
Structure-based design of haloperidol analogues as inhibitors of acetyltransferase Eis from Mycobacterium tuberculosis to overcome kanamycin resistance†
Ankita
Punetha‡
a,
Keith D.
Green‡
a,
Atefeh
Garzan‡
a,
Nishad
Thamban Chandrika
a,
Melisa J.
Willby
b,
Allan H.
Pang
 a,
Caixia
Hou
a,
Selina Y. L.
Holbrook
a,
Kyle
Krieger
c,
James E.
Posey
b,
Tanya
Parish
c,
Oleg V.
Tsodikov
*a and
Sylvie
Garneau-Tsodikova
*a
a,
Caixia
Hou
a,
Selina Y. L.
Holbrook
a,
Kyle
Krieger
c,
James E.
Posey
b,
Tanya
Parish
c,
Oleg V.
Tsodikov
*a and
Sylvie
Garneau-Tsodikova
*a
aDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone Street, Lexington, KY 40536, USA. E-mail: oleg.tsodikov@uky.edu; sylviegtsodikova@uky.edu
bLaboratory Branch, Division of Tuberculosis Elimination, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, Centers for Disease Control and Prevention, Atlanta, GA 30329, USA
cCenter for Global Infectious Disease Research, Seattle Children's Research Institute, 307 Westlake Avenue N, Seattle, WA 98109, USA
First published on 5th October 2021
Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a deadly bacterial disease. Drug-resistant strains of Mtb make eradication of TB a daunting task. Overexpression of the enhanced intracellular survival (Eis) protein by Mtb confers resistance to the second-line antibiotic kanamycin (KAN). Eis is an acetyltransferase that acetylates KAN, inactivating its antimicrobial function. Development of Eis inhibitors as KAN adjuvant therapeutics is an attractive path to forestall and overcome KAN resistance. We discovered that an antipsychotic drug, haloperidol (HPD, 1), was a potent Eis inhibitor with IC50 = 0.39 ± 0.08 μM. We determined the crystal structure of the Eis-haloperidol (1) complex, which guided synthesis of 34 analogues. The structure–activity relationship study showed that in addition to haloperidol (1), eight analogues, some of which were smaller than 1, potently inhibited Eis (IC50 ≤ 1 μM). Crystal structures of Eis in complexes with three potent analogues and droperidol (DPD), an antiemetic and antipsychotic, were determined. Three compounds partially restored KAN sensitivity of a KAN-resistant Mtb strain K204 overexpressing Eis. The Eis inhibitors generally did not exhibit cytotoxicity against mammalian cells. All tested compounds were modestly metabolically stable in human liver microsomes, exhibiting 30–60% metabolism over the course of the assay. While direct repurposing of haloperidol as an anti-TB agent is unlikely due to its neurotoxicity, this study reveals potential approaches to modifying this chemical scaffold to minimize toxicity and improve metabolic stability, while preserving potent Eis inhibition.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is currently the deadliest bacterial disease. In 2019, TB killed 1.2 million people, and 10 million new TB cases were reported.1 For many decades, the first-line therapeutic treatment of active TB disease has been a combination of four antibiotics: isoniazid, rifampin, ethambutol, and pyrazinamide. Because of the reliance on the same drugs, lack of new first-line anti-TB agents, inconsistent adherence to this regimen or use of inappropriate treatment regimens, and inadequate diagnosis and testing, drug-resistant Mtb strains evolved and spread globally. Drug-resistant TB is classified as multidrug-resistant TB (MDR TB) if it is resistant to the first-line drugs isoniazid and rifampin. In 2019, there were approximately 465![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases of rifamycin-resistant or MDR TB globally.1
000 new cases of rifamycin-resistant or MDR TB globally.1
Kanamycin (KAN) is an injectable drug that has been used to treat MDR TB. One of the clinically significant mechanisms of resistance to the commonly used MDR TB drug KAN is the upregulation of a versatile acetyltransferase known as enhanced intracellular survival (Eis) protein.2 The overexpression of Eis is a result of point mutations in the eis promoter or in the 5′ untranslated region of the gene that encodes transcription factor WhiB7, as observed in ∼40% of a broad set of KAN-resistant Mtb clinical isolates.2–5 Eis inactivates several aminoglycosides by transferring an acetyl group from acetyl coenzyme A (AcCoA) to one or more amino groups of an aminoglycoside,6 by a random sequential mechanism.7 The modified aminoglycoside is then unable to bind the 16S rRNA in the 30S ribosomal subunit, resulting in its failure to arrest protein synthesis in the bacterial cell. Structurally diverse aminoglycosides are accommodated in the large substrate binding site of Eis,8 which has evolved to acetylate the lysine-rich C-terminal region of a histone-like Mtb protein HupB.9–11 Acetylation and deacetylation of HupB change the chromosome compaction,9 thereby regulating gene expression. Given the broad selection of Eis substrates, developing a new aminoglycoside that would be efficacious against Mtb and would not succumb to Eis acetylation would likely be difficult.
An alternative approach to overcoming Eis-mediated KAN resistance is to develop Eis inhibitors that could serve as adjuvant therapeutics for aminoglycosides in the setting of drug-resistant TB. The inhibitors would block Eis, thereby preventing aminoglycoside inactivation and overcome KAN resistance of TB or reduce chances of development of such resistance. Our group has discovered and optimized several structural families of Eis inhibitors, generating compounds that exhibited high potency of Eis inhibition in vitro (some in nM range).12–18 Importantly, some of these compounds restored KAN sensitivity of the KAN-resistant Mtb strain K204.14,16Mtb K204 is an H37Rv strain variant that harbors a single, clinically observed mutation in the eis promoter (C-14T), which results in Eis upregulation and renders the cells resistant to KAN.2 The crystal structures of Eis-inhibitor complex obtained in these studies as well as analysis of inhibition kinetics revealed that these inhibitors were competitive with aminoglycosides.17,19 The crystal structures and a recent Eis mutagenesis study revealed inhibitor-Eis interactions critical for inhibitor binding.18
Our goal is the development of an aminoglycoside adjuvant anti-TB therapeutic that would overcome and prevent emergence of resistance to aminoglycosides. The process of bringing new chemical entities to the market is slow and expensive. Therefore, we decided to explore a possibility of repurposing agents that have other uses in clinic for their use against TB. Development of such compounds may be faster and cheaper than development of entirely novel chemical entities because this process can be informed by prior preclinical and clinical studies. Usually, extensive preclinical and clinical data are available on a drug in clinical use. Some preclinical data are likely to be directly relevant, if the same compound is considered for another indication. Other data would likely guide and streamline the modified testing process. In some cases, preclinical data on analogues of a drug are also available, which would aid development of new analogues. Orphan diseases and other rare conditions are examples of areas where research and development funds are limited and where drug repositioning could be a viable economical approach.20,21 Because of known toxicities of aminoglycosides, it is especially important not to add a toxic compound to a cocktail with an aminoglycoside. With that in mind, a potential outcome of our approach is an aminoglycoside adjuvant that may even lower the required therapeutic dose of an aminoglycoside, so that the cumulative toxic effects of the drug combination are not exacerbated further. In the pursuit of our goal, we discovered that haloperidol (1), an FDA-approved antipsychotic drug, was a potent Eis inhibitor. The study of the mechanism of Eis inhibition by haloperidol (1) and the initial development of haloperidol analogues as Eis inhibitors are presented herein.
Results
Discovery and validation of haloperidol (1) as an Eis inhibitor
To identify new Eis inhibitors, we used our previously established high-throughput screening (HTS) aminoglycoside acetylation activity assay12 with Mtb Eis to test ∼2500 compounds that were used in clinic or were already FDA-approved. The Z′ score of this HTS assay was 0.65, indicating that the assay was robust. Antipsychotic drugs haloperidol (1) and droperidol (DPD) were strong HTS hit with Z scores of 13 and 7, respectively. The structure of compound 1 (Fig. 1) differed from the structures of the previously identified Eis inhibitor scaffolds,12–19 prompting a detailed mechanistic characterization of inhibition by this compound. In order to validate 1 as an Eis inhibitor, we performed the same aminoglycoside acetylation assay as a dose–response using haloperidol (1) that we synthesized. Haloperidol (1) exhibited robust inhibition of Eis, with an IC50 = 0.39 ± 0.08 μM (Table 1). In comparison, DPD (Fig. 1), which had several structural differences from 1, exhibited weak Eis inhibition with an IC50 = 12.2 ± 0.9 μM. To test compound 1 as an Eis inhibitor in Mtb cells, we determined the effect of this compound on the MIC of KAN for the clinically relevant KAN-resistant strain Mtb K204. Mtb K204 is resistant to KAN, with MICKAN > 10 μg mL−1. When used at 100 μM, 1 increased the sensitivity of Mtb K204 to KAN more than 2-fold (MICKAN = 5 μg mL−1; Table 1), whereas the sensitivity of the parent strain H37Rv to KAN (MICKAN ≤ 1.25 μg mL−1) remained the same in the presence of 1. These observations validated 1 as an Eis inhibitor both in the test tube and in Mtb cells.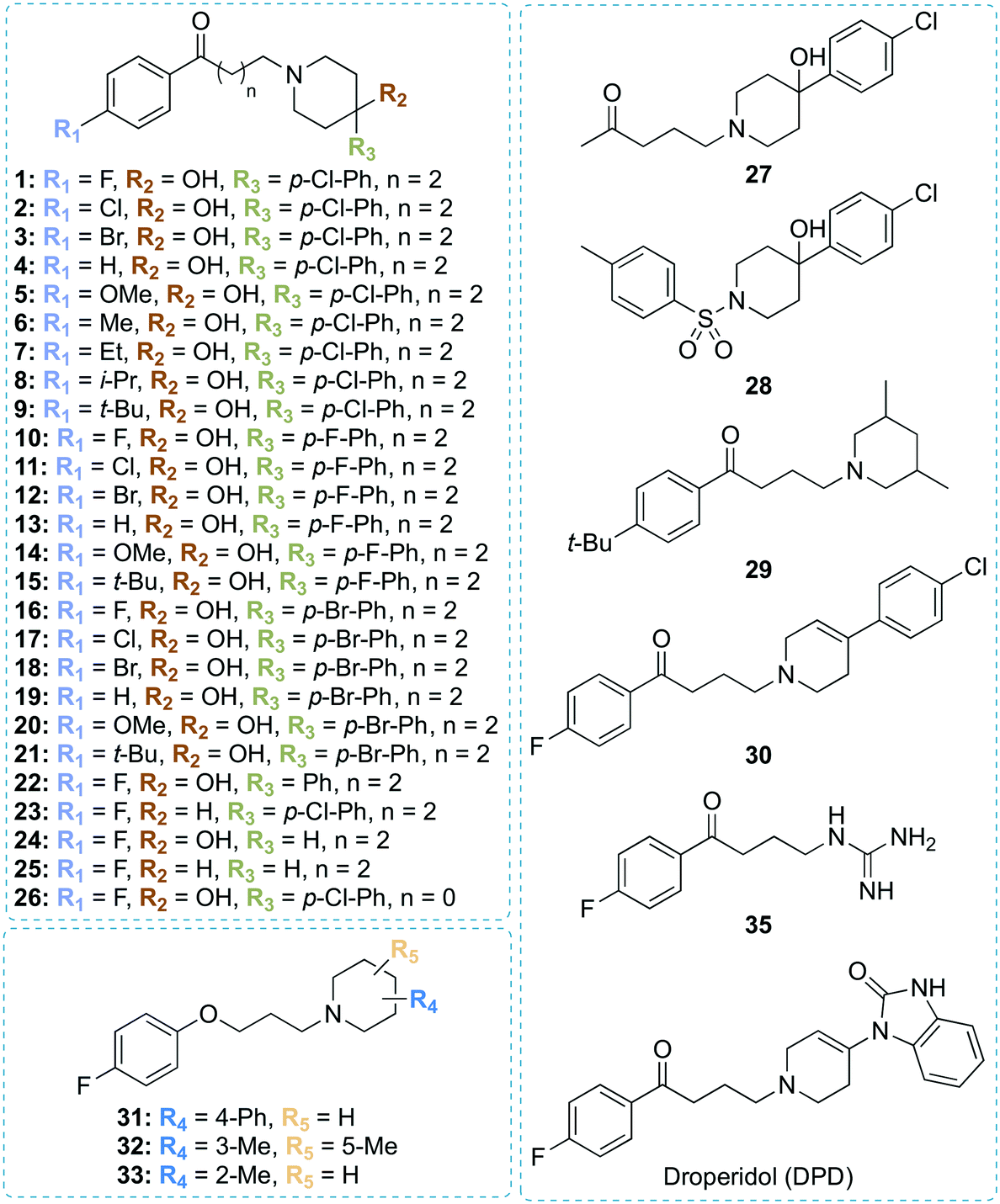 | ||
| Fig. 1 Chemical structures of all compounds synthesized and investigated in this study. | ||
| Cpd # | clogPa |
logDb |
R1 | R2 | R3 | n | IC50 (μM) against Eis | Mtb MICKAN (μg mL−1) | Concentration tested (μM) | |
|---|---|---|---|---|---|---|---|---|---|---|
| H37Rv | K204 | |||||||||
| a Values determined by ChemDraw 1.19 (or by Molinspiration). b As predicted at pH 7.0 by ChemAxon (https://disco.chemaxon.com/calculators/demo/plugins/logd/). c An estimate based on the fitting of the sigmoidal equation. d Measurements for the Mtb mc26230. e Measurements for the Mtb mc26230-K204 strain. f Toxic indicates that the growth of the Mtb strain tested was completely inhibited by the compound in the absence of KAN.Note: the R1, R2, and R3 groups represent those in Fig. 1. | ||||||||||
| 1 | 3.49 (4.30) | 2.31 | F | OH | p-Cl-Ph | 2 | 0.39 ± 0.08 | ≤1.25 | 5 | 100 |
| 2 | 3.89 (4.81) | 2.78 | Cl | OH | p-Cl-Ph | 2 | 5.1 ± 1.2 | |||
| 3 | 4.16 (4.94) | 2.94 | Br | OH | p-Cl-Ph | 2 | >200 | ≤1.25 | >10 | 100 |
| 4 | 3.34 (4.13) | 2.17 | H | OH | p-Cl-Ph | 2 | 5.3 ± 1.6 | ≤1.25 | >10 | 100 |
| 5 | 3.21 (4.19) | 2.01 | OMe | OH | p-Cl-Ph | 2 | ∼40c | ≤1.25 | >10 | 100 |
| 6 | 3.82 (4.58) | 2.68 | Me | OH | p-Cl-Ph | 2 | ∼100c | |||
| 7 | 4.24 (5.05) | 3.12 | Et | OH | p-Cl-Ph | 2 | ∼40c | |||
| 8 | 4.57 (5.64) | 3.41 | i-Pr | OH | p-Cl-Ph | 2 | >200 | |||
| 9 | 5.04 (5.84) | 3.71 | t-Bu | OH | p-Cl-Ph | 2 | >200 | Toxicf | Toxic | 100 |
| 10 | 3.09 (3.78) | 1.89 | F | OH | p-F-Ph | 2 | 0.76 ± 0.09 | ≤1.25 | 5 | 100 |
| 11 | 3.49 (4.30) | 2.36 | Cl | OH | p-F-Ph | 2 | 2.6 ± 0.5 | ≤1.25 | 10 | 100 |
| 12 | 3.76 (4.43) | 2.52 | Br | OH | p-F-Ph | 2 | 6.6 ± 0.3 | ≤1.25 | 10 | 100 |
| 13 | 2.94 (3.62) | 1.75 | H | OH | p-F-Ph | 2 | 3.1 ± 0.6 | ≤1.25 | 10 | 100 |
| 14 | 2.81 (3.67) | 1.58 | OMe | OH | p-F-Ph | 2 | ∼42c | ≤1.25 | >10 | 100 |
| 15 | 4.64 (5.32) | 3.29 | t-Bu | OH | p-F-Ph | 2 | ∼200 | |||
| 16 | 3.76 (4.43) | 2.46 | F | OH | p-Br-Ph | 2 | 0.52 ± 0.10 | ≤1.25 | 5 | 100 |
| 17 | 4.16 (4.94) | 2.92 | Cl | OH | p-Br-Ph | 2 | 1.1 ± 0.3 | ≤1.25 | 10 | 100 |
| 18 | 4.44 (5.07) | 3.09 | Br | OH | p-Br-Ph | 2 | >200 | ≤1.25 | >10 | 100 |
| 19 | 3.61 (4.26) | 2.32 | H | OH | p-Br-Ph | 2 | 2.2 ± 0.6 | ≤1.25 | 10 | 100 |
| 20 | 3.48 (4.32) | 2.15 | OMe | OH | p-Br-Ph | 2 | 5.6 ± 1.5 | ≤1.25 | >10 | 100 |
| 21 | 5.31 (5.97) | 3.85 | t-Bu | OH | p-Br-Ph | 2 | >200 | Toxic | Toxic | 100 |
| 22 | 2.94 (3.62) | 1.62 | F | OH | Ph | 2 | 0.85 ± 0.08 | ≤1.25d | >10e | 100 |
| 23 | 4.70 (5.34) | 3.09 | F | H | p-Cl-Ph | 2 | 1.1 ± 0.1 | Toxicd | Toxice | 100 |
| 24 | 1.32 (1.22) | −0.014 | F | OH | H | 2 | 0.69 ± 0.10 | ≤1.25d | >10e | 100 |
| 25 | 2.63 (3.10) | 0.88 | F | H | H | 2 | 0.62 ± 0.13 | ≤1.25d | >10e | 100 |
| 26 | 2.92 (3.75) | 3.06 | F | OH | p-Cl-Ph | 0 | ∼100c | |||
| 27 | 2.19 (2.53) | 0.83 | ∼40c | |||||||
| 28 | 3.40 (3.80) | 3.11 | >200 | |||||||
| 29 | 4.97 (5.36) | 2.71 | ∼200 | |||||||
| 30 | 4.37 (4.86) | 3.86 | 0.56 ± 0.05 | Toxicd | Toxice | 100 | ||||
| 31 | 4.42 (4.93) | 2.18 | 14 ± 5 | |||||||
| 32 | 3.70 (4.08) | 0.86 | ∼41c | |||||||
| 33 | 3.22 (3.70) | 0.83 | >200 | |||||||
| 35 | 1.14 (1.03) | −1.49 | 1.1 ± 0.1 | ≤1.25d | >10e | 100 | ||||
| DPD | 1.92 (3.40) | 1.40 | 12.2 ± 0.9 | |||||||
Crystal structure of the Eis-haloperidol complex
To understand the mechanism of inhibition of Eis by 1, we determined a crystal structure of the Eis-1 complex at the resolution of 2.03 Å (Fig. 2A and C and Table S1†). Strong omit difference (Fo–Fc) electron density for compound 1 enabled its unambiguous positioning (Fig. 2C). Haloperidol (1) was bound in the aminoglycoside substrate binding site, defined by the crystal structure of Eis-tobramycin complex (Fig. 2B; PDB ID: 4JD6).22 The N-linked p-fluorobutyrophenone moiety (ring A) attached to the piperidine ring (ring B) of compound 1 projected into a hydrophobic pocket lined by residues Trp13, Trp36, Val40, Leu63, Met65, Phe84, and the aliphatic stem of Arg37 (Fig. 2C). The only polar atom in the vicinity of the fluorine was the main chain nitrogen of Arg37, with the F–N distance of 3.5 Å. The other side of the molecule pointed towards the entrance of the binding site, with the p-chlorophenyl substituent (ring C) at position 4 of the piperidine ring protruding into the solvent and being mostly disordered. Our previously reported Eis inhibitors were also observed to be bound in the substrate binding site.12–19 For a representative comparison, we chose the crystal structure of Eis in complex with inhibitor SGT335 from a previous study (Fig. 2D; PDB ID: 6P3U18). The p-fluorobutyrophenone moiety of 1 interacted with Eis similarly to the fluoro-substituted acetophenone moiety of inhibitor SGT335. In contrast, the carbonyl oxygens of the two compounds were oppositely oriented: the oxygen of 1 formed a water-mediated hydrogen bond with the main chain amide nitrogen of Ile28, whereas the oxygen of SGT335 formed a direct hydrogen bond with the hydroxyl group of Ser83. This conformational difference was an apparent consequence of the unique chemical features of 1 and SGT335. The aliphatic portion of the linker between rings A and B of 1 made hydrophobic contacts with Ile28 and Trp36, and steric contacts with the hydroxyl group of Ser83 and of the C-terminal carboxyl group. The C-terminal carboxyl group of Eis was proposed to be involved directly in the acetyl transfer mechanism of Eis.6 The nitrogen of the piperidine ring (ring B) was bound in the electronegative environment of the C-terminal carboxyl group, Asp26 and Glu401. The hydroxy substitution at position 4 of ring B was not observed in the electron density, indicative of the rotational dynamics of this ring.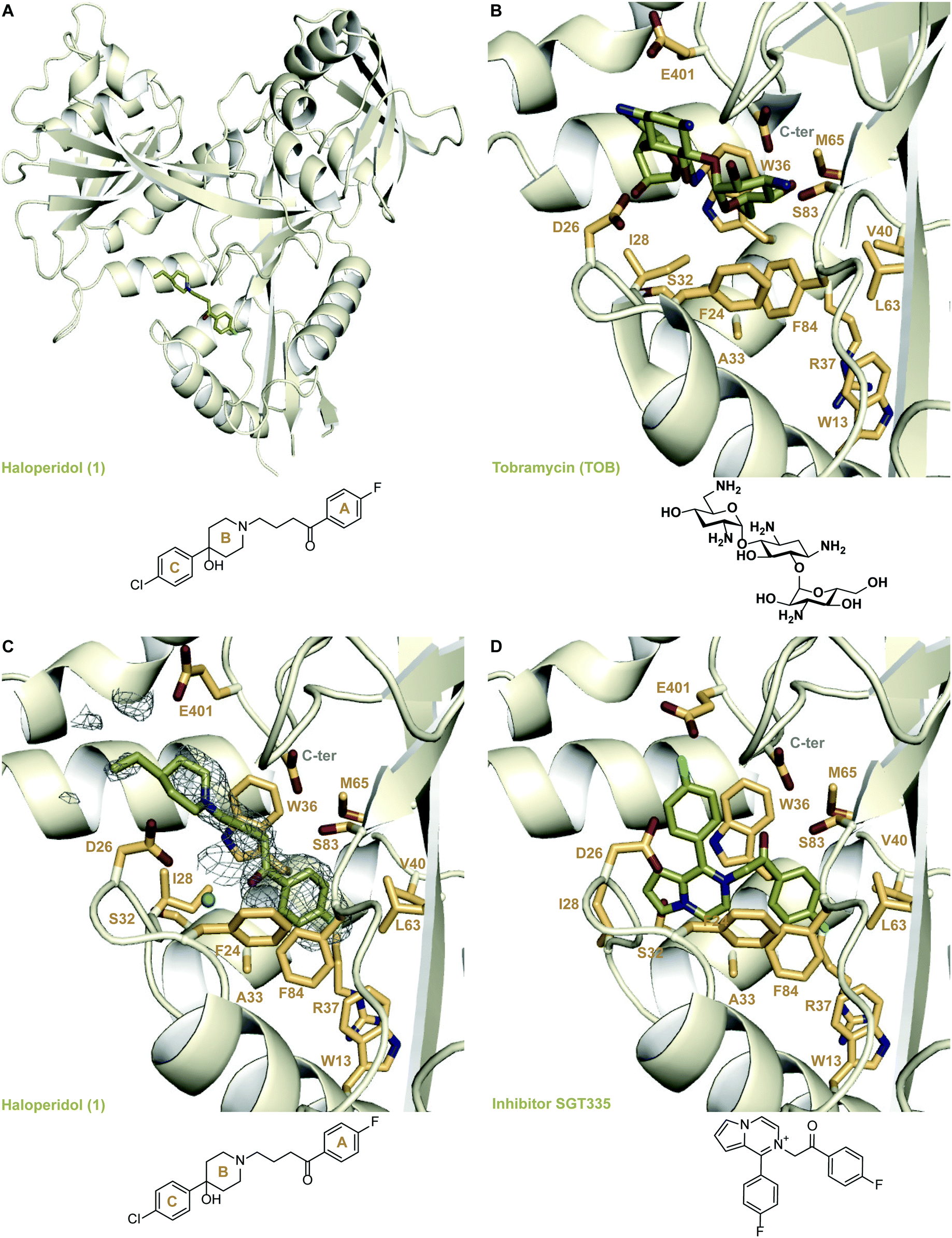 | ||
| Fig. 2 Crystal structures of Eis in complexes with its inhibitors and tobramycin. A. The crystal structure of Eis in complex with haloperidol (1) (PDB ID: 6X10). B. The crystal structure of Eis in complex with tobramycin (PDB ID: 4JD622); a zoomed-in view of the substrate binding site. C. A zoomed-in view of the Eis-inhibitor interface in the crystal structure of Eis-1 complex. The mesh is a polder mFo–DFc omit map contoured at 3.5σ. D. The zoomed-in view of the Eis-inhibitor interface in the crystal structure of Eis-SGT335 complex (PDB ID: 6P3U18). | ||
Design and chemical synthesis of new analogues
We used these observed interactions of compound 1 with Eis in our analogue design (Fig. 1). Based on the structure of the nonpolar pocket accommodating ring A of 1 and similar groups of our previously reported inhibitors, only small hydrophobic substitutions were possible at the R1 position of this ring. Therefore, fluoro, chloro, bromo, methoxy, methyl, ethyl, iso-propyl, and tert-butyl substituents were planned at the R1 position. Because 1 was already large enough to extend into the solvent, lengthening of the aliphatic linker was not planned. Ring C did not stably interact with the protein; therefore, we designed different chemical features that would replace ring C and/or the linker or eliminated ring C altogether. In these new analogues, a systematic variation of R1 groups was carried out for a specific R3 substituent. Compound 1 contains a hydroxyl group attached to ring B at position 4 (R2 position). Because the structure of Eis-1 complex indicated a dynamic character, as a result of which this hydroxyl group was not stably interacting with Eis, to test the contribution of this hydroxyl group, we did not introduce it in nine analogues (23, 25, 29–35). In 30, a double bond was introduced in ring B. Several analogues (24, 25, 29, 32–35) lacked ring C. In 29, 32, and 33, to optimize interaction in the binding pocket, methyl groups were introduced in ring B, while keeping the R2 and R3 positions unsubstituted. To examine the importance of the length of the linker, we shortened it in 26, while preserving all other features of 1. As a control, to investigate the likely key contribution of ring A to Eis binding, we synthesized analogue 27 without this ring. Our previously reported sulfonamide inhibitor SGT449 potently inhibited Eis.18 The crystal structure of Eis-SGT449 complex showed that the sulfonamide group forms a hydrogen bond with the main chain nitrogen of Ile28 (PDB ID: 6P3T18). To investigate an impact of introducing a sulfonamide group into the scaffold of 1, we synthesized analogue 28 in which the entire linker was replaced by a sulfonyl group. The entrance to the Eis substrate binding site is negatively charged. This prompted us to design analogues 34 (Scheme 1) and 35 that both contained ring A and either a terminal amino group or a guanidinium group in place of ring B, respectively. These positively charged groups were predicted to bind in the same electronegative environment as the amino group of 1 observed in the crystal structure. The carbonyl group was removed in three analogues (31–33). Analogue 31 was synthesized with a piperidine ring attached to an unsubstituted phenyl ring at the R4 position.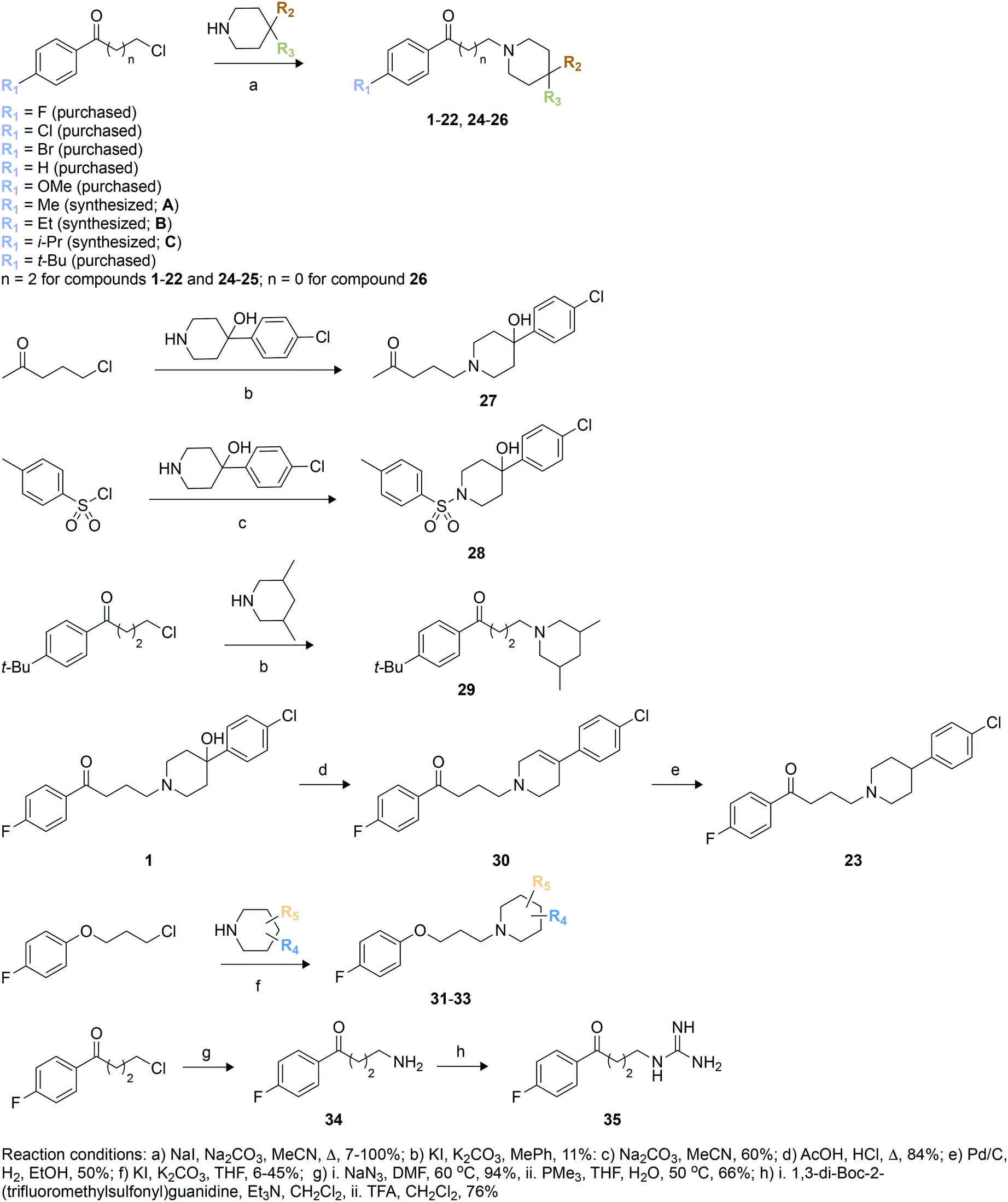 | ||
| Scheme 1 Synthetic schemes used to generate haloperidol and its analogues. | ||
To synthesize these compounds, we followed a general procedure reported for a similar molecule.23 The synthesis of most of the compounds involved formation of the individual butyrophenone and piperidine components, followed by their fusion by a simple substitution reaction (Scheme 1). For this, 4′-unsubstituted and substituted 4-chlorobutyrophenones or 4′-fluoro-2-chloroacetophenone were either purchased or constructed in a Friedel–Crafts reaction of unsubstituted and substituted benzene with 4-chlorobutyryl chloride. The reaction of 4-chlorobutyryl chloride with substituted benzene in presence of a Lewis acid (AlCl3) resulted in the formation of 4′-substituted-4-chlorobutyrophenones A–C in 62–77% yield. These butyrophenones and acetophenones were then reacted with substituted piperidines in the presence of a base to generate compounds 1–22 and 24–26 in 7–100% yield (Scheme 1; reaction conditions “a”). The reaction involved the use of a base (Na2CO3) and a catalytic amount of NaI in acetonitrile under reflux condition. The reaction of 5-chloro-2-pentanone with 4-(4-chlorophenyl)-4-hydroxypiperidine in the presence of K2CO3 and a catalytic amount of KI resulted in compound 27 in 11% yield (Scheme 1; reaction conditions “b”). Similarly, compound 29 was prepared by reacting 1-(4-(tert-butyl)phenyl)-3-chloropropan-1-one and 3,5-dimethylpiperidine in the presence of K2CO3 and a catalytic amount of KI mixture in 24% yield (Scheme 1; reaction conditions “b”). Compound 28 was synthesized in 60% yield with p-toluenesulfonyl chloride chosen as the electrophile and treated with 4-(4-chlorophenyl)-4-hydroxypiperidine in the presence of Na2CO3 (Scheme 1; reaction conditions “c”). The acid catalyzed dehydration reaction of compound 1 formed compound 30 in 84% yield (Scheme 1; reaction conditions “d”). A subsequent Pd/C mediated reduction of compound 30 yielded compound 23 in 50% yield (Scheme 1; reaction conditions “e”). Similarly to the synthesis of compounds 1–19, the reaction of various substituted piperidine nucleophiles with 3-(4-fluorophenoxy)propyl chloride in the presence of K2CO3 and KI resulted in compounds 31–33 in 6–45% yield (Scheme 1; reaction conditions “f”). A nucleophilic substitution reaction of 4′-fluoro-4-chlorobutyrophenone with NaN3 followed by reduction with PMe3 generated compound 34 in 66% yield (Scheme 1; reaction conditions “g”). The amine of 34 was treated with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine, followed by Boc deprotection by TFA to yield compound 35 in 76% yield (Scheme 1; reaction conditions “h”).
The structure–activity relationship study of haloperidol analogues
We examined the structure–activity relationship (SAR) for the rationally designed analogues of 1. The inhibitory potency of the analogues (IC50) was determined by measuring their dose–response effect on the Eis-catalyzed acetylation of KAN (Table 1 and Fig. S88 and S89†). For a rigorous interpretation of the data, we determined additional crystal structures of Eis in complex with three potent analogues (10, 30, 35) as well as DPD (Fig. 3 and 4 and Table S1†). | ||
| Fig. 3 Zoomed-in views of the Eis-inhibitor interfaces from crystal structures of Eis-10, Eis-30 complexes, and the inhibitor structures. A. The inhibitor binding site in the Eis-10 (SGT543) complex (PDB ID: 6X6I). B. The inhibitor binding site in the Eis-30 (SGT572) complex (PDB ID: 6X7A). In both panels, the mesh is a polder mFo–DFc omit map contoured at 3.5σ. Water molecules are indicated as turquoise balls. | ||
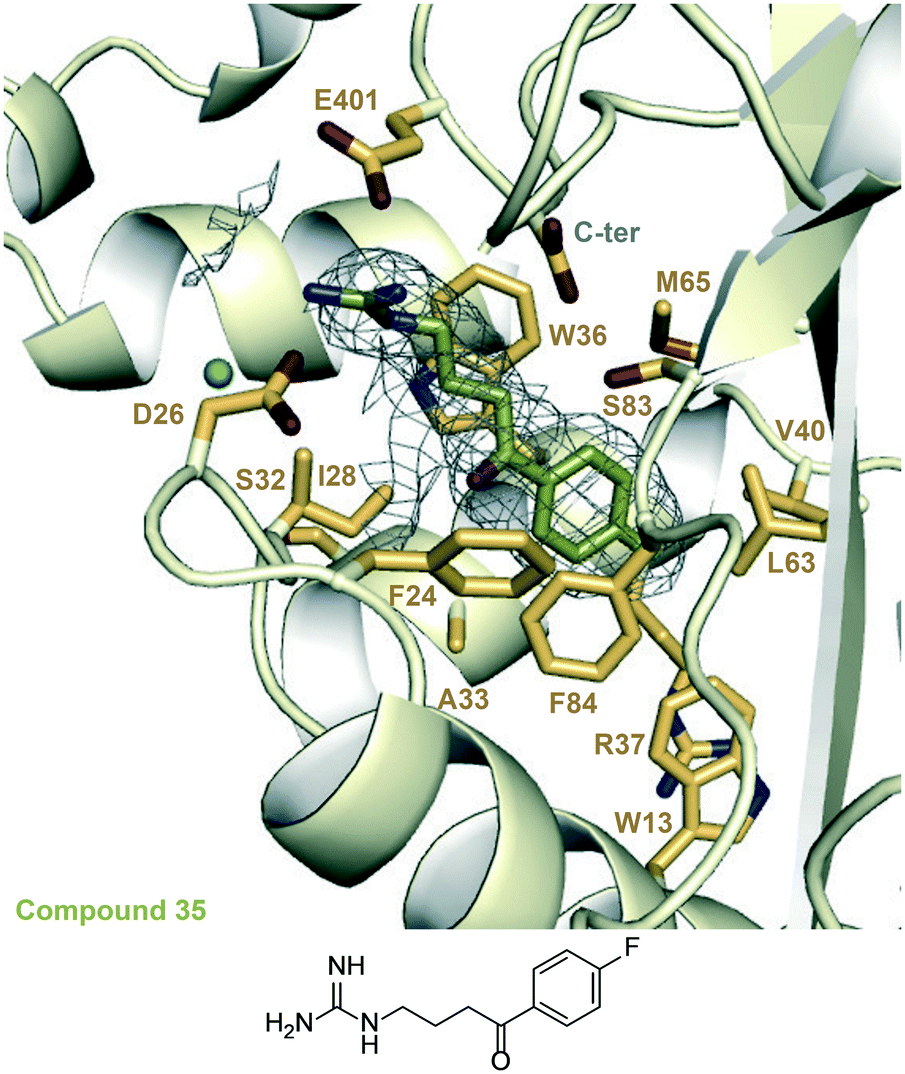 | ||
| Fig. 4 A zoomed-in view of the Eis-inhibitor interface in the crystal structure of the Eis-35 (SGT1264) complex (PDB ID: 6X6Y) and the structure of 35. The mesh is a polder mFo–DFc omit map contoured at 3.5σ. Water molecules are indicated as turquoise balls. | ||
In another set of analogues (10–15), we installed R3 = p-F-Ph, and varied R1. In this set, the fluorine (in 10) was also the most preferred substitution at the R1 position, although the IC50 value of 10 was 2-fold higher than that of 1. The bulkiest substituents at the R1 position tested, methoxy (in 14) and tert-butyl (in 15) groups, were not tolerated, as expected. Chlorine (in 11), hydrogen (in 13), and bromine (in 12) substituents lowered the potency 3–9-fold from that of 10, with chlorine being most favored and the bulky bromine least favored. The same SAR rationale as with set 1–9 likely applied. We determined a crystal structure of Eis in complex with 10 (Fig. 3A), also at a high resolution of 1.90 Å. Compound 10 was bound at the same site as 1, in the same conformation, but minor structural differences were observed. In the Eis-10 complex, the hydroxyl group was well resolved. This group formed water-mediated hydrogen bonds with Glu203 involving two ordered water molecules that were not observed in the Eis-1 complex due to the rotational dynamics of this region of the molecule. It appeared that this net conformational entropic penalty paid in the Eis-10 complex to establish these hydrogen bonds was not fully compensated by the enthalpic terms. The rotamer of Glu401 was different from that in the Eis-1 complex, possibly as a result of the limited conformational dynamics of ring B. As with the Eis-1 complex, the R3 substituent consisting of p-fluorophenyl was not well resolved and was omitted from the model.
In a third set, where R3 = p-Br-Ph (16–21), fluorine was also the most preferred substituent at R1 followed by chlorine, hydrogen, methoxy, bromine, and tert-butyl (Table 1). Here, a methoxy substituent was more tolerated than in the previous sets, whereas the large tert-butyl was not tolerated, as in the other sets. The preference of fluoro substitution at R1 in all three sets is apparently dictated by a combination of the size and polarity of this substitution in the confines of the mostly, but not entirely, hydrophobic binding pocket.
To assess the impact of ring C substituents, we synthesized compound 22, which had the same structure as 1, except that it contained unsubstituted ring C. Compound 22 showed two-fold higher potency compared to 1, with an IC50 value of 0.85 ± 0.08 μM, two-fold higher than for 1. In the crystal structure of Eis-1 complex ring C was largely disordered, consistent with the lack of a major effect of chlorine removal. Nevertheless, the chlorine likely somewhat favorably interacted with the protein.
Because substituents in ring C did not significantly change the potency of the inhibitors, we tested larger changes at R3. We synthesized compound 24 without ring C and 25, which additionally lacked the hydroxyl group at R2. The IC50 values of 24 and 25 were 0.69 ± 0.10 μM and 0.62 ± 0.13 μM, respectively. The similar potencies of 24, 25, and 1 were consistent with the crystal structures, where most of ring C was disordered, and indicated that this ring did not contribute significantly to protein binding.
Among our previously designed Eis inhibitors, sulfonamide inhibitors exhibited strong inhibitory potency.14 In analogue 28, the entire linker, including the carbonyl group, were replaced by a sulfonamide group. As with the shortening of the linker, this drastic modification abolished Eis inhibition.
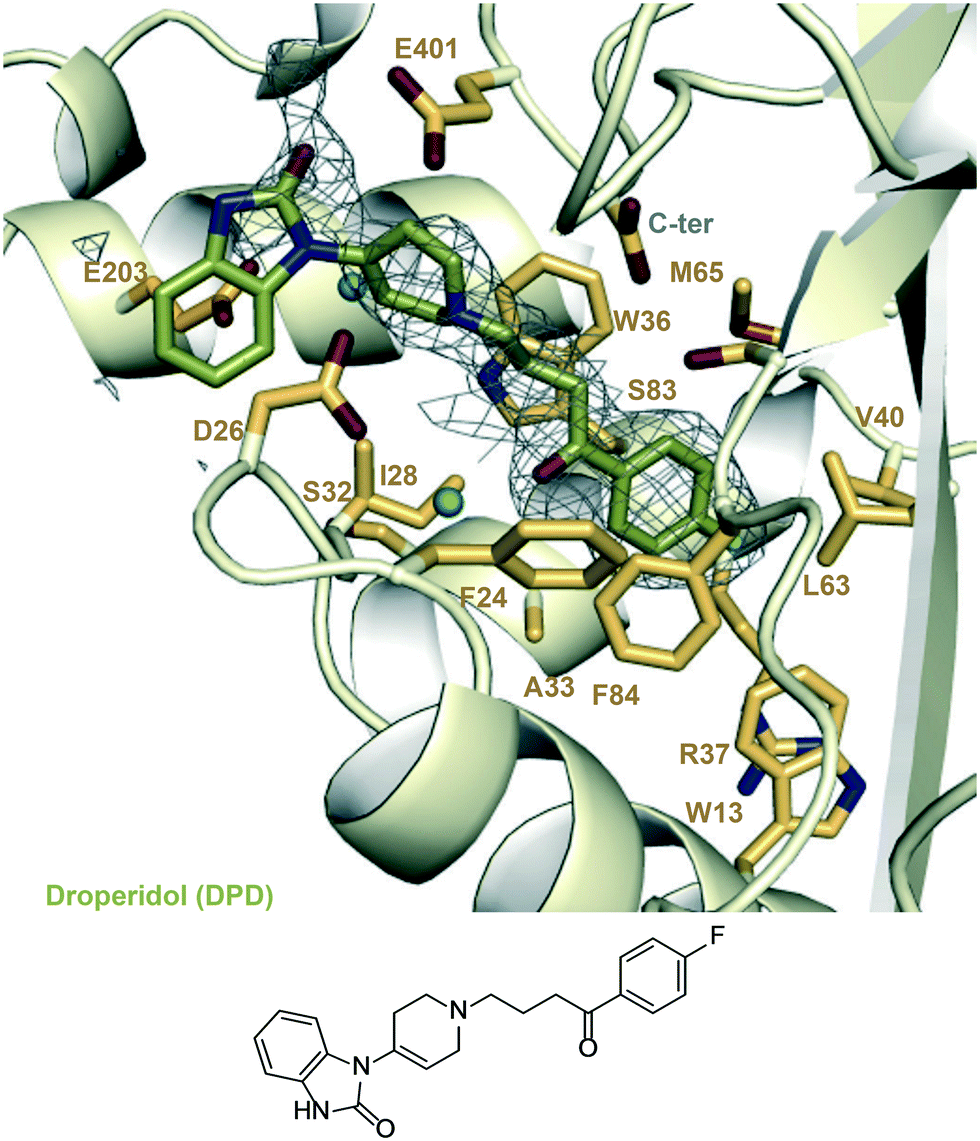 | ||
| Fig. 5 A zoomed-in view of the Eis-inhibitor interface from crystal structure of Eis-DPD complex (PDB ID: 6X6G) and the structure of DPD. The mesh is a polder mFo–DFc omit map contoured at 3.5σ. Water molecules are indicated as turquoise balls. | ||
Determining the mode of Eis inhibition and selectivity towards Eis by haloperidol analogues
To understand the mechanism of action of the haloperidol analogues, some of the submicromolar inhibitors, 1, 10, and 16, were tested in a series of kinetic experiments. The Michaelis–Menten kinetics of the Eis enzyme were determined in the presence of four concentrations of inhibitor (0, 0.25, 0.5, and 1 μM; Table S2,†Fig. 6A–C). The global analysis of these data, visualized by the Lineweaver–Burk plots in Fig. 6D–F indicated that these three inhibitors were competitive with KAN. This result was in excellent agreement with the crystal structure, which showed that the inhibitors occupied the aminoglycoside binding site. This analysis also yielded Ki values of 0.39 ± 0.02 μM, 0.91 ± 0.22 μM and 0.24 ± 0.02 μM for 1, 10 and 16, respectively.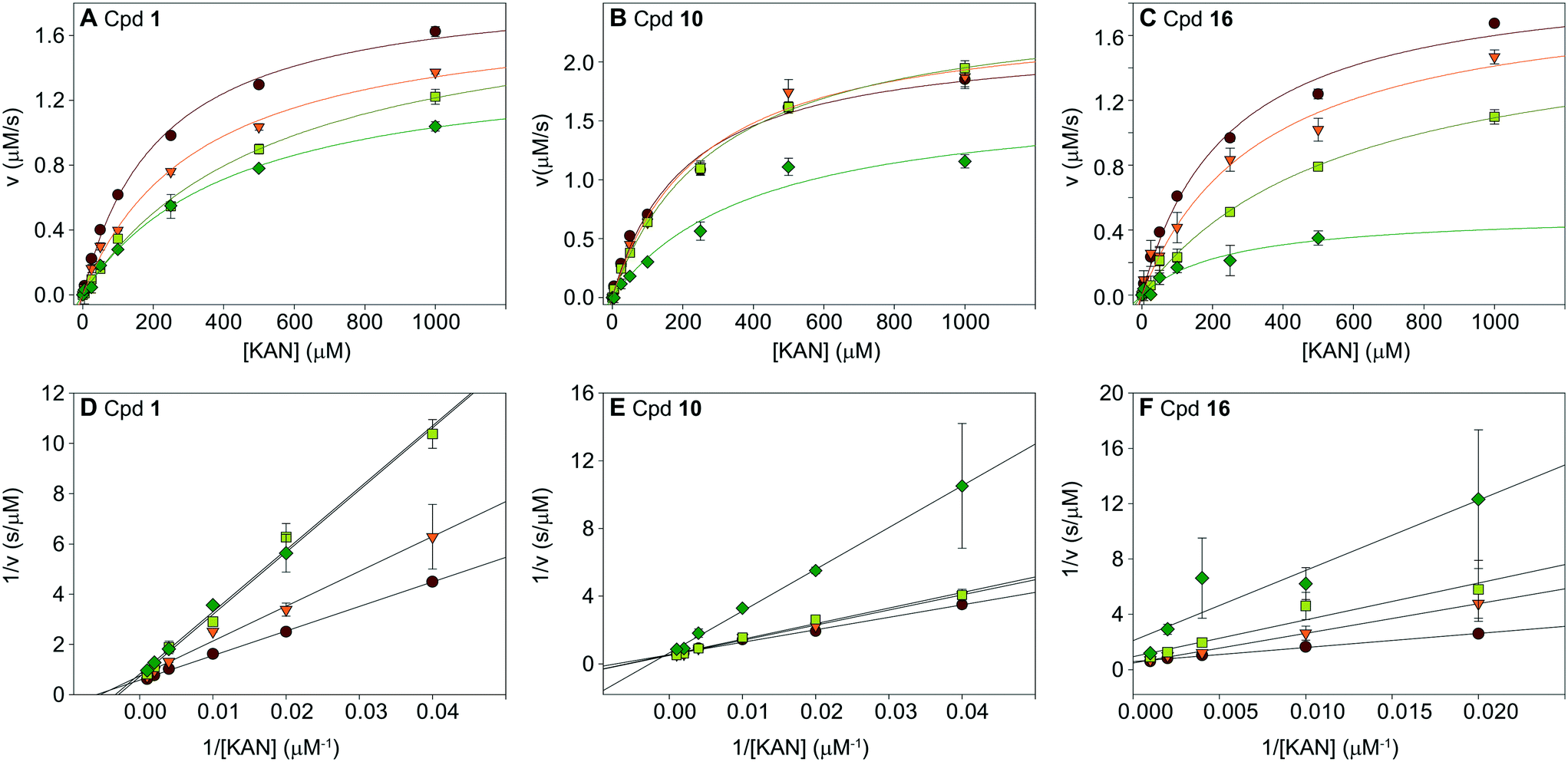 | ||
| Fig. 6 Analysis of inhibition kinetics for inhibitors 1, 10, and 16. Michaelis–Menten analysis is shown in A–C, and the Lineweaver–Burk representation of this analysis is in D–F. These data indicate that the molecules are competitive with KAN. The Ki values of inhibitors 1, 10, and 16 are 0.39 ± 0.02 μM, 0.91 ± 0.22 μM, and 0.24 ± 0.02 μM, respectively (Table S2†). | ||
To establish if the inhibitors were specific towards Eis or general to other AACs, we tested the specificity of compounds 1, 2, 3, 5, and 9, with three regiospecific AACs: AAC(6′)-Ie from the bifunctional AAC(6′)-Ie/APH(2′′)-Ia,24 AAC(3)-IV,24 and AAC(2′)-Ic.6 All compounds were found to be inactive at 200 μM against all enzymes tested, indicating that the haloperidol derivatives were selective towards Eis.
Overcoming Eis-mediated KAN resistance in the Mtb culture
In order to test whether haloperidol (1) and its analogues inhibit Eis in Mtb cells and abolish Eis-mediated resistance to KAN, we measured the effect of these compounds on the MIC of KAN (MICKAN) for strain K204. This strain was generated from the KAN-susceptible H37Rv (MICKAN ≤ 1.25 μg mL−1) by introducing a clinically relevant point mutation in the eis promoter.2 As a result of the mutation, the MICKAN of Mtb K204 is greater than 10 μg mL−1. We observed that inhibitors whose IC50 against Eis was low-μM had little to no effect on MICKAN of K204 (Table 1), whereas three out of six sub-μM inhibitors (1, 10, and 16) decreased resistance to KAN of K204, i.e., lowering its MICKAN to 5 μg mL−1, when used at 100 μM. None of these three compounds were toxic to Mtb when used without KAN. These results indicated that these three compounds indeed functioned by inhibiting Eis in Mtb cells. Notably, the three compounds were haloperidol (1) and its close analogues 10 and 16 that differed from 1 only by the identity of the halogen at R3. Other substituents that preserved the potency against Eis might have rendered those analogues unable to traverse the cell envelope of Mtb.One other sub-μM Eis inhibitor, compound 30, was toxic to Mtb in the absence of KAN, obscuring potential inhibition of Eis. Three other compounds (9, 21, and 23), two of which (9 and 21) were not Eis inhibitors and the other (23) was a low-μM inhibitor, were also toxic to Mtb without KAN, indicating that the mechanism of toxicity was not related to Eis inhibition. This mechanism remains to be investigated.
Antibacterial activity of haloperidol (1) analogues
Because some of the compounds were toxic to Mtb even in the absence of KAN, we tested all the compounds as single agents against seven additional mycobacterial strains (strains A–G; Table 2). Many of the compounds showed little to no effect on the mycobacterial strains tested (MIC = 31.3 to >62.5 μM). However, compounds 7, 8, 9, 15, 17, 21, 30, and 33 displayed good to moderate activity (MIC = 1 to 15.6 μM) against at least two mycobacterial strains. All the tested compounds performed better than haloperidol (1) (MIC = 31.3 to >62.5 μM) and bromperidol (16) (MIC ≥ 62.5 μM). Mycobacterium smegmatis (strain A) and Mycobacterium abscessus (strain B) were the most resistant to all compounds tested, while Mycobacterium bovis BCG (strain E) and Mtb H37Ra (strain F) were the most sensitive. Compounds 9 and 21, both with R1 = t-Bu, were found to have the lowest (best) overall MIC values against mycobacteria. The increase in potency (i.e., lower MIC values) from compound 6 (R1 = Me) to 7 (R1 = Et) to 8 (R1 = i-Pr) to 9 (R1 = t-Bu) indicated that an increase in bulkiness at the R1 position was favored. This fact means that the mechanism of action for these compounds against mycobacteria is different from their Eis inhibition, as these compounds are not active as Eis inhibitors. When making a pairwise comparison between compounds 1 (R2 = OH) and 23 (R2 = H) as well as between compounds 24 (R2 = OH) and 25 (R2 = H), we observed that the identity of the R2 group had no impact on the MIC values of these inactive compounds. When examining the effect of the R3 substituents by comparing the following sets of compounds active against mycobacteria: 2 (R3 = p-Cl-Ph) vs.11 (R3 = p-F-Ph) vs.17 (R3 = p-Br-Ph); 3vs.12vs.18; and 9vs.15vs.21; we observed that the para-bromophenyl group displayed the best MIC values followed by the para-chlorophenyl and then by the para-fluorophenyl group. Compounds 6, 7, 8, 9, 21, 22, 23, 30, and 35 were also tested against Mtb mc26230 (strain G) to check if the MIC results were consistent with those of Mtb H37Ra (strain F). Except for compound 30, all compounds were within a dilution factor of 2.| Cpd # | A | B | C | D | E | F | G |
|---|---|---|---|---|---|---|---|
| Bacterial strains: A = M. smegmatis mc2155, B = M. abscessus ATCC 19977, C = M. intracellulare ATCC 13950, D = M. avium ATCC 25921, E = M. bovis BCG ATCC 35734, F = M. tuberculosis H37Ra ATCC NRS22, G = M. tuberculosis mc26230. Abbreviations: HPD = haloperidol; BPD = bromperidol; DPD = droperidol; INH = isoniazid. Note: NT = not tested. | |||||||
| 1 (HPD) | >62.5 | >62.5 | 62.5 | 62.5 | 62.5 | 31.3 | NT |
| 2 | >62.5 | >62.5 | >62.5 | 62.5 | 62.5 | 15.6 | NT |
| 3 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | 31.3 | NT |
| 4 | >62.5 | >62.5 | >62.5 | 62.5 | 62.5 | 62.5 | NT |
| 5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | 62.5 | NT |
| 6 | 62.6 | >62.5 | >62.5 | >62.5 | 15.6 | 31.3 | 31.3 |
| 7 | 15.6 | 62.5 | 15.6 | 62.5 | 2 | 15.6 | 15.6 |
| 8 | 15.6 | 31.3 | 7.8 | >62.5 | 1 | 7.8 | 7.8 |
| 9 | 15.6 | >62.5 | 7.8 | 7.8 | 3.9 | 3.9 | 2 |
| 10 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 11 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 12 | >62.5 | >62.5 | 62.5 | 62.5 | 62.5 | 31.3 | NT |
| 13 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 14 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 15 | 62.5 | >62.5 | 31.3 | 31.3 | 15.6 | 15.6 | NT |
| 16 (BPD) | >62.5 | >62.5 | 62.5 | 62.5 | 62.5 | >62.5 | NT |
| 17 | 31.3 | >62.5 | 62.5 | >62.5 | 15.6 | 7.8 | NT |
| 18 | 31.3 | >62.5 | >62.5 | >62.5 | >62.5 | 7.8 | NT |
| 19 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | 62.5 | NT |
| 20 | >62.5 | >62.5 | >62.5 | 62.5 | 62.5 | 31.3 | NT |
| 21 | 15.6 | >62.5 | 7.8 | 7.8 | 3.9 | 3.9 | 4 |
| 22 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >64 |
| 23 | >62.5 | >62.5 | 31.3 | >62.5 | 62.5 | 62.5 | 64 |
| 24 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 25 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 26 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 27 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 28 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | >62.5 | NT |
| 29 | 62.5 | 62.5 | 62.5 | 62.5 | 31.3 | 31.3 | NT |
| 30 | >62.5 | >62.5 | 15.6 | 31.3 | 15.6 | 7.8 | 32 |
| 31 | >62.5 | 62.5 | 62.5 | >62.5 | >62.5 | 31.3 | NT |
| 32 | 62.5 | >62.5 | >62.5 | >62.5 | 15.6 | 62.5 | NT |
| 33 | 15.6 | >62.5 | 7.8 | 15.6 | 3.9 | 7.8 | NT |
| 35 | >62.5 | >62.5 | >62.5 | >62.5 | NT | 31.3 | >64 |
| DPD | >62.5 | >62.5 | >62.5 | >62.5 | NT | 31.3 | NT |
| INH | 3.9 | >62.5 | 3.9 | 62.5 | ≤1 | ≤1 | NT |
Some of the compounds with the best MIC values against the mycobacterial strains (7–9, 15, and 21) were also tested against eleven non-mycobacterial pathogens to determine the spectrum of their antibacterial activity (strains H–R; Table S3†). The compounds had little to no effect on the growth of these bacteria except for compounds 9 and 21 on E. coli MC1061 (strain M), however, this strain was designed to be more sensitive to drug treatment.
Synergy of haloperidol (1) analogues with anti-TB drugs
Previous studies showed that bromperidol (16) was synergistic with spectinomycin (SPT) against Mtb.25 Prompted by this observation, we tested first whether haloperidol (1) was synergistic with anti-TB drugs, aminoglycosides, macrolides, azoles, or other antibiotic compounds against Mycobacterium smegmatis (strain A; Table S4†). Haloperidol (1) displayed synergism with SPT and clarithromycin (CLR), but did not show any synergism or even showed antagonism with other compounds.Compounds 1, 2, 9, 10, 11, 15, 16, 17, and 21 were then tested against M. smegmatis (strain A; Table S5†) in combination with SPT and CLR, which displayed a 16-fold and 4-fold reduction in MIC values in the presence of 8 μg mL−1 of compound 1, respectively, and clofazimine (CLO) that displayed no change in MIC values when exposed to 1 in Table S4.† From these combinations it was found that compounds 2, 11, 16, and 17 showed synergy with CLR, as determined by FICI calculations. Interestingly, CLO and SPT were found to be synergistic with all nine haloperidol derivatives tested.
From the data gathered against M. smegmatis (strain A; Table S5†), we decided to examine compounds 2, 11, and 17, identical in R1 and R2, but representing the three main groups for R3 (p-chlorophenyl, p-fluorophenyl, and p-bromophenyl, respectively) for synergy with CLR, CLO, and SPT against the remaining mycobacteria. When tested against M. abscessus (strain B; Table S6†), compound 2 was additive with CLR and SPT, and synergistic with CLO. Compound 11 was additive with CLR and synergistic with CLO and SPT. Finally, compound 17 was synergistic with CLO and SPT. When tested against Mycobacterium intracellulare (strain C; Table S7†) and Mycobacterium avium (strain D; Table S8†) all combinations of antibiotics and haloperidol derivatives were additive. When tested against M. bovis BCG (strain E; Table S9†), compounds 2, 9, and 11 were additive with CLR and CLO. However, all three compounds were found to be synergistic with SPT. Mtb H37Ra (strain F; Table S10†) was only tested with SPT and the combinations with compounds 2, 11, and 17 were found to be additive. Altogether, these data indicated that the haloperidol derivatives, if not already toxic against mycobacteria, could have their effects amplified by co-treatment with SPT.
Analysis of cytotoxicity to mammalian cells
We measured the cytotoxic effect of six representative Eis inhibitors, 1, 2, 3, 5, 9, and 21, on three different mammalian cell lines: human adenocarcinoma (A549), human embryonic kidney (HEK-293), and mouse macrophage (J774A.1) (Fig. 7). Compound 1 was chosen as at 100 μM it partly restored the activity of KAN in Mtb K204 (Table 1). Compounds 2, 3, and 5 were selected to test if inhibition of Eis was correlated with cytotoxicity to mammalian cells. Compounds 9 and 21 were chosen to test if the toxicity to Mtb that these compounds displayed is correlated with mammalian cytotoxicity. Compounds 9 and 21 were toxic to all three cell lines at 25 μM. Compounds 1, 2, and 3 displayed no cytotoxic effect against HEK-293 cells at the highest concentration tested, 200 μM. Against A549 cells, 1 was very mildly cytotoxic in the range 50–200 μM with 80–70% cell survival, while 3 was less toxic, with 80–70% cell survival in the 100–200 μM range. In contrast, 2 showed no cytotoxic effect even at the highest concentration tested. Against J774A.1 cells, 1 showed cytotoxicity at 100 μM with 100% cell survival at 50 μM, while 2 exerted toxicity at 50 μM with 100% cell survival at 25 μM. Compound 3 was observed to have no cytotoxicity up to 100 μM with 100% cell survival. We observed that 5 was cytotoxic to J774A.1 cells at 100 μM, mildly toxic against A549 cells with approximately 78% cell survival at this concentration, and non-toxic against HEK-293 cells. These findings indicated that compounds 1, 2, 3, and 5 had better cytotoxicity profiles against various mammalian cells than 9 and 21. Toxicity to Mtb appeared to be associated with toxicity to the mammalian cells for these compounds, whereas Eis inhibition was not related to the cytotoxicity, as expected.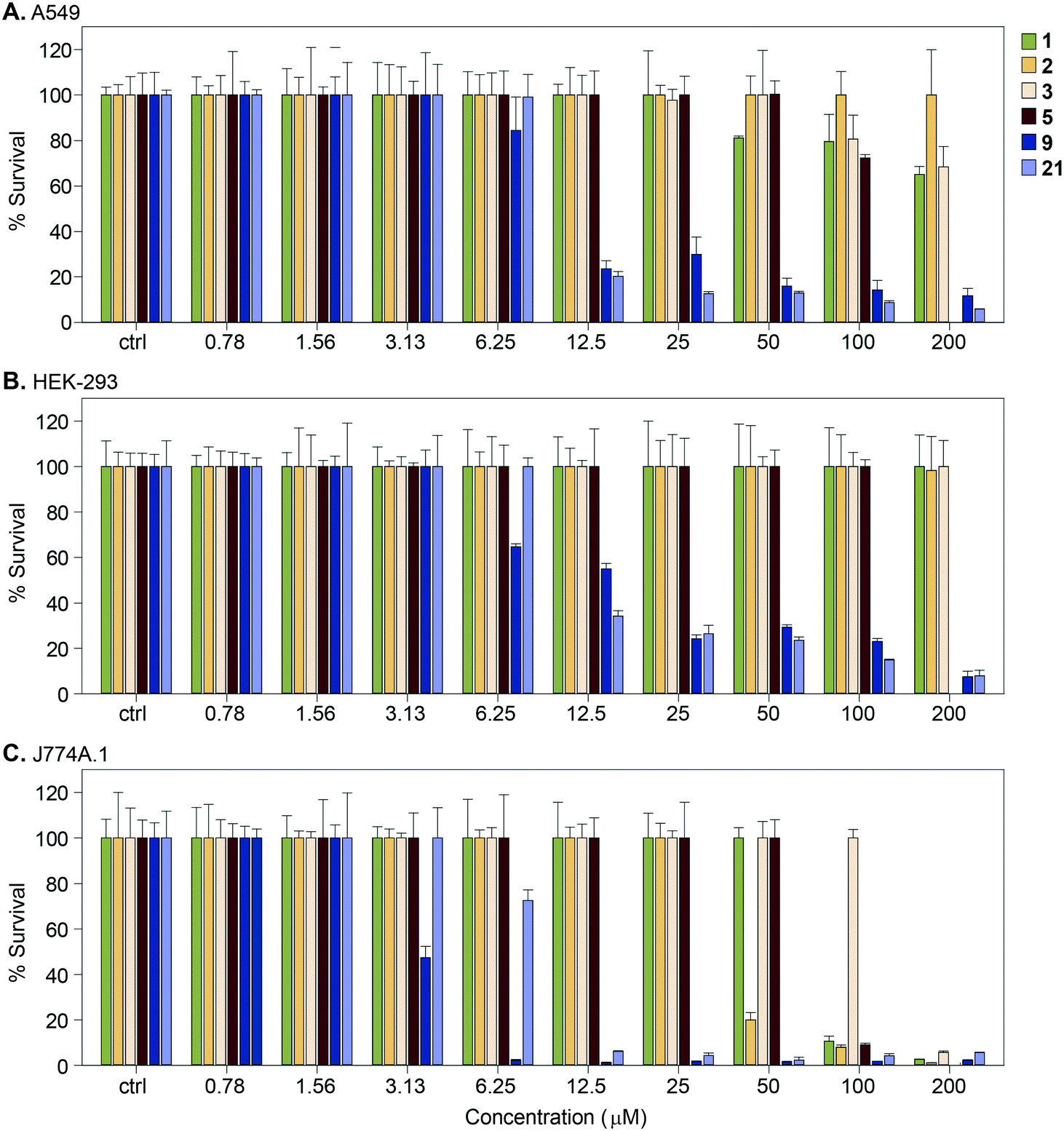 | ||
| Fig. 7 Graphs showing the cytotoxicity of selected compounds 1, 2, 3, 5, 9, and 21 against three mammalian cell lines normalized at 100%. A. A549, B. HEK-293, and C. J774A.1. | ||
Microsomal stability assessment
In the pipeline of any drug discovery, preclinical evaluation of drug metabolism plays a crucial role in drug optimization. We measured the metabolic stability of several haloperidol derivatives (9, 15, 17, 21, and 30) in human and mouse liver microsomes (Table 3). We chose analogues that were representative of the structural diversity among them and were predicted to be stable. These compounds displayed moderate to good stability in human microsomes and moderate to poor stability in mouse microsomes, with the exception of compound 15, which was moderately stable in both (50% and 48% of 15 metabolized, respectively). The electron donating vs. withdrawing nature of the moieties attached to the phenyl ring of these compounds appeared to play an important role in their microsomal stability. The stability in human microsomes was mainly affected by the nature of the R1 side chain. Compounds 9, 15, and 21 contain a t-Bu group present at R1 (i.e., the 4-position of the first phenyl ring), which could be prone to oxidation in both human and mouse liver microsomes (Table 3). The electron donating property of the t-Bu group at R1 results in an activated ring affecting its stability. These compounds could be metabolized by oxidation of the tert-butyl group. Compounds 17 and 30 with chloro and fluoro moieties at the R1 position exhibited high microsomal stability in humans, with 36% and 30% of the compound metabolized, respectively. This high stability is explained by the electron-withdrawing halogens deactivating the ring. The highest stability of 30 could be attributed to the electron withdrawing effect of the fluorine atom at the R1 position making the ring system highly electron deficient. The likely labile tertiary hydroxyl group at the R2 position is also absent in this compound. The elimination product is a double bond at the R2 position. The substituents with electron withdrawing effect seemed to stabilize both ends of the derivatives.Discussion
By rational structure-driven SAR, we dissected the molecular features of haloperidol-scaffold based Eis inhibitors that were responsible for Eis binding. Based on our chemical and structural analysis of Eis-haloperidol (1) and Eis-haloperidol analogue complexes, the important features were determined to be the linearly connected p-halo-phenyl moiety, the keto-alkyl moiety, and the piperidine moiety or a positively charged group in its place. These three structural features need to be appropriately spaced to establish the observed interactions with Eis.Even though haloperidol (1) and these first-generation derivatives were not highly potent in sensitizing Mtb K204 to KAN in the cell culture assays, likely due to poor penetration through the mycobacterial cell envelope, further tuning of the derivatives with the above SAR in mind could achieve improvement in cell-based activity. For launching a haloperidol derivative as an anti-TB drug, important aspects to consider are its metabolism and safety. Previous studies documented that haloperidol has high bioavailability whether that be orally (60–70%) or injected into the subcutaneous tissue (presumed 100%).26 Because of its lipophilicity, haloperidol is typically 90% bound to albumin, which gives the compound a high degree of distribution throughout the body.27,28 While binding to albumin could be considered a negative property, the Mtb in this study is grown in 1% bovine serum albumin and the compounds are still active, which is encouraging as the normal level of albumin in human blood is 3.5–5.5%. The pharmaceutical properties (i.e., logD) of compounds synthesized herein have not been experimentally determined, however, based on the predicted logD at pH 7.0, only a few compounds (24, 27, and 32–35) have drastically different logD values (Table 1), suggesting that at this pH, they will likely behave similarly to haloperidol. Haloperidol is well known to be extensively metabolized in the liver with its major biotransformation pathways having been extensively characterized.29–41 Its quaternary pyridinium metabolites were associated with neurotoxicity.37,42,43 As we showed here, the piperidine ring could be replaced by a positively charged group without a significant loss of potency. Microsomal stability assays demonstrated that liver metabolism of haloperidol could, in principle, be minimized. We also demonstrated that cytotoxicity of the derivatives to mammalian cells was not coupled to its Eis inhibition and, in principle, can be avoided. However, when considering further modifications, one needs to keep in mind that haloperidol (1) is a neuroactive molecule that uses its specific structural features to bind D-2-like dopamine receptors (such as DRD2) to exert its antipsychotic effect.44–49 The p-halo-phenyl and the keto-alkyl moieties of haloperidol (1) interact with the orthosteric binding pocket of the receptor. The protonated nitrogen in the piperidine ring of haloperidol forms a salt bridge with a conserved Asp114 of DRD2, and the hydroxyl group forms a hydrogen bond with Tyr408.49 The chlorobenzene moiety interacts with the residues in the secondary binding pocket of this receptor.49 Therapeutic effects of haloperidol (1) are achieved upon chronic administration with plasma levels in in the range of 2.7 nM to 93.1 nM, much lower than those that would be required for Eis inhibition in vivo. Because the same features of the haloperidol scaffold are responsible for binding Eis and DRD2, uncoupling Eis binding from dopamine receptor binding may not be possible. Does this mean that the approach of further development of haloperidol derivatives as a TB treatment is futile? Not necessarily so, if blood–brain barrier penetration of a haloperidol derivative could be blocked by introducing some substitution at R2 or R3, which would not perturb Eis binding. This hypothesis is yet to be investigated. As haloperidol can be used either orally or by injection, finding a suitable route of administration is not likely to be a major obstacle, but this issue will be evaluated in due course, when a suitable potent and safe analogue is identified.
Conclusion
Discovery of haloperidol (1) as an Eis inhibitor that was structurally distinct from our previously reported inhibitors led us to the identification of the regions of the molecule important for Eis binding, through crystal structure-aided SAR. Even though none of the analogues were more potent than 1, several analogues that showed similar submicromolar potency were much simpler structurally than 1. Some analogues partially restored KAN sensitivity of Mtb K204 overexpressing Eis, but the activity of these analogues in the Mtb cell was low, likely due to poor penetration through the cell envelope. The metabolic stability study indicated that phenyl ring substitutions controlled the metabolism. This study showed that direct repurposing of haloperidol (1) or these first-generation derivatives as anti-TB drugs was not feasible due to their poor activity in the cells and their neuroactive effects. Therefore, a future generation of haloperidol analogues would need to have improved mycobacterial cell envelope penetration and, conversely, poor penetration of the blood–brain barrier.Author contributions
Ankita Punetha, Allan H. Pang, Caixia Hou, and Oleg V. Tsodikov performed structural biology experiments. Atefeh Garzan and Nishad Thamban Chandrika performed chemical syntheses. Keith D. Green performed the biochemical experiments against purified enzymes, molecular biology and microbiology testing with purified enzymes against BSL-2 pathogens. Melisa J. Willby and James E. Posey performed testing against M. tuberculosis H37Rv. Selina Y. L. Holbrook performed the cytotoxicity experiments. Kyle Krieger and Tanya Parish performed the microsomal stability assessment. Oleg V. Tsodikov and Sylvie Garneau-Tsodikova were responsible for the overall design, supervision, completion of the work, and writing of the manuscript. All authors contributed to the writing of their respective experimental section(s). All authors have read and approved of the manuscript and supporting information.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by a grant from the National Institutes of Health (NIH) AI090048 (to S. G.-T.) and by startup funds from the College of Pharmacy at the University of Kentucky (to S. G.-T. and O. V. T.). We thank Abdelrahman S. Mayhoub, Matthew D. Demars, and Joseph T. Madak for preliminary synthesis of some of the molecules tested in this study. We thank Sultan Chowdhury for advice on metabolic stability. Use of trade names is for identification only and does not constitute endorsement by the U.S. Department of Health and Human Services, the U.S. Public Health Service, or the CDC. The findings and conclusions in this report are those of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention.References
- World Health Organization, Global tuberculosis report 2020, Geneva, 2020, Licence: CC BY-NC-SA 3.0 Igo Search PubMed.
- M. A. Zaunbrecher, R. D. Sikes, Jr., B. Metchock, T. M. Shinnick and J. E. Posey, Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20004–20009 CrossRef CAS PubMed.
- P. J. Campbell, G. P. Morlock, R. D. Sikes, T. L. Dalton, B. Metchock, A. M. Starks, D. P. Hooks, L. S. Cowan, B. B. Plikaytis and J. E. Posey, Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2011, 55, 2032–2041 CrossRef CAS PubMed.
- A. Sowajassatakul, T. Prammananan, A. Chaiprasert and S. Phunpruch, Overexpression of eis without a mutation in promoter region of amikacin- and kanamycin-resistant Mycobacterium tuberculosis clinical strain, Ann. Clin. Microbiol. Antimicrob., 2018, 17, 33 CrossRef PubMed.
- A. Z. Reeves, P. J. Campbell, R. Sultana, S. Malik, M. Murray, B. B. Plikaytis, T. M. Shinnick and J. E. Posey, Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7, Antimicrob. Agents Chemother., 2013, 57, 1857–1865 CrossRef CAS PubMed.
- W. Chen, T. Biswas, V. R. Porter, O. V. Tsodikov and S. Garneau-Tsodikova, Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9804–9808 CrossRef CAS PubMed.
- O. V. Tsodikov, K. D. Green and S. Garneau-Tsodikova, A random sequential mechanism of aminoglycoside acetylation by Mycobacterium tuberculosis Eis protein, PLoS One, 2014, 9, e92370 CrossRef PubMed.
- R. E. Pricer, J. L. Houghton, K. D. Green, A. S. Mayhoub and S. Garneau-Tsodikova, Biochemical and structural analysis of aminoglycoside acetyltransferase Eis from Anabaena variabilis, Mol. BioSyst., 2012, 8, 3305–3313 RSC.
- K. D. Green, T. Biswas, A. H. Pang, M. J. Willby, M. S. Reed, O. Stuchlik, J. Pohl, J. E. Posey, O. V. Tsodikov and S. Garneau-Tsodikova, Acetylation by Eis and deacetylation by Rv1151c of Mycobacterium tuberculosis HupB: Biochemical and structural insight, Biochemistry, 2018, 57, 781–790 CrossRef CAS PubMed.
- A. Sakatos, G. H. Babunovic, M. R. Chase, A. Dills, J. Leszyk, T. Rosebrock, B. Bryson and S. M. Fortune, Posttranslational modification of a histone-like protein regulates phenotypic resistance to isoniazid in mycobacteria, Sci. Adv., 2018, 4, eaao1478 CrossRef PubMed.
- S. Ghosh, B. Padmanabhan, C. Anand and V. Nagaraja, Lysine acetylation of the Mycobacterium tuberculosis HU protein modulates its DNA binding and genome organization, Mol. Microbiol., 2016, 100, 577–588 CrossRef CAS PubMed.
- K. D. Green, W. Chen and S. Garneau-Tsodikova, Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis, ChemMedChem, 2012, 7, 73–77 CrossRef CAS PubMed.
- M. J. Willby, K. D. Green, C. S. Gajadeera, C. Hou, O. V. Tsodikov, J. E. Posey and S. Garneau-Tsodikova, Potent inhibitors of acetyltransferase Eis overcome kanamycin resistance in Mycobacterium tuberculosis, ACS Chem. Biol., 2016, 11, 1639–1646 CrossRef CAS PubMed.
- A. Garzan, M. J. Willby, K. D. Green, C. S. Gajadeera, C. Hou, O. V. Tsodikov, J. E. Posey and S. Garneau-Tsodikova, Sulfonamide-based inhibitors of aminoglycoside acetyltransferase Eis abolish resistance to kanamycin in Mycobacterium tuberculosis, J. Med. Chem., 2016, 59, 10619–10628 CrossRef CAS PubMed.
- A. Garzan, M. J. Willby, K. D. Green, O. V. Tsodikov, J. E. Posey and S. Garneau-Tsodikova, Discovery and optimization of two Eis inhibitor families as kanamycin adjuvants against drug-resistant M. tuberculosis, ACS Med. Chem. Lett., 2016, 7, 1219–1221 CrossRef CAS PubMed.
- A. Garzan, M. J. Willby, H. X. Ngo, C. S. Gajadeera, K. D. Green, S. Y. Holbrook, C. Hou, J. E. Posey, O. V. Tsodikov and S. Garneau-Tsodikova, Combating enhanced intracellular survival (Eis)-mediated kanamycin resistance of Mycobacterium tuberculosis by novel pyrrolo[1,5-a]pyrazine-based Eis inhibitors, ACS Infect. Dis., 2017, 3, 302–309 CrossRef CAS PubMed.
- H. X. Ngo, K. D. Green, C. S. Gajadeera, M. J. Willby, S. Y. L. Holbrook, C. Hou, A. Garzan, A. S. Mayhoub, J. E. Posey, O. V. Tsodikov and S. Garneau-Tsodikova, Potent 1,2,4-triazino[5,6 b]indole-3-thioether Inhibitors of the kanamycin resistance enzyme Eis from Mycobacterium tuberculosis, ACS Infect. Dis., 2018, 4, 1030–1040 CrossRef CAS PubMed.
- K. D. Green, A. Punetha, C. Hou, S. Garneau-Tsodikova and O. V. Tsodikov, Probing the robustness of inhibitors of tuberculosis aminoglycoside resistance enzyme Eis by mutagenesis, ACS Infect. Dis., 2019, 5, 1772–1778 CrossRef CAS PubMed.
- A. Punetha, H. X. Ngo, S. Y. L. Holbrook, K. D. Green, M. J. Willby, S. A. Bonnett, K. Krieger, E. K. Dennis, J. E. Posey, T. Parish, O. V. Tsodikov and S. Garneau-Tsodikova, Structure-guided optimization of inhibitors of acetyltransferase Eis from Mycobacterium tuberculosis, ACS Chem. Biol., 2020, 15, 1581–1594 CrossRef CAS PubMed.
- T. T. Ashburn and K. B. Thor, Drug repositioning: Identifying and developing new uses for existing drugs, Nat. Rev. Drug Discovery, 2004, 3, 673–683 CrossRef CAS PubMed.
- C. R. Chong and D. J. Sullivan, Jr., New uses for old drugs, Nature, 2007, 448, 645–646 CrossRef CAS PubMed.
- J. L. Houghton, T. Biswas, W. Chen, O. V. Tsodikov and S. Garneau-Tsodikova, Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase Eis, ChemBioChem, 2013, 14, 2127–2135 CrossRef CAS PubMed.
- J. W. Hulshof, P. Casarosa, W. M. Menge, L. M. Kuusisto, H. van der Goot, M. J. Smit, I. J. de Esch and R. Leurs, Synthesis and structure-activity relationship of the first nonpeptidergic inverse agonists for the human cytomegalovirus encoded chemokine receptor US28, J. Med. Chem., 2005, 48, 6461–6471 CrossRef CAS PubMed.
- K. D. Green, W. Chen, J. L. Houghton, M. Fridman and S. Garneau-Tsodikova, Exploring the substrate promiscuity of drug-modifying enzymes for the chemoenzymatic generation of N-acylated aminoglycosides, ChemBioChem, 2010, 11, 119–126 CrossRef CAS PubMed.
- S. Ramon-Garcia, C. Ng, H. Anderson, J. D. Chao, X. Zheng, T. Pfeifer, Y. Av-Gay, M. Roberge and C. J. Thompson, Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen, Antimicrob. Agents Chemother., 2011, 55, 3861–3869 CrossRef CAS PubMed.
- L. G. Franken, B. C. de Winter, H. J. van Esch, L. van Zuylen, F. P. Baar, D. Tibboel, R. A. Mathot, T. van Gelder and B. C. Koch, Pharmacokinetic considerations and recommendations in palliative care, with focus on morphine, midazolam and haloperidol, Expert Opin. Drug Metab. Toxicol., 2016, 12, 669–680 CrossRef CAS PubMed.
- A. Forsman and R. Ohman, Studies on serum protein binding of haloperidol, Curr. Ther. Res. Clin. Exp., 1977, 21, 245–255 CAS.
- F. J. Rowell, S. M. Hui, A. F. Fairbairn and D. Eccleston, Total and free serum haloperidol levels in schizophrenic patients and the effect of age thioridazine and fatty acid on haloperidol-serum protein binding in vitro, Br. J. Clin. Pharmacol., 1981, 11, 377–382 CrossRef CAS PubMed.
- W. Soudijn, I. Van Wijngaarden and F. Allewijn, Distribution, excretion and metabolism of neuroleptics of the butyrophenone type. I. Excretion and metabolism of haloperidol and nine related butyrophenone-derivatives in the Wistar rat, Eur. J. Pharmacol., 1967, 1, 47–57 CrossRef CAS PubMed.
- J. Fang and J. W. Gorrod, High-performance liquid chromatographic method for the detection and quantitation of haloperidol and seven of its metabolites in microsomal preparations, J. Chromatogr., 1993, 614, 267–273 CrossRef CAS PubMed.
- J. W. Gorrod and J. Fang, On the metabolism of haloperidol, Xenobiotica, 1993, 23, 495–508 CrossRef CAS PubMed.
- S. J. Grossman, E. G. Herold, J. M. Drey, D. W. Alberts, D. R. Umbenhauer, D. H. Patrick, D. Nicoll-Griffith, N. Chauret and J. A. Yergey, CYP1A1 specificity of verlukast epoxidation in mice, rats, rhesus monkeys, and humans, Drug Metab. Dispos., 1993, 21, 1029–1036 CAS.
- J. Fang, G. B. Baker and R. T. Coutts, Determination of 4-(4-chlorophenyl)-4-hydroxypiperidine, a metabolite of haloperidol, by gas chromatography with electron-capture detection, J. Chromatogr. B: Biomed. Sci. Appl., 1996, 682, 283–288 CrossRef CAS PubMed.
- D. C. Goff, K. K. Midha, A. W. Brotman, M. Waites and R. J. Baldessarini, Elevation of plasma concentrations of haloperidol after the addition of fluoxetine, Am. J. Psychiatry, 1991, 148, 790–792 CrossRef CAS PubMed.
- K. K. Midha, J. K. Cooper, E. M. Hawes, J. W. Hubbard, E. D. Korchinski and G. McKay, An ultrasensitive method for the measurement of haloperidol and reduced haloperidol in plasma by high-performance liquid chromatography with coulometric detection, Ther. Drug Monit., 1988, 10, 177–183 CrossRef CAS PubMed.
- T. Someya, M. Shibasaki, T. Noguchi, S. Takahashi and T. Inaba, Haloperidol metabolism in psychiatric patients: Importance of glucuronidation and carbonyl reduction, J. Clin. Psychopharmacol., 1992, 12, 169–174 CrossRef CAS PubMed.
- B. Subramanyam, H. Rollema, T. Woolf and N. Castagnoli, Jr., Identification of a potentially neurotoxic pyridinium metabolite of haloperidol in rats, Biochem. Biophys. Res. Commun., 1990, 166, 238–244 CrossRef CAS PubMed.
- C. J. Van der Schyf, K. Castagnoli, E. Usuki, H. G. Fouda, J. M. Rimoldi and N. Castagnoli, Jr., Metabolic studies on haloperidol and its tetrahydropyridine analog in C57BL/6 mice, Chem. Res. Toxicol., 1994, 7, 281–285 Search PubMed.
- J. Fang, G. McKay, J. Song, A. Remillrd, X. Li and K. Midha, In vitro characterization of the metabolism of haloperidol using recombinant cytochrome p450 enzymes and human liver microsomes, Drug Metab. Dispos., 2001, 29, 1638–1643 CAS.
- K. M. Avent, J. J. DeVoss and E. M. Gillam, Cytochrome P450-mediated metabolism of haloperidol and reduced haloperidol to pyridinium metabolites, Chem. Res. Toxicol., 2006, 19, 914–920 Search PubMed.
- A. Forsman and M. Larsson, Metabolism of haloperidol, Curr. Ther. Res., 1978, 24, 567–568 CAS.
- J. Fang and J. W. Gorrod, Dehydration is the first step in the bioactivation of haloperidol to its pyridinium metabolite, Toxicol. Lett., 1991, 59, 117–123 CrossRef CAS PubMed.
- D. W. Eyles, H. R. McLennan, A. Jones, J. J. McGrath, T. J. Stedman and S. M. Pond, Quantitative analysis of two pyridinium metabolites of haloperidol in patients with schizophrenia, Clin. Pharmacol. Ther., 1994, 56, 512–520 CrossRef CAS PubMed.
- A. E. Moritz, R. B. Free and D. R. Sibley, Advances and challenges in the search for D2 and D3 dopamine receptor-selective compounds, Cell. Signalling, 2018, 41, 75–81 CrossRef CAS PubMed.
- J. Xiao, R. B. Free, E. Barnaeva, J. L. Conroy, T. Doyle, B. Miller, M. Bryant-Genevier, M. K. Taylor, X. Hu, A. E. Dulcey, N. Southall, M. Ferrer, S. Titus, W. Zheng, D. R. Sibley and J. J. Marugan, Discovery, optimization, and characterization of novel D2 dopamine receptor selective antagonists, J. Med. Chem., 2014, 57, 3450–3463 CrossRef CAS PubMed.
- B. Mannel, M. Jaiteh, A. Zeifman, A. Randakova, D. Moller, H. Hubner, P. Gmeiner and J. Carlsson, Structure-guided screening for functionally selective D2 dopamine receptor ligands from a virtual chemical library, ACS Chem. Biol., 2017, 12, 2652–2661 CrossRef PubMed.
- S. Lober, H. Hubner, N. Tschammer and P. Gmeiner, Recent advances in the search for D3- and D4-selective drugs: Probes, models and candidates, Trends Pharmacol. Sci., 2011, 32, 148–157 CrossRef PubMed.
- M. F. Zou, T. M. Keck, V. Kumar, P. Donthamsetti, M. Michino, C. Burzynski, C. Schweppe, A. Bonifazi, R. B. Free, D. R. Sibley, A. Janowsky, L. Shi, J. A. Javitch and A. H. Newman, Novel analogues of (R)-5-(methylamino)-5,6-dihydro-4H-imidazo[4,5,1-ij]quinolin-2(1H)-one (sumanirole) provide clues to dopamine D2/D3 receptor agonist selectivity, J. Med. Chem., 2016, 59, 2973–2988 CrossRef CAS PubMed.
- L. Fan, L. Tan, Z. Chen, J. Qi, F. Nie, Z. Luo, J. Cheng and S. Wang, Haloperidol bound D2 dopamine receptor structure inspired the discovery of subtype selective ligands, Nat. Commun., 2020, 11, 1074 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: PDB ID: Eis-haloperidol: 6X10, Eis-droperidol: 6X6G, Eis-SGT543: 6X6I, Eis-SGT572: 6X7A, Eis-SGT1264: 6X6Y. See DOI: 10.1039/d1md00239b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |