
Covalent PROTACs: the best of both worlds?
Neil P.
Grimster
 *
*
Oncology R&D, AstraZeneca, Waltham, USA. E-mail: neil.grimster@astrazeneca.com
First published on 15th July 2021
Abstract
Covalent PROTACs combine the cutting edge research areas of targeted covalent inhibitors (TCIs) and proteolysis targeting chimeras (PROTACs). This nascent field of research has already demonstrated several interesting findings, and holds an immense amount of potential to expand the druggable proteome. In this opinion, we present some of these intriguing early findings and discuss the potential advantages and disadvantages of this approach.
Targeted covalent inhibitors (TCIs)1,2 and proteolysis targeting chimeras (PROTACs)3 are two areas of research at the cutting edge of medicinal chemistry. Over the past decade, multiple TCIs have received approval, such as osimertinib for the inhibition of EGFR,4 the BTK inhibitor acalabrutinib,5 and most recently sotorasib, a covalent inhibitor of the whitest of oncology whales KRASG12C.6 Meanwhile, PROTACs have led to a paradigm shift in the pharmaceutical industry, and in some respects have upended the way we think about targeting proteins of interest (POI).7 Their ability to convert small molecule binders into degraders of the protein opens up a plethora of new opportunities. This includes previous high value targets where genetic validation was not recapitulated by small molecule inhibition, something which is often attributed to the small molecule's inability to remove the POI. Consequently, both TCIs and PROTACs have been labeled with the mantra of being able to “drug the undruggable”.8,9 As these two approaches are not mutually exclusive, this raises the possibility of combining both methodologies to generate covalent PROTACs, and accordingly a fertile field of investigation has emerged. We should, however, remember that it is not only the advantages that will be combined here, but also the complexities associated with both approaches, and these may not be merely additive, but possibly multiplicative. As with any new area of investigation, this combined approach may lead to new findings, both positive and negative, that represent more than the sum of their parts. There are already excellent reviews on the area of covalent PROTACs,10,11 here we highlight some of the exemplary work in this nascent area and identify some of the unexpected findings.
It should be stated that molecules that function via a covalent mechanism of action are not novel in the field of medicinal chemistry, indeed Nature is more than happy to utilize the formation of a covalent bond to fulfil its intended goal. Moreover, aspirin, one of the earliest pharmaceuticals, modulates its target through the formation of a covalent bond; although this was only elucidated decades after its discovery.12 However, fueled by the fear of idiosyncratic toxicities arising from indiscriminate modification of off-target proteins, the medicinal chemistry community shield away from employing covalent inhibition. Now, with a clearer understanding of the requirements governing covalent inhibition, the field has seen a “resurgence”.1,2 The careful tailoring of the reactivity of covalent warheads, partnered with an understanding of what factors affect the nucleophilicity of amino-acid side chains in binding pockets, has allowed medicinal chemists to thread the reactivity needle. In turn, this has led to the generation of exquisitely selective compounds that react with their intended target, and thus avoid anticipated toxicities resulting from off-target inhibition. To better understand reactivity, any successful covalent inhibitor program requires the ability to probe the different aspects of the covalent bond formation. Unlike typical non-covalent inhibitors, TCIs function through a two-step process (Scheme 1), wherein the compound must first bind non-covalently before undergoing the covalent binding event. To effectively optimize TCIs, specialist assays are required to tease apart Ki and kinact to allow for comprehension and optimization of their contribution to the overall potency.
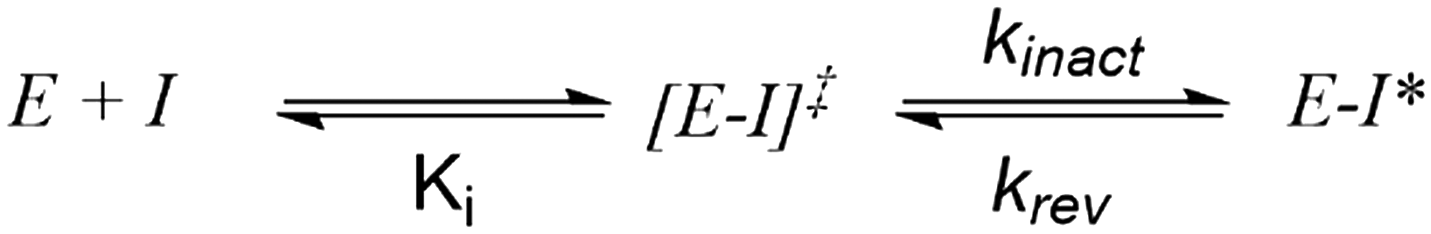 | ||
| Scheme 1 Model for two-stage covalent modification of a protein target (E) by a covalent substrate (I). [E–I] represents the initial non-covalent complex formed between E and I; E–I* represents the covalent adduct. | ||
PROTACS, conceived by Professor Craig Crews and co-workers in 2001,3 are heterobifunctional molecules capable of provoking protein degradation of a desired target (Scheme 2). This is accomplished by engaging both a POI and an E3 ligase; the resultant ternary complex allows for the ubiquitination and subsequent proteasomal degradation of the desired target. The result of this is that degradation is potentially catalytic, allowing the PROTACs to be “recycled” to degrade another molecule of the desired target, which can manifest as a large increase in apparent potency. In common with TCIs, there are a number of considerations that must be made when starting a PROTACs project. First, by their very nature, PROTACs require optimization of three, almost distinct, entities: the POI ligand, the linker and the E3 ligase ligand (Scheme 2). Attention has therefore focused on the generation, interrogation and crystallization of ternary complexes to aid with the design of PROTACs, efforts which have been further strengthened by the development of computational tools.13 Moreover, while simple biochemical assays and biophysical measurements can provide information about inhibition and binding of the desire target (whether E3, POI, or the ternary complex as a whole), the necessity to engage the entirety of the cellular machinery to observe protein degradation means that, in general, the designation of a PROTAC can only be made after carefully performed cellular experiments. As with TCIs, the resynthesis rate of the protein which is intended to be degraded must be considered. With PROTACs, however, the rate of degradation is also key, because the relationship between the two parameters determines the effectiveness of protein degradation. To date, the vast majority of PROTACs have used ligands against only two E3 ligases: cereblon (CRBN) and von Hippel–Lindau tumor suppressor (VHL); with prototypical ligands lenalidomide and pomalidomide targeting CRBN and VH-298 binding to VHL.7
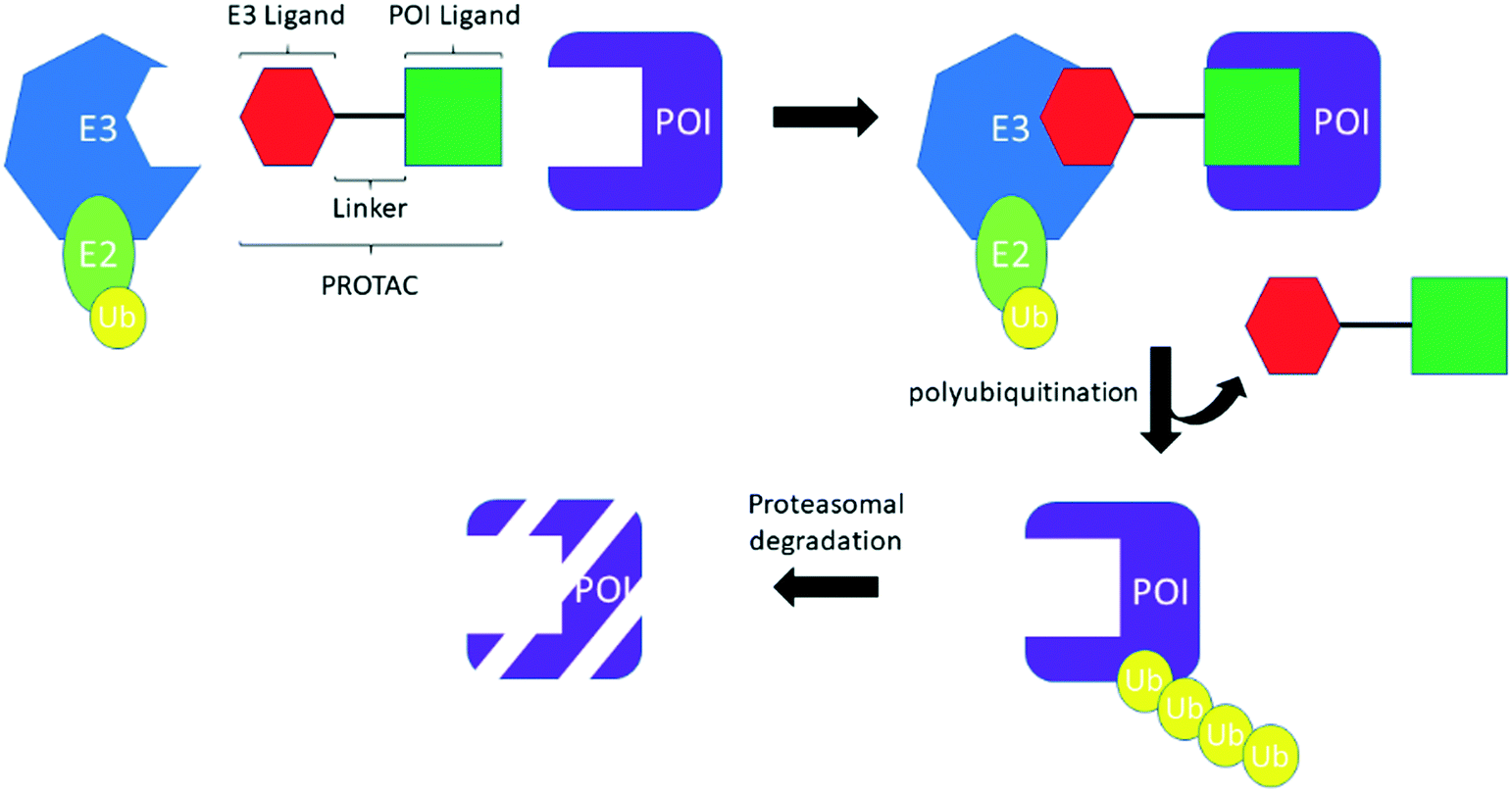 | ||
| Scheme 2 Schematic of the PROTACs mechanism. | ||
Covalent PROTACs are expected to benefit from all of the advantages associated with TCIs, for instance: increased selectivity from targeting non-conserved nucleophilic residues. Moreover, it has been demonstrated that the covalent bond formation of TCIs can effectively divorce the relationship between pharmacodynamics and pharmacokinetics, and thus the pharmacological activity of the target only returns upon protein resynthesis, and not clearance of the compound as is the case with non-covalent inhibitors. This means that covalent PROTACs may only require transient PK exposure to enable full target engagement, and thus reduce possible toxicities arising from prolonged systemic exposure. However, the promise of covalent PROTACs really derives from the possibility that they can address the challenges associated with the development of PROTACs as orally bioavailable drugs. By their very nature, PROTACs are significantly larger than most small molecule inhibitors, and operate in the so-called “beyond rule of 5” chemical space.14 In turn, this leads to challenges in optimization of physical chemical properties; impacts permeability; and makes PROTACs vulnerable to transporter mediated expulsion from the cell. Interestingly, the ability of a small molecule to make a covalent linkage with its desired protein target has been demonstrated to provide a disproportionate amount of binding affinity,15 and as a consequence much smaller and/or less complex TCIs can deliver high potencies. In the context of PROTACs, this suggests that potency can be maintained, while reducing molecular weight to improve physical chemical properties and cellular permeability; or permit the removal of hydrogen bond donors to reduce the recognition of the PROTACs by the efflux machinery. In addition, due to the increased affinity provided by the covalent warhead, covalent PROTACs could permit the targeting of less tractable allosteric or functionally silent binding sites. Because the PROTAC mechanism of action does not require a binding site to be functionally active, covalent PROTACs could greatly increase the number of druggable targets (Table 1). Finally, the reduction in complexity of PROTACs through addition of a covalent warhead may also make the desired molecules more synthetically tractable.
| POI ligand | E3 ligand | Potential advantages | Potential disadvantages | Ref. |
|---|---|---|---|---|
| Irreversible covalent | Reversible | Reduce size/complexity of POI ligand | Loss of PROTACs catalytic mechanism | 3, 16–18, 31, 32 |
| Increased selectivity | ||||
| Increased pool of tractable targets | ||||
| Reversible | Irreversible covalent | Reduce size/complexity of E3 ligand | Transient blockage of E3 machinery | 21, 22, 29, 33, 34 |
| Maintain PROTACs catalytic mechanism | ||||
| Increased selectivity | ||||
| Target intractable ligases | ||||
| Reversible covalent | Reversible | Reduce size/complexity of POI ligand | Characterization of kinetics challenging | 18, 27, 35, 36 |
| Increased pool of tractable POI targets | ||||
| Increased selectivity | ||||
| Maintain PROTACs catalytic mechanism | ||||
| Possible cellular concentration increases | ||||
| Reversible | Reversible covalent | Reduce size/complexity of E3 ligand | Characterization of kinetics challenging | 28 |
| Increase pool of tractable E3 targets | ||||
| Increased selectivity | ||||
| Maintain PROTACs catalytic mechanism |
Clearly, when compared to standard non-covalent small molecule binder/inhibitor programs, there are several complicating factors for both TCI and PROTAC based projects. However, it should be noted that some requirements do align, for instance both approaches would often favor a longer protein resynthesis rate. When initiating a covalent PROTAC program, researchers will need to build the appropriate assays to fully assess the compounds under investigation, including the ability to measure the rate of covalent bond formation and target occupancy, both biochemically and cellularly, as well as the rate and extent of protein degradation. Interestingly, the first PROTAC ever developed was based on a covalent POI ligand, the natural product ovalicin, and was demonstrated to degrade MetAP-2.3 However, since this initial disclosure the vast majority of PROTACs have utilized non-covalent binders of both the POI and E3 ligase to elicit their effects. When undertaking a covalent PROTAC project there are a number of possibilities when considering where to situate the covalent warhead. While PROTACs are typically constructed in three interconnected pieces, almost by definition installation of the covalent warhead on the linker region is unnecessary. This leaves three possibilities: a covalent POI ligand, a covalent E3 ligase ligand, and dual covalent POI and E3 ligase ligands. While it is conceivable that there are advantages to bis-covalent PROTACs, currently no examples have been reported, and thus far the majority of covalent PROTACs have focused on covalent POI ligands. This observation may be the result of the aforementioned resurgence in TCIs, which has provided a multitude of covalent inhibitors that can be readily converted into PROTACs via installation of a linker and E3 ligase ligand. However, there is a clear disadvantage to this approach; covalent warheads necessarily react in a 1 to 1 stoichiometry with the POI, which removes the possibility of engaging the catalytic PROTAC mechanism. Interestingly, as described above, the complexity of combining these two approaches means that deconvoluting the contribution of the two parts has proven difficult. In fact, in publications detailing the generation of covalent PROTACs targeting the oncogenic protein BTK, contradictory results have been reported. In one instance, Harling and co-workers observed no degradation of BTK using covalent PROTACs based on the known covalent BTK inhibitor ibrutinib, regardless of whether an IAP or CRBN binder was used as the E3 ligand.16 In contrast, their highly homologous reversible PROTACs were shown to successfully degrade the target. However, Xue et al. were able to show degradation of BTK using both CRBN and VHL based covalent POI PROTACs, some of which utilized ibrutinib as the covalent POI ligand.17 Considering these publications together, they suggest that degradation can be achieved for covalent POI PROTACs with appropriate optimization of the linker. However, in 2020 London and co-workers were successful in generating Ibrutinib based covalent BTK PROTACs, but the rate of covalent modification was thought to be slower than the rate of degradation, and the authors concluded that degradation was likely driven by non-covalent binding.18 As if to underline this point, the C481S mutant of BTK, which abrogates covalent binding of ibrutinib, was degraded by the “covalent” BTK PROTACs generated in the study. These publications together demonstrate the complexities of designing covalent PROTACs, and the need to fully understand the contribution of the covalent warhead. This militates for more thorough kinetic investigations of both covalent bond formation and degradation of the desired target. The other issue raised by these studies is that, due to their appropriation of known covalent ligands, they have not successfully utilized the covalent bond formation to reduce the molecular weight of the resultant PROTAC. Consequently, these compounds are expected to suffer from the same physical chemical properties and permeability issues observed for standard reversible PROTACs. Covalent POI PROTACs that de novo utilize the covalent bond formation to drive the discovery process, and optimize both covalent bond formation and degradation in parallel may generate more drug-like molecules. Moreover, utilizing covalent bond formation in the screening of fragments may identify non-functional binding sites on previously intractable proteins, which could subsequently be transformed into functional molecules through deployment of the PROTAC mechanism.
In the area of covalent E3 PROTACs, unlike covalent POI PROTACs, there is no repository of small molecule covalent E3 ligase binders, and so efforts have focused on utilizing the covalent mechanism to identify new E3 ligases to generate novel PROTACs. In fact, despite over 600 E3 ligases being encoded in the human genome, only 4 (VHL, CRBN, IAP and MDM2) have been widely utilized for PROTACs.7 To the best of our knowledge, there are no reports of known E3 ligase ligands being converted into covalent binders, likely due to the lack of reactive cysteines within their binding pockets. However, other covalent warheads have been successfully demonstrated to form covalent linkages with non-cysteine amino-acid side chains, e.g. tyrosine,19,20 and conceivably these could be utilized to generate covalent E3 ligands. Currently, efforts in the area of covalent E3 ligase PROTACs have focused on the use of activity-based protein profiling (ABPP) platforms to harness the power of proteomics to identify covalent ligands for novel E3 ligases, and subsequently transform them into PROTACs. Nomura and co-workers used just such a methodology to identify RNF114 as the target of the covalent natural product nimbolide, which reacts with a cysteine in a region that is predicted to be intrinsically disordered.21,22 Subsequently, linking known BRD4 binder JQ1 to nimbolide generated a PROTAC capable of degrading BRD4. In a follow up study, the Nomura lab embarked on a covalent fragment screen, via a gel-based competitive ABPP assay, against RNF114, and identified a moderately selective and synthetically more tractable ligand, EN219.21 Subsequently, they demonstrated that despite EN219's modest selectivity profile, it could be utilized to generate covalent PROTACs active against BRD4 and BCR-ABL. Interestingly, these BCR-ABL covalent PROTACs displayed selectivity against c-ABL, whereas previous BCR-ABL degraders showed the opposite selectivity.23
Concurrently, Cravatt and co-workers were utilizing reactive covalent “scout” fragments to identify new E3 ligases.24 Scouts are promiscuous covalent small molecules that engage a broad fraction of the targetable cysteines, which were discovered by chemo-proteomics experiments.25 By linking the scouts to SLF, a known binder of FKBP12, the team was able to identify a scout-SLF pair that degraded FBPK12. Through rigorous chemo-proteomics and genetic validation experiments, DCAF16 was identified as the E3 ligase responsible, and was further validated as a generalizable E3 ligase for PROTACs through the generation of covalent DCAF16-BRD4 PROTACs. Interestingly, DCAF16 is only present in the nuclear cellular fraction, and thus this methodology should allow for another level of control in protein degradation. Moreover, despite complete degradation of the POI, target engagement experiments demonstrated that only a fraction of DCAF16 had been covalently labelled by the warhead, suggesting that unreacted DCAF16 would still be available to perform its necessary functions, potentially abrogating any safety concerns associated with ligase depletion. All of the above examples have identified covalent bond formation via cysteine residues; however, as mentioned above, other covalent warheads have been demonstrated to react with nucleophilic non-cysteine amino-acid side chains. Despite this, there are currently no reports utilizing these warheads to broaden which E3 ligases can be targeted covalently. Moreover, we have demonstrated that sulfonyl fluoride (SF) warhead containing fragments can react with tyrosine residues to generate covalent bonds, but unlike cysteine targeting warheads the bond formation is driven by Ki not kinact.26 Therefore, in the case of SF fragments it may be possible to identify the E3 ligases through ABPP, and subsequently design out the sulfonyl fluoride warhead to generate non-covalent binders.
Finally, researchers have also investigated the use of reversible-covalent warheads on both the POI and E3 ligands to generate reversible-covalent PROTACs. Reversible covalent inhibitors provide the same advantages associated with TCIs, but unlike their irreversible counterparts, the covalent linkage breaks upon unfolding of the target protein and the small molecule is “recycled” to bind to another target. Due to the rapid reversible nature of the warhead, covalent bond formation is driven by the non-covalent interactions of the small molecule with the desired target, and indiscriminate protein modification is suppressed, potentially reducing toxicity risks. In addition, in the area of reversible covalent PROTACs the ability to break the covalent linkage means that the catalytic PROTAC mechanism can still function, because reversible covalent inhibitors do not adhere to the 1 to 1 stoichiometry of the irreversible covalent PROTACs described previously. Although other reversible covalent warheads exist, the majority of studies so far have focused on highly electron deficient olefins, which react with the sulfur of cysteine. Unfortunately, the reversible nature of these warheads complicates analysis, mainly due to the fact that the workhorse of covalent inhibitors, mass spectrometry, is rendered ineffective. Consequently, when reversible covalent inhibitors are used to generate PROTACs the results can be difficult to interpret. As with the covalent POI PROTACs, entry into this area is easiest via the conversion of known reversible covalent inhibitors into PROTACs, and currently two reversible covalent POI ligand PROTACs targeting BTK have been described.18,27 Possibly the most interesting finding from these studies was reported by Guo et al., who demonstrated that their reversible covalent POI PROTAC displayed intracellular accumulation 10 to 16 fold higher than their non-covalent and irreversible POI PROTACs, respectively.27 This observation was attributed to the rapid reversible nature of the warhead and its interactions with either intracellular glutathione, acting as a sink to enrich the PROTAC in the cell, or through interaction with free cysteine residues on the cell surface helping to mediate enhanced cellular uptake. The precise mechanism was beyond the scope of the work, but does suggest that other possible benefits could arise when deploying the reversible covalent warheads in the PROTACs space. It should be noted, however, that despite London and co-workers studying similar reversible-covalent BTK inhibitors no such accumulation was observed for their compounds,18 and thus this effect appears to be nongeneralizable.
So far, only a single example of reversible covalent E3 ligase ligands exists, in which bardoxolone was used to engage KEAP1 via its cyano-enone moiety, and when coupled to JQ1 the resultant PROTAC was demonstrated to degrade BRD4 in a dose and proteasome dependent manner.28 Unfortunately, the direct detection of all of bardoxolone's cellular targets has proven challenging, again demonstrating the extra complexities associated with the reversible covalent inhibitor space, and thus the authors could not definitively rule out that the observed degradation was due to another E3 ligase.
As we can see from the work outlined above, the area of covalent and reversible covalent PROTACs is nascent, but has demonstrated an immense amount of potential. Presently, the current examples have only partially delivered on the postulated promises for covalent PROTACs. For reversible and irreversible covalent POI PROTACs, initial studies have focused on the transformation of known ligands into PROTACs, and these studies have identified some of the complexities associated with covalent PROTACs. However, we believe that the true power of this approach will emerge when drug discovery programs are initiated with the concept of covalent POI PROTACs in mind from the start. For some targets, a covalent mode of action may be the only viable option for targeting “undruggable proteins”, which lack a well-defined binding site (e.g. protein–protein interactions, intrinsically disordered proteins, etc.). Consequently, hit finding strategies should be designed from the outset to discover covalent fragments or covalent lead-like compounds, which are later transformed into covalent PROTACs. In this way, the advantages of covalent bond formation can not only impact the overall properties of the molecule, but allow for the targeting of intractable proteins.
In contrast, the current covalent E3 PROTACs that have been reported are noticeably smaller in size, no doubt due to the fact that they were identified using fragment based ABPP approaches. However, it is as yet unclear if the reduced size of these molecules will have the desired beneficial effect on physical chemical properties and permeability due to the very early nature of these programs. Meanwhile, publications continue to emerge in the area of covalent E3 PROTACs that use covalent bond formation to discover new E3 ligase ligands as novel starting points for PROTACs.29 In the future, this approach may lead to the discovery of E3 ligases with tissue or, in the case of oncology, tumor restricted expression, and allow for the development of highly targeted therapies. In these cases, the covalent bond formation may serve two purposes, the first to permit the identification of the E3 through ABPP based approaches, and the second to allow targeting of the ligases which may be intractable to reversible ligands. For now, these methodologies are being used to identify other heterobifunctional molecules capable of recruiting different parts of the cellular machinery, e.g. deubiquitinases.30 Evidently, this field of research displays a huge amount of promise, and is so early in its journey that all the possible combinations have yet to be disclosed e.g. irreversible-covalent POI ligand reversible-covalent E3 ligase ligand PROTACs. In the end, only with more research and further optimization will we discover if these initial findings can be optimized into true clinical candidates like their non-covalent PROTAC cousins that have preceded them.
Conflicts of interest
There is no conflict of interest to declare.References
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, The resurgence of covalent drugs, Nat. Rev. Drug Discovery, 2011, 10(4), 307–317 CrossRef CAS PubMed
.
- E. Vita, 10 years into the resurgence of covalent drugs, Future Med. Chem., 2021, 13(2), 193–210 CrossRef CAS PubMed
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(15), 8554–8559 CrossRef CAS PubMed
- M. R. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. McFarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor, J. Med. Chem., 2014, 57(20), 8249–8267 CrossRef CAS PubMed
- T. Barf, T. Covey, R. Izumi, B. van de Kar, M. Gulrajani, B. van Lith, M. van Hoek, E. de Zwart, D. Mittag, D. Demont, S. Verkaik, F. Krantz, P. G. Pearson, R. Ulrich and A. Kaptein, Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile, J. Pharmacol. Exp. Ther., 2017, 363(2), 240–252 CrossRef CAS PubMed
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors, J. Med. Chem., 2020, 63(1), 52–65 CrossRef CAS
- S. Alabi and C. Crews, Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs, J. Biol. Chem., 2021, 100647 CrossRef CAS
- D. Kessler, M. Gmachl, A. Mantoulidis, L. J. Martin, A. Zoephel, M. Mayer, A. Gollner, D. Covini, S. Fischer, T. Gerstberger, T. Gmaschitz, C. Goodwin, P. Greb, D. Haring, W. Hela, J. Hoffmann, J. Karolyi-Oezguer, P. Knesl, S. Kornigg, M. Koegl, R. Kousek, L. Lamarre, F. Moser, S. Munico-Martinez, C. Peinsipp, J. Phan, J. Rinnenthal, J. Sai, C. Salamon, Y. Scherbantin, K. Schipany, R. Schnitzer, A. Schrenk, B. Sharps, G. Siszler, Q. Sun, A. Waterson, B. Wolkerstorfer, M. Zeeb, M. Pearson, S. W. Fesik and D. B. McConnell, Drugging an undruggable pocket on KRAS, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(32), 15823–15829 CrossRef CAS
- J. Rudolph, J. Settleman and S. Malek, Emerging Trends in Cancer Drug Discovery-From Drugging the “Undruggable” to Overcoming Resistance, Cancer Discovery, 2021, 11(4), 815–821 CrossRef CAS PubMed
- H. Kiely-Collins, G. E. Winter and G. J. L. Bernardes, The role of reversible and irreversible covalent chemistry in targeted protein degradation, Cell Chem. Biol., 2021, 1–17 Search PubMed
- R. Gabizon and N. London, The rise of covalent proteolysis targeting chimeras, Curr. Opin. Chem. Biol., 2021, 62, 24–33 CrossRef CAS PubMed
- G. J. Roth, N. Stanford and P. W. Majerus, Acetylation of prostaglandin synthase by aspirin, Proc. Natl. Acad. Sci. U. S. A., 1975, 72(8), 3073–3076 CrossRef CAS PubMed
- M. L. Drummond and C. I. Williams, In Silico Modeling of PROTAC-Mediated Ternary Complexes: Validation and Application, J. Chem. Inf. Model., 2019, 59(4), 1634–1644 CrossRef CAS PubMed
- S. D. Edmondson, B. Yang and C. Fallan, Proteolysis targeting chimeras (PROTACs) in 'beyond rule-of-five' chemical space: Recent progress and future challenges, Bioorg. Med. Chem. Lett., 2019, 29(13), 1555–1564 CrossRef CAS PubMed
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, The maximal affinity of ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(18), 9997–10002 CrossRef CAS PubMed
- C. P. Tinworth, H. Lithgow, L. Dittus, Z. I. Bassi, S. E. Hughes, M. Muelbaier, H. Dai, I. E. D. Smith, W. J. Kerr, G. A. Burley, M. Bantscheff and J. D. Harling, PROTAC-Mediated Degradation of Bruton's Tyrosine Kinase Is Inhibited by Covalent Binding, ACS Chem. Biol., 2019, 14(3), 342–347 CrossRef CAS PubMed
- G. Xue, J. Chen, L. Liu, D. Zhou, Y. Zuo, T. Fu and Z. Pan, Protein degradation through covalent inhibitor-based PROTACs, Chem. Commun., 2020, 56(10), 1521–1524 RSC
- R. Gabizon, A. Shraga, P. Gehrtz, E. Livnah, Y. Shorer, N. Gurwicz, L. Avram, T. Unger, H. Aharoni, S. Albeck, A. Brandis, Z. Shulman, B. Z. Katz, Y. Herishanu and N. London, Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs, J. Am. Chem. Soc., 2020, 142(27), 11734–11742 CrossRef CAS PubMed
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry, Angew. Chem., Int. Ed., 2014, 53(36), 9430–9448 CrossRef CAS PubMed
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, A study of the reactivity of S((VI))-F containing warheads with nucleophilic amino-acid side chains under physiological conditions, Org. Biomol. Chem., 2017, 15(45), 9685–9695 RSC
- M. Luo, J. N. Spradlin, L. Boike, B. Tong, S. M. Brittain, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function, Cell Chem. Biol., 2021, 28(4), 559–566 e15 CrossRef CAS PubMed
- B. Tong, J. N. Spradlin, L. F. T. Novaes, E. Zhang, X. Hu, M. Moeller, S. M. Brittain, L. M. McGregor, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL, ACS Chem. Biol., 2020, 15(7), 1788–1794 CrossRef CAS PubMed
- G. M. Burslem, A. R. Schultz, D. P. Bondeson, C. A. Eide, S. L. Savage Stevens, B. J. Druker and C. M. Crews, Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation, Cancer Res., 2019, 79(18), 4744–4753 CrossRef CAS PubMed
- X. Zhang, V. M. Crowley, T. G. Wucherpfennig, M. M. Dix and B. F. Cravatt, Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16, Nat. Chem. Biol., 2019, 15(7), 737–746 CrossRef CAS PubMed
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. Gonzalez-Paez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Proteome-wide covalent ligand discovery in native biological systems, Nature, 2016, 534(7608), 570–574 CrossRef CAS PubMed
- H. Mukherjee, N. Su, M. A. Belmonte, D. Hargreaves, J. Patel, S. Tentarelli, B. Aquila and N. P. Grimster, Discovery and optimization of covalent Bcl-xL antagonists, Bioorg. Med. Chem. Lett., 2019, 29(23), 126682 CrossRef CAS PubMed
- W. H. Guo, X. Qi, X. Yu, Y. Liu, C. I. Chung, F. Bai, X. Lin, D. Lu, L. Wang, J. Chen, L. H. Su, K. J. Nomie, F. Li, M. C. Wang, X. Shu, J. N. Onuchic, J. A. Woyach, M. L. Wang and J. Wang, Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry, Nat. Commun., 2020, 11(1), 4268 CrossRef PubMed
- B. Tong, M. Luo, Y. Xie, J. N. Spradlin, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Bardoxolone conjugation enables targeted protein degradation of BRD4, Sci. Rep., 2020, 10(1), 15543 CrossRef CAS PubMed
- N. J. Henning, A. G. Manford, J. N. Spradlin, S. M. Brittain, J. M. McKenna, J. A. Tallarico, M. Schirle, M. Rape and D. K. Nomura, Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications, bioRxiv, 2021, DOI:10.1101/2021.04.15.439993.
- N. J. Henning, L. Boike, J. N. Spradlin, C. C. Ward, B. Belcher, S. M. Brittain, M. Hesse, D. Dovala, L. M. McGregor, J. M. McKenna, J. A. Tallarico, M. Schirle and D. K. Nomura, Deubiquitinase-Targeting Chimeras for Targeted Protein Stabilization, bioRxiv, 2021, DOI:10.1101/2021.04.30.441959.
- M. Zeng, Y. Xiong, N. Safaee, R. P. Nowak, K. A. Donovan, C. J. Yuan, B. Nabet, T. W. Gero, F. Feru, L. Li, S. Gondi, L. J. Ombelets, C. Quan, P. A. Janne, M. Kostic, D. A. Scott, K. D. Westover, E. S. Fischer and N. S. Gray, Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C), Cell Chem. Biol., 2020, 27(1), 19–31 e6 CrossRef CAS PubMed
- M. J. Bond, L. Chu, D. A. Nalawansha, K. Li and C. M. Crews, Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs, ACS Cent. Sci., 2020, 6(8), 1367–1375 CrossRef CAS PubMed
- J. N. Spradlin, X. Hu, C. C. Ward, S. M. Brittain, M. D. Jones, L. Ou, M. To, A. Proudfoot, E. Ornelas, M. Woldegiorgis, J. A. Olzmann, D. E. Bussiere, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Harnessing the anti-cancer natural product nimbolide for targeted protein degradation, Nat. Chem. Biol., 2019, 15(7), 747–755 CrossRef CAS PubMed
- C. C. Ward, J. I. Kleinman, S. M. Brittain, P. S. Lee, C. Y. S. Chung, K. Kim, Y. Petri, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle and D. K. Nomura, Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications, ACS Chem. Biol., 2019, 14(11), 2430–2440 CrossRef CAS PubMed
- M. de Wispelaere, G. Du, K. A. Donovan, T. Zhang, N. A. Eleuteri, J. C. Yuan, J. Kalabathula, R. P. Nowak, E. S. Fischer, N. S. Gray and P. L. Yang, Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations, Nat. Commun., 2019, 10(1), 3468 CrossRef PubMed
- L. Peng, Z. Zhang, C. Lei, S. Li, Z. Zhang, X. Ren, Y. Chang, Y. Zhang, Y. Xu and K. Ding, Identification of New Small-Molecule Inducers of Estrogen-related Receptor alpha (ERRalpha) Degradation, ACS Med. Chem. Lett., 2019, 10(5), 767–772 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |