approaches that aim to develop non-covalent inhibitors against active RAS mutants.
The study also provided structural insights as to how RAS mutants G12V and G12C can be selectively targeted. There is a shallow pocket on the surface of monobody 12VC1 that directly recognizes certain small side chains at residue 12 of RAS. This finding provides a strategy for developing future inhibitors against RAS mutants or “undruggable” mutants alike, where a mutant protein that lacks a suitable druggable pocket can be recognize by a molecule containing a pocket that recognizes the mutation. For example, the structure may guide the design of “molecular glue” compounds.140
Effort to inhibit challenging targets such as RAS can benefit from having more than one mode of inhibition. This view called for the exploration of novel interfaces on RAS that can potentially be targeted for inhibiting its activity. A monobody termed NS1 has been developed that targets the α4–β6–α5 interface (a.a. residues 123–168) on RAS,56 which is away from the effector binding surface of RAS (Fig. 3b). NS1 show potent inhibition of RAS-mediated signaling and proliferation of RAS-driven cancer cells in both cell-based and animal models.141 The binding of NS1 to RAS blocked RAS–RAS association as well as CRAF–BRAF heterodimerization (Fig. 2c). The functional importance of the α4–β6–α5 interface had previously been unrecognized prior to this work, primarily due to a lack of a tool compound directed to this interface. The discovery of NS1 and its binding site has provided a new interface to target for inhibiting RAS function and inspired many follow-on studies.141,142
Interestingly, the potent inhibitory activities of NS1 has not been recapitulated by point mutations that are designed to disrupt the proposed mode of RAS self association.56 This discrepancy suggests that the size of monobody (10 kDa) may contribute to its efficacy. If this is the case, it may be challenging to translate the findings into small molecule inhibitors. Further studies are needed to better define the effect of the inhibitor size targeting this region of RAS.
Alternative modes of inhibition have also been explored with Bcr–Abl kinase, a drug target in myelogenous leukemia using the monobody technology.51,143 The study was motivated by the observation that small molecule drugs that bind the ATP-binding site of the kinase domain are prone to escape mutations and that the SH2 domain of Abl serves as an intramolecular allosteric activator of the kinase activity.143,144 Monobodies termed AS25 and AS27 were developed to target the SH2 domain on a surface that interacts with the kinase domain (Fig. 2d) and inhibited the kinase activity.51 These results have validated the kinase binding surface of SH2 as a druggable site of Bcr–Abl.
In another recent example, monobody has been used successfully for identifying a new interface for inhibiting mixed lineage kinase domain-like (MLKL).60 Phosphorylation of MLKL, a necrotic effector pseudokinase, is a critical step in the activation of necroptosis cell death pathway.145 The mis-regulation of necroptosis pathway can lead to inflammatory diseases. In the activation of necroptosis pathway, MLKL is phosphorylated by a protein complex involving RIPK1 and RIPK3 kinases, followed by oligomerization and membrane translocation, which leads to an eventual permeabilization of the cell membrane. The molecular interface of MLKL that are responsible for these signaling events and whether inhibition of any of these individual steps can block necroptosis are still poorly understood. The authors generated monobodies that bind to MLKL, which blocked ligand-induced necroptosis.60 Monobody, termed Mb33, inhibits necroptosis and blocks the translocation of MLKL to the cell membrane, but it does not block the oligomerization of MLKL. Thus, it helped define the order of these events. The crystal structure shows that this monobody binds to a previously uncharacterized surface, the α4 helix of the four-helical bundle domain (Fig. 3b). Mutation studies confirmed the action of Mb33 and validated the importance of its binding interface. In a follow-up study, the authors identified two additional monobodies that bind to two distinct conformations of MLKL during its activation.61 These monobodies helped dissect the conformation change of MLKL following its phosphorylation by RIPK3, shedding new light on how MLKL can be selectively targeted for controlling multiple events in the process of necroptosis.
Monobody as a starting point for drug design
The case studies above have demonstrated that monobodies are powerful reagents for validating potential drug target and discovering new druggable sites. The molecular interaction posed by the “loop” regions of monobodies, revealed by crystal structures of monobody-target complexes, have been recapitulated by other drug modalities, including small molecule compound and peptides in the following case studies.There have been intense efforts to develop compounds that allosterically control kinases, because of the difficulty in directly targeting the substrate binding site of a specific kinase.146,152 From a total of 84 monobodies binding to areas overlapping the site for an natural allosteric activator of Aurora Kinase A (AurA), monobody termed Mb2 was discovered to allosterically inhibit, rather than activate, the kinase activity.59 The crystal structure of Mb2 bound to AurA revealed that Mb2 binds to a hydrophobic pocket near αC-helix of AurA and stabilizes an inactive conformation. In particular, the placement of a tyrosine residue of the monobody in this pocket closely resembles that of small molecule inhibitors of a homologous kinase, PDK1, that was discovered using disulfide tethering and fragment-based screening,146 suggesting the potential to use the monobody structure as a starting point for designing ligand that target this region of AurA (Fig. 3c).
As described above, the monobody libraries currently utilize two designs (Fig. 1b).104 The FG loop of the monobody is the longest and the most flexible among the loops of the monobody scaffold, and it is the most extensively diversified region in these libraries. Therefore, it is probably not surprising that some monobodies are capable of binding to a target utilizing only the FG loop. Recently, a cyclic peptide was computationally designed from an antibody loop, which demonstrated the feasibility of generating a much more compact entity that would recapitulate the same binding interface as a single loop binder.153 This potential was implicated as early as 2001 from the development of a monobody that binds to Estrogen receptor alpha (ER-α).149 This monobody inhibited the activity of ER-α by blocking the binding of ER-α to its co-activator. The ER-α binding monobody bound to the co-activator binding site using a single FG loop, which presented itself as an α-helical conformation. Decades later, several α-helical peptides that mimicked this interaction has been developed targeting the same binding pocket (Fig. 3d).148,154 These peptides potently inhibited the transcriptional activity of ER-α. This example shows that developing peptidomimetics that utilize the same binding interface as single-loop binding monobodies is a realistic idea.
Resemblance of single-loop binding monobody to peptides and small molecules drug continues to be found. As described above,57 monobody S4 that binds to the WIN site of WDR5 closely resembles a natural peptide as well as a small molecule drug, that binds to the same pocket (Fig. 3d).150,151 A monobody was developed for isoprenylcysteine carboxyl methyltransferase (ICMT),58 a transmembrane protein that may act as potential drug target in cancers driven by RAS and other proteins that rely the modification of CaaX motif for membrane tethering. This ICMT monobody acts as both inhibitor and crystallization chaperone. Its FG loop reaches into a crevice between two transmembrane helical regions of ICMT and competes with a natural substrate. These studies show that monobodies that primarily use the FG loop for target binding may mimic a natural ligand and provide templates for small molecule design.
Other articles have also highlighted the possibility of converting from a protein–protein binding interface to a minimalistic peptide to protein binding interface.153,155 How to leverage from the existing plethora of structural information from monobody target complex should continue to be explored.
Monobody fusions for validating targeted protein degradation approach
Proteolysis-targeting chimera (PROTAC) has emerged as a new therapeutic modality.156 The PROTAC approach opened up new possibilities towards undruggable proteins and may offer improvements over existing therapeutics.157 PROTAC is advantageous over occupancy-based therapeutic in that an efficient PROTAC molecule is able to engage multiple copies of a target during its lifetime, which should effectively reduce the required concentration and affinity to the target inside the cell.157,158There are many intricacies involved in building an efficient PROTAC molecule to selectively degrade a target. First, its target-engaging moiety should have high selectivity with few off-targets, just as conventional inhibitors. Second, it should have an affinity window to the target that allows for efficient degradation. An efficient PROTAC should bind to the target just long enough to facilitate ubiquitination and to move on to the next target. However, current PROTACs are mostly constructed based on pre-existing inhibitors or known ligands, which have been developed to achieve the strongest possible affinity.159 The limited availability of ligands and a lack of understanding of optimal binding characteristics are both impediments to PROTAC development. Important questions include what is the optimal affinity or binding kinetic that a ligand should possess for efficient degradation of a target, and which epitope a PROTAC should bind to effectively ubiquitinate a target. The optimization and target validation of PROTAC are mostly performed using small-molecule based methods, such as dTAG or similar bifunctional small molecules.160–162 However, it is laborious to alter the binding affinity of small molecules in a controlled manner, and difficult to develop a panel of small molecule ligands that target a variety of binding epitopes. On the other hand, protein-based degraders can be readily designed in the form of expression vectors and produced intracellularly so that the effectiveness of many designs can be tested rapidly and systematically. Therefore, binding protein platforms, such as monobody, are ideally suited for studying factors affecting degradation-based drug modality. To validate whether PROTAC is an efficient strategy for a given therapeutic target, one can simply generate binders with a wide range of affinity and binders that target different epitopes, and genetically encode these binders as part of a degrader for intracellular expression. This way, the affinity window and epitope can be systematically optimized (Fig. 4a).
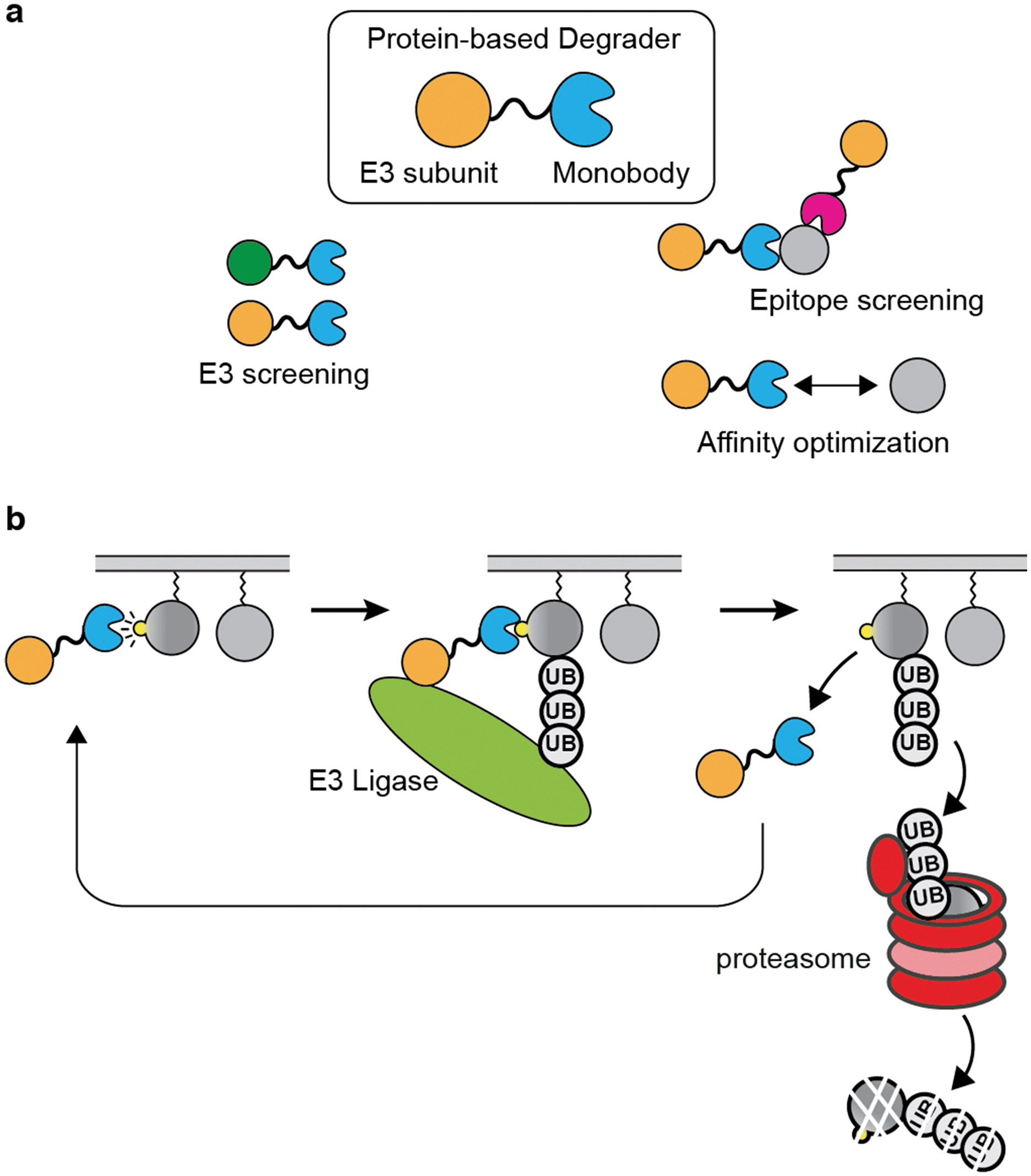 | ||
| Fig. 4 Development of protein-based degraders using monobody as a targeting ligand. (a) Modular design of degraders using monobodies. Diverse degraders can easily be developed by exploring different E3 ligase subunits, by using monobodies that bind to different epitopes, or by tuning the affinity of the monobody. (b) High selectivity of monobodies can be exploited to examine whether targeted degradation is feasible and potentially therapeutically effective by using a genetically encoded degrader. | ||
Genetically encoded protein-based degraders have become increasingly popular as a proof-of-concept tools for demonstrating the validity of degrading a drug target.48,163–165 In contrast to other binding protein technology, the monobody degraders undergo minimum degradation by itself.48,63 This may be due to the characteristic that the monobody scaffold is small and contains few lysine residues, which decrease its chance for being ubiquitinated. Recently, several studies have used monobodies developed by our group for generating protein-based degraders against several oncogenic targets by fusing a monobody directly to a subunit of E3 ligase.48,163–165
Sapkota et al. first demonstrated the utility of monobody-based degraders termed AdPROMs by fusing a monobody selective to SHP2 to the VHL domain.166 They demonstrated the feasibility of degrading endogenous RAS using NS1 monobody-based AdPROMs.164 Around the same time, a work done by the Rabbitts group showed that other intracellular biologics, such as DARPin, are also capable warheads of protein-based degraders. They generated degraders with DARPin that are selective to KRAS isoform, and demonstrated the selective degradation of KRAS in cells.48 They also touched on the idea that different E3 subunit when fused to the DARPin, provided different efficiency of degradation. Taking these ideas further, an interesting study conducted by the Partridge group compared the degradation of RAS by NS1 monobody and other RAS-binding proteins fused to SPOP, which was the most efficient E3 ligase subunit among those that they tested.163 They showed that the NS1 monobody fused to SPOP successfully degraded RAS in an isoform specific manner, which was in line with the expectation that NS1 binds to HRAS and KRAS but not to NRAS. The term bio-PROTAC was used in this study when referring to protein-based degraders. This study has demonstrated that both the E3 ligase subunit and the binding protein in a bio-PROTAC can be easily exchanged in order to screen for the desired selectivity and affinity of target depletion. Furthermore, the work demonstrated that protein-based degrader can be delivered and expressed in cells via mRNA technology,163 which may overcome the fundamental limitation of biologics directed to intracellular targets.
A recent work by our group harnessed the exceptional selectivity of the 12VC1 monobody to KRAS mutants in the active state (Fig. 3a and 4b).63 The affinity of 12VC1 was tuned by mutation. A variant with weaker affinity exhibited higher efficiency in degrading the RAS G12C mutant than the parent monobody. These results suggest that there is an optimal affinity window for achieving efficient target degradation. They also illustrate the importance of the ability to readily tune the affinity of a monobody by mutating residues at the binding interface.
Future directions
The examples presented in this review demonstrate that intracellular biologics such as the monobody technology offer impactful tools for the early drug discovery pipeline. The high success rate of monobody technology for generating highly selective and potent monobodies against challenging drug targets in a short period enables the development of tool biologics that represent the best-case scenario for selective target engagement. Overall, we envision that on-tissue efficacy data with intracellular monobodies should help address the target validation question early in a project (Fig. 1a).A major limitation of genetically encoded tool biologics in biological validation is the challenge in delivering the expression vectors to all cells of interest and in controlling the expression level of the encoded biologic reagent throughout the duration of the experiment.63 Cells can silence the expression of the encoded biologic reagent. It is still difficult to examine systemic toxicity and off-tissue effects in an animal study without developing transgenic animals. Developing a facile and robust method to deliver a biologic in either genetic or protein format, e.g., using mRNA, gene therapy, or protein delivery technologies, will substantially expand the utility and impact of these tools. We envision that monobodies and other synthetic binding proteins have the potential to directly act as a therapeutic both extracellularly and as an intracellular inhibitor or degrader.
The examination of protein–protein interaction interfaces between intracellular biologics and its target has the potential to facilitate small molecule drug discovery process in many ways beyond target validation. The Rabbitts group has demonstrated the possibility of incorporating intracellular biologics in screening assays of small molecules.167,168 In these assays, small molecules that are displaced by the binding of biologics to the target can reveal potential hits. In addition, monobodies that act as crystallization chaperones have contributed to successful determination of crystal structures of many potential drug targets.89 Future work should also exploit the rich structure database of monobody–drug target complexes for guiding the design of other therapeutic modalities, as exemplified by a recent work by Teng et al. that identified key components for selective recognition of RAS mutants.63
Author contributions
All authors wrote the manuscript.Conflicts of interest
KWT is a current employee of Merck & Co., Inc. SK is listed as an investor of patents covering aspects of the monobody technology (US9512199 B2 and related) filed by University of Chicago and Novartis AG. S. K. is a scientific advisory board member and holds equity in and receives consulting fees from Black Diamond Therapeutics and receives research funding from Puretech Health and Argenx BVBA.Acknowledgements
We would like to thank Drs. O. Hantschel and G. Salzman for critical reading of the manuscript. This work was supported in part by the American Cancer Society fellowship PF-18-180-01-TBE (KWT) and the National Institutes of Health grants R01 CA194864, R01 CA212608 and R21AI158997 (SK).References
- J. Frearson and P. Wyatt, Expert Opin. Drug Discovery, 2010, 5, 909–919 CrossRef PubMed.
- J. R. Everett, Expert Opin. Drug Discovery, 2015, 10, 937–944 CrossRef PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2019, 18, 85–89 CrossRef PubMed.
- K. Smietana, M. Siatkowski and M. Moller, Nat. Rev. Drug Discovery, 2016, 15, 379–380 CrossRef CAS PubMed.
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203–214 CrossRef CAS PubMed.
- M. Hay, D. W. Thomas, J. L. Craighead, C. Economides and J. Rosenthal, Nat. Biotechnol., 2014, 32, 40–51 CrossRef CAS PubMed.
- B. Munos, Nat. Rev. Drug Discovery, 2009, 8, 959–968 CrossRef CAS PubMed.
- F. Pammolli, L. Magazzini and M. Riccaboni, Nat. Rev. Drug Discovery, 2011, 10, 428–438 CrossRef CAS PubMed.
- J. W. Scannell, A. Blanckley, H. Boldon and B. Warrington, Nat. Rev. Drug Discovery, 2012, 11, 191–200 CrossRef CAS PubMed.
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3, 711–715 CrossRef CAS PubMed.
- M. E. Bunnage, Nat. Chem. Biol., 2011, 7, 335–339 CrossRef CAS PubMed.
- H. Dowden and J. Munro, Nat. Rev. Drug Discovery, 2019, 18, 495–496 CrossRef CAS PubMed.
- H. Matthews, J. Hanison and N. Nirmalan, Proteomes, 2016, 4, 28 CrossRef PubMed.
- A. T. Plowright, Target discovery and validation : methods and strategies for drug discovery, Wiley-VCH, Weinheim, 2020 Search PubMed.
- P. Mali, J. Aach, P. B. Stranges, K. M. Esvelt, M. Moosburner, S. Kosuri, L. Yang and G. M. Church, Nat. Biotechnol., 2013, 31, 833–838 CrossRef CAS PubMed.
- W. Y. Hwang, Y. Fu, D. Reyon, M. L. Maeder, S. Q. Tsai, J. D. Sander, R. T. Peterson, J. R. Yeh and J. K. Joung, Nat. Biotechnol., 2013, 31, 227–229 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- J. K. Joung and J. D. Sander, Nat. Rev. Mol. Cell Biol., 2013, 14, 49–55 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- P. Huang, A. Xiao, M. Zhou, Z. Zhu, S. Lin and B. Zhang, Nat. Biotechnol., 2011, 29, 699–700 CrossRef CAS PubMed.
- L. Tesson, C. Usal, S. Menoret, E. Leung, B. J. Niles, S. Remy, Y. Santiago, A. I. Vincent, X. Meng, L. Zhang, P. D. Gregory, I. Anegon and G. J. Cost, Nat. Biotechnol., 2011, 29, 695–696 CrossRef CAS PubMed.
- F. D. Urnov, E. J. Rebar, M. C. Holmes, H. S. Zhang and P. D. Gregory, Nat. Rev. Genet., 2010, 11, 636–646 CrossRef CAS PubMed.
- A. M. Geurts, G. J. Cost, Y. Freyvert, B. Zeitler, J. C. Miller, V. M. Choi, S. S. Jenkins, A. Wood, X. Cui, X. Meng, A. Vincent, S. Lam, M. Michalkiewicz, R. Schilling, J. Foeckler, S. Kalloway, H. Weiler, S. Menoret, I. Anegon, G. D. Davis, L. Zhang, E. J. Rebar, P. D. Gregory, F. D. Urnov, H. J. Jacob and R. Buelow, Science, 2009, 325, 433 CrossRef CAS PubMed.
- Y. Doyon, J. M. McCammon, J. C. Miller, F. Faraji, C. Ngo, G. E. Katibah, R. Amora, T. D. Hocking, L. Zhang, E. J. Rebar, P. D. Gregory, F. D. Urnov and S. L. Amacher, Nat. Biotechnol., 2008, 26, 702–708 CrossRef CAS PubMed.
- M. Bibikova, M. Golic, K. G. Golic and D. Carroll, Genetics, 2002, 161, 1169–1175 CrossRef CAS PubMed.
- P. Colas, B. Cohen, T. Jessen, I. Grishina, J. McCoy and R. Brent, Nature, 1996, 380, 548–550 CrossRef CAS PubMed.
- C. W. Xu and Z. Luo, Oncogene, 2002, 21, 5753–5757 CrossRef CAS PubMed.
- S. Schmidt, S. Diriong, J. Mery, E. Fabbrizio and A. Debant, FEBS Lett., 2002, 523, 35–42 CrossRef CAS PubMed.
- S. Shibasaki, M. Karasaki, T. Graslund, P. A. Nygren, H. Sano and T. Iwasaki, AMB Express, 2014, 4, 82 CrossRef PubMed.
- S. Grimm, E. Lundberg, F. Yu, S. Shibasaki, E. Vernet, M. Skogs, P. A. Nygren and T. Graslund, New Biotechnol., 2010, 27, 766–773 CrossRef CAS PubMed.
- S. Grimm, S. Salahshour and P. A. Nygren, New Biotechnol., 2012, 29, 534–542 CrossRef CAS PubMed.
- M. Summanen, N. Granqvist, R. K. Tuominen, M. Yliperttula, C. T. Verrips, J. Boonstra, C. Blanchetot and E. Ekokoski, PLoS One, 2012, 7, e35630 CrossRef CAS PubMed.
- K. Van Impe, J. Bethuyne, S. Cool, F. Impens, D. Ruano-Gallego, O. De Wever, B. Vanloo, M. Van Troys, K. Lambein, C. Boucherie, E. Martens, O. Zwaenepoel, G. Hassanzadeh-Ghassabeh, J. Vandekerckhove, K. Gevaert, L. A. Fernandez, N. N. Sanders and J. Gettemans, Breast Cancer Res., 2013, 15, R116 CrossRef PubMed.
- D. Chatterjee, M. Bhatt, D. Butler, E. De Genst, C. M. Dobson, A. Messer and J. H. Kordower, NPJ Parkinsons Dis., 2018, 4, 25 CrossRef PubMed.
- A. Steels, L. Vannevel, O. Zwaenepoel and J. Gettemans, Sci. Rep., 2019, 9, 12680 CrossRef PubMed.
- S. G. Rasmussen, H. J. Choi, J. J. Fung, E. Pardon, P. Casarosa, P. S. Chae, B. T. Devree, D. M. Rosenbaum, F. S. Thian, T. S. Kobilka, A. Schnapp, I. Konetzki, R. K. Sunahara, S. H. Gellman, A. Pautsch, J. Steyaert, W. I. Weis and B. K. Kobilka, Nature, 2011, 469, 175–180 CrossRef CAS PubMed.
- T. W. M. De Groof, N. D. Bergkamp, R. Heukers, T. Giap, M. P. Bebelman, R. Goeij-de Haas, S. R. Piersma, C. R. Jimenez, K. C. Garcia, H. L. Ploegh, M. Siderius and M. J. Smit, Nat. Commun., 2021, 12, 4357 CrossRef CAS PubMed.
- R. Irannejad, J. C. Tomshine, J. R. Tomshine, M. Chevalier, J. P. Mahoney, J. Steyaert, S. G. Rasmussen, R. K. Sunahara, H. El-Samad, B. Huang and M. von Zastrow, Nature, 2013, 495, 534–538 CrossRef CAS PubMed.
- S. Biocca, P. Pierandrei-Amaldi and A. Cattaneo, Biochem. Biophys. Res. Commun., 1993, 197, 422–427 CrossRef CAS PubMed.
- M. Kovaleva, I. Bussmeyer, B. Rabe, J. Grotzinger, E. Sudarman, J. Eichler, U. Conrad, S. Rose-John and J. Scheller, J. Virol., 2006, 80, 8510–8520 CrossRef CAS PubMed.
- M. Y. Koo, J. Park, J. M. Lim, S. Y. Joo, S. P. Shin, H. B. Shim, J. Chung, D. Kang, H. A. Woo and S. G. Rhee, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6269–6274 CrossRef CAS PubMed.
- A. Cardinale, I. Filesi, P. B. Singh and S. Biocca, Exp. Cell Res., 2015, 338, 70–81 CrossRef CAS PubMed.
- E. Tse, M. N. Lobato, A. Forster, T. Tanaka, G. T. Chung and T. H. Rabbitts, J. Mol. Biol., 2002, 317, 85–94 CrossRef CAS PubMed.
- L. Kummer, P. Parizek, P. Rube, B. Millgramm, A. Prinz, P. R. Mittl, M. Kaufholz, B. Zimmermann, F. W. Herberg and A. Pluckthun, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2248–E2257 CrossRef CAS PubMed.
- L. Pecqueur, C. Duellberg, B. Dreier, Q. Jiang, C. Wang, A. Pluckthun, T. Surrey, B. Gigant and M. Knossow, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12011–12016 CrossRef CAS PubMed.
- N. Bery, S. Legg, J. Debreczeni, J. Breed, K. Embrey, C. Stubbs, P. Kolasinska-Zwierz, N. Barrett, R. Marwood, J. Watson, J. Tart, R. Overman, A. Miller, C. Phillips, R. Minter and T. H. Rabbitts, Nat. Commun., 2019, 10, 2607 CrossRef PubMed.
- S. Guillard, P. Kolasinska-Zwierz, J. Debreczeni, J. Breed, J. Zhang, N. Bery, R. Marwood, J. Tart, R. Overman, P. Stocki, B. Mistry, C. Phillips, T. Rabbitts, R. Jackson and R. Minter, Nat. Commun., 2017, 8, 16111 CrossRef CAS PubMed.
- N. Bery, A. Miller and T. Rabbitts, Nat. Commun., 2020, 11, 3233 CrossRef CAS PubMed.
- Y. Wu, A. Honegger, A. Batyuk, P. R. E. Mittl and A. Pluckthun, J. Mol. Biol., 2018, 430, 2128–2138 CrossRef CAS PubMed.
- P. Parizek, L. Kummer, P. Rube, A. Prinz, F. W. Herberg and A. Pluckthun, ACS Chem. Biol., 2012, 7, 1356–1366 CrossRef CAS PubMed.
- J. Wojcik, A. J. Lamontanara, G. Grabe, A. Koide, L. Akin, B. Gerig, O. Hantschel and S. Koide, J. Biol. Chem., 2016, 291, 8836–8847 CrossRef CAS PubMed.
- J. Wojcik, O. Hantschel, F. Grebien, I. Kaupe, K. L. Bennett, J. Barkinge, R. B. Jones, A. Koide, G. Superti-Furga and S. Koide, Nat. Struct. Mol. Biol., 2010, 17, 519–527 CrossRef CAS PubMed.
- F. Sha, E. B. Gencer, S. Georgeon, A. Koide, N. Yasui, S. Koide and O. Hantschel, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14924–14929 CrossRef CAS PubMed.
- N. Nady, A. Gupta, Z. Ma, T. Swigut, A. Koide, S. Koide and J. Wysocka, eLife, 2015, 4, e10150 CrossRef PubMed.
- A. Bowman, A. Koide, J. S. Goodman, M. E. Colling, D. Zinne, S. Koide and A. G. Ladurner, Nucleic Acids Res., 2017, 45, 643–656 CrossRef CAS PubMed.
- R. Spencer-Smith, A. Koide, Y. Zhou, R. R. Eguchi, F. Sha, P. Gajwani, D. Santana, A. Gupta, M. Jacobs, E. Herrero-Garcia, J. Cobbert, H. Lavoie, M. Smith, T. Rajakulendran, E. Dowdell, M. N. Okur, I. Dementieva, F. Sicheri, M. Therrien, J. F. Hancock, M. Ikura, S. Koide and J. P. O'Bryan, Nat. Chem. Biol., 2017, 13, 62–68 CrossRef CAS PubMed.
- A. Gupta, J. Xu, S. Lee, S. T. Tsai, B. Zhou, K. Kurosawa, M. S. Werner, A. Koide, A. J. Ruthenburg, Y. Dou and S. Koide, Nat. Chem. Biol., 2018, 14, 895–900 CrossRef CAS PubMed.
- M. M. Diver, L. Pedi, A. Koide, S. Koide and S. B. Long, Nature, 2018, 553, 526–529 CrossRef CAS PubMed.
- A. Zorba, V. Nguyen, A. Koide, M. Hoemberger, Y. Zheng, S. Kutter, C. Kim, S. Koide and D. Kern, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13937–13942 CrossRef CAS PubMed.
- E. J. Petrie, R. W. Birkinshaw, A. Koide, E. Denbaum, J. M. Hildebrand, S. E. Garnish, K. A. Davies, J. J. Sandow, A. L. Samson, X. Gavin, C. Fitzgibbon, S. N. Young, P. J. Hennessy, P. P. C. Smith, A. I. Webb, P. E. Czabotar, S. Koide and J. M. Murphy, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8468–8475 CrossRef CAS PubMed.
- S. E. Garnish, Y. Meng, A. Koide, J. J. Sandow, E. Denbaum, A. V. Jacobsen, W. Yeung, A. L. Samson, C. R. Horne, C. Fitzgibbon, S. N. Young, P. P. C. Smith, A. I. Webb, E. J. Petrie, J. M. Hildebrand, N. Kannan, P. E. Czabotar, S. Koide and J. M. Murphy, Nat. Commun., 2021, 12, 2211 CrossRef CAS PubMed.
- G. La Sala, C. Michiels, T. Kukenshoner, T. Brandstoetter, B. Maurer, A. Koide, K. Lau, F. Pojer, S. Koide, V. Sexl, L. Dumoutier and O. Hantschel, Nat. Commun., 2020, 11, 4115 CrossRef CAS PubMed.
- K. W. Teng, S. T. Tsai, T. Hattori, C. Fedele, A. Koide, C. Yang, X. Hou, Y. Zhang, B. G. Neel, J. P. O'Bryan and S. Koide, Nat. Commun., 2021, 12, 2656 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- S. Fox, S. Farr-Jones, L. Sopchak, A. Boggs, H. W. Nicely, R. Khoury and M. Biros, J. Biomol. Screening, 2006, 11, 864–869 CrossRef PubMed.
- K. H. Bleicher, H. J. Bohm, K. Muller and A. I. Alanine, Nat. Rev. Drug Discovery, 2003, 2, 369–378 CrossRef CAS PubMed.
- M. Kostic and L. H. Jones, Trends Pharmacol. Sci., 2020, 41, 305–317 CrossRef CAS PubMed.
- R. P. Nowak and L. H. Jones, SLAS Discovery, 2021, 26, 474–483 CAS.
- H. Gille, M. Hulsmeyer, S. Trentmann, G. Matschiner, H. J. Christian, T. Meyer, A. Amirkhosravi, L. P. Audoly, A. M. Hohlbaum and A. Skerra, Angiogenesis, 2016, 19, 79–94 CrossRef CAS PubMed.
- R. Vazquez-Lombardi, T. G. Phan, C. Zimmermann, D. Lowe, L. Jermutus and D. Christ, Drug Discovery Today, 2015, 20, 1271–1283 CrossRef CAS PubMed.
- E. Sachdev, J. Gong, B. Rimel and M. Mita, Curr. Oncol. Rep., 2015, 17, 35 CrossRef PubMed.
- A. Pluckthun, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 489–511 CrossRef CAS PubMed.
- M. D. Diem, L. Hyun, F. Yi, R. Hippensteel, E. Kuhar, C. Lowenstein, E. J. Swift, K. T. O'Neil and S. A. Jacobs, Protein Eng., Des. Sel., 2014, 27, 419–429 CrossRef CAS PubMed.
- T. Wurch, A. Pierre and S. Depil, Trends Biotechnol., 2012, 30, 575–582 CrossRef CAS PubMed.
- L. Bloom and V. Calabro, Drug Discovery Today, 2009, 14, 949–955 CrossRef CAS PubMed.
- M. T. Stumpp, H. K. Binz and P. Amstutz, Drug Discovery Today, 2008, 13, 695–701 CrossRef CAS PubMed.
- S. Koide, in Protein Engineering and Design, ed. S. J. Park and J. R. Cochran, CRC Press, Boca Raton, FL, 2010, pp. 109–130 Search PubMed.
- S. S. Sidhu and S. Koide, Curr. Opin. Struct. Biol., 2007, 17, 481–487 CrossRef CAS PubMed.
- S. A. Gai and K. D. Wittrup, Curr. Opin. Struct. Biol., 2007, 17, 467–473 CrossRef CAS PubMed.
- A. Skerra, Curr. Opin. Biotechnol., 2007, 18, 295–304 CrossRef CAS PubMed.
- D. Lipovsek and A. Pluckthun, J. Immunol. Methods, 2004, 290, 51–67 CrossRef CAS PubMed.
- S. J. Park and J. R. Cochran, Protein engineering and design, CRC Press, Boca Raton, 2010 Search PubMed.
- S. Reverdatto, D. S. Burz and A. Shekhtman, Curr. Top. Med. Chem., 2015, 15, 1082–1101 CrossRef CAS PubMed.
- M. Crawford, R. Woodman and P. Ko Ferrigno, Briefings Funct. Genomics Proteomics, 2003, 2, 72–79 CrossRef CAS PubMed.
- P. Forrer, M. T. Stumpp, H. K. Binz and A. Pluckthun, FEBS Lett., 2003, 539, 2–6 CrossRef CAS PubMed.
- S. Al-Khodor, C. T. Price, A. Kalia and Y. Abu Kwaik, Trends Microbiol., 2010, 18, 132–139 CrossRef CAS PubMed.
- J. Li, A. Mahajan and M. D. Tsai, Biochemistry, 2006, 45, 15168–15178 CrossRef CAS PubMed.
- F. Sha, G. Salzman, A. Gupta and S. Koide, Protein Sci., 2017, 26, 910–924 CrossRef CAS PubMed.
- O. Hantschel, M. Biancalana and S. Koide, Curr. Opin. Struct. Biol., 2020, 60, 167–174 CrossRef CAS PubMed.
- C. Rothe and A. Skerra, BioDrugs, 2018, 32, 233–243 CrossRef CAS PubMed.
- F. C. Deuschle, E. Ilyukhina and A. Skerra, Expert Opin. Biol. Ther., 2021, 21, 509–518 CrossRef CAS PubMed.
- A. Richter, E. Eggenstein and A. Skerra, FEBS Lett., 2014, 588, 213–218 CrossRef CAS PubMed.
- S. Stahl, T. Graslund, A. E. Karlstrom, F. Y. Frejd, P. A. Nygren and J. Lofblom, Trends Biotechnol., 2017, 35, 691–712 CrossRef PubMed.
- F. Y. Frejd and K. T. Kim, Exp. Mol. Med., 2017, 49, e306 CrossRef CAS PubMed.
- J. Feldwisch and V. Tolmachev, Methods Mol. Biol., 2012, 899, 103–126 CrossRef CAS PubMed.
- P. Kunz, K. Zinner, N. Mucke, T. Bartoschik, S. Muyldermans and J. D. Hoheisel, Sci. Rep., 2018, 8, 7934 CrossRef PubMed.
- C. McMahon, A. S. Baier, R. Pascolutti, M. Wegrecki, S. Zheng, J. X. Ong, S. C. Erlandson, D. Hilger, S. G. F. Rasmussen, A. M. Ring, A. Manglik and A. C. Kruse, Nat. Struct. Mol. Biol., 2018, 25, 289–296 CrossRef CAS PubMed.
- E. Romao, F. Morales-Yanez, Y. Hu, M. Crauwels, P. De Pauw, G. G. Hassanzadeh, N. Devoogdt, C. Ackaert, C. Vincke and S. Muyldermans, Curr. Pharm. Des., 2016, 22, 6500–6518 CrossRef CAS PubMed.
- J. Yan, G. Li, Y. Hu, W. Ou and Y. Wan, J. Transl. Med., 2014, 12, 343 CrossRef PubMed.
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS PubMed.
- M. Dumoulin, K. Conrath, A. Van Meirhaeghe, F. Meersman, K. Heremans, L. G. Frenken, S. Muyldermans, L. Wyns and A. Matagne, Protein Sci., 2002, 11, 500–515 CrossRef CAS PubMed.
- J. Helma, M. C. Cardoso, S. Muyldermans and H. Leonhardt, J. Cell Biol., 2015, 209, 633–644 CrossRef CAS PubMed.
- A. Koide, C. W. Bailey, X. Huang and S. Koide, J. Mol. Biol., 1998, 284, 1141–1151 CrossRef CAS PubMed.
- A. Koide, J. Wojcik, R. N. Gilbreth, R. J. Hoey and S. Koide, J. Mol. Biol., 2012, 415, 393–405 CrossRef CAS PubMed.
- W. P. Verdurmen, M. Luginbuhl, A. Honegger and A. Pluckthun, J. Controlled Release, 2015, 200, 13–22 CrossRef CAS PubMed.
- X. Liao, A. E. Rabideau and B. L. Pentelute, ChemBioChem, 2014, 15, 2458–2466 CrossRef CAS PubMed.
- C. Gronwall, A. Jonsson, S. Lindstrom, E. Gunneriusson, S. Stahl and N. Herne, J. Biotechnol., 2007, 128, 162–183 CrossRef PubMed.
- A. S. Lo, Q. Zhu and W. A. Marasco, Handb. Exp. Pharmacol., 2008, 343–373, DOI:10.1007/978-3-540-73259-4_15.
- M. Visintin, E. Tse, H. Axelson, T. H. Rabbitts and A. Cattaneo, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11723–11728 CrossRef CAS PubMed.
- D. W. Colby, Y. Chu, J. P. Cassady, M. Duennwald, H. Zazulak, J. M. Webster, A. Messer, S. Lindquist, V. M. Ingram and K. D. Wittrup, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17616–17621 CrossRef CAS PubMed.
- K. Daniel, J. Icha, C. Horenburg, D. Muller, C. Norden and J. Mansfeld, Nat. Commun., 2018, 9, 3297 CrossRef PubMed.
- E. Caussinus, O. Kanca and M. Affolter, Nat. Struct. Mol. Biol., 2011, 19, 117–121 CrossRef PubMed.
- A. L. Marschall and S. Dubel, Comput. Struct. Biotechnol. J., 2016, 14, 304–308 CrossRef CAS PubMed.
- T. Kondo, Y. Iwatani, K. Matsuoka, T. Fujino, S. Umemoto, Y. Yokomaku, K. Ishizaki, S. Kito, T. Sezaki, G. Hayashi and H. Murakami, Sci. Adv., 2020, 6, eabd3916 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- M. A. Abdalla and L. J. McGaw, Molecules, 2018, 23, 2080 CrossRef PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- Z. Qian, P. G. Dougherty and D. Pei, Curr. Opin. Chem. Biol., 2017, 38, 80–86 CrossRef CAS PubMed.
- J. Zhou and C. Wild, Curr. Top. Med. Chem., 2019, 19, 1363–1364 CrossRef CAS PubMed.
- A. S. Hauser, M. M. Attwood, M. Rask-Andersen, H. B. Schioth and D. E. Gloriam, Nat. Rev. Drug Discovery, 2017, 16, 829–842 CrossRef CAS PubMed.
- R. H. Purcell and R. A. Hall, Annu. Rev. Pharmacol. Toxicol., 2018, 58, 429–449 CrossRef CAS PubMed.
- H. M. Stoveken, S. D. Larsen, A. V. Smrcka and G. G. Tall, Mol. Pharmacol., 2018, 93, 477–488 CrossRef CAS PubMed.
- H. M. Stoveken, L. L. Bahr, M. W. Anders, A. P. Wojtovich, A. V. Smrcka and G. G. Tall, Mol. Pharmacol., 2016, 90, 214–224 CrossRef CAS PubMed.
- B. Zhu, R. Luo, P. Jin, T. Li, H. C. Oak, S. Giera, K. R. Monk, P. Lak, B. K. Shoichet and X. Piao, J. Biol. Chem., 2019, 294, 19246–19254 CrossRef CAS PubMed.
- G. S. Salzman, S. Zhang, A. Gupta, A. Koide, S. Koide and D. Arac, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10095–10100 CrossRef CAS PubMed.
- J. H. Bushweller, Nat. Rev. Cancer, 2019, 19, 611–624 CrossRef CAS PubMed.
- M. Lambert, S. Jambon, S. Depauw and M. H. David-Cordonnier, Molecules, 2018, 23, 1479 CrossRef PubMed.
- J. E. Darnell, Nat. Med., 2005, 11, 595–596 CrossRef CAS PubMed.
- J. Bollrath, T. J. Phesse, V. A. von Burstin, T. Putoczki, M. Bennecke, T. Bateman, T. Nebelsiek, T. Lundgren-May, O. Canli, S. Schwitalla, V. Matthews, R. M. Schmid, T. Kirchner, M. C. Arkan, M. Ernst and F. R. Greten, Cancer Cell, 2009, 15, 91–102 CrossRef CAS PubMed.
- K. A. Dorritie, J. A. McCubrey and D. E. Johnson, Leukemia, 2014, 28, 248–257 CrossRef CAS PubMed.
- L. Bai, H. Zhou, R. Xu, Y. Zhao, K. Chinnaswamy, D. McEachern, J. Chen, C. Y. Yang, Z. Liu, M. Wang, L. Liu, H. Jiang, B. Wen, P. Kumar, J. L. Meagher, D. Sun, J. A. Stuckey and S. Wang, Cancer Cell, 2019, 36, 498–511 e417 CrossRef CAS PubMed.
- M. Wu and H. B. Shu, Chin. J. Cancer, 2011, 30, 240–246 CrossRef CAS PubMed.
- G. N. Hatzopoulos, T. Kukenshoner, N. Banterle, T. Favez, I. Fluckiger, V. Hamel, S. Andany, G. E. Fantner, O. Hantschel and P. Gonczy, Nat. Commun., 2021, 12, 3805 CrossRef CAS PubMed.
- B. Papke and C. J. Der, Science, 2017, 355, 1158–1163 CrossRef CAS PubMed.
- A. R. Moore, S. C. Rosenberg, F. McCormick and S. Malek, Nat. Rev. Drug Discovery, 2020, 19, 533–552 CrossRef CAS PubMed.
- I. A. Prior, F. E. Hood and J. L. Hartley, Cancer Res., 2020, 80, 2969–2974 CrossRef CAS PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed.
- M. R. Janes, J. Zhang, L. S. Li, R. Hansen, U. Peters, X. Guo, Y. Chen, A. Babbar, S. J. Firdaus, L. Darjania, J. Feng, J. H. Chen, S. Li, S. Li, Y. O. Long, C. Thach, Y. Liu, A. Zarieh, T. Ely, J. M. Kucharski, L. V. Kessler, T. Wu, K. Yu, Y. Wang, Y. Yao, X. Deng, P. P. Zarrinkar, D. Brehmer, D. Dhanak, M. V. Lorenzi, D. Hu-Lowe, M. P. Patricelli, P. Ren and Y. Liu, Cell, 2018, 172, 578–589 e517 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
- D. Gillingham, Chimia, 2021, 75, 439–441 CrossRef CAS PubMed.
- I. Khan, R. Spencer-Smith and J. P. O'Bryan, Oncogene, 2019, 38, 2984–2993 CrossRef CAS PubMed.
- C. Ambrogio, J. Kohler, Z. W. Zhou, H. Wang, R. Paranal, J. Li, M. Capelletti, C. Caffarra, S. Li, Q. Lv, S. Gondi, J. C. Hunter, J. Lu, R. Chiarle, D. Santamaria, K. D. Westover and P. A. Janne, Cell, 2018, 172, 857–868 e815 CrossRef CAS PubMed.
- F. Grebien, O. Hantschel, J. Wojcik, I. Kaupe, B. Kovacic, A. M. Wyrzucki, G. D. Gish, S. Cerny-Reiterer, A. Koide, H. Beug, T. Pawson, P. Valent, S. Koide and G. Superti-Furga, Cell, 2011, 147, 306–319 CrossRef CAS PubMed.
- N. P. Shah, J. M. Nicoll, B. Nagar, M. E. Gorre, R. L. Paquette, J. Kuriyan and C. L. Sawyers, Cancer Cell, 2002, 2, 117–125 CrossRef CAS PubMed.
- E. J. Petrie, J. J. Sandow, W. I. L. Lehmann, L. Y. Liang, D. Coursier, S. N. Young, W. J. A. Kersten, C. Fitzgibbon, A. L. Samson, A. V. Jacobsen, K. N. Lowes, A. E. Au, H. Jousset Sabroux, N. Lalaoui, A. I. Webb, G. Lessene, G. Manning, I. S. Lucet and J. M. Murphy, Cell Rep., 2019, 28, 3309–3319 e3305 CrossRef CAS PubMed.
- J. D. Sadowsky, M. A. Burlingame, D. W. Wolan, C. L. McClendon, M. P. Jacobson and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6056–6061 CrossRef CAS PubMed.
- T. J. Rettenmaier, J. D. Sadowsky, N. D. Thomsen, S. C. Chen, A. K. Doak, M. R. Arkin and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18590–18595 CrossRef CAS PubMed.
- T. E. Speltz, C. G. Mayne, S. W. Fanning, Z. Siddiqui, E. Tajkhorshid, G. L. Greene and T. W. Moore, Org. Biomol. Chem., 2018, 16, 3702–3706 RSC.
- A. Koide, S. Abbatiello, L. Rothgery and S. Koide, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1253–1258 CrossRef CAS PubMed.
- E. R. Aho, J. Wang, R. D. Gogliotti, G. C. Howard, J. Phan, P. Acharya, J. D. Macdonald, K. Cheng, S. L. Lorey, B. Lu, S. Wenzel, A. M. Foshage, J. Alvarado, F. Wang, J. G. Shaw, B. Zhao, A. M. Weissmiller, L. R. Thomas, C. R. Vakoc, M. D. Hall, S. W. Hiebert, Q. Liu, S. R. Stauffer, S. W. Fesik and W. P. Tansey, Cell Rep., 2019, 26, 2916–2928 e2913 CrossRef CAS PubMed.
- H. Karatas, Y. Li, L. Liu, J. Ji, S. Lee, Y. Chen, J. Yang, L. Huang, D. Bernard, J. Xu, E. C. Townsend, F. Cao, X. Ran, X. Li, B. Wen, D. Sun, J. A. Stuckey, M. Lei, Y. Dou and S. Wang, J. Med. Chem., 2017, 60, 4818–4839 CrossRef CAS PubMed.
- V. B. de Souza and D. F. Kawano, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129448 CrossRef CAS PubMed.
- A. M. Sevy, I. M. Gilchuk, B. P. Brown, N. G. Bozhanova, R. Nargi, M. Jensen, J. Meiler and J. E. Crowe Jr., Structure, 2020, 28, 1114–1123 e1114 CrossRef CAS PubMed.
- M. Xie, H. Zhao, Q. Liu, Y. Zhu, F. Yin, Y. Liang, Y. Jiang, D. Wang, K. Hu, X. Qin, Z. Wang, Y. Wu, N. Xu, X. Ye, T. Wang and Z. Li, J. Med. Chem., 2017, 60, 8731–8740 CrossRef CAS PubMed.
- M. G. Wuo and P. S. Arora, Curr. Opin. Chem. Biol., 2018, 44, 16–22 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- Q. Lu, X. Ding, T. Huang, S. Zhang, Y. Li, L. Xu, G. Chen, Y. Ying, Y. Wang, Z. Feng, L. Wang and X. Zou, Am. J. Transl. Res., 2019, 11, 5728–5739 CAS.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- S. An and L. Fu, EBioMedicine, 2018, 36, 553–562 CrossRef PubMed.
- B. Nabet, F. M. Ferguson, B. K. A. Seong, M. Kuljanin, A. L. Leggett, M. L. Mohardt, A. Robichaud, A. S. Conway, D. L. Buckley, J. D. Mancias, J. E. Bradner, K. Stegmaier and N. S. Gray, Nat. Commun., 2020, 11, 4687 CrossRef CAS PubMed.
- B. Nabet, J. M. Roberts, D. L. Buckley, J. Paulk, S. Dastjerdi, A. Yang, A. L. Leggett, M. A. Erb, M. A. Lawlor, A. Souza, T. G. Scott, S. Vittori, J. A. Perry, J. Qi, G. E. Winter, K. K. Wong, N. S. Gray and J. E. Bradner, Nat. Chem. Biol., 2018, 14, 431–441 CrossRef CAS PubMed.
- K. T. G. Samarasinghe, S. Jaime-Figueroa, M. Burgess, D. A. Nalawansha, K. Dai, Z. Hu, A. Bebenek, S. A. Holley and C. M. Crews, Cell Chem. Biol., 2021, 28, 648–661 e645 CrossRef CAS PubMed.
- S. Lim, R. Khoo, Y. C. Juang, P. Gopal, H. Zhang, C. Yeo, K. M. Peh, J. Teo, S. Ng, B. Henry and A. W. Partridge, ACS Cent. Sci., 2021, 7, 274–291 CrossRef CAS PubMed.
- S. Roth, T. J. Macartney, A. Konopacka, K. H. Chan, H. Zhou, M. A. Queisser and G. P. Sapkota, Cell Chem. Biol., 2020, 27, 1151–1163 e1156 CrossRef CAS PubMed.
- L. J. Fulcher, L. D. Hutchinson, T. J. Macartney, C. Turnbull and G. P. Sapkota, Open Biol., 2017, 7, 170066 CrossRef PubMed.
- L. J. Fulcher, T. Macartney, P. Bozatzi, A. Hornberger, A. Rojas-Fernandez and G. P. Sapkota, Open Biol., 2016, 6, 160255 CrossRef PubMed.
- N. Bery, C. J. R. Bataille, A. Russell, A. Hayes, F. Raynaud, S. Milhas, S. Anand, H. Tulmin, A. Miller and T. H. Rabbitts, Sci. Adv., 2021, 7, eabg1950 CrossRef CAS PubMed.
- C. E. Quevedo, A. Cruz-Migoni, N. Bery, A. Miller, T. Tanaka, D. Petch, C. J. R. Bataille, L. Y. W. Lee, P. S. Fallon, H. Tulmin, M. T. Ehebauer, N. Fernandez-Fuentes, A. J. Russell, S. B. Carr, S. E. V. Phillips and T. H. Rabbitts, Nat. Commun., 2018, 9, 3169 CrossRef PubMed.
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Present affiliation: Discovery Biologics, Merck & Co., Inc., Boston, MA 02115, USA. |
| This journal is © The Royal Society of Chemistry 2021 |