
Proteomics in idiopathic pulmonary fibrosis: the quest for biomarkers
Tila
Khan†
 ,
Sanjukta
Dasgupta†
,
Nilanjana
Ghosh
and
Koel
Chaudhury
*
,
Sanjukta
Dasgupta†
,
Nilanjana
Ghosh
and
Koel
Chaudhury
*
School of Medical Science and Technology, Indian Institute of Technology Kharagpur, 721302, India. E-mail: koel@smst.iitkgp.ac.in
First published on 19th October 2020
Abstract
Idiopathic pulmonary fibrosis (IPF) is a debilitating chronic progressive and fibrosing lung disease that culminates in the destruction of alveolar integrity and dismal prognosis. Its etiology is unknown and pathophysiology remains unclear. While great advances have been made in elucidating the pathogenesis mechanism, considerable gaps related to information on pathogenetic pathways and key protein targets involved in the clinical course of the disease exist. These issues need to be addressed for better clinical management of this highly challenging disease. Omics approach has revolutionized the entire area of disease understanding and holds promise in its translation to clinical biomarker discovery. This review outlines the contribution of proteomics towards identification of important biomarkers in IPF in terms of their clinical utility, i.e. prognosis, differential diagnosis, disease progression and treatment monitoring. The major dysregulated pathways associated with IPF are also discussed. Based on numerous proteomics studies on human and animal models, it is proposed that IPF pathogenesis involves complex interactions of several pathways such as oxidative stress, endoplasmic reticulum stress, unfolded protein response, coagulation system, inflammation, abnormal wounding, fibroblast proliferation, fibrogenesis and deposition of extracellular matrix. These pathways and their key path-changing mediators need further validation in large well-planned multi-centric trials at various geographical locations for successful development of clinical biomarkers of this confounding disease.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a specific form of chronic, progressive, irreversible fibrosing interstitial pneumonia of unknown origin that predominantly occurs in adults >60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) years of age.1 IPF is most common amongst the interstitial lung diseases (ILDs) and remains a lethal disease irrespective of treatment, with the median survival rate ranging from 3 to 5 years.2–4 The incidence and prevalence of the disease is not very well documented. Isolated reports from individual countries suggest an estimate of 15–40 cases per 100000 each year.3,5 A number of risk factors associated with IPF have been identified based on numerous epidemiological studies. Population-based retrospective studies on incidence, prevalence and survival of IPF patients conducted in Sweden,6 Korea,7 Canada8 and Italy9 have identified advanced age and male sex to be intrinsic risk factors for the development of IPF. Exposure to metal and wood dust, asbestos exposure, farming and history of raising birds, and air pollution are some of the occupational and environmental risk factors for IPF.7,10–13 The association between incidence of IPF and cigarette smoking is also suggested.13,14 Awareness of chronic environmental insult/lifestyle exposure and suitable control measures may enable better mitigation of risk and prevention of IPF.
years of age.1 IPF is most common amongst the interstitial lung diseases (ILDs) and remains a lethal disease irrespective of treatment, with the median survival rate ranging from 3 to 5 years.2–4 The incidence and prevalence of the disease is not very well documented. Isolated reports from individual countries suggest an estimate of 15–40 cases per 100000 each year.3,5 A number of risk factors associated with IPF have been identified based on numerous epidemiological studies. Population-based retrospective studies on incidence, prevalence and survival of IPF patients conducted in Sweden,6 Korea,7 Canada8 and Italy9 have identified advanced age and male sex to be intrinsic risk factors for the development of IPF. Exposure to metal and wood dust, asbestos exposure, farming and history of raising birds, and air pollution are some of the occupational and environmental risk factors for IPF.7,10–13 The association between incidence of IPF and cigarette smoking is also suggested.13,14 Awareness of chronic environmental insult/lifestyle exposure and suitable control measures may enable better mitigation of risk and prevention of IPF.
IPF is characterized by rapid infiltration of fibrotic tissues to the lungs resulting in irreversible thickening and scarring of lung parenchyma. There is no definitive treatment for IPF; treatment usually varies with the stage of the disease. In recent years, antifibrotic drugs, such as pirfenidone and nintedanib in combination with other immunosuppressive agents have generated considerable interest for the treatment of IPF subjects.15,16 Lung transplantation is strongly recommended as five-year survival rate is reported to be 50–56%.17
The intriguing pathophysiology and diagnostic challenges of IPF
IPF is a challenging disease for clinicians due to its idiopathic etiology and relies on a close collaboration between pathologists, radiologists and chest physicians to minimize diagnostic uncertainty.18 The pathogenesis of IPF is not completely understood, primarily due to the lack of a reliable animal model.19 In contrast to the earlier theory of excessive chronic inflammation of the lung epithelial cells, currently it is widely accepted that IPF results from injury to the alveolar epithelial cells (AECs) caused by an unknown trigger.20 AEC injury may arise due to genetic and epigenetic factors or environmental exposures such as cigarette smoke, dust, viral infections and others.21,22 In recent times, disturbances in the lung microenvironment including type 1 T helper (Th1)/type 2 T helper (Th2) cytokine imbalance and coagulation have been implicated in the pathogenesis of IPF.23It is challenging to establish a precise diagnosis of IPF. It is usually based on clinical, laboratory, radiological and histopathological evaluation based on the American Thoracic Society (ATS)/European Respiratory Journal (ERJ) guidelines.24 So far, no biochemical test exists that can rule out IPF and predict therapeutic responsiveness. In addition to abnormal pulmonary function tests (PFTs), high resolution computed tomography (HRCT) findings can best provide robust and characteristic radiological pattern which may often act as surrogate for lung biopsies. The presence of usual interstitial pneumonia (UIP) pattern in an HRCT image is the hallmark of IPF identification.25 However, precise identification of UIP pattern is not straightforward even amongst radiologists.2 If there is possible UIP or inconsistent pattern of UIP, lung biopsy becomes essential despite complications associated with surgical biopsy, such as triggering of the pneumothorax, pulmonary collapse, pneumonia and others.26,27
Misdiagnosis often results in delayed diagnosis of IPF due to frequent overlap of symptoms with more common conditions such as cardiac disease, asthma and chronic obstructive pulmonary disease (COPD).28,29 The presence of IPF is often suspected when a predominantly basal reticular pattern, with or without cystic air-spaces, appears on a chest X-ray accompanied by reduced lung volumes. However, these features can also appear in other fibrotic diseases including chronic hypersensitivity pneumonitis, systemic autoimmune disease-associated pulmonary fibrosis or stage IV sarcoidosis and in non-fibrotic diseases such as bullous emphysema, bronchiectasis, infections, etc.30 Also, certain diagnostic features, e.g. honeycombing on HRCT images of IPF patients are often subjective and significant inter-observer variability have been reported.31 Early and differential diagnosis of IPF is critical since this disease is unresponsive to standard therapies with majority of the patients succumbing within 5 years of diagnosis.32 The lung function would have declined considerably due to late referral to specialized clinicians. Timely intervention with anti-fibrotic drugs is critical to prevent progressive respiratory failure in IPF patients and prolong their survival.
Few diagnostic and prognostic biomarkers using serum and bronchoalveolar lavage (BAL) fluid have been reported for IPF; however, they are far from routine clinical use. While Krebs von den Lungen-6 (KL-6),33 surfactant proteins A and D,34 chemokine IL8 and chemokine (C-X-C motif) ligand 13 (CXCL13)35 and matrix metallopeptidase 28 (MMP28)36 are reported to be diagnostic serum biomarkers; the prognostic markers of IPF include matrix remodelling protein lysyl oxidase homolog 2 (LOXL2), periostin, chemokine CC chemokine ligand18 (CCL18), growth factors and adhesion molecules such as YKL-40, intracellular adhesion molecule-1 (ICAM-1) and vascular endothelial growth factor (VEGF).37 Recent reports also suggest that advanced glycation end-products and their receptors could be useful prognostic biomarkers of IPF.38 Serum and BAL matrix metalloproteinase have also been investigated but are not considered to be reliable biomarkers.39 Furthermore, nitric oxide, 8-isoprostane, hydrogen peroxide, nickel, chromium, silicon, cobalt, iron, copper, selenium, molybdenum, nitrite, nitrate, docosatetraenoyl lypophosphatidic acid, p-cymene, acetoin, isoprene, ethylbenzene and unidentified volatile organic components are also reported to be diagnostic and prognostic breath biomarkers of IPF.40
Clinical proteomics
Proteomics, a relatively well-established field of “omics”, refers to the study of proteins in terms of their expression, quantitation, localization, function, variations in their molecular forms, post-translational modifications (PTMs), conformations, chemical modifications and protein–protein interactions at a specific time and under specific condition.41 “Clinical proteomics” is a sub-discipline of proteomics that involves the application of proteomic technologies on clinical specimens such as tissues, cells and biofluids. This tool provides a snapshot of complex disease mechanisms and helps identify novel biomarkers for diagnosis, therapy, and prognosis.28 Advent of gel-based and gel-free proteomic techniques together with advancements in mass spectrometry (MS) has enabled rapid, unbiased, systematic and high-throughput identification and quantification of complex protein mixtures.27Bottom-up and top-down proteomics are the two broad approaches that are utilized in clinical proteomics. While bottom-up proteomics includes analysis of peptides, the top-down approach analyses intact proteins. The bottom-up approach, also known as ‘shotgun proteomics’, involves proteolytic digestion of crude protein extracts into smaller peptides which are separated using gel-based or gel-free methods and then fragmented and analysed using MS/MS for the identification of peptides and their back-tracking towards origin proteins.42 In the gel-based method, protein mixture is first separated using two-dimensional (2-DE) or 2D difference in-gel electrophoresis (2D-DIGE). The 2-DE workflow involves isoelectric focusing of proteins in the first dimension, and separation based on molecular weight in the second dimension, thus generating protein spots on the gel. Subsequently, proteins are extracted from the spots, subjected to proteolytic digestion and the resultant peptides analysed using MS. The 2D-DIGE method involves labeling of up to three different protein samples with size matched fluorescent cyanine-based dyes (Cy 2,3,5), followed by their co-separation on a single 2-DE gel,29 making spot comparison and protein quantitation more reliable by reducing inter-gel variability. The gel-free method or “quantitative proteomics” involves metabolic, enzymatic or chemical labeling of protein samples or can be a label-free technique. Here, the protein mixture is proteolytically digested directly into peptides which are separated using liquid chromatography (LC) and subjected to tandem MS analysis.43 The bottom-up method is most widely used due to its robustness, simplicity, high-resolution separation of peptides, high-throughput, reproducible identification and quantitation, and requirement of less sophisticated instrumentation and expertise. The major disadvantage of this approach is the issue with protein inference and limited protein sequence coverage which results in loss of functional information about molecules such as protein complexes, proteoforms, protein conformation, PTMs and alternative splice variants.
In the top-down approach, intact proteins are first purified from complex biological mixtures, separated using gel electrophoresis or gel free techniques, and directly introduced into MS/MS for fragmentation and identification.44,45 This ensures intactness, profiling and quantitation of all proteoforms, and provides an overview of complete protein sequence, alternative splicing, protein isoforms, protein subunits and locates PTMs with sequence variations. Despite immense potential of this approach, some challenges remain such as high cost involvement, lower sensitivity for high molecular weight proteins, low-throughput, problems associated with labeling and separation of some intact proteins, solubility, and is time consuming. To address these challenges, the top-down method is rapidly evolving with methodological and technological developments for successful applications in future, and alternative solutions such as the middle-down approach are also underway.46 The advancements in top-down proteomics is beyond the scope of the present review, and is well described in detail in some excellent reviews.47,48
It is important to mention that IPF is associated with extreme local production of functionally different proteins.22 Within such a context, the aim of this review is to provide a summary of the proteomic methods and samples used to investigate the proteomic profile of patients with IPF. Though limited, proteomic studies on animal models of IPF and cells cultured from lung tissues of patients with IPF are also discussed.
Search strategy
Literature search of PUBMED, MEDLINE and EMBASE database using the term “idiopathic pulmonary fibrosis” and “proteomics” showed 106 results. Out of these, 19 are review papers and the rest are original research articles. There are only 2 review papers published so far where proteomics approach (and not other omics technologies) in IPF is reported. One of them discusses the contribution of proteomics towards understanding of personalized medicine in a number of respiratory diseases. The other review paper provides an overview of proteomic approaches used for the study of COPD and different classes of ILDs. Fig. 1 shows the search strategy which highlights exclusion of unrelated studies and inclusion of 45 proteomics based original articles on IPF in humans (n = 38) and animals (n = 7). Two articles on proteome analysis of BALF and serum in IPF are in Chinese language and, therefore, not discussed. We have sub-divided all proteomics studies carried out on IPF patients under different types of biofluids analysed. A separate section on animal studies related to IPF is also provided. Table 1 summarizes the major dysregulated proteins and associated pathways in humans with IPF, animal models of pulmonary fibrosis and cells isolated from IPF patients.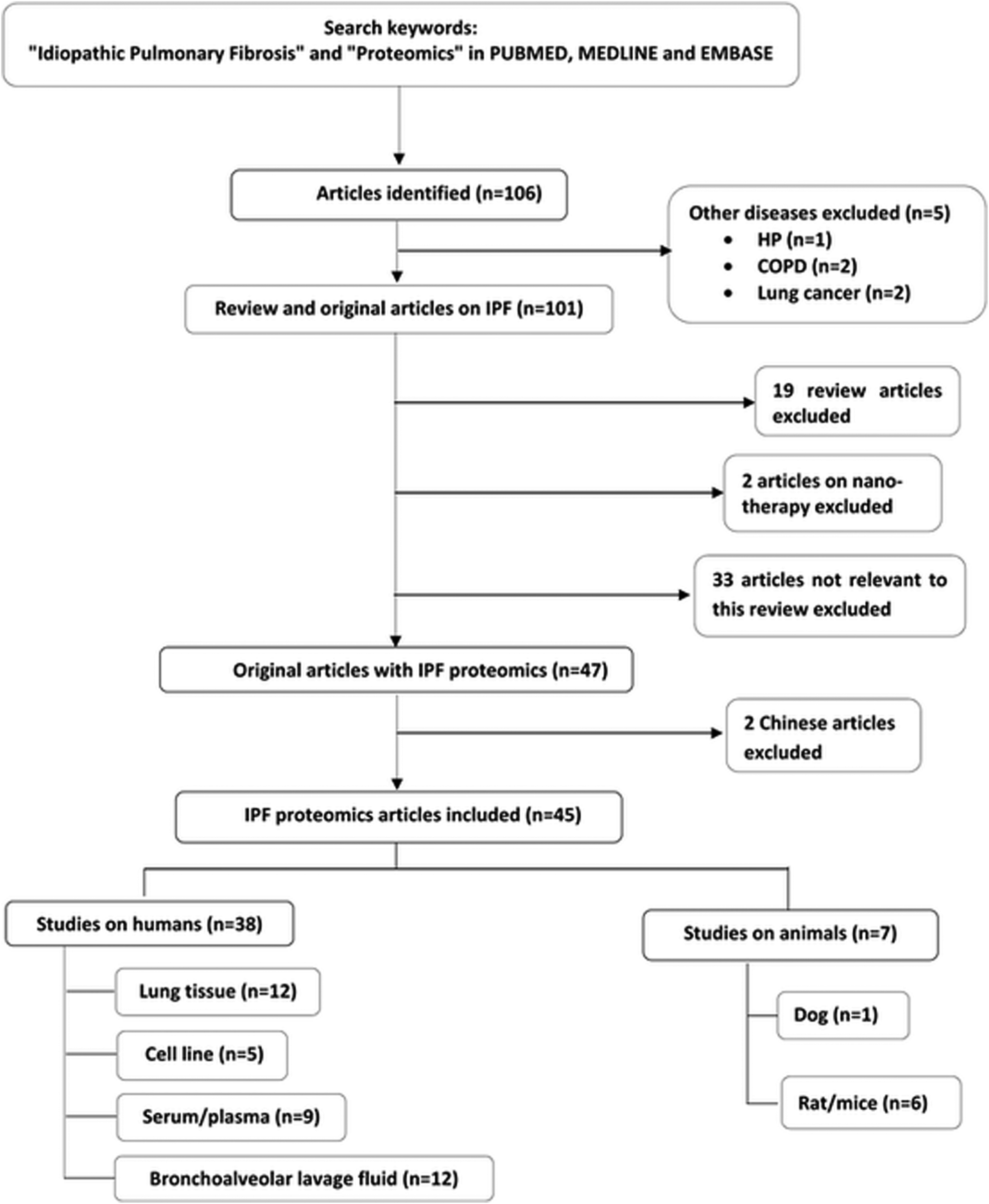 | ||
| Fig. 1 A flow diagram of literature search outlining inclusion and exclusion of articles. | ||
| Sample | Study design | Technique | Validation assay | Major proteins/pathways identified in IPF | Ref. |
|---|---|---|---|---|---|
| Abbreviations: BALF, Bronchoalveolar lavage fluid; IPF, Idiopathic pulmonary fibrosis; f-IPF, Familial IPF; s-IPF, Sporadic IPF; 2-DE, 2 dimensional electrophoresis; MS, Mass spectrometry; MALDI-ToF, Matrix assisted laser desorption/ionization time of flight; LC-ESI-IT-MS2, Liquid chromatography-electrospray ionization-iontrap mass spectrometry; SFTPA2, Surfactant protein A2; ER, Endoplasmic reticulum; OS, Oxidative stress; LC-MS/MS, Liquid chromatography tandem mass spectrometry; HDMSE, High definition MS data independent acquisition; MRM, Multiple reaction monitoring; MMP7, Matrix metallopeptidase 7; CCL24, Chemokine ligand 24; CXCL7, Chemokine ligand 7; CCL18, Chemokine ligand 18; NF-κB, Nuclear factor κB; PPARγ, Peroxisome proliferator activated receptorsγ; SS, Systemic sclerosis; PLCH, Pulmonary langerhans cell histiocytosis; S10A6, Calcyclin; GSTP1, Glutathione S transferase P1; NSIP, Non-specific interstitial pneumonia; COP, Cryptogenic organising pneumonia; CVD-IP, Collagen vascular disease associated interstitial pneumonia; fNSIP, Fibrotic NSIP; ELISA, Enzyme linked immunosorbent assay; IHC, Immunohistochemistry; ECM1, Extracellular matrix protein-1; S100A9, Calgranulin; PCR, Polymerase chain reaction; ApoAI., Apolipoprotein AI; LMW, Low molecular weight; MIF, Macrophage migration inhibitory factor; HP, Hypersensitivity pneumonitis; LASP1, LIM and SH3 protein 1; ILD, Interstitial lung disease; UIP, Usual interstitial pneumonia; 2D-DIGE, 2-D fluorescence difference gel electrophoresis; UPR, Unfolded protein response; HSP, Heat shock protein; COPD, Chronic obstructive pulmonary disease; RAGE, Receptor for advanced glycation end products; SOMAmer, 1129 analyte slow-off rate modified aptamer; FCN2, Ficolin-2; Cath-s, Cathepsin S; LGMN, Legumain; sVEGFR2, Soluble vascular endothelial growth factor receptor 2; ICOS, Inducible T cell costimulatory; Try, Trypsin 3; iTRAQ, Isobaric tag for relative and absolute quantification; AHSG, Alpha-2-HS-glycoprotein; AMBP, Alpha 1 microglobulin/bikunin precursor; CRP C, Reactive protein; KNG1, Kininogen-1; LCM-MS, Laser capture microdissection coupled to mass spectrometry; SWATH-MS, Sequential window acquisition of all theoretical mass spectra; Q-ToF MS, Quadrupole time of flight mass spectrometer; AHSG, Alpha-2-HS-glycoprotein; AMBP, Alpha 1 microglobulin/bikunin precursor; SPARC, Secreted protein acidic and rich in cysteine; EVs, Extracellular vesicles; tRNA, Transfer ribonucleic acid; AE-IPF, IPF patients with acute exacerbation; PI3K-Akt, Phosphatidylinositol 3-kinase, protein kinase B; RAP1B, Member Of RAS oncogene family; Gas6/TAM, Growth arrest specific gene 6/TYRO3 (tyrosine protein kinase); AXL, Anexelekto; MERTK, c-mer proto-oncogene tyrosine kinase. | |||||
| BALF | 18 stable IPF and 9 IPF patients with acute exacerbations | 2-DE and MALDI-ToF/ToF | Dot-blot analysis | AE-IPF: acute phase proteins involved in response signalling, clathrin-mediated endocytosis signalling and lung carcinogenesis | Carleo et al. (2020)48 |
| BALF | 10 f-IPF and 17 s-IPF patients | 2-DE, MALDI-ToF/ToF MS, nanoscale LC-ESI-IT-MS2 | None | fIPF: upregulated SFTPA2 isoforms, ER stress, wounding, coagulation, immune response, ion homeostasis proteins | Carleo et al. (2016)47 |
| sIPF: OS response protein | |||||
| BALF | 4 IPF patients and 5 controls | 2-DE, LC-MS/MS, HDMSE | MRM | Profibrotic proteins: osteopontin, MMP7, CCL24, CXCL7, CCL18 | Foster et al. (2015)46 |
| BALF | 7 IPF patients, 8 non-mokers and 10 smokers | 2-DE, MALDI-ToF/ToF MS, LC-ESI/MS-MS | Western blot | Functional hub: NF-κB, PPARγ and c-myc | Landi et al. (2014)45 |
| Pathways: angiotensin system, heme metabolism coagulation system OS, hypoxia response, iron transport | |||||
| BALF | 7 IPF, 9 sarcoidosis, 7 SS, 9 PLCH patients, 10 non-smokers and 8 smokers | 2-DE, MALDI-ToF/ToF MS, LC-MS/MS | Western blot | Proteins: plastin 2, annexin A3, 14-3-3ε, S10A6, GSTP1, peroxiredoxin 1 | Landi et al. (2013)44 |
| Pathways: alternative complement, protein folding, slit-robo signalling, coagulation | |||||
| BALF | 28 IPF, 15 NSIP, 20 COP, 35 CVD-IP patients and 23 controls | 2-DE, LCMS-IT-ToF | ELISA, IHC | Upregulated: S100A9 in IPF | Hara et al. (2012)41 |
| BALF | 14 IPF patients and 8 controls | 2-DE, nano LC-MS/MS | ELISA, IHC, PCR, animal studies | Downregulated: ApoA1 in IPF | Kim et al. (2010)40 |
| BALF | 30 IPF and 12 controls | 2-DE | ELISA, IHC | MIF | Bargagli et al. (2009)43 |
| BALF | 11 IPF, 9 sarcoidosis, 11 SS patients and 5 controls | 2-DE and MALDI-ToF | None | Calgranulin B | Bargagli et al. (2008)42 |
| BALF | 10 IPF, 11 sarcoidosis and 10 SS patients | 2-DE, MALDI-ToF-MS | None | Local LMW proteins, galectin 1, MIF, calgranulin B, p23, cyclophilin A, ubiquitin | Rottoli et al. (2005)39 |
| BALF | 16 sarcoidosis, 13 IPF, 10 SS patients and 5 controls | 2-DE, MALDI-ToF MS | ELISA, Western blot | Upregulated: carbonylated proteins in IPF, SS, sarcoidosis, protein targets of oxidation in IPF | Rottoli et al. (2005)38 |
| BALF | 6 IPF and 6 sarcoidosis patients | 2-DE, MALDI-ToF MS | Western blot | MIF, calgranulin, antioxidant peroxysomal enzyme and thioredoxin peroxidase 2 | Magi et al. (2002)37 |
| Pathways: antioxidant response, protease antiprotease balance, fibrosis | |||||
| Lung tissue | 3 IPF patients | LCM-MS | IHC | 1252 uniquely expressed proteins | Herrera et al. (2020)74 |
| Lung tissue | 4 IPF patients and 4 healthy donors | Matrisome classification system combined with LC-MS/MS | None | Upregulated: collagens, proteoglycans, and ECM glycoproteins, nidogen-2 | Rendin et al. (2019)70 |
| Downregulated: laminins and collagen IV | |||||
| Lung tissue | Lung fibroblasts from IPF patients, human lung epithelial fibroblasts and bleomycin-induced lung fibrosis model | LC-MS/MS | None | Eupatilin inhibits trans differentiation of pathogenic myofibroblasts | Kim et al. (2019)79 |
| Lung tissue | 20 IPF patients and 20 controls | LC-MS/MS and iTRAQ | Western blot and IHC | 662 significantly altered proteins (455 unregulated and 207 down regulated) | Tian et al. (2019)69 |
| Pathways: mainly associated with PI3K-Akt signalling, ECM-receptor interaction, focal adhesion and carbon metabolism | |||||
| Lung tissue | 6 IPF, 5 COPD patients and 5 controls | 2-DE and LC-MS/MS | IHC | Collagen VI and laminins | Ahrman et al. (2018)62 |
| Lung tissue | 10 IPF, 10 SS patients and 10 healthy donors | LC-MS | None | Fibrotic matrisome profile deposited by IPF and SS fibroblasts were distinct as compared to controls | Mullenbrock et al. (2018)71 |
| Lung tissue | 45 ILD tissue and 10 healthy donors | LC MS/MS quadrupole/orbitrap MS | Western blot, confocal imaging | Auto-antibodies in IPF lungs | Schiller et al. (2017)68 |
| Lung tissue | 8 UIP, 8 NSIP patients and 30 controls | 2D-DIGE, MALDI-ToF MS | Western blot, IHC | Differences in vimentin subtypes in NSIP and UIP | Ohara et al. (2014)67 |
| Lung tissue | 14 IPF, 8 fNSIP patients and 10 controls | 2D-DIGE, MALDI-ToF MS | IHC, BALF Western blot | ER and OS in both diseases. | Korfei et al. (2013)66 |
| Upregulated: stress response | |||||
| Downregulated: anti-apoptotic factors, antifibrotic molecules in both the diseases | |||||
| Lung tissue | 14 IPF patients and 10 controls | 2-DE, MALDI-ToF MS | IHC, Western blot | Upregulated: UPR, HSP, DNA damage stress markers | Korfei et al. (2011)63 |
| Downregulated: annexin family, antioxidants, structural epithelial proteins | |||||
| Lung tissue | 4 IPF, 4 COPD patients and 4 controls | 2-DE, MS | Western blot, IHC and morphometry | Downregulated: hemoglobin α and β and their complexes. No change in COPD | Ishikawa et al. (2010)59 |
| Lung tissue | 4 IPF patients and 4 controls | 2-DE, MS and MS/MS | Western blot, ELISA | 3 variants of RAGE | Ohlmeier et al. (2010)60 |
| Lung tissue and cell line | 3 IPF patients, 3 normal tissue samples and NCl-H441 cell line | LC-MS | Western blot | Pathway: SPARC mediated paracrine signalling pathway | Conforti et al. (2020)77 |
| Lung tissue and cell line | IPF and non-IPF fibroblasts isolated from patients and IMR90 human fetal lung diploid fibroblasts | SWATH-MS | None | Fibroblast-derived EVs carry fibronectin on the vesicular surface and promote fibroblast invasion | Chanda et al. (2019)76 |
| Lung tissue and cell line | Human samples from 4 IPF patients, 4 healthy donors and secondary lung fibroblast cell line | LC-MS/MS | None | Pathway: de novo serine synthesis pathway is necessary for collagen production | Nigdelioglu et al. (2016)72 |
| Lung tissue and cell line | 22 IPF patients and A549 human type II cells | Tandem MS | None | Autoantibodies against alanyl-tRNA synthetase | Takahashi et al. (2007)75 |
| Secondary cell line | Normal lung fibroblasts TIG7 and IPF lung fibroblasts (LF1 and LF2) | Q-ToF MS | 2-DE | Downregulated: Ser586 phosphorylation of very long chain acyl-CoA dehydrogenase in IPF | Kabuyama et al. (2010)81 |
| Plasma | 17 IPF patients and 19 controls | Label-free proteomics | None | Pathway: dysregulated complement activation system and oxidative damage | Saraswat et al. (2020)56 |
| Plasma | 300 IPF patients and 100 subjects without known lung disease | Aptamer based platform encompassing 1305 proteins | None | Altered: glycoproteins thrombospondin 1 and von Willebrand factor immune-related proteins C–C motif chemokine ligand 17 and bactericidal permeability-increasing protein | Todd et al. (2019)54 |
| Plasma | 10 IPF patients and 10 controls | iTRAQ | MRM | Upregulated: platelet basic protein | Moodley et al. (2019)55 |
| Downregulated: actin, cytoplasmic 2, antithrombin-III, ECM1 and fibronectin | |||||
| Plasma | 60 IPF patients and 21 controls | SOMAscan assay | None | Pathway: dysregulation in plasma proteins involved in defense response, wound healing and protein phosphorylation | O’Dwyer et al. (2017)53 |
| Plasma | 35 IPF progressors and 25 IPF non-progressors | Six SOMAmer assay | None | Upregulated: FCN2, Cath-S, LGMN, sVEGFR2. | Ashley et al. (2016)52 |
| Downregulated: ICOS, TRY3 | |||||
| Serum | 6 IPF patients | 2-DE and MALDI-ToF | ELISA | 13 protein species including haptoglobin increased significantly after one year of nintedanib treatment | Landi et al. (2020)58 |
| Serum | 20 IPF patients and 20 controls | iTRAQ coupled with 2DE and LC-MS/MS | ELISA | Biomarker: CRP, fibrinogen-α, haptoglobin, and kininogen-1 | Zhang et al. (2018)50 |
| Serum | 60 IPF patients and 60 controls | iTRAQ nano-LC-MS/MS | ELISA | Biomarkers: AHSG, AMBP, CRP, KNG1 | Niu et al. (2017)49 |
| Serum | 40 IPF patients and 40 controls | Immunoblot and MALDI-ToF | None | Circulating anti-periplakin antibodies | Taillé et al. (2011)51 |
| Blood and tissues | Rat model of pulmonary fibrosis | iTRAQ-coupled LC-MS/MS | None | Upregulated: fibronectin 1, LIM and SH3 protein 1 (LASP1), ras-related protein RAP1 B | Fukunaga et al.85 |
| Down-regulated: vinculin and aldehyde dehydrogenase 1 family member A1 | |||||
| Tissue | Mice model of pulmonary fibrosis | LC-MS | Western blot | PI3K/Akt and Wnt signalling pathways are the most significant profibrotic pathways | Kulkarni et al.86 |
| Tissue | Mice model of pulmonary fibrosis, 3 IPF patients and 3 organ donor individuals | LC-MS/MS | None | Isolevuglandins modification | Mont et al.87 |
| Tissue | Pulmonary fibrosis induced murine model and human IPF lung tissue | Quantitative proteomics | None | Gas6/TAM receptor contributes in IPF pathogenesis | Espindola et al.88 |
| Tissue | Nedd4-2−/− mice and human IPF lung tissue | Nano-flow LC coupled to quadrupole orbitrap MS | None | Lung proteome alterations seen due to pirfenidone treatment | Duerr et al.89 |
| Tissue | Mice model of pulmonary fibrosis and human lung IPF cell | LC-MS/MS | None | Secretome and exosome inhalation can regenerate normal alveolar structure and reduce fibrotic response | Dinh et al.90 |
| BALF | Canine model of pulmonary fibrosis | 2D-DIGE and LC-MS | Western blot | Upregulated: complement 3, α-1-antitrypsin, apolipoprotein 1, haptoglobin, β-actin and transketolase | Lilja-Maula et al.92 |
| Downregulated: lysozyme C | |||||
Bronchoalveolar lavage fluid (BALF)
Proteomics has been used to elucidate the pathophysiology of IPF for more than a decade now. Possible proteins and pathways linked to the pathophysiologic alterations in IPF and different ILDs are summarized in Table 1. Early in 2002, Magi et al. analysed differences in protein expression in BALF of patients with two forms of ILD, sarcoidosis and IPF using 2-DE and matrix assisted laser desorption ionization-time of flight MS (MALDI-ToF) mass fingerprinting. IPF was found to be associated with an increase in proteins related to oxidative stress.49 Rotolli et al. sought to analyse oxidative stress induced changes in BALF protein carbonyl content of patients with different diffuse lung diseases utilizing 2-DE and MALDI-ToF MS combined with immunoblotting. Their findings suggest that several oxidation-related proteins such as albumin, immunoglobulin heavy chains, complement C3, transferrin, immunoglobulin low chains and immunoglobulin A are present in IPF.50 In another study, the authors analysed the immunological profile (Th1 versus Th2) and BALF proteomic differences in sarcoidosis, systemic sclerosis (SS) and IPF patients. The 2-DE approach indicated that differences in BALF protein content were more apparent between sarcoidosis and IPF. IPF was characterized by low molecular weight proteins. SS represented an intermediary profile between IPF and sarcoidosis with some unique characteristics.51Kim et al. investigated protein expression in BALF of patients with IPF using 2-DE and LC-MS/MS. Low concentration of apolipoprotein A1 (ApoA1) in subjects with IPF was validated in BALF of IPF animal model, i.e. bleomycin (BLM) treated mice. Besides ApoA1, reduced levels of α1-antitrypsin, α1-antichymotrypsin, angiotensinogen, hemoglobin chain B, clusterin, protein disulphide isomerase A3, immunoglobulin, and complement C4A and increased haptoglobulin were also observed in BALF of patients with IPF. A variety of fibrotic interstitial pneumonias exist that resemble IPF such as idiopathic nonspecific interstitial pneumonia (I-NSIP) and collagen vascular disease-associated interstitial pneumonia (CVD-IP), but clinically they are superior in terms of their response to therapy and prognosis.52 Hara et al. used 2-DE and LC-MS to search for biomarkers for differentiating IPF from other fibrotic interstitial pneumonias. The usefulness of S100A9 (calgranulin B) as a discriminating biomarker of IPF from different fibrotic interstitial pneumonias was also explored using enzyme-linked immunosorbent assay (ELISA). The authors concluded that BALF S100A9 is a specific and sensitive biomarker in differentiating IPF from CVD-IP and I-NSIP. These findings suggest that serum S100A9 reflects systemic inflammatory conditions, whereas BALF S100A9 reveals local inflammation in the lung.53 In another similar kind of study, the concentration of calgranulin, assessed using 2-DE and MALDI-ToF, was found to be significantly elevated in IPF subjects as compared to sarcoidosis, SS and healthy controls. The authors hypothesize that dysregulation of calgranulin could be due to the activation of lymphocytes and neutrophils involved in the fibrosis process.54 The same group conducted another study to investigate the differential expression of macrophage migration inhibitory factor (MIF), a multi-function pleiotropic cytokine in BAL of patients with IPF, sarcoidosis, SS and healthy controls. MIF concentration, ascertained using 2-DE and validated with ELISA and immunohistochemistry (IHC), was found to be significantly higher in IPF patients as compared to controls and also correlated with BAL neutrophil concentration.55
Landi and co-workers investigated BALF of patients with four types of ILDs [sarcoidosis, IPF, pulmonary langerhans cell histiocytosis (PLCH), SS], smokers and non-smoker controls using 2-DE and MALDI-ToF/ToF technique. Several proteins such as plastin 2, annexin A3, 14-3-3ε and S10A6 (calcyclin) were identified and validated.56 The same group, in another study, explored changes in protein expression in BALF of IPF subjects, never-smoker healthy controls and smoker controls using proteomics and functional analysis. Furthermore, enrichment analysis suggested that angiotensin system, heme metabolism, coagulation system, oxidative stress, hypoxia response, iron transport pathway and renin–angiotensin–aldosterone system pathway were involved in IPF pathogenesis.57 Foster and his team (2015) conducted a gel-free proteomic analysis of BALF in patients with IPF. They found an enhanced expression of osteopontin, MMP7, CXCL7, CCL18, eosinophil- and neutrophil-derived proteins, in addition to proteins related to fibroblast foci in IPF as compared to controls.58
Genetic cause of IPF is demonstrated; when IPF occurs in at least 2 members of the same family, it is called familial IPF (f-IPF). Carleo and his team (2016) observed significant proteomic differences in BALF of patients with f-IPF versus sporadic IPF. Using 2-DE and MALDI-ToF/ToF, 22 differentially expressed unique proteins could be identified in f-IPF as compared to sporadic IPF. The authors suggest that endothelial reticulum (ER) stress possibly plays a central pathogenic role in f-IPF with the upregulated proteins involved in wounding and immune responses, coagulation system, and ion homeostasis. Oxidative stress response could be associated with the upregulated proteins in the sporadic IPF group.59 The same group, in another recent comparative study using gel-based proteomics and MALDI-ToF/ToF, has identified aberrantly expressed proteins in IPF patients with acute exacerbation (AE-IPF). AE-IPF was found to be associated with acute phase proteins involved in response signalling, clathrin-mediated endocytosis signalling and lung carcinogenesis. The authors also concluded that ApoA1, VEGFA, and interleukin-6 (IL-6) play a key role in the pathogenesis of IPF.60
Serum/plasma
It is well accepted that serum/plasma biomarkers, if identified for disease progression, will tremendously help clinicians in therapeutic management of IPF. With an aim to identify non-invasive biomarkers for the disease, Niu et al. (2017) used a coupled isobaric tags for relative and absolute quantitation (iTRAQ)-LC-MS/MS approach to examine the expression of proteins in serum of 60 IPF patients and compared with an equal number of healthy controls. ELISA was used to validate the candidate markers. The authors could successfully identify and validate alpha-2-HS-glycoprotein (AHSG), protein alpha 1 microglobulin/bikunin precursor (AMBP), C-reactive protein (CRP) and kininogen-1 (KNG1) as promising IPF specific biomarkers.61 The results seem to be robust since an independent validation patient cohort was recruited to confirm the findings. The same group later, in an identical study, demonstrated that the levels of CRP and fibrinogen-α chain are higher, and haptoglobin and KNG-1 are lower in IPF patients. However, the sample size of this study was limited to only 20 IPF patients and 20 healthy controls.62 The role of autoimmunity in IPF has been explored by Taillé et al. (2010). Using immunoblot and MALDI-ToF, the authors identified the presence of circulating anti-periplakin antibodies in the serum of patients with IPF. The antibodies could also be detected in BALF and were associated with disease severity.63A large number of new slow off – rate modified aptamers (SOMAmers) have been developed against protein targets with high binding affinity and specificity, and this technology has recently drawn considerable attention in the area of biomarker discovery. This proteomics platform measures 1129 analytes with increased sensitivity and is an exciting tool for the identification of low-abundance protein molecules. Ashley and co-workers (2016) conducted an observational prospective, multi-centric, longitudinal follow up proteomics study on 60 IPF patients undergoing treatment, and who completed 80 weeks of follow-up. A six-SOMAmer analyte index consisting of soluble vascular endothelial growth factor receptor 2 (VEGFsR2), ficolin-2 (FCN-2), legumain (LGMN), cathepsin-S (cath-S), inducible T cell costimulator (ICOS) and trypsin-3 (TRY-3) were identified in plasma and correlated with disease progression.64 Increased levels of analytes involved in immune function, proteolysis and angiogenesis were observed. Another similar type of study demonstrates the use of SOMAmer platform to identify differentially expressed plasma proteins in IPF patients as compared to normal controls. Using similar type of aptamer approach, O’Dwyer et al. (2017) identified 1100 proteins in the peripheral blood of IPF patients and normal volunteers. They identified dysregulation in plasma proteins involved in defense response, wound healing and protein phosphorylation of IPF subjects when compared to controls. Further enrichment analysis of the upregulated proteins suggested that amino acid phosphorylation, VEGF signalling and intracellular signalling cascade were significantly enriched. In contrast, defense response, anti-apoptosis and immune response were found to be most significantly enriched by analysing the downregulated protein profile.65
In another recent study, Todd et al. (2019) used aptamer-based platform covering 1305 proteins (SOMAscan) to compare protein expression in plasma of 300 IPF patients with 100 participants having unknown pulmonary disease. Glycoproteins (thrombospondin 1 and von Willebrand factor) and immune-related proteins (C–C motif chemokine ligand 17 and bactericidal permeability-increasing protein) were found to be most significantly altered in IPF subjects as compared to controls. The authors further suggest that nine proteins (ApoA1, complement C1r, MMP3, single-frequency network, CCL18, ICAM5, sonic hedgehog, oxidized low-density lipoprotein receptor 1, macrophage-capping protein), when considered collectively, can differentiate IPF patients from healthy controls with very high accuracy (area under the curve = 0.99).66
Non-targeted discovery proteomics using iTRAQ followed by targeted quantitation using multiple reaction monitoring (MRM) have been recently used by Moodley and co-workers to identify circulating plasma proteins in 10 IPF subjects. Five proteins were found to be differentially expressed, with platelet basic protein upregulated and actin, cytoplasmic 2, antithrombin-III, extracellular matrix protein-1 (ECM1) and fibronectin downregulated in IPF compared to healthy controls. The authors further observed that antithrombin-III correlated significantly with forced vital capacity (FVC) of the recruited patients and could reflect severity of the disease.67
In a recent study, to identify biomarkers that can diagnose outcome in IPF patients or their therapeutic response, Saraswat and coworkers (2020) have performed label-free plasma proteomics in 17 IPF cases and compared with 19 healthy controls. Rigorous data analysis suggested that haptoglobin-related protein (HPR) holds promise as a candidate biomarker for IPF. Furthermore, pathway analysis indicated dysregulated complement activation system and oxidative damage in these patients.68
Interaction of drugs with the human proteome is diverse and complex. Drugs are known to affect protein–protein, protein–nucleic acid, or PTMs, thereby leading to beneficial or detrimental actions in the human body. Pharmacoproteomics provides an opportunity to study drug mechanisms at the proteome level, helps to explore a drug's efficacy, toxicity and resistance and facilitates discovery of new drug targets with improved safety and tolerability.69 Recently, Landi et al. (2020) have explored the molecular mechanism of action of a tyrosine kinase inhibitor drug, nintedanib for the treatment in IPF. Six IPF patients were recruited and received nintedanib therapy at a dose of 150mg twice a day for one year. Serum samples were analysed using 2-DE before initiating treatment and following one year of therapy. The differentially expressed spots were identified using MALDI-ToF. The authors found 13 protein species to be upregulated following one year of nintedanib treatment. They further concluded that haptoglobin, validated by Western blot and ELISA, holds promise as a theragnostic marker of nintedanib action in IPF.70
Lung tissue
It is well agreed upon that lung tissue analysis in pulmonary research provides an insight into the pathways and molecular alterations associated with the pathogenesis of disease. However, ethical and regulatory restrictions often pose a challenge and such studies are not always possible. Nonetheless, there are quite a few interesting reports on proteomic analysis of lung tissue in IPF patients. Ishikawa et al. (2010) have compared lung protein expression profile of IPF patients with COPD and controls using 2-DE and Western blot analysis. Their findings suggest significantly low levels of hemoglobin (Hb) α and β monomers in patients with IPF. They could also detect Hbα and β as monomers in lung tissue and as tetramers in BALF and induced sputum. However, the exact role of Hb in the pathogenesis of IPF could not be underlined.71In the same year, this group conducted another study on lung tissues of patients with IPF, COPD, COPD with α-1-antitrypsin deficiency (AAT) and healthy smokers using 2-DE, MALDI-ToF/ToF and Western blot analysis.72 They could identify variation in the receptor of advanced glycation end products (RAGE) which was in agreement with their previous research findings.73 However, it is important to mention that both these studies consisted of low patient cohort (n < 10). In a similar type of study, Ahrman et al. (2018) compared IPF lung tissue with COPD lung to investigate disease specific proteins involved in lung ECM remodeling. LC-MS/MS and IHC were used for this purpose. The authors observed that proteins associated with cell adhesion, such as collagen VI and laminins were dysregulated and proteins involved in regulating endopeptidase activity were overexpressed in the lungs of IPF subjects.74
Korfei and co-workers used 2-DE and MALDI-ToF to compare lung proteomes of patients with sporadic IPF to that of controls. Their findings showed that IPF is characterized by massive series of events, particularly increased ER stress response in AECII, ER dysfunction, abnormal proliferation of bronchiolar basal cells, oxidative stress, increased fibrinogenesis and inflammation. However, the results need to be interpreted with caution since the highly abundant proteins were not depleted in this study.75 Amongst the idiopathic interstitial pneumonias (IIPs), IPF and NSIP are often clinically indistinguishable despite having distinct histological presentations, treatment and prognostic outcomes.76 Use of HRCT for differential diagnosis of IIPs remains contradictory due to large variations in the HRCT findings of NSIP.77 Therefore, Korfei and group, in another study, sought to compare the lung tissue proteome of patients with sporadic IPF, fibrotic NSIP (fNSIP) and controls. Their findings revealed a similar proteome profile between the two IIPs with respect to controls. Oxidative stress and epithelial ER stress emerged as key players in the pathogenesis of both the diseases. The authors attributed enhanced expression of antioxidant acting proteins in fNSIP to better prognostic outcome of fNSIP versus IPF. Western blot analysis of BALF samples collected from the recruited subjects further confirmed the findings.78 In another study comparing UIP and NSIP and controls, Ohara and co-workers (2014) found qualitative differences in the expression of vimentin subtype, thereby proposing vimentin to be a differentiating marker for both the diseases.79 This is the first report on IIPs from the Asian subcontinent.
Schiller and group have demonstrated common involvement of antibody mediated autoimmunity in the pathogenesis of IPF and fibrotic skin lesions. Using LC-MS/MS and Western blot techniques, the authors concluded that marginal zone B- and B1-cell-specific proteins were upregulated in IPF.80 In a recent study, Tian et al. have compared 20 IPF lung tissues with 20 controls using iTRAQ combined with LC-MS/MS and further validated using Western blot and IHC. The authors detected 662 significantly altered proteins (455 unregulated and 207 down regulated) in IPF tissue which generally belonged to the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt) signalling, ECM–receptor interaction, focal adhesion and carbon metabolism pathways.81
ECM is an extensive molecular network that determines cellular behaviours in healthy and diseased tissue. Fibroblasts are majorly responsible for ECM deposition and formation of fibrosis in IPF. Differentiation of fibroblasts to myofibroblasts and excessive collagen deposition are the defining features of IPF. Matrisomes consist of an ensemble of genes encoding ECM and ECM-associated proteins. Rendin and co-workers (2016) have used matrisome classification system combined with LC-MS/MS to explore the matrisome properties of scaffolds, i.e. decellularized tissue originating from sub-pleural IPF and healthy lungs. The authors observed that collagens, proteoglycans, and ECM glycoproteins were significantly increased in IPF scaffolds. Also, specific basement membrane proteins such as laminin, collagen IV and nidogen-2 were found to be altered. This novel approach suggests that matrisome protein signatures of IPF are distinct from healthy scaffolds.82
In another similar comprehensive study, Mullenbrock et al. (2018) used next-generation sequencing and MS to identify microribonucleic acid (miRNA), messenger RNA (mRNA) and deposited matrisome protein profiles in lung fibroblasts isolated from lung transplants of IPF and SS patients and healthy donors. A clear distinction between the fibrotic matrisome profile deposited by IPF and SS fibroblasts could be observed when compared with controls. LC-MS analysis also revealed that ECM patterns were altered which underline fibrogenic activity.83
It is well established that excessive collagen deposition mediated by transforming growth factor-β (TGF-β) is a characteristic feature of IPF. Nigdelioglu et al. (2016) have reported that TGF-β induces increased glycolytic flux and expresses enzymes associated with de novo serine synthesis pathway (phosphoglycerate dehydrogenase, phosphoserine aminotransferase 1, phosphoserine phosphatase) and de novo glycine synthesis (serine hydroxymethyltransferase). Their studies demonstrated that these enzymes are necessary for collagen synthesis. Furthermore, to ascertain whether collagen uptakes glucose derived carbon, human lung fibroblasts (HLFs) were cultured with media containing either 12C-glucose or uniformly labelled 13C-glucose and then treated with TGF-β. The lysates were separated by electrophoresis and collagen bands analysed using LC-MS/MS. The proteomics approach clearly evidenced uptake of glucose-derived glycine into collagen. The authors hypothesize that de novo synthesis of glycine from glucose through the de novo serine synthesis pathway could be a potential therapeutic target for treatment of diseases with organ fibrosis, e.g. IPF.84
It is well realized that considerable amount of tissue samples is required to perform comprehensive proteomic analysis. Protein diversity decreases in small samples, and under such conditions it is challenging to evaluate differences between conditions. Over the past decade, micro-proteomics has emerged as an exciting field for the analysis of protein diversity in small samples. Laser capture microdissection (LCM) – based micro-proteomics permits proteome studies of enriched specific cell types of interest acquired from a very small amount of tissue.85 Herrera and his team have investigated morphologically normal paraffin-embedded haematoxylin and eosin stained distal lung tissue of IPF subjects using LCM coupled to MS (LCM-MS). A total of 1252 uniquely expressed and 892 differentially expressed proteins between the regions are reported. They could also identify 137 ECM proteins. The authors further validated localization of laminin subunit beta-1 and tenascin in morphologically normal human lung alveoli and human lung blood vessels, respectively using IHC. It is successfully demonstrated that this tool can be applied to tissue volumes as low as 0.0125 mm3.86
It is suggested that immunological responses are involved in patients with IPF. Takahashi and co-workers have tested the hypothesis that a significant number of patients with IPF have circulating autoantibodies to type II alveolar pneumocytes. Serum samples obtained from 22 patients with IPF and 37 healthy controls were examined for immunostaining of human lung tissue and A549 human type II cells. The authors also attempted to identify the relevant antigen using immunoprecipitation and tandem MS. It was observed that a significant number of IPF subjects possess circulating autoantibodies against alanyl-tRNA synthetase, as evidenced by proteome analysis.87
Extracellular vesicles (EVs) are plasma membrane derived vesicles that facilitate intercellular communication by serving as vehicles for transfer between cytosolic RNA, lipids, proteins, and cell membranes. Chanda et al. have shown that fibroblasts isolated from lungs of IPF patients release significantly higher numbers of EVs as compared with healthy controls. The findings were found to be similar in IPF and non-IPF cell lines. Further, sequential window acquisition of all theoretical mass spectra (SWATH-MS) based proteomics approach and flow cytometric analyses indicated that fibroblast-derived EVs carry fibronectin on the vesicular surface, which promotes fibroblast invasion, a characteristic feature of IPF.88 Conforti et al., in a recent study, isolated AECs and co-cultured with IPF fibroblasts isolated from three tissue biopsy samples and compared with control fibroblasts. The authors hypothesize that the secreted protein acidic and rich in cysteine (SPARC) prevents restoration of epithelial barrier in pulmonary fibrosis. Furthermore, the authors suggest that SPARC mediated paracrine signalling pathway could be a potential target to treat IPF subjects as it enhances the reestablishment of lung epithelial homeostasis in fibrosis.89
The need for the development of powerful curative drugs for IPF is well realized. Pirfenidone and nintedanib are the two anti-fibrotic drugs used in IPF; however, both drugs are not curative. Chromone and its derivatives are present in flavones, which are members of the polyphenol family and have been widely explored in lung diseases owing to their anti-inflammatory and antioxidant properties.90 Kim and co-workers have explored the therapeutic efficacy of a chromone derivative, eupatilin on human lung fibroblasts taken from IPF patients, human lung epithelial fibroblasts and BLM-induced lung fibrosis model. Proteomic analysis and gene expression studies evidenced that eupatilin inhibits trans- differentiation of pathogenic myofibroblasts and, therefore, holds potential as an anti-fibrotic drug for IPF.91
Increased oxidative stress has been reported in culture cell lines obtained from lung tissue lesions of IPF patients, attributing the myofibroblasts to be potential reactive oxygen species (ROS) generators.92 Kabuyama et al. analysed cultured normal human lung fibroblasts TIG7 and IPF myofibroblasts (LF1 and LF2) to understand the exact mechanism of ROS generation in IPF. They found significantly high levels of free cholesterol and its peroxides in IPF cells as compared to normal TIG7 lung fibroblasts. Proteomic analysis using 2-DE and quadrupole time-of-flight MS (Q-ToF MS) was performed on mitochondrial proteins of IPF and TIG cells to comprehend the molecular basis underlying ROS generation. Ser586 phosphorylation of very long chain acyl-CoA dehydrogenase (VLCAD) was found to be significantly less in IPF cells. The authors suggest that this dysregulation of VLCAD contributes to the pathophysiology of IPF.93
Animal studies in IPF
There is no single experimental animal model that can reliably induce progressive fibrosis with characteristic UIP.94 Nevertheless, it is largely believed that BLM induced pulmonary fibrosis closely mimics IPF in humans and is considered to be a standard preclinical model for research purpose.95 Although IPF development does not seem to arise from chronic inflammation, BLM administration first induces inflammatory responses in the animal lung followed by fibrosis.96Limited proteomics-based studies are reported in animal model for understanding molecular mechanisms of the disease pathology. Fukunaga and co-workers utilized iTRAQ-coupled LC-MS/MS and miRNA microarray approach in BLM administered rats to identify crucial proteins and pathways involved in IPF. They could identify several upregulated proteins including fibronectin 1, LIM and SH3 protein 1 (LASP1), ras-related protein RAP1 B and down-regulated target proteins viz., vinculin and aldehyde dehydrogenase 1 family member A1 (ALDH1A1). This study suggests the predominance of inflammatory and fibrotic pathways following BLM administration. The authors concluded that proteomics and microarray data together provide new insights for better understanding of IPF pathogenesis.97
Another group has integrated lung proteomics (label-free LC-MS) and systems biology to explore the role of VEGF inhibitor as a therapeutic agent in inhibiting angiogenesis in BLM induced IPF mice model. They showed that BLM treatment increased the expression of mammalian target of rapamycin (mTOR) and extracellular signal regulated kinase (ERK), pro- and antifibrotic markers, which were downregulated following VEGF inhibitor treatment. The authors hypothesized that novel drug therapy can be identified against this lethal disease by targeting the altered proteins and signalling pathways.98 In the same year, proteomics was used to elucidate protein modification in IPF. Although a specific disease marker could not be established, the group could identify isolevuglandin-modified protein in the lungs as a result of oxidative stress from radiation induced pulmonary injury and IPF in mice.99
Gas6, also known as growth arrest-specific 6, binds to Tyro3, Axl, and Mertk (TAM) receptors, [TYRO3 (tyrosine protein kinase), AXL (Anexelekto) and MERTK (c-mer proto-oncogene tyrosine kinase)] and promotes fibroblast activation in fibrosis. Espindola and his team (2017) utilized quantitative genomic, proteomic, and functional analyses to investigate the role of Gas6/TAM receptors in IPF lung and pulmonary fibrosis induced murine model. The authors concluded that Gas6/TAM receptor targeted clinical trials warrant attention as they play a major role in fibrosis formation.100
In a recent study, Duerr et al. (2020) have demonstrated that ubiquitin ligase Nedd4-2 protein expression is significantly reduced in lung tissue of IPF patients as compared to age-matched controls. They also observed an altered expression of Nedd4-2 in lung epithelial cells of IPF induced mice model, based on which the authors concluded that conditional deletion of Nedd4-2 may cause rapid fibrosis in lungs of adult mice. Lung proteome signatures were further explored in lung tissue of IPF subjects and conditional Nedd4-2−/− mice, and were compared to their respective controls. A nano-flow LC coupled to quadrupole orbitrap MS was used for this purpose. Furthermore, therapeutic effect of orally administered anti-fibrotic drug, pirfenidone investigated in terms of lung proteome alterations in Nedd4-2−/− mice suggested reduced pulmonary fibrosis.101 In another recent study, Dinh and team used LC-MS/MS based proteomics approach to demonstrate that inhalation of lung spheroid cell-secretome and exosome has a therapeutic role in resolving fibrosis in mice model of silica and BLM-induced lung fibrosis. The author concluded that secretome and exosome inhalation can regenerate normal alveolar structure and reduce fibrotic response.102
Among large animal vertebrates, dogs are most vulnerable towards the development of IPF and chronic bronchitis, both of which are similar chronic and progressive pulmonary diseases; differentiating them usually requires invasive surgical procedures.103 IPF, in particular, disproportionately affects mid-aged terrier breeds. Lilja-Maula and co-workers utilized DIGE and LC-MS to identify IPF and chronic bronchitis specific markers in canine BAL. While a number of proteins including complement 3, α-1-antitrypsin, apolipoprotein 1, haptoglobin, β-actin and transketolase were found to be upregulated, only one protein, lysozyme C was observed to be significantly downregulated in both the disease groups, as compared to healthy dogs. β-actin, a general stress protein, was seen to be upregulated only in IPF dogs and not found to be dysregulated in dogs with chronic bronchitis.104 However, the authors could not identify a specific robust biomarker for canine IPF.
Proposed mechanism of IPF pathogenesis
Herein we summarize and propose a possible mechanism of the disease pathogenesis based on the differentially expressed proteins and dysregulated cellular pathways identified in IPF patients using proteomics approach. The proposed mechanism is schematically represented in Fig. 2. | ||
| Fig. 2 The proposed mechanism of IPF pathogenesis based on proteomics data. IPF pathogenesis involves an unknown exogenous injury (cigarette smoke, pollution, dust, virus, superoxide, reactive nitrogen species), or genetic change or autoimmune response or ageing related predisposal mediated alveolar epithelial cells (AEC) dysfunction. This leads to an activation of stress response (endoplasmic reticulum and oxidative stress, unfolded protein, heat shock protein and DNA damage response), inflammatory response [S100A9, eosinophil and neutrophil proteins, decreased apolipoprotein A2 (Apo A2), α-1 antitrypsin, receptor for advanced glycation end products (RAGE)], and downregulation of cell proliferative pathways, antioxidant [decreased peroxisomal enzyme and thiorperoxidase 2], and anti-apoptotic responses (anti-apoptotic factors). These processes result in the activation of apoptotic pathway in AEC. AEC apoptosis is accompanied by abnormalities in the coagulation system [suppressed kininogen, haemoglobin (Hb)] and activation of profibrotic pathways [transforming growth factor beta (TGF-β), tumor necrosis factor alpha (TNFα), CC chemokine ligand 24 (CCL24), decreased annexin VI] leading to the recruitment and proliferation of resident lung fibroblasts and formation of fibroblast foci aided by migration inhibitory factor (MIF). Fibroblasts further differentiate into contractile and secretory myofibroblasts with support of profibrotic mediators [TGF-β, osteopontin, fibronectin, matrix metallopeptidase 7 (MMP7), CCL18, CCL24; decreased ApoA1 and clusterin], Th2 cytokines [interleukin 4, interleukin 3 (IL-4, IL-3)) and mitogenesis mediators (CXCL7, vascular endothelial growth factor receptor 2 (VEGFsR2)], thus augmenting myofibroblast proliferation. The burgeoning fibroblasts and myofibroblasts form fibroblast foci aggregates, which is a characteristic of usual interstitial pneumonia (UIP). Angiotensin system is also involved in pulmonary fibrosis, though the exact role remains unclear. Myofibroblasts secrete extracellular matrix (ECM) (collagen, vimentin, CXCL7) excessively, embed within and remodel the matrix (cathepsin S, LOXL2, periostin), thereby resulting in damage of lung parenchyma, contraction and progression to terminal pulmonary fibrosis. | ||
It is well established that IPF originates from unknown microinjuries resulting from recurrent exposures of the lung epithelium. The exposures could be environmental such as cigarette smoke, pollution, dust, superoxide, reactive nitrogen species, microbial agents (virus), genetic changes, epigenetic changes, autoimmune responses, or ageing related predisposition.
All these exposures of the lung epithelium or AEC result in the dysfunction of AEC and generation of an aberrant repair mechanism. This leads to protein over-expression and ER stress response, associated with imbalanced protein synthesis. In order to restore normalcy in ER activity, the unfolded protein response (UPR) is initiated. UPR has serious consequences on the cells, leading to the activation of apoptotic pathways and downregulation of cell proliferative and anti-apoptotic factors. In addition, oxidative stress is generated in response to injuries as indicated by an increase in the oxidative stress proteins and decrease in antioxidant proteins, which lead to a redox imbalance, cellular death and contribute to the activation of profibrotic response. Oxidative stress also damages the DNA and induces DNA damage response. Further, in response to injuries and stress, heat shock protein response and inflammatory responses are initiated. The injuries also cause endothelial damage leading to dysfunctional alveolar-capillary barrier and vascular leak associated with increased VEGF which induces a profibrotic response.
These responses in AEC accompanied by the dysregulation of normal repair pathway, impaired re-epithelialization and aberrant wound healing result in the subsequent activation of apoptotic pathway. AEC apoptosis is accompanied by abnormalities in the coagulation system and activation of the profibrotic process, as observed by the increase in profibrotic mediators (TGF-β, tumor necrosis factor α, CCL24), leading to the recruitment and proliferation of resident lung fibroblasts and formation of fibroblast foci.
Fibroblasts further trans-differentiate into contractile and secretory myofibroblasts with support of profibrotic mediators secreted by activated fibroblasts, AECs and alveolar macrophages, Th2 cytokines and mitogenesis mediators (CXCL7, IL-6, VEGFsR2), thus augmenting myofibroblast proliferation. The burgeoning fibroblasts and myofibroblasts form fibroblast foci aggregates, a characteristic of UIP. Angiotensin system is also involved, though the exact role remains unclear. Myofibroblasts excessively secrete extracellular matrix via ECM proteins, embed within and remodel the matrix through remodeling proteins (cathepsin S, LOXL2, periostin). This persistent dysregulated matrix remodeling results in an increased deposition of collagen and fibronectin and increased matrix stiffness, resulting in progressive damage to the lung parenchyma, contraction, fibrosis and subsequently respiratory failure.
There is ample evidence to postulate that IPF pathogenesis can be categorized into three steps involving (i) exposure to stimuli or predisposition (ii) initiation of AEC dysfunction, fibroblast recruitment and proliferation and (iii) progression of fibrosis through fibroblast differentiation, matrix deposition and remodeling. This involves cross-talks between AECs, endothelium, mesenchymal cells, inflammatory cells, alveolar macrophages and fibroblasts orchestrated by different proteins generating a profibrotic milieu for progressive fibrosis.
Challenges and future scope
Although clinical proteomics has provided dramatic insights into IPF pathogenesis and led to the discovery of proteins in terms of prognosis, diagnosis and disease progression, limitations do exist. Most of the studies reported so far indicate inclusion of a limited number of patient population, which is not surprising given the fact that IPF is a debilitating disease with poor average life expectancy. Since low sample size is associated with statistical challenges and involves risks of over fitting and misleading calculations,105 power and sample size estimation though challenging, would be meaningful. Lack of a robust subject selection process could lead to marker identification not specific to IPF. The overlapping clinical manifestations of different ILDs with IPF remains a clinical challenge which is seen in terms of overlapping protein molecules, making them less reliable for distinguishing ILDs. Clinical characteristics such as physiological status, age, gender and treatment could influence the proteome since it is widely dynamic. Therefore, baseline characteristics of IPF patients and controls must be well matched and the subjects selected in a stringent manner.106 Considerable non-uniformity regarding inclusion of smokers and non-smokers population for comparison purposes exist. Also, only a few groups appear to have included healthy controls in their study design. This makes comparisons and conclusions without bias difficult. Most of the studies on IPF proteomics were not executed in a longitudinal scale for larger proteome coverage; it is, therefore, not prudent to consider the putative proteins identified as prognostic markers of the disease. In fact, the potential protein signatures, identified in the discovery patient cohort, require validation in a fresh cohort (validation cohort) to establish robustness of the markers. Furthermore, their target levels of sensitivity and specificity should be established. A few groups could not validate the candidate markers of IPF, which is critical for the discovery of biomarkers. In fact, lack of adequate validation studies is one of the major reasons why the numerous clinical biomarkers identified so far have not reached clinical practice.107Proteomics as an analytical technique involves multiple steps of experimentation and thus is not impervious to human errors. The standard operating procedures during sample collection, processing, handling and storage must be conserved across all studies. Non-uniformity in the procurement and processing of samples may lead to protein loss and degradation.107,108 Inconsistency in data acquisition methods and analysis may lead to variation in findings with lack of reproducibility. The proteomic techniques used to explore IPF across different studies are varied, with the majority groups having used conventional gel-based technique (2-DE). The findings, therefore, warrant cautious interpretation since the inherent weakness of 2-DE, including inability to detect low abundant, hydrophobic, highly acidic and highly basic proteins, and proteins with very low and very high molecular weights is well realized.109
Though the clinical utility of investigating BALF in lung diseases is well recognized, proteomic analysis of BALF remains a challenge. Processing of BALF requires desalting, concentration and depletion of high abundant proteins. Due to lack of standardized BALF collection protocols, proteomic profiling of the collected samples could be affected, making the results questionable.108,110 On the other hand, serum despite being an easily accessible sample, may not always reflect lung pathology due to its extreme diverse range and also requires depletion of high abundant proteins.111,112
Protein chips and arrays may be considered for multiplexing and enhancing throughput. Single cell proteomics has also emerged as a promising tool to identify biomarkers for disease diagnosis and prognosis. The multifaceted benefits offered by the various new MS tools may help overcome the existing bottlenecks and eventually lead to identification of robust and reproducible candidate markers of IPF. A replicative set of robust protein markers identified at multiple sites can be potentially used to develop biochemical and immunological tests. Finally, a multi-omics approach integrating the large-scale expression data could help in the accurate diagnosis of IPF and empower clinicians to implement suitable course of action for a particular individual in an efficient manner.
Conclusion
Considerable advances have been made in the past fifteen years in terms of understanding the pathophysiology of IPF, leading to the recognition of key pathways and potential protein biomarkers for prognosis, differential diagnosis, disease progression and treatment monitoring. This article provides a comprehensive review of all proteomics related studies conducted on IPF patients and IPF animal models till date. Numerous pathways have been highlighted and several candidate protein signatures identified. However, the area of biomarker discovery should be treated with caution. Rigorous characterization and validation, sensitivity and specificity assessment, and adequate evidence to justify the clinical relevance of the aberrantly expressed molecules is crucial. Large scale, well designed, multi-centric clinical trials in confirmed IPF patients and appropriate controls are needed for robust unbiased diagnosis of this deteriorating chronic lung disease. This could assist in the early diagnosis of IPF, monitoring disease progression, better clinical management of the disease and future drug development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely appreciate the support and clinical guidance of pulmonologists Dr Parthasarathi Bhattacharyya and Dr Sushmita Roy Chowdhury.References
- G. Raghu, M. Remy-Jardin, J. L. Myers, L. Richeldi, C. J. Ryerson, D. J. Lederer, J. Behr, V. Cottin, S. K. Danoff, F. Morell, K. R. Flaherty, A. Wells, F. J. Martinez, A. Azuma, T. J. Bice, D. Bouros, K. K. Brown, H. R. Collard, A. Duggal, L. Galvin, Y. Inoue, R. G. Jenkins, T. Johkoh, E. A. Kazerooni, M. Kitaichi, S. L. Knight, G. Mansour, A. G. Nicholson, S. N. J. Pipavath, I. Buendia-Roldan, M. Selman, W. D. Travis, S. Walsh, K. C. Wilson, American Thoracic Society, European Respiratory Society, Japanese Respiratory Society and Latin American Thoracic Society, Am. J. Respir. Crit. Care Med., 2018, 198, e44–e68 CrossRef.
- G. Raghu, H. R. Collard, J. J. Egan, F. J. Martinez, J. Behr, K. K. Brown, T. V. Colby, J. F. Cordier, K. R. Flaherty, J. A. Lasky, D. A. Lynch, J. H. Ryu, J. J. Swigris, A. U. Wells, J. Ancochea, D. Bouros, C. Carvalho, U. Costabel, M. Ebina, D. M. Hansell, T. Johkoh, D. S. Kim, T. E. King Jr, Y. Kondoh, J. Myers, N. L. Muller, A. G. Nicholson, L. Richeldi, M. Selman, R. F. Dudden, B. S. Griss, S. L. Protzko, H. J. Schunemann and ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis, Am. J. Respir. Crit. Care Med., 2011, 183, 788–824 CrossRef.
- V. Cottin, A. Schmidt, L. Catella, F. Porte, C. Fernandez-Montoya, K. Le Lay and S. Benard, PLoS One, 2017, 12, e0166462 CrossRef.
- Y. Kondoh, V. Cottin and K. K. Brown, Eur. Respir. Rev., 2017, 26, 175050 CrossRef.
- S. L. Barratt, A. Creamer, C. Hayton and N. Chaudhuri, J. Clin. Med., 2018, 7, 201 CrossRef CAS.
- G. Ferrara, L. Arnheim-Dahlstrom, K. Bartley, C. Janson, K. U. Kirchgassler, A. Levine and C. M. Skold, Pulm. Ther., 2019, 5, 55–68 CrossRef.
- S. H. Lee, D. S. Kim, Y. W. Kim, M. P. Chung, S. T. Uh, C. S. Park, S. H. Jeong, Y. B. Park, H. L. Lee, J. S. Song, J. W. Shin, N. S. Yoo, E. J. Lee, J. H. Lee, Y. Jegal, H. K. Lee and M. S. Park, Chest, 2015, 147, 465–474 CrossRef.
- R. B. Hopkins, N. Burke, C. Fell, G. Dion and M. Kolb, Eur. Respir. J., 2016, 48, 187–195 CrossRef.
- S. Harari, F. Madotto, A. Caminati, S. Conti and G. Cesana, PLoS One, 2016, 11, e0147072 CrossRef.
- G. Paolocci, I. Folletti, K. Toren, M. Ekstrom, M. Dell'Omo, G. Muzi and N. Murgia, BMC Pulm. Med., 2018, 18, 75 CrossRef.
- S. Conti, S. Harari, A. Caminati, A. Zanobetti, J. D. Schwartz, P. A. Bertazzi, G. Cesana and F. Madotto, Eur. Respir. J., 2018, 51, 1700397 CrossRef.
- J. W. Koo, J. P. Myong, H. K. Yoon, C. K. Rhee, Y. Kim, J. S. Kim, B. S. Jo, Y. Cho, J. Byun, M. Choi, H. R. Kim and E. A. Kim, Int. J. Tuberc. Lung Dis., 2017, 21, 107–112 CrossRef.
- M. J. Abramson, T. Murambadoro, S. M. Alif, G. P. Benke, S. C. Dharmage, I. Glaspole, P. Hopkins, R. F. Hoy, S. Klebe, Y. Moodley, S. Rawson, P. N. Reynolds, R. Wolfe, T. J. Corte, E. H. Walters and I. P. F. R. Australian, Thorax, 2020, 75, 864–869 CrossRef.
- M. Karkkainen, H. P. Kettunen, H. Nurmi, T. Selander, M. Purokivi and R. Kaarteenaho, Respir. Res., 2017, 18, 160 CrossRef.
- G. A. Margaritopoulos, A. Trachalaki, A. U. Wells, E. Vasarmidi, E. Bibaki, G. Papastratigakis, S. Detorakis, N. Tzanakis and K. M. Antoniou, BMC Pulm. Med., 2018, 18, 177 CrossRef CAS.
- P. Rivera-Ortega, C. Hayton, J. Blaikley, C. Leonard and N. Chaudhuri, Ther. Adv. Respir. Dis., 2018, 12, 1753466618800618 Search PubMed.
- A. Kumar, S. G. Kapnadak, R. E. Girgis and G. Raghu, Expert Rev. Respir. Med., 2018, 12, 375–385 CrossRef CAS.
- D. Castillo, S. Walsh, D. M. Hansell, M. Vasakova, V. Cottin, G. Altinisik, S. Palmucci, M. Sterclova, S. Harari, L. Richeldi, C. Vancheri and A. U. Wells, Lancet Respir. Med., 2018, 6, 88–89 CrossRef.
- X. Xu, H. Dai and C. Wang, Clin. Respir. J., 2016, 10, 133–141 CrossRef.
- M. Kasper and K. Barth, Biosci. Rep., 2017, 37, BSR20171301 CrossRef CAS.
- A. L. Olson, J. J. Swigris, D. C. Lezotte, J. M. Norris, C. G. Wilson and K. K. Brown, Am. J. Respir. Crit. Care Med., 2007, 176, 277–284 CrossRef.
- E. Renzoni, V. Srihari and P. Sestini, F1000Prime Rep., 2014, 6, 69 Search PubMed.
- M. P. Keane, Eur. Respir. Rev., 2008, 17, 151–156 CrossRef.
- C. C. Thomson, A. Duggal, T. Bice, D. J. Lederer, K. C. Wilson and G. Raghu, Ann. Am. Thorac. Soc., 2019, 16, 285–290 Search PubMed.
- A. U. Wells, Lancet Respir. Med., 2018, 6, 735–737 CrossRef.
- V. Tzilas, A. Tzouvelekis, S. Chrysikos, S. Papiris and D. Bouros, Front. Med., 2017, 4, 151 CrossRef.
- J. P. Hutchinson, A. W. Fogarty, T. M. McKeever and R. B. Hubbard, Am. J. Respir. Crit. Care Med., 2016, 193, 1161–1167 CrossRef CAS.
- A. Guenther, E. Krauss, S. Tello, J. Wagner, B. Paul, S. Kuhn, O. Maurer, S. Heinemann, U. Costabel and M. A. N. Barbero, Respir. Res., 2018, 19, 141 CrossRef.
- T. Hewson, T. M. McKeever, J. E. Gibson, V. Navaratnam, R. B. Hubbard and J. P. Hutchinson, Thorax, 2018, 73, 683–685 CrossRef.
- M. Aburto, I. Herráez, D. Iturbe and A. Jiménez-Romero, Med. Sci., 2018, 6, 73 Search PubMed.
- K. R. Flaherty, T. E. King Jr, G. Raghu, J. P. Lynch III, T. V. Colby, W. D. Travis, B. H. Gross, E. A. Kazerooni, G. B. Toews and Q. Long, Am. J. Respir. Crit. Care Med., 2004, 170, 904–910 CrossRef.
- T. J. Gross and G. W. Hunninghake, N. Engl. J. Med., 2001, 345, 517–525 CrossRef CAS.
- S. Ohshimo, N. Ishikawa, Y. Horimasu, N. Hattori, N. Hirohashi, K. Tanigawa, N. Kohno, F. Bonella, J. Guzman and U. Costabel, Respir. Med., 2014, 108, 1031–1039 CrossRef.
- B. Ley, K. K. Brown and H. R. Collard, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2014, 307, L681–691 CrossRef CAS.
- A. Prasse, C. Probst, E. Bargagli, G. Zissel, G. B. Toews, K. R. Flaherty, M. Olschewski, P. Rottoli and J. Muller-Quernheim, Am. J. Respir. Crit. Care Med., 2009, 179, 717–723 CrossRef CAS.
- M. Maldonado, I. Buendia-Roldan, V. Vicens-Zygmunt, L. Planas, M. Molina-Molina, M. Selman and A. Pardo, PLoS One, 2018, 13, e0203779 CrossRef.
- J. Guiot, C. Moermans, M. Henket, J. L. Corhay and R. Louis, Lung, 2017, 195, 273–280 CrossRef CAS.
- C. Machahua, A. Montes-Worboys, L. Planas-Cerezales, R. Buendia-Flores, M. Molina-Molina and V. Vicens-Zygmunt, Respir. Res., 2018, 19, 215 CrossRef CAS.
- A. Pardo, S. Cabrera, M. Maldonado and M. Selman, Respir. Res., 2016, 17, 23 CrossRef.
- C. Hayton, D. Terrington, A. M. Wilson, N. Chaudhuri, C. Leonard and S. J. Fowler, Respir. Res., 2019, 20, 7 CrossRef.
- P. Scherp, G. Ku, L. Coleman and I. Kheterpal, Methods Mol. Biol., 2011, 702, 163–190 CrossRef CAS.
- M. L. Fournier, J. M. Gilmore, S. A. Martin-Brown and M. P. Washburn, Chem. Rev., 2007, 107, 3654–3686 CrossRef CAS.
- C. Abdallah, E. Dumas-Gaudot, J. Renaut and K. Sergeant, Int J Plant Genomics, 2012, 2012, 494572 CrossRef.
- A. D. Catherman, O. S. Skinner and N. L. Kelleher, Biochem. Biophys. Res. Commun., 2014, 445, 683–693 CrossRef CAS.
- A. Tholey and A. Becker, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 2191–2199 CrossRef CAS.
- P. B. Pandeswari and V. Sabareesh, RSC Adv., 2019, 9, 313–344 RSC.
- B. Chen, K. A. Brown, Z. Lin and Y. Ge, Anal. Chem., 2018, 90, 14643 CrossRef CAS.
- K. A. Cupp-Sutton and S. Wu, Mol. Omics, 2020, 16, 91–99 RSC.
- B. Magi, L. Bini, M. G. Perari, A. Fossi, J. C. Sanchez, D. Hochstrasser, S. Paesano, R. Raggiaschi, A. Santucci and V. Pallini, Electrophoresis, 2002, 23, 3434–3444 CrossRef CAS.
- P. Rottoli, B. Magi, R. Cianti, E. Bargagli, C. Vagaggini, N. Nikiforakis, V. Pallini and L. Bini, Proteomics, 2005, 5, 2612–2618 CrossRef CAS.
- P. Rottoli, B. Magi, M. G. Perari, S. Liberatori, N. Nikiforakis, E. Bargagli, R. Cianti, L. Bini and V. Pallini, Proteomics, 2005, 5, 1423–1430 CrossRef CAS.
- T. H. Kim, Y. H. Lee, K. H. Kim, S. H. Lee, J. Y. Cha, E. K. Shin, S. Jung, A. S. Jang, S. W. Park, S. T. Uh, Y. H. Kim, J. S. Park, H. G. Sin, W. Youm, E. S. Koh, S. Y. Cho, Y. K. Paik, T. Y. Rhim and C. S. Park, Am. J. Respir. Crit. Care Med., 2010, 182, 633–642 CrossRef CAS.
- A. Hara, N. Sakamoto, Y. Ishimatsu, T. Kakugawa, S. Nakashima, S. Hara, M. Adachi, H. Fujita, H. Mukae and S. Kohno, Respir. Med., 2012, 106, 571–580 CrossRef.
- E. Bargagli, C. Olivieri, A. Prasse, N. Bianchi, B. Magi, R. Cianti, L. Bini and P. Rottoli, Inflammation, 2008, 31, 351–354 CrossRef CAS.
- E. Bargagli, C. Olivieri, N. Nikiforakis, M. Cintorino, B. Magi, M. G. Perari, C. Vagaggini, D. Spina, A. Prasse and P. Rottoli, Respir. Physiol. Neurobiol., 2009, 167, 261–267 CrossRef CAS.
- C. Landi, E. Bargagli, L. Bianchi, A. Gagliardi, A. Carleo, D. Bennett, M. G. Perari, A. Armini, A. Prasse, P. Rottoli and L. Bini, J. Proteomics, 2013, 83, 60–75 CrossRef CAS.
- C. Landi, E. Bargagli, A. Carleo, L. Bianchi, A. Gagliardi, A. Prasse, M. G. Perari, R. M. Refini, L. Bini and P. Rottoli, Proteomics: Clin. Appl., 2014, 8, 932–950 CAS.
- M. W. Foster, L. D. Morrison, J. L. Todd, L. D. Snyder, J. W. Thompson, E. J. Soderblom, K. Plonk, K. J. Weinhold, R. Townsend, A. Minnich and M. A. Moseley, J. Proteome Res., 2015, 14, 1238–1249 CrossRef CAS.
- A. Carleo, E. Bargagli, C. Landi, D. Bennett, L. Bianchi, A. Gagliardi, C. Carnemolla, M. G. Perari, G. Cillis, A. Armini, L. Bini and P. Rottoli, J. Breath Res., 2016, 10, 026007 CrossRef CAS.
- A. Carleo, C. Landi, A. Prasse, L. Bergantini, M. D’Alessandro, P. Cameli, S. Janciauskiene, P. Rottoli, L. Bini and E. Bargagli, Monaldi Arch. Chest Dis., 2020, 90 DOI:10.4081/monaldi.2020.1231.
- R. Niu, Y. Liu, Y. Zhang, Y. Zhang, H. Wang, Y. Wang, W. Wang and X. Li, PLoS One, 2017, 12, e0170741 CrossRef.
- Y. Zhang, Q. Xin, Z. Wu, C. Wang, Y. Wang, Q. Wu and R. Niu, Med. Sci. Monit., 2018, 24, 4146–4153 CrossRef CAS.
- C. Taille, S. Grootenboer-Mignot, C. Boursier, L. Michel, M. P. Debray, J. Fagart, L. Barrientos, A. Mailleux, N. Cigna, F. Tubach, J. Marchal-Somme, P. Soler, S. Chollet-Martin and B. Crestani, Am. J. Respir. Crit. Care Med., 2011, 183, 759–766 CrossRef CAS.
- S. L. Ashley, M. Xia, S. Murray, D. N. O'Dwyer, E. Grant, E. S. White, K. R. Flaherty, F. J. Martinez and B. B. Moore, PLoS One, 2016, 11, e0159878 CrossRef.
- D. N. O'Dwyer, K. C. Norman, M. Xia, Y. Huang, S. J. Gurczynski, S. L. Ashley, E. S. White, K. R. Flaherty, F. J. Martinez, S. Murray, I. Noth, K. B. Arnold and B. B. Moore, Sci. Rep., 2017, 7, 46560 CrossRef.
- J. L. Todd, M. L. Neely, R. Overton, K. Durham, M. Gulati, H. Huang, J. Roman, L. K. Newby, K. R. Flaherty, R. Vinisko, Y. Liu, J. Roy, R. Schmid, B. Strobel, C. Hesslinger, T. B. Leonard, I. Noth, J. A. Belperio, S. M. Palmer and IPF-PRO Registry investigators, Respir. Res., 2019, 20, 227 CrossRef.
- Y. P. Moodley, T. J. Corte, B. G. Oliver, I. N. Glaspole, A. Livk, J. Ito, K. Peters, R. Lipscombe, T. Casey and D. B. A. Tan, Respirology, 2019, 24, 1111–1114 CrossRef.
- M. Saraswat, S. Joenvaara, T. Tohmola, E. Sutinen, V. Vartiainen, K. Koli, M. Myllarniemi and R. Renkonen, Sci. Rep., 2020, 10, 7787 CrossRef CAS.
- A. B. Chambliss and D. W. Chan, Clin. Proteomics, 2016, 13, 25 CrossRef.
- C. Landi, L. Bergantini, P. Cameli, M. d'Alessandro, A. Carleo, E. Shaba, P. Rottoli, L. Bini and E. Bargagli, Sci. Rep., 2020, 10, 9378 CrossRef CAS.
- N. Ishikawa, S. Ohlmeier, K. Salmenkivi, M. Myllärniemi, I. Rahman, W. Mazur and V. L. Kinnula, Respir. Res., 2010, 11, 123 CrossRef.
- S. Ohlmeier, W. Mazur, K. Salmenkivi, M. Myllarniemi, U. Bergmann and V. L. Kinnula, Proteomics: Clin. Appl., 2010, 4, 97–105 CAS.
- M. A. Queisser, F. M. Kouri, M. Konigshoff, M. Wygrecka, U. Schubert, O. Eickelberg and K. T. Preissner, Am. J. Respir. Cell Mol. Biol., 2008, 39, 337–345 CrossRef CAS.
- E. Ahrman, O. Hallgren, L. Malmstrom, U. Hedstrom, A. Malmstrom, L. Bjermer, X. H. Zhou, G. Westergren-Thorsson and J. Malmstrom, J. Proteomics, 2018, 189, 23–33 CrossRef.
- M. Korfei, S. Schmitt, C. Ruppert, I. Henneke, P. Markart, B. Loeh, P. Mahavadi, M. Wygrecka, W. Klepetko, L. Fink, P. Bonniaud, K. T. Preissner, G. Lochnit, L. Schaefer, W. Seeger and A. Guenther, J. Proteome Res., 2011, 10, 2185–2205 CrossRef CAS.
- T. E. Hartman, S. J. Swensen, D. M. Hansell, T. V. Colby, J. L. Myers, H. D. Tazelaar, A. G. Nicholson, A. U. Wells, J. H. Ryu and D. E. Midthun, Radiology, 2000, 217, 701–705 CrossRef CAS.
- T. Johkoh, H. Itoh, N. L. Müller, K. Ichikado, H. Nakamura, J. Ikezoe, M. Akira and T. Nagareda, Radiology, 1999, 211, 155–160 CrossRef CAS.
- M. Korfei, D. von der Beck, I. Henneke, P. Markart, C. Ruppert, P. Mahavadi, B. Ghanim, W. Klepetko, L. Fink, S. Meiners, O. H. Kramer, W. Seeger, C. Vancheri and A. Guenther, J. Proteomics, 2013, 85, 109–128 CrossRef CAS.
- I. Ohara, S. Aida, H. Shimazaki, H. Kobayashi, H. Tsuda, T. Toda, K. Nakanishi and S. Tamai, Histol. Histopathol., 2014, 29, 377–386 CAS.
- H. B. Schiller, C. H. Mayr, G. Leuschner, M. Strunz, C. Staab-Weijnitz, S. Preisendorfer, B. Eckes, P. Moinzadeh, T. Krieg, D. A. Schwartz, R. A. Hatz, J. Behr, M. Mann and O. Eickelberg, Am. J. Respir. Crit. Care Med., 2017, 196, 1298–1310 CrossRef CAS.
- Y. Tian, H. Li, Y. Gao, C. Liu, T. Qiu, H. Wu, M. Cao, Y. Zhang, H. Ding, J. Chen and H. Cai, Clin. Proteomics, 2019, 16, 6 CrossRef.
- L. Elowsson Rendin, A. Lofdahl, E. Ahrman, C. Muller, T. Notermans, B. Michalikova, O. Rosmark, X. H. Zhou, G. Dellgren, M. Silverborn, L. Bjermer, A. Malmstrom, A. K. Larsson-Callerfelt, H. Isaksson, J. Malmstrom and G. Westergren-Thorsson, Int. J. Mol. Sci., 2019, 20, 4013 CrossRef.
- S. Mullenbrock, F. Liu, S. Szak, X. Hronowski, B. Gao, P. Juhasz, C. Sun, M. Liu, H. McLaughlin, Q. Xiao, C. Feghali-Bostwick and T. S. Zheng, Genes, 2018, 9, 588 CrossRef.
- R. Nigdelioglu, R. B. Hamanaka, A. Y. Meliton, E. O'Leary, L. J. Witt, T. Cho, K. Sun, C. Bonham, D. Wu, P. S. Woods, A. N. Husain, D. Wolfgeher, N. O. Dulin, N. S. Chandel and G. M. Mutlu, J. Biol. Chem., 2016, 291, 27239–27251 CrossRef CAS.
- H. B. Gutstein, J. S. Morris, S. P. Annangudi and J. V. Sweedler, Mass Spectrom. Rev., 2008, 27, 316–330 CrossRef CAS.
- J. A. Herrera, V. Mallikarjun, S. Rosini, M. A. Montero, C. Lawless, S. Warwood, R. O'Cualain, D. Knight, M. A. Schwartz and J. Swift, Clin. Proteomics, 2020, 17, 24 CrossRef CAS.
- T. Takahashi, I. Wada, Y. Ohtsuka, M. Munakata, Y. Homma and Y. Kuroki, Respirology, 2007, 12, 642–653 CrossRef.
- D. Chanda, E. Otoupalova, K. P. Hough, M. L. Locy, K. Bernard, J. S. Deshane, R. D. Sanderson, J. A. Mobley and V. J. Thannickal, Am. J. Respir. Cell Mol. Biol., 2019, 60, 279–288 CrossRef CAS.
- F. Conforti, R. Ridley, C. Brereton, A. Alzetani, B. Johnson, B. G. Marshall, S. V. Fletcher, C. H. Ottensmeier, L. Richeldi, P. Skipp, Y. Wang, M. G. Jones and D. E. Davies, Cell Death Discovery, 2020, 6, 54 CrossRef CAS.
- J. H. Lago, A. C. Toledo-Arruda, M. Mernak, K. H. Barrosa, M. A. Martins, I. F. Tiberio and C. M. Prado, Molecules, 2014, 19, 3570–3595 CrossRef.
- H. S. Kim, Y. M. Yoon, M. K. Meang, Y. E. Park, J. Y. Lee, T. H. Lee, J. E. Lee, I. H. Kim and B. S. Youn, EBioMedicine, 2019, 39, 484–496 CrossRef.
- Y. Kabuyama, K. Oshima, T. Kitamura, M. Homma, J. Yamaki, M. Munakata and Y. Homma, Genes Cells, 2007, 12, 1235–1244 CrossRef CAS.
- Y. Kabuyama, T. Suzuki, N. Nakazawa, J. Yamaki, M. K. Homma and Y. Homma, Am. J. Physiol.: Cell Physiol., 2010, 298, C107–113 CrossRef CAS.
- A. L. Degryse and W. E. Lawson, Am. J. Med. Sci., 2011, 341, 444–449 CrossRef.
- A. Moeller, K. Ask, D. Warburton, J. Gauldie and M. Kolb, Int. J. Biochem. Cell Biol., 2008, 40, 362–382 CrossRef CAS.
- M. A. Mouratis and V. Aidinis, Curr. Opin. Pulm. Med., 2011, 17, 355–361 CrossRef CAS.
- S. Fukunaga, A. Kakehashi, K. Sumida, M. Kushida, H. Asano, M. Gi and H. Wanibuchi, Toxicol. Appl. Pharmacol., 2015, 286, 188–197 CrossRef CAS.
- Y. M. Kulkarni, S. Dutta, A. K. V. Iyer, R. Venkatadri, V. Kaushik, V. Ramesh, C. A. Wright, O. J. Semmes, J. S. Yakisich and N. Azad, Proteomics, 2016, 16, 33–46 CrossRef CAS.
- S. Mont, S. S. Davies, L. J. Roberts II, R. L. Mernaugh, W. H. McDonald, B. H. Segal, W. Zackert, J. A. Kropski, T. S. Blackwell, K. R. Sekhar, J. J. Galligan, P. P. Massion, L. J. Marnett, E. L. Travis and M. L. Freeman, Sci. Rep., 2016, 6, 24919 CrossRef CAS.
- M. S. Espindola, D. M. Habiel, R. Narayanan, I. Jones, A. L. Coelho, L. A. Murray, D. Jiang, P. W. Noble and C. M. Hogaboam, Am. J. Respir. Crit. Care Med., 2018, 197, 1443–1456 CrossRef CAS.
- J. Duerr, D. H. W. Leitz, M. Szczygiel, D. Dvornikov, S. G. Fraumann, C. Kreutz, P. K. Zadora, A. Seyhan Agircan, P. Konietzke, T. A. Engelmann, J. Hegermann, S. Mulugeta, H. Kawabe, L. Knudsen, M. Ochs, D. Rotin, T. Muley, M. Kreuter, F. J. F. Herth, M. O. Wielputz, M. F. Beers, U. Klingmuller and M. A. Mall, Nat. Commun., 2020, 11, 2012 CrossRef CAS.
- P.-U. C. Dinh, D. Paudel, H. Brochu, K. D. Popowski, M. C. Gracieux, J. Cores, K. Huang, M. T. Hensley, E. Harrell and A. C. Vandergriff, Nat. Commun., 2020, 11, 1–14 Search PubMed.
- V. Johnson, B. Corcoran, P. Wotton, T. Schwarz and M. Sullivan, J. Small Anim. Pract., 2005, 46, 381–388 CrossRef CAS.
- L. I. Lilja-Maula, M. J. Palviainen, H. P. Heikkilä, M. R. Raekallio and M. M. Rajamäki, Am. J. Vet. Res., 2013, 74, 148–154 CrossRef CAS.
- B. Hernández, A. Parnell and S. R. Pennington, Proteomics, 2014, 14, 1587–1592 CrossRef.
- M. R. Macleod, A. Lawson McLean, A. Kyriakopoulou, S. Serghiou, A. de Wilde, N. Sherratt, T. Hirst, R. Hemblade, Z. Bahor, C. Nunes-Fonseca, A. Potluru, A. Thomson, J. Baginskaite, K. Egan, H. Vesterinen, G. L. Currie, L. Churilov, D. W. Howells and E. S. Sena, PLoS Biol., 2015, 13, e1002273 CrossRef.
- R. Savino, S. Paduano, M. Preianò and R. Terracciano, Int. J. Mol. Sci., 2012, 13, 13926–13948 CrossRef CAS.
- M. Bhargava, L. Higgins, C. H. Wendt and D. H. Ingbar, Clin. Transl. Med., 2014, 3, 1–11 Search PubMed.
- F. Ratjen, W. Havers and J. Braun, Thorax, 1999, 54, 432–436 CrossRef CAS.
- R. Terracciano, G. Pelaia, M. Preiano and R. Savino, Proteomics Clin Appl, 2015, 9, 203–220 CrossRef CAS.
- M. West-Nielsen, E. V. Høgdall, E. Marchiori, C. K. Høgdall, C. Schou and N. H. Heegaard, Anal. Chem., 2005, 77, 5114–5123 CrossRef CAS.
- A. Hakimi, J. Auluck, G. D. Jones, L. L. Ng and D. J. Jones, Proteomics, 2014, 14, 4–13 CrossRef CAS.
Footnote |
| † Both authors have contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |