
Metabolically engineered bacteria as light-controlled living therapeutics for anti-angiogenesis tumor therapy†
Xingang
Liu‡
a,
Min
Wu‡
a,
Meng
Wang
b,
Yukun
Duan
a,
ChiUyen
Phan
c,
Guobin
Qi
a,
Guping
Tang
 *c and
Bin
Liu
*ad
*c and
Bin
Liu
*ad
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Engineering Drive 4, 117585, Singapore. E-mail: cheliub@nus.edu.sg
bDepartment of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital, Zhejiang University, School of Medicine, Hangzhou 310003, China
cDepartment of Chemistry, Zhejiang University, Hangzhou 310028, China. E-mail: tangguping@zju.edu.cn
dJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 16th December 2020
Abstract
A living therapeutic system based on attenuated Salmonella was developed via metabolic engineering using an aggregation-induced emission (AIE) photosensitizer MA. The engineered bacteria could localize in the tumor tissues and continue to colonize and express exogenous genes. Under light irradiation, the encoded VEGFR2 gene was released and expressed in tumor tissues, which can suppress angiogenesis induced by a T cell-mediated autoimmune response and inhibit tumor growth.
New conceptsLiving bacteria therapeutic systems are living factories that autonomously self-replicate, generate and pump out therapeutics inside the body. The main limitation of living bacteria therapy is the inefficient release of therapeutics from bacteria, especially for bactofection delivery of therapeutic genes, which mostly depends on spontaneous lysis or exogenous molecules, making it difficult to realize on-demand control of the process. Herein, a living therapeutic system was developed based on attenuated Salmonella via metabolic engineering using an aggregation-induced emission (AIE) photosensitizer MA to realize light-controlled gene release for breast cancer therapy. The labeling of MA does not inhibit Salmonella reproduction so that the administered MA-engineered Salmonella carrying vascular endothelial growth factor receptor 2 (VEGFR2) plasmids can be localized in the tumor tissues and continue to colonize and express exogenous genes. Following an appropriate treatment schedule, the constructed plasmid could undergo controlled-release into the cytoplasm of the host cells under light irradiation. The designed expression of VEGFR2 proteins could then block the immunological tolerance to VEGFR2 and induce a T cell-mediated autoimmune antiangiogenic response. Through a series of in vitro and in vivo experiments, prominent tumor suppression performance was validated with the engineered living therapeutic system, demonstrating its great potential in precise tumor treatment. |
Introduction
Engineered living therapeutic systems are renowned as the next generation of therapeutics for a broad range of indications from treating and preventing infections to suppressing tumors and curing metabolic disorders.1 Compared with traditional systemic treatment such as gene delivery systems mediated by viral and non-viral vectors, living therapeutics serve as factories that autonomously self-replicate, generate and pump out therapeutics inside the body.2 The use of live bacteria as living therapeutics for cancer regression has been recognized and practiced, in which programmable attenuated bacterial strains express extraneous genes or generate products in vitro and in vivo like enzymes, proteins, and immunotoxins.3,4 Although bacteria have been used as therapeutic agents for many years, pathogenic effects and ineffective therapeutic drug release from intracellular bacteria are issues that remain to be addressed.Taking an intracellular live bacterium, attenuated Salmonella, as an example, it has many desirable properties, such as specific tumor targeting, and being able to proliferate inside tumor tissues and induce tumor regression.5–8 Genetically engineered Salmonella is able to deliver therapeutic proteins or produce therapeutic cytokines, tumor-specific antigens, and antibodies or transfer genes in eukaryotic vectors for bactofection.9 The main shortcoming of the Salmonella strategy is the ineffective therapeutic drug release from the intracellular bacteria, especially for bactofection delivery of therapeutics genes. Bactofection is a method of using bacteria as a vector to deliver genes directly into the target cells in vivo.10–12 After the bacteria enter the target cells, the plasmid encoding the therapeutic gene is released and finally transferred into the cell nucleus, where the therapeutic gene is expressed by the host cell's expression system. Currently, gene release from intracellular bacteria is mostly dependent on spontaneous bacterial lysis or exogenous molecules to trigger drug release by membrane degradation, which may cause undesirable side effects on the body and cannot be controlled on demand.13,14 Programed lysis of bacteria based on lysis genes is a promising solution, but it requires more complex genetic engineering of Salmonella, which adds time and cost for the process.1 Therefore, a simple and biocompatible system with controlled gene release is vital for living bactofection therapeutics.
Vascular endothelial growth factor receptor 2 (VEGFR2) is overexpressed in activated endothelial cells during angiogenesis in the tumor vasculature, which plays an important role in tumor growth, invasion and metastasis.15 Therefore, it can be applied as a potential therapeutic target for anti-angiogenesis and tumor growth suppression.16–19 In the present study, we construct a living therapeutic system based on attenuated Salmonella (strain VNP20009) with light controlled VEGFR2 gene release ability, which can suppress angiogenesis induced by a T cell-mediated autoimmune response against self-antigens expressed by proliferating endothelial cells. As shown in Scheme 1, VEGFR2 plasmid transduced live Salmonella can be engineered by a metabolic labeling strategy with MeTTPy-D-Ala (MA). MA exhibits low emission in aqueous media due to the hydrophilicity of pyridinium and D-Ala. The far-red/near-infrared (FR/NIR) fluorescence would turn on once the MA molecules were attached to bacterial peptidoglycan through metabolic labeling.20,21 The metabolically incorporated MA can also serve as a photosensitizer to generate ROS under light irradiation. VNP20009 is a bacterial strain genetically modified by depleting the purI genes, creating an auxotrophic mutant for adenine that bacteria can obtain from necrotic areas inside tumors, making bacteria replication more tumor-specific.22,23 As tumor-targeting bacteria, the intravenously administered MA-engineered Salmonella carrying VEGFR2 therapeutic plasmids can be localized in the tumor tissues and continue to colonize and express exogenous genes. Applying light irradiation would destruct the bacteria cell membrane by generating singlet oxygen (1O2) from MA to facilitate on-demand plasmid release into the host cell cytoplasm. The released plasmid could express the VEGFR2 protein inside the host cells, which is considered as a foreign antigen to break immunological tolerance to VEGFR2 and induce a T cell-mediated autoimmune antiangiogenic response leading to the suppression of tumor growth.24–26
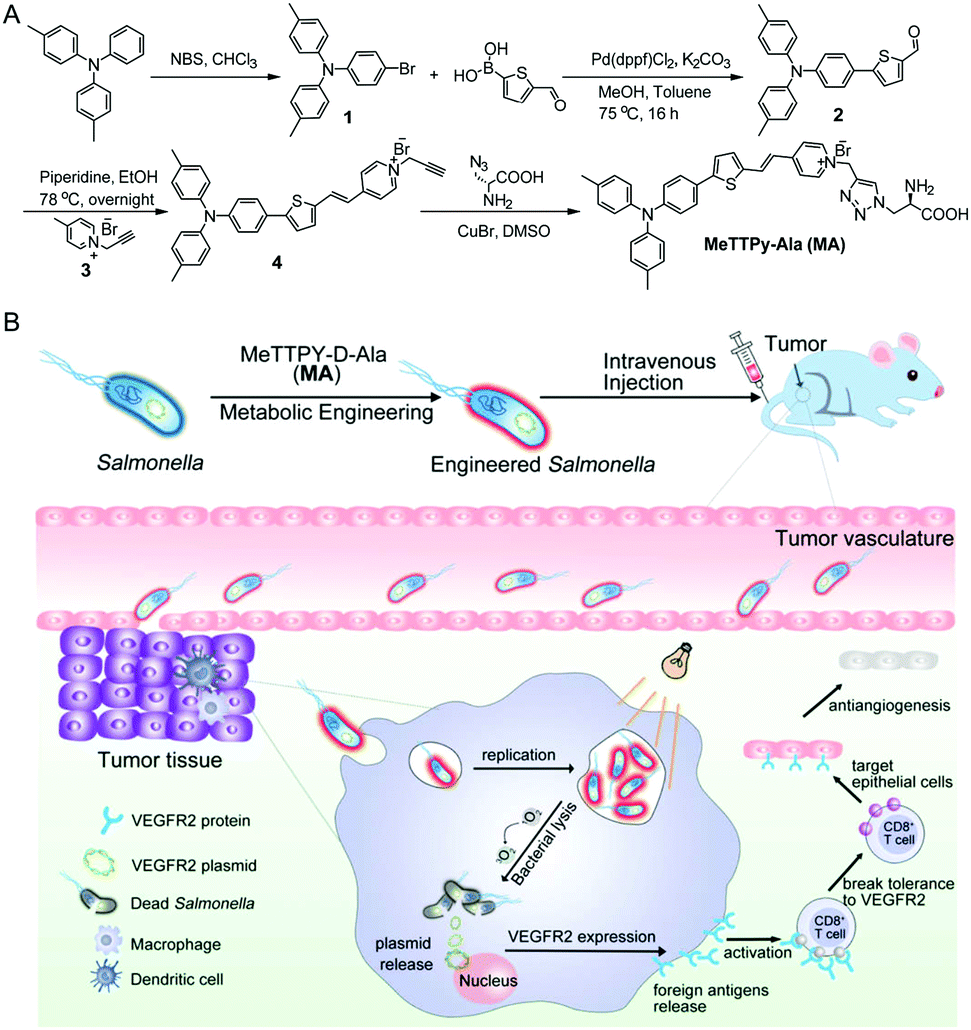 | ||
| Scheme 1 Schematic illustration of (A) the synthetic route to target compound MA and (B) Salmonella encoding the VEGFR2 plasmid engineered with MA and its anti-angiogenesis therapy of breast cancer in vivo. | ||
Results and discussion
The synthetic route to MA is shown in Scheme 1A. Briefly, 4,4′-dimethyltriphenylamine was brominated with N-bromosuccinimide (NBS) and further reacted with 5-formyl-2-thienylboronic acid via a Suzuki reaction to give compound 2 in 71% yield. A Knoevenagel condensation reaction between compounds 2 and 3 yielded the intermediate compound 4, which was further reacted with D-Ala via a copper(I)-catalyzed azide–alkyne cycloaddition reaction to give MA in 32% yield. The chemical structures of MA and the intermediates were well characterized by NMR and mass spectroscopy (Fig. S1–S6, ESI†) and the purity was verified by reverse-phase HPLC (Fig. S7, ESI†).MA has a donor–π–acceptor (D–π–A) structure comprised of methyl substituted triphenylamine (as the D), thiophene (as the D and π bridge), a carbon–carbon double bond (as the π bridge) and pyridinium (as the A), which shows an absorption peak at 480 nm and maximum emission located at 710 nm in DMSO/water (v/v = 1/99) solution (Fig. 1A). As shown in Fig. 1B, MA has low fluorescence when the fraction of poor solvent toluene is between 0 and 70% but becomes highly emissive when 90% (v/v) of toluene is added, which displays a typical AIE property. MA also shows photosensitizing capability to generate singlet oxygen (1O2).27 The ROS generation capacity of MA was evaluated using 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) as the indicator, which was further compared with commercial photosensitizer Rose Bengal. As shown in Fig. 1C and Fig. S8 (ESI†), the degradation rate of ABDA was about 15 nmol min−1 by MA and 10 nmol min−1 by Rose Bengal under the same light illumination (30 mW cm−2), revealing highly effective 1O2 production capability of MA.
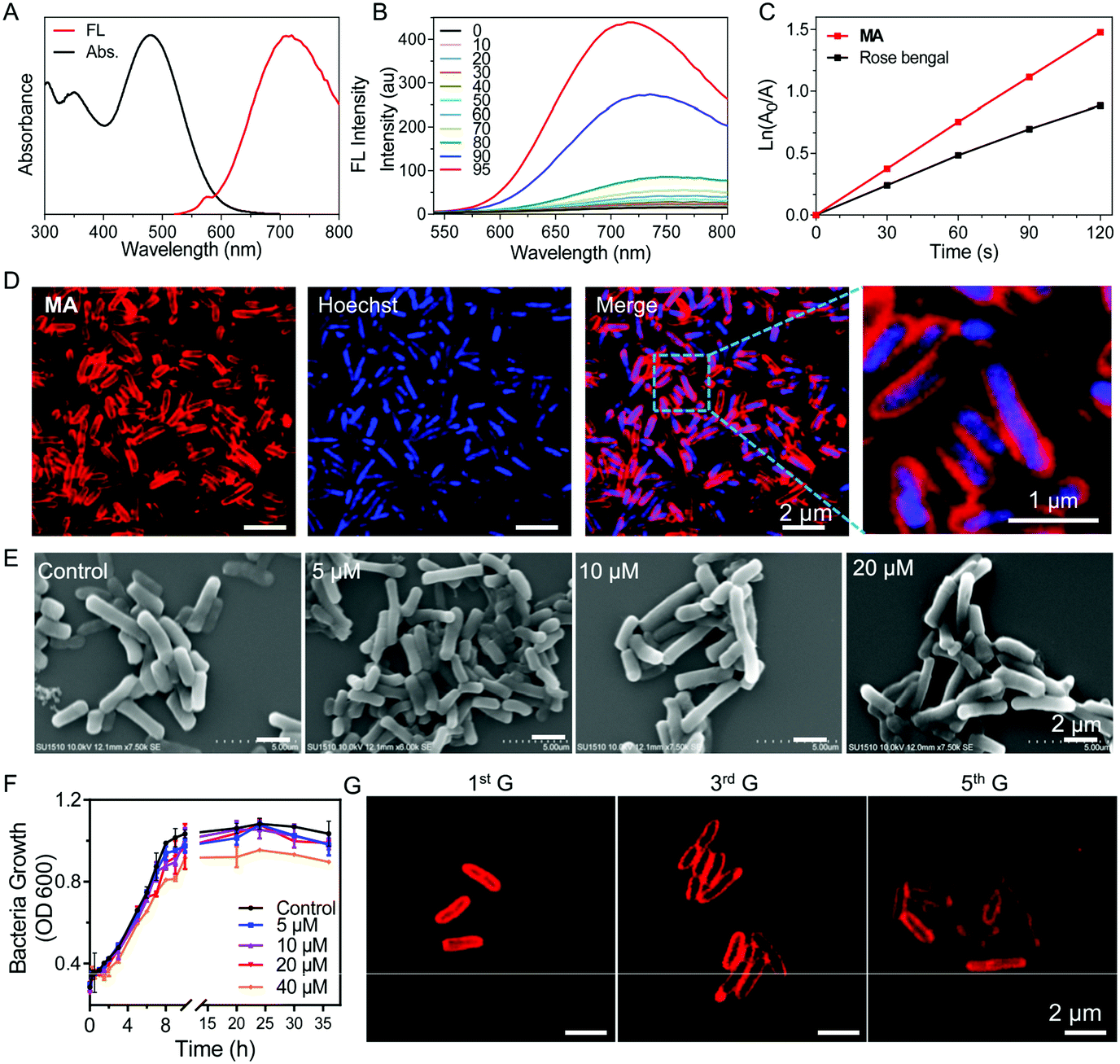 | ||
| Fig. 1 (A) Normalized absorption and fluorescence (FL) spectra of MA in DMSO and water (v/v = 1/99) solution. (B) FL spectra of MA (10 μM) in DMSO/toluene mixtures with different toluene fractions (v:v); λex: 488 nm. (C) Decomposition rates of ABDA induced by ROS generated from MA (10 μM) under white light irradiation (30 mW cm−2) in DMSO/water (v/v = 1/99) solution. [ABDA] = 5 × 10−5 M, time interval for recording the UV-vis spectra: 30 s. (D) Confocal microscopy images of metabolically labelled Salmonella (20 μM MA for 30 min). (E) Field emission scanning electron microscopy images of Salmonella labelled with various concentrations of MA. The control group is the untreated Salmonella. (F) The bacterial growth of MA labelled Salmonella in fresh LB solution. (G) Confocal microscopy images of MA labelled Salmonella at different cell generation cycles in fresh LB solution. | ||
We next investigated whether MA could be used to label bacteria through a metabolic pathway. After incubating Salmonella cells with MA (20 μM) for 30 min, almost all the bacteria were effectively labeled by MA without any washing procedure needed (Fig. 1D). The covalent ligation of MA in peptidoglycan was further validated by MALDI-TOF mass analysis of MA-treated Salmonella cell lysate (Fig. S9, ESI†). MA was anticipated to be included in the peptidoglycan biosynthesis process, during which MA would be inserted into bacterial peptidoglycan to emit strong fluorescence as a result of the restricted intramolecular motions.15 Interestingly, when compound 4 without the D-Ala segment was used as a control to label Salmonella, no light-up fluorescence was observed (Fig. S10, ESI†). MA release assay from MA@SV conducted in PBS at 37 °C also showed the excellent stability of MA@SV (Fig. S11, ESI†). These indicated that the red signal was produced from metabolically labeled bacteria but not by nonspecific interactions. When Salmonella was incubated with various concentrations of MA for 30 minutes, the confocal images (Fig. S12, ESI†) and flow cytometric analysis (Fig. S13, ESI†) results were observed to be dose dependent. As the Salmonella upon incubation with 20 μM of MA showed the highest brightness, 20 μM was selected as the optimal concentration to label Salmonella in the following experiments. Meanwhile, MA labeling of Salmonella was noticed to be time-dependent and most bacteria were stained effectively in 30 min (Fig. S14, ESI†).
To evaluate whether the metabolic labeling method would affect the functions of Salmonella, scanning electron microscopy (SEM) was used to observe the bacteria morphology after MA labeling. Salmonella displayed a typical rod form with a smooth membrane exterior when labeled with 5–20 μM MA, and no significant morphological changes were found in the SEM images compared with the control Salmonella (Fig. 1E). No nanoaggregates were found to adhere on the surface of MA-labeled Salmonella (Salmonella VEGFR2@MA, or, for short, SV@MA), indicating that MA dissolved well and existed as a monodisperse molecule at 5–20 μM in aqueous media.
As a living therapeutic system, the ability of the engineered Salmonella to autonomously self-replicate and produce the therapeutic inside the body is critical for disease treatment. Therefore, the effect of the MA labeling method on bacterial growth was studied with various concentrations of MA. As shown in Fig. 1F, the growth curve of Salmonella clearly described the lag, log and stationary phase at a concentration of 20 μM, demonstrating that MA exerted no significant inhibitory effects on the growth of Salmonella. After SV@MA growing in fresh LB solution for 5 generations, bright red fluorescence could still be observed on the cell membrane of each bacterium (Fig. 1G), indicating that MA metabolic labeling is a biocompatible and lasting engineering method for construction of a living therapeutic system.
The inefficient release of therapeutic genes from bacteria into the host cells is an obstacle faced in bactofection due to the weak membrane crossing of genes in the majority of cases. Recent papers revealed that the bacterial membrane can be damaged by ROS generated from membrane-bound photosensitizers.28–30 We next studied whether the MA engineering method could facilitate therapeutic gene release. Firstly, live/dead co-staining assays were performed. When labeled with 5–20 μM MA, most Salmonella remained alive in the dark (Fig. 2A), indicating good biocompatibility of the staining method. Under light irradiation (30 mW cm−2) for 10 min, substantial cell death was caused by the generated ROS from MA (Fig. 2A), which resulted in a high level of bacterial membrane weakening with 20 μM SV@MA (Fig. 2B). The live and dead assay results demonstrated that the ROS generation of intracellular SV@MA would not contribute to cytotoxic effects on the host cells (Fig. S15, ESI†). Since extensive bacterial membrane damage was verified, the supernatant absorbance at 260 nm was detected to allow quantification of the released DNA.31 Concentration-dependent release profiles were observed in DNA release (Fig. 2C), where the amount of DNA released in 20 μM labeled SV@MA was 1.5-fold higher than that of 5 μM labeled SV@MA and 9-fold higher than that of the control Salmonella, showing an effective light-controlled gene release capability. The cellular uptake of SV@MA by 4T1 cells (murine breast cancer cell line) was analyzed with fluorescence microscopy. Red signals were observed in the cytoplasm of 4T1 cells in the confocal images (Fig. 2D) and the transmission electron microscope (TEM) images of ultrathin cell slices showed that SV@MA was able to invade eukaryotic cells (Fig. 2E). Next, protein expression of VEGFR2 was confirmed by western blotting of transfected 4T1 cells (Fig. 2F). The SV@MA treated cells with light irradiation showed 4-fold higher VEGFR2 expression compared with the other groups (Fig. 2G). These results collectively demonstrate that SV@MA is able to enter the target cells and realize light-controlled plasmid DNA release and protein expression inside the target cells.
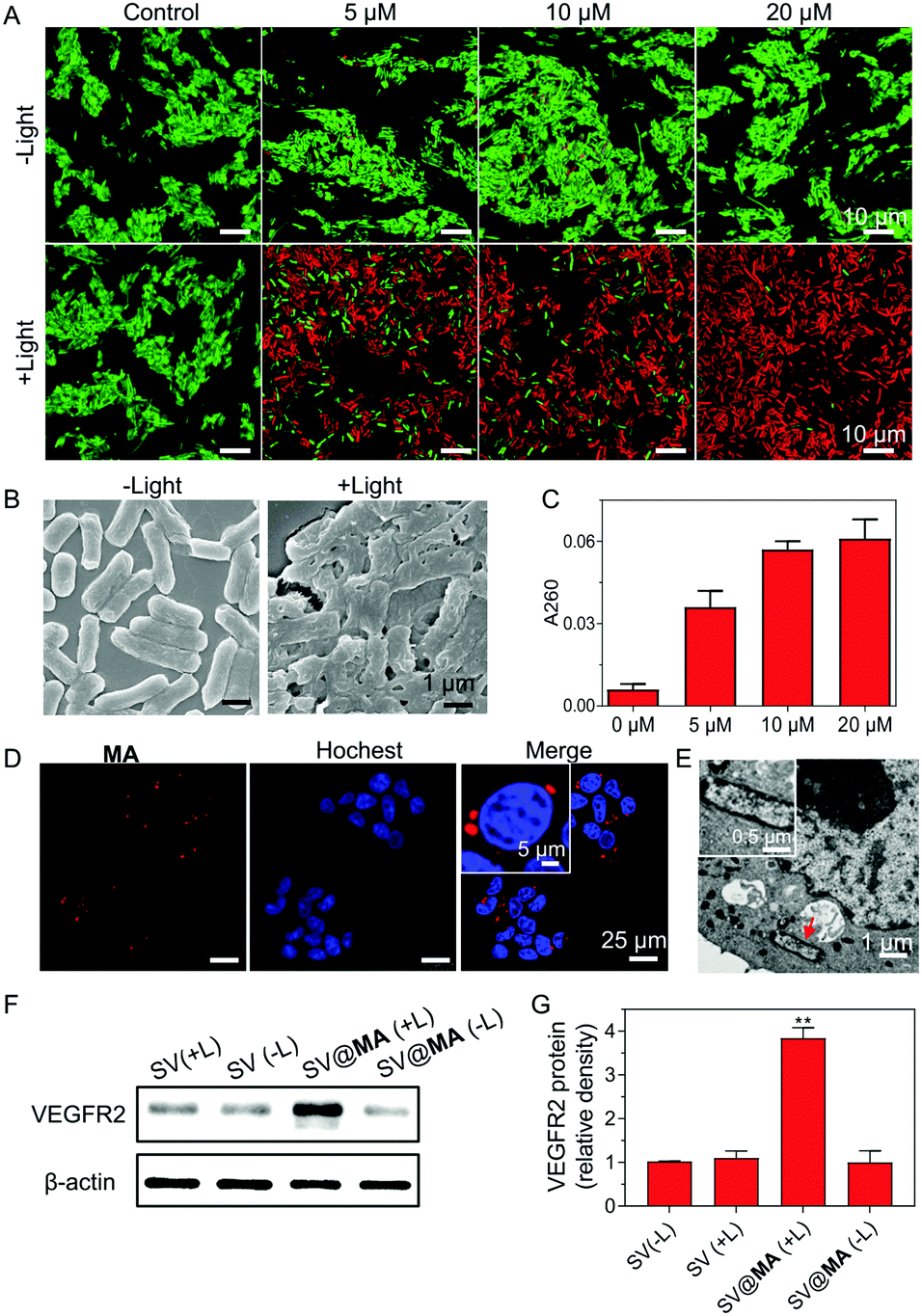 | ||
| Fig. 2 (A) Live and dead assays of Salmonella stained with different concentrations of MA with or without white light irradiation (30 mW cm−2, 10 min). (B) Field emission scanning electron microscopy images of SV@MA (20 μM) with or without light irradiation (30 mW cm−2, 10 min). (C) DNA release studies of Salmonella stained with different concentrations of MA with light irradiation (30 mW cm−2, 10 min). (D) Confocal fluorescence images of 4T1 cells incubated with SV@MA for 4 h. (E) TEM images showing the intracellular position of SV@MA in the ultrathin cell slices of 4T1 cells. (F) Western blot analysis of VEGFR2 expression in 4T1 cells after different transfection treatment. Light irradiation: 30 mW cm−2, 10 min. (G) Quantitative analysis of VEGFR2 protein expression in 4T1 cells after different treatment. | ||
Encouraged by the effective proliferation and light-controlled gene release capability of the living SV@MA therapeutic system in vitro, we then investigated whether the labeled Salmonella could be used as a bactofection vehicle in vivo. BALB/c mice were subcutaneously inoculated with 4T1 cells at the right abdomen, where tumors were allowed to grow to a volume of approximately 100 mm3. Fig. 3A shows the detailed treatment schedule. The tumor targeting efficacy of SV@MA was evaluated by monitoring the fluorescence signal of MA using an IVIS spectrum imaging system. As shown in Fig. 3B, MA showed unsatisfactory tumor targeting capacity, and was mainly distributed in the liver and kidneys. A notable fluorescence signal was observed in the tumor area 4 h post-injection of SV@MA (Fig. 3C and Fig. S16, ESI†), demonstrating the intrinsic tumor-targeting capability of MA-labeled Salmonella. Next, the amount of Salmonella in tumors was respectively counted at days 1, 3, 7 and 22 after intravenous injection of SV@MA. At day 3 before the 1st light irradiation, the bacterial concentration in tumor tissue was increased 2.6-fold compared to that on day 1 (Fig. 3D),32,33 indicating that the bacteria retain their proliferation characteristics in the target tissue. After the 2nd light irradiation treatment, the bacterial concentration in tumors at day 7 was reduced dramatically as compared with that of day 3 and subsequently became undetectable at day 22 (Fig. 3E). This was due to the bacterial death caused by the light-generated ROS followed by body clearance. However, the bacteria in tumors of the SV@MA(−L) and SV(+L) treated mice multiplied to significantly higher bacterial contents at day 7 (Fig. S17, ESI†). The above results indicate that SV@MA could preferentially accumulate and proliferate to a high level in tumor tissues, and the bacterial growth could be inhibited in a light-controlled manner.
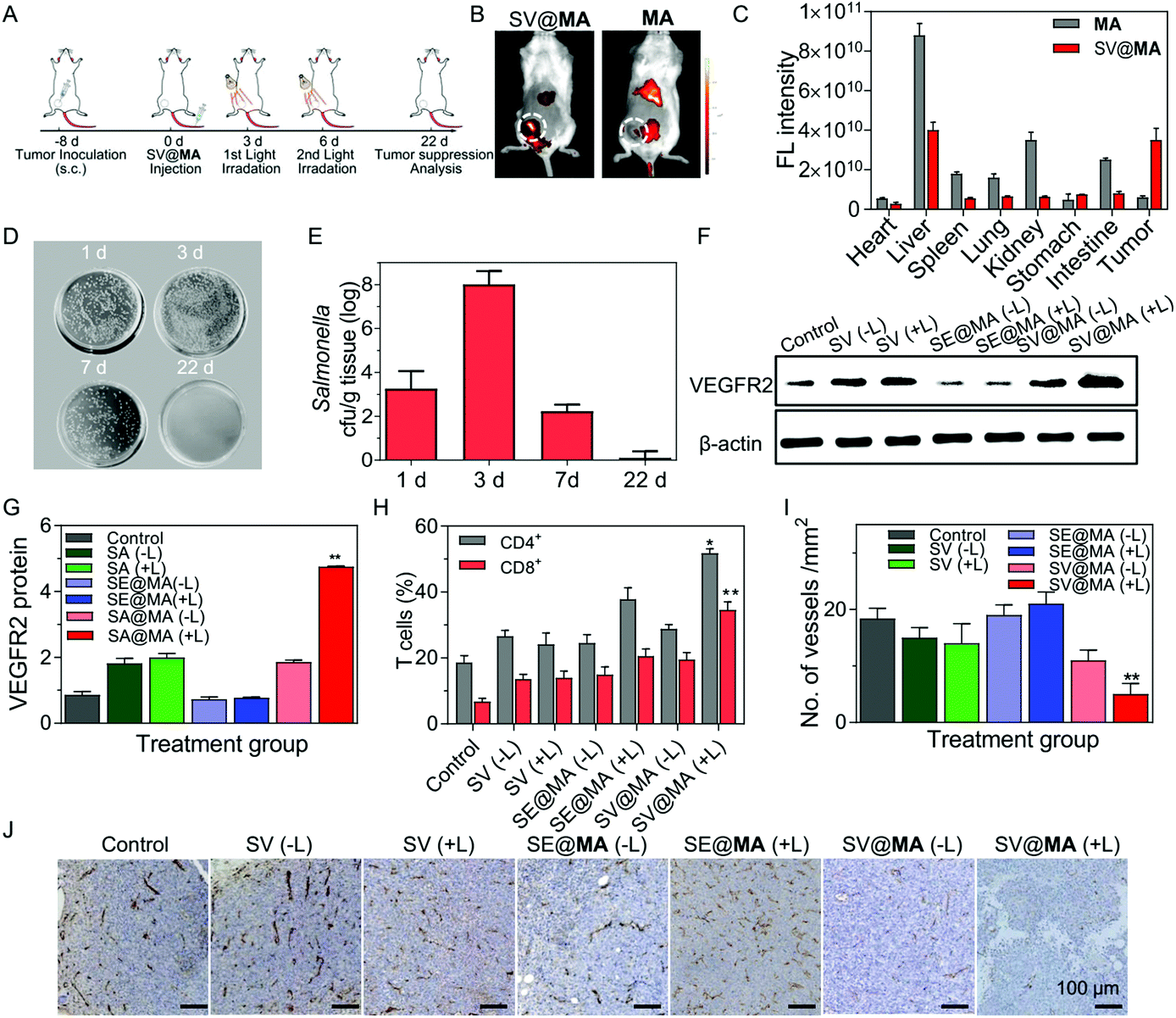 | ||
| Fig. 3 (A) Schematic illustration of the treatment schedule for anti-angiogenesis tumor therapy mediated by SV@MA in a 4T1 tumor model. (B) Fluorescence imaging of the SV@MA and MA distributions in 4T1 tumor-bearing BALB/c mice in vivo. Fluorescence images were taken 4 h after the mice were injected with SV@MA or MA. (C) Average fluorescence intensities in main organs and tumor tissues 4 h after intravenous injection of SV@MA or MA (n = 3). (D) Selective Salmonella growth and (E) Salmonella counts in tumor tissues. Tumor tissue homogenates of SV@MA(+L) at different days were cultured on solid LB agar at 37 °C for 24 h. 1 d and 3 d represent the 1st and 3rd day after injecting SV@MA. 7 d and 22 d represent the 1st and 14th day after the second light irradiation. (F) Western blot analysis and (G) quantification of VEGFR2 protein expression in tumor tissue after treatment with different formulations (n = 5). (H) Quantitative flow cytometry analysis of CD4+ and CD8+ T cells in 4T1 tumor model BALB/c mice immunized with different formulations. The cells were isolated from the blood and stained with FITC-CD4 and PE-CD8 antibodies. (I) Quantification and (J) immunohistochemical analysis of CD31 in the tumor slices of the 4T1 tumor model at day 22 after treating with different formulations. | ||
Under light irradiation, the ROS generated from SV@MA was expected to destroy the bacterial membrane, and cause the release of therapeutic plasmids into the host cells.34 Western blotting was used to analyse the protein expression of the released plasmid inside the host cells (Fig. 3F). There was an increase in VEGFR2 expression in SV@MA(+L) treated tumors, which was 5.0-fold and 1.6-fold higher than the PBS and SV@MA(−L) treated group (Fig. 3G), respectively, indicating the high-efficiency of controlled gene release and expression under light irradiation.
Normally, the tumor vascular epithelial cells are immune tolerant to T cell killing due to the down-regulation of major histocompatibility (MHC) antigens required for T cell-mediated antitumor responses in tumors.35,36 Several preclinical studies have shown that xenogenic VEGFR2 as a foreign antigen can break the immunological tolerance against VEGFR2 and evoke an antiangiogenic response by inducing a T cell-mediated immune response against VEGFR2 overexpressed endothelial cells in the tumor vasculature.24–26,37 Here, we explored the potential of expressed VEGFR2 proteins as a foreign antigen to stimulate T cell responses including polyfunctional cytokine-secreting CD4+ and CD8+ T cells. As shown in Fig. 3H, significant infiltration of CD8+ T cells (34.6%) was found inside the serum of SV@MA(+L) treated mice, much higher than the control mice treated with PBS (6.75%). In the case of CD4+ T cells, the population of CD4+ T cells in the serum of mice treated with PBS was only 18.6%, while 51.8% CD4+ T cells were observed when the mice were administered with SV@MA(+L) (Fig. S18, ESI†). The above results showed that SV@MA with light irradiation can break the immunological tolerance against VEGFR2 and activate the CD4+ and CD8+ T cells, which can induce a T cell-mediated autoimmune response against VEGFR2 overexpressed in proliferating endothelial cells. The impact of the activated CD4+ and CD8+ T cells on the suppression of angiogenesis through T cell-mediated killing of endothelial cells was further studied. Immunohistochemical staining with an antibody reactive to CD31 was used to identify the microvessels in 4T1 tumors. The number of microvessels in the SV@MA (+L) group was observed to be significantly less than the SV@MA (−L) and SV (+L) groups (Fig. 3I and J), indicating the important role of the light-controlled gene release ability in anti-angiogenesis. All the above results showed that SV@MA could simultaneously remain and proliferate in the tumor tissue and suppress angiogenesis effectively by a T cell-mediated autoimmune response under light irradiation.
The in vivo tumor inhibition effect of SV@MA was assessed in a 4T1 tumor-bearing BALB/c mouse model by monitoring the tumor volumes. As expected, SV@MA(+L) showed excellent efficiency for tumor inhibition as indicated by the best tumor suppression outcome based on the tumor size (Fig. 4A) from mice on day 22. The tumor growth trends of the SV(−L), SE@MA(−L), SV@MA(−L) and SV(+L) groups were similar to that of the PBS control, suggesting a poor anti-tumor effect during the treatment. Furthermore, the immumohistochemical staining results, such as terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (Fig. 4B and D), Ki67 assay staining (Fig. 4C and D) and hematoxylin–eosin (H&E) (Fig. 4D), of tumor tissues from mice after the specified treatment further confirmed the prominent anti-tumor performance of SV@MA(+L), in which significant cell apoptosis and proliferation inhibition were found as compared with the other treatment groups.
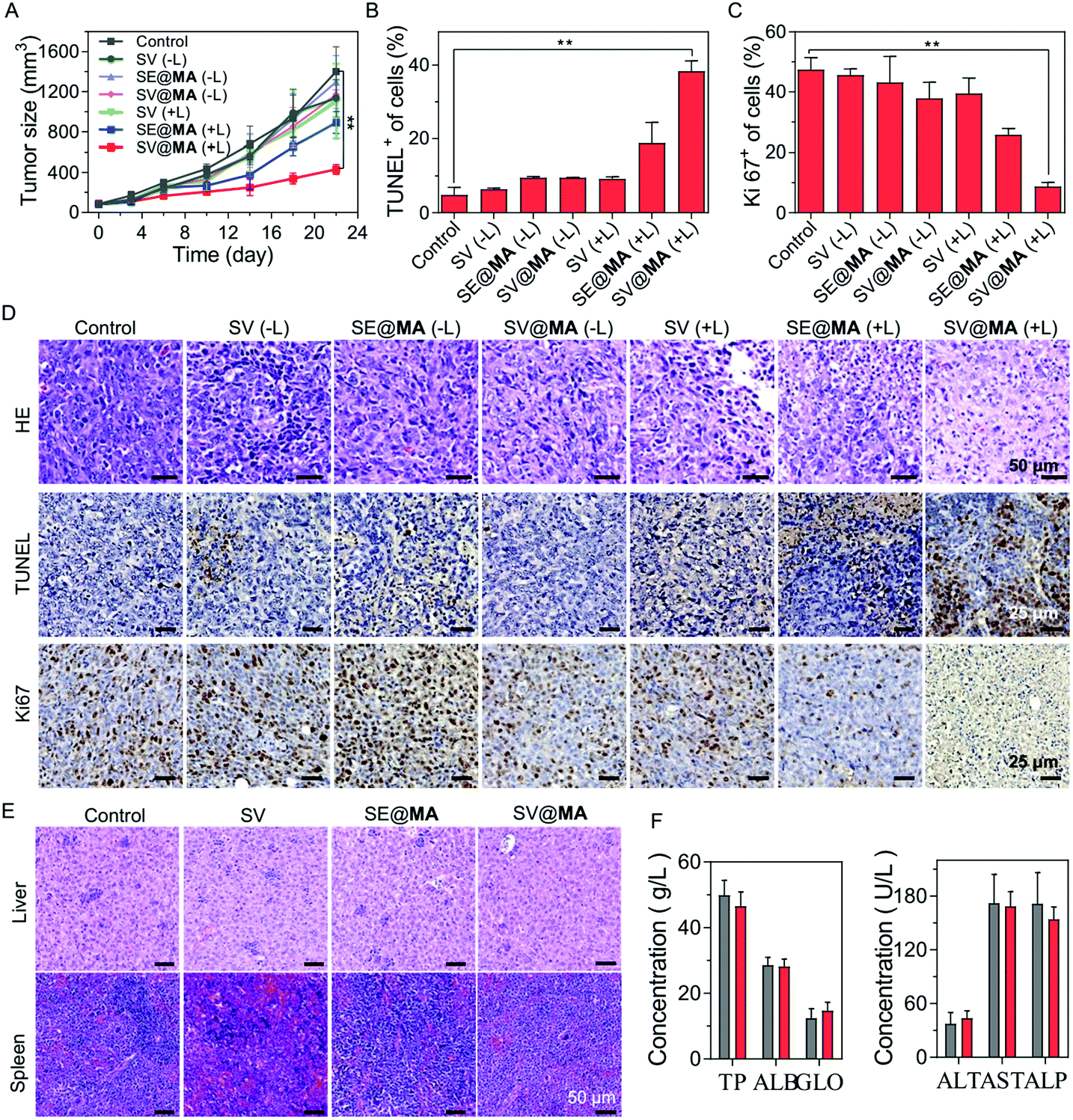 | ||
| Fig. 4 (A) Tumor growth curve of BALB/c mice with 4T1 xenografts after treatment with different formulations (n = 5). Quantification analyses of (B) TUNEL and (C) Ki-67 of tumor tissues from BALB/c mice after different treatments. (D) HE, TUNEL and Ki-67 analyses of tumor tissues of BALB/c mice bearing 4T1 tumors after different treatments. (E) HE analyses of liver and spleen tissues of BALB/c mice bearing 4T1 tumors after different treatments. (F) Liver and renal function results of mice administered with PBS (gray) or SV@MA (red) (n = 3). | ||
As a bacteria-based therapeutic system, biosafety-related concerns may be raised due to the administered Salmonella. Therefore, a series of biosafety tests were conducted. Fig. S19 (ESI†) shows that Salmonella in the blood can be effectively cleared within one week. Meanwhile, at day 1, there were bacteria distributed in the liver and spleen via blood circulation, but the bacterial concentrations in the liver and spleen were found to be continuously decreasing until undetectable at day 7, showing good biosafety of such an approach. Notably, no obvious body weight loss was observed in all the treatment groups throughout the observation period (Fig. S20, ESI†), indicating low systemic toxicity of SV@MA treatment. This was further confirmed by the negligible toxicity found in H&E staining of major organs (the heart, liver, spleen, lungs, and kidneys) (Fig. 4E and Fig. S21, ESI†) and the negligible changes observed in blood biochemistry analysis including liver functions and renal functions (Fig. 4F and Fig. S22, ESI†) of mice on the 22nd day. All the biosafety results demonstrate that the bacteria-based therapy is well-tolerated by animals with excellent biocompatibility as a living bacteria therapeutic system.
Conclusion
In conclusion, we developed a living therapeutic system based on attenuated Salmonella via a metabolic engineering method using an AIE photosensitizer to realize light-controlled gene release for breast cancer therapy. The MA labeling method had no inhibitory effects on Salmonella reproduction, and thus the administered MA-engineered Salmonella carrying VEGFR2 plasmids was able to be localized in the tumor tissues and continue to colonize and express exogenous genes. Following the treatment schedule, the constructed plasmids could be released into the cytoplasm of the host cells under light irradiation. The expression of the released plasmid was then demonstrated to be capable of blocking immunological tolerance to VEGFR2 and inducing a T cell-mediated autoimmune antiangiogenic response. Through in vitro and in vivo experiments, prominent tumor suppression performance was validated with our designed living therapeutic system, which is encouraging for broad therapeutic biomedical research fields, especially for cancer treatment. It's noteworthy that the excitation wavelength of MA was in the visible region, which limited its light penetration in in vivo applications. Future work will focus on the improvement of the metabolic engineering method using two-photon photosensitizers or NIR photosensitizers to further extend the theranostic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Singapore National Research Foundation (R279-000-483-281) and NUS (R279-000-482-133) for financial support. This work was also partially supported by the National Natural Science Foundation of China (grant no. 51873185).Notes and references
- D. B. Pedrolli, N. V. Ribeiro, P. N. Squizato, V. N. de Jesus, D. A. Cozetto, R. B. Tuma, A. Gracindo, M. B. Cesar, P. J. Freire and A. F. da Costa, Trends Biotechnol., 2019, 37, 100–115 CrossRef CAS PubMed.
- A. Maxmen, Nat. Med., 2017, 23, 5–7 CrossRef CAS PubMed.
- S. Chowdhury, S. Castro, C. Coker, T. E. Hinchliffe, N. Arpaia and T. Danino, Nat. Med., 2019, 25, 1057–1063 CrossRef CAS PubMed.
- N. Bernardes, R. Seruca, A. M. Chakrabarty and A. M. Fialho, Bioengineered, 2010, 1, 178–190 CrossRef PubMed.
- Q. Hu, M. Wu, C. Fang, C. Cheng, M. Zhao, W. Fang, P. K. Chu, Y. Ping and G. Tang, Nano Lett., 2015, 15, 2732–2739 CrossRef CAS.
- W. Chen, Z. Guo, Y. Zhu, N. Qiao, Z. Zhang and X. Sun, Adv. Funct. Mater., 2020, 30, 1906623 CrossRef CAS.
- V. Singh, P. Schwerk and K. Tedin, Gut Pathog., 2018, 10, 33 CrossRef PubMed.
- S. Castanheira and F. García-del Portillo, Front. Cell. Infect. Microbiol., 2017, 7, 432 CrossRef PubMed.
- E. M. Camacho, B. Mesa-Pereira, C. Medina, A. Flores and E. Santero, Sci. Rep., 2016, 6, 30591 CrossRef CAS PubMed.
- R. Palffy, R. Gardlik, J. Hodosy, M. Behuliak, P. Reško, J. Radvánský and P. Celec, Gene Ther., 2006, 13, 101–105 CrossRef CAS PubMed.
- S. Pilgrim, J. Stritzker, C. Schoen, A. Kolb-Mäurer, G. Geginat, M. J. Loessner, I. Gentschev and W. Goebel, Gene Ther., 2003, 10, 2036–2045 CrossRef CAS PubMed.
- N. Souders, T. Verch and Y. Paterson, DNA Cell Biol., 2006, 25, 142–151 CrossRef CAS PubMed.
- N. Bernardes, A. M. Chakrabarty and A. M. Fialho, Appl. Microbiol. Biotechnol., 2013, 97, 5189–5199 CrossRef CAS PubMed.
- S. Zhou, C. Gravekamp, D. Bermudes and K. Liu, Nat. Rev. Cancer, 2018, 18, 727–743 CrossRef CAS PubMed.
- B. Kim, S. Suvas, P. P. Sarangi, S. Lee, R. A. Reisfeld and B. T. Rouse, J. Immunol., 2006, 177, 4122–4131 CrossRef CAS.
- W. Song, Q. Sun, Z. Dong, D. Spencer, G. Nunez and J. Nör, Gene Ther., 2005, 12, 320–329 CrossRef CAS.
- Y. Li, M. Wang, H. Li, K. D. King, R. Bassi, H. Sun, A. Santiago, A. T. Hooper, P. Bohlen and D. J. Hicklin, J. Exp. Med., 2002, 195, 1575–1584 CrossRef CAS.
- J. Liu, Y. Wei, L. Yang, X. Zhao, L. Tian, J. Hou, T. Niu, F. Liu, Y. Jiang and B. Hu, Blood, 2003, 102, 1815–1823 CrossRef CAS PubMed.
- Y. Wei, Q. Wang, X. Zhao, L. Yang, L. Tian, Y. Lu, B. Kang, C. Lu, M. Huang and Y. Lou, Nat. Med., 2000, 6, 1160–1166 CrossRef CAS PubMed.
- F. Hu, G. Qi, D. Mao, S. Zhou, M. Wu, W. Wu and B. Liu, Angew. Chem., 2020, 59, 9288–9292 CrossRef CAS.
- D. Wang, M. M. Lee, G. Shan, R. T. Kwok, J. W. Lam, H. Su, Y. Cai and B. Z. Tang, Adv. Mater., 2018, 30, 1802105 CrossRef PubMed.
- J. H. Zheng and J.-J. Min, Chonnam Med. J., 2016, 52, 173–184 CrossRef CAS PubMed.
- S. Rius-Rocabert, F. Llinares Pinel, M. J. Pozuelo, A. García and E. Nistal-Villan, FEMS Microbiol. Lett., 2019, 366, fnz136 CrossRef CAS.
- A. G. Niethammer, R. Xiang, J. C. Becker, H. Wodrich, U. Pertl, G. Karsten, B. P. Eliceiri and R. A. Reisfeld, Nat. Med., 2002, 8, 1369–1375 CrossRef CAS.
- H. Zhou, Y. Luo, M. Mizutani, N. Mizutani, R. A. Reisfeld and R. Xiang, Blood, 2005, 106, 2026–2032 CrossRef CAS PubMed.
- J. Lyons, B. Sheahan, S. Galbraith, R. Mehra, G. Atkins and M. Fleeton, Gene Ther., 2007, 14, 503–513 CrossRef CAS PubMed.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS PubMed.
- H. Su, C. Chou, D. Hung, S. Lin, I. Pao, J. Lin, F. Huang, R. Dong and J. Lin, Biomaterials, 2009, 30, 5979–5987 CrossRef CAS.
- H. Jia, Y. Zhu, Z. Chen and F. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 15943–15951 CrossRef CAS PubMed.
- M. Wu, W. Wu, Y. Duan, X. Li, G. Qi and B. Liu, Chem. Mater., 2019, 31, 7212–7220 CrossRef CAS.
- C. H. Jones, S. Rane, E. Patt, A. Ravikrishnan, C. Chen, C. Cheng and B. A. Pfeifer, Mol. Pharmaceutics, 2013, 10, 4301–4308 CrossRef CAS.
- S. Xie, L. Zhao, X. Song, M. Tang, C. Mo and X. Li, J. Controlled Release, 2017, 268, 390–399 CrossRef CAS PubMed.
- C. Clairmont, K. Lee, J. Pike, M. Ittensohn, K. Low, J. Pawelek, D. Bermudes, S. Brecher, D. Margitich and J. Turnier, J. Infect. Dis., 2000, 181, 1996–2002 CrossRef CAS.
- M. Wu, X. Liu, H. Bai, L. Lai, Q. Chen, G. Huang, B. Liu and G. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 9850–9859 CrossRef CAS PubMed.
- R. Heidenreich, A. Kappel and G. Breier, Cancer Res., 2000, 60, 6142–6147 CAS.
- D. J. Hicklin, F. M. Marincola and S. Ferrone, Mol. Med. Today, 1999, 5, 178–186 CrossRef CAS.
- X. Lu, X. Jiang, R. Liu and S. Zhang, Vaccine, 2008, 26, 5352–5357 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mh01582b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |